
Regulation of pro-apoptotic leucocyte granule serine proteinases by intracellular serpins
11
Introduction Proteolysis and apoptosis Many biological functions require proteolysis limited to a particular tissue or temporal compartment. For example, proper functioning of the immune system requires specific proteolysis to drive processes such as complement fixation, phagocytosis, diapedesis, cytotoxicity, and cytokine matura- tion and release. The blood coagulation pathway (review 1 ) exemplifies the role of proteolysis in a complex biological system and pro- vides a conceptual framework that can be applied to other proteolytic pathways. (Indeed there are many similarities between the blood coagulation and complement pathways, and they are functionally linked through the serine protein- ase, kaillikrein, and the serpin, C1 inhibitor.) Coagulation begins with a signal (typically injury to the blood vessel wall) that leads to the conversion by proteolysis of a pre-existing proteinase precursor (zymogen) into an active proteinase. In a process that also depends on the presence of specific co- factors, the activated proteinase in turn cleaves and activates another zymogen, which then activates another. This sequen- tial activation of zymogens, or cascade, amplifies the origi- nal signal so that enough thrombin is eventually generated to effect formation of the blood clot. Amplification is also achieved via a positive feedback loop in which thrombin pro- teolytically activates two key coagulation cofactor proteins. Of equal importance to the initiation and promulgation of the coagulation cascade are the systems that regulate it: the mechanisms that ensure that it is only triggered in reponse to a bona fide signal, that it is contained to the site of injury, and that proteinase activation is terminated once the clot is fully formed. How a balance is maintained between these anti- coagulant systems and the opposing procoagulant systems is not completely understood, but thrombin can participate in a negative feedback loop and halt the cascade by activating a specific proteinase that destroys the two key coagulation cofactors (review 2 ), and a cofactor upstream in the cascade is also proteolytically inactivated in a separate feedback loop. In addition, circulating inhibitors belonging to the serine pro- teinase inhibitor (serpin) family can bind to and inactivate most of the coagulation proteinases. The key concepts, therefore, that arise from the blood coagulation system are: (i) pre-existing quiescent proteinases (zymogens) that are proteolytically activated in response to a specific signal; (ii) signal amplification via a cascade of proteinase activation; (iii) cofactors that potentiate proteinase or inhibitor function; (iv) positive and negative feedback loops; and (v) inhibitors of activated proteinases. During the past few years it has been appreciated that apoptosis or programmed cell death is essential for the correct development and functioning of the immune system (review 3 ). Similarities to the blood coagulation system include the proteolytic activation of intracellular zymogens (caspases) in response to a specific initiating signal, amplifi- cation of the signal by sequential (cascading) activation of these enzymes, and the existence of positive and negative effectors of caspase activation (exemplified by the Bcl-2/ Bcl-X/Bax system 4 ). Although the components of the pro- apoptotic signalling and effector pathways are beginning to Immunology and Cell Biology (1999) 77, 47–57 Special Feature Regulation of pro-apoptotic leucocyte granule serine proteinases by intracellular serpins PI BIRD Department of Medicine, Monash Medical School, Box Hill Hospital, Box Hill, Victoria, Australia Summary Caspase activation and apoptosis can be initiated by the introduction of serine proteinases into the cytoplasm of a cell. Cytotoxic lymphocytes have evolved at least one serine proteinase with specific pro-apoptotic activity (granzyme B), as well as the mechanisms to deliver it into a target cell, and recent evidence suggests that other leucocyte granule proteinases may also have the capacity to kill if released into the interior of cells. For example, the monocyte/granulocyte proteinase cathepsin G can activate caspases in vitro, and will induce apop- tosis if its entry into cells is mediated by a bacterial pore-forming protein. The potent pro-apoptotic activity of granzyme B and cathepsin G suggests that cells producing these (or other) proteinases would be at risk from self- induced death if the systems involved in packaging, degranulation or targeting fail and allow proteinases to enter the host cell cytoplasm. The purpose of the present review is to describe recent work on a group of intracellular serine proteinase inhibitors (serpins) which may function in leucocytes to prevent autolysis induced by the granule serine proteinases. Key words: apoptosis, cathepsin G, cytotoxicity, granule, granzyme B, leucocyte, M/NEI, phagocytosis, PI-6, PI-9, serpin. Correspondence: Dr P Bird, Department of Medicine, Box Hill Hospital, Box Hill, Vic. 3128, Australia. Email: <[email protected]> Received 13 October 1998; accepted 13 October 1998.
Transcript of Regulation of pro-apoptotic leucocyte granule serine proteinases by intracellular serpins

Introduction
Proteolysis and apoptosis
Many biological functions require proteolysis limited to aparticular tissue or temporal compartment. For example,proper functioning of the immune system requires specificproteolysis to drive processes such as complement fixation,phagocytosis, diapedesis, cytotoxicity, and cytokine matura-tion and release.
The blood coagulation pathway (review1) exemplifies therole of proteolysis in a complex biological system and pro-vides a conceptual framework that can be applied to otherproteolytic pathways. (Indeed there are many similaritiesbetween the blood coagulation and complement pathways,and they are functionally linked through the serine protein-ase, kaillikrein, and the serpin, C1 inhibitor.) Coagulationbegins with a signal (typically injury to the blood vessel wall)that leads to the conversion by proteolysis of a pre-existingproteinase precursor (zymogen) into an active proteinase. Ina process that also depends on the presence of specific co-factors, the activated proteinase in turn cleaves and activatesanother zymogen, which then activates another. This sequen-tial activation of zymogens, or cascade, amplifies the origi-nal signal so that enough thrombin is eventually generated toeffect formation of the blood clot. Amplification is alsoachieved via a positive feedback loop in which thrombin pro-teolytically activates two key coagulation cofactor proteins.
Of equal importance to the initiation and promulgation ofthe coagulation cascade are the systems that regulate it: themechanisms that ensure that it is only triggered in reponse toa bona fide signal, that it is contained to the site of injury, andthat proteinase activation is terminated once the clot is fullyformed. How a balance is maintained between these anti-coagulant systems and the opposing procoagulant systems isnot completely understood, but thrombin can participate in anegative feedback loop and halt the cascade by activating aspecific proteinase that destroys the two key coagulationcofactors (review2), and a cofactor upstream in the cascade isalso proteolytically inactivated in a separate feedback loop. Inaddition, circulating inhibitors belonging to the serine pro-teinase inhibitor (serpin) family can bind to and inactivatemost of the coagulation proteinases.
The key concepts, therefore, that arise from the bloodcoagulation system are: (i) pre-existing quiescent proteinases(zymogens) that are proteolytically activated in response to aspecific signal; (ii) signal amplification via a cascade ofproteinase activation; (iii) cofactors that potentiate proteinaseor inhibitor function; (iv) positive and negative feedbackloops; and (v) inhibitors of activated proteinases.
During the past few years it has been appreciated thatapoptosis or programmed cell death is essential for thecorrect development and functioning of the immune system(review3). Similarities to the blood coagulation systeminclude the proteolytic activation of intracellular zymogens(caspases) in response to a specific initiating signal, amplifi-cation of the signal by sequential (cascading) activation ofthese enzymes, and the existence of positive and negativeeffectors of caspase activation (exemplified by the Bcl-2/Bcl-X/Bax system4). Although the components of the pro-apoptotic signalling and effector pathways are beginning to
Immunology and Cell Biology (1999) 77, 47–57
Special Feature
Regulation of pro-apoptotic leucocyte granule serine proteinases byintracellular serpins
PI BIRD
Department of Medicine, Monash Medical School, Box Hill Hospital, Box Hill, Victoria, Australia
Summary Caspase activation and apoptosis can be initiated by the introduction of serine proteinases into thecytoplasm of a cell. Cytotoxic lymphocytes have evolved at least one serine proteinase with specific pro-apoptoticactivity (granzyme B), as well as the mechanisms to deliver it into a target cell, and recent evidence suggests thatother leucocyte granule proteinases may also have the capacity to kill if released into the interior of cells. Forexample, the monocyte/granulocyte proteinase cathepsin G can activate caspases in vitro, and will induce apop-tosis if its entry into cells is mediated by a bacterial pore-forming protein. The potent pro-apoptotic activity ofgranzyme B and cathepsin G suggests that cells producing these (or other) proteinases would be at risk from self-induced death if the systems involved in packaging, degranulation or targeting fail and allow proteinases to enterthe host cell cytoplasm. The purpose of the present review is to describe recent work on a group of intracellularserine proteinase inhibitors (serpins) which may function in leucocytes to prevent autolysis induced by the granuleserine proteinases.
Key words: apoptosis, cathepsin G, cytotoxicity, granule, granzyme B, leucocyte, M/NEI, phagocytosis, PI-6, PI-9, serpin.
Correspondence: Dr P Bird, Department of Medicine, Box HillHospital, Box Hill, Vic. 3128, Australia. Email: <[email protected]>
Received 13 October 1998; accepted 13 October 1998.

be understood in broad detail, less is known about how theyare regulated. From studies of the anti-apoptotic products ofvarious viruses and their cellular counterparts, it has becomeclear that apoptosis can be negatively regulated at severalpoints. The best-characterized negative regulators includedecoy receptors that compete for or inhibit binding of ‘deathligands’,5 intracellular caspase analogues (FLICE inhibitoryproteins (FLIPs)) that bind to the death receptor complex andprevent caspase activation,6 analogues of Bcl-2 that preventcaspase activation,4 and proteins (inhibitors of apoptosis(IAP); cytokine response modifier A (CrmA)) that directlybind and inhibit activated caspases.7,8
Apoptosis can be induced by a wide variety of stresses orspecific signals, but in the case of the immune system ittypically occurs in response to a death ligand or cytokineacting through a dedicated receptor,9 or to a proteinase actingon a specific target.10,11 The best-characterized pro-apoptoticproteinase of the immune system is granzyme B, which isused by cytotoxic lymphocytes to bypass death receptors andinduce target cell death by entering the cytoplasm anddirectly activating one or more caspases.12,13 However, thereis evidence that other, normally non-cytotoxic, proteinasescan also induce apoptosis if they reach the cytoplasm.14 Thisindicates that the apoptotic pathways are relatively easy totrigger by intracellular proteolysis, and raises the question ofhow immune cells, particularly those producing cytotoxicproteinases, are protected from apoptosis initiated by expo-sure to misdirected or non-specific proteinases.
In principle, protection could be achieved by producing an inhibitor of the initiating proteinase or by producinginhibitors of activated caspases like IAP. In the present reviewI will discuss the regulation of the leucocyte serineproteinases granzyme B and cathepsin G by members of arecently described group of intracellular serpins, and developthe idea that immune cells such as cytotoxic lymphocytes andphagocytes are protected by these serpins from apoptosisinduced by their resident proteinases.
Leucocyte granule proteinases and apoptosis
Cytotoxic T cells, natural killer cells, monocytes,macrophages, granulocytes and mast cells contain a varietyof serine proteinases (review15). As summarized in Table 1,these include the granzymes of cytotoxic cells, theazurophilic granule proteinases of monocytes and granulo-cytes, and mast cell tryptase and chymase. These enzymes arestructurally related, have similar gene organizations and chro-mosomal locations, and on the basis of substrate preferenceare classified as tryptic (granzyme A, tryptases), chymo-tryptic (chymase, cathepsin G) or elastinolytic (elastase,proteinase 3). Although they are related to chymotrypsin,granzymes B and M (Met-ase) have quite narrow substratespecificities and are essentially restricted to cleaving proteinsafter acidic amino acids or Met, respectively.
The physiological roles and regulation of most leucocyteproteinases have not been fully elucidated. Unlike the situa-tion for the serine proteinases functioning in the coagulationand complement pathways, where the study of naturallyoccurring mutants or acquired dysfunctional variants hasyielded great insight into their biological roles, no hereditaryloss of expression of a gene encoding any single member of
the leucocyte serine proteinase group has been described.With the advent of gene targeting technology this situation isslowly being addressed as mice deficient in specific pro-teinases are generated. To date granzyme A-deficient,17
granzyme B-deficient18 and neutrophil elastase-deficient19
mice have been described, illustrating the central role ofgranzymes in cytotoxic lymphocyte function and implicatingelastase in the clearance of Gram-negative bacteria.
Possible roles of the granule proteinases have thereforemainly been inferred from in vitro identification of potentialprotein substrates, and from the analysis of proteinase levelsand distribution in pathophysiological situations (Table 1). Asdescribed in the following section, two granule proteinases,granzyme B and cathepsin G, have been implicated in theinduction of apoptosis.
Granzyme B
The granule serine proteinases produced by cytotoxic T cells and natural killer cells are collectively termed‘granzymes’ (review11). Like many other serine proteinases,granzymes are synthesized as zymogens that are activated atthe time of packaging into granules. One or more of thesesynergize with the granule pore-forming protein perforin topromote the calcium-dependent destruction of abnormal orvirally infected cells. Humans have two major granzymes(A and B) and two minor granzymes (H and Met-ase),whereas rodents have several additional granzymes. Thisprobably reflects differing evolutionary pressures on thehuman and rodent immune systems through exposure todifferent pathogens.
Extensive studies have shown that target cell destructionby cytotoxic lymphocytes depends critically on perforin andgranzyme B. Perforin mediates access of granzyme B to thetarget cell cytoplasm, and once inside granzyme B inducesapoptosis by directly activating caspases (reviews20,21).Studies by Froelich et al. and Pinkoski et al. have suggestedthat granzyme B is internalized by receptor-mediated endo-cytosis, and that the role of perforin is to mediate release ofgranzyme B from endocytic vesicles. Indeed, perforin can bereplaced by other vesicle-disrupting factors such as thoseproduced by adenovirus.13,22
The role of the other granzymes is presently unclear, butas indicated by the incomplete loss of cytotoxic activity inmice lacking granzyme B, they may serve to reinforce orreplace granzyme B in situations where its function is sup-pressed by viral inhibitors. Alternatively, they may have func-tions unrelated to target cell destruction. For example, it hasbeen suggested that granzyme A activates cytokines, or func-tions to modulate viral infectivity by cleaving viral surfacereceptors.23
Cathepsin G
Mononuclear phagocytes and neutrophils play key roles inthe elimination of bacterial and fungal pathogens via thegeneration of reactive oxygen species and secretion ofazurophilic granule cytotoxins into phagocytic vesicles.24–26
Among the azurophilic granule cytotoxins are the neutralserine proteinases cathepsin G, proteinase 3 and elastase,which participate in the destruction of phagocytosed targets.
PI Bird48

Serpins 49
Like the granzymes, these enzymes are synthesized as zymo-gens that are activated during packaging into granules.27
Recent work has shown that mice lacking elastase have animpaired ability to kill Gram-negative bacteria,19 and onefeature of Chediak–Higashi syndrome, a membrane-sortingdisorder associated with immune deficiency, is abnormalchemotaxis and killing of micro-organisms due to defectiveneutrophil granule formation and lack of cathepsin G andelastase (review28).
The physiological impact of the azurophilic granuleproteinases is probably not limited to phagocytosis. There is evidence that they are sometimes secreted and can affectthe extracellular matrix, cytokine processing,29 chemotaxis,30
and platelet activation.31 Examples of extracellular targets of the azurophilic granule proteinases include connectivetissue components such as fibronectin, elastin, proteoglycans,and collagen types III and IV,32–35 and they have been impli-cated in cell and tissue injury in several acute and chronicdiseases, including emphysema, cystic fibrosis, asthma,arthritis and psoriasis.36–39 The pathological effects of theseproteinases probably result from a localized imbalancebetween their levels and the levels of their major seruminhibitors, α1-proteinase inhibitor (α1-antitrypsin) and α1-antichymotrypsin. This is supported by the observation
that α1-proteinase inhibitor deficiency is associated with thedevelopment of emphysema.40
As well as roles in phagocytosis, chemotaxis and extra-cellular matrix modification, cathepsin G may also have theability to induce apoptosis. It has been recently demonstratedthat cathepsin G can proteolytically activate the pro-apoptoticcaspase, caspase-7,41 and we have shown that cathepsin Gintroduced into the cytoplasm of cells via the bacterial pore-forming protein, streptolysin O, will induce morphologicalchanges characteristic of apoptosis (C Bird, and P Bird,unpubl. data, 1998). At present it is unclear how cathepsin Gwould access the interior of a cell in a physiological setting,or what type of cell it would target. It would presumably needan accessory molecule analagous to perforin to mediate itsuptake, but several candidates exist, such as perforin itself,complement, or pore-forming proteins of bacteria or viruses.
Regulation of the pro-apoptotic functions of granzymeB and cathepsin G
Both granzyme B and cathepsin G have the capacity toinduce apoptosis by direct activation of caspases. The factthat cell death can also be triggered by introducing relativelynon-specific proteinases such as chymotrypsin, trypsin or
Table 1 Properties and expression of leucocyte granule serine proteinases
Compiled from11, 15, 16. P1 residue indicates the preferred cleavage point of the proteinase.Baso, basophil; Cat, cathepsin; Cyto, cytotoxic T cell; Macro, macrophage; Mast, mast cell; Mono, monocyte; Mr, molecular mass; NK,
natural killer cell; Neut, netrophil; Pro, proteinase.
Mr Expression patternProteinase (kDa) Cyto NK Neut Mono Macro Mast Baso P1 residue Chromosome Proposed function(s)
Granzyme A 60 ++ ++ Arg, Lys 5q11–12 Cytotoxicity, cytokineactivation, receptorcleavage
Tryptase-2 30 + + Arg, Lys 5q11–12Granzyme B 29 ++ + Asp, Glu 14q11–12 Cytotoxicity, DNA
degradation, caspaseactivation
Granzyme H 30 + + 14q11–12Met-ase 30 ± ++ Met 19p13.3 CytotoxicityElastase 29 ++ + ± Val, Ala 19pter Phagocytosis, secretion,
connective tissuedegradation
Pro. 3 25 ++ + ± Val, Ala 19pter Phagocytosis, connectivetissue degradation
Cat G 23 ++ + ± ++ Lys, Phe, 14q11–12 Phagocytosis, chemotaxis,Arg, Met, antimicrobial defense,Leu platelet activation,
connective tissuedegradation, caspaseactivation
Chymase 30 ++ Phe, Tyr 14q11–12 Secretion, connective tissuedegradation
Tryptases 140 +++ ± Lys, Arg 16 Bronchoconstriction,mitogenesis,anticoagulation,connective tissuedegradation

proteinase K into the cytoplasm14 suggests that other granuleproteinases besides cathepsin G and granzyme B are likely tohave pro-apoptotic potential. This poses a particular problemfor cells synthesizing and secreting granule proteinases: toavoid self-induced death, active enzyme must be preventedfrom entering or acting in the cytoplasm.
It is likely that several mechanisms exist to protect cellsagainst their own proteinases. These include storage in dedi-cated compartments (granules), synthesis as zymogens, and activation only during or after packaging in granules.However, such measures would not protect the cell fromactive proteinase leaking from a faulty storage granule. Norwould they prevent proteinase leaking into the cytoplasm orre-entering the cell via endocytosis during the normal processof degranulation, which involves fusion of granules with theplasma membrane or phagocytic vesicle. To prevent theinduction of apoptosis the cell would require a means ofrapidly inhibiting the proteinase as it enters the cytoplasm. Asdescribed in the following section, we believe that cytotoxiccells and phagocytic cells contain cytoplasmic inhibitors ofgranule serine proteinases that fulfil this function.
Regulation of proteolysis by serpins
The physiological regulation of serine proteinases is achievedmainly through interactions with members of a second classof protein, the serine proteinase inhibitors (serpins).42
Serpins form a very large protein superfamily with more than300 members identified in the higher eukaryotes. Althoughmost apparently function as proteinase inhibitors, a numberhave evolved different roles as peptide hormone precursors,hormone transporters, chaperones and chromatin-bindingproteins. This diversity of function is a reflection of the extra-ordinary flexibility of serpins at the molecular level, whichhas become evident through detailed structural and kineticanalysis of functional and non-functional family members.The importance of serpins to homeostasis is illustrated by theconsequences of loss or dysfunction (review43). For example,loss or mutation of α1-proteinase inhibitor causes emphy-sema and liver cirrhosis; loss or mutation of antithrombin IIIunderlies thrombosis; and loss or mutation of C1 inhibitorleads to angioedema.
The interaction between a serpin and its target proteinasetypically leads to the formation of an essentially irreversible1:1 complex, a property that gives rise to the description ofserpins as ‘suicide substrates’.44 The molecular events under-lying complex formation are being intensively studied, anddepend on specific sequences and structures within bothserpin and proteinase. One of the key regions in the serpin is the carboxy-terminal inhibitory domain. This inhibitorydomain, or reactive centre, contains a sequence that variesfrom serpin to serpin and resembles the natural substrate ofthe target proteinase. It is flanked by two highly conservedsequences known as the hinge domains.
On binding to the serpin, the proteinase attempts to cleavea scissile peptide bond between two residues in the reactivecentre, designated P1 and P1′. The P1 residue primarilydefines the specificity of the serpin and dictates the type ofproteinase it will inhibit. (The classic example of the impor-tance of the P1 residue to specificity is the Pittsburgh variantof α1-proteinase inhibitor which has a Met to Arg mutation
at this position, converting it from an inhibitor of neutrophilelastase to a potent inhibitor of thrombin43). Although theserpin reactive centre sequence is usually very similar tothose found in the natural substrate of the target proteinase,hydrolysis of the P1–P1′ bond does not proceed efficiently,and the proteinase and serpin become trapped in a complexthat resembles an acyl- or tetrahedral intermediate of the pro-teinase–substrate reaction.
Biochemical evidence suggests that the complex forma-tion follows a kinetic pathway in which serpin–proteinasebinding results in a reversible Michaelis complex that is con-verted rapidly to the irreversible locked complex (inhibitorypathway).44,45 In some cases, a locked complex does notform, leading to breakdown of the Michaelis complex andrelease of active proteinase and inactive, cleaved serpin (sub-strate pathway). The latter pathway usually results from aninteraction between a serpin and non-cognate proteinasebecause the exposed nature of the reactive centre makes itsensitive to non-specific proteolysis (cleavage other thanbetween the P1–P1′ bond). However, some reactions betweenserpins and cognate proteinases appear to involve competi-tion between the inhibitory and substrate pathways, suggest-ing that stable complex formation requires rapidisomerization of an intermediate between the initialMichaelis and final locked complexes. The rate of thecomplex formation, and its stability, can be modulated by theparticipation of cofactors. One of the best examples isheparin, which modulates the interaction of the serpinsantithrombin III, heparin cofactor II and protease nexin-Iwith their target proteinases. In the case of the interactionbetween antithrombin III and thrombin, the rate of complexformation is increased 1000-fold in the presence of heparin.46
Although several crystal structures of serpins and cognateproteinases exist, to date a structure for a serpin–proteinasecomplex has not been determined. Much of what we knowabout the formation of the complex has been inferred fromanalysis of functional serpins, and comparison to either nat-urally occurring or deliberately mutated non-functionalserpins (review47). From these studies it is apparent thatserpins are essentially globular molecules consisting of threeβ-sheets and nine α-helices. A distinguishing feature is theexposed nature of the reactive centre which extrudes as aflexible polypeptide loop (Fig. 1). It is also clear that serpinsundergo a marked conformational change on interaction witha target proteinase that includes a re-positioning of thereactive centre loop so that all or part of it is inserted betweentwo strands of one of the β-sheets. This is dependent on otherserpin domains, particularly the two flanking hinges, and isprobably accompanied by structural alterations to the pro-teinase that distort the active site.48,49
Ovalbumin (ov) serpins Most serpins are glycoproteins thatregulate extracellular serine proteinases; however, it hasrecently become clear that some function within cells. Thebest characterized example of an intracellular serpin is thecowpox virus product CrmA which inhibits both granzymeB, and the caspases involved in cytokine maturation andapoptosis.50,51
CrmA belongs to a subgroup of serpins that resemblechicken ovalbumin in structure (Table 2). These ‘ovalbumin’(ov) serpins are unusual because they lack an amino terminal
PI Bird50

Serpins 51
signal peptide normally required to initiate extracellularsecretion of the protein, although several appear to exist inboth intracellular and extracellular forms.52 A second unusualfeature is the potential to inhibit proteinases of two distinctclasses (cross-class inhibition). This was first described forCrmA, which inhibits caspases (cysteine proteinases) andgranzyme B (serine proteinase). It is probably not limited toCrmA because a mammalian ov-serpin, squamous cell carcin-oma antigen-1 (SCCA-1), can inhibit cathepsins S, L and K(cysteine proteinases unrelated to caspases) and chymo-trypsin (serine proteinase).53
As indicated in Table 2, the present human members ofthe ov-serpin group include plasminogen activator inhibitor
2 (PAI-254), SCCA-1 and -2,55,56 monocyte/neutrophil elas-tase inhibitor (M/NEI57), maspin,58 proteinase inhibitor 6 (PI-659), proteinase inhibitor 8 (PI-860), proteinase inhibitor 9 (PI-960,61), bomapin,62 and megakaryocyte maturationfactor63 (Table 2). Their genes are clustered at two distinctchromosomal loci. The first group at 18q21–23 encodes PAI-2, maspin, SCCA-1, SCCA-2, PI-8, and bomapin.52,56,64
The second group of human ov-serpin genes is located at6p25, and encodes PI-6, PI-9 and M/NEI.65–69 They havehighly conserved gene structures that are essentially ident-ical, except that genes at 6p25 have one less intron.70
The physiological roles of the ov-serpins are poorlyunderstood, but some may function in processes associated
Figure 1 Structure and functionof serpins. Stereo view of a modelof a serpin in the initial stages ofan interaction with a protease. Thediagram shows human granzymeB (top ribbon structure, modelledon rat mast cell protease) bindingto the intracellular serpin PI-9(lower ribbon structure, modelledon antithrombin III). Note theexposed, flexible serpin reactivecentre loop inserted into the activesite cleft of the proteinase. Subse-quent stages in the interactionrequire reactive centre loop mobil-ity and result in conformationalchanges to both serpin and pro-teinase that lock the complex.48,49
Image produced by Dr J Whisstockusing a program developed by DrA Lesk (University of Cambridge).
Table 2 Properties of the human ovalbumin (ov) serpins
(f), facultative secretion; MMF, megakaryocyte maturation factor; M/NEI, monocyte/neutrophil elastase inhibitor; PAI-2, plasminogen acti-vator inhibitor 2; PI, proteinase inhibitor; SCCA, squamous cell carcinoma antigen; t-PA, tissue plasminogen activator; u-PA, urokinase plas-minogen activator. Source references are cited in the text.
Mr Target GeneSerpin (kDa) Expression proteinase(s) Secretion Chromosome structure Possible function(s)
PAI-2 47 Monocytes, u-PA + (f) 18q21 8 exons Control of fibrinolysis and matrix re-modelling,epithelia promotes cell survival
SCCA-1 44 Epithelia Cat. K, L, S + 18q21 8 exons Regulation of lysosomal proteinasesSCAA-2 44 Epithelia Cat. G, chymase 18q21 8 exonsMaspin 42 Epithelia t-PA + 18q21 Regulation of cell–matrix interactions, tumour
suppressorBomapin 45 Marrow 18q21 Lymphocyte differentiationMMF 43 Megakaryocytes + Megakaryocyte differentiation
dendritic cellsPI-8 43 Heart, marrow Furin convertase – 18q21 7 exons Regulation of protein processingM/NEI 42 Monocytes, Elastase, Pro. 3, – 6p25 7 exons Regulation of monocyte/granulocyte proteinases
granulocytes Cat. GPI-6 42 Epithelia, Cat. G – 6p25 7 exons Regulation of monocyte/granulocyte proteinases
monocytes,granulocytes
PI-9 42 Cytotoxic cells Gra. B – 6p25 7 exons Anti-apoptosis: regulation of granzyme B

with tumorigenesis and inflammation. For example, loss ofmaspin correlates with increased malignancy and metastasisof breast carcinomas;58 increases in the levels and release ofSCCA occur in squamous cell carcinomas;55 and increasedexpression and release of PAI-2 occurs from monocytesduring inflammation.71 As described in the following section,recent evidence suggests that M/NEI, PI-6, and PI-9 are reg-ulators of leucocyte granule serine proteinases.
Regulation of granzyme B by PI-9
Target cell apoptosis induced by cytotoxic lymphocytes is akey defence against viral infection. Many viruses have there-fore evolved proteins that inhibit apoptosis and prolonginfection.72 Poxviruses produce an array of proteins designedto circumvent or suppress host defences.73 These includeCrmA, which is essential for suppression of the acuteinflammatory response during poxvirus infection. Theprimary target of CrmA in infected cells is caspase-1, whichcleaves pro-IL-1β after Asp,74,75 but many studies haveshown that it is also capable of inhibiting death receptor-mediated apoptosis by inhibiting pro-apoptotic caspases.Thus CrmA may also offer the virus the means to suppressFas-mediated apoptosis resulting from contact with a cyto-toxic lymphocyte.
Although first identified as a caspase rather than serineproteinase inhibitor, CrmA is obstensibly a functional serpinwith a conserved proximal hinge domain and an unusual P1
Asp in the reactive centre (Fig. 2). This suggested that itmight also inhibit a serine proteinase with a preference forcleaving after Asp, and efficient inhibition of granzyme B(the only known serine proteinase with this specificity) byCrmA has been demonstrated in vitro.76 However, three inde-pendent studies investigating apoptosis induced by cytotoxiccells have demonstrated that CrmA blocks Fas-mediated butnot granule-mediated apoptosis.77–79 Thus the physiologicalrole of CrmA appears to be to suppress caspases involved incytokine processing and Fas-mediated apoptosis, rather thaninhibiting granzyme B-mediated apoptosis.
Some of the viral proteins that inhibit apoptosis haveevolved by recruitment and modification of host genes, asindicated by several cellular regulators of apoptosis that havebeen identified through the analysis of viruses.72 Thereforethe identification of CrmA and the demonstration of its anti-apoptotic function raised the possibility that an endogenouscounterpart may exist, prompting a search that resulted in thediscovery of PI-9.
PI-9 is an efficient inhibitor of granzyme B in vivo and invitro PI-9 is a 42-kDa human serpin, also known as cyto-plasmic antiproteinase 3 or granzyme B inhibitor, that wasoriginally isolated on the basis of homology to PI-6.60,61 Ourinitial sequence analysis revealed striking similarities toCrmA in the reactive centre including an acidic P1 residue,which in PI-9 is Glu rather than Asp (Fig. 2). Knowing thatgranzyme B can also cleave after Glu,80 we predicted anddemonstrated that PI-9 inhibits granzyme B, and it proved tobe a better inhibitor than CrmA (association rate constantsfor complex formation: PI-9, 1.7 × 106 per mol/L per s;CrmA, 2.9 × 105 per mol/L per s). We also showed that pre-incubation with PI-9 inhibits purified granzyme B and per-forin-induced apoptosis of mouse FDC-P1 cells, that it isintracellular and that it is expressed in immune tissue, par-ticularly in cells producing granzyme B.61 Taken together,these observations led to the hypothesis that PI-9 is producedby cytotoxic cells to protect against ectopic granzyme B.
To test this hypothesis we have recently studied the func-tion of PI-9 in greater detail.81 In particular, we have testedthe key prediction that intracellular PI-9 should prevent apop-tosis mediated by granzyme B. We found that FDC-P1 cellsexpressing intracellular PI-9 resist apoptosis induced by puri-fied granzyme B and perforin, and the degree of protectionis proportional to the amount of PI-9 expressed. Protection isalso observed when Fas-negative human MCF-7 cellsexpressing PI-9 are exposed to cytotoxic lymphocytes, sug-gesting that PI-9 is protective even when other cytotoxins areoperating. (This is consistent with the view that granzyme Bis the primary proteinase in granule-mediated apoptosis.)
We also found that one striking difference between PI-9and CrmA is that PI-9 does not protect cells against Fas-mediated apoptosis. Consistent with this observation, wetested nine of the 10 known human caspases and showed thatPI-9 is not an effective caspase inhibitor.81 This functionaldistinction between PI-9 and CrmA is due to their different P1
residues. The fact that PI-9 with a P1 Glu is a better granzymeB inhibitor than CrmA with a P1 Asp contradicts serpindogma that the P1 residue should reflect the substrate prefer-ence of the target proteinase (in this case, Asp). However, it is consonant with the observation that granzyme B alsocleaves peptide substrates after Glu though less efficientlythan after Asp.80 Mutation of the P1 Glu to Asp converts PI-9from an efficient granzyme B inhibitor but poor caspaseinhibitor into a poor granzyme B inhibitor (100-fold lessefficient) but effective caspase inhibitor.81 In addition, the P1
Asp mutant is less protective against granzyme B-mediated
PI Bird52
Figure 2 Comparison of PI-6, M/NEI, PI-9 and CrmA C-terminal inhibitory domains comprising the hinge (solid lines) and variablereactive centre (double line) sequences. Spaces introduced to optimize sequence alignment are indicated by single dashes. The scissileP1-P1′ bond is indicated by a double dash.
apoptosis but efficiently protects cells against Fas-mediatedapoptosis. Taken together, these results suggest that the P1
Glu makes PI-9 specific for granzyme B, and prevents aninteraction with caspases.
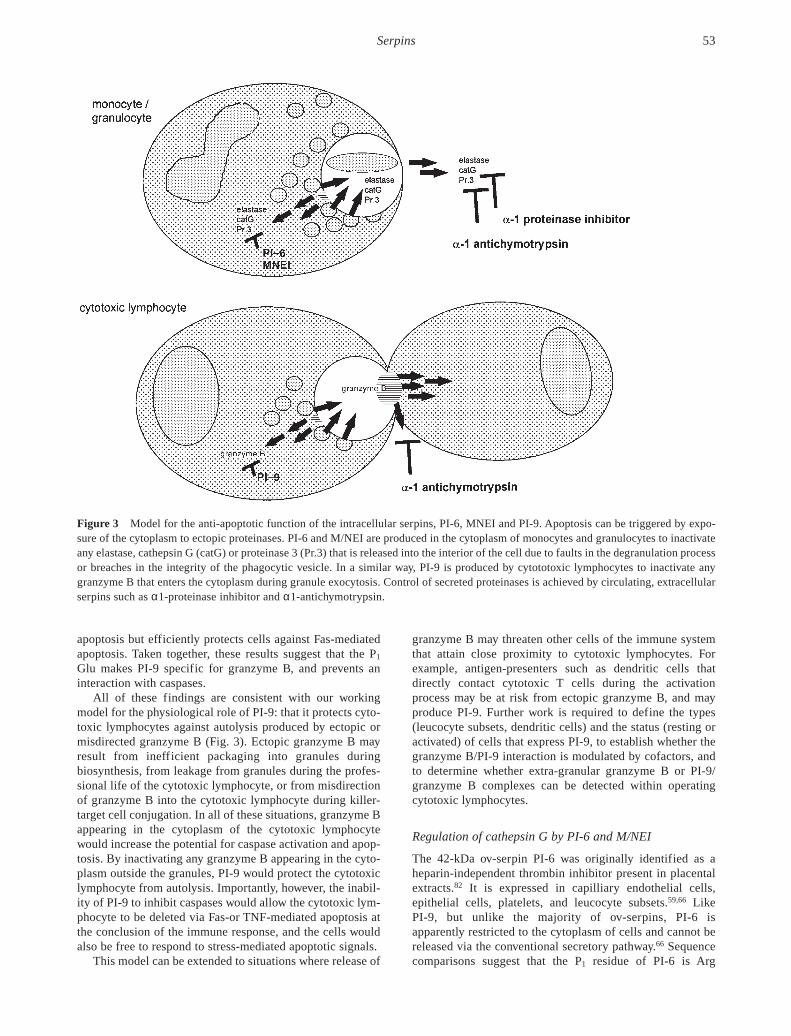
All of these findings are consistent with our workingmodel for the physiological role of PI-9: that it protects cyto-toxic lymphocytes against autolysis produced by ectopic ormisdirected granzyme B (Fig. 3). Ectopic granzyme B mayresult from inefficient packaging into granules duringbiosynthesis, from leakage from granules during the profes-sional life of the cytotoxic lymphocyte, or from misdirectionof granzyme B into the cytotoxic lymphocyte during killer-target cell conjugation. In all of these situations, granzyme Bappearing in the cytoplasm of the cytotoxic lymphocytewould increase the potential for caspase activation and apop-tosis. By inactivating any granzyme B appearing in the cyto-plasm outside the granules, PI-9 would protect the cytotoxiclymphocyte from autolysis. Importantly, however, the inabil-ity of PI-9 to inhibit caspases would allow the cytotoxic lym-phocyte to be deleted via Fas-or TNF-mediated apoptosis atthe conclusion of the immune response, and the cells wouldalso be free to respond to stress-mediated apoptotic signals.
This model can be extended to situations where release of
granzyme B may threaten other cells of the immune systemthat attain close proximity to cytotoxic lymphocytes. Forexample, antigen-presenters such as dendritic cells thatdirectly contact cytotoxic T cells during the activationprocess may be at risk from ectopic granzyme B, and mayproduce PI-9. Further work is required to define the types(leucocyte subsets, dendritic cells) and the status (resting oractivated) of cells that express PI-9, to establish whether thegranzyme B/PI-9 interaction is modulated by cofactors, andto determine whether extra-granular granzyme B or PI-9/granzyme B complexes can be detected within operatingcytotoxic lymphocytes.
Regulation of cathepsin G by PI-6 and M/NEI
The 42-kDa ov-serpin PI-6 was originally identified as aheparin-independent thrombin inhibitor present in placentalextracts.82 It is expressed in capilliary endothelial cells,epithelial cells, platelets, and leucocyte subsets.59,66 Like PI-9, but unlike the majority of ov-serpins, PI-6 is apparently restricted to the cytoplasm of cells and cannot bereleased via the conventional secretory pathway.66 Sequencecomparisons suggest that the P1 residue of PI-6 is Arg
Serpins 53
Figure 3 Model for the anti-apoptotic function of the intracellular serpins, PI-6, MNEI and PI-9. Apoptosis can be triggered by expo-sure of the cytoplasm to ectopic proteinases. PI-6 and M/NEI are produced in the cytoplasm of monocytes and granulocytes to inactivateany elastase, cathepsin G (catG) or proteinase 3 (Pr.3) that is released into the interior of the cell due to faults in the degranulation processor breaches in the integrity of the phagocytic vesicle. In a similar way, PI-9 is produced by cytototoxic lymphocytes to inactivate anygranzyme B that enters the cytoplasm during granule exocytosis. Control of secreted proteinases is achieved by circulating, extracellularserpins such as α1-proteinase inhibitor and α1-antichymotrypsin.

(Fig. 2), which explains the interaction of PI-6 with throm-bin.59 Subsequent analyses have shown that PI-6 is only amoderately effective thrombin inhibitor, and that it inhibitstrypsin and plasmin far more efficiently.83,84 Of particularinterest is a recent finding that PI-6 is also a very goodchymotrypsin inhibitor due to the presence of Met at the P2
position in the reactive centre.85 Thus PI-6 has the capacity toinhibit multiple serine proteinases by utilizing overlappingreactive centres.
The broad distribution, intracellular localization and dualinhibitory capacity of PI-6 suggests that it fulfils an impor-tant regulatory or protective role associated with proteolysiswithin cells, and that it is not a physiological thrombininhibitor. To identify potential targets of PI-6, we examinedits distribution in leucocyte subsets in more detail.85 Weshowed that it is expressed in monocytes and granulocytesbut not lymphocytes, and that an SDS-stable complex isformed between PI-6 and a membrane-associated proteinaseon lysis of primary monocytes or myelomonocytic cell lines.Using antibodies to the major granule serine proteinasesproduced by these cells (elastase, cathepsin G, proteinase 3),we established that the complex contains cathepsin G. ThePI-6–cathepsin G interaction was then confirmed using puri-fied components, and the kinetic parameters were deter-mined. Inhibition of cathepsin G by PI-6 is very rapid (ka
> 107 per mol/L per s) and is optimal at a stoichiometry of 1.This rate is at the upper end of serpin–proteinase interactions,which, taken with the co-compartmentalization of PI-6 andcathepsin G in monocytes and granulocytes, strongly sug-gests that this a physiologically relevant serpin–proteinasepair. However, PI-6 is present in many cells that do notproduce cathepsin G, and it is also a good inhibitor of otherserine proteinases in vitro.66 Thus it is possible that the roleof PI-6 extends beyond the control of cathepsin G, and that italso regulates one or more proteinases that have yet to beidentified.
In considering the physiological role of PI-6, it should benoted that monocytes and neutrophils also produce M/NEI,which is an efficient inhibitor of elastase and to a lesserextent proteinase 3 and cathepsin G.57,87,88 Thus, betweenthem, PI-6 and M/NEI have the potential to complement the extracellular serpins α1-proteinase inhibitor and α1-antichymotrypsin in vivo, and to efficiently regulate the threemajor monocyte/granulocyte granule proteinases.
Why should both extracellular and intracellular inhibitorsof elastase, proteinase 3 and cathepsin G exist? The answer tothis question probably lies in the dual functions of theseproteinases: as enzymes acting within phagocytic vesicles todestroy microbes or other ingested material and as extra-cellular modulators of the inflammatory response. Wepropose that α1-proteinase inhibitor and α1-antichymotrypsinact to contain extracellular collateral damage mediated by thegranule proteinases, while PI-6 and M/NEI act to containintracellular damage occurring as microbes or tissue debrisare ingested and destroyed. If the vacuole integrity isbreached during phagocytosis, any proteinases released intothe interior of the cell would be rapidly inactivated by PI-6and M/NEI (Fig. 3).
Is it likely that the granule proteinases leak into thecytoplasm during phagocytosis? Although no studies havebeen done on monocytes or granulocytes, it is known that
oxidative stress such as exposure to hydrogen peroxide caninduce lysosomal rupturing in cultured fibroblasts, leading tothe release of lysosomal contents, including proteases, intothe cell.89 Although cells can apparently tolerate and repairlysosomal ruptures to a certain degree, as the amount ofdamage increases cells may undergo apoptosis (moderatedamage) or necrosis (substantial damage). Given thatmyeloperoxidase is a key component of azurophilic granules,and that phagocytic vesicles and lysosomes have manystructural similarities, some leakage of phagocytic vesiclecontents due to oxidative stress is likely to occur in mono-cytes, macrophages and neutrophils. To avoid death andremain functional these cells would require vesicle repairmechanisms similar to those described in fibroblasts,89 aswell as the means of countering the potentially cytotoxiccomponents of leaking vesicles, such as cathepsin G andelastase. In addition to countering proteinases leaking fromphagocytic vesicles, PI-6 and M/NEI could also inactivateany proteinases that are misdirected into the cytoplasmduring biosynthesis or degranulation.
Alternatively, PI-6 and M/NEI may form an intracellularpool of inhibitors that can be rapidly released under specificconditions to operate at high local concentrations in the extra-cellular milieu and supplement α1-proteinase inhibitor andα1-antichymotrypsin. However, active release of M/NEIfrom monocytes or neutrophils has not been demonstrated,while we have shown that PI-6 cannot be detected in mediaconditioned by U937 and other cells, and that it cannot bereleased from cells through the classical secretory pathway.66
Perspectives
The potential of most proteinases to hydrolyse a wide varietyof substrates necessitates opposing regulatory mechanisms tomaintain tissue homeostasis. In systems such as the bloodcoagulation pathway, active serine proteinases are inhibitedby serpins. Leucocyte granule serine proteinases are less wellunderstood but can apparently act inside or outside cells. Atleast one granule proteinase, granzyme B, has evolved to actwithin the cytoplasm of target cells to induce apoptosis, butothers also have the potential to induce apoptosis if they enterthe cytoplasm of a cell.
Outside cells the action of the leucocyte granule serineproteinases is limited by circulating inhibitors such as α2-macroglobulin, α1-proteinase inhibitor and α1-antichy-motrypsin. We have identified a family of intracellularov-serpins produced by leucocytes, and propose that theyfunction inside cells to inactivate granule proteinases thatenter the host cell cytoplasm, thus preventing non-specificapoptosis and autolysis. Interestingly, the genes encodingthese intracellular serpins (PI-6, M/NEI, PI-9) are part of acluster on human chromosome 6p25,70 suggesting a closeevolutionary relationship and similarities or redundancy infunction. They are mirrored by the granule proteinase geneswhich have also evolved in clusters.
The concept of intracellular serpins existing to regulatemiscompartmentalized or misdirected proteinase can beextended to suggest roles for other ov-serpins. For example,it is evident that SCCA-1 is an inhibitor of lysosomal cathep-sins,53 suggesting that under situations of stress it may protectcells from the effects of lysosomal breakdown and release of
PI Bird54

these proteinases into the cytoplasm. Likewise, intracellularPAI-2 may protect the interior of monocytes from deleteriouseffects of urokinase, which is known to be efficiently inter-nalized by endocytosis (review90), and potentially releasedinto the cytoplasm.
A key feature of our model that remains to be directlytested is the idea that processes such as granule formation,fusion and exocytosis are not perfect, and that a proportion ofthe granule contents will enter the host cell cytoplasm.However, the existence of PI-9 is consistent with circum-stantial evidence that cytotoxic lymphocytes possess mecha-nisms that protect against granule cytotoxins. This includesthe presence of extra-granular granzyme B in cytotoxiccells,91 and their intrinsic resistance to cytolysis.92–96
We also propose that destruction of ingested material in aphagocytic vesicle involves stresses that may lead to breachesin the integrity of the surrounding membrane. These stressesmay include endogenously generated reactive oxygenspecies, or exposure to endogenous defensins or microbialpore-forming proteins that may rupture vesicles, leading tothe release of their contents into the cytoplasm. We see theintracellular serpins as complementing membrane repairsytems and radical-scavengers in maintaining the viabilityand functionality of immune effector cells. These repair andprotective mechanisms in leucocytes have received very littleattention in the past, and are likely to provide a fruitful areafor future investigation.
Acknowledgements
The author is supported by the National Health and MedicalResearch Council. I thank the present and past members of mylaboratory who have contributed to the work on intracellularserpins, in particular Dr J Sun, Ms C Bird, Ms F Scott, Ms CHirst and Dr P Coughlin. I am also grateful to Dr J Trapani and Dr V Sutton (Austin Research Institute), Dr SKumar (Hanson Centre), Dr S Bottomley and Dr J Whisstock(Monash University), and Dr J Ragoussis (University ofLondon), for their continuing collaborative support andencouragement.
References
1 Furie B, Furie BC. Molecular and cellular biology of bloodcoagulation. N. Engl. J. Med. 1992; 326: 800–6.
2 Bird PI. Activation and function of protein C and protein S occuras part of a complex network regulating hemostasis and inflam-mation. Hematol. Rev. 1996; 9: 251–74.
3 Vaux DL, Strasser A. The molecular biology of apoptosis. Proc.Natl Acad. Sci.USA 1996; 93: 2239–44.
4 Yang E, Korsmeyer SJ. Molecular thanatopsis: A discourse onthe BCL2 family and cell death. Blood 1996; 88: 386–401.
5 Sheridan JP, Marsters SA, Pitti RM et al. Control of TRAIL-induced apoptosis by a family of signaling and decoy receptors.Science 1997; 277: 818–21.
6 Irmler M, Thome M, Hahne M et al. Inhibition of death recep-tor signals by cellular FLIP. Nature 1997; 388: 190–5.
7 Zhou Q, Snipas S, Orth K, Muzio M, Dixit V, Salvesen G. Targetprotease specificity of the viral serpin CrmA. J. Biol. Chem.1997; 272: 7797–800.
8 Roy N, Deveraux QL, Takahashi R, Salvesen GS, Reed JC. Thec-IAP-1 and c-IAP-2 proteins are direct inhibitors of specific
caspases. EMBO J. 1997; 16: 6914–25.9 Nagata S. Apoptosis by death factor. Cell 1997; 88: 355–65.
10 Smyth MJ, Trapani JA. Granzymes: Exogenous proteinases thatinduce target cell apoptosis. Immunol. Today 1995; 16: 202–6.
11 Trapani JA. Dual mechanisms of apoptosis induction by cyto-toxic lymphocytes. Int. Rev. Cytol. 1998; 182: 111–92.
12 Darmon AJ, Ley TJ, Nicholson DW, Bleackley RC. Cleavage ofCPP32 by granzyme B represents a critical role for granzyme Bin the induction of target cell DNA fragmentation. J. Biol. Chem.1996; 271: 21709–12.
13 Froelich CJ, Orth K, Turbov J et al. New paradigm for lympho-cyte granule mediated cytotoxicity: Target cells bind and inter-nalize granzyme B but an endosomolytic agent is necessary forcytosolic delivery and subsequent apoptosis. J. Biol. Chem.1996; 271: 29073–9.
14 Williams MS, Henkart PA. Apoptotic cell death induced byintracellular proteolysis. J. Immunol. 1994; 153: 4247–55.
15 Caughey GH. Serine proteinases of mast cell and leukocytegranules. A league of their own. Am. J. Respir. Crit. Care Med.1994; 150: S138–42.
16 Polanowska J, Krokosynska I, Czapinska H, Watorek W, DadlezM, Otlewski J. Specificity of human cathepsin G. Biochim.Biophys. Acta 1998; 1386: 189–98.
17 Ebnet K, Hausmann M, Lehmann-Grube F et al. Granzyme A-deficient mice retain potent cell-mediated cytotoxicity.EMBO J. 1995; 14: 4230–9.
18 Heussel JW, Wesselschmidt RL, Shresta S, Russell JH, Ley TJ.Cytotoxic lymphocytes require granzyme B for the rapidinduction of DNA fragmentation and apoptosis in allogeneictarget cells. Cell 1994; 76: 977–87.
19 Belaaouaj A, McCarthy R, Baumann M et al. Mice lackingneutrophil elastase reveal impaired host defense against gramnegative bacterial sepsis. Nature Med. 1998; 4: 615–18.
20 Nicholson DW, Thornberry NA. Caspases: Killer proteases.Trends Biochem. Sci. 1997; 22: 299–306.
21 Darmon AJ, Bleackley RC. Proteases and cell-mediated toxicity.Crit. Rev. Immunol. 1998; 18: 255–73.
22 Pinkoski MJ, Hobman M, Heibein JA et al. Entry and traffick-ing of granzyme B in target cells during granzyme B-perforin-mediated apoptosis. Blood 1998; 92: 1044–54.
23 Mullbacher A, Ebnet K, Blanden RV et al. Granzyme A iscritical for recovery of mice from infection with the naturalcytopathic viral pathogen, ectromelia. Proc. Natl Acad. Sci. USA1996; 93: 5783–7.
24 Borregaard N, Cowland JB. Granules of the human neutrophilicpolymorphonuclear leukocyte. Blood 1997; 89: 3503–21.
25 Spitznagel JK. Antibiotic proteins of human neutrophils. J. Clin.Invest. 1990; 86: 1381–6.
26 Lehrer RI, Ganz T. Antimicrobial polypeptides of humanneutrophils. Blood 1990; 76: 2169–81.
27 Rao NV, Rao GV, Marshall BC, Hoidal JR. Biosynthesis andprocessing of proteinase 3 in U937 cells. J. Biol. Chem. 1996;271: 2972–8.
28 Malech HL, Nauseef WM. Primary inherited defects in neu-trophil function: Etiology and treatment. Semin. Hematol. 1997;34: 279–90.
29 Padrines M, Wolf M, Walz A, Baggiolini M. Interleukin-8processing by neutrophil elastase, cathepsin G and proteinase 3.FEBS Lett. 1994; 352: 231–5.
30 Chertov O, Ueda H, Xu LL et al. Identification of human neu-trophil-derived cathepsin G and azurocidin/CAP37 as chemo-attractants for mononuclear cells and neutrophils. J. Exp. Med.1997; 186: 739–47.
31 Evangelista V, Rajtar G, de Gaetano G, White JG, Cerletti C.
Serpins 55

Platelet activation by fMLP-stimulated polymorphonuclearleukocytes: The activity of cathepsin G is not prevented byantiproteinases. Blood 1991; 77: 2379–88.
32 Janoff A, Feinstein G, Malemud CJ, Elias JM. Degradation ofcartilage proteoglycan by human leukocyte granule neutral pro-teinases: A model of joint injury. I. Penetration of enzyme intorabbit articular cartilage and release of 35SO4-labeled materialfrom the tissue. J. Clin. Invest. 1976; 57: 615–24.
33 McDonald JA, Kelley DG. Degradation of fibronectin byhuman leukocyte elastase. J. Biol. Chem. 1980; 255: 8848–58.
34 Gadek JE, Fells GA, Wright DG, Crystal RG. Human neutrophilelastase functions as a type III collagen ‘collagenase’. Biochem.Biophys. Res. Commun. 1980; 95: 1815–22.
35 Mainardi CL, Dixit SN, Kang AH. Degradation of type IV(basement membrane) collagen by a proteinase isolated fromhuman polymorphonuclear leukocyte granules. J. Biol. Chem.1980; 255: 5435–41.
36 Janoff A. Elastase in tissue injury. Ann. Rev. Med. 1985; 36:2207–16.
37 Lee CT, Fein AM, Lippman M, Holtzman H, Kimbel P, Wein-baum G. Elastolytic activity in pulmonary lavage fluid frompatients with adult respiratory distress syndrome. N. Engl. J.Med. 1981; 304: 192–6.
38 Bruce MC, Poncz L, Klinger JD, Stern RC, Tomashefski JF,Dearborn DG. Biochemical and pathologic evidence forproteolytic destruction of lung connective tissue in cysticfibrosis. Am. Rev. Respir. Dis. 1985; 132: 529–35.
39 Weiss SJ. Tissue destruction by neutrophils. N. Engl. J. Med.1989; 320: 365–76.
40 Laurell C-B, Eriksson S. The electrophoretic alpha1-globulinpattern of serum in alpha1-antitrypsin deficiency. Scand. J. Clin.Lab. Invest. 1963; 15: 132–40.
41 Zhou Q, Salvesen GS. Activation of pro-caspase-7 by serineproteases includes a non-canonical specificity. Biochem. J.1997; 324: 361–4.
42 Potempa J, Korzus E, Travis J. The serpin superfamily ofproteinase inhibitors: Structure, function and regulation. J. Biol.Chem. 1994; 269: 15 957–60.
43 Stein PE, Carrell RW. What do dysfunctional serpins tell us aboutmolecular mobility and disease. Struct. Biol. 1995; 2: 96–113.
44 Patston PA, Gettins PGW, Schapira M. Serpins are suicidesubstrates: Implications for the regulation of proteolytic path-ways. Semin. Thromb. Hemost. 1994; 20: 410–16.
45 Travis J, Salveson GS. Human plasma proteinase inhibitors.Ann. Rev. Biochem. 1983; 52: 655–709.
46 Jin L, Abrahams JP, Skinner R, Petitou M, Pike RN, Carrell RW.The anticoagulant activation of antithrombin by heparin. Proc.Natl Acad. Sci. USA 1998; 94: 14 683–8.
47 Whisstock J, Skinner R, Lesk AM. An atlas of serpin conform-ations. Trends Biochem. Sci. 1998; 23: 63–7.
48 Wilczynska M, Fa M, Karolin J, Ohlsson P-I, Johansson LB-A,Ny T. Structural insights into serpin-protease complexes revealthe inhibitory mechanism of serpins. Nat. Struct. Biol. 1997; 4:354–7.
49 Stratikos E, Gettins PW. Major proteinase movement uponstable serpin-proteinase complex formation. Proc. Natl Acad.Sci. USA 1997; 94: 453–8.
50 Gagliardini V, Fernandez P-A, Lee RK et al. Prevention ofvertebrate neuronal cell death by the crmA gene. Science 1994;263: 826–8.
51 Miura M, Zhu H, Rotello R, Hartwieg EA, Yuan J. Induction ofapoptosis in fibroblasts by IL-1 beta-converting enzyme, amammalian homologue of the C.elegans cell death gene ced-3.Cell 1993; 75: 653–60.
52 Remold-O’Donnell E. The ovalbumin family of serpin proteins.FEBS Lett. 1993; 315: 105–8.
53 Schick C, Pemberton PA, Shi G-P et al. Cross-class inhibition ofthe cysteine proteinases cathepsins K, L, and S by the serpinsquamous cell carcinoma antigen 1: A kinetic analysis.Biochemistry 1998; 37: 5258–66.
54 Ye RD, Ahern SM, Le Beau MM, Lebo RV, Sadler JE. Structureof the gene for human plasminogen activator inhibitor-2. Thenearest mammalian homologue of chicken ovalbumin. J. Biol.Chem. 1989; 264: 5495–502.
55 Suminami Y, Kishi F, Sekiguchi K, Kato H. Squamous cellcarcinoma antigen is a new member of the serine proteaseinhibitors. Biochem. Biophys. Res. Commun. 1991; 181: 51–8.
56 Schneider SS, Schick C, Fish KE et al. A serine proteinaseinhibitor locus at 18q21.3 contains a tandem duplication of thehuman squamous cell carcinoma antigen gene. Proc. Natl Acad.Sci. USA 1995; 92: 3147–51.
57 Remold-O’Donnell E, Chin J, Alberts M. Sequence and molec-ular characterisation of human monocyte/neutrophil elastaseinhibitor. Proc. Natl Acad. Sci. USA 1992; 89: 5635–9.
58 Zou Z, Anisowicz A, Hendrix MJC et al. Maspin, a serpin withtumor-suppressing activity in human mammary epithelial cells.Science 1994; 263: 526–9.
59 Coughlin P, Sun J, Cerruti L, Salem HH, Bird P. Cloning andmolecular characterization of a human intracellular proteinaseinhibitor. Proc. Natl Acad. Sci. USA 1993; 90: 9417–21.
60 Sprecher CA, Morgenstern KA, Mathewes S et al. Molecularcloning, expression, and partial characterization of two novelmembers of the ovalbumin family of serine proteinaseinhibitors. J. Biol. Chem. 1995; 270: 29 854–61.
61 Sun J, Bird CH, Sutton V et al. A cytosolic granzyme B inhibitorrelated to the viral apoptotic regulator cytokine responsemodifier A is present in cytotoxic lymphocytes. J. Biol. Chem.1996; 271: 27 802–9.
62 Riewald M, Schleef RR. Molecular cloning of bomapin (pro-tease inhibitor 10), a novel human serpin that is expressedspecifically in the bone marrow. J. Biol. Chem. 1995; 270: 26754–7.
63 Tsujimoto M, Tsuruoka N, Ishida N et al. Purification, cDNAcloning, and characterization of a new serpin with megakaryo-cyte maturation activity. J. Biol. Chem. 1997; 272: 15 373–80.
64 Bartuski AJ, Kamachi Y, Schick C, Overhauser J, Silverman GA.Cytoplasmic antiproteinase 2 (PI8) and bomapin (PI10) map tothe serpin cluster at 18q21.3. Genomics 1997; 43: 321–8.
65 Coughlin P, Nicholl J, Sun J, Salem H, Bird P, Sutherland G.Chromosomal mapping of the human proteinase inhibitor 6(PI6) gene to 6p25 by fluorescence in situ hybridization.Genomics 1995; 26: 431–3.
66 Scott FL, Coughlin PB, Bird C, Cerruti L, Hayman JA, Bird P.Proteinase inhibitor 6 cannot be secreted, which suggests it is anew type of cellular serpin. J. Biol. Chem. 1996; 271: 1605–12.
67 Ooms L, Nicholl J, Bird P, Sutherland GR. Localization of thehuman monocyte/neutrophil elastase inhibitor gene to chromo-some 6p25. Chromosome Res. 1995; 3: 447.
68 Evans E, Cooley J, Remold-O’Donnell E. Characterization andchromosomal localization of ELANH2, the gene encodinghuman monocyte/neutrophil elastase inhibitor. Genomics 1995;28: 235–40.
69 Eyre HJ, Sun J, Sutherland GR, Bird P. Chromosomal mappingof the gene (PI9) encoding the intracellular serpin proteinaseinhibitor 9–6p25 by fluorescence in situ hybridization.Genomics 1996; 37: 406–8.
70 Sun J, Stephens R, Mirza G, Kanai H, Ragoussis J, Bird PI. Aserpin gene cluster on human chromosome 6p25 contains PI6,
PI Bird56

Serpins 57
PI9 and ELANH2 which have a common structure almost iden-tical to the 18q21 ovalbumin serpin genes. Cytogenet. CellGenet. 1998 (in press).
71 Belin D. Biology and facultative secretion of plasminogenactivator inhibitor-2. Thromb. Haemost. 1993; 70: 144–7.
72 Tschopp J, Thome M, Hofman K, Meinl E. The fight of virusesagainst apoptosis. Curr. Opin. Genet. Dev. 1998; 8: 82–7.
73 Pickup DJ. Poxviral modifiers of cytokine responses to infec-tion. Infect. Agents Dis. 1994; 3: 116–27.
74 Ray CA, Black RA, Kronheim SR et al. Viral inhibition ofinflammation: Cowpox virus encodes an inhibitor of the inter-leukin-1-beta converting enzyme. Cell 1992; 69: 597–604.
75 Komiyama T, Ray CA, Pickup DJ et al. Inhibition of interleukin-1beta converting enzyme by the cowpox virus serpin crmA. J.Biol. Chem. 1994; 269: 19 331–7.
76 Quan LT, Caputo A, Bleackley RC, Pickup DJ, Salvesen GS.Granzyme B is inhibited by the cowpox virus serpin cytokineresponse modifier A. J. Biol. Chem. 1995; 270: 10 377–9.
77 Tewari M, Dixit VM. Fas- and tumor necrosis factor-inducedapoptosis is inhibited by the poxvirus crmA gene product. J.Biol. Chem. 1995; 270: 3255–60.
78 Macen JL, Garner RL, Musy PY et al. Differential induction ofthe Fas- and granule-mediated cytolysis pathways by theorthopoxvirus cytokine response modifierA/SPI-2 and SPI-1protein. Proc. Natl Acad. Sci. USA 1996; 93: 9108–13.
79 Atkinson EA, Barry M, Darmon AJ et al. Cytotoxic T lympho-cyte-assisted suicide. Caspase 3 activation is primarily the resultof the direct action of granzyme B. J. Biol. Chem. 1998; 273: 21261–6.
80 Odake S, Kam CM, Narasimhan L et al. Human and murinecytotoxic T lymphocyte serine proteases: Subsite mapping withpeptide thioester substrates and inhibition of enzyme activityand cytolysis by isocoumarins. Biochemistry 1991; 30: 2217–27.
81 Bird CH, Sutton VR, Sun J et al. Selective regulation of apop-tosis: The cytotoxic lymphocyte serpin proteinase inhibitor 9protects against granzyme B-mediated apoptosis without per-turbing the Fas cell death pathway. Mol. Cell. Biol. 1998; 18:6387–98.
82 Coughlin PB, Tetaz T, Salem HH. Identification and purificationof a novel serine proteinase inhibitor. J. Biol. Chem. 1993; 268:9541–7.
83 Sun J, Coughlin P, Salem H, Bird P. Production and characteri-zation of recombinant human proteinase inhibitor 6 expressed inPichia pastoris. Biochim. Biophys. Acta 1995; 1252: 28–34.
84 Morgenstern KA, Sprecher C, Holth L et al. Complementary
DNA cloning and kinetic characterization of a novel intra-cellular serine proteinase inhibitor: Mechanism of action withtrypsin and factor Xa as model proteinases. Biochemistry 1994;33: 3432–41.
85 Scott FL, Hirst CE, Sun J, Bottomley SP, Bird CH, Bird PI. Theintracellular serpin proteinase inhibitor 6 (PI-6) is expressed inmonocytes and granulocytes and is a potent inhibitor of theazurophilic granule protease, cathepsin G. Blood (in press).
86 Riewald M, Schleef RR. Human cytoplasmic antiproteinaseneutralizes rapidly and efficiently chymotrypsin and trypsin-likeproteinases utilizing distinct reactive site residues. J. Biol.Chem. 1996; 271: 14 526–32.
87 Remold-O’Donnell E, Nixon JC, Rose RM. Elastase inhibitor.Characterization of the human elastase inhibitor moleculeassociated with monocytes, macrophages, and neutrophils. J.Exp. Med. 1989; 169: 1071–86.
88 Sugimori T, Cooley J, Hoidal JR, Remold-O’Donnell E.Inhibitory properties of recombinant monocyte/neutrophilelastase inhibitor. Am. J. Respir. Cell Mol. Biol. 1995; 13:314–22.
89 Brunk UT, Dalen H, Roberg K, Hellquist HB. Photo-oxidativedisruption of lysosomal membranes causes apoptosis of culturedhuman fibroblasts. Free Radic. Biol. Med. 1997; 23: 616–26.
90 Dear AE, Medcalf RL. The cellular and molecular biology ofplasminogen activator inhibitor type-2. Fibrinolysis 1995; 9:321–30.
91 Trapani JA, Smyth MJ, Apostolidis VA, Dawson M, Browne K.Granule serine proteinases are normal nuclear constituents ofnatural killer cells. J. Biol. Chem. 1994; 269: 18 359–65.
92 Nagler-Anderson C, Verret CR, Firmenich AA, Berne M, EisenHN. Resistance of primary CD8+ cytotoxic T lymphocytes tolysis by cytotoxic granules from cloned T cell lines. J. Immunol.1988; 141: 3299–305.
93 Golstein P. Sensitivity of cytotoxic T cells to T-cell mediatedcytotoxicity. Nature 1974; 252: 81–3.
94 Kranz DM, Eisen HN. Resistance of cytotoxic lymphocytes tolysis by a clone of cytotoxic T lymphocytes. Proc. Natl Acad.Sci. USA 1987; 84: 3375–9.
95 Blakely A, Gorman K, Ostergaard H et al. Resistance of clonedcytotoxic T lymphocytes to cell-mediated cytotoxicity. J. Exp.Med. 1987; 166: 1070–83.
96 Liu C-C, Jiang S, Persechini PM, Zychlinsky A, Kaufmann Y,Young JD. Resistance of cytolytic lymphocytes to perforin-mediated killing. Induction of resistance correlates with increasein cytotoxicity. J. Exp. Med. 1989; 169: 2211–25.


















