
Response of the Bacteriophage T4 Replisome to Noncoding Lesions and Regression of a Stalled...
14
Response of the Bacteriophage T4 Replisome to Noncoding Lesions and Regression of a Stalled Replication Fork Scott W. Nelson 1 ⁎ and Stephen J. Benkovic 2 1 Department of Biochemistry, Biophysics, and Molecular Biology, 4112 Molecular Biology Building, Iowa State University, Ames, IA 50011, USA 2 Department of Chemistry, 414 Wartik Laboratory, The Pennsylvania State University, University Park, PA 16802, USA Received 5 March 2010; received in revised form 13 June 2010; accepted 15 June 2010 Available online 25 June 2010 DNA is constantly damaged by endogenous and exogenous agents. The resulting DNA lesions have the potential to halt the progression of the replisome, possibly leading to replication fork collapse. Here, we examine the effect of a noncoding DNA lesion in either leading strand template or lagging strand template on the bacteriophage T4 replisome. A damaged base in the lagging strand template does not affect the progression of the replication fork. Instead, the stalled lagging strand polymerase recycles from the lesion and initiates the synthesis of a new Okazaki fragment upstream of the damaged base. In contrast, when the replisome encounters a blocking lesion in the leading strand template, the replication fork only travels approximately 1 kb beyond the point of the DNA lesion before complete replication fork collapse. The primosome and the lagging strand polymerase remain active during this period, and an Okazaki fragment is synthesized beyond the point of the leading strand lesion. There is no evidence for a new priming event on the leading strand template. Instead, the DNA structure that is produced by the stalled replication fork is a substrate for the DNA repair helicase UvsW. UvsW catalyzes the regression of a stalled replication fork into a “chicken-foot” structure that has been postulated to be an intermediate in an error-free lesion bypass pathway. © 2010 Elsevier Ltd. All rights reserved. Edited by M. Gottesman Keywords: T4 DNA replication; lesion bypass; fork regression; UvsW Introduction The bacteriophage T4 (T4) replisome has proven to be an excellent model system for the study of replisome-dependent coupled DNA replication. 1 Despite the variation in the number and the nature of individual proteins between replication systems, the components that constitute an operative repli- cation fork are faithfully represented in other more complex replication systems. 2 Therefore, what has been learned from the T4 replisome can be extended to the replisomes of other organisms, including higher eukaryotes. 3 The T4 replisome is made up of eight proteins, which are thought of as three subcomplexes. The leading strand and lagging strand holoenzymes make up two of the subcomplexes. The holoenzyme is composed of the polymerase (gp43) and the clamp protein (gp45). 4 The trimeric clamp protein encircles the DNA duplex behind the polymerase to increase its processivity from just a few bases incorporated per single binding event to several thousands. 5 The holoenzyme can be assembled through multiple routes via the action of the clamp loader protein, which uses the energy of ATP hydrolysis to chaperone the assembly process. 6–8 The third sub- complex is known as the primosome, which moves along the lagging strand DNA template in the 5′-to- 3′ direction. 9 The primosome is composed of a hexameric helicase (gp41) and an oligomeric pri- mase (gp61). The helicase is loaded onto the replication fork by the action of the hexameric helicase loader protein (gp59). 10,11 The helicase loader also acts to coordinate the assembly of the primosome with the initiation of leading strand synthesis by directly interacting with and inhibiting the activity of the leading strand polymerase. 12–14 Once the helicase is loaded, gp59–polymerase interaction is broken, and gp59 is displaced from *Corresponding author. E-mail address: [email protected]. Abbreviations used: T4, bacteriophage T4; ssDNA, single-stranded DNA; dsDNA, double-stranded DNA; EDTA, ethylenediaminetetraacetic acid. doi:10.1016/j.jmb.2010.06.027 J. Mol. Biol. (2010) 401, 743–756 Available online at www.sciencedirect.com 0022-2836/$ - see front matter © 2010 Elsevier Ltd. All rights reserved.
-
Upload
scott-w-nelson -
Category
Documents
-
view
213 -
download
0
Transcript of Response of the Bacteriophage T4 Replisome to Noncoding Lesions and Regression of a Stalled...

doi:10.1016/j.jmb.2010.06.027 J. Mol. Biol. (2010) 401, 743–756
Available online at www.sciencedirect.com
Response of the Bacteriophage T4 Replisome toNoncoding Lesions and Regression of a StalledReplication Fork
Scott W. Nelson1⁎ and Stephen J. Benkovic2
1Department of Biochemistry,Biophysics, and MolecularBiology, 4112 Molecular BiologyBuilding, Iowa State University,Ames, IA 50011, USA2Department of Chemistry,414 Wartik Laboratory,The Pennsylvania StateUniversity, University Park,PA 16802, USA
Received 5 March 2010;received in revised form13 June 2010;accepted 15 June 2010Available online25 June 2010
*Corresponding author. E-mail addrAbbreviations used: T4, bacteriop
single-stranded DNA; dsDNA, doubEDTA, ethylenediaminetetraacetic a
0022-2836/$ - see front matter © 2010 E
DNA is constantly damaged by endogenous and exogenous agents. Theresulting DNA lesions have the potential to halt the progression of thereplisome, possibly leading to replication fork collapse. Here, we examinethe effect of a noncoding DNA lesion in either leading strand template orlagging strand template on the bacteriophage T4 replisome. A damagedbase in the lagging strand template does not affect the progression of thereplication fork. Instead, the stalled lagging strand polymerase recyclesfrom the lesion and initiates the synthesis of a new Okazaki fragmentupstream of the damaged base. In contrast, when the replisome encountersa blocking lesion in the leading strand template, the replication fork onlytravels approximately 1 kb beyond the point of the DNA lesion beforecomplete replication fork collapse. The primosome and the lagging strandpolymerase remain active during this period, and an Okazaki fragment issynthesized beyond the point of the leading strand lesion. There is noevidence for a new priming event on the leading strand template. Instead,the DNA structure that is produced by the stalled replication fork is asubstrate for the DNA repair helicase UvsW. UvsW catalyzes the regressionof a stalled replication fork into a “chicken-foot” structure that has beenpostulated to be an intermediate in an error-free lesion bypass pathway.
© 2010 Elsevier Ltd. All rights reserved.
Edited by M. Gottesman
Keywords: T4 DNA replication; lesion bypass; fork regression; UvsWIntroduction
The bacteriophage T4 (T4) replisome has provento be an excellent model system for the study ofreplisome-dependent coupled DNA replication.1
Despite the variation in the number and the natureof individual proteins between replication systems,the components that constitute an operative repli-cation fork are faithfully represented in other morecomplex replication systems.2 Therefore, what hasbeen learned from the T4 replisome can be extendedto the replisomes of other organisms, includinghigher eukaryotes.3
The T4 replisome is made up of eight proteins,which are thought of as three subcomplexes. Theleading strand and lagging strand holoenzymes
ess: [email protected] T4; ssDNA,le-stranded DNA;cid.
lsevier Ltd. All rights reserve
make up two of the subcomplexes. The holoenzymeis composed of the polymerase (gp43) and the clampprotein (gp45).4 The trimeric clamp protein encirclesthe DNA duplex behind the polymerase to increaseits processivity from just a few bases incorporatedper single binding event to several thousands.5 Theholoenzyme can be assembled through multipleroutes via the action of the clamp loader protein,which uses the energy of ATP hydrolysis tochaperone the assembly process.6–8 The third sub-complex is known as the primosome, which movesalong the lagging strand DNA template in the 5′-to-3′ direction.9 The primosome is composed of ahexameric helicase (gp41) and an oligomeric pri-mase (gp61). The helicase is loaded onto thereplication fork by the action of the hexamerichelicase loader protein (gp59).10,11 The helicaseloader also acts to coordinate the assembly of theprimosome with the initiation of leading strandsynthesis by directly interacting with and inhibitingthe activity of the leading strand polymerase.12–14Once the helicase is loaded, gp59–polymeraseinteraction is broken, and gp59 is displaced from
d.

744 Stalling of the T4 Replisome and Fork Regression
the front of the replication fork prior to the initiationof DNA synthesis.15 The fate of gp59 has beensomewhat controversial, with several lines of dataindicating that gp59 remains part of the replicationfork, interacting with gp32 [single-stranded DNA(ssDNA) binding protein]-coated ssDNA on thelagging strand template.16,17 However, the impor-tance of this interaction is not well understood, assevere dilution of gp59 following replisome assem-bly has little effect on the rates of leading strand andlagging strand syntheses or on the overall sizedistribution of Okazaki fragments.18
The T4 replisome appears to be the most dynamicamong the characterized replisomes, with the heli-case being the only protein that does not exchangewith solution during coupled leading strand andlagging strand syntheses. The clamp, clamp loader,and single-stranded binding proteins enter andleave the replisome during each cycle of Okazakifragment synthesis.19,20 The primase displays mod-erate processivity that is at least partially controlledby the efficiency of primer handoff.21 The leadingstrand and lagging strand polymerases are ‘dynam-ically processive’ in that they participate in an activeexchange mechanism with polymerases fromsolution.18 This active exchange appears to bemediated through an interaction between the C-terminus of the polymerase and the clamp protein.These dynamic processes are very likely related tothe mechanism of Okazaki fragment synthesis,which is initiated every 2–10 s.22
The opposite polarities of duplex DNA requirethe leading strand and lagging strand polymerasesto move in opposing directions relative to theirrespective templates. However, because the laggingstrand template loops back upon itself, the laggingstrand polymerase remains coupled to the replica-tion fork and travels in the same apparent directionas the leading strand polymerase.16,17,23 Since thelagging strand polymerase remains with thereplication fork, it must repeatedly release thelagging strand template and recycle to a newlysynthesized primer during each cycle of Okazakifragment synthesis. There are two mechanisms bywhich the lagging strand polymerase can bereleased from its position on the lagging strandtemplate. The first mechanism is termed the‘collision model.’ In the collision model, the laggingstrand polymerase is released from its DNAtemplate after completely copying the availabletemplate and colliding with the 5′ end of thepreviously synthesized Okazaki fragment.24 Thesecond mechanism is termed the ‘signalingmodel.’25,26 In the signaling model, the polymerasedoes not need to reach the end of the availablessDNA template. If the synthesis of the RNA primerand the subsequent loading of the clamp onto thatprimer precede the collision of the polymerase withthe previous Okazaki fragment, the polymerase willrelease its template and leave an ssDNA gapbetween the point of release and the previousOkazaki fragment.27 It is assumed that the gapwill be filled in by a polymerase recruited from
solution, possibly during the normal Okazaki frag-mentmaturation process (i.e., followingRNAprimerremoval by RNase H1).In vivo, the replisome is constantly faced with
potential replisome-stalling agents such as DNAlesions or other DNA-bound proteins.28 DNAlesions can be generated through intrinsic mutagenssuch as reactive oxygen species or extrinsic muta-gens such as UV light, infrared, and a variety ofmutagenic chemicals. These lesions or blocks havethe potential to disrupt the forward progress of thereplication fork. Stalled replication forks havedemonstrated the potential to cause illegitimaterecombination and cellular dysfunction. When theEscherichia coli replisome encounters a blocking lesionin the leading strand template, the polymerasesuncouple, and the lagging strand polymeraseremains active long enough to synthesize an Okazakifragment past the blocking lesion.29–31 On the otherhand, a lagging strand blocking lesion does notimpede the forward progress of the E. coli replisomedue to the recycling of the stalled lagging standpolymerase to a newly synthesized primer.30–32 Inthis report, we test the response of the T4 replisometo noncoding lesions locating in either the leadingstrand template or the lagging strand template. Alesion in the leading strand template stalls theleading strand polymerase, but the helicase con-tinues to unwind duplex DNA, and the primasesynthesizes at least one RNA primer beyond thepoint of the DNA lesion. Compared to the coupledreplisome, the uncoupled replisome is destabilizedwith a disassembly rate of N2 min− 1. The DNAstructure produced when the T4 replisome encoun-ters a leading strand lesion is a substrate for theDNA repair helicase UvsW. UvsW catalyzes theATP-dependent regression of a well-defined DNAfork that mimics that produced by the replisome.The regression product, often termed a chicken foot,is likely a necessary intermediate in the bypass and/or repair of the leading strand lesion. In contrast to aleading strand lesion, a lesion in the lagging strandtemplate does not impede the forward progress ofthe replisome. The stalled lagging strand polymer-ase simply recycles to a new RNA primer, likely viathe signaling mechanism, to reinitiate Okazakifragment synthesis upstream of the lesion.
Results
Replisome-mediated DNA synthesis with alesion located in the leading strand template
To observe the response of the T4 replisome to alesion located in the leading strand template, wemust use large DNA substrates because the assem-bly of the primosome is slow, and the leading strandholoenzyme must travel an average of 0.5–1 kbbefore the lagging strand initiates the synthesis ofthe first Okazaki fragment.33 For this reason, wehave employed a plasmid-based substrate con-
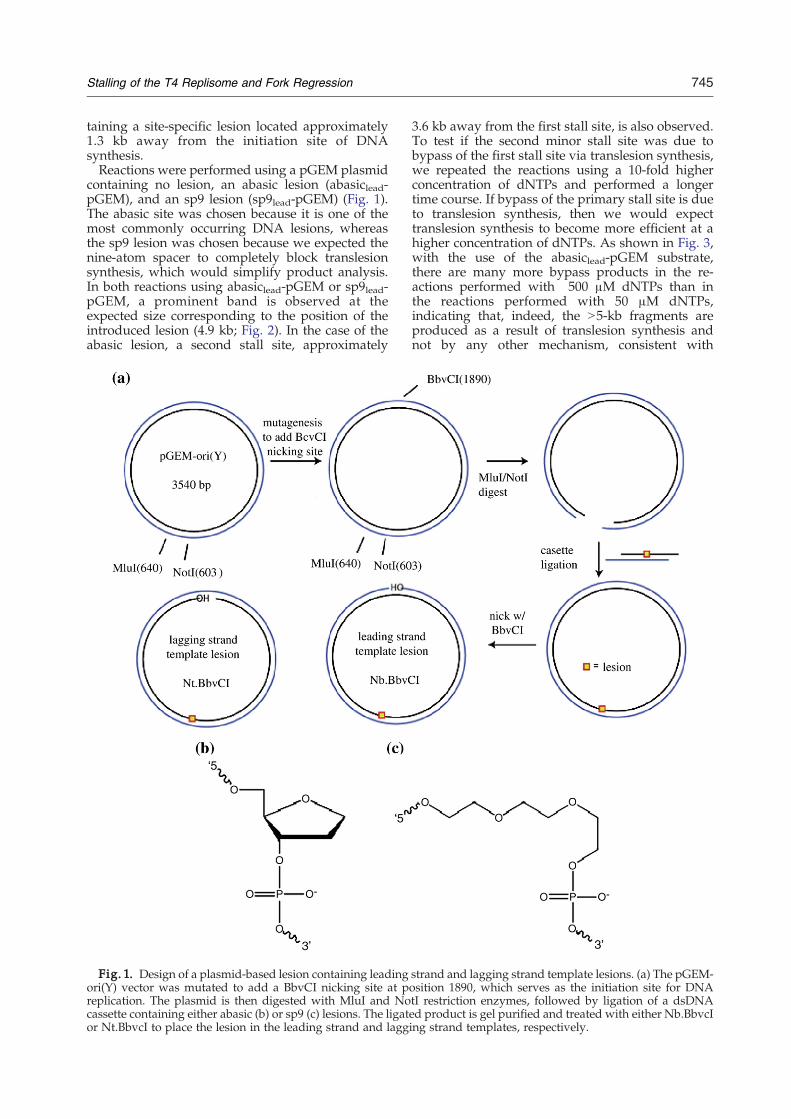
745Stalling of the T4 Replisome and Fork Regression
taining a site-specific lesion located approximately1.3 kb away from the initiation site of DNAsynthesis.Reactions were performed using a pGEM plasmid
containing no lesion, an abasic lesion (abasiclead-pGEM), and an sp9 lesion (sp9lead-pGEM) (Fig. 1).The abasic site was chosen because it is one of themost commonly occurring DNA lesions, whereasthe sp9 lesion was chosen because we expected thenine-atom spacer to completely block translesionsynthesis, which would simplify product analysis.In both reactions using abasiclead-pGEM or sp9lead-pGEM, a prominent band is observed at theexpected size corresponding to the position of theintroduced lesion (4.9 kb; Fig. 2). In the case of theabasic lesion, a second stall site, approximately
Fig. 1. Design of a plasmid-based lesion containing leadingori(Y) vector was mutated to add a BbvCI nicking site at preplication. The plasmid is then digested with MluI and Nocassette containing either abasic (b) or sp9 (c) lesions. The ligator Nt.BbvcI to place the lesion in the leading strand and lagg
3.6 kb away from the first stall site, is also observed.To test if the second minor stall site was due tobypass of the first stall site via translesion synthesis,we repeated the reactions using a 10-fold higherconcentration of dNTPs and performed a longertime course. If bypass of the primary stall site is dueto translesion synthesis, then we would expecttranslesion synthesis to become more efficient at ahigher concentration of dNTPs. As shown in Fig. 3,with the use of the abasiclead-pGEM substrate,there are many more bypass products in the re-actions performed with 500 μM dNTPs than inthe reactions performed with 50 μM dNTPs,indicating that, indeed, the N5-kb fragments areproduced as a result of translesion synthesis andnot by any other mechanism, consistent with
strand and lagging strand template lesions. (a) The pGEM-osition 1890, which serves as the initiation site for DNAtI restriction enzymes, followed by ligation of a dsDNAed product is gel purified and treated with either Nb.BbvcIing strand templates, respectively.

Fig. 2. DNA replication products with lesions located in the leading strand template. The synthesis products areseparated via alkaline agarose electrophoresis. Reactions performed with the nicked pGEM plasmid containing no lesion(pGEM), an abasic lesion (abasiclead-pGEM), and an sp9 lesion (sp9lead-pGEM) are shown. The four lanes for each reactionare time points of 0.5, 1, 1.5, and 2 min. Due to the rolling circle nature of the non-lesion-containing substrate, many moreDNA synthesis products are produced; therefore, the signal is much higher, and the exposure has been decreasedcompared to the abasic and sp9 substrates.
746 Stalling of the T4 Replisome and Fork Regression
previous observations.34 There are at least fivediscrete stall sites observed, representing at leastfour cycles of stalling, translesion synthesis, andstalling again, leading to a product 19.3 kb long(close to the resolution limit of the gel). Importantly,the sp9 lesion completely stalls the leading strandpolymerase with no translesion synthesis observedafter 12 min at either concentration of dNTPs.It is clear in both reactions containing an abasic
lesion or an sp9 lesion that lagging strand fragmentsare produced (running as a smear of b3.6 kb).Control reactions indicate that ribonucleotide tri-phosphates and/or primase omission results in thedisappearance of these products (data not shown).This indicates that the primosome has fully assem-bled and that the lagging strand polymerase hasstarted to produce Okazaki fragments prior to theencounter of the lesion by the leading strandpolymerase. Quantitation of the leading strand andlagging strand products in Fig. 2 reveals that withboth abasic and sp9 lesions, there are more laggingstrand products than leading strand products(Fig. 4b). In standard reactions performed withnonlesion substrates, the leading strand and lagging
strand polymerases are coupled, and the amountsof leading and lagging strands are nearly equi-valent21,27,33 (Fig. 4a). This differential increase inlagging strand products over leading strand pro-ducts indicates that the lagging strand DNAsynthesis has gone beyond the point of the DNAlesion (i.e., the leading strand and lagging strandpolymerases have become uncoupled). Since thepositions of the replication initiation site and theleading strand sp9 lesion are known, we candetermine the average number of nucleotides thathave been incorporated into the leading and laggingstrands. The number of bases incorporated into theleading strand is the number of bases between theinitiation site and the sp9 lesion (i.e., 1277 bases).Since the leading strand makes up 35.25% of thetotal replication products, the total number of basesreplicated is 3622 (1277/0.3525). The lagging strandtotal is then 2345 bases, 1068 of which come after thepoint of the DNA lesion (2345−1277), which isapproximately the length of a single Okazakifragment. The above calculation assumes that allreplication forks contain both leading strand andlagging strand holoenzymes, which appears to be

Fig. 3. DNA replication pro-ducts with lesions located in theleading strand template at normaland high concentrations of dNTPs.The replication reactions withabasiclead-pGEM and sp9lead-pGEM DNA substrates were car-ried out using the dNTP concentra-tions indicated on the figure at timepoints of 3,6, 9, and 12 min.
747Stalling of the T4 Replisome and Fork Regression
valid based on the equivalent amounts of leadingstrand and lagging strand products when a lesion-free substrate is used (Fig. 4a).To confirm replisomal uncoupling, we probed the
structure of the DNA products produced during areplication reaction using the sp9lead-pGEM sub-strate. We utilized two restriction sites located oneither side of the introduced DNA lesion (SapI andMfeI). Since both strands are labeled by theincorporation of dGTP α-32P, we can visualize thedigestion products of the newly synthesized leadingand lagging strands (Fig. 5a). The synthesis of anOkazaki fragment beyond the point of the leadingstrand lesion will produce a unique band that is1.74 kb in length when theMfeI restriction enzyme isused, whereas if lagging strand synthesis is stronglycoupled with the leading strand synthesis, then onlya fragment N3.6 kb will be observed. As shown inFig. 5b, a band approximately ∼1.74 kb in length isreadily apparent in MfeI-digested products, un-equivocally demonstrating that an Okazaki frag-ment has been synthesized beyond the position ofthe leading strand sp9 lesion.To determine the stability of the replisome stalled
at a DNA lesion, we took advantage of the ability ofthe T4 polymerase to perform translesion synthesisacross an abasic lesion. We performed a reactionwhere the replisome was allowed to encounter theabasic lesion under normal conditions and thenperformed a rapid 10-fold dilution into a buffercontaining all components, except for the DNA
helicase. Control reactions indicate that, under ourconditions, reducing the concentration of helicase tob100 nM eliminates the formation of new replicationforks (data not shown). The stability of the replisomecan be measured by the appearance of translesionsynthesis products after dilution. As seen in Fig. 6,dilution of the helicase virtually eliminates theappearance of translesion synthesis products, indi-cating that the stalled replisome is not stable enoughto resume DNA synthesis following translesionsynthesis by the polymerase without the need forreassembly. From these data, we can estimate thatthe half-life for replisome stability is b30 s and that,for each translesion synthesis event, the replisomemust reassemble before replicating around thecircular plasmid to encounter the lesion again.
Replisome-mediated DNA synthesis with lesionlocated in the lagging strand template
We next investigated the effect of an abasic lesionor an sp9 lesion on the lagging strand template. Asshown in Fig. 7, neither the abasic lesion nor the sp9lesion causes a significant amount of stalling in theleading strand or lagging strand polymerase. This isconsistent with a previous study that placed anabasic site near the end of the template for the firstOkazaki fragment.34 The majority of the leadingstrand products are longer than the resolution of thegel, and the amounts of lagging strand productssynthesized are comparable (i.e., leading strand and
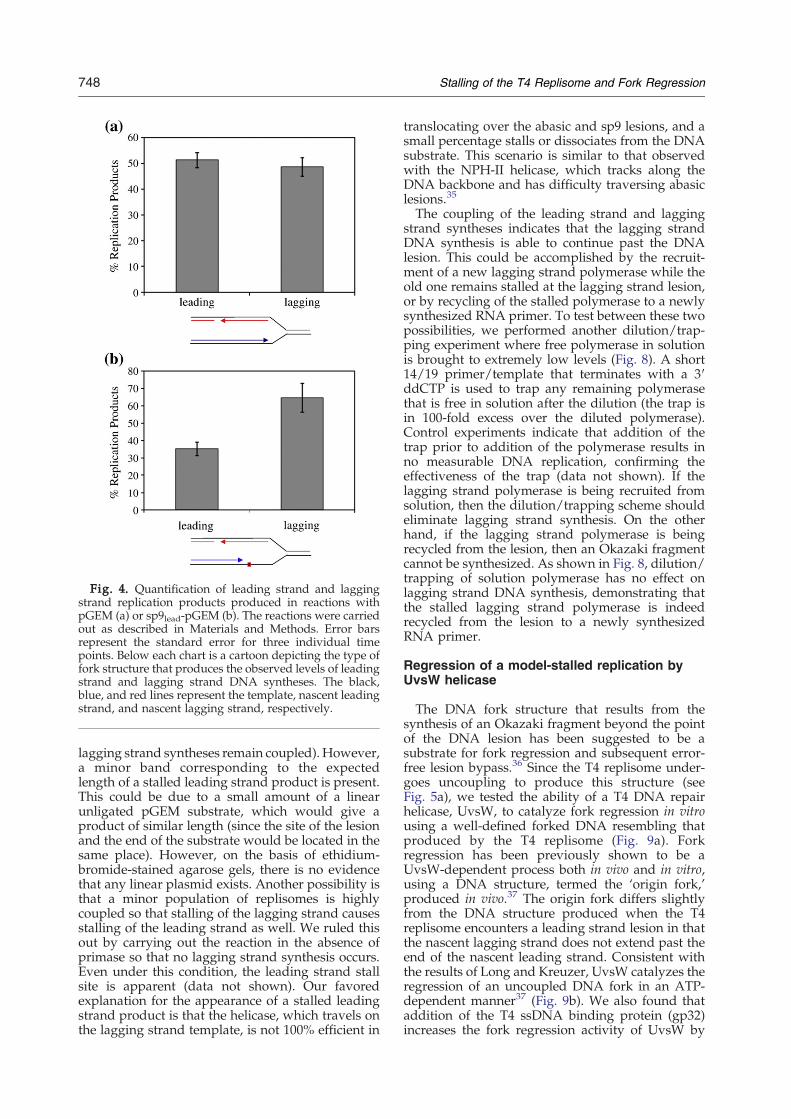
Fig. 4. Quantification of leading strand and laggingstrand replication products produced in reactions withpGEM (a) or sp9lead-pGEM (b). The reactions were carriedout as described in Materials and Methods. Error barsrepresent the standard error for three individual timepoints. Below each chart is a cartoon depicting the type offork structure that produces the observed levels of leadingstrand and lagging strand DNA syntheses. The black,blue, and red lines represent the template, nascent leadingstrand, and nascent lagging strand, respectively.
748 Stalling of the T4 Replisome and Fork Regression
lagging strand syntheses remain coupled). However,a minor band corresponding to the expectedlength of a stalled leading strand product is present.This could be due to a small amount of a linearunligated pGEM substrate, which would give aproduct of similar length (since the site of the lesionand the end of the substrate would be located in thesame place). However, on the basis of ethidium-bromide-stained agarose gels, there is no evidencethat any linear plasmid exists. Another possibility isthat a minor population of replisomes is highlycoupled so that stalling of the lagging strand causesstalling of the leading strand as well. We ruled thisout by carrying out the reaction in the absence ofprimase so that no lagging strand synthesis occurs.Even under this condition, the leading strand stallsite is apparent (data not shown). Our favoredexplanation for the appearance of a stalled leadingstrand product is that the helicase, which travels onthe lagging strand template, is not 100% efficient in
translocating over the abasic and sp9 lesions, and asmall percentage stalls or dissociates from the DNAsubstrate. This scenario is similar to that observedwith the NPH-II helicase, which tracks along theDNA backbone and has difficulty traversing abasiclesions.35
The coupling of the leading strand and laggingstrand syntheses indicates that the lagging strandDNA synthesis is able to continue past the DNAlesion. This could be accomplished by the recruit-ment of a new lagging strand polymerase while theold one remains stalled at the lagging strand lesion,or by recycling of the stalled polymerase to a newlysynthesized RNA primer. To test between these twopossibilities, we performed another dilution/trap-ping experiment where free polymerase in solutionis brought to extremely low levels (Fig. 8). A short14/19 primer/template that terminates with a 3′ddCTP is used to trap any remaining polymerasethat is free in solution after the dilution (the trap isin 100-fold excess over the diluted polymerase).Control experiments indicate that addition of thetrap prior to addition of the polymerase results inno measurable DNA replication, confirming theeffectiveness of the trap (data not shown). If thelagging strand polymerase is being recruited fromsolution, then the dilution/trapping scheme shouldeliminate lagging strand synthesis. On the otherhand, if the lagging strand polymerase is beingrecycled from the lesion, then an Okazaki fragmentcannot be synthesized. As shown in Fig. 8, dilution/trapping of solution polymerase has no effect onlagging strand DNA synthesis, demonstrating thatthe stalled lagging strand polymerase is indeedrecycled from the lesion to a newly synthesizedRNA primer.
Regression of a model-stalled replication byUvsW helicase
The DNA fork structure that results from thesynthesis of an Okazaki fragment beyond the pointof the DNA lesion has been suggested to be asubstrate for fork regression and subsequent error-free lesion bypass.36 Since the T4 replisome under-goes uncoupling to produce this structure (seeFig. 5a), we tested the ability of a T4 DNA repairhelicase, UvsW, to catalyze fork regression in vitrousing a well-defined forked DNA resembling thatproduced by the T4 replisome (Fig. 9a). Forkregression has been previously shown to be aUvsW-dependent process both in vivo and in vitro,using a DNA structure, termed the ‘origin fork,’produced in vivo.37 The origin fork differs slightlyfrom the DNA structure produced when the T4replisome encounters a leading strand lesion in thatthe nascent lagging strand does not extend past theend of the nascent leading strand. Consistent withthe results of Long and Kreuzer, UvsW catalyzes theregression of an uncoupled DNA fork in an ATP-dependent manner37 (Fig. 9b). We also found thataddition of the T4 ssDNA binding protein (gp32)increases the fork regression activity of UvsW by

Fig. 5. Restriction digest assay to determine uncoupling of replication fork. (a) A schematic showing the position ofrestriction sites in relation to the leading strand sp9 lesion. The black, blue, and red lines represent the template, nascentleading strand, and nascent lagging strand, respectively. If the replisome becomes uncoupled and the nascent laggingstrand extends past the nascent leading strand, then treatment of the replicated product with the MfeI restriction enzymeshould produce two fragments with apparent mobilities of 1.8 and N3.6 kb. If the replisome is coupled and the nascentlagging strand stops before the leading strand or at the same point as the leading strand, then treatment of the replicatedproduct with the MfeI restriction enzyme will only produce a single product with an apparent mobility of N3.6 kb.Treatment with SapI should produce 0.9-kb and 3.6-kb fragments for both coupled and uncoupled DNA replicationproducts. (b) Native gel electrophoresis of MfeI and SapI restriction digestion products. The open-circle, linear, 1.8-kb,and 0.9-kb fragments are encircled.
749Stalling of the T4 Replisome and Fork Regression
approximately 2-fold (Fig. 9c). This result is surpris-ing, since gp32 has been shown to inhibit theannealing of complementary ssDNA strands,38 andthe ssDNA binding rate of gp32 to ssDNA is
extremely rapid39 (1×1010 to 1×1011 M−1 s−1). Theenhancement of fork regression activity by gp32suggests that the two arms of the nascent leadingand lagging strands are being unwound and

Fig. 6. The stability of the replisome with leading strand polymerase stalled at an abasic lesion. Reactions were carriedout as described in Materials and Methods using the abasiclead-pGEM DNA substrate. (a) A time course of replicationproducts with andwithout dilution. (b) Quantification of the data shown in (a). The ratio of translesion synthesis productsto the first stalled product is plotted versus time for the undiluted reaction (closed diamonds) and for the diluted reaction(closed squares).
750 Stalling of the T4 Replisome and Fork Regression
annealed in a coupled manner (i.e., gp32 is beingexcluded from binding to the ssDNA intermediate).In this scenario, the function of gp32 is to increasethe affinity of UvsW for the DNA substrate, perhapswhile bound to the ssDNA in front of the leadingstrand lesion.
Discussion
DNA replisomes of all organisms constantlyencounter DNA lesions, which may lead to stallingand replication fork collapse.28 Here, we haveshown that the T4 replisome reacts differently tolesions located in the leading strand template andlesions located in the lagging strand template. Aleading strand lesion stalls the leading strandpolymerase and causes replication fork collapse(Fig. 10a), whereas a lagging strand lesion poses nothreat to the forward movement of the replisome,and the stalled lagging strand polymerase rapidlyrecycles to a newly synthesized RNA primer toinitiate another round of Okazaki fragment synthe-sis (Fig. 10b). The stalling of the leading strandpolymerase does not immediately inhibit the primo-some, since the helicase continues to unwind
double-stranded DNA (dsDNA), the primasesynthesizes an RNA primer, and the lagging strandpolymerase produces at least one Okazaki fragmentbeyond the point of the leading strand lesion beforereplication fork collapses. The DNA structure thatresults from this temporary uncoupling is a sub-strate for the DNA repair helicase UvsW, whichcatalyzes the ATP-dependent regression into a“chicken-foot” intermediate.It is well-known that the leading strand polymer-
ase and the replicative helicase are functionallycoupled so that the presence of a fully activepolymerase increases the rate of the helicase by upto 10-fold.40,41 The mere physical presence of thepolymerase at the replication fork is not enoughfor this rate enhancement—the polymerase mustincorporate dNTPs behind the moving helicase.40
With a minicircle substrate, reducing the rate of theleading strand polymerase (by a reduction in dGTPconcentration) reduces the rate of the helicase by asimilar degree, reaching a value of ∼40 bp/s at thelowest dNTP concentration tested (Nelson andBenkovic, unpublished observations). The unwind-ing rate of the helicase alone has been determined tobe 30 bp/s,42 suggesting that the unwinding rate ofthe helicase with a stalled polymerase behind it is

Fig. 7. DNA replication products with lesions locatedin the lagging strand template. Reactions were carried outusing standard conditions as described in Materials andMethods. Reactions performed with the abasiclag-pGEMand sp9lag-pGEM DNA substrates are shown (for thepGEM reaction, see Fig. 2). The four lanes for each reactionare time points of 3, 6, 9, and 12 min.
751Stalling of the T4 Replisome and Fork Regression
between 30 and 40 bp/s. The data presented hereindicate that at least one Okazaki fragment isproduced beyond the point of the DNA lesion,
Fig. 8. Recycling of lagging strand polymerase. (a) A schemdetermine the recycling properties of the lagging strand polymtext. (b) Reactions were carried out as described in Materials anand sp9lag-pGEM substrates were analyzed using alkaline agtime points of 3, 6, 9, and 12 min.
demonstrating that the primosome is able to functionat a much less optimal rate (30–40 bp/s). However,the replisome as a whole is much more unstablewhen the leading strand polymerase is stalled at alesion, indicating that the primosome cannot contin-ue indefinitely without an actively replicatingleading strand polymerase. We cannot unequivocal-ly conclude if a particular component or the entirereplisome disassembles after reaching a leadingstrand lesion, but Hacker and Alberts have shownthat the holoenzyme in isolation is extremely stablewhen stalled due to nucleotide omission.43 Addi-tionally, single-molecule assays indicate that the T4helicase alone has an average processivity rangingbetween 100 and 800 bp,42 similar to the number ofbases incorporated into the lagging strand beyondthe leading strand lesion determined here. These twoobservations lead us to conclude that it is very likelythat the helicase is the first component of the stalled/uncoupled replisome to disassemble. Moreover, it isprobable that it is this instability that limits contin-ued lagging strand synthesis to only a singleOkazakifragment beyond the DNA lesion.In contrast to the T4 replisome studied here, the E.
coli replisome has been shown to be highly stablewhen stalled at a leading strand lesion.29 A possiblereason for this difference may lie in the differingways that E. coli and T4 bypass DNA lesions. E. coliuses a specialized translesion polymerase to bypassnoncoding lesions such as abasic sites.44 In recentyears, a tool-belt model has emerged where thereplicative and translesion polymerases simulta-neously bind to the sliding clamp during normalDNA replication.45,46 When a lesion is encountered,
atic illustrating the assembly and dilution scheme used toerase. “Q” represents quench. Details can be found in thed Methods. The products of DNA replication using pGEMarose electrophoresis. The four lanes for each reaction are

Fig. 9. ATP-dependent UvsW-catalyzed DNA fork regression. (a) A schematic of the DNA substrate used to monitorfork regression. The asterisk represents a 5′ 32P label. The black, blue, and red lines represent the template, nascent leadingstrand, and nascent lagging strand, respectively. (b) A native PAGE gel of the fork regression reactions. Reactions werecarried out as described in Materials and Methods. Lanes 1 through 4 contain ATP/EcoRI, UvsW/ATP/EcoRI, UvsW/EcoRI, and UvsW/ATP, respectively. (c) A plot of percent forks regressed versus the concentration of UvsW without T4SSB (gp32) (black diamonds) and with T4 SSB (gp32) (black squares).
752 Stalling of the T4 Replisome and Fork Regression
the translesion polymerase switches places with thereplicative polymerase and incorporates a verylimited number of nucleotides into the site of theDNA lesion. After translesion synthesis, the high-fidelity replicative polymerase trades places with thetranslesion polymerase and, presumably due to thehigh stability of the replisome, coupled leadingstrand DNA synthesis and lagging strand DNAsynthesis resume. T4 codes for a single DNApolymerase (gp43), and there is no evidence forhost cell translesion polymerases participating in T4phage genomic replication.47 As a consequence ofthis, T4 phage is likely to be completely dependenton recombination-based pathways for the bypass
and restart of replisome-dependent DNA synthesis(e.g., UvsW-catalyzed DNA fork regression). Withthis being considered, it would be advantageous tohave a relatively rapid replisome disassembly whenthe leading strand polymerase stalls at a DNA lesion.The noncoding sp9 lesion used here results in
absolutely no leading strand DNA synthesis beyondthe lesion. This result is in contrast to what would beexpected if, like the E. coli replisome, the T4replisome can restart DNA synthesis on the leadingstrand through primosome-mediated priming.48,49
It is unclear why the T4 replisome does not have thisability; however, it may be related to the verydifferent situations that accompany the replication
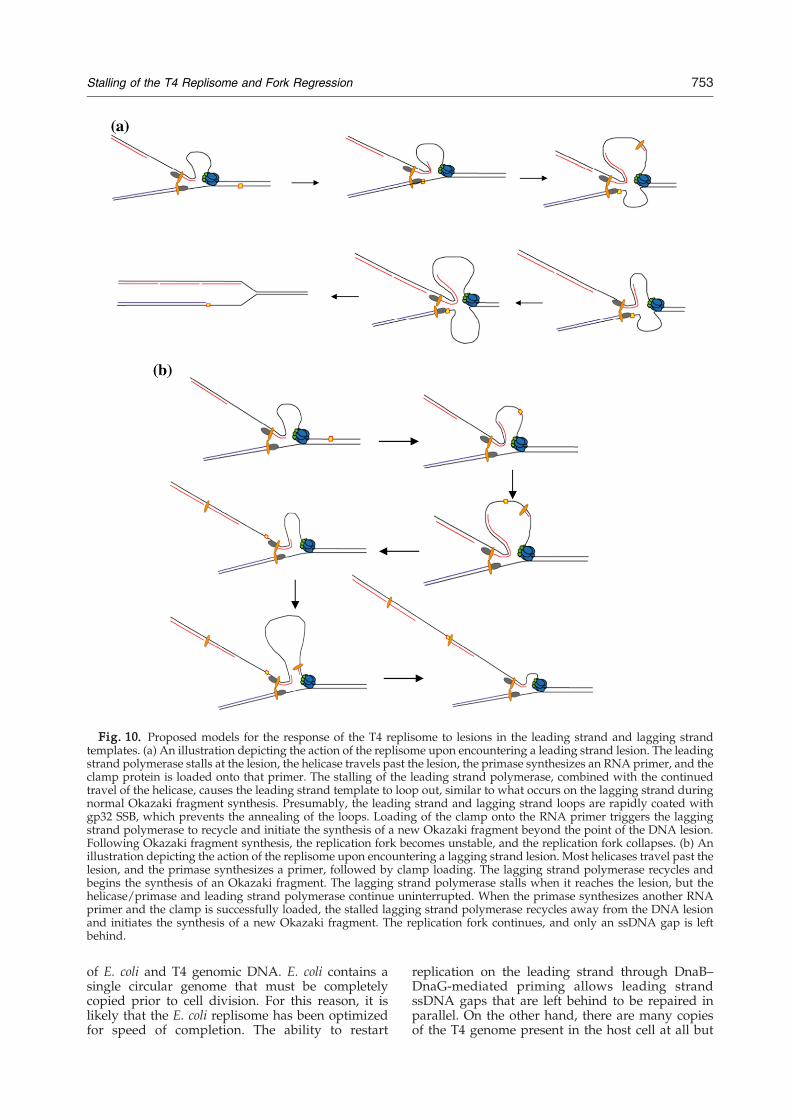
Fig. 10. Proposed models for the response of the T4 replisome to lesions in the leading strand and lagging strandtemplates. (a) An illustration depicting the action of the replisome upon encountering a leading strand lesion. The leadingstrand polymerase stalls at the lesion, the helicase travels past the lesion, the primase synthesizes an RNA primer, and theclamp protein is loaded onto that primer. The stalling of the leading strand polymerase, combined with the continuedtravel of the helicase, causes the leading strand template to loop out, similar to what occurs on the lagging strand duringnormal Okazaki fragment synthesis. Presumably, the leading strand and lagging strand loops are rapidly coated withgp32 SSB, which prevents the annealing of the loops. Loading of the clamp onto the RNA primer triggers the laggingstrand polymerase to recycle and initiate the synthesis of a new Okazaki fragment beyond the point of the DNA lesion.Following Okazaki fragment synthesis, the replication fork becomes unstable, and the replication fork collapses. (b) Anillustration depicting the action of the replisome upon encountering a lagging strand lesion. Most helicases travel past thelesion, and the primase synthesizes a primer, followed by clamp loading. The lagging strand polymerase recycles andbegins the synthesis of an Okazaki fragment. The lagging strand polymerase stalls when it reaches the lesion, but thehelicase/primase and leading strand polymerase continue uninterrupted. When the primase synthesizes another RNAprimer and the clamp is successfully loaded, the stalled lagging strand polymerase recycles away from the DNA lesionand initiates the synthesis of a new Okazaki fragment. The replication fork continues, and only an ssDNA gap is leftbehind.
753Stalling of the T4 Replisome and Fork Regression
of E. coli and T4 genomic DNA. E. coli contains asingle circular genome that must be completelycopied prior to cell division. For this reason, it islikely that the E. coli replisome has been optimizedfor speed of completion. The ability to restart
replication on the leading strand through DnaB–DnaG-mediated priming allows leading strandssDNA gaps that are left behind to be repaired inparallel. On the other hand, there are many copiesof the T4 genome present in the host cell at all but

754 Stalling of the T4 Replisome and Fork Regression
the earliest stages of phage infection. A delay inreplicating a single genomic copy should not pose agreat constraint on the success of phage genomicreplication when multiple copies are present.In contrast to a lesion in the leading strand
template, a lagging strand template lesion has noapparent effect on the leading strand polymerase oron continued DNA synthesis by the lagging strandpolymerase. The data presented here indicate thatthe lagging strand polymerase simply recycles fromthe lagging strand lesion to a newly synthesizedRNA primer to initiate another round of Okazakifragment synthesis. While the mechanism forrecycling the stalled lagging polymerase from thelesion is not completely certain, it very likely uses amechanism identical to what is referred to as the‘signaling mechanism’ (Fig. 10b). In T4 phage, thesignaling mechanism relies on the loading of theclamp protein onto the newly synthesized RNAprimer to signal the release of the lagging strandpolymerase and its recycling to the clamp-loadedprimer. We propose that one of the features of thesignaling mechanism is to enable the lagging strandpolymerase, which normally would have a highdegree of stability at a noncoding lesion, to bereleased from the site of the DNA lesion and tocontinue to travel with the replication fork as usual.The presence of an Okazaki fragment beyond the
point of the leading strand lesion leaves open thepossibility of an intramolecular “template switch”mechanism for lesion bypass.36,38 It was for thisreason that we tested the ability of UvsW to catalyzefork regression on a large substrate designed tomimic the uncoupled DNA fork produced by the T4replisome. It was recently shown by Long andKreuzer that UvsW promotes fork regression usinga coupled replication fork where the nascent leadingstrand was extended past the position of the nascentlagging strand.37 Here, we demonstrate that thesubstrate specificity of UvsW is flexible enough tohandle both types of stalled replication fork.However, based on recent results from the Kreuzerlaboratory, while fork regression appears to be thedominant mechanism for the processing of a stalledreplication fork, it is unlikely that the chicken-footstructure is used to directly bypass the lesion.Instead, the chicken foot is a substrate for nucleolyticdegradation by either T4 Mre11/Rad50 homologs(gp46/47) or the Holliday junction endonuclease(gp49). In this scenario, the purpose of forkregression is to move the DNA lesion back intoduplex DNA, where it can be repaired throughconventional pathways (e.g., base and nucleotideexcision repair).In general, the results reported here using the T4
system are in accordance with those obtained usingthe E. coli replisome.29–32 This similarity in mecha-nism highlights the conservation of fundamentalproperties between the replisomes of all organismsand supports the notion that the T4 replisome is anexcellent model for the investigation of DNAreplication. Through this work and others' work, ithas become clear that the T4 replisome is a dynamic
system with several proteins entering and leavingthe complex during active DNA replication. It islikely that this flexibility has evolved, at least in part,to deal with DNA lesions and road blocks thatwould potentially interfere with the completion ofDNA replication.
Materials and Methods
[α-32P]dGTP was purchased from Perkin-Elmer LifeSciences. Unlabeled ribonucleotides were purchased fromRoche Applied Science. T4 replication proteins gp41, gp61,gp43 exo−, gp44/62, gp45, and gp32, and the primase trapprotein were purified as described previously.50,51 Thesequences of the oligonucleotides used to make the lesion-containing cassettes were 5′-GGCCTAGCGATCAXT-GAATCAG-3′ (top strand, where X denotes either theabasic lesion or the sp9 lesion) and 5′-GCGCCTGATT-CACTGATCGCTA-3′ (bottom strand).
Construction of DNA lesion containing substrates
A large dsDNA substrate containing either an abasiclesion or an sp9 lesion was constructed via cassetteligation (Fig. 1). One milligram of pGEM_nick plasmid14
was digested with 200 U each of MluI and NotI enzymesin 50 mM Tris–HCl (pH 7.5), 10 mM MgCl2, 1 mM ATP,and 10 mMDTT in a volume of 1 ml for 15 h at 37 °C (a 10-fold overdigest). The digestion reaction was heat dena-tured at 65 °C for 30 min, followed by a 50-fold dilutioninto a ligation buffer containing a 10-fold molar excess ofthe 5′ phosphorylated dsDNA cassette containing eitherabasic or sp9 lesions and T4 DNA ligase (100 Weiss units).The ligation reaction was allowed to proceed for 24 h atroom temperature. The ligation reaction was then con-centrated in a YM-30 Centricon and loaded onto a 1% TAEagarose gel containing 10 mg/ml ethidium bromide (topromote supercoiling of the ligated plasmid). The super-coiled band was gel extracted using a Qiagen gelextraction kit. Finally, commercially available nickingenzymes (New England Biolabs) were used to createinitiation sites for DNA synthesis. The enzyme Nb.BbCvIintroduces a nick such that the lesion is located in theleading strand template, whereas the lesion is located inthe lagging strand template when Nt.BbCvI is used. Thenicking site is approximately equidistant from the site ofthe DNA lesion in either direction. The overall yield for thelesion containing substrate synthesis was 2.1%.
Replisome-mediated DNA synthesis
Replication reactions were carried out in a standardreplication buffer [25 mM Tris acetate (pH 7.8), 125 mMKOAc, and 10 mM Mg(OAc)2] at 37 °C. The standardconditions used to assemble and initiate the replicationreaction consisted of 2 nM lesion substrate; 200 nM each ofgp43, gp45 (as trimer), and gp44/62; 600 nM each ofmonomer gp41, gp61, and gp59; 4 μM gp32; 100 μM eachof CTP, GTP, and UTP; 2 mM ATP; and 50 μM each ofdATP, dGTP, dCTP, and dTTP with 25 μCi of [α-32P]dGTP, in a typical reaction volume of 45 μl. In thepolymerase dilution/trapping experiment (Fig. 8), thereaction was diluted 10-fold into a buffer containing allcomponents at standard concentrations, except for thehelicase and polymerase with a 14/19 trap concentrationof 2 μM. Ten-microliter aliquots were removed at the

755Stalling of the T4 Replisome and Fork Regression
indicated time points and quenched with a half volume of0.5 M ethylenediaminetetraacetic acid (EDTA). Thequenched reactions were analyzed with alkaline agarosegel electrophoresis. The quenched reactions were run at40 V for 24 h in 30 mMNaOH and 1mMEDTA buffer. Thegel was dried and exposed overnight to a phosphorimagerplate and analyzed using a Molecular Dynamics Storm800 Phosphorimager system (Amersham Biosciences).To determine the stability of the stalled replisome, we
initiated a replication reaction using the pGEM_nicksubstrate with an abasic lesion in the leading strandtemplate under standard conditions; however, at 20 s afterthe start of the reaction, the reaction was diluted 10-foldinto a buffer containing all components, except for gp41.Control reactions indicate that at 60 nM helicase, no newreplication forms (data not shown). Reactions were pro-cessed and analyzed as described above, except that thevolume loaded onto the alkaline agarose gel was doubled.
Restriction enzyme digest to determine the degree ofreplisome uncoupling
DNA synthesis reactions under standard conditionswere carried out using the pGEM_nick plasmid contain-ing an sp9 lesion in the leading strand template. After thereaction had been allowed to proceed for 12 min, it wasquenched in Qiagen PB buffer and purified using aQiaquick PCR purification column. The purified reactionwas then treated with either SapI or MfeI for 60 min at37 °C. Reactions were quenched with EDTA and loadedonto a 1% TBE agarose gel. The gel was run for 4 h at 80 V,dried onto DEAE paper, and exposed to the storagephosphor screen.
UvsW-catalyzed regression of a model replication fork
The model-uncoupled replication fork was constructedas described previously.52 The pG68 and pG46 plasmidswere generously provided by Dr. LeonardWu (Universityof Oxford, UK). The basis of this assay is the movement ofEcoRI restriction sites from the plasmid arms to theregressed arm of the DNA fork, allowing regression to bemonitored by the appearance of an 86-bp digestionproduct (see Fig. 9). The fork regression reactions werecarried out at 37 °C in standard replication buffer con-taining 0.2 mM ATP, 2 nM DNA, and UvsW concentra-tions indicated in the figure legend. The reaction wasinitiated by the addition of UvsW and allowed to proceedfor 15 min before quenching by the addition of 1 mMATPγS and 10 μM ssM13 DNA (bases). EcoRI restrictionenzyme was then added to the quenched reaction, and thedigest was carried out for 1 h at 37 °C. The digestionproducts were analyzed using 10% polyacrylamideelectrophoresis in 1× TBE (35 mA for 2 h), exposed to aphosphorimager plate overnight, and analyzed using aMolecular Dynamics Storm 800 Phosphorimager system(Amersham Biosciences).
Acknowledgements
This work was supported by National Institutes ofHealth grant GM013306 (to S.J.B.). S.W.N. was aFellow of the Jane Coffin Childs Memorial Fund forMedical Research during a portion of this work.
References
1. Benkovic, S. J., Valentine, A. M. & Salinas, F. (2001).Replisome-mediated DNA replication. Annu. Rev.Biochem. 70, 181–208.
2. Nossal, N. G. (1992). Protein–protein interactions at aDNA replication fork: bacteriophage T4 as a model.FASEB J. 6, 871–878.
3. Nelson, S. W., Zhuang, Z., Spiering, M. M. & Benkovic,S. J. (2009). T4phage replisome.Viral GenomeReplication,337–364; http://dx.doi.org/10.1007/b135974_16.
4. Shamoo, Y. & Steitz, T. A. (1999). Building a replisomefrom interacting pieces: sliding clamp complexed to apeptide from DNA polymerase and a polymeraseediting complex. Cell, 99, 155–166.
5. Munn, M. M. & Alberts, B. M. (1991). The T4 DNApolymerase accessory proteins form an ATP-depen-dent complex on a primer–template junction. J. Biol.Chem. 266, 20024–20033.
6. Kaboord, B. F. & Benkovic, S. J. (1996). Dual role of the44/62 protein as a matchmaker protein and DNApolymerase chaperone during assembly of the bacte-riophage T4 holoenzyme complex. Biochemistry, 35,1084–1092.
7. Smiley, R. D., Zhuang, Z., Benkovic, S. J. & Hammes,G. G. (2006). Single-molecule investigation of the T4bacteriophage DNA polymerase holoenzyme: multi-ple pathways of holoenzyme formation. Biochemistry,45, 7990–7997.
8. Zhuang, Z., Berdis, A. J. & Benkovic, S. J. (2006). Analternative clamp loading pathway via the T4 clamploader gp44/62–DNA complex. Biochemistry, 45,7976–7989.
9. Hinton, D. M. & Nossal, N. G. (1987). Bacteriophage T4DNA primase–helicase. Characterization of oligomersynthesis by T4 61 protein alone and in conjunctionwith T4 41 protein. J. Biol. Chem. 262, 10873–10878.
10. Barry, J. & Alberts, B. (1994). Purification andcharacterization of bacteriophage T4 gene 59 protein.A DNA helicase assembly protein involved in DNAreplication. J. Biol. Chem. 269, 33049–33062.
11. Jones, C. E., Mueser, T. C., Dudas, K. C., Kreuzer, K.N. & Nossal, N. G. (2001). Bacteriophage T4 gene 41helicase and gene 59 helicase-loading protein: aversatile couple with roles in replication and recom-bination. Proc. Natl Acad. Sci. USA, 98, 8312–8318.
12. Dudas, K. C. & Kreuzer, K. N. (2005). BacteriophageT4 helicase loader protein gp59 functions as gate-keeper in origin-dependent replication in vivo. J. Biol.Chem. 280, 21561–21569.
13. Xi, J., Zhuang, Z., Zhang, Z., Selzer, T., Spiering,M. M., Hammes, G. G. & Benkovic, S. J. (2005).Interaction between the T4 helicase-loading protein(gp59) and the DNA polymerase (gp43): a lockingmechanism to delay replication during replisomeassembly. Biochemistry, 44, 2305–2318.
14. Nelson, S. W., Yang, J. & Benkovic, S. J. (2006). Site-directed mutations of T4 helicase loading protein(gp59) reveal multiple modes of DNA polymeraseinhibition and the mechanism of unlocking by gp41helicase. J. Biol. Chem. 281, 8697–8706.
15. Xi, J., Zhang, Z., Zhuang, Z., Yang, J., Spiering, M. M.,Hammes, G. G. & Benkovic, S. J. (2005). Interactionbetween the T4 helicase loading protein (gp59) andthe DNA polymerase (gp43): unlocking of the gp59–gp43–DNA complex to initiate assembly of a fullyfunctional replisome. Biochemistry, 44, 7747–7756.
16. Chastain, P. D., Makhov, A. M., Nossal, N. G. &Griffith, J. (2003). Architecture of the replication

756 Stalling of the T4 Replisome and Fork Regression
complex and DNA loops at the fork generated bythe bacteriophage T4 proteins. J. Biol. Chem. 278,21276–21285.
17. Nossal, N. G., Makhov, A. M., Chastain, P. D., Jones,C. E. & Griffith, J. D. (2007). Architecture of thebacteriophage T4 replication complex revealed withnanoscale biopointers. J. Biol. Chem. 282, 1098–1108.
18. Yang, J., Zhuang, Z., Roccasecca, R. M., Trakselis,M. A. & Benkovic, S. J. (2004). The dynamic proces-sivity of the T4 DNA polymerase during replication.Proc. Natl Acad. Sci. USA, 101, 8289–8294.
19. Kadyrov, F. A. & Drake, J. W. (2001). Conditionalcoupling of leading-strand and lagging-strand DNAsynthesis at bacteriophage T4 replication forks. J. Biol.Chem. 276, 29559–29566.
20. Trakselis, M. A., Roccasecca, R. M., Yang, J., Valentine,A. M. & Benkovic, S. J. (2003). Dissociative propertiesof the proteins within the bacteriophage T4 replisome.J. Biol. Chem. 278, 49839–49849.
21. Nelson, S. W., Kumar, R. & Benkovic, S. J. (2008). RNAprimer handoff in bacteriophage T4 DNA replication:the role of single-stranded DNA-binding protein andpolymerase accessory proteins. J. Biol. Chem. 283,22838–22846.
22. Spiering, M. M., Nelson, S. W. & Benkovic, S. J. (2008).Repetitive lagging strand DNA synthesis by thebacteriophage T4 replisome.Mol. Biosyst. 4, 1070–1074.
23. Alberts, B. M., Barry, J., Bedinger, P., Formosa, T.,Jongeneel, C. V. & Kreuzer, K. N. (1983). Studies onDNA replication in the bacteriophage T4 in vitro system.Cold Spring Harb. Symp. Quant. Biol. 47, 655–668.
24. Yuzhakov, A., Kelman, Z. & O'Donnell, M. (1999).Trading places on DNA—a three-point switch under-lies primer handoff from primase to the replicativeDNA polymerase. Cell, 96, 153–163.
25. Tougu, K. & Marians, K. J. (1996). The interactionbetween helicase and primase sets the replication forkclock. J. Biol. Chem. 271, 21398–21405.
26. Li, X. & Marians, K. J. (2000). Two distinct triggers forcycling of the lagging strand polymerase at thereplication fork. J. Biol. Chem. 275, 34757–34765.
27. Yang, J., Nelson, S. W. & Benkovic, S. J. (2006). Thecontrol mechanism for lagging strand polymeraserecycling during bacteriophage T4 DNA replication.Mol. Cell, 21, 153–164.
28. Cox, M. M., Goodman, M. F., Kreuzer, K. N., Sherratt,D. J., Sandler, S. J. & Marians, K. J. (2000). Theimportance of repairing stalled replication forks.Nature, 404, 37–41.
29. McInerney, P. & O'Donnell, M. (2007). Replisome fateupon encountering a leading strand block and clear-ance from DNA by recombination proteins. J. Biol.Chem. 282, 25903–25916.
30. Pagès, V. & Fuchs, R. P. (2003). Uncoupling of leading-and lagging-strand DNA replication during lesionbypass in vivo. Science, 300, 1300–1303.
31. Higuchi, K., Katayama, T., Iwai, S., Hidaka, M.,Horiuchi, T. &Maki, H. (2003). Fate of DNA replicationfork encountering a single DNA lesion during oriCplasmid DNA replication in vitro. Genes Cells, 8,437–449.
32. McInerney, P. & O'Donnell, M. (2004). Functionaluncoupling of twin polymerases: mechanism ofpolymerase dissociation from a lagging-strand block.J. Biol. Chem. 279, 21543–21551.
33. Yang, J., Trakselis, M. A., Roccasecca, R. M. &Benkovic, S. J. (2003). The application of a minicirclesubstrate in the study of the coordinated T4 DNAreplication. J. Biol. Chem. 278, 49828–49838.
34. Kadyrov, F. A. & Drake, J. W. (2002). Characterizationof DNA synthesis catalyzed by bacteriophage T4replication complexes reconstituted on syntheticcircular substrates. Nucleic Acids Res. 30, 4387–4397.
35. Kawaoka, J., Jankowsky, E. & Pyle, A. M. (2004).Backbone tracking by the SF2 helicase NPH-II. Nat.Struct. Mol. Biol. 11, 526–530.
36. Atkinson, J. & McGlynn, P. (2009). Replication forkreversal and the maintenance of genome stability.Nucleic Acids Res. 37, 3475–3492.
37. Long, D. T. & Kreuzer, K. N. (2009). Fork regression isan active helicase-driven pathway in bacteriophageT4. EMBO Rep. 10, 394–399.
38. Nelson, S. W. & Benkovic, S. J. (2007). The T4 phageUvsW protein contains both DNA unwinding andstrand annealing activities. J. Biol. Chem. 282, 407–416.
39. Pant, K., Karpel, R. L., Rouzina, I. & Williams, M. C.(2005). Salt dependent binding of T4 gene 32 proteinto single and double-stranded DNA: single moleculeforce spectroscopy measurements. J. Mol. Biol. 349,317–330.
40. Delagoutte, E. & von Hippel, P. H. (2001). Molecularmechanisms of the functional coupling of the helicase(gp41) and polymerase (gp43) of bacteriophage T4within the DNA replication fork. Biochemistry, 40,4459–4477.
41. Stano, N. M., Jeong, Y. J., Donmez, I., Tummalapalli,P., Levin, M. K. & Patel, S. S. (2005). DNA synthesisprovides the driving force to accelerate DNA un-winding by a helicase. Nature, 435, 370–373.
42. Lionnet, T., Spiering, M. M., Benkovic, S. J., Bensimon,D. & Croquette, V. (2007). Real-time observation ofbacteriophage T4 gp41 helicase reveals an unwindingmechanism. Proc. Natl Acad. Sci. USA, 104, 19790–19795.
43. Hacker, K. J. & Alberts, B. M. (1994). The slow disso-ciation of the T4 DNA polymerase holoenzyme whenstalled by nucleotide omission. An indication of a highlyprocessive enzyme. J. Biol. Chem. 269, 24209–24220.
44. Jarosz, D. F., Beuning, P. J., Cohen, S. E. & Walker,G. C. (2007). Y-family DNA polymerases in Escherichiacoli. Trends Microbiol. 15, 70–77.
45. Indiani, C., McInerney, P., Georgescu, R., Goodman,M. F. & O'Donnell, M. (2005). A sliding-clamp toolbeltbinds high- and low-fidelity DNA polymerasessimultaneously. Mol. Cell, 19, 805–815.
46. Friedberg, E. C., Lehmann, A. R. & Fuchs, R. P. P.(2005). Trading places: how do DNA polymerasesswitch during translesion DNA synthesis? Mol. Cell,18, 499–505.
47. Miller, E. S., Kutter, E.,Mosig, G., Arisaka, F., Kunisawa,T. & Rüger, W. (2003). Bacteriophage T4 genome.Microbiol. Mol. Biol. Rev. 67, 86–156; table of contents.
48. Lehmann, A. R. & Fuchs, R. P. (2006). Gaps and forksin DNA replication: rediscovering old models DNA.Repair (Amsterdam), 5, 1495–1498.
49. Heller, R. C. & Marians, K. J. (2006). Replication forkreactivation downstream of a blocked nascent leadingstrand. Nature, 439, 557–562.
50. Trakselis, M. A., Roccasecca, R.M., Yang, J., Valentine,A. M. & Benkovic, S. J. (2003). Dissociative propertiesof the proteins within the bacteriophage T4 replisome.J. Biol. Chem. 278, 49839–49849.
51. Yang, J., Zhuang, Z., Roccasecca, R. M., Trakselis,M. A. & Benkovic, S. J. (2004). The dynamic proces-sivity of the T4 DNA polymerase during replication.Proc. Natl Acad. Sci. USA, 101, 8289.
52. Ralf, C. (2006). The bloom's syndrome helicase canpromote the regression of a model replication fork.J. Biol. Chem. 281, 22839–22846.


















