Retrovirus-Mediated Gene Therapy For Farber Disease · Retrovirus-Mediated Gene Therapy For Farber...
160
Retrovirus-Mediated Gene Therapy For Farber Disease by Shobha Ramsubir A thesis submitted in conformity with the requirements for the degree of Doctor of Philosophy Graduate Department of Medical Biophysics University of Toronto © Copyright by Shobha Ramsubir (2008)
Transcript of Retrovirus-Mediated Gene Therapy For Farber Disease · Retrovirus-Mediated Gene Therapy For Farber...

Retrovirus-Mediated Gene Therapy For Farber Disease
by
Shobha Ramsubir
A thesis submitted in conformity with the requirements
for the degree of Doctor of Philosophy
Graduate Department of Medical Biophysics
University of Toronto
© Copyright by Shobha Ramsubir (2008)

ii
Retrovirus-mediated Gene Therapy for Farber Disease
Doctor of Philosophy, 2008
Graduate Department of Medical Biophysics
University of Toronto
Shobha Ramsubir
Abstract
Farber disease is a rare lysosomal storage disease (LSD) caused by a deficiency of
acid ceramidase (AC). Patients show a classic triad of symptoms including subcutaneous
granulomas, laryngeal involvement and painful swollen joints. The most common and severe
form has neurological manifestations and patients typically die by the age of two. Current
treatment consists of symptomatic supportive care and allogeneic bone marrow
transplantation (BMT). However, BMT has shown limited success. Gene therapy has
previously been shown to be a promising treatment strategy for monogenetic diseases and
has the potential to treat the underlying cause of the disease. Presented here is the first report
of in vivo testing of retrovirus-mediated gene therapy strategies for the treatment of Farber
disease. Retroviral vectors were engineered to overexpress AC and a cell surface marker,
human CD25. Transduction with these viral vectors corrected the enzymatic defect in Farber
patient cells and in vivo administration of the lentiviral vector led to long-term expression of
the marking transgene as well as increased AC expression in the liver. To determine the
effect of over-expression of AC, human CD34+ cells were transduced and transplanted into
NOD/SCID animals. It was found that transgene-expressing cells could reconstitute the host.

iii
To address the neurological manifestations of Farber disease, vascular endothelial growth
factor (VEGF) was investigated as an agent to transiently open the blood brain barrier for
entry of lentivirus. It was found that in addition to increasing the amount of therapeutic virus
in the brain, VEGF treatment also increased transduction in other organs. Further, to address
the concerns of insertional mutagenesis associated with using integrating vectors, an
immunotoxin-based strategy was developed as a safety system to clear transduced cells. It
was found that a CD25-targeted immunotoxin could eliminate both transduced hematopoietic
cells as well as tumor cells over-expressing CD25. This strategy can be employed following
gene therapy should an unwanted proliferative event occur. Together, these studies represent
considerable advances towards the development of a cure for Farber disease, demonstrating
both therapeutic potential and also containing a built-in safety system.

iv
Acknowledgements
This journey has been one of incredible growth for me and I have learnt so much
about persistence and perseverance. I am so grateful for the friendship and support of all of
my colleagues throughout the years. Washing away failed experiments with pints at the pub
with all of you kept me hanging on.
My heartfelt thanks go to Dr. Makoto Yoshimitsu for his mentorship, encouragement
and sense of humour. I am grateful to Dr. Koji Higuchi and Dr. Takeya Sato for their
continuous support and valuable advice over the years. Thanks to Renee Head for always
being willing to pick up the slack, to Gillian Sleep for her help and patience in dealing with
the many mice of my career and to Vanessa Rasaiah for helping with manuscripts, my thesis
and things too many to mention.
To the ladies that I began this journey with - Julie Symes and Miriam Mossoba - we
have shared many laughs and a few tears over the years and I wish you both the best of luck
in all your future endeavors. To my fellow graduate students Greg Rampersad, Sean Devine
and Anton Neschadim – all the chats and laughs made coming to the lab a pleasure. You are
all destined for great things. I am also grateful to the post-doctoral fellows Dr. Takahiro
Nonaka, Dr. Josh Silvertown, Dr. Chris Siatskas, Dr. Nobuo Mizue, Dr. Jagdeep Singh-
Walia, Dr. Severine Meyer and Dr. Chyang-Jang Lee for their helpful ideas and numerous
discussions about my project and science in general.
I would like to thank the individuals under whose guidance I truly grew as a scientist.
Thanks to my supervisor Dr. Jeffrey Medin for the opportunity to work on this project and

v
for all his help throughout the years. I am thankful to Dr. Thierry Levade for welcoming me
into his lab, for teaching me and for his assistance with biochemical assays. Dr. Joe Clarke
gave me the opportunity to meet a patient affected with Farber Disease and this experience
truly gave me greater perspective on the importance of the research that I and other scientists
do. Dr. Hans Messner always made time to review my research and progress and provided
much helpful advice. I am also grateful to Dr. David Rose for his candor, mentorship and
support. I am also grateful to him for his help in preparing for my defense.
Finally, I would like to thank my family for their unconditional support and love. I
could not have gotten here without you and I am eternally indebted. Thanks especially to my
mother for always going the extra mile, to my father for his support and to my sister Lesley
for always believing in me. I dedicate this thesis to you all.

vi
Table of Contents
ABSTRACT.............................................................................................................................II
ACKNOWLEDGEMENTS ................................................................................................. IV
LIST OF FIGURES AND TABLES................................................................................. VIII
LIST OF ABBREVIATIONS .............................................................................................. IX
CHAPTER 1: INTRODUCTION 1.1 LYSOSOMAL STORAGE DISEASES ...................................................................................................2
1.1.1 Overview ...........................................................................................................................................2 1.1.2 Treatment of Lysosomal Storage Diseases .......................................................................................3 1.1.3 Examples of Common Lysosomal Storage Diseases ........................................................................5
1.2 FARBER DISEASE..................................................................................................................................7
1.1.2 Disease Overview..............................................................................................................................7 1.2.2 Treatment of Farber Disease .............................................................................................................9 1.2.3 Mouse Model of Farber Disease .....................................................................................................11
1.3 ACID CERAMIDASE ............................................................................................................................12 1.3.1 Gene, Structure and Biochemistry ..................................................................................................12 1.3.2 Mutations in Farber Disease............................................................................................................13 1.3.3 Other Ceramidases ..........................................................................................................................13
1.4 CERAMIDE............................................................................................................................................14
1.4.1 Structure and Physiological Function .............................................................................................14 1.4.2 Ceramide Signaling and Apoptosis .................................................................................................16
1.5 GENE THERAPY...................................................................................................................................18
1.5.1 Methods of Gene Transfer...............................................................................................................18 1.5.2 Treatment Modalities with Viral Vectors........................................................................................21 1.5.3 Gene Therapy for Farber Disease....................................................................................................21 1.5.4 Retroviral Genotoxicity...................................................................................................................24
1.6 MARKING OF TRANSDUCED CELLS ..............................................................................................26
1.6.1 Structure and Function of CD25 .....................................................................................................26 1.6.2 Use as a Pre-selective Marker .........................................................................................................27 1.6.3 Aberrant Expression in Cancer .......................................................................................................27
1.7 CURRENT STUDY OBJECTIVES .......................................................................................................28

vii
CHAPTER 2: IN VIVO DELIVERY OF HUMAN ACID CERAMIDASE VIA CORD BLOOD TRANSPLANTATION AND DIRECT INJECTION OF LENTIVIRUS AS NOVEL APPROACHES FOR THE TREATMENT OF FARBER DISEASE
2.1 ABSTRACT............................................................................................................................................35 2.2 INTRODUCTION ..................................................................................................................................36 2.3 MATERIALS AND METHODS............................................................................................................38 2.4 RESULTS ...............................................................................................................................................45 2.5 DISCUSSION .........................................................................................................................................50
CHAPTER 3: ADMINISTRATION OF VEGF PRIOR TO LENTIVIRUS DELIVERY INCREASES TRANSDUCTION OF MULTIPLE ORGANS IN MICE TREATED AS NEONATES
3.1 ABSTRACT............................................................................................................................................62 3.2 INTRODUCTION ..................................................................................................................................63 3.3 MATERIALS AND METHODS............................................................................................................65 3.4 RESULTS ...............................................................................................................................................68 3.5 DISCUSSION .........................................................................................................................................71
CHAPTER 4: ANTI-CD25 TARGETED KILLING OF BICISTRONICALLY TRANSDUCED CELLS: A NOVEL SAFETY MECHANISM AGAINST RETROVIRAL GENOTOXICITY
4.1 ABSTRACT............................................................................................................................................81 4.2 INTRODUCTION ..................................................................................................................................82 4.3 MATERIALS AND METHODS............................................................................................................85 4.4 RESULTS ...............................................................................................................................................90 4.5 DISCUSSION .........................................................................................................................................96
CHAPTER 5: CONCLUSIONS AND FUTURE DIRECTIONS ................................ 107
REFERENCES.................................................................................................................... 116

viii
List of Figures and Tables Chapter 1 Figure 1.1: Schematic of the sphingomyelin pathway showing some of the major lipids and
enzymes involved. Figure 1.2: Schematic of CD25 clearance strategy. Figure 1.3: Schematic of vector systems. Table 1: Reported mutations and polymorphisms of the ASAH gene in Farber disease Chapter 2 Figure 2.1: huCD25 expression on transduced, immortalized Farber patient cells. Figure 2.2: AC activity in transduced Farber patient cells. Figure 2.3: Ceramide content of transduced Farber patient cells. Figure 2.4: Metabolic co-operativity demonstrated by uptake of secreted AC by non-
transduced Farber fibroblasts. Figure 2.5: Infection of human HSPCs from multiple sources. Figure 2.6: Transgene expression following direct LV delivery to neonatal mice. Table 2: Engraftment of LV/enGFP- or LV/AC/huCD25-transduced human CD34+ cells
into NOD/SCID recipients. Chapter 3 Figure 3.1: Whole body luminescence imaging of mice showing long-term luciferase
expression. Figure 3.2: Luc expression in the brain following treatment with LV/luc and VEGF. Figure 3.3: Luc expression in organs following treatment with LV/luc and VEGF. Figure 3.4: Luciferase activity assays of organ homogenates. Figure 3.5: Identification of the transduced cell types in the brain. Figure 3.6: Identification of the transduced cell types in the heart. Chapter 4 Figure 4.1: In vitro clearance of C1498 cells expressing a broad concentration range of
huCD25 molecules by ATS. Figure 4.2: In vitro clearance of a C1498/CD25 clone by ATS. Figure 4.3: The in vivo effect of different antibody doses on plasma huCD25 levels. Figure 4.4: ATS and AT treatment in a huCD25-expressing myeloid leukemia model. Figure 4.5: Bone marrow transplantation model. Figure 4.6: Clearance of retrovirally-transduced bone marrow-derived cells by ATS and AT. Figure 4.7: Systemic effect of ATS treatment on α-gal activity.

ix
List of Abbreviations °C degree Celsius γ gamma α-gal A alpha-galactosidase A AC acid ceramidase APC allophycocyanine AT anti-Tac ATP adenosine triphosphate ATS anti-Tac-saporin BBB blood-brain barrier BM bone marrow BMT bone marrow transplantation bp base pair BSA bovine serum albumin CAPK ceramide-activated protein kinase CAPP ceramide-activated protein phosphatase CB (umbilical) cord blood CD cluster of differentiation CNS central nervous system cDNA complementary DNA CO2 carbon dioxide CoA co-enzyme A Da Dalton DAG diacylglycerol E embryonic day EC Enzyme Commission ELISA enzyme-linked immunosorbent assay enGFP enhanced green fluorescence protein enYFP enhanced yellow fluorescent protein ERK extracellularly regulated protein kinase ERT enzyme replacement therapy FBS fetal bovine serum DNA deoxyribonucleic acid FACS fluorescence-activated cell sorting GvHD graft-versus-host disease G-CSF granulocyte colony stimulating factor Gy Gray HCl hydrochloric acid HPLC high-performance liquid chromatography HSPC hematopoietic stem/progenitor cells hu human IP infectious particles

x
i.p. intraperitoneal i.v. intravenous IL interleukin INF-α interferon alpha IRES internal ribosomal entry site JNK c-Jun N-terminal kinase LDH lactate dehydrogenase LSD lysosomal storage disease LNGFR low affinity nerve growth factor LV lentivirus; lentivector LTR long terminal repeat Luc luciferase mAb monoclonal antibody MACS magnetic-activated cell sorting MSC mesenchymal stem cell MTT 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide LSD lysosomal storage disease MAPK mitogen-activated protein kinase MNC mononuclear cell MOI multiplicity of infection NaCl sodium chloride NaOH sodium hydroxide NHP non-human primate NOD/SCID non-obese diabetic/severe combined immunodeficiency PB peripheral blood PBS phosphate-buffered saline PE phycoerythrin qPCR quantitative polymerase chain reaction RNA ribonucleic acid RV oncoretrovirus SAP saporin SAPK stress-activated protein kinase SCF stem cell factor SD standard deviation SDS sodium dodecyl sulfate SEM standard error of the mean SIN self-inactivating SM sphingomyelin SMase sphingomyelinase sr sterian Tac T cell activation antigen TLC thin-layer chromatography TNF-α tumor necrosis factor alpha TPO thrombopoietin TUNEL terminal deoxynucleotidyl transferase biotin-dUTP nick end labeling

xi
U unit VEGF vascular endothelial growth factor VSV-g vesicular stomatitis virus glycoprotein VVO vesicular-vacuolar organelle WBLI whole body luminescence imaging WPRE woodchuck hepatitis virus post-transcriptional regulatory element

1
Chapter 1: Introduction

2
1.1 LYSOSOMAL STORAGE DISEASES
1.1.1 Overview
Lysosomal storage disorders (LSDs) are a group of over 40 distinct metabolic
conditions resulting from deficient activity of proteins associated with the lysosome, an
acidic membrane-bound compartment within the cell [1]. While individually their prevalence
is low, as a group they can occur at high frequencies (up to 1 in 7,700) in some populations
[2-4]. These diseases are characterized by lysosomal accumulation of macromolecules
including sphingolipids, oligosaccharides, gangliosides, glycosaminoglycans and sulfatides
[5]. LSDs are monogenetic and can result from deficiencies in acid hydrolases (eg.
sphingolipidoses, mucopolysacaridoses and glycoproteinoses), deficiencies in co-factors
involved in activating lysosomal hydrolases (eg. prosaposin deficiency), defects in lysosomal
transporters (eg. cystinosis), defects in lysosomal trafficking (eg. mucolipidosis I and III) or
defects in the lysosomal membrane (eg. Danon disease) [5, 6]. The pathogenesis of LSDs and
the link between substrate accumulation and disease symptoms have not been fully
elucidated for most of the diseases [7].
Clinical symptoms of LSDs generally occur along a spectrum of severity that varies
both within and among the different diseases. Infantile forms are generally very severe with
neurological involvement and patients typically succumb to the disease at an early age [8].
Adult or late-onset forms of LSDs are generally milder and mostly involve peripheral
symptoms such as hepatosplenomegaly, cardiac and renal damage and muscle atrophy [8].
The juvenile forms are intermediate in severity between the infantile and adult forms. It has

3
been proposed that the differences in the age of onset and symptom severity are correlated to
differences in residual enzyme activity [9]. In this theory, Conzelmann and Sandhoff suggest
that there is a critical threshold for enzyme activity below which substrate accumulation
occurs, so that even small decreases in enzyme activity can lead to disease [9]. In general, the
lower the residual activity of the enzyme, the earlier the age of onset and the more severe are
the symptoms.
1.1.2 Treatment of Lysosomal Storage Diseases
Enzyme Replacement Therapy
One of the first treatments investigated for patients with LSDs was enzyme replacement
therapy (ERT), where purified enzyme is injected into patients [10]. It was first proposed as a
treatment for LSDs in 1964 by de Duve [11] and then later by Roscoe Brady as treatment for
sphingolipidoses. [12]. To date, ERT has been used successfully to treat patients with the
non-neuropathic form of Gaucher disease [13] and Fabry disease [13, 14]. The efficacy of
ERT is also being evaluated for the treatment of Pompe disease [15], Hurler syndrome [14],
Maroteaux-Lamy syndrome [16] and Hunter syndrome [17]. It has shown some success in
reducing stored material and relieving visceral symptoms, but in many cases the disease still
progresses [18, 19]. The use and efficacy of ERT are limited by a number of factors. First,
none of the recombinant enzymes can cross the blood brain barrier and as such, ERT has not
been effective in treating patients with neurological involvement [20]. Another concern is
that some patients develop neutralizing antibodies against the recombinant enzyme that
reduce the clinical efficacy of the treatment [21]. Patients can, however, develop tolerance to

4
the enzyme [22]. Finally, the use of ERT can be limiting for patients since this type of
therapy is very expensive, costing between $70 000-$500 000/year/patient depending on the
enzyme, the dosing regime and the weight of the patient [23, 24].
Small Molecules
Carbohydrate-based inhibitors are also being investigated for the treatment of LSDs.
One such molecule is miglustat (N-butyldeoxynojirimycin), which is an imino-sugar
inhibitor of ceramide-specific glucosyltransferase, the enzyme that catalyzes the initial
committed step in glycosphingolipid synthesis [25]. The rational is that a reduction in the rate
of glycosphingolipid biosynthesis could reduce the level of substrate to a level where the
residual enzyme activity could prevent storage [26]. This type of therapy, termed substrate
reduction therapy, is mostly used for the treatment of Gaucher disease where it has shown
some benefit [27]. Its use in combination with ERT for the treatment of Gaucher disease has
also been tested but has yet to show any increased benefit [28]. Miglustat is also being
investigated for its efficacy in treating Niemann-Pick disease type C [29], late-onset Tay-
Sachs disease [30] and Sandhoff disease [31]. Screening is also ongoing for other molecules
that act as pharmacological chaperones by stabilizing the mutant protein, thus allowing for its
exit from the lumen of the endoplasmic reticulum and trafficking to the lysosome. For
instance, in the case of Tay-Sach’s disease, it is thought that inhibitors of beta-
hexosaminidase A could enhance any residual enzyme activity and reduce lipid storage [32].
Other molecules with active-site-specific chaperone activity that increase the activity of

5
hydrolases are also being investigated for disease such as Gaucher disease [33] and Fabry
disease [34].
Cell Therapy
Cell-based therapies are routinely used in the treatment of LSDs and most commonly
involve the transplantation of hematopoietic stem/progenitor cells (HSPCs) since these cells
can provide a systemic source of enzyme. It was first attempted for Hurler’s disease in 1981
[35] and has since been used to treat other LSDs such as other mucopolysaccaridoses and
metachromatic leukodystrophy [36]. This has been done for Gaucher disease using both
allogeneic donor cells [37, 38] and using genetically modified cells [39]. In general,
hematopoietic cell transplantation has showed success in relieving the visceral symptoms of
disease but has shown limited efficacy in patients with neurological involvement [36].
1.1.3 Examples of Common Lysosomal Storage Diseases
Gaucher Disease
Gaucher Disease, the most common LSD, results from a deficiency in the enzyme
glucocerbrosidase that hydrolyses the breakdown of glucosylceramide into glucose and
ceramide [40]. Glucosylceramide is a component of the cell membrane of red and white
blood cells, which are cleared from the body by macrophages [41]. As a result, these cells
accumulate glucosylceramide and become “Gaucher cells” that can be found in tissues such
as spleen, liver, kidneys, lungs, brain and bone marrow [41]. Symptoms may include, but are
not limited to, splenomegaly, hepatomegaly/liver malfunction, skeletal disorders and bone

6
lesions, severe neurologic complications, and swelling of lymph nodes [41]. Type 1 Gaucher
disease is the non-neuropathic form while types 2 and 3 are characterized by acute central
nervous system (CNS) complications [41]. One of the earliest treatments for Gaucher
involved complete or partial splenectomy to relieve pain, hypersplenism and problems
arising from splenomegaly [42]. However, the risk of sepsis and the advent of ERT have
decreased the use of this type of treatment [43-45]. As previously mentioned, a number of
treatment options with varying degrees of efficacy are available for Gaucher patients
including ERT using imiglucerase [46], substrate reduction therapy using miglustat [27] and
BMT [37, 38]. These have all shown varying degrees of success depending on the severity of
the symptoms. For instance, while there is clear benefit for using ERT in patients with type 1
Gaucher disease [46], improved outcomes in treating type 3 Gaucher patients have yet to be
demonstrated [47].
Fabry Disease
Fabry disease is an X-linked disorder characterized by a deficiency in the enzyme
alpha-galactosidase A (α-gal A) and the accumulation of glycosphingolipids, especially
globotriaosylceramide (Gb3) [48, 49]. It occurs in about 1 in 40, 000 males and affected
organs include the vascular endothelium, kidneys, heart, brain and peripheral and central
nervous system [49-51]. Gb3 accumulation within these tissues impairs normal function.
Common symptoms of the disease include angiokeratomas, burning pain in the extremities,
impaired ability to sweat, and severe renal insufficiency/failure, often leading to end stage
renal disease [49]. In addition, there have been reports of cases of Fabry disease where the
sole manifestation is left ventricular hypertrophy, termed “cardiac” Fabry disease [52]. Fabry

7
disease is most commonly treated with ERT using recombinant α-gal A (either agalsidase
alpha [53] or agalsidase beta [54]). Renal insufficiency often requires dialysis and in some
cases, kidney transplantation [55, 56].
1.2 FARBER DISEASE
1.1.2 Disease Overview
Farber disease was first identified by the pediatric pathologist Sidney Farber in 1952
as a lipogranulomatosis [57]. The biochemical defect was later identified as deficient activity
of ceramidase and stored ceramide was implicated in the development of some of the
ultrastructural abnormalities observed, including “elongated membranes”, “zebra bodies”,
comma-shaped curvilinear tubules called Farber bodies, and spindle-shaped bodies that can
be detected in fibroblasts, histiocytes, and endothelial cells [58, 59].
The genetic basis for the disease is a mutation of the gene ASAH which encodes the
enzyme acid ceramidase (AC; N-acylsphingosine deacylase; EC 3.5.1.23) [60, 61]. It is an
autosomal recessive disorder characterized by the accumulation of ceramide in the lysosomal
compartment of cells [62]. Farber disease can be diagnosed by the measurement of AC
activity in cultured skin fibroblasts, white blood cells, or cultured amniocytes [63].
Symptoms can appear as early as two weeks of age and include subcutaneous granulomas,
progressive hoarseness, painful swollen joints, psychomotor retardation, respiratory
insufficiency, and poor weight gain; with affected tissues showing massive infiltrations of
granulocytes and lipid-laden macrophages [62]. Since its elucidation, the Farber disease

8
phenotype has been divided into seven different sub-groups that differ in the age of onset, the
severity of the symptoms, and the tissues affected [62]. While studies have shown that the
level of stored ceramide in the lysosomes is significantly correlated with the
neurodegenerative course of Farber disease and the age of death of the patient [64], there
appears to be no correlation between the levels of residual AC activity and the degree of
ceramide accumulation or symptom severity [65]. In addition, the pathogenesis of the disease
and the mechanisms by which stored ceramide results in granulomatous inflammation and in
the development of the symptoms seen in Farber patients have yet to be fully elucidated.
Type 1 Farber disease is the classical form of the disorder that affects the majority of
patients (~50% of reported cases) [62]. In addition to having severe classical symptoms,
affected patients show signs of nervous system dysfunction that include impaired
psychomotor development, mild retardation, and peripheral nerve involvement [62, 66].
These patients typically succumb to the disease by the age of two [62]. Patients with Type 2
and 3 Farber disease exhibit minimal to no symptoms of CNS disease but are still severely
affected with granulomatous inflammation that results in the formation of subcutaneous
nodules, joint pain and contractures, hoarseness and respiratory insufficiency [62]. Type 4
Farber disease involves severe neurological deterioration, extreme hepatosplenomegaly at
birth, and granulomatous infiltrations in the liver, spleen, lymphoid tissue, thymus and lungs
[67]. Type 5 Farber disease is characterized by progressive CNS dysfunction starting within
the first 2 years of life and patients present with loss of speech, seizures, mental retardation,
tetraplegia and myoclonia [68]. Type 6 Farber disease is actually a single report of a patient
with a combination of Farber disease and Sandhoff disease (an LSD caused by deficiency of
hexosaminidase A and B) [69]. This patient showed hoarseness, stridor (noisy breathing),

9
scattered skin nodules, painful swelling of hand joints and ankles, and cherry-red macular
spots [69]. Type 7 is a single report of a patient with a mutation in the prosaposin gene whose
protein products enhance the activities of lysosomal enzymes [70]. As a result, this patient
showed combined deficiency of glucocerebrosidase, galactocerebrosidase and ceramidase
[70].
1.2.2 Treatment of Farber Disease
Currently there is no treatment for Farber disease and most patients succumb to the
disorder at a very young age. Treatment consists primarily of palliative care such as
corticosteroids for the pain, tracheostomy to relieve respiratory difficulties, and surgery to
remove the granulomas [62, 71]. Allogeneic bone marrow transplantation (BMT) has been
attempted for some Farber patients based on the reasoning that a population of cells with
normal enzyme activity could ameliorate the effects of the deficient enzyme [36, 66, 72, 73].
In transplants of four mildly-affected Farber patients (Type 2/3), granulomatous
infiltrations were reduced, the hoarseness disappeared, and joint mobility improved [72, 73].
Pre-conditioning for all patients was busulfan-based myeloablation and all patients achieved
donor chimerism of >90% post-transplant [73]. The first patient was a female aged 3 years
and 11 months who received bone marrow mononuclear cells (BMMNCs) from her HLA-
identical sister. Examination 450 days post-transplant showed that the number of
subcutaneous nodules had decreased from 58 to 8 and the number of joints with restricted
motion had decreased from 26 to 2 [72]. In addition, her erythrocyte sedimentation rate
normalized and hoarseness improved [72]. The second patient, was similar in age and

10
received matched unrelated donor bone marrow. This patient showed a reduction in the
number of subcutaneous nodules from 39 to 14 and the number of affected joints had
decreased from 24 to 4 [72]. A 2 year-old and 21-year old were also transplanted with
matched related bone marrow. The two year old showed similar improvements in the number
of nodules and in joint mobility [73]. The 21 year-old patient was only mildly affected but
was transplanted to improve mobility in her legs [73]. Later follow-up 3 to 6 years post-
transplant showed that patients still had donor chimerisms of >90% and were all still alive
[74].
Earlier transplants on patients with the more common Type 1 Farber disease showed
limited success. In these studies, BMT lessened the peripheral symptoms but there was no
improvement in neurological function and patients died soon after transplant [66, 75]. In the
first case, the patient was 18 months at the time of transplant. While the granulomas
regressed, the patient died 6 months post-transplant with progressive neurological
deterioration [75]. In the second instance, a 9.5 month-old Farber patient with 6% residual
enzyme activity in the peripheral blood leukocytes received bone marrow from her HLA-
identical heterozygous sister. Within 6 weeks of transplant, AC activity in the leukocytes had
increased to the heterozygote donor level of 44% of normal activity [66]. Subcutaneous
nodules and hoarseness resolved within 2 months post-transplant and by 6 months, the joint
pain and contractures had also resolved [66]. A progressive loss of donor chimerism was
seen and by 21 months post-transplant, chimerism was <1%. The patient’s neurological
status deteriorated over time and the infant died 28 months post-transplant at the age of 37.5
months [66]. These limited outcomes mirror those observed after BMTs in other lysosomal
storage diseases with neurological involvement [76]. Further, ERT is not currently available

11
for Farber disease and with the relatively smaller Farber patient population, there is little
economic incentive for the development of ERT for Farber disease. Therefore, the
development of improved alternative treatment modalities remains important.
1.2.3 Mouse Model of Farber Disease
In 2002, Li et al. attempted to produce a mouse model of Farber disease by targeted
disruption of the gene encoding murine AC, Asah1 [77]. They obtained a clone that
contained three tandem insertions of the targeting vector in exon 12. In both the F2 and F3
generations, no Asah-/- mice were found. Examination of the embryos of F3 mice revealed
that beginning at embryonic day (E) 8.5, there were no homozygotes and evidence suggested
that these embryos were resorbed. It was found that in normal embryos, AC mRNA is
upregulated beginning at E7 and remains high throughout embryonic development [77].
Later studies showed that AC plays a critical role in early embryo survival by removing
ceramide, thus inhibiting apoptosis of the cells of the embryo [78]. These studies support the
hypothesis that a complete lack of AC activity results in death of the developing embryo and
suggest a crucial role for AC in the apoptotic facet of ceramide metabolism. While F2+/- mice
showed some pathological abnormalities, no overt clinical symptoms were observed [77].
Therefore, a suitable model to study the pathogenesis of the disease and possible treatments
remains to be developed.

12
1.3 ACID CERAMIDASE
1.3.1 Gene, Structure and Biochemistry
Human AC, also known as N-acylsphingosine amidohydrolase, is encoded by the
ASAH gene located in chromosomal region 8p21.3-p22 [61]. The gene spans approximately
30 kb and contains 14 exons. The mRNA is expressed mainly as a 2.4 kb transcript but minor
transcripts of 1.7 and 1.2 kb are also detectable in some tissues [61]. The mRNA has a 17 bp
5’-untranslated sequence, an open reading frame of 1185 bp, a 3’-untranslated sequence of
1110 bp, and an 18 bp poly-(A) tail [79].
AC is expressed as a single precursor polypeptide of ~53-55 kDa that is processed in
the lysosome into the mature, heterodimeric protein with α and β subunits of 13 and 40 kDa,
respectively [80]. It has been shown that this cleavage is mediated by autocatalytic activity of
the precursor protein. It has been proposed that this cleavage is mediated by the residues
Cys143, Arg159 and Asp162 and occurs between Cys143 and Ile 142 [81]. This results in the
exposure of the nucleophilic Cys at the N-terminal side of the β subunit, which acts as the
catalytic site of the enzyme [81]. The heterodimeric AC protein has a half-life of >20 h [80]
and appears to be held together by 3 disulfide bonds: C10-C319, C122-C271 and C367-C371
[82]. It has also been determined that the mature enzyme contains mannose-6-phosphate
residues and 6 possible sites for N-glycosylation (N152, N174, N238, N265, N321, N327)
[82] in the β subunit, five of which are used [80].
The biological function of AC is to catalyze the hydrolysis of ceramide into
sphingosine and a free fatty acid [83]. Using N-laurylsphingosine as the substrate, AC was

13
found to have a Km of 149 µM and a Vmax of 136 nmol/h/mg, with optimum activity at pH 4.5
[84]. AC has also been shown to catalyze ceramide synthesis from sphingosine and a fatty
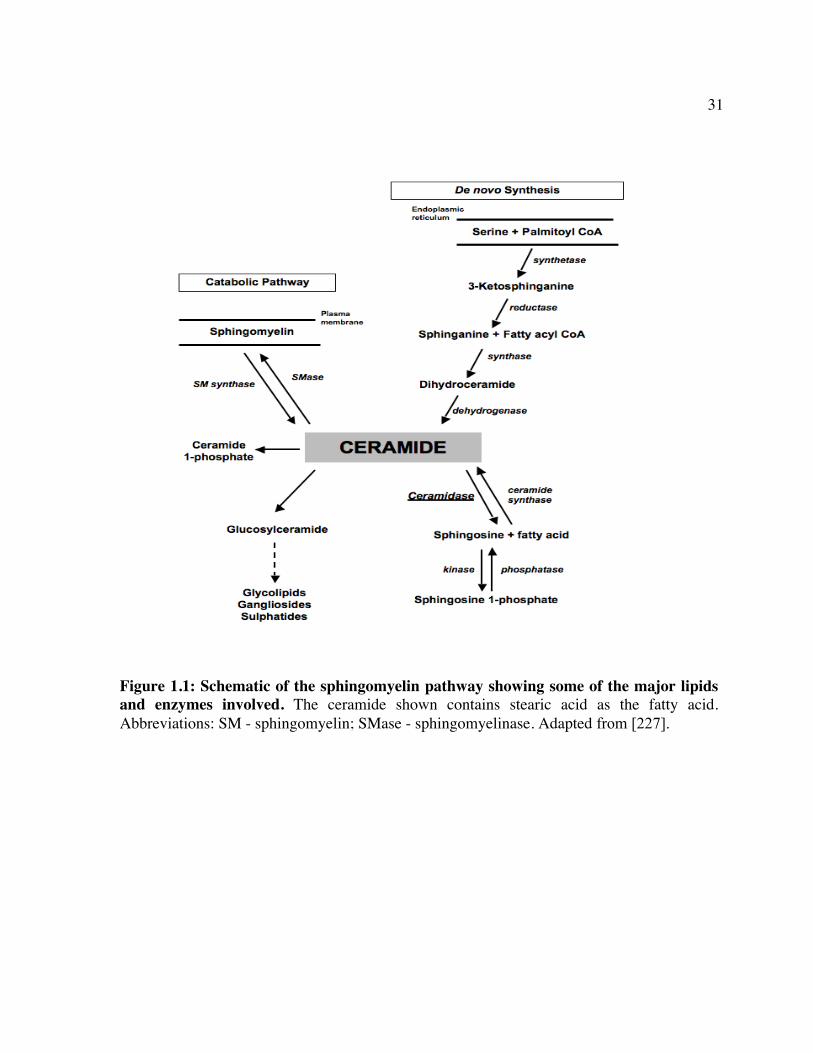
acid in a reverse reaction with a pH optimum of 5.5 [85, 86] (Figure 1.1). Located in the
lysosomal of cells, it is expressed at high levels in heart, lung, kidney, placenta and lungs and
at lower levels in the brain, liver, pancreas, skeletal muscle and throughout the
gastrointestinal tract [87].
1.3.2 Mutations in Farber Disease
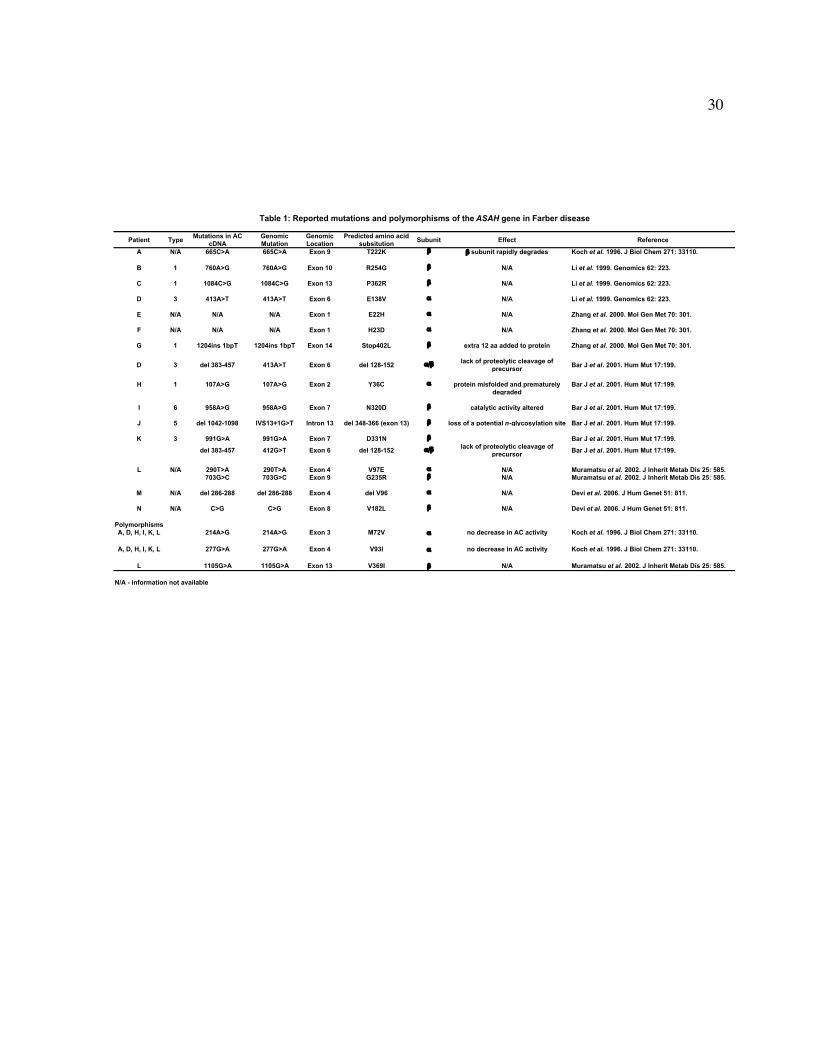
To date, 17 different mutations and 16 polymorphisms have been identified in the
ASAH gene of Farber patients [61, 79, 87-91]. These mutations are distributed along all 14
exons and affects both subunits with the absence of any apparent ‘hot-spots’ (Table 1). Most
of these mutations are point mutations that result in amino acid changes while the others
result in small deletions or insertions. Though the effect of each mutation has not been
characterized, the majority of Farber patients tested showed less than 6% of normal AC
activity as measured in a variety of tissues [62]. Deficient enzyme activity can be caused by
changes in catalytic activity, lack of processing of the AC precursor protein, or by premature
degradation of misfolded protein (Table 1) [89].
1.3.3 Other Ceramidases
In humans, both neutral and alkaline ceramidases have been identified. The first
human neutral ceramidase reported was shown to be located primarily in the mitochondria
and to catalyze ceramide hydrolysis with a pH optimum between 7.5 and 9.5 [92]. It was

14
shown to be ubiquitously expressed with the highest levels being detected in the kidney,
skeletal muscle, and heart [92]. Later reports showed that this protein was truncated and that
the full length enzyme was located primarily in the plasma membrane [93] and that it could
also catalyze the synthesis of ceramide in the reverse reaction [94]. It has also been found in
the intestinal tract and is thought to be released into the intestinal lumen where it catabolizes
dietary ceramides [94]. Sequence and phylogenetic analysis revealed that there is no
significant homology between neutral and acid ceramidases and that they belong to a
completely different family in both mouse and humans [95]. A ubiquitously expressed
human alkaline ceramidase was also identified that localized to the Golgi and endoplasmic
reticulum [96]. It was found to have ceramidase activity, in particular phytoceramidase
activity, with a pH optimum of 9.5 but does not show any reverse activity [96]. In addition, a
murine alkaline ceramidase has also been isolated that is localized in the endoplasmic
reticulum and is abundantly expressed in the skin [97]. However, a human homologue has
not yet been isolated.
1.4 CERAMIDE
1.4.1 Structure and Physiological Function
Ceramides are a family of sphingolipids comprised of sphingosine or a related long-
chain base and a fatty acid (usually containing 2-28 carbon atoms) [98]. They are
components of a number of complex sphingolipids such as sphingomyelin, cerebrosides, and
gangliosides and play a central role in sphingolipid biosynthesis [98]. Ceramides can be
synthesized via three pathways: de novo synthesis from serine and palmitoyl CoA, synthesis

15
from sphingosine and a fatty acid by the reverse action of AC, or by degradation of
sphingomyelin [86, 99] (Figure 1.1).
Most ceramides contain fatty acyl chains of greater than 16 carbon atoms. These are
among the most hydrophobic lipids found in the cell membrane [98]. As a result of this
hydrophobicity, free ceramides do not exist in biological fluids such as the cytosol and thus,
ceramides exert their biological effects at the membrane level [98]. Indeed, it appears that
ceramide is abundant in sphingolipid-rich regions within membranes, such as caveolae [100]
and rafts [101, 102], which serve as important components of signaling microdomains within
the cell [103].
Ceramide signaling is induced by stimuli such as TNF-α, Fas, IL-1β, INF-α, CD28,
complement, serum deprivation, γ-irradiation, heat shock, ultraviolet light, and
chemotherapeutic drugs [104]. Ceramide and its metabolites have been found to be important
mediators in cell processes such as signaling [105], stress responses [106], growth [107,
108], senescence [109], and apoptosis [110]. The exact mechanisms through which ceramide
participates in this diverse array of biological effects remain controversial but it appears that
activation of various kinases and phosphatases play a role [103].
Due to its diverse biological effects, abnormal ceramide metabolism has been
implicated in a number of disorders. For instance, ceramide and other sphingolipids are
important components of the stratum corneum of the skin and ceramide has been shown to be
involved in the pathogenesis of skin disorders such as psoriasis and atopic dermatitis [111],
as well as in aging of the skin [112]. Ceramide has also been shown to be involved in other
processes such autophagy [113], cytokine signaling [114] and insulin resistance leading to
diabetes due to its inhibitory effect on protein kinase B signaling [115, 116]. Its regulation by

16
acid sphingomyelinase has also resulted in ceramide being a key regulator in liver cirrhosis
associated with Wilson’s disease [117], the formation of pulmonary edema in acute lung
injury [118], and susceptibility to viral and bacterial infections [119, 120].
1.4.2 Ceramide Signaling and Apoptosis
While the outcome of ceramide signaling is cell type-dependent, most often the
results are antagonistic to growth and survival [103]. It appears that the balance between the
levels of ceramide and other related molecules, such as sphingosine-1 phosphate, is an
important determinant in cell fate. Together they act as a “sphingolipid rheostat” that
determines whether or not a cell undergoes apoptosis [103, 106, 121].
Following stress stimuli, ceramide activates a number of kinases and phosphatases
whose downstream effects drive cells towards apoptosis. Ceramide kinases appear to target
stress-activated protein kinases (SAPKs), the Jun N-terminal kinases (JNK), [122] protein
kinase C zeta (PKC zeta) [123], and kinase suppressor of Ras (KSR) [124]. Activation of
these pathways not only induce apoptosis, but also suppresses proliferation and promotes cell
cycle arrest [103]. Ceramide also activates a number of protein phosphatases (PP) including
PP2A [125] and PP1 [126]. PP2A has been shown to inhibit the activity of both pro-growth
kinases, such as PKC alpha [127] and Akt [128], and anti-apoptotic molecules like Bcl2
[129] and Bad [130]. Activated PP1 dephosphorylates and inactivates the retinoblastoma
gene (Rb), leading to growth arrest [131]. The mechanisms by which ceramide activates
these kinases and phosphatases is not known.

17
Ceramide’s involvement in regulating apoptosis has been shown to be important in
male and female fertility [132, 133], Alzheimer’s disease [134, 135], embryo survival [78],
and in resistance of malignant cells to apoptosis [136-138]. However, studies on the
apoptotic response to ceramide accumulation in Farber patient cells have resulted in
discrepant results. For instance, it has been shown that in Farber fibroblasts and lymphocytes,
lysosomal ceramide pools do not mediate stress-induced apoptosis [65, 139, 140]. In contrast,
Farina et al. (2000) demonstrated by TUNEL staining that colonocytes from Farber patients
undergo increased apoptotic cell death [141]. Overexpression of AC has also resulted in
different outcomes. It has been found that overexpression of AC had no effect on the
apoptotic response due to TNF and CD40L in Farber patient fibroblasts [140]. Yet Strelow et
al. found that overexpression of AC protected L929 cells, which are TNF-sensitive, from
TNF-mediated cell death [142]. What is not clear, however, is whether ceramide is a direct
effector or a second-messenger in the apoptotic pathway, or how the turnover between
membrane and lysosomal ceramide affects the outcome of signaling [65, 143].
Other players in the sphingomyelin/ceramide pathway also determine cell fate. For
instance, sphingosine-1 phosphate can promote cell survival and proliferation by activation
of the extracellularly regulated protein kinase 1/2 (ERK1/2) signaling pathway [144] or by
negatively regulating pro-apoptotic Bcl2 proteins such as Bax and Bid [145]. Ceramide 1-
phosphate also promotes proliferation via the ERK1/2, JNK or the protein kinase B pathway
[146]. Sphingosine can also promote an apoptotic phenotype by inhibiting anti-apoptotic
proteins such as Bcl-x(L) [147]. Therefore, the sphingomyelin/ceramide pathway is a key
target for strategies where modulation of apoptosis is required as in the case of cancer
therapy.

18
1.5 GENE THERAPY
1.5.1 Methods of Gene Transfer
Several non-viral and viral methods are currently employed for delivery of genes to
cells. The use of non-viral methods of gene transfer, such as electroporation of DNA and
liposomes, is often limited by inefficient gene transfer and the transient nature of transgene
expression [148]. A number of viral vectors for gene delivery have been used in the
laboratory and in clinical gene therapy trials, including those based on adenoviruses, adeno-
associated viruses (AAVs), herpesviruses, poxviruses, polyomaviruses and retroviruses, each
having their advantages and disadvantages [149]. The most commonly used viral systems in
gene therapy are recombinant adenoviruses, AAVs and retroviruses.
Adenoviruses are non-enveloped, double-stranded DNA viruses that infect cells via
the coxsackie-adenovirus receptor [150]. They have a cloning capacity of up to ~8 kb of
DNA [151] and exist in a number of different serotypes that allows for efficient targeting of a
wide range of cell types [152]. However, they are limited by the fact that they can generate
host immune responses and they do not integrate into the host genome, which often makes
transgene expression transient [152].
AAVs are single-stranded DNA viruses that require a helper virus for replication
(usually an adenovirus or herpesvirus). AAVs can infect non-replicating cells, transgene
expression can be long-term and different serotypes offer the advantage of a broad host
range, although each serotype is tissue-type specific [153]. The use of AAVs is limited by a
small cloning capacity (~4-5 kb), the presence of contaminating wild-type adenovirus in

19
preparations of AAV and the fact that a host response can be mounted against the virus
capsid protein [154]. In addition, natural infections with wild-type AAV results in the
presence of neutralizing antibodies against the vector.
Retroviruses are a large family of single-stranded RNA viruses that include
oncoretroviruses, lentiviruses and spumaviruses (foamy viruses). Retroviral vector systems
offer the advantages of stable integration into host genomes, the ability to transduce a wide
variety of cells types, high levels of transgene expression, lower immunogenicity compared
to other viral vectors and the ability to transfer large inserts (8-10 kb)[155]. In addition, the
tropism of retroviruses can be manipulated by changing the envelope glycoprotein that
encapsulates the virus in a process called pseudotyping [156]. Therefore, they can be
engineered to transduce a wide range of cell types. The receptor for the virus depends on the
viral envelope used, however, the mechanism of uptake is generally by membrane fusion and
deposition of the viral genome into the cytoplasm [156]. Disadvantages of retroviruses
include their potential to generate replication-competent retroviruses [157], the occurrence of
methylation and silencing of the LTRs [158] and the potential risk of oncogenesis from the
random pattern of integration (discussed later).
Oncoretroviruses are simple retroviruses that encode gag, pol and env proteins. The
genome is flanked by long-terminal repeats (LTRs) that mediate viral integration and also
contains a packaging signal that allows the virus to be encapsidated [156]. They have been
generated from a number of different oncoretroviruses, including murine leukemia virus
(MLV), spleen necrosis virus, Rous sarcoma virus, and avian leukosis virus [152].

20
Recombinant oncoretroviruses have all of the viral protein genes deleted and replaced with
gene expression cassettes that may or may not contain exogenous promoters.
Lentiviruses (LVs) also contain the gag, pol and env proteins as well as additional
proteins that are involved in virus replication and infection [155]. In addition to vectors
derived from human infectious virus (HIV-1), the vectors can also be built from the
backbones of Simian, Equine and Feline lentiviruses [159]. While oncoretroviruses require
cell division for integration, lentiviruses have been shown to transduce slower-dividing cells
due to its unique pre-integration complex [160, 161]. It should be noted that while mitosis is
not required for transduction by lentiviruses, transduction rates are ten-fold higher if the
target cells are in the G1b or S/G2/M phases of the cell cycle [162].
A number of safety features have been developed within both oncoretroviral and
lentiviral vectors that minimize the risk of replication competent viruses being formed. In
both systems, many of the viral accessory genes have been removed and those that remain
have been separated into multiple plasmids [163]. In addition, self-inactivating long terminal
repeats (SIN LTRs) in lentiviral systems reduce the risk that full-length viral transcripts can
be formed [164]. SIN LTRs contain a deletion of the U3 region of the 3’ LTR and following
integration, the deletion is copied into the 5’ LTR. The integrated provirus is thus unable to
produce a full-length viral transcript or to replicate [164, 165].
Retroviruses are produced by transfection of cells with plasmids that encode the viral
genes and sequences necessary for producing an infectious viral particle. These include the
structural gag and pol genes, an encapsidation signal on the gene transfer vector, an envelope
gene, and any other additional proteins required, for example tat or nef in LV systems [155].

21
In the case of oncoretroviruses, there are a number of packaging lines that are stably
transfected with plasmids that encode the gag and pol genes, as well as an envelope gene
[166]. Lentiviruses are produced by transient co-transfection of plasmids into 293T cells
[155]. Recently, stable packaging lines for generating lentiviruses have been developed that
overcome the issue of toxicity of the commonly used vesicular stomatitis virus glycoprotein
(VSV-g) by using an inducible promotor to drive its expression [167].
1.5.2 Treatment Modalities with Viral Vectors
Viral vectors have been used therapeutically in a number of ways. They can either be
delivered directly in vivo or they can be used to transduce cells ex vivo, which are then
transplanted into patients. Delivery in vivo involves administration of virus either to the
bloodstream [168] or to tissues such as the brain [169, 170], heart [171], or tumors [172]. Ex
vivo strategies include, but are not limited to, transduction of hematopoietic stem/progenitor
cells (HSPCs) prior to transplantation [173, 174] and transduction of other cell types that are
used directly for transplant to provide a source for the therapeutic transgene product(s) [175].
Each of these methods has qualities that make them more or less suitable for use depending
on the disease symptoms in question.
1.5.3 Gene Therapy for Farber Disease
Farber disease is an attractive target for gene therapy for a number of reasons: it is
caused by a single gene defect; the cDNA has been subcloned; and the enzyme is fairly well
characterized. Most importantly, it has been shown that cells transduced with a recombinant

22
oncoretrovirus carrying the human AC cDNA over-express and secrete the enzyme [176].
This secreted AC can subsequently be taken up into non-transduced cells through receptor-
mediated endocytosis involving the mannose-6-phosphate receptor, and subsequently restore
enzyme activity in non-transduced cells [176]. This phenomenon, termed metabolic
cooperativity, can allow for a small number of transduced cells to exert a larger therapeutic
effect in vivo since secretion from transduced cells can exert systemic correction once the
enzyme is taken up into non-transduced cells.
Hematopoietic cells are good targets for gene therapy since they facilitate systemic
delivery of the gene-augmented cells and their progeny as they circulate throughout the body.
In addition, if HSPCs are targeted and transduced, these cells can be a continuous source of
the transgene product due to their ability to self-renew and differentiate into all blood cell
lineages such as T cells, monocytes, macrophages and others [177]. It has also been shown
that cells that are engineered to over-express lysosomal enzymes secrete a considerable
amount of the enzyme [178]. As such, it is expected that transplantation of transduced
HSPCs will result in better systemic correction of enzyme-deficient cells in comparison to
cells from a normal donor. In addition, gene therapy allows the use of autologous
hematopoietic cells for transplantation. This obviates the need to find a matched donor and
reduces the morbidity that is associated with allogeneic transplantation [179]. Indeed,
approaches similar to BMT using lentivirally-transduced HSPCs have been tested in animal
models for a number of LSDs such as the mucopolysaccharidoses, Gaucher disease, and
Niemann-Pick disease with promising results [36, 180-182].

23
One major target cell population for therapy for Farber disease is human umbilical
cord blood (CB)-derived CD34+ cells. CD34+ cells are self-renewing and pluripotent, and are
thought to represent a portion of the HSPC population. It has been shown that HSPCs derived
from CB have greater repopulating ability than do adult BM-derived HSPCs cells [183], due
in part to the fact that cord blood contains higher proportion of more primitive sub-
populations [184]. Furthermore, a greater degree of HLA mismatch is acceptable when
selecting donor cells for transplantation since the lymphocytes contained in CB are more
immature than those found in other sources of HSPCs and as such are less likely to initiate an
immune response against the host [185]. CB-derived HSPCs are thus an ideal target for
correction of Farber disease since these cells can provide a long-term systemic source of the
deficient enzyme with reduced risk of graft-versus-host disease (GvHD).
Gene therapy for disorders that have manifestations affecting the central nervous
system (CNS), such as Farber disease, often requires a strategy to get the transgene itself or
its protein product across the blood-brain barrier (BBB) for metabolic correction. While the
BBB prevents large circulating molecules from entering the brain, lymphocytes and myelo-
monocytic cells are able to enter the CNS via numerous routes including the lepto-meninges,
choroids plexus, and perivascular area surrounding small vessels [186]. A variety of methods
have been employed for getting viral particles into the CNS including injection directly to
different regions of the brain [187], into the lateral cerebral ventricles [188] or into the
cerebrospinal fluid [189]. In addition, agents such as vascular endothelial factor (VEGF) or
bradykinin [190] have been tested for their ability to permeabilize the BBB and allow access
of therapeutic molecules to the brain [191].

24
Transplantation of transduced HSPCs may also contribute to correction of the
neurological manifestations of Farber disease since they can make their way into the brain.
Microglial cells are macrophage-like cells that account for 5-20% of the entire cell
population in the CNS and it has been shown that they are able to cross the BBB [192].
Recent evidence has supported the long-debated view that microglial cells arise from two
main sources: macrophages derived from mesenchymal progenitor cells, and circulating
monocytes [193]. Therefore, these cells are potential vehicles for delivering therapeutic
genes to the CNS since their precursors can be transduced and transplanted with relative
ease. Indeed, several studies have recently shown that genetically modified hematopoietic
cells enter the brain and differentiate into microglial cells throughout the brain [194, 195].
Therefore, transplantation of transduced HSPC is a promising therapeutic option for the
treatment of Farber disease.
1.5.4 Retroviral Genotoxicity
Integration of retroviruses into the host genome, while desirable for long-term gene
expression, presents the risk of initiation of oncogenesis through aberrant integration events.
A most striking example is the development of leukemia by four X-linked severe combined
immunodeficiency patients in a recent gene therapy clinical trial using a retroviral vector
[196-198] (a fourth was reported at the 33rd Annual Meeting of the European Group for
Blood and Marrow Transplantation in Lyon, France on March 25–28, 2007). It has been
reported that two of these patients developed leukemia characterized by insertion of the
retroviral vector into the LMO-2 oncogene [199] while the other two patients show insertions

25
into the LYL1 and c-Jun oncogenes [200].
A variety of theories for this outcome have been proposed such as the viral enhancer
may have activated the LMO-2 oncogene or that there were other chromosomal
abnormalities that contributed to the oncogenic event [199]. It has also been proposed that
the over-expression of the common γ chain, both singly [201] and in co-operation with LMO-
2 [202], gives cells a proliferative advantage that leads to oncogenesis. However, the exact
mechanism of leukemogenesis has remained unresolved since no other clinical trials have
reported this type of adverse event [199, 203]. Therefore, the development of improved
vectors and safety strategies is exceedingly important and timely.
A number of studies have been undertaken to characterize the insertion sites of both
oncoretroviral and lentiviral vectors. It has been shown that while oncoretroviral vectors
integrate into promotor-proximal regions and near transcriptional start sites (within ~5 kb
either upstream or downstream), HIV-based lentiviral vectors integrate throughout active
transcriptional units [204-206]. In patients with chronic granulomatous disease, mice and
rhesus macaques, transplantation of HSPCs transduced with oncoretroviral vectors has
shown that integration is non-random, with a high frequency of insertion (10-4 to 10-5 per
transduced Lin- cell) observed in the MDS/Evi1 locus [207-209]. This locus has been
implicated in the development of human myeloid leukemias. To date, the only study that has
shown genotoxicity associated with lentiviral vectors has been a study in which T-
lymphoblastic leukemia developed in mice when human factor XI was delivered for
treatment of hemophilia B [210]. It was found that these leukemias were probably caused by
the irradiation protocol used and that exposure to high doses of lentivirus did not lead to
leukemias [210].

26
Despite the risk of insertional mutagenesis associated with these vectors, retroviral
gene therapy continues because of the conceptual effectiveness of the treatment and the fact
that gene therapy is the only potential cure available for many disorders such as SCID and
LSDs. In addition, in over 300 clinical trials using retroviral vectors, adverse events such as
those seen in the clinical trial for X-SCID have not been reported [211].
1.6 MARKING OF TRANSDUCED CELLS
If hematopoietic cell engraftment is limited by the availability of “space” in
recipients, then transplantation of a higher percentage of genetically-corrected cells may
allow more effective occupation of that space. Therefore, the ability to enrich for transduced
cells ex vivo may enhance the therapeutic effects of hematopoietic cell-based gene therapy.
One strategy to enrich transduced cells within the population of transduced cells is to
engineer the expression of a factor, such as a cell surface marker, that can be used to pre-
select transduced cells prior to transplantation. A number of markers have been used in this
context. These include, but are not limited to, tyrosine kinase receptors such as low affinity
nerve growth factor receptor (LNGFR) [212], cytokine receptors such as the erythropoietin
receptor [213] and other cell surface antigens such as CD24 [214].
1.6.1 Structure and Function of CD25
Human CD25, the α-chain of the interleukin-2 (IL-2) receptor, is the ‘low affinity
receptor’ for IL-2 [215]. It is a membrane protein with a small (13 aa) cytoplasmic region

27
and is incapable of mediating IL-2 internalization or signaling by itself, however, in tandem
with the β chain of the receptor, it forms the ‘high-affinity’ receptor for IL-2 [215].
Expression of CD25 is absent on resting T cells, B cells, monocytes, and CD34+-enriched
cells but can be induced upon activation or by stimulation with IL-2, IL-4, IL-5 or IL-10
[216, 217].
1.6.2 Use as a Pre-selective Marker
In previous studies done by the Medin lab, no aberrant proliferation has been
observed in vitro or in vivo by over-expressing CD25 on HSPCs [218], unlike that seen with
truncated LNGFR [207], for example. The CD25 marker was used in our vectors to allow for
the immuno-affinity enrichment of transduced cells. In previous studies using this marker for
pre-selection of transduced cells, higher percentages of multilineage, gene-corrected cells in
the circulation of transplanted Fabry animals along with corresponding increases in enzyme
activity in relevant organs were observed [218]. More recently, we have also focused on the
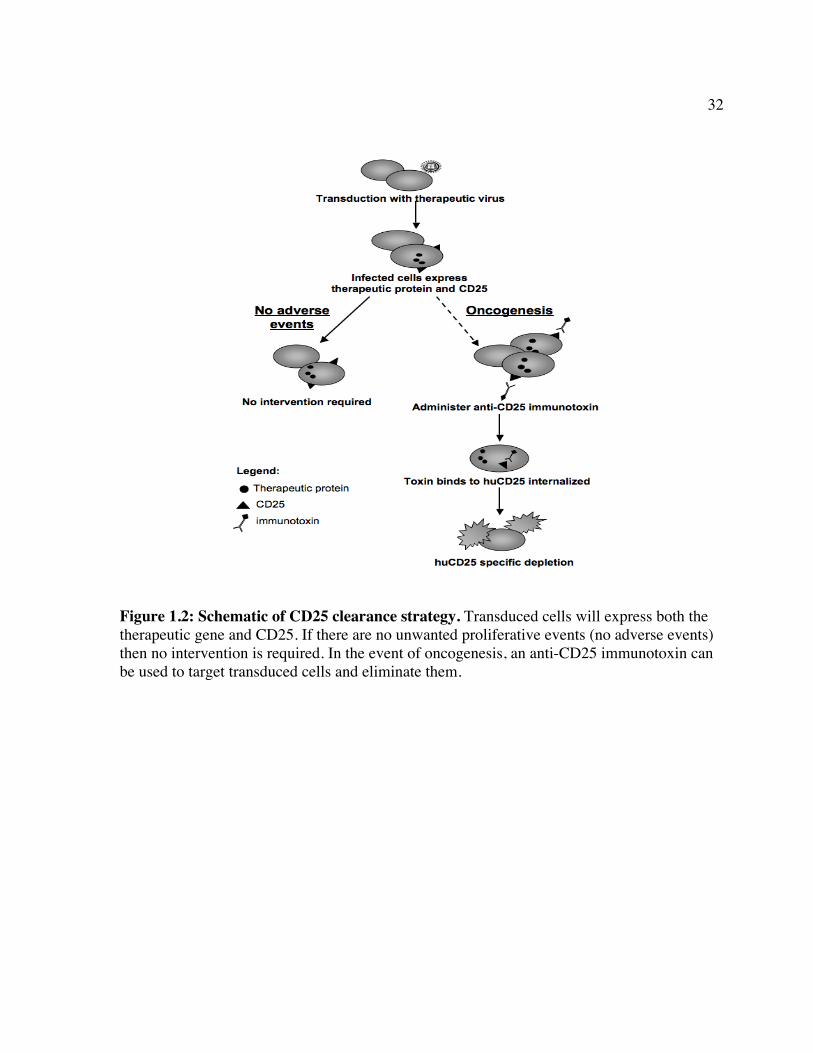
use of huCD25 as a cell surface marker that may also allow for the possibility of removal of
transduced cells, using clinically approved α-CD25 antibodies or newer, highly potent Ab-
toxin conjugates [219], should an unwanted proliferative defect occur (Fig. 1.2).
1.6.3 Aberrant Expression in Cancer
Aberrant levels of CD25 expression characterizes numerous disorders such as adult T
cell leukemia/lymphoma, Hodgkin’s lymphoma, hairy cell leukemias and true histiocytic
lymphomas [219]. Treatment of these diseases using antibodies against CD25, as well as

28
newer recombinant immunotoxins, has resulted in complete and partial remissions in patients
[219-221]. Currently, anti-CD25 antibodies are widely used for the prevention of renal graft
rejection and in some cases for prophylactic treatment against GvHD [222, 223]. Further,
studies have shown that when anti-CD25 antibodies are used to deplete CD4+CD25+
regulatory T cells, anti-tumor immunity is enhanced [224-226]. These findings provided the
rationale for using anti-CD25 toxin-conjugated antibodies to target CD25.
1.7 CURRENT STUDY OBJECTIVES
The aim of the studies presented in this thesis is to develop and test improved gene
therapy strategies for the treatment of Farber disease using novel recombinant retroviral
vectors. Vectors were engineered to express human AC and a selectable cell surface marker,
huCD25 that can be used to enrich for and track transduced cells (Figure 1.3). The aim was
to show that retrovirus-mediated overexpression of AC has no untoward effects in vivo and
as such, that this method is a viable option for the treatment of Farber disease. To this end,
vectors were tested in HSPCs to determine the effect of AC over-expression on
hematopoiesis. Since early treatment of Farber patients could prevent the harmful effects of
ceramide accumulation, the LV was also delivered to neonates and the persistence and
efficacy of the vector were assessed. To address the CNS manifestations of the disease, the
ability of VEGF to increase delivery of virus to the brain was explored. Due to the ability of
retroviral vectors to integrate into the host genome, there is risk of insertional mutagenesis.
Therefore, a novel safety strategy is proposed that utilizes the huCD25 marker that is already
included in our viral vectors as a tool for selective removal of transduced cells. This strategy

29
employs an antibody against the CD25 marker and can be used following gene transfer if an
oncogenic event occurs (Fig. 1.2). Together, these studies are the first to address the
development and pre-clinical testing of a retroviral gene therapy strategy for the treatment of
Farber disease. In addition, this is also the first report of an antibody-based safety strategy for
viral vectors.
30
Patient Type Mutations in AC cDNA
Genomic Mutation
Genomic Location
Predicted amino acid subsitution Subunit Effect Reference
A N/A 665C>A 665C>A Exon 9 T222K β β subunit rapidly degrades Koch et al. 1996. J Biol Chem 271: 33110.
B 1 760A>G 760A>G Exon 10 R254G β N/A Li et al. 1999. Genomics 62: 223.
C 1 1084C>G 1084C>G Exon 13 P362R β N/A Li et al. 1999. Genomics 62: 223.
D 3 413A>T 413A>T Exon 6 E138V α N/A Li et al. 1999. Genomics 62: 223.
E N/A N/A N/A Exon 1 E22H α N/A Zhang et al. 2000. Mol Gen Met 70: 301.
F N/A N/A N/A Exon 1 H23D α N/A Zhang et al. 2000. Mol Gen Met 70: 301.
G 1 1204ins 1bpT 1204ins 1bpT Exon 14 Stop402L β extra 12 aa added to protein Zhang et al. 2000. Mol Gen Met 70: 301.
D 3 del 383-457 413A>T Exon 6 del 128-152 α/β lack of proteolytic cleavage of precursor Bar J et al. 2001. Hum Mut 17:199.
H 1 107A>G 107A>G Exon 2 Y36C α protein misfolded and prematurely degraded
Bar J et al. 2001. Hum Mut 17:199.
I 6 958A>G 958A>G Exon 7 N320D β catalytic activity altered Bar J et al. 2001. Hum Mut 17:199.
J 5 del 1042-1098 IVS13+1G>T Intron 13 del 348-366 (exon 13) β loss of a potential n-glycosylation site Bar J et al. 2001. Hum Mut 17:199.
K 3 991G>A 991G>A Exon 7 D331N β Bar J et al. 2001. Hum Mut 17:199.
del 383-457 412G>T Exon 6 del 128-152 α/β lack of proteolytic cleavage of precursor Bar J et al. 2001. Hum Mut 17:199.
L N/A 290T>A 290T>A Exon 4 V97E α N/A Muramatsu et al. 2002. J Inherit Metab Dis 25: 585.703G>C 703G>C Exon 9 G235R β N/A Muramatsu et al. 2002. J Inherit Metab Dis 25: 585.
M N/A del 286-288 del 286-288 Exon 4 del V96 α N/A Devi et al. 2006. J Hum Genet 51: 811.
N N/A C>G C>G Exon 8 V182L β N/A Devi et al. 2006. J Hum Genet 51: 811.
PolymorphismsA, D, H, I, K, L 214A>G 214A>G Exon 3 M72V α no decrease in AC activity Koch et al. 1996. J Biol Chem 271: 33110.
A, D, H, I, K, L 277G>A 277G>A Exon 4 V93I α no decrease in AC activity Koch et al. 1996. J Biol Chem 271: 33110.
L 1105G>A 1105G>A Exon 13 V369I β N/A Muramatsu et al. 2002. J Inherit Metab Dis 25: 585.
N/A - information not available
Table 1: Reported mutations and polymorphisms of the ASAH gene in Farber disease
31
Figure 1.1: Schematic of the sphingomyelin pathway showing some of the major lipids and enzymes involved. The ceramide shown contains stearic acid as the fatty acid. Abbreviations: SM - sphingomyelin; SMase - sphingomyelinase. Adapted from [227].
32
Figure 1.2: Schematic of CD25 clearance strategy. Transduced cells will express both the therapeutic gene and CD25. If there are no unwanted proliferative events (no adverse events) then no intervention is required. In the event of oncogenesis, an anti-CD25 immunotoxin can be used to target transduced cells and eliminate them.

33
Figure 1.3: Schematic of vector systems. (A) Oncoretroviral gene transfer vector that uses the Moloney Murine Leukemia Virus as the backbone and encodes human AC, the internal ribosomal entry site (IRES) and the human CD25 marker gene. (B) The second generation lentiviral packaging system comprises three vectors: the HIV-1 derived gene transfer vector, the gag-pol construct (pCMV ∆R8.91) and the envelope construct (pMD.G) that encodes the Vesicular Stomatitis Virus glycoprotein (VSV-g).

34
Chapter 2: In vivo delivery of human acid ceramidase via cord blood transplantation and direct injection of lentivirus as novel approaches for the treatment of Farber disease A version of this chapter has been submitted for publication to the journal Human Gene Therapy. Ramsubir S et al. In vivo delivery of human acid ceramidase via cord blood transplantation and direct injection of lentivirus as novel approaches for the treatment of Farber disease (manuscript under review)

35
2.1 ABSTRACT
Farber disease is a rare lysosomal storage disorder (LSD) caused by a deficiency of
acid ceramidase (AC) activity and subsequent accumulation of ceramide. Currently, there is
no treatment for Farber disease beyond palliative care and most patients succumb to the
disorder at a very young age. Previously, our group showed that gene therapy using
oncoretroviral vectors (RV) has the potential to provide a lasting cure for the disease. The
studies described here used novel RV and lentiviral (LV) vectors that engineered co-
expression of AC and a cell surface marking transgene product, human CD25 (huCD25). It
was shown that transduction of Farber patient fibroblasts and B cells with these vectors
resulted in overexpression of AC and led to a 90% and 50% reduction in the accumulation of
ceramide, respectively. In a xenotransplantation model using NOD/SCID mice, it was shown
that human CD34+ cord blood cells transduced with an LV engineering expression of AC and
huCD25 were able to repopulate recipient animals. The effect of delivering LV expressing
AC and huCD25 directly to neonatal animals was also investigated. Up to 14 weeks post-
injection, soluble CD25 was detected in the plasma and increased AC activity was present in
the livers, suggesting the persistence of vector and long-term transgene expression in these
mice. To our knowledge, this is the first report of in vivo testing of direct therapies for Farber
disease.

36
2.2 INTRODUCTION
Farber disease is an autosomally inherited LSD caused by mutation of the gene
(ASAH1) encoding N-acylsphingosine deacylase (acid ceramidase, AC; EC 3.5.1.23), a
protein that catabolizes the hydrolysis of ceramide into sphingosine and free fatty acids [62,
89]. While the phenotype of the disease varies, most patients present with a characteristic
triad of symptoms: subcutaneous granulomas, a hoarse cry, and painful swollen joints [62].
In the classic and most severe type of Farber disease, patients also show progressive
neurological deterioration and patients typically die by the age of two [62].
While there is currently no cure for this disorder, allogeneic bone marrow
transplantation (BMT) has been attempted for some Farber patients. The rationale was that
the introduction of a population of cells with normal AC activity would ameliorate the
consequences of the enzymatic deficiency in these patients. While BMT in those cases did
resolve the granulomas and other peripheral symptoms, it did not relieve the neurological
manifestations that are seen in the majority of Farber patients. Furthermore, they still
succumbed to the disease [62, 66, 72, 74].
Farber disease is an attractive target for gene therapy since it is caused by a single
gene defect, and the cDNA of AC has been cloned [61]. In addition, the enzyme is fairly
well-characterized [80, 85, 86]. Previous gene therapy studies from the Medin laboratory
directed towards this disorder have shown that the enzymatic deficiency in immortalized
Farber patient cells could be corrected by transduction with an oncoretrovirus (RV) that
engineers overexpression of human AC. Importantly, that study showed that enzyme secreted

37 from transduced cells could be endocytosed by non-transduced cells through the mannose-6-
phosphate receptor pathway and restore enzyme activity [176]. This phenomenon, known as
‘metabolic cooperativity’ or ‘cross-correction’ is important for gene therapy applications as it
allows for a lower level of functionally transduced cells to have greater therapeutic effect.
There are a number of approaches that have been used to introduce therapeutic genes
in vivo. Viral methods using vectors such as retroviruses offer the advantage of long-term
gene transfer since the viral DNA can integrate into the host genome and can be transmitted
to progeny cells [149]. Our lab and others have shown that transduction of bone marrow-
derived cells with a viral vector engineered to overexpress a therapeutic transgene can
potentially provide a systemic, circulating source of enzyme [180, 228, 229]. In addition, it
has been shown that lentiviral vectors (LV) directly delivered to neonatal Fabry mice can
lead to sustained transgene expression in multiple organs, including the brain [168], an organ
that is of particular importance in Farber disease.
Here novel retroviral vectors were constructed to engineer expression of human AC
and a cell surface marker, human CD25, which can be used to enrich and track transduced
cells [218]. These studies demonstrated that AC expression mediated by transduction with
these viral vectors can restore enzyme activity in Farber patient cells. They also showed the
potential efficacy of using these vectors in vivo in both a cord blood transplantation model
and as a direct viral delivery agent to neonatal mice. The results of these studies demonstrate
that these approaches are promising and potentially curative treatments for Farber disease.

38
2.3 MATERIALS AND METHODS
Vector Construction. The oncoretroviral vector (RV) employed in this study was made by
subcloning the AC cDNA from pG1-ACER [176] into the cloning shuttle vector
pSV/IRES/huCD25 and subsequently subcloning the AC/IRES/huCD25 sequence into the
pUMFG backbone [228] as follows: mutagenesis was performed using the QuikChange®
Site-Directed Mutagenesis Kit (Stratagene, Cedar Creek, TX) to remove an Nco I site from
position 1634 of the pG1-ACER vector (primers: 5’- GGT GCA GTT CCC TGG TAC ACC
ATA AAT C - 3’; 5’- GAT TTA TGG TGT ACC AGG GAA CTG CAC C - 3’). Both pG1-
ACER and pSV/IRES/huCD25 were digested with Sal I and Nco I (New England Biolabs
(NEB), Ipswich, MA). The 1239 bp AC fragment was then ligated into the digested
pSV/IRES/huCD25 vector to yield the new vector pSV/AC/IRES/huCD25. This vector was
then digested with Nco I and Not I (NEB) to isolate the 2668 bp AC/IRES/huCD25 fragment
that was then ligated into the pUMFG backbone to produce the vector
pUMFG/AC/IRES/huCD25 (RV/AC/huCD25).
To construct the lentiviral vector (LV), the AC/IRES/huCD25 fragment from the
pSV/AC/IRES/huCD25 shuttle vector was ligated into the pHR’ lentiviral backbone [163].
The shuttle vector was mutated to introduce a Bam HI site at position 644 by site-directed
mutagenesis (Stratagene) (primers: 5’ - ACT CAC TAT AGG GAT CCG CCA TGG CGG
GC - 3’; 5’ - GCC CGC CAT GGC GGA TCC CTA TAG TGA GT - 3’). Next, a Bam HI
site was removed at position 1842 (5’ - AGG TTG GTG AGG GCG AAT CCC CCG GGC
TGC - 3’; 5’ - CGA GCC CGG GGG ATT CGC CCT CAC CAA CCT - 3’). The

39 pSV/AC/IRES/huCD25 shuttle vector was digested with Bam HI and Dra I (NEB) to isolate
the 2654 bp AC/IRES/huCD25 fragment. The lentiviral vector pHR’EF-GW-SIN
(LV/enGFP; provided by Robert Hawley, American Red Cross, Rockville, MD) was
prepared by digesting with Bam HI to remove the enhanced green florescent protein (enGFP)
cDNA. The AC/IRES/huCD25 fragment was then ligated into the pHR backbone to produce
the vector pHR’EF-AC-IRES-huCD25-W-SIN (LV/AC/huCD25).
Oncoretrovirus production. The pUMFG/AC/IRES/huCD25 vector was transfected into the
FLYRD18 packaging cell line [230] by calcium phosphate-mediated transfection using 10 µg
of the plasmid and 1 µg of the pGTN28 plasmid that carries the neomycin-resistance cDNA.
Stable transfectants were selected using 0.8 mg/ml G418 (Sigma, Oakville, ON, Canada).
These cells were stained with a phycoerythrin-conjugated anti-human CD25 antibody (BD
Bioscience Canada, Mississauga, ON, Canada) and then sorted by flow cytometry to derive
an enriched pool of producer cells and a single cell-derived clone. Viral titer was determined
by transduction of HeLa cells with serial dilutions of supernatant from producer cells,
followed by measurement of downstream huCD25 expression by flow cytometry. The clone
with the highest titer, clone #17, was then used to produce viral supernatant for all
experiments that employed the oncoretroviral vector RV/AC/huCD25, except where
otherwise indicated. The viral titer of supernatant collected from this clone was typically ~4
x 106 IU/ml.
Lentivirus production. Recombinant LV/AC/huCD25 virus was produced by transient
transfection of 293T cells with three plasmids: pMD.G (VSV-g envelope), pCMV ΔR8.91

40 (packaging) and either LV/AC/huCD25 or LV/enGFP. Transfections of 293T cells were
performed using calcium phosphate as previously described [168]. Viral supernatants were
concentrated 300-fold by centrifugation at 50,000 x g for 2 h at 4°C. Viral titer was
determined as previously described [168] and was typically in the range of 1-3 x 108 IP/ml
after concentration.
Viral transduction of Farber cells. Immortalized Farber patient skin fibroblasts and B cells
[176] were maintained in DMEM and RMPI 1640 (both Sigma), respectively, supplemented
with 10% fetal calf serum (PAA Laboratories, Rexdale, ON, Canada), 2 mM L-glutamine,
1% sodium pyruvate, and 1% penicillin-streptomycin (all Sigma). Using RV/AC/huCD25,
Farber fibroblasts were transduced three times with supernatant from an enriched pool of
producer cells (MOI of 20) while B cells were transduced once with supernatant from clone
#17 (MOI of 8). B cell transductions were performed in fibronectin-coated (5 µg/cm2; Roche,
Mississauga, ON, Canada) plates. For experiments employing the LV vector, Farber
fibroblasts were transduced once with unconcentrated virus at an MOI of ~1, while the B
cells were transduced once with concentrated virus at an MOI of 50. All transductions were
performed in the presence of 8 µg/ml protamine sulfate (Sigma). The transduced B cell pools
were then enriched by magnetic-activated cell sorting (MACS) using magnetic beads
conjugated to the anti-CD25 antibody and MS+ columns (both from Miltenyi Biotech,
Auburn, CA).
AC activity assays. AC activity was determined for transduced cells, non-transduced (NT)
Farber cells, and normal controls as previously described [231]. Briefly, cells were pulsed

41 with [3H-ceramide]-sphingomyelin and 48 h later, lipids were extracted and resolved by
analytical thin layer chromatography (TLC). The distribution of the radioactivity on the plate
was analyzed using a Berthold LB 2832 radiochromatoscan. Additionally, ceramide and
other lipid fractions were scraped off the plate for direct liquid scintillation counting. The
percentage of labeled sphingomyelin that was metabolized to ceramide was calculated from
the radioactivity of each fraction.
In mice treated by direct virus injection, AC activity was assessed using a modified,
fluorescence-based HPLC method. Organs were first homogenized in 0.25 M sucrose. Then,
equal volumes of organ lysate and substrate buffer (0.2 M citrate/phosphate (pH 4.5), 0.2
mM Bodipy-labeled C5-ceramide (Molecular Probes, Burlington, ON), 0.5% sodium
taurocholate (Sigma), 0.2% Igepal CA-630 (Sigma), 0.1% BSA, 0.3 M NaCl) were co-
incubated for 6 h at 37°C. Samples were then evaluated as previously described [232].
Ceramide quantitation. Intracellular ceramide levels were measured using E. coli
diacylglycerol (DAG) kinase and [γ32P]ATP as described [176]. Briefly, lipids were extracted
from cell lysates by the Folch method [233] and subjected to mild alkaline hydrolysis using
NaOH for 2 h at 37°C. The solution was then neutralized and lipids extracted using
chloroform/methanol (2:1, v/v). Lipids were then incubated with sn-l,2-diacylglycerol kinase
and [γ32P]ATP as described [234]. Lipids were resolved by TLC as above. Ceramide-[32P]-1-
phosphate was quantified by scraping the band from the TLC plate and analyzing the fraction
by liquid scintillation counting.

42 Metabolic cooperativity studies. Non-transduced Farber patient fibroblasts were cultured for
48 h in filtered media harvested from the following cell lines: non-transduced Farber
fibroblasts, RV- or LV-transduced Farber fibroblasts, and normal fibroblasts. Cells were then
pulsed with [3H-ceramide]-sphingomyelin for 24 h and the enzyme activity in each cell
population was determined as described above (see AC activity assay).
Infection of HSPCs. CD34+ cells were obtained from AllCells, LLC (Berkeley, CA). Cells
were pre-stimulated for 12 h in StemSpan SFEM medium (Stem Cell Technologies,
Vancouver, BC, Canada) supplemented with recombinant human SCF, Flt3-L,
thrombopoietin (all at 50 ng/ml), and IL-6 (20 ng/ml). All cytokines were obtained from
R&D Systems Inc. (Minneapolis, MN). Cells were then transduced with virus in the presence
of cytokines and 8 µg/ml protamine sulfate. Transductions were performed in 6-well plates
coated with RetroNectin CH296 (10 µg/cm2; Takara Shuzo, Otsu, Japan) in a total volume of
2 ml/well at a concentration of 0.5 x 106 cells/ml. For parallel comparison of infections on
different sources of HSPCs, cells were infected with LV/AC/huCD25 at an MOI of 40. The
cord blood-derived cells used for in vivo transplantation were infected with LV/AC/huCD25
or LV/enGFP at an MOI of 2 and 10 respectively. Cells were then washed twice in PBS and
either re-cultured in StemSpan supplemented with cytokines, plated into Methocult™ H4434
(Stem Cell Technologies), or re-suspended in PBS for transplantation.
Human cord blood transplantation in NOD/SCID mice.
Mice were obtained from Taconic (New York, NY) and were bred and maintained at
the UHN Animal Resource Centre. Animal experimentation protocols were approved by the

43 UHN Animal Care Committee. Mice were irradiated at 350 cGy and injected with 200 µg of
anti-CD122 antibody into the intraperitoneal cavity 24h prior to transplantation [235]. The
anti-CD122 monoclonal antibody was used to clear any residual natural killer cells in vivo
[236] and was purified from the TM-β1 hybridoma cell line (a gift from Dr T. Tanaka, Osaka
University Medical Center, Osaka, Japan), using the High Trap Protein G Column (GE
Healthcare). Transduced CD34+ cells were then injected into the animals via the tail vein
(1.65 x 106 and 2.4 x 106 cells/mouse for AC and enGFP groups respectively). Six weeks
later, peripheral blood (PB) was collected and the mice were sacrificed. Bone marrow (BM)
was flushed from the tibia and femur of both hind legs and splenocytes were harvested by
crushing the spleen through a nylon mesh. Engraftment of human cells was assessed by flow
cytometry of living nucleated cells from the PB, BM and spleen; dead cells were excluded by
staining with 7-amino-actinomycin D (Sigma). Cells were stained with antibodies against
human CD45 (BD Bioscience Canada) and functional transgene expression was assessed by
flow cytometry for either huCD25 or enGFP.
Assessment of vector positive cells. Cells were taken from methylcellulose colonies by
aspiration using a micropipettor. Cells were lysed in 10 µl of lysis buffer (50 mM KCl, 10
mM Tris-HCl (pH 8.3), 1.5 mM MgCl2, 0.1% gelatin, 0.45% Tween-20, 0.45% Nonidet-P40,
125 µg/mL proteinase K) and incubated at 55 °C for 2-16 h. 1 µl of lysate was then used for
PCR using the primers specific for the viral vector (ACnest-FP: 5' - gaaacttacctgcgggactg - 3'
and ACnest-RP: 5' - acaccggccttattccaag - 3') or for the human GAPDH gene (huGAPDH-
FP: 5' - accgtcaaggctgagaaacgg - 3' and huGAPDH-RP: 5' - acgtactcagcgccagcatc - 3').
Cycling conditions were as follows: 94 °C for 45 s, 63°C for 30 s, 72 °C for 15 s.

44
Delivery of LV to neonatal mice. One- to two-day-old C57BL/6 mice (The Jackson
Laboratories, Bar Harbor, MI) were injected via the superficial temporal vein with either
LV/enGFP (6.65 x 107 IP/5,000 pg p24/mouse) or LV/AC/huCD25 (4.75 x 10
7 IP/10,000 pg
p24/mouse) in a volume of 100 µl of sterile PBS. Mice were analyzed at weeks 7, 10 and 14
for the expression of soluble human CD25 (sCD25) in the plasma. Plasma was isolated from
mouse PB samples by centrifugation at 16,000 x g for 20 min. The level of sCD25 was
measured by a direct ELISA using the BD OptEIA™ Human IL-2 sRα ELISA Set (BD
Bioscience Canada) as per the manufacturer’s instructions. Each sample was measured in
triplicate. Mice were sacrificed at 14 weeks post-virus injection, organs were harvested and
AC activity was measured as above.

45
2.4 RESULTS
RV- and LV-transduced Farber patient cells express human CD25 and have restored AC
activity.
We first tested the efficacy of the novel recombinant viral vectors to transduce
immortalized Farber patient fibroblasts and B cells. Cells from a single patient were
transduced. No huCD25 expression could be detected on non-transduced (NT) Farber patient
fibroblasts, however, cells transduced three times with the recombinant RV and once with
LV were ~100% and 87% positive for huCD25, respectively (Fig. 2.1a) and stably expressed
the marker over time. Non-transduced B cells showed low levels of huCD25 expression
(2.5%) but once transduced with RV/AC/huCD25, approximately 26% of the cells were
positive for huCD25 (data not shown). Sorting of this pool enabled enrichment of the
huCD25-expressing population to ~88% (Fig. 2.1b). Similar results were obtained when cells
were transduced with LV/AC/huCD25 (Fig. 2.1b).
In order to determine if functional AC is expressed after transduction, ceramide
degradation was then measured. Cells were pulsed with [3H-ceramide]-sphingomyelin and 48
h later, lipids were isolated from the cells and analyzed by TLC. In non-transduced Farber
fibroblasts, ceramide comprised ~84% of the sphingomyelin metabolites; normal fibroblasts
contain 26%. Those values reflect previous data [64, 176, 237]. Transduction with the
recombinant RV and LV reduced the ceramide fraction to 11% and 16%, respectively (Fig.
2.2a). In previous studies, mock transduction of these cells did not result in any changes in
the ceramide levels [176]. Similar results were seen in the Farber B cells; ceramide levels

46 were reduced from 76% to 16% and 33% in RV/AC/huCD25- and LV/AC/huCD25-
transduced pools, respectively (Fig. 2.2b).
In another assay, intracellular ceramide levels were also measured in transduced and
control cells using E. coli diacylglycerol (DAG) kinase and [γ32P]ATP. DAG phosphorylates
ceramide to produce ceramide-1-phosphate. In the presence of [γ32P]ATP, the ceramide-1-
phosphate is radiolabeled and can be quantified by liquid scintillation counting. As seen in
Figure 2.3, while non-transduced Farber fibroblasts store large amounts of ceramide, there
was ~90% reduction in ceramide storage in RV- and LV-transduced fibroblasts. Transduction
of Farber B cells resulted in a 50% reduction of ceramide storage (Fig. 2.3). These results
collectively demonstrate that transduction with the recombinant retroviral vectors results in
restoration of the AC activity in Farber patient cells.
Metabolic cooperativity occurs between AC-transduced and NT Farber patient fibroblasts
We next tested the ability of AC activity derived from therapeutically transduced cells
to be taken up by non-transduced cells. This phenomenon is an important concept in gene
therapy, especially as directed towards LSDs since it enables a smaller number of transduced
cells to have wide therapeutic effect through metabolic co-operativity. Non-transduced
Farber fibroblasts were cultured for 48 h in conditioned media harvested from the following
cell lines: non-transduced Farber fibroblasts, Farber fibroblasts transduced with RV and LV,
and normal fibroblasts. Cells were then pulsed with [3H-ceramide]-sphingomyelin and the
ceramide content in each cell population was determined. While media from normal
fibroblasts reduced ceramide levels from 80% to 62%, incubation with media from the RV
and LV-transduced cells reduced ceramide content to approximately 20% of sphingomyelin

47 metabolites (Fig. 2.4), a level that is comparable to that observed in normal fibroblasts (see
Fig. 2.2a).
Transplantation of LV/AC/huCD25-transduced CD34+ cells can re-populate recipient
animals
We next evaluated LV/AC/huCD25 in vivo in a model representative of a BMT in a
Farber patient. For this, the immuno-deficient NOD/SCID mouse [238] was used so that the
effect of transduction and AC-overexpression in human hematopoietic cells, as would be
used in a patient, could be evaluated. CD34+ cells derived from human umbilical cord blood,
BM and mobilized peripheral blood (mPB) were transduced with LV/AC/huCD25 (MOI
~40) since these are all potential sources of HSPCs that can be used for transplantation.
Following transduction, CD25 expression was assessed by flow cytometry and it was found
that all cells were transduced with similar efficiency (Fig. 2.5). The ability of these cells to
generate hematopoietic colonies was tested by seeding the cells in methylcellulose. It was
found that all populations generated colonies. When granulocyte/macrophage colonies were
assessed by PCR to determine the presence of vector, it was found that vector-positive
colonies were more abundant in the cord blood-derived population (14/46; 30%) compared to
the cells derived from the bone marrow (5/70; 7%) and mPB (0/52; 0%). This is likely due to
the higher percentage of more primitive HSPCs found in cord blood [183, 184]. As a result,
CD34+ cells derived from human umbilical cord blood were chosen for in vivo transplants.
Cord-blood derived CD34+ cells were transduced with either LV/AC/huCD25 or
LV/enGFP and transplanted into sub-lethally irradiated NOD/SCID recipient mice. Six
weeks post-transplantation, BM, splenocytes, and PB were harvested. Human cell

48 engraftment was assessed by flow cytometry to detect the human panhematopoietic marker
CD45. It was found that, on average, human chimerism in the BM was 65% and 49% for the
LV/AC/huCD25 and LV/enGFP-transduced groups, respectively (Table 2). Further, in mice
transplanted with LV/AC/huCD25, it was found that ~0.41% of the human cells expressed
the downstream marking transgene huCD25. Expression of this surrogate marker has been
shown to be correlated with functional transgene expression [168, 229]. It is also important to
note that this marker allows an evaluation of transgene expression even in enzymatically
normal cells in which changes above background AC activity may be hard to detect. In
addition, colony-forming assays of BM harvested from these mice showed normal
proportions of each lineage of hematopoietic cells (data not shown). Therefore, it appears
that CD34+ cells transduced with an LV that engineers overexpression of AC are able to
reconstitute an irradiated recipient and can give rise to all lineages of hematopoietic cells,
suggesting that the repopulation ability of these cells is not impaired.
Neonatal LV delivery engineers persistent expression of downstream marking transgene
Treatment of neonatal recipients offers several advantages that include exploiting an
incompletely formed blood-brain barrier and an immature immune system, as well as the
possibility of administering treatment before irreversible organ and neurological damage has
occurred as a result of the disease. In proof-of-principle experiments, one- to three-day-old
C57BL/6 mice were treated with either LV/AC/huCD25 or LV/enGFP. No adverse effects
were seen in treated animals and mice developed normally. PB was harvested at weeks 7, 10
and 14 post-viral delivery and plasma levels of sCD25 were measured by ELISA. As shown
in Fig. 2.6a, at 7 weeks post-treatment all mice treated with LV/AC/huCD25 showed high

49 levels of sCD25 in the plasma and three of six mice showed persistent levels of sCD25 up to
14 weeks post-viral delivery. As expected, untreated mice and animals treated with
LV/enGFP showed no detectable levels of sCD25.
In addition, it was found that the livers of mice treated with LV/AC/huCD25 showed
increased AC activity over both non-treated mice and mice treated with LV/enGFP (Fig.
2.6b). AC activity in other organs (such as the lung, brain, kidney, spleen and heart) was
assessed but significantly increased AC activity over normal background levels was not
observed (data not shown). This proof-of-principle experiment shows the potential utility of
treating Farber disease shortly after birth, a time that may be critical to preventing
irreversible organ and neurological damage.

50
2.5 DISCUSSION
Current treatment for Farber disease consists mainly of symptomatic supportive care
since enzyme replacement therapy is not available as it is for some other LSDs [10]. While
BMT can relieve some of the symptoms of the disease, it is only available to patients with
matched donors and it does not relieve the progressive neurological deterioration that is seen
in the majority of patients affected with this disease [62, 66, 72]. Therefore, the prognosis for
Farber patients remains poor and the development of treatments remains important.
Virus-mediated gene therapy offers the potential for a one time curative treatment
since integrating vectors such as retroviruses have the ability to persist long-term in
transduced cells and their progeny. It has previously been shown in vitro that transduction of
Farber patient cells with a RV engineering expression of AC could restore enzymatic activity
[176]. Here similar in vitro results are shown using novel recombinant RV and LV and those
results are expanded to the testing of the viral vectors in vivo. As a complete knock-out for
the AC gene is embryonic lethal [77], there is currently no mouse model of Farber disease.
Therefore surrogate models for in vivo testing of gene therapy strategies for the treatment of
Farber disease were developed. To our knowledge, this is the first report of such studies for
treatment strategies for Farber disease.
The recombinant RV used in these experiments is based on a viral backbone that has
been shown to result in higher transgene expression [239] than the pG1-ACER vector used
previously [176]. The virus is also pseudotyped with a more clinically relevant envelope that
is not inactivated by human sera [230]. Although they are not widely used in humans as RVs

51 are, a recombinant LV was developed because of its ability to transduce more quiescent cell
populations. Also, the broad tropism offered by the VSV-g envelope make our LV a useful
vehicle to target cells such as hematopoietic stem/progenitor cells (HSPCs) and certain neural
cells [240]. Both vectors included the huCD25 cell surface marker that has been used in
previous studies by the Medin laboratory to enrich for and track transduced cells [168, 218].
To confirm that the viral vectors constructed could transduce cells and produce
functional enzyme, Farber patient B cells and fibroblasts were transduced. High levels of
huCD25 expression from transduced cells could be detected and the huCD25 marker was
used to enrich the pool of transduced B cells. Measurement of AC activity showed that
transduced cells had significantly increased enzyme activity and decreased ceramide storage
following transduction. It was also shown that functional enzyme was secreted and could be
taken up and utilized by non-transduced cells. Previous studies have shown that this uptake is
mediated by the mannose-6-phosphate receptor and can be blocked by co-incubation with
mannose-6-phosphate [241]. These studies are preliminary since the data shown here are
from infection of cells obtained from only one Farber patient. Follow-up studies will further
confirm the in vitro efficacy by infection of cells from multiple Farber patients to compare
the effect of transduction. In addition, Farber fibroblasts will be infected at lower MOIs to
determine the minimum level of transduction required to see a therapeutic effect. Further
experiments that demonstrate metabolic cooperativity will also be performed.
The transduction of HSPCs is an attractive option for treating a number of LSDs since
these cells can provide a circulating source of the therapeutic factor. Moreover, HSPCs self-
renew and cells of the hematopoietic lineage also reside in key organs such as the brain and
liver [36, 195]. In order to test the effect of transduction of human cells on engraftment, the

52 NOD/SCID xenotransplantation model [242] was used since it is routinely used to assess
human cell engraftment. As mentioned, CD34+ cells derived from human umbilical cord
blood was used since it contains higher proportions of CD34+ cells and it is thought that the
HSPCs are more primitive than those derived from the bone marrow [183, 184]. In addition,
when using cord blood for transplants, a greater degree of HLA mismatch can be tolerated as
compared to BMT and there is a lower risk of causing graft-versus-host disease [185]. These
cells are a prime target for treatment of patients with Farber disease. CD34+ cells were
transduced with LV/AC/huCD25 and transplanted into irradiated NOD/SCID mice. High
levels of human cell engraftment were achieved as assessed by measurement of CD45
expression and it was found that, on average, 0.41% of engrafted cells expressed the huCD25
marking gene in the bone marrow. This low level of transgene expression is most likely due
to the fact that huCD25 is the downstream gene and expression driven by the IRES element
is not as efficient as expression from the viral promoter itself - as seen in the mice
transplanted with LV/enGFP.
The effect of delivering an LV expressing AC and huCD25 directly to neonatal
animals was also investigated. At the neonatal stage, the blood-brain barrier is not fully
formed and the immune system is not fully developed [243]. These physiological properties
may increase the efficacy of treatment administered at this stage, since delivery to the brain
would be increased and tolerance to the transgene can be induced. In addition, when
treatment is administered before symptoms appear it may prevent irreversible neurological
damage from occurring. In this study, normal neonatal mice were injected with
LV/AC/huCD25 and it was found that up to 14 weeks post-injection, sCD25 was detected in
the plasma, suggesting the persistence of vector and long-term transgene expression. Non-

53 treated mice did not have any detectable levels of sCD25 (data not shown). Therapeutic
transgene expression was further evidenced by the increased AC activity found in the livers
of mice treated with LV/AC/huCD25 as compared to wild-type mice. Increased AC activity
was not seen in other organs and may be due to limits in the sensitivity of the assay for
detecting small increases over the high background of enzyme activity found in normal mice.
The results of these studies suggest that the use of viral vectors that overexpress AC
has the potential to provide a curative treatment for Farber disease. These therapeutic vectors
can be delivered in a number of ways. The use of transduced HSPCs can provide a
circulating source of enzyme and the progeny can differentiate into cells that reside
throughout the body, such as microglia in the brain [186]. Neonatal delivery of virus via the
temporal vein also been shown to result in transduction of numerous cell types. It is
hypothesized that this treatment strategy may result in better transduction in the brain to
ameliorate the neurological effects of the disease. Further, improved delivery to the brain
could possibly be achieved by using vascular endothelial growth factor to further
permeabilize the blood brain barrier as previously reported [191].
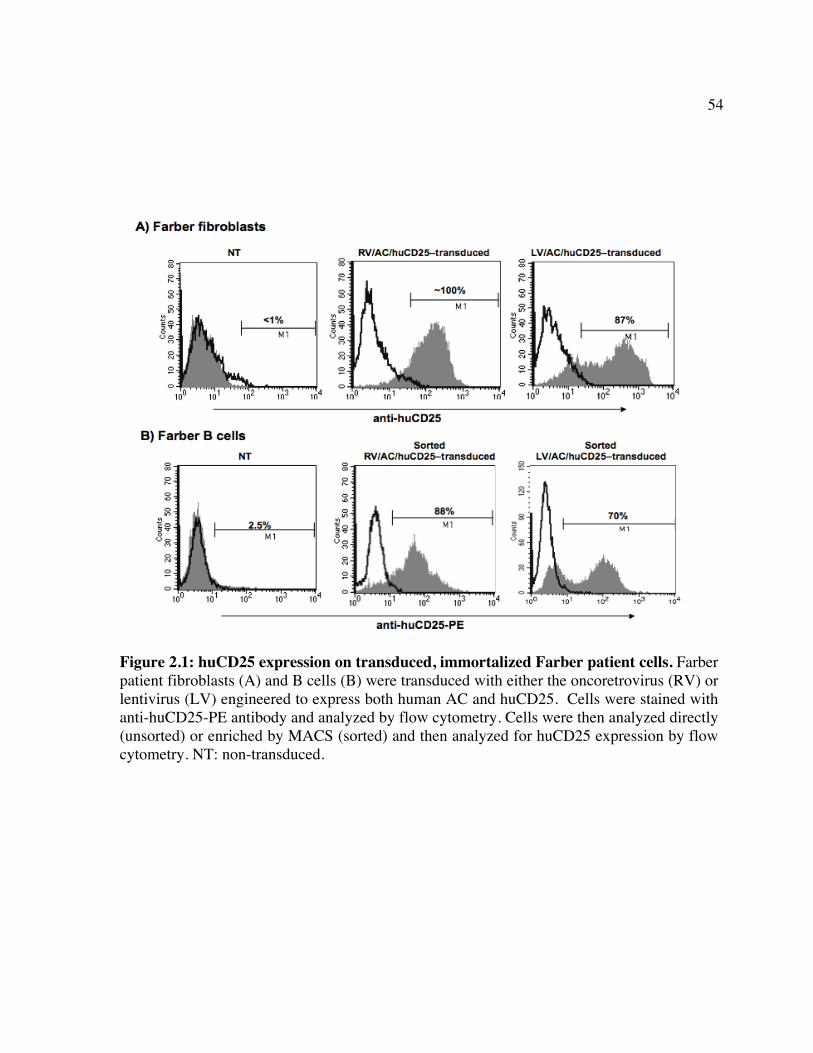
54
Figure 2.1: huCD25 expression on transduced, immortalized Farber patient cells. Farber patient fibroblasts (A) and B cells (B) were transduced with either the oncoretrovirus (RV) or lentivirus (LV) engineered to express both human AC and huCD25. Cells were stained with anti-huCD25-PE antibody and analyzed by flow cytometry. Cells were then analyzed directly (unsorted) or enriched by MACS (sorted) and then analyzed for huCD25 expression by flow cytometry. NT: non-transduced.
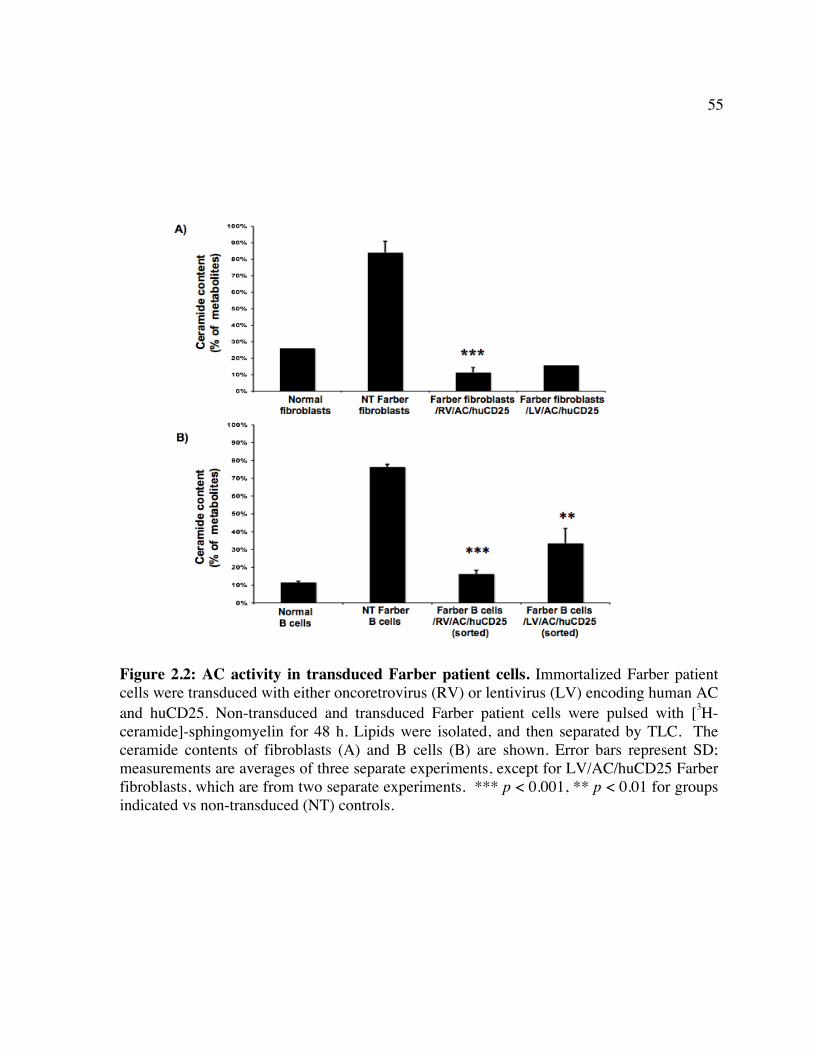
55
Figure 2.2: AC activity in transduced Farber patient cells. Immortalized Farber patient cells were transduced with either oncoretrovirus (RV) or lentivirus (LV) encoding human AC and huCD25. Non-transduced and transduced Farber patient cells were pulsed with [3H-ceramide]-sphingomyelin for 48 h. Lipids were isolated, and then separated by TLC. The ceramide contents of fibroblasts (A) and B cells (B) are shown. Error bars represent SD; measurements are averages of three separate experiments, except for LV/AC/huCD25 Farber fibroblasts, which are from two separate experiments. *** p < 0.001, ** p < 0.01 for groups indicated vs non-transduced (NT) controls.
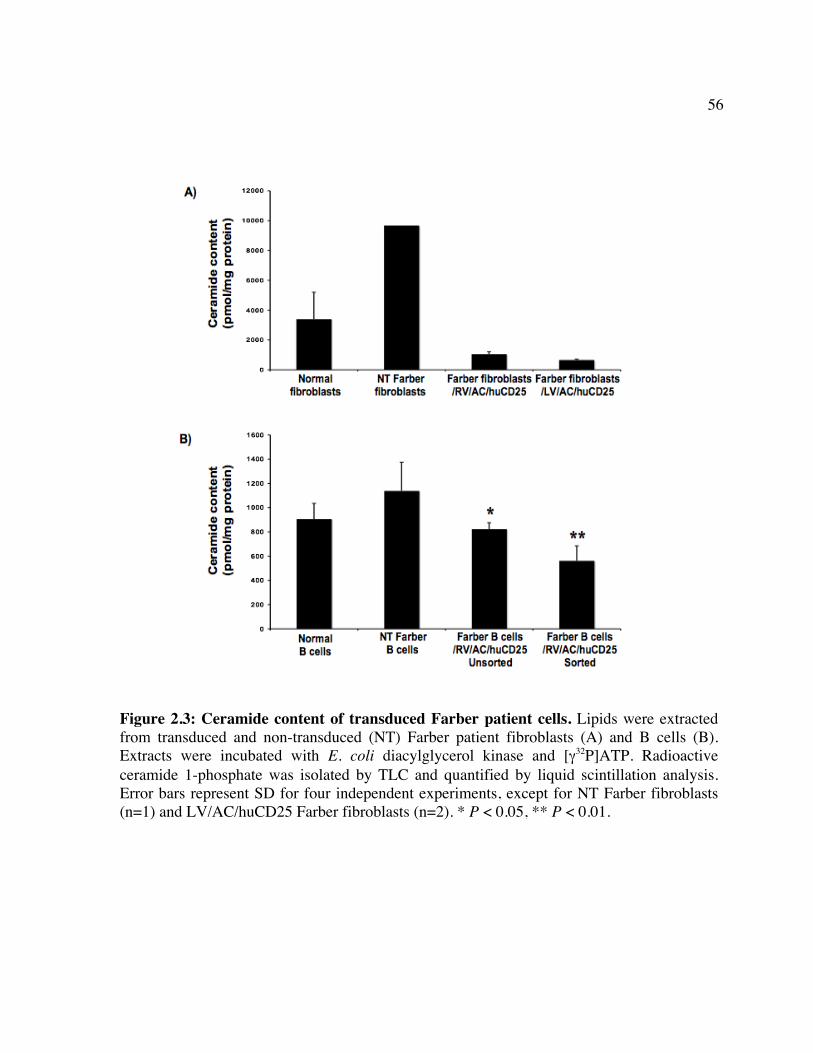
56
Figure 2.3: Ceramide content of transduced Farber patient cells. Lipids were extracted from transduced and non-transduced (NT) Farber patient fibroblasts (A) and B cells (B). Extracts were incubated with E. coli diacylglycerol kinase and [γ32P]ATP. Radioactive ceramide 1-phosphate was isolated by TLC and quantified by liquid scintillation analysis. Error bars represent SD for four independent experiments, except for NT Farber fibroblasts (n=1) and LV/AC/huCD25 Farber fibroblasts (n=2). * P < 0.05, ** P < 0.01.
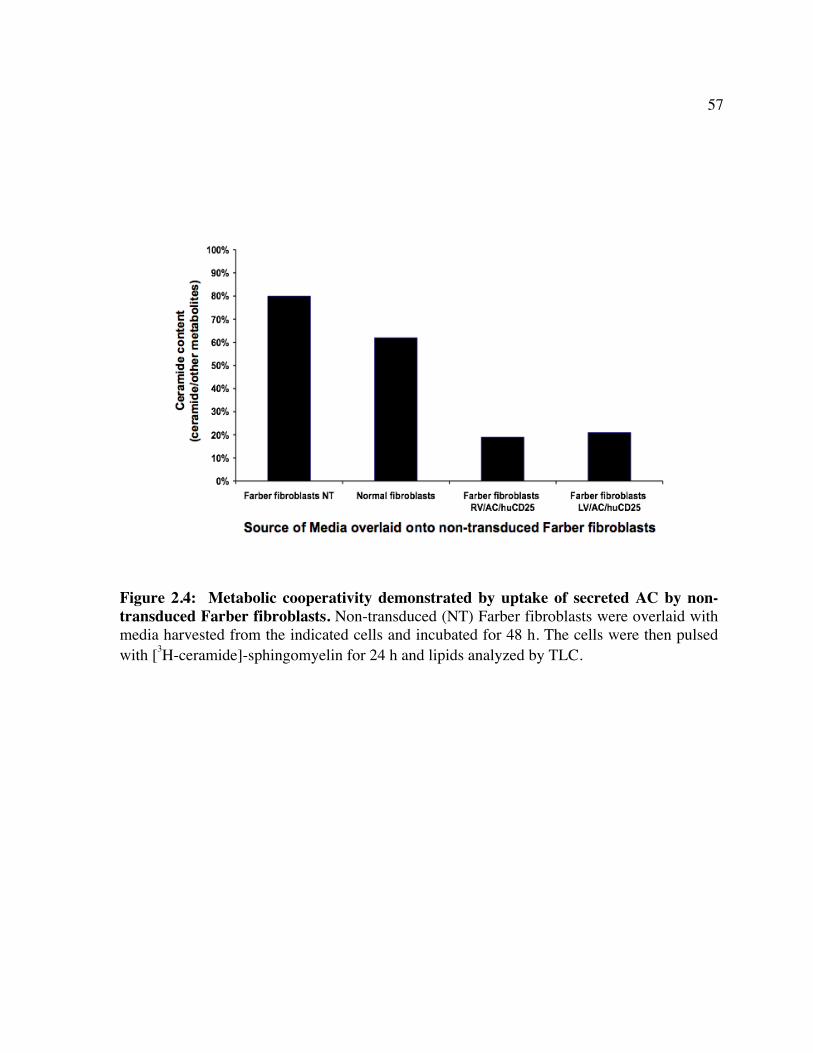
57
Figure 2.4: Metabolic cooperativity demonstrated by uptake of secreted AC by non-transduced Farber fibroblasts. Non-transduced (NT) Farber fibroblasts were overlaid with media harvested from the indicated cells and incubated for 48 h. The cells were then pulsed with [3H-ceramide]-sphingomyelin for 24 h and lipids analyzed by TLC.
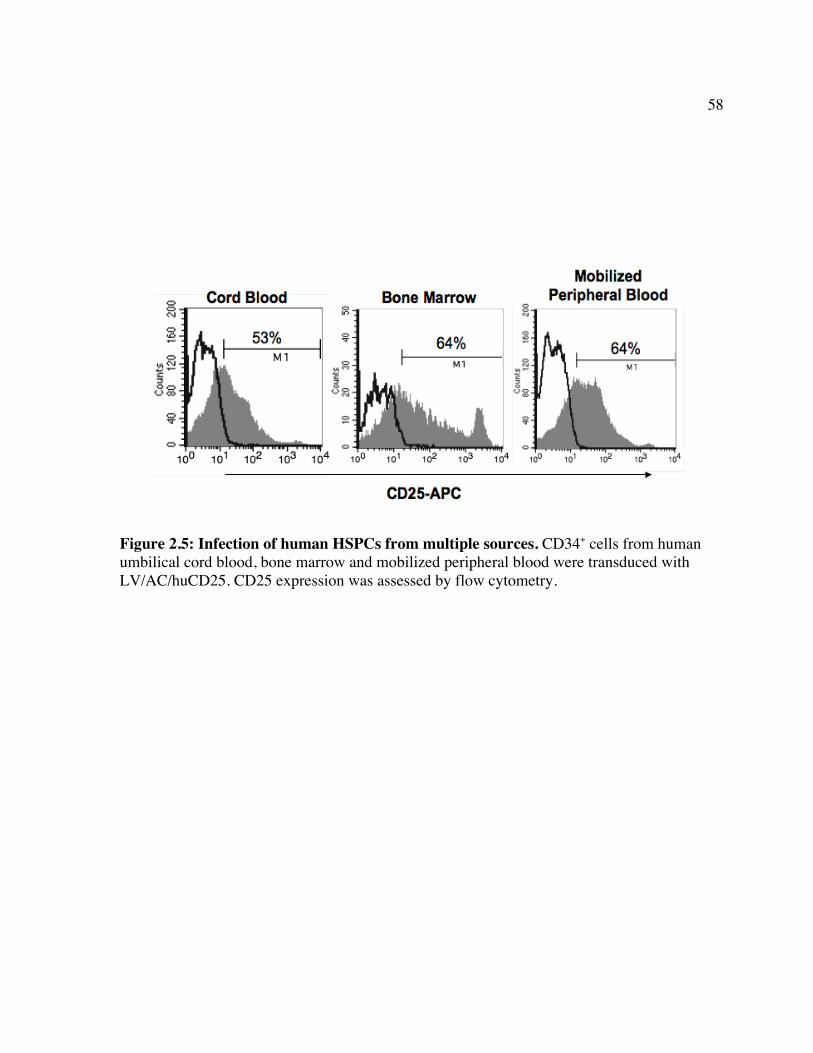
58
Figure 2.5: Infection of human HSPCs from multiple sources. CD34+ cells from human umbilical cord blood, bone marrow and mobilized peripheral blood were transduced with LV/AC/huCD25. CD25 expression was assessed by flow cytometry.
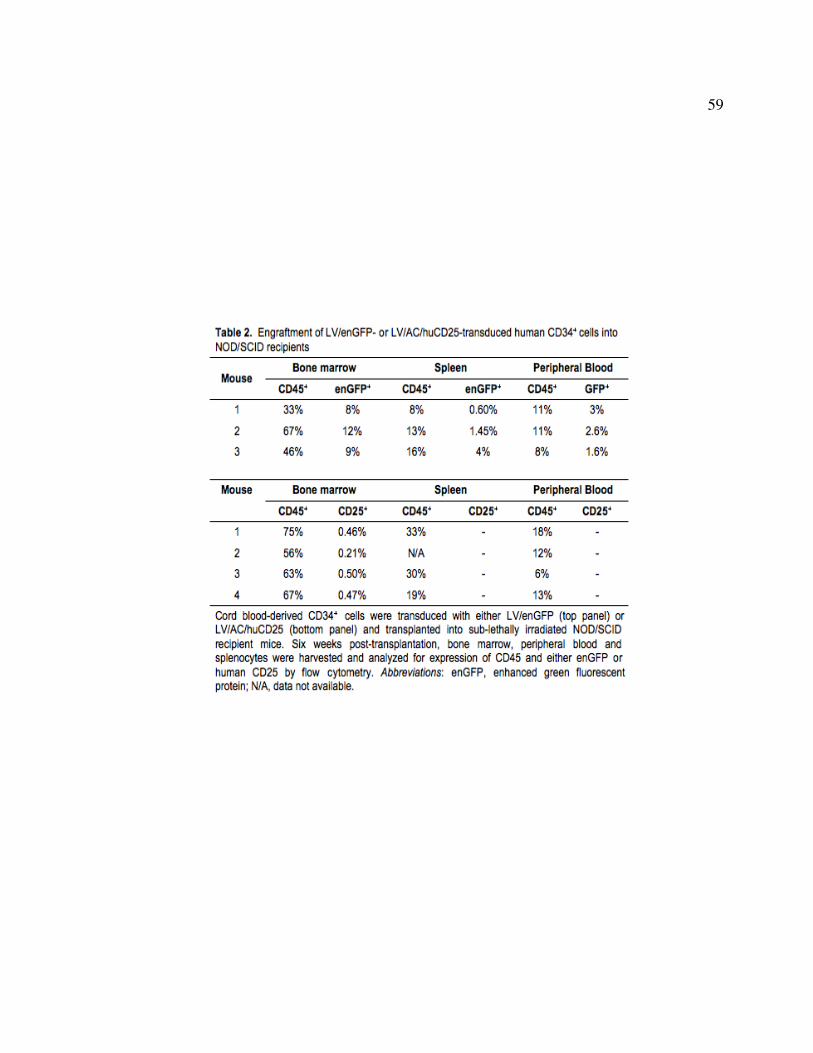
59
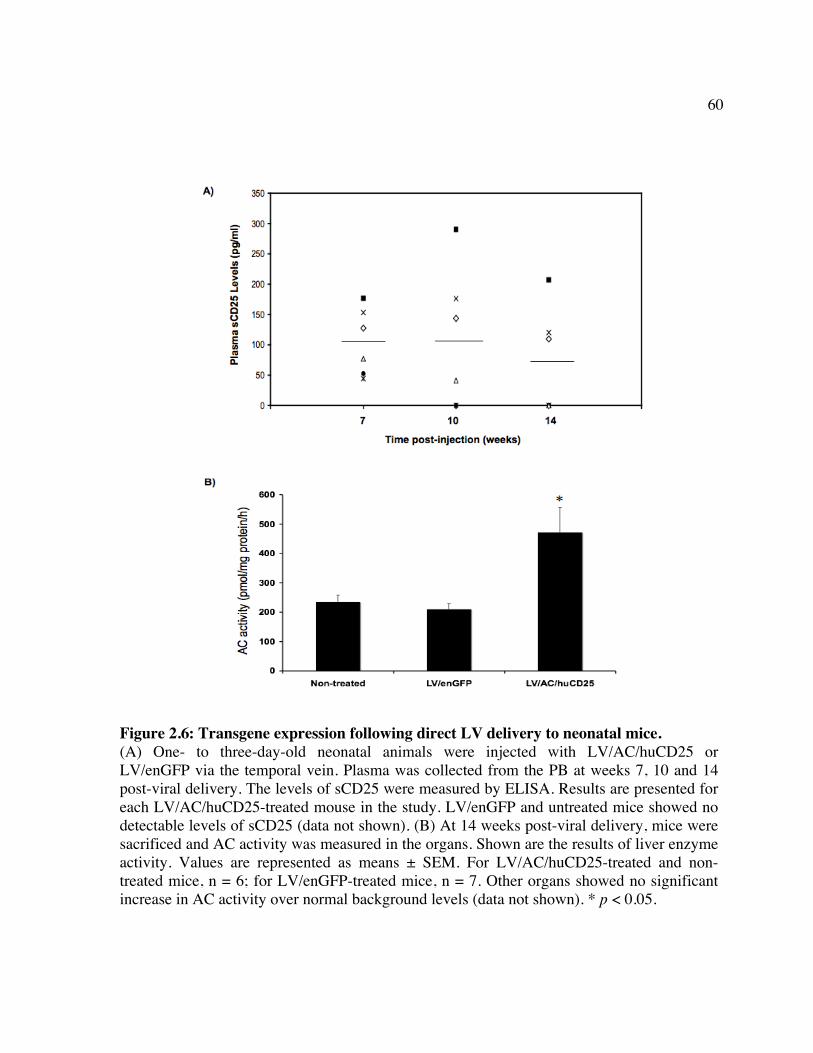
60
Figure 2.6: Transgene expression following direct LV delivery to neonatal mice. (A) One- to three-day-old neonatal animals were injected with LV/AC/huCD25 or LV/enGFP via the temporal vein. Plasma was collected from the PB at weeks 7, 10 and 14 post-viral delivery. The levels of sCD25 were measured by ELISA. Results are presented for each LV/AC/huCD25-treated mouse in the study. LV/enGFP and untreated mice showed no detectable levels of sCD25 (data not shown). (B) At 14 weeks post-viral delivery, mice were sacrificed and AC activity was measured in the organs. Shown are the results of liver enzyme activity. Values are represented as means ± SEM. For LV/AC/huCD25-treated and non-treated mice, n = 6; for LV/enGFP-treated mice, n = 7. Other organs showed no significant increase in AC activity over normal background levels (data not shown). * p < 0.05.

61
Chapter 3: Administration of VEGF prior to lentivirus delivery increases transduction of multiple organs in mice treated as neonates

62
3.1 ABSTRACT
Current treatment of Farber disease by allogeneic BMT has not been successful in
resolving the neurological manifestations of this disease. Studies of other LSDs have shown
that transplantation of genetically-modified hematopoietic stem/progenitor cells (HSPCs)
alone may not result in complete alleviation of neurological symptoms. The delivery of
recombinant enzyme is not a clinical option for Farber disease at this time since enzyme
replacement therapy has yet to be developed for this LSD. The lack of viable treatment
options for addressing the neurological manifestations of Farber disease makes the
development of a minimally invasive means of getting enzyme and other therapeutic particles
into the brain and across the blood-brain barrier (BBB) both timely and important. Here the
effect of pre-treatment with vascular endothelial growth factor (VEGF) on the ability of
lentivirus (LV) engineering expression of firefly luciferase (luc) to cross the BBB of neonatal
mice was investigated. LV/luc was delivered to VEGF-treated neonatal mice via the temporal
vein. Whole-body luminescence imaging (WBLI) of luc expression showed that VEGF pre-
treatment does not diminish transgene expression since it remained steady for up to 12
weeks. Ex vivo imaging of the organs showed that VEGF pre-treatment resulted in
significantly increased luc expression not only in the brain, but also in the heart, lung and
kidney. This study shows that VEGF may have therapeutic importance not only for delivery
of virus to the brain, but also to other organs of interest.

63
3.2 INTRODUCTION
Many LSDs affect the central nervous system, including metachromatic
leukodystrophy [244], mucopolysaccharidoses [245, 246] and Sandhoff disease [247].
Therapies for LSDs are mainly aimed at treating the visceral symptoms but in many cases, as
in Farber disease, patients succumb to the neurological manifestations of the disease, even
when intervention by BMT is attempted [66, 75]. A number of methods have been employed
to address the CNS manifestations of LSDs including the injection of cells [248], enzymes
[249] or therapeutic viral vectors [250] into the brain. Most often, these techniques are
invasive and pathology is only corrected in the vicinity of the injection site [250-252].
Therefore, the development of a means to achieve more widespread delivery of therapeutic
particles throughout the brain would be beneficial.
Vascular endothelial growth factor (VEGF) plays a role in both vasculogenesis,
angiogenesis and even lymphangiogenesis [253, 254]. It initiates signaling cascades by
binding to tyrosine kinase receptors on the cell surface. It acts primarily on endothelial cells
but also on hematopoietic cells [255], kidney epithelium [256] and neural cells [257]. VEGF
also has the ability to induce vascular permeability [253] and it has been shown that VEGF
can induce permeability of the blood brain barrier (BBB) [258]. It appears that VEGF
enhances the activity of an organelle called the vesicular-vacuolar organelle (VVO) that is
found intermittently throughout the endothelial cells (ECs) lining small blood vessels [259].
These organelles are clusters of vesicles and vacuoles that are interconnected with each other

64
and the plasma membrane of the ECs by means of fenestrae that open and close to
allow/prevent the flow of macromolecules through the vesicles and into the tissue [260].
Studies in the mouse model of globoid cell leukodystrophy have shown that in
neonates, administration of VEFG prior to delivery of transduced bone marrow cells or
lentivirus (LV) expressing β-glucuronidase (GUS-B) led to increased numbers of GUS-B-
expressing cells in the brain [191, 261]. In studies where LV was injected, examination of the
brain showed LV-transduced cells present in all areas of the brain and also found that
neurons, glial and endothelial cells were all transduced [191]. Neonatal gene transfer offers
the advantages of administering therapeutic vector before permanent organ and neurological
damage has occurred. It also offers the potential to tolerize patients to the therapeutic protein
expressed from the vector since the immune system of neonates is still relatively immature
[243]. Thus, since neonatal gene transfer combined with VEGF treatment has the potential to
treat both systemic and neurological manifestation early in life, it is a promising therapeutic
option for Farber disease.
In the present study, a recombinant LV expressing luc was used to track LV-derived
gene expression. LV was injected into the temporal vein of neonatal mice, with or without
VEGF, and luc expression was monitored over 12 weeks. Ex vivo imaging of the organs
showed that VEGF pretreatment increased expression of luc in the brain, lung, kidney and
heart compared to the mice that received LV alone. These studies are the first to show the
systemic effect of VEGF pretreatment and provide encouraging evidence that this therapy
can be of therapeutic benefit for the treatment of Farber disease.

65
3.3 MATERIALS AND METHODS
LV production and determination of titer. The lentiviral vector pHR’cppt-EF-luciferase
(LV/luc) has previously been described [168]. VSV-g-pseudotyped LV was produced by co-
transfection of 293T cells with LV/luc and the accessory plasmids pMD.G and
pCMVΔR8.91, using the polyethyleneimine-transfection procedure [262-264]. Cell culture
medium was changed 16 h post-transfection. Viral supernatants were harvested after 48 h and
concentrated by ultracentrifugation at 50,000 x g for 2 h. The concentrated virus was
suspended in sterile phosphate buffered saline (PBS) and stored at -80°C until use. The level
of p24 antigen in the LV/luc virus preparation was determined using an HIV-1 p24 ELISA
kit (PerkinElmer Canada Inc., Vaudreuil-Dorian, QC) and was found to be 3,100 ng p24/ml.
Animal procedures. The animal experimentation procedures described here were performed
under protocols approved by the University Health Network (UHN) Animal Care Committee.
Balb/c mice were maintained at the animal facility of the UHN. Two hours prior to virus
injection, 1.7 ng of recombinant mouse VEGF164 (R&D Systems, Minneapolis, MN) was
administrated to one to three-day-old neonatal mice through the superficial temporal vein in a
volume of 100 µl. Control mice received 100 µl of PBS. Concentrated LV (300 ng p24 in
100 µl PBS) was then injected via the superficial temporal vein.
In vivo and ex vivo bioluminescent imaging. In vivo bioluminescent imaging (BLI) was
performed at the Advanced Optical Microscopy Facility (AOMF) at the UHN with an IVIS

66
Imaging System (Xenogen, Alameda, CA) comprised of a CCD camera mounted in a light-
tight camera box. Image and measurements of bioluminescent signals were acquired and
analyzed using Living Image software (Xenogen). For whole body luminescence imaging,
mice were anesthetized, administered D-luciferin (Molecular Imaging Products, Ann Arbor,
MI) at 100 mg/kg in PBS by i.p. injection and then imaged 10 min later. For ex vivo organ
imaging, 2 min after receiving D-luciferin, mice were sacrificed and organs were collected
and washed with PBS. Images were immediately acquired (5 min exposure time). Following
imaging, the organs were cut in half. One half of each organ was immersed in optimal cutting
temperature (OCT) compound (Pelco International, Redding, CA). The other half was
transferred into a microcentrifuge tube, frozen on dry ice, and then stored at -80°C until use.
Measurement of organ luciferase activity. Sections of each organ were minced and
homogenized using a microfuge pestle in 1X Cell Culture Lysis Reagent (Promega Corp.,
Madison, WI). Lysates were then spun at 12,000 x g for 5 min at 4 °C. The supernatants were
transferred to a microcentrifuge tube and luciferase activity was measured using the
Luciferase Assay System from Promega, as per manufacturer’s instructions. Protein
concentrations were measured using the Bio-Rad DC Protein Assay (Bio-Rad Laboratories,
Mississagua, ON) as per manufacturer’s instructions.
Immunohistochemistry. Following ex vivo imaging, sections of each organ were
cryopreserved in OCT compound and stored at -80 °C. The specimens were cryo-sectioned to
a 5 µm thickness. The sections were mounted on glass slides, air dried for 1 hour at room
temperature, washed with PBS containing 0.02 M sodium phosphate and 0.15 M NaCl, and

67
then post-fixed in 4% buffered formalin in 0.1 M sodium phosphate buffer, pH 7.4. Slides
were washed with PBS and then incubated in PBS containing 0.1% (V/V) Triton X-100 for
15 minutes, the samples were treated with 5% (V/V) normal donkey serum in PBS for 30
minutes. The sections were sequentially reacted with primary antibody solution (1:100
dilution in PBS) at 4°C overnight, followed by incubation in PBS-containing secondary
antibody (1:500 dilution in PBS) labeled with either Alexa488 or Alexa546 for 3 hours at
room temperature. Antibodies used in this study were as follows: goat anti-luciferase
antibody (Chemicon International Inc., Temecula, CA), rat monoclonal anti-mouse CD31
antibody (BD Pharmingen, San Diego, CA), rabbit anti-GATA4 antibody (Santa Cruz
Biotechnology, Inc., Santa Cruz, CA), rabbit anti-doublecortin (Abcam, Cambridge, MA),
rabbit anti-glial fibrillary acidic protein (GFAP) (Lab Vision, Fremont, CA), Alexa488-
labeled anti-rabbit or anti-rat IgG antibody (Molecular Probes, Inc., Eugene, OR), and
Alexa546-labeled anti-goat IgG antibody (Molecular Probes). Fluorescence signals were
analyzed using a confocal laser-scanning microscope LSM-5 and LSM System version 3.98
(Carl Zeiss, Oberkochen, Germany).

68
3.4 RESULTS
Whole body luminescence imaging (WBLI).
To determine the effect of VEGF administration on the transduction pattern of LV in
vivo, one- to three-day old neonatal Balb/c mice were treated with VEGF two hours prior to
injection of LV/luc [VEGF (+)]. Control VEGF (-) mice received only virus. No adverse
events from VEGF treatment were observed in this and other studies [191, 261]. Transgene
expression was assessed by whole body luminescence imaging (WBLI) following injection
of D-luciferin as a substrate for luciferase. In both groups of animals, luminescent signals
could be detected beginning at 4 weeks (data not shown) and persisted to similar levels up to
12 weeks (Fig. 3.1). This pattern is similar to that observed in a previous study [168]. These
results indicate that VEGF administration does not grossly affect expression of the viral gene.
VEGF pre-treatment increases luc expression in the organs.
Following WBLI at week 12, mice were sacrificed and the organs were imaged ex
vivo. The luminescent signal intensity was measured and it was found that compared to
VEGF (-) mice, VEGF (+) mice had increased signal intensity in the brain when observed
from both the top and bottom views (Fig. 3.2). The level of expression in the brain is lower
than in our previous experiments [168] though this is likely due to lower functional titer of
the batch of virus used in this experiment. Low titer virus was used here to reduce the
possibility of signal saturation that may arise from higher levels of transduction, thus
allowing us to better detect differences between groups. By ex vivo imaging, it was also

69
found that the lung, heart and kidney from VEGF (+) mice showed increased signal intensity
over organs from VEGF (-) mice, while there was no apparent benefit to VEGF pre-treatment
in the liver and spleen (Fig. 3.3). However, this may be due to the high level of luc
expression in both organs that may have saturated the captured signal, despite the use of low
titer virus as these organs appear to be the most readily penetrated and transduced by LV as
seen in our previous studies [168].
To further quantify the actual amount of luc in the organs, sections of each organ
were homogenized and the luc activity in each sample was measured. It was found that
organs from LV/luc-treated mice showed high luc activity while untreated mice showed only
background levels (Fig. 3.4). VEGF pre-treatment showed a tendency to increase luc activity
in all organs with the increase in activity in the heart being significantly higher in VEGF-
treated mice (p < 0.05) (Fig. 3.4). These finding are of particular significance for diseases
that have multiple organ involvement and especially important for diseases with cardiac
involvement like the cardiac variant of Fabry disease and other metabolic disorders [265-
267]. Increased luc activity in the other organs of VEGF-treated mice as measured by this
assay could not be determined (Fig. 3.4). Since gross sections were made for this assay, it is
possible here that the luc protein was diluted in the background of non-transduced tissue and
that this impacted the activity calculations in contrast to the ex vivo imaging data.
Injection of LV via the temporal vein leads to transduction various cell types
Tissue sections from brain were immunostained to identify the cell types transduced
by the LV/luc vector using either an anti-doublecortin antibody as a neuronal cell marker or
an anti-GFAP antibody as a glial cell marker. Immunoreactive luciferase was detected in

70
neuronal cells as shown by staining with the anti-doublecortin antibody (Fig. 3.5a). It also
appears that in mice treated with VEGF, Purkinje cells of the cerebellum were transduced.
No immunoreactivity was observed against luc in glial cells of the brain (Fig. 3.5b). In the
heart, it was found that immunoreactive luc co-localized with both vascular endothelial cells
(Fig 3.6a) and with myocardial cells (Fig 3.6b). Similar results were also seen in the liver
where both endothelial and parenchymal cells showed immunoreactivity against luc (data not
shown). These results indicate that the luc detected by imaging and by direct luc activity
assays resulted from transduction of the cells of the organ and not only from transduction of
the endothelial cells of the vessels.

71
3.5 DISCUSSION
The treatment of both the neurological and visceral symptoms of Farber disease is of
utmost importance since the majority of Farber patients present with progressive neurological
failure in addition to the classic symptoms [62]. Previous studies by the Medin laboratory
have shown that neonatal gene transfer could provide long-term systemic correction the
alpha-galatosidase A activity of Fabry mice [168]. Studies by another group found that in
neonates, administration of VEGF prior to injection of LV resulted in increased numbers of
cells transduced in the brain [191]. While examination of the organs showed that VEGF had
no overt effects on organ development and did not cause tumor development in any of the
organs, there was no examination of the effect on transgene expression specifically in the
organs themselves [191]. In the present study, a similar delivery approach using luc as a
marking transgene was taken to determine the effects of VEGF administration on the
transduction of the major internal organs.
Transgene expression in LV/luc-treated mice was monitored monthly for three
months by WBLI and it was found that expression remained steady in both VEGF (+) and
VEGF (-) mice. Quantification of transgene expression by ex vivo imaging of the luminescent
signal showed significantly increased luc in the brain, heart, lung and kidney of VEGF-
treated mice compared to mice that were not pre-treated with VEGF. While there appeared to
be no benefit to VEGF pre-treatment in the spleen and liver, luc expression was very high in
both organs. We may have reached the saturation point of the assay, and as such, differences
between groups would be harder to detect. This finding is not surprising since the liver is

72
often the most highly transduced organ when virus is delivered directly to the bloodstream
[168, 191, 268]. Thus it is possible that VEGF treatment may have a negligible effect on
already highly transduced organs like the liver.
Measurement of luc activity in tissue homogenates showed a significant increase in
luc expression in the hearts of VEGF (+) mice while no significant increases could be
detected in other organs. These differences in results from the two methods of analysis are
likely caused by a number of factors including the architecture and vascularization of the
organ. For instance, it was surprising that little difference in luc activity was seen in the lungs
since macroscopic observation of the images obtained by ex vivo imaging showed a larger
area of luminescence in the lungs of VEGF (+) mice compared to those from VEGF (-) mice.
However, the areas of the lungs that were transduced by LV/luc in VEGF (-) mice have a
higher intensity signal than the areas transduced in the VEGF (+) mice. These observations
may be due to the highy vascularized nature of the lung that allowed a more diffuse
distribution of the virus after administration of VEGF, whereas in the VEGF (-) mice the
virus appears to have remained concentrated in a smaller area. Gross sections of each organ
were used for the activity assay, which may account for the lack of differences measured,
despite differences in luminescence observed by imaging in organs. In the kidney, for
example, only small concentrated areas of the tissue exhibited luminescence in the VEGF-
treated animals. Since random sections were used for the luc activity assay, the transduced
areas may have been inadvertently excluded from the analyses and may explain the lack of
observed differences in this organ.
Despite the differences in sensitivity of the two methods of transgene detection used
in this study, it is clear that pre-administration of VEGF increases the efficacy of treatment

73
with recombinant lentiviruses. This protocol can potentially be of therapeutic benefit for the
treatment of diseases like Farber disease. Preliminary results from immunohistochemistry
show that neuronal cells of the brain and myocardial cells in the heart are transduced by
LV/luc and provide evidence that the organs themselves are transduced. Future studies will
include the staining of tissues from other organs to determine the cell types transduced and
the pattern of transduction in all organs. To determine the effect of VEGF on virus delivery
to the liver and spleen, similar studies should be performed using lower titre virus to allow
for the detection of differences between groups. In addition, testing in a relevant disease
model will facilitate the evaluation of phenotypical correction and immunological responses
to the therapeutic enzyme. It also still remains to be determined if the human BBB is as
immature as that of the mouse at an analogous stage of development and would as such be
affected in the same way by VEGF treatment and viral administration.
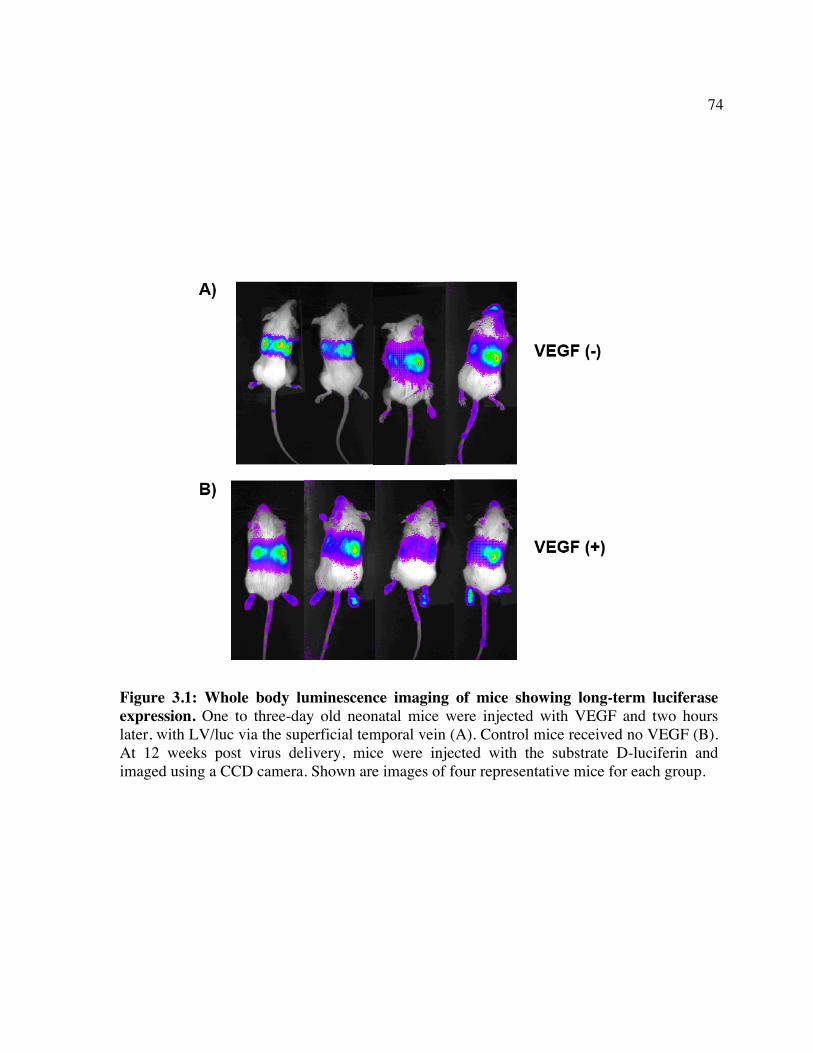
74
Figure 3.1: Whole body luminescence imaging of mice showing long-term luciferase expression. One to three-day old neonatal mice were injected with VEGF and two hours later, with LV/luc via the superficial temporal vein (A). Control mice received no VEGF (B). At 12 weeks post virus delivery, mice were injected with the substrate D-luciferin and imaged using a CCD camera. Shown are images of four representative mice for each group.

75
Figure 3.2: Luc expression in the brain following treatment with LV/luc and VEGF. Following WBLI, mice were sacrificed and the brain was removed and imaged from both the top (A) and bottom (B) sides. Shown are images of four representative mice from each group.
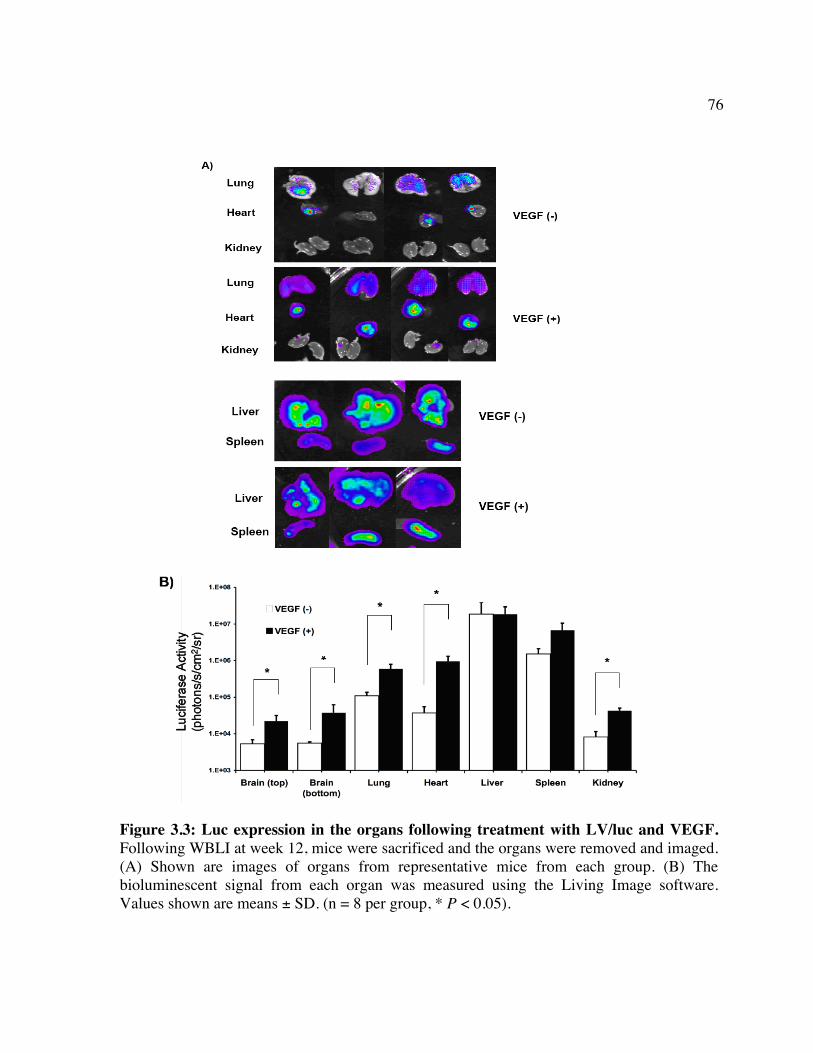
76
Figure 3.3: Luc expression in the organs following treatment with LV/luc and VEGF. Following WBLI at week 12, mice were sacrificed and the organs were removed and imaged. (A) Shown are images of organs from representative mice from each group. (B) The bioluminescent signal from each organ was measured using the Living Image software. Values shown are means ± SD. (n = 8 per group, * P < 0.05).
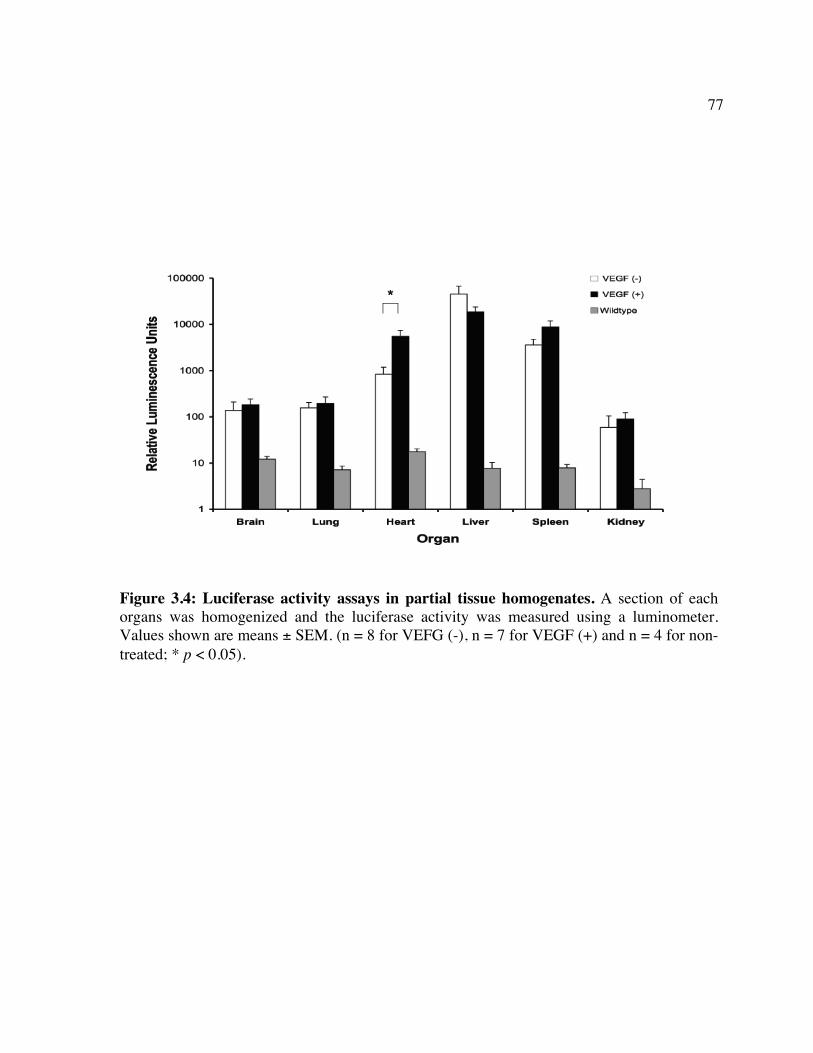
77
Figure 3.4: Luciferase activity assays in partial tissue homogenates. A section of each organs was homogenized and the luciferase activity was measured using a luminometer. Values shown are means ± SEM. (n = 8 for VEFG (-), n = 7 for VEGF (+) and n = 4 for non-treated; * p < 0.05).
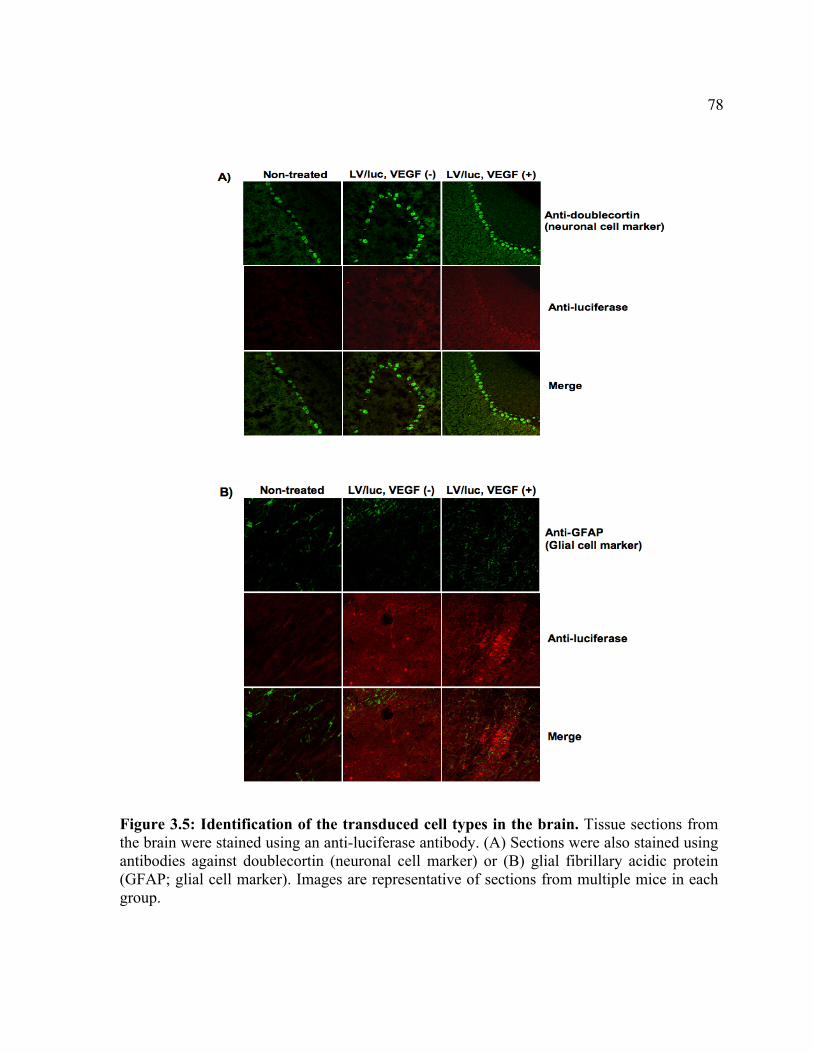
78
Figure 3.5: Identification of the transduced cell types in the brain. Tissue sections from the brain were stained using an anti-luciferase antibody. (A) Sections were also stained using antibodies against doublecortin (neuronal cell marker) or (B) glial fibrillary acidic protein (GFAP; glial cell marker). Images are representative of sections from multiple mice in each group.
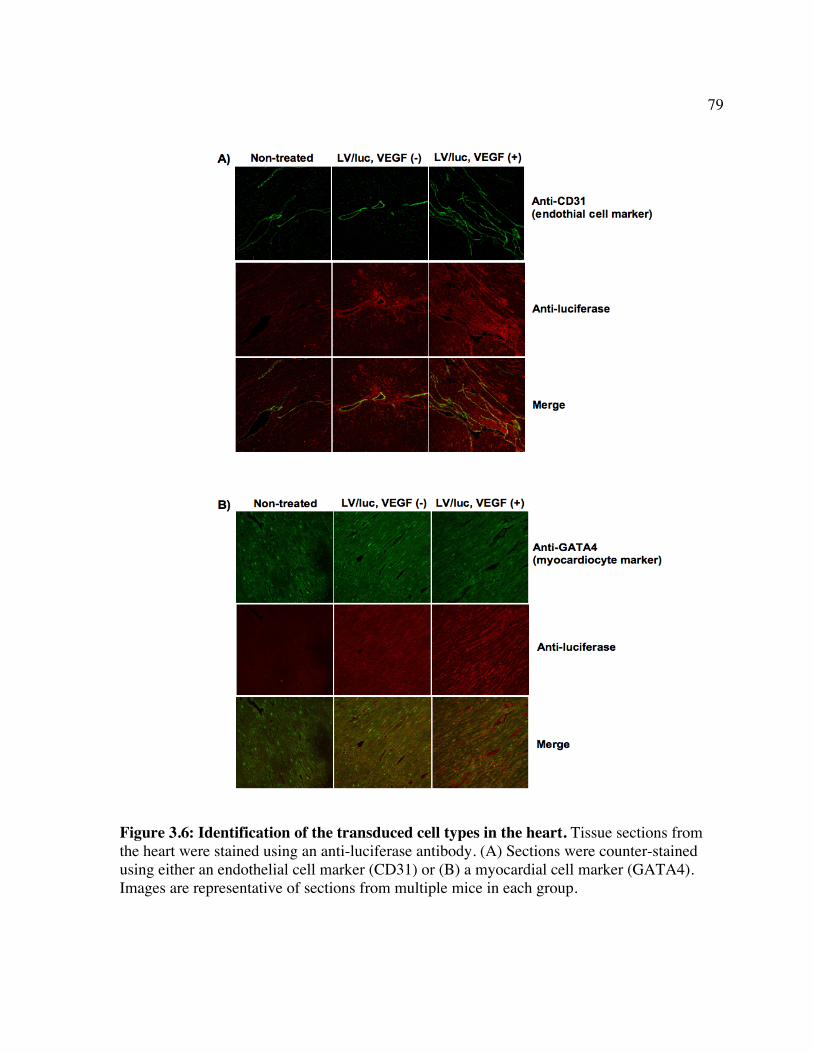
79
Figure 3.6: Identification of the transduced cell types in the heart. Tissue sections from the heart were stained using an anti-luciferase antibody. (A) Sections were counter-stained using either an endothelial cell marker (CD31) or (B) a myocardial cell marker (GATA4). Images are representative of sections from multiple mice in each group.

80
Chapter 4: Anti-CD25 targeted kil l ing of bicistronically transduced cells: a novel safety mechanism against retroviral genotoxicity A version of this chapter has been published. Ramsubir S, Yoshimitsu M and Medin JA. 2007. Anti-CD25 targeted Killing of bicistronically transduced cells: a novel safety mechanism against retroviral genotoxicity. Mol Ther. 15:1174-81.

81
4.1 ABSTRACT
Gene therapy for LSDs has the potential to provide a lasting cure with a single
treatment. Despite modifications to existing vectors, concerns have arisen regarding the risk
of genotoxicity associated with the use of retroviruses. To address safety concerns, it is
proposed that co-expression of a cell surface protein, human CD25, in a bicistronic format
with any therapeutic gene, can provide a target that can be used to selectively kill transduced
cells should transformative events occur. It was shown that an anti-CD25 antibody and
immunotoxin could specifically target and eliminate leukemic cells transduced to express
CD25. In a murine leukemia model, antibody treatment reduced tumor burden 32-fold and
increased survival compared to non-treated mice. Further, in another model employing bone
marrow transplantation of therapeutically transduced cells into Fabry mice, antibody
treatment reduced the number of retrovirally-transduced, human CD25-expressing cells in
the peripheral blood. A depletion of transduced cells with functional consequences was also
evident in the liver and spleen. This proof-of-principle study demonstrates that a targeted
antibody can reduce tumor burden and selectively clear bicistronically transduced
hematopoietic cells that express a target antigen, thus acting as a built-in safety mechanism
for the gene therapy vectors developed here.

82
4.2 INTRODUCTION
Gene therapy has been used to successfully treat a number of inherited disorders [269,
270] and remains the most promising option for Farber disease since BMT has only resulted
in limited success. While many viral and non-viral gene delivery alternatives exist, retroviral
vectors offer the advantages of stable integration into host genomes, the ability to transduce a
wide variety of cell types, and relatively high levels of transgene expression [271]. However,
concerns regarding the safety of integrating vectors have been prompted by the development
of leukemia in four X-linked severe combined immunodeficiency (X-linked SCID) patients
in a recent clinical trial using an oncoretroviral vector [196-198]. A variety of explanations
for this outcome have been proposed but the exact mechanism of leukemogenesis has
remained unresolved since no other clinical trials have reported this type of adverse event
[199, 203]. Despite this outcome, the use of retroviral gene therapy continues because of the
conceptual effectiveness of the treatment and the fact that gene therapy is the only potential
cure available for many disorders such as X-linked SCID. Therefore, the development of
improved vectors and viable alternative safety strategies is exceedingly important and timely.
These studies were aimed at developing a safety system that can be used in the event
of an oncogenic event following gene transfer. For this proof-of-principle study, the mouse
model of another related lysosomal storage disorder (LSD) was used since a mouse model of
Farber disease is not currently available. The Medin laboratory has been developing various
retrovirus-based gene therapy approaches for Fabry disease, a disorder resulting from a
deficiency of α-galactosidase A (α-gal A) activity [48]. Using retroviral gene transfer

83
strategies, long-term enzymatic correction and corresponding lipid reduction have been
achieved in a mouse model of Fabry disease by bone marrow transplantation (BMT) of
transduced cells [229, 272, 273] and by direct delivery of lentivirus into neonates [168].
Thus, it is a good surrogate model for testing of a safety strategy for Farber disease.
Retroviral vectors that engineer expression of both α-gal A and human CD25
(huCD25) in a bicistronic format have been previously developed and utilized by the Medin
laboratory [218] and is similar to the vectors constructed for the treatment of Farber disease
in this thesis. CD25, also known as the T-cell activation antigen (Tac) and the IL-2 receptor
alpha chain (IL-2Rα) [215], is incapable of mediating IL-2 internalization or signaling by
itself, however, in tandem with the β chain of the receptor and the γc chain, it forms the
‘high-affinity’ receptor for IL-2 [274]. Though it can be induced upon activation, expression
of CD25 is absent on resting T cells, B cells, monocytes, and CD34+-enriched cells [216,
217]. Thus, its limited expression pattern and lack of ability to mediate signaling makes it a
good choice as a cell surface marking protein in bicistronic vectors. In our previous studies,
human CD25 expression was used to functionally assess viral titers, for the enrichment of
transgene positive cells prior to BMT, and for tracking transduced cells post-BMT [218].
Since it is also cleaved from the IL-2 receptor complex on the cell surface and can be
detected as soluble CD25 (sCD25) in the plasma [275], sCD25 has previously been used as a
surrogate marker to evaluate the level of transgene expression in an experimental setting
[168].
In this study, the use of huCD25 expressed from the bicistronic retroviral vector
constructs was extended into the development and application of a built-in safety mechanism
within the gene therapy context. It was proposed that if an unwanted proliferative

84
abnormality occurs following retroviral gene transfer, huCD25 could act as a target antigen
to selectively eliminate transduced cells using either clinically approved anti-CD25
antibodies, or newer highly potent antibody-toxin conjugates (immunotoxins). Of the many
immunotoxins that are approved for use in humans, the murine anti-Tac (AT) monoclonal
antibody [276] fused to saporin (SAP) [277], a toxin that irreversibly damages ribosomes by
cleaving adenine molecules from ribosomal RNA [278], was used in these studies. Here, it
has been demonstrated both in vitro and in vivo that the anti-Tac-SAP (ATS) complex can
specifically target and kill retrovirally-transduced cells that express huCD25. Importantly,
enzymatic correction in a mouse model of Fabry disease using a bicistronic vector was
achieved and it was then possible to remove transduced cells using both ATS and AT. Thus,
this model of using a cell surface antigen such as huCD25 in a bicistronic gene expression
cassette is proposed as a novel safety mechanism for retroviral vectors.

85
4.3 MATERIALS AND METHODS
Cells lines. The cell lines C1498 (C57BL/6 derived), 293T (obtained by MTA from Michele
Calos of Stanford University), 3T3 and HeLa cells (American Type Culture Collection,
Manassas, VA) were maintained in DMEM supplemented with 10% FCS (PAA, Rexdale,
ON), 2 mM L-glutamine, 1 mM sodium pyruvate, 100 U/ml penicillin, and 100 mg/ml
streptomycin (all from Sigma, Oakville, ON) at 37 °C in a humidified incubator with 5%
CO2.
Vector constructs and viral vector production. The lentiviral vector pHR’cppt-EF-α-gal A-
IRES-huCD25-W-SIN (LV/α-gal A/huCD25) was previously constructed in our laboratory
[168]. Virus was produced by co-transfection of the LV with accessory plasmids pMD.G and
pCMVΔR8.91 into 293T cells using FuGENE 6 transfection reagent (Roche, Mississauga,
ON, Canada) and titered on HeLa cells as previously described [279].
The ecotropic oncoretroviral packaging cell line E86/pMFG/α-gal A/IRES/huCD25
clone 21 (RV/α-gal A/huCD25) was constructed to produce virus engineered to express both
α-gal A and huCD25 as previously described [218]. The control vector used was
E86/pUMFG/enYFP (RV/enYFP), which has the same vector backbone and expresses
enhanced yellow fluorescent protein (enYFP) [241]. 4 x 106 producer cells were seeded in
15-cm dishes and media containing virus was harvested after 72 h. Viral titer was determined
by transduction of 3T3 cells. Transduced cells were then analyzed 72 h later by flow
cytometry to detect either huCD25 or enYFP. huCD25 expression was detected using a

86
phycoerythrin (PE) conjugated antibody against CD25 (α-CD25-PE; BD Bioscience Canada,
Mississauga, ON) while enYFP expression was measured directly. Flow cytometry was
performed using the FACSCalibur and analyzed using the CELLQuest™ software (BD
Bioscience).
Establishment of human CD25 expressing murine leukemia cell line. C1498 cells were
transduced with LV/α-gal A/huCD25 at a multiplicity of infection (MOI) of 10 productively
infectious particles (IP)/cell. Cells were re-suspended in filtered viral supernatant
supplemented with 8 µg/ml protamine sulfate and overlaid onto plates coated with
fibronectin (Roche). Transduced C1498 cells were sorted by magnetic activated cell sorting
into pools and by flow cytometry based on expression of huCD25 into single cell clones
(C1498/huCD25).
In vitro clearance of retrovirally transduced cells. Transduced C1498 cell pools,
C1498/huCD25 or non-transduced C1498 cells (C1498 NT) were plated in triplicate at a
density of 1 x 104 cells/well in a 96-well plate in volumes of 100 µl. C1498/huCD25 cells
were incubated with increasing concentrations (0.1 nM - 10 nM) of one of the following
reagents: anti-Tac antibody (AT), anti-Tac conjugated to saporin (SAP) (ATS), control IgG
conjugated to SAP (IgG-SAP) or SAP (kindly provided by Advanced Targeting Systems,
San Diego, CA). C1498 NT cells were treated with ATS at the same concentrations. Cells
were incubated at 37°C and growth inhibition and cell death were then assessed. All
treatments were tested in at least 2 independent experiments.

87
To assess growth inhibition, 10 µl of 5 mg/ml of MTT (3-(4,5-Dimethyl-2-thiazolyl)-
2,5-diphenyl-2H-tetrazolium bromide) labeling reagent (Sigma) was added to each well 72 h
after seeding cells. Plates were incubated for 4 h at 37°C in a humidified incubator with 5%
CO2. 100 µl of solubilizing solution (10% SDS, 0.01 M HCl) was then added and plates were
incubated at 37°C overnight. Cell death was assessed 48 h after seeding by the measurement
of lactate dehydrogenase (LDH) release using the CytoTox 96® Non-Radioactive
Cytotoxicity Assay Kit (Promega Corp., Madison, WI) as per the manufacturer’s
instructions.
Establishment of in vivo leukemia model. C1498/huCD25 cells were used to generate a
leukemia model in Fabry mice [280]. Mice were lethally irradiated (11 Gy) and 4 h later, 1 x
106 C1498/huCD25 cells were injected into the tail vein along with 1 x 106 fresh bone
marrow mononuclear cells (BMMNCs) that were isolated by flushing the femurs and tibias
of syngeneic donor Fabry mice. Control mice were injected with 1 x 106 C1498 NT cells and
BMMNCs. All recipient mice were then treated with 5 µg ATS or equimolar (24.4 pmol)
amounts of either AT or IgG-SAP on days 2, 4 and 6 post-cell transplantation, by injection
into the intraperitoneal (i.p.) cavity in a volume of 200 µl. Mice were monitored daily for
evidence of disease or distress in compliance with standards set by the Animal Care
Committee of the UHN.
In vivo clearance of gene-corrected cells in a bone marrow transplantation model. Donor
Fabry mice were treated with 150 mg/kg 5-fluorouracil (Sigma). Three days later, BM was
isolated by flushing the femurs and tibias of treated donor Fabry mice. Mononuclear cells

88
were isolated by centrifugation on Nycoprep® and stimulated for 12 h in DMEM
supplemented with 10% FCS, 2 mM L-glutamine, 1 mM sodium pyruvate, 100 U/ml
penicillin, 100 mg/ml streptomycin, 50 ng/mL stem cell factor (SCF), 20 ng/mL Flt3 ligand
(Flt3L) and 20 ng/mL interleukin-6 (IL-6). All cytokines were obtained from R&D Systems
(Minneapolis, MN, USA). Cells were transduced twice (at 12 h intervals) using supernatant
from RV/α-gal A/huCD25 or RV/enYFP producer cell lines [218] at MOIs of ~3 and 1
IP/cell, respectively. Transductions were performed on plates coated with fibronectin
(Roche) and viral supernatant was supplemented with the same cytokine cocktail above plus
8 µg/ml protamine sulfate (Sigma).
Recipient Fabry mice were irradiated (11 Gy) and 4 h later, transduced cells were
injected via the tail vein. Cell doses were 0.4 x 106 cells/mouse and 0.3 x 106 cells/mouse for
the groups transplanted with cells transduced with RV/α-gal A/huCD25 and RV/enYFP,
respectively. Beginning at 4 weeks post-transplant, PB cells were monitored to detect
engraftment every 4 weeks. Eight weeks after transplantation, mice were treated i.p. with
three doses of 5 µg ATS or equimolar amounts of either AT or IgG-SAP. Doses were
administered every two days. At ten weeks post-transplant, PB was analyzed for response to
the immunotoxins. A fourth dose of immunotoxin was administered, as before, 11 weeks
after transplant and the animals were sacrificed 12 weeks after transplant.
Soluble human CD25 ELISA. Plasma was isolated from PB of mice by centrifugation at
16000 x g for 20 min. The level of soluble CD25 was measured by a direct ELISA using the
BD OptEIA™ Human IL-2 sRα ELISA Set (BD Bioscience Canada) as per the
manufacturer’s instructions. Each sample was measured in triplicate.

89
α-gal A Activity Assay. α-gal A activity was measured by a microtiter plate-based
fluorometric assay using 5 mM 4-methylumbelliferyl α-D-galactopyranoside (Research
Products International, Mt Prospect, IL, USA) as the substrate for α-gal A, and 0.1 M N-
acetyl-D-galactosamine (Sigma) as an inhibitor of α-N-acetylgalactosaminidase, as
previously described [272]. Plasma was added directly to the plate in triplicate repeats for
each analysis. For measurement of organ enzyme activity, frozen tissue samples were
homogenized and lysates prepared as previously described [168]. Plates were read on a
fluorescence microtiter plate reader (Dynex, Chantilly, VA) against nine independent
dilutions of a 4-methylumbelliferone standard (Sigma). The protein concentrations of tissue
samples were determined using the BCA Protein Assay Kit (Pierce, Rockford, IL).
Statistical analysis. Data presented represent means of triplicate determinations for each
sample and are representative of results obtained from independent experiments that
produced similar relative results. Differences between groups for enzyme assays and ELISAs
were assessed using Student t-tests. The Kaplan Meier product-limit method was used to
assess the survival of mice and the log-rank statistic was used to test differences between
groups (Excel, Microsoft Corporation). Values of P < 0.05 were considered to be statistically
significant.

90
4.4 RESULTS
In vitro effect of targeting human CD25 with a specific immunotoxin.
We first wanted to determine the specificity and efficacy of the huCD25-targeted
immunotoxin, ATS. A murine myeloid leukemia cell line, C1498, was transduced with a
lentiviral vector (LV) pHR’cPPT-EF-α-gal A-IRES-huCD25-W-SIN (LV/α-gal A/huCD25)
that is engineered to express both human α-gal A and huCD25 [168]. Transduced pools
were enriched for expression of huCD25 by magnetic activated cell sorting. Two populations
of cells that have a broad spectrum of huCD25 expression were tested with 5 nM of each
reagent: ATS, AT, control IgG Ab conjugated to SAP (IgG-SAP), or SAP only. These
populations, shown in Figs. 4.1a and b, were 90% and 45% positive for huCD25 expression,
respectively. MTT assays showed that both populations of cells treated with ATS showed
reduced proliferation (Figs. 4.1c,d) and increased cell death as measured by lactate
dehydrogenase (LDH) release (Figs. 4.1e,f) compared to cells treated with other reagents.
Non-transduced cells did not show any inhibition of proliferation or increased cytotoxicity
when treated with ATS (data not shown).
Next, the ability of ATS to clear a clonal population of transduced cells was tested. A
single cell clone expressing huCD25 (C1498/huCD25) was isolated from the transduced pool
of cells by flow cytometry-based sorting (Fig. 4.2a). Both C1498/huCD25 and C1498 non-
transduced (C1498 NT) cells were incubated with increasing concentrations of each reagent.
The effects on cellular proliferation and cell killing were then measured. As shown in Fig.
4.2b, inhibition of cellular proliferation was significantly higher (p < 0.001) when cells were

91
treated with ATS than when cells were treated with control reagents. This effect was specific
to cells expressing huCD25, since C1498 NT cells treated with ATS did not show impaired
growth. Similar results were obtained from an LDH assay, where at low doses (< 1 nM), cell
killing was higher in cells treated with ATS than in cells treated with control reagents (p <
0.001) (Fig. 4.2c).
Clearance of huCD25-expressing cells in vivo:
Leukemia model.
Towards determining whether treatment with a CD25 antibody or immunotoxin could
clear huCD25-expressing leukemic cells in the mouse model of Fabry disease, the dose of
C1498 leukemia cells to use in this strain was optimized. Increasing doses (1 x 103 to 1 x
106) of C1498 NT cells were injected into Fabry mice and the effects were monitored. While
leukemic cells were not present in the peripheral blood (PB), mice showed systemic
subcutaneous invasion, splenomegaly, and lymphoadenopathy, which mimics some leukemic
phenotypes (data not shown). For cell doses of 1 x 103 and 1 x 104 cells/mouse, it was found
that 100% and 70% of mice, respectively, survived the challenge (data not shown). For
higher cell doses of 1 x 105 and 1 x 106 cells/mouse, 100% of the mice succumbed to the
leukemia within 60 days and 30 days, respectively (data not shown). To obtain a clinically
relevant leukemia model, a cell dose of 1 x 106 cells/mouse was chosen for future studies
since at this higher cell dose the phenotype of the transplanted mice progressed to the disease
state more quickly and aggressively.
As no previous in vivo studies have been done with murine ATS and most studies
using other AT derivatives use receptor-saturating doses of antibody [281], it was next

92
necessary to determine an effective dose of immunotoxin. Two different doses of ATS were
tested for their ability to eliminate huCD25-expressing cells in Fabry mice challenged with
C1498/huCD25 leukemia. Mice were lethally irradiated and injected with 1 x 106
C1498/huCD25 cells and supportive syngeneic BM cells. At days two, four, and six post-
leukemic transplant, animals were injected i.p. with either 5 µg ATS or 20 µg ATS, SAP
only, or were left untreated (n = 3 per group). Eleven days post-challenge, blood was
sampled and plasma analyzed for levels of sCD25 by ELISA. Evaluation of sCD25 levels is a
common method used in the clinical setting to monitor tumor burden and treatment response
in patients with CD25-expressing lymphoma and leukemia [282]. This method also allows a
sensitive detection of the presence of CD25-positive cells for these studies as it can also
reflect contributions from concealed populations. As shown in Fig. 4.3, treatment with ATS
significantly reduced sCD25 (p < 0.05) levels compared to animals treated with the control
reagent SAP and those that were left untreated. Since treatment with the lower dose of 5 µg
of ATS had a similar effect as the 20 µg dose (Fig. 4.3), the lower dose of ATS was used in
future experiments since this was more cost-effective and might lower the risk of secondary
or non-specific toxicities.
To further test the efficacy of the CD25-targeting approach, a larger experiment using
5 µg ATS was next performed. Mice were lethally irradiated and injected with 1 x 106
C1498/huCD25 or C1498 NT cells along with supportive syngeneic BM cells via the tail
vein. Mice transplanted with C1498/huCD25 cells were then treated with equimolar amounts
of ATS, AT, or IgG-SAP. Mice transplanted with C1498 NT cells were treated with 5 µg
ATS as a control. All animals were bled on days 7, 11, and 18 post-transplant and levels of
sCD25 in the plasma were measured by ELISA. As shown in Fig. 4.4a, in mice challenged

93
with C1498/huCD25 cells, average plasma sCD25 levels at 18 days post-transplant were
significantly lower in animals treated with ATS (474 pg/ml) and AT (848 pg/ml) compared
to mice that were treated with IgG-SAP (4,762 pg/ml; p < 0.01) or that were not treated
(15,450 pg/ml; p < 0.05). This indicates a lower tumor burden in mice treated with both
CD25-targeted reagents, ATS and AT.
The inherent α-gal A deficiency of Fabry mice and the fact that the transplanted
tumor cells were engineered to express α-gal A meant that differences in α-gal A activity
itself could be used as another surrogate marker of tumor burden. Therefore, plasma α-gal A
activity was measured and it was found that α-gal A activity was lowest in mice treated with
ATS (16 nmol/hr/ml) and AT (21 nmol/h/ml) (Fig. 4.4b). These levels were significantly
lower than in mice that received IgG-SAP (47 nmol/h/ml; p < 0.001 and p < 0.01, versus
ATS and AT respectively) or that were left untreated (77 nmol/h/ml; p < 0.05). Therefore,
both ATS and AT are able to de-bulk tumor burden in this huCD25-expressing leukemia
model.
To further determine the ability of anti-CD25 antibodies to impact survival, animals
were monitored daily and a Kaplan-Meier representation of survival was prepared in Fig.
4.4c. In mice treated with ATS, the median survival duration was 29 days. This was
significantly higher (P < 0.01) than that seen in mice that were not treated (median survival =
23 days). Increased survival was also seen in mice treated with AT (median survival of 30
days, p < 0.05 versus non-treated mice). Therefore, even in the context of a very high
leukemic burden, treatment with CD25-targeted antibodies was able to increase survival
compared to control treatments. Note that these results are representative of two independent
experiments.

94
BMT model.
The next step was to test the clearance strategy in the context of a therapeutic BMT
model. BMT is a common gene therapy approach [283] and incorporation of a cell surface
protein that can be targeted can improve the safety of the system. Murine bone marrow
mononuclear cells (BMMNCs) were isolated and transduced twice with one of two ecotropic
oncoretroviral vectors, either E86/pMFG/α-gal A/IRES/huCD25 clone 21 (RV/α-gal
A/huCD25) or E86/pUMFG/enYFP (RV/enYFP) [218]. Flow cytometry analysis of these
transduced BMMNCs showed that cells transduced with RV/α-gal A/huCD25 were ~30%
positive for expression of huCD25 (Fig. 4.5a) and cells transduced with RV/enYFP were
~20% positive for enYFP expression (Fig. 4.5b). Cells were then injected into lethally-
irradiated Fabry mice that were monitored monthly for engraftment.
At eight weeks post-transplant, plasma from recipient Fabry mice was analyzed for α-
gal A activity and for levels of sCD25. Average plasma α-gal A activity in mice transplanted
with BMMNCs transduced with RV/α-gal A/huCD25 was 65 nmol/h/ml, approximately six-
fold higher than in both control Fabry mice and mice transplanted with RV/enYFP-
transduced BMMNCs (Fig. 4.5c). This indicates that therapeutic correction of α-gal A
activity in Fabry animals was achieved at levels approximately two-fold above normal
C57BL/6 mice (Fig. 4.5c). At this time, the average level of sCD25 in the plasma of Fabry
mice transplanted with BMMNCs transduced with RV/α-gal A/huCD25 was 1212 ± 370
pg/ml. In contrast, sCD25 was undetectable in mice transplanted with RV/enYFP-transduced
cells, in wild-type C57BL/6 mice and untouched Fabry mice (data not shown).

95
Mice were then treated with either ATS, AT, or IgG-SAP as was previously done in
the leukemia model (see above). Seven days after the third dose of immunotoxin, plasma was
sampled to determine the effect of treatment. Comparisons were made to pre-treatment
values collected for each mouse at eight weeks post-transplant. As shown in Fig. 4.6a,
treatment with ATS resulted in lower plasma sCD25 levels than in mice that were treated
with IgG-SAP or mice that were not treated (P < 0.05). In addition, analysis of huCD25
expression on PB mononuclear cells (PBMNCs) by flow cytometry showed that mice treated
with ATS had significantly reduced numbers of huCD25-expressing PBMNCs than mice
treated with IgG-SAP (P < 0.01) or non-treated mice (P < 0.05) (Fig. 4.6b). Similar effects
were also observed in mice treated with AT, further supporting the conceptual ability of
targeted anti-CD25 antibodies to eliminate retrovirally-transduced donor hematopoietic cells
in vivo. Expression of enYFP was monitored before and after treatment with ATS and it was
found that levels remained stable over the course of the experiment (Fig. 4.6c),
demonstrating the specificity of the immunotoxin for cells expressing huCD25.
To examine the effect of a later administration of antibody or immunotoxin, one final
dose was administered and then mice were sacrificed. Enzyme activity was measured in
various tissues to determine the systemic effect of each reagent. PBMNCs from mice that
were treated with ATS showed significantly lower (P < 0.05) α-gal A activity than mice
treated with IgG-SAP (Fig. 4.7a). Similarly, α-gal A activity in the livers of mice treated
with ATS showed significantly lower (P < 0.05) enzyme activity than in livers of mice
treated with AT, IgG-SAP or non-treated mice (Fig. 4.7b). Likewise, in the spleens of mice
treated with ATS, there was significantly lower (P < 0.01) α-gal A activity than in IgG-SAP-
treated or non-treated mice (Fig. 4.7c).

96
4.5 DISCUSSION
Gene therapy is the most promising curative treatment for monogenetic diseases such
as LSDs [8]. While considerable advances have been made towards the development of
retrovirus-based gene therapy strategies for LSDs, concerns remain regarding the safety of
integrating vectors. Since the viral systems developed in this thesis involve such vectors,
these studies aimed at addressing this issue. It is proposed that a cell surface marker such as
huCD25 can act as an effective built-in safety mechanism in the event of insertional
genotoxicity by facilitating the clearance of transduced cells with a specifically targeted
immunotoxin. The Medin laboratory has previously used huCD25 in combination with α-gal
A in studies evaluating the efficacy of retroviral gene therapy for Fabry disease [168, 218].
We have not observed any untoward effects of exogenously expressing this protein nor have
we observed altered therapeutic effects of this surface antigen on α-gal A-mediated
correction in vivo.
Monoclonal antibodies (mAb) have been successfully used in the clinic for many
years to treat hematological malignancies, with minimal toxicity [284, 285]. For instance,
rituximab, an anti-CD20 antibody, has been used to treat a variety of lymphoid malignancies
[284, 286-288]. In addition, a strategy for clearing transduced hematopoietic cells in vivo
using an anti-CD20 Ab was proposed for the treatment of graft-versus-host disease (GvHD)
[289]. The premise is that T cells can be transduced with a viral vector carrying the cDNA
for CD20 prior to BMT and if GvHD occurs, then anti-CD20 antibodies can be used to

97
eliminate the donor T cells. These studies have shown promising results in vitro, however, no
studies have been done to demonstrate efficacy in vivo [290, 291].
As previously mentioned, numerous malignancies are characterized by aberrant
expression of CD25 [219] and both partial and complete remissions in patients have been
achieved by treating with antibodies against CD25, as well as newer recombinant
immunotoxins [219, 220]. In addition to being used as treatment for leukemia and
lymphomas, anti-CD25 antibodies are used in the clinic to modulate the effects of regulatory
T cells in settings such as preventing renal graft rejection or GvHD [222, 223] and to
enhance anti-tumor activity by depleting CD4+CD25+ regulatory T cells [224, 226]. These
findings provided the rationale for using anti-CD25 toxin-conjugated antibodies to target
huCD25.
In the present study it has been shown, both in vitro in cell culture and in vivo in a
Fabry mouse model, that a CD25 targeted treatment can specifically and effectively kill
leukemia cells that express both a therapeutic transgene, α-gal A, and huCD25 following
transduction with a retroviral vector. In the leukemia model using human CD25-expressing
C1498 leukemia cells, measurement of sCD25 levels and α-gal A activity following ATS
treatment showed a 32- and 5-fold reduction over non-treated mice, respectively. Similar
results were obtained when mice were treated with AT. In addition, treatment with either
ATS or AT extended survival by approximately 26% over mice that were not treated. It was
not unexpected that despite this increase in survival time, these mice still succumbed to the
leukemia, since a very high tumor dose was administered. As has been observed in clinical
trials for the treatment of naturally occurring CD25-expressing leukemias, it is expected that
better outcomes could be achieved with multi-modal therapy [292-294]. In addition, since it

98
is outside the scope of this study, the optimal dose of immunotoxin or administration regime
to use in this setting was not systematically determined but it may be possible to achieve
greater differences in response to treatment with different doses of ATS or AT. However,
proof-of-principle that this clearance strategy can de-bulk tumor burden and extend survival
has been shown.
It is further proposed that this clearance strategy can be used as a safety mechanism
against retroviral-induced genotoxicity in hematopoietic stem/progenitor cells. Thus, the
system was evaluated in a BMT setting in a mouse model of Fabry disease. In this therapy-
oriented experiment, an oncoretroviral vector previously used by the Medin laboratory was
used as the gene delivery vehicle [218]. An oncoretroviral vector was used here since
lentiviral vectors are not yet approved for use in non-HIV transduced humans. In addition,
studies have shown that oncoretroviral vectors have a greater propensity for integrating near
transcriptional start sites, proto-oncogenes and cell cycle regulatory genes than do lentiviral
vectors [204-206], perhaps making them more likely to cause dysregulation in gene
expression leading to leukemias [196], for example.
Following a standard gene transfer and BMT protocol in Fabry mice, supra-
physiological levels of α-gal A activity was observed in the plasma of transplanted mice.
Anti-CD25 targeted treatment of transplanted mice decreased levels of α-gal A activity in
PBMNCs as well as decreased expression of huCD25 on these cells, indicating clearance of
the transduced cell population itself. As expected, a corresponding decrease in the level of
sCD25 in the PB was seen. This corresponds well with previous data from the Medin
laboratory showing a positive correlation between levels of sCD25 and α-gal A activity in
the PB of mice treated with LV/α-gal A/huCD25 [168]. In this present study, it was also

99
found that ATS treatment was more effective at clearing transduced cells from the organs
than AT. ATS treatment resulted in a systemic decrease in organ α-gal A activity, indicating
that there was widespread elimination of transduced cells. It further provides evidence that
ATS is not merely clearing circulating sCD25 directly but that it is targeting and killing the
CD25-expressing cells.
To our knowledge, the study presented here is the first report of an antibody-mediated
clearance strategy being applied to gene therapy in the context of a therapeutic BMT. As
previously mentioned, while it is not expected that this system can cure a patient that
develops leukemia due to insertional mutagenesis, it is believed that it can help to de-bulk
tumor burden and thus increase the efficacy of other therapies such as chemotherapy. Further
work in this area will involve more rigorous in vivo testing to determine any off-target effects
and testing in other animal models such as non-human primates. Incidentally, the Medin
laboratory has recently cloned the cDNA for CD25 from the rhesus macaque which will
facilitate this endeavor [295]. It is believed that this novel strategy has great potential, as a
variety of cell surface proteins can be incorporated into various retroviral vectors in
combination with any therapeutic transgene such as AC. Using this system will add another
safety mechanism to current and future retroviral gene transfer systems such as the one
developed in this thesis for the treatment of Farber disease.
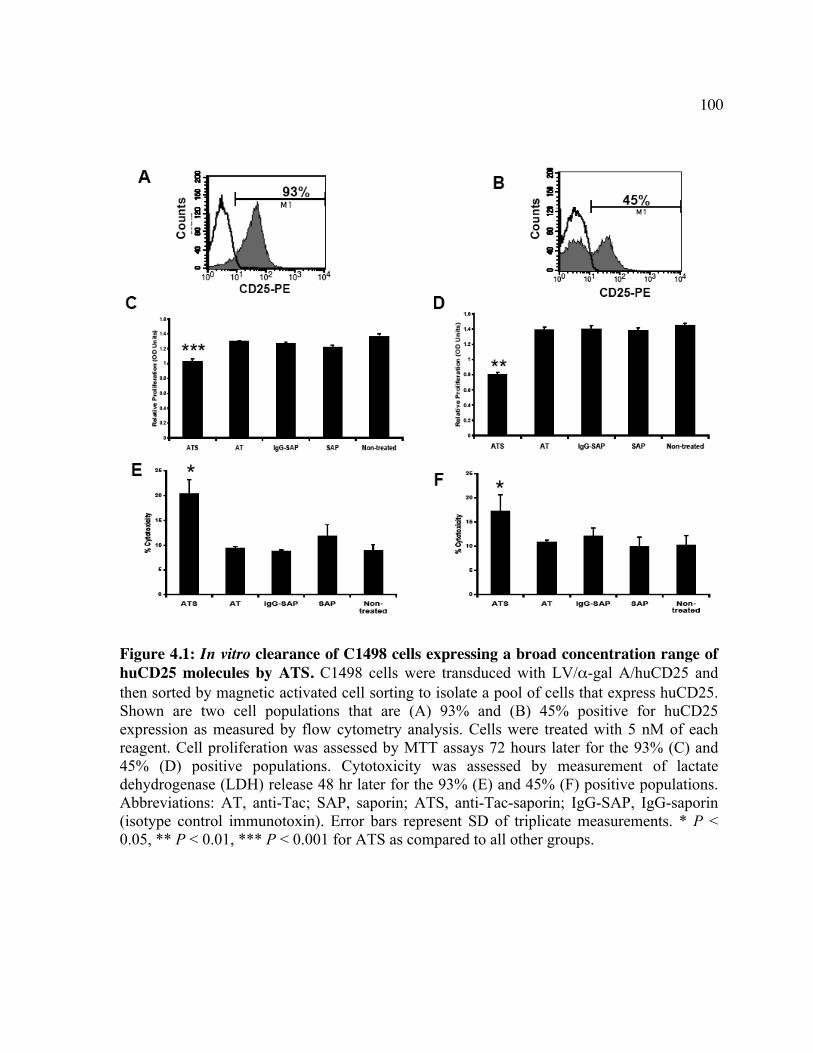
100
Figure 4.1: In vitro clearance of C1498 cells expressing a broad concentration range of huCD25 molecules by ATS. C1498 cells were transduced with LV/α-gal A/huCD25 and then sorted by magnetic activated cell sorting to isolate a pool of cells that express huCD25. Shown are two cell populations that are (A) 93% and (B) 45% positive for huCD25 expression as measured by flow cytometry analysis. Cells were treated with 5 nM of each reagent. Cell proliferation was assessed by MTT assays 72 hours later for the 93% (C) and 45% (D) positive populations. Cytotoxicity was assessed by measurement of lactate dehydrogenase (LDH) release 48 hr later for the 93% (E) and 45% (F) positive populations. Abbreviations: AT, anti-Tac; SAP, saporin; ATS, anti-Tac-saporin; IgG-SAP, IgG-saporin (isotype control immunotoxin). Error bars represent SD of triplicate measurements. * P < 0.05, ** P < 0.01, *** P < 0.001 for ATS as compared to all other groups.
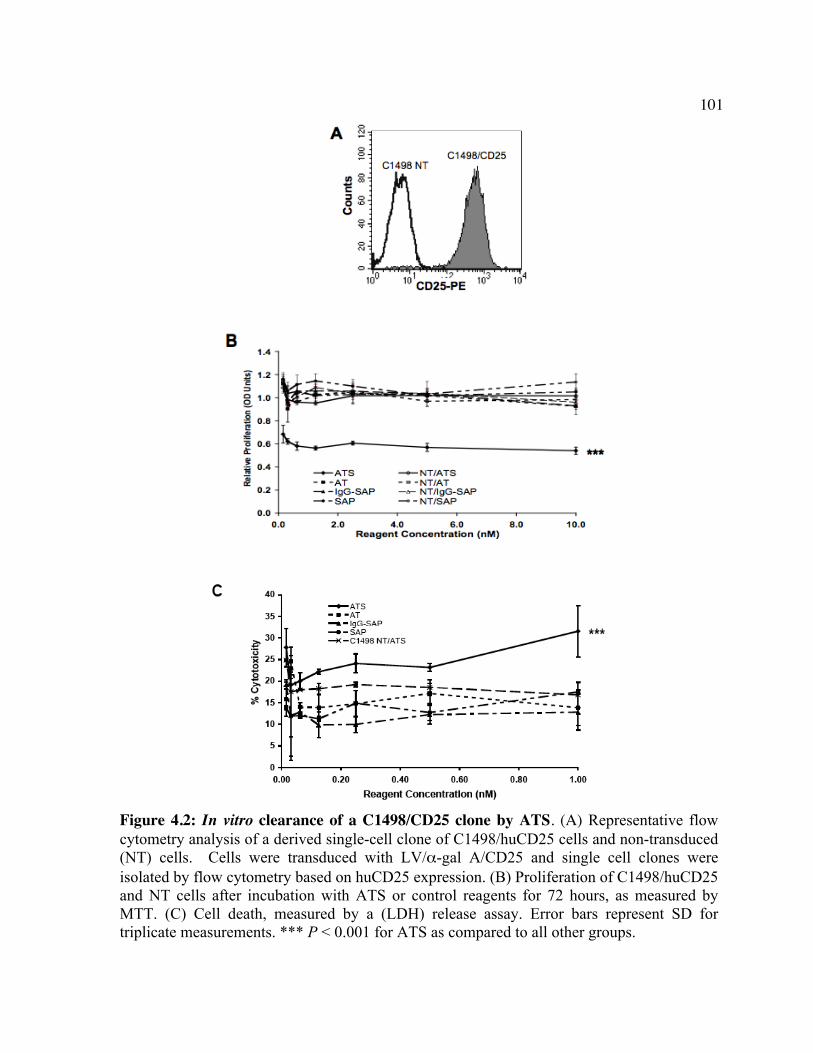
101
Figure 4.2: In vitro clearance of a C1498/CD25 clone by ATS. (A) Representative flow cytometry analysis of a derived single-cell clone of C1498/huCD25 cells and non-transduced (NT) cells. Cells were transduced with LV/α-gal A/CD25 and single cell clones were isolated by flow cytometry based on huCD25 expression. (B) Proliferation of C1498/huCD25 and NT cells after incubation with ATS or control reagents for 72 hours, as measured by MTT. (C) Cell death, measured by a (LDH) release assay. Error bars represent SD for triplicate measurements. *** P < 0.001 for ATS as compared to all other groups.
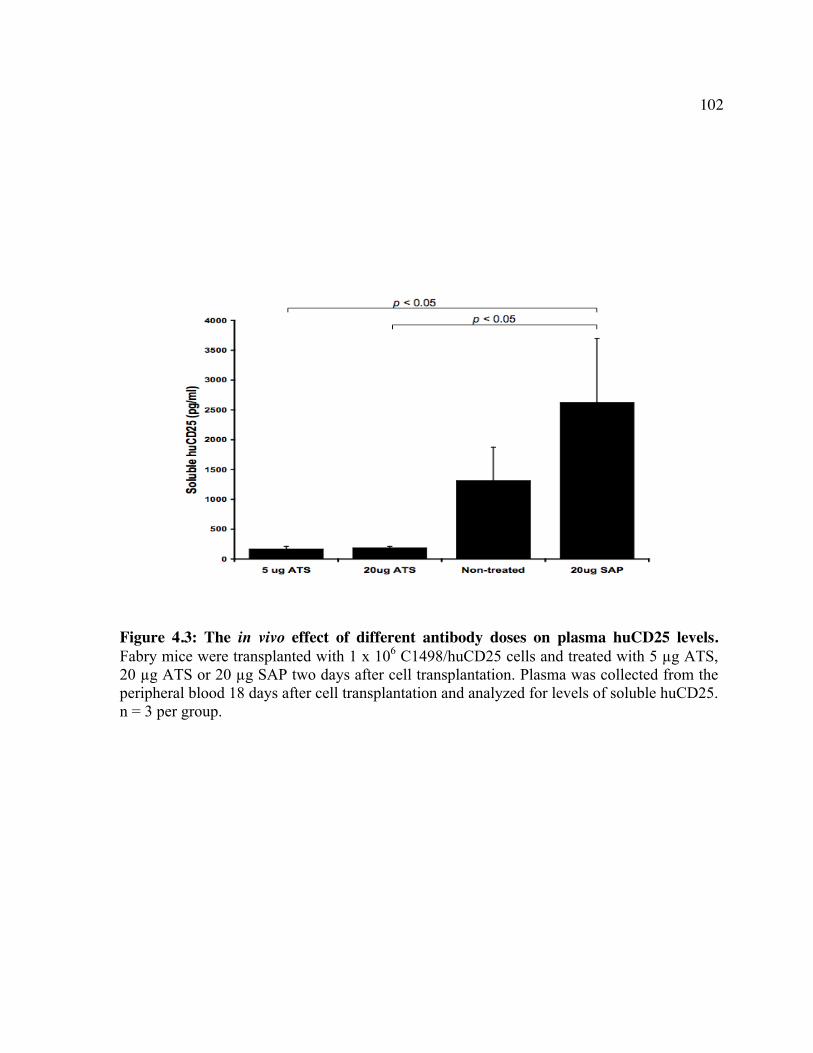
102
Figure 4.3: The in vivo effect of different antibody doses on plasma huCD25 levels. Fabry mice were transplanted with 1 x 106 C1498/huCD25 cells and treated with 5 µg ATS, 20 µg ATS or 20 µg SAP two days after cell transplantation. Plasma was collected from the peripheral blood 18 days after cell transplantation and analyzed for levels of soluble huCD25. n = 3 per group.
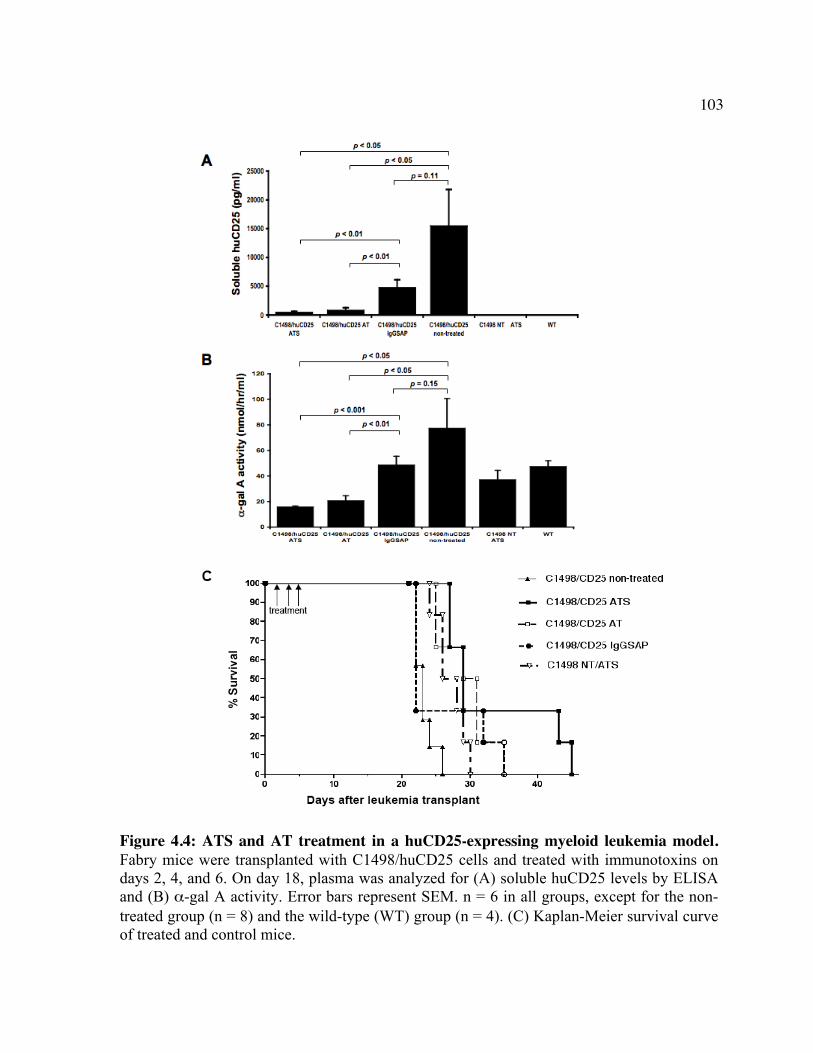
103
Figure 4.4: ATS and AT treatment in a huCD25-expressing myeloid leukemia model. Fabry mice were transplanted with C1498/huCD25 cells and treated with immunotoxins on days 2, 4, and 6. On day 18, plasma was analyzed for (A) soluble huCD25 levels by ELISA and (B) α-gal A activity. Error bars represent SEM. n = 6 in all groups, except for the non-treated group (n = 8) and the wild-type (WT) group (n = 4). (C) Kaplan-Meier survival curve of treated and control mice.
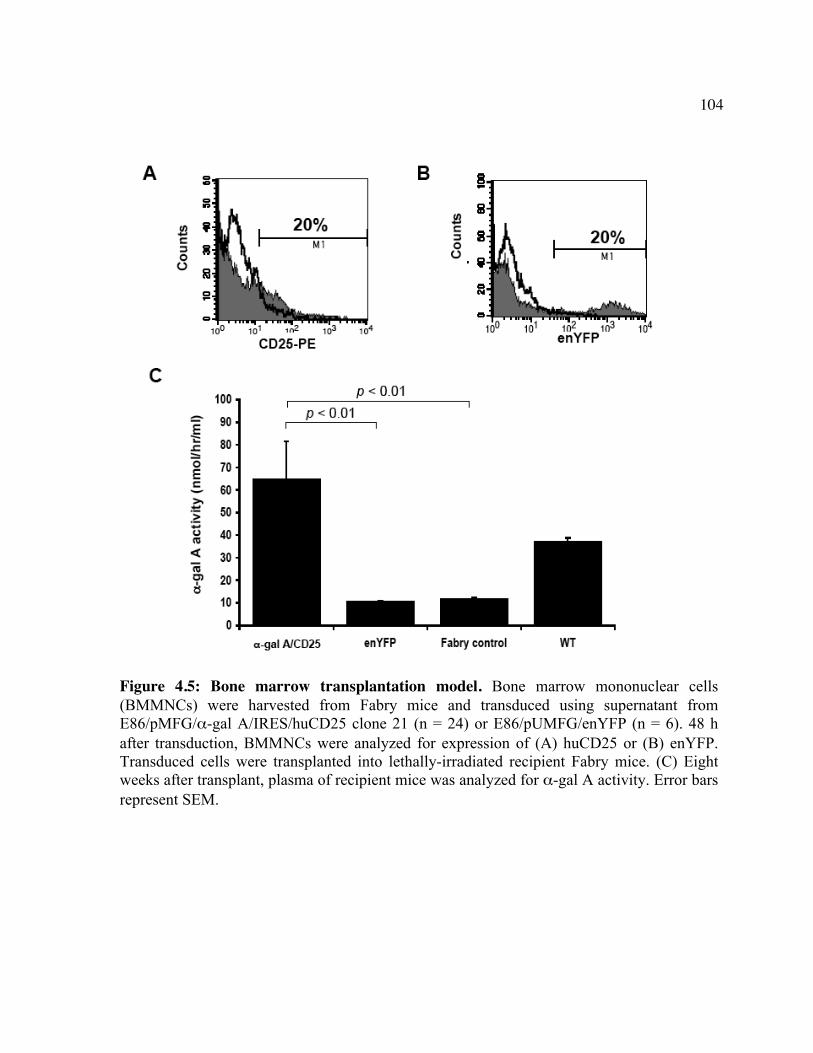
104
Figure 4.5: Bone marrow transplantation model. Bone marrow mononuclear cells (BMMNCs) were harvested from Fabry mice and transduced using supernatant from E86/pMFG/α-gal A/IRES/huCD25 clone 21 (n = 24) or E86/pUMFG/enYFP (n = 6). 48 h after transduction, BMMNCs were analyzed for expression of (A) huCD25 or (B) enYFP. Transduced cells were transplanted into lethally-irradiated recipient Fabry mice. (C) Eight weeks after transplant, plasma of recipient mice was analyzed for α-gal A activity. Error bars represent SEM.
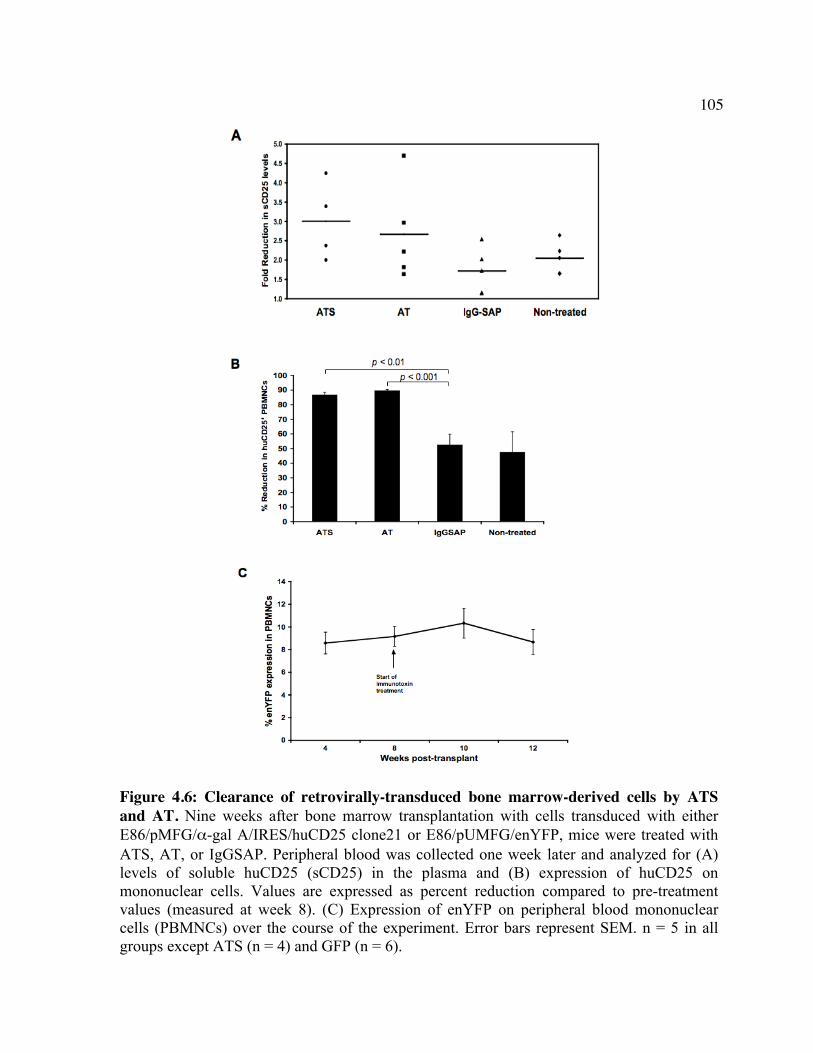
105
Figure 4.6: Clearance of retrovirally-transduced bone marrow-derived cells by ATS and AT. Nine weeks after bone marrow transplantation with cells transduced with either E86/pMFG/α-gal A/IRES/huCD25 clone21 or E86/pUMFG/enYFP, mice were treated with ATS, AT, or IgGSAP. Peripheral blood was collected one week later and analyzed for (A) levels of soluble huCD25 (sCD25) in the plasma and (B) expression of huCD25 on mononuclear cells. Values are expressed as percent reduction compared to pre-treatment values (measured at week 8). (C) Expression of enYFP on peripheral blood mononuclear cells (PBMNCs) over the course of the experiment. Error bars represent SEM. n = 5 in all groups except ATS (n = 4) and GFP (n = 6).
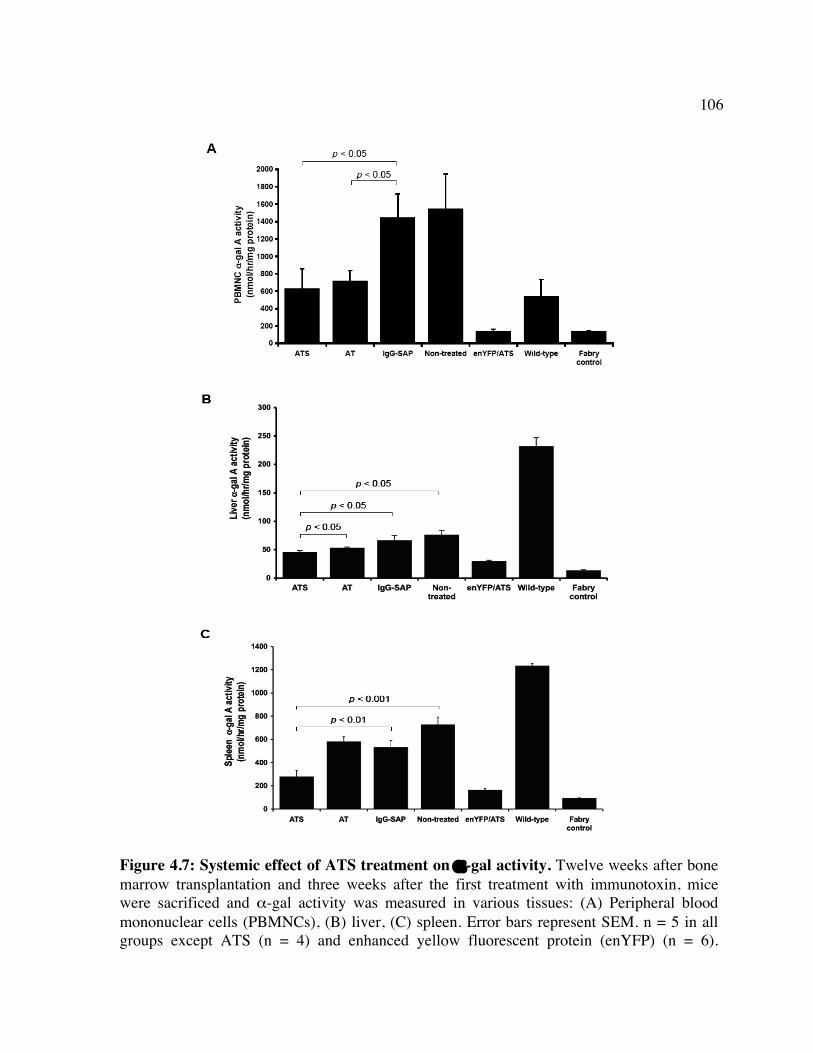
106
Figure 4.7: Systemic effect of ATS treatment on α-gal activity. Twelve weeks after bone marrow transplantation and three weeks after the first treatment with immunotoxin, mice were sacrificed and α-gal activity was measured in various tissues: (A) Peripheral blood mononuclear cells (PBMNCs), (B) liver, (C) spleen. Error bars represent SEM. n = 5 in all groups except ATS (n = 4) and enhanced yellow fluorescent protein (enYFP) (n = 6).

107
Chapter 5: Conclusions and Future Directions

108
The development of better treatment modalities for Farber disease remains important
since to date, allogeneic BMT has shown only limited success in treating the severe and most
common form of the disease [36, 66, 72, 73]. Gene therapy using retroviral vectors has the
potential to provide a lasting cure with a single treatment. Farber disease is a good candidate
for gene therapy since it is caused by a single gene defect for which the cDNA has been
cloned and the enzyme is well-characterized [79, 84]. Previous work towards the
development of a gene therapy strategy for Farber disease has come from the Medin
laboratory in 1999 in which an oncoretroviral vector (RV) was constructed to engineer
expression of human AC [176]. In that study, it was shown that transduction with the vector
could correct the enzymatic deficiency in immortalized Farber patient cells and that
transduced cells exhibited metabolic co-operativity effects whereby transduced cells secreted
AC that could be utilized by neighboring naïve cells [176].
The work presented in this thesis extends these preliminary studies and furthers the
development of gene therapy strategies for Farber disease. The vectors used here are more
clinically relevant since they incorporate a number of features that make them more suitable
for use in the clinic than the vector used in the 1999 study. The oncoretroviral backbone
contains splice donor and splice acceptor sites that result in the ability of higher titer virus to
be produced [239]. The virus is also pseudotyped with the RD114 envelope protein that is
resistant to inactivation by human sera and is stable during ultracentrifugation, allowing the
virus to be concentrated to further increase titers [230]. In addition to the RV, a lentiviral
vector (LV) was also constructed and tested since it is better at targeting more slowly
dividing cells such as HSPCs and neural cells. In addition, recent evidence suggests that LVs
may be safer than RV since it has been shown that RVs have a tendency to integrate near

109
transcriptional start sites and proto-oncogenes while LVs do not [204, 205]. The LV used is
based on a second generation HIV-based lentiviral system that incorporates a number of
safety features. It contains a self-inactivating long terminal repeat (LTR), a number of viral
genes have been removed and the remaining viral genes are separated onto multiple
plasmids, all of which reduce the risk of homologous recombination resulting in a wild-type
virus being formed [163]. It also pseudotyped with the VSVg envelope protein that allows
transduction of a wide variety of cell types and the concentration of the virus by
ultracentrifugation [279, 296, 297]. Further, both of the viruses constructed herein contain a
marking transgene, huCD25, that can be used to enrich for and track transduced cells and
also as a safety tag as was demonstrated here.
The efficacy of the viral vectors was first tested by transducing immortalized Farber
patient fibroblasts and B cells. This resulted in increased AC activity and decreased ceramide
storage in both cell lines. It was also shown that transduced Farber fibroblasts secrete AC that
can be taken up and utilized by non-transduced cells. This phenomenon of metabolic co-
operativity is important for diseases such as Farber disease where systemic correction is
required since it is virtually impossible to achieve lasting transduction in all cells.
To test the efficacy of the vectors in vivo, two surrogate models were developed since
a complete knockout of AC results in embryonic lethality in mice [77]. In order to test the
effect of transduction on human HSPCs cells, CB-derived CD34+ cells were transduced with
LV/AC/CD25 and then transplanted into irradiated NOD/SCID mice. Results show that
huCD25-expressing cells could engraft in the BM, indicating that AC overexpression did not
affect the ability of transduced cells to repopulate the hematopoietic system. These findings
suggest that HSPC transplantation using cells augmented to over-express AC is a viable

110
treatment option for the treatment of Farber disease. Indeed, HSPCs from other sources such
as the BM and mobilized PB would also be equally amenable to this approach.
In the second in vivo model, the direct delivery of virus to neonates was tested. Farber
disease typically claims the lives of victims by the age of two. As such, the administration of
gene therapy at the neonatal stage of life could reduce ceramide storage before irreversible
organ damage occurs and could be more effective. In addition, there is evidence in mice that
the BBB is not fully formed in neonates [243]. This may make it more permissive to the
entry of viral particles and as such, the neurological manifestations of Farber disease may be
better treated. The relatively immature immune system of neonates also makes tolerization
to the therapeutic protein a possibility, thus reducing the risk of the protein being eliminated
from the body by an antibody response. Therefore, a neonatal treatment approach was tested
using LV/AC/CD25. Virus was injected into the temporal vein of neonatal mice and
expression of soluble CD25 was monitored as a surrogate marker for tracking transgene
expression. It was found that mice were still transgene positive up to 14 weeks of age.
Measurement of AC activity in the organs showed that the livers of mice treated with
LV/AC/CD25 had increased AC activity compared to mice treated with LV/enGFP or
untreated mice. These findings suggest that AC expression can be restored systemically by
administration of viral vectors at the neonatal stage and offers another treatment regime for
Farber disease.
Another major concern for Farber disease is the treatment of the neurological
manifestations associated with the disease since this aspect has not been resolved using
traditional BMT. Thus, based on findings from a previous study in which treatment with
VEGF prior to administration of LV resulted in increased numbers of transduced cells in the

111
brain and improved functional outcomes [191], this approach was tested using a marking LV
expressing luc. In particular, the effect of VEGF pre-treatment on the ability of LV to infect
the organs was examined. Neonatal mice were injected with LV/luc following treatment with
VEGF. Expression of luc was monitored by WBLI for 12 weeks and it was found that luc
expression remained steady over the course of the experiment. Ex vivo imaging of the organs
showed that VEGF treatment increased the level of luc expression in organs compared to
organs from mice that were not treated with VEGF. These findings, combined with those
from the neonatal study using LV/AC/CD25, provide evidence that VEGF can be of
therapeutic benefit for increasing delivery of virus to the brain and other organs. This is
important for a disease such as Farber disease that has both visceral and neurological
manifestations.
The use of integrating vectors has attracted some negative attention for their potential
to cause insertional mutagenesis in transduced cells [196-198]. Therefore, the development
of improved viral vectors and gene therapy strategies is vital for successful translation to the
clinic. Here the CD25 marking transgene, previously used to enrich and track transduced
cells, was used in an antibody-based targeting strategy. The principle of this system is that
transduced cells will express CD25 from the gene expression cassette of the viral vector. If
integration causes a mutagenic event, it is expected that the leukemic cells will continue to
express CD25. Thus, it is proposed that a CD25 antibody or immunotoxin such as AT or
ATS can be used to target the CD25-expressing cells. As mentioned previously, this strategy
can be used to debulk tumour burden and can be combined with other anti-leukemic
strategies such as chemotherapy.

112
The efficacy of the strategy was first tested in vitro using a mouse leukemic cell line
over-expressing CD25 and it was found that treatment with both AT and ATS specifically
killed CD25-expressing leukemic cells. These reagents were also able to decrease the
leukemic burden and increase survival of mice with CD25-expressing leukemias. The safety
strategy that is proposed for the clinic was tested in a murine model of a related lysosomal
storage disease model. Fabry mice were transplanted with virus engineered to express the
relevant therapeutic transgene α-gal A and the huCD25 marker. Enzymatic activity was
restored in transplanted mice as determined by an increase in α-gal A activity. Following
treatment with ATS and AT, there was a reduction in circulating α-gal A activity as well as a
decrease in the level of expression of CD25 on PBMNCs. In addition, α-gal A activity was
reduced in the liver and spleen, indicating a CD25-specific clearance of cells. This strategy
can be implemented in the clinic in the event of the onset of tumorigenesis following gene
therapy using a clinically approved CD25 antibody such as daclizumab and basiliximab [298-
300].
The ultimate goal is that the gene therapy strategies presented in this thesis be
translated into the clinic for the treatment of Farber disease. Currently, allogeneic BMT is
used to treat Farber patients with some success in mildly affected patients [36, 66, 72, 73].
The transplantation of transduced HSPCs from sources such as CB, BM, or mobilized PB can
be implemented in a similar manner and augmenting them to over-express AC by
transduction with our vectors would be ideal. The ability to restore AC activity by
transduction with viral vectors such as those constructed here also allows for the possibility
to use autologous cells, which reduces the risk of GvHD and morbidity associated with
allogeneic transplantation [301]. Importantly, it also allows for transplantation in patients

113
who lack a matched donor. Future studies will involve transduction of CD34+ cells from CB,
BM and mobilized PB to compare the transduction efficiency and cell growth properties of
each transduced population since all of these are relevant for this type of therapy.
Ideally, administration of LV engineered to express AC would occur at a young age
in order to provide the most therapeutic effect. In addition, as demonstrated by the results
presented in this thesis, VEGF can be used to permeabilize the BBB to increase the efficacy
of viral delivery at the neonatal stage of development. Further, the Medin laboratory and
others have been investigating the possibility of performing in utero gene transfer [302],
representing yet another delivery approach that may be of therapeutic benefit. For these to be
possible, pre-natal or neonatal diagnosis of a Farber patient would be necessary. Efforts are
being made to implement neonatal screening for lysosomal storage diseases since their early
detection and treatment offers the best chance to prevent some of the irreversible organ
damage from occurring [303, 304], thus reducing both morbidity and mortality.
It also important to test the proposed gene therapy strategy in a relevant animal model
such as an AC-deficient mouse. However, it was found that homozygous knock-out of the
AC gene resulted in embryonic lethality [77]. While the heterozygous mouse survived, it
does not show any of the clinical symptoms of the disease. Thus, a better model is required.
Studies are currently underway in the Medin laboratory to develop mouse models of Farber
disease. In one method AC activity will be knocked down using shRNAs against murine AC.
This method has shown success in achieving gene knockdown using LVs engineered to
express the siRNA [305] and is similar to the one that will be undertaken by the Medin lab.
Gene trapping will also be used to replacing the wild-type AC gene with one that contains a
mutation found in a Farber patient having ~4% residual AC activity. In both of these designs,

114
it is hoped that the residual AC activity will allow the embryo to survive to birth. The
creation of a mouse model will allow for a more accurate evaluation of the efficacy of the
gene therapy strategy and will allow the evaluation of the immune response to the transgene
introduced by gene therapy.
Another important step towards the clinical application of the gene therapy strategies
is testing for safety in a large animal model. This is currently being done by the Medin lab in
non-human primates (NHP) using the LV/AC/CD25 vector. In these animals, toxicity of viral
delivery will be assessed and animals will be monitored for adverse events such as
tumorigenesis and abnormal hematopoiesis. In this protocol, PBMNCs from a rhesus
macaque were mobilized using G-CSF and were then transduced with LV/AC/CD25 and
transplanted back into the irradiated animal. The first animal has survived the transplant
crisis and it has been shown by both standard and real-time PCR by myself and others that
cells in the PB contain integrated LV up to 1 year post-transplant (data not shown). A second
animal has been successfully transplanted and a third is planned for 2008.
Together, the studies presented in this thesis have examined numerous aspects of
gene therapy for Farber disease. The viral vectors constructed and the studies presented
represent significant progress towards the treatment of Farber disease. Outside of traditional
BMT, which is available only to those with a matched donor, there is no viable treatment for
this debilitating disorder. As shown, there are a number of treatment regimes that can address
the underlying cause of the disease by engineering over-expression of human AC.
Transplantation of transduced HSPCs may be more suitable for older patients whereas direct
delivery of LV, either alone or in combination with VEGF, would be more suitable for
newborns. Regardless of the delivery method used, the LV contains a built-in safety system

115
that can be used in the event of insertional mutagenesis to debulk tumor burden. Combined
with the studies that are underway in the Medin laboratory, these studies represent significant
advances towards the development of gene therapy for Farber disease.

116
References

117
1. de Duve C. (1975). Exploring cells with a centrifuge. Science 189: 186-194.
2. Meikle PJ, Hopwood JJ, Clague AE, Carey WF. (1999). Prevalence of lysosomal storage disorders. JAMA 281: 249-254.
3. Pinto R, Caseiro C, Lemos M, Lopes L, Fontes A, Ribeiro H, et al. (2004). Prevalence of lysosomal storage diseases in Portugal. Eur. J. Hum. Genet. 12: 87-92.
4. Poorthuis BJ, Wevers RA, Kleijer WJ, Groener JE, de Jong JG, van Weely S, et al. (1999). The frequency of lysosomal storage diseases in The Netherlands. Hum. Genet. 105: 151-156.
5. Desnick RJ, Schuchman EH. (2002). Enzyme replacement and enhancement therapies: lessons from lysosomal disorders. Nat. Rev. Genet. 3: 954-966.
6. Beck M. (2007). New therapeutic options for lysosomal storage disorders: enzyme replacement, small molecules and gene therapy. Hum. Genet. 121: 1-22.
7. Vellodi A. (2005). Lysosomal storage disorders. Br. J. Haematol. 128: 413-431.
8. Futerman AH, van Meer G. (2004). The cell biology of lysosomal storage disorders. Nat. Rev. Mol. Cell Biol. 5: 554-565.
9. Conzelmann E, Sandhoff K. (1991). Biochemical basis of late-onset neurolipidoses. Dev. Neurosci. 13: 197-204.
10. Brady RO. (2006). Enzyme replacement for lysosomal diseases. Annu. Rev. Med. 57: 283-296.
11. de Duve C. (1964). From Cytases to Lysosomes. Fed. Proc. 23: 1045-1049.
12. Brady RO. (1966). The sphingolipidoses. N. Engl. J. Med. 275: 312-318.

118
13. Weinreb NJ, Charrow J, Andersson HC, Kaplan P, Kolodny EH, Mistry P, et al. (2002). Effectiveness of enzyme replacement therapy in 1028 patients with type 1 Gaucher disease after 2 to 5 years of treatment: a report from the Gaucher Registry. Am. J. Med. 113: 112-119.
14. Wraith JE, Clarke LA, Beck M, Kolodny EH, Pastores GM, Muenzer J, et al. (2004). Enzyme replacement therapy for mucopolysaccharidosis I: a randomized, double-blinded, placebo-controlled, multinational study of recombinant human alpha-L-iduronidase (laronidase). J. Pediatr. 144: 581-588.
15. Van den Hout JM, Kamphoven JH, Winkel LP, Arts WF, De Klerk JB, Loonen MC, et al. (2004). Long-term intravenous treatment of Pompe disease with recombinant human alpha-glucosidase from milk. Pediatrics 113: e448-57.
16. Harmatz P, Giugliani R, Schwartz I, Guffon N, Teles EL, Miranda MC, et al. (2006). Enzyme replacement therapy for mucopolysaccharidosis VI: a phase 3, randomized, double-blind, placebo-controlled, multinational study of recombinant human N-acetylgalactosamine 4-sulfatase (recombinant human arylsulfatase B or rhASB) and follow-on, open-label extension study. J. Pediatr. 148: 533-539.
17. Muenzer J, Wraith JE, Beck M, Giugliani R, Harmatz P, Eng CM, et al. (2006). A phase II/III clinical study of enzyme replacement therapy with idursulfase in mucopolysaccharidosis II (Hunter syndrome). Genet. Med. 8: 465-473.
18. Lidove O, Joly D, Barbey F, Bekri S, Alexandra JF, Peigne V, et al. (2007). Clinical results of enzyme replacement therapy in Fabry disease: a comprehensive review of literature. Int. J. Clin. Pract. 61: 293-302.
19. Burrow TA, Hopkin RJ, Leslie ND, Tinkle BT, Grabowski GA. (2007). Enzyme reconstitution/replacement therapy for lysosomal storage diseases. Curr. Opin. Pediatr. 19: 628-635.
20. Brady RO, Schiffmann R. (2004). Enzyme-replacement therapy for metabolic storage disorders. Lancet Neurol. 3: 752-756.
21. Germain DP, Kaneski CR, Brady RO. (2001). Mutation analysis of the acid beta-glucosidase gene in a patient with type 3 Gaucher disease and neutralizing antibody to

119
alglucerase. Mutat. Res. 483: 89-94.
22. Rosenberg M, Kingma W, Fitzpatrick MA, Richards SM. (1999). Immunosurveillance of alglucerase enzyme therapy for Gaucher patients: induction of humoral tolerance in seroconverted patients after repeat administration. Blood 93: 2081-2088.
23. Beutler E, Garber AM. (1994). Alglucerase for Gaucher's disease: dose, costs and benefits. Pharmacoeconomics 5: 453-459.
24. Moore DF, Ries M, Forget EL, Schiffmann R. (2007). Enzyme replacement therapy in orphan and ultra-orphan diseases: the limitations of standard economic metrics as exemplified by Fabry-Anderson disease. Pharmacoeconomics 25: 201-208.
25. Pastores GM. (2006). Miglustat: substrate reduction therapy for lysosomal storage disorders associated with primary central nervous system involvement. Recent. Patents CNS Drug Discov. 1: 77-82.
26. Aerts JM, Hollak CE, Boot RG, Groener JE, Maas M. (2006). Substrate reduction therapy of glycosphingolipid storage disorders. J. Inherit. Metab. Dis. 29: 449-456.
27. Pastores GM, Barnett NL, Kolodny EH. (2005). An open-label, noncomparative study of miglustat in type I Gaucher disease: efficacy and tolerability over 24 months of treatment. Clin. Ther. 27: 1215-1227.
28. Elstein D, Dweck A, Attias D, Hadas-Halpern I, Zevin S, Altarescu G, et al. (2007). Oral maintenance clinical trial with miglustat for type I Gaucher disease: switch from or combination with intravenous enzyme replacement. Blood 110: 2296-2301.
29. Patterson MC, Vecchio D, Prady H, Abel L, Wraith JE. (2007). Miglustat for treatment of Niemann-Pick C disease: a randomised controlled study. Lancet Neurol. 6: 765-772.
30. Bembi B, Marchetti F, Guerci VI, Ciana G, Addobbati R, Grasso D, et al. (2006). Substrate reduction therapy in the infantile form of Tay-Sachs disease. Neurology 66: 278-280.

120
31. Glaros EN, Kim WS, Quinn CM, Wong J, Gelissen I, Jessup W, et al. (2005). Glycosphingolipid accumulation inhibits cholesterol efflux via the ABCA1/apolipoprotein A-I pathway: 1-phenyl-2-decanoylamino-3-morpholino-1-propanol is a novel cholesterol efflux accelerator. J. Biol. Chem. 280: 24515-24523.
32. Tropak MB, Blanchard JE, Withers SG, Brown ED, Mahuran D. (2007). High-throughput screening for human lysosomal beta-N-Acetyl hexosaminidase inhibitors acting as pharmacological chaperones. Chem. Biol. 14: 153-164.
33. Chang HH, Asano N, Ishii S, Ichikawa Y, Fan JQ. (2006). Hydrophilic iminosugar active-site-specific chaperones increase residual glucocerebrosidase activity in fibroblasts from Gaucher patients. FEBS J. 273: 4082-4092.
34. Shimotori M, Maruyama H, Nakamura G, Suyama T, Sakamoto F, Itoh M, et al. (2008). Novel mutations of the GLA gene in Japanese patients with Fabry disease and their functional characterization by active site specific chaperone. Hum. Mutat. 29: 331.
35. Hobbs JR, Hugh-Jones K, Barrett AJ, Byrom N, Chambers D, Henry K, et al. (1981). Reversal of clinical features of Hurler's disease and biochemical improvement after treatment by bone-marrow transplantation. Lancet 2: 709-712.
36. Malatack JJ, Consolini DM, Bayever E. (2003). The status of hematopoietic stem cell transplantation in lysosomal storage disease. Pediatr. Neurol. 29: 391-403.
37. Ringden O, Groth CG, Erikson A, Backman L, Granqvist S, Mansson JE, et al. (1988). Long-term follow-up of the first successful bone marrow transplantation in Gaucher disease. Transplantation 46: 66-70.
38. Ringden O, Groth CG, Erikson A, Granqvist S, Mansson JE, Sparrelid E. (1995). Ten years' experience of bone marrow transplantation for Gaucher disease. Transplantation 59: 864-870.
39. Dunbar CE, Kohn DB, Schiffmann R, Barton NW, Nolta JA, Esplin JA, et al. (1998). Retroviral transfer of the glucocerebrosidase gene into CD34+ cells from patients with Gaucher disease: in vivo detection of transduced cells without myeloablation. Hum. Gene Ther. 9: 2629-2640.

121
40. Brady RO, Kanfer JN, Bradley RM, Shapiro D. (1966). Demonstration of a deficiency of glucocerebroside-cleaving enzyme in Gaucher's disease. J. Clin. Invest. 45: 1112-1115.
41. Beutler E, Grabowski GA (2001). Gaucher disease. In: Scriver CR, Beaudet AL, Sly WS and Valle D (eds). The Metabolic and Molecular Basis of Inherited Disease, 8th edn. McGraw-Hill: New York, pp 3635-3668.
42. Thomas WE, Winfield DA. (1991). Partial splenectomy for massive splenomegaly secondary to Gaucher's disease. Postgrad. Med. J. 67: 1072-1074.
43. Zer M, Freud E. (1992). Subtotal splenectomy in Gaucher's disease: towards a definition of critical splenic mass. Br. J. Surg. 79: 742-744.
44. Freud E, Cohen IJ, Neuman M, Mor C, Zer M. (1997). Should repeated partial splenectomy be attempted in patients with hematological diseases? Technical pitfalls and causes of failure in Gaucher's disease. J. Pediatr. Surg. 32: 1272-1276.
45. Ein SH, Shandling B, Simpson JS, Stephens CA, Bandi SK, Biggar WD, et al. (1977). The morbidity and mortality of splenectomy in childhood. Ann. Surg. 185: 307-310.
46. Pastores GM, Sibille AR, Grabowski GA. (1993). Enzyme therapy in Gaucher disease type 1: dosage efficacy and adverse effects in 33 patients treated for 6 to 24 months. Blood 82: 408-416.
47. Davies EH, Erikson A, Collin-Histed T, Mengel E, Tylki-Szymanska A, Vellodi A. (2007). Outcome of type III Gaucher disease on enzyme replacement therapy: review of 55 cases. J. Inherit. Metab. Dis. 30: 935-942.
48. Brady RO, Gal AE, Bradley RM, Martensson E, Warshaw AL, Laster L. (1967). Enzymatic defect in Fabry's disease. Ceramidetrihexosidase deficiency. N. Engl. J. Med. 276: 1163-1167.
49. Desnick RJ, Ioannou YA and Eng CM (2001). Fabry disease. In: Scriver CR, Beaudet AL, Sly WS and Valle D (eds). The Metabolic and Molecular Bases of Inherited Disease, 8th edn. McGraw-Hill: New York, pp 3733-3774.

122
50. Clavelou P, Besson G, Elziere C, Ferrier A, Pinard JM, Hermier M, et al. (2006). Neurological aspects of Fabry's disease. Rev. Neurol. (Paris) 162: 569-580.
51. Fellgiebel A, Muller MJ, Ginsberg L. (2006). CNS manifestations of Fabry's disease. Lancet Neurol. 5: 791-795.
52. von Scheidt W, Eng CM, Fitzmaurice TF, Erdmann E, Hubner G, Olsen EG, et al. (1991). An atypical variant of Fabry's disease with manifestations confined to the myocardium. N. Engl. J. Med. 324: 395-399.
53. Ries M, Clarke JT, Whybra C, Mehta A, Loveday KS, Brady RO, et al. (2007). Enzyme replacement in Fabry disease: pharmacokinetics and pharmacodynamics of agalsidase alpha in children and adolescents. J. Clin. Pharmacol. 47: 1222-1230.
54. Germain DP, Waldek S, Banikazemi M, Bushinsky DA, Charrow J, Desnick RJ, et al. (2007). Sustained, long-term renal stabilization after 54 months of agalsidase beta therapy in patients with Fabry disease. J. Am. Soc. Nephrol. 18: 1547-1557.
55. Erten Y, Ozdemir FN, Demirhan B, Karakayali H, Demirag A, Akkoc H. (1998). A case of Fabry's disease with normal kidney function at 10 years after successful renal transplantation. Transplant. Proc. 30: 842-843.
56. Dziemianko I, Jezior D, Boratynska M, Patrzalek D, Kuzniar J, Szyber P, et al. (2007). Kidney transplantation and enzyme alpha-galactosidase A therapy in patient with Fabry disease: a case report. Transplant. Proc. 39: 2925-2927.
57. Farber S. (1952). A lipid metabolic disorder: disseminated lipogranulomatosis; a syndrome with similarity to, and important difference from, Niemann-Pick and Hand-Schuller-Christian disease. AMA Am. J. Dis. Child. 84: 499-500.
58. Farber S, Cohen J, Uzman LL. (1957). Lipogranulomatosis; a new lipo-glycoprotein storage disease. J. Mt. Sinai Hosp. N. Y. 24: 816-837.
59. Schmoeckel C, Hohlfed M. (1979). A specific ultrastructural marker for disseminated lipogranulomatosis (Faber). Arch. Dermatol. Res. 266: 187-196.

123
60. Sugita M, Dulaney JT, Moser HW. (1972). Ceramidase deficiency in Farber's disease (lipogranulomatosis). Science 178: 1100-1102.
61. Li CM, Park JH, He X, Levy B, Chen F, Arai K, et al. (1999). The human acid ceramidase gene (ASAH): structure, chromosomal location, mutation analysis, and expression. Genomics 62: 223-231.
62. Moser HW (2001). Acid ceramidase deficiency: Farber lipogranulomatosis. In: Scriver CR, Beaudet AL, Sly WS and Valle D (eds). McGraw-Hill: New York, pp 3573-3588.
63. Dulaney JT, Milunsky A, Sidbury JB, Hobolth N, Moser HW. (1976). Diagnosis of lipogranulomatosis (Farber disease) by use of cultured fibroblasts. J. Pediatr. 89: 59-61.
64. Levade T, Moser HW, Fensom AH, Harzer K, Moser AB, Salvayre R. (1995). Neurodegenerative course in ceramidase deficiency (Farber disease) correlates with the residual lysosomal ceramide turnover in cultured living patient cells. J. Neurol. Sci. 134: 108-114.
65. van Echten-Deckert G, Klein A, Linke T, Heinemann T, Weisgerber J, Sandhoff K. (1997). Turnover of endogenous ceramide in cultured normal and Farber fibroblasts. J. Lipid Res. 38: 2569-2579.
66. Yeager AM, Uhas KA, Coles CD, Davis PC, Krause WL, Moser HW. (2000). Bone marrow transplantation for infantile ceramidase deficiency (Farber disease). Bone Marrow Transplant. 26: 357-363.
67. Antonarakis SE, Valle D, Moser HW, Moser A, Qualman SJ, Zinkham WH. (1984). Phenotypic variability in siblings with Farber disease. J. Pediatr. 104: 406-409.
68. Eviatar L, Sklower SL, Wisniewski K, Feldman RS, Gochoco A. (1986). Farber lipogranulomatosis: an unusual presentation in a black child. Pediatr. Neurol. 2: 371-374.
69. Fusch C, Huenges R, Moser HW, Sewell AC, Roggendorf W, Kustermann-Kuhn B, et al. (1989). A case of combined Farber and Sandhoff disease. Eur. J. Pediatr. 148: 558-

124
562.
70. Schnabel D, Schroder M, Furst W, Klein A, Hurwitz R, Zenk T, et al. (1992). Simultaneous deficiency of sphingolipid activator proteins 1 and 2 is caused by a mutation in the initiation codon of their common gene. J. Biol. Chem. 267: 3312-3315.
71. Haraoka G, Muraoka M, Yoshioka N, Wakami S, Hayashi I. (1997). First case of surgical treatment of Farber's disease. Ann. Plast. Surg. 39: 405-410.
72. Vormoor J, Ehlert K, Groll AH, Koch HG, Frosch M, Roth J. (2004). Successful hematopoietic stem cell transplantation in Farber disease. J. Pediatr. 144: 132-134.
73. Ehlert K, Roth J, Frosch M, Fehse N, Zander N, Vormoor J. (2006). Farber's disease without central nervous system involvement: bone-marrow transplantation provides a promising new approach. Ann. Rheum. Dis. 65: 1665-1666.
74. Ehlert K, Frosch M, Fehse N, Zander A, Roth J, Vormoor J. (2007). Farber disease: clinical presentation, pathogenesis and a new approach to treatment. Pediatr. Rheumatol. Online J. 5: 15.
75. Souillet G, Guibaud P, Fensom AH, Maire I and Zabot MT (1989). Outcome of displacement bone marrow transplantation in Farber's disease: a report of a case. In: Hobbs JR (eds). Correction of Certain Genetic Diseases by Transplantation, COGENT: London, pp 137-141.
76. Hoogerbrugge PM, Brouwer OF, Bordigoni P, Ringden O, Kapaun P, Ortega JJ, et al. (1995). Allogeneic bone marrow transplantation for lysosomal storage diseases. The European Group for Bone Marrow Transplantation. Lancet 345: 1398-1402.
77. Li CM, Park JH, Simonaro CM, He X, Gordon RE, Friedman AH, et al. (2002). Insertional mutagenesis of the mouse acid ceramidase gene leads to early embryonic lethality in homozygotes and progressive lipid storage disease in heterozygotes. Genomics 79: 218-224.
78. Eliyahu E, Park JH, Shtraizent N, He X, Schuchman EH. (2007). Acid ceramidase is a novel factor required for early embryo survival. FASEB J. 21: 1403-1409.

125
79. Koch J, Gartner S, Li CM, Quintern LE, Bernardo K, Levran O, et al. (1996). Molecular cloning and characterization of a full-length complementary DNA encoding human acid ceramidase. Identification Of the first molecular lesion causing Farber disease. J. Biol. Chem. 271: 33110-33115.
80. Ferlinz K, Kopal G, Bernardo K, Linke T, Bar J, Breiden B, et al. (2001). Human acid ceramidase: processing, glycosylation, and lysosomal targeting. J. Biol. Chem. 276: 35352-35360.
81. Shtraizent N, Eliyahu E, Park JH, He X, Shalgi R, Schuchman EH. (2008). Autoproteolytic cleavage and activation of human acid ceramidase. J. Biol. Chem. .
82. Schulze H, Schepers U, Sandhoff K. (2007). Overexpression and mass spectrometry analysis of mature human acid ceramidase. Biol. Chem. 388: 1333-1343.
83. Gatt S. (1963). Enzymic Hydrolysis and Synthesis of Ceramides. J. Biol. Chem. 238: 3131-3133.
84. Bernardo K, Hurwitz R, Zenk T, Desnick RJ, Ferlinz K, Schuchman EH, et al. (1995). Purification, characterization, and biosynthesis of human acid ceramidase. J. Biol. Chem. 270: 11098-11102.
85. He X, Okino N, Dhami R, Dagan A, Gatt S, Schulze H, et al. (2003). Purification and characterization of recombinant, human acid ceramidase. Catalytic reactions and interactions with acid sphingomyelinase. J. Biol. Chem. 278: 32978-32986.
86. Okino N, He X, Gatt S, Sandhoff K, Ito M, Schuchman EH. (2003). The reverse activity of human acid ceramidase. J. Biol. Chem. 278: 29948-29953.
87. Zhang Z, Mandal AK, Mital A, Popescu N, Zimonjic D, Moser A, et al. (2000). Human acid ceramidase gene: novel mutations in Farber disease. Mol. Genet. Metab. 70: 301-309.
88. Seelan RS, Qian C, Yokomizo A, Bostwick DG, Smith DI, Liu W. (2000). Human acid ceramidase is overexpressed but not mutated in prostate cancer. Genes Chromosomes

126
Cancer 29: 137-146.
89. Bar J, Linke T, Ferlinz K, Neumann U, Schuchman EH, Sandhoff K. (2001). Molecular analysis of acid ceramidase deficiency in patients with Farber disease. Hum. Mutat. 17: 199-209.
90. Muramatsu T, Sakai N, Yanagihara I, Yamada M, Nishigaki T, Kokubu C, et al. (2002). Mutation analysis of the acid ceramidase gene in Japanese patients with Farber disease. J. Inherit. Metab. Dis. 25: 585-592.
91. Devi AR, Gopikrishna M, Ratheesh R, Savithri G, Swarnalata G, Bashyam M. (2006). Farber lipogranulomatosis: clinical and molecular genetic analysis reveals a novel mutation in an Indian family. J. Hum. Genet. 51: 811-814.
92. El Bawab S, Roddy P, Qian T, Bielawska A, Lemasters JJ, Hannun YA. (2000). Molecular cloning and characterization of a human mitochondrial ceramidase. J. Biol. Chem. 275: 21508-21513.
93. Hwang YH, Tani M, Nakagawa T, Okino N, Ito M. (2005). Subcellular localization of human neutral ceramidase expressed in HEK293 cells. Biochem. Biophys. Res. Commun. 331: 37-42.
94. Ohlsson L, Palmberg C, Duan RD, Olsson M, Bergman T, Nilsson A. (2007). Purification and characterization of human intestinal neutral ceramidase. Biochimie 89: 950-960.
95. Tani M, Okino N, Mori K, Tanigawa T, Izu H, Ito M. (2000). Molecular cloning of the full-length cDNA encoding mouse neutral ceramidase. A novel but highly conserved gene family of neutral/alkaline ceramidases. J. Biol. Chem. 275: 11229-11234.
96. Mao C, Xu R, Szulc ZM, Bielawska A, Galadari SH, Obeid LM. (2001). Cloning and characterization of a novel human alkaline ceramidase. A mammalian enzyme that hydrolyzes phytoceramide. J. Biol. Chem. 276: 26577-26588.
97. Mao C, Xu R, Szulc ZM, Bielawski J, Becker KP, Bielawska A, et al. (2003). Cloning and characterization of a mouse endoplasmic reticulum alkaline ceramidase: an enzyme that preferentially regulates metabolism of very long chain ceramides. J. Biol. Chem.

127
278: 31184-31191.
98. Kolesnick RN, Goni FM, Alonso A. (2000). Compartmentalization of ceamide signalling: Physical foundations and biological effects. J. Cell. Physiol. 184: 285-300.
99. Merrill AHJ, Schmelz EM, Dillehay DL, Spiegel S, Shayman JA, Schroeder JJ, et al. (1997). Sphingolipids--the enigmatic lipid class: biochemistry, physiology, and pathophysiology. Toxicol. Appl. Pharmacol. 142: 208-225.
100. Liu P, Anderson RG. (1995). Compartmentalized production of ceramide at the cell surface. J. Biol. Chem. 270: 27179-27185.
101. Simons K, Ikonen E. (1997). Functional rafts in cell membranes. Nature 387: 369-372.
102. Kolesnick RN. (2002). The therapeutic potential of regulating the ceramide/sphingomyelin pathway. J. Clin. Invest. 110: 3-8.
103. Ruvolo PP. (2003). Intracellular signal transduction pathways activated by ceramide and its metabolites. Pharmacol. Res. 47: 383-392.
104. Dbaibo GS, Hannun YA. (1998). Signal transduction and the regulation of apoptosis: roles of ceramide. Apoptosis 3: 317-334.
105. Perry DK, Hannun YA. (1998). The role of ceramide in cell signaling. Biochim. Biophys. Acta 1436: 233-243.
106. Hannun YA. (1996). Functions of ceramide in coordinating cellular responses to stress. Science 274: 1855-1859.
107. Spiegel S, Merrill AH, Jr. (1996). Sphingolipid metabolism and cell growth regulation. FASEB J. 10: 1388-1397.
108. Schwarz A, Futerman AH. (1997). Distinct roles for ceramide and glucosylceramide at different stages of neuronal growth. J. Neurosci. 17: 2929-2938.

128
109. Venable ME, Lee JY, Smyth MJ, Bielawska A, Obeid LM. (1995). Role of ceramide in cellular senescence. J. Biol. Chem. 270: 30701-30708.
110. Zhang P, Liu B, Jenkins GM, Hannun YA, Obeid LM. (1997). Expression of neutral sphingomyelinase identifies a distinct pool of sphingomyelin involved in apoptosis. J. Biol. Chem. 272: 9609-9612.
111. Holleran WM, Takagi Y, Uchida Y. (2006). Epidermal sphingolipids: metabolism, function, and roles in skin disorders. FEBS Lett. 580: 5456-5466.
112. Jensen JM, Forl M, Winoto-Morbach S, Seite S, Schunck M, Proksch E, et al. (2005). Acid and neutral sphingomyelinase, ceramide synthase, and acid ceramidase activities in cutaneous aging. Exp. Dermatol. 14: 609-618.
113. Scarlatti F, Bauvy C, Ventruti A, Sala G, Cluzeaud F, Vandewalle A, et al. (2004). Ceramide-mediated macroautophagy involves inhibition of protein kinase B and up-regulation of beclin 1. J. Biol. Chem. 279: 18384-18391.
114. Sakata A, Ochiai T, Shimeno H, Hikishima S, Yokomatsu T, Shibuya S, et al. (2007). Acid sphingomyelinase inhibition suppresses lipopolysaccharide-mediated release of inflammatory cytokines from macrophages and protects against disease pathology in dextran sulphate sodium-induced colitis in mice. Immunology 122: 54-64.
115. Summers SA, Garza LA, Zhou H, Birnbaum MJ. (1998). Regulation of insulin-stimulated glucose transporter GLUT4 translocation and Akt kinase activity by ceramide. Mol. Cell. Biol. 18: 5457-5464.
116. Schmitz-Peiffer C, Craig DL, Biden TJ. (1999). Ceramide generation is sufficient to account for the inhibition of the insulin-stimulated PKB pathway in C2C12 skeletal muscle cells pretreated with palmitate. J. Biol. Chem. 274: 24202-24210.
117. Lang PA, Schenck M, Nicolay JP, Becker JU, Kempe DS, Lupescu A, et al. (2007). Liver cell death and anemia in Wilson disease involve acid sphingomyelinase and ceramide. Nat. Med. 13: 164-170.

129
118. Goggel R, Winoto-Morbach S, Vielhaber G, Imai Y, Lindner K, Brade L, et al. (2004). PAF-mediated pulmonary edema: a new role for acid sphingomyelinase and ceramide. Nat. Med. 10: 155-160.
119. Grassme H, Jendrossek V, Riehle A, von Kurthy G, Berger J, Schwarz H, et al. (2003). Host defense against Pseudomonas aeruginosa requires ceramide-rich membrane rafts. Nat. Med. 9: 322-330.
120. Grassme H, Riehle A, Wilker B, Gulbins E. (2005). Rhinoviruses infect human epithelial cells via ceramide-enriched membrane platforms. J. Biol. Chem. 280: 26256-26262.
121. Spiegel S, Kolesnick R. (2002). Sphingosine 1-phosphate as a therapeutic agent. Leukemia 16: 1596-1602.
122. Verheij M, Bose R, Lin XH, Yao B, Jarvis WD, Grant S, et al. (1996). Requirement for ceramide-initiated SAPK/JNK signalling in stress-induced apoptosis. Nature 380: 75-79.
123. Fox TE, Houck KL, O'Neill SM, Nagarajan M, Stover TC, Pomianowski PT, et al. (2007). Ceramide recruits and activates protein kinase C zeta (PKC zeta) within structured membrane microdomains. J. Biol. Chem. 282: 12450-12457.
124. Zhang Y, Yao B, Delikat S, Bayoumy S, Lin XH, Basu S, et al. (1997). Kinase suppressor of Ras is ceramide-activated protein kinase. Cell 89: 63-72.
125. Dobrowsky RT, Kamibayashi C, Mumby MC, Hannun YA. (1993). Ceramide activates heterotrimeric protein phosphatase 2A. J. Biol. Chem. 268: 15523-15530.
126. Chalfant CE, Kishikawa K, Mumby MC, Kamibayashi C, Bielawska A, Hannun YA. (1999). Long chain ceramides activate protein phosphatase-1 and protein phosphatase-2A. Activation is stereospecific and regulated by phosphatidic acid. J. Biol. Chem. 274: 20313-20317.
127. Lee JY, Hannun YA, Obeid LM. (1996). Ceramide inactivates cellular protein kinase Calpha. J. Biol. Chem. 271: 13169-13174.

130
128. Schubert KM, Scheid MP, Duronio V. (2000). Ceramide inhibits protein kinase B/Akt by promoting dephosphorylation of serine 473. J. Biol. Chem. 275: 13330-13335.
129. Ruvolo PP, Clark W, Mumby M, Gao F, May WS. (2002). A functional role for the B56 alpha-subunit of protein phosphatase 2A in ceramide-mediated regulation of Bcl2 phosphorylation status and function. J. Biol. Chem. 277: 22847-22852.
130. Chiang CW, Harris G, Ellig C, Masters SC, Subramanian R, Shenolikar S, et al. (2001). Protein phosphatase 2A activates the proapoptotic function of BAD in interleukin- 3-dependent lymphoid cells by a mechanism requiring 14-3-3 dissociation. Blood 97: 1289-1297.
131. Dbaibo GS, Pushkareva MY, Jayadev S, Schwarz JK, Horowitz JM, Obeid LM, et al. (1995). Retinoblastoma gene product as a downstream target for a ceramide-dependent pathway of growth arrest. Proc. Natl. Acad. Sci. U. S. A. 92: 1347-1351.
132. Perez GI, Jurisicova A, Matikainen T, Moriyama T, Kim MR, Takai Y, et al. (2005). A central role for ceramide in the age-related acceleration of apoptosis in the female germline. FASEB J. 19: 860-862.
133. Furland NE, Zanetti SR, Oresti GM, Maldonado EN, Aveldano MI. (2007). Ceramides and sphingomyelins with high proportions of very long-chain polyunsaturated fatty acids in mammalian germ cells. J. Biol. Chem. 282: 18141-18150.
134. Huang Y, Tanimukai H, Liu F, Iqbal K, Grundke-Iqbal I, Gong CX. (2004). Elevation of the level and activity of acid ceramidase in Alzheimer's disease brain. Eur. J. Neurosci. 20: 3489-3497.
135. Satoi H, Tomimoto H, Ohtani R, Kitano T, Kondo T, Watanabe M, et al. (2005). Astroglial expression of ceramide in Alzheimer's disease brains: a role during neuronal apoptosis. Neuroscience 130: 657-666.
136. Alesse E, Zazzeroni F, Angelucci A, Giannini G, Di Marcotullio L, Gulino A. (1998). The growth arrest and downregulation of c-myc transcription induced by ceramide are related events dependent on p21 induction, Rb underphosphorylation and E2F sequestering. Cell Death Differ. 5: 381-389.

131
137. Kimura K, Markowski M, Edsall LC, Spiegel S, Gelmann EP. (2003). Role of ceramide in mediating apoptosis of irradiated LNCaP prostate cancer cells. Cell Death Differ. 10: 240-248.
138. Saad AF, Meacham WD, Bai A, Anelli V, Elojeimy S, Mahdy AE, et al. (2007). The Functional Effects of Acid Ceramidase Overexpression in Prostate Cancer Progression and Resistance to Chemotherapy. Cancer. Biol. Ther. 6: .
139. Tohyama J, Oya Y, Ezoe T, Vanier MT, Nakayasu H, Fujita N, et al. (1999). Ceramide accumulation is associated with increased apoptotic cell death in cultured fibroblasts of sphingolipid activator protein-deficient mouse but not in fibroblasts of patients with Farber disease. J. Inherit. Metab. Dis. 22: 649-662.
140. Segui B, Bezombes C, Uro-Coste E, Medin JA, Andrieu-Abadie N, Auge N, et al. (2000). Stress-induced apoptosis is not mediated by endolysosomal ceramide. FASEB J. 14: 36-47.
141. Farina F, Cappello F, Todaro M, Bucchieri F, Peri G, Zummo G, et al. (2000). Involvement of caspase-3 and GD3 ganglioside in ceramide-induced apoptosis in Farber disease. J. Histochem. Cytochem. 48: 57-62.
142. Strelow A, Bernardo K, Adam-Klages S, Linke T, Sandhoff K, Kronke M, et al. (2000). Overexpression of acid ceramidase protects from tumor necrosis factor-induced cell death. J. Exp. Med. 192: 601-612.
143. Burek C, Roth J, Koch HG, Harzer K, Los M, Schulze-Osthoff K. (2001). The role of ceramide in receptor- and stress-induced apoptosis studied in acidic ceramidase-deficient Farber disease cells. Oncogene 20: 6493-6502.
144. Maupas-Schwalm F, Auge N, Robinet C, Cambus JP, Parsons SJ, Salvayre R, et al. (2004). The sphingomyelin/ceramide pathway is involved in ERK1/2 phosphorylation, cell proliferation, and uPAR overexpression induced by tissue-type plasminogen activator. FASEB J. 18: 1398-1400.
145. Betito S, Cuvillier O. (2006). Regulation by sphingosine 1-phosphate of Bax and Bad activities during apoptosis in a MEK-dependent manner. Biochem. Biophys. Res.

132
Commun. 340: 1273-1277.
146. Gangoiti P, Granado MH, Wang SW, Kong JY, Steinbrecher UP, Gomez-Munoz A. (2008). Ceramide 1-phosphate stimulates macrophage proliferation through activation of the PI3-kinase/PKB, JNK and ERK1/2 pathways. Cell. Signal. 20: 726-736.
147. Shirahama T, Sakakura C, Sweeney EA, Ozawa M, Takemoto M, Nishiyama K, et al. (1997). Sphingosine induces apoptosis in androgen-independent human prostatic carcinoma DU-145 cells by suppression of bcl-X(L) gene expression. FEBS Lett. 407: 97-100.
148. Lechardeur D, Lukacs GL. (2002). Intracellular barriers to non-viral gene transfer. Curr Gene Ther 2: 183-194.
149. Medin JA, Karlsson S. (1997). Viral vectors for gene therapy of hematopoietic cells. Immunotechnology 3: 3-19.
150. Bergelson JM, Cunningham JA, Droguett G, Kurt-Jones EA, Krithivas A, Hong JS, et al. (1997). Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5. Science 275: 1320-1323.
151. Bett AJ, Haddara W, Prevec L, Graham FL. (1994). An efficient and flexible system for construction of adenovirus vectors with insertions or deletions in early regions 1 and 3. Proc. Natl. Acad. Sci. U. S. A. 91: 8802-8806.
152. Verma IM, Weitzman MD. (2005). Gene therapy: twenty-first century medicine. Annu. Rev. Biochem. 74: 711-738.
153. Kootstra NA, Verma IM. (2003). Gene therapy with viral vectors. Annu. Rev. Pharmacol. Toxicol. 43: 413-439.
154. Chirmule N, Xiao W, Truneh A, Schnell MA, Hughes JV, Zoltick P, et al. (2000). Humoral immunity to adeno-associated virus type 2 vectors following administration to murine and nonhuman primate muscle. J. Virol. 74: 2420-2425.

133
155. Srinivasakumar N. (2001). HIV-1 vector systems. Somat. Cell Mol. Genet. 26: 51-81.
156. Palu G, Parolin C, Takeuchi Y, Pizzato M. (2000). Progress with retroviral gene vectors. Rev. Med. Virol. 10: 185-202.
157. Kappes JC, Wu X. (2001). Safety considerations in vector development. Somat. Cell Mol. Genet. 26: 147-158.
158. Zentilin L, Qin G, Tafuro S, Dinauer MC, Baum C, Giacca M. (2000). Variegation of retroviral vector gene expression in myeloid cells. Gene Ther. 7: 153-166.
159. Curran MA, Nolan GP. (2002). Nonprimate lentiviral vectors. Curr. Top. Microbiol. Immunol. 261: 75-105.
160. Popov S, Rexach M, Zybarth G, Reiling N, Lee MA, Ratner L, et al. (1998). Viral protein R regulates nuclear import of the HIV-1 pre-integration complex. EMBO J. 17: 909-917.
161. Haffar OK, Popov S, Dubrovsky L, Agostini I, Tang H, Pushkarsky T, et al. (2000). Two nuclear localization signals in the HIV-1 matrix protein regulate nuclear import of the HIV-1 pre-integration complex. J. Mol. Biol. 299: 359-368.
162. Sutton RE, Reitsma MJ, Uchida N, Brown PO. (1999). Transduction of human progenitor hematopoietic stem cells by human immunodeficiency virus type 1-based vectors is cell cycle dependent. J. Virol. 73: 3649-3660.
163. Zufferey R, Nagy D, Mandel RJ, Naldini L, Trono D. (1997). Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat. Biotechnol. 15: 871-875.
164. Miyoshi H, Blomer U, Takahashi M, Gage FH, Verma IM. (1998). Development of a self-inactivating lentivirus vector. J. Virol. 72: 8150-8157.
165. Zufferey R, Dull T, Mandel RJ, Bukovsky A, Quiroz D, Naldini L, et al. (1998). Self-inactivating lentivirus vector for safe and efficient in vivo gene delivery. J. Virol. 72: 9873-9880.

134
166. Danos O, Mulligan RC. (1988). Safe and efficient generation of recombinant retroviruses with amphotropic and ecotropic host ranges. Proc. Natl. Acad. Sci. U. S. A. 85: 6460-6464.
167. Kafri T, van Praag H, Ouyang L, Gage FH, Verma IM. (1999). A packaging cell line for lentivirus vectors. J. Virol. 73: 576-584.
168. Yoshimitsu M, Sato T, Tao K, Walia JS, Rasaiah VI, Sleep GT, et al. (2004). Bioluminescent imaging of a marking transgene and correction of Fabry mice by neonatal injection of recombinant lentiviral vectors. Proc. Natl. Acad. Sci. U. S. A. 101: 16909-16914.
169. Kaplitt MG, Feigin A, Tang C, Fitzsimons HL, Mattis P, Lawlor PA, et al. (2007). Safety and tolerability of gene therapy with an adeno-associated virus (AAV) borne GAD gene for Parkinson's disease: an open label, phase I trial. Lancet 369: 2097-2105.
170. Oldfield EH, Ram Z, Culver KW, Blaese RM, DeVroom HL, Anderson WF. (1993). Gene therapy for the treatment of brain tumors using intra-tumoral transduction with the thymidine kinase gene and intravenous ganciclovir. Hum. Gene Ther. 4: 39-69.
171. Stewart DJ, Hilton JD, Arnold JM, Gregoire J, Rivard A, Archer SL, et al. (2006). Angiogenic gene therapy in patients with nonrevascularizable ischemic heart disease: a phase 2 randomized, controlled trial of AdVEGF(121) (AdVEGF121) versus maximum medical treatment. Gene Ther. 13: 1503-1511.
172. Kaufman HL, Cohen S, Cheung K, DeRaffele G, Mitcham J, Moroziewicz D, et al. (2006). Local delivery of vaccinia virus expressing multiple costimulatory molecules for the treatment of established tumors. Hum. Gene Ther. 17: 239-244.
173. Dunbar CE, Kohn DB, Schiffmann R, Barton NW, Nolta JA, Esplin JA, et al. (1998). Retroviral transfer of the glucocerebrosidase gene into CD34+ cells from patients with Gaucher disease: in vivo detection of transduced cells without myeloablation. Hum. Gene Ther. 9: 2629-2640.
174. Becker S, Wasser S, Hauses M, Hossle JP, Ott MG, Dinauer MC, et al. (1998). Correction of respiratory burst activity in X-linked chronic granulomatous cells to therapeutically relevant levels after gene transfer into bone marrow CD34+ cells. Hum.

135
Gene Ther. 9: 1561-1570.
175. Mavilio F, Pellegrini G, Ferrari S, Di Nunzio F, Di Iorio E, Recchia A, et al. (2006). Correction of junctional epidermolysis bullosa by transplantation of genetically modified epidermal stem cells. Nat. Med. 12: 1397-1402.
176. Medin JA, Takenaka T, Carpentier S, Garcia V, Basile JP, Segui B, et al. (1999). Retrovirus-mediated correction of the metabolic defect in cultured Farber disease cells. Hum. Gene Ther. 10: 1321-1329.
177. Uchida N, Fleming WH, Alpern EJ, Weissman IL. (1993). Heterogeneity of hematopoietic stem cells. Curr. Opin. Immunol. 5: 177-184.
178. Ioannou YA, Bishop DF, Desnick RJ. (1992). Overexpression of human alpha-galactosidase A results in its intracellular aggregation, crystallization in lysosomes, and selective secretion. J. Cell Biol. 119: 1137-1150.
179. Souillet G, Guffon N, Maire I, Pujol M, Taylor P, Sevin F, et al. (2003). Outcome of 27 patients with Hurler's syndrome transplanted from either related or unrelated haematopoietic stem cell sources. Bone Marrow Transplant. 31: 1105-1117.
180. Jin HK, Schuchman EH. (2003). Ex vivo gene therapy using bone marrow-derived cells: combined effects of intracerebral and intravenous transplantation in a mouse model of Niemann-Pick disease. Mol. Ther. 8: 876-885.
181. Di Domenico C, Villani GR, Di Napoli D, Reyero EG, Lombardo A, Naldini L, et al. (2005). Gene therapy for a mucopolysaccharidosis type I murine model with lentiviral-IDUA vector. Hum. Gene Ther. 16: 81-90.
182. Enquist IB, Nilsson E, Ooka A, Mansson JE, Olsson K, Ehinger M, et al. (2006). Effective cell and gene therapy in a murine model of Gaucher disease. Proc. Natl. Acad. Sci. U. S. A. 103: 13819-13824.
183. Hao QL, Shah AJ, Thiemann FT, Smogorzewska EM, Crooks GM. (1995). A functional comparison of CD34 + CD38- cells in cord blood and bone marrow. Blood 86: 3745-3753.

136
184. Wang JC, Doedens M, Dick JE. (1997). Primitive human hematopoietic cells are enriched in cord blood compared with adult bone marrow or mobilized peripheral blood as measured by the quantitative in vivo SCID-repopulating cell assay. Blood 89: 3919-3924.
185. Gluckman E, Koegler G, Rocha V. (2005). Human leukocyte antigen matching in cord blood transplantation. Semin. Hematol. 42: 85-90.
186. Krivit W, Sung JH, Shapiro EG, Lockman LA. (1995). Microglia: the effector cell for reconstitution of the central nervous system following bone marrow transplantation for lysosomal and peroxisomal storage diseases. Cell Transplant. 4: 385-392.
187. Deroose CM, Reumers V, Gijsbers R, Bormans G, Debyser Z, Mortelmans L, et al. (2006). Noninvasive monitoring of long-term lentiviral vector-mediated gene expression in rodent brain with bioluminescence imaging. Mol. Ther. 14: 423-431.
188. Geraerts M, Eggermont K, Hernandez-Acosta P, Garcia-Verdugo JM, Baekelandt V, Debyser Z. (2006). Lentiviral vectors mediate efficient and stable gene transfer in adult neural stem cells in vivo. Hum. Gene Ther. 17: 635-650.
189. Fedorova E, Battini L, Prakash-Cheng A, Marras D, Gusella GL. (2006). Lentiviral gene delivery to CNS by spinal intrathecal administration to neonatal mice. J. Gene Med. 8: 414-424.
190. Barnett FH, Rainov NG, Ikeda K, Schuback DE, Elliott P, Kramm CM, et al. (1999). Selective delivery of herpes virus vectors to experimental brain tumors using RMP-7. Cancer Gene Ther. 6: 14-20.
191. Young PP, Fantz CR, Sands MS. (2004). VEGF disrupts the neonatal blood-brain barrier and increases life span after non-ablative BMT in a murine model of congenital neurodegeneration caused by a lysosomal enzyme deficiency. Exp. Neurol. 188: 104-114.
192. Dobrenis K. (1998). Microglia in cell culture and in transplantation therapy for central nervous system disease. Methods 16: 320-344.

137
193. Kaur C, Hao AJ, Wu CH, Ling EA. (2001). Origin of microglia. Microsc. Res. Tech. 54: 2-9.
194. Priller J, Flugel A, Wehner T, Boentert M, Haas CA, Prinz M, et al. (2001). Targeting gene-modified hematopoietic cells to the central nervous system: use of green fluorescent protein uncovers microglial engraftment. Nat. Med. 7: 1356-1361.
195. Vallieres L, Sawchenko PE. (2003). Bone marrow-derived cells that populate the adult mouse brain preserve their hematopoietic identity. J. Neurosci. 23: 5197-5207.
196. Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P, et al. (2003). LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science 302: 415-419.
197. Check E. (2005). Gene therapy put on hold as third child develops cancer. Nature 433: 561.
198. Marshall E. (2003). Gene therapy. Second child in French trial is found to have leukemia. Science 299: 320.
199. McCormack MP, Rabbitts TH. (2004). Activation of the T-cell oncogene LMO2 after gene therapy for X-linked severe combined immunodeficiency. N. Engl. J. Med. 350: 913-922.
200. Pike-Overzet K, van der Burg M, Wagemaker G, van Dongen JJ, Staal FJ. (2007). New insights and unresolved issues regarding insertional mutagenesis in X-linked SCID gene therapy. Mol. Ther. 15: 1910-1916.
201. Woods NB, Bottero V, Schmidt M, von Kalle C, Verma IM. (2006). Gene therapy: therapeutic gene causing lymphoma. Nature 440: 1123.
202. Dave UP, Jenkins NA, Copeland NG. (2004). Gene therapy insertional mutagenesis insights. Science 303: 333.
203. Baum C, von Kalle C, Staal FJ, Li Z, Fehse B, Schmidt M, et al. (2004). Chance or necessity? Insertional mutagenesis in gene therapy and its consequences. Mol. Ther. 9:

138
5-13.
204. De Palma M, Montini E, Santoni de Sio FR, Benedicenti F, Gentile A, Medico E, et al. (2005). Promoter trapping reveals significant differences in integration site selection between MLV and HIV vectors in primary hematopoietic cells. Blood 105: 2307-2315.
205. Wu X, Li Y, Crise B, Burgess SM. (2003). Transcription start regions in the human genome are favored targets for MLV integration. Science 300: 1749-1751.
206. Montini E, Cesana D, Schmidt M, Sanvito F, Ponzoni M, Bartholomae C, et al. (2006). Hematopoietic stem cell gene transfer in a tumor-prone mouse model uncovers low genotoxicity of lentiviral vector integration. Nat. Biotechnol. 24: 687-696.
207. Li Z, Dullmann J, Schiedlmeier B, Schmidt M, von Kalle C, Meyer J, et al. (2002). Murine leukemia induced by retroviral gene marking. Science 296: 497.
208. Calmels B, Ferguson C, Laukkanen MO, Adler R, Faulhaber M, Kim HJ, et al. (2005). Recurrent retroviral vector integration at the Mds1/Evi1 locus in nonhuman primate hematopoietic cells. Blood 106: 2530-2533.
209. Modlich U, Bohne J, Schmidt M, von Kalle C, Knoss S, Schambach A, et al. (2006). Cell-culture assays reveal the importance of retroviral vector design for insertional genotoxicity. Blood 108: 2545-2553.
210. Siapati EK, Bigger BW, Kashofer K, Themis M, Thrasher AJ, Bonnet D. (2007). Murine leukemia following irradiation conditioning for transplantation of lentivirally-modified hematopoietic stem cells. Eur. J. Haematol. 78: 303-313.
211. Edelstein ML, Abedi MR, Wixon J. (2007). Gene therapy clinical trials worldwide to 2007--an update. J. Gene Med. 9: 833-842.
212. Ng YY, Bloem AC, van Kessel B, Lokhorst H, Logtenberg T, Staal FJ. (2002). Selective in vitro expansion and efficient retroviral transduction of human CD34+ CD38- haematopoietic stem cells. Br. J. Haematol. 117: 226-237.

139
213. Nagashima T, Ueda Y, Hanazono Y, Kume A, Shibata H, Ageyama N, et al. (2004). In vivo expansion of gene-modified hematopoietic cells by a novel selective amplifier gene utilizing the erythropoietin receptor as a molecular switch. J. Gene Med. 6: 22-31.
214. Medin JA, Migita M, Pawliuk R, Jacobson S, Amiri M, Kluepfel-Stahl S, et al. (1996). A bicistronic therapeutic retroviral vector enables sorting of transduced CD34+ cells and corrects the enzyme deficiency in cells from Gaucher patients. Blood 87: 1754-1762.
215. Taniguchi T, Minami Y. (1993). The IL-2/IL-2 receptor system: a current overview. Cell 73: 5-8.
216. Hanisch UK, Quirion R. (1995). Interleukin-2 as a neuroregulatory cytokine. Brain Res. Brain Res. Rev. 21: 246-284.
217. David D, Bani L, Moreau JL, Demaison C, Sun K, Salvucci O, et al. (1998). Further analysis of interleukin-2 receptor subunit expression on the different human peripheral blood mononuclear cell subsets. Blood 91: 165-172.
218. Qin G, Takenaka T, Telsch K, Kelley L, Howard T, Levade T, et al. (2001). Preselective gene therapy for Fabry disease. Proc. Natl. Acad. Sci. U. S. A. 98: 3428-3433.
219. Kreitman RJ, Wilson WH, White JD, Stetler-Stevenson M, Jaffe ES, Giardina S, et al. (2000). Phase I trial of recombinant immunotoxin anti-Tac(Fv)-PE38 (LMB-2) in patients with hematologic malignancies. J. Clin. Oncol. 18: 1622-1636.
220. Kreitman RJ, Wilson WH, Robbins D, Margulies I, Stetler-Stevenson M, Waldmann TA, et al. (1999). Responses in refractory hairy cell leukemia to a recombinant immunotoxin. Blood 94: 3340-3348.
221. Schnell R, Vitetta E, Schindler J, Borchmann P, Barth S, Ghetie V, et al. (2000). Treatment of refractory Hodgkin's lymphoma patients with an anti-CD25 ricin A-chain immunotoxin. Leukemia 14: 129-135.
222. Chen HR, Ji SQ, Wang HX, Yan HM, Zhu L, Liu J, et al. (2003). Humanized anti-CD25 monoclonal antibody for prophylaxis of graft-vs-host disease (GVHD) in haploidentical bone marrow transplantation without ex vivo T-cell depletion. Exp. Hematol. 31: 1019-

140
1025.
223. Lin M, Ming A, Zhao M. (2006). Two-dose basiliximab compared with two-dose daclizumab in renal transplantation: a clinical study. Clin. Transplant. 20: 325-329.
224. Onizuka S, Tawara I, Shimizu J, Sakaguchi S, Fujita T, Nakayama E. (1999). Tumor rejection by in vivo administration of anti-CD25 (interleukin-2 receptor alpha) monoclonal antibody. Cancer Res. 59: 3128-3133.
225. Fecci PE, Sweeney AE, Grossi PM, Nair SK, Learn CA, Mitchell DA, et al. (2006). Systemic anti-CD25 monoclonal antibody administration safely enhances immunity in murine glioma without eliminating regulatory T cells. Clin. Cancer Res. 12: 4294-4305.
226. Knutson KL, Dang Y, Lu H, Lukas J, Almand B, Gad E, et al. (2006). IL-2 immunotoxin therapy modulates tumor-associated regulatory T cells and leads to lasting immune-mediated rejection of breast cancers in neu-transgenic mice. J. Immunol. 177: 84-91.
227. Hannun YA, Luberto C. (2000). Ceramide in the eukaryotic stress response. Trends Cell Biol. 10: 73-80.
228. Takenaka T, Qin G, Brady RO, Medin JA. (1999). Circulating alpha-galactosidase A derived from transduced bone marrow cells: relevance for corrective gene transfer for Fabry disease. Hum. Gene Ther. 10: 1931-1939.
229. Yoshimitsu M, Higuchi K, Ramsubir S, Nonaka T, Rasaiah VI, Siatskas C, et al. (2007). Efficient correction of Fabry mice and patient cells mediated by lentiviral transduction of hematopoietic stem/progenitor cells. Gene Ther. 14: 256-265.
230. Cosset FL, Takeuchi Y, Battini JL, Weiss RA, Collins MK. (1995). High-titer packaging cells producing recombinant retroviruses resistant to human serum. J. Virol. 69: 7430-7436.
231. Levade T, Leruth M, Graber D, Moisand A, Vermeersch S, Salvayre R, et al. (1996). In situ assay of acid sphingomyelinase and ceramidase based on LDL-mediated lysosomal targeting of ceramide-labeled sphingomyelin. J. Lipid Res. 37: 2525-2538.

141
232. He X, Li CM, Park JH, Dagan A, Gatt S, Schuchman EH. (1999). A fluorescence-based high-performance liquid chromatographic assay to determine acid ceramidase activity. Anal. Biochem. 274: 264-269.
233. Folch J, Lees M, Sloane Stanley GH. (1957). A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 226: 497-509.
234. Bielawska A, Perry DK, Hannun YA. (2001). Determination of ceramides and diglycerides by the diglyceride kinase assay. Anal. Biochem. 298: 141-150.
235. McKenzie JL, Gan OI, Doedens M, Dick JE. (2005). Human short-term repopulating stem cells are efficiently detected following intrafemoral transplantation into NOD/SCID recipients depleted of CD122+ cells. Blood 106: 1259-1261.
236. Tanaka T, Kitamura F, Nagasaka Y, Kuida K, Suwa H, Miyasaka M. (1993). Selective long-term elimination of natural killer cells in vivo by an anti-interleukin 2 receptor beta chain monoclonal antibody in mice. J. Exp. Med. 178: 1103-1107.
237. Chatelut M, Harzer K, Christomanou H, Feunteun J, Pieraggi MT, Paton BC, et al. (1997). Model SV40-transformed fibroblast lines for metabolic studies of human prosaposin and acid ceramidase deficiencies. Clin. Chim. Acta 262: 61-76.
238. Greiner DL, Hesselton RA, Shultz LD. (1998). SCID mouse models of human stem cell engraftment. Stem Cells 16: 166-177.
239. Krall WJ, Skelton DC, Yu XJ, Riviere I, Lehn P, Mulligan RC, et al. (1996). Increased levels of spliced RNA account for augmented expression from the MFG retroviral vector in hematopoietic cells. Gene Ther. 3: 37-48.
240. Naldini L, Blomer U, Gage FH, Trono D, Verma IM. (1996). Efficient transfer, integration, and sustained long-term expression of the transgene in adult rat brains injected with a lentiviral vector. Proc. Natl. Acad. Sci. U. S. A. 93: 11382-11388.
241. Medin JA, Brandt JE, Rozler E, Nelson M, Bartholomew A, Li C, et al. (1999). Ex vivo expansion and genetic marking of primitive human and baboon hematopoietic cells. Ann.

142
N. Y. Acad. Sci. 872: 233-40; discussion 240-2.
242. Lapidot T, Fajerman Y, Kollet O. (1997). Immune-deficient SCID and NOD/SCID mice models as functional assays for studying normal and malignant human hematopoiesis. J. Mol. Med. 75: 664-673.
243. Sarzotti M. (1997). Immunologic tolerance. Curr. Opin. Hematol. 4: 48-52.
244. Tylki-Szymanska A, Berger J, Loschl B, Lugowska A, Molzer B. (1996). Late juvenile metachromatic leukodystrophy (MLD) in three patients with a similar clinical course and identical mutation on one allele. Clin. Genet. 50: 287-292.
245. Jones MZ, Alroy J, Rutledge JC, Taylor JW, Alvord EC,Jr, Toone J, et al. (1997). Human mucopolysaccharidosis IIID: clinical, biochemical, morphological and immunohistochemical characteristics. J. Neuropathol. Exp. Neurol. 56: 1158-1167.
246. Mut M, Cila A, Varli K, Akalan N. (2005). Multilevel myelopathy in Maroteaux-Lamy syndrome and review of the literature. Clin. Neurol. Neurosurg. 107: 230-235.
247. Barness LA, Henry K, Kling P, Laxova R, Kaback M, Gilbert-Barness E. (1991). A 7-year old white-male boy with progressive neurological deterioration. Am. J. Med. Genet. 40: 271-279.
248. Taylor RM, Wolfe JH. (1997). Decreased lysosomal storage in the adult MPS VII mouse brain in the vicinity of grafts of retroviral vector-corrected fibroblasts secreting high levels of beta-glucuronidase. Nat. Med. 3: 771-774.
249. Savas PS, Hemsley KM, Hopwood JJ. (2004). Intracerebral injection of sulfamidase delays neuropathology in murine MPS-IIIA. Mol. Genet. Metab. 82: 273-285.
250. Brooks AI, Stein CS, Hughes SM, Heth J, McCray PM,Jr, Sauter SL, et al. (2002). Functional correction of established central nervous system deficits in an animal model of lysosomal storage disease with feline immunodeficiency virus-based vectors. Proc. Natl. Acad. Sci. U. S. A. 99: 6216-6221.

143
251. Hennig AK, Levy B, Ogilvie JM, Vogler CA, Galvin N, Bassnett S, et al. (2003). Intravitreal gene therapy reduces lysosomal storage in specific areas of the CNS in mucopolysaccharidosis VII mice. J. Neurosci. 23: 3302-3307.
252. Haskell RE, Hughes SM, Chiorini JA, Alisky JM, Davidson BL. (2003). Viral-mediated delivery of the late-infantile neuronal ceroid lipofuscinosis gene, TPP-I to the mouse central nervous system. Gene Ther. 10: 34-42.
253. Bates DO, Lodwick D, Williams B. (1999). Vascular endothelial growth factor and microvascular permeability. Microcirculation 6: 83-96.
254. Nagy JA, Vasile E, Feng D, Sundberg C, Brown LF, Detmar MJ, et al. (2002). Vascular permeability factor/vascular endothelial growth factor induces lymphangiogenesis as well as angiogenesis. J. Exp. Med. 196: 1497-1506.
255. Casella I, Feccia T, Chelucci C, Samoggia P, Castelli G, Guerriero R, et al. (2003). Autocrine-paracrine VEGF loops potentiate the maturation of megakaryocytic precursors through Flt1 receptor. Blood 101: 1316-1323.
256. Karihaloo A, Karumanchi SA, Cantley WL, Venkatesha S, Cantley LG, Kale S. (2005). Vascular endothelial growth factor induces branching morphogenesis/tubulogenesis in renal epithelial cells in a neuropilin-dependent fashion. Mol. Cell. Biol. 25: 7441-7448.
257. Nishijima K, Ng YS, Zhong L, Bradley J, Schubert W, Jo N, et al. (2007). Vascular endothelial growth factor-A is a survival factor for retinal neurons and a critical neuroprotectant during the adaptive response to ischemic injury. Am. J. Pathol. 171: 53-67.
258. Proescholdt MA, Heiss JD, Walbridge S, Muhlhauser J, Capogrossi MC, Oldfield EH, et al. (1999). Vascular endothelial growth factor (VEGF) modulates vascular permeability and inflammation in rat brain. J. Neuropathol. Exp. Neurol. 58: 613-627.
259. Qu-Hong, Nagy JA, Senger DR, Dvorak HF, Dvorak AM. (1995). Ultrastructural localization of vascular permeability factor/vascular endothelial growth factor (VPF/VEGF) to the abluminal plasma membrane and vesiculovacuolar organelles of tumor microvascular endothelium. J. Histochem. Cytochem. 43: 381-389.

144
260. Dvorak HF, Brown LF, Detmar M, Dvorak AM. (1995). Vascular permeability factor/vascular endothelial growth factor, microvascular hyperpermeability, and angiogenesis. Am. J. Pathol. 146: 1029-1039.
261. Young PP, Hofling AA, Sands MS. (2002). VEGF increases engraftment of bone marrow-derived endothelial progenitor cells (EPCs) into vasculature of newborn murine recipients. Proc. Natl. Acad. Sci. U. S. A. 99: 11951-11956.
262. Boussif O, Lezoualc'h F, Zanta MA, Mergny MD, Scherman D, Demeneix B, et al. (1995). A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc. Natl. Acad. Sci. U. S. A. 92: 7297-7301.
263. Baekelandt V, Claeys A, Cherepanov P, De Clercq E, De Strooper B, Nuttin B, et al. (2000). DNA-Dependent protein kinase is not required for efficient lentivirus integration. J. Virol. 74: 11278-11285.
264. Sharp TV, Wang HW, Koumi A, Hollyman D, Endo Y, Ye H, et al. (2002). K15 protein of Kaposi's sarcoma-associated herpesvirus is latently expressed and binds to HAX-1, a protein with antiapoptotic function. J. Virol. 76: 802-816.
265. Linhart A, Lubanda JC, Palecek T, Bultas J, Karetova D, Ledvinova J, et al. (2001). Cardiac manifestations in Fabry disease. J. Inherit. Metab. Dis. 24 Suppl 2: 75-83; discussion 65.
266. Shah JS, Hughes DA, Sachdev B, Tome M, Ward D, Lee P, et al. (2005). Prevalence and clinical significance of cardiac arrhythmia in Anderson-Fabry disease. Am. J. Cardiol. 96: 842-846.
267. Wilcken DE. (2003). Overview of inherited metabolic disorders causing cardiovascular disease. J. Inherit. Metab. Dis. 26: 245-257.
268. Carbonaro DA, Jin X, Petersen D, Wang X, Dorey F, Kil KS, et al. (2006). In vivo transduction by intravenous injection of a lentiviral vector expressing human ADA into neonatal ADA gene knockout mice: a novel form of enzyme replacement therapy for ADA deficiency. Mol. Ther. 13: 1110-1120.

145
269. Aiuti A, Slavin S, Aker M, Ficara F, Deola S, Mortellaro A, et al. (2002). Correction of ADA-SCID by stem cell gene therapy combined with nonmyeloablative conditioning. Science 296: 2410-2413.
270. Gaspar HB, Parsley KL, Howe S, King D, Gilmour KC, Sinclair J, et al. (2004). Gene therapy of X-linked severe combined immunodeficiency by use of a pseudotyped gammaretroviral vector. Lancet 364: 2181-2187.
271. Robbins PD, Ghivizzani SC. (1998). Viral vectors for gene therapy. Pharmacol. Ther. 80: 35-47.
272. Medin JA, Tudor M, Simovitch R, Quirk JM, Jacobson S, Murray GJ, et al. (1996). Correction in trans for Fabry disease: expression, secretion and uptake of alpha-galactosidase A in patient-derived cells driven by a high-titer recombinant retroviral vector. Proc. Natl. Acad. Sci. U. S. A. 93: 7917-7922.
273. Takenaka T, Murray GJ, Qin G, Quirk JM, Ohshima T, Qasba P, et al. (2000). Long-term enzyme correction and lipid reduction in multiple organs of primary and secondary transplanted Fabry mice receiving transduced bone marrow cells. Proc. Natl. Acad. Sci. U. S. A. 97: 7515-7520.
274. Gaffen SL. (2001). Signaling domains of the interleukin 2 receptor. Cytokine 14: 63-77.
275. Rubin LA, Kurman CC, Fritz ME, Biddison WE, Boutin B, Yarchoan R, et al. (1985). Soluble interleukin 2 receptors are released from activated human lymphoid cells in vitro. J. Immunol. 135: 3172-3177.
276. Uchiyama T, Broder S, Waldmann TA. (1981). A monoclonal antibody (anti-Tac) reactive with activated and functionally mature human T cells. I. Production of anti-Tac monoclonal antibody and distribution of Tac (+) cells. J. Immunol. 126: 1393-1397.
277. Lappi DA, Esch FS, Barbieri L, Stirpe F, Soria M. (1985). Characterization of a Saponaria officinalis seed ribosome-inactivating protein: immunoreactivity and sequence homologies. Biochem. Biophys. Res. Commun. 129: 934-942.

146
278. Barbieri L, Valbonesi P, Bonora E, Gorini P, Bolognesi A, Stirpe F. (1997). Polynucleotide:adenosine glycosidase activity of ribosome-inactivating proteins: effect on DNA, RNA and poly(A). Nucleic Acids Res. 25: 518-522.
279. Naldini L, Blomer U, Gallay P, Ory D, Mulligan R, Gage FH, et al. (1996). In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 272: 263-267.
280. Ohshima T, Murray GJ, Swaim WD, Longenecker G, Quirk JM, Cardarelli CO, et al. (1997). alpha-Galactosidase A deficient mice: a model of Fabry disease. Proc. Natl. Acad. Sci. U. S. A. 94: 2540-2544.
281. Zhang Z, Zhang M, Garmestani K, Talanov VS, Plascjak PS, Beck B, et al. (2006). Effective treatment of a murine model of adult T-cell leukemia using 211At-7G7/B6 and its combination with unmodified anti-Tac (daclizumab) directed toward CD25. Blood 108: 1007-1012.
282. Perez-Encinas M, Villamayor M, Campos A, Gonzalez S, Bello JL. (1998). Tumor burden and serum level of soluble CD25, CD8, CD23, CD54 and CD44 in non-Hodgkin's lymphoma. Haematologica 83: 752-754.
283. Boelens JJ. (2006). Trends in haematopoietic cell transplantation for inborn errors of metabolism. J. Inherit. Metab. Dis. 29: 413-420.
284. Dillman RO. (2001). Monoclonal antibody therapy for lymphoma: an update. Cancer Pract. 9: 71-80.
285. Gokbuget N, Hoelzer D. (2004). Treatment with monoclonal antibodies in acute lymphoblastic leukemia: current knowledge and future prospects. Ann. Hematol. 83: 201-205.
286. McLaughlin P, Grillo-Lopez AJ, Link BK, Levy R, Czuczman MS, Williams ME, et al. (1998). Rituximab chimeric anti-CD20 monoclonal antibody therapy for relapsed indolent lymphoma: half of patients respond to a four-dose treatment program. J. Clin. Oncol. 16: 2825-2833.

147
287. Grillo-Lopez AJ, Hedrick E, Rashford M, Benyunes M. (2002). Rituximab: ongoing and future clinical development. Semin. Oncol. 29: 105-112.
288. Cvetkovic RS, Perry CM. (2006). Rituximab: a review of its use in non-Hodgkin's lymphoma and chronic lymphocytic leukaemia. Drugs 66: 791-820.
289. Introna M, Barbui AM, Bambacioni F, Casati C, Gaipa G, Borleri G, et al. (2000). Genetic modification of human T cells with CD20: a strategy to purify and lyse transduced cells with anti-CD20 antibodies. Hum. Gene Ther. 11: 611-620.
290. van Meerten T, Claessen MJ, Hagenbeek A, Ebeling SB. (2006). The CD20/alphaCD20 'suicide' system: novel vectors with improved safety and expression profiles and efficient elimination of CD20-transgenic T cells. Gene Ther. 13: 789-797.
291. Serafini M, Manganini M, Borleri G, Bonamino M, Imberti L, Biondi A, et al. (2004). Characterization of CD20-transduced T lymphocytes as an alternative suicide gene therapy approach for the treatment of graft-versus-host disease. Hum. Gene Ther. 15: 63-76.
292. Zhang M, Zhang Z, Goldman CK, Janik J, Waldmann TA. (2005). Combination therapy for adult T-cell leukemia-xenografted mice: flavopiridol and anti-CD25 monoclonal antibody. Blood 105: 1231-1236.
293. Jaeger G, Neumeister P, Brezinschek R, Hofler G, Quehenberger F, Linkesch W, et al. (2002). Rituximab (anti-CD20 monoclonal antibody) as consolidation of first-line CHOP chemotherapy in patients with follicular lymphoma: a phase II study. Eur. J. Haematol. 69: 21-26.
294. Claviez A, Eckert C, Seeger K, Schrauder A, Schrappe M, Henze G, et al. (2006). Rituximab plus chemotherapy in children with relapsed or refractory CD20-positive B-cell precursor acute lymphoblastic leukemia. Haematologica 91: 272-273.
295. Silvertown JD, Walia JS, Medin JA. (2005). Cloning, sequencing and characterization of lentiviral-mediated expression of rhesus macaque (Macaca mulatta) interleukin-2 receptor alpha cDNA. Dev. Comp. Immunol. 29: 989-1002.

148
296. Schlegel R, Tralka TS, Willingham MC, Pastan I. (1983). Inhibition of VSV binding and infectivity by phosphatidylserine: is phosphatidylserine a VSV-binding site? Cell 32: 639-646.
297. Coil DA, Miller AD. (2004). Phosphatidylserine is not the cell surface receptor for vesicular stomatitis virus. J. Virol. 78: 10920-10926.
298. Waldmann TA. (2007). Daclizumab (anti-Tac, Zenapax) in the treatment of leukemia/lymphoma. Oncogene 26: 3699-3703.
299. Waldmann TA. (2007). Anti-Tac (daclizumab, Zenapax) in the treatment of leukemia, autoimmune diseases, and in the prevention of allograft rejection: a 25-year personal odyssey. J. Clin. Immunol. 27: 1-18.
300. Schmidt-Hieber M, Fietz T, Knauf W, Uharek L, Hopfenmuller W, Thiel E, et al. (2005). Efficacy of the interleukin-2 receptor antagonist basiliximab in steroid-refractory acute graft-versus-host disease. Br. J. Haematol. 130: 568-574.
301. Laughlin MJ, Eapen M, Rubinstein P, Wagner JE, Zhang MJ, Champlin RE, et al. (2004). Outcomes after transplantation of cord blood or bone marrow from unrelated donors in adults with leukemia. N. Engl. J. Med. 351: 2265-2275.
302. Tarantal AF, Lee CI, Ekert JE, McDonald R, Kohn DB, Plopper CG, et al. (2001). Lentiviral vector gene transfer into fetal rhesus monkeys (Macaca mulatta): lung-targeting approaches. Mol. Ther. 4: 614-621.
303. Li Y, Scott CR, Chamoles NA, Ghavami A, Pinto BM, Turecek F, et al. (2004). Direct multiplex assay of lysosomal enzymes in dried blood spots for newborn screening. Clin. Chem. 50: 1785-1796.
304. Meikle PJ, Grasby DJ, Dean CJ, Lang DL, Bockmann M, Whittle AM, et al. (2006). Newborn screening for lysosomal storage disorders. Mol. Genet. Metab. 88: 307-314.
305. Tiscornia G, Singer O, Ikawa M, Verma IM. (2003). A general method for gene knockdown in mice by using lentiviral vectors expressing small interfering RNA. Proc.

149
Natl. Acad. Sci. U. S. A. 100: 1844-1848.