
Perfil transcriptómico y proteómico de MCF7, CCD18 y Jurkat .
Upload
frank-schmidtCategory
view
217download
4
Frank Schmidt1
Hanne K. Hustoft1
Margarita Strozynski1
Christiane Dimmler2
Thomas Rudel2
Bernd Thiede1
1The BiotechnologyCentre of Oslo,University of Oslo,Oslo, Norway
2Max Planck Institutefor Infection Biology,Berlin, Germany
Received February 16, 2007Revised May 7, 2007Accepted July 24, 2007
Research Article
Quantitative proteome analysis ofcisplatin-induced apoptotic Jurkat T cellsby stable isotope labeling withamino acids in cell culture, SDS-PAGE,and LC-MALDI-TOF/TOF MS
Quantitative proteome analysis of cisplatin-induced apoptosis in total Jurkat Tcell lysates wasperformed in order to identify modified proteins. Proteins were labeled in cell culture withstable isotopes of arginines, and fractionated by SDS-PAGE. Subsequently, tryptic peptideswere analyzed by nano-LC coupled offline to MALDI-TOF/TOF-MS as an alternative tocommonly used online LC-ESI-MS. As a result, 26 proteins were found with a relative abun-dance higher than 1.5, thereof 19 already known and seven unknown to be involved inapoptosis (adenine phosphoribosyltransferase, microsomal signal peptidase 25 kDa sub-unit, phosphomevalonate kinase, probable rRNA processing protein EBP2, RNA-bindingprotein 4, transmembrane protein 33, and tetratricopeptide repeat domain 9C). Immuno-blotting of core-binding factor beta and elongation factor 2 revealed similar quantitativechanges as detected by the SILAC-based proteomics approach. Strikingly, 8 of 26 identifiedapoptosis-modified proteins contained at least one RNA-binding motif. Three caspase cleav-age sites of the 54 kDa nuclear RNA-binding protein (p54nrb) were mapped at DQLD231;D,DQVD286;R, and MMPD422;G by applying caspase-3 to the in vitro translated protein andmutation analysis. The determined caspase cleavage sites were located C-terminal to the twoRNA-binding motifs and one (DQLD231;D) within the NOPS domain of p54nrb. Concisely,quantitative protein data generated by offline LC-MALDI-MS were shown to be particularlyaccurate. Furthermore, only regulated peptides were selected in a result-dependent mannerfor MS/MS analyses and revealed novel apoptosis-modified proteins.
Keywords:
Apoptosis / Cisplatin / LC-MALDI / p54nrb / Stable isotope labeling with aminoacids in cell culture DOI 10.1002/elps.200700119
Electrophoresis 2007, 28, 4359–4368 4359
1 Introduction
DNA damage and subsequent induction of apoptosis is pos-sibly the primary cytotoxic mechanism induced by cisplatinand other DNA-binding anticancer drugs [1]. Apoptosis is aform of programmed cell death and can be triggered in a cell
through either the extrinsic or the intrinsic pathway. Theextrinsic pathway is initiated by the binding of a ligand to adeath receptor. For example, interaction of the CD95 (Fas/Apo-1) receptor with CD95 ligand (FasL/Apo-1L) triggers theformation of the death-inducing signaling complex (DISC)that includes the adaptor protein FADD, which in turnrecruits procaspase-8. Aggregation of procaspase-8 releasesactive caspase-8 and this results in the activation of eithereffector caspases or the mitochondrial pathway via Bid cleav-age [2]. The intrinsic apoptotic pathway can be induced as aresponse to stress such as heat shock and DNA damage. Cen-tral to the intrinsic apoptotic pathway is the mitochondria andthe Bcl-2 protein family which function as regulator of cas-pase activation by both proapoptotic and mitochondria-asso-ciated antiapoptotic proteins [3]. Proapoptotic Bcl-2 familymembers in the cytoplasm, e.g., Bax and Bid, bind to the outermembrane of the mitochondria and promote the transloca-tion of cytochrome c into the cytoplasm. Following the release,
Correspondence: Dr. Bernd Thiede, The Biotechnology Centre ofOslo, University of Oslo, Gaustadalleen 21, P.O. Box 1125 Blin-dern, 0317 Oslo, NorwayE-mail: [email protected]: 147-22840501
Abbreviations: hnRNP, heterogeneous nuclear ribonucleopro-tein; NOPS, C-terminal domain of NONA and PSP1 proteins;PARP, poly(ADP-ribose) polymerase; RNP, ribonucleoprotein;SILAC, stable isotope labeling with amino acids in cell culture;Z-VAD-fmk, Z-Val-Ala-DL-Asp-fluoromethylketone
© 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.electrophoresis-journal.com

4360 F. Schmidt et al. Electrophoresis 2007, 28, 4359–4368
cytochrome c forms a complex in the cytoplasm with Apaf-1,dATP, and procaspase-9, the apoptosome, which activatescaspase-9 and enables activation of effector caspases. Fur-thermore, other proteins are translocated from the mito-chondria to the cytoplasm during activation of the intrinsicpathway as Smac/Diablo to block the inhibitor of apoptosisproteins (IAP). Cellular responses to nuclear DNA damageare either cell cycle arrest to facilitate DNA repair or inductionof apoptosis [4]. These processes are induced through a net-work of interacting signal transduction pathways [5]. NuclearDNA damage can be signaled to the mitochondria by p53,histone H1.2, Nurr77, Daxx, FADD, and caspase-2 activation[1]. Certainly, the tumor suppressor p53 plays a fundamentalrole for the cellular response to DNA damage [6].
Cis-diamminedichloroplatinum(II) (cisplatin) is one ofthe most potent anticancer agents used in human che-motherapy [7]. The antitumor activity of cisplatin is generallyattributed to the generation of DNA damage by intrastrandand interstrand cross-links [8]. The DNA adducts of cisplatinare thought to mediate their cytotoxic effects by inhibitingDNA replication and transcription and ultimately lead to celldeath by apoptosis [1]. However, the mechanism by whichcisplatin-induced DNA damage leading to the characteristiccytotoxic effects is not fully understood. Furthermore, cis-platin has a number of side effects, such as neurotoxicity andnephrotoxicity, and presence or acquisition of resistancelimits its use [9]. Thus, several proteome analyses with dif-ferent cell lines were performed to identify proteins asso-ciated with resistance to cisplatin [10–14].
Several proteomic approaches were applied to study pro-teins involved in apoptosis [15]. The proteome analysesenabled the identification of more than 100 different apop-tosis-modified proteins using different apoptosis-inducingagents, cells, and proteome approaches. Thereby, mainlyprotein degradation was observed beside translocation,altered synthesis rates and other modifications. Most of theidentified proteins were unknown to be involved in apopto-sis. However, cleavage of many of these proteins by caspaseswas determined by COFRADIC [16] or after in vitro transla-tion and treatment with recombinant caspase-3 [17].
MS-based methods using stable isotopes for quantitativeproteome analysis have been developed significantly inrecent years [18]. Stable-isotope labeling with amino acids incell culture (SILAC) has proven to be a simple and powerfulapproach for metabolic labeling [19]. SILAC involves grow-ing of one cell population in normal medium (light) andanother in medium containing specific heavy isotopic aminoacids. These cell populations can then be subjected to differ-ent stimuli and combined prior to further purification. Dur-ing analysis by MS, relative peak intensities between unla-beled and labeled peptides are quantitative, enabling to eval-uate the effects of specific treatment on a large number ofproteins within a single experiment. Apoptosis was alreadystudied by SILAC-based quantitative proteome analysis ofCD95 (Fas/Apo-1)-induced apoptosis in combination with2-DE and MALDI-MS of total cell lysates [20] and by SDS-
PAGE and m-LC-MS/MS of mitochondrial and nuclear pro-teins [21, 22].
In this report, apoptotic cell death was induced by cisplatinin arginine-labeled proteins of Jurkat Tcells. Quantitative pro-teome analysis of control versus apoptotic cells was performedby SDS-PAGE, tryptic digestion of 56 gel lanes, separation ofthe generated peptides by nano-LC which were spotted onto aMALDI target, and their relative quantification and identifica-tion by MALDI-TOF/TOF-MS. In addition, immunoblottingwas performed to confirm quantitative values received by theSILAC approach. One of the identified proteins, p54nrb (alsoknown as NonO and nuclear matrix protein NMT55), is anRNA-binding protein with multiple functions [23]. Pre-dominately RNA-binding proteins were identified in proteomeanalyses of CD95 (Fas/Apo1)-induced apoptosis [16, 17, 24].Recently, p54nrb was identified in the TLE1 corepressor com-plex together with actin, DNA topoisomerase II, heat shockprotein 70, myosin heavy chain 10, nucleolin, nucleophosmin,PARP-1, and Rad50 [25]. Most of these proteins are known tobe involved in apoptosis [26] and many of them were identifiedby proteome analysis of CD95 (Fas/Apo-1)-induced apoptosis[27]. Therefore, p54nrb was studied in more detail to deter-mine caspase cleavage sites precisely.
2 Materials and methods
2.1 Cell culture, SILAC, and induction of apoptosis
The Jurkat T cell line E6 was maintained in MEM tissue cul-ture medium without arginine (PromoCell, Heidelberg,Germany) supplemented with 10% dialyzed fetal calf serum(Gibco BRL, Karlsruhe, Germany), penicillin (100 U/mL)/streptomycin (100 mg/mL) (Gibco BRL) at 377C in 5.0% CO2
and 126 mg/L mg of arginine-12C6 monohydrochloride(light) or arginine-13C6 monohydrochloride (heavy) (euriso-top GmbH, Saarbrücken, Germany). Four population dou-blings of the cells were performed to check the incorporationof arginine by SDS-PAGE and PMF of five gel bands. Afterfive cell population doublings, apoptosis was induced in26106 Jurkat T cells grown in heavy-arginine containingmedium for 16 h at 377C in 5.0% CO2 by 60 mM cisplatin(Sigma–Aldrich, Steinheim, Germany).
2.2 SDS-PAGE
Total lysates of control and apoptotic cells containing equalamounts of protein were pooled. SDS-PAGE was performedwith 15% w/v acrylamide and 0.2% bisacrylamide for proteinseparation and was stained with CBB G-250 (Serva, Heidel-berg, Germany).
2.3 2-DE
Total lysates of control and apoptotic cells containing equalamounts of protein were pooled. Subsequently, a large-gel
© 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.electrophoresis-journal.com

Electrophoresis 2007, 28, 4359–4368 Proteomics and 2-DE 4361
2-DE technique (gel size 30 cm623 cm) was performed forprotein separation as described [28]. Briefly, IEF rod gelswere used for the first dimension with a diameter of 0.9 mmfor analytical gels and 1.5 mm for preparative gels. SDS-PAGE gels with 15% w/v acrylamide and 0.2% bisacrylamidewere used for the second dimension. Preparative gels werestained with CBB G-250 (Serva) and analytical gels werestained with silver nitrate.
2.4 LC-MALDI-TOF/TOF MS
The CBB G-250-stained single gel lanes were excised for in-gel digestion with 0.1 mg of trypsin (Promega, Madison, WI,USA) in 20 mL 25 mM ammonium bicarbonate, pH 7.8 at377C for 16 h. The purified and dried peptides were dissolvedin 6 mL 0.1% TFA in water. An UltiMate™ 3000 nano-LC sys-tem (LC-Packings/Dionex, CA, USA) equipped with a FLM3X00 (integrated flow manager) and microvacuum degasserwas used for peptide separation. The peptides were loadedonto a C18 PepMap 100 column (3.0 mm, 150 mm675 mm)for gradient elution (eluent A, 0.05% TFA in water and 5%ACN; eluent B: 0.05% TFA in 95% ACN) from 0 to 50% elu-ent B within 40 min and for further 5 min from 50 to 90%eluent B. After 15 min, the eluted sample fractions werecontinuously diluted with 0.5 mL/min a-cyano-4-hydro-xycinnamic acid (CHCA) and spotted onto a MALDI targetusing a Probot (LC-Packings/Dionex) with an interval of 30 sresulting in 60 fractions for each gel slice. MALDI-TOF-MSspectra were acquired using an ULTRAFLEX II MALDI-TOF/TOF (Bruker Daltonics, Bremen, Germany) after exter-nal calibration. Peak lists were created from the raw data byFlexAnalysis 2.4 (Bruker Daltonics) using the following peakfilter settings: mass-range 900–4000 Da (MS), and minimalS/N 5. Only MS/MS spectra of preselected peaks (out ofpeaks pairs with a mass difference of 6.02 or 12.04 Da, and arelative ratio exceeding factor 1.5) after smoothing and base-line subtractions were generated automatically by theWARP-LC 1.0 program (Bruker Daltonics). The MS/MSpeptide identifications were achieved by database compar-isons (SwissProt database, 20050621, human, 12359sequences) using BioTools 3.0 (Bruker Daltonics) in con-junction with the MASCOT in-house version 2.0.0. A toler-ance of 100 ppm was used for the precursor mass and 0.4 Dafor MS/MS fragments. Furthermore, trypsin was selected asenzyme considering up to one missed cleavage site and vari-able protein modifications were allowed, such as SILAC-Arg-13C6, SILAC-Pro-13C5, Met-oxidation, and N-acetylation.
2.5 MALDI-MS-based quantitative proteome analysis
The summary report of MASCOT search results were parsedby the WARP-LC program and combined with the ratio cal-culations. The ratios between labeled and unlabeled peptideswith a shift of 6.02 Da for arginine and 5.02 Da for proline-containing peptides (as a side product of arginine biosyn-thesis) were calculated with a tolerance of 0.04 Da. Proteins
were considered to be modified if at least two tryptic peptidepairs with an ion score of at least 20 and a relative ratioexceeding factor 1.5 was found with a total MASCOTscore ofat least 53. All MS data of modified proteins were verifiedmanually.
2.6 Immunoblotting
Protein samples were separated on SDS-PAGE and trans-ferred to PVDF membranes (Immobilon P, Millipore, Oslo,Norway) using a Mini Trans-Blot cell (BioRad, Munich, Ger-many). Affinity-purified mouse or rabbit antibodies againstPARP-1 (MedProbe, Oslo, Norway), core-binding factor beta(BD Norge AS, Trondheim, Norway), and elongation factor 2(MedProbe) were used at 1/2000, 1/500, and 1/1000 dilution,respectively. Anti-GAPDH rabbit mAb (MedProbe) was usedat 1/10 000 dilution after stripping of the PVDF membraneswith 0.2 M glycin/HCl pH 2.4, 0.1% SDS, and 1% Tween 20and washing twice with 0.02% Tween 20 in TBS to confirmloading of SDS-PAGE lanes. Blots were exposed to horse-radish peroxidase (HRP) conjugated anti-mouse or anti-rabbitIgG (Promega), diluted 1/10 000, and developed by Super-Signal West Pico Chemiluminescent (Diagen AS, Rygge,Norway). The gel image software program TopSpot (freedownload at http://web.mpiib-berlin.mpg.de/pdbs/2d-page/downloads.html) was employed to quantify protein signals.
2.7 Preparation of cDNAs and cloning
The cDNAs were prepared by a combination of reverse tran-scription and PCR techniques (RT-PCR). Reverse transcrip-tion was performed on poly(A)1 RNA from the human Jur-kat T cell line using oligo-dT primer. The reverse transcrip-tion products were then used as templates for PCR usingspecific primers according to the Di-/Trinucleotide sticky end(DI/TRISEC) cloning method (Roche Diagnostics GmbH,Mannheim, Germany). Primer sequences of p54nrb were 5’-primer TCGAGAGTAATAAAACTTTTAAC, and 3’-primerCTGGTATCGGCGACGTTTGTTTGGG. The PCR productsas well as the cloning vector pET28c (Calbiochem-Nova-biochem GmbH, Bad Soden, Germany) were treated accord-ing to the Di-/Trinucleotide sticky end (DI/TRISEC) cloningmethod as described by the manufacturer (Roche Diag-nostics GmbH) and then ligated. The cloned cDNAs weresequenced with T3 and T7 sequencing primers.
2.8 In vitro mutagenesis
The p54nrb plasmid was used as template in a PCR per-formed with the mutation primers D422N, 5’-primer P-AACGGAACTTTGGGATTGACCCCA, and 3’-primer P-TTCGGCATCATAGTGGCAGGTCCT; D286N, 5’-primer P-AACCGCAACATCAAGGAGGCTCGTGA, and 3’-primer P-TTCACTTGGTCCTGCTGCTGCTTCT; D231N, 5’-primer P-AACGATGAAGAGGGACTTCCAGAGA, and 3’-primer P-TTCAACTGGTCCATGGGCTTCACAGT. The PCR frag-
© 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.electrophoresis-journal.com

4362 F. Schmidt et al. Electrophoresis 2007, 28, 4359–4368
ment ends were trimmed to short complementary ends bythe controlled 3’–5’exonuclease activity of T4 DNA poly-merase in the presence of stopping nucleotides, and thenreligated and cloned.
2.9 In vitro translation and cleavage assay
The cDNAs were in vitro translated using 35S-labeledmethionine with the TNT® coupled reticulocyte lysate systemaccording to the instruction of the manufacturer (Promega,Mannheim, Germany). One microliter of the translationproducts was cleaved by 20 U caspase-3 (Biomol, Hamburg,Germany) in 20 mL cleavage buffer (25 mM HEPES pH 7.5,1 mM DTT, 1 mM EDTA and the protease inhibitors pefa-bloc, pepstatin, leupeptin, and aprotinin) for 1.5 h at 377C.For inhibition experiments, 1 mL 5 mM Z-Val-Ala-DL-Asp-fluoromethylketone (Z-VAD-fmk) was added. The cleavagemixture was supplemented with 5 mL loading buffer (1 mLglycerol, 1 mL 10% SDS, 0.25 mL 2-mercaptoethanol,0.075 mg Tris-base, and 0.125 mg bromophenol blue) andapplied to a 10% SDS-PAGE gel. The gel was washed, dried,covered with a BioMax™ MR film (Kodak, Chalon-sur-Saone,France) over night. Thereafter, the film was developed.
3 Results
3.1 Identification and quantification of cisplatin-
induced modified proteins
Poly(ADP-ribose) polymerase (PARP) is cleaved by caspase-3from the native 116 to 85 kDa and is a hallmark of apoptosis.Immunoblot analysis of PARP was performed to determinethe time point for full activation of apoptosis. Completecleavage of PARP was detected 16 h after addition of cisplatin(Fig. 1A). This time point was chosen for further experi-ments to analyze cisplatin-induced apoptotic Jurkat T cells.
In order to differentially label proteins of control andapoptotic Jurkat T cells, control cells were grown in mediumwith arginine-12C6 (light) whereas cells destined to undergoapoptosis were grown in medium containing arginine-13C6
(heavy). Proteins of control and apoptotic cells were com-bined and separated by SDS-PAGE and stained with Coo-massie Blue G250. First, five gel slices were investigated toexamine the incorporation rate and average ratio of the pro-teins. Arginine-13C6 (heavy) was fully incorporated into theproteins, and additional peaks with a mass shift of 5 Da weredetected for proline-containing peptides. Therefore, the cor-rect peak intensity of the heavy state was the sum of both thearginine-13C6 and the proline-13C5 mass peaks.
In total, 56 lanes were digested with trypsin and furtheranalyzed by nano-LC coupled offline to a MALDI-TOF/TOFmass spectrometer. As a result, 5497 peptide pairs with amass shift of 6.02 or 12.04 Da were determined by theWARP-LC software, whereof 572 were chosen for MS/MSfragmentation due to the fulfilled criteria for MS/MS selec-
Figure 1. Time course immunoblot analysis of PARP, core-bind-ing factor beta, and elongation factor 2. Full cleavage of PARPwas observed 16 h after additon of cisplatin to Jurkat T cells (A).The amount of core-binding factor beta (CBF-beta) was reducedafter 16 h (B), whereas an increased amount was observed foreukaryotic elongation factor 2 (eEF2) (C). GAPDH was used ascontrol and displayed intensities were determined using Top-Spot.
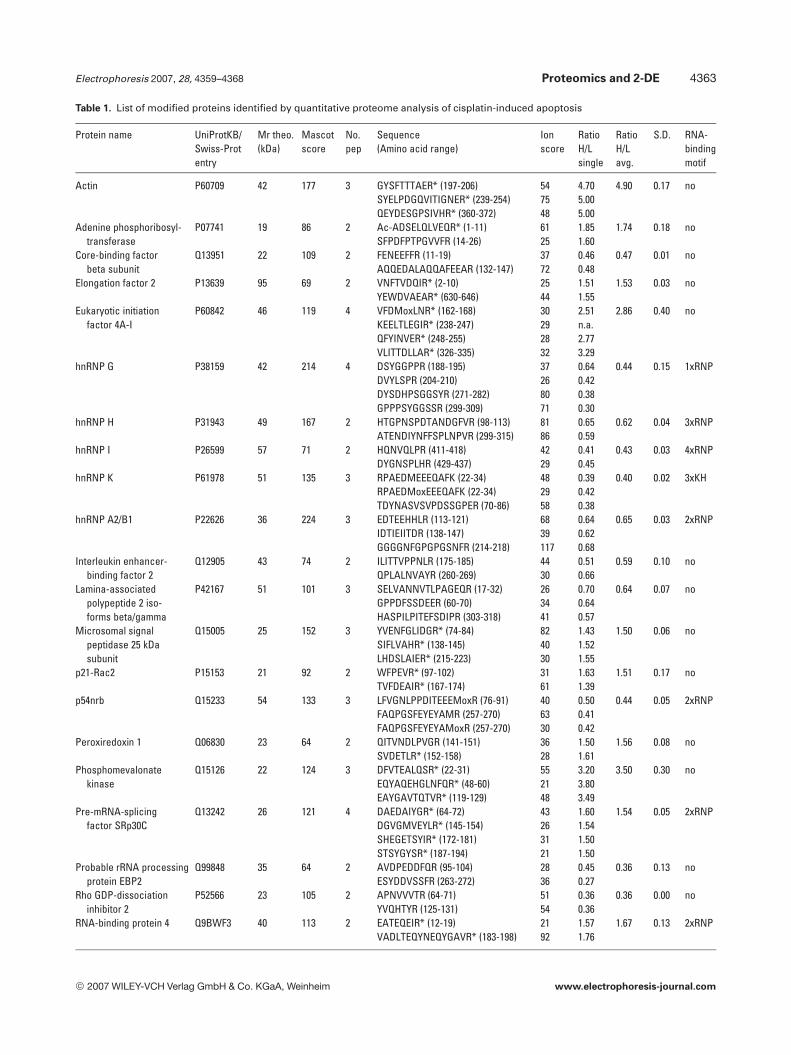
tion (ratio , 0.66 or . 1.5, peak intensity . 1000 counts,S/N . 5). Thereby, 223 peptides were identified by databasesearch using MASCOT. The majority of proteins were iden-tified by single peptide hits. However, 26 proteins wereidentified with relative ratios exceeding 1.5 with at least twopeptides fragmented by MS/MS (Table 1). As an example, thequantification and identification of phosphomevalonatekinase is shown in Fig. 2. Three tryptic peptides were detec-ted in MS mode derived from a single SDS-PAGE slice withan average ratio heavy-to-light of 3.50 (60.30) and wereselected for further fragmentation by MS/MS (Fig. 2). Allthree MS/MS spectra were matched to tryptic peptides ofphosphomevalonate kinase (Fig. 2). Obviously, the presenceof C-terminal arginine-13C6 could be confirmed in all MS/MSspectra by the y1-ion at 181 Da (Fig. 2). Notably, a clear pre-cursor ion selection of corresponding peptides labeled witharginine-12C6 arginine-13C6 was frequently not fully achieved.Thus, y-ion series due to C-terminal arginine could be deter-mined by considering a mass difference of 6 Da by manualinspection of MS/MS spectra. This useful information couldnot be exploited for database searches using MASCOT. Bycontrary, only one mass (light or heavy, respectively) can beconsidered by database searches of MS/MS spectra withMASCOT and resulted in relatively low scores due to thesignificant influence of the fragment coverage into the prob-ability score. Nevertheless, 14 proteins displayed a heavy-to-light ratio . 1.5 and 12 proteins were identified with a heavy-to-light ratio , 0.66 (Table 1). The identified proteins can beclassified into nine nuclear RNA-binding (heterogeneousnuclear ribonucleoproteins (hnRNPs) A2/B1, hnRNP G,hnRNP H, hnRNP I, hnRNP K, p54nrb, pre-mRNA-splicingfactor SRp30c, and RNA-binding protein 4) or RNA-proces-sing (probable rRNA processing protein EBP2) proteins,
© 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.electrophoresis-journal.com
Electrophoresis 2007, 28, 4359–4368 Proteomics and 2-DE 4363
Table 1. List of modified proteins identified by quantitative proteome analysis of cisplatin-induced apoptosis
Protein name UniProtKB/Swiss-Protentry
Mr theo.(kDa)
Mascotscore
No.pep
Sequence(Amino acid range)
Ionscore
RatioH/Lsingle
RatioH/Lavg.
S.D. RNA-bindingmotif
Actin P60709 42 177 3 GYSFTTTAER* (197-206)SYELPDGQVITIGNER* (239-254)QEYDESGPSIVHR* (360-372)
547548
4.705.005.00
4.90 0.17 no
Adenine phosphoribosyl-transferase
P07741 19 86 2 Ac-ADSELQLVEQR* (1-11)SFPDFPTPGVVFR (14-26)
6125
1.851.60
1.74 0.18 no
Core-binding factorbeta subunit
Q13951 22 109 2 FENEEFFR (11-19)AQQEDALAQQAFEEAR (132-147)
3772
0.460.48
0.47 0.01 no
Elongation factor 2 P13639 95 69 2 VNFTVDQIR* (2-10)YEWDVAEAR* (630-646)
2544
1.511.55
1.53 0.03 no
Eukaryotic initiationfactor 4A-I
P60842 46 119 4 VFDMoxLNR* (162-168)KEELTLEGIR* (238-247)QFYINVER* (248-255)VLITTDLLAR* (326-335)
30292832
2.51n.a.2.773.29
2.86 0.40 no
hnRNP G P38159 42 214 4 DSYGGPPR (188-195)DVYLSPR (204-210)DYSDHPSGGSYR (271-282)GPPPSYGGSSR (299-309)
37268071
0.640.420.380.30
0.44 0.15 1xRNP
hnRNP H P31943 49 167 2 HTGPNSPDTANDGFVR (98-113)ATENDIYNFFSPLNPVR (299-315)
8186
0.650.59
0.62 0.04 3xRNP
hnRNP I P26599 57 71 2 HQNVQLPR (411-418)DYGNSPLHR (429-437)
4229
0.410.45
0.43 0.03 4xRNP
hnRNP K P61978 51 135 3 RPAEDMEEEQAFK (22-34)RPAEDMoxEEEQAFK (22-34)TDYNASVSVPDSSGPER (70-86)
482958
0.390.420.38
0.40 0.02 3xKH
hnRNP A2/B1 P22626 36 224 3 EDTEEHHLR (113-121)IDTIEIITDR (138-147)GGGGNFGPGPGSNFR (214-218)
6839117
0.640.620.68
0.65 0.03 2xRNP
Interleukin enhancer-binding factor 2
Q12905 43 74 2 ILITTVPPNLR (175-185)QPLALNVAYR (260-269)
4430
0.510.66
0.59 0.10 no
Lamina-associatedpolypeptide 2 iso-forms beta/gamma
P42167 51 101 3 SELVANNVTLPAGEQR (17-32)GPPDFSSDEER (60-70)HASPILPITEFSDIPR (303-318)
263441
0.700.640.57
0.64 0.07 no
Microsomal signalpeptidase 25 kDasubunit
Q15005 25 152 3 YVENFGLIDGR* (74-84)SIFLVAHR* (138-145)LHDSLAIER* (215-223)
824030
1.431.521.55
1.50 0.06 no
p21-Rac2 P15153 21 92 2 WFPEVR* (97-102)TVFDEAIR* (167-174)
3161
1.631.39
1.51 0.17 no
p54nrb Q15233 54 133 3 LFVGNLPPDITEEEMoxR (76-91)FAQPGSFEYEYAMR (257-270)FAQPGSFEYEYAMoxR (257-270)
406330
0.500.410.42
0.44 0.05 2xRNP
Peroxiredoxin 1 Q06830 23 64 2 QITVNDLPVGR (141-151)SVDETLR* (152-158)
3628
1.501.61
1.56 0.08 no
Phosphomevalonatekinase
Q15126 22 124 3 DFVTEALQSR* (22-31)EQYAQEHGLNFQR* (48-60)EAYGAVTQTVR* (119-129)
552148
3.203.803.49
3.50 0.30 no
Pre-mRNA-splicingfactor SRp30C
Q13242 26 121 4 DAEDAIYGR* (64-72)DGVGMVEYLR* (145-154)SHEGETSYIR* (172-181)STSYGYSR* (187-194)
43263121
1.601.541.501.50
1.54 0.05 2xRNP
Probable rRNA processingprotein EBP2
Q99848 35 64 2 AVDPEDDFQR (95-104)ESYDDVSSFR (263-272)
2836
0.450.27
0.36 0.13 no
Rho GDP-dissociationinhibitor 2
P52566 23 105 2 APNVVVTR (64-71)YVQHTYR (125-131)
5154
0.360.36
0.36 0.00 no
RNA-binding protein 4 Q9BWF3 40 113 2 EATEQEIR* (12-19)VADLTEQYNEQYGAVR* (183-198)
2192
1.571.76
1.67 0.13 2xRNP
© 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.electrophoresis-journal.com

4364 F. Schmidt et al. Electrophoresis 2007, 28, 4359–4368
Table 1. Continued
Protein name UniProtKB/Swiss-Protentry
Mr theo.(kDa)
Mascotscore
No.pep
Sequence(Amino acid range)
Ionscore
RatioH/Lsingle
RatioH/Lavg.
S.D. RNA-bindingmotif
Spectrin alpha chain Q13813 284 155 4 DLASVQALLR (286-295)AQLADSFHLQQFFR (567-580)SLQQLAEER (1217-1225)LLEAQSHFR (2079-2087)
44662421
0.180.210.240.21
0.21 0.03 no
Tetratricopeptide repeatdomain 9C
Q8N5M4 20 109 2 AGVAFFHLQDYDQAR (114-128)YLQLTQSELSSYHR* (147-160)
3277
2.702.50
2.60 0.14 no
Transmembraneprotein 33
P57088 28 79 2 ALLANALTSALR* (58-69)TLFNELR* (206-212)
4336
1.931.75
1.84 0.13 no
Tubulin alpha-ubiquitouschain
P68363 50 98 2 EDAANNYAR* (97-105)EIIDLVLDR* (113-121)
5444
2.742.67
2.71 0.05 no
Tubulin beta P07437 50 311 4 GHYTEGAELVDSVLDVVR* (104-121)YLTVAAVFR* (310-318)ISEQFTAMFR* (381-390)ISEQFTAMoxFR* (381-390)
166705124
2.002.191.942.01
2.04 0.11 no
The theoretical molecular mass of the proteins is displayed according to the UniProtKB/Swiss-Prot entry (Mr theo (kDa)). The relative pro-tein ratio derived from apoptotic to control cells was determined by the intensity of peptides with heavy-to-light labeled arginines (H/L) forthe single mass pairs and in average (avg.) and the SD of these peptides was calculated. MASCOTscore, number of peptides (No. pep), MS/MS sequences (Sequence), MS/MS ion scores (Ion score), of the identified proteins are shown. N-Terminal acetylation (Ac), methionineoxidation (Mox), and arginine-13C6 (R*) are indicated.
seven cytoskeletal (actin, spectrin alpha chain, tubulin alpha-ubiquitous chain, and tubulin beta chain) or cytoskeletal-associated proteins (lamina-associated polypeptide 2 iso-forms beta/gamma, p21-Rac2, and rho GDP-dissociation in-hibitor 2), three enzymes (adenine phosphoribosyltransfer-ase, peroxiredoxin 1, and phosphomevalonate kinase), twomembrane proteins (microsomal signal peptidase 25 kDasubunit, and transmembrane protein 33), two proteinsinvolved in transcription (core-binding protein factor betasubunit, and interleukin enhancer-binding factor 2), twoproteins essential in translation (elongation factor 2, eukar-yotic initiation factor 4A-I), and a protein with a tetratricopeptide repeat domain, which is a structural motif present ina wide range of proteins (tetratricopeptide repeat domain9C).
3.2 Immunoblot analysis of core-binding factor beta
and elongation factor 2 confirmed quantification
by the SILAC-based approach
Immunoblotting was performed to verify quantitative dataobtained by proteome analysis for core-binding factor betaand elongation factor 2. For exact quantitative calculation,the same blot was stripped and immunoblotted again withGAPDH as a control and analyzed by the image softwareprogram TopSpot. For core-binding factor beta, a value of0.64 was determined (Fig. 1B) in comparison to 0.47 by theSILAC approach (Table 1). A factor of 1.58 was observed forelongation factor 2 (Fig. 1C), well in accordance with 1.53revealed by SILAC-based proteome analysis (Table 1). Nota-
bly, no significant quantitative alterations were found forthese two proteins after adding cisplatin to the cells for 2 and6 h (Figs. 1B and C).
3.3 Caspase-3 cleavage site prediction of p54nrb
The RNA-binding protein p54nrb was in vitro translated toexamine whether caspase-3 contributes to the cleavage of theprotein. Five clear fragments of p54nrb were generated bycaspase-3 (Fig. 3). The fragments at 32 plus 22 kDa and 28plus 26 kDa resulted in the mass of the 54 kDa protein. Thecorresponding fragment to 48 kDa was not detected due tothe low mass of 6 kDa. The caspase inhibitor Z-VAD-fmkprohibited the appearance of the apoptotic fragments con-firming that p54nrb was cleaved by caspase-3 (Fig. 3).
The sequence of p54nrb was analyzed to predict caspase-3 mediated cleavage sites by the software program GraBCas[29]. Suitable sequences for caspase-3 and caspase-7 cleavagewere found at DQLD231;D and DQVD286;R of p54nrb. Themolecular masses of cleavage products at DQLD231;D (26and 28 kDa), and at DQVD286;R (33 and 21 kDa) are well inaccordance with the observed fragments by the cleavageassay. However, the remaining cleavage to produce the48 kDa could not be explained by a DXXD sequence (X, anyamino acid). Considering only aspartic acids as cleavage sitesfrom the amino- and carboxy-terminal end, two putative cas-pase cleavage sites could be calculated at aspartic acid 58 and422. However, p54nrb was found in CD95 (Fas/Apo-1)-induced apoptosis based on 2-DE [17]. In control cells,p54nrb was identified at 56 kDa with an pI of 8.5 (Fig. 4A),
© 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.electrophoresis-journal.com

Electrophoresis 2007, 28, 4359–4368 Proteomics and 2-DE 4365
Figure 2. Quantification andidentification of phosphomeva-lonate kinase by MALDI-TOF/TOF-MS. Three peptides with anaverage ratio of 3.5 (high to low)determined by nano-LC-MALDI-MS were automatically selectedby the software WARP-LC andphosphomevalonate kinase wasidentified by MALDI-MS/MSanalysis and database searchusing MASCOT.
approximately corresponding with the theoretical values54 kDa and 9.0. After induction of apoptosis, p54nrb wasidentified with a lower mass of 52 kDa with pI of 8.1 (Fig. 4B).Cleavage at the two potential aspartic acids of p54nrb atposition 58 and 422 would result in fragments of 48 kDa withpI 8.9 and 49 kDa with pI 8.4, respectively. Therefore, thesignificant change in pI of p54nrb due to apoptosis indicateda caspase cleavage at MMPD422;G. Moreover, the samemasses were detected by PMF for the two p54nrb spots incontrol and apoptotic cells, with the exception that thedominant mass peak of m/z 1228.58 Da was found only inthe mass spectrum of control cells. This mass correspondedto the amino acids 457–468 nearby the C-terminal end ofp54nrb (in total 471 amino acids), supporting that the 48 kDafragment derived from caspase cleavage near the carboxy-terminal end.
3.4 Mapping of p54nrb caspase-3 cleavage sites
Three constructs of p54nrb were generated with one, two,and three mutations of the predicted aspartic acids to aspar-agines. In the first construct, p54nrb was mutated atMMPD422;G, in addition at DQVD286;R in the second con-struct and finally supplementary at DQLD231;D. The muta-tion of MMPD422;G to MMPN422;G inhibited the generation
of the 48 kDa protein fragment by caspase-3 (Fig. 3A vs. B).The exchange of DQVD286;R to DQVN286;R prevented theformation of the 32 and 22 kDa fragments of p54nrb (Fig. 3Bvs. C). Finally, the additional mutation of DQLD231;D toDQLN231;D impeded the creation of the 28 and 26 kDaproducts (Fig. 3C vs. D). The caspase-3 cleavages of thesethree constructs and control experiments with the caspaseinhibitor Z-VAD-fmk confirmed the predicted cleavage sites(Fig. 3). Obviously, all three caspase cleavage sites were loca-ted C-terminal to the ribonucleoprotein (RNP) motifs ofp54nrb and cleavage at DQLD231;D occurred within the C-terminal domain of NONA and PSP1 proteins (NOPS) do-main (Fig. 5).
4 Discussion
Online LC-ESI-MS is typically preferred to offline LC-MALDI-MS for quantitative proteome analysis, because it isfaster and more MS/MS spectra can be generated. However,more time is still required on data analysis than performingLC runs. Furthermore, quantitative protein data generated byLC-MALDI-MS are easier to evaluate because only singlecharged peptide ions are generated and has been shown to beparticularly accurate [30]. In addition, limited datasets are
© 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.electrophoresis-journal.com

4366 F. Schmidt et al. Electrophoresis 2007, 28, 4359–4368
Figure 3. Caspase-3 cleavage of p54nrb and mapping of threecleavage sites. The caspase-3 cleavage pattern of p54nrb as wildtype (wt) (A), with one mutation (B), two mutations (C), and threemutations (D) are shown. In addition, the caspase inhibitor Z-VAD-fmk marked with 1 was added to control experiments. Thearrows indicate the loss of p54nrb fragments due to the gener-ated mutations.
produced by the LC-MALDI approach because peptides areselected for fragmentation in a result-dependent mannerwith a defined level of quantitative differences. Using ourapproach, peptides were detected in one to four fractionswithin a single LC run using a gradient of 30 min with spot-ting of the tryptic peptides every 30 s. As a result, this relativeslow spotting speed enabled that the quantification could beeasily validated manually using LC-MALDI-MS, in contrastto online LC-ESI-MS which requires more sophisticatedsoftware tools to combine all recorded data for single pep-tides for quantitative proteome analyses. Furthermore, thesample on MALDI targets is not fully used and can be rea-nalyzed. The MALDI-based approach can certainly beimproved by either development of better ion selection forMS/MS, or by considering the specific sequence informationof the MS/MS fragmentation spectra for database searches.In addition, complementary results can be expected usingLC-MALDI and LC-ESI [31].
Cisplatin is one of the most powerful and effective drugsfor treating a range of cancers. However, cisplatin has severeside effects and resistance to this drug can be developed,ultimately limiting its benefits for cancer patients. However,the mechanisms of the cytotoxic effects of cisplatin are notfully understood. Connections between DNA damage andapoptosis play certainly a crucial role of cisplatin action.Quantitative proteome analysis of cisplatin-induced apopto-sis provides an opportunity to identify proteins related to themechanism of cisplatin function. In total, 26 proteins wereidentified with significant different ratios in control andapoptotic cells by LC-MALDI-based analysis of metaboliclabeled proteins. However, relatively similar protein classeswere found in comparison to other proteome analyses of
Figure 4. Section of the 2-DE pattern including protein spots ofp54nrb.The 2-DE protein spot pattern of control (A) and apoptotic(B) Jurkat T cells in the range of 50–60 kDa, and pI 8.0–9.0,respectively, are depicted. Protein spots identified as p54nrb areindicated by arrows.
apoptosis using other agents than cisplatin. Particularly,many cytoskeletal and RNA-binding proteins are known tobe modified during apoptosis [15].
Notably, seven proteins were identified (adenine phos-phoribosyltransferase, microsomal signal peptidase 25 kDasubunit, phosphomevalonate kinase, probable rRNA proces-sing protein EBP2, RNA-binding protein 4, transmembraneprotein 33, and tetratricopeptide repeat domain 9C) whichwere not yet reported to be involved in apoptosis. Adeninephosphoribosyltransferase catalyze the Mg21-dependentreaction that transforms an adenine base into its corre-sponding nucleotide. Microsomal signal peptidase 25 kDasubunit is a membrane-bound endoproteinase that removessignal peptides from nascent proteins as they are translo-cated into the lumen of the endoplasmic reticulum. Phos-phomevalonate kinase is a cytosolic protein which catalyzesthe formation of mevalonate 5-diphosphate and ADP out ofmevalonate 5-diphosphate and ATP in isoprenoid/sterol bio-synthesis [32]. RNA-binding protein 4, also known as hLark,is a splicing regulator [33]. Nonetheless, seven of the eightidentified RNA-binding proteins except RNA-binding pro-tein 4 were already identified by proteome analysis of CD95(Fas/Apo-1)-induced apoptosis [17]. In addition, probablerRNA processing protein EBP2 can be classified into thisgroup of proteins although it does not contain an RNA-binding motif because it is required for the processing of the27S pre-rRNA [34]. Transmembrane protein 33 and tetra-tricopeptide repeat domain 9C were identified by DNA se-quencing projects and were classified according to theirstructural features.
RNP complexes are the functional forms in which pre-mRNAs and mRNAs exist in cells. RNA-binding proteinsparticipate in pre-mRNA processing and mRNA transport,localization, translation, and stability [35]. RNPs are highlydynamic because of associations or dissociations of RNA-binding proteins in specific processes, such as pre-mRNAsplicing [35]. The 54 kDa nuclear RNA-binding proteinp54nrb contains two RNA binding motifs and shows exten-sive homology to the human polypyrimidine-tract-binding-protein-associated splicing factor PSF [36]. PSF and p54nrbmay mediate contacts between RNA polymerase II and
© 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.electrophoresis-journal.com

Electrophoresis 2007, 28, 4359–4368 Proteomics and 2-DE 4367
snRNPs during coupling of transcription and splicing pro-cess [37]. Interaction of p54nrb was demonstrated for theETS-related transcription factor Spi-1/PU.1 and DNAtopoisomerase I. The transcription factor Spi-1/PU.1impedes the binding of p54nrb to RNA and alters thesplicing process [38]. The RNA-splicing factors PSF andp54nrb control the DNA topoisomerase I activity and sti-mulate the jumping of DNA topoisomerase I betweenseparate DNA helices [39]. Furthermore, p54nrb showed astrong preference for inosine-containing RNAs as bindingsubstrate [40]. Recently, p54nrb was found in a complexwith PSF, neuronal Wiskott-Aldrich syndrome protein (N-WASP), nuclear actin, and RNA polymerase II to regulatetranscription [41]. In addition, the TLE1 corepressor com-plex was identified which included actin, DNA topoisome-rase II, heat shock protein 70 (hsp70), myosin heavy chain,nucleolin, nucleophosmin, PARP-1, RAD50, and p54nrb[25]. Actin, DNA topoisomerase II, myosin heavy chain,nucleolin, PARP-1, and p54nrb were already identified ascaspase substrates beside RAD21 and RAD51 [26]. More-over, hsp70 and nucleophosmin are known antiapoptoticproteins [42, 43]. Furthermore, alternative splicing can be acrucial level of regulation of apoptosis. Alternative splicingof some genes acting at different steps in the apoptoticprocess were already identified, e.g., caspases, death recep-tors, and bcl-2 family members [44]. Thus, p54nrb wasstudied in detail after molecular cloning of the cDNA and invitro translation of the protein to prove cleavage of p54nrbby caspases and to map cleavage sites. The identification ofcaspase targets and the consequences of the cleavage arecrucial to understand the molecular mechanism of apopto-sis. A functional impact of cleavage was already demon-strated for some proteins [26]. Here, the caspase-3 mediatedcleavage of p54nrb was demonstrated. P54nrb contains twoRNP motifs, also known as RBD or RRM, the most widelyfound RNA-binding protein sequence motif [45]. The RNPmotif is composed of about 70–90 amino acids with twoconserved sequence motifs, RNP1 and RNP2. However,caspase-3 cleavage has occurred C-terminally to the RNPmotifs. Furthermore, the NOPS domain was cleaved bycaspase-3 (Fig. 5). NOPS was identified as a domain ofnucleolar proteins [46]. Paraspeckle protein 1 (PSP1), anucleolar protein which accumulates in paraspeckles, dis-plays high sequence similarity with p54nrb and also con-tains two RNP motifs and a NOPS domain. Interaction ofPSP1 and p54nrb was shown to be dependent on the NOPSdomain and independent of the RNP motifs [47].
Figure 5. Diagram of motifs according to Pfam and identifiedcaspase-3 cleavage sites of p54nrb.
Proteome analyses of apoptosis were performed withdifferent inducers of apoptosis and several proteomicapproaches. Variations in experimental design revealedsome similarities but also differences in identified apoptosis-modified proteins even with the same apoptosis-inducingagent [15]. Therefore, the specificity of the seven novel pro-teins to the action of cisplatin must be further evaluated.However, SILAC has been evolved as method of choice forquantitative proteome analyses using cell culture, usually incombination with SDS-PAGE and online LC-ESI-MS/MS.Here, offline LC-MALDI was shown as complementarymethod with high accuracy and straightforward evaluation ofquantitative data, but it is more time consuming and lesssequence data are generated with the current instrumenta-tion.
This work was supported by grant 0313029A from the Bun-desministerium für Bildung und Forschung (BMBF) to T.R.
5 References
[1] Norbury, C. J., Zhivotovsky, B., Oncogene 2004, 23, 2797–2808.
[2] Debatin, K. M., Krammer, P. H., Oncogene 2004, 23, 2950–2966.
[3] Cory, S., Adams, J. M., Nat. Rev. Cancer 2002, 2, 647–656.
[4] Rich, T., Allen, R. L., Wyllie, A. H., Nature 2000, 407, 777–783.
[5] Zhou, B. B., Elledge, S. J., Nature 2000, 408, 433–439.
[6] Slee, E. A., O’Connor, D. J., Lu, X., Oncogene 2004, 23, 2809–2818.
[7] Wang, D., Lippard, S. J., Nat. Rev. Drug Discov. 2005, 4, 307–320.
[8] Jamieson, E. R., Lippard, S. J., Chem. Rev. 1999, 99, 2467–2498.
[9] Siddik, Z. H., Oncogene 2003, 22, 7265–7279.
[10] Sinha, P., Poland, J., Kohl, S., Schnolzer, M. et al., Electro-phoresis 2003, 24, 2386–2404.
[11] Castagna, A., Antonioli, P., Astner, H., Hamdan, M. et al.,Proteomics 2004, 4, 3246–3267.
[12] Chekhun, V. F., Lukyanova, N. Y., Urchenko, O. V., Kulik, G. I.,Exp. Oncol. 2005, 27, 191–195.
[13] Stewart, J. J., White, J. T., Yan, X., Collins, S. et al., Mol. Cell.Proteomics 2006, 5, 433–443.
[14] LeMoguen, K., Lincet, H., Deslandes, E., Hubert-Roux, M. etal., Proteomics 2006, 6, 5183–5192.
[15] Thiede, B., Rudel, T., Mass Spectrom. Rev. 2004, 23, 333–349.
[16] Van Damme, P., Martens, L., Van Damme, J., Hugelier, K. etal., Nat. Methods 2005, 2, 771–777.
[17] Thiede, B., Dimmler, C., Siejak, F., Rudel, T., J. Biol. Chem.2001, 276, 26044–26050.
[18] Ong, S. E., Mann, M., Nat. Chem. Biol. 2005, 1, 252–262.
[19] Ong, S. E., Blagoev, B., Kratchmarova, I., Kristensen, D. B. etal., Mol. Cell. Proteomics 2002, 1, 376–386.
© 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.electrophoresis-journal.com

4368 F. Schmidt et al. Electrophoresis 2007, 28, 4359–4368
[20] Thiede, B., Kretschmer, A., Rudel, T., Proteomics 2006, 6,614–622.
[21] Rezaul, K., Wu, L., Mayya, V., Hwang, S. I., Han, D., Mol. Cell.Proteomics 2005, 4, 169–181.
[22] Hwang, S. I., Lundgren, D. H., Mayya, V., Rezaul, K. et al.,Mol. Cell. Proteomics 2006, 5, 1131–1145.
[23] Shav-Tal, Y., Zipori, D., FEBS Lett. 2002, 531, 109–114.
[24] Thiede, B., Siejak, F., Dimmler, C., Rudel, T., Proteomics2002, 2, 996–1006.
[25] Ju, B. G., Solum, D., Song, E. J., Lee, K. J. et al., Cell 2004,119, 815–829.
[26] Fischer, U., Janicke, R. U., Schulze-Osthoff, K., Cell DeathDiffer. 2003, 10, 76–100.
[27] Machuy, N., Thiede, B., Rajalingam, K., Dimmler, C. et al.,Mol. Cell. Proteomics 2005, 4, 44–55.
[28] Thiede, B., Siejak, F., Dimmler, C., Jungblut, P. R., Rudel, T.,Electrophoresis 2000, 21, 2713–2720.
[29] Backes, C., Kuentzer, J., Lenhof, H. P., Comtesse, N., Meese,E., Nucleic Acids Res. 2005, 33, W208–W213.
[30] Schmidt, F., Dahlmann, B., Janek, K., Kloss, A. et al., Prote-omics 2006, 6, 4622–4632.
[31] Creaser, C. S., Green, P. S., Kilby, P. M., Ratcliffe, L., RapidCommun. Mass Spectrom. 2006, 20, 829–836.
[32] Hogenboom, S., Tuyp, J. J., Espeel, M., Koster, J. et al., J.Lipid Res. 2004, 45, 697–705.
[33] Lai, M. C., Kuo, H. W., Chang, W. C., Tarn, W. Y., EMBO J.2003, 22, 1359–1369.
[34] Huber, M. D., Dworet, J. H., Shire, K., Frappier, L., McAlear,M. A., J. Biol. Chem. 2000, 275, 28764–28773.
[35] Dreyfuss, G., Kim, V. N., Kataoka, N., Nat. Rev. Mol. Cell Biol.2002, 3, 195–205.
[36] Dong, B., Horowitz, D. S., Kobayashi, R., Krainer, A. R.,Nucleic Acids Res. 1993, 21, 4085–4092.
[37] Kameoka, S., Duque, P., Konarska, M. M., EMBO J. 2004, 23,1782–1791.
[38] Hallier, M., Tavitian, A., Moreau-Gachelin, F., J. Biol. Chem.1996, 271, 11177–11181.
[39] Straub, T., Knudsen, B. R., Boege, F., Biochemistry 2000, 39,7552–7558.
[40] Zhang, Z., Carmichael, G. G., Cell 2001, 106, 465–475.
[41] Wu, X., Yoo, Y., Okuhama, N. N., Tucker, P. W. et al., Nat. CellBiol. 2006, 8, 756–763.
[42] Beere, H. M., J. Cell Sci. 2004, 117, 2641–2651.
[43] Ye, K., Cancer Biol. Ther. 2005, 4, 918–923.
[44] Schwerk, C., Schulze-Osthoff, K., Mol. Cell 2005, 19, 1–13.
[45] Birney, E., Kumar, S., Krainer, A. R., Nucleic Acids Res. 1993,21, 5803–5816.
[46] Staub, E., Fiziev, P., Rosenthal, A., Hinzmann, B., Bioessays2004, 26, 567–581.
[47] Fox, A. H., Bond, C. S., Lamond, A. I., Mol. Biol. Cell 2005, 16,5304–5315.
© 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.electrophoresis-journal.com


















