
Purification of a Sarcoplasmic Reticulum Protein That Binds Ca2 ...
11
THE JOURNAL 0 1989 by The American Society for Biochemistry and Molecular Biology, Inc. OF BIOLOGICAL CHEMISTRY Vol. 264, No. 14, Issue of May 15, pp. 8260-8270,1989 Printed in U.S.A. Purification of a Sarcoplasmic Reticulum Protein That Binds Ca2’ and Plasma Lipoproteins* (Received for publication, December 2, 1988) Sandra L. Hofmann$$,Michael S. Brown$, Ethan LeeST, Ravindra K. PathakII, Richard G. W. Anderson11 , and Joseph L. Goldstein$ From the Departments of $Molecular Genetics, $Internal Medicine, and IICell Biology, University of Texas Southwestern Medical Center, Dallas, Texas 75235 A protein in the sarcoplasmic reticulum of rabbit skeletal and cardiac muscle was identified because of its ability to bind ‘251-labeled low density lipoprotein (LDL) with high affinity aftersodium dodecyl sulfate- polyacrylamide gel electrophoresis. This protein, re- ferred to as the 165-kDa protein, is restricted to striated muscle. It was not detected in 14 other tissues, including several that contain smooth muscle, but it appears in rat L6 myoblasts when they differentiate into myocytes. Immunofluorescence and immunoelec- tron microscopic studies revealed that the protein is present throughout the sarcoplasmic reticulum and the terminal cisternae. It binds 4sCa2+ on nitrocellulose blots and stains metachromatically with Stains-all, a cationic dye that stains Ca2+-binding proteins. It does not appear to be a glycoprotein, and it appears slightly larger than the 160-kDa glycoprotein previously de- scribed in sarcoplasmic reticulum. The 165-kDa pro- tein binds LDL, &migrating very low density lipopro- tein, and a cholesterol-induced high density lipoprotein particle that contains apoprotein E as its sole apopro- tein with much higher affinity than it binds high den- sity lipoprotein. The protein is stable to boiling and to treatment with sodium dodecylsulfate, but it becomes sensitive to these treatments when its cystine residues are reduced and alkylated. The protein was purified 1300-fold to apparent homogeneity from rabbit skel- etal muscle membranes. It differs from the cell surface LDL receptor in that 1) its apparentmolecular weight is not changed by reduction and alkylation; 2) it is present in Watanabe-heritable hyperlipidemic rabbits, which lack functional LDL receptors; 3) binding of lipoproteins is not inhibited byEDTA; and 4) it is located within the lumen of the sarcoplasmic reticulum where it has no access to plasma lipoproteins. It is unlikely that this protein ever binds lipoproteins in vivo; however, its lipoprotein binding activity has fa- cilitated its purification to homogeneity and suggests that this protein hasunusual structural features. The role of the 165-kDa protein in Ca2+ homeostasis in the sarcoplasmic reticulum, if any, remains to be deter- mined. * This work was supported in part by Grant HL 20948 from the National Institutes of Health and a research grant from the Perot Family Foundation. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “aduertisement” in accordance with 18 U.S.C. Section 1734 solelyto indicate this fact. § Recipient of American Heart Association-Squibb Clinician Sci- entist Award 86-412. ll An M.D./Ph.D. student supported by a Medical Scientist Train- ing Grant GM 08014 from the National Institutes of Health. The sarcoplasmic reticulum of striated muscle is an exten- sive network of membrane tubules and cisternae which stores Ca2+and releases it rapidly into the cytoplasm upon mem- brane depolarization. The sarcoplasmic reticulum contains terminal cisternae that form close contacts with the trans- verse tubules, which are invaginations of the plasma mem- brane (1). The terminal cisternae contain an abundant soluble protein, calsequestrin, that binds Ca2+and releases it upon depolarization (1). The other identified components of the sarcoplasmic reticulum are all attached to the membrane. These include a CaZ+-ATPase (Mr 110,000) that pumps Ca2+ into the lumen during the resting phase (2) and a 450,000- dalton protein that binds ryanodine, a plant alkaloid (3). The latter is believed to form a Ca2+ channel in the membrane of the sarcoplasmic reticulum. The ryanodine-binding protein differs from the dihydropyridine-binding protein, which is located in the membrane of the transverse tubule and consti- tutes a voltage-sensitive Ca2+ channel (4). In the current paper, we report the identification and pu- rification of a new protein of the sarcoplasmic reticulum. This protein was identified because of an unusual property: it specifically binds plasma lipoproteins that contain apoprotein B and apoprotein E. These properties are similar to those of the cell surface low density lipoprotein (LDL)‘ receptor (5). Like the LDL receptor (6), the sarcoplasmic reticulum protein has an apparent molecular weight of 160,000-170,000 when subjected to electrophoresis under reducing conditions on sodium dodecyl sulfate (SDS)-polyacrylamide gels. Both pro- teins retain the ability to bind LDL after boiling and after SDS-polyacrylamide gel electrophoresis as long as the cystine residues are not reduced or alkylated. The sarcoplasmic retic- ulum protein (referred to as the 165-kDa muscle protein) differs from the LDL receptor in that binding is not sensitive to EDTA. Immunofluorescence and immunoelectron micros- copy studies demonstrate that this protein is located within the lumen of the sarcoplasmic reticulum where it has no access to plasma lipoproteins. It is therefore unlikely to func- tion as a lipoprotein receptor. Nevertheless, its lipoprotein binding properties have permitted the characterization of this protein and its purification from skeletal muscle. EXPERIMENTALPROCEDURES Materials-We obtained phenylmethylsulfonyl fluoride, leupeptin, pepstatin, aprotinin, cyclohexanedione, hydroxylamine, heparin (cat- alog no. H3125), bovine serum albumin (Fraction V, catalog no. A4503), neuraminidase, and Stains-all dye from Sigma; dextran sul- ’ The abbreviations used are: LDL, low density lipoprotein; SDS, sodium dodecyl sulfate; (3-VLDL, (3-migrating very low density lipo- protein; apo, apoprotein; HDL, high density lipoprotein; apoE-HDL,, a cholesterol-induced HDL particle that contains apoE as its sole apoprotein. 8260
Transcript of Purification of a Sarcoplasmic Reticulum Protein That Binds Ca2 ...

THE JOURNAL 0 1989 by The American Society for Biochemistry and Molecular Biology, Inc.
OF BIOLOGICAL CHEMISTRY Vol. 264, No. 14, Issue of May 15, pp. 8260-8270,1989 Printed in U.S.A.
Purification of a Sarcoplasmic Reticulum Protein That Binds Ca2’ and Plasma Lipoproteins*
(Received for publication, December 2, 1988)
Sandra L. Hofmann$$, Michael S . Brown$, Ethan LeeST, Ravindra K. PathakII, Richard G . W. Anderson11 , and Joseph L. Goldstein$ From the Departments of $Molecular Genetics, $Internal Medicine, and IICell Biology, University of Texas Southwestern Medical Center, Dallas, Texas 75235
A protein in the sarcoplasmic reticulum of rabbit skeletal and cardiac muscle was identified because of its ability to bind ‘251-labeled low density lipoprotein (LDL) with high affinity after sodium dodecyl sulfate- polyacrylamide gel electrophoresis. This protein, re- ferred to as the 165-kDa protein, is restricted to striated muscle. It was not detected in 14 other tissues, including several that contain smooth muscle, but it appears in rat L6 myoblasts when they differentiate into myocytes. Immunofluorescence and immunoelec- tron microscopic studies revealed that the protein is present throughout the sarcoplasmic reticulum and the terminal cisternae. It binds 4sCa2+ on nitrocellulose blots and stains metachromatically with Stains-all, a cationic dye that stains Ca2+-binding proteins. It does not appear to be a glycoprotein, and it appears slightly larger than the 160-kDa glycoprotein previously de- scribed in sarcoplasmic reticulum. The 165-kDa pro- tein binds LDL, &migrating very low density lipopro- tein, and a cholesterol-induced high density lipoprotein particle that contains apoprotein E as its sole apopro- tein with much higher affinity than it binds high den- sity lipoprotein. The protein is stable to boiling and to treatment with sodium dodecyl sulfate, but it becomes sensitive to these treatments when its cystine residues are reduced and alkylated. The protein was purified 1300-fold to apparent homogeneity from rabbit skel- etal muscle membranes. It differs from the cell surface LDL receptor in that 1) its apparent molecular weight is not changed by reduction and alkylation; 2) it is present in Watanabe-heritable hyperlipidemic rabbits, which lack functional LDL receptors; 3) binding of lipoproteins is not inhibited by EDTA; and 4) it is located within the lumen of the sarcoplasmic reticulum where it has no access to plasma lipoproteins. It is unlikely that this protein ever binds lipoproteins in vivo; however, its lipoprotein binding activity has fa- cilitated its purification to homogeneity and suggests that this protein has unusual structural features. The role of the 165-kDa protein in Ca2+ homeostasis in the sarcoplasmic reticulum, if any, remains to be deter- mined.
* This work was supported in part by Grant HL 20948 from the National Institutes of Health and a research grant from the Perot Family Foundation. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “aduertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
§ Recipient of American Heart Association-Squibb Clinician Sci- entist Award 86-412.
ll An M.D./Ph.D. student supported by a Medical Scientist Train- ing Grant GM 08014 from the National Institutes of Health.
The sarcoplasmic reticulum of striated muscle is an exten- sive network of membrane tubules and cisternae which stores Ca2+ and releases it rapidly into the cytoplasm upon mem- brane depolarization. The sarcoplasmic reticulum contains terminal cisternae that form close contacts with the trans- verse tubules, which are invaginations of the plasma mem- brane (1). The terminal cisternae contain an abundant soluble protein, calsequestrin, that binds Ca2+ and releases it upon depolarization (1). The other identified components of the sarcoplasmic reticulum are all attached to the membrane. These include a CaZ+-ATPase (Mr 110,000) that pumps Ca2+ into the lumen during the resting phase (2) and a 450,000- dalton protein that binds ryanodine, a plant alkaloid (3). The latter is believed to form a Ca2+ channel in the membrane of the sarcoplasmic reticulum. The ryanodine-binding protein differs from the dihydropyridine-binding protein, which is located in the membrane of the transverse tubule and consti- tutes a voltage-sensitive Ca2+ channel (4).
In the current paper, we report the identification and pu- rification of a new protein of the sarcoplasmic reticulum. This protein was identified because of an unusual property: it specifically binds plasma lipoproteins that contain apoprotein B and apoprotein E. These properties are similar to those of the cell surface low density lipoprotein (LDL)‘ receptor (5). Like the LDL receptor (6), the sarcoplasmic reticulum protein has an apparent molecular weight of 160,000-170,000 when subjected to electrophoresis under reducing conditions on sodium dodecyl sulfate (SDS)-polyacrylamide gels. Both pro- teins retain the ability to bind LDL after boiling and after SDS-polyacrylamide gel electrophoresis as long as the cystine residues are not reduced or alkylated. The sarcoplasmic retic- ulum protein (referred to as the 165-kDa muscle protein) differs from the LDL receptor in that binding is not sensitive to EDTA. Immunofluorescence and immunoelectron micros- copy studies demonstrate that this protein is located within the lumen of the sarcoplasmic reticulum where it has no access to plasma lipoproteins. It is therefore unlikely to func- tion as a lipoprotein receptor. Nevertheless, its lipoprotein binding properties have permitted the characterization of this protein and its purification from skeletal muscle.
EXPERIMENTAL PROCEDURES
Materials-We obtained phenylmethylsulfonyl fluoride, leupeptin, pepstatin, aprotinin, cyclohexanedione, hydroxylamine, heparin (cat- alog no. H3125), bovine serum albumin (Fraction V, catalog no. A4503), neuraminidase, and Stains-all dye from Sigma; dextran sul-
’ The abbreviations used are: LDL, low density lipoprotein; SDS, sodium dodecyl sulfate; (3-VLDL, (3-migrating very low density lipo- protein; apo, apoprotein; HDL, high density lipoprotein; apoE-HDL,, a cholesterol-induced HDL particle that contains apoE as its sole apoprotein.
8260

Lipoprotein-binding Protein from Sarcoplasmic Reticulum 8261
fate from Pharmacia LKB Biotechnology Inc.; suramin from Mobay Chemical Corp.; Triton X-100, endoglycosidase F (from Flauobacter- ium meningosepticum), and glycopeptidase F (from F. meningosepti- cum) from Boehringer Mannheim; endo-a-N-acetylgalactosamini- dase (0-glycanase) from Genzyme Corp.; nitrocellulose (BA85) from Schleicher & Schuell; CNBr-activated Sepharose from Pharmacia; 1,4-diazobicyclo[2.2.2]octane from Aldrich; rabbit anti-sheep IgG conjugated to fluorescein isothiocyanate and affinity-purified rabbit anti-mouse IgG from Zymed (San Francisco, CA); goat anti-rabbit IgG labeled with 10-nm gold particles from EBTEC (Agawam, MA); rabbit anti-sheep IgG from Cappell (Malvern, PA); and 45CaC12 (38 Ci/g) from Du Pont-New England Nuclear. Cultured L6 rat myoblasts (7) were kindly provided by Woodring Wright (University of Texas Southwestern Medical Center, Dallas, TX). Human LDL (d 1.019- 1.063 g/ml) (8) , human HDL3 (d 1.125-1.215 g/ml) (81, and rabbit p - VLDL (d < 1.006 g/ml) (9) were prepared by ultracentrifugation as described in the indicated reference and radiolabeled by the iodine monochloride method (8). Cyclohexanedione was covalently attached to Iz5I-LDL and released with hydroxylamine as described previously (10). Canine '251-apoE-HDL, was kindly provided by Thomas L. Innerarity (Gladstone Research Foundation Laboratories, San Fran- cisco, CA). Bovine LDL receptor was purified as described previously (11). New Zealand White rabbits (2.5-4 kg) were purchased from Hickory Hill Rabbitry (Flint, TX). Homozygous Watanabe-heritable hyperlipidemic rabbits (2.5-4 kg) were raised in Dallas, TX (12). Mice (C57BL X SJL) were purchased from Jackson Laboratories. A mouse hybridoma secreting monoclonal antibody MB47 directed against the receptor binding domain of human apoB (13) was kindly provided by Linda K. Curtiss (Research Institute of Scripps Clinic, La Jolla, CA) and Joseph L. Witztum (University of California, San Diego, La Jolla, CA). IgG fractions of MB47 and monoclonal antibody 2001 (directed against an irrelevant antigen) (14) were isolated from ascites fluid on protein A-Sepharose columns (14).
Detergent Solubilization of Membranes-This procedure was per- formed essentially as described by Schneider et al. (15) except that Triton X-100 was used instead of octylglucoside and the following protease inhibitors were present throughout the homogenization and solubilization: 1 mM phenylmethylsulfonyl fluoride, 0.1 mM leupeptin, 1 pg/ml pepstatin, 1 mM 1,lO-phenanthroline, and 0.5 pg/ml apro- tinin. Briefly, one rabbit of either sex was killed by lethal injection of sodium pentobarbital, and the hind legs and back muscles were removed. The muscle (200 g) was dissected free of adherent fatty and connective tissue on ice and homogenized in 1 liter of ice-cold buffer containing 20 mM Tris-HC1 at pH 8.0, 1 mM CaC12, 150 mM NaCl, and the protease inhibitors listed above in a Brinkmann homogenizer (Brinkmann Instruments) for 30 s on setting 5 and for 30 s on setting 7 using a large probe. The homogenate was then centrifuged at 4,000 rpm (2,000 X g) for 10 min at 4 'C in a Sorvall Superspeed centrifuge in an SA600 rotor. The supernatant fraction was filtered through wet Miracloth (Calbiochem) and centrifuged at 30,000 rpm (100,000 X g) for 1 h at 4 "C in a Sorvall A641 rotor. The pellets were resuspended in 108 ml of buffer containing 250 mM Tris maleate at pH 6.0, 2 mM CaCIz, and protease inhibitors by repeated aspiration through a 22- gauge needle. The membranes were solubilized by the addition of 8.64 ml of 4 M NaCl followed sequentially by brief sonication and the addition of 56.2 ml of distilled water and 43.2 ml of 5% (v/v) Triton X-100. The extract was stirred for 10 min on ice and centrifuged at 30,000 rpm (100,000 X g) for 1 h at 4 "C. The supernatant fraction was then quick-frozen in liquid nitrogen. lZ5I-LDL binding activity of this preparation was stable for at least 1 week at -70 "C.
Heat Treatment and Chromatography of Membrane Extract-The membrane extract was thawed, adjusted to pH 8.0 with 1 M Tris base, and divided into several 40-ml aliquots in 50-ml polypropylene tubes. Each tube was placed in boiling water for 15 min, cooled on ice, and then centrifuged at 12,000 rpm (16,800 X g) for 10 min at 4 "C in a Sorvall Superspeed centrifuge. The supernatant was concentrated to 12-15 ml in an Amicon concentrator using a PM-30 membrane. The concentrated material was spun at 12,000 rpm for 10 min at 4 "C to remove particulate material and then applied to a 1-ml Mono Q 5/5 column (Pharmacia) that had been equilibrated in buffer containing 20 mM Tris-HC1 at pH 8.0, 1% (v/v) Triton X-100, 2 mM CaCl2, and 0.3 M NaCl in the absence of protease inhibitors. The column was washed with at least 5 column volumes of this buffer and eluted with a 20-ml NaCl gradient to a final concentration of 0.6 M NaCl. Fractions (0.5 ml) were collected and assayed for protein content (16) and lZ5I-LDL binding in the ligand blotting assay (see below). Active fractions were frozen in liquid nitrogen and stored at -70 "C for several months without loss of binding activity.
Ligand Blotting Assay-Ligand blotting with ''51-lipoproteins was performed as described previously (6) with several modifications. Briefly, SDS-polyacrylamide gel electrophoresis was carried out in 7% slab gels containing 0.1% (w/v) SDS. Just prior to electrophoresis, an equal volume of buffer containing 125 mM Tris-HCI at pH 6.8, 4.5% (w/v) SDS, 20% (v/v) glycerol, and 0.1% (w/v) bromphenol blue was added to the samples. The samples were not exposed to reducing agent and were not heated. Electrophoresis was carried out at 4 "C at 30 mA/gel for 3 h. Proteins were transferred from SDS slab gels to nitrocellulose paper as described (17) in a buffer containing 10 mM NaHC03 and 3 mM NaZCO3 at pH 9.9 in 20% (v/v) methanol. Transfers were typically carried out overnight at 4 "C. The filters were incubated with '"I-LDL at 5 pg of protein/ml or other lZ5I- labeled ligands as indicated in the figure legends. Binding was per- formed at 37 "C in a binding buffer containing 50 mM Tris-HC1 at pH 8.0, 1 mM potassium iodide, and 50 mg/ml bovine serum albumin and supplemented with either 2 mM CaC12 or 5 mM disodium EDTA. The binding reaction was carried out for 1 h with gentle agitation. The filters were then washed twice with binding buffer in the absence of 1'51-ligand for 15 min at room temperature and once briefly in binding buffer without albumin, dried, and exposed to Kodak XAR film as described in the figure legends. The following marker proteins were used to calibrate molecular weights: myosin, 205,000; p-galac- tosidase, 116,000; phosphorylase b, 97,500; bovine serum albumin, 68,000; and ovalbumin, 45,000.
Zmmunoblotting Assay-Nitrocellulose filters containing proteins transferred from SDS-polyacrylamide slab gels were prepared as described for the ligand blotting assay (see above). The filters were incubated for 1 h at room temperature with 1 pg/ml affinity-purified anti-165-kDa muscle protein antibody or 1 pg/ml preimmune anti- body in a blotting buffer as described (14), washed, and then probed with '251-labeled rabbit anti-sheep IgG (14), washed, and exposed to film as indicated in the legend to Fig. 12.
Preparation of Affinity-purified PolyclonalAntibodies-A sheep was immunized with 2.2 mg of the purified 165-kDa muscle protein in Freund's complete adjuvant, boosted 1 month later with another 2.2 mg of the purified protein in Freund's incomplete adjuvant, and bled 1 month later. A y-globulin fraction was prepared from preimmune or immune serum by sequential ammonium sulfate precipitation (18), and the immune antibody was further purified by specific absorption to a column consisting of the purified 165-kDa muscle protein coupled to CNBr-activated Sepharose (5 mg of 165-kDa protein/ml gel), which was prepared according to the directions supplied by the manufacturer except that 0.5% Triton X-100 was present in the coupling buffer. A total of 91 mg of crude antibody (y-globulin fraction) was passed over 0.5 ml of gel, and the flow-through was collected for use as the nonimmune antibody control. The bound antibody was then washed on the column with 10 mM Tris-HC1 a t pH 7.7, 150 mM NaCI, 1 mM EDTA, eluted in 1 ml of 0.1 M ammonium hydroxide, pH 10.7, neutralized with glacial acetic acid, and immediately lyophilized to yield 5 mg of antibody of protein. Two mg of this protein was further purified by absorption against rabbit calsequestrin-Sepharose (1 mg/ 0.3 ml) prepared as above, yielding 1 mg of antibody protein that did not immunoblot rabbit calsequestrin.' This final antibody is referred to below as the "affinity-purified antibody."
Immunofluorescence Microscopy-Rabbits were perfused through the aortic arch with Ca2+- and MF-free Dulbecco's phosphate- buffered saline at a flow rate of -50 ml/min for 30 min followed by 3% paraformaldehyde, 6 mM trinitrophenol, 4 mM KCI, and 2 mM MgCL in 37.5 mM sodium phosphate buffer at pH 8. Tissue samples of cardiac (atrium) and skeletal (triceps) muscle were removed and fixed overnight in a modified methacarn fixative (19) that contained 2% (v/v) trichloroacetic acid, 8% (v/v) glacial acetic acid, 30% (v/v) l,l,l-trichloroethane, 60% (v/v) absolute methanol, dehydrated in isopropyl alcohol, cleared in xylenes, and embedded in paraffin. Sections of -6-8 pm were cut using a stainless steel knife, mounted on glass slides, dried overnight in a 50 "C oven, removed from paraffin using xylenes, and rehydrated in distilled water. Indirect immunoflu- orescence was carried out with the following sequential incubations using antibodies diluted to the indicated concentration with buffer A
Purified calsequestrin was obtained during the purification of the 165-kDa muscle protein. A fraction eluting from the Mono Q column at 0.5 M NaCl (see "Experimental Procedures") contained a protein that migrated as a single band on SDS-polyacrylamide gel electro- phoresis ( M , -63,000). This protein was shown to be authentic rabbit calsequestrin (26) by amino acid sequence analysis of the N terminus (40 residues) and five tryptic peptides (17-40 residues in length).
8262 Lipoprotein-binding Protein from Sarcoplasmic Reticulum
(20 mM Tris-HC1 at pH 8.5, 200 mM NaC1, 0.1% (w/v) bovine serum albumin, 0.05% (w/v) saponin, and 0.01% (w/v) NaN3): 1) a 30-min incubation with no antibodies in buffer A; 2) overnight incubation with either 1 pg/ml affinity-purified antibody directed against the 165-kDa muscle protein, 25 pg/ml nonimmune antibody (flow- through from affinity column), or 25 pg/ml preimmune antibody; and 3) a 2-h incubation with 25 pg/ml rabbit anti-sheep IgG conjugated to fluorescein isothiocyanate. Sections were washed three times for 5
1251 - apoE H DLc
+ Ca++
+EDTA
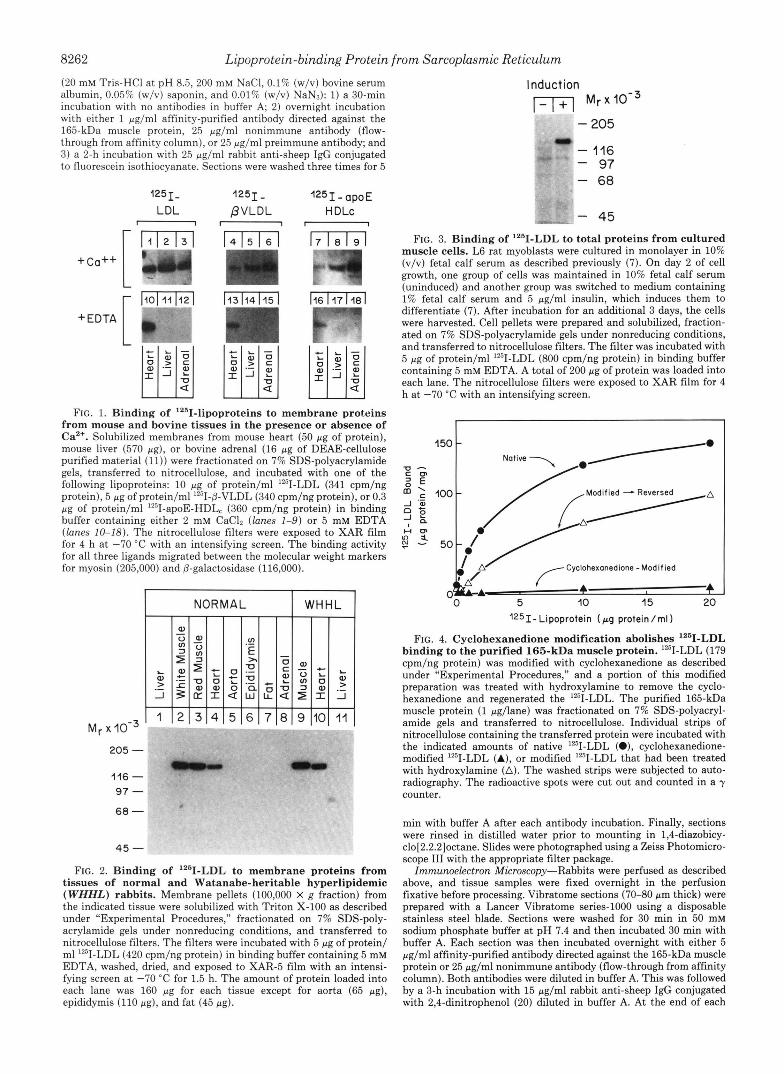
FIG. 1. Binding of 1251-lipoproteins to membrane proteins from mouse and bovine tissues in the presence or absence of Ca2+. Solubilized membranes from mouse heart (50 pg of protein), mouse liver (570 pg), or bovine adrenal (16 pg of DEAE-cellulose purified material (11)) were fractionated on 7% SDS-polyacrylamide gels, transferred to nitrocellulose, and incubated with one of the following lipoproteins: 10 pg of protein/ml lZ5I-LDL (341 cpm/ng protein), 5 pg of protein/m11Z51-@-VLDL (340 cpm/ng protein), or 0.3 pg of protein/ml 1251-apoE-HDL, (360 cpm/ng protein) in binding buffer containing either 2 mM CaClZ (lanes 1-9) or 5 mM EDTA (lanes 10-18). The nitrocellulose filters were exposed to XAR film for 4 h at -70 “C with an intensifying screen. The binding activity for all three ligands migrated between the molecular weight markers for myosin (205,000) and @-galactosidase (116,000).
NORMAL I W H H L 1
205 -
116 - 97 - 60 -
45 - FIG. 2. Binding of 12&I-LDL to membrane proteins from
tissues of normal and Watanabe-heritable hyperlipidemic (WHHL) rabbits. Membrane pellets (100,000 X g fraction) from the indicated tissue were solubilized with Triton X-100 as described under “Experimental Procedures,” fractionated on 7% SDS-poly- acrylamide gels under nonreducing conditions, and transferred to nitrocellulose filters. The filters were incubated with 5 pg of protein/ ml lZ5I-LDL (420 cpm/ng protein) in binding buffer containing 5 mM EDTA, washed, dried, and exposed to XAR-5 film with an intensi- fying screen at -70 “C for 1.5 h. The amount of protein loaded into each lane was 160 pg for each tissue except for aorta (65 pg), epididymis (110 pg), and fat (45 pg).
Induction 1 - 1 + 1 M,XIO-~
- 205 - 116 - 97 - 68
.I)
- 45 FIG. 3. Binding of ‘261-LDL to total proteins from cultured
muscle cells. L6 rat myoblasts were cultured in monolayer in 10% (v/v) fetal calf serum as described previously (7). On day 2 of cell growth, one group of cells was maintained in 10% fetal calf serum (uninduced) and another group was switched to medium containing 1% fetal calf serum and 5 pg/ml insulin, which induces them to differentiate (7). After incubation for an additional 3 days, the cells were harvested. Cell pellets were prepared and solubilized, fraction- ated on 7% SDS-polyacrylamide gels under nonreducing conditions, and transferred to nitrocellulose filters. The filter was incubated with 5 pg of protein/ml lZ5I-LDL (800 cpm/ng protein) in binding buffer containing 5 mM EDTA. A total of 200 pg of protein was loaded into each lane. The nitrocellulose filters were exposed to XAR film for 4 h at -70 “C with an intensifying screen.
1
I5O t Native -
kA Cyclohexanedione -Modified
A 0 ” c
0 5 10 15 20 1251-Lipoprotein (pg protein/rnI)
FIG. 4. Cyclohexanedione modification abolishes I2’1-LDL binding to the purified 165-kDa muscle protein. lZ5I-LDL (179 cpm/ng protein) was modified with cyclohexanedione as described under “Experimental Procedures,” and a portion of this modified preparation was treated with hydroxylamine to remove the cyclo- hexanedione and regenerated the lZ5I-LDL. The purified 165-kDa muscle protein (1 pg/lane) was fractionated on 7% SDS-polyacryl- amide gels and transferred to nitrocellulose. Individual strips of nitrocellulose containing the transferred protein were incubated with the indicated amounts of native lZ5I-LDL (0). cyclohexanedione- modified lZ5I-LDL (A), or modified lZ5I-LDL that had been treated with hydroxylamine (A). The washed strips were subjected to auto- radiography. The radioactive spots were cut out and counted in a y counter.
min with buffer A after each antibody incubation. Finally, sections were rinsed in distilled water prior to mounting in 1,4-diazobicy- clo[2.2.2]octane. Slides were photographed using a Zeiss Photomicro- scope 111 with the appropriate filter package.
Immunoelectron Microscopy-Rabbits were perfused as described above, and tissue samples were fixed overnight in the perfusion fixative before processing. Vibratome sections (70-80 pm thick) were prepared with a Lancer Vibratome series-1000 using a disposable stainless steel blade. Sections were washed for 30 min in 50 mM sodium phosphate buffer a t pH 7.4 and then incubated 30 min with buffer A. Each section was then incubated overnight with either 5 pg/ml affinity-purified antibody directed against the 165-kDa muscle protein or 25 pg/ml nonimmune antibody (flow-through from affinity column). Both antibodies were diluted in buffer A. This was followed by a 3-h incubation with 15 pg/ml rabbit anti-sheep IgG conjugated with 2,4-dinitrophenol (20) diluted in buffer A. At the end of each

Lipoprotein-binding Protein from Sarcoplasmic Reticulum 8263
TABLE I Purification of the 165-kDa protein from rabbit skeletal muscle
The starting homogenate was prepared from 200 g of skeletal muscle obtained from one rabbit. The data shown are from one preparation and are similar to results obtained in three other experiments. Although LDL binding activity was detectable in the crude homogenate, it could not be accurately quantified until after the 2000 X g centrifugation step. The resulting 2000 X g supernatant fraction is considered to be the starting material for the calculations presented in the table.
Fraction Total Total activity Specific protein activity Purification Yield
mg pg LDL bound -fold %
Homogenate 26,500 2000 X g supernatant 7,200 216 0.03 1 100 Solubilized membranes 1,760 312 0.18 5.9 144 Boiled and concentrated membranes 20 129 6.5 220 60 Mono Q chromatography 2.8 107 39.2 1,310 51
pg LDL bound1 mg protein
Mono Q Fractions
M r x I A I B I C 1 D 10142143144145(46147148
A- 205- 1 116 - 97 - 68 -
I I I I 1 1 1 1 1 l l l 1 B. 205-
116 - 97 - 68 -
45 -
FIG. 5. Purification of the 165-kDa protein from rabbit skeletal muscle. At various stages of purification (see Table I), aliquots of protein were subjected to electrophoresis on 7% SDS- polyacrylamide gels under nonreducing conditions. The proteins were either transferred to nitrocellulose for blotting with 5 pg of protein/ ml lz5I-LDL (664 cpm/ng protein) as described under "Experimental Procedures" ( A ) or stained directly with Coomassie Brilliant Blue ( B ) . The nitrocellulose filter was exposed to XAR-5 film for 1 h a t -70 "C with an intensifying screen. Lane A , 220 pg of protein of crude homogenate; lane B, 100 pg of the low speed supernatant; lane C, 88 pg of solubilized membrane extract; lane D , -20 pg of boiled and concentrated membrane extract. Also shown are the indicated frac- tions ( 2 4 aliquots) from Mono Q chromatography of the boiled and concentrated membrane extracts. The 2-4 aliquot from fraction 45 that was loaded onto the lane contained 2 pg of protein. Arrows denote the purified protein.
incubation, sections were washed three times for 5 min in buffer A. After the final wash, sections were rinsed twice in 50 mM sodium phosphate buffer at pH 7.4 and fixed with 1.33% glutaraldehyde in the same buffer for 2 h. The tissue was postfixed with osmium tetroxide and embedded in Eponate. Immunogold labeling of plastic thin sections was carried out as described previously (20). Ultrathin sections were cut, transferred to grids, and etched in saturated sodium metaperiodate for 1 h at room temperature. The sections were incu- bated sequentially with the following antibodies in buffer A at room temperature: 1) 0.5 pg/ml of a mouse monoclonal anti-2,4-dinitro- phenol IgG (20) overnight; 2) 5 pg/ml unlabeled rabbit anti-mouse IgG for 2 h; and 3) goat anti-rabbit IgG labeled with 10-nm gold particles for 1 h. The concentration of the gold particles was adjusted to an absorbance of 0.2 at 520 nm. The sections were rinsed in
165-kDa Protein LDL Receptor
1 2 3 4 5 6 7 M r x,o-3
I t-
- 205
- 116 - 97
Additions
FIG. 6. Inhibition of lZ6I-LDL binding to the 165-kDa mus- cle protein and LDL receptor by anionic compounds. The purified 165-kDa muscle protein (1 pg) or purified bovine LDL receptor (0.7 pg) were subjected to electrophoresis on SDS gels under nonreducing conditions and transferred to nitrocellulose filters. The filters were incubated with 5 pg of protein/ml of Y - L D L (395 cpm/ ng protein) in the presence of 2 mM CaC12 and one of the following compounds: heparin (10 pg/ml), dextran sulfate (10 pglml), or sura- min (10 mM). The nitrocellulose filters were exposed to XAR-5 film for 2 h at -70 "C with an intensifying screen. The bovine LDL receptor migrates as a 130-kDa protein under these nonreducing conditions.
distilled water, air-dried, stained, and analyzed with a JEOL lOOCX electron microscope (20).
RESULTS
Biochemical Characterization of the 165-kDa Muscle Pro- tein-Fig. 1 shows ligand blots performed with membrane extracts from mouse heart, mouse liver, and bovine adrenal. The extracts were subjected to SDS-polyacrylamide gel elec- trophoresis, transferred to nitrocellulose, and incubated with one of three 1251-labeled lipoproteins that are known to bind to the LDL receptor: human LDL, (3-VLDL from cholesterol- fed rabbits, and apoE-HDL, from cholesterol-fed dogs. The latter two ligands contain apoE, which binds to LDL receptors with higher affinity than does the apoB-100 of LDL (21). The blotting was performed in the presence of Ca2+ (upperpanels) or EDTA (lower panels). In the presence of Ca2+, all three tissues showed a binding activity that recognized LDL, (3- VLDL, and apoE-HDL,. The mobility of the binding protein under nonreducing conditions corresponded to an apparent molecular weight in the range of 130,000-165,000 (see below). EDTA abolished the binding of the three ligands to the proteins from liver and adrenal, as would be expected for
8264 Lipoprotein-binding Protein from Sarcoplasmic Reticulum
"0 50 100 150 Unlabeled Lipoprotein (Fg protein/ml)
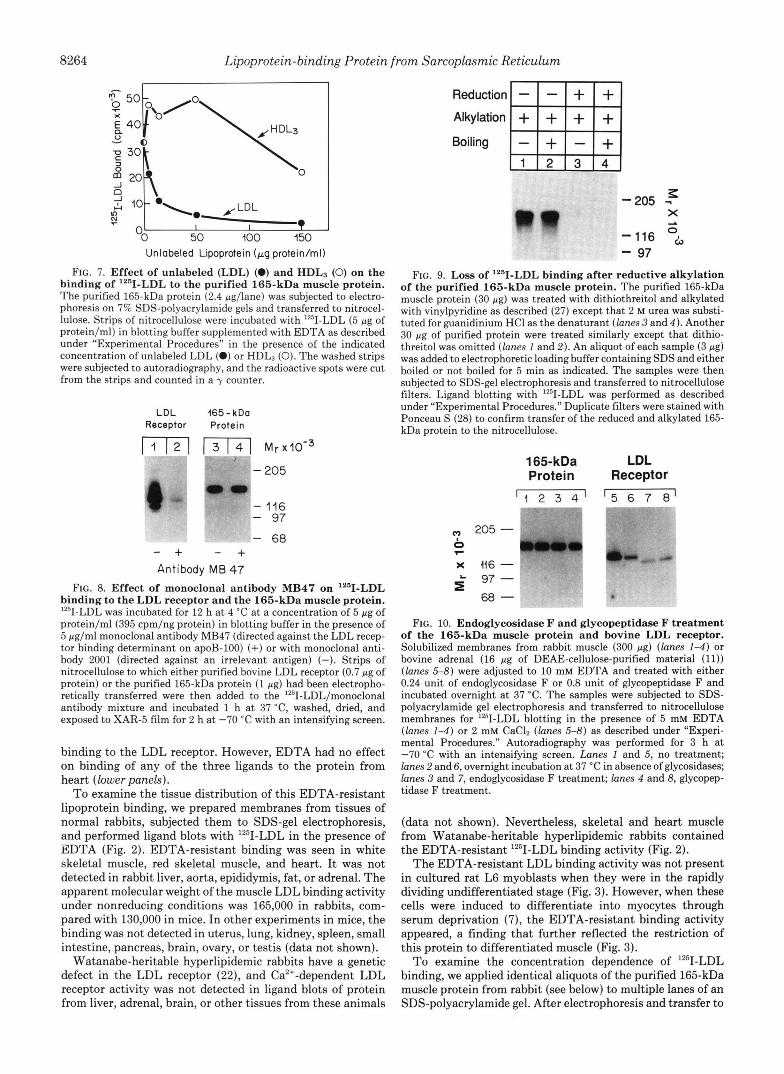
FIG. 7. Effect of unlabeled (LDL) (0) and HDLs (0) on the binding of '"1-LDL to the purified 165-kDa muscle protein. The purified 165-kDa protein (2.4 pg/lane) was subjected to electro- phoresis on 7% SDS-polyacrylamide gels and transferred to nitrocel- lulose. Strips of nitrocellulose were incubated with '*'I-LDL (5 pg of protein/ml) in blotting buffer supplemented with EDTA as described under "Experimental Procedures" in the presence of the indicated concentration of unlabeled LDL (0) or HDL3 (0). The washed strips were subjected to autoradiography, and the radioactive spots were cut from the strips and counted in a y counter.
LDL 165 - kDo Receptor Protein [m 1 3 1 4 1 M~ x10-3
w m F " - 205
1
- 116 - 97
- 68 - + - + Antibody MB 47
FIG. 8. Effect of monoclonal antibody MB47 on lZ6I-LDL binding to the LDL receptor and the 165-kDa muscle protein. '*'I-LDL was incubated for 12 h at 4 "C at a concentration of 5 pg of protein/ml(395 cpm/ng protein) in blotting buffer in the presence of 5 pg/ml monoclonal antibody MB47 (directed against the LDL recep- tor binding determinant on apoB-100) (+) or with monoclonal anti- body 2001 (directed against an irrelevant antigen) (-). Strips of nitrocellulose to which either purified bovine LDL receptor (0.7 pg of protein) or the purified 165-kDa protein (1 pg) had been electropho- retically transferred were then added to the '2SI-LDL/monoclonal antibody mixture and incubated 1 h at 37 "C, washed, dried, and exposed to XAR-5 film for 2 h at -70 "C with an intensifying screen.
binding to the LDL receptor. However, EDTA had no effect on binding of any of the three ligands to the protein from heart (lower panels).
To examine the tissue distribution of this EDTA-resistant lipoprotein binding, we prepared membranes from tissues of normal rabbits, subjected them to SDS-gel electrophoresis, and performed ligand blots with '251-LDL in the presence of EDTA (Fig. 2). EDTA-resistant binding was seen in white skeletal muscle, red skeletal muscle, and heart. It was not detected in rabbit liver, aorta, epididymis, fat, or adrenal. The apparent molecular weight of the muscle LDL binding activity under nonreducing conditions was 165,000 in rabbits, com- pared with 130,000 in mice. In other experiments in mice, the binding was not detected in uterus, lung, kidney, spleen, small intestine, pancreas, brain, ovary, or testis (data not shown).
Watanabe-heritable hyperlipidemic rabbits have a genetic defect in the LDL receptor (22), and Ca2'-dependent LDL receptor activity was not detected in ligand blots of protein from liver, adrenal, brain, or other tissues from these animals
Reduction
Alkylation + + + + Boiling
-205 - X A
-116 o~ - 97 FIG. 9. Loss of '"I-LDL binding after reductive alkylation
of the purified 165-kDa muscle protein. The purified 165-kDa muscle protein (30 pg) was treated with dithiothreitol and alkylated with vinylpyridine as described (27) except that 2 M urea was substi- tuted for guanidinium HC1 as the denaturant (lanes 3 and 4 ) . Another 30 pg of purified protein were treated similarly except that dithio- threitol was omitted (lanes 1 and 2). An aliquot of each sample (3 pg) was added to electrophoretic loading buffer containing SDS and either boiled or not boiled for 5 min as indicated. The samples were then subjected to SDS-gel electrophoresis and transferred to nitrocellulose filters. Ligand blotting with '*'II-LDL was performed as described under "Experimental Procedures." Duplicate filters were stained with Ponceau S (28) to confirm transfer of the reduced and alkylated 165- kDa protein to the nitrocellulose.
165-kDa LDL Protein Receptor
' I 2 3 4 ' ' 5 6 7 8'
z 205 - I
7
x 416 - 5 97 -
68 -
' CC- ._ .&
FIG. 10. Endoglycosidase F and glycopeptidase F treatment of the 165-kDa muscle protein and bovine LDL receptor. Solubilized membranes from rabbit muscle (300 pg) (lanes 1-4) or bovine adrenal (16 pg of DEAE-cellulose-purified material (11)) (lanes 5-8) were adjusted to 10 mM EDTA and treated with either 0.24 unit of endoglycosidase F or 0.8 unit of glycopeptidase F and incubated overnight at 37 "C. The samples were subjected to SDS- polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes for '*'II-LDL blotting in the presence of 5 mM EDTA (lanes 1-4) or 2 mM CaC12 (lanes 5-8) as described under "Experi- mental Procedures." Autoradiography was performed for 3 h at -70 "C with an intensifying screen. Lanes 1 and 5, no treatment; lanes 2 and 6, overnight incubation a t 37 "C in absence of glycosidases; lanes 3 and 7, endoglycosidase F treatment; lanes 4 and 8, glycopep- tidase F treatment.
(data not shown). Nevertheless, skeletal and heart muscle from Watanabe-heritable hyperlipidemic rabbits contained the EDTA-resistant '251-LDL binding activity (Fig. 2).
The EDTA-resistant LDL binding activity was not present in cultured rat L6 myoblasts when they were in the rapidly dividing undifferentiated stage (Fig. 3). However, when these cells were induced to differentiate into myocytes through serum deprivation (7), the EDTA-resistant binding activity appeared, a finding that further reflected the restriction of this protein to differentiated muscle (Fig. 3).
To examine the concentration dependence of '"1-LDL binding, we applied identical aliquots of the purified 165-kDa muscle protein from rabbit (see below) to multiple lanes of an SDS-polyacrylamide gel. After electrophoresis and transfer to

Lipoprotein-binding Protein from Sarcoplasmic Reticulum 8265
Stains-all 45Ca*+
5-75-7 205 -
45 -
FIG. 11. Stains-all staining and Ca2+ blotting of skeletal muscle membranes and the purified 165-kDa muscle protein. Solubilized rabbit muscle membranes (130 pg) (lanes I and 3 ) and the purified 165-kDa protein (5 pg) (lanes 2 and 4 ) were prepared as described under "Experimental Procedures," subjected to electropho- resis on 7% SDS-polyacrylamide slab gels, and either stained with Stains-all (24) or transferred to nitrocellulose for blotting with 45CaC12 as indicated. Calcium blotting was performed exactly as described (29). Briefly, nitrocellulose filters were incubated a t room temperature in buffer containing 10 mM imidazole-HC1 at pH 6.8, 60 mM KCl, 5 mM MgC12 for 1 h, followed by 1 pCi/ml of 4'CaC12 in the same buffer for 10 min. Filters were washed with 50% ethanol for 5 min, dried, and exposed directly to XAR-5 film overnight at room temperature. The prominent Stains-all binding protein and 45CaC12-binding protein a t 63 kDa co-migrated with authentic rabbit calsequestrin in an adjacent lane (data not shown). The arrows for lanes 1 and 2 denote blue-staining proteins.
1 2 3 M~ X 10-3
-D
FIG. 12. Immunoblotting of crude skeletal muscle homoge- nate with an affinity-purified antibody directed against the 165-kDa muscle protein. Rabbit skeletal muscle was homogenized with a Polytron homogenizer as described under "Experimental Pro- cedures." An aliquot of the total homogenate (200 pg of protein) was subjected to electrophoresis on 7% SDS-polyacrylamide slab gels and either transferred to nitrocellulose for immunoblotting (lanes 1 and 2 ) or stained with Coomassie Brilliant Blue (lane 3 ) . The nitrocellu- lose filters were incubated with 1 pg/ml of either affinity-purified polyclonal sheep antibody against the 165-kDa protein (lane I ) or preimmune antibody (lane 2 ) followed by 0.5 pg/ml '251-labeled rabbit anti-sheep IgG (-1 X lo7 cpm/pg). The washed and dried filters were exposed to XAR-5 film for 16 h at room temperature. The open arrow denotes the position of migration of the 165-kDa protein; the closed arrow denotes the position of migration of calsequestrin in an adjacent lane (data not shown).
nitrocellulose, the strips were cut out and incubated with varying concentrations of lZ5I-LDL. The radioactive bands were then cut out from the nitrocellulose and subjected to y counting. As shown in Fig. 4, binding of lZ5I-LDL rose with saturation kinetics as the concentration of '251-LDL was in- creased. Half-maximal binding was observed at an "'I-LDL concentration of approximately 4 pg of protein/ml. When the arginine residues of "'I-LDL were modified with cyclohex- anedione, binding was obliterated. About half of the binding returned when the cyclohexanedione was removed by treat- ment with hydroxylamine. Thus, the binding of LDL to the
- 205 - 116 - 97
- 68 f
- 45
muscle protein, as to the LDL receptor (lo), requires intact arginine residues on the LDL.
The 165-kDa LDL-binding protein was purified from rabbit skeletal muscle in a series of steps as outlined in Table I and illustrated in Fig. 5. A crude homogenate of rabbit skeletal muscle contained the LDL binding activity (Fig. 5A, lune A). The activity was present in the supernatant after a spin at 2,000 X g (lune B ) and was pelleted at 100,000 x g (lune C). Lune D shows the binding activity after the membranes were solubilized with Triton X-100, boiled, and spun at 16,800 X g for 10 min. The protein remained in the supernatant, and it retained full activity. Together, the membrane isolation, sol- ubilization, boiling, and centrifugation steps led to a 220-fold purification of the binding activity with a 60% yield (Table I). The protein could then be purified to near homogeneity by chromatography on a Mono Q ion exchange resin using a fast protein liquid chromatography system. The binding activity emerged from the column in fractions 44-46 (Fig. 5A). Coo- massie Blue staining revealed that these fractions contained a single visible protein with an apparent molecular weight of approximately 165,000 (Fig. 5B). The fractions immediately preceding the peak fraction contained LDL binding activities with faster mobilities than the 165-kDa protein. These bind- ing activities were not present in crude extracts (lune A) nor in the fraction prior to Mono Q chromatography (lane D). They are likely to be fragments of the 165-kDa protein gen- erated during the isolation procedure. Overall, this series of steps led to a 1,310-fold purification of the LDL-binding activity with a 51% yield (Table I). The absolute binding activity of the purified protein (39.2 pg of LDL/mg protein) is a substantial underestimate because it is unlikely that every molecule subjected to electrophoresis retains binding activity.
As reported previously (11, 23), binding of "'I-LDL to the LDL receptor is inhibited by polyanionic compounds such as dextran sulfate and suramin (Fig. 6, lunes 6 and 7). Similarly, the binding of "'I-LDL to the 165-kDa protein was also inhibited by dextran sulfate (lane 3 ) , suramin (lane 4 ) , and heparin (lune 2). Thus, it is likely that this binding involves strong ionic interactions.
Binding of lipoproteins to the 165-kDa muscle protein appeared to be specific for apoB-100 or apoE-containing lipoproteins because HDL3, which contains apoA-I, did not effectively compete for LDL binding to the purified 165-kDa protein (Fig. 7). However, the binding region of apoB-100 appeared to differ from the region that binds to the LDL receptor. Binding of LDL to the LDL receptor is known to be inhibited by a monoclonal antibody, MB47, which is directed at an epitope on apoB-100 that is involved in the binding (13). Fig. 8 shows that this antibody prevented binding of lZ5I- LDL to the purified LDL receptor on ligand blots (Fig. 8, lane 2 ) . In the same experiment this antibody did not inhibit the binding of "'I-LDL to the 165-kDa muscle protein (Fig. 8, lunes 3 and 4).
The binding activity of the LDL receptor is abolished when the disulfide bonds in the ligand binding domain are reduced (3). Fig. 9 shows that cystine residues are also crucial for the binding activity of the 165-kDa muscle protein. When this protein was reduced and alkylated, its ability to bind '251-LDL was lost (Fig. 9, lunes 3 and 4 ) . However, in contrast to the LDL receptor whose apparent molecular mass increases from 130 to 160 kDa upon reduction (6), the 165-kDa muscle protein did not undergo a change in apparent molecular mass upon reduction and alkylation, as revealed by staining with Coomassie Blue (data not shown).
Digestion of the 165-kDa protein with glycosidases caused no change in electrophoretic mobility as determined by ligand
8266 Lipoprotein-binding Protein from Sarcoplasmic Reticulum
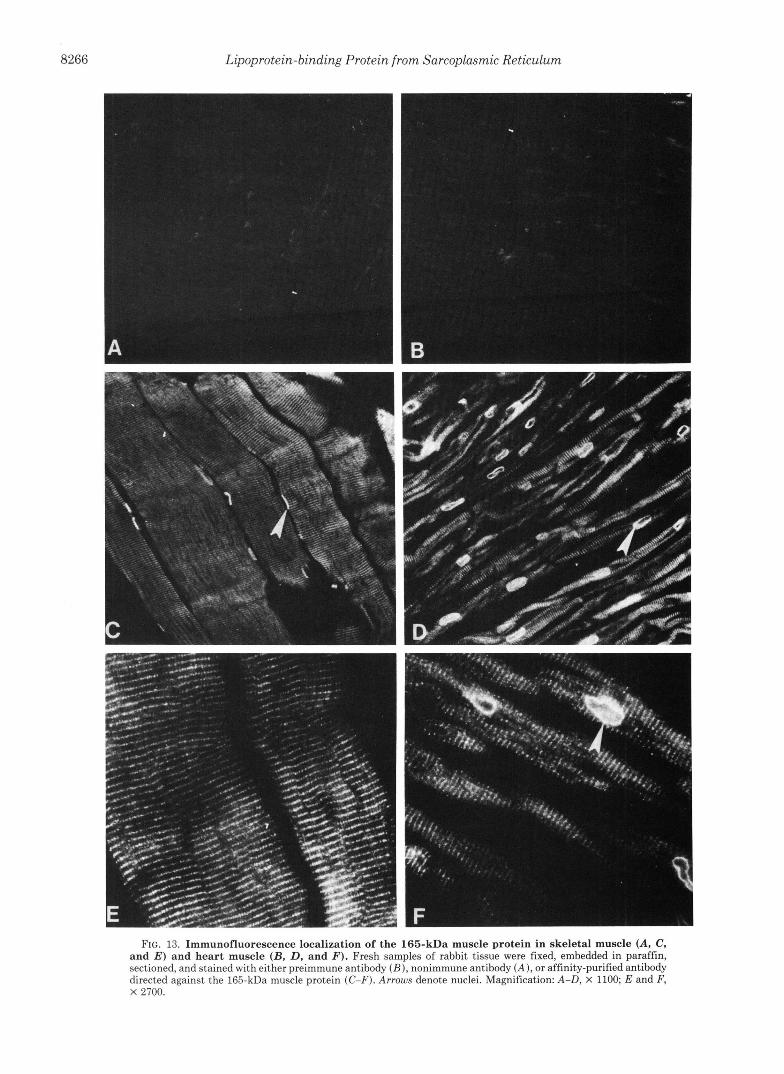
FIG. 13. Immunofluorescence localization of the 165-kDa muscle protein in skeletal muscle (A, C, and E ) and heart muscle (B, D, and 3'). Fresh samples of rabbit tissue were fixed, embedded in paraffin, sectioned, and stained with either preimmune antibody ( B ) , nonimmune antibody ( A ) , or affinity-purified antibody directed against the 165-kDa muscle protein (C-F). Arrows denote nuclei. Magnification: A-D, X 1100; E and F, X 2700.
Lipoprotein-binding Protein from Sarcoplasmic Reticulum 8267
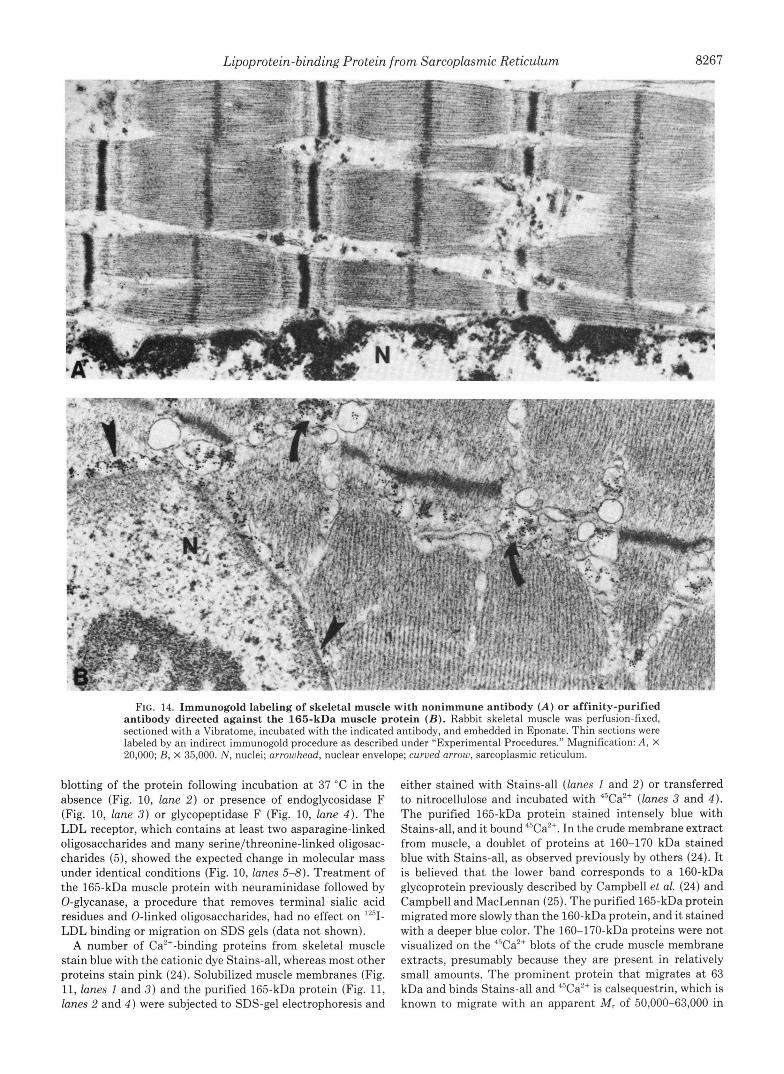
FIG. 14. Immunogold labeling of skeletal muscle with nonimmune antibody ( A ) or affinity-purified antibody directed against the 165-kDa muscle protein ( B ) . Rabbit skeletal muscle was perfusion-fixed, sectioned with a Vibratome, incubated with the indicated antibody, and embedded in Eponate. Thin sections were labeled by an indirect immunogold procedure as described under "Experimental Procedures." Magnification: A , X 20,000; B, X 35,000. N , nuclei; arrowhead, nuclear envelope; curved arrow, sarcoplasmic reticulum.
blotting of the protein following incubation at 37 "C in the absence (Fig. 10, lune 2) or presence of endoglycosidase F (Fig. 10, lune 3 ) or glycopeptidase F (Fig. 10, lune 4 ) . The LDL receptor, which contains a t least two asparagine-linked oligosaccharides and many serine/threonine-linked oligosac- charides (5), showed the expected change in molecular mass under identical conditions (Fig. 10, lunes 5-8). Treatment of the 165-kDa muscle protein with neuraminidase followed by 0-glycanase, a procedure that removes terminal sialic acid residues and 0-linked oligosaccharides, had no effect on "'1- LDL binding or migration on SDS gels (data not shown).
A number of Ca2'-binding proteins from skeletal muscle stain blue with the cationic dye Stains-all, whereas most other proteins stain pink (24). Solubilized muscle membranes (Fig. 11, lunes 1 and 3 ) and the purified 165-kDa protein (Fig. 11, lunes 2 and 4 ) were subjected to SDS-gel electrophoresis and
either stained with Stains-all (lunes 1 and 2) or transferred to nitrocellulose and incubated with 45Ca2+ (lunes 3 and 4 ) . The purified 165-kDa protein stained intensely blue with Stains-all, and it bound 4sCa2+. In the crude membrane extract from muscle, a doublet of proteins at 160-170 kDa stained blue with Stains-all, as observed previously by others (24). It is believed that the lower band corresponds to a 160-kDa glycoprotein previously described by Campbell et ul. (24) and Campbell and MacLennan (25). The purified 165-kDa protein migrated more slowly than the 160-kDa protein, and it stained with a deeper blue color. The 160-170-kDa proteins were not visualized on the 45Cazc blots of the crude muscle membrane extracts, presumably because they are present in relatively small amounts. The prominent protein that migrates at 63 kDa and binds Stains-all and 4sCa2+ is calsequestrin, which is known to migrate with an apparent M , of 50,000-63,000 in
6
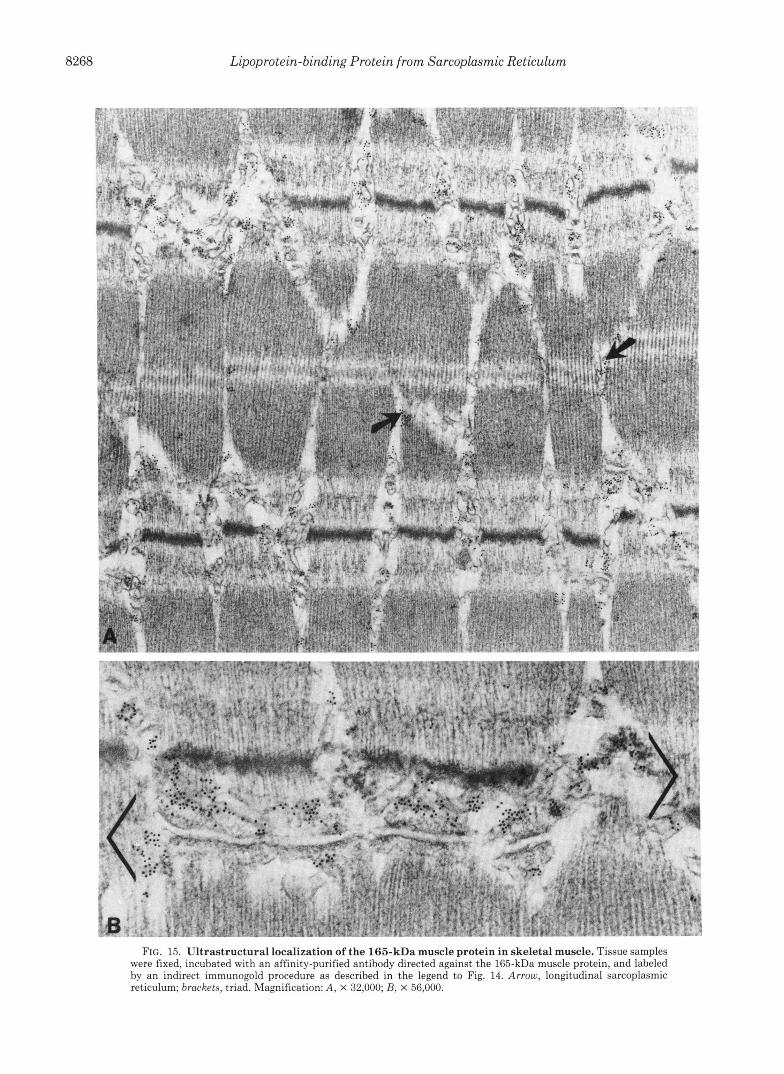
FIG. 15. Ultrastructural localization of the 165-kDa muscle protein in skeletal muscle. Tissue samples were fixed, incubated with an affinity-purified antibody directed against the 165-kDa muscle protein, and labeled by an indirect immunogold procedure as described in the legend to Fig. 14. Arrow, longitudinal sarcoplasmic reticulum; brackets, triad. Magnification: A, X 32,000; B, X 56,000.

Lipoprotein-binding Protein from Sarcoplasmic Reticulum 8269
SDS gels, despite its true protein M, of 42,435, as deduced from its cDNA sequence (26). The 160-170-kDa proteins appear to be much less abundant than calsequestrin in the crude membrane extracts (Fig. 11).
Localization of the 165-kDa Muscle Protein in Sarcoplasmic Reticulum by Immunocytochemistry-The purified rabbit 165- kDa muscle protein was injected into sheep, and the resultant antibody was purified by retention on a column containing the 165-kDa protein. Any activity that cross-reacted with calsequestrin was removed by passage through a column con- taining calsequestrin. When reacted with crude homogenates of skeletal muscle, this antibody stained only the 165-kDa protein and did not stain calsequestrin (Fig. 12, lane 1 ). The 165-kDa protein is estimated to comprise -0.5% of the total protein in the crude muscle homogenate and was not visible on Coomassie-stained gels (Fig. 12, lane 3) .
The affinity-purified antibody was used to determine the localization of the 165-kDa protein in sections of rabbit skel- etal and cardiac muscle (Fig. 13). Antibody fractions of non- immune serum (i.e. the fraction that was not retained on the 165-kDa affinity column) and preimmune serum from the immunized sheep (Fig. 13, A and B, respectively) failed to give significant immunofluorescence signals. The affinity- purified antibody produced periodic fluorescent bands con- sistent with the localization of the 165-kDa protein in a single position within each sarcomere. In addition, bright fluores- cence surrounded each nucleus (arrows in Fig. 13, C, D, and F ) . The pattern was similar in skeletal (Fig. 13, C and E ) and cardiac muscle (Fig. 13, D and F ) .
To determine the location of the 165-kDa muscle protein by electron microscopy, we prepared Vibratome sections of formaldehyde-fixed skeletal muscle. The tissue was permea- bilized with saponin and incubated with the affinity-purified sheep antibody followed by a rabbit anti-sheep IgG conjugated with 2,4-dinitrophenol. The thick sections were then em- bedded in plastic, sliced into thin sections, and incubated with mouse monoclonal anti-2,4,dinitrophenol IgG, followed by a rabbit anti-mouse IgG, followed by goat anti-rabbit IgG la- beled with gold. As a control, we replaced the affinity-purified anti-165-kDa antibody with nonimmune antibody, which was the y-globulin fraction of immune serum that did not adhere to the 165-kDa affinity column. The nonimmune antibody gave no specific immunogold staining (Fig. 14A). The affinity- purified antibody gave specific labeling of the sarcoplasmic reticulum (curved arrows in Fig. 14B) and the nuclear enve- lope (arrowheads in Fig. 14B). In the sarcoplasmic reticulum the immunogold appeared to be located within the lumen and not in direct juxtaposition to the membrane.
lmmunogold labeling was present in both the terminal cisternae (Fig. 15B, enclosed in brackets) and longitudinal sarcoplasmic reticulum (Fig. 15A, arrows). There was no labeling of the transverse tubule membrane or its contents. Biochemical data support the localization of the 165-kDa protein to the lumen of the sarcoplasmic reticulum and sug- gest that it is unlikely to be an intrinsic membrane protein. The protein is released in soluble form by sodium carbonate treatment (data not shown). In addition, once the membranes are solubilized by extraction with Triton X-100 and the 165- kDa protein is bound to the Mono Q ion exchange resin, the detergent can be removed and the protein eluted in soluble form in the absence of detergent (data not shown).
DISCUSSION
In the current paper, we describe a striated muscle-specific protein that binds '*'I-LDL with high affinity. This 165-kDa protein was not detected in smooth muscle or in any of 14
o&e'r tissues that were analyzed. The protein is localized to the sarcoplasmic reticulum where it is unlikely to bind to plasma lipoproteins. Although the lipoprotein binding prop- erty of the 165-kDa protein is of no physiological significance, it did provide a marker that allowed it to be specifically followed during purification.
The lipoprotein binding reaction was characterized in some detail because it provides insight into the physical properties of the 165-kDa protein. Binding of LDL to the 165-kDa muscle protein was prevented when positively charged argi- nine residues on LDL were modified with cyclohexanedione. Moreover, binding was abolished in the presence of polyan- ionic compounds. These data suggest that the binding involves an ionic interaction between positively charged residues on LDL and, by implication, negatively charged residues on the binding protein. However, the binding is not produced by a simple attraction of oppositely charged amino acids: confor- mation of the 165-kDa protein is also important. Thus, bind- ing was abolished when the cystine residues of the 165-kDa protein were reduced and alkylated. This reaction should not affect the negatively charged amino acids, but it is likely to have changed their conformation in such a way that binding activity was destroyed. The arginine residues on LDL that participate in binding to the 165-kDa muscle protein do not seem to be the same as the ones that interact with the LDL receptor because monoclonal antibody MB47, which abolishes LDL binding to the LDL receptor, did not affect binding to the 165-kDa protein.
The 165-kDa muscle protein is a member of a family of Ca2+-binding proteins of sarcoplasmic reticulum. It binds Ca2+ on ligand blots and stains intensely blue with Stains-all. These properties are shared by calsequestrin and a family of previously described calsequestrin-like proteins in the molec- ular weight range of 160,000-170,000 (24, 25). One of these 160-170-kDa proteins is a glycoprotein that has been char- acterized by Campbell et al. (24) and Campbell and Mac- Lennan (25). The currently described 165-kDa protein does not appear to be the same as this 160-kDa glycoprotein for two reasons: 1) two monoclonal antibodies directed against the 160-kDa glycoprotein (kindly provided by D. H. Mac- Lennan, Charles H. Best Institute, University of Toronto, Toronto, Canada) do not cross-react with the 165-kDa pro- tein; 2) the purified 160-kDa glycoprotein does not bind lZ5I- LDL in ligand blots (data not shown); and 3) the 165-kDa protein does not appear to be a glycoprotein because it is not digested with endoglycosidase F, glycopeptidase F, neuramin- idase, or 0-glycanase (Fig.
I t is highly unlikely that the 165-kDa protein functions as a binding protein for LDL or any other plasma lipoprotein. Its location in the sarcoplasmic reticulum would preclude access to the plasma. It may play a role, however, in Ca2+ homeostasis in the sarcoplasmic reticulum. Further insight into the function of the 165-kDa protein should occur when its amino acid sequence is obtained.
Acknowledgments-Sharon Tucker and Shirley Hall provided ex- cellent technical assistance. We thank Kim Orth and Clive Slaughter for determining the partial amino acid sequence of calsequestrin. We thank Dr. David H. MacLennan for his generous gift of the purified 160-kDa muscle glycoprotein and monoclonal antibodies directed against it.
REFERENCES
1. MacLennan, D. H., Campbell, K. P., and Reithmeier, R. A. (1983) in Calcium and Cell Function (Cheung, W. Y., ed) Vol. IV, pp.
S. L. Hofmann, M. S. Brown, and J. L. Goldstein, unpublished observations.

8270 Lipoprotein-binding Protein from Sarcoplasmic Reticulum 151-173, Academic Press, New York
(1985) Nature 316 , 696-700 2. MacLennan, D. H., Brandl, C. J., Korczak, B., and Green, N. M.
3. Fill, M., and Coronado, R. (1988) Trends Neurosci. 11, 453-457 4. Catterall, W. A,, Seager, M. J., and Takahashi, M. (1988) J. Biol.
Chem. 263,3535-3538 5. Goldstein, J. L., Brown, M. S., Anderson, R. G. W., Russell, D.
W., and Schneider, W. J. (1985) Annu. Reu. Cell Biol. 1 , 1-39 6. Daniel, T. O., Schneider, W. J., Goldstein, J. L., and Brown, M.
S. (1983) J. Biol. Chem. 258,4606-4611 7. Wright, W. E. (1984) J. Cell Biol. 9 8 , 436-443 8. Goldstein, J. L., Basu, S. K., and Brown, M. S. (1983) Methods
Enzymol. 9 8 , 241-260 9. Kovanen, P. T., Brown, M. S., Basu, S. K., Bilheimer, D. W., and
Goldstein, J. L. (1981) Proc. Natl. Acad. Sci. U. S. A. 78 , 1396- 1400
10. Innerarity, T. L., Pitas, R. E., and Mahley, R. W. (1986) Methods Enzymol. 129,542-565
11. Schneider, W. J., Beisiegel, U., Goldstein, J. L., and Brown, M. S. (1982) J. Biol. Chem. 2 5 7 , 2664-2673
12. Kita, T., Brown, M. S., Watanabe, Y., and Goldstein, J. L. (1981) Proc. Natl. Acad. Sci. U. S. A. 78,2268-2272
13. Young, S. G., Witztum, J. L., Casal, D. C., Curtiss, L. K., and Bernstein, S. (1986) Arteriosclerosis 6 , 178-188
14. Beisiegel, U., Schneider, W. J., Brown, M. S., and Goldstein, J. L. (1982) J. Biol. Chem. 2 5 7 , 13150-13156
15. Schneider, W. J., Basu, S. K., McPhaul, M. J., Goldstein, J. L.,
16. 17. 18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
Bradford, M. M. (1976) Anal. Biochem. 7 2 , 248-254 Dunn, S. D. (1986) Anal. Biochem. 157 , 144-153 Garvey, J. S., Cremer, N. E., and Sussdorf, D. H. (eds) (1977) in
Methods in Immunology, 3rd Ed., pp. 218-219, Benjamin/Cum- mings Publishing Co., Reading, MA
Mitchell, D., Ibrahim, S., and Gusterson, B. A. (1985) J. Histo- chem. Cytochem. 33,491-495
Pathak, R. A., and Anderson, R. G. W. (1989) J. Histochem. Cytochem. 37,69-74
Mahley, R. W., and Innerarity, T. L. (1983) Biochim. Biophys.
Yamamoto, T., Bishop, R. W., Brown, M. S., Goldstein, J. L.,
Goldstein, J. L., Basu, S. K., Brunschede, G . Y., and Brown, M.
Campbell, K. P., MacLennan, D. H., and Jorgensen, A. 0. (1983)
Campbell, K. P., and MacLennan, D. H. (1981) J. Biol. Chem.
Fliegel, L., Ohnishi, M., Carpenter, M. R., Khanna, V. K., Reith- meier, R. A. F., and MacLennan, D. H. (1987) Proc. Natl. Acad. Sci. U. S. A. 8 4 , 1167-1171
Tarr, G. E. (1986) in Methods of Protein Microcharacterization: A Practical Handbook (Shivelv. J. E.. ed) DD. 162-163. Humana
Acta 737 , 197-222
and Russell, D. W. (1986) Science 232,1230-1237
S. (1976) Cell 7,85-95
J. Biol. Chem. 2 5 8 , 11267-11273
256,4626-2632
Press, Clifton, NJ " I . . "
Aebersold, R. H.. Leavitt. J.. Saavedra. R. A,. Hood. L. E.. and Kent, SI B. H. (1987) Proc.' Natl. Acad. Sci. U . S. A; 84,6970- 6974
and Brown, M. S. (1979) Proc. Natl. Acad. Sci. U. S. A. 76 , 29. Maruyama, K., Mikawa, T., and Ebashi, S. (1984) J. Biochem. 5577-5581 (Tokyo) 95,511-519


















