pVACtools: a computational toolkit to identify and ......Jan 04, 2020 · review, and interpret...
45
pVACtools: a computational toolkit to identify and visualize cancer neoantigens pVACtools: toolkit for tumor neoantigen characterization Jasreet Hundal 1+ , Susanna Kiwala 1+ , Joshua McMichael 1 , Christopher A. Miller 1,2,4 , Huiming Xia 1 , Alexander T. Wollam 1 , Connor J. Liu 1 , Sidi Zhao 1 ,Yang-Yang Feng 1 , Aaron P. Graubert 1 , Amber Z. Wollam 1 , Jonas Neichin 1 , Megan Neveau 1 , Jason Walker 1 , William E Gillanders 4,5 , Elaine R. Mardis 3 , Obi L. Griffith 1,2,4,6, * , Malachi Griffith 1,2,4,6, * Affiliations (1) McDonnell Genome Institute, Washington University School of Medicine, St. Louis, MO, USA (2) Division of Oncology, Department of Medicine, Washington University School of Medicine, St. Louis, MO, USA (3) Institute for Genomic Medicine, Nationwide Children’s Hospital, 575 Children’s Crossroad, Columbus OH, USA (4) Siteman Cancer Center, Washington University School of Medicine, St. Louis, MO, USA (5) Department of Surgery, Washington University School of Medicine, St. Louis, MO, USA (6) Department of Genetics, Washington University School of Medicine, St. Louis, MO, USA + These authors contributed equally to this work * Corresponding authors. [email protected], [email protected] on May 15, 2021. © 2020 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 6, 2020; DOI: 10.1158/2326-6066.CIR-19-0401
Transcript of pVACtools: a computational toolkit to identify and ......Jan 04, 2020 · review, and interpret...

pVACtools: a computational toolkit to identify and visualize
cancer neoantigens
pVACtools: toolkit for tumor neoantigen characterization
Jasreet Hundal1+
, Susanna Kiwala1+
, Joshua McMichael1, Christopher A. Miller
1,2,4, Huiming
Xia1, Alexander T. Wollam
1, Connor J. Liu
1, Sidi Zhao
1 ,Yang-Yang Feng
1, Aaron P. Graubert
1,
Amber Z. Wollam1, Jonas Neichin
1, Megan Neveau
1, Jason Walker
1, William E Gillanders
4,5,
Elaine R. Mardis3, Obi L. Griffith
1,2,4,6,*, Malachi Griffith
1,2,4,6,*
Affiliations
(1) McDonnell Genome Institute, Washington University School of Medicine, St. Louis, MO,
USA
(2) Division of Oncology, Department of Medicine, Washington University School of Medicine,
St. Louis, MO, USA
(3) Institute for Genomic Medicine, Nationwide Children’s Hospital, 575 Children’s Crossroad,
Columbus OH, USA
(4) Siteman Cancer Center, Washington University School of Medicine, St. Louis, MO, USA
(5) Department of Surgery, Washington University School of Medicine, St. Louis, MO, USA
(6) Department of Genetics, Washington University School of Medicine, St. Louis, MO, USA
+ These authors contributed equally to this work
* Corresponding authors. [email protected], [email protected]
on May 15, 2021. © 2020 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 6, 2020; DOI: 10.1158/2326-6066.CIR-19-0401

Abstract
Identification of neoantigens is a critical step in predicting response to checkpoint
blockade therapy and design of personalized cancer vaccines. This is a cross-
disciplinary challenge, involving genomics, proteomics, immunology, and computational
approaches. We have built a computational framework called pVACtools that, when
paired with a well-established genomics pipeline, produces an end-to-end solution for
neoantigen characterization. pVACtools supports identification of altered peptides from
different mechanisms including point mutations, in-frame or frameshift insertions and
deletions, and gene fusions. Prediction of peptide:MHC binding is accomplished by
supporting an ensemble of MHC Class I and II binding algorithms, within a framework
designed to facilitate the incorporation of additional algorithms. Prioritization of predicted
peptides occurs by integrating diverse data including mutant allele expression, peptide
binding affinities, and determination whether a mutation is clonal or subclonal.
Interactive visualization via a web interface allows clinical users to efficiently generate,
review, and interpret results, selecting candidate peptides for individual patient vaccine
designs. Additional modules support design choices needed for competing vaccine
delivery approaches. One such module optimizes peptide ordering to minimize
junctional epitopes in DNA-vector vaccines. Downstream analysis commands for
synthetic long peptide vaccines are available to assess candidates for factors that
influence peptide synthesis. All of the aforementioned steps are executed via a modular
workflow consisting of tools for neoantigen prediction from somatic alterations
(pVACseq and pVACfuse), prioritization, and selection using a graphical web-based
interface (pVACviz), and design of DNA vector-based vaccines (pVACvector) and
synthetic long peptide vaccines. pVACtools is available at pvactools.org.
on May 15, 2021. © 2020 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 6, 2020; DOI: 10.1158/2326-6066.CIR-19-0401

Introduction
The increasing use of cancer immunotherapies has spurred interest in identifying and
characterizing predicted neoantigens encoded by a tumor’s genome. The facility and
precision of computational tools for predicting neoantigens has become increasingly
important(1), and several resources aiding in these efforts have been published(2–4).
Typically, these tools start with a list of somatic variants (in Variant Call Format (VCF) or
other formats such as Browser Extensible Data (BED)) with annotated protein changes,
and predict the strongest MHC binding peptides (8-11-mer for class I MHC and 13-25-
mer for class II) using one or more prediction algorithms(5–7). The predicted
neoantigens are then filtered and ranked based on defined metrics including
sequencing read coverage, variant allele fraction (VAF), gene expression, and
differential binding compared to the wildtype peptide (agretopicity index score(8)).
However, of the prediction tools overviewed in Supp. Table S1 (Vaxrank(3), MuPeXI(2),
CloudNeo(4), FRED2(9), Epi-Seq(8), and ProTECT(10)) most lack key functionality,
including predicting neoantigens from gene fusions, aiding optimized vaccine design for
DNA cassette vaccines, and explicitly incorporating nearby germline or somatic
alterations into the candidate neoantigens(11). In addition, none of these tools offer an
intuitive graphical user interface for visualizing and efficiently selecting the most
promising candidates— a key feature for facilitating involvement of clinicians and other
researchers in the process of neoantigen prediction and evaluation.
To address these limitations, we created a comprehensive and extensible toolkit for
computational identification, selection, prioritization, and visualization of neoantigens—
on May 15, 2021. © 2020 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 6, 2020; DOI: 10.1158/2326-6066.CIR-19-0401

‘pVACtools’, that facilitates each of the major components of neoantigen identification
and prioritization. This computational framework can be used to identify neoantigens
from a variety of somatic alterations, including gene fusions and insertion/deletion
frameshift variants, both of which potentially create strong immunogenic
neoantigens(12–14). pVACtools can facilitate both MHC class I and II predictions and
provides an interactive display of predicted neoantigens for review by the end user.
Materials and Methods
TCGA data pre-processing
Aligned (build GRCh38) tumor and normal BAMs from BWA(15) (version 0.7.12-r1039)
as well as somatic variant calls from VarScan2(16,17) (in VCF format) were
downloaded from the Genomic Data Commons (GDC, https://gdc.cancer.gov/). Since
the GDC does not provide germline variant calls for TCGA data, we used GATK’s(18)
HaplotypeCaller to perform germline variant calling using default parameters. These
calls were refined using VariantRecalibrator in accordance with GATK Best
Practices(19). Somatic and germline missense variant calls from each sample were
then combined using GATK’s CombineVariants, and the variants were subsequently
phased using GATK’s ReadBackedPhasing algorithm.
Phased Somatic VCF files were annotated with RNA depth and expression information
using VAtools (http://vatools.org). We restricted our analysis to only consider ‘PASS’
variants in these VCFs as these are higher confidence than the raw set, and the
variants were annotated using the “--pick” option in VEP (Ensembl version 88).
on May 15, 2021. © 2020 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 6, 2020; DOI: 10.1158/2326-6066.CIR-19-0401

Existing in silico HLA typing information was obtained from The Cancer Immunome
Atlas (TCIA) database(20).
Neoantigen prediction
The VEP-annotated VCF files were then analyzed with pVACseq using all eight Class I
prediction algorithms for neoantigen peptide lengths 8-11. The current MHC Class I
algorithms supported by pVACseq are NetMHCpan(21), NetMHC(7,21),
NetMHCcons(22), PickPocket(23), SMM(24), SMMPMBEC(25), MHCflurry(26), and
MHCnuggets(27). The four MHC Class II algorithms that are supported are
NetMHCIIpan(28), SMMalign(29), NNalign(30), and MHCnuggets(31). For the
demonstration analysis, we limited our prediction to only MHC Class I alleles due to
availability of HLA typing information from TCIA, though binding predictions for Class II
alleles can also be generated using pVACtools.
Ranking of Neoantigens
To help prioritize neoantigens, a rank is assigned to all neoantigens that pass initial
filters where each of the following four criteria are assigned a rank-ordered value (where
the best is assigned rank = 1):
B = Rank of binding affinity
F = Rank of Fold Change between MT and WT alleles
M = Rank of mutant allele expression, calculated as (rank of Gene expression * rank of
on May 15, 2021. © 2020 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 6, 2020; DOI: 10.1158/2326-6066.CIR-19-0401

Mutant allele RNA Variant allele fraction)
D = Rank of DNA Variant allele fraction
A final ranking is based on a score obtained from combining these values:
B+F+(M*2)+(D/2).
Whereas agretopicity is considered in ranking, we do not recommend filtering
candidates solely based on mutant versus wildtype binding value (default fold change
value is 0). A previous study on the binding properties of known neoantigen candidates
showed that most changes reside in T cell receptor contact regions and not in anchor
residues(32). To help end users make an informed decision while selecting candidates,
we also provide the position of the variant in the final report. We are not comfortable
relying on hard rules about “anchor positions” (e.g. position 2 and C-terminal) because
these may vary by MHC allele and the peptide length. As we gain more empirical results
and validation data, we hope that future versions of pVACtools may include more
options for prioritizing based on these factors.
The rank is not meant to be a definitive metric of peptide suitability for vaccines, but was
designed to be a useful first step in the peptide selection process.
Implementation
pVACtools is written in Python3. The individual tools are implemented as separate
command line entry points that can be run using the `pvacseq`, `pvacfuse`,
on May 15, 2021. © 2020 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 6, 2020; DOI: 10.1158/2326-6066.CIR-19-0401

`pvacvector`, `pvacapi`, and `pvacviz` commands for each respective tool.
pVACapi is required to run pVACviz so both the `pvacapi` and `pvacviz` commands
need to be executed in separate terminals.
The code test suite is implemented using the Python unittest framework and GitHub
integration tests are run using travis-ci (travis-ci.org). Code changes are integrated
using GitHub pull requests (https://github.com/griffithlab/pVACtools/pulls). Feature
additions, user requests, and bug reports are managed using the GitHub issue tracking
(https://github.com/griffithlab/pVACtools/issues). User documentation is written using
the reStructuredText markup language and the Sphinx documentation framework
(sphinx-doc.org). Documentation is hosted on Read the Docs (readthedocs.org) and
can be viewed at pvactools.org.
The pymp-pypi package was used to add support for parallel processing. The number
of processes is controlled by the --n-threads parameter.
pVACseq
For pVACseq, the pyvcf package is first used for parsing the input VCF file and
extracting information about the supported missense, inframe insertion, inframe
deletion, and frameshift variants into TSV format.
This output is then used to determine the wildtype peptide sequence by extracting a
region around the somatic variant according to the --peptide-sequence-length
on May 15, 2021. © 2020 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 6, 2020; DOI: 10.1158/2326-6066.CIR-19-0401

specified by the user. The variant’s amino acid change is incorporated in this peptide
sequence to determine the mutant peptide sequence. For frameshift variants, the new
downstream protein sequence calculated by VEP is reported from the variant position
onward. The number of downstream amino acids to include is controlled by the --
downstream-sequence-length parameter. If a phased VCF with proximal variants
is provided, proximal missense variants that are in phase with the somatic variant of
interest are incorporated into the mutant and wildtype peptide sequences, as
appropriate. The mutant and wildtype sequences are stored in a FASTA file. The
FASTA file is then submitted to the individual prediction algorithms for binding affinity
predictions. For algorithms included in IEDB, we either use the IEDB API or a
standalone installation, if an installation path is provided by the user (--iedb-
install-directory). The mhcflurry and mhcnuggets packages are used to run
the MHCflurry(26) and MHCnuggets(27) prediction algorithms, respectively.
The predicted mutant antigens are then parsed into a TSV report format, and, for each
mutant antigen, the closest wildtype antigen is determined and reported. Predictions for
each mutant antigen/neoantigen from multiple algorithms are aggregated into the “best”
(lowest) and median binding scores. The resulting TSV is processed through multiple
filtering steps (as described below in results: Comparison of filtering criteria): (1) Binding
filter: this filter selects the strongest binding candidates based on the mutant binding
score and the fold change (WT score / MT score). Depending on the --top-score-
metric parameter setting, this filter is either applied to the median score across all
chosen prediction algorithms (default) or the best (lowest) score amongst the chosen
on May 15, 2021. © 2020 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 6, 2020; DOI: 10.1158/2326-6066.CIR-19-0401

prediction algorithms. (2) Coverage filter: this filter accepts VAF and coverage
information from the tumor DNA, tumor RNA, and normal DNA if these values are
available in the input VCF. (3) Transcript-support-level (TSL) filter: this filter evaluates
each transcript’s support level if this information was provided by VEP in the VCF. (4)
Top-score filter: the filter picks the top mutant peptide for each variant using the binding
affinity as the determining factor. This filter is implemented to only select the best
candidate from amongst multiple candidates that could result from a single variant due
to different peptide lengths, variant registers, transcript sequences, and HLA alleles.
The result of these filtering steps is reported in a filtered report TSV. The remaining
neoantigens are then annotated with cleavage site and stability predictions by NetChop
and NetMHCStabPan, respectively, and a relative rank (as described in the materials
and methods: Ranking of Neoantigens) is assigned. The rank-ordered final output is
reported in a condensed file. The pandas package is used for data management while
filtering and ranking the neoantigen candidates.
pVACvector
When running pVACvector with a pVACseq output file, the original input VCF must also
be provided (--input-vcf parameter). The VCF is used to extract a larger peptide
sequence around the target neoantigen (length determined by the --input-n-mer
parameter). Alternatively, a list of target peptide sequences can be provided in a FASTA
file. The set of peptide sequences are then combined in all possible pairs, and an
ordering of peptides for the vector is produced as follows:
on May 15, 2021. © 2020 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 6, 2020; DOI: 10.1158/2326-6066.CIR-19-0401

To determine the optimal order of peptide-spacer-peptide combinations, binding
predictions are made for all peptide registers overlapping the junction. A directed graph
is then constructed, with nodes defined as target peptides, and edges representing
junctions. The score of each edge is defined as the lowest binding score of its junctional
peptides (a conservative metric). Edges with scores below the threshold are removed,
and if heuristics indicate that a valid graph may exist, a simulated annealing procedure
is used to identify a path through the nodes that maximizes junction scores (preserving
the weakest overall predicted binding for junctional epitopes). If no valid ordering is
found, additional “spacer” amino acids are added to each junction, binding affinities are
re-calculated, and a new graph is constructed and tested, setting edge weights equal to
that of the best performing (highest binding score) peptide-junction-spacer combination.
The spacers used for pVACvector are set by the user with the --spacers parameter.
This parameter defaults to
None,AAY,HHHH,GGS,GPGPG,HHAA,AAL,HH,HHC,HHH,HHHD,HHL,HHHC, where
None is the placeholder for testing junctions without a spacer sequence. Spacers are
tested iteratively, starting with the first spacer in the list, and adding subsequent spacers
if no valid path is found. If the user wishes to specify a custom spacer such as furin
cleavage site (e.g. R-X-(R/K)-R) they can do so (e.g. --spacers RKKR).
If no result is found after testing with the full set of spacers, the ends of “problematic”
peptides, where all junctions contain at least one well-binding epitope, will be clipped by
removing one amino acid at a time, then repeating the above binding and graph-building
on May 15, 2021. © 2020 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 6, 2020; DOI: 10.1158/2326-6066.CIR-19-0401

process. This clipping may be repeated up to the number of times specified in the --
max-clip-length parameter.
It may be necessary to explore the parameter space when running pVACvector.
As binding predictions for some sites vary substantially across algorithms, the
most conservative settings may result in no valid paths, often due to one
"outlier" prediction. Carefully selecting which predictors to run may help
ameliorate this issue as well. In general, setting a higher binding threshold (e.g. 1000
nM) and using the median binding value (--top-score-metric median) will lead to
greater possibility of a design, while more conservative settings of 500 nM and
lowest/best binding value (--top-score-metric lowest) will give more confidence
that there are no junctional neoepitopes.
Our current recommendation is to run pVACvector several different ways, and
choose the path resulting from the most conservative set of parameters that produces a
vector design containing all candidate neoantigens.
pVACapi and pVACviz
pVACapi is implemented using the Python libraries Flask and Bokeh. The pVACviz
client is written in TypeScript using the Angular web application framework, the Clarity
UI component library, and the ngrx library for managing application state.
Data availability
on May 15, 2021. © 2020 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 6, 2020; DOI: 10.1158/2326-6066.CIR-19-0401

Data from 100 cases each of melanoma, hepatocellular carcinoma and lung squamous
cell carcinoma were obtained from TCGA and downloaded via the Genomics Data
Commons (GDC). This data can be accessed under dbGaP study accession
phs000178. Data for demonstration and analysis of fusion neoantigens was
downloaded from the Github repo for Integrate
(https://github.com/ChrisMaherLab/INTEGRATE-Vis/tree/master/example).
Software availability
The pVACtools codebase is hosted publicly on GitHub at
https://github.com/griffithlab/pVACtools and https://github.com/griffithlab/BGA-interface-
projects (pVACviz). User documentation is available at pvactools.org. This project is
licensed under the Non-Profit Open Software License version 3.0 (NPOSL-3.0,
https://opensource.org/licenses/NPOSL-3.0). pVACtools has been packaged and
uploaded to PyPI under the “pvactools” package name and can be installed on Linux
systems by running the `pip install pvactools` command. Installation requires a Python
3.5 environment which can be emulated by using Conda. Versioned Docker images are
available on DockerHub (https://hub.docker.com/r/griffithlab/pvactools/). Releases are
also made available on GitHub (https://github.com/griffithlab/pVACtools/releases).
Example pipeline for creation of pVACtools input files
pVACtools was designed to support a standard VCF variant file format and thus should
be compatible with many existing variant calling pipelines. However, as a reference, we
provide the following description of our current somatic and expression analysis pipeline
(manuscript in preparation) which has been implemented using docker, CWL(33), and
on May 15, 2021. © 2020 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 6, 2020; DOI: 10.1158/2326-6066.CIR-19-0401

Cromwell(34). The pipeline consists of workflows for alignment of exome DNA- and
RNA-Seq data, somatic and germline variant detection, RNA-Seq expression
estimation, and HLA typing.
This pipeline begins with raw patient tumor exome and cDNA capture(35) or RNAseq
data and leads to the production of annotated VCFs for neoantigen identification and
prioritization with pVACtools. Our pipeline consists of several modular components:
DNA alignment, HLA typing, germline and somatic variant detection, variant annotation,
and RNAseq analysis. More specifically, we use BWA-MEM(15) for aligning the
patient’s tumor and normal exome data. For germline variant calling, the output BAM
then undergoes merging (merging of separate instrument data) (Samtools(36) Merge),
query name sorting (Picard SortSam), duplicate marking (Picard MarkDuplicates),
position sorting (Picard SortSam), and base quality recalibration (GATK
BaseRecalibrator). GATK’s HaplotypeCaller(18) is used for germline variant calling and
the output variants were annotated using VEP(37) and filtered for coding sequence
variants.
For somatic variant calling, the pipeline is consistent with above except that the variant
calling step combines the output of four variant detection algorithms- Mutect2(38),
Strelka(39), Varscan(16,17) and Pindel(40). The combined variants were normalized
using GATK’s LeftAlignAndTrimVariants to left-align the indels and trim common bases.
Vt(41) was used to split multi-allelic variants. Several filters such as gnomAD allele
frequency (maximum population allele frequency), percentage of mapq0 reads, as well
as pass-only variants are applied prior to annotation of the VCF using VEP. We used a
on May 15, 2021. © 2020 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 6, 2020; DOI: 10.1158/2326-6066.CIR-19-0401

combination of custom and standard plugins for VEP annotation (parameters: --format
VCF --plugin Downstream --plugin Wildtype --symbol --term SO --transcript_version --tsl
--coding_only --flag_pick --hgvs). Variant coverageis assessed using bam-readcount
(https://github.com/genome/bam-readcount) for both the tumor and normal DNA exome
data, and this information is also annotated into the VCF output using VAtools
(http://vatools.org).
Our pipeline also generates a phased-VCF file by combining both the somatic and
germline variants and running the sorted combined variants through GATK
ReadBackedPhasing.
For RNAseq data, the pipeline first trims adapter sequences using flexbar(42) and
aligns the patient’s tumor RNAseq data using HISAT2(43). Two different methods,
Stringtie(44) and Kallisto(45), are employed for evaluating both the transcript and gene
expression values. Additionally, coverage support for variants in RNAseq data is
determined with bam-readcount. This information is added to the VCF using VAtools
and serves as an input for neoantigen prioritization with pVACtools.
Optionally, our pipeline can also run HLA-typing in silico using OptiType(46) when
clinical HLA typing is not available.
on May 15, 2021. © 2020 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 6, 2020; DOI: 10.1158/2326-6066.CIR-19-0401

We aim to support various versions of these pipelines (via the use of standardized file
formats), and are also actively developing an end-to-end public CWL workflow starting
from sequence reads to pVACseq output
(https://github.com/genome/analysis-
workflows/blob/master/definitions/pipelines/immuno.cwl).
Results
The pVACtools workflow (Fig. 1) is divided into modular components that can be run
independently. The main tools in the workflow are: (a) pVACseq: a significantly
enhanced and reengineered version of our previous tool(47) for identifying and
prioritizing neoantigens from a variety of tumor-specific alterations, (b) pVACfuse: a tool
for detecting neoantigens resulting from gene fusions, (c) pVACviz: a graphical user
interface web client for process management, visualization, and selection of results from
pVACseq, (d) pVACvector: a tool for optimizing design of neoantigens and nucleotide
spacers in a DNA vector that prevents high-affinity junctional neoantigens, and (e)
pVACapi: an OpenAPI HTTP REST interface to the pVACtools suite.
pVACseq
pVACseq(47) has been re-implemented in Python3 and extended to include many new
features since our initial report of its release. pVACseq no longer requires a custom
input format for variants, and now uses a standard VCF file annotated with VEP(37). In
our own neoantigen identification pipeline, this VCF is obtained by merging results from
multiple somatic variant callers and RNA expression tools (as described in the materials
on May 15, 2021. © 2020 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 6, 2020; DOI: 10.1158/2326-6066.CIR-19-0401

and methods section above: Example pipeline for creation of pVACtools input files).
Information that is not natively available in the VCF output from somatic variant callers
(such as coverage and variant allele fractions for RNA and DNA, as well as gene and
transcript expression values) can be added to the VCF using VAtools
(http://vatools.org), a suite of accessory routines that we created to accompany
pVACtools. pVACseq queries these features directly from the VCF, enabling
prioritization and filtering of neoantigen candidates based on sequence coverage and
expression information. In addition, pVACseq now makes use of phasing information
taking into account variants proximal to somatic variants of interest. Since proximal
variants can change the peptide sequence and affect neoantigen binding predictions,
this is important for ensuring that the selected neoantigens correctly represent the
individual’s genome(11). We have also expanded the supported variant types for
neoantigen predictions to include in-frame indels and frameshift variants. These
capabilities expand the potential number of targetable neoantigens several-fold in many
tumors(12,14)(Supplementary Fig. S2).
To prioritize neoantigens, pVACseq now offers support for eight different MHC Class I
antigen prediction algorithms and four MHC Class II prediction algorithms. The tool
does this in part by leveraging the Immune Epitope Database (IEDB)(48) and their suite
of six different MHC class I prediction algorithms, as well as three MHC Class II
algorithms (as described in the materials and methods: Neoantigen prediction).
pVACseq supports local installation of these tools for high-throughput users, access
through a docker container (https://hub.docker.com/r/griffithlab/pvactools), and ready-to-
on May 15, 2021. © 2020 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 6, 2020; DOI: 10.1158/2326-6066.CIR-19-0401

go access via the IEDB RESTful web interface. In addition, pVACseq now contains an
extensible framework for supporting new neoantigen prediction algorithms that has
been used to add support for two new non-IEDB algorithms—MHCflurry(26) and
MHCnuggets(27). By creating a framework that integrates many tools, we allow for (a) a
broader ensemble approach than IEDB, and (b) a system that other users can leverage
to develop improved ensemble ranking, or to integrate proprietary or not-yet-public
prediction software. This framework enables non-informatics-expert users to predict
neoantigens from sequence variant data sets.
Once neoantigens have been predicted, binding affinity values from the selected
prediction algorithms are aggregated into a median binding score and a report file is
generated. Finally, a rank is calculated that can be used to prioritize the predicted
neoantigens. The rank takes into account gene expression, sequence read coverage,
binding affinity predictions, and agretopicity (as described in the materials and methods:
Ranking of Neoantigens). The pVACseq rank allows users to pick the exemplar peptide
for each variant from a large number of lengths, registers, alternative transcripts, and
HLA alleles. In addition to applying strict binding affinity cutoffs, the pipeline also offers
support for MHC allele-specific cutoffs(49). The allele-specific binding filter incorporates
allele-specific thresholds from IEDB for the 38 most common HLA-A and HLA-B alleles,
representative of the nine major supertypes, instead of the 500 nM cutoff. Since these
allele-specific thresholds only include cutoffs for HLA-A and HLA-B alleles, we
recommend evaluating binding predictions for HLA-C allele separately with a less
stringent cutoff (such as 1,000 nM) and merging results when picking final candidates.
on May 15, 2021. © 2020 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 6, 2020; DOI: 10.1158/2326-6066.CIR-19-0401

We also offer cleavage position predictions via optional processing through
NetChop(50) as well as stability predictions made by NetMHCstabPan(51).
pVACfuse
Previous studies show that the novel protein sequences produced by known driver gene
fusions frequently produce neoantigen candidates(52). pVACfuse provides support for
predicting neoantigens from such gene fusions. One possible input to pVACfuse is
annotated fusion file from AGFusion(53), thus enabling support for a variety of fusion
callers such as STAR-Fusion(54), FusionCatcher(55), JAFFA(56) and TopHat-
Fusion(57). pVACfuse also supports annotated BEDPE format from any fusion caller
which can, for example, be generated using INTEGRATE-Neo(52). These variants are
then assessed for presence of fusion neoantigens using predictions from any of the
pVACseq-supported binding prediction algorithms.
pVACviz and pVACapi
Implementing cancer vaccines in a clinical setting requires multidisciplinary teams,
which may not include informatics experts. To support this growing community of users,
we developed pVACviz, which is a browser-based user interface that assists in
launching, managing, reviewing, and visualizing the results of pVACtools processes.
Instead of interacting with the tools via terminal/shell commands, the pVACviz client
provides a modern web-based user experience. Users complete a pVACseq (Fig. 2)
process setup form that provides helpful documentation and suggests valid values for
on May 15, 2021. © 2020 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 6, 2020; DOI: 10.1158/2326-6066.CIR-19-0401

inputs. The client also provides views showing ongoing processes, their logs, and
interim data files to aid in managing and troubleshooting. After a process has
completed, users may examine the results as a filtered data table or as a scatterplot
visualization—allowing them to curate results and save them as a CSV file for further
analysis. Extensive documentation of the visualization interface can be found in the
online documentation (https://pvactools.readthedocs.io/en/latest/pvacviz.html).
To support informatics groups that want to incorporate or build upon the pVACtools
features, we developed pVACapi, which provides an HTTP REST interface to the
pVACtools suite. Currently, it provides the API that pVACviz uses to interact with the
pVACtools suite. Advanced users could develop their own user interfaces or use the
API to control multiple pVACtools installations remotely over an HTTP network.
pVACvector
Once a list of neoantigen candidates has been prioritized and selected, the pVACvector
utility can be used to aid in the construction of DNA-based cancer vaccines. The input is
either the output file from pVACseq or a FASTA file containing peptide sequences.
pVACvector returns a neoantigen sequence ordering that minimizes the effects of
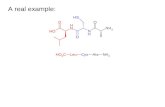
junctional peptides (which may create novel antigens) between the sequences (Fig. 3).
This is accomplished by using the core pVACseq module to predict the binding scores
for each junctional peptide and by modifying junctions with spacer(58,59) amino acid
sequences, or by trimming the ends of the peptides in order to reduce reactivity. The
final vaccine ordering is achieved through a simulated annealing procedure(60) that
on May 15, 2021. © 2020 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 6, 2020; DOI: 10.1158/2326-6066.CIR-19-0401

returns a near-optimal solution, when one exists (details can be found in the materials
and methods: Implementation).
As many prediction algorithms are CPU-intensive, pVACseq, pVACfuse, and
pVACvector also support using multiple cores to improve runtime. Using this feature,
calls to IEDB and other prediction algorithms are made in parallel over a user-defined
number of processes (details can be found in the materials and methods:
Implementation).
pVACtools has been used to predict and prioritize neoantigens for several
immunotherapy studies(61–63) and cancer vaccine clinical trials (e.g. NCT02348320,
NCT03121677, NCT03122106, NCT03092453 and NCT03532217). We also have a
large external user community that has been actively evaluating and using these
packages for their neoantigen analysis and has aided in subsequent refinement of
pVACtools through extensive feedback. The original ‘pvacseq’ package has been
downloaded over 41,000 times from PyPI, the ‘pvactools’ package has been
downloaded over 18,000 times, and the ‘pvactools’ docker image has been pulled over
30,000 times.
Analysis of TCGA data using pVACtools
To demonstrate the utility and performance of the pVACtools package, we downloaded
exome sequencing and RNA-Seq data from The Cancer Genome Atlas (TCGA)(19)
on May 15, 2021. © 2020 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 6, 2020; DOI: 10.1158/2326-6066.CIR-19-0401

from 100 cases each of melanoma, hepatocellular carcinoma, and lung squamous cell
carcinoma and used patient-specific MHC Class I alleles (Supplemental Fig. S1) to
determine neoantigen candidates for each patient. There were a total of 64,422 VEP-
annotated variants reported across 300 samples, with an average of 214 variants per
sample. Of these, 61,486 were single nucleotide variants (SNVs), 479 were inframe
insertions and deletions, and 2,465 were frameshift mutations (Supplemental Fig. S2).
We used this annotated list of variants as input to the pVACseq component of
pVACtools to predict neoantigenic peptides. pVACseq reported 14,599,993 unfiltered
neoantigen candidates. The original version of pVACseq(20) reported 10,284,467
neoantigens. This demonstrated that, by extending support for additional variant types
as well as prediction algorithms (due to support for additional alleles), we produced 42%
more raw candidate neoantigens.
After applying our default median binding affinity cutoff of 500 nM across all eight MHC
Class I prediction algorithms, there were 96,235 predicted strong binding neoantigens,
derived from 34,552 somatic variants (32,788 missense SNVs, 1,603 frameshift
variants, and 131 in-frame indels). This set of strong binders was further reduced by
filtering out mutant peptides with median predicted binding affinities (across all
prediction algorithms) greater than that of the corresponding wildtype peptide (i.e.
mutant/wildtype binding affinity fold change > 1), resulting in 70,628 neoantigens from
28,588 variants (26,880 SNVs, 1,583 frameshift, and 125 in-frame indels).
This set was subsequently filtered by evaluation of exome sequencing data coverage
and our recommended defaults as follows: by applying the default criteria of variant
on May 15, 2021. © 2020 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 6, 2020; DOI: 10.1158/2326-6066.CIR-19-0401

allele fraction (VAF) cutoff of > 25% in tumors and < 2% in normal samples, with at least
10X tumor coverage and at least 5X normal coverage, 10,730 neoantigens from 4,891
associated variants (4,826 SNVs, 56 frameshift, and 9 in-frame indels) were obtained,
with an average of 36 neoantigens predicted per case. Since RNAseq data also were
available, the filtering criteria included RNA-based coverage filters (tumor RNA VAF >
25% and tumor RNA coverage > 10X) as well as a gene expression filter (FPKM > 1).
To condense the results even further, only the top ranked neoantigen was selected per
variant (‘top-score filter’) across all alleles, lengths, and registers (position of amino acid
mutation within peptide sequence), resulting in 4,891 total neoantigens with an average
of 16 neoantigens per case. This list was then processed with pVACvector to determine
the optimum arrangement of the predicted high quality neoantigens for a DNA-vector
based vaccine design (Fig. 3).
Correlations and bias among binding prediction tools
Since we offer support for as many as eight different class I binding prediction tools, we
assessed agreement in binding affinity predictions (IC50) between these algorithms
from a random subset of 100,000 neoantigen peptides from our TCGA analysis,
described above (Fig. 4). The highest correlation was observed between the two
algorithms that are based on a stabilization matrix method (SMM)— SMM and
SMMPMBEC. The next best correlation was observed between NetMHC and
MHCflurry, possibly due to both being allele-specific predictors employing neural
network-based models. Overall the correlation between prediction algorithms is low
(mean correlation of 0.388 and range of 0.18-0.89 between all pairwise comparisons of
on May 15, 2021. © 2020 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 6, 2020; DOI: 10.1158/2326-6066.CIR-19-0401

algorithms).
We also evaluated if there were any biases among the algorithms to predict strong
binding epitopes (i.e. binding affinity < = 500 nM) by plotting the number of predicted
peptides based on their respective strong-predicting algorithms (Fig. 5 and
Supplemental Fig. S3). We found that MHCnuggets (v2.2) predicted the highest number
of strong-binding candidates alone (Fig. 5A). Of the total number of strong binding
candidates predicted, 64.7% of these candidates were predicted by a single algorithm
(any one of the eight algorithms), 35.2% were predicted as strong-binders by two to
seven algorithms, and only 1.8% of the strong-binding candidates were predicted as
strong binders by the combination of all eight algorithms (Supplemental Fig. S3). In fact,
as shown in Figure 6, even if one (or more) algorithms predict a peptide to be a strong
binder, often another algorithm will disagree by a large margin, in some cases predicting
that same peptide as a very weak binder. This remarkable lack of agreement
underscores the potential value of an ensemble approach that considers multiple
algorithms.
Next we determined if the number of human HLA alleles supported by these eight
algorithms differed. As shown (Supplemental Fig. S4), MHCnuggets supports the
highest number of human HLA alleles.
Comparison of filtering criteria
Since pVACtools offers a multitude of ways to filter a list of predicted neoantigens, we
evaluated and compared the effect of each filter for selecting high quality neoantigen
on May 15, 2021. © 2020 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 6, 2020; DOI: 10.1158/2326-6066.CIR-19-0401

candidates. There were 14,599,993 unfiltered neoantigen candidates predicted by
pVACseq resulting from 64,422 VEP-annotated variants reported across 300 samples.
We first compared the effect of running pVACtools with the commonly used standard
binding score cutoff of <= 500 nM (parameters: -b 500) to the newly added allele-
specific score filter (parameters: -a). These cutoffs were, by default, applied to the
“median” binding score of all prediction algorithms. In total, 96,235 neoantigens
(average 320.8 per patient) were predicted using the 500 nM binding score cutoff
compared to 94,068 neoantigens (average 313.6 per patient) using allele-specific filters.
About 79% of neoantigens were shared between the two sets.
We also further narrowed these sets to include only those predictions where the default
“median” predicted binding affinities (<= 500 nM) were lower than each corresponding
median wildtype peptide affinity (parameters: -b 500 -c 1) (i.e. a binding affinity
mutant/wildtype ratio or differential agretopicity value indicating that the mutant version
of the peptide is a stronger binder) (21). Using the agretopicity value filter, 70,628
neoantigens (average 235.43 per patient) were predicted versus the previously reported
96,235 neoantigens without this filter.
We also evaluated the effect of the agretopicity value filter when applied to the set of
peptides filtered on “lowest” binding score of <= 500 nM (parameters: -b 500 -c 1 -m
lowest). This filter limits peptides to those where at least one of the algorithms predicts a
strong binder, instead of calculating a median score and requiring that to meet the 500
nM threshold (Figure 6). Using the lowest binding score filter resulted in an 11-fold
on May 15, 2021. © 2020 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 6, 2020; DOI: 10.1158/2326-6066.CIR-19-0401

increase in the number of candidates predicted (827,423 candidates, average 2,758.08
per patient). This approach may be useful for finding candidates in tumors with low
mutation burden (but may also presumably lead to a higher rate of false positives).
Using the median (default) binding affinity filtering criteria, we next applied coverage
and expression-based filters. First, we filtered using the recommended defaults, i.e.
greater than 5X normal DNA coverage, less than 2% normal VAF, greater than 10X
tumor RNA and DNA coverage and greater than 25% tumor RNA and DNA VAF, along
with FPKM > 1 for transcript level expression (parameters: --normal-cov 5 --tdna-cov 10
--trna-cov 10 --normal-vaf 0.02 --tdna-vaf 0.25 --trna-vaf 0.25 --expn-val 1). A total of
10,730 neoantigens were shortlisted across all samples with an average of 35
neoantigens per case. We then compared this set with slightly more stringent criteria
using tumor DNA and RNA VAF of 40% (parameters: --tdna-vaf 0.40 --trna-vaf 0.40).
This shortened our list of predicted neoantigens to 4,073 candidates with an average of
13 candidates per patient.
Demonstration of neoantigen analysis using pVACfuse
To demonstrate the potential of neoantigens resulting from gene fusions, we analyzed
TCGA prostate cancer RNA-seq data from 302 patients. This dataset was previously
used as a demonstration set for the fusion neoantigen prediction supported by
INTEGRATE-Neo(22). We wanted to assess the difference (if any) in neoantigen
candidates reported by INTEGRATE-Neo using the one MHC Class I prediction
on May 15, 2021. © 2020 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 6, 2020; DOI: 10.1158/2326-6066.CIR-19-0401

algorithm it supports (NetMHC) versus an ensemble of eight Class I prediction
algorithms supported in pVACfuse.
Using 1,619 gene fusions across 302 samples as input, pVACfuse reported 2,104
strong binding neoantigens (binding affinity <= 500nM) resulting from 739 gene fusions.
On average, there were about 7 neoantigens per sample resulting from an average of 2
fusions per case. This is an eightfold increase in the number of strong binding
neoantigens predicted by pVACfuse versus those reported by INTEGRATE-Neo alone,
which reported 261 neoantigens across 210 fusions.
Validation of pVACtools results
To assess the real-world validity of pVACtools results, we reanalyzed raw sequencing
data from published studies that had performed immunological validation of candidate
neoantigens (Supplemental Tables S2, S3). Analysis of a melanoma patient (pt3713)
(68) showed that pVACtools was able to recapitulate eight (80%) of the positively
validated peptides in the filtered list of neoantigens using our suggested filtering
methodology (median binding predicted IC50 < 500 nM, > 5x DNA & RNA coverage, >
10% DNA & RNA VAF, > 1 TPM gene expression). Two of the positively validated
peptides were filtered out due to low RNA depth & VAF values but had median binding
affinity predictions of 13.707 nM and 4.799 nM. We were also able to eliminate 68% of
negatively validated peptides using these filters. In an additional analysis of a chronic
lymphocytic leukemia patient (pt5002) (69), we were able to recapitulate two (100%) of
the positively validated peptides in our filtered list of neoantigens using our suggested
on May 15, 2021. © 2020 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 6, 2020; DOI: 10.1158/2326-6066.CIR-19-0401

filtering methodology (median binding predicted IC50 < 500 nM, > 5x DNA coverage, >
10% DNA VAF). No RNA data was available for this sample. We were also able to
eliminate 57% of negatively validated peptides using these filters.
Discussion
As reported from our demonstration analysis, a typical tumor has too many possible
neoantigen candidates to be practical for a vaccine. There is therefore a critical need for
a tool that takes in the input from a tumor sequencing analysis pipeline and reports a
filtered and prioritized list of neoantigens. pVACtools enables a streamlined, accurate,
and user-friendly analysis of neoantigenic peptides from NGS cancer datasets. This
suite offers a complete and easily configurable end-to-end analysis, starting from
somatic variants and gene fusions (pVACseq and pVACfuse respectively), through
filtering, prioritization, and visualization of candidates (pVACviz), and determining the
best arrangement of candidates for a DNA vector vaccine (pVACvector). By supporting
additional classes of variants as well as gene fusions, we predict an increased number
of predicted neoantigens which may be critical for low mutational burden tumors.
Finally, by extending support for multiple binding prediction algorithms, we allow for a
consensus approach. The need for this integrated approach is made abundantly clear
by the disagreement between the candidate neoantigens identified by these algorithms,
as observed in our demonstration analyses.
To support a wider range of variant events not easily representable in VCF format such
as alternatively spliced transcripts that may create tumor antigens(65), upcoming
on May 15, 2021. © 2020 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 6, 2020; DOI: 10.1158/2326-6066.CIR-19-0401

versions of pVACtools will add a new tool, pVACbind, which can be used to execute our
prediction pipeline from sequences contained in a FASTA input file. Another major
planned feature is the addition of reference proteome similarity assessment of predicted
peptides in order to exclude peptides that are not true neoantigens because they occur
in other parts of the proteome. Upcoming versions will also include the calculation of
various metrics for peptide manufacturability for use in synthetic long peptide-based
vaccines. In addition, we are planning on adding binding affinity percentile ranks to our
prediction outputs for those prediction algorithms that support it. This would also allow
us to support additional IEDB prediction algorithms, such as Combinatorial Library(66),
and Sturniolo(67). The IEDB software added support for various class II epitope lengths
starting with version 2.22. We plan on updating pVACtools in an upcoming version to
take advantage of this new functionality. Lastly, we are working on a common workflow
language (CWL) immunotherapy pipeline that will execute all steps from alignment,
somatic and germline variant calling, HLA-typing, and RNAseq analysis including fusion
calling, followed by running pVACseq and pVACfuse. This will support users that would
like a full end-to-end solution not supported by pVACtools itself.
The results from pVACtools analyses are already being used in dozens of cancer
immunology studies, including studying the relationship between tumor mutation burden
and neoantigen load to predict response to immunotherapies(63,70,71), and the design
of cancer vaccines in ongoing clinical trials (e.g. NCT02348320, NCT03121677,
NCT03122106, NCT03092453, and NCT03532217). Whether the predicted
neoantigens will induce T-cell responses remains an open question for the field(72–74).
Pipelines like pVACtools are meant to assist these groups to obtain a prioritized set of
on May 15, 2021. © 2020 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 6, 2020; DOI: 10.1158/2326-6066.CIR-19-0401

neoantigen candidates from amongst a myriad of possible epitope predictions. Pursuant
to that, they should perform additional functional validations according to the standards
in this field(73). Whereas it may not be possible to evaluate a de novo response to all
predicted candidate neoantigens prior to vaccination, we highly recommend adding a
similar validation step as part of the clinical workflow to determine those candidates that
have a pre-existing immune response. Validation data from the studies currently using
pVACtools will be incorporated into our toolkit as they become available, and we hope
that over time this will improve the prioritization process. We anticipate that pVACtools
will make such analyses more robust, reproducible, and facile as these efforts continue.
Acknowledgements
We thank the patients and their families for donation of their samples and participation
in clinical trials. We also thank our growing user community for testing the software and
providing useful input, critical bug reports, as well as suggestions for improvement and
new features. We are grateful to Drs. Robert Schreiber, Gavin Dunn and Beatriz
Carreno for their expertise and guidance on foundational work on cancer immunology
using neoantigens and suggestions on improving the software. Obi Griffith was
supported by the National Cancer Institute (NCI) of the National Institutes of Health
(NIH) under Award Numbers U01CA209936, U01CA231844 and U24CA237719.
Malachi Griffith was supported by the National Human Genome Research Institute
(NHGRI) of the NIH under Award Number R00HG007940 and the V Foundation for
Cancer Research under Award Number V2018-007.
on May 15, 2021. © 2020 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 6, 2020; DOI: 10.1158/2326-6066.CIR-19-0401

Author contributions
JH and SK were involved in all aspects of this study including designing and developing
the methodology, and writing the manuscript, with help from CAM, ERM, OLG and MG.
JH analyzed and interpreted data with input from HX, CJL, SZ and Y-YF. SK wrote the
software code with help from ATW, APG, AZW, MN and JW. JM developed pVACviz
and pVACapi with assistance from APG and ATW. JN, MN, and CAM developed
pVACvector. WEG provided input about clinical needs for vaccine design to help
improve the tool’s features. OG and MG supervised the project and revised the paper.
All authors read and approved the final manuscript.
Competing interests
The authors declare no competing interests.
References
1. Liu XS, Shirley Liu X, Mardis ER. Applications of Immunogenomics to Cancer. Cell. 2017;168:600–12.
2. Bjerregaard A-M, Nielsen M, Hadrup SR, Szallasi Z, Eklund AC. MuPeXI: prediction of neo-epitopes from tumor sequencing data. Cancer Immunol Immunother [Internet]. 2017; Available from: http://dx.doi.org/10.1007/s00262-017-2001-3
3. Rubinsteyn A, Hodes I, Kodysh J, Hammerbacher J. Vaxrank: A Computational Tool For Designing Personalized Cancer Vaccines [Internet]. 2017. Available from: http://dx.doi.org/10.1101/142919
4. Bais P, Namburi S, Gatti DM, Zhang X, Chuang JH. CloudNeo: a cloud pipeline for identifying patient-specific tumor neoantigens. Bioinformatics. 2017;33:3110–2.
5. Andreatta M, Nielsen M. Gapped sequence alignment using artificial neural networks: application to the MHC class I system. Bioinformatics. 2016;32:511–7.
6. Jurtz V, Paul S, Andreatta M, Marcatili P, Peters B, Nielsen M. NetMHCpan-4.0: Improved
on May 15, 2021. © 2020 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 6, 2020; DOI: 10.1158/2326-6066.CIR-19-0401

Peptide–MHC Class I Interaction Predictions Integrating Eluted Ligand and Peptide Binding Affinity Data. The Journal of Immunology. 2017;199:3360–8.
7. Nielsen M, Lundegaard C, Worning P, Lauemøller SL, Lamberth K, Buus S, et al. Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci. 2003;12:1007–17.
8. Duan F, Duitama J, Al Seesi S, Ayres CM, Corcelli SA, Pawashe AP, et al. Genomic and bioinformatic profiling of mutational neoepitopes reveals new rules to predict anticancer immunogenicity. J Exp Med. 2014;211:2231–48.
9. Schubert B, Walzer M, Brachvogel H-P, Szolek A, Mohr C, Kohlbacher O. FRED 2: an immunoinformatics framework for Python. Bioinformatics. 2016;32:2044–6.
10. Rao AA, Madejska AA, Pfeil J, Paten B, Salama SR, Haussler D. ProTECT – Prediction of T-cell Epitopes for Cancer Therapy [Internet]. bioRxiv. 2019 [cited 2019 Dec 10]. page 696526. Available from: https://www.biorxiv.org/content/10.1101/696526v1.abstract
11. Hundal J, Kiwala S, Feng Y-Y, Liu CJ, Govindan R, Chapman WC, et al. Accounting for proximal variants improves neoantigen prediction. Nat Genet [Internet]. 2018; Available from: http://dx.doi.org/10.1038/s41588-018-0283-9
12. Turajlic S, Litchfield K, Xu H, Rosenthal R, McGranahan N, Reading JL, et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: a pan-cancer analysis. Lancet Oncol. 2017;18:1009–21.
13. Zamora AE, Crawford JC, Allen EK, Guo X-ZJ, Bakke J, Carter RA, et al. Pediatric patients with acute lymphoblastic leukemia generate abundant and functional neoantigen-specific CD8+ T cell responses. Sci Transl Med [Internet]. 2019;11. Available from: http://dx.doi.org/10.1126/scitranslmed.aat8549
14. Yang W, Lee K-W, Srivastava RM, Kuo F, Krishna C, Chowell D, et al. Immunogenic neoantigens derived from gene fusions stimulate T cell responses. Nat Med. 2019;25:767–75.
15. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60.
16. Koboldt DC, Larson DE, Wilson RK. Using VarScan 2 for Germline Variant Calling and Somatic Mutation Detection. Current Protocols in Bioinformatics. 2013. page 15.4.1–15.4.17.
17. Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, et al. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–76.
18. DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–8.
19. Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best
on May 15, 2021. © 2020 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 6, 2020; DOI: 10.1158/2326-6066.CIR-19-0401

practices pipeline. Curr Protoc Bioinformatics. 2013;43:11.10.1–33.
20. Charoentong P, Finotello F, Angelova M, Mayer C, Efremova M, Rieder D, et al. Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep. 2017;18:248–62.
21. Hoof I, Peters B, Sidney J, Pedersen LE, Sette A, Lund O, et al. NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics. 2009;61:1–13.
22. Karosiene E, Lundegaard C, Lund O, Nielsen M. NetMHCcons: a consensus method for the major histocompatibility complex class I predictions. Immunogenetics. 2011;64:177–86.
23. Zhang H, Lund O, Nielsen M. The PickPocket method for predicting binding specificities for receptors based on receptor pocket similarities: application to MHC-peptide binding. Bioinformatics. 2009;25:1293–9.
24. Peters B, Sette A. 10.1186/1471-2105-6-132 [Internet]. BMC Bioinformatics. 2005. page 132. Available from: http://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-6-132
25. Kim Y, Sidney J, Pinilla C, Sette A, Peters B. Derivation of an amino acid similarity matrix for peptide: MHC binding and its application as a Bayesian prior. BMC Bioinformatics. 2009;10:394.
26. O’Donnell TJ, Rubinsteyn A, Bonsack M, Riemer AB, Laserson U, Hammerbacher J. MHCflurry: Open-Source Class I MHC Binding Affinity Prediction. Cell Syst. 2018;7:129–32.e4.
27. Bhattacharya R, Sivakumar A, Tokheim C, Guthrie VB, Anagnostou V, Velculescu VE, et al. Evaluation of machine learning methods to predict peptide binding to MHC Class I proteins [Internet]. 2017. Available from: http://dx.doi.org/10.1101/154757
28. Nielsen M, Lundegaard C, Blicher T, Peters B, Sette A, Justesen S, et al. Quantitative Predictions of Peptide Binding to Any HLA-DR Molecule of Known Sequence: NetMHCIIpan [Internet]. PLoS Computational Biology. 2008. page e1000107. Available from: http://dx.doi.org/10.1371/journal.pcbi.1000107
29. Nielsen M, Lundegaard C, Lund O. Prediction of MHC class II binding affinity using SMM-align, a novel stabilization matrix alignment method. BMC Bioinformatics. BioMed Central; 2007;8:238.
30. Nielsen M, Lund O. NN-align . An artificial neural network-based alignment algorithm for MHC class II peptide binding prediction. BMC Bioinformatics. BioMed Central; 2009;10:1–10.
31. Shao XM, Bhattacharya R, Huang J, Sivakumar IKA, Tokheim C, Zheng L, et al. High-throughput prediction of MHC Class I and Class II neoantigens with MHCnuggets [Internet]. Available from: http://dx.doi.org/10.1101/752469
32. Fritsch EF, Rajasagi M, Ott PA, Brusic V, Hacohen N, Wu CJ. HLA-binding properties of tumor neoepitopes in humans. Cancer Immunol Res. 2014;2:522–9.
33. Peter Amstutz, Michael R. Crusoe, Nebojša Tijanić (editors), Brad Chapman, John Chilton,
on May 15, 2021. © 2020 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 6, 2020; DOI: 10.1158/2326-6066.CIR-19-0401

Michael Heuer, Andrey Kartashov, Dan Leehr, Hervé Ménager, Maya Nedeljkovich, Matt Scales, Stian Soiland-Reyes, Luka Stojanovic. Common Workflow Language, v1.0. 2016; Available from: http://dx.doi.org/10.6084/m9.figshare.3115156.v2
34. Voss K GJAV der AG. Full-stack genomics pipelining with GATK4 + WDL + Cromwell [version 1; not peer reviewed]. F1000Res [Internet]. 2017; Available from: http://dx.doi.org/10.7490/f1000research.1114631.1
35. Cabanski CR, Magrini V, Griffith M, Griffith OL, McGrath S, Zhang J, et al. cDNA hybrid capture improves transcriptome analysis on low-input and archived samples. J Mol Diagn. 2014;16:440–51.
36. Li H E al. The Sequence Alignment/Map format and SAMtools. - PubMed - NCBI [Internet]. [cited 2018 Dec 19]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19505943
37. McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GRS, Thormann A, et al. The Ensembl Variant Effect Predictor [Internet]. 2016. Available from: http://dx.doi.org/10.1101/042374
38. Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31:213–9.
39. Saunders CT, Wong WSW, Swamy S, Becq J, Murray LJ, Cheetham RK. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics. 2012;28:1811–7.
40. Ye K, Schulz MH, Long Q, Apweiler R, Ning Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009;25:2865–71.
41. Tan A, Abecasis GR, Kang HM. Unified representation of genetic variants. Bioinformatics. 2015;31:2202–4.
42. Roehr JT, Dieterich C, Reinert K. Flexbar 3.0 - SIMD and multicore parallelization. Bioinformatics. 2017;33:2941–2.
43. Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12:357–60.
44. Pertea M, Pertea GM, Antonescu CM, Chang T-C, Mendell JT, Salzberg SL. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33:290–5.
45. Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol. 2016;34:525–7.
46. Szolek A, Schubert B, Mohr C, Sturm M, Feldhahn M, Kohlbacher O. OptiType: precision HLA typing from next-generation sequencing data. Bioinformatics. 2014;30:3310–6.
47. Hundal J, Carreno BM, Petti AA, Linette GP, Griffith OL, Mardis ER, et al. pVAC-Seq: A genome-guided in silico approach to identifying tumor neoantigens. Genome Med. 2016;8:11.
on May 15, 2021. © 2020 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 6, 2020; DOI: 10.1158/2326-6066.CIR-19-0401

48. Vita R, Zarebski L, Greenbaum JA, Emami H, Hoof I, Salimi N, et al. The Immune Epitope Database 2.0. Nucleic Acids Res. 2009;38:D854–62.
49. Paul S, Weiskopf D, Angelo MA, Sidney J, Peters B, Sette A. HLA Class I Alleles Are Associated with Peptide-Binding Repertoires of Different Size, Affinity, and Immunogenicity. The Journal of Immunology. 2013;191:5831–9.
50. Keşmir C, Nussbaum AK, Schild H, Detours V, Brunak S. Prediction of proteasome cleavage motifs by neural networks. Protein Eng. 2002;15:287–96.
51. Rasmussen M, Fenoy E, Harndahl M. Pan-Specific Prediction of Peptide–MHC Class I Complex Stability, a Correlate of T Cell Immunogenicity. The Journal of [Internet]. Am Assoc Immnol; 2016; Available from: http://www.jimmunol.org/content/197/4/1517.short
52. Zhang J, Mardis ER, Maher CA. INTEGRATE-neo: a pipeline for personalized gene fusion neoantigen discovery. Bioinformatics. 2017;33:555–7.
53. Murphy C, Elemento O. AGFusion: annotate and visualize gene fusions [Internet]. bioRxiv. 2016 [cited 2019 Sep 6]. page 080903. Available from: https://www.biorxiv.org/content/10.1101/080903v1.abstract
54. Haas BJ, Dobin A, Stransky N, Li B, Yang X, Tickle T, et al. STAR-Fusion: Fast and Accurate Fusion Transcript Detection from RNA-Seq [Internet]. bioRxiv. 2017 [cited 2019 Sep 25]. page 120295. Available from: https://www.biorxiv.org/content/10.1101/120295v1.abstract
55. Nicorici D, Şatalan M, Edgren H, Kangaspeska S, Murumägi A, Kallioniemi O, et al. FusionCatcher – a tool for finding somatic fusion genes in paired-end RNA-sequencing data [Internet]. bioRxiv. 2014 [cited 2019 Sep 25]. page 011650. Available from: https://www.biorxiv.org/content/10.1101/011650v1.abstract
56. Davidson NM, Majewski IJ, Oshlack A. JAFFA: High sensitivity transcriptome-focused fusion gene detection [Internet]. Available from: http://dx.doi.org/10.1101/013698
57. Kim D, Salzberg SL. TopHat-Fusion: an algorithm for discovery of novel fusion transcripts. Genome Biol. 2011;12:R72.
58. Schubert B, Kohlbacher O. Designing string-of-beads vaccines with optimal spacers. Genome Med. 2016;8:9.
59. Negahdaripour M, Nezafat N, Eslami M, Ghoshoon MB, Shoolian E, Najafipour S, et al. Structural vaccinology considerations for in silico designing of a multi-epitope vaccine. Infect Genet Evol. 2018;58:96–109.
60. Kirkpatrick S, Gelatt CD Jr, Vecchi MP. Optimization by simulated annealing. Science. 1983;220:671–80.
61. Miller A, Asmann Y, Cattaneo L, Braggio E, Keats J, Auclair D, et al. High somatic mutation and neoantigen burden are correlated with decreased progression-free survival in multiple myeloma. Blood Cancer J. Nature Publishing Group; 2017;7:e612.
62. Balachandran VP, Łuksza M, Zhao JN, Makarov V, Moral JA, Remark R, et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature. Nature
on May 15, 2021. © 2020 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 6, 2020; DOI: 10.1158/2326-6066.CIR-19-0401

Publishing Group; 2017;551:512.
63. Formenti SC, Rudqvist N-P, Golden E, Cooper B, Wennerberg E, Lhuillier C, et al. Radiotherapy induces responses of lung cancer to CTLA-4 blockade. Nat Med. Nature Publishing Group; 2018;24:1845.
64. Cancer Genome Atlas Research Network, Weinstein JN, Collisson EA, Mills GB, Shaw KRM, Ozenberger BA, et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet. 2013;45:1113–20.
65. Kahles A, Lehmann K-V, Toussaint NC, Hüser M, Stark SG, Sachsenberg T, et al. Comprehensive Analysis of Alternative Splicing Across Tumors from 8,705 Patients. Cancer Cell. 2018;34:211–24.e6.
66. Sidney J, Peters B, Frahm N, Brander C, Sette A. HLA class I supertypes: a revised and updated classification. BMC Immunol. 2008;9:1.
67. Sturniolo T, Bono E, Ding J, Raddrizzani L, Tuereci O, Sahin U, et al. Generation of tissue-specific and promiscuous HLA ligand databases using DNA microarrays and virtual HLA class II matrices. Nat Biotechnol. 1999;17:555–61.
68. Prickett TD, Crystal JS, Cohen CJ, Pasetto A, Parkhurst MR, Gartner JJ, et al. Durable Complete Response from Metastatic Melanoma after Transfer of Autologous T Cells Recognizing 10 Mutated Tumor Antigens [Internet]. Cancer Immunology Research. 2016. page 669–78. Available from: http://dx.doi.org/10.1158/2326-6066.cir-15-0215
69. Rajasagi M, Shukla SA, Fritsch EF, Keskin DB, DeLuca D, Carmona E, et al. Systematic identification of personal tumor-specific neoantigens in chronic lymphocytic leukemia. Blood. 2014;124:453–62.
70. Johanns TM, Miller CA, Dorward IG, Tsien C, Chang E, Perry A, et al. Immunogenomics of Hypermutated Glioblastoma: A Patient with Germline POLE Deficiency Treated with Checkpoint Blockade Immunotherapy [Internet]. Cancer Discovery. 2016. page 1230–6. Available from: http://dx.doi.org/10.1158/2159-8290.cd-16-0575
71. Lauss M, Donia M, Harbst K, Andersen R, Mitra S, Rosengren F, et al. Mutational and putative neoantigen load predict clinical benefit of adoptive T cell therapy in melanoma. Nat Commun. 2017;8:1738.
72. Linette GP, Carreno BM. Neoantigen Vaccines Pass the Immunogenicity Test. Trends Mol. Med. 2017. page 869–71.
73. Vitiello A, Zanetti M. Neoantigen prediction and the need for validation [Internet]. Nature Biotechnology. 2017. page 815–7. Available from: http://dx.doi.org/10.1038/nbt.3932
74. The problem with neoantigen prediction [Internet]. Nature Biotechnology. 2017. page 97–97. Available from: http://dx.doi.org/10.1038/nbt.3800
Figure legends
on May 15, 2021. © 2020 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 6, 2020; DOI: 10.1158/2326-6066.CIR-19-0401

Figure 1: Overview of pVACtools workflow: The pVACtools workflow is highly
modularized and is divided into flexible components that can be run independently. The
main tools under the workflow include pVACseq for identifying and prioritizing
neoantigens from a variety of somatic alterations (red inset box); pVACfuse (green) for
detecting neoantigens resulting from gene fusions; pVACviz (blue) for process
management, visualization, and selection of results; and pVACvector (orange) for
optimizing design of neoantigens and nucleotide spacers in a DNA vector. All of these
tools interact via the pVACapi (purple), an OpenAPI HTTP REST interface to the
pVACtools suite.
Figure 2: pVACviz GUI client: pVACtools offers a browser-based graphical user
interface, pVACviz, which provides an intuitive means to launch pipeline processes,
monitor their execution, and analyze, export, or archive their results. To launch a
process, users navigate to the Start Page (A) and complete a form containing all of the
relevant inputs and settings for a pVACseq process. Each form field includes help text
and provides typeahead completion where applicable. For instance, the Alleles field
provides a typeahead dropdown menu that filters available alleles to only display those
alleles that match the text that the user has entered. Once a process is launched, a user
may monitor its progress on the Manage Page (B), which lists all running, stopped, and
completed processes. The Details Page (C) shows a process’ current log, attributes,
and any results files as well as provides controls for stopping, restarting, exporting, and
archiving the process. The results of pipeline processes may be analyzed on the
Visualize Page (D), which displays a customizable scatterplot of a file's rows. The X and
on May 15, 2021. © 2020 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 6, 2020; DOI: 10.1158/2326-6066.CIR-19-0401

Y axis may be set to any column in the result set, and filters may be applied to values in
any column. Additionally, points may be selected on the scatter plot or data grid (not
visible in this figure) for further analysis or export as CSV files.
Figure 3: An example pVACvector output showing the optimum arrangement of
candidate neoantigens for a DNA-vector based vaccine design. The figure depicts
a circularized DNA insert carrying 10 encoded neoantigenic peptide sequences to be
synthesized and encoded/cloned into a DNA plasmid. DNA sequences encoding each
peptide are ordered (with use of spacer sequences where needed) to ensure there are
no strong-binding junctional epitopes. Each neoantigenic peptide candidate is shown in
Blue, Green, Red, Orange, Purple, and Brown. In order to minimize junctional epitope
affinity, pVACvector adds spacer sequences where needed. These are depicted in
Black, along with the binding affinity value of the junctional epitope. Labels represent
the Gene Name and Amino Acid Change for each candidate.
Figure 4: Correlation between prediction algorithms: Spearman Correlation
between prediction values from all 8 class I prediction algorithms (MHCflurry,
MHCnuggets, NetMHC, NetMHCcons, NetMHCpan, PickPocket, SMM, and
SMMPMBEC) generated from a random subsample of 100,000 peptides.
Figure 5: Intersection of peptide sequences predicted by different algorithms
(MHCflurry, MHCnuggets, NetMHC, NetMHCcons, NetMHCpan, PickPocket, SMM,
and SMMPMBEC): The y-axis displays the number of overlapping unique neoantigenic
on May 15, 2021. © 2020 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 6, 2020; DOI: 10.1158/2326-6066.CIR-19-0401

peptides predicted for each combination of algorithm depicted on the x-axis. Each
collection of connected circles shows the set contained in an exclusive intersection (i.e.
the identity of each algorithm), while the light gray circles represent the algorithm(s) that
do not participate in this exclusive intersection. (A) Upset plot for the top 20 algorithm
combinations ranked by the number of peptides predicted to be a good binder (mutant
IC50 score < 500 nM). The combination of all eight algorithms (highlighted orange)
ranks 8th highest; (B) Upset plot for algorithm combinations where at least six
algorithms agree on predicting a peptide to be a good binder (MT IC50 score < 500 nM).
The combination of all eight algorithms (highlighted orange) ranks the highest.
Figure 6: Overall distribution of binding affinity scores (nM) for peptides where at
least one of the algorithms predicted a strong binder. Out of 14,599,993 peptides
predicted, 126,648 peptides were predicted to be strong binders by at least one of the
following algorithms: MHCflurry, MHCnuggets, NetMHC, NetMHCcons, NetMHCpan,
PickPocket, SMM, or SMMPMBEC. To define the set of peptides that were strong
binders according to at least one algorithm, HLA allele subtype-specific thresholds were
applied when available, otherwise the default cutoff binding affinity of 500 nM was used.
The peptides were further filtered using the default coverage-based filters (Normal
coverage > 5x and VAF < 2%, Tumor DNA coverage > 10x and VAF > 25%, Tumor
RNA coverage > 10x and VAF > 25%, Transcript expression > 1 fpkm). Peptides with
predicted MT IC50 scores lower than their respective cutoff scores are highlighted in
orange, while those higher are plotted in grey. The median MT IC50 scores of each
algorithm’s prediction (purple line) and the 500 nM binding affinity cutoff (red dotted line)
are indicated for reference.
on May 15, 2021. © 2020 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 6, 2020; DOI: 10.1158/2326-6066.CIR-19-0401

on May 15, 2021. © 2020 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 6, 2020; DOI: 10.1158/2326-6066.CIR-19-0401

on May 15, 2021. © 2020 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 6, 2020; DOI: 10.1158/2326-6066.CIR-19-0401

on May 15, 2021. © 2020 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 6, 2020; DOI: 10.1158/2326-6066.CIR-19-0401

on May 15, 2021. © 2020 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 6, 2020; DOI: 10.1158/2326-6066.CIR-19-0401

on May 15, 2021. © 2020 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 6, 2020; DOI: 10.1158/2326-6066.CIR-19-0401

on May 15, 2021. © 2020 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 6, 2020; DOI: 10.1158/2326-6066.CIR-19-0401

Published OnlineFirst January 6, 2020.Cancer Immunol Res Jasreet Hundal, Susanna Kiwala, Joshua McMichael, et al. cancer neoantigenspVACtools: a computational toolkit to identify and visualize
Updated version
10.1158/2326-6066.CIR-19-0401doi:
Access the most recent version of this article at:
Material
Supplementary
http://cancerimmunolres.aacrjournals.org/content/suppl/2020/01/04/2326-6066.CIR-19-0401.DC1
Access the most recent supplemental material at:
Manuscript
Authoredited. Author manuscripts have been peer reviewed and accepted for publication but have not yet been
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://cancerimmunolres.aacrjournals.org/content/early/2020/01/04/2326-6066.CIR-19-0401To request permission to re-use all or part of this article, use this link
on May 15, 2021. © 2020 American Association for Cancer Research. cancerimmunolres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 6, 2020; DOI: 10.1158/2326-6066.CIR-19-0401