
QUANTIFYING NATURAL AND ANTHROPOGENIC …s3.amazonaws.com/zanran_storage/€¦ · ·...
119
COOPERATIVE RESEARCH CENTRE FOR COAL IN SUSTAINABLE DEVELOPMENT Established and supported under the Australian Government’s Cooperative Research Centres Program QUANTIFYING NATURAL AND ANTHROPOGENIC SOURCED MERCURY EMISSIONS FROM AUSTRALIA IN 2001 -A local scale modelling assessment of transport and deposition patterns for anthropogenic mercury air emissions RESEARCH REPORT 46 Authors: Christian Peterson Peter Nelson Anthony Morrison Macquarie University April 2004 QCAT Technology Transfer Centre, Technology Court Pullenvale Qld 4069 AUSTRALIA Telephone (07) 3871 4400 Facsimile (07) 3871 4444 Email: [email protected]
Transcript of QUANTIFYING NATURAL AND ANTHROPOGENIC …s3.amazonaws.com/zanran_storage/€¦ · ·...

COOPERATIVE RESEARCH CENTRE FOR COAL IN SUSTAINABLE DEVELOPMENT Established and supported under the Australian Government’s Cooperative Research Centres Program
QUANTIFYING NATURAL AND ANTHROPOGENIC SOURCED
MERCURY EMISSIONS FROM AUSTRALIA IN 2001
-A local scale modelling assessment of transport and deposition patterns for anthropogenic mercury air emissions
RESEARCH REPORT 46
Authors:
Christian Peterson Peter Nelson
Anthony Morrison
Macquarie University
April 2004 QCAT Technology Transfer Centre, Technology Court
Pullenvale Qld 4069 AUSTRALIA Telephone (07) 3871 4400 Facsimile (07) 3871 4444
Email: [email protected]

This page is intentionally left blank

DISTRIBUTION LIST CCSD Chairman; Chief Executive Officer; Research Manager, Manager Technology; Files Industry Participants Australian Coal Research Limited ............................................................. Mr Ross McKinnon BHP Billiton Minerals - Coal .................................................................... Mr Ross Willims ................................................................................................................ Mr Alan Davies CNA Resources.......................................................................................... Mr Ashley Conroy CS Energy .................................................................................................. Dr Chris Spero Delta Electricity ......................................................................................... Mr Steve Saladine Queensland Natural Resources, Mines and Energy................................... Mr Bob Potter Rio Tinto (TRPL)....................................................................................... Mr David Cain .................................................................................................................... Dr Jon Davis Stanwell Corporation ................................................................................. Dr Paul Simshauser Tarong Energy ........................................................................................... Mr Burt Beasley The Griffin Coal Mining Co Pty Ltd ......................................................... Mr Jim Coleman Wesfarmers Premier Coal Ltd ................................................................... Mr Peter Ashton Western Power ........................................................................................... Mr Keith Kirby Xstrata Coal Australia Pty Ltd................................................................... Mr Barry Isherwood Research Participants CSIRO ……............................................................................................... Dr David Brockway Curtin University of Technology ............................................................... Dr Barney Glover Macquarie University ................................................................................ Prof Jim Piper The University of Newcastle ..................................................................... Prof Adrian Page The University of New South Wales ......................................................... Prof David Young The University of Queensland ................................................................... Prof Don McKee

This page is intentionally left blank

Cooperative Research Centre for Coal in Sustainable Development
QCAT Technology Transfer Centre Technology Court
Pullenvale, Qld 4069 Telephone: (07) 3871 4400
Fax: (07) 3871 4444
Feedback Form To help us improve our service to you may I ask you for five minutes of your time to complete this
questionnaire. Please fax or mail it back to me, or, if you would prefer, give me a call.
ATTENTION MANAGER TECHNOLOGY TRANSFER FAX NO 07 3871 4444 DATE ............................................................................... FROM NAME: .................................................................................................................................. COMPANY:............................................................................................................................ REPORT TITLE: QUANTIFYING NATURAL AND ANTHROPOGENIC SOURCED MERCURY EMISSIONS FROM
AUSTRALIA IN 2001 AUTHORS: CHRISTIAN PETERSON, PETER NELSON, ANTHONY MORRISON Would you please rate our performance in the following areas by ticking the appropriate box:
The Research Agree Neutral Disagree Don’t Know
This work has achieved its research objectives
This work has delivered to agreed milestones
This work is relevant to my organisation
Communication of progress has been timely & useful
The Report
The report is well presented
The context of the report is clear
The content of the report is substantial
The report is clearly written and understandable
This product is good value
Any further comments which would assist us in better serving your future requirements?: …………………………….……………………………………………………………………………………………
…………………………………………………………………………….……………………………………………
Are there any people you would like to add to our distribution list?:
………………………………………………………………………………………………………….………………
………………………………………………………………………………………………………………….…....... Thank you for your response

This page is intentionally left blank

Quantifying natural and anthropogenic sourced mercury emissions from Australia
in 2001
-A local scale modelling assessment of transport and deposition patterns for anthropogenic mercury air emissions
Christian Peterson, Peter Nelson, Anthony Morrison
Mount Piper PS
Maldon CW
BHP Steel PKW
Orica Chlorine P SYDNEY
Vales Point PSEraring PS
PasmincoComsteel
Lidell PSBayswater PS

i
SUMMARY Mercury is continuously released both directly and indirectly to the atmosphere from anthropogenic and natural sources (i.e. oceans, land and vegetation) by emission and re-emission. The atmosphere also constantly deposits mercury by a variety of mechanisms to receiving natural surfaces. Thus, mercury is continually cycled between the air and the natural environment until it is finally stored in soil and sediments (or alternatively converted to methyl-Hg). Elevated levels of mercury are today found in sediment and fish tissue around the world. Although mercury is naturally occurring, the total amount of mercury in the environment has increased by a factor of two to five compared to pre industrial levels. Due to its global mobility it is suggested that a significant proportion of the children born each year are at risk of adverse neurological effects caused by mercury.As the pre-eminent source of anthropogenic mercury is fuel combustion, particularly coal, there is a need to understand its role as a mercury source. A number of studies have estimated that the yearly total global input of mercury to the atmosphere ranges between 5800-7000 tonnes. Of these emissions, somewhere between 35-60 % originates from anthropogenic sources. However, if re-emission of anthropogenic mercury previously deposited on natural surfaces is taken into account, the anthropogenic portion of the total global mercury emissions may be as high as 75 percent. Calculations performed as part of this study have estimated that mercury emissions from natural sources in Australia are in the range 130-270 tonnes/yr. However, these estimates were based on a number of simplifying assumptions and the result should be treated with some caution. Because of its physio-chemical properties mercury is used in a broad variety of manufacturing industries and products, although this use is diminishing. The processing of mineral resources at high temperatures such as roasting and smelting of ores, combustion of fossil fuels, kiln operation in the cement industry, and waste incineration all release significant amounts of mercury to the atmosphere. Global anthropogenic sources are estimated to have emitted 1900 tonnes of mercury in 1995. The most significant source of global anthropogenic mercury is the stationary combustion of fossil fuels (mainly coal), which accounts for 77 percent of total emissions. Of the approximately 10.2 tonnes of anthropogenic Hg released annually in Australia, it is estimated that about 9.9 tonnes is emitted into the atmosphere, with the remaining 0.3 tonnes distributed between the water and land compartments. Of the mercury emitted into the atmosphere it was calculated that 4.75 tonnes (48 %) are in the form of elemental mercury (Hg0), 1.30 tonnes is in the form of divalent mercury (13 %), and 3.88 tonnes (39 %) is particulate mercury. This elemental mercury becomes part of the global atmospheric mercury pool, and the Australian contribution constitutes about 0.5 percent of the annual increase in the global mercury pool. Even though the estimated emissions from Australia are only minor, because of a dependence on a resource based economy, the country is a significant per capita emitter (0.51 g Hgtot/person), compared to the global average (0.36 Hgtot/person). It is apparent that mercury emission inventories are subjected to large uncertainties. According to the latest global emission inventory, Australia was claimed to annually emit more than 110.9 tonnes of anthropogenic mercury. This value is nearly 11 times more than that estimated by the Australian National Pollution Inventory. The report demonstrates that the former value of 110.9 tonnes is unrealistically high and that the discrepancy between the calculated values is predominantly due to the use of inappropriate emission factors when calculating the mercury emitted from the combustion of coal. The dispersion and deposition of mercury from ten significant industrial point source emitters in the central, coastal parts of NSW was investigated using a three-dimensional, regional

ii
scale, Eulerian air quality model (TAPM). Since the model was primarily developed to investigate the air quality of an airshed in relation to SOx, NOx, and photochemical smog, mercury was modelled as an inert pollutant (tracer) where chemical transformation processes, as well as, wet deposition processes are omitted For simplicity a facility emission cutoff of 20 kg/yr was used which ensured that more than 90 % (1282 kg/yr) of the total anthropogenic point emissions in NSW (NPI, 2003a) were embraced by the simulation. The sources of Hg emissions simulated include: combustion of coal (5 power plants), basic iron and steel manufacturing (2 sources), cement manufacturing (1 source), Cu/Ag/Pb/Zn smelter (1 source), and chemical production (1 source). A simulation was conducted for the January 2001 period. The model used 25×25 grids in the outer domain with a grid size of 30×30 km. In order to obtain a finer resolution for concentration simulations, an outer and an inner pollution grid domain was used with 97×97 grids in the horizontal plane, with grid sizes of 7.5 × 7.5 km and 2.5 × 2.5 km. Vertically, there were 25 non-uniform layers in the model, with the finest resolution near the surface (10 m). The top of the modeling domain was 8 km. The mercury species considered in the simulation were Hg0 and Hg(II)/Hgp (combined) and the background concentration of Hg0 was set to zero. Even though deposition of mercury was not included in TAPM, dry deposition fluxes were calculated by post-processing hourly-simulated grid concentration outputs from TAPM. Hourly dry deposition fluxes were derived from each grid cell using default values for the deposition velocities. The deposition velocity for Hg(II)/Hgp was set to be 0.5 cm/s during the day and zero cm/s during the night, corresponding values for Hg0 were respectively, 0.03 and zero cm/s. The simulation calculated that the maximum ambient ground level mercury concentration was 3.1 ng/m3. Even if the background concentration of Hg0 is added to this value the total was well below (i) the US EPA determined reference concentration of Hg vapor of 0.3 µg/m3 for the general population, (ii) the limit value for exposure in Europe of 0.05µg/m3, and (iii) the air quality objectives set in Victoria, Australia, of 9.4 µg/m3, for inorganic mercury,. The simulated total average mercury deposition flux in the inner domain varied between 0.2 and 1.4 µg/m2/yr (at the 10th and 90th percentile level, respectively). In occasional cases, close to emitting sources (1-2 grid cells away from the source), the deposition flux of Hgtot was calculated to reach levels of 50-60 µg/m2/yr. A number of further calculations were performed to investigate the area average deposition flux of mercury and the percentage of total mercury deposited at various distances from the emitting sources. The general trend observed from these calculations was that the area average deposition flux of mercury, when expressed as a percentage of the total mercury emitted, is relatively small, (0.1-9.4 %, depending on the distance to the source). Thus it appears that, a significant part of the mercury emitted from the facilities investigated would be transported away from the domain. By integrating the average simulated deposition rate in the outer domain over the area of study, it was calculated that approximately 105 kg of mercury would be deposited annually within the entire domain. This constitutes about 8 % of the total mercury emissions in the simulation. The numerous mercury species present in the atmosphere have differing atmospheric residence times, which affect the distance they can be transported before being deposited to the surface. Atmospheric transformation/interaction processes, which determine the speciation of mercury, are therefore important to include if models are to accurately simulate mercury transportation and deposition. In order to obtain more accurate data in future simulations, mercury transformation/interaction and deposition processes should be integrated into the TAPM model.

iii
The study has allowed the establishment of an initial methodological framework for assessing the environmental impact of mercury (and in the future other trace elements) from power stations and other major emission sources.

iv
TABLE OF CONTENTS
Page
ABSTRACT i
TABLE OF CONTENTS iii
LIST OF TABLES vi
LIST OF FIGURES viii
1. INTRODUCTION 1
1.1 Background 1
1.2 The purpose and scope of this report 5
2. ATMOSPHERIC CHEMISTRY AND RESIDENCE TIME 6
2.1 The properties of atmospheric mercury species 6
2.2 Chemical reaction and interaction in the atmosphere 9
3. EMISSION OF MERCURY 12
3.1 Definition 12
3.2 The global atmospheric Hg cycle 12
3.3 Natural mercury emission 16
3.3.1 Background 16
3.3.2 Bi-directional exchange of mercury 17
3.3.3 Estimated natural emissions of mercury from Australia 21
3.4 Anthropogenic mercury emissions 25
3.4.1 Global anthropogenic emissions 26
3.4.2 2001 Australian mercury emission inventory 32
3.4.2.1 Atmospheric emission from point sources 32
3.4.2.2 Atmospheric emission from area sources 34
3.4.2.3 Total Australian anthropogenic emission 35
3.4.2.4 Accuracy of emission estimates 35
3.4.2.5 Estimation of mercury speciation 40

v
TABLE OF CONTENTS (cont)
Page
4. DEPOSITION OF MERCURY 43
4.1 Dry deposition 44
4.2 Deposition patterns of mercury 46
4.3 Model simulation 48
4.3.1 The Model 48
4.3.2 Simulation procedure 48
4.3.2.1 Simulation domain and period 48
4.3.2.2 Meteorological conditions 50
4.3.2.4 Mercury emission data 50
4.3.2.4 Initial and boundary conditions 50
4.3.2.5 Deposition 50
4.3.3 Simulation result 51
4.3.3.1 The simulation 51
4.3.3.2 Ambient mercury concentrations 51
4.3.3.3 Dry deposition of mercury 53
5. THE CHEMISTRY OF ATMOSPHERIC MERCURY 61
5.1 Chemical transformations in the aqueous phase 63
5.1.1 Oxidation 63
5.1.1.1 Oxidation of Hg0 by O3 63
5.1.1.2 Oxidation of Hg0 by ·OH 64
5.1.1.3 Oxidation of Hg0 by chlorine (HOCL/OCL-) 65
5.1.2 Reduction 67
5.1.2.1 Reduction of Hg(ΙΙ) by S(ΙV) 67
5.1.2.2 Photoreduction of Hg(ΙΙ) 69
5.1.2.3 Reduction of Hg(ΙΙ) by HO2 69
5.2 Chemical transformations in the gaseous phase 70
5.2.1 Oxidation of Hg0 by O3 70
5.2.2 Oxidation of Hg0 by ·OH 71
5.2.3 Oxidation of Hg0 by NO3· 72
TABLE OF CONTENTS (cont)

vi
Page 5.2.4 Oxidation of Hg0 by H2O2
72
5.2.5 Dimethyl mercury reactions 73
5.2.5.1 Reaction with nitrate radical 73
5.2.5.2 Reaction with other species 74
5.3 Equilibria tables 75
5.4 Summary of half lives and residence times for elemental and
divalent mercury 76
6. CONCLUSIONS 77
7. ACKNOWLEDGEMENTS 79
8. REFERENCES 80
APPENDIX A Estimated Hg emissions by point source in Australia 2001 A1
APPENDIX B Estimation of Hg emission from area sources related to
the Pacyna and Pacyna (2002) study B1
APPENDIX C Input data to TAPM C1
APPENDIX D Simulation result from TAPM D1
CONTACT DETAILS

vii
LIST OF TABLES
Page
Table 1 Post and pre-industrial mercury fluxes recorded in lake sediment cores 3
Table 2 Chemical transformations in the aqueous phase 10
Table 3 Chemical transformations in the gaseous phase 11
Table 4 Estimated global emissions (tonnes/yr) (Mason et al., 1994) 14
Table 5 Estimated global emissions (tonnes/yr) (Bergan and Rohde, 2001) 15
Table 6 Summary of estimated global emissions (tonnes/yr) 16
Table 7 Emission rates of mercury from different natural surfaces 19
Table 8 Global natural emission of mercury from forests (Lindberg et al., 1998) 20
Table 9 Estimated emission of mercury from natural land surfaces in Australia 24
Table 10 Global atmospheric emissions of total mercury from major anthropogenic
sources in 1995 (tonnes) 26
Table 11 Estimated 1995 mercury emissions from area and point sources
in various countries (tonnes) 29
Table 12 Distribution of total estimated Australian anthropogenic mercury (kg)
for 2001 35
Table 13 Country-by-country comparison based on anthropogenic atmospheric
Hgtot/capita 39
Table 14 Emission speciation (fraction of the total) of mercury from anthropogenic
sources 41
Table 15 Estimates of Australian atmospheric mercury emission rates by source (2001) 42
Table 16 Dry deposition velocity of mercury species (cm/s) 46
Table 17 Percentile analysis of simulated ambient mercury concentration 52
Table 18 Percentile analysis of simulated dry deposition fluxes of mercury species 53
Table 19 Area average mercury deposition rates around each facility (Run 10) 58
Table 20 Percent of total mercury dry deposited around each facility (Run 10) 58
Table 21 Area average mercury deposition rates around each facility (Run 20) 59
Table 22 Area average mercury deposition rates around each facility (Run 30) 59
Table 23 Percent of total mercury dry deposited around each facility (Run 20) 60
Table 24 Percent of total mercury dry deposited around each facility (Run 30) 60
Table 25 Oxidation of DMM with different oxidants 74

viii
LIST OF TABLES (continued)
Page
Table 26 Equilibria for aqueous phase Hg(II) speciation 75
Table 27 Solid-liquid equlibria of mercury compounds 75
Table 28 Gas/aqueous equlibria of Hg and some of its compounds 75
Table 29 Summary chemical transformations in the aqueous phase 76
Table 30 Summary chemical transformations in the gaseous phase 76

ix
LIST OF FIGURES
Page
Figure 1 Mercury oxidation, reduction and mass transfer processes in the atmosphere 8 Figure 2 The global atmospheric mercury cycle 13 Figure 3 Geographical distribution of mercury emissions over Australia and South-East Asia (tonnes/yr) 28 Figure 4 Estimated mercury emissions (point sources) from Australian States and Territories (2001) (kg/yr) 33 Figure 5 Estimated mercury to air (point sources) by source category in Australia, 2001 (kg/yr) 33 Figure 6 Geographical distribution of mercury emitting point sources in Australia 34 Figure 7 Estimates made using NPI (1999b, 2003b) emission factors of Australian anthropogenic atmospheric mercury emissions from fuel and coal combustion compared with emissions which arise from combustion during electricity generation 37 Figure 8 Geographical distribution of point sources include in the TAPM simulation 49 Figure 9 Contour plot of simulates of dry deposition of divalent /particulate Hg (unit: µg/m3) 55 Figure 10 Contour plot of simulates of dry deposition of elemental Hg(unit: µg/m3) 56 Figure 11 The magnitude of dry deposition fluxes from TAPM simulation(unit: µg/m3) 57 Figure 12 Atmospheric mercury chemistry 61

1
1. INTRODUCTION
1.1 Background
Mercury (Hg) is among the most bio-concentrated trace metal in the food chain, especially in
fish tissue. Consumption of fish with elevated concentrations of Hg may lead to adverse
health affects and can in some cases even be lethal. There are several historical examples of
severe poisoning disasters. For instance, in Minimata Bay (Japan), in the 1950s, a large
number of people were severely poisoned by eating fish (their primary source of food)
polluted with Hg (methyl mercury) by local industries. Methyl mercury was accumulated in
marine organisms in the bay over time until the level of concentration in fish tissue exceeded
a healthy dose. Many people died and others were faced with a variety of neurological
problems. In particular, children, who had been exposed to Hg in the womb, suffered serious
developmental deficits (Goldfrank et al,. 1990). This is a somewhat unusual and extreme
example, however, elevated levels of Hg are today found in sediment and fish tissue around
the world. Even if the concentration is modest, long-term "exposure" can present a significant
risk to humans and wildlife. Accumulated levels of Hg in the human body can cause, as
mentioned in the example above, developmental distortion in features, as well as permanent
damage to the kidneys and the central nervous system (WHO, 1990 and 1991). According to
the US Centre for Disease Control and Prevention, 10 percent of American women already
have so much Hg in their blood that if they become pregnant, it would pose a threat to the
developing fetus (US CDC, 2001). It has been estimated that at least 60 000 children born
each year in the US are at risk of adverse neurological effects from Hg (US NAS, 2000).
Since the Hg exposure pathway of the greatest concern is consumption of fish contaminated
with methyl mercury (US EPA, 1997)1, many countries have issued fish consumption
advisories for waterbodies with elevated levels of methyl mercury, as well as, introduced
information campaigns addressed to women which aim to increase awareness about the risks
of eating fish during their pregnancy (Schroeder and Munthe, 1998). Although Hg is naturally
occurring, the total amount of Hg in the environment has increased by a factor of two to five
compared to pre-industrial levels (Mason et al., 1994). As the amount of Hg is increasing in
the environment so is the risk of exposure to methyl mercury (US EPA, 1997).
1 Vaporised elemental mercury is also of concern when inhaled. Even at low levels, mercury can cause permanent damage to the brain and central nervous system (EPA, 2000). However, ambient air concentrations of elemental Hg are approximately in the range of 2 - 10 ngm-3. Compared to the US EPA reference concentration for elemental Hg of 0.3 µgm-3 for the general population, ambient air exposure of elemental Hg are unlikely to pose a significant risk to human health (US EPA, 1997). In Europe, the annual average limit of Hg exposure is 0.05 µgm-3 (Pirrone et al., 2001b).

2
Due to the seriousness of the health effects of Hg it is one of the most studied elements in the
world and a very large number of scientific articles have been published that address different
issues related to Hg. In an attempt to summarise existing information about Hg, including its
emission sources, its chemistry, its transportation and deposition pathways, the production
and use of Hg, prevention and control techniques, and health effects, a number of
international studies and assessments have been conducted (EC, 2001; UNEP, 2002; US EPA,
1997). The overall aims of these studies are to address the global adverse impacts of Hg and
to reduce the risks to human health and the environment. Thus, an increase in the general
knowledge about Hg, especially among decision makers, will hopefully lead to a restriction of
releases of this toxic metal in the future. It is, however, difficult to decrease the emissions
from Hg, since Hg is a trace metal found in many raw materials such as coals and ores. Coal
is of particular concern since not only is it one of the major sources of energy for electricity
generation in the world, it is also by far the largest source of global atmospheric Hg (Pacyna
and Pacyna, 2002).
Mercury is a naturally occurring metal found in small quantities throughout the environment;
in the atmosphere and in aquatic and terrestrial compartments. It is continuously released,
transported, transformed and stored in and between these compartments. The atmosphere is
considered to be the dominant transport media of Hg in the environment (Fitzgerald et al.,
1991; Lindquist et al., 1991), Hg enters the atmosphere through natural sources (e.g.
volcanoes, surface emissions and forest fires) as well as through anthropogenic sources (e.g.
fossil fuel and waste combustion, mining and mineral processing, and from different
commercial products). Once released, Hg is transported in the atmosphere where it is
subjected to a number of chemical and physical processes before being deposited by wet
(precipitation scavenging) and/or dry (gravitational settling) processes to environmental
surfaces. A large proportion of the deposited Hg is vaporised (through chemical, physical and
biological processes) and re-emitted to the atmosphere. The rest of the deposited Hg is cycled
in the terrestrial/aquatic environment where it is finally stored in soil and lake, stream and
ocean sediments. In these sediments some of the Hg is biologically transformed via bacteria
to methyl mercury, which is partitioned between the sediment and the water phase. Living
aquatic organisms adsorb some of the methyl mercury from the water, resulting in an
increasingly high concentration of methyl mercury along the food chain, particularly in fish
tissue2 (ie. bioaccumulation) (Schroeder and Munthe, 1998).
2 Methyl mercury has the ability to bio-concentrate up to a million times in the aquatic food chain (Schroeder and Munthe, 1998).

3
In contrast to the Japanese example where the polluting source of Hg was local industries,
elevated Hg levels are also found in sediment and fish tissues from lakes far away from
industrial areas. Analysis of lake sediment cores from widely separated regions of the
Northern Hemisphere show a three fold increase in Hg fluxes since the start of the industrial
revolution (Table 1) (Landers et al., 1998). The origin of the Hg in these sediment cores is of
some debate. Some argue that natural geological sources are the main contributor of Hg in
these remote areas (Rasmussen, 1994). However, there is today a global consensus among the
world's researchers that Hg can be transported vast distances through the atmosphere from
emitting sources, and consequently that the Hg deposited in remote sensitive areas interacts
with the local environment, where some methylates, and hence bioaccumulates in aquatic
organisms.
A number of review reports that have summarised published data concerning the long-range
atmospheric transportation of Hg from industrial areas, conclude that there is scientific
evidence of the linkage between anthropogenic Hg emissions and elevated Hg concentrations
in remote areas (Fitzgerald et al., 1998; Jackson, 1997). For instance, in Sweden, scientists
have gathered a large amount of data that demonstrates the existence of a north-south gradient
with high Hg concentrations in environmental compartments (ie. soil, sediment, peat bogs and
rainwater) in the southern part and low levels in the northern part of the country.
Table 1 Post and pre-industrial mercury fluxes recorded in lake sediment cores
(Landers et al., 1998) Location Lake name Pre Hg Flux
(µg/m2/yr) Post Hg Flux
(µg/m2/yr) Source
Finland Iso-Iehm atampl 3.2 28.7 Verta (1989) Finland Vekea Kotinen 15.3 49.5 Verta (1989) Finland Sonnanen 2.7 42.1 Verta (1989) Finland Vakeinen 4.0 4.8 Verta (1989) Sweden Tussjon 2.5 17.0 Johansson (1985) Sweden Skarvsjon 1.9 14.3 Johansson (1985) Sweden Bjorken 4.0 9.0 Johansson (1985) Sweden Uggsjon 4.8 11.0 Johansson (1985) Russia Nyagome 23.3 30.2 Landers (1995) Russia Khuyudaturka 6.6 7.1 Landers (1995) W. Canada Ela 7.4 21.3 Lockhart (1995) W. Canada Kusawa 5.3 8.5 Lockhart (1995) W.Canada Amituk 7.0 28.4 Lockhart (1995) Quebec Jobert 18.9 33.9 Lucotte (1995) Quebec La Cabane 11.1 30.1 Lucotte (1995) USA Wonder 2.5 3.3 Landers (1995) USA Toolik 20.3 23.2 Landers (1995) USA Little Rock 10.0 40.3 Swain (1992) USA Kjostad 2.3 74.1 Swain (1992)

4
The same trend applies throughout Scandinavia, which excludes local Hg emitting sources in
Sweden as an explanation for the high concentration in these southern parts. Since the
prevailing winds in this part of the world are from southwest to northeast (ie. from areas in
Europe that are heavily industrialised), the conclusion that Hg is transported into Sweden and
the rest of Scandinavia by Hg polluted air masses from major emitting sources in Europe, is
supported. In addition, an improvement of the air quality in Sweden has been linked with
reduction of Hg emission from sources in Europe. Similar observations have occurred in
North America (Jackson, 1997). Furthermore, computer simulations (which investigate the
source-receptor relationship) (e.g. Pai et al., 1997; Petersen et al., 1995, 2001; Xu et al.,
2000a & b) as well as measurements of Hg concentrations in ambient air across Europe
(Wängberg et al., 2001) support the conclusion that Hg deposited in remote areas may
originate from anthropogenic sources far away. Thus, Hg is a global problem not only
affecting local areas that are heavily industrialised, but also remote areas far away from
emitting sources (e.g. Antarctic).
One of the most efficient means to determine how atmospheric Hg is transported, transformed
and deposited is through the use of numerical computer simulation, using so called air quality
models (AQM). The strength of these models are that they can be used to link emission
sources with deposition at receptors. Thus they can identify which sources are contributing
most intensively to an area and also investigate how deposition fluxes might vary across
regions. These models incorporate surface conditions, meteorological information, the
physics and chemistry known to affect atmospheric processes, along with estimated
anthropogenic emission data, to predict, spatially and temporally, ambient Hg concentrations
as well as deposition fluxes to environmental surfaces.
There are an increasing number of published simulation studies that have studied Hg
transportation, transformation and deposition on regional scales (e.g. Bullock, 2000a & b;
Bullock and Brehme, 2002; US EPA, 1997; Ilyn et al., 2001; Lee et al., 2001; Pai et al., 1997,
2000 a & b; Petersen et al., 1995, 2001; Xu et al., 2000a & b) as well as on an global scale
(e.g. Bergan et al., 1999, Bergan and Rohde, 2001; Seigneur et al., 2001; Travnikov and
Ryaboshapko, 2002). However, even though the accuracy of these models has increased over
the years many uncertainties still remain. In particular, a general lack of knowledge about Hg
emissions, their transformation and deposition processes create uncertainties when the
existing information about these processes is incorporated in the models (Bullock, 2000b).

5
1.2 The purpose and scope of this report
The purpose of this study is (i) to quantify the emissions of Hg from natural and
anthropogenic sources in Australia, and (ii) to conduct a dispersion and deposition simulation
of Hg in the central, coastal parts of New South Wales, using a three-dimensional regional
scale Eulerian air quality model (The Air Pollution Model (TAPM)). The obtained results will
be compared to existing publicised data related to the findings in this study.
The scope of this report concerns the atmospheric Hg transportation and transformation
processes, the sources of Hg to the atmosphere, and the deposition pathways of Hg to aquatic
and terrestrial compartments. Thus, transformation /transportation processes of Hg in and
between the oceans and terrestrial compartments are not included in this study and neither are
health issues related to exposure of Hg. Pollution abatement techniques and other emission
reducing action are not considered.
There are, as mentioned above, an extremely large number of scientific studies concerning
this subject and it is beyond the scope of this report to attempt a comprehensive review of the
published literature. Instead, based on selected published studies and data, an estimation of
anthropogenic Hg emissions, both from point and specific area sources, is performed, as well
as, an estimation of emissions from Australian natural sources. An Hg simulation study in
which secondary emission data is used as input data in the model is also performed. Since the
knowledge of the atmospheric transformation processes of Hg is essential for modelling its
atmospheric transportations, concentrations and deposition patterns, a general overview of the
different transformation processes are also presented in this paper.
The report consists of six sections. The atmospheric Hg chemistry is presented in Section 2
and 5. In Section 3, emissions from natural and anthropogenic sources are quantified. Section
4 describes the model and modelling approach utilised in this paper, as well as, the result
obtained from the Hg simulation. Finally, in Section 6, some conclusions from the results
presented in the report are drawn.

6
2. ATMOSPHERIC CHEMISTRY AND RESIDENCE TIME
In addition to presenting a summary of a more detailed description of chemical reactions in
section 5, this section gives a brief presentation of the different species of Hg that are present
in the atmosphere, their properties, their chemical and physical transformations, and their
deposition mechanisms.
2.1 The properties of atmospheric mercury species
Mercury is released to the atmosphere in three main forms; elemental Hg (Hg0), divalent Hg
(Hg(II)) and particulate phase mercury (Hgp))3 (EC, 2001) (Figure 1). The three different Hg
species have, due to differences in physical and chemical properties, different atmospheric
behaviour and residence times.
The prevailing Hg species in the atmosphere is elemental Hg (ca 98 %) (Lindquist et al.,
1991). Due to its substantial vapour pressure it exists predominantly in the gaseous phase4
(Schroeder et al., 1991). Background concentration of Hg0 in ambient air is approximately
1.3-1.5 ngm-3 in the Northern Hemisphere and 0.9-1.2 ngm-3 in the Southern Hemisphere (EC,
2001). Elemental Hg is relative unreactive (reacts slowly with atmospheric oxidants), it is
mainly transported back to the surface through dry deposition at a very low rate, and it is
highly insoluble which prevents it from being removed efficiently through wet deposition
(Lin and Pehkonen, 1999; Schroeder et al., 1991). All these properties combined lead to a
global distribution and an atmospheric residence time of approximately one-year (Bergan et
al., 1999)5. In addition, elemental Hg may be removed from the atmosphere by being oxidised
to divalent Hg or adsorbed onto particulate matter (EC, 2001; Lindquist et al., 1991) (Figure
1).
Divalent and particulate Hg, which are present in ambient air at concentration of less than 2
percent of Hg0, are much more water-soluble (at least 105 times more so than Hg0 (Linberg
3 Mercury also exists in a monovalent form Hg(I) (e.g. Hg2Cl2). However, it is extremely unstable and will rapidly disportionate to form Hg(II) and Hg0 (McElroy and Munthe, 1991). It is therefore assumed to have a minor importance in atmospheric mercury chemistry (Schroeder and Munthe, 1998). In addition to these species, methyl mercury is also believed to be emitted (mainly from industrial process), however, in much smaller quantities (US EPA, 1997). Natural sources are assumed to emit mainly elemental Hg (Lindquist et al., 1991). 4 The vaporisation rate of Hg approximately doubles each 10 0C increase in temperature. The saturation level of Hg in air increases logarithmically with increasing temperature. Thus, seasonal, daily and latitudinal changes in ambient air levels occur (Mitra, 1986). 5 Hg0 can be transported over long distances, up to 10 000 km, and hence enter the global Hg cycle (Porcella et al., 1996).

7
and Stratton, 1998) and are readily removed after emission on local to regional scales via wet
and dry processes6 (Lindquist et al., 1991; Slemr et al., 1985; Schroeder and Munthe, 1998).
These two inorganic Hg forms have residence times of a few hours to several months
(Lindquist et al., 1991). However, some fine particles can approach the residence time of
elemental Hg even after precipitation has occurred indicating that these may also be
distributed on a global scale (Porcella et al., 1996). Furthermore, particulate Hg is
exceptionally abundant in the atmosphere over polluted areas (eg. industrial sites) where it
may reach levels of 50 percent of the total Hg concentration (Schroder et al., 1991; Keeler et
al,. 1995; Pirrone et al., 1996).
Divalent Hg, frequently referred to as reactive gaseous mercury (RGM), can react with a
number of different ligands (OH-, Cl-, Br-, I-, SO32- and CN-) to form relatively stable
inorganic complexes (e.g. HgCl2 and Hg(OH)2) (Seigneur et al., 1994; Travnikov and
Ryaboshapko, 2002). In addition, divalent mercury may interact with organic molecules both
through chemical processes and by micro-organisms such as bacteria in aquatic systems
forming organic Hg compounds such as monomethyl mercury (MMM) (e.g. CH3HgCl,
CH3HgOH, CH3HgBr) and dimethyl mercury (DMA) (e.g. Hg(CH3)2) (Seigneur et al., 1994).
MMM is extremely toxic and of great environmental importance because of its ability to bio-
concentrate in, for instance, fish tissues, which in turn effect human health (especially the
central nervous system) following consumption (WHO 1990, 1991). DMM is highly volatile
and is rapidly released through the water phase to the atmosphere where it interacts with other
chemical species (see Section 5) (US EPA, 1997).
Particulate Hg is formed when divalent Hg complexes such as Hg(OH)2, HgCl2, HgSO3 and
Hg(NO3)2 are adsorbed onto particles particularly within atmospheric water droplets (Pleije
and Munthe 1995a,b; Seigneur et al., 1994). In a study by Seigneur et al (1998), it is
suggested that up to 35 % of the dissolved divalent Hg species can be adsorbed onto
particulate matter. In the gaseous phase, particulate divalent Hg consists mainly of solid
compounds such as HgO and HgS (Seigneur et al 1998; Travnikov and Ryaboshapko, 2002).
These compounds have a low solubility and are primarily removed via dry deposition,
however, approximateley 50 % of the Hg in atmospheric rainwater is represented by insoluble
compounds (Brosset and Lord, 1991), which indicates that a significant proportion
6 A significant part of the emissions from these two species may be deposited approximately 50 km from the emission point, although Hg(IIp) can be transported long distance if at high altitude (Porcella et al., 1996). According to the US EPA (1997) approximately 7-45 percent of the total Hg emitted is deposited within 50 km from the source. The two main factors determining the amount deposited are the source characteristics such as stack height and plume rise, and the speciation (the distribution between the Hg forms) of the Hg emitted (US EPA, 1997).

8
can be scavenged into the atmospheric aqueous phase.
Figure 1 Mercury oxidation, reduction and mass transfer processes in the atmosphere
Hg0(g)
Hg0(aq)
Hg0(ads)
Hg(II)(aq)
Hg(II)(g)
Hg(II)(ads)
Ant
hrop
ogen
ic so
urce
s
Nat
ural
sour
ces
Ant
hrop
ogen
ic so
urce
s
Ant
hrop
ogen
ic so
urce
s
Ant
hrop
ogen
ic so
urce
s Natural sources (including re-emission of previous deposited Hg) are also emitting Hgp (represented in the figure as Hg(ads)) and Hg(II) but in small quantities. The idea for the figure came from the front page of 2001 Special Issue of Atmospheric Environment (vol.35, no.17).
Although elemental Hg is present as a vapour in the atmosphere, it may also adsorb onto
particles and is hence subject to wet and dry deposition (EC, 2001). The amount that is
adsorbed is dependent upon the composition of the particle and the gas phase concentration of
Hg. The adsorption is more likely to occur when the particulate matter is rich in elemental
carbon (soot), since soot particles possess the highest sorption capacity (i.e. the adsorption
coefficient for Hg on soot is high) (Petersen et al., 1998; Pirrone et al., 2000). Another source
that incorporates Hg to particulate matter is combustion of fossil fuels where some of the Hg
present in the fuel is emitted bound to particulate matter. This type of bound Hg is not
released or engaged in any further reactions and is therefore deposited together with the
particle (EC, 2001).

9
2.2 Chemical reactions and interactions in the atmosphere
From the brief description above, it is clear that the speciation of atmospheric Hg forms is
critical to removal rates and transportation distance from emission sources. Near-source
contamination is most likely related to the emission of divalent and particulate form of Hg,
while the effects at some distance from the source are associated with elemental Hg. To
evaluate the global cycling of Hg and its effects in the environment it is important to
understand the different transformation processes, including transitions between the gaseous,
aqueous and soil phases, and chemical reactions in the gaseous and aqueous environment. In
the following paragraphs a brief description of partitioning mechanisms and different
atmospheric reactions is presented7.
In the atmosphere, the different Hg species and other substances will partition between the
liquid (eg rain and cloud droplets) and vapour phase under equilibrium conditions8 (illustrated
in Figure 12, section 5). The magnitude and the direction of the flux of a substance (eg.
elemental Hg) across the air/water interface is dependent on the concentration of the
substance in air and water relative to the Henry’s law constant (Table 21). The driving force
across the interface is either aqueous oxidation of elemental Hg to the more water soluble
divalent Hg form, which will lead to a transportation of Hg from the air to the raindrop, or
aqueous reduction of divalent Hg which will transport elemental Hg in the opposite direction9
(the upper part of Figure 1) (Pleijel and Munthe, 1995a,b; Schroeder et al., 1991). The other
reactants (oxidants and reducing agents) partitioning between the air/water interface are also
important since their concentration in the water phase determines the rate of reaction of Hg
and hence the Hg concentration in the droplet (Lin and Pehkonen, 1998a; Seigneur et al.
1994). It is these processes along with a number of factors, including temperature and
barometric pressure, which determine the amount of divalent Hg, and to some extent
elemental Hg that is removed from the atmosphere through wet deposition (Schroeder and
Munthe, 1998). The gaseous reactions where divalent Hg is formed are also of interest with
regard to aqueous chemistry of Hg since some of the Hg will also dissolve into the raindrop
(Lin and Pehkonen, 1997, 1998a; Pleijel and Munthe, 1995a,b). Thus, it will also affect the
concentration of Hg in the water droplet (right-hand side of Figure 1).
7 The exchange of mercury between atmospheric, marine and terrestrial compartments is dealt with separately in section 3.3. 8 The same process is applicable to air and marine, lake or other water surfaces. 9 Divalent Hg, due to its greater solubility and lower volatility, does not generally outgas from the aqueous phase to the atmosphere (Hedgecock and Pirrone, 2001).

10
In the atmospheric aqueous phase there are, as previously mentioned, two simultaneous
actions – oxidation of elemental Hg and reduction of divalent Hg. Important oxidants are (i)
ozone, (ii) hydroxyl radical and (iii) chlorine (Table 2, R1, R2 and R3, respectively). As
shown in table 2, the rate coefficient (k) of the hydroxyl radical reaction is significant higher
compared to the other reactions. However, depending on factors such as the individual
concentration of a substance, its degree of solubility, the pH and the temperature of the water
phase, either oxidation path can be dominant. For instance, in a relative polluted atmosphere
with an ozone concentration exceeding 20 ppb the radical reaction only contributes to 10 % of
the total oxidation (Travnikov and Ryaboshapko, 2002). Chlorine, which compared to other
oxidants is present at much lower concentration in ambient air, can also cause considerable
oxidation, due to its higher solubility10 (Lin and Pehkonen, 1998b). This is especially the case
in the marine boundary layer where chlorine is produced by the presence of sea-salt particles
(Keene et al., 1993; Oum et al., 1998). Temporal variability also occurs, where chlorine is
dominant during the night when the concentration of both ozone and the hydroxyl radical is
greatly decreased due to the absence of sunlight (Lin and Pehkonen, 1999b).
Reducing agents are primarily (i) sulfite complexes and (ii) hydroperoxide radical (Table 2,
R4 and R6, respectively). Photo-reduction of divalent Hg complexes (eg Hg(OH)2) does also
occur, although at a much lower rate (R5). Depending on the prevailing conditions in the
aquatic solution, divalent Hg forms complexes with different constituents. For instance, in the
presence of high chloride concentrations, Hg(II) is mostly
Table 2 Chemical transformations in the aqueous phase
No
Reaction
k (M-1s-1 or else indicated)
t1/2
Reference
R1 Hg0(aq) + O3(aq) → Hg2+
(aq) + OH- (aq) + O2(aq) (4.7±2.2) ×107 40 s Munthe, 1992
R2 Hg0(aq) + · OH(aq) → Hg+
(aq) + OH- (aq)
Hg+(aq) + · OH(aq) → Hg2+
(aq) + OH- (aq)
Hg0
(aq) + · OH(aq) → · HgOH(aq)
· HgOH(aq) + O2(aq) → Hg(OH)2(aq) + H+(aq) + O-
2(aq
2.0 ×109
(2.4±0.3)×109
350 s
290 s
Lin and Pehkonen, 1998
Gårdfeldt et al,
2001 R3 HOCl(aq) + Hg0
(aq) → Hg2+(aq) + Cl-
(aq) + OH- (aq)
OCl-(aq) + Hg0
(aq) → Hg2+(aq) + Cl-
(aq) + OH- (aq)
(2.09±0.06)×106
(1.99±0.05)×106 - -
Lin and Pehkonen, 1998
R4 HgSO3(aq) → Hg0(aq) + products
HgSO3(aq) → Hg0
(aq) + S(VI)
0.6 s-1
(0.0106±0.0009) s-1
1 s
65 s
Munthe et al., 1991
Van Loon et al, 2000
R5 Hg(OH)2(aq) → Hg0(aq) + products 3×10-7s-1 600 h Xiao et al., 1994
R6 HO·2(aq) + Hg(II)(aq) → Hg(I)(aq) + O2(aq) + H+
(aq) HO·
2(aq) + Hg(I)(aq) → Hg0(aq) + O2(aq) + H+
(aq) 1.7 ×104 1 h Lin and
Pehkonen, 1998 The calculation of the half-life (t1/2) is presented in section 5.
10 The solubility, which depends on both pH and the chloride concentration, is governed by the effective Henry law constant (Lin and Pehkonen, 1999a & b) (section 5.1.1.3).

11
present as HgCl2 (Lin and Pehkonen, 1999b). It is not until the chloride concentration is below
5×10-6 M, that the sulfite reduction starts to become significant (Ryaboshapko et al., 2001).
In Europe the chloride concentration in atmospheric water is always above 2×10-6 M, which
indicates that the contribution from S(IV) reduction is, under these conditions, small (Ilyin et
al., 2001).
In the atmospheric gaseous phase, there are a number of chemical substances that are capable
of oxidising elemental Hg. Those mostly referred to are (i) ozone, (ii) hydroxyl radical, (iii)
nitrate radical and (iv) hydrogen peroxide (Table 3, R7, R8, R9 and R10, respectively). The
gaseous reaction rate is significant slower than the reactions in the aqueous phase. However,
the two rates are comparable due to the relatively low liquid water content in the atmosphere
along with the low solubility of elemental Hg (EC, 2001). Since divalent Hg (the product of
the reactions) is less volatile than Hg0 it tends to condense onto atmospheric particulate matter
which is either scavenged into atmospheric water droplets or dry deposited to marine or
terrestrial surfaces (EC, 2001). It may also, as above mentioned, dissolve into precipitation
elements. Gaseous reactions between DMM and other species are presented in section 5.
Table 3 Chemical transformations in the gaseous phase
No. Reaction k (cm3molec.-1s-1) t1/2 τ
Reference
R7 Hg0(g) + O3(aq) → Hg2+
(g) + O2(g) (3±2)×10-20 1.2 yr 1.7 yr Hall, 1995 R8 Hg0
(g) + · OH(g) → · HgOH(g) · HgOH(g) + O2(g) → HgO(g) + HO·
2(g) (8.7±2.8)×10-14 0.25 yr 0.4 yr Sommar et
al., 2001 R9 Hg0
(g) + NO·2(g) → HgO(g) + NO2(g) 4 ×10-15 20 d 30 d Sommar et
al., 1997 R10 Hg0
(g) + H2O2(g) → Hg(OH)2(g) 4.0×10-16 24 yr 34 yr Tokos et al., 1998
The calculation of the half-life (t1/2) and the residence time (τ) is presented in section 5.

12
3. EMISSION OF MERCURY
3.1 Definition
The main sources of emissions of Hg to the atmosphere are defined as follows (EPAMP,
1994):
• Natural mercury emissions refer to the mobilisation and release of geologically bound
Hg by natural processes, with mass transfer of Hg to the atmosphere.
• Re-emission of mercury is the mass transfer of Hg to the atmosphere by biologic and
geologic processes drawing from a pool of Hg that was deposited to earth’s surface
after initial mobilisation by either anthropogenic or natural activities.
• Anthropogenic mercury emissions refer to the mobilisation and release of
geologically bound Hg by human activities, with mass transfer of Hg to the
atmosphere.
Thus, the total amount of Hg in the atmosphere is from a mix of emission from natural,
anthropogenic and re-emission sources.
3.2 The global atmospheric mercury cycle
There are a number of studies that have quantified the major fluxes of Hg on a global scale to
and from the atmosphere. These studies are either based on calculations using mass balances
or models. Depending on the study (e.g. which assumptions are being made, what is included
in the model etc.) quite different results are often achieved, reflecting the complexity and
uncertainties surrounding these flux estimates. Despite the existing uncertainties, those
studies suggest that 5800 - 7000 tonnes of Hg are released annually from the combination of
anthropogenic and natural sources.
Anthropogenic and natural sources (i.e. oceans, land and vegetation) are, in the broadest
sense, continuously releasing Hg, both directly and indirectly via re-emission to the
atmosphere11. The atmosphere is on the other hand also constantly depositing Hg via different
mechanisms to receiving natural surfaces12 (Schroeder and Munthe, 1998). Thus, Hg is being
11 The mentioned sources are also releasing Hg directly to land and water but those processes are not included in this study. 12 The emission/re-emission and deposition from/to natural surfaces are also called bi-directional exchange of Hg, section 3.3.2.

13
Figure 2 The global atmospheric mercury cycle
The global atmospheric Hg pool (Consists mainly of Hg0, with a residence time of ~ 1 yr)
Ant
hrop
ogen
ic e
mis
sion
Nat
ural
em
issi
on
Re-
emis
sion
from
nat
ural
su
rfac
es
Glo
bal d
epos
ition
to
natu
rals
urfa
ces
Atmosphere
Ant
hrop
ogen
ic
emis
sion
Loca
l/reg
iona
l de
posi
tion
Dry Wet
Emissions of Hg0 are entering the global Hg pool while divalent and particulate Hg is deposited to local and regional areas (left-hand part of Figure 2).
cycled between the earth and the atmosphere, which is presented in Figure 2. As described in
Section 2, different Hg species have different properties, which consequently effect their
atmospheric residence time. Elemental Hg is in most global distribution studies, assumed to
have a residence time of one year (Bergan et al., 1999). Due to its substantial atmospheric
lifetime, Hg0 enters what is referred to as the global atmospheric Hg pool where it circulates
with the prevailing winds (Porcella et al., 1996). Thus, Hg0 released from a local source can
due to its volatile nature be cycled through the global environment until it is finally stored in
soil or sediments (or alternatively converted to methyl-Hg) far away from the source.
Compared to Hg0, divalent and particulate Hg have relatively short atmospheric residence
times, which hinder their entrance into the global pool of Hg (Figure 2).
In a study by Mason et al (1994), it is suggested that 5000 tonnes of Hg0 enter the global
atmospheric Hg pool each year. Of these emissions, 2000 tonnes are emitted from
anthropogenic sources, 1000 tonnes from terrestrial surfaces and 2000 tonnes from the oceans
(including re-emission) (Table 4). An additional 2000 tonnes are, according to the study,
released from anthropogenic sources. However, these amounts are deposited locally and do
not enter the global distribution. Thus, the total anthropogenic emission and the total Hg flux
to the atmosphere is 4000 (59% of total emissions) and 7000 tonnes/yr, respectively. The pre-
industrial flux is estimated to 1600 tonnes annually (Table 4); 1000 tonnes of which was
released from terrestrial surfaces and 600 tonnes from the oceans.

14
Table 4 Estimated global emissions (tonnes/yr) (Mason et al., 1994)
Source Hg0 Hg(II), Hgp Total Anthropogenic 2000 (0) 2000 (0) 4000 (0) Oceana 2000 (600) 0 (0) 2000 (600) Land 1000 (1000) 0 (0) 1000 (1000) Total 5000 (1600) 2000 (0) 7000 (1600)
Figures within brackets are estimated pre-industrial emissions. a Of the 2000 tonnes, 1400 tonnes is anthropogenically derived Hg previously deposited to the oceans. The speciation of Hg in table 4 is based on knowledge of different Hg species atmospheric properties, which affects their dispersion and deposition patterns. Thus, 50 percent of the anthropogenic emissions is in the form of elemental Hg and the other 50 percent are a mix of divalent and particulate Hg. Natural sources are mainly emitting elemental Hg (section 3.3). Of the 2000 tonnes Hg emitted from the oceans each year 1400 tonnes is, according to the
study, re-emission of previously deposited anthropogenic Hg (ie. 70 % of the emissions). As a
consequence, the annual anthropogenic contribution (direct and indirect) to the global Hg
pool is 3400 tonnes/yr or 68 percent of the Hg flux to the atmosphere. However, if the 2000
tonnes deposited locally is included in the equation anthropogenic sources accounts for 77
percent of the global input each year (5400 tonnes/yr).
Based on the assumption that elemental Hg has a residence time of one year, the study by
Mason et al (1994) concludes that of the 5000 tonnes Hg deposited each year, 2000 tonnes
enter the oceans and 3000 tonnes the terrestrial surfaces. The authors also estimate the total
anthropogenic Hg contribution to the atmosphere over the past century to be approximately
200 000 tonnes of which 95 percent has accumulated in terrestrial soil, 3.6 percent is present
in ocean surface water, and 1.7 percent is left in the atmosphere.
In a later study by Hudson et al (1995), which is a revision of the model by Mason et al
(1994), a similar distribution between natural and anthropogenic sources is calculated with the
result that approximately 2200 and 4600 tonnes of Hg (including 600 tonnes of re-emission
from the oceans and 2000 tonnes which is deposited locally) is believed to be emitted each
year(Table 6). Thus, the result from these two mentioned studies suggest that the
anthropogenic flux of elemental Hg to the global atmosphere is in the range of 2700-3400
tonnes/yr (or 4700-5400 tonnes/yr including emissions of divalent and particulate Hg).
In contrast to the suggested quantities of anthropogenic Hg released annually (Mason et al.,
1994; Hudson et al., 1995), Pirrone et al (1996) estimate the total global anthropogenic Hg

15
emissions during the period of 1983-1992 to be in the range of 1900-2200 tonnes/yr, with a
mean of 2100 tonnes/yr13. These estimated emission fluxes, which include both
Table 5 Estimated global emissions (tonnes/yr) (Bergan and Rohde, 2001)
Emission source Hg0 Hg(II) Total Anthropogenic 1286 (1300) 857 (850) 2143 (2150) Re-emission 1260 (2000) 0 1260 (2000) Natural land 1320 (500) 0 1320 (500) Natural sea 1100 (1400) 0 1100 (1400) Total 4966 (5200) 857 (850) 5823 (6050)
The model simulates the global distribution of Hg0 and divalent mercury compounds Hg(II). Figures within brackets are estimates from a previous study (Bergan et al., 1999). The speciation of the anthropogenic emission is 60 percent Hg0 and 40 percent Hg(II), which is similar to other studies (Table 14).
elemental and divalent Hg species, amount to approximately half of the emissions estimated
by Mason et al (1994) and Hudson et al (1995). According to Pirrone at al (1996) one-third of
the global atmospheric burden of Hg is due to direct anthropogenic output, the other two-
thirds are equally due to natural sources and re-emission of Hg. Thus, the total estimated
emission of Hg is approximately 6000 tonnes/yr (Table 6).
In a model simulation by Bergan and Rohde (2001) (Table 5), which is derived from a
previous study by Bergan et al 1999, 5823 tonnes of Hg is estimated to be released each year.
Of these emissions, 4966 tonnes/yr enter the global distribution while 857 tonnes/yr is
deposited both locally and regionally. In Table 5, the direct anthropogenic emissions amount
to 2143 tonnes/yr, which is similar to the emission levels suggested by Pirrone et al. 1996.
Compared to the investigation by Bergan et al (1999), the result from the simulation study by
Bergan and Rohde (2001) show a significantly different amount of estimated Hg emissions.
For example, the emission of Hg from land base sources has increased by a factor of 3.
Based on the result from the four studies presented (Mason et al., 1994; Hudson et al., 1995;
Pirrone et al., 1996; Bergan and Rohde, 2001) (listed in Table 6) the following points can be
summarized:
• Natural sources including re-emission emits approximately 2800–4000 tonnes of Hg0
annually. Re-emission accounts for 35–50 percent of this value.
• The anthropogenic output varies between 2000–4000 tonnes Hg/yr, which is 35–60
percent of the total annual emissions.
13 Pacyna and Pacyna (2002) presented similar estimations with a total anthropogenic emission of 2100 and 1900 tonnes/yr during 1990 and 1995, respectively (Table 10, section 3.4.1).

16
• The atmospheric Hg pool receives approximately 5000 tonnes of Hg0 each year. Based on
the assumption of a residence time of one year for Hg0, the same amount of Hg (5000
tonnes) is suggested to be deposited to natural surfaces annually and globally.
Table 6 Summary of estimated global emissions (tonnes/yr)
Emission source Mason et al. 1994
Hudson et al., 1995
Pirrone et al, 1996
Bergan and Rohde, 2001
Anthropogenic 4000 4000 2000 2143 Re-emission 1400 600 2000 1260 Natural 1600 2200 2000 2420 Natural land 1000 900 - 1320 Natural sea 600 1300 - 1100 Total 7000 6800 6000 5823
3.3 Natural mercury emission
3.3.1 Background
Mercury, mainly in the form of elemental Hg, is released from natural sources (Lindquist et
al., 1991)14. The magnitude of the emissions of Hg released depends on a number of
biological, chemical, physical, and meteorological factors of which few are fully understood.
There are, however, an increasing number of studies conducted on this topic and the more
important of these are presented in this section. Based on the results from these studies a
crude calculation of emissions from natural sources in Australia is performed and presented.
It is, however, difficult to verify the result of the calculation since there is no published data
regarding emissions of this sort in Australia. There is nevertheless an accordance between the
estimated emissions from natural sources in this study and the estimated average global
emissions from natural sources (Mason et al., 1994; Lindberg et al., 1998), indicating that a
reasonable estimation has been made.
Mercury is primarily present in the earth’s crust and mantle. It occurs naturally in
hydrothermal deposits in rocks as various minerals (eg. cinnabar, HgS), in coal, and in some
sedimentary rocks, especially shales of high organic and sulfide content (Schroeder and
Munthe, 1998). Areas that are geologically and naturally enriched in Hg (i.e. areas that have
natural elevated Hg levels) are located globally in three broad belts. One of these belts starts
in eastern Australia, continues via New Zealand, Indonesia, Philippines and ends in Japan
14 Dimethyl mercury is also released from natural sources, however, in much smaller quantities than elemental Hg (Schroeder and Munthe, 1998; US EPA, 1997). Other sources such as forest fires in addition to elemental Hg, also emit divalent and particulate Hg (Porcella, 1996).

17
(Gustin et al., 2000). The world’s largest Hg deposit is found in the Mediterranean region
(Ferrara et al., 1998a,b). Mercury also exists as a trace element in numerous secondary
sources in terrestrial environments (eg. soil and vegetation) and in the ocean (Jackson, 1996).
Divalent Hg, originating from both natural and anthropogenic sources, is the predominant
form of Hg deposited to the earth (Linberg and Stratton, 1998; Bergan et al., 1999). After
deposition some of the Hg is reduced chemically and bio-chemically to elemental Hg which
due to its volatile nature is re-emitted back to the atmosphere. This bi-directional exchange
(deposition-to-emission) of Hg across the air-surface interface makes it difficult to distinguish
between emissions from a “pure” natural source and re-emission of previously deposited Hg.
As described in the previous section, a large part (up to 50 percent) of the Hg that is emitted
from natural sources is actually of anthropogenic origin (Mason et al., 1994). The only
emissions that by definition are natural and hence undisturbed by anthropogenic influences
are eruptions from volcanoes (one of the major "natural" Hg sources), emissions of Hg from
the earth's subsurface crust and degassing from mineralised soil. Evaporation of Hg from the
ocean's surface, emission of Hg from soil, vegetation and the release of Hg in forest fires, are
consequently a mix of natural and re-emitted Hg. From this brief review it is clear that care
needs to be taken when referring to natural emissions since the term "natural" in this context
maybe somewhat misleading. Failure to take this ambiguous terminology into consideration
may lead either to an overestimation or an underestimation of the contribution from a specific
source. For instance, direct anthropogenic emission to the atmosphere is not the same as the
total anthropogenic input, which also include Hg recycled from secondary sources in the
natural environment. In the following pages "natural emissions" will by definition also
include re-emissions.
3.3.2 Bi-directional exchange of mercury
The bi-directional exchange of Hg across the air/water, air/land and air/vegetation interface is
governed by emission and deposition mechanisms along with physical, chemical and
biological interactions in the media. In the following paragraphs a short description of each
exchange process is given along with a table of emission fluxes from different surfaces (Table
7).
The air-sea exchange is considered to be one of the major natural processes to release Hg to
the atmosphere (Mason et al., 1994). The efficiency by which this evaporation occurs depends
upon parameters such as (i) the intensity of the solar radiation, (ii) the temperature of the air
parcel above the seawater, (iii) the water temperature, and (iv) the concentration of Hg in the

18
surface water (Ferrara et al., 2000). The concentration of Hg in the water phase is partially
determined by the amount of Hg that enters the sea (eg. via wet deposition), and partially on
chemical and physical processes occurring across the air-water interface, as described in
section 2. The magnitude of the evaporation shows a clear diurnal trend with maximum fluxes
during days when the temperature and the level of solar radiation is at its highest, and
minimum fluxes during nights when respective level is at their lowest. In addition, seasonal
patterns affect the Hg flux with minimum flux values during winter times and maximum flux
values during the summer (Ferrara et al., 2000).
The air-soil exchange processes are less well known, however, there are a number of
investigations where the Hg flux was measured over different types of soils using dynamic
flux chamber techniques (Table 7). From the results of these studies it is apparent that some
parameters affect not only the temporal trends but also the magnitude of the Hg flux. The
emission of Hg from soil is driven by (i) the intensity of solar radiation (positive correlation,
pc), (ii) soil temperature (strong pc), (iii) the level of soil moisture (strong pc), (iv) the level
of Hg concentration in the soil (strong pc), (v) barometric pressure (pc), and (vi) the
turbulence of the air parcel above the surface (pc) (Gustin et al., 1996, 2000; Gillis and
Miller, 2000a; Kim et al., 2002; Lindberg et al. 1998; Poissant and Casmir, 1998; Xu et al.,
1999). The Hg flux is characterised by the same diurnal and seasonal patterns as the air-water
exchange with high emissions during summer periods and days (Kim et al., 2002; Poissant
and Casmir, 1998). Moreover, it is estimated that the Hg flux from the soil-air exchange is 6-8
times greater than that of the water-air exchange (Poissant and Casimir, 1998).
The relative importance of the different parameters (mentioned above) is, however, not
clearly understood. Data from different studies suggest though that the Hg concentration in
Hg enriched soil determine the magnitude of the emissions of Hg from the soil to the
atmosphere (Engle et al., 2001; Gustin et al., 2000). In natural soil, which has a low Hg
content, the air-soil exchange is highly dependent on the soil temperature and the Hg
concentration gradient between the soil and the air in the vicinity of the soil surface (Gillis
and Miller, 2000a). Thus, soil has the potential to be a source or sink of Hg depending on
these parameters. According to the study by Gillis and Miller (2000a), Hg emission from the
soil occurs when the soil concentration [Hgsoil] is greater than the [Hgair], and adsorption
occurs when [Hgair]> [Hgsoil]. Furthermore, even if there is a strong positive correlation
between the Hg flux and the soil temperature (Ts), there is no correlation between [Hgair] and
Ts. Thus, the Ts and the Hg concentration gradient should according to the study be treated as
independent variables affecting the Hg flux rate.

19
The air-vegetation exchange is in most studies measured over forest canopies. There is
however a lack of data, which to some extent is explained by the difficulties in measuring at
tenths of metres above the ground as these studies require. In Table 7, two different flux
studies are presented, of which one is based on a model and the other on field measurements.
Table 7 Emission rates of Hg from different natural surfaces
Country
Surface type
Period
Day/ Night
Hg flux rate ngm-2h-1
Reference
NTSa (polluted coastal zone)
Sea surface Summer D N
11.25 2.4
Ferrara et al., 2000
NTS (unpoll. costal zone)
Sea surface Summer
Winter
D N D
0.7-10.1 1
0.7-2.0
Ferrara et al., 2000
NTS (off shore) Sea surface Summer D N
2.5 1.16
Ferrara et al., 2000
Sweden Lake surface - - 2.05-20.5 Schroeder et al., 1989. Xiao et al., 1991.
North Sea Sea surface - - 1.6-2.5 Cossa et al., 1996 South Europe Top soil Summer D
N 4-5 1
Pirrone et al., 2000
Oak Ridge, Tennessee
Open field soil Deciduous forest
soil
April-August
- -
12-45 2-7
Carpi and Lindberg, 1998
Quebecb
Rural grassy site
Lake surface
July
July
D
D
0.62-8.3; 2.95 (mean)
1
Poissant and Casimir, 1998
Minnesotac Top soil May D/N 9.67 (mean) Cobos et al., 2002 WBWd, Tennessee
Deciduous forest canopy
July-Sept D 7-290 100±80 (mean)
Linberg et al., 1998
Northeast USe Deciduous and mixed forest canopy
Sea surface Crop land
July
July July
D/N
D/N D/N
22 (mean)g
2.6 (mean)g 32 (mean)g
Xu et al., 1999
Greenhousef Mixed tree canopy - D/N 1.7-5.5 (mean) Hanson et al., 1995 All studies except one (Northeast US) are based on field measurements via flux chambers. The study in Northeast US is based on a newly developed simulation model. a NTS: Northern-Tyrrenian Sea (Italy). b The site is surrounded by farms and some wooded area. c The site has been continuously cropped in corn, soybeans, and alfalfa for at least two decades. d Walker Branch Watershed, located 3 km from a former weapons plant and 20 km from two large coal-fired power plants. e The study/model covered a region (34×41 grids in the horizontal direction, with grid size of (12 km)2) of the Northeast US and a part of the Atlantic Ocean. Six basic cover types were included in the calculations: urban, agriculture, deciduous and mixed forest, and water. f Measurements of Hg0 exchange with white oak, red maple, Norway spruce and yellow-popular under controlled conditions in a greenhouse. g The emissions are net emission, ie. emission-dry deposition.

20
Table 8 Global natural emission of Hg from forests (Lindberg et al., 1998)
Forest type Q Range (ngm-2h-1) Area km2 Emission t/yr Boreal forests 0.08 - 0.8 1.37×107 10 - 100 Temperate forests 3.3 - 7.7 1.04×107 300 - 700 Tropical forests 3.3 - 7.7 1.76×107 540 - 1200 Total 4.17×107 940 - 2000
All data is based on measured fluxes, which are then scaled up, temporally and spatially, to represent the total emission from each type of forest. The study based on field measurements has scaled up individual Hg fluxes to represent
emissions from three different types of forests covers (Lindberg at al., 1998). The results are
presented in Table 8. From the data presented it is clear that temperate and tropical forests are
the major Hg emitters, which according to the study is to be expected considering the
favourable climatic conditions in these kinds of forest (see below) (Linberg et al., 1998).
Forests are in general believed to act as dynamic exchange surfaces for Hg, which can
function as sources or sinks of elemental Hg depending on factors such as (i) leaf
temperature, (ii) leaf surface conditions (wet vs. dry), (iii) level of atmospheric oxidants, (iv)
temporal fluctuation (day/night), and (v) biological factors (Hanson et al., 1995; Lindberg et
al., 1998). Depending on the balance between these factors, which is refereed to as a
compensation point, the forest is either a net emitter or receiver (via dry deposition) of
elemental Hg. For instance, at an atmospheric concentration of elemental Hg of <1.5 ngm-3
the foliage is releasing Hg to the atmosphere. It is not until the air concentration is between
10-20 ngm-3 that dry deposition becomes significant (Hanson et al., 1995; Lindberg et al.,
1998). In addition, both deposition and emission follows diurnal pattern with high values
during the day (Xu et al., 1999), and both mechanisms experience reduced fluxes during
drought (Hanson et al, 1995; Lindberg et al., 1998).
In addition to a function in the exchange processes of Hg, forests can in the context of forest
fires emit significant amounts of Hg. The Australian National Pollution Inventory (NPI)
estimates that 1.5-3 g Hg/ha is released to the atmosphere in these fires (NPI, 1999a)15.
15 Sydney, and other parts of Australia, experiences each year large bushfires. In the end of 2001 (24 December 2002 – 16 January 2002), 733 000 ha of bush was burnt around Sydney (The Sun-Herald, December 8, 2002; www.bushfire.nsw.gov.au). Assuming a fuel load corresponding to that of a forest, these fires emitted approximately 1.1 - 2.2 tonnes of Hg.

21
3.3.3 Estimated natural emissions of mercury from Australia
The Australian continent covers approximately 7.7 million square kilometres with a total
population of 19.6 million (ABS, 2002a & c). The majority of the population is concentrated
along the eastern and southeastern coasts. Australia's land cover is diverse and complex, it
includes temperate, tropical, sub-tropical environments along with deserts bush- and
grasslands, alpine areas and arable land. It is estimated that grazing lands cover 57 percent of
Australia, forests 21 percent and cropland 3 percent. The remaining part of the land cover is
included in a category named "other", which might range from deserts to urban areas) (ABS,
2002d; NFI, 2001). The country is rich in natural resources such as minerals and fossil fuels
(ABS, 2002b,d).
In order to derive estimates of annual natural Hg flux from Australia, individual flux
measurements must be integrated over space and time. This process is complex from three
aspects. Firstly, all available measurements of Hg emissions originate from the Northern
Hemisphere, which has a different type of climate, vegetation and a higher population density
to that of Australia16. Since emission from natural sources also include re-emission of
anthropogenic Hg it would be unreasonable to assume similar fluxes, on an average, for
Australia as for the countries represented in Table 7. According to the calculations presented
in Table 12, Australia emits approximately 0.54 g Hgtot/capita/year which can be compared to
figures from Sweden (0.1 g Hgtot/capita/yr), the UK (0.21), Europe (0.43), the US (0.62), and
Canada (1.07). As the country-by-country comparison shows, Australia appears to be a
significant global per capita emitter. However, if 40 percent of the total anthropogenic Hg
emission from the previously stated countries is deposited and equally divided over their
landmasses, each country receives a deposition flux of 1.4, 0.8, 51.9, 25.2, 18.3, and 3.3 g
Hgkm-2yr-1, respectively (or 0.15, 0.09, 5.9, 1.1, 0.84, and 0.15 ngm-2h-1)17. Thus, the land
cover in Australia receives on average 2 - 40 (excluding Sweden) times less Hg per year and
km2 from its own Hg emitting sources than received in the listed countries. These deposition
fluxes of Hg refer only to one part of the overall deposited Hg and do not say anything
directly about the magnitude of the emissions from natural sources. However, these
16 There is to the extent of the literature search conducted in this study no flux measurements from natural surfaces in climates similar to that of Australia (eg. temperate forests, rainforests and deserts). 17 In the investigation by Bergan and Rohde, 2001, Pacyna and Payna 2002, approximately 40 % of the total anthropogenic emissions were divalent and particulate Hg. Since these two Hg species have a relative short atmospheric residence time they tend to deposit on a local to regional scale. Thus, it is assumed that all emitted divalent/particulate Hg is deposited within respective countries borders. This assumption is based on the EPA (1997) study which concluded that about 98 % of the deposited anthropogenic Hg (to the 48 states included in the model) was emitted in the form of divalent and particulate Hg.

22
deposition fluxes are a reasonable indicator of how much Hg, proportionally, might be re-
emitted back to the atmosphere and hence enter the global Hg pool. This is also verified by
the previously presented studies which showed that the Hg content in the emitting media is
positively correlated with the emission flux. The same emission trend is demonstrated by the
data in Table 7 where local anthropogenic sources seem to have a significant impact on the
magnitude of the emissions from natural areas. For instance, in Tennessee, forest canopies
close to local pollution sources, emit significantly more elemental Hg than is estimated to be
emitted over a large part of the Northeastern US. Similar differences in Hg emissions can be
observed from soil where measurements range between 0.62 and 45 ng/m2/h. Thus, emissions
of Hg from natural areas in Australia are on an average most likely smaller than emissions
from similar areas in the countries of the Northern Hemisphere.
The second aspect which makes the integration of individual flux measurements over
Australia difficult to conduct is the large number of variables (eg. temperature, solar
radiation, Hg content in the emitting media, precipitation, diurnal and seasonal trends) which
affect the magnitude of Hg flux across the air-surface interface. To consider all possible
combinations of these variables is beyond the bounds of possibility for this study. A diurnal
trend is however included in the performed calculations.
A third aspect concerns the difficulty of having only a few studies (~10) conducted during a
specific time range (several hours up to days) and over a small land area (< 1 m2) to represent
the complexity of a continent. This kind of mathematical integration is subject to many
uncertainties and the result needs therefore to be interpreted accordingly. Moreover, most of
the studies presented in Tables 7 and 8 have used dynamic flux chamber techniques to
investigate the Hg flux. This methodology has several limitations; firstly, the flux chamber
alters the environment of the area of the study by affecting wind speed, turbulence and solar
radiation. The alterations of these environmental factors have an accentuating effect on the
derived Hg flux, as the measurement times are extended (Cobos et al., 2002). Thus, studies
that are only conducted during a short time period (hours) will experience a less pronounced
effect on the derived flux than measurements conducted for days. Secondly, there is
dependence between the rate of the sample flow through the chamber and the measured flux
rate. Altering the sample flow rate in any significant way may introduce great uncertainties to
the derived flux rate (Cobos et al., 2002). Thirdly, there is a significant correlation between
the wind speed and the measured flux rate despite the fact that most of the wind is excluded
from the chamber (Wallschlager et al., 1999). A wind speed of 1 ms-1 and 2 ms-1 decreases the
measured flux rates by 40 and 90 percent, respectively (Gillis and Miller, 2000b).

23
It is apparent that in order to scale up natural source emissions to represent Australia some
sort of crude assumption has to be made concerning the appropriate Hg fluxes for the area of
study. The following points are therefore considered:
(i) Even if the flux results from the studies in Tables 7 and 8 are based upon types of forest
and soil that do not exist in Australia, the fluxes, are as such, assumed to be
representative for Australia (the choice of a specific flux rate, see (ii)). Forests and
lakes are the two categories, which are easiest to distinguish from the Australian land
cover and hence appoint appropriate fluxes. The rest of the land cover is more diverse
and complex which makes the selection process more complicated. Since deserts, bush,
and grasslands cover a large part of Australia, the derived flux for the rest of the
country is from measurements over topsoil and rural grassy sites. Thus, the selected
flux represents an average emission factor for all possible surfaces within the area.
(ii) As discussed earlier the fluxes shown in Table 7 lie most likely in the upper region of
that emitted from natural areas in Australia. The calculated annual natural Hg emission
from Australia is therefore based on fluxes from the lower end of the range presented in
Table 9. A minimum and maximum flux is determined based on the assumption
presented and an upper and lower emission flux from natural surfaces in Australia is
calculated. The results are listed in Table 9.
(iii) Most of the studies in Table 7 were conducted during the summer and the Hg fluxes
presented are therefore assumed to represent the average climatic conditions in
Australia18. To account for the diurnal trends with low emissions at nights, fluxes are
calculated for a 12-hour day (i.e. zero flux during the night, which is represented in the
calculations by dividing the Hg fluxes by 2).
Using the Hg flux data presented in Table 9, a total annual emission of Hg from natural
sources (including urban areas) to the atmosphere is estimated to be in the range of 130 - 270
tonnes/yr (with a mean of 200 tonnes/yr), representing an emission rate of 17 - 35 µgm-2yr-1
(or 2 - 4 ngm-2h-1). Since there have been no similar investigations performed in Australia it is
difficult to verify this result for the Australian situation. If the result in this study is compared
to the average annual natural Hg emissions from Europe of approximately 250-300 tonnes/yr
(25-30 µgm-2yr-1or 2.9-3.4 ngm-2h-1)19 (Axenfeld et al., 1991; Pacyna et al., 2001) and to the
18 The magnitude of some of the variables affecting the flux is most likely significantly important due to the Australian climate with a high average temperature and level of solar radiation. 19 The area of Europe is 9 892 923 km2 (Geoscience Australia, 2002, http://www.auslig.gov.au/facts).

24
estimated emission from the earth's total landmasses of 1000 - 3200 tonnes/yr (7.7 - 24 µgm-
2yr-1 or 0.8 - 2.7 ngm-2h-1)20,21(Mason et al., 1994; Lindberg et al., 1998), the upper end of the
calculated range of Hg emissions for Australia is somewhat high. If the average emission rate
from the earth's landmasses is used instead of the fluxes listed in Table 9 the average emission
from land sources in Australia is within the range of 54 - 182 t/yr, with a mean of 118 t/yr.
Thus, an emission rate of 130 tonnes/yr is therefore probably the more reasonable initial
assumption based on these estimates.
Table 9 Estimated emission of Hg from natural land surfaces in Australia
Land cover Q range (ngm-2h-1)
Qmin (ngm-2h-1)
Qmax (ngm-2h-1)
Area km2
Mmin ton/yr
Mmax ton/yr
Foresta 7-290, 22, Table 8 3.5 7.7 1644120 50.4 111 Lakeb 1, 2.05-20.5 0.5 10 15267 0.1 1.5 Rural grassy sites/Soil/Otherc
1,4-5,0.62-8.3(2.95), 9.67, 32, 12-45
1.5 3 6032573 80 156
Totald 7691960 130.5 268.5
a BRS, National Forest Inventory, 2001. http://www.brs.gov.au/npi/. 3.5 ngm-2h-1is the mean of 7, however, it is also close to the average estimates of 3.3 ngm-2h-1 from forests presented in table 8. The flux of 7.7 is from table 8. b Geoscience Australia, 2002. (Area of major lakes). http://www.auslig.gov.au/facts/landforms/. The two estimated fluxes are the average of the presented range. c Area by difference. There is no flux measurement from eg, urban and alpine areas. The flux is regarded as a mean for all thinkable surfaces other than the two previously listed. Qmin is the mean of 2.95 and Qmax is the mean of 1 + 5 ngm-2h-1which is measured in South Europe. d The total area is from Geoscience Australia, 2002. http://www.auslig.gov.au/facts/dimensions/.
As discussed previously in this section, emissions from natural sources include re-emissions
of previously deposited Hg. Based on the results presented in section 3.2, which conclude that
re-emission of Hg accounts for approximately 35-50 % of the natural Hg emissions; this
suggests that approximately 45-65 tonnes of the Hg emitted from Australian natural sources
originate from previously deposited Hg.
From the discussion above it can be seen that estimations of emissions from natural sources
are complicated to conduct and subjected to large uncertainties. These factors may explain
why there is so little published data regarding emissions from natural sources on regional
levels. However, there are a number of investigations which have scaled up emission fluxes
from in situ measurements to local areas using models which include variables controlling the
flux in conjunction with a Geographic Information System (GIS) (Engle et al., 2001; Gustin
20 The total global landmass (excluding the Antarctic continent) is 135 774 000 km2 (ABS, 2002a). 21 There is a large variation concerning emission from land base sources, for instance, Bergan et al., 1999 estimated the emission from land sources in a model simulation to be 500 tonnes/yr. However,

25
et al, 2000). Beyond these investigations a linear dependency between Hg content in soil
(ppm) and the Hg flux to the atmosphere has been established indicating that the soil
concentration of Hg is a dominant variable controlling the flux. This linear relationship
suggests that it is possible, assuming that the Hg content in the soil is known, and that it
exceeds at least 100 ppb, to scale up fluxes to local and regional areas without actually
performing any in situ measurements. However, it is not possible to conduct this kind of
estimation in Australia since most soils contain a much lower Hg concentration, of the order
of 5-50 ppb (Carr and Wilmshurst, 2000; Carr et al., 1986).
From the calculations, estimations and data presented in this and the previous section (3.2), it
is obvious that natural sources (including re-emission of Hg) have an important role in the
overall emissions of Hg to the global atmospheric Hg pool. Whereas anthropogenic emission
inventories for Hg sources are measured and updated regularly in most industrialised
countries, this is not the case for natural emissions. Thus, estimations of emission from
natural sources are highly uncertain, even if the numbers of flux studies are increasing.
3.4 Anthropogenic mercury emission
A large proportion of the Hg present in the global atmosphere today is due to anthropogenic
activities. These activities have increased the overall Hg levels in the atmosphere by roughly a
factor of three (Munthe at al. 2001). As previously discussed, direct anthropogenic emissions
account for 2000-4000 tonnes/yr, which is approximately 35-60 percent of the annually total
Hg emissions. However, if the indirect Hg emissions are taken into account, the
anthropogenic portion of the yearly total global input to the atmosphere may be as high as 75
percent (Mason et al. 1994).
Although the level of use is decreasing Hg is used in a broad variety of manufacturing
industries and products due to its particular physio-chemical properties (i.e. high specific
gravity, low electrical resistance, constant volume of expansion), (CRC, 1998; Volland,
1991)22. Its toxic properties also see it used in different medications, antiseptics, and
pesticides (US EPA, 1997). The production, use and disposal of these products, along with
when the study was revised two years later the corresponding value had risen to 1320 tonnes/yr, i.e. roughly by a factor of 3 (Bergan and Rohde, 2001). 22 For instance, thermometers, barometers, thermostats, batteries, switches, fluorescent lamps, mercury boiler, mercury salts, mirrors, catalysts for oxidation of organic compounds, gold and silver extraction from ores, rectifiers, cathodes in electrolysis, use in the generation of chlorine and caustic paper processing, in dental amalgams, as laboratory reagent, lubricants, in dyes, wood preservatives, floor wax, furniture polish, fabric softeners, and chlorine bleach (CRC, 1998; Volland, 1991).

26
Hg released by other manufacturing processes (eg papermaking) liberates Hg to the
environment. In particular, processing of mineral resources at high temperatures such as
roasting and smelting of ores, combustion of fossil fuels, kiln operation in the cement
industry, as well as waste incineration all release substantial amounts of Hg (EC, 2001).
Globally, the stationary combustion of fossil fuels (mainly coal) is the most significant single
source of Hg and accounted for 77 percent of global Hg emissions in 1995 (Pacyna and
Pacyna, 2002).
The following sections aim to quantify the anthropogenic Hg emission from Australia in
2001, distribute it among the three different Hg species according to a source profile, and
compare the results from these steps with corresponding studies from other countries, as well
as with overall global emissions. The first part describes the global emissions of Hg whereas
the second part estimates the anthropogenic contribution from Australia.
3.4.1 Global anthropogenic emissions
An estimate of total global emissions of Hg from anthropogenic sources for 1995 are
summarised in Table 10 (Pacyna and Pacyna, 2002). As Table 10 shows, approximately 1900
tonnes of anthropogenic Hg were estimated to be emitted, which is an apparent decrease of 10
percent since 1990. In considering these values it should be noted that 325 tonnes of Hg
emissions from gold production were excluded from the total as they were regarded as highly
speculative. If this Hg were included in the approximation, the estimated amount of global
emissions would have increased by 5 percent between 1990 and 1995 rather than appearing to
decrease.
Table 10 Global atmospheric emissions of mercury from major anthropogenic sources in 1995 (tonnes)a
Continent
Stationary combustion
Non-ferrous metal
production
Pig iron & steel
production
Cement
production
Waste
disposal
Total Europe 185.5 15.4 10.2 26.2 12.4 249.7 Africa 197.0 7.9 0.5 5.2 210.6 Asia 860.4 87.4 12.1 81.8 32.6 1 074.3 North America 104.8 25.1 4.6 12.9 66.1 213.5 South America 26.9 25.4 1.4 5.5 59.2 Australia 97.0 4.4 0.3 0.7 0.1 102.5 Oceania 2.9 0.1 3.0 TOTAL 1995 1 474.5 165.6 29.1 132.4 111.2 1 912.8b TOTAL 1990c 1 295.1 394.4 28.4 114.5 139.0 2 143.1d
a Table from Pacyna and Pacyna, 2002 and personal communication with the authors. b 325 tonnes of Hg emissions from gold production is not included (>50% assumed to occur in Africa). c Estimates of maximum values, which are regarded as close to the best value. d The total emission estimate for 1990 includes also 171.7 tonnes of Hg emission from chloralkali production and other less significant sources.

27
When the emission data from 1990 and 1995 are compared (Table 10), the contribution of Hg
from non-ferrous metal production appears inexplicably to have decreased by 60 percent in
the time period of 1990 to 1995. In the same time period Hg emissions from stationary
combustion have also apparently increased by 15 percent and by 1995 constituted 77 percent
of global Hg emissions. It is suggested that this latter increase has occurred because fossil
fuels, particularly coal, have been increasingly relied upon for the production of electricity
and heat in a majority of the countries around the world. Hence, even though Hg is a minor
constituent of coal, the vast amount of coal consumed globally each year makes it a
significant source of Hg in the environment.
Asian countries, were estimated to have released about 1000 tonnes of Hg annually (or 56
percent of the total atmospheric input of Hg in 1995) and have increased their use of fossil
fuels, with 80 % of the total emissions from these countries due to stationary combustion.
China23 and India, for example, are estimated to emit respectively, 495 and 117 tonnes Hg /yr
due to the combustion of fossil fuels, which when combined is approximately one-third of the
total global emissions (26% and 6 %) (Table 11). If all sources of Hg are considered in the
two countries they contribute 40 % of the total global emission of Hg. The geographical
distribution of estimated Hg emissions from South-East Asia and Australia are presented in
Figure 3.
The total Hg emission from Europe (13 % of the global emission) appears to have decreased
by 45 % in 1995 when compared to 1990 (Pacyna et al., 2001). Combustion of coal in power
plants and residential heat furnaces generates more than 50 % of European (including the
European part of Russia) emissions of Hg (342 tonnes). Emissions of Hg from combustion of
fossil fuels have not changed significantly over the last decade (Pacyna et al., 2001), which
may be largely due to the continuing use of coal in both Western and Eastern Europe as the
major source of energy. The decrease of total Hg emissions in Europe during the 1990-1995
time period has been primarily attributed to the closure of chlor-alkali plants, and to changes
in production technology (Pacyna et al., 2001).
23 More than 75 percent of the energy requirements in China are fulfilled by coal (Daniel, 1994).

28
Figure 3 Geographic distribution of mercury emissions over Australia and South-East Asia (tonnes/yr)
Figure received from J Pacyna, personal communication.
An inventory of anthropogenic emissions in North America estimated the total Hg flux to
amount to 272 tonnes in 1990 (Pai et al., 2000b). Thus, the apparent 1995 emission flux of Hg
(Table 10) has declined by 22 % when compared to 1990. This apparent reduction of Hg
emissions can be explained by the continuing installation of abatement technologies (Pacyna
and Pacyna, 2002). As is the case in both Asia and Europe, combustion of fossil fuels is also
the most important source category of Hg to the environment in North America.
In the investigation by Pacyna and Pacyna (2002) the total amount of emission of Hg globally
was also divided among the three different Hg species according to the suggested chemical
speciation of the Hg released. Of the 1900 tonnes believed to have been emitted, 1000 tonnes
are estimated to be elemental Hg (53 %), 700 tonnes divalent Hg (37 %), and 200 tonnes
particulate Hg (10 %). A similar distribution was found in Europe, in 1995, where elemental,
divalent, and particulate Hg accounted for about 61, 32, and 7 percent of the total estimated
emission of Hg (Pacyna et al., 2001).

29
Australia, according to the study by Pacyna and Pacyna (2002) emits >102.5 tonnes Hg/yr of
which 97 tonnes is derived from stationary combustion (~5 % of estimated total global
anthropogenic emissions) (Tables 10 and 11). The Pacyna and Pacyna emission estimate is
significantly different to that which was produced by the Australian National Pollutant
Inventory (NPI, 2003a), which estimated that the total annual Australian anthropogenic Hg
emission for the years between 1999- 2002 was around 10 tonnes. The discrepancy between
these estimations will be discussed in detail in Section 3.4.2.4.
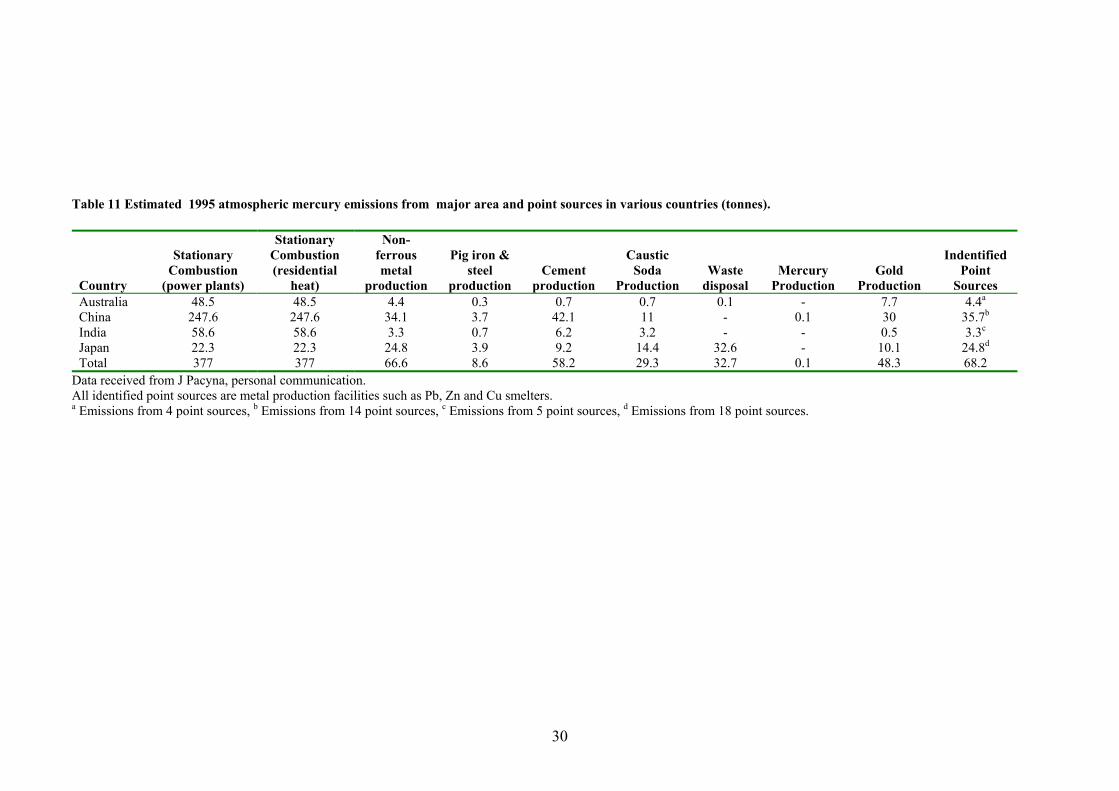
30
Table 11 Estimated 1995 atmospheric mercury emissions from major area and point sources in various countries (tonnes).
Country
Stationary
Combustion (power plants)
Stationary Combustion (residential
heat)
Non-ferrous metal
production
Pig iron &
steel production
Cement production
Caustic
Soda Production
Waste disposal
Mercury Production
Gold Production
Indentified
Point Sources
Australia 48.5 48.5 4.4 0.3 0.7 0.7 0.1 - 7.7 4.4a China 247.6 247.6 34.1 3.7 42.1 11 - 0.1 30 35.7b India 58.6 58.6 3.3 0.7 6.2 3.2 - - 0.5 3.3c Japan 22.3 22.3 24.8 3.9 9.2 14.4 32.6 - 10.1 24.8d Total 377 377 66.6 8.6 58.2 29.3 32.7 0.1 48.3 68.2
Data received from J Pacyna, personal communication. All identified point sources are metal production facilities such as Pb, Zn and Cu smelters. a Emissions from 4 point sources, b Emissions from 14 point sources, c Emissions from 5 point sources, d Emissions from 18 point sources.

31
3.4.2 2001 Australian mercury emission inventory
The Australian continent is rich in natural resources such as bauxite, coal, iron ore, copper, tin,
uranium, nickel, tungsten, mineral sands, lead, zinc, diamonds, natural gas, and petroleum (ABS,
2002b & d). The economy is strongly resource based and commodities such as fossil fuels,
minerals, metals, and agriculture products account for 57 percent of the value of the total export
earnings (ABS, 2002d & e). Anthropogenic Hg emission levels are enhanced in the process of
extraction and treatment of natural resources, such as metal production, as well as by reliance on
fossil fuel combustion for electricity generation. Since fossil fuel combustion accounts for
approximately 90 percent of Australian electricity generation capacity and plays an important role
in servicing the needs of energy intensive commodity production such as aluminium, steel and
iron (ABS, 2002d & e), a large proportion of Australian anthropogenic Hg emissions originate
from these types of activities.
The following Hg emission inventory in Australia is divided into two parts. The first part deals
with point sources (i.e. for a defined industry locations with known latitude and longitude
coordinates for each Hg source), where raw data for Hg emissions has been provided to the
Australian National Pollution Inventory (NPI, 2003a). In the second part, emissions from area
sources (i.e. Hg sources with no specific location), which were calculated as part of the NPI, on
the basis of emission factors (mass Hg per unit product) and statistical data concerning amount
consumed/produced of each product are discussed. Thus, the total Hg emission from Australia
becomes the sum of Hg from these two sources. The total emissions from each source category
are proportioned according to a source profile between the three different Hg species emitted.
Table 12 summarises the estimated Hg emission rates by source type from Australia in
2000/2001.
3.4.2.1 Atmospheric emission from point sources
Identified Australian point sources were estimated to emit about 7.0 tonnes Hgtot/yr (or ~70% of
the estimated total anthropogenic atmospheric emissions in 2000/2001, Table 12, Figure 4). Three
States; Western Australia (WA), Queensland (QLD), and New South Wales (NSW) account for
more than 83 percent of the total Hg emission from point sources to the atmosphere. Point sources
in these States emit, respectively, 2.3 tonnes (33%), 2.0 tonnes (29%) and 1.4 tonnes (21%). The
other 17 percent of the estimated Hg emissions are divided between Victoria (VIC) (6.9 %),
Tasmania (TAS) (4.8 %), South Australia (SA) (3.0 %), Australian Capital Territory (ACT) (1.3
%), and the Northern Territory (NT) (0.5 %) (Figure 4 and Appendix A)(NPI, 2003a).

32
Distribution of Australian anthropogenic Hg emissions geographically and between source
categories are shown in Figures 5 and 6.
Figure 4 Estimated mercury emissions to air (point sources) from States and Territories in Australia 2000/2001 (kg/yr).
Figure 5 Estimated mercury to air (point sources) by source category in Australia, 2001 (kg/yr).
Electricity generation is a significant component of identified point sources of Hg. The NPI (2003a) estimates that 1.9tonnes (~19% of Australian anthropogenic atmospheric emissions) emanates from this industry.
Point source emission of Hg from Australian States and Territories
0.0
500.0
1000.0
1500.0
2000.0
2500.0
WA QLD NSW VIC TAS SA ACT NT
kg Hg(tot)kg Hg0kg Hg(II)kg Hgp
Emission of Hg by point source type
0
500
1000
1500
2000
Elec
trici
tySu
pply
Alu
min
iaPr
oduc
tion
Stee
l and
met
al
Oth
erIn
dust
ry
Was
teD
ispo
sal
Min
ing
Che
mic
alIn
dust
ry
Oil
and
gas
Hg(total) Hg0Hg(II)Hgp

33
Figure 6 Geographical distribution of mercury emitting point sources in Australia 2001 (NPI, 2003a).
Figure downloaded from the NPI(2003a) website.
3.4.2.2 Atmospheric emission from areal sources
Estimates of atmospheric emissions from areal sources (i.e. where it is not possible to define a
source location) have also been made as part of the NPI. Examples of areal sources are diverse
and include emissions from electric lamp breakage, fossil fuel combustion (industrial /commercial
/residential), diffuse area and mobile sources, such as domestic combustion for heating and motor
vehicles (Bullock, 2000a; Pai et al., 2000b). Estimates of anthropogenic emissions of Hg from
areal sources suggest that a further 3.2 tonnes is emitted from disperse or mobile sources (Table
12, NPI, 2003a). This estimate might be increased by a further 2.6 tonnes were Hg emissions
included, from combustion of vegetation in wildfires (often as a result of human intervention),
burning as part of fuel reduction/regeneration programs and burning carried out as part of land
clearing and agriculture.

34
3.4.2.3 Total Australian anthropogenic emission
The total anthropogenic Hg emission from Australia in 2000/2001, including point and area
sources (but excluding burning of vegetation), going to air, land and water was estimated to be
about 10.2 tonnes (Table 12), (NPI, 2003a). Of this 9.9 tonnes is emitted to the atmosphere and
comprises about 0.5 percent of the estimated global anthropogenic atmospheric Hg emissions24.
There is a significant difference apparent between the NPI (2003a) estimate of total annual
Australian anthropogenic atmospheric emissions of 9.9 tonnes for 2000/2001 and the 1995
estimate made by Pacyna and Pacyna (2002) of 110.9 tonnes (Table 11).
Table 12 Distribution of total estimated Australian anthropogenic mercury (kg) for 2001 (NPI 2003a)
Total Air Land Water Identified point sources 6959 6701 54 204 Areal Sources 3210 3210 Total Area and Point Sources 10169 9911 54 204 Burning (including wildfires) 2600 2600 Total all possible sources 12769 12511 54 204
3.4.2.4 Accuracy of emission estimates
A number of scientific studies have attempted to quantify anthropogenic Hg emissions on both
regional (Bullock, 2000a; US EPA, 1997; Pacyna et al., 2001; Pai et al., 2000b; Pilgrim et al.,
2000; Lee et al., 2001) and global scales (Pacyna and Pacyna, 2002; Pirrone et al., 1996, 2001b).
Although there have been an increasing number of Hg emission inventories published, there are
still many uncertainties surrounding estimates of anthropogenic emissions (Bullock, 2000b). As
stated previously, in the latest published study on global emissions, the total atmospheric Hg flux
from Australia in 1995 was estimated to be at least 110.9 tonnes/yr (Table 11) (Pacyna and
Pacyna, 2002). However, according to an earlier study, the emission rate of Hg in Australia was
suggested to be 35 tonnes/yr (based on 1992) (Pirrone et al., 1996), i.e. one third of the later
estimate. Moreover, if the emission of Hg in the Pacyna and Pacyna (2002) study is compared to
the latest NPI (2003a) figures the difference in estimated emission fluxes is even larger (by a
factor of nearly 11).
The large variances in suggested emission fluxes between different studies appear, amongst other
things, to be due to the fact that most of the investigations conducted use emission factors (EF) for
each source category in terms of mass of Hg emitted per unit of fuel consumed or product
produced. These emission factors may be either based on direct measurements of gases
discharged, or on expert judgments, both of which need take into account the Hg content in the

35
raw material, the technology of the industrial process, and the type of abatement technology.
Assuming that suitable representative sample techniques, sampling locations (i.e. allowing typical
sample output in the stack to be determined) and appropriate analytical techniques are applied,
emission factors based on direct measurements are considered to be more accurate than emission
factors derived from expert judgments (Pacyna et al., 2001; Pai et al., 2000b). However, a major
uncertainty associated with direct measured-emission factors can be caused by extrapolation of
measurements conducted at a limited number of facilities with specific production conditions to
generalize across an industry. For instance, for an inventory in the US, an emission factor based
on 10 measurements was used to estimate emissions from 200 different waste incineration plants
that burnt different kind of fuels (Pai et al., 2000b).
Since in many cases, it is impossible to carry out in situ measurements some other sort of
assumption and validation/judgement needs to be made to estimate individual emissions. As
always, assumptions can introduce substantial biases in the estimated Hg emission from a specific
facility or source category, which consequently affects the end result. The US EPA (1997) for
example estimated the Hg emission from fuel oil combustion in the US to be 10 tonnes/yr.
According to Wilhelm (2001) this value was based on an emission factor that overestimates the
Hg concentration in fuel oils by a factor of 3 - 10. Thus, the actual Hg emission is, according to
Wilhelm (2001), probably in the range of 1 - 3 tonnes/yr.
In the study by Pacyna and Pacyna (2002) and Pacyna et al., (2001) a range of emission factors
were used. They concluded that the following accuracy of emission estimates can be assigned to
the different source categories:
• stationary fossil fuel combustion: ± 25 %
• non-ferrous metal production: ± 30 %
• iron and steel production: ± 30 %
• cement production: ± 30 %
• waste disposal: a factor of up to 5
A further source of uncertainty associated with emission inventories, is error by omission, which
occurs due to lack of specific information of particular source categories. For example, in the
inventory of anthropogenic Hg emissions in North America, the category "non-ferrous metal
smelting" for the US, a potential major Hg source, was excluded due to lack of data (Pai et al.,
24 Assuming an average global anthropogenic emission of total mercury of approximately 2 000 tonnes/yr (Bergan et al., 1999; Pacyna and Pacyna, 2002).

36
2000). The US EPA (1997) concluded that if the missing emission sources were added to the
calculations in their study, they could increase the total estimated amount of emitted Hg by as
much as 20 percent.
As previously mentioned there are significant differences between the data collected in the
NPI (2003a) and the estimation by Pacyna and Pacyna (2002) (PP) of anthropogenic atmospheric
Hg emissions from Australia. The most obvious differences lie in estimates of Hg emissions from
combustion of fossil fuels. PP suggest that the total emission of Hg from stationary combustion of
fossil fuels (mostly coal) in Australia is 97 tonnes/yr (1995). An alternative estimate of Hg
emissions from total fuel combustion using data for all Australian fuel sources (ABARE, 2003)
and the emission factors used in the NPI (for differing energy sources, 1999b, 2003b) calculates
that this value should be a maximum of 7.0 tonnes for the year 1995 (Figure 7). By comparison
the calculated emissions using the same data and the PP emission factors for coal gives an
estimate of total Hg emission from all fuel combustion of 111.7 tonne.
Figure 7 Estimates made using NPI (1999b, 2003b) emission factors of Australian anthropogenic atmospheric mercury emissions from fuel and coal combustion compared with emissions which arise from combustion during electricity generation
0
1
2
3
4
5
6
7
8
9
1990 1992 1994 1996 1998 2000 2002 Reporting Year
Mer
cury
Em
issi
on (t
onne
s)
All fuel sources
Electricity generation
All coal and coal products
The difference in the 1995 values can be explained as being simply due to the use of differing
emission factors during calculation. For example, PP used an emission factor of

37
1.0E-03(tonne/ktonne) or 1000ppb for coal (Pacyna and Pacyna, 2002), whereas the NPI(1999b)
uses 4.2E-05 (tonne/ktonne)(42ppb). The use of the latter value can be justified on the basis of
the work of Dale(1999), who found that the trace Hg content of Australian coals was in the range
16-76 ppb, with a mean of 44 ppb. It is also known that the actual level of emissions may be also
be further diminished during the combustion process as 5 - 30 percent of the Hg can be captured
by the fly ash and is consequently not emitted to the atmosphere (Levin (EPRI), 2001). The Hg
concentration in the flue gas may be even further reduced due to the application of various
abatement technologies, although power plants in Australia currently have no such systems
installed. Thus it is possible to calculate that the total Hg emissions from coal combustion for
1995 could have been as low as 3.2 tonnes ( assuming 30% of coal emissions were trapped in the
flyash), but it is inconceivable that it could have been as high as 97 tonnes.
Using the NPI (1999b, 2003b) emission factors, the value calculated for Hg emitted from all fossil
fuel combustion for the 2000/2001 year is 8.4 tonnes (Figure 7). This result compares favorably
with the values calculated in the NPI (2003a) for Hg emissions in that year (Table12), given that
fossil fuel combustion, in various forms, is the major mechanism for anthropogenic Hg release.
Even so there remain apparent discrepancies between the calculations of Hg emissions using
Australian national consumption figures (for example fuel used in electricity generation) and
those generated in the NPI (2003a) which uses the sum of values calculated at the individual plant
(point source) level. For the 2001 reporting year these results were 5.1 tonnes and 1.9 tonnes
respectively. This discrepancy is being investigated as part of the ongoing CCSD project on
emissions.
As further evidence to support the view that the lower NPI calculated emission rate is more
reasonable, a country-by-country comparison, has been conducted. In this comparison the
calculated anthropogenic emission flux of Hg in each country was divided by their respective
population. The Australian per capita emission of Hg is, depending on whether the emission flux
is 110.9 (PP) or 9.9 tonnes/yr (NPI, 2003a), 5.66 or 0.51 g Hgtot/capita/yr, respectively. The latter
value compares favorably to values for other countries which range from 1.46 (Czech Republic)
to 0.10 g Hgtot/capita/yr (Sweden) (Table 13). Thus, the emission rate of 110.9 tonnes/yr as
suggested by PP seems again, based on the results from the per capita emission comparison,
unrealistically high.

38
Table 13 Country-by-country comparison based on anthropogenic atmospheric Hgtot/capita.
Country
Hg (total) (tonnes/yr)
Populationa (million)
g Hgtot/capita
Reference
Czech Republic 15 10.3 1.46 Pacyna et al., 2001 Japan 139.6 127.3 1.09 Pacyna and Pacyna, 2002 Canada 33.2 31.0 1.07 Bullock, 2000a Romania 23 22.4 1.03 Pacyna et al., 2001 Poland 33.6 38.6 0.87 Pacyna et al., 2001 Ukraine 36 49.1 0.73 Pacyna et al., 2001 USA 176 285.9 0.62 Pai et al., 2000b Russia 87.7 144.7 0.61 Pacyna et al., 2001 Australia 9.9 19.6 0.51 NPI 2003a Australiab 110.9 19.6 5.66 Pacyna and Pacyna, 2002 China 616.0 1285 0.48 Pacyna and Pacyna, 2002 Europe 249.7 582.2 0.43 Pacyna and Pacyna, 2002 Germany 31.3 82 0.38 Pacyna et al., 2001 Global averagec 2200 6134 0.36 This study France 17.6 59.5 0.30 Pacyna et al., 2001 UK 127 59.5 0.21 Lee et al., 2001 India 131.1 1025 0.13 Pacyna and Pacyna, 2002 Sweden 0.9 8.8 0.10 Pacyna et al., 2001
a From the State of the World, 2001. http://www.unfpa.org/swp/2001/english/indicators/indicators2.html. b This figure is from the investigation of Pacyna and Pacyna 2002, indicating the unrealistic high emission that is estimated from Australia. c Emission based on Bergan and Rhode, 2001; Pacyna and Pacyna, 2002
The PP study also estimates the combined Hg emissions from non-ferrous metal production (Cu,
Pb, Zn), pig iron/steel production and cement production in Australia (1995) to be approximately
5.4 tonnes/yr (Table 11), which is a factor of about three higher than that estimated by the NPI
(2003a) [1.7 tonnes/yr for (2000/2001)](Table 14). This figure from PP also appears to be in
error, given that the economic mix has not changed substantively in the period. It should however
be noted that the absolute difference in these values is relatively small when compared to the
difference between estimates of Hg emissions from coal combustion.
To further investigate these differences the emission rates of Hg from the three source categories
(non-ferrous metal production (Cu, Pb, Zn), pig iron/steel production and cement production)
were calculated using the EF in the PP study (Pacnya and Pacyna 2002) and statistical production
data from Australia (2001). The calculations are shown in Appendix B. The results show that if
the calculated emissions, (using the PP emission factors), from the three source categories are
compared to the NPI emission data, 11.1 tonnes rather than 2.0 tonnes of Hg is calculated to be
released annually from Australia from these sources. Thus it is apparent that depending on which
emission estimation technique is being used, including the choice of EF and statistical data,
significant discrepancies can occur between calculated emission results.

39
3.4.2.5 Estimation of mercury speciation
As discussed previously the speciation of Hg (i.e. the fraction of Hg0, Hg(II) and Hgp in the total
emission) strongly affects its transportation, atmospheric residence time and deposition pathways
through the atmosphere. In order to investigate the potential environmental effects of Hg, the
dispersion and deposition, and the contribution of elemental Hg to the global atmospheric pool,
the estimated total Hg emission from Australia was divided between the three different species
(Hg0, Hg(II), Hgp). This division is achieved using an estimation of the speciation of the Hg
emitted from various sources and processes. In Table 14, a number of speciation profiles are listed
based on source categories. Thus, depending on the source a speciation profile is assigned and the
distribution between the species can be calculated (Table 15).
Of the approximately 9.9 tonnes of anthropogenic Hg released to the atmosphere annually in
Australia, it is estimated that about 9.9 tonnes is emitted into the atmosphere, it is calculated that
4.75 tonnes (48 %) are in the form of elemental Hg, 1.30 tonnes in the form of divalent Hg
(13 %), and 3.88 tonnes (39 %) is in the form of particulate Hg. The Hg0 is therefore available to
enter the global atmospheric pool each year, this constitutes about 0.5 percent of the increase in
the annual global anthropogenic Hg pool, based on an increasing pool of about 1000 tonnes per
annum (Pacyna and Pacyna, 2002). The remaining emissions of Hg (~5.0 tonnes) are assumed
to be deposited on a local to regional scale. Deposition is discussed in more detail in Section 4.

40
Table 14 Emission speciation (fraction of the total) of mercury from anthropogenic sources Sources Hg0 Hg(II) Hgp Reference
Chemical production: 0.8 0.1 0.1 1 Industrial chemicals (general) Chlor-alkali production: 0.7 0.3 0 1 Industrial chemicals (general) Electric utilities: 0.5 0.3 0.2 1 Electric power generation Iron and steel production: 0.8 0.1 0.1 1 Coke and gas Iron and steel production Iron ore mining and beneficiation Steel foundries Pig & iron 0.8 0.15 0.05 2 Non-ferrous metal smelting: 0.8 0.1 0.1 1 Copper smelting & refining (base metal smelting) Copper smelting & refining (mining, milling and conc.) Non-ferrous smelting and refining (miscellaneous) Lead smelting & refining Zinc smelting & refining Lead and Zinc 0.8 0.15 0.05 2 Cement manufacturing: 0.8 0.1 0.1 1 Cement and concrete industry Cement production 0.8 0.15 0.05 Waste combustion: 0.2 0.6 0.2 1 Commercial incineration Municipal incineration Waste incineration 0.2 0.6 0.2 2 Fossil fuel combustion: 0.5 0.3 0.2 1 Commercial fuel combustion Electric power generation Residential combustion Coal and oil combustion 0.5 0.4 0.1 2 Medical waste incineration: 0.02 0.73 0.25 1 Biomedical waste incineration Non-medical waste incineration: 0.2 0.6 0.2 1 Commercial incineration Municipal incineration Sewage sludge incineration High temperature fabrication: 0.8 0.1 0.1 1 Aluminum oxides (abrasives manufacturing) Ferro-alloys manufacturing Glass manufacturing Non-ferrous smelting and refining misc. Caustic soda 0.7 0.3 0 2 Average of all sources, Europe 1995 0.64 0.285 0.075 2 Average of all sources, Global, 1995 0.53 0.37 0.10 4 Average of all sources, Northern Hemisphere, 2002 0.58 0.33 0.09 3 Ref. 1: Bullock et al., 2000; Ref. 2: Pacyna et al., 2000; Ref. 3:Pacyna and Pacyna, 2002; Ref.4: Travnikov and Ryaboshapko, 2002.

41
Table 15 Estimates of Australian atmospheric mercury emission rates by source 2001 (NPI, 2003a) Identified Point Sources Hg(total) % of Hg0 Hg(II) Hgp
kg/yr total kg/yr kg/yr kg/yr Alumina Production 1789.0 26.7 1431.2 178.9 178.9 Aluminium Smelting 5.3 0.1 4.2 0.5 0.5 Basic Iron and Steel Manufacturing 386.1 5.8 308.8 38.6 38.6 Basic Non-Ferrous Metal Manufacturing n.e.c. 358.4 5.3 286.7 35.8 35.8 Bauxite Mining 9.9 0.1 7.9 1.0 1.0 Black Coal Mining 88.7 1.3 71.0 8.9 8.9 Cement and Lime Manufacturing 290.4 4.3 232.3 29.0 29.0 Chemical Product Manufacturing n.e.c. 125.1 1.9 100.1 12.5 12.5 Clay Brick Manufacturing 17.9 0.3 14.3 1.8 1.8 Copper Ore Mining 7.6 0.1 6.1 0.8 0.8 Copper, Silver, Lead and Zinc Smelting, Refining 966.9 14.4 773.5 96.7 96.7 Dairy Product Manufacturing n.e.c. 66.0 1.0 52.8 6.6 6.6 Electricity Supply 1903.0 28.4 951.5 570.9 380.6 Fertiliser Manufacturing 2.1 0.03 1.7 0.2 0.2 Glass and Glass Product Manufacturing 9.1 0.1 7.3 0.9 0.9 Gold Ore Mining 17.7 0.3 14.2 1.8 1.8 Hospitals (Except Psychiatric Hospitals) 20.5 0.3 16.4 2.0 2.0 Inorganic Industrial Chemical Manufacturing n.e.c. 70.4 1.1 49.3 21.1 0.0 Iron Ore Mining 29.2 0.4 23.3 2.9 2.9 Meat Processing 3.0 0.0 2.4 0.3 0.3 Milk and Cream Processing 2.0 0.0 1.6 0.2 0.2 Mining n.e.c. 15.3 0.2 12.2 1.5 1.5 Oil and Gas Extraction 14.2 0.2 11.3 1.4 1.4 Organic Industrial Chemical Manufacturing n.e.c. 4.3 0.1 3.5 0.4 0.4 Paper Product Manufacturing n.e.c. 2.1 0.0 1.6 0.2 0.2 Paper Stationery Manufacturing 7.0 0.1 5.6 0.7 0.7 Petroleum and Coal Product Manufacturing n.e.c. 3.7 0.1 2.9 0.4 0.4 Petroleum Refining 68.6 1.0 54.9 6.9 6.9 Plaster Product Manufacturing 3.0 0.0 2.4 0.3 0.3 Pulp, Paper and Paperboard Manufacturing 27.4 0.4 21.9 2.7 2.7 Silver-Lead-Zinc Ore Mining 77.7 1.2 62.1 7.8 7.8 Sugar Manufacturing 18.5 0.3 14.8 1.8 1.8 Waste Disposal Services 271.9 4.1 54.4 163.1 54.4 Other sources (n.e.c.) 19.6 0.3 15.7 2.0 2.0 Total (Point source, Air only) 6701 100 4620 1201 881
Anthropogenic Areal Sources Hg(total) % of Hg0 Hg(II) Hgp kg/yr total kg/yr kg/yr kg/yr
Paved/Unpaved roads (particulates) 2800 87.2 0 0 2800 Fuel combustion (sub reporting threshold) 180 5.6 90 54 36 Windblown particulates 140 4.4 0 0 140 Motor Vehicles 66 2.1 33 19.8 13.2 Domestic fuel combustion 7.9 0.2 3.95 2.37 1.58 Other 16.1 0.5 8.05 4.83 3.22 Total of Areal Sources (to Air) 3210 100 135 81 2994 Total Area & Point Sources ( to Air) 9911 4755 1282 3875 The data in column 1 is proportioned between the three different Hg species according to the speciation profile listed in Table 13.

42
4. DEPOSITION OF MERCURY
The most important delivery process of Hg to terrestrial and water compartments is deposition
from air via wet and dry processes (Fitzgerald et al., 1991; Lindquist et al,. 1991; Slemr et al.,
1985). The relative importance of these processes depends on the following factors: (i) the
chemical form of the pollutant (assumed in this study: to be Hg0/Hg(II)/Hgp), (ii) the solubility of
the pollutant in water (iii) the amount of precipitation in the region, and (iv) the characteristics of
the land and surface cover (Seinfeld and Pandis, 1998).
Dry deposition of gaseous species and particles refers to atmospheric removal processes, which
do not include precipitation. The factors that, in general, govern the dry deposition processes are
(i) the physio-chemical properties of the deposited pollutant, (ii) the characteristics of the surface,
and (iii) the level of atmospheric turbulence. The level of atmospheric turbulence effects the rate
at which a pollutant is transported through the atmosphere. This is particularly the case in the air
layer closest to the ground. For gaseous species, properties such as solubility and reactivity affect
their interaction with other species and media in the atmosphere, as well as uptake at the receiving
surface. For particles; shape, size and density affect their transportation via the atmosphere and
whether or not they are adhering to a surface. The shape and cover of the surface itself is also a
factor that affects the process of dry deposition. For instance, a non-reactive surface does not
promote dry deposition since it resists adsorption/absorption of a gaseous species. Furthermore, a
smooth surface may prevent the capturing of particles. However, most natural surfaces, generally,
promote dry deposition (Seinfeld and Pandis, 1998).
Wet deposition of atmospheric contaminants refers to the removal process associated with
precipitation (i.e., when contaminants are incorporated into precipitation elements (clouds, rain
droplets, and aerosols), which subsequently fall to the ground). The wet deposition process is
regarded as highly complex. Due to its complexity, wet deposition is not included in this study.
In Section, 4.1, the theory behind dry deposition will is reviewed, Section 4.2 looks at the
deposition patterns of Hg species, and in Section 4.3, a dispersion and deposition study conducted
over the central, coastal part of NSW is presented.

43
4.1 Dry Deposition
Dry deposition is usually described with a single parameter; the deposition velocity (vd) (ms-1),
which consists of two components; diffusion and gravitational settling. Gaseous species are
assumed to have zero gravitational settling velocity due to their negligible molecular weight,
whereas particles, in general, have both diffusion and gravitational settling components. In the
following paragraphs, two equations which are used in calculating the deposition velocity are
presented; one for gaseous pollutants, and one for particles. References to this section can be
found in Finlayson-Pitts and Pitts (2000), and Seinfeld and Pandis (1998).
In modelling dry deposition using vd, it is assumed that the rate of deposition per unit area; F
(µgm-2s-1) is proportional to the concentration of the deposited species, at a reference height (z)
above the surface:
F = - vd×C(x,y,z) (1)
By using a single parameter (vd) to describe the whole variety of physical and chemical processes,
whereby pollutants may be transported and removed from the atmosphere to a surface, the model
simulations are simplified. Thus, as long as the deposition velocity and the concentration of the
pollutant are known, the deposition flux can be calculated in any geographical location. The
disadvantage with excluding chemical and physical processes that affects the interactions between
atmospheric constituents and media is that it becomes difficult to choose a representative
deposition velocity. Most studies, however, use default values for vd (Table 16).
The process of dry deposition of a pollutant is generally described in three steps: (i) aerodynamic
transport down through the atmosphere to the vicinity of the surface, (ii) crossing of a stagnant
sublayer, called the quasi-laminar sublayer, via either molecular (for gases) or Brownian (for
particles) diffusion, and finally (iii) uptake at the surface. Each of the described steps determines
the value of the deposition velocity.
As the pollutant is transported from the atmosphere to the surface (via step i-iii) it experiences a
number of so-called resistances which affect the magnitude of its deposition rate and hence its
deposition fluxes. Assuming that the deposition flux is independent of height, a resistance, r, can
be defined for each of the above mentioned steps, that is, for each zone that the pollutant has to
cross:

44
r(z2,z1) = C(z2) – C (z1) (2)
F
The deposition velocity for gaseous species is defined as:
vd = 1/r (3)
where r can be broken down into three resistances:
r = ra + rb + rs (4)
1. ra is the aerodynamic resistance determined by the vertical eddy diffusivity, which in turn
depends on the meteorology and the surface roughness. Except in the case of large particles
where gravitational settling must be taken into consideration (see below), ra is independent of
the physical form of the pollutant.
2. rb is the sub-layer resistance (i.e., the resistance to cross the thin layer in the vicinity of the
surface). A variety of mechanisms such as the physical form of the species and the
characteristics of the surface affect the transport across this layer. The resistance ra, together
with rb, are effected by wind speed, vegetation height and atmospheric stability. The sum of
the two resistances tend, in general terms, to decrease with increasing wind speed and
vegetation height. Thus, the resistance is lower (i.e., vd is higher) over forests than over grassy
areas. In daytime, ra is relatively low whereas it is considerably higher and often the
controlling factor for vd during the night, when turbulent mixing is reduced.
3. rs is the surface resistance which is, as for rb, determined by the characteristics of the
receiving surface as well as by the properties of the pollutant. The resistance that is offered by
different surfaces to dry deposition generally depends on the level of moisture and pH of the
surface, and on the solubility and reactivity of the gaseous species.

45
Table 16 Dry deposition velocity of mercury species (cm/s)
Hg0 Hg(II) Hgp Reference 0 0.5 0.025 Petersen et al. 1995a 0.03 (May-Oct) 0.01 (Rest of the year)
0.5 - Ryaboshapko et al. 2000b
0.03d (warm season) 0.01d (Temp. 8-10 0C)
0.3 (Day) 0.15 (Night)
- Bergan et al. 1999c, Bergan and Rohde, 2001d
0.06 2.9 (average, day) 0.3 (night)
0.2 (day) 0.02 (night)
US EPA, 1997e
a No temporal or spatial variability of vd. b Used in a regional Eulerian model (Europe) where dry deposition is only included over land. If the surface air temperature is negative, no dry deposition of Hg0 is assumed. The deposition velocity of Hg(II) is also assumed to be independent of season and surface cover. c A study of the global distribution of Hg0 and Hg(II) using a climatic transport model. Dry deposition is mainly assumed to affect Hg(II). No seasonal variation of vd is included. d This work is revision of the study by Bergan et al. 1999. The same dry deposition rate of Hg(II) is used. Dry deposition of Hg0 is added to the reference case in order to investigate the sensitivity of the model. e A comprehensive assessment concerning Hg where, among other things, dry deposition is modeled using a Lagrangian-type simulation model (RELMAP) (regional basis, dry deposition of Hg0 is excluded) as well as a so-called ISC3 model for predicting deposition 50 km from the emission source (including dry deposition of Hg0).
The deposition velocity of particles is similar to that of gaseous compounds except that particles
are also subjected to a gravitational settling velocity (vs). Thus, by rearranging Equation (3)
(which is described in more detail in Seinfeld and Pandis (1998)), assuming rs = 0 (particles
adhere upon surface contact) the following vd is derived:
vd =(1/(ra + rb + rarbvs)) + vs (5)
Thus, since the deposition velocity is the reciprocal of resistance, each individual resistance term
sets a boundary on the rate of deposition. However, as previously mentioned, many studies use
default values for vd. In Table 16, a number of vd of Hg species are listed.
4.2 Deposition patterns of mercury
Until recently it was assumed that dry deposition of elemental Hg was only a minor portion of the
total amount of Hg deposited. In recent published simulation studies, however, dry deposition of
elemental Hg is often included in the calculations. The reason is that, even if the deposition
velocity of Hg0 is low (Table 16), the significant amount of Hg0 present in the atmosphere may
still have an important effect on the overall deposition flux of Hg to environmental surfaces. For

46
instance, in a comprehensive data simulation study in the US, it was concluded that approximately
25 % of the total deposition of Hg is in the form of dry deposition of Hg0 (Xu et al., 2000a,b)25.
The different Hg species deposition pathways are described in more detail in Sections 2 and 5. In
essence, Hg(II)(g) is removed from the atmosphere in three ways (i) it may condense onto
particulate material, which are either scavenged by atmospheric water droplets or dry deposited to
the surface, (ii) it may, in the presence of precipitation, dissolve in the aqueous phase, and hence
undergo wet deposition, and/or (iii) it may dry deposit. Particulate Hg is, as Hg(II), readily
removed from the atmosphere via both wet and dry deposition. Elemental Hg, on the other hand,
is, due to its low solubility and high vapour pressure, not as efficiently removed from the
atmosphere. Even if elemental Hg is not as efficiently removed as Hg(II) and Hgp, it is, still
removed from the atmosphere, either by, (1) transformation to divalent Hg (subjected to (i)-(iii))
and/or (2) dry deposition, which may be a significant removal pathway for Hg0 from the
atmosphere.
Divalent Hg is the predominant form of Hg deposited to the surface (Lindquist et al., 1991). It had
been assumed that wet deposition was the main removal mechanism of Hg(II) from the
atmosphere. However, more recent studies have shown that dry deposition of Hg(II) may be an
equally important (or more important) removal mechanism, even in areas which have relative
high levels of precipitation (Rea et al., 2000; Vette et al., 2002; Landis et al., 2002a,b). A similar
result was obtained in the study by Xu et al (2000a,b), where dry and wet deposition of Hg(II)
constituted 35 and 30 % of the total simulated deposition of Hg, respectively.
These results indicate that a major part of the total deposited Hg may consist of dry deposited
elemental and divalent Hg.
25 The simulation study by Xu et al. (2000a,b) is based on a comprehensive three-dimensional regional scale Eulerian air quality model that incorporates mercury chemistry, in-cloud transformation processes, and air-surface exchange processes. The bi-directional air-surface exchange was use to model emission of Hg0 and dry deposition of Hg from/to natural surfaces. The simulation include Hg0, Hg(II), and Hgp. A background concentration was assigned to each of the three Hg species.

47
4.3 Model simulation
4.3.1 The model
TAPM (The Air Pollution Model) (developed by CSIRO Atmospheric Research, Australia) is a
three-dimensional regional scale Eulerian air quality model, connected to databases for
meteorology, terrain and vegetation. Its dispersion module (which is used in this study) includes
not only an Eulerian grid module, but also an optional Lagrangian particle module for predicting
concentrations of various species near the source (the combination of the two modules are used in
this simulation). Emission data is added either as point or area sources. Information about
TAPM's general theoretical consideration, reference studies and technical descriptions can be
found in Hurley (2002a, b) and Hurley et al., 2002.
Since TAPM is primarily developed to investigate the air quality of an airshed in relation to SOx,
NOx and photochemical smog (including chemical transformations and deposition mechanisms
related to these pollutants), Hg is modeled as an inert pollutant (so-called tracer) where chemical
transformation processes, as well as, deposition processes are omitted. Thus, none of the chemical
reactions/interactions described in Section 2 are included in the model.
Even though deposition of Hg is not included in TAPM, dry deposition fluxes are calculated by
post-processing hourly-simulated grid concentration outputs from TAPM according to
Equation (6). Thus, an hourly dry deposition flux is derived from each grid cell using a default
value for the deposition velocity, as described in section 4.1.
F = - vd×C(x,y,10 m) (6)
The deposition velocity for Hg(II)/Hgp is set to be 0.5 cm/s during the day and zero cm/s during
the night. Corresponding values for Hg0 are 0.03 and zero cm/s, respectively.
4.3.2 Simulation procedure
4.3.2.1 Simulation domain and period
The simulation was conducted to model the period of 1st to the 31st of January 2001. The
simulation domain covered a portion of the central, coastal part of NSW, as shown in Figure 8.
There are two meteorological grid domains; the outer has 25×25 grids in the horizontal plane,

48
with grid size of 30 × 30 km, while the inner grid domain has the same number of grids but a grid
size of 10 × 10 km.
In order to obtain a finer resolution with regard to concentration simulations, an outer and an
inner pollution grid domain is used with 97×97 grids in the horizontal plane, with grid size 7.5 ×
7.5 km and 2.5 × 2.5 km, respectively.
The vertically resolution consists of 25 non-uniform layers, with the finest resolution near the
surface (located 10 m above the ground). The top of the modeling domain is 8 km.
Depending on the stack height of the source, emissions of Hg enter somewhere between the first
and the seventh model layer
Figure 8 Geographical distribution of point sources included in TAPM simulation
Number of grid cells The frame of the figure represents the outer grid domain.
2 4 6 8 10 12 14 16 18 20 22 24
2
4
6
8
10
12
14
16
18
20
22
24
Mount Piper PS
Maldon CW
BHP Steel PKW
Orica Chlorine P SYDNEY
Vales Point PSEraring PS Pasminco
Comsteel
Lidell PSBayswater PS

49
4.3.2.2 Meteorological conditions
Meteorological data used in the simulation is from 2001. January is a summer month in Australia
with high average temperatures. Inclusion of rain is an optional parameter in TAPM, but since
wet deposition is excluded from the simulation, so is rain. The prevailing wind direction during
the simulation period was east/northeast.
4.3.2.3 Mercury emission data
For simplicity a facility emission cutoff of 20 kg/yr was used which ensured that more than 90 %
(1282 kg/yr) of the total anthropogenic point emissions in NSW (NPI, 2003a) were embraced by
the simulation. These emissions were divided between elemental and divalent/particulate Hg
according to the source profiles, previously described in Section 3.4.2.3. The emission rate of Hg
is assumed to be constant over every hour of the year. In order to simulate the concentrations and
deposition fluxes of the different Hg species, a specific "tracer" is assigned to each of the two
groups (Hg0 and Hg(II)/Hgp) . Each tracer can then be analysed.
Emissions from natural sources and area sources are not included in the simulations.
The sources of Hg emissions simulated include: combustion of coal (5 power plants), basic iron
and steel manufacturing (2 sources), cement manufacturing (1 source), Cu/Ag/Pb/Zn smelter (1
source), and chemical production (1 source) (Figure 8, Appendix C).
4.3.2.4 Initial and boundary conditions
Background tracer (Hg) concentrations, as well as, boundary conditions (i.e., the amount of Hg
imported from the global Hg pool to the model domain) are set to be zero. Thus, the ambient Hg
concentration and deposition flux derived from the study originates only from the 10 facilities
investigated.
4.3.2.5 Deposition
Wet deposition is not included in the model simulation whereas dry deposition is calculated by
post-processing hourly simulated grid concentrations of Hg, according to Equation (1). By using
default values for the deposition velocity, an hourly deposition flux is calculated for respective
grid cell and Hg species in the entire domain. Based on the result, an average hourly deposition

50
flux for each grid cell is calculated. These average hourly deposition fluxes are then integrated
over year 2001.
The choice of deposition velocity for each Hg species is based on the information shown in Table
15. Since divalent and particulate Hg are, due to practical considerations, combined in one
category, the chosen velocity represents a weighted average of Hg(II) and Hgp rates. The
deposition velocity of divalent/particulate Hg is assumed to be 0.5 cm/s during day time (12
hours) while at night, due to reduced turbulent mixing (high ra, Section 4.1), it reduces to zero.
Similarly, the deposition velocity of elemental Hg during the day and night is assumed to be 0.03
and zero cm/s, respectively.
4.3.3 Simulation results
4.3.3.1 The simulation
Three simulations are presented in the current report. The first simulation (Run 10), which
includes all ten Hg emitting facilities (Figure 8), was performed to investigate the ambient ground
Hg concentrations (the first vertical layer in the model; 10 m) (4.3.3.2), as well as the deposition
flux of respective Hg species (4.3.3.3, Table 17) in the entire domain. In addition, a number of
mass balances are calculated to determine the average amount of Hg, as well as the percent of
total Hg that would be deposited at different distances around each facility (and in some case
facilities) (each distance is represented by a square box where the source/sources is/are
approximately in the center of the box).
In the Run 10 model simulation, the real stack height of 5 of the sources is unknown
(Appendix C). In order to investigate possible deposition fluxes of Hg at (i) different stack
heights, and at (ii) different distances around respective source (as in Run 10), two additional
simulations (Runs 20 and 30) were conducted (for the 5 sources). When calculating area average
deposition fluxes around some of the facilities in Run 10, some of the grid cells surrounding the
facilities were, at certain distances, overlapping. In order to separate the different facilities and Hg
species, different tracers were assigned to each source as well as to each Hg species.
4.3.3.2 Ambient Hg concentrations
Table 17 shows a percentile analysis of the simulated Hg concentration results from the grid cells
over the entire domain. Based on the result, it is clear that the contribution of Hg from the 10
facilities included in the simulation is relatively small compared to (i) the assumed background
concentration of elemental Hg in Australia of 1.3-1.4 ng/m3 (Bergan and Rodhe, 2001), and (ii)

51
the measured average total Hg concentration in Northwest Europe and in the Mediterranean
region of 1.6-2.4 ng/m3 (Wängberg et al., 2001).
In order to evaluate the contribution of particulate and divalent Hg from the 10 sources included
in the TAPM simulation, a number of published investigations are presented. In a study conducted
by US EPA (1997), the simulated total concentration of divalent and particulate Hg (over the
lower 48 States) at the 10th, 50th and 90th percentile level was 1.0, 5.6, and 23 pg/m3,
respectively26. Moreover, measurements conducted in Tennessee (US) show that divalent Hg
concentrations vary between 50-200 pg/m3 depending on the distance to the emitting sources
(Linberg and Stratton, 1998). In Europe, measured concentrations of particulate Hg ranges
between 10 (Sweden) and 50 (Germany) pg/m3 (Wängberg et al., 2001). Thus, the simulated
concentrations in this study seem to be in general agreement with the published data.
In Table 17, the minimum concentration level of Hg is zero. A zero level of Hg occurs mostly
over the ocean, both in the inner and outer domain, reflecting the dispersion of Hg with the
prevailing winds (the dispersion is represented in Figure 9 and 10).
Table 17 Percentile Analysis of simulated ambient mercury concentrations
Grid domain
Species
Min pg/m3
10th pg/m3
50th pg/m3
90th pg/m3
Max ng/m3
Inner Hg0 0 7 15 44 2.5 Hg(II)/Hgp 0 2 6 15 0.6 Hgtot 0 9 21 59 3.1 Outer Hg0 0 0 2 14 0.9 Hg(II)/Hgp 0 0 1 5 0.2 Hgtot 0 0 3 19 1.1
The maximum simulated ground level ambient Hg concentration (3.1 ng/m3) is (even if the
background concentration of Hg0 is added), well below (i) the US EPA determined reference
concentration of Hg vapor of 0.3 µg/m3 for the general population (US EPA, 1997), (ii) the limit
value for exposure in Europe of 0.05µg/m3 (Pirrone et al., 2001a)27, and (iii) the proposed air
quality objectives set in Victoria, Australia, of 1.8 µg/m3, for inorganic Hg (VIC EPA, 2002).
26 This study (US EPA (1997)) is, among other things, based on a Lagrangian-type simulation model (RELMAP) as well as a so-called ISC3 model for predicting Hg deposition 50 km from the emission source (including deposition of Hg0). RELMAP is used to investigate the emission, transport and fate of airborne Hg (Hg0, Hg(II), Hgp) over continental US. It incorporates some of the chemistry known to affects the Hg speciation, as well as wet and dry deposition (dry deposition of Hg0 is excluded from the simulation) mechanisms. The background concentration of Hg0 is included in the simulation. 27 According to WHO (2003), a guideline for inhalation of inorganic Hg vapor is established, as an annual average value, in Europe of 1 µg/m3. Thus, it is somewhat higher compared to the limit value reported by (Pirrone et al., 2001a).

52
4.3.3.3 Dry deposition of mercury
Table 18 shows a percentile analysis of simulated dry deposition rates from the grid cells over the
area of study. The simulated total average deposition flux in the inner domain varies between 0.2
and 1.4 µg/m2/yr (at the 10th and 90th percentile level, respectively). In occasional cases, close to
emitting sources (1-2 grid cells away from the source), the deposition flux of Hgtot may reach
levels of 50-60 µg/m2/yr. Corresponding values for the outer domain are smaller as the domain
includes relatively large areas that are unaffected by emitting Hg sources. Similarly, the average
deposition rate for the inner and outer domain is 0.8 and 0.2 µg/m2/yr, i.e. a difference of a factor
of 4.
In order to verify the obtained deposition rates of Hg species in this study, a comparison is made
with simulated dry deposition rates in an earlier US EPA (1997) study. The US study shows that
the simulated total dry deposition flux (excluding elemental Hg) for the lower 48 States in the
USA is, for the 10th, 50th, 90th percentile levels,: minimum 0.05, 0.18, 0.89 and maximum, 5.6 and
62.6 µg/m2/yr, respectively. Thus, the deposition fluxes of Hg in the US study are, compared to
the results in this study, significant higher. One obvious reason for the discrepancy in the
deposition rates of Hg species, is that the lower 48 States of the USA cover approximately the
same land area as Australia, but emit roughly 20 times as much Hg as Australia. A further reason
for the differences in simulated deposition rates may be that the US study used significantly
higher deposition velocities for the Hg species investigated than those used this study (Table 16).
Table 18 Percentile analysis of simulated dry deposition fluxes of mercury species
Grid domain
Species
Min µg/m2/yr
10th µg/m2/yr
50th µg/m2/yr
90th µg/m2/yr
Max µg/m2/yr
Inner Hg0 0.00025 0.033 0.070 0.210 11.6 Hg(II)/Hgp 0.0007 0.155 0.454 1.152 48.4 Hgtot 0.001 0.20 0.55 1.4 60 Outer Hg0 0 0 0.06 0.43 28.7 Hg(II)/Hgp 0 0 0.07 0.41 18.6 Hgtot 0 0 0.14 0.85 48
The outer grid domain includes parts of the ocean, which due to the prevailing wind direction does not experience any dry deposition.
Of the total anthropogenic Hg mass predicted to be deposited to the surface in the model domain,
85 % is estimated to come from divalent/particulate Hg emissions and 15 % from elemental Hg.
In the study by Xu et al (2000a,b), it is estimated that 57 % of the total amount of Hg that is dry
deposited in the model domain came from Hg(II), 6 % from Hgp, and 37 % from Hg0. Thus, the
contribution of Hg0 calculated as a percentage of the total deposition is, compared to this study,

53
somewhat higher. This may be explained by the fact that the Xu et al (2000a,b) simulation
includes the background concentration of Hg0.
If the average simulated deposition rate in the outer domain of 0.2 µg/m2/yr is integrated over the
area of study, approximately 105 kg (~ 8 % of the total emissions from the facilities investigated)
of Hg are deposited annually within the entire domain. The rest of the Hg is transported outside of
the domain and deposited elsewhere.
Since the simulated tracer (Hg) is dispersed with the prevailing winds, which, during this
simulation comes from east/northeast, the major part of the deposition fluxes of Hg can be found
over the mainland, as is shown in Figures 9 and 10. Moreover, it is clear from the contour plots
that the deposition fluxes of Hg are significant higher adjacent to the emitting source, than they
are at a greater distance from the source. This deposition trend is also illustrated in Figure 11,
where the deposition fluxes of Hg are concentrated around specific point sources. Thus, from the
information provided in Figure 11, it is obvious that the area around Comsteel, Pasminco CCS,
Vales Point PS and Eraring PS, would experience the greatest amount of deposited Hg in the
domain. The second and third areas that receive relatively large simulated masses of deposited Hg
are the Orica Chlorine plant and BHP Steel PKW.
In order to investigate (i) the aggregated average amount of Hg, and (ii) the percent of total
emitted Hg that is deposited at different distances around each facility, a number of deposition
calculations are presented. These calculations are based on simulated hourly grid deposition
fluxes of Hg around each facility, as well as on the emission rates of Hg from respective facility.
The result is presented in Tables 19-24 and Appendix D.
It is well-known that the differences in simulated air concentrations and deposition fluxes across a
model domain are mainly dependent on factors such as stack height (real height), the exit velocity
(momentum) of the plume from the stack, and the emission rate of Hg28. Thus, these
factors determine the height to which the plume rises from the stack top (the so-called effective
stack height), and consequently the distance the pollutant is carried before deposition (Cartwright,
1993)29. For example, power plants, which have relatively tall stacks, high exit velocities and
28 Additional factors that determine the effective stack height is wind speed, diameter of the stack, gas exit temperature, ambient temperature, and the atmospheric stability condition. (Cartwright, 1993) 29 The speciation of Hg species is, as previously described, crucial for the deposition patterns of Hg. Thus, atmospheric transformation/interaction processes, which determine the speciation of Hg, are important to include in models aiming at simulating Hg transportation and deposition. In this study, however, none of these processes/interactions are included. Thus, the "only" parameters effecting the dispersion and deposition of Hg, in this study, are the physical characteristics of the emission source and the atmospheric conditions.

54
emission rates, disperse the emitted pollutant further away from the emission point than sources
with the opposite conditions (as will be seen in the following paragraphs). In the model simulation
the actual stack height of five of the ten sources was unknown (Appendix C). In order to
investigate possible deposition fluxes at different stack heights and distances around each facility,
two additional simulations were conducted (Runs 20 and 30). The result of the simulations and
calculations are presented in Tables 19-24 and in Appendix D.
Figure 9 Contour plot of simulates of dry deposition of divalent/particulate mercury (unit: µg/h)
2 4 6 8 10 12 14 16 18 20 22 24
2
4
6
8
10
12
14
16
18
20
22
24
Mount Piper PS
Maldon CW
BHP Steel PKW
Orica Chlorine P SYDNEY
Vales Point PSEraring PS Pasminco
Comsteel
Lidell PSBayswater PS
Number of grid cells
In Tables 19 and 21-22, the area average deposition flux of Hg (12.5×12.5 km) ranges between
0.6-12.4 µg/m2/yr. The highest area (12.5×12.5 km) deposition flux of Hg are to be found around
Comsteel (12.4 µg/m2/yr), Pasminco CCS (12 µg/m2/yr), and the Orica Chlorine plant (8.8
µg/m2/yr). If these area deposition fluxes are calculated as a percentage of the total Hg emission
from respective facility, each Hg deposition flux represents 0.7, 1.7 and 1.1 % of the total Hg
emitted, respectively. However, the stack height of each of these facilities needs to be increased

55
by a factor of 5 (Table 21-22), to decrease the previously calculated fluxes by a factor of
approximately 2.
Figure 10 Contour plot of simulates of dry deposition of elemental Hg (unit: µg/h)
2 4 6 8 10 12 14 16 18 20 22 24
2
4
6
8
10
12
14
16
18
20
22
24
Mount Piper PS
Maldon CW
BHP Steel PKW
Orica Chlorine P SYDNEY
Vales Point PSEraring PS Pasminco
Comsteel
Lidell PSBayswater PS
Number of grid cells
The only source that does not experience a reduction in deposition fluxes of Hg as the height of
the stack is increased, is the Maldon cement works. The reason for this is unclear. One reason
may be that the facility is located in the inland and that it is subject to other atmospheric
conditions than those sources located closer to the coastline.
The highest emission rate of Hg in the model simulation comes from Bayswater power station,
which emits 301 kg/yr. Since Lidell power station (46 kg/yr) is located close to Bayswater power
station, the two sources are included, together, in the area-deposition calculations. The average
deposition flux of Hg in each of the three areas (12.5×12.5 km, 37.5 ×37.5 km and 52.5× 52.5
km), is 1.7, 2.0 and 0.8 µg/m2/yr, respectively (which constitute 0.1, 0.4 and 0.7 % of the total Hg

56
emitted). Thus, as the simulation shows, a significant amount of the emitted Hg is transported
away from their source with the prevailing winds.
Figure 11 The magnitude of dry deposition fluxes from TAPM simulation (unit: µg/h) The height of the plot is proportionally to the deposition flux of divalent/particulate Hg. The western corner in Figure 11 corresponds (since the Figure is tilted) to the northwest corner in Figures 8,9 and 10. The first, second, third and fourth highest deposition flux of Hg corresponds to source/sources (i) Comsteel, Pasminco CCS, Vales Point PS, Eraring PS, (ii) Orica Chlorine plant, (iii) BHP Steel PKW, and (iv) Lidell PS and Bayswater PS, respectively.
As the calculations presented in Tables 19-24 show, the area average deposition flux of Hg,
expressed as a percentage of the total Hg emitted, was in general, relatively small. This suggests
that a significant part of the Hg emitted from the facilities investigated would be transported away
from the domain.

57
Table 19 Area Average Mercury Deposition Rates for each Facility/Facilities
Run 10 Area Average Values of Dry Deposition
Emission Speciation of emission 12.5*12.5
km 37.5*37.5
km 52.5*52.5
km
Facility Name No. Stack height Hg(tot) Hg0 Hg(II)/Hgp Total Hg0 Hg(II)/Hgp Total Hg0 Hg(II)/Hgp Total Hg0 Hg(II)/Hgp
m kg/yr kg/yr kg/yr ugm-2yr-1 ugm-2yr-1 ugm-2yr-1 ugm-2yr-1 ugm-2yr-1 ugm-2yr-1 ugm-2yr-1 ugm-2yr-1 ugm-2yr-1 Mount Piper PS 2 250 45 22.5 22.5 0.6 0.06 0.5 1.0 0.1 0.9 0.5 0.1 0.4 Liddell PS 3 168 46 23 23 Bayswater PS 4 250} 301 150 150 1.7a 0.10 1.6 2.0 0.20 1.8 0.8 0.1 0.7 Comsteel 5 10 282 226 56 12.4 2.4 10.0 Pasminco CCS 6 74 110 88 22 7.8 1.5 6.3 3.3 0.6 2.7 Vales Point PS 7 178 86 43 43 2.1 0.30 1.8 Eraring PS 8 200 164 82 82 3.0 0.50 2.5 Maldon CW 9 10 23 18.4 4.6 2.0 0.3 1.7 2.8 0.4 2.4 N/A N/A N/A BHP Steel PKW 10 10 102 82 20 N/A N/A N/A 3.6 0.6 3.0 N/A N/A N/A Orica Chlorine P 11 10 124 87 37 5.2 1.0 4.2 4.2 0.5 3.7 1.7 0.2 1.5
a Source 3 & 4 is included in each area The deposition rate is simulated in TAPM during January 2001. The deposition flux is then scaled up to cover the whole year In the area 12.5*12.5 km, source 5,6,7 and 8 is located close to each other and some of the grid cells are therefore overlapping when the deposition is calculated. The same occurs for source 9 & 10 in area 37.5*37.5 km. More accurate data concerning these sources can be found in Run 20 and 30. Table 20 Percent of Total Mercury Dry Deposited around each Facility/Facilities
Run 10 Percent of Total Mercury Emissions Dry Deposited Within Each Area
Emission Speciation of emission 12.5*12.5
km 37.5*37.5
km 52.5*52.5
km
Facility Name No. Stack height Hg(tot) Hg0 Hg(II)/Hgp Total Hg0 Hg(II)/Hgp Total Hg0 Hg(II)/Hgp Total Hg0 Hg(II)/Hgp
m kg/yr kg/yr kg/yr % % % % % % % % % Mount Piper PS 2 250 45 22.5 22.5 0.2 0.02 0.2 1.6 0.2 1.4 3.1 0.4 2.7 Liddell PS 3 168 46 23 23 Bayswater PS 4 250} 301 150 150 0.1a 0.01 0.1 0.4 0.05 0.4 0.7 0.1 0.6 Comsteel 5 10 282 226 56 0.7 0.1 0.6 Pasminco CCS 6 74 110 88 22 1.1 0.2 0.9 1.4 0.3 1.1 Vales Point PS 7 178 86 43 43 0.4 0.06 0.3 Eraring PS 8 200 164 82 82 0.3 0.05 0.2 Maldon CW 9 10 23 18.4 4.6 1.4 0.2 1.2 9.4 1.5 7.9 N/A N/A N/A BHP Steel PKW 10 10 102 82 20 N/A N/A N/A 2.7 0.5 2.2 N/A N/A N/A Orica Chlorine P 11 10 124 87 37 0.7 0.1 0.5 2.6 0.3 2.2 3.9 0.5 3.4
a Source 3 & 4 is included in each area
Sources 5,6,7, and 8 are included in this area
Sources 5,6,7, and 8 are included in this area

58
Table 21 Area Average Mercury Deposition Rates for each Facility
Run 20 Area Average Values of Dry Deposition Emission Speciation of emission 12.5*12.5 km 37.5*37.5 km 52.5*52.5 km
Facility Name No. Stack height Hg(tot) Hg0 Hg(II)/Hgp Total Hg0 Hg(II)/Hgp Total Hg0 Hg(II)/Hgp Total Hg0 Hg(II)/Hgp
m kg/yr kg/yr kg/yr ugm-2yr-
1 ugm-2yr-1 ugm-2yr-1 ugm-2yr-
1 ugm-2yr-1 ugm-2yr-1 ugm-2yr-
1 ugm-2yr-1 ugm-2yr-1
Comsteel 5 10 282 226 56 11.8 2.3 9.5 3.4 0.60 2.8 3.0 0.60 2.4 Pasminco CCS 6 10 110 88 22 12.0 2.3 9.7 1.4 0.20 1.2 1.4 0.2 1.2
Maldon CW 9 10 23 18.4 4.6 1.3 0.2 1.1 0.8 0.1 0.7 0.6 0.1 0.5 BHP Steel PKW 10 10 102 82 20 2.3 0.8 1.5 1.6 0.3 1.3 1.8 0.3 1.5 Orica Chlorine P 11 10 124 87 37 8.8 1.1 7.7 2.1 0.3 1.8 1.5 0.2 1.3 The deposition rate is simulated in TAPM during January 2001. The deposition flux is then scaled up to cover the whole year. Table 22 Area Average Mercury Deposition Rates for each Facility
Run 30 Area Average Values of Dry Deposition Emission Speciation of emission 12.5*12.5 km 37.5*37.5 km 52.5*52.5 km
Facility Name No. Stack height Hg(tot) Hg0 Hg(II)/Hgp Total Hg0 Hg(II)/Hgp Total Hg0 Hg(II)/Hgp Total Hg0 Hg(II)/Hgp
m kg/yr kg/yr kg/yr ugm-2yr-
1 ugm-2yr-1 ugm-2yr-1 ugm-2yr-
1 ugm-2yr-1 ugm-2yr-1 ugm-2yr-
1 ugm-2yr-1 ugm-2yr-1
Comsteel 5 50 282 226 56 6.2 1.2 5.0 2.1 0.40 1.7 2.1 0.40 1.7 Pasminco CCS 6 74 110 88 22 2.4 0.5 1.9 0.7 0.10 0.6 1.0 0.1 0.9 Maldon CW 9 50 23 18.4 4.6 1.0 0.2 0.8 0.7 0.1 0.6 0.6 0.1 0.5 BHP Steel PKW 10 50 102 82 20 0.9 0.3 0.6 1.1 0.2 0.9 1.4 0.2 1.2 Orica Chlorine P 11 50 124 87 37 4.2 0.5 3.7 1.4 0.2 1.2 1.0 0.1 0.9 The deposition rate is simulated in TAPM during January 2001. The deposition flux is then scaled up to cover the whole year.

59
Table 23 Percent of Total Mercury Dry Deposited around each Facility
Run 20 Percent of Total Mercury Emissions Dry Deposited Within Each Area Emission Speciation of emission 12.5*12.5 km 37.5*37.5 km 52.5*52.5 km
Facility Name No. Stack height Hg(tot) Hg0 Hg(II)/Hgp Total Hg0 Hg(II)/Hgp Total Hg0 Hg(II)/Hgp Total Hg0 Hg(II)/Hgp
m kg/yr kg/yr kg/yr % % % % % % % % %
Comsteel 5 10 282 226 56 0.6 0.1 0.5 1.7 0.30 1.4 3.0 0.60 2.4 Pasminco CCS 6 10 110 88 22 1.7 0.3 1.4 1.8 0.30 1.5 3.7 0.6 3.1 Maldon CW 9 10 23 18.4 4.6 1.0 0.2 0.8 5.3 0.8 4.5 7.6 1.2 6.4 BHP Steel PKW 10 10 102 82 20 0.3 0.1 0.2 2.2 0.4 1.8 4.7 0.8 3.9 Orica Chlorine P 11 10 124 87 37 1.1 0.1 1.0 2.3 0.3 2.0 3.3 0.4 2.9 The deposition rate is simulated in TAPM during January 2001. The deposition flux is then scaled up to cover the whole year.
Table 24 Percent of Total Mercury Dry Deposited around each Facility
Run 30 Percent of Total Mercury Emissions Dry Deposited Within Each Area Emission Speciation of emission 12.5*12.5 km 37.5*37.5 km 52.5*52.5 km
Facility Name No. Stack height Hg(tot) Hg0 Hg(II)/Hgp Total Hg0 Hg(II)/Hgp Total Hg0 Hg(II)/Hgp Total Hg0 Hg(II)/Hgp
m kg/yr kg/yr kg/yr % % % % % % % % %
Comsteel 5 50 282 226 56 0.4 0.1 0.3 1.1 0.20 0.9 2.1 0.40 1.7 Pasminco CCS 6 74 110 88 22 0.4 0.1 0.3 1.0 0.20 0.8 2.5 0.3 2.2 Maldon CW 9 50 23 18.4 4.6 0.6 0.1 0.5 4.4 0.7 3.7 6.6 1.0 5.6 BHP Steel PKW 10 50 102 82 20 0.2 0.1 0.1 1.4 0.2 1.2 3.7 0.5 3.2 Orica Chlorine P 11 50 124 87 37 0.6 0.1 0.5 1.5 0.2 1.3 2.2 0.3 1.9 The deposition rate is simulated in TAPM during January 2001. The deposition flux is then scaled up to cover the whole year.

60
5. The Chemistry of Atmospheric Mercury
A knowledge of Hg reactions in the atmosphere is critical for modelling its transportation,
transformation, and concentrations throughout the atmosphere as well as for estimating its
deposition fluxes to environmental surfaces. The following section describes some of the
chemical reactions, which take place in the atmosphere along with kinetic data for Hg. In
addition, the residence time (τ) and/or half-life (t1/2) of Hg, which are convenient measures of the
reaction rate, were calculated and are presented for most of the reactions. In the most general
terms, the chemistry of atmospheric Hg involves aqueous phase (e.g., a raindrop or a fog droplet)
and gaseous phase reactions, transitions of elemental Hg (Hg0) and divalent Hg (Hg(II)) species
between the aqueous and gaseous phase, the solid and aqueous phase and also between the solid
and gaseous phase. These reactions and interactions between different atmospheric reactants and
media determine the speciation of Hg, its removal by processes such as wet and dry deposition, as
well as its atmospheric residence time (Schroeder and Munthe, 1998).
Section 5.1 deals with individual chemical reactions in the aqueous phase. In this phase, for Hg,
two simultaneous reactions occur; oxidation of elemental Hg and reduction of divalent Hg. In
addition, different Hg species and other atmospheric reactants partition themselves between the
air and water phases under equilibrium conditions (Figure 12).
Figure 12 Atmospheric mercury chemistry
X - oxidants, Y - reducing agents. Some of the divalent Hg in the water droplet is also adsorbed onto particles.
Hg(II)(g) Hg(II)(aq) Hg0(aq) Hg0
(g)
X(g)
X(aq)
Y(g)
Y(aq)
Wet Deposition
Atmosphere
Environmental surfaces
H2O
Hg0(g) Hg0
(ads)
Hg(II)(ads) Dry Deposition
Dry
/wet
dep
ositi
on
Dry
/wet
dep
ositi
on
Dry/wet deposition
Hg(II)(s)

61
The driving force and the direction of the Hg flux across the interface, which determine the Hg
concentration in the water phase, are partly governed by the redox reactions taking place in the
aqueous and gaseous phase and partly by the Henry's law constant (Pleijel and Munthe, 1995a,b;
Schroeder et al., 1991). For instance, when divalent Hg (Hg(II)(aq)) is reduced to elemental Hg
(Hg0(aq)) it will, due to its lower solubility, partition back into the gas phase. Thus, the Hg
concentration decrease in the water droplet is less and is hence subjected to wet deposition, and
vice versa. This is illustrated in Figure 12.
As shown in Figure 12, reducing agent (Y), oxidant (X) and elemental Hg are divided across the
air/water surface (in equilibrium). Unlike these three groups of reactants, divalent Hg, due to its
low volatility and high solubility, does not generally partition back into the atmosphere
(Hedgecock and Pirrone, 2001).. Thus, Hg(II)(aq) has to be reduced to Hg0(aq)
in order to reach the
atmosphere. In the water droplet, which is sometimes referred to as a chemical reactor (Lin and
Pehkonen, 1998b), reactions between Hg0/Hg(II) and X/Y occurs as well as reactions between X
and Y (and between different reaction products). Depending on the concentration of individual
chemical species, the pH, the liquid water content in the atmosphere, and other meteorological
factors (e.g. temperature and barometric pressure) different reactions temporarily dominate over
others (Lin and Pehkonen, 1997; Seigneur et al., 1994). Thus, the outcome of the reactions is
determined by the prevailing conditions in the water droplet and in the atmosphere. The aqueous
oxidants are (i) ozone, (ii) hydroxyl radical and (iii) chlorine, and the reducing agents are: (iv)
sulfite complexes, (v) hydroperoxide radicals, and (vi) photoreduction of divalent Hg complexes.
Section 5.2 discusses the oxidation of elemental Hg to divalent Hg in the gas phase (right-hand
side of the figure). The following atmospheric oxidants are present - (i) ozone, (ii) hydroxyl
radical, (iii) nitrate radical, and (iv) hydrogen peroxide30. The products of these reactions
(Hg(II)(g)) are removed from the atmosphere in three ways31: (i) they may condense onto
particulate materials, which are either scavenged by atmospheric water droplets or dry deposited
to marine or terrestrial surfaces, (ii) it may, in the presence of precipitation, dissolve into the
aqueous phase (as described above) and/or (iii) it may dry deposit to the earth surface. Thus,
elemental Hg is removed from the atmosphere by transforming to divalent Hg, either by
partitioning into the aqueous phase where it interacts with different constituents (see aqueous
phase reactions), or by being oxidised in the gaseous phase as described in this paragraph.
Furthermore, it may be directly adsorbed onto particles which are either wet or dry deposited, or
30 In addition, reactions with DMM are also summarised in section 5.2.5. 31 Hg(II)(g) enters also the atmosphere from different emitting sources and are consequently subjected to the same removal processes.

62
Hg0(g) is simply dry deposited (at a very low rate) to the ground (left-hand side of Figure 12) (EC,
2001; Porcella et al., 1996; Seigneur et al., 1994, 1998; Schroeder and Munthe, 1998).
The atmospheric chemistry of Hg is diverse and complex, and the review that follows will
therefore necessarily be simplified. Furthermore, it should be mentioned that the scientific
experiments presented are subject to a degree of uncertainty particularly when it comes to the
derivation of kinetic data in association with gaseous oxidation reactions (some of the Hg0 is lost
during the experiments at the walls of reactor vessels, in so called heterogeneous reactions). In
order to exemplify these uncertainties, often more than one study is included in the description of
a reaction.
5.1 Chemical transformations in the aqueous phase32
5.1.1 Oxidation
5.1.1.1 Oxidation of Hg0 by O3
An investigation of aqueous phase oxidation of elemental Hg by ozone (O3) was performed by
Munthe (1992a) using a relative rate technique. In the experiment the relative consumption of Hg0
and S(IV) was measured in the presence of O3. The rate constant of the aqueous oxidation of Hg0
with O3 was then estimated by comparing the measured data with the rate constant of the reaction
between S(IV) and O3. Based on the result the average rate constant (k7) was estimated to be
(4.7±2.2) ×107 M-1s-1. The experiments were also performed at different temperatures and pH and
the result showed that the reaction is independent of pH (5.2-6.2) and temperature (5-35 0C).
Pleije and Munthe (1995a) suggested the following reaction scheme:
)(2)(2
)()(3)( aqaqaqH
aqoaq OOHHgOHg ++→+ −++ (7)
[ ] [ ] [ ])(3)(7)(
7 aqoaq
oaq OHgk
dtHgd
R ×=−= (8)
Ozone is, in broad terms, a product of a rather complex photochemical reaction that occurs in the
presence of sunlight, nitrogen oxides (NOx) and volatile organic compounds (VOCs) (Finlayson-
Pitts and Pitts, 1986). Atmospheric gaseous ozone scavenges the aqueous phase where it can
interact with, amongst other things, elemental Hg. The expected half-life of Hg0 (t1/2 = ln2 k7-
32 The structure of this part and 5.2 is from Lin and Pehkonen (1999a).

63
1[O3(aq)]-1) is estimated to be approximately 40 s at an ozone concentration of 4×10-10 M (Seinfeld
and Pandis, 1997).
5.1.1.2 Oxidation of Hg0 by ·OH
Lin and Pehkonen (1997) studied the kinetics of aqueous-phase oxidation of Hg0 by the hydroxyl
radical(·OH) using a steady-state technique employing nitrate photolysis as the radical source
and benzene as the radical scavenger. The experiments were performed in the absence of oxygen.
In the proposed chemical scheme elemental Hg is first oxidised to Hg+(reaction (9)), which in turn
is oxidised to divalent mercury (reaction (10)). In the study conducted by Lin and Pehkonen
(1997), the rate constant of reaction (9) was calculated to be k9 = 2.0 ×109 M-1s-1. The rate
constant of reaction (10) was determined in a previous study to be approximately k10 = 1010 M-1s-1
(Buxton et al., 1988). Thus, the rate-determining step is the oxidation of elemental Hg and the
overall rate expression can therefore be written as (11).
−+ +→⋅+ )()()(
0)( aqaqaqaq OHHgOHHg (9)
−++ +→⋅+ )(2
)()()( aqaqaqaq OHHgOHHg (10)
[ ] [ ] [ ])(0
)(9
0)(
9 aqaqaq OHHgk
dtHgd
R ⋅×=−= (11)
In a recent study of the Hg0 +·OH reaction (Gårdfeldt et al, 2001), a new rate constant was
determined using a relative rate technique with methyl mercury as a reference compound. The
following reaction scheme was proposed:
)()()(0
aqaqaq HgOHOHHg ⋅→⋅+ (12)
( ) )(2)()(2)(2)(2)( aqaqaqaqaqaq OHOHHgOHOHgOH −+ ++→++⋅ (13)
[ ] [ ] [ ])(0
)(12
0)(
12 aqaqaq OHHgk
dtHgd
R ⋅×=−= (14)
where k12 = (2.4±0.3)×109 M-1s-1. The oxidation of elemental Hg by OH radical forms a ·HgOH
intermediate that is assumed to be the dominant Hg(І) species in the aqueous phase. In the
presence of dissolved oxygen, the intermediate will be rapidly oxidised to Hg(ІІ) (Gårdfeldt et al,
2001).

64
Compared to the rate constant of ozone, the OH radical reaction is theoretically faster. However,
depending on the individual concentration of ·OH and O3 in atmospheric waters, either oxidation
path can be dominant. When the aqueous-phase concentration of ozone is less than 5×10-12 M, the
predominant pathway of Hg0 is by hydroxyl radicals, providing a [·OH] of 10-12 M (Lin and
Pehkonen, 1997). However, in a more polluted atmosphere, with an ozone concentration
exceeding 20 ppb, the contribution of the radical reaction is only about 10 % (Travnikov and
Ryaboshapko, 2002).
The hydroxyl radical (·OH) is a highly reactive species produced by a number of photochemical
processes in the atmosphere (Jacob, 1986). The radical has a relatively high solubility and can be
scavenged into atmospheric droplets (Lin and Pehkonen, 1997). Studies have also shown that
·OH can be readily formed within droplets through photolysis of ferric-hydroxide complexes
(H2O2, HNO3, HONO) (Faust and Hoigne, 1990; Faust and Allen, 1993). Since [·OH] is a
daytime oxidant it reaches its peak levels around midday (Lin and Pehkonen, 1999b). Assuming a
midday [·OH] of 10-12 M in atmospheric waters (Seinfeld and Pandis, 1997), the half-life of Hg0
(t1/2 = ln2 k9-1[·OH(aq)]-1) is estimated to be approximately 6 minutes.
5.1.1.3 Oxidation of Hg0 by chlorine (HOCL/OCL-)
Lin and Pehkonen (1998b) have investigated the kinetics of aqueous-phase oxidation of elemental
Hg by chlorine (HOCl/OCL-) using chloramine as a novel reservoir. In the presence of water
droplets, Cl2 will partition into the aqueous phase and form HOCL (Cl2 + H2O → HOCL + H++
Cl-). Depending on pH, chlorine exists in two different forms: the protonated hypochlorous acid
(HOCl) and the deprotonated hypochlorite ion (OCl-), (HOCl ↔ OCl- + H+) (Lin and Pehkonen,
1998b). Both chlorine species can oxidise Hg0:
−−+ ++→+ )()(2
)(0
)()( aqaqaqaqaq OHClHgHgHOCl (15)
−−++− ++→+ )()(2
)(0
)()( aqaqaqH
aqaq OHClHgHgOCl (16)
According to Lin and Pehkonen (1998b), the distribution of HOCl and OCl- can be expressed as
α0[Cl2(aq)]act and α1[Cl2(aq)]act, where α0 and α1 are the fraction of [HOCL] and [OCl-], respectively,
compared to [Cl2(aq)]act.where [Cl2(aq)]act represents the actual free chlorine concentration in the
aqueous phase, that is, the summation of [HOCL] and [OCl-]. The overall rate expression can then
be expressed as:

65
[ ] [ ] [ ]actaqaq
aq ClHgkdt
HgdR )(2
0)(
0)(
1615 ×=−=+ (17)
where k15 = (2.09±0.06)×106 M-1s-1, k16 = (1.99±0.05)×106 M-1s-1, k = k15α0+ k16α1, α0 =
[H+]/([H+]+Ka,HOCl) and α1 = Ka,HOCl/([H+]+Ka,HOCl) (Lin and Pehkonen, 1998b).
Atmospheric chlorine may be either produced in the marine boundary layer through the photolysis
of ozone in the presence of sea-salt particles (Oum et al., 1998), or volatilised from sea-salt
aerosol generated by wave breaking on ocean surfaces (Kenne et al., 1996). Reactive chlorine has
the highest levels just before sunrise in the marine boundary layer and in coastal regions (Keene et
al., 1993). During daytime, its levels decrease due to the photolysis of HOCl and Cl2 (Impey et al.,
1995).
The solubility of gaseous chlorine in water changes considerably with variations in pH and the
chloride concentration. According to Lin and Pehkonen (1998b), the solubility of chlorine in
atmospheric waters is governed by the effective Henry law constant Heff:
[ ] [ ]( ) [ ] [ ]( ))10101( 2
8.103.3
int +−
−
+−
−
×+
×+=
HClHClHH eff (18)
where Hint is the intrinsic Henry’s Law constant for chlorine, Hint = 7.61×10-2 M atm-1. Another
study by Lin and Pehkonen (1999b) showed that increasing pH and decreasing [Cl-] increases the
divalent Hg concentration in atmospheric droplets due to the increase in [Cl2](aq).
Due to the prevalence of marine environment over a large proportion of the earth’s surface,
chlorine is an important oxidant in the chemistry of the atmosphere. Although it is present at
much lower concentrations in ambient air compared to the other oxidants, its higher solubility
leads to much higher [Cl2](aq) compared to O3 and ·OH, and it therefore causes considerable
oxidation of Hg0 in the water droplet (Lin and Pehkonen, 1999b). Since the chlorine concentration
in water droplets is determined by a rather complex mechanism, the calculation of the half-life of
Hg0 is not attempted.

66
5.1.2 Reduction
5.1.2.1 Reduction of Hg(ΙΙ) by S(ΙV)
Munthe et al. (1991) investigated the kinetics of the reduction of divalent Hg (Hg(ΙΙ)) by sulfite
(in excess) using UV-spectrometry. Divalent mercury forms a number of complexes depending on
the ions present in the aqueous phase. The reaction between sulfite S(IV) ions and divalent Hg
forms a highly unstable complex, HgSO3, and a relative stable complex Hg(SO3)22-. According to
proposed reduction mechanism, the unstable complex decomposes to Hg+, which in turn is rapidly
reduced to Hg0 (Reaction (13)). The stable complex does not form Hg0 (Munthe, 1994). In the
experiments conducted by Munthe et al (1991), HgSO3 was not directly observed. The rate of its
decomposition is instead estimated from a composite rate constant, assuming HgSO3 to be a
steady-state intermediate. The proposed overall chemical scheme for the reduction is as follows
(Pleijel and Munthe, 1995a):
productsHgHgSO aqaq +→ 0)()(3 (19)
[ ] [ ])(319)(
19
)(aq
aq HgSOkdtIIHgd
R =−= (20)
where k19 = 0.6 s-1. The half-life (t1/2=ln2k19-1) of Hg(II) is approximately 1 s.
However, in a recent study (Van Loon et al, 2000), the rate coefficient of the decomposition of
HgSO3 was directly measured using UV-spectrometry.
)(0)()(3 Ι+ → VSHgHgSO aq
decompaq (21)
[ ] [ ])(321)(
21
)(aq
aq HgSOkdtIIHgd
R =−= (22)
According to the study, the rate coefficient k15 is independent of [Hg2+], [HSO3-]. [O2(aq)], and
ionic strength. It is highly temperature dependant and it roughly quadruples with each 10 0C
increase in temperature. At pH 3 and 25 0C, the value of the rate constant k21 is
(0.0106±0.0009) s-1, i.e. 55 times slower than previously reported. The rate constant can be
calculated for different temperatures:

67
TTTk 12595)971.31()/ln( 21
−×= (23)
Based on Equation 23, the half-life (t1/2 = ln2k211) of Hg(II) is 65 s and 3600 s at 25 0C and 0 0C,
respectively. The major role of sulfite in the cloud chemistry of Hg is to reduce divalent Hg to
elemental Hg, which partitions back into the gas phase. However, if the air masses cools,
reduction of divalent Hg becomes much slower, i.e. more Hg will be present in the water droplet
and can consequently undergo wet deposition (Van Loon et al., 2000). In a recent study by Van
Loon et al., (2001), it is suggested that the product of reaction (15) may be Hg·SO2, instead of
Hg0(aq), due to direct reaction of Hg0 and SO2(g) or aqueous hydrogen sulfite, or by reduction of
Hg(ΙΙ) in the presence of excess S(IV). This may also have an effect on the total Hg concentration
in the water phase, since the Hg·SO2 complex is at least three orders of magnitude more soluble
than Hg0(aq). These results appear to have important implications for the partitioning of
atmospheric Hg across the water/gas interface prior to its wet deposition (Van Loon et al., 2001).
Sulfite (in the form of H2SO3, HSO3- and SO3
2-) is produced by the scavenging of sulphur dioxide,
SO2(g), into atmospheric water droplets. In the droplet, S(IV) exists in equilibrium with SO2(g). The
solubility of SO2(g) is highly dependant on the pH value of the aqueous phase and the amount of
dissolved S(IV) increases with increasing pH (Seinfeld and Pandis, 1997). In the water phase
S(IV) interacts with divalent Hg and forms two different complexes. In previous investigations, it
has been suggested that the stable Hg(SO3)22- molecule is the major form of mercury-sulfite
complex in the water phase (Lin and Pehkonen, 1998a). However, in a recent study HgSO3 has
been shown to be more abundant in clouds than Hg(SO3)22- under virtually all atmospheric
conditions, even in highly polluted air masses with up to 10 ppb SO2(g) (Van Loon et al., 2001).
Depending on the concentration of the constituents of the aqueous phase different complexes are
formed. In the presence of high chloride concentrations, Hg(ΙΙ) is mostly present as HgCl2 (Lin
and Pehkonen, 1999b). It is not until the chloride concentration is below 5×10-6 M, that the sulfite
reduction becomes significant (Ryaboshapko et al., 2001). However, in Europe, the chloride
concentration in atmospheric water is always above 2×10-6 M (Ilyin et al., 2001). Thus, the
contribution from S(IV) reduction is, under these conditions, small.
Sulphur dioxide SO2(g) is a pollutant directly discharged into the atmosphere from different
emission sources and cannot be formed through atmospheric processes. Despite its lifetime of less
than a week, SO2(g) can be horizontally and vertically distributed over large areas. The highest

68
concentrations of SO2(g) is observed near industrial areas especially in the northern hemisphere
(Lelieveld, 1997).
5.1.2.2 Photoreduction of Hg(ΙΙ)
Xiao et al (1994) studied the photochemical behaviour of Hg(OH)2 and HgS22- in aqueous
solutions. Their results indicate that when HgS22- was subjected to broadband light (wavelength
>290 nm) it produced both Hg0 and HgS, with HgS as the major form. Hg(OH)2, which was found
to be the most photoreactive species, produced Hg0 when it was irradiated with the same type of
light as HgS22-. The kinetic data for the latter photoreduction was estimated using the following
chemical scheme:
productsHgOHHg aqhv
aq +→ 0)()(2)( (24)
[ ] [ ])(224)(
24 )()(
aqaq OHHgk
dtIIHgd
R =−= (25)
where k24 = 3×10-7s-1. This reaction is approximately 2 to 4 times faster than the formation of Hg0
from HgS22-. The rate constant corresponds to a half-life (t1/2 = ln2 k24
-1) of 600 h, which indicates
that this pathway is unlikely to be significant in atmospheric processes (Xiao et al., 1994).
5.1.2.3 Reduction of Hg(ΙΙ) by HO2·
The kinetics of divalent Hg reduction by hydroperoxyl radicals (HO2·) have been studied by
Pehkonen and Lin (1998) using a FEP Teflon reactor employing oxalate photolysis as the radical
source. The reaction scheme is initiated by the reduction of divalent Hg, forming Hg+, which is
then rapidly reduced to elemental Hg:
+⋅ ++Ι→ΙΙ+ )()(2)()()(2 )()( aqaqaqaqaq HOHgHgHO (26)
+⋅ ++→Ι+ )()(20
)()()(2 )( aqaqaqaqaq HOHgHgHO (27)
[ ] [ ] [ ]⋅×ΙΙ=ΙΙ
−= )(2)(26)(
26 )()(
aqaqaq HOHgk
dtHgd
R (28)

69
where k26= 1.7 ×104 M-1s-1. Since Reaction (26) is assumed to be the rate-determining step, the
overall rate expression can be written as Equation (28). However, when chlorine is present it
affects the rate constant k26 which decreases to 1.1 ×104 M-1s-1. The reduction is probably caused
by the presence of the stable complex HgCl20 (Pehkonen and Lin (1998).
The hydroperoxyl radical [HO2·] is, like [·OH], a highly reactive species formed by a number of
different photochemical processes in the atmosphere (Jacob, 1986). Due to its high solubility it
can be efficiently scavenged into atmospheric droplets (Lin and Pehkonen, 1997). Studies have
also shown that [HO2·] can be formed within water droplets (Schwartz, 1984; Arakaki et al.,
1995). A major sink of [HO2·] is the formation of hydrogen peroxide (H2O2), (2 HO2· → H2O2)
(Faust et al., 1993), which is considered to be the main oxidant of S(IV) in the liquid phase at low
pH values (Möller and Mausersberger, 1995).
Assuming a [HO2·] concentration of 10-8 M (Finlayson-Pitts and Pitts, 1986), the estimated half-
life of Hg (ΙΙ) (t1/2 = ln2 k26-1[HO2(aq)·]-1) is approximately 1 hour.
5.2 Chemical transformations in the gaseous phase
5.2.1 Oxidation of Hg0 by O3
Hall (1995) has examined the kinetics of the gas-phase oxidation of Hg0 with ozone using a FEP
Teflon reactor. The experiments were performed in sunlight, in darkness, at different
temperatures, and with reactors of different surface-to-volume ratios. The following oxidation
scheme was suggested:
)(2),()(30
)( gsggg OHgOOHg +→+ (29)
` [ ] [ ] [ ])(3
0)(23
0)(
23 ggg OHgk
dtHgd
R ×=−= (30)
where k29 = (3±2)×10-20 cm3molec.-1s-1 at 20 0C, assuming first order O3 dependence. According
to the experiment, the order of the reaction with respect to ozone is slightly below 1.0 (0.81). The
reason for this deviation was not clear. However, if the obtained order (0.81) is used instead, the
rate coefficient (k29) is calculated to be 4.1×10-20 cm3molec.-1s-1. The reaction rate is also found to
be six times faster in sunlight than in darkness (Hall, 1995). Based on experimental parameters, its
temperature dependence can be expressed as follows:

70
]/1203exp[101.2 1829 Tk −××= − (cm3molec.-1s-1) (31)
Background concentrations of atmospheric ozone are about 20-30 ppb, but can be several hundred
ppb in polluted air (Finlayson-Pitts and Pitts, 1986). Assuming a ozone concentration of 30 ppb
and a troposphere temperature of 0 0C (k29=2.56 ×10-20), the expected half-life (t1/2 = ln2 k29-
1[O3(g)]-1) and residence time (τ = k26-1[O3(g)]-1) of Hg0 is approximately 1.2 yr and 1.7 yr,
respectively. However, in a more polluted air mass, with an ozone concentration of 100 ppb,
corresponding values are 0.35 yr and 0.5 yr.
In a recent study by Bergan and Rodhe (2001) it was suggested that the reaction rate reported by
Hall (1995) was slower than actually occurs, and that the reaction with O3 is not, as previously
thought, the major atmospheric oxidant (see next section).
5.2.2 Oxidation of Hg0 by·OH
The kinetics of gas-phase oxidation between elemental Hg and hydroxyl radical (·OH) has been
studied using a relative rate technique with cyclohexane as the reference compound (Sommar et
al, 2001). OH radicals were produced by photolysis of methyl nitrate. The rate coefficient is at
(295±2 K), measured to be k32 = (8.7±2.8)×10-14cm3s-1, using the following chemical scheme:
)()(0
)( ggg HgOHOHHg ⋅→⋅+ (32)
)(2)()(2)( gggg HOHgOOHgOH ⋅+→+⋅ (33)
[ ] [ ] [ ])(0
)(32
0)(
32 ggg OHHgk
dtHgd
R ⋅×=−= (34)
The OH radical is considered to be a daytime active species, and its concentration is dependent of
the presence of other pollutants in the atmosphere that can either produce OH radicals or react
with them. According to a simulation by Bergan and Rodhe (2001), OH radicals rather than O3
may have a significant part in the removal of Hg0 from the troposphere. The OH radical may also
be a key player for initiating oxidation of trace compounds in the atmosphere, such as dimethyl
mercury (Niki et al., 1983b) (see 2.5.2). Using the average concentration of the atmospheric
hydroxyl radical of 106 molecules cm-3 (Tranikov and Ryaboshapko, 2002), the expected half-life
(t1/2 = ln2 k32-1[·OH]-1
(g)) and residence time (τ = k32-1[·OH]-1
(g)) of Hg0 are calculated to be
approximately 0.25 and 0.4 yrs, respectively.

71
5.2.3 Oxidation of Hg0 by NO3·
The gas-phase rate coefficient for the reaction between elemental Hg and the nitrate radical
(NO3·) has been investigated using a fast flow-discharge technique (Sommar et al, 1997). In the
study, nitrate radicals was generated by reaction of fluorine atoms in an excess of nitric acid. The
following reaction scheme was assumed:
)(2)()(30
)( gggg NOHgONOHg +→+ ⋅ (35)
[ ] [ ] [ ]⋅×=−= )(30
)(35
0)(
35 ggg NOHgk
dtHgd
R (36)
where k35 = 4 ×10-15 cm3molec.-1s-1. However, since the obtained rate constant was subjected to
statistical errors it should be regarded as an upper limit (Sommar et al., 1997).
The most significant source of NO3·in the atmosphere is the reaction of ozone and nitrogen
dioxide. The radical reaction of Hg0 + NO3· is assumed to influence the atmospheric chemistry
only at night, since NO3(g)· is photolyzed by solar radiation (Finlayson-Pitts and Pitts, 1986).
Assuming a NO3(g)·concentration of 1×108 molecules cm-3 (Finlayson-Pitts and Pitts, 1986) the
expected half-life (t1/2 = ln2 k35-1[NO3(g)·]-1) and residence time (τ = k35
-1[NO3(g)·]-1) of Hg0 are
approximately 20 d and 30 d, respectively.
5.2.4 Oxidation of Hg0 by H2O2
Tokos et al (1998) have studied the kinetics of the gas-phase oxidation of elemental Hg and
hydrogen peroxide (H2O2) using a FEP Teflon reactor. Since the results obtained from the
experiment were at or below the detection limit of the equipment, the rate constant should,
according to the study, be viewed as an upper limit. The constant was measured to be k37 =
8.5×10-19 cm3molec.-1s-1. The following chemical scheme was suggested by Seigneur et al.
(1994):
)(2)(220
)( )( ggg OHHgOHHg →+ (37)

72
[ ] [ ] [ ])(220
)(37
0)(
37 ggg OHHgk
dtHgd
R ×=−= (38)
In the study by Seigneur et al. in 1994, the same rate constant was estimated to be k37 = 4.0×10-16
cm3molec. -1s-1, i.e. three orders of magnitude larger than the above mentioned study. The reason
for this is not obvious, however, the rate constant used by Seigneur et al. (1994), was deduced
from experimental studies of the gas-phase oxidation of elemental Hg and Cl2, i.e. an entirely
different reaction (Schroder et al. 1991).
The reaction rate is strongly dependent on temperature and Tranikov and Ryaboshapko (2002)
derived the following Ahrrenius expression using the activation energy of 75 kJ/mole:
k37 = 8.4×10-6 ×exp[-9021/T] (cm3molec. -1s-1) (39)
Hydrogen peroxide (H2O2) is a daytime active oxidant, produced by photooxidation of
formaldehyde and hydrocarbons in the presence of NOx (Finlayson-Pitts and Pitts, 1986).
Assuming a H2O2(g) concentration of 1 ppb (Tokos et al, 1998) and a tropospheric temperature of 0 0C (k37=3.7 ×10-20), the expected half-life (t1/2 = ln2 k37
-1[H2O2(g)]-1) and residence time (τ = k37-
1[H2O2(g)]-1) of Hg0 is approximately 24 yr and 34 yr, respectively. Hence, this reaction
mechanism is likely to be insignificant compared to the other reactions.
5.2.5 Dimethyl mercury reactions
5.2.5.1 Reaction with nitrate radical
The rate constant of the reaction between dimethyl mercury (DMM) ((CH3)2Hg) and nitrate
radical (NO3·) has been investigated using a fast flow-discharge technique (Sommar et al, 1996).
In the study, nitrate radicals was produced by the reaction of fluorine atoms (formed in a
microwave discharge) and nitric acid (in excess). The proposed overall chemical scheme is as
follows:
productsNOHgCHCH gg →+ •)(3)(33 (40)
[ ] [ ] [ ]•×=−= )(3)(3340)(33
40 ggg NOHgCHCHk
dtHgCHCHd
R (41)

73
where k40 = 8.7×10-14 cm3molec. -1s-1 at 25 0C. The dominant reaction product was HgO although
minor quantities of elemental Hg (~2%) were also observed (Sommar et al., 1997). The study also
showed that all carbon in DMM was transformed into gas-phase organic compounds, such as
formaldehyde, methanol and methyl peroxynitrate.
The rate of reaction is temperature dependent and the Arrhenius equation is:
]/)4001760(exp[102.3 1140 Tk ±−××= − (cm3molec. -1s-1) (42)
The expected half-life (t1/2 = ln2 k40-1[NO3(g)·]-1) and residence time (τ = k40
-1[NO3(g)·]-1) of DMM
at a tropospheric temperature of 0 0C (k36 = 5.1×10-14 cm3molec. -1s-1) is approximately 4 h and
5.5 h, respectively, assuming a [NO3(g)·] of 1×109 molecules cm-3 (Finlayson-Pitts and Pitts,
1986).
5.2.5.2 Reaction with other species
Table 25 gives a summary of kinetic data and residence time for DMM with different oxidants.
Table 25 Oxidation of DMM with different oxidantsa
Oxidant
k, cm3molec.-1s-1
Reported products
Concentration (molec.-1 cm3)
τ
Ref.
O3 ≤ 1×10-21 None detected 7.5×1011 42 yr 2 ·OH (1.97±0.23)×10-11 None detected 1×106 14.1 h 2 Cl atom (2.75±0.30)×10-10 CH3HgCl 2×103 21 d 1 O(3P) (2.5±0.2)×10-11 HgO - - 3 18F· (4.7±0.5)×10-10 None detected - - 4
a From Sommar et al. (1997). References: 1.Niki et al., (1983a); 2. Niki et al, (1983b); 3. Lund Thomsen and Egsgaard et al., (1986); 4. McKeown et al, (1983). Concentration: O3-(30 ppb), ·OH and Cl atom from Sommar et al., (1996).

74
5.3 Equilibria Tables
Tables 26-28 give the different equilibrium reactions which can occur between Hg and other
constituents in the aqueous phase, across the solid/aqueous phase, and across the gaseous/aqueous
phase.
Table 26 Equilibria for aqueous phase Hg(II) speciationa
No Equilibrium Log(Keq) E1 H2O·SO4 ↔ H+ + HSO3
- -1.91 E2 HSO3
- ↔ H+ + SO32- -7.18
E3 H2C2O4 ↔ H+ + HC2O4- -1.10
E4 HC2O4- ↔ H+ + C2O4
2- -3.85 E5 Hg2+ + OH- ↔ Hg(OH)+ 10.63 E6 Hg2+ + 2 OH- ↔ Hg(OH)2 22.24 E7 Hg2+ + SO3
2- ↔ HgSO3 12.7 E8 Hg2+ + 2 SO3
2- ↔ Hg(SO3)22- 24.1
E9 Hg2+ + OH- + Cl- ↔ HgOHCl 18.25 E10 Hg2+ + Cl- ↔ HgCl+ 7.30 E11 Hg2+ + 2 Cl- ↔ HgCl2 14.0 E12 Hg2+ + 3 Cl- ↔ HgCl3
- 15.0 E13 Hg2+ + 4 Cl- ↔ HgCl4
2- 15.6 E14 Hg2+ + C2O4
2- ↔ HgC2O4 9.66 a Table from Lin and Pehkonen (1999)
Table 27 Solid-liquid equlibria of mercury compoundsa
No. Equilibrium Log (K) SL1 HgO(s) + H2O ↔ Hg2+ + 2OH- -25.44 SL2 HgS(s) + 2 H+ ↔ Hg2+ + H2S- -31.7 SL3 HgCl2(s) ↔ Hg2+ + 2 Cl- -14.57 SL4 Hg(OH)2(s) ↔ Hg2+ + 2 OH- -24.96 SL5 Hg2Cl2(s) ↔ Hg2+ + 2 Cl- -17.91 SL6 Hg2SO4(s) ↔ 2 Hg2+ + SO4
2- -6.13 a Table from Lin and Pehkonen (1999).
Table 28 Gas/aqueous equlibria of Hg and some of its compoundsa
No. Equilibrium H (Matm-1) GL1 Hg0
(g) ↔ Hg0(aq) 0.11
GL2 Hg(0H)2(g) ↔ Hg(0H)2(aq) 1.2 × 104 GL3 HgCl2(g) ↔ HgCl2(aq) 1.4 × 106 GL4 CH3HgCl(g) ↔ CH3HgCl(aq) 2.2 × 103 GL5 CH3HgCH3(g) ↔ CH3HgCH(aq) 0.13
a Table from Lin and Pehkonen (1999).

75
5.4 Summary of half lives and residence times for elemental and divalent mercury
Tables 29, 30 give a summary of half-lives and residence times of elemental and divalent Hg.
Table 29 Summary of chemical reactions in the aqueous phase
Reaction Reaction rate Temperature Concentration of reactant Half-life (t1/2) Oxidation of Hg0 by O3 (4.7±2.2) ×107 M-1s-1 - 4×10-10 M 40 s Oxidation of Hg0 by ·OH (2.4±0.3)×109 M-1s-1 - 10-12 M 6 min Oxidation of Hg0 by chlorine (HOCL/OCL-)
See 5.1.1.3 - - -
Reduction of Hg(ΙΙ) by S(ΙV) ln((0.0106/T)-12595)/T) s-1 0 0C - 3600 s Photoreduction of Hg(ΙΙ) 3×10-7s-1 - - 600 h Reduction of Hg(ΙΙ) by HO2·
1.7 ×104 M-1s-1 - 10-8 M 1 h
Table 30 Summary of chemical reaction in the gaseous phase
Reaction Reaction rate Temperature Concentration of reactant Half-life (t1/2) Residence time (τ) Oxidation of Hg0 by O3 2.1 ×1018 × [-1203/T]
cm3molec. -1s-1 0 0C 0 0C
30 ppb 100 ppb
1.2 yr 0.35 yr
1.7 yr 0.5 yr
Oxidation of Hg0 by ·OH
(8.7±2.8)×10-14cm3s-1 - 106 molecules cm-3 0.25 yr 0.4 yr
Oxidation of Hg0 by NO3·
4 ×10-15 cm3molec.-1s-1 - 108 molecules cm-3 20 d 30 d
Oxidation of Hg0 by H2O2
8.4×10-6 × exp[-9021/T] cm3molec. -1s-1
0 0C 1 ppb 24 yr 34 yr

77
6. CONCLUSIONS
The total anthropogenic Hg emission from Australia is estimated to be 10.2 tonnes/yr
(excluding burning of vegetation). Of this total emission about 9.9 tonnes is emitted to air
(∼4.76 tonnes Hg0, 1.28 tonnes Hg(II), 3.88 tonnes Hgp) and this comprises about 0.5 percent
of the estimated global anthropogenic Hg emissions to the atmosphere. Since the Australian
economy is heavily resource based, a large part of the anthropogenic point source Hg
emissions (>98%) are due to activities in this sector such as; electricity production (mainly
using coal) (28.4%), alumina production (26.7%), steel and metal production (25.5%), and
other industry/waste disposal/mining/ chemical industry/oil and gas production/combustion
(17.4%). Even though the estimated emissions from Australia are a small percentage of global
emissions, the country is a significant per capita emitter with 0.51 g.Hgtot.,/capita compared to
the global average of 0.36 g Hgtot/capita.
Emission inventories of Hg are subjected to large uncertainties. According to the latest
published global emission inventory, Australia is suggested to emit 110.9 tonnes Hg/yr, which
is nearly 11 times more than that estimated by the National Pollution Inventory. It has been
demonstrated that the higher figure of 110.9 tonnes is not credible and arises by the
application of incorrect emission factors (particularly for coal combustion) during the
calculation of Hg emissions.
According to the calculations performed in this study, emissions of Hg from natural sources
in Australia are estimated to be around 130-270 tonnes/yr. However, these emission estimates
are based on relatively crude assumptions and the result should therefore be interpreted with
caution..
In order to investigate the dispersion and deposition of Hg from 10 facilities in the central,
coastal parts of NSW, TAPM a three-dimensional regional scale Eulerian air quality model
was used. The model was set with 25×25 grids in the outer domain with the grid size 30×30
km. In order to obtain a finer resolution for the concentration simulations, an outer and an
inner pollution grid domain was used with 97×97 grids in the horizontal plane, with grid sizes
7.5 × 7.5 km and 2.5 × 2.5 km, respectively. Vertically, the model has 25 non-uniform layers,
with the finest resolution near the surface (10 m). The top of the modeling domain is 8 km.
Atmospheric Hg transformation processes are not included in the simulation neither is wet
deposition. The Hg species considered in this simulation are Hg0 and Hg(II)/Hgp (combined)
The background concentration of Hg0 was set to zero and the deposition velocity for

78
Hg(II)/Hgp was set to be 0.5 cm/s during the day and zero cm/s during the night.
Corresponding values for Hg0 are 0.03 and 0 cm/s, respectively.
The simulation shows that the maximum simulated ground level ambient Hg concentration
(3.1 ng/m3) is (even if the background concentration of Hg0 of 1.3-1.4 ng/m3 is added), well
below (i) the US EPA determined reference concentration of Hg vapor of 0.3 µg/m3 for the
general population, (ii) the limit value for exposure in Europe of 0.05µg/m3, and (iii) the air
quality objectives set in Victoria, Australia, of 9.4 µg/m3, for inorganic Hg.
Simulated total average deposition flux in the inner domain varies between 0.2 and 1.4
µg/m2/yr (at the 10th and 90th percentile level, respectively). In occasional cases, close to
emitting sources (1-2 grid cells away from the source), the deposition flux of Hgtot may reach
levels of 50-60 µg/m2/yr.
To investigate the area average deposition flux of Hg, as well as, the percentage of total Hg
that is deposited at various distances around the facilities, a number of calculations were
performed. The general observed trend is that the area average deposition flux of Hg,
expressed as a percentage of the total Hg emitted, is relatively small (0.1-9.4 %, depending on
the distance to the source). This suggests that a significant part of the Hg emitted from the
facilities investigated is transported away from the domain. If the average simulated
deposition rate in the outer domain of 0.2 µg/m2/yr is integrated over the study area, it was
calculated that around 105 kg of Hg are deposited annually within the entire domain. This
constitutes about 8 % of the total Hg emissions in the simulation.
The various Hg species present in the atmosphere have different atmospheric residence times,
which affect the distance they are transported before being deposited to the surface.
Atmospheric transformation/interaction processes, which determine the speciation of Hg, are
therefore important to include in models which aim at simulating Hg transportation and
deposition. In order to obtain more accurate data in future Hg simulations using TAPM,
transformation/interaction and deposition processes for Hg should be integrated in the model.

79
7. ACKNOWLEDGEMENTS
The authors wish to acknowledge the financial support provided by the Cooperative
Research Centre for Coal in Sustainable Development, which is funded in part by the
Cooperative Research Centres Program of the Commonwealth Government of
Australia. They would also like to thank Josef and Elizabeth Pacyna who generously
made available data beyond that accessible from their published papers.

80
8. REFERENCES
ABARE (Australian Bureau of Agriculture and Resource Economics), 2001. Australian
energy outlook to 2019-20. ABARE Conference paper 2001.27. Prepared by: Dickson, A.,
Thorpe, S., Harman, J., Donaldson, K., Tedesco, L. Australian Institute of Energy, National
conference; "Energy 2001 - Exploring Australia's Energy Future", Sydney.
ABARE, 2003. Energy Statistics-Full Historical Dataset-All States 2002. Australian
Bureau of Agricultural and Resource Economics, Canberra, ACT.
ABS (Australian Bureau of Statistics), 2002a. Geography of Australia - Position and area.
http//:www.abs.gov.au/Ausstats
ABS, 2002b. Mineral resources and geology. http//:www.abs.gov.au/Ausstats
ABS, 2002c. 3101.1 Australian Demographic Statistics. http//:www.abs.gov.au/Ausstats
ABS, 2002d. 4613.0 Australia's Environment: Issues and trends. http//:www.abs.gov.au
/Ausstats
ABS, 2002e. Year Book Australia 2002. http//:www.abs.gov.au/Ausstats
Arakaki, T., Anastasio, C., Shu, P.G., Faust, B.C., 1995. Aqueous-phase photoproduction of
hydrogen peroxide in authentic cloud waters: Wavelength dependence, and the effects of
filtration and freeze-thaw cycles. Atmospheric Environment 29, (14), 1697-1703.
Axenfeld, F., Munthe, J., Pacyna, J.M., 1991. Europaische Test-Emissiondatenbasis von
Quecksilber-komponenten fur Modellrechnungen. Umweltforschungsplan des
Bundesministers fur Umwelt Naturschutz und Reaktoricherheit, Luftreinhaltung: 104 02
726, Friedrichshaften, Germany.
Bergan, T., Gallardo, L., Rodhe, H., 1999. Mercury in the global troposphere: a three-
dimensional model study. Atmospheric Environment 33, 1575-1585.
Bergan, T., Rodhe,H., 2001. Oxidation of elemental mercury in the atmosphere; Constraints
imposed by Global Scale Modelling. Journal of Atmospheric Chemistry 40, 191-212.
Binham, M.K., 1990. Field detection and implications of Mercury in natural gas. SPE
Production Engineering, May 1990, 120-124.www.gaschem.com/mercury
Brosset, C., Lord, E., 1991. Mercury in precipitation and ambient air - a new scenario. Water,
Air and Soil Pollution 56, 493-506.
Bullock, O.R. Jr., 2000a. Modeling assessment of transport and deposition patterns of
anthropogenic mercury air emissions in the United States and Canada. The Science of the
Total Environment 259, 145-157.
Bullock, O.R. Jr., 2000b. Current methods and research strategies for modeling atmospheric
mercury. Fuel Processing Technology 65-66, 459-471.

81
Bullock, O.R. Jr., Brehme, K.A., 2002. Atmospheric mercury simulation using the CMAQ
model: formulation description and analysis of wet deposition results. Atmospheric
Environment 36, 3987-3997.
Buxton, G.V., Greenstock, C.L., Helman, W.P., Ross, A.B., 1988. Critical review of rate
constants of reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals in
aqueous solutions. Journal of Physical Chemistry Reference Data 17, 513-780.
Capri, A., Linberg, K.A., 1998. Application of a Teflon dynamic flux chamber for quantifying
soil mercury flux: tests and results over background soil. Atmospheric Environment 32 (5),
873-882.
Carr, G.R., Wilmshurst, J.R., Ryall, W.R., 1986. Evaluation of mercury pathfinder
techniques: Base metal and uranium deposits. Journal of Geochemical Exploration 26 (1), 1
– 117.
Carr, G.R., Wilmshurst, J.R., 2000. Chapter 12, Mercury. In: Hale, M. (Ed.), Geochemical
Remote Sensing of the Subsurface. Handbook of Exploration Geochemistry, vol 7, 395-437.
CRC, 1998. CRC Handbook of Chemistry and Physics. Lide, D.R. (Ed). 78th edition, 1997-
1998. Cleveland, Ohio: Chemical Rubber Pub. Co.
Cartwright, J Timothy, 1993. Modeling the World in a Spreedsheet; Environmental
Simulation on a Microcumputer. John Hopkins University Press.
Cobos, D.R., Baker, J.M., Nater, E.A., 2002. Conditional sampling of mercury vapor fluxes.
Atmospheric Environment 36, 4309-4321.
Cossa, D., Coquery, M., Gobeil, C., Martin, J-M., 1996. Mercury fluxes at the ocean margins.
In:. Watras, C.J and Huckabee, J.W. (Eds.)Mercury is a global pollutant: Towards
Integration and Synthesis. Lewis Publisher, 229-247.
Dale, L., 1999. Investigation Report CET/IR215 Mechanism for trace element partitioning in
Australian coals, Co-operative Research Centre for Black Coal Utilisation.
Daniel, M., 1994. Chinese coal prospects to 2010. IEA Coal Research. IEAPER/11.
http//;www.caer.uky.edu/iea/ieaper18.htm.
EC (European Communities), 2001. Ambient Air Pollution by Mercury (Hg). Position Paper.
Prepared by the Working Group On Mercury. Luxembourg: Office for Official Publication
of the European Communities. http//:www.europa.eu.int.
EDMC (Energy Data Modelling Centre), 2001. Australia - Energy Overview.
http//:www.ieej.or.jp/apec/database/
Engle,M.A, Gaustin, M.S., Zhang, H., 2001. Quantifying natural source mercury emissions
from the Ivanhoe Mining District, North-Central Nevada, USA. Atmospheric Environment
35, 2135-2146.
Expert Panel on Mercury Atmospheric Processes (EPMAP), 1994. Mercury Atmospheric
Processes: A Synthesis Report. Electric Power Research Institute. Report No. TR-104214.

82
Faust, B.C., Allen, J.M., 1993. Aqueous-phase photochemical formation of hydroxyl radical
in authentic cloudwaters and fogwaters. Environmental Science and Technology 27, 1221-
1224.
Faust, B.C, Hoigne, J., 1990. Photolysis of Fe(III)-hydroxide complexes as sources of OH
radicals in clouds, fog, and rain. Atmospheric Environment 24A, 79-89.
Faust, B. C., Anastasio, C., Allen, J. M. and Arakaki, T, 1993. Aqueous-phase photochemical
formation of peroxides in authentic cloud and fog waters, Science 260,3-75.
Ferrara, R., Maserti, B.E., Andersson, M., Edner, H., Ragnarsson, P., Svanberg, S.,
Hernandez, A., 1998a. Atmospheric mercury concentrations and fluxes in the Almden
District (Spain). Atmospheric Environment 32, 3897-3904.
Ferrara, R., Mazzolai, B., Edner, H., Svanberg, S., Wallinder, E., 1998b. Atmospheric
mercury sources in the Mt. Amiata area, Italy. The Science of the Total Environment 213,
13-23.
Ferrara, R., Mazzolai, B., Lanzillotto, e., Nucaro, E., Pirrone, N., 2000. Temporal trends in
gaseous mecury evasion from the Mediterranean seawaters. The Science of the Total
Environment 259, 183-190.
Finlayson-Pitts, B.J., Pitts, Jr., J.N., 1986. Atmospheric Chemistry: Fundamentals and
Experimental Techniques. New york: Wiley.
Finlayson-Pitts, B.J., Pitts, Jr., J.N., 2000. Chemistry of the Upper and Lower Atmosphere.
New York: Academic Press.
Fitzgerald, W.F, Mason, R.P, Vandal, G.M, 1991. Atmospheric cycle and air-water exchange
of mercury over midcontinental lacustrine regions. Water, Air, and Soil Pollution 56, 745-
767.
Fitzgerald, W.F., Engstrom, D.R., Mason, R.P., Nater, E.A., 1998. The case for atmospheric
mercury contamination in remote area. Environmental Science and Technology 32 (1)1-7.
Fthenakis, V.M., Lipfert, F.W., Moskowitz, P.D., Saroff, L., 1995. An assessment of mercury
emissions and health risks from a coal-fired power plant. Journal of Hazardous Materials
44, 267-283.
Galbreath, K.C., Zygarlicke, J., 1996. Mercury Speciation in Coal Combustion and
Gasification Flue gases. Environmental Science and Technology 30, No. 8, 2421-2426.
Gillis, A.A., Miller, D.R., 2000a. Some local environmental effects on mercury emission and
adsorption at a soil surface. The Science of the Total Environment 260, 191-200.
Gillis, A.A., Miller, D.R., 2000b. Some potential errors in the measurement of mercury gas
exchange at the soil surface using a dynamic flux chamber. The Science of the Total
Environment 260, 181-189.

83
Goldfrank, L., Bresnitz, E., Howland, M., Weisman, R., 1990. Mercury. In Goldfrank, L.,
Flomenbaum, N., Lewis, N., (Eds.). Goldfrank's Toxicologic Emergencies. Norwalk, CT:
Appelton & Lange; 641-648.
Guentzel, J.L., Landing, W.M., Gill, G.A., Pollman, C.D., 2001. Processes influencing
rainfall deposition of mercury in Florida. Environmental Science and Technology 35, 863-
873.
Gustin, M.S., Lindberg, S.E., Austin, K., Coolbaugh, M., Vette, A., Zhang, H., 2000.
Assessing the contribution of natural sources to regional atmospheric mercury budgets. The
Science of the Total Environment 259, 61-71.
Gustin, M.S., Taylor, G.E., Leonard, T.L, 1996. Atmospheric mercury concentrations
associated with geologically and anthropogenically enriched sites in Central Western
Nevada. Environmental Science and Technology 30, No. 8, 2572-2579.
Gårdfeldt, K., Sommar, J., Strombeig, D., Feng, X. 2001. Oxidation of atomic mercury by
hydroxyl radicals and photoinduced decomposition of methylmercury specie in the aqueous
phase. Atmospheric Environment 35, 3039-3047.
Hall, B., 1995. The gas phase oxidation of elemental mercury by ozone. Water, Air and Soil
Pollution 80, 301-315.
Hanson, P.J., Lindberg, S.E., Tabberer, T.A., Owens, J.G., Kim, K-H., 1995. Foliar exchange
of mercury vapour: evidence for a compensation point. Water, Air and Soil Pollution 80,
373-382.
Hedgecock, I., Pirrone, N., 2001. Mercury and photochemistry in the marine boundary layer -
modelling studies suggest the in situ production of reactive gas phase mercury. Atmospheric
Environment 35, 3055-3062.
Hennico, B., Barthel, Y., Cosyns, J., Courty, P., 1991. Mercury and arsenic removal in natural
gas, refining and petrochemical industries. Oil and Gas European Magazine 17, 36-38.
Hudson, R.J.M., Gherini, S.A., Fitzgerald, W.F., Porcella, D.B., 1995. Anthropogenic
influences on the global mercury cycle: A model-based analysis. Water, Air and Soil
Pollution 80, 265-272.
Hurley, P.J., 2002a. The air Pollution model (TAPM) Version 2. Part 1: Technical
description. CSIRO Atmospheric Research Technical Paper No. 55.
Hurley, P.J., 2002b. The air Pollution model (TAPM) Version 2. User manual. CSIRO
Atmospheric Research Technical Paper No. 25.
Hurley, P.J., Physick, W.L., Luhar, A.K., 2002. The air Pollution model (TAPM) Version 2.
Part 2: Summary of some verification studies. CSIRO Atmospheric Research Technical
Paper No. 57.

84
Ilyin I., Ryaboshapko, A., Afinogenova, O., Berg, T., Hjellebrekke, A-G., 2001. Evaluation of
transboundary transport of heavy metals in 1999. Trend analysis. EMEP/MSC-E Technical
Report 3/2001, Meteorolgical Synthesizing Centre - East, Moscow, Russia.
Impey, G.A., Shepson, P. B., Hastie, D. R., Barrie L. A., Anlauf, K. G., 1995. Measurement
of photolyzable chlorine and bromine during the polar sunrise experiment, Journal of
Geophysical Research 102, 16005-16010.
Jacob, D. J., 1986. Chemistry of OH in remote clouds and its role in the production offormic
acid and peroxymonosulfate. Journal of Geophysical Research 91; 9807-9826.
Jackson, T.A., 1997. Long-range atmospheric transport of mercury to ecosystems, and the
importance of anthropogenic emission- a critical review and evaluation of the published
evidence. Environmental review 5, 90-120.
Keeler, G., Glinsorn, G., Pirrone, N., 1995. Particulate mercury in the atmosphere: its
significance, transport, transformation and sources. Water, Air and Soil Pollution 80, 156-
168.
Keene, W.C., Maben, A.A, Pszenny, A.A.P., Galloway, J.N., 1993. Measurement technique
for inorganic chlorine gas in the marine boundary layer. Environmental Science and
Technology 27, 866-874.
Keene, W.C., Jacob, D.J., Fan, S.-M., 1996. Reactive chlorine: a potential sink for
dimethylsulfide and hydrocarbons in the marine boundary layer. Atmospheric Environment
30, No 6, i-iii.
Kim, K-H., Kim, M-Y., Kim, J., Lee, G., 2002. The concetrations and fluxes of total gaseous
nmercury in a western coastal area of Korea during late March 2001. Atmospheric
Environment 36, 3413-3427.
Landers, D.H., Gubala, C., Verta, M., Lucotte, M., Johansson, K., Vlasova, T., Lockhart,
W.L., 1998. Using lake sediment mercury flux ratios to evaluate the regional and
continental dimensions of mercury deposition in Artic and boreal ecosystems. Atmospheric
Environment 35, (5), 919-928.
Landis, M.S., Keeler, G.J., 2002a. Atmospheric mercury deposition to Lake Michigan during
the Lake Michigan Mass Balance Study. Environmental Science and Technology 36, 4518-
4524.
Landis, M.S., Vette, A.F., Keeler, G.J., 2002b. Atmospheric mercury in the Lake Michigan
Basin: Influence of the Chicago/Gary urban area. Environmental Science and Technology
36, 4508-4517.
Lee, D.S., Nemitz, E., Fowler, D., Kingdon, R.D., 2001. Modeling atmospheric mercury
transport and deposition across Europe and the UK. Atmospheric Environment 35, 5455-
5461.

85
Lelieveld, J., Roelofs, G.-J., Ganzeveld, L., Feichter, J. and Rodhe, H.,1997. Terrestrial
ources and distribution of atmospheric sulfar. Phil. Trans. R. Soc. Lond. B 352, 149-158.
Levin, L., (EPRI), 2001. Gambling in Latin: Uncertainties in Managing Environmental
Mercury. The A&WMA Specialty Conference on Mercury Emission: fate, effects and
control. Chicago, Illinois, USA.
Lin, C.-J., Pehkonen, S.O., 1997. Aqueous free radical chemistry of mercury in the presence
of iron oxides and ambient aerosol. Atmospheric Environment 31 (24), 4125-4137.
Lin, C.-J., Pehkonen, S.O., 1998a. Two-phase model of mercury chemistry in the atmosphere.
Atmospheric environment 32 (14/15), 2543-2558.
Lin, C.-J., Pehkonen, S.O., 1998b. Oxidation of elemental mercury by aqueous chlorine
(HOC1/OCL-): implications for troposphere mercury chemistry. Journal of Geophysical
Research 103 (D21), 28093-28102.
Lin, C.-J., Pehkonen, S.O., 1999a. The chemistry of atmospheric mercury: a review.
Atmospheric Environment 33,2067-2079.
Lin, C.-J., Pehkonen, S.O., 1999b. Aqueous phase reactions of mercury with free radicals and
chlorine: Implications for atmospheric mercury chemistry. Chemosphere 38, No 6, 1253-
1263.
Lindberg, S.E., Hanson, P.J., Meyers, T.P., Kim, K-H., 1998. Air/surface exchange of
mercury vapor over forests - The need for a reassessment of continental biogenic emissions.
Atmospheric Environment 35, (5), 895-908.
Lindberg, S.E., Stratton, W.J., 1998. Atmospheric mercury speciation: concentration and
behavior of reactive gaseous mercury in ambient air. Environmental Science and
Technology 32, 49-57.
Lindquist, O., Johansson, K., Aastrup, M., Andersson, A., Bringmark, L., Hovsenius, G,
Håkansson, L, Iverfeldt, Å., Meili, M., Timm, B., 1991. Water, Air and Soil Pollution 55, 1-
261.
Lund Thomsen, E., Egsgaard, H., 1986. Rate reaction od dimethylmercury with oxygen atoms
in the gas phase. Chem. Phys. Lett. 125, 378-382.
Mason, R.P., Fitzgerald, W.F., and Morel, F.M.M., 1994, The biogeochemical cycling of
elemental mercury: Anthropogenic influences: Geochemica Cosmochimica Acta, v. 58,
3191-3198.
McElroy, W.J., Munthe, J., 1991. The oxidation of mercury (I) by ozone in acidic aqueous
solution. Acta Chemica Scandinavica 45, 254-257.
McKeown, F.P., Subramonia, I.R., Rowland, F.S., 1983. Methyl fluoride formation from
thermal fluorine-18 reaction with dimethylmercury. Journal of Physical Chemistry 87,
3972-3975.
Mitra, S., 1986. Mercury in the Ecosysytem. Switzerland: Trans Tech Publications Ltd.

86
Munthe, J., 1994. The atmospheric chemistry of mercury: kinetic studies of redox reactions.
In:Watras, C.J. , Huckabee, J.W., (Eds.), Mercury as a Global Pollutant - Integration and
Synthesis, Lewis Publisher, 273-279.
Munthe, J., 1992. Aqueous oxidation of elemental Hg by O3. Atmospheric Environment 26A,
1461-1468.
Munthe, J., McElroy, W.J., 1992. Some aqueous reactions of potential importance in the
atmospheric chemistry of mercury. Atmospheric Environment 26A, 553-557.
Munthe. J., Wängberg, I., Iverfeldt, Å., Petersen, G., Ebinghaus, R., Schmolke, S., Bahlmann,
E., Lindquist, O., Strömberg, D., Sommar, J., Gårdfeldt, K., Feng, X., Larjava, K., Siemens,
V., 2001. Mercury species over Europe (MOE). Relative importance of depositional
methylmercury fluxes to various ecosystems. Final report to the European Commission,
Directorate General XII.
Munthe, J., Xiao, Z.F., Lindqvist, O., 1991. The aqueous reduction of divalent mercury by
sulfite. Water, Air and Soil Pollution 56, 621-630.
Möller, D., Mauersberger, G., 1995. An aqueous phase chemical reaction mechanism. In:
Cloudsmodels and mechanism, EUROTRAC Int. Sci. Secr. Gramisch-Partenkirchen, 77-93.
Nelson, P., 2002. Personal communication. Macquarie University. Graduate School of the
Environment. Sydney, Australia. Email: [email protected]
NFI (National Forest Inventory), 2001. Department of Agriculture Fisheries & Forestry -
Australia. http//:www.affa.gov.au/index
Niki, H., Maker, P.S., Savage, C.M., Breitenbach, L.P., 1983a. An FTIR study of the kinetics
and mechanism for the reaction Cl + CH3HgCH3. Journal of Physical Chemistry 87, 722-
3724.
Niki, H., Maker, P.S., Savage, C.M., Breitenbach, L.P., 1983b. A long-path FTIR study of the
kinetics and mechanism for the reaction · OH initiated oxidation of dimethylmercury.
Journal of Physical Chemistry 87, 4978-4981.
NPI, 2003a. National pollution inventory. http://www.npi.gov.au/. Site last accessed
27th August 2003.
NPI, 2003b. Emission estimation technique manual for combustion in boilers
(Version 1.2). Pub. Environment Australia, Canberra, ACT.
NPI, 1999a. Emissions Estimation Technique Manual for Aggregated Emissions from
Prescribed Burning and Wildfires. Pub. Environment Australia, Canberra, ACT.
NPI, 1999a. Emission estimation technique manual for fossil fuel electric power
generation. Pub. Environment Australia, Canberra, ACT.

87
NPI, 1999b. Emissions estimation technique manual for aggregated emissions from
paved and unpaved roads, Pub. Environment Australia, Canberra, ACT.
Oum, K.W., Lakin, M.J., Dehaan, D.O., Brauers, T., Finlayson- Pitts, B.J., 1998. Formation
of Cl2 from the photolysis of O3 and aqueous sea-salt particles. Science 279, 74-77.
Pacyna, E.G., Pacyna, J.M., Pirrone, N., 2001. European emissions of atmospheric mercury
from anthropogenic sources in 1995. Atmospheric Environment 35, 2987-2996.
Pacyna, E.G., Pacyna, J.M., 2002. Global emission of mercury from anthropogenic sources in
1995. Water, Air and Soil Pollution 137, 149-165.
Pacyna, J.M. (Personal communication). Norwegian Institute for Air Research. P.O. Box 100,
2027 Kjeller, Norway. Email: [email protected]
Pai, P., Karamchandani, P., Seigneur, C., 1997. Simulation of the regional atmospheric
transport and fate of mercury using comprehensive Eulerian model. Atmospheric
Environment 31, 2717-2732.
Pai, P., Karamchandani, P., Seigneur, C., 2000a. On artificial dilution of point source mercury
emissions in a regional atmospheric model. The Science of the Total Environment 259, 159-
168.
Pai, P., Niemi, D. and Powers, W., 2000b. A North American inventory of anthropogenic
mercury emissions. Fuel Processing Technology 65-66:101-115.
Pehkonen, S.O., Lin, C.-J., 1998. Aqueous photochemistry of divalent mercury with organic
acids. Journal of AWMA 48, 144-150.
Petersen, G., Iverfeldt, A., Munthe, J., 1995. Atmospheric mercury species over central and
northem Europe. Model calculations and comparison with observations from the Nordic Air
and Precipitation Network for 1987 and 1988. Atmospheric Environment 29 (1), 47-67.
Petersen, G., Munthe, J., Pleijel, K., Bloxam, R., Vinod Kumar, A., 1998. A comprehensive
Eulerian modeling framework for airborne mercury species: Development and testing of the
tropospheric chemistry module (TCM). Atmospheric Environment, 29, 829-843.
Petersen, G., Bloxam, R., Wong, S., Munthe, J., Krüger, O., Schmolke, S.R., Vinod Kumar,
A., 2001. A comprehensive eulerian modeling framework for airborne mercury species:
model development and application in Europe. Atmospheric Environment 35, 3063-3074.
Pilgrim, W., Poissant, L., Trip, L., 2000. The Northeast States and Eastern Canadian
Provinces mercury study: a framework for action: summary of the Canadian chapter. The
Science of the Total Environment 261, 177-184.
Pirrone, N., 2001a. Mercury Research in Europe: Towards the preparation of the New Air
Quality Directive. Atmospheric Environment 35, 2979-2986.

88
Pirrone, N., Costa, P., Pacyna, J.M., Ferrara, R., 2001b. Mercury emissions to the atmosphere
from natural and anthropogenic sources in the Mediterranean region. Atmospheric
Environment 35, 2997-3006.
Pirrone, N., Hedgecock, I.M., Forlano, L., 2000. Role of the Ambient Aerosol in the
Atmospheric Processing of semi-volatile contaminants: A parameterized numerical model
(GASPAR). Journal of Geophysical Research 105 (D8), 9773-9790.
Pirrone, N., Keeler, G.J., Nriagu, J., 1996. Regional differences in worldwide emissions of
mercury to the atmosphere. Atmospheric Environment 30, 2981-2987.
Pirrone, N., Pacyna, J.M., Mamane, Y., Munthe, J., Ferrara, R., 2000. Mediterranean
Atmospheric Mercury Cycle System (MAMCS). Technical final Report, European
Commission (Contract No. ENV4-CT97-0593).
Pleijel, K., Munthe, J., 1995a. Modeling the atmospheric mercury cycle-chemistry in fog
droplets. Atmospheric Environment 29 (12), 1441-1457.
P1eijel, K., Munthe, J., 1995b. Modeling the atmospheric Hg chemistry - the importance of a
detailed description of the chemistry of cloud water. Water, Air and Soil Pollution 80, 317-
324.
Poissant, L., Casimir, A., 1998. Water-air and soil-air exchange rate of total gaseous mercury
measured at background sites. Atmospheric Environment 32, 883-893.
Porcella, D.B., Chu, P., Alla, M.A., 1996. Inventory of North America Hg Emissions to the
Atmosphere; Relationship to the Global Hg Cycle. In; Global and Regional Mercury
Cycles: Sources, Fluxes, and Mass Balances, 179-190.
Rasmussen, P.E., 1994. Current methods of estimating atmospheric mercury fluxes in remote
areas. Environmental Science and Technology 28, 2233-2241.
Rea, A.W., Linberg, S.E., Keller, G.J, 2000. Assessment of dry deposition and foliar leaching
of mercury and selected trace elements based on washed foliar and surrogate surfaces.
Environmental Science and Technology 34, 2418-2425.
Ryaboshapko, A., Ilyin, I., Bullock, R., Ebinghaus, R., Lohman, K., Munthe, J., Petersen, G.,
Segneur, C., Wängberg, I., 2001. Intercomparison study of numerical models for long-range
atmospheric transport of mercury. Stage 1: Comparison of chemical modules for mercury
transformations in a cloud/fog environment. EMEP/MSC-E Technical Report 2/2001,
Meteorolgical Synthesizing Centre - East, Moscow, Russia.
Schroeder, W.H., Munthe, J., Lindquist, O., 1989. Cycling of mercury between water, air and
soil components of the environment. Water, Air and Soil Pollution 80, 317-324.
Schroeder, W.H., Munthe, J., 1998. Atmospheric mercury - an overview. Atomospheric
Environment 32, 809-822.

89
Schroeder, W.H., Yarwood, G., Niki, H., 1991. Transformation processes involving Hg
species in atmosphere – results from a literature survey. Water, Air and Soil Pollution 56,
653-666.
Schwartz, S.E., 1984. Gas- and aqueous-phase chemistry of HO2 in liquid water cloud.
Journal of Geophysical Research 89; 11589-11598.
Seigneur, C., Abeck, H., Chia, G., Reinhard, M., Bloom, N.S., Prestbo, E., Saxena, P., 1998.
Mercury adsorption to elemental carbon (soot) particles and atmospheric particulate matter.
Atmospheric Environment 32 (14/15), 2649-2657.
Seigneur, C., Karamchandani, P., Lohman, K., Vijayaraghavan, K., Shia, R-L., 2001.
Multiscale modeling of the atmospheric fate and transport of mercury. Journal of
Geophysical Research 106 (D21), 27795-27809.
Seigneur, C., Wrobel, J., Constantinou, E., 1994. A chemical kinetic mechanism for
atmospheric inorganic mercury. Environmental Science and Technology 28, 1589-1597.
Seinfeld, J.H., Pandis, S.N., 1997. Atmospheric Chemistry and Physics. New York: Wiley-
Interscience.
Seinfeld, J.H., Pandis, S.N., 1998. Atmospheric chemistry and physics: From air pollution to
climate change. New York: John Wiley & Sons, Inc.
Slemr, F., Schuster, G., Seiler, W., 1985. Distribution, speciation and budget of atmospheric
mercury. Journal of Atmospheric Chemistry 3, 407-434.
Sommar, J., Gårdfeldt, K., Strömberg, D., Feng, X. 2001. A kinetic study of the as-phase
reaction between the hydroxyl radical and atomic mercury. Atmospheric Environment 35,
3049-3054.
Sommar, J., Hallquist, M., Ljungström, E., 1996. Rate of reaction between the nitrate radical
and dimethylmercury in the gas phase. Chemical Physical Letters 257, 434-438.
Sommar, J., Hallquist, M., Ljungström, E., Lindqvist, O., 1997. On the gaseous reaction
between volatile mercury species and the nitrate radical. Journal of Atmospheric Chemistry
27,233-247.
Tokos, J.J., Hall, B., Calhoun, J.A., Prestbo, E.M., 1998. Homogeneous gas-phase reaction of
Hg° with H2O2, O3, CH3I, and (CH3)2S: implications for atmospheric Hg cycle. Atmospheric
Environment 32, 823-827.
Travnikov, O., Ryaboshapko, A., 2002. Modelling of mercury hemispheric transport and
depositions. EMEP/MSC-E Technical Report 6/2002, Meteorological Synthesising Centre-
East, Moscow, Russia.
UNEP, 2002. Global Mercury Assessment - revised draft of 4 October 2002, incorporating
comments from WG meeting, 9-13 September 2002.
http//:www.chem.unep.ch/mercury/default.

90
US CDC, 2001. CDC National Health and Nutrition Examination Survey (NHANES) IV.
http//:www.cdc.gov/nchs/nhanes.
US EPA, 1997. Mercury Study Report to the Congress. Fate and the transport of mercury in
the environment, Vol, III. EPA-452/R-97-005, US Environmental Protection Agency, US
Government Printing Office, Washington, DC.
US NAS (National Academy of Science), 2000. Toxicological effects of Mewthylmercury.
Washington, DC: National Academy Press.
Van Loon, L., Mader, E., Scott, S.L., 2000. Reduction of the Aqueous Mercuric Ion by
Sulfite: UV Spectrum of HgS03 and Its Intramolecular Redox Reaction. Journal of Physical
Chemistry A, 104, 1621-1626.
Van Loon, L., Mader, E., Scott, S.L., 2001. Sulfite Stabilization and Reduction of the
Aqueous Mercuric lon: Kinetic Determination of Sequential Formation Constants. Journal
of Physical Chemistry A, 105, 3190-3195.
Vette, A.F., Landis, M.S., Keeler, G.J., 2002. Deposition and emission of gaseous mercury to
and from Lake Michigan during the Lake Michigan Mass Balance Study (July, 1994-
October, 1995). Environmental Science and Technology 36, 4525-4532.
VIC EPA (Victoria Environmental Protection Agency), 2003. Indicators for air quality
management and criteria for assessment. http://www.epa.vic.gov.au
Volland, C.S., 1991. Mercury Emissions from Municipal Solid Waste Combustion.
Presentation at the 84th Annual Air and Waste Management Association Meeting and
Exhibition, Vancouver, BC. http//:www.sfei.org/rmp/reports/mercury/mercury.
Wallschlager, D.L., Turner, R.R., London, J., Ebinghaus, R., Kock, H.H., Sommar, J., Xiao,
Z., 1999. Factors affecting the measurement of mercury emissions from soil with flux
chambers. Journal of Geophysical Research 104 (D17), 21859-21871.
WHO, 1990. Methyl Mercury. Environmental Health Criteria 101, World Health
Organisation, Geneva.
WHO, 1991. Inorganic Mercury. Environmental Health Criteria 118, World Health
Organisation, Geneva.
WHO (World Health Organization) Regional office for Europe, Copenhagen, 2003. Air
quality guidelines for Europe. WHO Regional Publications, European Series, No. 91.
Second edition. http://www.euro.who.int/document/e71922.pdf
Wilhelm, S.M., 2001. Estimate of mercury emissions of the atmosphere from petroleum.
Environmental Science and Technology 35, 4704-4710.
Wängberg, I., Munthe, J., Pirrone, N., Iverfeldt, Å., Bahlman, E., Costa, P., Ebinghaus, R.,
Feng, X., Ferrara, R., Gårdfeldt, K., Kock, H., Lanzillotta, E., Mamane, Y., Mas, F.,
Melamed, E., Osnat, Y. , Prestbo E., Sommar, J., Spain, G., Sprovieri, F., Tuncel, G., 2001.

91
Atmospheric Mercury Measurements in Europe during MAMCS and MOE. Atmospheric
Environment 35, 3019-3025.
Xiao, Z.F., Munthe, J., Stromberg, D., Lindqvist, O., 1994. Photochemical behavior of
inorganic Hg compounds in aqueous solution. In: Watras C.J., Huckabee, J.W. (Eds.),
Mercury as a Global Pollutant - Integration and Synthesis., Lewis Publishers, pp. 581-592.
Xiao, Z., Munthe, J., Schroeder, W.H.Lindquist, O., 1991. Vertical fluxes of mercury over
forest soil and lake surfaces in Sweden. Tellus, 43B, 267-279.
Xu, X., Yang, X., Miller, D.R, Helble, J.J, Carley, R.J, 1999. Formulation of bi-directional
air-surface exchange of elemental mercury. Atmospheric Environment 33 (27), 4345.
Xu, X., Yang, X., Miller, D.R., Helble, J. J, Carley, R.J., 2000a. A regional scale modeling
study of atmospheric transport and transformation of mercury. I. Model development and
evaluation. Atmospheric Environment 34, 4933-4944.
Xu, X., Yang, X., Miller, D.R., Helble, J. J, Carley, R.J., 2000b. A regional scale modeling
study of atmospheric transport and transformation of mercury. II. Simulation results.
Atmospheric Environment 34, 4945-4955.
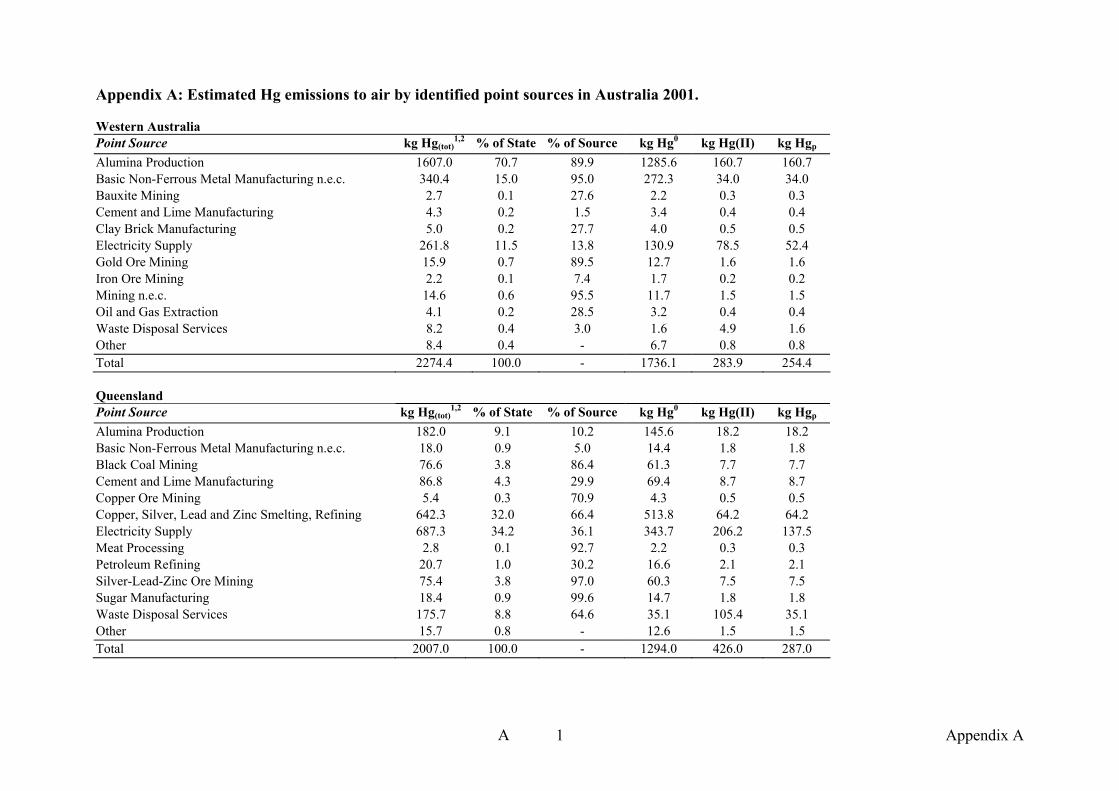
Appendix A 1A
Appendix A: Estimated Hg emissions to air by identified point sources in Australia 2001. Western Australia Point Source kg Hg(tot)
1,2 % of State % of Source kg Hg0 kg Hg(II) kg Hgp Alumina Production 1607.0 70.7 89.9 1285.6 160.7 160.7 Basic Non-Ferrous Metal Manufacturing n.e.c. 340.4 15.0 95.0 272.3 34.0 34.0 Bauxite Mining 2.7 0.1 27.6 2.2 0.3 0.3 Cement and Lime Manufacturing 4.3 0.2 1.5 3.4 0.4 0.4 Clay Brick Manufacturing 5.0 0.2 27.7 4.0 0.5 0.5 Electricity Supply 261.8 11.5 13.8 130.9 78.5 52.4 Gold Ore Mining 15.9 0.7 89.5 12.7 1.6 1.6 Iron Ore Mining 2.2 0.1 7.4 1.7 0.2 0.2 Mining n.e.c. 14.6 0.6 95.5 11.7 1.5 1.5 Oil and Gas Extraction 4.1 0.2 28.5 3.2 0.4 0.4 Waste Disposal Services 8.2 0.4 3.0 1.6 4.9 1.6 Other 8.4 0.4 - 6.7 0.8 0.8 Total 2274.4 100.0 - 1736.1 283.9 254.4 Queensland Point Source kg Hg(tot)
1,2 % of State % of Source kg Hg0 kg Hg(II) kg Hgp Alumina Production 182.0 9.1 10.2 145.6 18.2 18.2 Basic Non-Ferrous Metal Manufacturing n.e.c. 18.0 0.9 5.0 14.4 1.8 1.8 Black Coal Mining 76.6 3.8 86.4 61.3 7.7 7.7 Cement and Lime Manufacturing 86.8 4.3 29.9 69.4 8.7 8.7 Copper Ore Mining 5.4 0.3 70.9 4.3 0.5 0.5 Copper, Silver, Lead and Zinc Smelting, Refining 642.3 32.0 66.4 513.8 64.2 64.2 Electricity Supply 687.3 34.2 36.1 343.7 206.2 137.5 Meat Processing 2.8 0.1 92.7 2.2 0.3 0.3 Petroleum Refining 20.7 1.0 30.2 16.6 2.1 2.1 Silver-Lead-Zinc Ore Mining 75.4 3.8 97.0 60.3 7.5 7.5 Sugar Manufacturing 18.4 0.9 99.6 14.7 1.8 1.8 Waste Disposal Services 175.7 8.8 64.6 35.1 105.4 35.1 Other 15.7 0.8 - 12.6 1.5 1.5 Total 2007.0 100.0 - 1294.0 426.0 287.0

Appendix A 2A
New South Wales Point Source kg Hg(tot)
1,2 % of State % of Source kg Hg0 kg Hg(II) kg Hgp Basic Iron and Steel Manufacturing 385.4 27.2 99.8 308.3 38.5 38.5 Black Coal Mining 11.9 0.8 13.4 9.5 1.2 1.2 Cement and Lime Manufacturing 68.5 4.8 23.6 54.8 6.8 6.8 Chemical Product Manufacturing n.e.c. 125.0 8.8 100.0 100.0 12.5 12.5 Clay Brick Manufacturing 3.6 0.3 20.1 2.8 0.4 0.4 Copper, Silver, Lead and Zinc Smelting, Refining 110.8 7.8 11.5 88.6 11.1 11.1 Electricity Supply 679.7 48.0 35.7 339.9 203.9 135.9 Glass and Glass Product Manufacturing 4.5 0.3 49.5 3.6 0.4 0.4 Organic Industrial Chemical Manufacturing n.e.c. 4.0 0.3 93.0 3.2 0.4 0.4 Paper Stationery Manufacturing 7.0 0.5 100.0 5.6 0.7 0.7 Petroleum Refining 6.0 0.4 8.7 4.8 0.6 0.6 Other 8.9 0.5 - 7.1 0.9 0.9 Total 1415.1 100.0 - 928.2 277.5 209.5 Victoria Point Source kg Hg(tot)
1,2 % of State % of Source kg Hg0 kg Hg(II) kg Hgp Aluminium Smelting 4.8 1.2 90.6 3.8 0.5 0.5 Cement and Lime Manufacturing 5.1 1.2 1.7 4.0 0.5 0.5 Clay Brick Manufacturing 3.5 0.9 19.5 2.8 0.3 0.3 Electricity Supply 266.7 65.5 14.0 133.3 80.0 53.3 Glass and Glass Product Manufacturing 2.3 0.6 24.9 1.8 0.2 0.2 Inorganic Industrial Chemical Manufacturing n.e.c. 70.1 17.2 99.6 49.1 21.0 0.0 Oil and Gas Extraction 2.4 0.6 16.9 1.9 0.2 0.2 Petroleum Refining 39.0 9.6 56.9 31.2 3.9 3.9 Other 13.5 2.9 - 10.9 1.4 1.4 Total 407.4 100.0 - 238.9 108.1 60.4

Appendix A 3A
Tasmania
Point Source kg Hg(tot)1,2 % of State % of Source kg Hg0 kg Hg(II) kg Hgp
Cement and Lime Manufacturing 121.8 40.5 41.9 97.4 12.2 12.2 Copper, Silver, Lead and Zinc Smelting, Refining 92.3 30.7 9.5 73.8 9.2 9.2 Dairy Product Manufacturing n.e.c. 65.0 21.6 98.5 52.0 6.5 6.5 Pulp, Paper and Paperboard Manufacturing 16.9 5.6 61.8 13.5 1.7 1.7 Other 4.9 1.6 - 3.9 0.5 0.5 Total 300.9 100.0 - 240.7 30.1 30.1 South Australia Point Source kg Hg(tot)
1,2 % of State % of Source kg Hg0 kg Hg(II) kg Hgp Cement and Lime Manufacturing 4.0 2.3 1.4 3.2 0.4 0.4 Clay Brick Manufacturing 4.6 2.6 25.7 3.7 0.5 0.5 Copper, Silver, Lead and Zinc Smelting, Refining 120.0 68.4 12.4 96.0 12.0 12.0 Electricity Supply 5.6 3.2 0.3 2.8 1.7 1.1 Iron Ore Mining 27.0 15.4 92.5 21.6 2.7 2.7 Oil and Gas Extraction 4.1 2.3 28.9 3.2 0.4 0.4 Pulp, Paper and Paperboard Manufacturing 7.2 4.1 26.3 5.8 0.7 0.7 Other 3.0 0.7 - 2.4 0.3 0.3 Total 175.5 100.0 - 138.7 18.7 18.1 Australian Capital Territory Point Source kg Hg(tot)
1,2 % of State % of Source kg Hg0 kg Hg(II) kg Hgp Waste Disposal Services 87.46 100 32.2 17.5 52.5 17.5 Total 87.46 100 - 17.5 52.5 17.5

Appendix A 4A
Northern Territory Point Source kg Hg(tot)
1,2 % of State % of Source kg Hg0 kg Hg(II) kg Hgp Bauxite Mining 6.0 17.8 60.6 4.8 0.6 0.6 Hospitals (Except Psychiatric Hospitals) 19.9 59.1 97.0 0.4 14.5 5.0 Oil and Gas Extraction 2.2 6.4 15.3 1.7 0.2 0.2 Plaster Product Manufacturing 3.0 8.9 100.0 2.4 0.3 0.3 Other 2.6 7.8 - 2.0 0.3 0.3 Total 33.7 100.0 - 11.4 15.9 6.3 1 Data from NPI (2003a). 2 Sources that emit 2 kg/yr or less are summed in the category "Other".

Appendix B B 1
APPENDIX B Estimation of Hg emission from sources related to the Pacyna and Pacyna (2002) study Source category Production
Tonnes/yr Emission factor for Hgtot
g/t producedd Total emission
tonnes/yr NPI Point emission
tonnes/yre Non-ferrous metal productiona: - Cu 687 000 5.6 3.85 - Pb 300 000 3.0 0.9 - Zn 684 000 7.6 5.2 Total Non-Ferrous - - 9.95 1.33 Pig iron and steel productionb 7 012 000 0.04 0.28 0.36 Cement productionc 8 438 000 0.1 0.84 0.29 Total 11.07 1.98
a The production figures are from ABS, 2002. Manufacturing production (8301.0). March Quarter, 2002. www.abs.gov.au The EF applied for the smelter emissions in Europe/North America/Australia are lower than smelters in Asia/Africa/South America due to the assumption that more advanced control equipment is in place in the developed countries. b The production figure is from the Department of Industry, Tourism and Resources. www.industry.gov.au c The production figure is from ABS, 2002. Manufacturing production (8301.0). March Quarter, 2002. www.abs.gov.au d From Pacyna and Pacyna, 2002. e From NPI, 2003a.

Appendix C C -1
APPENDIX C
Input data to TAPM Run 10g Run 20 Run 30 Stack Exit Stack Exit Stack Exit
Hg(tot)a Hg0 Hg(II)/Hgp Latitudea Longitudea height Temperature velocity height Temperature velocity height Temperature velocity
No. Facility Name kg/yr kg/yr kg/yr Decimal degrees Decimal degrees m °K m/s m °K m/s m °K m/s
2 Mount Piper PS 45 22.5 22.5 -33.3554 150.0512 250 403 23 3 Liddell PS 46 23 23 -32.3719 150.9783 168 396 22.2 4 Bayswater PS 300 150 150 -32.3940 150.9500 250 403 23 5 Comsteelb 282 226 56 -32.8901 151.7263 10 373 15 10 373 15 50 373 15 6 Pasminco CCSc 110 88 22 -32.9465 151.6236 74 373 15 10 373 15 74 373 15 7 Vales Point PS 86 43 43 -33.1611 151.5417 178 384 20 8 Eraring PS 164 82 82 -33.0623 151.5214 200 403 23 9 Maldon CWd 23 18 5 -34.1898 150.6345 10 373 15 10 373 15 50 373 15
10 BHP Steel PKWe 102 82 20 -34.4634 150.8877 10 373 15 10 373 15 50 373 15 11 Orica Chlorine Pf 124 87 37 -33.9561 151.2209 10 373 15 10 373 15 50 373 15
a From NPI, 2003a. PS - Power Station b Basic Iron/Steel Manuf. c Cu, Ag, Pb, Zn Smelt/Ref. g Data from PS as well as the stack height of Pasminco are provided by CSIRO - EnergyTechnology - Newcastle, NSW. The rest of the data in respective Run is based on assumptions. Grid Centre Coordinates for Run 10: Lat. -31deg 15 min Long.:150 deg 50.5min Grid Centre Coordinates for Run 20 and 30: Lat. -33 deg 39.5 min Long.:150 deg 49.5min

Appendix D D-1
APPENDIX D Simulation: Run 10 (Inner domain) Dry deposition of Hg0 (Inner domain)
Run 10 Total 7.5*7.5 km 12.5*12.5 km 27.5*27.5 km 47.5*47.5 km
Emission Emission Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep
source g/h ug/h g/h % ug/h g/h % ug/h g/h % ug/h g/h %
Mount Piper PS 5.14 389 0.0004 0.01 1019 0.0010 0.02 4597 0.0046 0.09 13698 0.0137 0.27 Bayswater/Lidell PS 39.58 N/A N/A N/A 2590 0.0026 0.01 9622 0.0096 0.02 24651 0.0247 0.06
Comsteel 32.19 26092 0.0261 0.08 42693 0.0427 0.13
Pasminco CCS 12.57 11178 0.0112 0.09 26750 0.0267 0.21 167638 0.1676 0.23 Vales Point PS 9.82 2250 0.0022 0.02 6128 0.0061 0.06 Eraring PS 18.72 3277 0.0033 0.02 9050 0.0090 0.05 Maldon CW 2.63 2757 0.0028 0.11 6077 0.0061 0.23 22628 0.0226 0.86 N/A N/A N/A BHP Steel PKW 11.64 N/A N/A N/A N/A N/A N/A N/A N/A N/A N/A N/A N/A Orica Chlorine P 14.16 11433 0.0114 0.08 17849 0.0178 0.13 37013 0.0370 0.26 64916 0.0649 0.46
The first column within each area is derived from the post-processing of hourly simulated grid concentrations from TAPM. Source BHP Steel PKW is not included in the grid domain. Since sources: Comsteel, Pasminco CCS, Vales Point PS, and Eraring PS are located close to each other, some of the grid cells are overlappning when the deposition rate is calculated for the area 12.5*12.5 km. Dry deposition of Hg(II)/Hgp (Inner domain)
Run 10 Total 7.5*7.5 km 12.5*12.5 km 27.5*27.5 km 47.5*47.5 km
Emission Emission Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep
source g/h ug/h g/h % ug/h g/h % ug/h g/h % ug/h g/h %
Mount Piper PS 5.14 3810 0.0038 0.07 9522 0.0095 0.19 39798 0.0398 0.77 111696 0.1117 2.17 Bayswater/Lidell PS 39.58 N/A N/A N/A 29111 0.0291 0.07 85267 0.0853 0.22 182321 0.1823 0.46
Comsteel 32.19 108635 0.1086 0.34 178011 0.1780 0.55
Pasminco CCS 12.57 46995 0.0470 0.37 112549 0.1125 0.90 747696 0.7477 1.02 Vales Point PS 9.82 11747 0.0117 0.12 31395 0.0314 0.32 Eraring PS 18.72 17063 0.0171 0.09 45183 0.0452 0.24 Maldon CW 2.63 13603 0.0136 0.52 31168 0.0312 1.19 121573 0.1216 4.63 N/A N/A N/A BHP Steel PKW 11.64 N/A N/A N/A N/A N/A N/A N/A N/A N/A N/A N/A N/A Orica Chlorine P 14.16 5418 0.0054 0.04 75481 0.0755 0.53 219977 0.2200 1.55 402499 0.4025 2.84 N/A - Not Available, a term, which is used in this appendix, when the deposition rate has not been able to be derived from TAPM. The reason for this may be that (i) the sources are located to close to each other which makes it difficult to distinguish the deposition fluxes apart, (ii) the source/sources is/are located close to the domain border.
Sources 5,6,7, and 8 is included in this area
Sources 5,6,7, and 8 is included in this area

Appendix D D-2
Simulation: Run 10 (Outer domain) Dry deposition of Hg0 (Outer domain)
Run 10 Total 22.5*22.5 km 37.5*37.5 km 52.5*52.5 km 82.5*82.5 km
Emission Emission Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep
source g/h ug/h g/h % ug/h g/h % ug/h g/h % ug/h g/h %
Mount Piper PS 5.14 3665 0.0037 0.07 9948 0.0099 0.19 19576 0.0196 0.38 50032 0.0500 0.97 Bayswater/Lidell PS 39.58 7896 0.0079 0.02 17855 0.0179 0.05 31093 0.0311 0.08 Comsteel 32.19 N/A N/A N/A Pasminco CCS 12.57 N/A N/A N/A 188696 0.1887 0.26 265088 0.2651 0.36 Vales Point PS 9.82 N/A N/A N/A Eraring PS 18.72 N/A N/A N/A Maldon CW 2.63 16879 0.0169 0.64 38436 0.0384 1.46 N/A N/A N/A N/A N/A N/A BHP Steel PKW 11.64 31472 0.0315 0.27 55793 0.0558 0.48 N/A N/A N/A N/A N/A N/A Orica Chlorine P 14.16 23464 0.0235 0.17 47017 0.0470 0.33 72794 0.0728 0.51 N/A N/A N/A The first column within each area is derived from the post-processing of hourly simulated grid concentrations from TAPM. Source BHP Steel PKW and Orica Chlorine P are located close to each other and some of their gridcells are overlapping in the area 37.7*37.5 km.
Dry deposition of Hg(II)/Hgp (Outer domain)
Run 10 Total 22.5*22.5 km 37.5*37.5 km 52.5*52.5 km 82.5*82.5 km
Emission Emission Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep
source g/h ug/h g/h % ug/h g/h % ug/h g/h % ug/h g/h %
Mount Piper PS 5.14 28672 0.0287 0.56 73606 0.0736 1.43 140591 0.1406 2.74 348201 0.3482 6.78 Bayswater/Lidell PS 39.58 78605 0.0786 0.20 153041 0.1530 0.39 243134 0.2431 0.61 N/A N/A N/A Comsteel 32.19 N/A N/A N/A Pasminco CCS 12.57 N/A N/A N/A 842348 0.8423 1.15 1218264 1.2183 1.66 Vales Point PS 9.82 N/A N/A N/A Eraring PS 18.72 N/A N/A N/A Maldon CW 2.63 87598 0.0876 3.34 207862 0.2079 7.92 N/A N/A N/A N/A N/A N/A BHP Steel PKW 11.64 140024 0.1400 1.20 260964 0.2610 2.24 N/A N/A N/A N/A N/A N/A Orica Chlorine P 14.16 161220 0.1612 1.14 315565 0.3156 2.23 474383 0.4744 3.35 N/A N/A N/A N/A-Not available
Sources 5,6,7, and 8 is included in this area
Sources 5,6,7, and 8 is included in this area

Appendix D D-3
Simulation: Run 20 (Inner domain) Dry deposition of Hg0 (Inner domain)
Run 20 Total 7.5*7.5 km 12.5*12.5 km 27.5*27.5 km 47.5*47.5 km Emission Emission Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep
source g/h ug/h g/h % ug/h g/h % ug/h g/h % ug/h g/h %
Comsteel 32.19 26516 0.0265 0.08 40741 0.0407 0.13 75531 0.0755 0.23 113272 0.1133 0.35 Pasminco CCS 12.57 35320 0.0353 0.28 41019 0.0410 0.33 55226 0.0552 0.44 70906 0.0709 0.56 Maldon CW 2.63 2204 0.0022 0.08 4156 0.0042 0.16 13082 0.0131 0.50 30287 0.0303 1.15 BHP Steel PKW 11.64 9151 0.0092 0.08 14962 0.0150 0.13 30661 0.0307 0.26 48857 0.0489 0.42 Orica Chlorine P 14.16 13077 0.0131 0.09 19514 0.0195 0.14 37296 0.0373 0.26 61684 0.0617 0.44 The first column within each area is derived from the post-processing of hourly simulated grid concentrations from TAPM.
Dry deposition of Hg(II)/Hgp (Inner domain) Run 20 Total 7.5*7.5 km 12.5*12.5 km 27.5*27.5 km 47.5*47.5 km
Emission Emission Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep source g/h ug/h g/h % ug/h g/h % ug/h g/h % ug/h g/h %
Comsteel 32.19 110162 0.1102 0.34 169252 0.1693 0.53 313761 0.3138 0.97 470456 0.4705 1.46 Pasminco CCS 12.57 141592 0.1416 1.13 173167 0.1732 1.38 N/A N/A N/A N/A N/A N/A Maldon CW 2.63 10257 0.0103 0.39 20312 0.0203 0.77 68870 0.0689 2.62 167349 0.1673 6.37 BHP Steel PKW 11.64 20374 0.0204 0.17 26478 0.0265 0.23 N/A N/A N/A N/A N/A N/A Orica Chlorine P 14.16 92717 0.0927 0.66 137442 0.1374 0.97 256474 0.2565 1.81 408345 0.4083 2.88 N/A - Not Available

Appendix D D-4
Simulation: Run 20 (Outer domain) Dry deposition of Hg0 (Outer domain)
Run 20 Total 22.5*22.5 km 37.5*37.5 km 52.5*52.5 km 82.5*82.5 km Emission Emission Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep
source g/h ug/h g/h % ug/h g/h % ug/h g/h % ug/h g/h %
Comsteel 32.19 65427 0.0654 0.20 99187 0.0992 0.31 177992 0.1780 0.55 257581 0.2576 0.80 Pasminco CCS 12.57 25500 0.0255 0.20 38591 0.0386 0.31 69458 0.0695 0.55 100098 0.1001 0.80 Maldon CW 2.63 10373 0.0104 0.40 21728 0.0217 0.83 N/A N/A N/A N/A N/A N/A BHP Steel PKW 11.64 27555 0.0276 0.24 43111 0.0431 0.37 86999 0.0870 0.75 141428 0.1414 1.21 Orica Chlorine P 14.16 29704 0.0297 0.21 48308 0.0483 0.34 N/A N/A N/A N/A N/A N/A The first column within each area is derived from the post-processing of hourly simulated grid concentrations from TAPM. N/A - Not Available
Dry deposition of Hg(II)/Hgp (Outer domain) Run 20 Total 22.5*22.5 km 37.5*37.5 km 52.5*52.5 km 82.5*82.5 km
Emission Emission Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep source g/h ug/h g/h % ug/h g/h % ug/h g/h % ug/h g/h %
Comsteel 32.19 300386 0.3004 0.93 445736 0.4457 1.38 770047 0.7700 2.39 1101919 1.1019 3.42 Pasminco CCS 12.57 117213 0.1172 0.93 186249 0.1862 1.48 392031 0.3920 3.12 694447 0.6944 5.52 Maldon CW 2.63 53229 0.0532 2.03 117984 0.1180 4.49 N/A N/A N/A N/A N/A N/A BHP Steel PKW 11.64 125202 0.1252 1.08 204637 0.2046 1.76 458433 0.4584 3.94 885500 0.8855 7.60 Orica Chlorine P 14.16 148421 0.1484 1.05 282652 0.2827 2.00 N/A N/A N/A N/A N/A N/A N/A - Not Available

Appendix D D-5
Simulation: Run 30 (Inner domain) Dry deposition of Hg0 (Inner domain)
Run 30 Total 7.5*7.5 km 12.5*12.5 km 27.5*27.5 km 47.5*47.5 km Emission Emission Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep
source g/h ug/h g/h % ug/h g/h % ug/h g/h % ug/h g/h %
Comsteel 32.19 12734 0.0127 0.04 21255 0.0213 0.07 45513 0.0455 0.14 74930 0.0749 0.23 Pasminco CCS 12.57 5860 0.0059 0.05 8305 0.0083 0.07 15567 0.0156 0.12 25286 0.0253 0.20 Maldon CW 2.63 1250 0.0012 0.05 2681 0.0027 0.10 10126 0.0101 0.39 25842 0.0258 0.98 BHP Steel PKW 11.64 3066 0.0031 0.03 6238 0.0062 0.05 16344 0.0163 0.14 30125 0.0301 0.26 Orica Chlorine P 14.16 5453 0.0055 0.04 9453 0.0095 0.07 22373 0.0224 0.16 42430 0.0424 0.30 The first column within each area is derived from the post-processing of hourly simulated grid concentrations from TAPM. N/A - Not Available
Dry deposition of Hg(II)/Hgp (Inner domain) Run 30 Total 7.5*7.5 km 12.5*12.5 km 27.5*27.5 km 47.5*47.5 km
Emission Emission Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep source g/h ug/h g/h % ug/h g/h % ug/h g/h % ug/h g/h %
Comsteel 32.19 52898 0.0529 0.16 88295 0.0883 0.27 189033 0.1890 0.59 311127 0.3111 0.97 Pasminco CCS 12.57 24369 0.0244 0.19 33288 0.0333 0.26 N/A N/A N/A N/A N/A N/A Maldon CW 2.63 6256 0.0063 0.24 14101 0.0141 0.54 56146 0.0561 2.14 147331 0.1473 5.61 BHP Steel PKW 11.64 6307 0.0063 0.05 10126 0.0101 0.09 N/A N/A N/A N/A N/A N/A Orica Chlorine P 14.16 38219 0.0382 0.27 65501 0.0655 0.46 149888 0.1499 1.06 271773 0.2718 1.92 N/A - Not Available

Appendix D D-6
Simulation: Run 30 (Outer domain)
Dry deposition of Hg0 (Outer domain) Run 30 Total 22.5*22.5 km 37.5*37.5 km 52.5*52.5 km 82.5*82.5 km
Emission Emission Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep source g/h ug/h g/h % ug/h g/h % ug/h g/h % ug/h g/h %
Comsteel 32.19 36261 0.0363 0.11 60461 0.0605 0.19 125672 0.1257 0.39 198303 0.1983 0.62 Pasminco CCS 12.57 11284 0.0113 0.09 19020 0.0190 0.15 41146 0.0411 0.33 66823 0.0668 0.53 Maldon CW 2.63 7490 0.0075 0.29 17367 0.0174 0.66 N/A N/A N/A N/A N/A N/A BHP Steel PKW 11.64 15683 0.0157 0.13 26943 0.0269 0.23 61583 0.0616 0.53 112537 0.1125 0.97 Orica Chlorine P 14.16 17340 0.0173 0.12 31921 0.0319 0.23 N/A N/A N/A N/A N/A N/A The first column within each area is derived from the post-processing of hourly simulated grid concentrations from TAPM. N/A - Not Available
Dry deposition of Hg(II)/Hgp (Outer domain) Run 30 Total 22.5*22.5 km 37.5*37.5 km 52.5*52.5 km 82.5*82.5 km
Emission Emission Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep Dry Dep source g/h ug/h g/h % ug/h g/h % ug/h g/h % ug/h g/h %
Comsteel 32.19 171009 0.1710 0.53 278027 0.2780 0.86 549930 0.5499 1.71 851505 0.8515 2.65 Pasminco CCS 12.57 57770 0.0578 0.46 104283 0.1043 0.83 273613 0.2736 2.18 555814 0.5558 4.42 Maldon CW 2.63 40414 0.0404 1.54 97526 0.0975 3.71 N/A N/A N/A N/A N/A N/A BHP Steel PKW 11.64 81889 0.0819 0.70 145499 0.1455 1.25 370325 0.3703 3.18 767352 0.7674 6.59 Orica Chlorine P 14.16 91785 0.0918 0.65 185480 0.1855 1.31 N/A N/A N/A N/A N/A N/A N/A - Not Available

CONTACT DETAILS
Christian Peterson Graduate School of the Environment
Macquarie University NSW 2109, Australia
Ph 02 9850 7988 Fax 02 9850 7972 Email: [email protected]
Professor Peter Nelson
Graduate School of the Environment Macquarie University NSW 2109, Australia
Ph 02 9850 6958 Fax 02 9850 7972
Email: [email protected]
Anthony Morrison Graduate School of the Environment
Macquarie University NSW 2109, Australia
Ph 02 9850 7869 Fax 02 9850 7972
Email: [email protected]


















