
RegulationofHspb7byMEF2andAP …jcs.biologists.org/content/joces/129/21/4076.full.pdf ·...
15
RESEARCH ARTICLE Regulation of Hspb7 by MEF2 and AP-1: implications for Hspb7 in muscle atrophy Stephanie Wales Tobin 1,2,3 , Dabo Yang 4 , John Girgis 4 , Ali Farahzad 1,2,3 , Alexandre Blais 4 and John C. McDermott 1,2,3,5, * ABSTRACT Mycocyte enhancer factor 2 (MEF2) and activator protein 1 (AP-1) transcription complexes have been individually implicated in myogenesis, but their genetic interaction has not previously been addressed. Using MEF2A, c-Jun and Fra-1 chromatin immunoprecipitation sequencing (ChIP-seq) data and predicted AP- 1 consensus motifs, we identified putative common MEF2 and AP-1 target genes, several of which are implicated in regulating the actin cytoskeleton. Because muscle atrophy results in remodelling or degradation of the actin cytoskeleton, we characterized the expression of putative MEF2 and AP-1 target genes (Dstn, Flnc, Hspb7, Lmod3 and Plekhh2) under atrophic conditions using dexamethasone (Dex) treatment in skeletal myoblasts. Heat shock protein b7 (Hspb7) was induced by Dex treatment and further analyses revealed that loss of MEF2A using siRNA prevented Dex- regulated induction of Hspb7. Conversely, ectopic Fra-2 or c-Jun expression reduced Dex-mediated upregulation of Hspb7 whereas AP-1 depletion enhanced Hspb7 expression. In vivo, expression of Hspb7 and other autophagy-related genes was upregulated in response to atrophic conditions in mice. Manipulation of Hspb7 levels in mice also impacted gross muscle mass. Collectively, these data indicate that MEF2 and AP-1 confer antagonistic regulation of Hspb7 gene expression in skeletal muscle, with implications for autophagy and muscle atrophy. KEY WORDS: Myogenesis, MEF2, AP-1, Autophagy INTRODUCTION Muscle atrophy is a phenomenon associated with reduced muscle fibre number and size caused by increased proteolysis and decreased protein synthesis (Romanick et al., 2013). In the elderly, muscle wasting is referred to as sarcopenia (Morley et al., 2001); in patients with cancer, AIDS or other chronic diseases, muscle atrophy is referred to as cachexia (Kotler, 2000). Improving or maintaining muscle mass in these populations has a profound impact on the ‘health span’ of individuals. For example, there is evidence that cachexia in cancer patients directly affects the time to tumour progression and disease recurrence (Kadar et al., 2000; Prado et al., 2009). In the ubiquitin proteasome pathway, the forkhead box protein (FoxO) family of transcription factors activates muscle atrophy through induction of two E3 ubiquitin ligases, MAFbx/atrogin-1 and MuRF1 (also known as TRIM63) (Sandri et al., 2004). Current treatment programs for muscle atrophy include activating the serine/threonine protein kinase (Akt) pathway, which induces muscle hypertrophy by inactivating FoxO proteins (Stitt et al., 2004). However, Akt can be inhibited by myostatin, a member of the transforming growth factor β (TGF-β) superfamily (Trendelenburg et al., 2009), superseding Akt activation as a treatment option. A new antibody recently characterized to bind to both members (A and B) of the myostatin/activin type II receptor (ActRII) induces hypertrophy in a muscle wasting model in vivo (Lach-Trifilieff et al., 2014). Additionally, targeting ActRIIB in cancer cachexia models can prevent atrophy, which results in prolonged survival without tumour manipulation (Zhou et al., 2010). The autophagy pathway is an alternative mechanism of protein degradation that has also been implicated in muscle wasting. Foxo3, unlike other members of the FoxO family, is able to regulate autophagy in addition to the ubiquitin–proteasome pathway (Mammucari et al., 2007; Zhao et al., 2007). Several possible autophagy pathways have been identified in muscle, two of which are macroautophagy and chaperone-mediated autophagy (CMA). Although both processes ultimately lead to protein degradation in the lysosome, they achieve this through different mechanisms. In CMA, heat shock cognate 70 (Hsc70) targets proteins directly to the lysosome (Chiang et al., 1989). Macroautophagy requires de novo synthesis of autophagosomes in a multistep process that involves autophagy-related protein (Atg) family members. Autophagy is required for muscle homeostasis, as demonstrated by the muscle atrophy in mouse knockout models that lack proteins involved in autophagosome formation, such as Atg5 and Atg7 (Masiero et al., 2009; Raben et al., 2008). Aged muscle shows decreased autophagy and therefore build-up of protein aggregates (Demontis and Perrimon, 2010). LC3B (Map1lc3b) is an Atg protein that provides a useful readout for autophagy because it is post- translationally modified as it becomes part of the autophagosome (Kabeya et al., 2000). First, pro-LC3B is cleaved by Atg4 to form cytosolic LC3B-I. Atg7 then lipidates LC3B-I to form LC3B-II, which can form part of the autophagosome. Using samples from various atrophic mouse models, LC3B was shown to be strongly upregulated (Lecker et al., 2004). Furthermore, Foxo3 can directly regulate several autophagy-related genes, including that encoding LC3B (Mammucari et al., 2007; Zhao et al., 2007). A form of autophagy termed chaperone-assisted selective autophagy (CASA) merges the chaperone-mediated and macroautophagy pathways. In CASA, Hsc70 forms a complex with Bag3, Hspb8 and E3 ubiquitin ligase CHIP to identify protein aggregates and target them to the autophagosome (Arndt et al., 2010). Received 29 March 2016; Accepted 8 September 2016 1 Department of Biology, York University, 4700 Keele Street, Toronto, Ontario, Canada M3J 1P3. 2 Muscle Health Research Centre (MHRC), York University, 4700 Keele Street, Toronto, Ontario, Canada M3J 1P3. 3 Centre for Research in Biomolecular Interactions (CRBI), 4700 Keele Street, Toronto, Ontario, Canada M3J 1P3. 4 Ottawa Institute of Systems Biology, University of Ottawa, Health Sciences Campus, 451 Smyth Road, Ottawa, Ontario, Canada K1H 8M5. 5 Centre for Research in Mass Spectrometry (CRMS), York University, 4700 Keele Street, Toronto, Ontario, Canada M3J 1P3. *Author for correspondence ( [email protected]) J.C.M., 0000-0001-9696-8929 4076 © 2016. Published by The Company of Biologists Ltd | Journal of Cell Science (2016) 129, 4076-4090 doi:10.1242/jcs.190009 Journal of Cell Science
Transcript of RegulationofHspb7byMEF2andAP …jcs.biologists.org/content/joces/129/21/4076.full.pdf ·...

RESEARCH ARTICLE
Regulation of Hspb7 by MEF2 and AP-1: implications for Hspb7 inmuscle atrophyStephanie Wales Tobin1,2,3, Dabo Yang4, John Girgis4, Ali Farahzad1,2,3, Alexandre Blais4 andJohn C. McDermott1,2,3,5,*
ABSTRACTMycocyte enhancer factor 2 (MEF2) and activator protein 1 (AP-1)transcription complexes have been individually implicated inmyogenesis, but their genetic interaction has not previously beenaddressed. Using MEF2A, c-Jun and Fra-1 chromatinimmunoprecipitation sequencing (ChIP-seq) data and predicted AP-1 consensus motifs, we identified putative common MEF2 and AP-1target genes, several of which are implicated in regulating the actincytoskeleton. Because muscle atrophy results in remodelling ordegradation of the actin cytoskeleton, we characterized theexpression of putative MEF2 and AP-1 target genes (Dstn, Flnc,Hspb7, Lmod3 and Plekhh2) under atrophic conditions usingdexamethasone (Dex) treatment in skeletal myoblasts. Heat shockprotein b7 (Hspb7) was induced by Dex treatment and furtheranalyses revealed that loss of MEF2A using siRNA prevented Dex-regulated induction of Hspb7. Conversely, ectopic Fra-2 or c-Junexpression reduced Dex-mediated upregulation of Hspb7 whereasAP-1 depletion enhanced Hspb7 expression. In vivo, expression ofHspb7 and other autophagy-related genes was upregulated inresponse to atrophic conditions in mice. Manipulation of Hspb7levels in mice also impacted gross muscle mass. Collectively, thesedata indicate that MEF2 and AP-1 confer antagonistic regulation ofHspb7 gene expression in skeletal muscle, with implications forautophagy and muscle atrophy.
KEY WORDS: Myogenesis, MEF2, AP-1, Autophagy
INTRODUCTIONMuscle atrophy is a phenomenon associated with reduced musclefibre number and size caused by increased proteolysis anddecreased protein synthesis (Romanick et al., 2013). In theelderly, muscle wasting is referred to as sarcopenia (Morleyet al., 2001); in patients with cancer, AIDS or other chronicdiseases, muscle atrophy is referred to as cachexia (Kotler, 2000).Improving or maintaining muscle mass in these populations has aprofound impact on the ‘health span’ of individuals. For example,there is evidence that cachexia in cancer patients directly affects thetime to tumour progression and disease recurrence (Kadar et al.,
2000; Prado et al., 2009). In the ubiquitin proteasome pathway, theforkhead box protein (FoxO) family of transcription factorsactivates muscle atrophy through induction of two E3 ubiquitinligases, MAFbx/atrogin-1 and MuRF1 (also known as TRIM63)(Sandri et al., 2004). Current treatment programs for muscleatrophy include activating the serine/threonine protein kinase (Akt)pathway, which induces muscle hypertrophy by inactivating FoxOproteins (Stitt et al., 2004). However, Akt can be inhibited bymyostatin, a member of the transforming growth factor β (TGF-β)superfamily (Trendelenburg et al., 2009), superseding Aktactivation as a treatment option. A new antibody recentlycharacterized to bind to both members (A and B) of themyostatin/activin type II receptor (ActRII) induces hypertrophyin a muscle wasting model in vivo (Lach-Trifilieff et al., 2014).Additionally, targeting ActRIIB in cancer cachexia models canprevent atrophy, which results in prolonged survival withouttumour manipulation (Zhou et al., 2010).
The autophagy pathway is an alternative mechanism of proteindegradation that has also been implicated in muscle wasting. Foxo3,unlike other members of the FoxO family, is able to regulateautophagy in addition to the ubiquitin–proteasome pathway(Mammucari et al., 2007; Zhao et al., 2007). Several possibleautophagy pathways have been identified in muscle, two of whichare macroautophagy and chaperone-mediated autophagy (CMA).Although both processes ultimately lead to protein degradation inthe lysosome, they achieve this through different mechanisms. InCMA, heat shock cognate 70 (Hsc70) targets proteins directly to thelysosome (Chiang et al., 1989). Macroautophagy requires de novosynthesis of autophagosomes in a multistep process that involvesautophagy-related protein (Atg) family members. Autophagy isrequired for muscle homeostasis, as demonstrated by the muscleatrophy in mouse knockout models that lack proteins involved inautophagosome formation, such as Atg5 and Atg7 (Masiero et al.,2009; Raben et al., 2008). Aged muscle shows decreased autophagyand therefore build-up of protein aggregates (Demontis andPerrimon, 2010). LC3B (Map1lc3b) is an Atg protein thatprovides a useful readout for autophagy because it is post-translationally modified as it becomes part of the autophagosome(Kabeya et al., 2000). First, pro-LC3B is cleaved by Atg4 to formcytosolic LC3B-I. Atg7 then lipidates LC3B-I to form LC3B-II,which can form part of the autophagosome. Using samples fromvarious atrophic mouse models, LC3B was shown to be stronglyupregulated (Lecker et al., 2004). Furthermore, Foxo3 can directlyregulate several autophagy-related genes, including that encodingLC3B (Mammucari et al., 2007; Zhao et al., 2007). A form ofautophagy termed chaperone-assisted selective autophagy (CASA)merges the chaperone-mediated and macroautophagy pathways. InCASA, Hsc70 forms a complex with Bag3, Hspb8 and E3 ubiquitinligase CHIP to identify protein aggregates and target them to theautophagosome (Arndt et al., 2010).Received 29 March 2016; Accepted 8 September 2016
1Department of Biology, York University, 4700 Keele Street, Toronto, Ontario,Canada M3J 1P3. 2Muscle Health Research Centre (MHRC), York University, 4700Keele Street, Toronto, Ontario, Canada M3J 1P3. 3Centre for Research inBiomolecular Interactions (CRBI), 4700 Keele Street, Toronto, Ontario, CanadaM3J1P3. 4Ottawa Institute of Systems Biology, University of Ottawa, Health SciencesCampus, 451 Smyth Road, Ottawa, Ontario, Canada K1H 8M5. 5Centre forResearch in Mass Spectrometry (CRMS), York University, 4700 Keele Street,Toronto, Ontario, Canada M3J 1P3.
*Author for correspondence ( [email protected])
J.C.M., 0000-0001-9696-8929
4076
© 2016. Published by The Company of Biologists Ltd | Journal of Cell Science (2016) 129, 4076-4090 doi:10.1242/jcs.190009
Journal
ofCe
llScience

Myocyte-enhancer factor 2 (MEF2) is a member of the MADS-box family of transcription factors found in many tissues, includingskeletal and cardiac muscle (Edmondson et al., 1994; Lin et al.,1997). MEF2 functions in a homo- or heterodimer complex withfour different MEF2 isoforms in vertebrates (MEF2A–MEF2D),which bind to the consensus sequence [C/TTA(A/T)4TAG/A].Previously we have shown that MEF2A can target a shared subset ofgenes in C2C12myoblasts, an in vitromodel of skeletal myogenesis,and in primary cardiomyocytes (Wales et al., 2014). Gene ontology(GO) analysis contained terms enriched for actin cytoskeletonorganization and actin filament-based processes. In addition, manyof these cytoskeletal MEF2 target genes were enriched for activatorprotein 1 (AP-1) cis elements, suggesting the possibility ofcombinatorial control. AP-1, consisting of Jun homodimer or Jun–Fos family heterodimers, recognizes the consensus sequence TGAG/CTCA (Angel et al., 1987). Global analysis of myoblastdetermination protein (MyoD) target genes in skeletal myoblastslikewise showed that AP-1 motifs are prominent in neighbouringsequences (Blais et al., 2005; Cao et al., 2010). NeighbouringMEF2and AP-1 cis elements have recently been shown to be enriched inmacrophages and neurons (Ma and Telese, 2015; Nagy et al., 2013).Several AP-1 subunits have been implicated in myogenesis. Inparticular, c-Jun antagonizes MyoD transcriptional activity in vitro(Bengal et al., 1992; Li et al., 1992). Using high throughput data,Blum et al. (2012) reported that c-Jun and MyoD coordinate muscleenhancers, indicating a more complex role for AP-1 in muscle thanpreviously anticipated (Blum et al., 2012). Additionally, the Fosfamily member Fra-2 is thought to play a role in maintenance of theskeletal muscle satellite cell population (Alli et al., 2013).Although MEF2 and AP-1 have individually been shown to
function in the myogenic program, their potential interaction has notbeen documented. Additionally, although loss of MEF2 and AP-1have been implicated in loss of sarcomere integrity duringdevelopment and satellite cell-mediated muscle regeneration(Hinits and Hughes, 2007; Potthoff et al., 2007; Windak et al.,2013), the combined role of these factors in muscle atrophy has notbeen investigated. Here, we document that MEF2 and AP-1 regulateseveral genes associated with the actin cytoskeleton. Among thegene products, the small heat shock protein, Hspb7 is implicated inmuscle atrophy.
RESULTSIdentification of biological processes associated withMEF2A and AP-1 recruitmentMEF2 and AP-1 are transcription complexes involved in myoblastproliferation and differentiation, yet whether they regulate commonor non-overlapping target genes during differentiation has not beenthoroughly explored. To address this, we utilized a previouslyreported MEF2A dataset from chromatin immunoprecipitationcombined with exonuclease digestion (ChIP-exo) fordifferentiating C2C12 myogenic cells (48 h in differentiationmedium; DM) (Wales et al., 2014) and compared these bindingevents with data from c-Jun (Blum et al., 2012) and Fra-1 (Woldgroup, ENCODE) ChIP in combination with sequencing (ChIP-seq), which was likewise carried out in myogenic cells. Fra-1 isprimarily associated with bone development (Eferl et al., 2004;Fleischmann, 2000) and c-Jun has been implicated in many tissues,including muscle (Bengal et al., 1992; Blum et al., 2012; Li et al.,1992). Both the c-Jun and Fra-1 ChIP-seq data were completed inC2C12 myoblasts in growth medium (GM), making these datacomparable with our previously generated MEF2A dataset. The c-Jun dataset contained 9778 binding events and the Fra-1 dataset
contained 6507. To determine the percentage of shared binding sitesacross these datasets we used a MEF2A-centric analysis (Fig. 1A).From this analysis we observed that the majority of MEF2A bindingsites (69%) were independent of c-Jun or Fra-1 recruitment, yet 17%of MEF2A-bound DNA also contained c-Jun recruitment and 12%contained both c-Jun and Fra-1. Fra-1 andMEF2A alone shared fewbinding sites (2%).
Functional roles for shared MEF2A and AP-1 binding sites wereidentified using the Genomics Regions Enrichment of AnalysisTool (GREAT), which revealed enriched GO terms for biologicalprocesses, the top ten of which are depicted in Fig. 1B. The GOterms were ranked by binomial raw P-value; the number of geneswithin each GO term is indicated. DNA enriched for MEF2A-alonewas associated with the better-known functions of MEF2 such asactin-filament-based processes and skeletal muscle tissuedevelopment, but also with cell development (blue). There wereno GO terms identified for MEF2A and Fra-1. However, MEF2Aand c-Jun had GO terms for striated muscle development, vasculardevelopment and heart morphogenesis (purple). The complete listof GO terms is given in Supplementary Dataset S1. Fewer GO termswere enriched for genes associated with all three factors and thesewere related to the actin cytoskeleton and negative regulation ofsmooth muscle cell proliferation (black).
Because the MEF2A dataset was obtained from differentiatingmyoblasts, and given that several other AP-1 components apart fromc-Jun and Fra-1 could be associated with MEF2 target genes, wedetermined the location of AP-1 consensus sequences containing thesequence TGAGTCA using cisGenome and allowing zeromismatches. From the mm9 genome, this search identified 264,537AP-1 consensus sites. We focused on putative MEF2A and AP-1binding sites within ±10 kb of the transcription start site (TSS) of thesingle nearest gene and observed that 11 out of these 76 genes areassociated with the molecular function ‘cytoskeleton protein binding’(Table S1). We then assessed the recruitment of MEF2A, c-Jun orFra-1and the presence of AP-1 consensus sequences near five of thesegenes, encoding the proteins destrin (Dstn), filamin C (Flnc), heatshock protein family, member 7 (Hspb7), leiomodin 3 (Lmod3) andpleckstrin homology domain containing family H, member 2(Plekhh2). Dstn is an actin-depolymerizing protein (Carlier et al.,1997) and FlInc, a muscle-specific filamin (Thompson et al., 2000)that promotes the cross-linking of actin. We had also previouslyidentified the genes encoding Hspb7 (a small heat shock protein) andLmod3 (tropomodulin family member) as MEF2 target genes incardiac muscle (Wales et al., 2014). Human nemaline myopathy hasbeen associated with mutations in Lmod3 (Cenik et al., 2015; Garget al., 2014; Yuen et al., 2014). Hspb7 expression is higher in mdx (agenetic model of muscular dystrophy) mice than in normal mice andis also enhanced in ageing muscle (Doran et al., 2006, 2007).Recently, a skeletal muscle-specific conditional Hspb7 knockoutmodel was reported to result in progressive myopathy (Juo et al.,2016). The role of Plekhh2 appears to be to stabilize the cortical actincytoskeleton (Perisic et al., 2012). The recruitment pattern ofMEF2A,c-Jun, Fra-1 and anyAP-1 consensus sequences is indicated in imagesfrom UCSC (Fig. S1). The overall trends demonstrate several points.First, MEF2A was performed using ChIP-exo, which involvesexonuclease digestion prior to sequencing, therefore MEF2A peaksare more defined. This indicates one of the advantages of ChIP-exoover conventional ChIP-seq. Second, the scale of c-Jun and Fra-1recruitment differ dramatically, which could indicate differentialantibody affinities or distinct DNA binding affinities of AP-1 familymembers. Third,MEF2A and c-Jun or Fra-1 show similar recruitmentpatterns to a subset of putative target genes. Of these five MEF2A
4077
RESEARCH ARTICLE Journal of Cell Science (2016) 129, 4076-4090 doi:10.1242/jcs.190009
Journal
ofCe
llScience

target genes, c-Jun was recruited to an overlapping or neighbouringbinding event near Flnc, Hspb7 and Plekhh2 (Fig. S1). Fra-1 andMEF2A recruitment only overlapped at the promoter ofFlnc, and Fra-1 was detected within the second intron of Hspb7, where c-Jun alsoshowed recruitment (Fig. S1). This in silico analysis identifies apotential interplay between AP-1 and MEF2 in the transcriptionalcontrol of target genes involved in muscle stability.
Actin cytoskeletal genes are regulated by MEF2A and AP-1Aside from c-Jun and Fra-1, AP-1 has many family members thatare expressed in muscle, including Fra-2, JunD, c-Fos and several
others. We assessed the expression pattern of MEF2A and AP-1family members during C2C12 differentiation (Fig. 2A). Asmyogenin and MEF2A levels increased during myogenesis, onlyone AP-1 subunit (Fra-2) also increased. c-Jun, JunD and, mostdramatically, Fra-1 levels were reduced in DM. To move forward indetermining a role for MEF2 and AP-1 in myogenesis, we focusedon Fra-2 and c-Jun because they are expressed in myoblasts andhave been shown to have a role in myogenesis (Alli et al., 2013;Bengal et al., 1992; Blum et al., 2012; Li et al., 1992). Additionally,the Fra-2–c-Jun heterodimer is one of the main AP-1 bindingcomplexes present in C2C12 differentiation (Andreucci et al.,
B
A
17%
2%
69%
12%
MEF2A (2783)
MEF2A with c-Jun only
MEF2A with Fra-1 only
MEF2A alone
MEF2A with c-Jun and Fra-1
GO Biological Processes-log10(Binomial p -value)
0 1 2 3 4 5 6
posi�ve regula�on of apopto�c process (22)small GTPase mediated signal transduc�on (25)
response to molecule of bacterial origin (18)heart morphogenesis (25)
Ras protein signal transduc�on (11)striated muscle �ssue development (28)
regula�on of vasculature development (18)hemopoiesis (30)
posi�ve regula�on of cell death (23)muscle �ssue development (30)
MEF2A with c-Jun
0 1 2 3 4 5 6 7
mesoderm development (11)regula�on of cell growth (15)
segment specifica�on (4)middle ear morphogenesis (5)
nega�ve regula�on of transport (18)ac�n filament-based process (18)
nega�ve regula�on of smooth muscle cell prolifera�on (5)
MEF2A with c-Jun and Fra-1
0 2 4 6 8 10 12
skeletal muscle �ssue development (32)posi�ve regula�on of myeloid cell differen�a�on (25)
regula�on of B cell apopto�c process (9)regula�on of leukocyte migra�on (21)
ac�n filament organiza�on (29)posi�ve regula�on of apopto�c signaling pathway (24)
nega�ve regula�on of cysteine-type endopep�dase ac�vity (16)nega�ve regula�on of cysteine-type endopep�dase ac�vity involved in apopto�c process (16)
regula�on of myeloid cell differen�a�on (42)ac�n filament-based process (71)
MEF2A alone
Fig. 1. Comparison of MEF2A and AP-1 target genes in skeletal muscle. (A) Percentage overlap between MEF2A, c-Jun, and Fra-1. The total number ofMEF2AChIP-seq binding events is 2783. All datasets were converted to themm9 genome and then overlapping peaks were identified from individual bed files forMEF2A, c-Jun and Fra-1 using theUCSC intersect function. (B) Functional roles for MEF2A alone, MEF2A and c-Jun, andMEF2Awith c-Jun and Fra-1. Using thedatasets from 1A, GREATanalysis revealed GO terms for biological processes. The total number of genes per GO term are indicated to the right of each GO term.
4078
RESEARCH ARTICLE Journal of Cell Science (2016) 129, 4076-4090 doi:10.1242/jcs.190009
Journal
ofCe
llScience
A B
D
48 hr DM
scr
siM
EF2A
siFr
a-2
sic-
Jun
MEF2A
Fra-2
c-Jun
Ac�n
C
GM 24 hr 48 hr
MEF2A
c-Jun
MyoG
Ac�n
Fra-2
Fra-1
JunD
0.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
1.6
0 hr DM 24 hr DM 72 hr DM
Fold
Cha
nge
Dstn
0
5
10
15
20
25
30
35
40
45
0 hr DM 24 hr DM 72 hr DM
Fold
Cha
nge
Plekhh2
Control
Tam67
E
0 hr 24 hr 72 hr 0 hr 24 hr 72 hr
GFP
Ac�n
F
0
20
40
60
80
100
120
Dstn Flnc Hspb7 Lmod3 Plekhh2
Fold
Cha
nge
0 hr
48 hr
*
*
*
*
pcDNA3 GFP-Tam67
100
300
500
Hspb7
Fold
Cha
nge
0.0
2.0
4.0
0 hr DM 24 hr DM 72 hr DM
0.0
2.0
4.0
0 hr DM 24 hr DM 72 hr DM
100
600
Lmod3
0
5
0 hr DM 24 hr DM 72 hr DM
20
70
120
Flnc
0.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
1.6
1.8
Dstn Flnc Hspb7 Lmod3 Plekhh2 MEF2A Fra-2 c-Jun
Fold
Cha
nge
siMEF2A
siFra-2
sic-Jun
*****
*
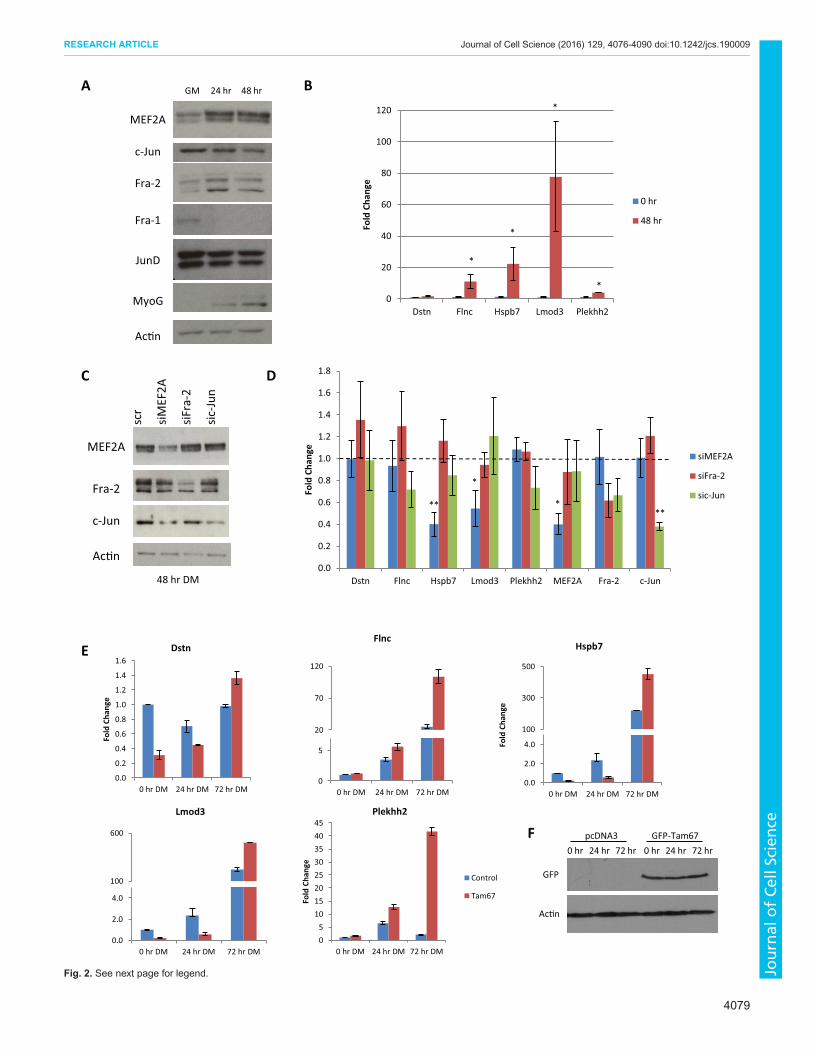
Fig. 2. See next page for legend.
4079
RESEARCH ARTICLE Journal of Cell Science (2016) 129, 4076-4090 doi:10.1242/jcs.190009
Journal
ofCe
llScience

2002). Fra-2 is present in different isoforms and subject to post-translational modification by ERK (Alli et al., 2013; Andreucciet al., 2002). To date, Fra-2 ChIP-seq data is not available.The expression of cytoskeletal genes during myogenesis was
determined using quantitative reverse transcription (qRT)-PCR inGM (myoblasts) and at 48 h in DM (myocytes) (Fig. 2B). DuringC2C12 differentiation, the expression of each gene except Dstnincreased. To confirm MEF2A recruitment to Dstn, Flnc, Hspb7,Lmod3 and Plekhh2, ChIP-qPCR was performed in growthconditions (GM) and during differentiation (48 h DM). Duringdifferentiation, MEF2Awas recruited to each gene, compared with acontrol region upstream of SMA (Fig. S2). MEF2A recruitment toLmod3 and Hspb7 was the most significant and reflects the ChIP-seq data (Fig. S1). Fra-2 and c-Jun recruitment were also assessed atthe same regions asMEF2A (Figs S1 and S2). c-Jun was moderatelyrecruited to Flnc, Hspb7 and Plekhh2 in both GM and 48 h DMequally but was not detected near Dstn or Lmod3. Fra-2 enrichmentwas detected for all genes at 48 h DM,with the strongest recruitmentobserved on Flnc (Fig. S2).To determine whether actin cytoskeletal genes are sensitive to the
loss of AP-1 and MEF2 we utilized siRNA-mediated genesilencing. MEF2A is the predominant MEF2 subunit indifferentiating myogenic cells and our previous ChIP-exo datawas completed using a MEF2A antibody; therefore, we initiallyused siRNA targeting MEF2A. Because AP-1 could function inJun–Fos or Jun–Jun homodimers and because in muscle c-Jun andFra-2 have been documented to be crucial factors in regulatingmyoblast proliferation (Alli et al., 2013; Bengal et al., 1992), weused siRNA targeting c-Jun and Fra-2 (Fig. 2C). Depletion ofMEF2A resulted in downregulation ofHspb7 and Lmod3 (Fig. 2D).Dstn, Flnc and Plekhh2 showed MEF2A recruitment (Fig. S2) butno differential expression with MEF2A knockdown, possiblyreflecting some level of redundancy in the MEF2 family. Areduction in c-Jun did not significantly affect target gene expression,although decreases in Flnc, Plekhh2 and Fra-2 were observed.Similarly, although Fra-2 expression was partially reduced, this didnot result in dramatic changes in gene expression, potentiallyindicating AP-1 functional redundancy.To account for partial knockdown with siRNA technology or the
involvement of other AP-1 family members in expression of thesegenes, we exogenously expressed GFP–Tam67, a potent dominantnegative form of c-Jun that lacks the transactivation domain butcontains the DNA binding and dimerization domains. Previously,Tam67 has been used as a dominant negative of the AP-1 complexand, as such, is a useful tool to use when redundancy of the complexAP-1 subunits is suspected (Hennigan and Stambrook, 2001). Weassessed expression of the five target genes in growth conditions(0 h DM) and after 24 and 72 h in DM. Tam67 expression had
marginal effects on Dstn, Flnc, Hspb7, Lmod3 and Plekhh2expression at 0 and 24 h in DM (Fig. 2E). However, after 72 h inDM, all five genes were robustly upregulated by Tam67 expression.Expression of GFP–Tam67 is depicted in Fig. 2F. Together thesedata indicate that a subset of MEF2A target genes aretranscriptionally regulated by AP-1.
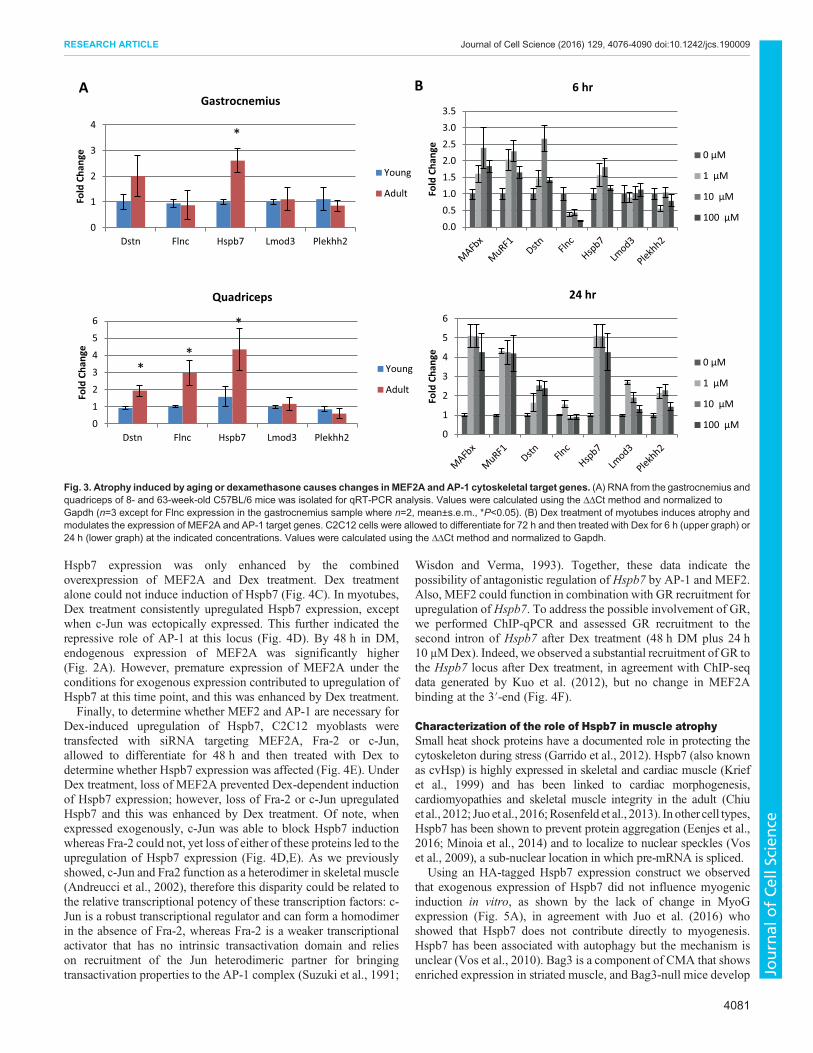
Atrophy induced by age or dexamethasone modulatesexpression of MEF2A and AP-1 cytoskeletal target genesThe cytoskeleton is integrally linked to the contractile unit of themyofibril, the sarcomere, which is mainly comprised of α-actin andmyosin. During muscle atrophy (a phenomenon observed insarcopenia, cachexia and various genetic diseases) thecytoskeleton and components of the sarcomere become degraded,resulting in overall muscle loss and weakness. Because these fiveproteins (Dstn, Flnc, Hspb7, Lmod3 and Plekhh2) are involved inthe actin cytoskeleton, to varying degrees, we next determinedwhether expression of the corresponding MEF2 and AP-1 targetgenes changes under atrophic conditions. To determine whetherthese genes were differentially expressed in growing postnatalmuscle compared with mature adult muscle we isolated RNA fromthe gastrocnemius and quadriceps of 8- and 63-week-old mice(Fig. 3A). In the gastrocnemius,Hspb7was upregulated with age. Inthe quadriceps, Dstn, Flnc and Hspb7 were upregulated.
In cell culture, muscle atrophy can be replicated by treatment withDex, a synthetic glucocorticoid. To model glucocorticoid-inducedatrophy, C2C12 myoblasts were allowed to differentiate for 72 h inDM, and then treated with Dex (Fig. 3B). In this analysis weincluded two E3 ubiquitin ligases, MAFbx and MuRF1, which areassociated with muscle atrophy and serve as positive controls for theprocess. These E3 ligases promote atrophy and ubiquitinate proteinsfor degradation. In muscle, MuRF1 directly targets myosin andmyosin binding proteins for degradation, contributing to sarcomereloss (Clarke et al., 2007; Cohen et al., 2009). After 6 or 24 h oftreatment with Dex, MAFbx and MuRF1 were upregulated.Treatment with Dex for 6 h increased Dstn and Hspb7 expressionand decreased Flnc. Expression of Lmod3 and Plekhh2 wasunchanged. After 24 h treatment with Dex these trends were similar;however, Hspb7 was upregulated fivefold, equivalent to the degreeof MAFbx and MuRF1 induction.
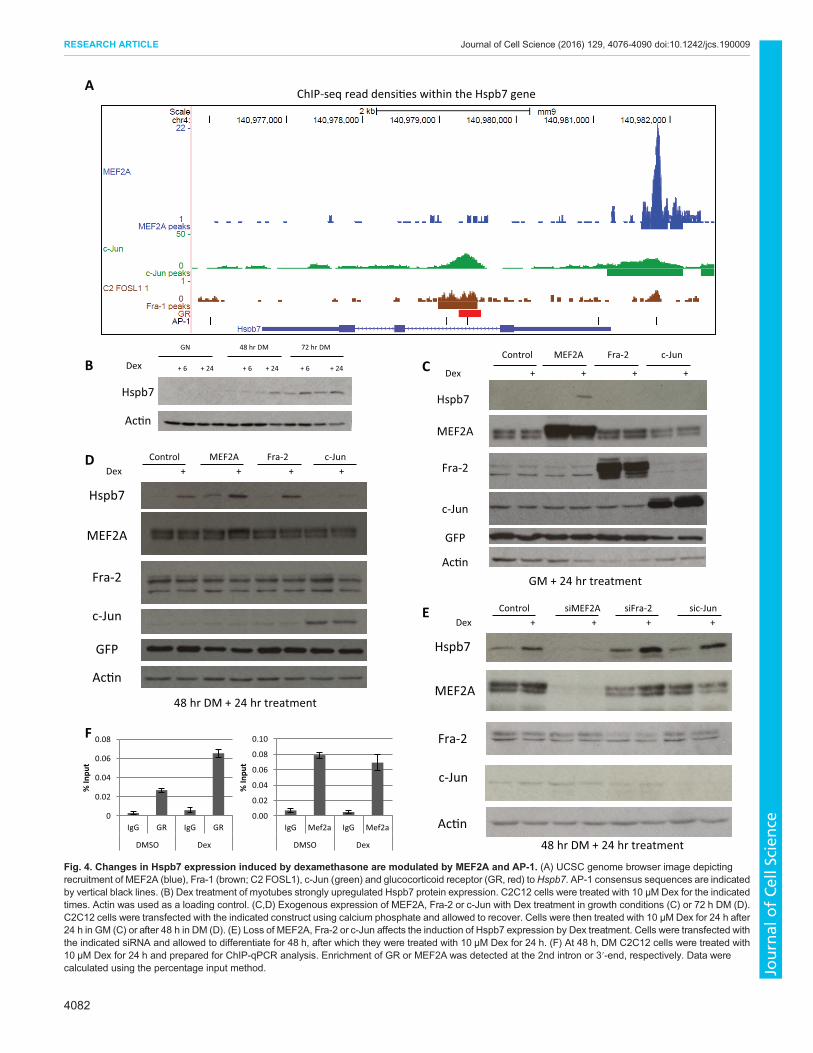
MEF2A and c-Jun and Fra-2 regulate atrophy-induced Hspb7expressionHspb7 was the most dynamically regulated gene during ageing inskeletal muscle and was also highly inducible in response to Dextreatment; therefore, we pursued the regulation of this gene byMEF2A, c-Jun and Fra-2 in more detail. A previous study hadidentified target genes of the glucocorticoid receptor (GR) in Dex-treated C2C12 myotubes (Kuo et al., 2012). Interestingly, this studyfound GR recruitment to an intron of Hspb7. Using the UCSCbrowser we plotted the MEF2A, c-Jun, Fra-1 and AP-1 peaks andcompared them with GR recruitment reported by Kuo et al. (2012)within the Hspb7 gene (Fig. 4A). GR was recruited following Dextreatment to within the second intron of Hspb7, which contained c-Jun and Fra-1 enrichment peaks. We utilized Dex treatment tofurther determine how MEF2A, AP-1 and GR might contribute toHspb7 expression.
At the protein level, Dex treatment upregulated Hspb7 expressionin the late stages of differentiation but not under growth conditions(Fig. 4B). Next, the effect of exogenous expression of MEF2A, Fra-2 or c-Jun in combination with Dex treatment was assessed ingrowth conditions and in 72 h myotubes. Under growth conditions,
Fig. 2. MEF2A and AP-1 regulation of actin cytoskeletal genes. (A) MEF2Aand AP-1 protein expression during myogenesis. C2C12 cells were allowed todifferentiate frommyoblasts (GM) to 48 h DM. (B) Expression pattern of MyoG,Dstn, Flnc, Hspb7, Lmod3 and Plekhh2 during C2C12 differentiation in GMand 48 h DM. Values were calculated using the ΔΔCt method and normalizedto β-actin (n=3, mean±s.e.m., *P<0.05, **P<0.01). (C) Efficiency of knockdownof MEF2A, Fra-2 and c-Jun in C2C12 myocytes (48 h DM) using siRNA-mediated gene silencing. C2C12 cells were transfected with siRNA andallowed to differentiate for 48 h before western blot analysis. scr indicates ascrambled siRNA control. (D) siRNA-mediated knockdown of MEF2A, Fra-2and c-Jun at 48 h DM was carried out to assess changes in target geneexpression. Data were analysed as in B. (E) C2C12 cells were transfected withGFP–Tam67 using calcium phosphate. RNAwas isolated at the indicated timepoints and analysed via qRT-PCR as in B. (F) Western blot depictingexpression of GFP-tagged Tam67.
4080
RESEARCH ARTICLE Journal of Cell Science (2016) 129, 4076-4090 doi:10.1242/jcs.190009
Journal
ofCe
llScience
Hspb7 expression was only enhanced by the combinedoverexpression of MEF2A and Dex treatment. Dex treatmentalone could not induce induction of Hspb7 (Fig. 4C). In myotubes,Dex treatment consistently upregulated Hspb7 expression, exceptwhen c-Jun was ectopically expressed. This further indicated therepressive role of AP-1 at this locus (Fig. 4D). By 48 h in DM,endogenous expression of MEF2A was significantly higher(Fig. 2A). However, premature expression of MEF2A under theconditions for exogenous expression contributed to upregulation ofHspb7 at this time point, and this was enhanced by Dex treatment.Finally, to determine whether MEF2 and AP-1 are necessary for
Dex-induced upregulation of Hspb7, C2C12 myoblasts weretransfected with siRNA targeting MEF2A, Fra-2 or c-Jun,allowed to differentiate for 48 h and then treated with Dex todetermine whether Hspb7 expression was affected (Fig. 4E). UnderDex treatment, loss of MEF2A prevented Dex-dependent inductionof Hspb7 expression; however, loss of Fra-2 or c-Jun upregulatedHspb7 and this was enhanced by Dex treatment. Of note, whenexpressed exogenously, c-Jun was able to block Hspb7 inductionwhereas Fra-2 could not, yet loss of either of these proteins led to theupregulation of Hspb7 expression (Fig. 4D,E). As we previouslyshowed, c-Jun and Fra2 function as a heterodimer in skeletal muscle(Andreucci et al., 2002), therefore this disparity could be related tothe relative transcriptional potency of these transcription factors: c-Jun is a robust transcriptional regulator and can form a homodimerin the absence of Fra-2, whereas Fra-2 is a weaker transcriptionalactivator that has no intrinsic transactivation domain and relieson recruitment of the Jun heterodimeric partner for bringingtransactivation properties to the AP-1 complex (Suzuki et al., 1991;
Wisdon and Verma, 1993). Together, these data indicate thepossibility of antagonistic regulation of Hspb7 by AP-1 and MEF2.Also, MEF2 could function in combination with GR recruitment forupregulation of Hspb7. To address the possible involvement of GR,we performed ChIP-qPCR and assessed GR recruitment to thesecond intron of Hspb7 after Dex treatment (48 h DM plus 24 h10 μMDex). Indeed, we observed a substantial recruitment of GR tothe Hspb7 locus after Dex treatment, in agreement with ChIP-seqdata generated by Kuo et al. (2012), but no change in MEF2Abinding at the 3′-end (Fig. 4F).
Characterization of the role of Hspb7 in muscle atrophySmall heat shock proteins have a documented role in protecting thecytoskeleton during stress (Garrido et al., 2012). Hspb7 (also knownas cvHsp) is highly expressed in skeletal and cardiac muscle (Kriefet al., 1999) and has been linked to cardiac morphogenesis,cardiomyopathies and skeletal muscle integrity in the adult (Chiuet al., 2012; Juo et al., 2016;Rosenfeld et al., 2013). In other cell types,Hspb7 has been shown to prevent protein aggregation (Eenjes et al.,2016; Minoia et al., 2014) and to localize to nuclear speckles (Voset al., 2009), a sub-nuclear location in which pre-mRNA is spliced.
Using an HA-tagged Hspb7 expression construct we observedthat exogenous expression of Hspb7 did not influence myogenicinduction in vitro, as shown by the lack of change in MyoGexpression (Fig. 5A), in agreement with Juo et al. (2016) whoshowed that Hspb7 does not contribute directly to myogenesis.Hspb7 has been associated with autophagy but the mechanism isunclear (Vos et al., 2010). Bag3 is a component of CMA that showsenriched expression in striated muscle, and Bag3-null mice develop
BA
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
Fold
Cha
nge
6 hr
0 μM
1 μM
10 μM
100 μM
0
1
2
3
4
5
6
Fold
Cha
nge
24 hr
0 μM
1 μM
10 μM
100 μM
0
1
2
3
4
Dstn Flnc Hspb7 Lmod3 Plekhh2
Fold
Cha
nge
Gastrocnemius
Young
Adult
*
0
1
2
3
4
5
6
Dstn Flnc Hspb7 Lmod3 Plekhh2
Fold
Cha
nge
Quadriceps
Young
Adult
**
*
Fig. 3. Atrophy induced by aging or dexamethasone causes changes inMEF2A and AP-1 cytoskeletal target genes. (A) RNA from the gastrocnemius andquadriceps of 8- and 63-week-old C57BL/6 mice was isolated for qRT-PCR analysis. Values were calculated using the ΔΔCt method and normalized toGapdh (n=3 except for Flnc expression in the gastrocnemius sample where n=2, mean±s.e.m., *P<0.05). (B) Dex treatment of myotubes induces atrophy andmodulates the expression of MEF2A and AP-1 target genes. C2C12 cells were allowed to differentiate for 72 h and then treated with Dex for 6 h (upper graph) or24 h (lower graph) at the indicated concentrations. Values were calculated using the ΔΔCt method and normalized to Gapdh.
4081
RESEARCH ARTICLE Journal of Cell Science (2016) 129, 4076-4090 doi:10.1242/jcs.190009
Journal
ofCe
llScience
E
Hspb7
MEF2A
Ac�n
GFP
c-Jun
Fra-2
Dex + +++
Control MEF2A Fra-2 c-JunC
48 hr DM + 24 hr treatment
GM + 24 hr treatment
Dex
48 hr DM
+ 6 + 24
Ac�n
Hspb7
Dex + +++Control MEF2A Fra-2 c-Jun
c-Jun
Fra-2
Ac�n
GFP
MEF2A
Hspb7
72 hr DMGN
+ 6 + 24+ 6 + 24
Hspb7
MEF2A
c-Jun
Fra-2
Ac�n
Dex + +++Control siMEF2A siFra-2 sic-Jun
48 hr DM + 24 hr treatment
ChIP-seq read densi�es within the Hspb7 gene
0.00
0.02
0.04
0.06
0.08
0.10
IgG Mef2a IgG Mef2a
DMSO Dex
% In
put
0
0.02
0.04
0.06
0.08
IgG GR IgG GR
DMSO Dex
% In
put
A
D
B
F
Fig. 4. Changes in Hspb7 expression induced by dexamethasone are modulated by MEF2A and AP-1. (A) UCSC genome browser image depictingrecruitment of MEF2A (blue), Fra-1 (brown; C2 FOSL1), c-Jun (green) and glucocorticoid receptor (GR, red) toHspb7. AP-1 consensus sequences are indicatedby vertical black lines. (B) Dex treatment of myotubes strongly upregulated Hspb7 protein expression. C2C12 cells were treated with 10 μMDex for the indicatedtimes. Actin was used as a loading control. (C,D) Exogenous expression of MEF2A, Fra-2 or c-Jun with Dex treatment in growth conditions (C) or 72 h DM (D).C2C12 cells were transfected with the indicated construct using calcium phosphate and allowed to recover. Cells were then treated with 10 μMDex for 24 h after24 h in GM (C) or after 48 h in DM (D). (E) Loss of MEF2A, Fra-2 or c-Jun affects the induction of Hspb7 expression by Dex treatment. Cells were transfected withthe indicated siRNA and allowed to differentiate for 48 h, after which they were treated with 10 μM Dex for 24 h. (F) At 48 h, DM C2C12 cells were treated with10 μM Dex for 24 h and prepared for ChIP-qPCR analysis. Enrichment of GR or MEF2A was detected at the 2nd intron or 3′-end, respectively. Data werecalculated using the percentage input method.
4082
RESEARCH ARTICLE Journal of Cell Science (2016) 129, 4076-4090 doi:10.1242/jcs.190009
Journal
ofCe
llScience

myopathies (Homma et al., 2006). Interestingly, Hspb7 and Bag3single nucleotide polymorphisms (SNPs) have been associated withheart failure (Garnier et al., 2015). Hspb7 has also been shown tointeract with Hspb8 (Sun et al., 2004), an autophagy-related proteinthat interacts with Bag3 (Carra et al., 2008). Therefore, weinvestigated whether Hspb7 could affect autophagy by using theconversion of LC3B-I to LC3B-II as a molecular readout for thisprocess. We observed that LC3B-II levels were not significantlyaffected by Hspb7 overexpression; however, exogenous Hspb7protein was rapidly degraded within 24 h whereas GFP proteinlevels were maintained (Fig. 5A). We hypothesized that the turnoverof Hspb7 protein could indicate its involvement in autophagy andsubsequent degradation in the autolysosome. To confirm whetherthe loss of exogenous Hspb7 demonstrated in Fig. 5A could be viaautophagy we monitored endogenous Hspb7 proteins levels inresponse to treatment with rapamycin, an mTOR inhibitor.Endogenous Hspb7 was reduced after 24 h of rapamycintreatment (Fig. 5B), indicating that Hspb7 could be degradedwithin the autophagosome.
To investigate whether Hspb7 associates with autophagosomes,we generated an EYFP–Hspb7 construct and determined the co-localization of Hspb7 with LC3B under rapamycin treatment inmyoblasts (Fig. 5C). After 6 h of rapamycin treatment, LC3B wasdetected in puncta throughout the cell and in large aggregates, whereEYFP–Hspb7 was also observed.
Because rapamycin affected endogenous Hspb7 protein levelsand Hspb7 and LC3B co-localize, we hypothesized that Hspb7could be directly involved in autophagic flux. Related familymember Hspb8 was able to induce LC3B-II accumulation inprevious studies, but Hspb7 had no effect (Vos et al., 2010) eventhough these studies were carried out without pharmacologicalactivation of autophagy and without lysosome inhibitors, which areprobably necessary to interpret autophagic flux. Therefore, wetreated cells with rapamycin in the presence or absence of Hspb7–HA (Fig. 5D). It was observed that rapamycin alone induced LC3B-II levels but this was reduced with Hspb7–HA expression. Changesin LC3B-II levels without lysosomal blockade are invoked by eitherincreased or decreased autophagic flux; therefore, we next used
AB
D
LC3B
Hspb7
HA
MyoG
GFP
Ac�n
GM 24 hr DM 48 hr DM
pCAGGSnHC
Hspb7-HA + + +
+ + +
LC3B
Ac�n
Hspb7
6 hr 24 hr+ +Rapa
III
I
II
LC3B
HA
Hspb7
Ac�n
+ +Rapa
pCAGGSnHC Hspb7-HA
I
II
C
E
pCAGGSnHC
Hspb7-HA
LC3B
HA
Hspb7
Ac�n
III
BafADMSO BafA+Rapa
+ + ++ + +
LC3B EYFP Merge
DM
SORa
pa
0.0
0.5
1.0
1.5
2.0
2.5
3.0
DMSO BafA BafA+Rapa
LC3B
II/A
c�n
(Arb
itra
ry U
nits
)
pCAGGSnHC Hspb7-HA
F
Fig. 5. Role of Hspb7 in skeletal muscle atrophy. (A) Overexpression of Hspb7 in differentiating C2C12 myoblasts. Cells were transfected with Hspb7–HA andallowed to differentiate for the indicated times. Extracts were prepared for western blot. (B) Rapamycin treatment decreases Hspb7 expression. Myotubes (72 h)were treated with rapamycin (10 μg/ml) for the indicated times. Protein extracts were then prepared for western blot analysis. (C) Myoblasts transfected withEYFP–Hspb7 were treated with rapamycin (10 μg/ml) for 6 h and then assessed for LC3B co-localization (red). Hoechst stain was used to visualize nuclei.(D) C2C12 cells were transfected with Hspb7–HA or control and treated with rapamycin (10 μg/ml) for 6 h in GM, 48 h post-transfection. Protein extracts were thenprepared for western blot. (E) C2C12 cells were prepared as in D. Cells were treated with either BafA alone (200 nM) or BafA plus rapamaycin (10 μg/ml) for 6 h.(F) Quantification of LC3B-II expression was relative to actin. Values were quantified in ImageJ.
4083
RESEARCH ARTICLE Journal of Cell Science (2016) 129, 4076-4090 doi:10.1242/jcs.190009
Journal
ofCe
llScience
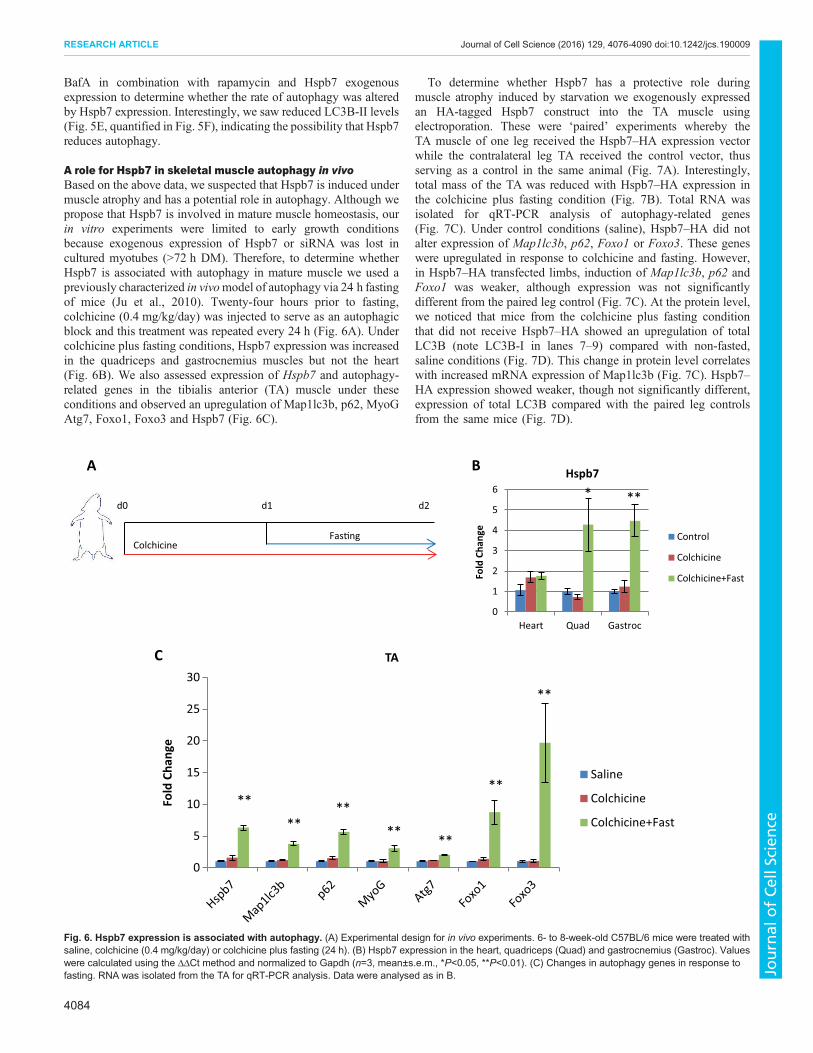
BafA in combination with rapamycin and Hspb7 exogenousexpression to determine whether the rate of autophagy was alteredby Hspb7 expression. Interestingly, we saw reduced LC3B-II levels(Fig. 5E, quantified in Fig. 5F), indicating the possibility that Hspb7reduces autophagy.
A role for Hspb7 in skeletal muscle autophagy in vivoBased on the above data, we suspected that Hspb7 is induced undermuscle atrophy and has a potential role in autophagy. Although wepropose that Hspb7 is involved in mature muscle homeostasis, ourin vitro experiments were limited to early growth conditionsbecause exogenous expression of Hspb7 or siRNA was lost incultured myotubes (>72 h DM). Therefore, to determine whetherHspb7 is associated with autophagy in mature muscle we used apreviously characterized in vivomodel of autophagy via 24 h fastingof mice (Ju et al., 2010). Twenty-four hours prior to fasting,colchicine (0.4 mg/kg/day) was injected to serve as an autophagicblock and this treatment was repeated every 24 h (Fig. 6A). Undercolchicine plus fasting conditions, Hspb7 expression was increasedin the quadriceps and gastrocnemius muscles but not the heart(Fig. 6B). We also assessed expression of Hspb7 and autophagy-related genes in the tibialis anterior (TA) muscle under theseconditions and observed an upregulation of Map1lc3b, p62, MyoGAtg7, Foxo1, Foxo3 and Hspb7 (Fig. 6C).
To determine whether Hspb7 has a protective role duringmuscle atrophy induced by starvation we exogenously expressedan HA-tagged Hspb7 construct into the TA muscle usingelectroporation. These were ‘paired’ experiments whereby theTA muscle of one leg received the Hspb7–HA expression vectorwhile the contralateral leg TA received the control vector, thusserving as a control in the same animal (Fig. 7A). Interestingly,total mass of the TA was reduced with Hspb7–HA expression inthe colchicine plus fasting condition (Fig. 7B). Total RNA wasisolated for qRT-PCR analysis of autophagy-related genes(Fig. 7C). Under control conditions (saline), Hspb7–HA did notalter expression of Map1lc3b, p62, Foxo1 or Foxo3. These geneswere upregulated in response to colchicine and fasting. However,in Hspb7–HA transfected limbs, induction of Map1lc3b, p62 andFoxo1 was weaker, although expression was not significantlydifferent from the paired leg control (Fig. 7C). At the protein level,we noticed that mice from the colchicine plus fasting conditionthat did not receive Hspb7–HA showed an upregulation of totalLC3B (note LC3B-I in lanes 7–9) compared with non-fasted,saline conditions (Fig. 7D). This change in protein level correlateswith increased mRNA expression of Map1lc3b (Fig. 7C). Hspb7–HA expression showed weaker, though not significantly different,expression of total LC3B compared with the paired leg controlsfrom the same mice (Fig. 7D).
C
A B
d0 d1 d2
ColchicineFas�ng
0
1
2
3
4
5
6
Heart Quad Gastroc
Fold
Cha
nge
Hspb7
Control
Colchicine
Colchicine+Fast
* **
0
5
10
15
20
25
30
Fold
Cha
nge
TA
Saline
Colchicine
Colchicine+Fast
**
******
**
**
**
Fig. 6. Hspb7 expression is associated with autophagy. (A) Experimental design for in vivo experiments. 6- to 8-week-old C57BL/6 mice were treated withsaline, colchicine (0.4 mg/kg/day) or colchicine plus fasting (24 h). (B) Hspb7 expression in the heart, quadriceps (Quad) and gastrocnemius (Gastroc). Valueswere calculated using the ΔΔCt method and normalized to Gapdh (n=3, mean±s.e.m., *P<0.05, **P<0.01). (C) Changes in autophagy genes in response tofasting. RNA was isolated from the TA for qRT-PCR analysis. Data were analysed as in B.
4084
RESEARCH ARTICLE Journal of Cell Science (2016) 129, 4076-4090 doi:10.1242/jcs.190009
Journal
ofCe
llScience

To further define the role of Hspb7, we utilized a tandemly taggedLC3 plasmid containing both GFP and mRFP (Kimura et al., 2007).GFP becomes degraded in the acidic lysosomal compartmentand, therefore, the difference in fluorescence corresponds toautophagosomes or autolysosomes, which appear yellow (GFP+
and RFP+) or red (GFP− and RFP+), respectively. Weelectroporated this plasmid into the TA muscle of mice withHspb7–HA and initiated a protocol of colchicine plus fasting, asdepicted in Fig. 6A. Compared with saline, mice treated withcolchicine plus fasting showed upregulation of LC3 puncta with the
Saline Colchicine
+ ++
Gapdh
Hspb7
B
G
A
0
10
20
30
40
50
scr siHspb7 scr siHspb7
Saline Colchicine
wei
ght
(mg)
*
d0 d3 d5
Electropora�onof TA
Fas�ng
d4
ColchicineHspb7-HA pCAGGSnHC
05
1015202530354045
Control Hspb7-HA Control Hspb7-HA
Saline Colchicine+Fast
wei
ght
(mg)
**
Hspb7-HA
Saline Colchicine+Fast
+ ++
HA
Gapdh
C
LC3B
D E
F
H
d0 d2 d4
Electropora�onof mouse TA with siRNA
Colchicinesi-Hspb7 scr
si-Hspb7 + ++
+ ++
GFP RFP Merge
Salin
e
Cont
rol
Cont
rol
Hsp
b7H
spb7
Colc
hici
ne+F
ast
III
0.0
0.5
1.0
1.5
2.0
2.5
3.0
Map1lc3 p62 Foxo1 Foxo3
Fold
Cha
nge
Saline Control
Saline Hspb7-HA
Colchicine+Fast Control
Colchicine+Fast Hspb7-HA
* *
****
Fig. 7. Hspb7 regulates grossmusclemass and LC3B expression. (A) Experimental design for in vivo experiments. Hspb7–HAwas electroporated into the TAmuscle while the contralateral TA muscle received the control plasmid (pCAGGSnHC). Mice were then treated with saline or colchicine plus fasting for theindicated times (colchicine, 0.4 mg/kg/day; fasting, 24 h). (B) Total TA muscle mass under the indicated experimental conditions (n=6; mean±s.e.m., **P<0.01).(C) In vivo gene expression during colchicine plus fasting with exogenous expression of Hspb7–HA or control in paired leg experiment. Saline, n=6; colchicineplus fasting, n=5; mean±s.e.m.; *P<0.05, **P<0.01 compared with saline control. Values were calculated using the ΔΔCt method and normalized to the geometricmean of β-actin, Gapdh, Tbp and Rps26. (D) Samples from C were analysed using western blot to determine level of exogenous expression of Hspb7–HA andLC3B. (E) TA muscles were electroporated with tandem LC3 and Hspb7–HA or tandem LC3 and control plasmid and allowed to recover for 3 days before beingtreated with saline or colchicine plus fasting, as depicted in A. Transverse sections of the muscle were then prepared for analysis of tandem LC3 localization.(F) Experimental design of siRNA experiments. Three independent siRNAs targeting Hspb7 were pooled into a final concentration of 450 pmol (150 pmol of each)and electroporated into the TAmuscle. Control siRNA (scr) was electroporated into the contralateral control TA muscle. At 2 days post-electroporation, colchicinewas administered daily for a further 2 days. (G) Expression of Hspb7 protein levels in the TAmuscle after siRNA knockdown. (H) Total TA muscle mass under theindicated experimental conditions (n=3; mean±s.e.m., *P<0.05).
4085
RESEARCH ARTICLE Journal of Cell Science (2016) 129, 4076-4090 doi:10.1242/jcs.190009
Journal
ofCe
llScience

control plasmid (Fig. 7E). True LC3 puncta corresponding toautophagosomes or autolysosomes can be difficult to determineusing this method because exogenous LC3 is prone to aggregation(Kuma et al., 2007) and, indeed, we did see minimal aggregation atthe periphery of myofibres with saline and control plasmids. Hspb7has been shown to prevent protein aggregation in vitro (Eenjes et al.,2016), therefore we predicted that Hspb7 could also reduce LC3aggregation. Interestingly, we observed an overall trend whereby co-expression of Hspb7–HA with tandem-LC3 resulted in enhancedaggregation of LC3 (Fig. 7E).We next utilized siRNA to knockdown Hspb7 in vivo, in
combination with 2 days of colchicine treatment (Fig. 7F). Loss ofHspb7 protein was detected most clearly with colchicine treatment(Fig. 7G). Interestingly, conditions with a significant reduction ofHspb7 protein levels were associated with an increase in musclemass (Fig. 7H). Collectively, these data suggest a possibleinvolvement of Hspb7 in regulation of overall muscle mass andautophagic flux. Further studies are required to fully characterize thecomplex function of Hspb7 in muscle in vivo.
DISCUSSIONFrom our previous study, in which we identified novel MEF2Atarget genes in skeletal and cardiac muscle (Wales et al., 2014), weobserved that AP-1 consensus cis elements were enriched byMEF2A binding events and, based on GO term analysis, there wasindication that MEF2A regulates the actin cytoskeleton (Potthoffet al., 2007;Wales et al., 2014). In thework presented here, we showthrough bioinformatic and biochemical analyses thatMEF2 andAP-1 share a number of putative common target genes related to theestablishment and maintenance of the actin cytoskeleton, some ofwhich was validated using ChIP-qPCR and knockdown analysis. Inparticular, we focused onHspb7 under conditions of muscle atrophyand observed that AP-1 and MEF2 antagonistically regulateexpression of this gene. Moreover, experiments in mice and incultured muscle cells suggest a role for Hspb7 in autophagy, havingpossible implications for muscle atrophy.
MEF2 and AP-1 regulation of the actin cytoskeleton andmuscle atrophyMEF2 and AP-1 are ubiquitous transcription factors yet theirpotential interaction at the transcriptional level has not beenstudied. Based on our results, MEF2 and AP-1 appear to inverselyregulate Hspb7, wherein MEF2 promotes expression and AP-1represses it. We also identified several other potential commontarget genes that MEF2 and AP-1 might co-operatively orcompetitively regulate. Based on GO term analysis, Fra-1 andMEF2A do not share a significant number of target genescompared with MEF2A and c-Jun (Fig. 1). This could indicatethat Fra-1 and MEF2 have fundamentally different functions andalso that Fra-1 associates with another Jun family member such asJunB or JunD to target differential AP-1 target genes underproliferative conditions. Interestingly, MEF2 and c-Jun wereenriched for several muscle-related GO terms. This could reflectthe role of c-Jun in priming muscle-specific enhancers with MyoD(Blum et al., 2012) and possibly MEF2. Fra-1 and c-Jun exclusivetargets were mainly associated with angiogenesis and the responseto bacterium, the latter being associated with inflammation andcytokine production, one of the traditional roles of AP-1 (Hesset al., 2004; Shaulian and Karin, 2002). The bioinformatic analysisdemonstrates that analysis of differential transcription factors canreveal distinct roles of cooperative and exclusive biologicalfunctions.
A striking feature of Fra-1 and c-Jun recruitment is that the anti-inflammatory GR also binds within the same location of the secondintron of Hspb7 (Fig. 4A). GR and AP-1 competitive regulation isnot a new phenomenon and was observed several decades ago onthe gene encoding collagenase I (Jonat, 1990). Subsequently, manyother genes have been shown to be regulated by GR and AP-1.Interestingly, GR cooperates with a Jun homodimer but inhibitsJun–Fos heterodimers (Adcock and Caramori, 2001; Herrlich,2001). Additionally, AP-1 has been shown to potentiate GRrecruitment by promoting accessible chromatin in epithelial cells(Biddie et al., 2011); however, in the case ofHspb7 expression, AP-1 and GR do not synergize. In the case of Hspb7, we observed thatloss of c-Jun or Fra-2 induces Hspb7 expression upon Dextreatment, indicating that a Jun–Fos dimer is involved inregulation of this gene. Moreover, our studies implicate awidespread level of cooperativity between AP-1 and MEF2 thatwarrants further investigation.
In striated muscle, the actin cytoskeleton stabilizes the sarcomerein concert with the costamere, which tethers the Z-line of thesarcomere to the sarcolemma (Ervasti and Campbell, 1993). Thisfacilitates sarcomere stabilization and connects the actincytoskeleton to the extracellular matrix via the costamere, but italso has roles in other cell processes including migration, adhesionand gene expression (Zheng et al., 2009). Therefore, identifyingpathways that mediate actin cytoskeletal gene expression hasimplications for different types of muscle disease. MEF2 has awell-established role in sarcomere organization, because it regulateskey target genes associated with the costamere and sarcomericproteins (Ewen et al., 2011; Hinits and Hughes, 2007; Potthoff et al.,2007). There are fewer studies that have investigated the role of AP-1in sarcomere integrity; however, in cardiomyocytes c-Jun has beenshown to have an important role in promoting sarcomere geneexpression and sarcomere integrity (Windak et al., 2013).Interestingly, destabilization of the actin cytoskeleton triggers c-Jun activity in vitro and represses glucocorticoid receptor activity(Oren et al., 1999). The myofibres in mouse models of cachexia areassociated with a defective sarcolemma, similar to that seen inmuscular dystrophies (Acharyya et al., 2005); therefore, commonstructural and cytoskeletal defects could contribute to variouspathologies. In cancer cachexia, expression of a dominant negativeAP-1 factor Tam67 or AP-1 and NF-κB double inhibitor can preventloss of muscle mass (Moore-Carrasco et al., 2006, 2007). Bycontrast, JunB was found to be universally downregulated in modelsof atrophy (Lecker et al., 2004) and loss of AP-1 factors indenervation-induced muscle atrophy prevents upregulation ofMAFbx and MURF1 (Choi et al., 2012). Dysregulation of AP-1 incancer could therefore not onlymodulate proliferation andmetastasis(Eferl and Wagner, 2003) but also impact muscle health, which isknown to be modulated in cancer cachexia (Dodson et al., 2011).
A role for Hspb7 in muscle disease and sarcopeniaTo date, it has been thought that the role of small heat shockproteins, including Hspb7, is stabilization of the cytoskeleton understress conditions. Hspb7 can interact with α-filamin (Krief et al.,1999), filamin C (Juo et al., 2016), the cytoskeleton in tachypacedcardiomyocytes (Ke et al., 2011) and myofibrils in response tocardiac ischemia (Golenhofen et al., 2004). However, based onstudies by Vos and colleagues, it appears that Hspb7 could have arole in autophagy and protein degradation, making its role moreextensive than previously thought (Vos et al., 2009, 2010).Recently, Hspb7 has been shown to prevent aggregate formation,and not to contribute to aggregate clearance (Eenjes et al., 2016).
4086
RESEARCH ARTICLE Journal of Cell Science (2016) 129, 4076-4090 doi:10.1242/jcs.190009
Journal
ofCe
llScience

Our study confirms that Hspb7 is upregulated with age, and also inthe context of GR-mediated atrophy and fasting conditions in vivo.These observations agree with the previously stated idea that Hspb7has a protective role (Doran et al., 2007); however, our data indicatethat after rapamycin treatment in vitro the exogenous expression ofHspb7 localizes with LC3B and decreases autophagic flux. The rateof autophagy is considered to be a delicate balance because toomuch or too little results in different molecular deficiencies;therefore, the ability of Hspb7 to modulate this process in musclecould have pivotal consequences in the context of sarcopenia andother forms of muscle atrophy.Because impairment of autophagy is believed to be associated
with sarcopenia (Rubinsztein et al., 2011), our findings could haveimportant implications in future therapies that seek to targetautophagy in muscle wasting. Deficits in satellite cell numbershas also recently been associated with decreased autophagy (García-Prat et al., 2016), which could implicate a role for Hspb7 in satellitecell biology. Although Hspb7 is primarily associated withautophagy (Vos et al., 2010) it could have uncharacterized rolesin proteasomal degradation pathways in other tissues. Additionally,the induction of Hspb7 via glucocorticoids could result in asubsequent maladaptive role for Hspb7 if signalling is prolonged.Skeletal muscles deficient in Hspb7 do not show defects inmyogenesis but still develop myopathies postnatally, indicating thatHspb7 has a potentially crucial role in skeletal muscle integrity inadults (Juo et al., 2016). It would be interesting to further investigatethis in the context of ageing or fasting. Hspb7 is highly expressed inthe heart, and a crucial role for Hspb7 in heart development has beenshown in zebrafish (Rosenfeld et al., 2013). Mutations in Hspb7 arecorrelated with cardiomyopathies and, co-incidentally, Hspb7 SNPswithin the second intron (where GR and AP-1 recruitment wasobserved) are associated with cardiomyopathies (Garnier et al.,2015; Matkovich et al., 2010). In our studies, although Hspb7 wasinduced in skeletal muscles in response to fasting it was notupregulated in the heart (Fig. 7). This could indicate that Hspb7 isinduced to protect tissues in response to different forms of stress.The importance of MEF2, AP-1 and GR in Hspb7 regulation in theheart could also be related to cardiac development and it would beinteresting to determine whether Hspb7, and the GR–MEF2–AP-1signaling axis as highlighted here, has a role in cardiac hypertrophyor other cardiomyopathies.In conclusion, MEF2 and AP-1 have historically been assigned
fairly distinct functions in tissue-specific gene expression andgrowth control, respectively. Here, we implicate them together inthe antagonistic or cooperative control of cytoskeletal genes such asHspb7 in skeletal muscle. These studies also highlight the role of theactin cytoskeleton and its regulation by MEF2 and AP-1transcriptional regulators, which may be of particular importancefor muscle function and pathology.
MATERIALS AND METHODSCell cultureC2C12 myoblasts were obtained from American Type Culture Collection(ATCC). Cells were maintained in Dulbecco’s modified Eagle’s medium(DMEM) with high glucose and L-glutamine (Hyclone) supplemented with10% foetal bovine serum (HyClone) and 1% penicillin/streptomycin(Invitrogen). C2C12 myoblasts were induced to differentiate indifferentiation medium containing DMEM/high glucose/L-glutaminesupplemented with 2% horse serum (Hyclone) and 1% penicillin/streptomycin for the indicated times. Cells were maintained in anhumidified, 37°C incubator at 5% CO2 and replenished with freshmedium every 24 h. Pharmacological drug treatments were completed forthe indicated times.
TransfectionsC2C12 myoblasts were transfected using the calcium phosphateprecipitation method. Cells were then harvested at 48 h post-transfectionor the medium was changed to DM.
PlasmidsExpression plasmids for pMT2–MEF2A, pCMV–c-Jun, pcDNA3.1-Fra-2,pCMV–dsRed2, pcDNA–GFP, GFP–Tam67 have been describedpreviously (Alli et al., 2013; Hennigan and Stambrook, 2001; Perry et al.,2009). pCAGGSnHC–HSPB7-HA was described by Lin et al. (2014).EYFP–Hspb7 was sub-cloned from Hspb7–HA into EYFP–pcDNA3 usingXhoI and EcoRI.
Antibodies and reagentsRabbit polyclonal MEF2A antibody has been previously described (Coxet al., 2003). The following antibodies were purchased from Santa CruzBiotechnology: actin (sc-1616), dsRed (sc-33354), MEF2A (sc-313X; usedin ChIP), donkey anti-goat IgG-HRP (sc-2020), Fra-2 (sc-604), c-Jun (sc-1694), GFP (sc-9996), MCK (sc-365046) and MyoD (sc-304). Anti-LC3Bwas from Cell Signaling (2775). Myogenin and HA monoclonal antibodieswere obtained from the Developmental Studies Hybridoma Bank. Theremaining antibodies were as follows: Hspb7 (Abcam, ab150390),glucocorticoid receptor (Abcam, ab3579), rabbit IgG (Millipore, 12-370).Dexamethasone (sc-29059) was used at a concentration of 10 μM, unlessotherwise indicated. DMSO was used as a volume control. Rapamycin(10 μg/ml) and BafA1 (200 nM) were purchased from Santa CruzBiotechnology (sc-3504 and sc-201550).
siRNA transfection of C2C12 myoblastsKnockdown of target genes was carried out using siRNAs obtained fromSigma-Aldrich; targets are listed in Table S2. In C2C12 myoblasts, siRNAwas transfected at the following concentrations: MEF2A (30 nM), c-Jun(50 nM) and Fra-2 (50 nm).
ImmunoblotsCells were washed with 1× PBS and lysed in NP-40 lysis buffer (50 mMTris, 150 mM NaCl, 0.5% NP-40, 2 mM EDTA, 100 mM NaF and 10 mMNa pyrophosphate) containing protease inhibitor cocktail (Sigma-Aldrich),1 mM phenylmethylsulfonyl fluoride (Sigma-Aldrich) and 1 mM sodiumorthovanadate (Bioshop). Protein concentrations were determined byBradford assay (Bio-Rad). Twenty micrograms of total protein wereresolved on 10% SDS–PAGE and then transferred onto Immobilon-FLPVDF membrane (Millipore) for 1 h or overnight. Nonspecific binding siteswere blocked using 5% milk in PBS or TBST. Membranes were incubatedwith primary antibodies overnight at 4°C in 5% milk in PBS or 5% BSA inTBST. Horseradish peroxidase-conjugated secondary antibody was addedfor 1 h at room temperature. Protein was detected with ECLchemiluminescence reagent (Pierce). For cyto-nuclear fractionation, theThermo Scientific NE-PER Nuclear and Cytoplasmic Extraction Kit(78833) was used.
Chromatin immunoprecipitationMethods were carried out as described previously described (Wales et al.,2014) with the addition of a third immunoprecipitation wash buffer (IP washbuffer III; 20 mM Tris pH 8.1, 250 mM LiCl, 1% NP-40, 1% deoxycholate,1 mM EDTA).
RNA extractionTotal RNAwas extracted from C2C12 myoblasts using the RNeasy Plus kit(Qiagen) and Qiashredder (Qiagen). RNA isolated from tissue was extractedusing Trizol (Invitrogen). RNA was converted to cDNA using SuperscriptIII (Invitrogen) according to the manufacturer’s instructions.
Quantitative PCRSybrGreen (BioRad or ABM) was combined with 2.5 μl gDNA or cDNAand 500 nM primers in a final volume of 20 μl. cDNAwas diluted 1:10 priorto use. Each sample was prepared in triplicate and analysed using Rotor-
4087
RESEARCH ARTICLE Journal of Cell Science (2016) 129, 4076-4090 doi:10.1242/jcs.190009
Journal
ofCe
llScience

Gene Q (Qiagen). Parameters for qRT-PCR using BioRadwere were 30 s95°C, [5 s 95°C, 30 s 60°C]×40 cycles. Parameters for qRT-PCR usingABM were 10 m 95°C, [3 s 95°C, 30 s 60°C]×35 cycles. Parameters forChIP-qPCR were 5 min 95°C, [5 s 95°C, 15 s 60°C]×40 cycles. Foldenrichment (ChIP-qPCR) and fold change (qRT-PCR) were quantifiedusing the ΔΔCt method. Primers used in qRT-PCR and ChIP-qPCR arelisted in Tables S3 and S4, respectively.
BioinformaticsAP-1 consensus sequences were mapped using cisGenome. GREAT(default settings) identified GO terms based on DNA sequences obtainedfrom available datasets: GSE61207 for MEF2A; ENCSR000AIK for Fra-1;and GSE37525 for c-Jun. All datasets were converted to mm9 using UCSCLiftOver. Overlapping binding sites were determined using the UCSCTable Browser Intersect function using default settings.
Animal careFor ageing experiments, 63- and 8-week-old C57BL/6 male mice wereobtained from Jackson Lab or Charles River, respectively. Mice weresacrificed using cervical dislocation in accordance with the InstitutionalAnimal Care and Use Committee of York University. For autophagyexperiments, 6- to 8-week-old C57BL/6 male mice were sacrificed usingcervical dislocation in accordance with University of Ottawa Animal Careand Use Committee. Autophagic flux was induced using a combination ofdaily intraperitoneal colchicine administration (0.4 mg/kg/day) and fastingas described by Ju et al. (2010) with the following changes: (1) mice wereplaced on a 24 h fast; (2) 3 days of recovery were allowed after ptfLC3,Hspb7–HA or pCAGSSnHC electroporation.
Electroporation of plasmid DNA into mouse tissuePlasmid DNAwas prepared as follows: twenty-five micrograms of Hspb7–HA or empty vector (pCAGGSnHC) in sterile half saline were injected intothe right and left TA muscles of 6- to 8-week-old C57BL/6 male mice,respectively. Co-electroporation of tfLC3 and Hspb7–HA or empty vectorwas carried out using equal amounts of 12.5 μg. After injection, muscleswere subject to electrical stimulation using an electroporation system (ECM830, BTX) with electroporation array needle (model no.533, BTX)programmed at 80 V, 6 pulses, 50 ms/pulse and 500 ms interval betweenpulses. Tissues were harvested immediately after cervical dislocation andhomogenized with 1 ml Trizol in a Lysing Matrix D tube (6913100, MPBiomedical) with MagNA Lyser (03358968001, Roche) programmed at17,000 rpm. Three homogenizations of 20 s were carried out, betweenwhich the samples were chilled on ice for 30 s. Tissue homogenates werethen prepared for RNA and protein extraction using the Trizol protocol(Invitrogen).
Electroporation of siRNA into mouse tissuePrior to injection of siRNA, the right TA muscle was injected with 25 μl of0.4 unit/μl hyaluronidase (LS005477, Worthington) and digested for 1 h.siRNA targeting Hspb7 was pooled at 150 pmol to give a final amount of450 pmol. The paired leg received 450 pmol of non-targeting scrambledsiRNA (scr). The siRNAs used are listed in Table S2. At 2 days post-electroporation, saline or colchicine (0.4 mg/kg/day) was administeredintraperitoneally and then every 24 h for a further 2 days.
ImmunofluorescenceC2C12 myoblasts were fixed and permeabilized using 90% methanol at−20°C for 6 min. The protocol used for immunofluorescence has beenpreviously described (Ehyai et al., 2015). Anti-LC3B (Cell Signaling, 2775)was used at a 1:200 dilution. Hoechst 33342 stain was added for 30 min tovisualize nuclei (1:10,000; Sigma-Aldrich). Images were captured using aCarl Zeiss Axio Observer.Z1 (Photometrics Evolve 512 EMCCD camera).Cells were imaged under a 63× (Plan-Apochromat; 1.40 NA in oil)objective, using ZEN image acquisition software 2.0. Immunofluorescentsections of TAmuscle were prepared as follows: TAmuscles were harvestedand frozen within Cryomatrix (6769006, Thermo Scientific) in liquidnitrogen-chilled isopentane; 10 μm sections of TA muscles were affixed to
positive charged slides and fixed with 4% paraformaldehyde for 10 min atroom temperature. Sections were rinsed with PBS and mounted withProlong Gold anti-fade with DAPI. Images were acquired using a ZeissObserver Z1 microscope.
StatisticsData are presented as mean±s.e.m. Statistical analysis was carried out usingtwo-tailed unpaired Student’s t-test.
AcknowledgementsWewould like to thank G. Sweeney (York University, Toronto, Canada) for providingthe tandem LC3 plasmid (tflc3) and Y. Nakamura (University of Chicago, Chicago,USA) for providing Hspb7–HA.
Competing interestsThe authors declare no competing or financial interests.
Author contributionsS.W.T. completed most of the data acquisition, analysis and interpretation. D.Y. andJ.G. completed in vivo experiments, which were then analysed by S.W.T. A.B.assisted in bioinformatic analysis. A.F. assisted with in vitro immunofluorescenceand imaging, ChIP-qPCR and Tam67 experiments. S.W.T., A.B. and J.C.M.designed experiments and wrote the manuscript.
FundingThis work was supported by a grant from the Canadian Institutes of Health Researchto J.C.M.
Supplementary informationSupplementary information available online athttp://jcs.biologists.org/lookup/doi/10.1242/jcs.190009.supplemental
ReferencesAcharyya, S., Butchbach, M. E. R., Sahenk, Z., Wang, H., Saji, M., Carathers, M.,
Ringel, M. D., Skipworth, R. J. E., Fearon, K. C. H., Hollingsworth, M. A. et al.(2005). Dystrophin glycoprotein complex dysfunction: a regulatory link betweenmuscular dystrophy and cancer cachexia. Cancer Cell 8, 421-432.
Adcock, I. M. and Caramori, G. (2001). Cross-talk between pro-inflammatorytranscription factors and glucocorticoids. Immunol. Cell Biol. 79, 376-384.
Alli, N. S., Yang, E. C., Miyake, T., Aziz, A., Collins-Hooper, H., Patel, K. andMcDermott, J. C. (2013). Signal-dependent fra-2 regulation in skeletal musclereserve and satellite cells. Cell Death Dis. 4, e692.
Andreucci, J. J., Grant, D., Cox, D.M., Tomc, L. K., Prywes, R., Goldhamer, D. J.,Rodrigues, N., Bedard, P.-A. and McDermott, J. C. (2002). Composition andfunction of AP-1 transcription complexes during muscle cell differentiation. J. Biol.Chem. 277, 16426-16432.
Angel, P., Imagawa, M., Chiu, R., Stein, B., Imbra, R. J., Rahmsdorf, H. J., Jonat,C., Herrlich, P. and Karin, M. (1987). Phorbol ester-inducible genes contain acommon cis element recognized by a TPA-modulated trans-acting factor. Cell 49,729-739.
Arndt, V., Dick, N., Tawo, R., Dreiseidler, M., Wenzel, D., Hesse, M., Furst, D. O.,Saftig, P., Saint, R., Fleischmann, B. K. et al. (2010). Chaperone-assistedselective autophagy is essential for muscle maintenance. Curr. Biol. 20, 143-148.
Bengal, E., Ransone, L., Scharfmann, R., Dwarki, V. J., Tapscott, S. J.,Weintraub, H. and Verma, I. M. (1992). Functional Antagonism between C-Junand Myod Proteins: a Direct Physical Association. Cell 68, 507-519.
Biddie, S. C., John, S., Sabo, P. J., Thurman, R. E., Johnson, T. A., Schiltz, R. L.,Miranda, T. B., Sung, M.-H., Trump, S., Lightman, S. L. et al. (2011).Transcription factor AP1 potentiates chromatin accessibility and glucocorticoidreceptor binding. Mol. Cell 43, 145-155.
Blais, A., Tsikitis, M., Acosta-Alvear, D., Sharan, R., Kluger, Y. and Dynlacht,B. D. (2005). An initial blueprint for myogenic differentiation. Genes Dev. 19,553-569.
Blum, R., Vethantham, V., Bowman, C., Rudnicki, M. and Dynlacht, B. D. (2012).Genome-wide identification of enhancers in skeletal muscle: the role of MyoD1.Genes Dev. 26, 2763-2779.
Cao, Y., Yao, Z., Sarkar, D., Lawrence, M., Sanchez, G. J., Parker, M. H.,MacQuarrie, K. L., Davison, J., Morgan, M. T., Ruzzo, W. L. et al. (2010).Genome-wide MyoD binding in skeletal muscle cells: a potential for broad cellularreprogramming. Dev. Cell 18, 662-674.
Carlier, M.-F., Laurent, V., Santolini, J., Melki, R., Didry, D., Xia, G.-X., Hong, Y.,Chua, N.-H. and Pantaloni, D. (1997). Actin depolymerizing factor (ADF/cofilin)enhances the rate of filament turnover: implication in actin-based motility. J. CellBiol. 136, 1307-1322.
Carra, S., Seguin, S. J. and Landry, J. (2008). HspB8 and Bag3: a new chaperonecomplex targeting misfolded proteins to macroautophagy. Autophagy 4, 237-239.
4088
RESEARCH ARTICLE Journal of Cell Science (2016) 129, 4076-4090 doi:10.1242/jcs.190009
Journal
ofCe
llScience

Cenik, B. K., Garg, A., McAnally, J. R., Shelton, J. M., Richardson, J. A., Bassel-Duby, R., Olson, E. N. and Liu, N. (2015). Severe myopathy in mice lacking theMEF2/SRF-dependent gene leiomodin-3. J. Clin. Invest. 125, 1569-1578.
Chiang, H. L., Terlecky, S. R., Plant, C. P. and Dice, J. F. (1989). A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins.Science 246, 382-385.
Chiu, T.-F., Li, C.-H., Chen, C.-C., Chen, C.-H., Cheng, C.-J., Yan, Y.-T. and Yang,R.-B. (2012). Association of plasma concentration of small heat shock protein B7with acute coronary syndrome. Circ. J. 76, 2226-2233.
Choi, M.-C., Cohen, T. J., Barrientos, T., Wang, B., Li, M., Simmons, B. J., Yang,J. S., Cox, G. A., Zhao, Y. and Yao, T.-P. (2012). A direct HDAC4-MAP kinasecrosstalk activates muscle atrophy program. Mol. Cell 47, 122-132.
Clarke, B. A., Drujan, D., Willis, M. S., Murphy, L. O., Corpina, R. A., Burova, E.,Rakhilin, S. V., Stitt, T. N., Patterson, C., Latres, E. et al. (2007). The E3 ligaseMuRF1 degrades myosin heavy chain protein in dexamethasone-treated skeletalmuscle. Cell Metab. 6, 376-385.
Cohen, S., Brault, J. J., Gygi, S. P., Glass, D. J., Valenzuela, D. M., Gartner, C.,Latres, E. and Goldberg, A. L. (2009). During muscle atrophy, thick, but not thin,filament components are degraded by MuRF1-dependent ubiquitylation. J. CellBiol. 185, 1083-1095.
Cox, D. M., Du, M., Marback, M., Yang, E. C. C., Chan, J., Siu, K. W. M. andMcDermott, J. C. (2003). Phosphorylation motifs regulating the stability andfunction of myocyte enhancer factor 2A. J. Biol. Chem. 278, 15297-15303.
Demontis, F. and Perrimon, N. (2010). FOXO/4E-BP signaling in Drosophilamuscles regulates organism-wide proteostasis during aging. Cell 143, 813-825.
Dodson, S., Baracos, V. E., Jatoi, A., Evans, W. J., Cella, D., Dalton, J. T. andSteiner, M. S. (2011). Muscle wasting in cancer cachexia: clinical implications,diagnosis, and emerging treatment strategies. Annu. Rev. Med. 62, 265-279.
Doran, P., Martin, G., Dowling, P., Jockusch, H. and Ohlendieck, K. (2006).Proteome analysis of the dystrophin-deficient MDX diaphragm reveals a drasticincrease in the heat shock protein cvHSP. Proteomics 6, 4610-4621.
Doran, P., Gannon, J., O’Connell, K. and Ohlendieck, K. (2007). Aging skeletalmuscle shows a drastic increase in the small heat shock proteins alpha B-crystallin/HspB35 and cvHsp/HspB7. Eur. J. Cell Biol. 86, 629-640.
Edmondson, D. G., Lyons, G. E., Martin, J. F. and Olson, E. N. (1994). Mef2Gene-ExpressionMarks theCardiac and Skeletal-Muscle Lineages duringMouseEmbryogenesis. Development 120, 1251-1263.
Eenjes, E., Dragich, J. M., Kampinga, H. H. and Yamamoto, A. (2016).Distinguishing aggregate formation and aggregate clearance using cell-basedassays. J. Cell Sci. 129, 1260-1270.
Eferl, R. and Wagner, E. F. (2003). AP-1: a double-edged sword in tumorigenesis.Nat. Rev. Cancer 3, 859-868.
Eferl, R., Hoebertz, A., Schilling, A. F., Rath, M., Karreth, F., Kenner, L., Amling,M. andWagner, E. F. (2004). The Fos-related antigen Fra-1 is an activator of bonematrix formation. EMBO J. 23, 2789-2799.
Ehyai, S., Dionyssiou, M. G., Gordon, J. W., Williams, D., Siu, K. W. M. andMcDermott, J. C. (2015). A p38 MAPK-regulated MEF2–β-catenin interactionenhances canonical Wnt signalling. Mol. Cell. Biol. 36, 330-346.
Ervasti, J. M. and Campbell, K. P. (1993). A role for the dystrophin-glycoproteincomplex as a transmembrane linker between laminin and actin. J. Cell Biol. 122,809-823.
Ewen, E. P., Snyder, C. M., Wilson, M., Desjardins, D. and Naya, F. J. (2011). TheMef2A transcription factor coordinately regulates a Costamere gene program incardiac muscle. J. Biol. Chem. 286, 29644-29653.
Fleischmann, A. (2000). Fra-1 replaces c-Fos-dependent functions in mice.GenesDev. 14, 2695-2700.
Garcıa-Prat, L., Martınez-Vicente, M., Perdiguero, E., Ortet, L., Rodrıguez-Ubreva, J., Rebollo, E., Ruiz-Bonilla, V., Gutarra, S., Ballestar, E., Serrano,A. L. et al. (2016). Autophagy maintains stemness by preventing senescence.Nature 529, 37-42.
Garg, A., O’Rourke, J., Long, C., Doering, J., Ravenscroft, G., Bezprozvannaya,S., Nelson, B. R., Beetz, N., Li, L., Chen, S. et al. (2014). KLHL40 deficiencydestabilizes thin filament proteins and promotes nemaline myopathy. J. Clin.Invest. 124, 3529-3539.
Garnier, S., Hengstenberg, C., Lamblin, N., Dubourg, O., De Groote, P.,Fauchier, L., Trochu, J.-N., Arbustini, E., Esslinger, U., Barton, P. J. et al.(2015). Involvement of BAG3 andHSPB7 loci in various etiologies of systolic heartfailure: results of a European collaboration assembling more than 2000 patients.Int. J. Cardiol. 189, 105-107.
Garrido, C., Paul, C., Seigneuric, R. and Kampinga, H. H. (2012). The small heatshock proteins family: the long forgotten chaperones. Int. J. Biochem. Cell Biol.44, 1588-1592.
Golenhofen, N., Perng, M. D., Quinlan, R. A. and Drenckhahn, D. (2004).Comparison of the small heat shock proteins alphaB-crystallin, MKBP, HSP25,HSP20, and cvHSP in heart and skeletal muscle. Histochem. Cell Biol. 122,415-425.
Hennigan, R. F. and Stambrook, P. J. (2001). Dominant negative c-jun inhibitsactivation of the cyclin D1 and cyclin E kinase complexes. Mol. Biol. Cell 12,2352-2363.
Herrlich, P. (2001). Cross-talk between glucocorticoid receptor and AP-1.Oncogene 20, 2465-2475.
Hess, J., Angel, P. and Schorpp-Kistner, M. (2004). AP-1 subunits: quarrel andharmony among siblings. J. Cell Sci. 117, 5965-5973.
Hinits, Y. and Hughes, S. M. (2007). Mef2s are required for thick filament formationin nascent muscle fibres. Development 134, 2511-2519.
Homma, S., Iwasaki, M., Shelton, G. D., Engvall, E., Reed, J. C. and Takayama,S. (2006). BAG3 deficiency results in fulminant myopathy and early lethality.Am. J. Pathol. 169, 761-773.
Jonat, C., Rahmsdorf, H. J., Park, K.-K., Cato, A. C. B., Gebel, S., Ponta, H. andHerrlich, P. (1990). Antitumor promotion and antiinflammation: down-modulationof AP-1 (Fos/Jun) activity by glucocorticoid hormone. Cell 62, 1189-1204.
Ju, J.-S., Varadhachary, A. S., Miller, S. E. andWeihl, C. C. (2010). Quantitation of“autophagic flux” in mature skeletal muscle. Autophagy 6, 929-935.
Juo, L.-Y., Liao, W.-C., Shih, Y.-L., Yang, B.-Y., Liu, A.-B. and Yan, Y.-T. (2016).HSPB7 interacts with dimerized FLNC and its absence results in progressivemyopathy in skeletal muscles. J. Cell Sci. 129, 1661-1670.
Kabeya, Y., Mizushima, N., Ueno, T., Yamamoto, A., Kirisako, T., Noda, T.,Kominami, E., Ohsumi, Y. and Yoshimori, T. (2000). LC3, a mammalianhomologue of yeast Apg8p, is localized in autophagosome membranes afterprocessing. EMBO J. 19, 5720-5728.
Kadar, L., Albertsson, M., Areberg, J., Landberg, T. and Mattsson, S. (2000).The prognostic value of body protein in patients with lung cancer. Ann. N. Y. Acad.Sci. 904, 584-591.
Ke, L., Meijering, R. A. M., Hoogstra-Berends, F., Mackovicova, K., Vos, M. J.,Van Gelder, I. C., Henning, R. H., Kampinga, H. H. and Brundel, B. J. J. M.(2011). HSPB1, HSPB6, HSPB7 and HSPB8 protect against RhoA GTPase-induced remodeling in tachypaced atrial myocytes. PLoS ONE 6, e20395.
Kimura, S., Noda, T. and Yoshimori, T. (2007). Dissection of the autophagosomematuration process by a novel reporter protein, tandem fluorescent-tagged LC3.Autophagy 3, 452-460.
Kotler, D. P. (2000). Cachexia. Ann. Intern. Med. 133, 622-634.Krief, S., Faivre, J.-F., Robert, P., Le Douarin, B., Brument-Larignon, N., Lefrere,
I., Bouzyk, M. M., Anderson, K. M., Greller, L. D., Tobin, F. L. et al. (1999).Identification and characterization of cvHsp. A novel human small stress proteinselectively expressed in cardiovascular and insulin-sensitive tissues. J. Biol.Chem. 274, 36592-36600.
Kuma, A., Matsui, M. and Mizushima, N. (2007). LC3, an autophagosomemarker,can be incorporated into protein aggregates independent of autophagy: caution inthe interpretation of LC3 localization. Autophagy 3, 323-328.
Kuo, T., Lew, M. J., Mayba, O., Harris, C. A., Speed, T. P. andWang, J.-C. (2012).Genome-wide analysis of glucocorticoid receptor-binding sites in myotubesidentifies gene networks modulating insulin signaling. Proc. Natl. Acad. Sci. USA109, 11160-11165.
Lach-Trifilieff, E., Minetti, G. C., Sheppard, K., Ibebunjo, C., Feige, J. N.,Hartmann, S., Brachat, S., Rivet, H., Koelbing, C., Morvan, F. et al. (2014). Anantibody blocking activin type II receptors induces strong skeletal musclehypertrophy and protects from atrophy. Mol. Cell. Biol. 34, 606-618.
Lecker, S. H., Jagoe, R. T., Gilbert, A., Gomes, M., Baracos, V., Bailey, J., Price,S. R., Mitch, W. E. and Goldberg, A. L. (2004). Multiple types of skeletal muscleatrophy involve a common program of changes in gene expression. FASEB J. 18,39-51.
Li, L., Chambard, J. C., Karin, M. and Olson, E. N. (1992). Fos and Jun represstranscriptional activation by myogenin and MyoD: the amino terminus of Jun canmediate repression. Genes Dev. 6, 676-689.
Lin, Q., Schwarz, J., Bucana, C. andOlson, E. N. (1997). Control of mouse cardiacmorphogenesis and myogenesis by transcription factor MEF2C. Science. 276,1404-1407.
Lin, J., Deng, Z., Tanikawa, C., Shuin, T., Miki, T., Matsuda, K. and Nakamura, Y.(2014). Downregulation of the tumor suppressor HSPB7, involved in the p53pathway, in renal cell carcinoma by hypermethylation. Int. J. Oncol. 44,1490-1498.
Ma, Q. and Telese, F. (2015). Genome-wide epigenetic analysis of MEF2A andMEF2C transcription factors in mouse cortical neurons. Commun. Integr. Biol. 8,e1087624.
Mammucari, C., Milan, G., Romanello, V., Masiero, E., Rudolf, R., Del Piccolo,P., Burden, S. J., Di Lisi, R., Sandri, C., Zhao, J. et al. (2007). FoxO3 controlsautophagy in skeletal muscle in vivo. Cell Metab. 6, 458-471.
Masiero, E., Agatea, L., Mammucari, C., Blaauw, B., Loro, E., Komatsu, M.,Metzger, D., Reggiani, C., Schiaffino, S. and Sandri, M. (2009). Autophagy isrequired to maintain muscle mass. Cell Metab. 10, 507-515.
Matkovich, S. J., Van Booven, D. J., Hindes, A., Kang, M. Y., Druley, T. E.,Vallania, F. L. M., Mitra, R. D., Reilly, M. P., Cappola, T. P. and Dorn, G. W.(2010). Cardiac signaling genes exhibit unexpected sequence diversity insporadic cardiomyopathy, revealing HSPB7 polymorphisms associated withdisease. J. Clin. Invest. 120, 280-289.
Minoia, M., Grit, C. and Kampinga, H. H. (2014). HSPA1A-independentsuppression of PARK2 C289G protein aggregation by human small heat shockproteins. Mol. Cell. Biol. 34, 3570-3578.
4089
RESEARCH ARTICLE Journal of Cell Science (2016) 129, 4076-4090 doi:10.1242/jcs.190009
Journal
ofCe
llScience

Moore-Carrasco, R., Garcıa-Martınez, C., Busquets, S., Ametller, E., Barreiro,E., Lopez-Soriano, F. J. and Argiles, J. M. (2006). The AP-1/CJUN signalingcascade is involved in muscle differentiation: implications in muscle wastingduring cancer cachexia. FEBS Lett. 580, 691-696.
Moore-Carrasco, R., Busquets, S., Almendro, V., Palanki, M., Lopez-Soriano,F. J. and Argiles, J. M. (2007). The AP-1/NF-kappaB double inhibitor SP100030can revert muscle wasting during experimental cancer cachexia. Int. J. Oncol. 30,1239-1245.
Morley, J. E., Baumgartner, R. N., Roubenoff, R., Mayer, J. and Nair, K. S.(2001). Sarcopenia. J. Lab. Clin. Med. 137, 231-243.
Nagy, G., Daniel, B., Jonas, D., Nagy, L. and Barta, E. (2013). A novel method topredict regulatory regions based on histone mark landscapes in macrophages.Immunobiology 218, 1416-1427.
Oren, A., Herschkovitz, A., Ben-Dror, I., Holdengreber, V., Ben-Shaul, Y., Seger,R. and Vardimon, L. (1999). The cytoskeletal network controls c-Jun expressionand glucocorticoid receptor transcriptional activity in an antagonistic and cell-type-specific manner. Mol. Cell. Biol. 19, 1742-1750.
Perisic, L., Lal, M., Hulkko, J., Hultenby, K., Onfelt, B., Sun, Y., Duner, F.,Patrakka, J., Betsholtz, C., Uhlen, M. et al. (2012). Plekhh2, a novel podocyteprotein downregulated in human focal segmental glomerulosclerosis, is involvedin matrix adhesion and actin dynamics. Kidney Int. 82, 1071-1083.
Perry, R. L. S., Yang, C., Soora, N., Salma, J., Marback, M., Naghibi, L., Ilyas, H.,Chan, J., Gordon, J. W. andMcDermott, J. C. (2009). Direct interaction betweenMyocyte Enhancer Factor 2 (MEF2) and protein phosphatase 1 alpha repressesMEF2-dependent gene expression. Mol. Cell. Biol. 29, 3355-3366.
Potthoff, M. J., Arnold, M. A., McAnally, J., Richardson, J. A., Bassel-Duby, R.and Olson, E. N. (2007). Regulation of skeletal muscle sarcomere integrity andpostnatal muscle function by Mef2c. Mol. Cell. Biol. 27, 8143-8151.
Prado, C. M. M., Baracos, V. E., McCargar, L. J., Reiman, T., Mourtzakis, M.,Tonkin, K., Mackey, J. R., Koski, S., Pituskin, E. and Sawyer, M. B. (2009).Sarcopenia as a determinant of chemotherapy toxicity and time to tumorprogression in metastatic breast cancer patients receiving capecitabinetreatment. Clin. Cancer Res. 15, 2920-2926.
Raben, N., Hill, V., Shea, L., Takikita, S., Baum, R., Mizushima, N., Ralston, E.and Plotz, P. (2008). Suppression of autophagy in skeletal muscle uncovers theaccumulation of ubiquitinated proteins and their potential role inmuscle damage inPompe disease. Hum. Mol. Genet. 17, 3897-3908.
Romanick, M., Thompson, L. V. and Brown-Borg, H. M. (2013). Murine models ofatrophy, cachexia, and sarcopenia in skeletal muscle. Biochim. Biophys. Acta1832, 1410-1420.
Rosenfeld, G. E., Mercer, E. J., Mason, C. E. and Evans, T. (2013). Small heatshock proteins Hspb7 andHspb12 regulate early steps of cardiacmorphogenesis.Dev. Biol. 381, 389-400.
Rubinsztein, D. C., Marin o, G. andKroemer, G. (2011). Autophagy and aging.Cell146, 682-695.
Sandri, M., Sandri, C., Gilbert, A., Skurk, C., Calabria, E., Picard, A., Walsh, K.,Schiaffino, S., Lecker, S. H. and Goldberg, A. L. (2004). Foxo transcriptionfactors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletalmuscle atrophy. Cell 117, 399-412.
Shaulian, E. and Karin, M. (2002). AP-1 as a regulator of cell life and death. Nat.Cell Biol. 4, E131-E136.
Stitt, T. N., Drujan, D., Clarke, B. A., Panaro, F., Timofeyva, Y., Kline, W. O.,Gonzalez, M., Yancopoulos, G. D. and Glass, D. J. (2004). The IGF-1/PI3K/Aktpathway prevents expression of muscle atrophy-induced ubiquitin ligases byinhibiting FOXO transcription factors. Mol. Cell 14, 395-403.
Sun, X., Fontaine, J.-M., Rest, J. S., Shelden, E. A., Welsh, M. J. and Benndorf,R. (2004). Interaction of human HSP22 (HSPB8) with other small heat shockproteins. J. Biol. Chem. 279, 2394-2402.
Suzuki, T., Okuno, H., Yoshida, T., Endo, T., Nishina, H. and Iba, H. (1991).Difference in transcriptional regulatory function between c-Fos and Fra-2. NucleicAcids Res. 19, 5537-5542.
Thompson, T. G., Chan, Y.-M., Hack, A. A., Brosius, M., Rajala, M., Lidov,H. G.W., McNally, E. M.,Watkins, S. andKunkel, L. M. (2000). Filamin 2 (FLN2):a muscle-specific sarcoglycan interacting protein. J. Cell Biol. 148, 115-126.
Trendelenburg, A. U., Meyer, A., Rohner, D., Boyle, J., Hatakeyama, S. andGlass, D. J. (2009). Myostatin reduces Akt/TORC1/p70S6K signaling, inhibitingmyoblast differentiation and myotube size. Am. J. Physiol. Physiol. 296,C1258-C1270.
Vos, M. J., Kanon, B. and Kampinga, H. H. (2009). HSPB7 is a SC35 speckleresident small heat shock protein. Biochim. Biophys. Acta Mol. Cell Res. 1793,1343-1353.
Vos, M. J., Zijlstra, M. P., Kanon, B., van Waarde-Verhagen, M. A. W. H., Brunt,E. R. P., Oosterveld-Hut, H. M. J., Carra, S., Sibon, O. C. M. and Kampinga,H. H. (2010). HSPB7 is the most potent polyQ aggregation suppressor within theHSPB family of molecular chaperones. Hum. Mol. Genet. 19, 4677-4693.
Wales, S., Hashemi, S., Blais, A. and McDermott, J. C. (2014). Global MEF2target gene analysis in cardiac and skeletal muscle reveals novel regulation ofDUSP6 by p38MAPK-MEF2 signaling. Nucleic Acids Res. 42, 11349-11362.
Windak, R., Muller, J., Felley, A., Akhmedov, A., Wagner, E. F., Pedrazzini, T.,Sumara, G. and Ricci, R. (2013). The AP-1 transcription factor c-Jun preventsstress-imposed maladaptive remodeling of the heart. PLoS ONE 8, e73294.
Wisdon, R. and Verma, I. M. (1993). Transformation by Fos proteins requires a C-terminal transactivation domain. Mol. Cell. Biol. 13, 7429-7438.
Yuen, M., Sandaradura, S. A., Dowling, J. J., Kostyukova, A. S., Moroz, N.,Quinlan, K. G., Lehtokari, V.-L., Ravenscroft, G., Todd, E. J., Ceyhan-Birsoy,O. et al. (2014). Leiomodin-3 dysfunction results in thin filament disorganizationand nemaline myopathy. J. Clin. Invest. 124, 4693-4708.
Zhao, J., Brault, J. J., Schild, A., Cao, P., Sandri, M., Schiaffino, S., Lecker, S. H.and Goldberg, A. L. (2007). FoxO3 coordinately activates protein degradation bythe autophagic/lysosomal and proteasomal pathways in atrophying muscle cells.Cell Metab. 6, 472-483.
Zheng, B., Han, M., Bernier, M. and Wen, J.-K. (2009). Nuclear actin and actin-binding proteins in the regulation of transcription and gene expression. FEBS J.276, 2669-2685.
Zhou, X., Wang, J. L., Lu, J., Song, Y., Kwak, K. S., Jiao, Q., Rosenfeld, R., Chen,Q., Boone, T., Simonet, W. S. et al. (2010). Reversal of cancer cachexia andmuscle wasting by ActRIIB antagonism leads to prolonged survival. Cell 142,531-543.
4090
RESEARCH ARTICLE Journal of Cell Science (2016) 129, 4076-4090 doi:10.1242/jcs.190009
Journal
ofCe
llScience












![Metformin alters skeletal muscle transcriptome adaptations …...loss of skeletal muscle mass, a condition known as age-associated muscle atrophy or sarcopenia [1–3]. This is usually](https://static.fdocuments.us/doc/165x107/60e5197499ee7611e91e8f6e/metformin-alters-skeletal-muscle-transcriptome-adaptations-loss-of-skeletal.jpg)





