
Progress in Polymer Science - Clips · Progress in Polymer Science 39 (2014) 1–42 Contents lists...
42
Progress in Polymer Science 39 (2014) 1–42 Contents lists available at ScienceDirect Progress in Polymer Science journa l h om epa ge: www.elsevier.com/locate/ppolysci Fundamental water and salt transport properties of polymeric materials Geoffrey M. Geise, Donald R. Paul, Benny D. Freeman ∗ The University of Texas at Austin, Department of Chemical Engineering, Texas Materials Institute and Center for Energy and Environmental Resources, 1 University Station, Mail Code: C0400, Austin, TX 78712, USA a r t i c l e i n f o Article history: Received 5 November 2012 Received in revised form 20 June 2013 Accepted 27 June 2013 Available online 13 July 2013 Keywords: Membrane Transport Ionomer Water permeability Salt permeability Desalination Separation a b s t r a c t Fundamental water and salt transport properties of polymers are critical for applications such as reverse osmosis (RO), nanofiltration (NF), forward osmosis (FO), pressure-retarded osmosis (PRO), and membrane capacitive deionization (MCDI) that require controlled water and salt transport. Key developments in the field of water and salt transport in polymer membranes are reviewed, and a survey of polymers considered for such applications is pro- vided. Many polymers considered for such applications contain charged functional groups, such as sulfonate groups, that can dissociate in the presence of water. Water and ion trans- port data from the literature are reviewed to highlight the similarities and differences between charged and uncharged polymers. Additionally, the influence of other polymer structure characteristics, such as cross-linking and morphology in phase separated systems, on water and salt transport properties is discussed. The role of free volume on water and salt transport properties is discussed. The solution–diffusion model, which describes the transport of water and ions in nonporous polymers, is used as a framework for discussing structure/property relations in polymers related to water and salt transport properties. Areas where current knowledge is limited and opportunities for further research are also noted. © 2013 Elsevier Ltd. All rights reserved. Contents 1. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2 1.1. Solution–diffusion model for transport in desalination polymers . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3 1.2. Experimental verification of the solution–diffusion model . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6 1.3. Water/salt tradeoff relationship in swollen polymers . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 8 1.4. Effect of free volume on solution diffusion and permeation properties . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 9 1.5. Ion size in hydrated polymers . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10 1.6. Influence of charge in desalination polymers on transport properties . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 12 2. Uncharged polymers . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 15 2.1. Water uptake . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16 2.2. Salt sorption . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16 2.3. Dependence of water and salt permeability and diffusion on water uptake . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17 2.4. Influence of ion size on salt transport . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 18 2.5. Influence of cross-linking on water uptake and water/salt selectivity . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 19 ∗ Corresponding author. Tel.: +1 512 232 2803; fax: +1 512 232 2807. E-mail address: [email protected] (B.D. Freeman). 0079-6700/$ – see front matter © 2013 Elsevier Ltd. All rights reserved. http://dx.doi.org/10.1016/j.progpolymsci.2013.07.001
Transcript of Progress in Polymer Science - Clips · Progress in Polymer Science 39 (2014) 1–42 Contents lists...

Fp
GTE
ARRAA
KMTIWSDS
C
0h
Progress in Polymer Science 39 (2014) 1– 42
Contents lists available at ScienceDirect
Progress in Polymer Science
journa l h om epa ge: www.elsev ier .com/ locate /ppolysc i
undamental water and salt transport properties ofolymeric materials
eoffrey M. Geise, Donald R. Paul, Benny D. Freeman ∗
he University of Texas at Austin, Department of Chemical Engineering, Texas Materials Institute and Center for Energy andnvironmental Resources, 1 University Station, Mail Code: C0400, Austin, TX 78712, USA
a r t i c l e i n f o
rticle history:eceived 5 November 2012eceived in revised form 20 June 2013ccepted 27 June 2013vailable online 13 July 2013
eywords:embrane
ransportonomer
ater permeabilityalt permeability
a b s t r a c t
Fundamental water and salt transport properties of polymers are critical for applicationssuch as reverse osmosis (RO), nanofiltration (NF), forward osmosis (FO), pressure-retardedosmosis (PRO), and membrane capacitive deionization (MCDI) that require controlled waterand salt transport. Key developments in the field of water and salt transport in polymermembranes are reviewed, and a survey of polymers considered for such applications is pro-vided. Many polymers considered for such applications contain charged functional groups,such as sulfonate groups, that can dissociate in the presence of water. Water and ion trans-port data from the literature are reviewed to highlight the similarities and differencesbetween charged and uncharged polymers. Additionally, the influence of other polymerstructure characteristics, such as cross-linking and morphology in phase separated systems,on water and salt transport properties is discussed. The role of free volume on water and
esalinationeparation
salt transport properties is discussed. The solution–diffusion model, which describes thetransport of water and ions in nonporous polymers, is used as a framework for discussingstructure/property relations in polymers related to water and salt transport properties.Areas where current knowledge is limited and opportunities for further research are also
noted.© 2013 Elsevier Ltd. All rights reserved.
ontents
1. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21.1. Solution–diffusion model for transport in desalination polymers . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 31.2. Experimental verification of the solution–diffusion model . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 61.3. Water/salt tradeoff relationship in swollen polymers . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 81.4. Effect of free volume on solution diffusion and permeation properties . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 91.5. Ion size in hydrated polymers . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 101.6. Influence of charge in desalination polymers on transport properties . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 12
2. Uncharged polymers. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 152.1. Water uptake . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16
2.2. Salt sorption . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .2.3. Dependence of water and salt permeability and diffusion o2.4. Influence of ion size on salt transport . . . . . . . . . . . . . . . . . . . . . .2.5. Influence of cross-linking on water uptake and water/salt
∗ Corresponding author. Tel.: +1 512 232 2803; fax: +1 512 232 2807.E-mail address: [email protected] (B.D. Freeman).
079-6700/$ – see front matter © 2013 Elsevier Ltd. All rights reserved.ttp://dx.doi.org/10.1016/j.progpolymsci.2013.07.001
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16n water uptake . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 18selectivity . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 19

2 G.M. Geise et al. / Progress in Polymer Science 39 (2014) 1– 42
3. Charged polymers . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 203.1. Water uptake and water permeability of charged polymers . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 203.2. Salt sorption in charged polymers . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 213.3. Salt permeability of charged polymers . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 233.4. Permeability of salts other than sodium chloride . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 243.5. Salt diffusion in charged polymers . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 263.6. Mixed ion transport and sorption in charged polymers . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 273.7. Influence of processing conditions on transport properties . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 28
4. Characterization of thin-film composite membrane active layers . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 295. Transport of other penetrants . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 306. Summary . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 31
Acknowledgements . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 32Appendix A. Relating hydraulic water permeability to diffusive water permeability and the average water diffusioncoefficient . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 32A.1. Polymers that sorb relatively little water . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 34A.2. Highly swollen polymers . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 34A.3. Summary . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 35A.4. An example using a cross-linked hydrogel . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 35
. . . . . . . .
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .1. Introduction
Two inexorably intertwined, pressing global challengesare providing: (1) sufficient clean water to satisfy agricul-tural, industrial, and municipal needs, and (2) reliable andefficient access to clean energy to support ever-increasingdemands [1–7]. These issues are highly interconnectedbecause thermoelectric power generation, which accountsfor 89% of the energy produced in power plants in theU.S. [8], requires large volumes of purified water [4,9],and energy is invariably required to purify water. In 2005,approximately 50% of all water used in the United Stateswas for power generation at thermoelectric power sta-tions [10]. Future thermoelectric power plants in the U.S.are projected to require as much as an additional 2.6 bil-lion gallons of water per day by 2030 (a ∼40% increasefrom 2010) [11]. Furthermore, production of oil and nat-ural gas through operations such as hydraulic fracturingcan consume millions of gallons of water per well and gen-erate substantial amounts of flowback water, which mustbe purified or otherwise managed [9,11,12]. Additionally,as population growth continues and fresh water sourcesbecome more scarce, we will rely more heavily on desali-nated water from seawater as well as increasingly salineinland sources to generate sufficient purified water to sat-isfy human consumption as well as agricultural and otherneeds [2,3,5]. Consequently, improvements in technologiesfor energy-efficient water purification are of great inter-est from scientific, technological, and social viewpoints[1].
Today, polymer membranes play a key role in address-ing these needs since reverse osmosis (RO) membranesare the dominant technology for desalination due, inpart, to their low energy requirement relative to othertechnologies [2,5,7,13–17]. Desalination has traditionallybeen accomplished using thermal processes to vaporize
water from a saline source and then condense this vapor torecover pure water [2,18,19]. The specific energy requiredto desalinate seawater by thermal technologies suchas multiple effect distillation (MED) and multiple stage. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 36
flash (MSF) is approximately 18–30 and 24–37 kW h m−3,respectively, though these estimates vary widely in the lit-erature [18,20]. In contrast, the specific energy required formembrane seawater desalination is less than 4 kW h m−3
[3,14,18,20], so membranes require significantly lessenergy than conventional thermal technologies. Conse-quently, membranes are used worldwide to produce over50% of all desalinated seawater [3].
The scale of water desalination is extraordinarily large.Membrane desalination plants having more than 106 m2
of membrane surface area are in operation [21]. In 2010,approximately 16 billion gallons (∼61 billion liters) ofdesalinated water were produced worldwide per day [5].To place the extraordinary size of this figure in perspec-tive, the worldwide production of crude oil (another fluidprocessed in large amounts) in 2010 was 3.7 billion gal-lons (∼14 billion liters) per day [22], so the productionrate of desalination of water is more than four times thatof oil. With a theoretical minimum energy requirementto desalinate seawater at 75% recovery of 1.29 kW h m−3
[3], improving the separation properties of membranesmay contribute to even lower energy costs to desalinatewater.
Technologies such as membrane capacitive deioniza-tion (MCDI) [23–31], electrodialysis (ED) [19,32–37], andforward osmosis (FO) [38–46], which all could furtherenhance the role of polymer membranes in desalination,are being actively explored. Additionally, membrane-basedrenewable energy technologies (e.g., reverse electrodial-ysis (RED) [47–56] and pressure retarded osmosis (PRO)[45,47,50,57–62]) can generate electrical energy by har-nessing the chemical energy inherently released whenstreams of different salt concentrations are mixed, forexample where a freshwater river flows into the sea. Thegeneration of energy from the sea by methods such as REDand PRO is often called “blue energy”. Because the concen-
trate produced by a seawater reverse osmosis plant hasan even higher salt concentration than that of seawater, aPRO plant could be fed with this highly saline water andseawater, thereby providing a membrane-based plant
n Polymer Science 39 (2014) 1– 42 3
pe
oPwmasfmbtp
msr3rt(mro
mBabauwsmctmsub
PtawfttttautgeToibd
Fig. 1. Hydrostatic pressure, p, chemical potential, �, and concentration, c,profiles in a dense, non-porous polymer film of thickness L during water(A), subscript w, and salt (B), subscript s, permeation. The direction ofwater and salt flux, n, is indicated. The superscripts s and m refer to the
transport of water and ions through such a membraneoccurs by the penetrants (i.e., water and ions) first molec-
G.M. Geise et al. / Progress i
roducing both purified water by RO and clean, renewablenergy via PRO [46,47].
All of these applications depend on controlling ratesf ion and water transport across polymer membranes.ractical membrane-based desalination became possibleith the development of asymmetric cellulose acetate (CA)embranes produced via nonsolvent-induced phase sep-
ration [63,64]. While cellulose acetate membranes aretill used for commercial desalination today [65–68], inter-acially polymerized, cross-linked aromatic polyamide
embranes have come to dominate the market due to theiretter combination of properties [14,69–73]. The struc-ures of both CA and these cross-linked polyamides areresented in Table 1.
When they were first discovered, interfacially poly-erized aromatic polyamide membranes introduced a
tep-change in water flux and salt rejection performanceelative to other membranes. Interfacially polymerized FT-0 aromatic polyamide membranes had 99.6–99.7% NaClejection with water fluxes ranging from 39 L m−2 h−1
o 55 L m−2 h−1 (5 wt% NaCl feed at 25 ◦C and 41.4 bar600 psi) feed pressure) [70]. In contrast, cellulose acetate
embranes had 92–97% NaCl rejection with water fluxesanging from 16 L m−2 h−1 to 38 L m−2 h−1 at the sameperating conditions [70].
The interfacially polymerized aromatic polyamideembranes, however, also introduced a conundrum.
ecause these membranes could not be readily prepareds freestanding films of well-defined thickness, it haseen very difficult to characterize fundamental waternd salt transport properties (e.g., water and salt sol-bility, diffusivity, and permeability) in such materials,hich rendered the generation of systematic, fundamental
tructure/property correlations challenging. Consequently,uch remains unknown about the influence of polymer
hemical and morphological structure on the fundamen-als of water and salt transport properties in desalination
embranes, frustrating the development of systematictructure/property correlations that have been so widelysed to guide the development of gas separation mem-ranes [91–98].
Different membrane applications (e.g., RO, FO, RED,RO, etc.) require specific combinations of water and saltransport properties and physical membrane structures,nd one objective of this review is to summarize existingater and salt transport structure–property relationships
or polymers in order to highlight general opportunities forailoring polymer structure to access polymer propertieshat would be useful in a variety of membrane applica-ions. Water and ion transport properties are sensitiveo, among other variables, water uptake by the polymer,nd polymer structure has a strong influence on waterptake [13,99–103]. Some of the polymers in Table 1 con-ain charged groups, because introduction of such chargedroups into the polymer backbone is one means to influ-nce water and ion sorption into the polymer [13,104,105].his review will discuss the influence of polymer chargen water and salt transport properties. In addition, thenfluence of polymer structural characteristics, other than
ackbone charge, on water and salt transport will also beiscussed.external solution and polymer (i.e., membrane) phases, respectively, andthe subscripts 0 and L refer to the feed and permeate faces of the film,respectively.
1.1. Solution–diffusion model for transport indesalination polymers
The impact of polymer structure on water and salt trans-port properties is codified in terms of the parameters ofthe solution–diffusion model, which is the accepted frame-work describing mass transport in nonporous polymers[13,106,107]. Fig. 1 provides a schematic of the chem-ical potential, concentration, and pressure gradients ina solution–diffusion membrane. As shown in this figure,
ularly dissolving into a polymer matrix at the high chemicalpotential face of the membrane. Afterwards, they diffuse

4 G.M. Geise et al. / Progress in Polymer Science 39 (2014) 1– 42
Table 1Examples of polymers considered for desalination.
Polymer Structure
Cross-linked aromatic polyamide [14,70,71,74]
Cellulose acetate [64,75,76]
Aromatic polyimide [77]
Straight-chain aromatic polyamide [77,78]
Poly(benzimi-dazopyrrolone) [77]
Poly(amide-hydrazide) [77]
Cross-linked polypiperazine-amide [14,72]
Di-sulfonated polysulfone [79–82]
Post-polymerization sulfonated polysulfone[83,84]
Post-polymerization sulfonated polyphenyleneoxide [85]
Sulfonated perfluorinated polymer (Nafion®) [86]
Sulfonated polystyrene cross-linked withdivinylbenzene [87–90]

n Polym
ttdccsrfvmc[ooa
P
t
monimw
n
wtfiVioafifi
n
wfiis
wbh[tti[
ecttgd
G.M. Geise et al. / Progress i
hrough the polymer down a chemical (or, for ions, elec-rochemical) potential gradient and finally desorb from theownstream side of the polymer film, and in both cases, thehemical or electrochemical potential gradient produces aoncentration gradient across the film [106]. The secondtep in this process, diffusion through the polymer, is theate limiting step. Furthermore, the rate limiting step of dif-usion is the opening and closing of transient gaps (i.e., freeolume elements) in the polymer matrix, due to the ther-ally stimulated, local segmental dynamics of the polymer
hains that permit penetrants to execute diffusional jumps95,108–110]. At steady state, the diffusive permeabilityf a penetrant i, Pi, can be written in terms of a sorption,r partition, coefficient, Ki, and an effective, concentrationveraged diffusion coefficient, Di [106]
i = Ki × Di (1)
This relationship describes both water and ion transporthrough polymers.
Merten [111] presented the solution–diffusionodel for desalination membranes, based on notions
f irreversible thermodynamics, to describe the desali-ation properties of cellulose acetate, which had been
dentified by Reid et al. [112,113] as a highly selective poly-er for desalination. In Merten’s model, the volumetricater flux through the film is given as [114]
w = cmw0Dw
�wL
Vw
RT(�p − ��) (2)
here nw is the steady state volumetric flux of water, Dw ishe effective, concentration-averaged water diffusion coef-cient, �w is the density of water, L is the film thickness,w is the molar volume of water, R is the gas constant, T
s absolute temperature, �p and �� are the pressure andsmotic pressure differences, respectively, across the film,nd cm
w0, is the mass concentration of water sorbed in thelm at the upstream face. The mass flux of salt through thelm is given as [114]
s = KsDs
L�cs
s (3)
here ns is the mass flux of salt, Ks is the salt sorption coef-cient, Ds is the average salt diffusion coefficient, and �cs
ss the salt concentration difference between the externalolutions (i.e., �cs
s = css0 − cs
sL).Lonsdale et al. used Merten’s model to describe
ater and salt transport in cellulose acetate mem-ranes [64]. The basic equations set forth by Mertenave become the standard equations for reverse osmosis64,106,107,111,115,116]. Paul re-derived Merten’s equa-ions as a special limiting case of a general theory developedo describe solution–diffusion transport of liquids and ionsn polymers for desalination and organic solvent transport107].
Basically, application of a hydrostatic pressure differ-nce across a nonporous polymer film establishes a wateroncentration gradient inside the film that drives water
ransport by Fickian diffusion. The relationship betweenhe hydrostatic pressure difference, water concentrationradient, and the water flux will be considered below and isescribed in more detail in Appendix A. It is the presence ofer Science 39 (2014) 1– 42 5
a difference in salt concentration on either side of the poly-mer film which causes a difference in salt electrochemicalpotential on either side of the film, establishing a salt con-centration gradient in the polymer that, in turn, drives salttransport by Fickian diffusion. Unlike water transport, thesalt flux in dense non-porous polymers is not significantlyaffected by the applied hydrostatic pressure gradient, butrather, the salt concentration gradient arises from the dif-ference in salt concentration across the film [107,115,117].The salt permeability, Ps, is readily derived by integratingFick’s law at steady state across the polymer film [64,107]
ns = KsDs
L�cs
s = Ps
L�cs
s = B�css (4)
where Ps is the salt permeability and B is the saltpermeance, which is commonly reported by membranemanufacturers [107,115]. In this review, we speak of thediffusion of salt through a polymer membrane because,in the absence of an applied electric field, if one imposesa concentration difference in, for example, NaCl, across apolymer film, electroneutrality considerations guaranteethat for every sodium ion diffusing through the film, a chlo-ride must also diffuse with it [13,104]. In cases where anelectric field is applied across the polymer, the discussionof salt transport becomes more complicated as ion trans-port can occur without satisfying the salt pair conditiondescribed above, and these electric potential effects canbe measured and accounted for in models as describedelsewhere [104,118–123]. The effect of frictional couplingor convective flow on salt transport is typically negligiblein desalination applications. For example, coupled trans-port of salt and water in cellulose acetate membranes wasshown to have a negligible effect on observed water andsalt transport for desalination applications [107].
In the Paul et al. approach, boundary conditions derivedfrom thermodynamic considerations are combined withFick’s law to describe water transport in desalinationmembranes. Unlike the situation in a porous polymerfilm, where the pressure of the fluid within the poresdecreases continuously through the film, mechanical equi-librium requires the hydrostatic pressure in a nonporouspolymer film to be constant throughout the film’s thick-ness and equal to the upstream pressure [107,124,125].Consequently, there is a discontinuity in pressure atthe downstream face of the polymer film, with watermolecules dissolved in the polymer being exposed to thefeed pressure, p0, and water molecules in the solutioncontiguous to the downstream face of the polymer beingexposed to the permeate pressure, pL [106,126–128]. Thisconcept is shown in Fig. 1. This discontinuity in pressureestablishes a water concentration gradient in the film thatdrives diffusion of water through the film, and Appendix Adescribes this phenomenon in more detail. Thus, the waterflux equation can be written as follows:
nw = Dw
LVw�cm
w = PHw
L(�p − ��) = A (�p − ��) (5)
where PHw is the hydraulic water permeability, Vw is the spe-
cific volume of water, �cmw , which is equal to
(cm
w0 − cmwL
),
is the mass concentration difference of water sorbed inthe film on the upstream and downstream sides, and A

n Polym
6 G.M. Geise et al. / Progress iis the water permeance, which is commonly reported bymembrane manufacturers [107,115]. For many of the saltsof interest in the applications considered in this review,the osmotic pressure is related to the salt concentration inthe solutions on either side of the polymer film using anapproximation for ideal solutions [129]
�k = RT∑
j
Csjk (6)
where Csjk
is the molar concentration of ion j in the externalsolution on side k of the film (i.e., feed side, 0, or permeateside, L) and the summation is performed over all ions in thesolution. For solutions where Eq. (6) does not apply, e.g.,concentrated brines and/or some multicomponent solu-tions, experimental data or empirical models, such as thePitzer model [130], may be used. Comparison of the Pitzermodel and Eq. (6), however, reveals for sodium chloridesolutions that the osmotic pressure calculated using Eq.(6) at 2000 mg(NaCl) L−1 (i.e., brackish water conditions) isless than 5% greater than that calculated using the Pitzermodel, and at 35,000 mg(NaCl) L−1 (i.e., seawater condi-tions), the osmotic pressure calculated using Eq. (6) is lessthan 8% greater than that calculated using the Pitzer model,so Eq. (6) is a reasonable approximation for many situationsof interest. The osmotic pressure difference is written as�� = �0 − �L.
The hydraulic water permeability, PHw , which is typically
measured experimentally, is related to the diffusive waterpermeability, Pw, which appears in Eq. (1). In the simplestcase, for materials that sorb relatively little water, the diff-usive water permeability is related to the hydraulic waterpermeability by [131]
Pw = PHw
RT
Vw(7)
Typical units for PHw are L �m m−2 h−1 bar−1, and typical
units for Pw are cm2 s−1. At high levels of water sorption, Eq.(7) must be replaced with more complex expressions thataccount for so-called frame of reference (i.e., convection)effects. Appendix A provides further details regarding therange of validity of Eq. (7) and alternative expressions touse when Eq. (7) is not valid.
Water uptake, wu, defined as the mass of water sorbedby the polymer divided by the mass of dry polymer, is alsocommonly used to report polymer water sorption. Wateruptake is related to the volume fraction of water in theswollen polymer, �w , as follows [81]:
�w = wu
wu + �w/�p(8)
where �p and �w are the densities of dry polymer andwater, respectively. This result is based on an assump-tion of volume additivity, which is typically reasonable inpolymers of interest for the applications addressed in thisreview.
In the solution–diffusion model, the amount of watersorbed by a polymer is characterized by the water sorptioncoefficient, Kw, as introduced in Eq. (1). The units of Kw are[g(H2O)/cm3 (swollen polymer)]/[g(H2O)/cm3(solution)]
er Science 39 (2014) 1– 42
[114,132]. The water sorption coefficient is related to �w
by [81]
Kw = �wMw
cswVw
(9)
where Mw is the molecular weight of water, and csw is
the mass concentration of water in the external solution.When cs
w is approximately equal to the density of purewater, which is often the case in desalination applications,the water sorption coefficient is essentially equal to thevolume fraction of water dissolved in the polymer (i.e.,Kw ≈ �w) [81,133]. According to Eq. (1), polymers that sorbmore water generally exhibit higher water permeabilityvalues than those polymers that sorb less water, in part dueto the appearance of Kw in the expression for Pw [13,107].Additionally, water content also strongly influences waterand salt diffusion coefficients, as will be discussed in moredetail later. Generally, non-porous polymers for watertreatment applications (e.g., RO or FO) need to be at leastsomewhat hydrophilic to achieve adequate water per-meability [13,107]. Synthetic methods, such as additionof sulfonated groups to the backbone of an otherwisehydrophobic polymer, for increasing hydrophilicity willbe discussed in more detail later in this review.
A common measure of a polymer membrane’s abilityto separate two components is the permeability selectiv-ity, ˛, defined as the ratio of the permeability of the morepermeable penetrant to that of the less permeable pen-etrant. Permeability selectivity is commonly viewed as amaterial property [107,132]. The permeability selectivity,˛, for water/salt separation, can be written as follows [132]
≡ Pw
Ps= Kw
Ks× Dw
Ds(10)
where the so-called sorption, or solubility, selectivity isKw/Ks, and the so-called diffusivity, or mobility, selectiv-ity is Dw/Ds. In the desalination literature, salt rejectionis often reported as a measure of a membrane’s selec-tivity. Rejection describes the removal of a given soluteunder specified operating conditions, so it is not a mate-rial property per se [106,132]. The apparent salt rejection,R, is defined by the salt concentrations in the feed and per-meate solutions, and it is related to selectivity as follows[107,132]
R = 1 − cssL
css0
= ˛ VwRT (�p − ��)
1 + ˛ VwRT (�p − ��)
(11)
Salt passage is defined as 1 – R, and both salt rejectionand passage are typically expressed as a percentage. Eq.(11) illustrates the dependence of salt rejection on filtrationconditions (e.g., �p and ��).
1.2. Experimental verification of the solution–diffusionmodel
Experimental results have validated the
solution–diffusion mechanism of transport in non-porous polymer films (e.g., the active layers of RO or FOmembranes). A key distinction between pore flow andsolution–diffusion transport is the concentration profile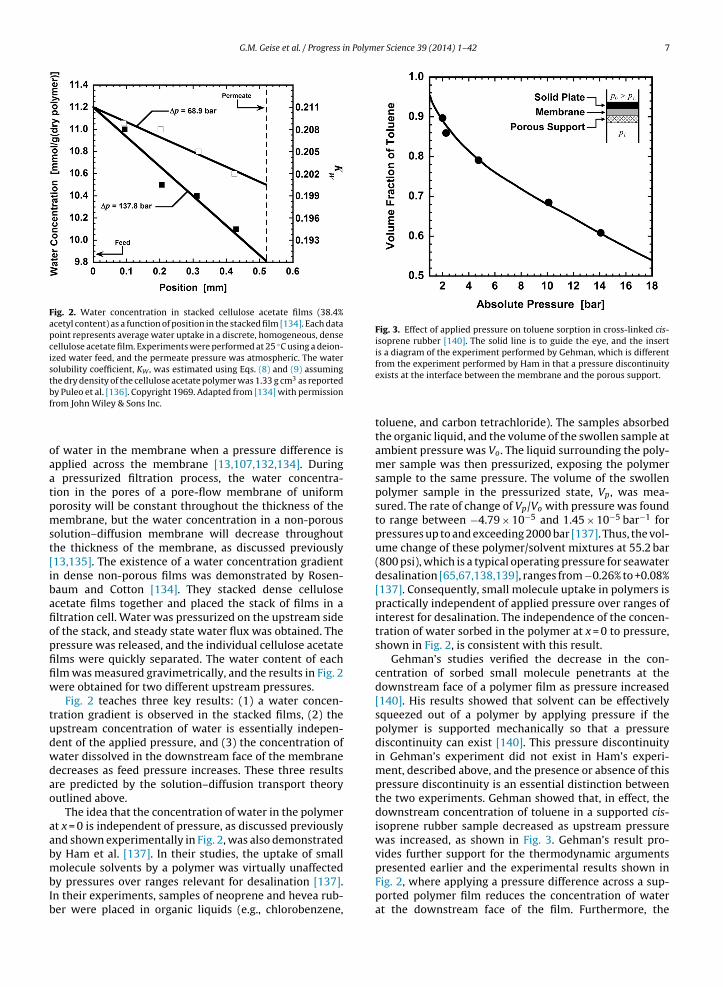
G.M. Geise et al. / Progress in Polymer Science 39 (2014) 1– 42 7
Fig. 2. Water concentration in stacked cellulose acetate films (38.4%acetyl content) as a function of position in the stacked film [134]. Each datapoint represents average water uptake in a discrete, homogeneous, densecellulose acetate film. Experiments were performed at 25 ◦C using a deion-ized water feed, and the permeate pressure was atmospheric. The watersolubility coefficient, KW , was estimated using Eqs. (8) and (9) assumingthe dry density of the cellulose acetate polymer was 1.33 g cm3 as reportedbf
oaatpmst[ibafiopfifiw
tudwdao
aabmbIb
Fig. 3. Effect of applied pressure on toluene sorption in cross-linked cis-isoprene rubber [140]. The solid line is to guide the eye, and the insertis a diagram of the experiment performed by Gehman, which is differentfrom the experiment performed by Ham in that a pressure discontinuity
y Puleo et al. [136]. Copyright 1969. Adapted from [134] with permissionrom John Wiley & Sons Inc.
f water in the membrane when a pressure difference ispplied across the membrane [13,107,132,134]. During
pressurized filtration process, the water concentra-ion in the pores of a pore-flow membrane of uniformorosity will be constant throughout the thickness of theembrane, but the water concentration in a non-porous
olution–diffusion membrane will decrease throughouthe thickness of the membrane, as discussed previously13,135]. The existence of a water concentration gradientn dense non-porous films was demonstrated by Rosen-aum and Cotton [134]. They stacked dense cellulosecetate films together and placed the stack of films in altration cell. Water was pressurized on the upstream sidef the stack, and steady state water flux was obtained. Theressure was released, and the individual cellulose acetatelms were quickly separated. The water content of eachlm was measured gravimetrically, and the results in Fig. 2ere obtained for two different upstream pressures.
Fig. 2 teaches three key results: (1) a water concen-ration gradient is observed in the stacked films, (2) thepstream concentration of water is essentially indepen-ent of the applied pressure, and (3) the concentration ofater dissolved in the downstream face of the membraneecreases as feed pressure increases. These three resultsre predicted by the solution–diffusion transport theoryutlined above.
The idea that the concentration of water in the polymert x = 0 is independent of pressure, as discussed previouslynd shown experimentally in Fig. 2, was also demonstratedy Ham et al. [137]. In their studies, the uptake of small
olecule solvents by a polymer was virtually unaffectedy pressures over ranges relevant for desalination [137].n their experiments, samples of neoprene and hevea rub-er were placed in organic liquids (e.g., chlorobenzene,
exists at the interface between the membrane and the porous support.
toluene, and carbon tetrachloride). The samples absorbedthe organic liquid, and the volume of the swollen sample atambient pressure was Vo. The liquid surrounding the poly-mer sample was then pressurized, exposing the polymersample to the same pressure. The volume of the swollenpolymer sample in the pressurized state, Vp, was mea-sured. The rate of change of Vp/Vo with pressure was foundto range between −4.79 × 10−5 and 1.45 × 10−5 bar−1 forpressures up to and exceeding 2000 bar [137]. Thus, the vol-ume change of these polymer/solvent mixtures at 55.2 bar(800 psi), which is a typical operating pressure for seawaterdesalination [65,67,138,139], ranges from −0.26% to +0.08%[137]. Consequently, small molecule uptake in polymers ispractically independent of applied pressure over ranges ofinterest for desalination. The independence of the concen-tration of water sorbed in the polymer at x = 0 to pressure,shown in Fig. 2, is consistent with this result.
Gehman’s studies verified the decrease in the con-centration of sorbed small molecule penetrants at thedownstream face of a polymer film as pressure increased[140]. His results showed that solvent can be effectivelysqueezed out of a polymer by applying pressure if thepolymer is supported mechanically so that a pressurediscontinuity can exist [140]. This pressure discontinuityin Gehman’s experiment did not exist in Ham’s experi-ment, described above, and the presence or absence of thispressure discontinuity is an essential distinction betweenthe two experiments. Gehman showed that, in effect, thedownstream concentration of toluene in a supported cis-isoprene rubber sample decreased as upstream pressurewas increased, as shown in Fig. 3. Gehman’s result pro-vides further support for the thermodynamic arguments
presented earlier and the experimental results shown inFig. 2, where applying a pressure difference across a sup-ported polymer film reduces the concentration of waterat the downstream face of the film. Furthermore, the
8 G.M. Geise et al. / Progress in Polymer Science 39 (2014) 1– 42
Fig. 4. Permeability selectivity versus water permeability for a variety ofpolymers [132]. The symbols are defined in Table 2, and the solid lineis drawn empirically based on the experimental data. To be consistentwith previously reported results, the diffusive water permeability valuesplotted here were calculated from hydraulic permeability measurementsusing Eq. (7). The symbol (©) corresponds to a hypothetical membranecomposed of water; the water permeability of such a membrane is takento be the product of the self-diffusion coefficient of water and the watersorption coefficient for pure water (i.e., unity), and the permeability selec-tivity is taken as the ratio of the water permeability to the permeabilityof salt in pure water (i.e., the product of the diffusion coefficient of NaClin water and the salt sorption coefficient in pure water, which is unity).
Table 2Legend for tradeoff relationship plots.
Name Symbol References
Polyimide � [77]Aromatic polyamide �,� [77,78]Poly(benzimi-dazopyrrolone) � [77]Poly(amide-hydrazide) � [77]Di-sulfonated poly(arylene
ether sulfone)� [80,81,141]
Cellulose acetate × [64,76]Cross-linked poly(ethylene
glycol diacrylate)� [101]
Poly(2-hydroxylethyl acrylate) ♦ [102]Poly(acrylic acid) � [102]Hydrogels from Yasuda et al. + [99]Methacrylate-based
copolymers [142]
Poly(hydroxylethylmethacrylamide)-basedhydrogels
[142]
Hypothetical membrane ©
Copyright 2010. Reproduced from [132] with permission from ElsevierLtd.
observations made with water in polymers are not anoma-lies related specifically to water. Rather, they are generalconsequences of the thermodynamics of sorption of anysmall molecule into a polymer.
1.3. Water/salt tradeoff relationship in swollen polymers
For many membrane applications, polymers withcombinations of high permeability and high selectivityproperties are desirable. Therefore, it is useful to developstructure/property relationships in terms of permeabil-ity and permeability selectivity. Recently, an empiricaltradeoff of water permeability, sorption, and diffusiv-ity with water/salt permeability, sorption, and diffusivityselectivity has been recognized [132]. In this relation-ship, water/sodium chloride permeability, sorption, anddiffusivity selectivities decrease with increasing waterpermeability, sorption, and diffusivity for a variety ofdesalination materials. The relationship for water/sodiumchloride permeability selectivity and water permeability,shown as Fig. 4, results from sorption and diffusivity trade-off relationships that will be discussed in this section. Thetradeoff between diffusion and diffusivity selectivity, how-ever, is the dominant factor giving rise to the behavior inFig. 4 (Table 2).
Tradeoffs between throughput and separation perfor-mance are observed in many areas of membrane science,including gas separation membranes [96–98], fuel cellmembranes [143,144], and ultrafiltration (UF) membranes
composed of water
[145], despite the fact that the polymers, solutes of interestand even the molecular transport mechanism, at least in thecase of UF membranes, is different in these various cases.Thin-film composite polyamide membranes follow a sim-ilar tradeoff relationship, though their water permeability(as reported on the horizontal axis in Fig. 4) depends on anassumed effective active layer thickness as the active layersin thin-film composite membranes are likely not uniform[146,147]. While subsequent sections of this review focus,in more detail, on the differences between un-chargedand charged desalination polymers, this section introducesgeneral concepts that appear to apply to a wide spectrumof polymer materials.
The tradeoff relationship between the average waterdiffusion coefficient and water/NaCl diffusion selectiv-ity is shown in Fig. 5 [132]. Similar to the permeabilitytradeoff relationship discussed previously, polymers thathave large average water diffusion coefficients tend tohave small water/salt diffusion selectivity and vice versa[93,96,97,132]. For a few polymers that sorb considerableamounts of water, the apparent water diffusion coefficientexceeds the value of the water self-diffusion coefficient.Apparent water diffusion coefficients in polymers greaterthan the self-diffusion coefficient of water are a resultof convective frame of reference effects in the polymersthat are described in more detail in Appendix A. The exis-tence of this tradeoff relationship is qualitatively consistentwith free volume theory as will be discussed subse-quently.
Fig. 6 presents the salt sorption selectivity, definedas Kw/Ks, as a function of water uptake for a variety ofpolymers. A tradeoff between the water sorption coef-ficient and water/salt sorption selectivity is apparentin Fig. 6. Sorption, or partitioning, of salt into a poly-mer matrix generally depends on polymer water uptake
[99,101,102,148]. However, the sorption selectivity valuesshown in Fig. 6 vary over roughly an order of magnitude atequivalent water uptake indicating that polymer proper-ties influence salt sorption in materials that have the same
G.M. Geise et al. / Progress in Polymer Science 39 (2014) 1– 42 9
Fig. 5. Diffusivity selectivity versus the effective water diffusion coeffi-cient for a variety of polymers [132]. The symbols are defined in Table 2,and the solid line is drawn empirically based on the experimental data. Thesymbol (©) corresponds to a hypothetical membrane composed of water;the self-diffusion coefficient of water and the diffusion coefficient of NaClin water were used to determine the position of this symbol. Copyright2010. Reproduced from [132] with permission from Elsevier Ltd.
wmsscpq
FoaTww
Fig. 7. Sodium chloride diffusion coefficients for a variety of hydrogels (�)[99,101,102] plotted against 1/Kw as motivated by Yasuda et al. [99]. The
◦ −1
ater uptake [132]. While many polymer–salt interactionechanisms could contribute to the water/salt sorption
electivity, whether the polymer is charged or not has atrong impact on salt sorption properties under certainircumstances [13,104,105,142,148,149]. The influence ofolymer charge on salt sorption is discussed in a subse-uent section.
ig. 6. Sorption selectivity versus water sorption coefficient for a varietyf polymers [64,101,102,132,142]. The symbols are defined in Table 2,nd the solid line is drawn empirically based on the experimental data.he symbol (©) corresponds to a hypothetical membrane composed ofater; in such a membrane, both the water sorption coefficient and theater/salt selectivity values are equal to 1.
NaCl diffusion coefficient in water (i.e., Kw = 1) at 25 C and 0.01 mol L(1.5 × 10−6 cm2 s−1) is shown as (�) [152].
1.4. Effect of free volume on solution diffusion andpermeation properties
Free volume theory relates the diffusion coefficient ofpenetrant i, Di, to the polymer’s average free volume
⟨vf
⟩as follows [150]:
Di = ai × exp
[− bi⟨
vf
⟩]
(12)
where ai and bi are adjustable constants. The constant biis proportional to the size of penetrant i [150]. The dif-fusivity tradeoff relationship shown in Fig. 5 is consistentwith Eq. (12) in that, as a polymer’s average free volumeincreases, the diffusion coefficients of the penetrants willincrease while, simultaneously, the diffusion selectivity ofthe smaller penetrant over the larger will decrease becausebi is proportional to penetrant size [132].
Yasuda et al. suggested that the average free volume,⟨vf
⟩, in swollen hydrogels would increase in proportion to
the volume fraction of water in, or the water sorption coef-ficient of, the hydrated polymer, i.e.,
⟨vf
⟩∼Kw [99]. Support
for Yasuda et al.’s suggestion is shown in Fig. 7 where aver-age NaCl diffusion coefficients in a variety of polymers arecorrelated with 1/Kw as motivated by Eq. (12). Gratifyingly,the data for a wide range of polymers correlate with 1/Kw
and intersect the diffusion coefficient of NaCl in water at1/Kw = 1 (a membrane with Kw = 1 would be a hypotheti-cal membrane composed of water), suggesting that watersorption has a strong impact on free volume in a variety ofswollen polymers [99,101,151]. However, there is scatter
about the trend line through the data, suggesting that fac-tors other than simply average water uptake also influencesalt transport.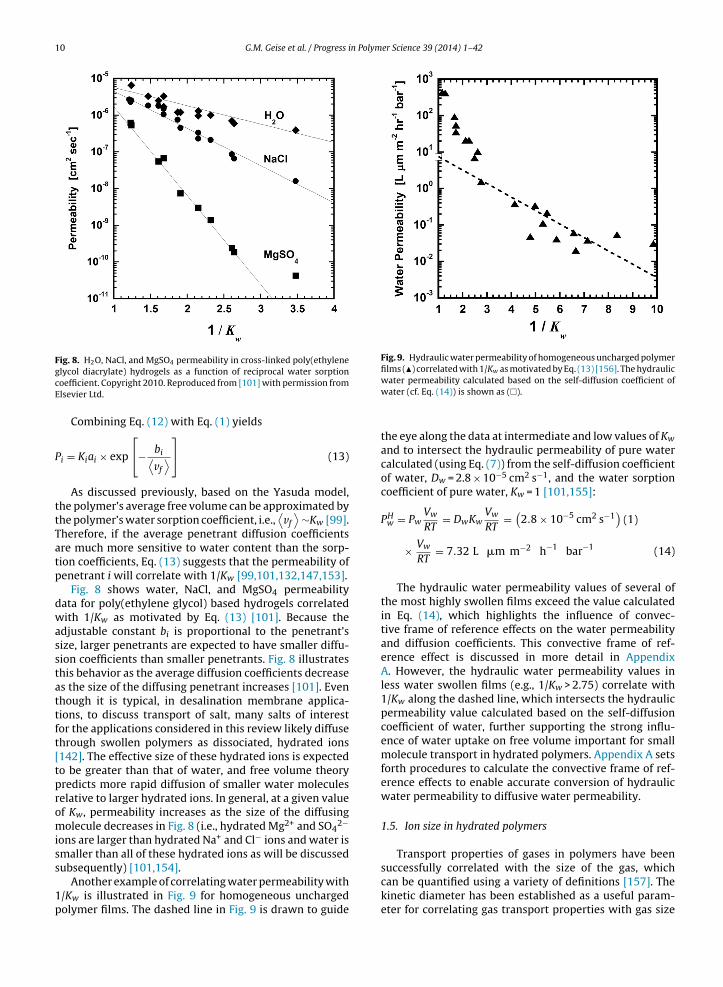
10 G.M. Geise et al. / Progress in Polymer Science 39 (2014) 1– 42
Fig. 8. H2O, NaCl, and MgSO4 permeability in cross-linked poly(ethylene Fig. 9. Hydraulic water permeability of homogeneous uncharged polymerfilms (�) correlated with 1/Kw as motivated by Eq. (13) [156]. The hydraulic
glycol diacrylate) hydrogels as a function of reciprocal water sorptioncoefficient. Copyright 2010. Reproduced from [101] with permission fromElsevier Ltd.
Combining Eq. (12) with Eq. (1) yields
Pi = Kiai × exp
[− bi⟨
vf
⟩]
(13)
As discussed previously, based on the Yasuda model,the polymer’s average free volume can be approximated bythe polymer’s water sorption coefficient, i.e.,
⟨vf
⟩∼Kw [99].
Therefore, if the average penetrant diffusion coefficientsare much more sensitive to water content than the sorp-tion coefficients, Eq. (13) suggests that the permeability ofpenetrant i will correlate with 1/Kw [99,101,132,147,153].
Fig. 8 shows water, NaCl, and MgSO4 permeabilitydata for poly(ethylene glycol) based hydrogels correlatedwith 1/Kw as motivated by Eq. (13) [101]. Because theadjustable constant bi is proportional to the penetrant’ssize, larger penetrants are expected to have smaller diffu-sion coefficients than smaller penetrants. Fig. 8 illustratesthis behavior as the average diffusion coefficients decreaseas the size of the diffusing penetrant increases [101]. Eventhough it is typical, in desalination membrane applica-tions, to discuss transport of salt, many salts of interestfor the applications considered in this review likely diffusethrough swollen polymers as dissociated, hydrated ions[142]. The effective size of these hydrated ions is expectedto be greater than that of water, and free volume theorypredicts more rapid diffusion of smaller water moleculesrelative to larger hydrated ions. In general, at a given valueof Kw, permeability increases as the size of the diffusingmolecule decreases in Fig. 8 (i.e., hydrated Mg2+ and SO4
2−
ions are larger than hydrated Na+ and Cl− ions and water issmaller than all of these hydrated ions as will be discussed
subsequently) [101,154].Another example of correlating water permeability with1/Kw is illustrated in Fig. 9 for homogeneous unchargedpolymer films. The dashed line in Fig. 9 is drawn to guide
water permeability calculated based on the self-diffusion coefficient ofwater (cf. Eq. (14)) is shown as (�).
the eye along the data at intermediate and low values of Kw
and to intersect the hydraulic permeability of pure watercalculated (using Eq. (7)) from the self-diffusion coefficientof water, Dw = 2.8 × 10−5 cm2 s−1, and the water sorptioncoefficient of pure water, Kw = 1 [101,155]:
PHw = Pw
Vw
RT= DwKw
Vw
RT=(
2.8 × 10−5 cm2 s−1)
(1)
× Vw
RT= 7.32 L �m m−2 h−1 bar−1 (14)
The hydraulic water permeability values of several ofthe most highly swollen films exceed the value calculatedin Eq. (14), which highlights the influence of convec-tive frame of reference effects on the water permeabilityand diffusion coefficients. This convective frame of ref-erence effect is discussed in more detail in AppendixA. However, the hydraulic water permeability values inless water swollen films (e.g., 1/Kw > 2.75) correlate with1/Kw along the dashed line, which intersects the hydraulicpermeability value calculated based on the self-diffusioncoefficient of water, further supporting the strong influ-ence of water uptake on free volume important for smallmolecule transport in hydrated polymers. Appendix A setsforth procedures to calculate the convective frame of ref-erence effects to enable accurate conversion of hydraulicwater permeability to diffusive water permeability.
1.5. Ion size in hydrated polymers
Transport properties of gases in polymers have been
successfully correlated with the size of the gas, whichcan be quantified using a variety of definitions [157]. Thekinetic diameter has been established as a useful param-eter for correlating gas transport properties with gas size
n Polymer Science 39 (2014) 1– 42 11
[tstucmin
sefatwitftdsmo[
lspctantthtTdtts
mtsnTsstcss
ifptpo
Table 3Measures of ion size [154].
Ion Crystal(pauling)radius [Å]
Stokesradius [Å]
Hydratedradius [Å]
Aluminum Al3+ 0.50 4.39 4.75Lithium Li+ 0.60 2.38 3.82Iron(III) Fe3+ 0.60 4.06 4.57Magnesium Mg2+ 0.65 3.47 4.28Copper Cu2+ 0.72 3.25 4.19Zinc Zn2+ 0.74 3.49 4.30Iron(II) Fe2+ 0.75 3.44 4.28Sodium Na+ 0.95 1.84 3.58Calcium Ca2+ 0.99 3.10 4.12Potassium K+ 1.33 1.25 3.31Ammonium NH4
+ 1.48 1.25 3.31Chloride Cl− 1.81 1.21 3.32Bromide Br− 1.95 1.18 3.30Nitrate NO3
− 2.64 1.29 3.35Carbonate CO3
2− 2.66 2.66 3.94
G.M. Geise et al. / Progress i
157]. Comparatively little is known about the best parame-ers for correlating ion transport properties with ion size inwollen polymer membranes [132]. This section addresseshe influence of ion size on salt transport properties ofncharged polymers, but additional studies (both theoreti-al and experimental) are needed to fully understand whateasure of ion size is most appropriate for correlation with
on transport properties in materials of interest for desali-ation applications.
According to activated state diffusion theory, the diffu-ion coefficient of a penetrant depends on the activationnergy, as defined by an Arrhenius relationship, requiredor that penetrant to execute a diffusion step [158]. Thisctivation energy is a function of the effective size ofhe diffusing molecule and has been suggested to scaleith the square of the effective diameter of the diffus-
ng molecule (e.g., the kinetic diameter for gas diffusionhrough polymers).[95] Therefore, the activation energyor diffusion generally increases as the size of the pene-rant increases, and that penetrant’s diffusion coefficientecreases (i.e., large molecules diffuse more slowly thanmall molecules) [95,158]. Ion diffusion through polymersay be governed by similar physics, but the effective size
f the diffusing ion is not as well defined as that for gases132].
The effective size of an ion diffusing through a polymerikely will exist between two extreme limits. In aqueousolutions, ions hydrate due to favorable interactions witholar water molecules [159]. At least some of these so-alled waters of hydration are strongly associated withhe ion, acting to increase the effective size of the ions it diffuses through the solution [159,160]. While theumber of water molecules that hydrate an ion in solu-ion and the affinity of those water molecules for the ionhat they hydrate are still debated in the literature, ionydration increases the effective size of the ion relative tohe ion’s crystallographic (i.e., unhydrated) size [159,160].herefore, the upper limit for the effective size of an ioniffusing through a polymer may be thought of as beinghe size of the fully hydrated ion (i.e., the extent of hydra-ion observed when the ion is present in dilute aqueousolution).
An ion’s extent of hydration while sorbed in a polymeray be different from that observed in a dilute solu-
ion containing that ion. When the ionic strength (i.e.,alt concentration) of an aqueous solution increases, theumber of waters of hydration often decreases [161].herefore, extent of ion hydration in a polymer may beomewhat less than that observed in a dilute aqueousolution due to influences from elevated salt concentra-ion and/or the polymer itself (perhaps particularly forharged polymers), causing hydrated ions to be effectivelyomewhat smaller in a polymer than in dilute aqueousolution.
The lower size limit is the case where the ion shedsts waters of hydration, upon sorbing into the polymer, inavor of interactions with the polymer matrix. An exam-
le of such a situation is found in the selective filter ofhe potassium channels used to control potassium trans-ort through cell walls in nature [162]. Thus, the radiusf the unhydrated ion may be considered to be a lowerSulfate SO42− 2.90 2.30 3.79
Note: The radius of a water molecule is reported as 1.38 A [154].
limit on its effective size when diffusing through a poly-mer.
Additionally, an intermediate size could be consideredwhere the ion sheds its waters of hydration, upon sorb-ing into the polymer, and pairs (or un-dissociates) with anoppositely charged ion that is also sorbed in the polymermatrix (e.g., Na+ + Cl− → NaCl) [142]. Such behavior wouldbe most likely to occur in low dielectric constant polymers[142]. This un-dissociated salt would be effectively largerthan each of the individual dissociated unhydrated ions andcould be effectively smaller than the dissociated hydratedions [142,163]. Therefore, an un-dissociated salt could dif-fuse more rapidly through a polymer than its effectivelylarger dissociated and hydrated ions [142]. While ion pair-ing in solution has been discussed extensively [163], it isdifficult to experimentally identify ion pairing in hydratedpolymers [142].
Table 3 lists the crystal, Stokes, and hydrated radii ofseveral ions of interest for water treatment applications asthese radii are often used to report ion size. The crystalradii, often reported using the definition of Pauling [164],which defines the crystal radius as the length between theion’s nucleus and the outer-most electrons, describes theion’s size in a crystal lattice (e.g., the radius of sodium orchloride in a dry sodium chloride crystal) [159]. The Stokesradii are calculated using the Stokes–Einstein equationand ion diffusion coefficients measured in dilute aque-ous solutions [154,159,160]. The Stokes–Einstein equationapplied in this manner, however, tends to yield a radius(the Stokes radius) that is believed to be smaller than thetrue hydrated radius of the ion because the Stokes–Einsteinmodel assumes that the diffusing species is sphericalin nature while diffusing through a continuous medium[154,159]. Because of the molecular length scales relevantfor ion transport through water, it may be inappropri-ate to consider water as a continuous medium through
which the ion diffuses [154,159,160]. To account for thisviolation of the assumptions of the Stokes–Einstein equa-tion, a correction can be applied to the Stokes radii tocalculate the effective hydrated radii reported in Table 3.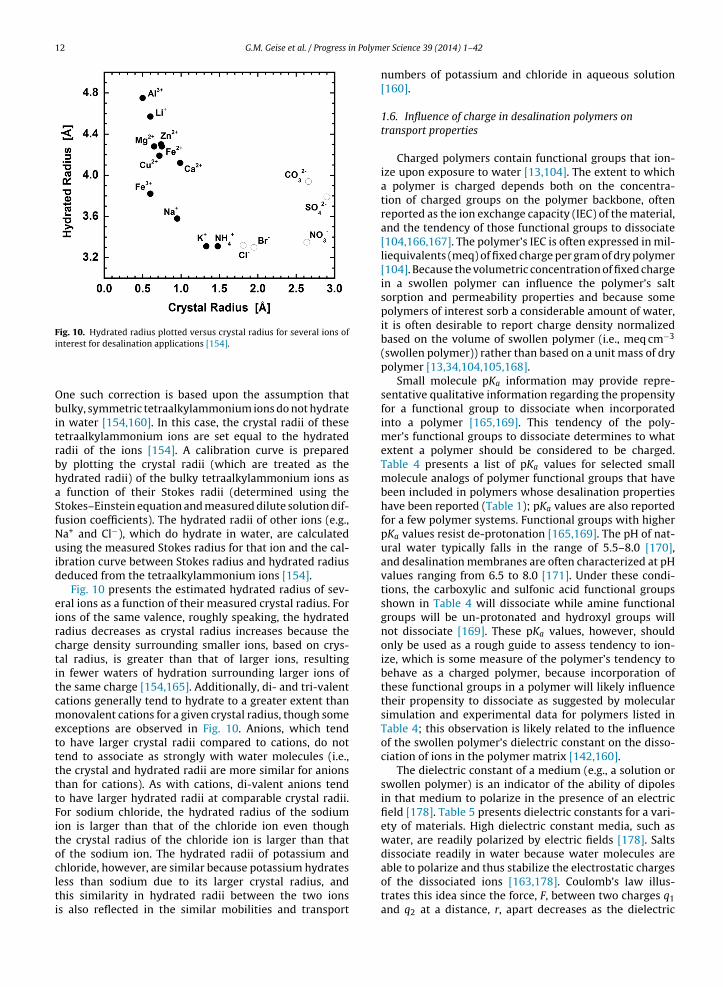
12 G.M. Geise et al. / Progress in Polym
Fig. 10. Hydrated radius plotted versus crystal radius for several ions of
able to polarize and thus stabilize the electrostatic chargesof the dissociated ions [163,178]. Coulomb’s law illus-
interest for desalination applications [154].
One such correction is based upon the assumption thatbulky, symmetric tetraalkylammonium ions do not hydratein water [154,160]. In this case, the crystal radii of thesetetraalkylammonium ions are set equal to the hydratedradii of the ions [154]. A calibration curve is preparedby plotting the crystal radii (which are treated as thehydrated radii) of the bulky tetraalkylammonium ions asa function of their Stokes radii (determined using theStokes–Einstein equation and measured dilute solution dif-fusion coefficients). The hydrated radii of other ions (e.g.,Na+ and Cl−), which do hydrate in water, are calculatedusing the measured Stokes radius for that ion and the cal-ibration curve between Stokes radius and hydrated radiusdeduced from the tetraalkylammonium ions [154].
Fig. 10 presents the estimated hydrated radius of sev-eral ions as a function of their measured crystal radius. Forions of the same valence, roughly speaking, the hydratedradius decreases as crystal radius increases because thecharge density surrounding smaller ions, based on crys-tal radius, is greater than that of larger ions, resultingin fewer waters of hydration surrounding larger ions ofthe same charge [154,165]. Additionally, di- and tri-valentcations generally tend to hydrate to a greater extent thanmonovalent cations for a given crystal radius, though someexceptions are observed in Fig. 10. Anions, which tendto have larger crystal radii compared to cations, do nottend to associate as strongly with water molecules (i.e.,the crystal and hydrated radii are more similar for anionsthan for cations). As with cations, di-valent anions tendto have larger hydrated radii at comparable crystal radii.For sodium chloride, the hydrated radius of the sodiumion is larger than that of the chloride ion even thoughthe crystal radius of the chloride ion is larger than thatof the sodium ion. The hydrated radii of potassium andchloride, however, are similar because potassium hydratesless than sodium due to its larger crystal radius, and
this similarity in hydrated radii between the two ionsis also reflected in the similar mobilities and transporter Science 39 (2014) 1– 42
numbers of potassium and chloride in aqueous solution[160].
1.6. Influence of charge in desalination polymers ontransport properties
Charged polymers contain functional groups that ion-ize upon exposure to water [13,104]. The extent to whicha polymer is charged depends both on the concentra-tion of charged groups on the polymer backbone, oftenreported as the ion exchange capacity (IEC) of the material,and the tendency of those functional groups to dissociate[104,166,167]. The polymer’s IEC is often expressed in mil-liequivalents (meq) of fixed charge per gram of dry polymer[104]. Because the volumetric concentration of fixed chargein a swollen polymer can influence the polymer’s saltsorption and permeability properties and because somepolymers of interest sorb a considerable amount of water,it is often desirable to report charge density normalizedbased on the volume of swollen polymer (i.e., meq cm−3
(swollen polymer)) rather than based on a unit mass of drypolymer [13,34,104,105,168].
Small molecule pKa information may provide repre-sentative qualitative information regarding the propensityfor a functional group to dissociate when incorporatedinto a polymer [165,169]. This tendency of the poly-mer’s functional groups to dissociate determines to whatextent a polymer should be considered to be charged.Table 4 presents a list of pKa values for selected smallmolecule analogs of polymer functional groups that havebeen included in polymers whose desalination propertieshave been reported (Table 1); pKa values are also reportedfor a few polymer systems. Functional groups with higherpKa values resist de-protonation [165,169]. The pH of nat-ural water typically falls in the range of 5.5–8.0 [170],and desalination membranes are often characterized at pHvalues ranging from 6.5 to 8.0 [171]. Under these condi-tions, the carboxylic and sulfonic acid functional groupsshown in Table 4 will dissociate while amine functionalgroups will be un-protonated and hydroxyl groups willnot dissociate [169]. These pKa values, however, shouldonly be used as a rough guide to assess tendency to ion-ize, which is some measure of the polymer’s tendency tobehave as a charged polymer, because incorporation ofthese functional groups in a polymer will likely influencetheir propensity to dissociate as suggested by molecularsimulation and experimental data for polymers listed inTable 4; this observation is likely related to the influenceof the swollen polymer’s dielectric constant on the disso-ciation of ions in the polymer matrix [142,160].
The dielectric constant of a medium (e.g., a solution orswollen polymer) is an indicator of the ability of dipolesin that medium to polarize in the presence of an electricfield [178]. Table 5 presents dielectric constants for a vari-ety of materials. High dielectric constant media, such aswater, are readily polarized by electric fields [178]. Saltsdissociate readily in water because water molecules are
trates this idea since the force, F, between two charges q1and q2 at a distance, r, apart decreases as the dielectric

G.M. Geise et al. / Progress in Polymer Science 39 (2014) 1– 42 13
Table 4pKa values reported for selected polymer systems and several small molecules that contain functional groups that are similar to those found in desalinationpolymers.
Functional group/compound Structure pKa*
Alkyl hydroxyl group [172] –OH 15–16
Phenol [172] 9.99
m-Phenylenediamine [172] 5.112.50
1,3,5-Benzenetricarboxylic (trimesic) acid [173] 4.73.93.1
Aniline [172] 4.87
1,3-Benzenedicarboxylic (isophthalic) acid [172] 4.603.70
Cyclohexanecarboxylic acid [172] 4.91
Benzoic acid [172] 4.20
Aromatic polyamide primary amine group [174] (Molecular simulation) 3.97Aromatic polyamide carboxyl group [174] (Molecular simulation) 3.74
Perfluoropropanoic acid [175] 2.5
Perfluorinated carboxylated polymer [176] 1.9
Acetanilide [172] 0.5
Perfluorinated sulfonated polymer [176] <1
Benzene sulfonic acid [177] −6.65
* The reported pKa values correspond to the conjugate acid form of the listed structure.

14 G.M. Geise et al. / Progress in Polymer Science 39 (2014) 1– 42
Table 5Dielectric constants for various substances at 25 ◦C (unless indicated otherwise) [172].
Medium Structure Dielectric constant, εr
Vacuum 1 (by definition)Dry air (CO2 free) at 1 atm 1.0005360Carbon dioxide at 1 atm O=C=O 1.0009217
Benzene 2.28
Polystyrene 2.6
Poly(methyl methacrylate) at 27 ◦C 3.12
Polyacrylonitrile 5.5
1-octanol 10.30
[120,166,183]. Cross-linked poly(ethylene glycol diacry-late) (XLPEGDA) hydrogels are examples of un-charged
Table 6Dielectric constants of water vapor equilibrated Nafion® 117(IEC = 0.909 meq g−1 (dry polymer)) at 30 ◦C as a function of watersorption [180].
Number of water moleculesper sulfonate group
Kw Dielectric constant
13 0.277 206 0.150 133 0.081 82 0.056 5
Water
constant (or relative permittivity) of the medium, εr,increases [179]
F = 14�ε0εr
q1q2
r2(15)
where ε0 is the permittivity of free space(8.85 × 10−12 C V−1 m−1). In general, as the dielectricconstant of a medium increases, the tendency of ionsto dissociate increases, and electrostatic forces betweencharged species are reduced [163]. As illustrated forNafion® 117 in Table 6 [180], increased sorption of waterin a polymer correlates with an increase in the dielectricconstant of the polymer/water mixture. Thus, ion dis-sociation is typically favored in a more highly swollenpolymer than in a less highly swollen polymer with a lowerdielectric constant, and ion dissociation is most favored inwater (i.e., with no polymer present). Therefore, functionalgroups in a polymer may be less likely to dissociate thansmall molecule analogs of the same functional groups inan aqueous solution [142,160].
Cellulose acetate (Table 1), which is typically employedin desalination applications as ∼40% acetylated celluloseacetate (i.e., the degree of acetylation is typically around2.7) [115], is an example of a polymer expected to be
80.1
relatively un-charged because this polymer’s hydroxylgroups are not expected to dissociate under typical desali-nation conditions. However, some experimental evidencesuggests that residual carboxylic acid functionality existsin cellulose acetate, and these charged groups may causecellulose acetate to behave as though it is weakly charged
1 0.029 4
Note: The water sorption coefficient was calculated using the water con-tent data in this table, the IEC, dry density of Nafion® 117 (1.8 g cm−3
[181,182]), and Eq. (8).

n Polymer Science 39 (2014) 1– 42 15
pt
pbcsca[f[wif
mtTmmtotftdcropc
ass[wwapccFfinwspmcmOmtatciXm
Fig. 11. Salt sorption in an uncharged polymer (left) and a cation exchangecharged polymer containing fixed negative charge groups, A− (right)such as sulfonate groups. In the uncharged polymer, the concentrationof sorbed cations is equal to the concentration of sorbed anions (i.e.,
G.M. Geise et al. / Progress i
olymers because they do not contain functional groupshat dissociate in water [99,101].
As dissociating functional groups are added to aolymer’s structure, the IEC increases, and the polymerecomes more charged [13,104,105]. Ionomers, such asarboxylated and sulfonated polymers, are typically con-idered to be charged polymers [13,104,105]. The extent ofharge depends on the concentration of functional groupsnd the tendency of the functional groups to dissociate120]. For example, based upon the pKa data in Table 4, sul-onic acids dissociate far more readily than carboxylic acids165,169]. Carboxylated polymers are often referred to aseakly charged polymers because, at the pH values used
n many water treatment applications, the carboxylic acidunctional groups are not strongly dissociated [120,176].
An ideal charged polymer can be defined, from aodeling perspective, as having a uniform electric poten-
ial throughout the polymer’s volume [13,104,184–186].he uniform electric potential of an ideal charged poly-er, described above, is highly idealized because theolecular-scale distribution of fixed charge groups on
he polymer backbone inherently introduce some levelf molecular-scale heterogeneity in the charge distribu-ion [184–186]. Additionally, dissociation of some chargedunctional groups, such as carboxyl groups, depends onhe pH of the solution in contact with the film [120]. Theielectric constant of the swollen film and the sorbed saltoncentration may affect the effective IEC of the mate-ial and, therefore, charge [120,142,166,167]. The extentf charge and/or the uniformity of the charge within theolymer influence ion sorption and diffusion properties ofharged polymers [104,133,148].
In sulfonated polymers considered for desalinationpplications, polymer charge is believed to influencealt transport by, among other things, influencingalt sorption into and diffusion through the polymer13,85,104,105,120,133,148,186–188], but the extent tohich this effect can be leveraged to prepare materialsith high salt rejection is not fully understood. Addition-
lly, polymer charge has been reported to influence theolymer’s propensity for fouling, which is a significanthallenge for membrane design [79,189–192]. Polymerharge influences individual ion sorption as illustrated byig. 11, which depicts an uncharged and a charged polymerlm immersed in a salt solution, M+X−, of infinite volume ateutral pH (note: if M+X− were an acid or base, then the pHould be not be neutral) [13,148]. On the left side of Fig. 11,
alt sorption in an uncharged polymer proceeds by a simpleartitioning mechanism whereby ions sorb into the poly-er matrix as mobile salt (i.e., each equivalent of sorbed
ations is accompanied by an equivalent of sorbed anions toaintain electro-neutrality within the film) [13,104,148].n the right side of Fig. 11, ion sorption in a cation exchangeaterial proceeds by both ion exchange and simple par-
itioning mechanisms [13,104,148]. In the absence of anpplied electric field, electro-neutrality must be main-ained at all times, so fixed charge groups associate with
ounter-ions [34,104,105,168]. Cations, M+, act as counter-ons to both the fixed charge groups, A−, and sorbed anions,−. Therefore, cation exchange polymers generally sorbore cations than anions on an equivalent basis (i.e., CmM >
CmM
= CmX
), but in the cation exchange polymer, the concentration of sorbedcations is greater than the concentration of sorbed anions (i.e., Cm
M> Cm
X)
[148].
CmX ) [104,188,193]. This difference in anion and cation con-
centration in the polymer is balanced electrostatically bythe polymer’s fixed charge groups, and this phenomenonis typically referred to as Donnan exclusion [104,187]. Aswill be discussed in more detail subsequently, the con-centration of mobile salt in a cation exchange polymeris characterized by the concentration of anions sorbed inthe polymer because a salt pair (e.g., M+X−) must con-tain an anion to transport through the cation exchangepolymer, which contains an excess of cations. In contrast,in uncharged polymers, the concentration of mobile saltis characterized by the concentration of either cations oranions [148].
Charged polymers are often characterized by theircounter-ion form [33,104]. An acid form polymer is onewhere the counter-ions are protons. In contrast, a cationexchange polymer would be said to be in the sodium form ifall of the fixed charge groups were associated with sodiumions [13,104]. Ion exchange processes govern the polymer’scounter-ion form, and these will be discussed subsequentlyin more detail [104].
The implications of the differences between unchargedand charged polymers will be discussed later in this review.The extent to which polymer charge influences water andsalt transport properties is important since many chargedmaterials are either used or being considered for use indesalination applications. This review presents informa-tion that is available on these systems, while comparingcharged polymer results to those of uncharged polymers.
2. Uncharged polymers
First we will describe the water and salt transportproperties of hydrophilic uncharged polymers as thesematerials are, in many ways, less complex than chargedpolymers. Perhaps the best known uncharged desalina-
tion polymer is cellulose acetate because of its centralrole in the early years of the membrane-based desalina-tion field [63,64]. Other hydrophilic polymers, however,are of interest for developing fundamental understanding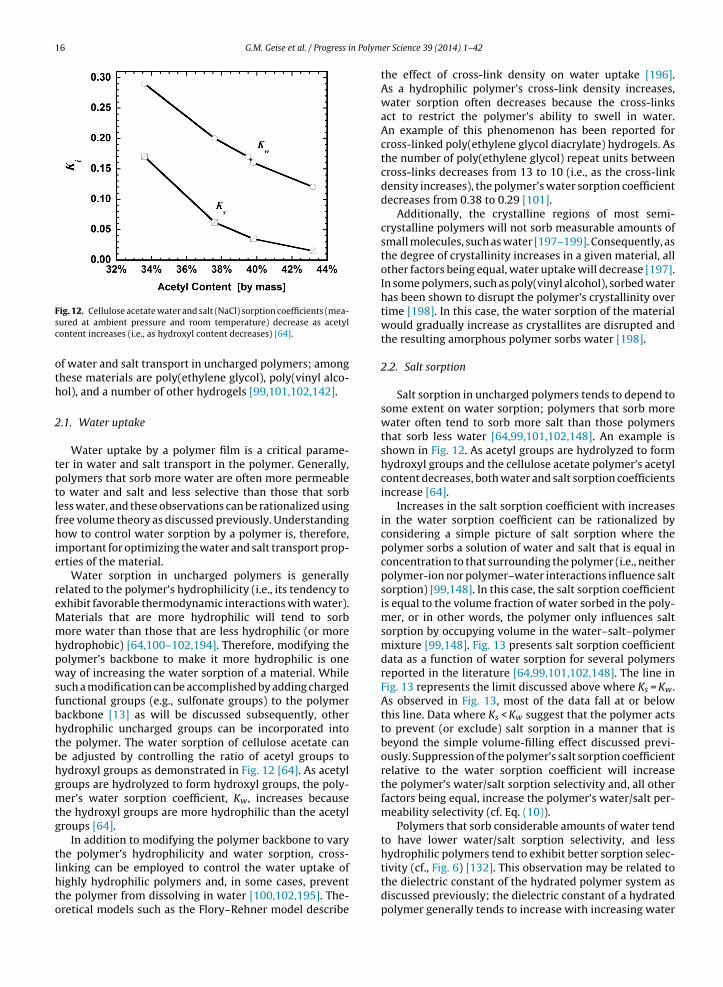
16 G.M. Geise et al. / Progress in Polym
Fig. 12. Cellulose acetate water and salt (NaCl) sorption coefficients (mea-sured at ambient pressure and room temperature) decrease as acetylcontent increases (i.e., as hydroxyl content decreases) [64].
of water and salt transport in uncharged polymers; amongthese materials are poly(ethylene glycol), poly(vinyl alco-hol), and a number of other hydrogels [99,101,102,142].
2.1. Water uptake
Water uptake by a polymer film is a critical parame-ter in water and salt transport in the polymer. Generally,polymers that sorb more water are often more permeableto water and salt and less selective than those that sorbless water, and these observations can be rationalized usingfree volume theory as discussed previously. Understandinghow to control water sorption by a polymer is, therefore,important for optimizing the water and salt transport prop-erties of the material.
Water sorption in uncharged polymers is generallyrelated to the polymer’s hydrophilicity (i.e., its tendency toexhibit favorable thermodynamic interactions with water).Materials that are more hydrophilic will tend to sorbmore water than those that are less hydrophilic (or morehydrophobic) [64,100–102,194]. Therefore, modifying thepolymer’s backbone to make it more hydrophilic is oneway of increasing the water sorption of a material. Whilesuch a modification can be accomplished by adding chargedfunctional groups (e.g., sulfonate groups) to the polymerbackbone [13] as will be discussed subsequently, otherhydrophilic uncharged groups can be incorporated intothe polymer. The water sorption of cellulose acetate canbe adjusted by controlling the ratio of acetyl groups tohydroxyl groups as demonstrated in Fig. 12 [64]. As acetylgroups are hydrolyzed to form hydroxyl groups, the poly-mer’s water sorption coefficient, Kw, increases becausethe hydroxyl groups are more hydrophilic than the acetylgroups [64].
In addition to modifying the polymer backbone to varythe polymer’s hydrophilicity and water sorption, cross-
linking can be employed to control the water uptake ofhighly hydrophilic polymers and, in some cases, preventthe polymer from dissolving in water [100,102,195]. The-oretical models such as the Flory–Rehner model describeer Science 39 (2014) 1– 42
the effect of cross-link density on water uptake [196].As a hydrophilic polymer’s cross-link density increases,water sorption often decreases because the cross-linksact to restrict the polymer’s ability to swell in water.An example of this phenomenon has been reported forcross-linked poly(ethylene glycol diacrylate) hydrogels. Asthe number of poly(ethylene glycol) repeat units betweencross-links decreases from 13 to 10 (i.e., as the cross-linkdensity increases), the polymer’s water sorption coefficientdecreases from 0.38 to 0.29 [101].
Additionally, the crystalline regions of most semi-crystalline polymers will not sorb measurable amounts ofsmall molecules, such as water [197–199]. Consequently, asthe degree of crystallinity increases in a given material, allother factors being equal, water uptake will decrease [197].In some polymers, such as poly(vinyl alcohol), sorbed waterhas been shown to disrupt the polymer’s crystallinity overtime [198]. In this case, the water sorption of the materialwould gradually increase as crystallites are disrupted andthe resulting amorphous polymer sorbs water [198].
2.2. Salt sorption
Salt sorption in uncharged polymers tends to depend tosome extent on water sorption; polymers that sorb morewater often tend to sorb more salt than those polymersthat sorb less water [64,99,101,102,148]. An example isshown in Fig. 12. As acetyl groups are hydrolyzed to formhydroxyl groups and the cellulose acetate polymer’s acetylcontent decreases, both water and salt sorption coefficientsincrease [64].
Increases in the salt sorption coefficient with increasesin the water sorption coefficient can be rationalized byconsidering a simple picture of salt sorption where thepolymer sorbs a solution of water and salt that is equal inconcentration to that surrounding the polymer (i.e., neitherpolymer-ion nor polymer–water interactions influence saltsorption) [99,148]. In this case, the salt sorption coefficientis equal to the volume fraction of water sorbed in the poly-mer, or in other words, the polymer only influences saltsorption by occupying volume in the water–salt–polymermixture [99,148]. Fig. 13 presents salt sorption coefficientdata as a function of water sorption for several polymersreported in the literature [64,99,101,102,148]. The line inFig. 13 represents the limit discussed above where Ks = Kw.As observed in Fig. 13, most of the data fall at or belowthis line. Data where Ks < Kw suggest that the polymer actsto prevent (or exclude) salt sorption in a manner that isbeyond the simple volume-filling effect discussed previ-ously. Suppression of the polymer’s salt sorption coefficientrelative to the water sorption coefficient will increasethe polymer’s water/salt sorption selectivity and, all otherfactors being equal, increase the polymer’s water/salt per-meability selectivity (cf. Eq. (10)).
Polymers that sorb considerable amounts of water tendto have lower water/salt sorption selectivity, and lesshydrophilic polymers tend to exhibit better sorption selec-
tivity (cf., Fig. 6) [132]. This observation may be related tothe dielectric constant of the hydrated polymer system asdiscussed previously; the dielectric constant of a hydratedpolymer generally tends to increase with increasing water
G.M. Geise et al. / Progress in Polymer Science 39 (2014) 1– 42 17
Fig. 13. Salt sorption coefficient, Ks , data as a function of the waters[
sscdatw
doatinuaaiwcdbhiccos
mmmataltt
Fig. 14. Cellulose acetate (�) [99] (©) [64], HPMA (�) [99], and cross-linked poly(ethylene glycol diacrylate) (n = 13) (�) [148] NaCl sorptionisotherms. The (©) data were measured using a 39.8 wt% acetylated cel-lulose acetate film[64]. The degree of acetylation was not reported in[99]. The corresponding salt sorption coefficients, Ks , are shown for each
−1 −1
As discussed previously, a polymer’s water uptake canhave an important influence on a polymer’s water and
Fig. 15. Sodium (♦) and chloride (�) sorption as a function of the externalNaCl solution concentration in a cross-linked poly(ethylene glycol diacry-
orption coefficient, Kw , for several polymers reported in the literature64,99,101,102,148].
orption, as shown in Table 6 [142,160,180]. Salts dis-olve to a greater extent in high dielectric constant mediaompared to lower dielectric constant media because theissociated ion’s electrostatic charge is better stabilized in
media that polarizes more readily [163]. Thus, salt sorp-ion may be less energetically favored in polymers with lowater uptake [160,163].
During the early development of cellulose acetateesalination membranes, Lonsdale, Merten, and Rileybserved that the salt sorption coefficient in cellulosecetate was essentially independent of salt concentra-ion [64]. That is, the salt concentration in the CA matrixncreased linearly with the salt concentration in the exter-al solution surrounding the polymer. Data for otherncharged polymers are also shown in Fig. 14, and theylso display a linear increase in sorbed salt in the polymers external salt solution concentration increases, resultingn essentially constant Ks values. Charged polymers, which
ill be discussed in the next section, exhibit ion sorptionoefficients that can depend strongly on salt concentrationue to the presence of fixed charges on the polymer back-one. While some reports suggest that cellulose acetateas some fixed charge character [121,166,183,200], the
ndependence of salt sorption on external solution salt con-entration suggests that the presence of any ion exchangeapacity in cellulose acetate is not as significant as thosebserved in charged materials that will be discussed sub-equently [13].
As indicated previously, ions sorb into uncharged poly-ers as ‘mobile’ salt. For sodium chloride (or otheronovalent-ion containing salt) sorption, uncharged poly-ers sorb equivalent molar concentrations of cations and
nions [13,148]. Fig. 15 presents experimental evidence ofhis behavior. In Fig. 15, sodium and chloride ions sorb inton uncharged cross-linked poly(ethylene glycol diacry-
ate) hydrogel in equivalent molar concentrations overwo orders of magnitude in external salt solution concen-ration [148]. Thus, for monovalent salt sorption into anpolymer in units of g(NaCl) L (swollen polymer)/(g(NaCl) L (externalsolution)).
uncharged polymer, both sodium and chloride sorptionrepresent mobile salt sorption in the material [148].
2.3. Dependence of water and salt permeability anddiffusion on water uptake
late) hydrogel (Kw = 0.34 in pure water) [148]. Molar units are used here,as opposed to mass units in Fig. 14, in order to properly compare theamounts of sodium and chloride ions sorbed in the polymer. Copyright2012. Reproduced from [148] with permission from Elsevier Ltd.
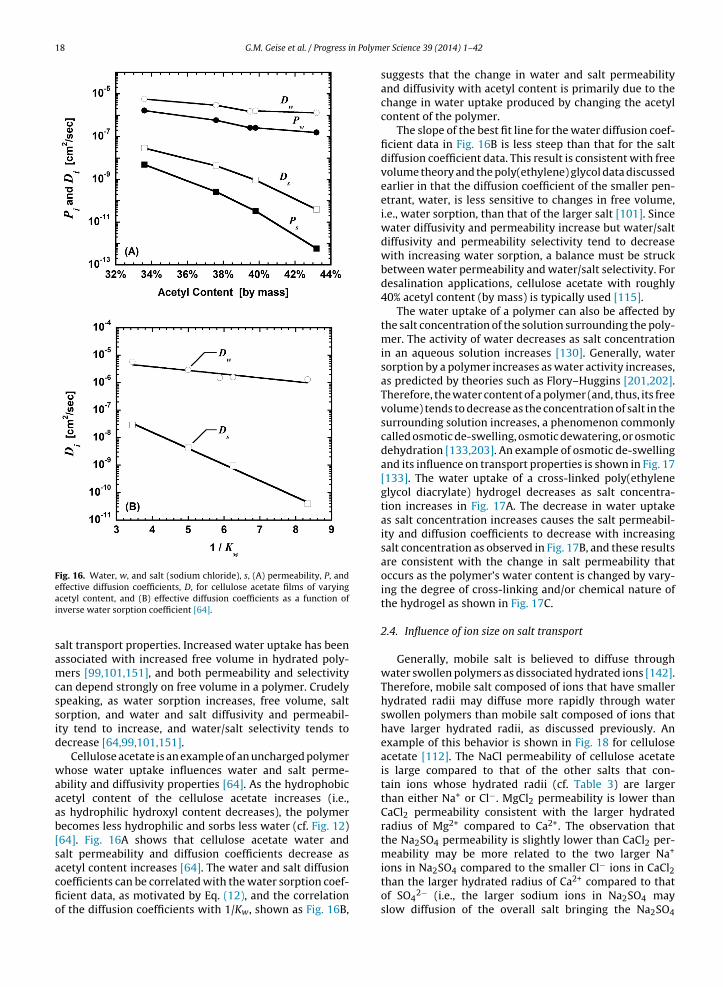
18 G.M. Geise et al. / Progress in Polym
Fig. 16. Water, w, and salt (sodium chloride), s, (A) permeability, P, and
−
effective diffusion coefficients, D, for cellulose acetate films of varyingacetyl content, and (B) effective diffusion coefficients as a function ofinverse water sorption coefficient [64].
salt transport properties. Increased water uptake has beenassociated with increased free volume in hydrated poly-mers [99,101,151], and both permeability and selectivitycan depend strongly on free volume in a polymer. Crudelyspeaking, as water sorption increases, free volume, saltsorption, and water and salt diffusivity and permeabil-ity tend to increase, and water/salt selectivity tends todecrease [64,99,101,151].
Cellulose acetate is an example of an uncharged polymerwhose water uptake influences water and salt perme-ability and diffusivity properties [64]. As the hydrophobicacetyl content of the cellulose acetate increases (i.e.,as hydrophilic hydroxyl content decreases), the polymerbecomes less hydrophilic and sorbs less water (cf. Fig. 12)[64]. Fig. 16A shows that cellulose acetate water andsalt permeability and diffusion coefficients decrease as
acetyl content increases [64]. The water and salt diffusioncoefficients can be correlated with the water sorption coef-ficient data, as motivated by Eq. (12), and the correlationof the diffusion coefficients with 1/Kw, shown as Fig. 16B,er Science 39 (2014) 1– 42
suggests that the change in water and salt permeabilityand diffusivity with acetyl content is primarily due to thechange in water uptake produced by changing the acetylcontent of the polymer.
The slope of the best fit line for the water diffusion coef-ficient data in Fig. 16B is less steep than that for the saltdiffusion coefficient data. This result is consistent with freevolume theory and the poly(ethylene) glycol data discussedearlier in that the diffusion coefficient of the smaller pen-etrant, water, is less sensitive to changes in free volume,i.e., water sorption, than that of the larger salt [101]. Sincewater diffusivity and permeability increase but water/saltdiffusivity and permeability selectivity tend to decreasewith increasing water sorption, a balance must be struckbetween water permeability and water/salt selectivity. Fordesalination applications, cellulose acetate with roughly40% acetyl content (by mass) is typically used [115].
The water uptake of a polymer can also be affected bythe salt concentration of the solution surrounding the poly-mer. The activity of water decreases as salt concentrationin an aqueous solution increases [130]. Generally, watersorption by a polymer increases as water activity increases,as predicted by theories such as Flory–Huggins [201,202].Therefore, the water content of a polymer (and, thus, its freevolume) tends to decrease as the concentration of salt in thesurrounding solution increases, a phenomenon commonlycalled osmotic de-swelling, osmotic dewatering, or osmoticdehydration [133,203]. An example of osmotic de-swellingand its influence on transport properties is shown in Fig. 17[133]. The water uptake of a cross-linked poly(ethyleneglycol diacrylate) hydrogel decreases as salt concentra-tion increases in Fig. 17A. The decrease in water uptakeas salt concentration increases causes the salt permeabil-ity and diffusion coefficients to decrease with increasingsalt concentration as observed in Fig. 17B, and these resultsare consistent with the change in salt permeability thatoccurs as the polymer’s water content is changed by vary-ing the degree of cross-linking and/or chemical nature ofthe hydrogel as shown in Fig. 17C.
2.4. Influence of ion size on salt transport
Generally, mobile salt is believed to diffuse throughwater swollen polymers as dissociated hydrated ions [142].Therefore, mobile salt composed of ions that have smallerhydrated radii may diffuse more rapidly through waterswollen polymers than mobile salt composed of ions thathave larger hydrated radii, as discussed previously. Anexample of this behavior is shown in Fig. 18 for celluloseacetate [112]. The NaCl permeability of cellulose acetateis large compared to that of the other salts that con-tain ions whose hydrated radii (cf. Table 3) are largerthan either Na+ or Cl−. MgCl2 permeability is lower thanCaCl2 permeability consistent with the larger hydratedradius of Mg2+ compared to Ca2+. The observation thatthe Na2SO4 permeability is slightly lower than CaCl2 per-meability may be more related to the two larger Na+
ions in Na2SO4 compared to the smaller Cl ions in CaCl2than the larger hydrated radius of Ca2+ compared to thatof SO4
2− (i.e., the larger sodium ions in Na2SO4 mayslow diffusion of the overall salt bringing the Na2SO4

G.M. Geise et al. / Progress in Polymer Science 39 (2014) 1– 42 19
Fig. 17. (A) Osmotic de-swelling in a cross-linked poly(ethylene gly-col diacrylate) hydrogel (XLPEGDA). (B) NaCl permeability and diffusioncoefficients for XLPEGDA as a function of salt concentration at 25 ◦C. (C)NaCl permeability plotted versus 1/Kw according to Yasuda et al.’s free vol-ume theory where the polymer’s water sorption was varied by changingeither the salt concentration in the external solution (�) or either chang-ing the cross-link density or incorporating a co-monomer (©). (B) and (C)Copyright 2013. Reproduced from [133] with permission from ElsevierLtd.
Fig. 18. Salt permeability of cellulose acetate (43 wt% acetylated) fordifferent salts measured at upstream salt concentrations ranging from
0.0054 mol L−1 to 0.11 mol L−1 at ambient temperature [112].permeability below that of CaCl2). As will be discussed sub-sequently, the relationship between hydrated radii and saltpermeability in charged polymers is influenced by elec-trostatic interactions between the diffusing ions and thecharged polymer.
The results shown above for cellulose acetate sug-gest that the hydrated ion radius should be used tocorrelate transport properties with ion size. However,the previously mentioned example regarding transportof unhydrated potassium through biological ion channels[162] and reports of ion diffusion coefficients in Nylon-6that decrease with increasing crystal radius [204] high-light the need for further experimental and theoreticalstudy in this area. Most of the salt transport data indesalination polymers is for sodium chloride; studies ondifferent ions are needed to correlate transport proper-ties with ion size and determine if ion size values canbe determined that provide useful correlations of ion sizewith ion transport through various desalination polymers[132].
2.5. Influence of cross-linking on water uptake andwater/salt selectivity
Cross-linking can be used to vary the diffusion charac-teristics of a polymer [205]. Typically, cross-linking reducesswelling [195], which will generally tend to decreasethe effective water and salt diffusion coefficients in thematerial [64,99,100,102]. As swelling is restricted and thediffusion coefficients decrease as a result of cross-linking,free volume theory and the tradeoff relationship (Fig. 5) dis-cussed earlier suggest that the polymer’s selectivity shouldtend to increase [99,101,132,151]. Fig. 19A shows an exam-
ple where cross-linking poly(hydroxyethyl methacrylate)(HEMA), using trimethylol propane trimethacrylate (TPT),results in an increase in the water/salt diffusivity selectiv-ity of the material [205]. As the amount of the cross-linking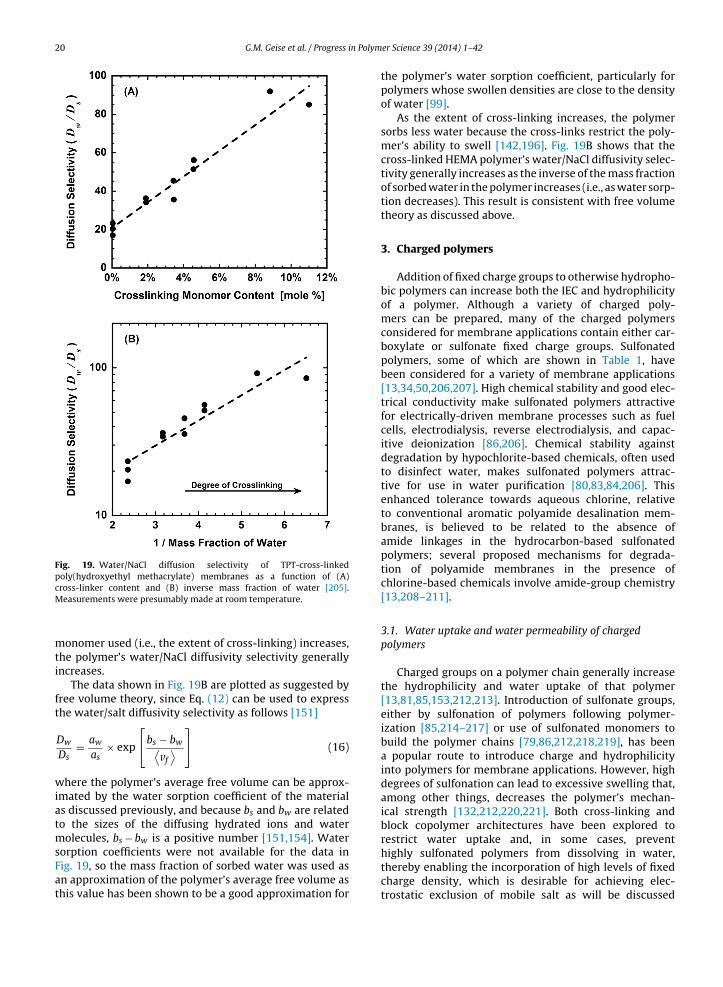
20 G.M. Geise et al. / Progress in Polym
Fig. 19. Water/NaCl diffusion selectivity of TPT-cross-linkedpoly(hydroxyethyl methacrylate) membranes as a function of (A)
highly sulfonated polymers from dissolving in water,
cross-linker content and (B) inverse mass fraction of water [205].Measurements were presumably made at room temperature.
monomer used (i.e., the extent of cross-linking) increases,the polymer’s water/NaCl diffusivity selectivity generallyincreases.
The data shown in Fig. 19B are plotted as suggested byfree volume theory, since Eq. (12) can be used to expressthe water/salt diffusivity selectivity as follows [151]
Dw
Ds= aw
as× exp
[bs − bw⟨
vf
⟩]
(16)
where the polymer’s average free volume can be approx-imated by the water sorption coefficient of the materialas discussed previously, and because bs and bw are relatedto the sizes of the diffusing hydrated ions and watermolecules, bs − bw is a positive number [151,154]. Watersorption coefficients were not available for the data in
Fig. 19, so the mass fraction of sorbed water was used asan approximation of the polymer’s average free volume asthis value has been shown to be a good approximation forer Science 39 (2014) 1– 42
the polymer’s water sorption coefficient, particularly forpolymers whose swollen densities are close to the densityof water [99].
As the extent of cross-linking increases, the polymersorbs less water because the cross-links restrict the poly-mer’s ability to swell [142,196]. Fig. 19B shows that thecross-linked HEMA polymer’s water/NaCl diffusivity selec-tivity generally increases as the inverse of the mass fractionof sorbed water in the polymer increases (i.e., as water sorp-tion decreases). This result is consistent with free volumetheory as discussed above.
3. Charged polymers
Addition of fixed charge groups to otherwise hydropho-bic polymers can increase both the IEC and hydrophilicityof a polymer. Although a variety of charged poly-mers can be prepared, many of the charged polymersconsidered for membrane applications contain either car-boxylate or sulfonate fixed charge groups. Sulfonatedpolymers, some of which are shown in Table 1, havebeen considered for a variety of membrane applications[13,34,50,206,207]. High chemical stability and good elec-trical conductivity make sulfonated polymers attractivefor electrically-driven membrane processes such as fuelcells, electrodialysis, reverse electrodialysis, and capac-itive deionization [86,206]. Chemical stability againstdegradation by hypochlorite-based chemicals, often usedto disinfect water, makes sulfonated polymers attrac-tive for use in water purification [80,83,84,206]. Thisenhanced tolerance towards aqueous chlorine, relativeto conventional aromatic polyamide desalination mem-branes, is believed to be related to the absence ofamide linkages in the hydrocarbon-based sulfonatedpolymers; several proposed mechanisms for degrada-tion of polyamide membranes in the presence ofchlorine-based chemicals involve amide-group chemistry[13,208–211].
3.1. Water uptake and water permeability of chargedpolymers
Charged groups on a polymer chain generally increasethe hydrophilicity and water uptake of that polymer[13,81,85,153,212,213]. Introduction of sulfonate groups,either by sulfonation of polymers following polymer-ization [85,214–217] or use of sulfonated monomers tobuild the polymer chains [79,86,212,218,219], has beena popular route to introduce charge and hydrophilicityinto polymers for membrane applications. However, highdegrees of sulfonation can lead to excessive swelling that,among other things, decreases the polymer’s mechan-ical strength [132,212,220,221]. Both cross-linking andblock copolymer architectures have been explored torestrict water uptake and, in some cases, prevent
thereby enabling the incorporation of high levels of fixedcharge density, which is desirable for achieving elec-trostatic exclusion of mobile salt as will be discussed
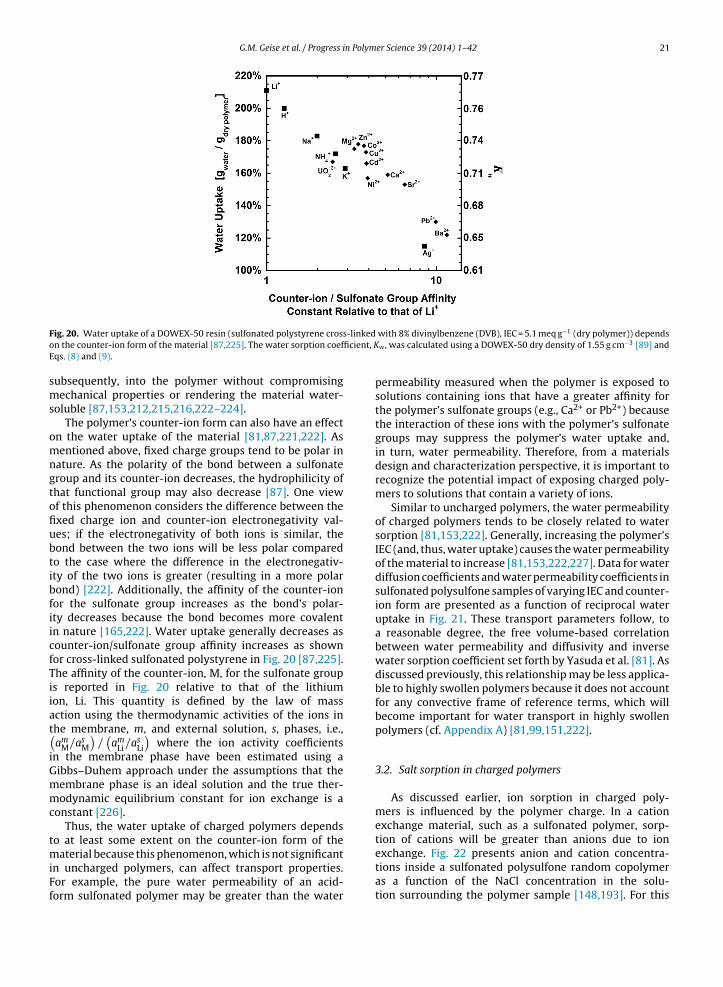
G.M. Geise et al. / Progress in Polymer Science 39 (2014) 1– 42 21
F -linkedo ficient, KE
sms
omngtofiubtibfiicfTiiat(iGmmc
tmiFf
exchange. Fig. 22 presents anion and cation concentra-
ig. 20. Water uptake of a DOWEX-50 resin (sulfonated polystyrene crossn the counter-ion form of the material [87,225]. The water sorption coefqs. (8) and (9).
ubsequently, into the polymer without compromisingechanical properties or rendering the material water-
oluble [87,153,212,215,216,222–224].The polymer’s counter-ion form can also have an effect
n the water uptake of the material [81,87,221,222]. Asentioned above, fixed charge groups tend to be polar in
ature. As the polarity of the bond between a sulfonateroup and its counter-ion decreases, the hydrophilicity ofhat functional group may also decrease [87]. One viewf this phenomenon considers the difference between thexed charge ion and counter-ion electronegativity val-es; if the electronegativity of both ions is similar, theond between the two ions will be less polar comparedo the case where the difference in the electronegativ-ty of the two ions is greater (resulting in a more polarond) [222]. Additionally, the affinity of the counter-ionor the sulfonate group increases as the bond’s polar-ty decreases because the bond becomes more covalentn nature [165,222]. Water uptake generally decreases asounter-ion/sulfonate group affinity increases as shownor cross-linked sulfonated polystyrene in Fig. 20 [87,225].he affinity of the counter-ion, M, for the sulfonate groups reported in Fig. 20 relative to that of the lithiumon, Li. This quantity is defined by the law of massction using the thermodynamic activities of the ions inhe membrane, m, and external solution, s, phases, i.e.,am
M/asM
)/(
amLi/as
Li
)where the ion activity coefficients
n the membrane phase have been estimated using aibbs–Duhem approach under the assumptions that theembrane phase is an ideal solution and the true ther-odynamic equilibrium constant for ion exchange is a
onstant [226].Thus, the water uptake of charged polymers depends
o at least some extent on the counter-ion form of theaterial because this phenomenon, which is not significant
n uncharged polymers, can affect transport properties.or example, the pure water permeability of an acid-orm sulfonated polymer may be greater than the water
with 8% divinylbenzene (DVB), IEC = 5.1 meq g−1 (dry polymer)) dependsw , was calculated using a DOWEX-50 dry density of 1.55 g cm−3 [89] and
permeability measured when the polymer is exposed tosolutions containing ions that have a greater affinity forthe polymer’s sulfonate groups (e.g., Ca2+ or Pb2+) becausethe interaction of these ions with the polymer’s sulfonategroups may suppress the polymer’s water uptake and,in turn, water permeability. Therefore, from a materialsdesign and characterization perspective, it is important torecognize the potential impact of exposing charged poly-mers to solutions that contain a variety of ions.
Similar to uncharged polymers, the water permeabilityof charged polymers tends to be closely related to watersorption [81,153,222]. Generally, increasing the polymer’sIEC (and, thus, water uptake) causes the water permeabilityof the material to increase [81,153,222,227]. Data for waterdiffusion coefficients and water permeability coefficients insulfonated polysulfone samples of varying IEC and counter-ion form are presented as a function of reciprocal wateruptake in Fig. 21. These transport parameters follow, toa reasonable degree, the free volume-based correlationbetween water permeability and diffusivity and inversewater sorption coefficient set forth by Yasuda et al. [81]. Asdiscussed previously, this relationship may be less applica-ble to highly swollen polymers because it does not accountfor any convective frame of reference terms, which willbecome important for water transport in highly swollenpolymers (cf. Appendix A) [81,99,151,222].
3.2. Salt sorption in charged polymers
As discussed earlier, ion sorption in charged poly-mers is influenced by the polymer charge. In a cationexchange material, such as a sulfonated polymer, sorp-tion of cations will be greater than anions due to ion
tions inside a sulfonated polysulfone random copolymeras a function of the NaCl concentration in the solu-tion surrounding the polymer sample [148,193]. For this

22 G.M. Geise et al. / Progress in Polymer Science 39 (2014) 1– 42
Fig. 21. Diffusive water permeability and effective water and salt (NaCl)diffusion coefficients for sulfonated polysulfone random copolymers plot-ted according to Eqs. (12) and (13) using the water sorption coefficient asa proxy for the polymer’s free volume [81]. The NaCl diffusion coefficients
Fig. 23. Equilibrium salt sorption coefficients (i.e., anion partitioncoefficients) at 25 ◦C for sulfonated polysulfone (�, IEC 1.2 meq g−1 (drypolymer)) and sulfonated styrenic pentablock copolymers (� and �, IEC1.5 and 2.0 meq g−1 (dry polymer), respectively) as a function of external
were measured at 25 ◦C using an upstream salt concentration of 1 mol L−1.
particular sample, the vast majority of the sorbed sodiumions are associated with the polymer’s sulfonate groups,and as a result, sodium sorption appears to change littlewith external NaCl concentration on the log-log plot inFig. 22 [148].
In contrast, mobile salt sorption, characterized by anionsorption in cation exchange materials (e.g., sulfonatedpolymers) [148], increases as salt concentration increases.Unlike the uncharged polymers discussed previously, salt
sorption coefficients of charged polymers depend on exter-nal solution salt concentration [13,104,120,184,185,193].The salt sorption coefficient, Ks, calculated and shownFig. 22. Equilibrium sorption of sodium and chloride ions into a sul-fonated polysulfone random copolymer (IEC 1.2 meq g−1 (dry polymer))at 25 ◦C and ambient pressure as a function of the NaCl concentration inthe neutral pH solution surrounding the polymer. Copyright 2012. Repro-duced from [148] with permission from Elsevier Ltd.
NaCl concentration. The solid lines are determined from a salt sorptionmodel. Copyright 2012. Reproduced from [148] with permission fromElsevier Ltd.
in Fig. 23, using the chloride sorption data shown inFig. 22, increases by roughly a factor of 8 as the exter-nal NaCl concentration increases from 0.01 mol L−1 to1.0 mol L−1 [148]. An example of the dependence ofcharged polymer Ks values on external solution salt con-centration is shown in Fig. 23 [148]. In contrast, the saltsorption coefficients of several uncharged polymers arerelatively independent of salt concentration as discussedpreviously (cf. Fig. 14) [64,148]. The sorption of anions,and thus, mobile salt in charged polymers decreases asexternal salt solution concentration decreases, and thisphenomenon is commonly referred to as Donnan exclusion[13,104,185,187].
The exclusion of co-ions (i.e., mobile salt) from acharged polymer via Donnan exclusion (or other means)is desirable for desalination membranes as, all otherfactors being equal, suppression of the salt sorptioncoefficient would contribute to reduction of the poly-mer’s salt permeability, perhaps improving the polymer’swater/salt separation properties [13]. As will be discussedsubsequently, Donnan exclusion is generally observedto varying extents in charged polymers. Polymers thatbehave more like ideal Donnan materials compared touncharged polymers, however, may be advantageous fordesalination applications due to the strong electrostaticexclusion of mobile salt that such materials could offer[13,148].
Donnan exclusion theory has been discussed exten-sively in the literature [13,104,184,185,187,228]. Accord-ing to ideal Donnan exclusion theory, the sorbed co-ionconcentration (i.e., mobile salt concentration) shouldincrease with the square of the external salt solution con-centration [13,104]. Real materials, however, do not strictly
obey the homogeneous charge distribution assumption ofDonnan theory [148,184,185]. Fig. 24 shows that the powerlaw slopes of co-ion sorption isotherms for several charged
G.M. Geise et al. / Progress in Polym
Fig. 24. Equilibrium co-ion concentration in the polymer (sorbed) as afunction of the external salt solution concentration for ACI (�, IEC 1.9,co-ion: Na+ [185]) and Permaplex A-20 (�, IEC 2.2, co-ion: Na+ [185])anion exchange membranes and ACI (�, IEC 2.3, co-ion: Br− [185]), BPSH-32 (�, IEC 1.2, co-ion Cl− [148]), sPBC (� and �, IEC 1.5 and 2.0, co-ionCl− [148]). Permaplex A-20 is a heterogeneous polyethylene matrix con-taining quaternary ammonium functionalized particles [185,230]. The ACI(Asahi Chemical Industries) cation and anion exchange membranes areacid- or base-functionalized polystyrene cross-linked with divinylben-zene [185]. BPSH-32 is a sulfonated polysulfone random copolymer, andsPBC is a sulfonated styrenic pentablock copolymer [148]. IEC values arereported in units of meq g−1 (dry polymer), and the slopes calculated usingis
pnsCpcsDTmmsaiticmittbw
m(cnFc
Aand K∞ are constants, then salt permeability should scale
deal Donnan exclusion theory and expected for uncharged polymers arehown for reference.
olymers are less than the slope calculated from ideal Don-an theory (i.e., a slope of 2). The slopes of the co-ionorption isotherms in Fig. 24 vary in the range of 1.2–1.5.onsidering that the power-law slope of an unchargedolymer’s ion sorption isotherm is expected to be unity, theharged polymers reported in Fig. 24 exhibit mobile saltorption properties intermediate between those of idealonnan charged materials and uncharged polymers [148].his observation has been used to develop ion sorptionodels that can be applied to real (i.e., not ideal Donnan)aterials [148,185,229]. The results of one such model are
hown as the solid curves in Fig. 23 [148]. The modelsnd theory suggest that co-ion (i.e., mobile salt) exclusionncreases as fixed charge concentration (e.g., the concentra-ion of sulfonated groups in a swollen sulfonated polymer)ncreases and the electric potential resulting from the fixedharge groups becomes more uniform [13,104,148]. Asany fixed charge groups are quite hydrophilic, achiev-
ng a high fixed charge concentration requires increasinghe polymer’s degree of functionalization while simul-aneously restricting polymer swelling via, for example,lock copolymerization or cross-linking, that would other-ise dilute the polymer’s fixed charge concentration [148].
Donnan theory also predicts that reduction of a poly-er’s fixed charge concentration will result in less co-ion
i.e., mobile salt) exclusion [13,104,148]. In the extremease, if the fixed charge concentration is effectively elimi-
ated, the polymer will behave as an uncharged polymer.ixed charge groups can be effectively neutralized byhemical or electrostatic means, and such reduction in theer Science 39 (2014) 1– 42 23
polymer’s effective fixed charge concentrations will tendto decrease the effects of Donnan exclusion [13,104,120].
Sulfonate groups can be neutralized using electronega-tive ions, such as aluminum [222]. When triethylaluminumis added to a sulfonated polymer, aluminum cross-linkscan form because aluminum can bond with an oxygenatom in up to 3 different sulfonate groups, and such bondsare often quite strong (i.e., they have a highly covalentcharacter) [222]. Aluminum neutralization of a sulfonatedstyrenic pentablock copolymer decreased water/salt solu-bility selectivity by 42% despite a reduction in the polymer’swater content by a factor of 5.5 upon neutralization [222].As discussed with regard to Fig. 6, water/salt sorptionselectivity generally increases as water sorption decreases,so the observed behavior is opposite to that shown ingeneral in Fig. 6 [132]. However, complete aluminum neu-tralization of the polymer’s sulfonate groups decreasedthe effective number of ionizable, charged groups in thepolymer, which decreased water sorption and stronglydecreased Donnan exclusion of mobile salt, resulting inan overall reduction of the polymer’s water/salt sorptionselectivity.
3.3. Salt permeability of charged polymers
In the simple case where salt transport of a monova-lent salt, e.g., NaCl, is considered, the introduction of fixedcharge groups into a polymer matrix affects the depend-ence of salt permeability on upstream salt concentration[153,231,232]. This effect can be understood within theframework of Donnan theory [104]. Salt permeability, Ps, inuncharged polymers is given by the following expression[13,107]
Ps = Ks
[DMDX
DM + DX
(d ln am
s
d ln Cms
)]= KsDs (17)
where DJ is the average diffusion coefficient of cations(M) or anions (X) through the swollen polymer, am
s is theactivity of salt in the membrane phase, Cm
s is the molarconcentration of salt in the membrane phase, Ks is the saltsorption coefficient, and Ds is the concentration averagedeffective salt diffusion coefficient. Alternatively, in chargedpolymers when the concentration of charged groups in thepolymer is much greater than the concentration of salt inthe external solution (which is often the case in membraneapplications), the salt permeability is expressed using Don-nan theory by [13]
Ps =[
DX
(d ln am
MamX
d ln Cms
)]Cs
s0Cm
A
(K∞)2 (18)
where amJ is the activity of ion J in the membrane phase,
CmA is the volume-based concentration of fixed charge
groups in the polymer matrix, Css0 is the molar concen-
tration of salt in the external upstream solution, and K∞is the salt sorption coefficient in the limit where Cm
s �Cm. If the term enclosed in square brackets in Eq. (18)
linearly with the concentration of salt in the externalupstream salt solution, Cs
s0 [13,184]. Fig. 25 shows exam-ples where the salt permeability of several sulfonated

24 G.M. Geise et al. / Progress in Polymer Science 39 (2014) 1– 42
Fig. 25. NaCl permeability as a function of concentration for: (A)sulfonated styrenic pentablock copolymers (IEC 1.5 and 2.0 meq g−1
(dry polymer)) (�) [153], Nafion® 117 (IEC 0.909 meq g−1 (dry poly-mer)) (�) [133], sulfonated poly(arylene ether sulfone) (IEC 1.2 meq g−1
(dry polymer)) (♦) [133] and (B) Nafion® 115 (IEC 0.909 meq g−1 (drypolymer)) (�,©) and MF-4SC (a Nafion®-like perfluorinated polymer,IEC = 0.9 meq g−1 (dry polymer) [233]) (�,�); filled symbols representsamples boiled in water and open symbols represent samples that werenot boiled in water [232]. The best fit lines were determined by a leastsquares fit of the data to a power law equation.
Fig. 26. (A) Salt permeability values for an initially acid counter-ion form 35% di-sulfonated poly(arylene ether sulfone), i.e., BPSH-35,(IEC = 1.52 meq g−1 (dry polymer) [80]) measured at an upstream saltconcentration of 1.0 mol L−1 at T = 25 ◦C [79]. (B) Ion passage val-ues for an ESPA2 brackish water RO membrane as reported by themanufacturer (approximate feed ionic strength = 1500 ppm, applied pres-sure difference = 10.3 bar (150 psi), temperature = 25 ◦C, permeate flow
of the RO membrane because the salt permeability cannotbe determined explicitly owing to uncertainty in the active
polymers increases with increasing upstream salt concen-tration. As many practical polymers will not rigorouslyobey Donnan theory because they do not strictly obey theequi-potential volume assumption discussed previously,the slope of Log(Ps) versus Log(Cs
s0) is often observed tobe less than unity [184,185,229]. The increase in salt per-meability with upstream salt concentration for chargedpolymers is important because characterizing the salt per-meability of the polymer at a single salt concentration is notsufficient to describe salt transport in the polymer over thewide range of salt concentrations experienced in desalina-
tion applications.rate = 34 m3 day−1 (9000 gpd), recovery = 15%, pH = 6.5 − 7) [236].
3.4. Permeability of salts other than sodium chloride
While sodium chloride is certainly the salt most stud-ied for desalination applications, natural water sourcescontain a variety of ions [170], and it is important to under-stand whether transport of ions (particularly multivalentions) other than sodium and chloride behaves differently[234,235]. The salt permeability of an initially acid counter-ion form sulfonated polysulfone material to a variety ofsalts is shown in Fig. 26A [79]. For comparison, ion passagevalues for a brackish water RO membrane are reported inFig. 26B, and these ion passage values, which are calculatedas 100%—salt rejection, approximate the salt permeability
layer thickness.

n Polymer Science 39 (2014) 1– 42 25
mdUhi[iMNgbtp
ccatFlttpwmTbwcF
tmahtsoSmmt(iCeg
hispatrtgico
Fig. 27. NaCl and MgCl2 permeability of sulfonated styrenic pentablockcopolymers (IEC 1.46: � and ©, IEC 1.6: � and ♦, and IEC 2.0: � and�) where NaCl permeability is represented by filled symbols and MgCl2permeability is represented by open symbols [237]. NaCl permeability isalso shown for an aluminum neutralized sulfonated styrenic pentablockcopolymer (�). IEC values are reported in units of meq g−1 (dry polymer)[222]. Copyright 2011. Reproduced from [237] with permission from the
G.M. Geise et al. / Progress i
In an uncharged cellulose acetate polymer, salt per-eability largely decreased as the hydrated radii of the
iffusing ions increased as discussed previously (cf. Fig. 18).nlike the uncharged polymer, the permeability of theighly charged sulfonated polysulfone to salts contain-
ng divalent cations, which have larger hydrated radii154], is greater than the permeability of salts contain-ng monovalent cations. The observation that the larger
gCl2 and CaCl2 salts are more permeable than the smalleraCl and KCl salts in the sulfonated polysulfone sug-ests that the effective size of the diffusing ions maye less important than electrostatic interactions betweenhe diffusing ions and the charged polymer on salt trans-ort.
In comparison, the passage of Mg2+ and Ca2+ for theross-linked polyamide RO membrane, which containsarboxylate ionic groups, which may be largely dissoci-ted as suggested by the pKa value in Table 4, is lowerhan that of the monovalent cations Na+ and K+ (cf.ig. 26B). Thus, in the less charged RO membrane, thearger hydrated radii of the divalent ions may contributeo their low passage relative to the monovalent ions, andhe greater passage of the smaller hydrated K+ ion com-ared to that of the larger Na+ ion is also consistentith a scenario where ion size is important in deter-ining ion passage through a polymer membrane [154].
he ion passage data for the RO membrane appears toe more similar to data reported for cellulose acetate,hich is essentially uncharged, (cf. Fig. 18) than to the
harged sulfonated polysulfone whose data are shown inig. 26A.
The permeability of the sulfonated polysulfone polymero salts containing divalent anions is less than the per-
eability of that material to salts containing monovalentnions. This result is qualitatively consistent with both theydrated radii of the ions involved and Donnan exclusionheory in that exclusion of di-valent co-ions (such as theulfate ion in this case) is expected to be stronger than thatf mono-valent co-ions (such as chloride) [104]. Since theO4
2− ion is large and co-ion exclusion ultimately affectsobile salt transport, as discussed previously, the salt per-eability values for Na2SO4 and MgSO4 are lower than
hose for NaCl and KCl. The data for the RO membraneFig. 26B) are similar in that the larger SO4
2− ion passages lower than that of the Cl− ion, and the lower passage ofl− compared to the larger Na+ ion may be a result of somextent of charge exclusion due to the polymer’s carboxylroups.
In Fig. 26A, salts containing divalent cations tend toave higher salt permeability values than salts contain-
ng monovalent cations. This result can be ascribed too-called charge screening or partial neutralization of theolymer’s fixed charge as a result of the larger bindingffinity of divalent ions (compared to monovalent ions)o the sulfonate groups, as shown in Fig. 20, and theseesults are at least qualitatively consistent with Donnanheory [13,83,87,104]. This larger divalent ion/sulfonate
roup binding affinity, compared to that for monovalentons, could act to reduce the polymer’s effective fixedharge concentration, thereby reducing the effectivenessf Donnan exclusion of mobile salt and increasing saltAmerican Chemical Society.
permeability [13,148]. The fact that, in charged polymers,salts containing divalent cations tend to be more permeablethan salts containing monovalent cations and the reversalof this behavior in uncharged or weakly charged poly-mers is an important difference between these classes ofpolymers. Uncharged polymers that have high NaCl/waterselectivity will likely have even greater ability to separatelarger ions from water, so NaCl may be a good indicator ofan uncharged polymer’s separation performance in a com-plex multi-salt filtration application. On the other hand, incharged polymers, other salts (containing ions such as Ca2+
or Mg2+) may be more permeable than NaCl, so chargedpolymers need to also be characterized using salts otherthan NaCl.
The variation of salt permeability with upstream saltconcentration depends on the salt used in the mea-surement. Fig. 27 shows that, for a sulfonated styrenicpentablock copolymer, the influence of upstream salt con-centration on the salt permeability is stronger for NaClthan for MgCl2 [237]. As discussed previously, divalentions such as magnesium typically bind more strongly tosulfonate groups than monovalent ions such as sodium[87], and this stronger affinity may effectively reduce sul-fonate group ionization, or in other words, divalent ionsmay reduce the polymer’s fixed charge concentration, sothat the polymer behaves more like an uncharged poly-mer in the presence of the divalent salt [237]. Additionally,the smaller slopes for MgCl2 permeability versus concen-tration compared to those for NaCl in Fig. 27 are at leastqualitatively consistant with Donnan theory though noquantitative tests of this explanation are available at the
current time. From a separations perspective, the result ofthis loss of Donnan exclusion is higher salt permeabilityand likely lower water/salt selectivity, particularly at low
26 G.M. Geise et al. / Progress in Polymer Science 39 (2014) 1– 42
Fig. 28. NaCl diffusion coefficients for sulfonated polysulfone polymerscorrelate (according to Eq. (12)) with the inverse average free volumeelement volume, determined using PALS, Copyright 2011. Adapted from
Fig. 29. NaCl diffusion coefficient data at 25 ◦C for sulfonated polysulfone(�, IEC 1.2 meq g−1 (dry polymer)) and two sulfonated styrenic pentablockcopolymers (� and �, IEC 1.5 meq g−1 and 2.0 meq g−1 (dry polymer),respectively) plotted as a function of (A) upstream salt concentration and(B) inverse water sorption coefficient. In (B), NaCl diffusion coefficientdata for sulfonated polysulfone measured at a single salt concentration(1 mol L−1) are shown as (×) for reference, and the arrows point in the
[151] with permission from the American Chemical Society. NaCl diffusioncoefficients were measured using a kinetic desorption experiment at 25 ◦Cwhere the polymers were initially equilibrated in 1 mol L−1 NaCl [81].
salt concentrations, than in cases where Donnan exclusionis not suppressed.
Further support for this conclusion can be found byobserving that the salt permeability of an aluminum-neutralized sulfonated styrenic pentablock copolymer isless sensitive to upstream salt concentration than othernon-neutralized sulfonated polymers [222]. As discussedpreviously, the aluminum neutralization process reducesthe polymer’s fixed charge concentration by formingstrong bonds between the sulfonate groups and alu-minum atoms [222]. Therefore, the aluminum-neutralizedsulfonated polymer behaves more like an unchargedpolymer than non-aluminum neutralized sulfonated poly-mers.
3.5. Salt diffusion in charged polymers
In applications where steady-state salt diffusion is ofinterest and the volume of processed solution at a givensalt concentration is much greater than the polymer’s vol-ume (i.e., neglecting ion exchange effects that may occurinitially), diffusion of salts in sulfonated polymers appearto be generally qualitatively consistent with free volumetheory [81,151,153]. As discussed previously for unchargedpolymers, salt diffusion, and thus, the logarithm of per-meability is expected to scale with 1/Kw in the Yasudaet al. free volume model [99]. Fig. 21 shows salt diffusionin sulfonated polysulfone following the empirical trendestablished by Yasuda et al. [81,151]. Furthermore, simi-lar results are observed for salt permeability in sulfonatedstyrenic pentablock copolymer films when the influenceof the block copolymer architecture is considered.[153]The free volume of sulfonated polymers can be probed
using positron annihilation lifetime spectroscopy (PALS)[151]. Fig. 28 shows that the average volume of free vol-ume elements (i.e., void space between polymer chains,as measured using PALS) in sulfonated polysulfone filmsdirection of increasing salt concentration from 0.01 mol L−1 to 1.0 mol L−1
NaCl. Copyright 2013. Reproduced from [133] with permission from Else-vier Ltd.
correlates well with salt transport properties, indicat-ing that the free volume element size in these materialsis representative of the polymer’s average free volume[151].
As discussed above, the incorporation of charged groupson the polymer backbone affects ion sorption in chargedpolymers because ion–polymer interactions are more sig-nificant when the polymer contains charged groups [148].These interactions may also affect the dependence of thesalt diffusion coefficient on upstream salt concentration[133]. For uncharged polymers, changing the salt concen-tration in the external solution affects salt permeability
through osmotic de-swelling in a manner consistent withfree volume theory (cf. Fig. 17) [133]. Fig. 29, however,shows that the variation of the NaCl diffusion coefficientsin sulfonated polymers is quite different from that of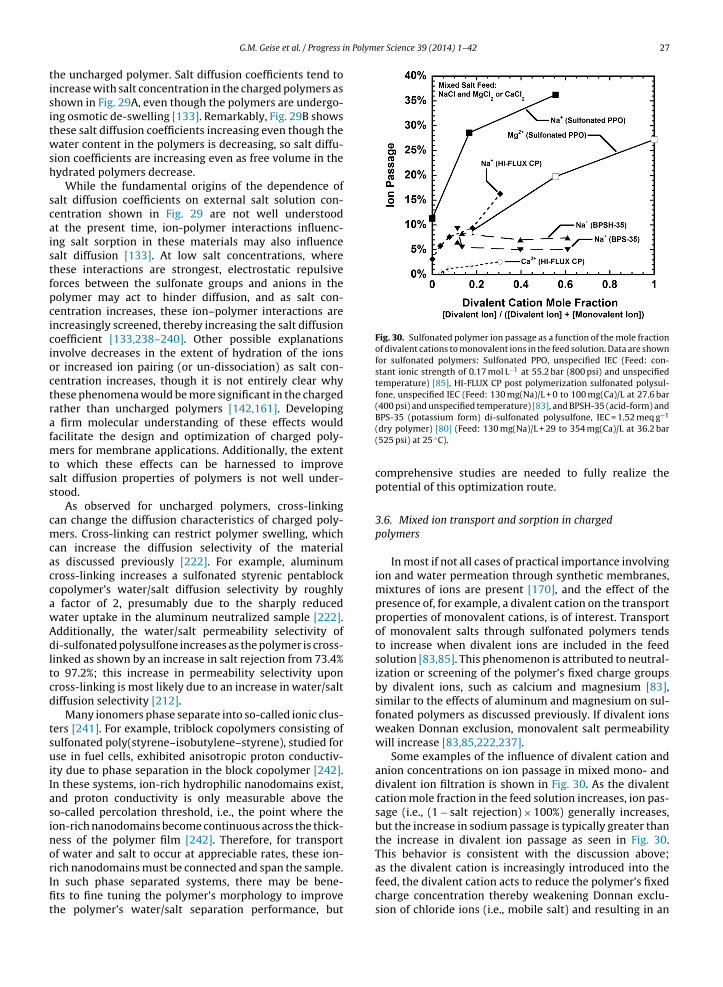
n Polymer Science 39 (2014) 1– 42 27
tisitwsh
scaistfpcicioctrafmtss
cmcaccawAdltcd
tsuiIasinorIfit
Fig. 30. Sulfonated polymer ion passage as a function of the mole fractionof divalent cations to monovalent ions in the feed solution. Data are shownfor sulfonated polymers: Sulfonated PPO, unspecified IEC (Feed: con-stant ionic strength of 0.17 mol L−1 at 55.2 bar (800 psi) and unspecifiedtemperature) [85], HI-FLUX CP post polymerization sulfonated polysul-fone, unspecified IEC (Feed: 130 mg(Na)/L + 0 to 100 mg(Ca)/L at 27.6 bar(400 psi) and unspecified temperature) [83], and BPSH-35 (acid-form) and
−1
G.M. Geise et al. / Progress i
he uncharged polymer. Salt diffusion coefficients tend toncrease with salt concentration in the charged polymers ashown in Fig. 29A, even though the polymers are undergo-ng osmotic de-swelling [133]. Remarkably, Fig. 29B showshese salt diffusion coefficients increasing even though theater content in the polymers is decreasing, so salt diffu-
ion coefficients are increasing even as free volume in theydrated polymers decrease.
While the fundamental origins of the dependence ofalt diffusion coefficients on external salt solution con-entration shown in Fig. 29 are not well understoodt the present time, ion-polymer interactions influenc-ng salt sorption in these materials may also influencealt diffusion [133]. At low salt concentrations, wherehese interactions are strongest, electrostatic repulsiveorces between the sulfonate groups and anions in theolymer may act to hinder diffusion, and as salt con-entration increases, these ion–polymer interactions arencreasingly screened, thereby increasing the salt diffusionoefficient [133,238–240]. Other possible explanationsnvolve decreases in the extent of hydration of the ionsr increased ion pairing (or un-dissociation) as salt con-entration increases, though it is not entirely clear whyhese phenomena would be more significant in the chargedather than uncharged polymers [142,161]. Developing
firm molecular understanding of these effects wouldacilitate the design and optimization of charged poly-
ers for membrane applications. Additionally, the extento which these effects can be harnessed to improvealt diffusion properties of polymers is not well under-tood.
As observed for uncharged polymers, cross-linkingan change the diffusion characteristics of charged poly-ers. Cross-linking can restrict polymer swelling, which
an increase the diffusion selectivity of the materials discussed previously [222]. For example, aluminumross-linking increases a sulfonated styrenic pentablockopolymer’s water/salt diffusion selectivity by roughly
factor of 2, presumably due to the sharply reducedater uptake in the aluminum neutralized sample [222].dditionally, the water/salt permeability selectivity ofi-sulfonated polysulfone increases as the polymer is cross-
inked as shown by an increase in salt rejection from 73.4%o 97.2%; this increase in permeability selectivity uponross-linking is most likely due to an increase in water/saltiffusion selectivity [212].
Many ionomers phase separate into so-called ionic clus-ers [241]. For example, triblock copolymers consisting ofulfonated poly(styrene–isobutylene–styrene), studied forse in fuel cells, exhibited anisotropic proton conductiv-
ty due to phase separation in the block copolymer [242].n these systems, ion-rich hydrophilic nanodomains exist,nd proton conductivity is only measurable above theo-called percolation threshold, i.e., the point where theon-rich nanodomains become continuous across the thick-ess of the polymer film [242]. Therefore, for transportf water and salt to occur at appreciable rates, these ion-
ich nanodomains must be connected and span the sample.n such phase separated systems, there may be bene-ts to fine tuning the polymer’s morphology to improvehe polymer’s water/salt separation performance, butBPS-35 (potassium form) di-sulfonated polysulfone, IEC = 1.52 meq g(dry polymer) [80] (Feed: 130 mg(Na)/L + 29 to 354 mg(Ca)/L at 36.2 bar(525 psi) at 25 ◦C).
comprehensive studies are needed to fully realize thepotential of this optimization route.
3.6. Mixed ion transport and sorption in chargedpolymers
In most if not all cases of practical importance involvingion and water permeation through synthetic membranes,mixtures of ions are present [170], and the effect of thepresence of, for example, a divalent cation on the transportproperties of monovalent cations, is of interest. Transportof monovalent salts through sulfonated polymers tendsto increase when divalent ions are included in the feedsolution [83,85]. This phenomenon is attributed to neutral-ization or screening of the polymer’s fixed charge groupsby divalent ions, such as calcium and magnesium [83],similar to the effects of aluminum and magnesium on sul-fonated polymers as discussed previously. If divalent ionsweaken Donnan exclusion, monovalent salt permeabilitywill increase [83,85,222,237].
Some examples of the influence of divalent cation andanion concentrations on ion passage in mixed mono- anddivalent ion filtration is shown in Fig. 30. As the divalentcation mole fraction in the feed solution increases, ion pas-sage (i.e., (1 − salt rejection) × 100%) generally increases,but the increase in sodium passage is typically greater thanthe increase in divalent ion passage as seen in Fig. 30.This behavior is consistent with the discussion above;
as the divalent cation is increasingly introduced into thefeed, the divalent cation acts to reduce the polymer’s fixedcharge concentration thereby weakening Donnan exclu-sion of chloride ions (i.e., mobile salt) and resulting in an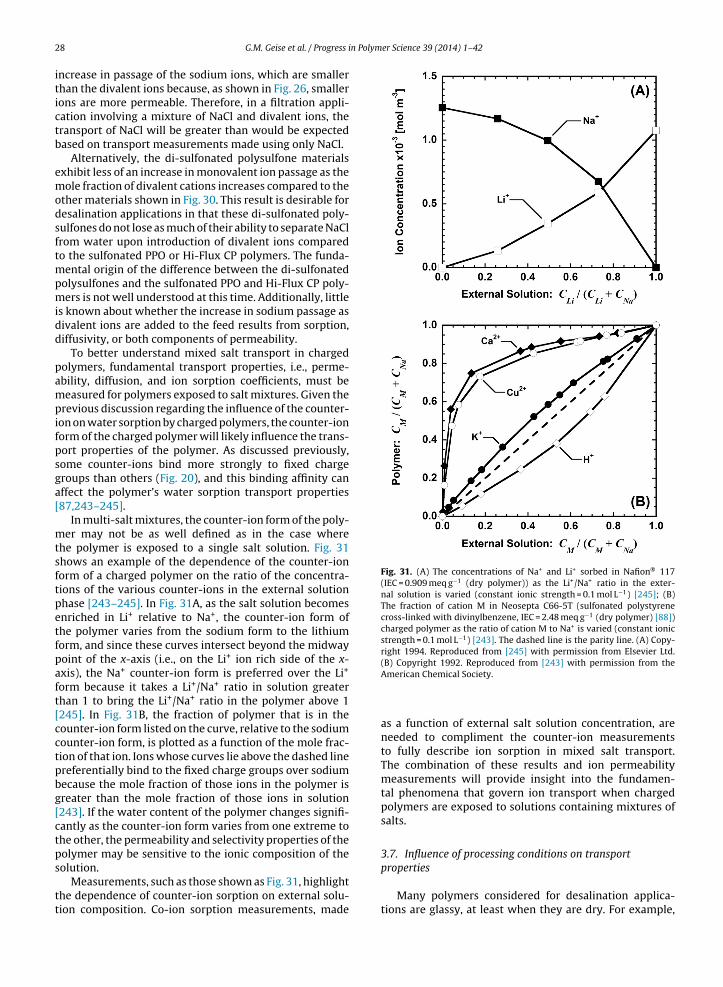
n Polymer Science 39 (2014) 1– 42
Fig. 31. (A) The concentrations of Na+ and Li+ sorbed in Nafion® 117(IEC = 0.909 meq g−1 (dry polymer)) as the Li+/Na+ ratio in the exter-nal solution is varied (constant ionic strength = 0.1 mol L−1) [245]; (B)The fraction of cation M in Neosepta C66-5T (sulfonated polystyrenecross-linked with divinylbenzene, IEC = 2.48 meq g−1 (dry polymer) [88])charged polymer as the ratio of cation M to Na+ is varied (constant ionicstrength = 0.1 mol L−1) [243]. The dashed line is the parity line. (A) Copy-right 1994. Reproduced from [245] with permission from Elsevier Ltd.(B) Copyright 1992. Reproduced from [243] with permission from the
28 G.M. Geise et al. / Progress i
increase in passage of the sodium ions, which are smallerthan the divalent ions because, as shown in Fig. 26, smallerions are more permeable. Therefore, in a filtration appli-cation involving a mixture of NaCl and divalent ions, thetransport of NaCl will be greater than would be expectedbased on transport measurements made using only NaCl.
Alternatively, the di-sulfonated polysulfone materialsexhibit less of an increase in monovalent ion passage as themole fraction of divalent cations increases compared to theother materials shown in Fig. 30. This result is desirable fordesalination applications in that these di-sulfonated poly-sulfones do not lose as much of their ability to separate NaClfrom water upon introduction of divalent ions comparedto the sulfonated PPO or Hi-Flux CP polymers. The funda-mental origin of the difference between the di-sulfonatedpolysulfones and the sulfonated PPO and Hi-Flux CP poly-mers is not well understood at this time. Additionally, littleis known about whether the increase in sodium passage asdivalent ions are added to the feed results from sorption,diffusivity, or both components of permeability.
To better understand mixed salt transport in chargedpolymers, fundamental transport properties, i.e., perme-ability, diffusion, and ion sorption coefficients, must bemeasured for polymers exposed to salt mixtures. Given theprevious discussion regarding the influence of the counter-ion on water sorption by charged polymers, the counter-ionform of the charged polymer will likely influence the trans-port properties of the polymer. As discussed previously,some counter-ions bind more strongly to fixed chargegroups than others (Fig. 20), and this binding affinity canaffect the polymer’s water sorption transport properties[87,243–245].
In multi-salt mixtures, the counter-ion form of the poly-mer may not be as well defined as in the case wherethe polymer is exposed to a single salt solution. Fig. 31shows an example of the dependence of the counter-ionform of a charged polymer on the ratio of the concentra-tions of the various counter-ions in the external solutionphase [243–245]. In Fig. 31A, as the salt solution becomesenriched in Li+ relative to Na+, the counter-ion form ofthe polymer varies from the sodium form to the lithiumform, and since these curves intersect beyond the midwaypoint of the x-axis (i.e., on the Li+ ion rich side of the x-axis), the Na+ counter-ion form is preferred over the Li+
form because it takes a Li+/Na+ ratio in solution greaterthan 1 to bring the Li+/Na+ ratio in the polymer above 1[245]. In Fig. 31B, the fraction of polymer that is in thecounter-ion form listed on the curve, relative to the sodiumcounter-ion form, is plotted as a function of the mole frac-tion of that ion. Ions whose curves lie above the dashed linepreferentially bind to the fixed charge groups over sodiumbecause the mole fraction of those ions in the polymer isgreater than the mole fraction of those ions in solution[243]. If the water content of the polymer changes signifi-cantly as the counter-ion form varies from one extreme tothe other, the permeability and selectivity properties of thepolymer may be sensitive to the ionic composition of the
solution.Measurements, such as those shown as Fig. 31, highlightthe dependence of counter-ion sorption on external solu-tion composition. Co-ion sorption measurements, made
American Chemical Society.
as a function of external salt solution concentration, areneeded to compliment the counter-ion measurementsto fully describe ion sorption in mixed salt transport.The combination of these results and ion permeabilitymeasurements will provide insight into the fundamen-tal phenomena that govern ion transport when chargedpolymers are exposed to solutions containing mixtures ofsalts.
3.7. Influence of processing conditions on transport
propertiesMany polymers considered for desalination applica-tions are glassy, at least when they are dry. For example,

G.M. Geise et al. / Progress in Polymer Science 39 (2014) 1– 42 29
Table 7Influence of processing conditions on a sulfonated polysulfone random copolymer [82].
Sample Kw Hydraulic waterpermeability[L �m m−2 h−1 bar−1]
Saltpermeability[cm2 s−1]
Water/saltpermeabilityselectivity
Films cast in the potassium counter-ion formAs cast 0.20 0.25 0.59 × 10−9 510Acidified at room temperature 0.24 0.65 1.4 × 10−9 460Acidified at Reflux Conditions 0.36 2.75 30 × 10−9 49Films cast in the acid counter-ion formAs cast 0.27 0.36 0.70 × 10−9 430Ion exchanged to the potassium counter-ion form 0.26 0.38 0.76 × 10−9 450Acidified at room temperature 0.29 0.48 0.91 × 10−9 410
N re watep
dptgWpsom[
cfibcem[tpfatcbTmpmlppa[
ssa[mppwct
technique is around 12% [260].
ote: Water sorption and transport measurements were made using puressure and 25 ◦C using a 0.1 mol(NaCl) L−1 upstream solution.
ry cellulose acetate, sulfonated polysulfone, aromaticolyamide, and sulfonated polystyrene have glass transi-ion temperatures that are greater and, in some cases, muchreater than room temperature [82,136,232,246,247].hile water sorption can depress the glass transition tem-
erature of these polymers, the inherent non-equilibriumtate of these dry polymers provides an opportunity forptimizing transport properties by conditioning the poly-ers and/or varying the details of the preparation protocols
82,248–250].Fig. 25B shows an example of the effect of thermal
onditioning on salt permeability; boiling Nafion® 115lms increases salt permeability [232]. Similar results haveeen reported for di-sulfonated polysulfone, and differentombinations of thermal treatment, acidification, and ionxchange can be used to optimize the water and salt per-eability, sorption, and diffusion properties of the polymer
82]. An example of the influence of processing condi-ions on sulfonated polysulfone water and salt permeabilityroperties is shown as Table 7. In these experiments, a sul-onated polymer was prepared in both the potassium andcid counter-ion forms, and these films were ion exchangedo the other counter-ion form and back (i.e., potassiumounter-ion samples were acidified and then convertedack to the potassium counter-ion from). As observed inable 7, water uptake, water permeability, and salt per-eability are sensitive to the counter-ion form of the
olymer, but these properties are most sensitive to theanner in which the polymer is brought to a particu-
ar counter-ion form. In particular, the water uptake andermeability properties increase considerably when theolymer is refluxed in 0.5 mol L−1 sulfuric acid, which isttributed to an increase in the polymer’s free volume82].
Additionally, block copolymers containing highlywollen hydrophilic micro-domains that phaseeparate from hydrophobic glassy micro-domainsre being considered for membrane applications79,133,148,153,215,216,237,251–254]. In these poly-
ers, the glassy hydrophobic blocks and the overallhase separated morphology likely will be sensitive torocessing conditions. In all of these cases, the extent to
hich varying processing and preparation parametersould be used to optimize a polymer’s water and saltransport properties is not currently well understood
r at ambient temperature. Salt permeability was measured at ambient
but could be useful for designing improved membranematerials.
4. Characterization of thin-film compositemembrane active layers
Characterizing fundamental water and salt permeabil-ity properties of thin-film composite membrane activelayers is challenging because the active layer thicknessis often not well known and difficult to measure [70].Typically the active layer thickness of a cross-linked thin-film composite polyamide membrane is reported to beapproximately 100 nm [13,255]. An additional challenge tomeasuring fundamental properties of thin-film compositemembrane active layers is the presence of gradients in com-position and degree of cross-linking, which result from theinterfacial polymerization reaction, throughout the thick-ness of the active layer [256,257]. However, interest inbetter understanding fundamental transport properties inthese materials has driven the development of several tech-niques for characterizing these membrane active layers[258].
The porous support layer used in thin film compos-ite membranes introduces another challenge in measuringtransport properties in such membranes because masstransfer resistance in the support layer is often not insignif-icant relative to the active layer [39,40,59,119]. In caseswhere the active layer of a composite membrane can beisolated from the support membrane without damagingthe active layer, AFM techniques have been developed tomeasure the thickness, swelling, and glass transition tem-perature of these thin films [147,247]. These techniques,performed on NF and RO membranes, indicate that themass fraction of water in the active layers of RO membranesis typically below 10%, and the mass fraction of water in theactive layers of NF membranes is typically greater (reportedvalues are in the range of 15–50%) [147,259]. Additionally,a quartz crystal microbalance technique has been used tomeasure water uptake in different commercial RO mem-brane active layers; the water uptake reported using this
These results further support the view that swellingmust be controlled to prepare membrane materials withhigh water/salt selectivity [13]. These results are also
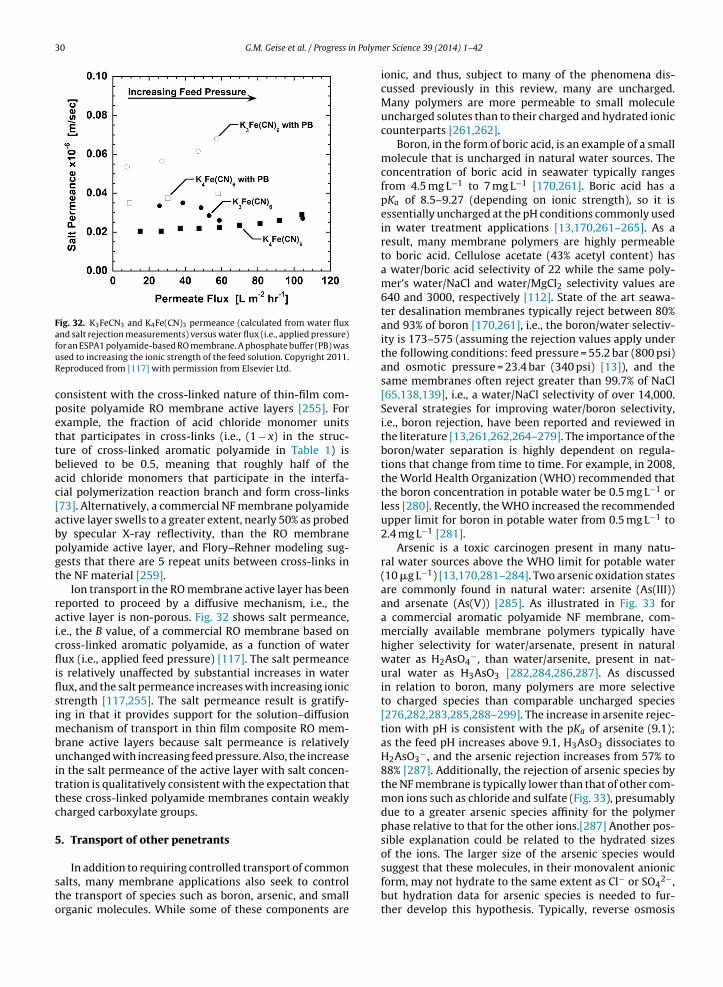
30 G.M. Geise et al. / Progress in Polym
Fig. 32. K3FeCN3 and K4Fe(CN)3 permeance (calculated from water fluxand salt rejection measurements) versus water flux (i.e., applied pressure)
for an ESPA1 polyamide-based RO membrane. A phosphate buffer (PB) wasused to increasing the ionic strength of the feed solution. Copyright 2011.Reproduced from [117] with permission from Elsevier Ltd.consistent with the cross-linked nature of thin-film com-posite polyamide RO membrane active layers [255]. Forexample, the fraction of acid chloride monomer unitsthat participates in cross-links (i.e., (1 − x) in the struc-ture of cross-linked aromatic polyamide in Table 1) isbelieved to be 0.5, meaning that roughly half of theacid chloride monomers that participate in the interfa-cial polymerization reaction branch and form cross-links[73]. Alternatively, a commercial NF membrane polyamideactive layer swells to a greater extent, nearly 50% as probedby specular X-ray reflectivity, than the RO membranepolyamide active layer, and Flory–Rehner modeling sug-gests that there are 5 repeat units between cross-links inthe NF material [259].
Ion transport in the RO membrane active layer has beenreported to proceed by a diffusive mechanism, i.e., theactive layer is non-porous. Fig. 32 shows salt permeance,i.e., the B value, of a commercial RO membrane based oncross-linked aromatic polyamide, as a function of waterflux (i.e., applied feed pressure) [117]. The salt permeanceis relatively unaffected by substantial increases in waterflux, and the salt permeance increases with increasing ionicstrength [117,255]. The salt permeance result is gratify-ing in that it provides support for the solution–diffusionmechanism of transport in thin film composite RO mem-brane active layers because salt permeance is relativelyunchanged with increasing feed pressure. Also, the increasein the salt permeance of the active layer with salt concen-tration is qualitatively consistent with the expectation thatthese cross-linked polyamide membranes contain weaklycharged carboxylate groups.
5. Transport of other penetrants
In addition to requiring controlled transport of commonsalts, many membrane applications also seek to controlthe transport of species such as boron, arsenic, and smallorganic molecules. While some of these components are
er Science 39 (2014) 1– 42
ionic, and thus, subject to many of the phenomena dis-cussed previously in this review, many are uncharged.Many polymers are more permeable to small moleculeuncharged solutes than to their charged and hydrated ioniccounterparts [261,262].
Boron, in the form of boric acid, is an example of a smallmolecule that is uncharged in natural water sources. Theconcentration of boric acid in seawater typically rangesfrom 4.5 mg L−1 to 7 mg L−1 [170,261]. Boric acid has apKa of 8.5–9.27 (depending on ionic strength), so it isessentially uncharged at the pH conditions commonly usedin water treatment applications [13,170,261–265]. As aresult, many membrane polymers are highly permeableto boric acid. Cellulose acetate (43% acetyl content) hasa water/boric acid selectivity of 22 while the same poly-mer’s water/NaCl and water/MgCl2 selectivity values are640 and 3000, respectively [112]. State of the art seawa-ter desalination membranes typically reject between 80%and 93% of boron [170,261], i.e., the boron/water selectiv-ity is 173–575 (assuming the rejection values apply underthe following conditions: feed pressure = 55.2 bar (800 psi)and osmotic pressure = 23.4 bar (340 psi) [13]), and thesame membranes often reject greater than 99.7% of NaCl[65,138,139], i.e., a water/NaCl selectivity of over 14,000.Several strategies for improving water/boron selectivity,i.e., boron rejection, have been reported and reviewed inthe literature [13,261,262,264–279]. The importance of theboron/water separation is highly dependent on regula-tions that change from time to time. For example, in 2008,the World Health Organization (WHO) recommended thatthe boron concentration in potable water be 0.5 mg L−1 orless [280]. Recently, the WHO increased the recommendedupper limit for boron in potable water from 0.5 mg L−1 to2.4 mg L−1 [281].
Arsenic is a toxic carcinogen present in many natu-ral water sources above the WHO limit for potable water(10 �g L−1) [13,170,281–284]. Two arsenic oxidation statesare commonly found in natural water: arsenite (As(III))and arsenate (As(V)) [285]. As illustrated in Fig. 33 fora commercial aromatic polyamide NF membrane, com-mercially available membrane polymers typically havehigher selectivity for water/arsenate, present in naturalwater as H2AsO4
−, than water/arsenite, present in nat-ural water as H3AsO3 [282,284,286,287]. As discussedin relation to boron, many polymers are more selectiveto charged species than comparable uncharged species[276,282,283,285,288–299]. The increase in arsenite rejec-tion with pH is consistent with the pKa of arsenite (9.1);as the feed pH increases above 9.1, H3AsO3 dissociates toH2AsO3
−, and the arsenic rejection increases from 57% to88% [287]. Additionally, the rejection of arsenic species bythe NF membrane is typically lower than that of other com-mon ions such as chloride and sulfate (Fig. 33), presumablydue to a greater arsenic species affinity for the polymerphase relative to that for the other ions.[287] Another pos-sible explanation could be related to the hydrated sizesof the ions. The larger size of the arsenic species would
suggest that these molecules, in their monovalent anionicform, may not hydrate to the same extent as Cl− or SO42−,but hydration data for arsenic species is needed to fur-ther develop this hypothesis. Typically, reverse osmosis
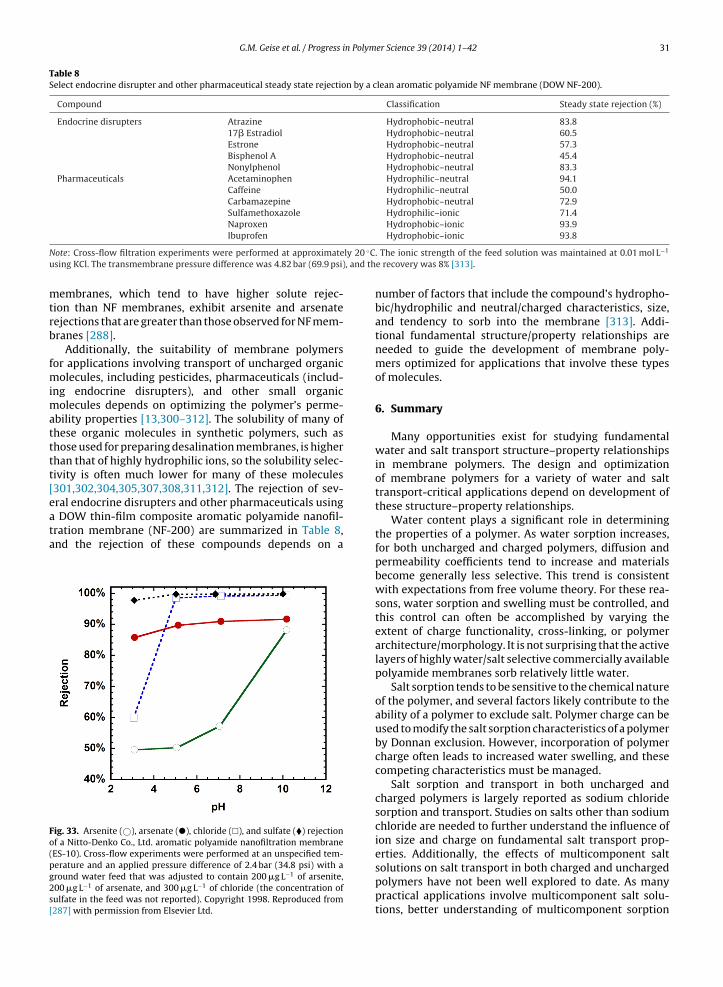
G.M. Geise et al. / Progress in Polymer Science 39 (2014) 1– 42 31
Table 8Select endocrine disrupter and other pharmaceutical steady state rejection by a clean aromatic polyamide NF membrane (DOW NF-200).
Compound Classification Steady state rejection (%)
Endocrine disrupters Atrazine Hydrophobic–neutral 83.817� Estradiol Hydrophobic–neutral 60.5Estrone Hydrophobic–neutral 57.3Bisphenol A Hydrophobic–neutral 45.4Nonylphenol Hydrophobic–neutral 83.3
Pharmaceuticals Acetaminophen Hydrophilic–neutral 94.1Caffeine Hydrophilic–neutral 50.0Carbamazepine Hydrophobic–neutral 72.9Sulfamethoxazole Hydrophilic–ionic 71.4Naproxen Hydrophobic–ionic 93.9
N ly 20 ◦Cu , and th
mtrb
fmimatttt[eata
Fo(pg2s[
Ibuprofen
ote: Cross-flow filtration experiments were performed at approximatesing KCl. The transmembrane pressure difference was 4.82 bar (69.9 psi)
embranes, which tend to have higher solute rejec-ion than NF membranes, exhibit arsenite and arsenateejections that are greater than those observed for NF mem-ranes [288].
Additionally, the suitability of membrane polymersor applications involving transport of uncharged organic
olecules, including pesticides, pharmaceuticals (includ-ng endocrine disrupters), and other small organic
olecules depends on optimizing the polymer’s perme-bility properties [13,300–312]. The solubility of many ofhese organic molecules in synthetic polymers, such ashose used for preparing desalination membranes, is higherhan that of highly hydrophilic ions, so the solubility selec-ivity is often much lower for many of these molecules301,302,304,305,307,308,311,312]. The rejection of sev-ral endocrine disrupters and other pharmaceuticals using
DOW thin-film composite aromatic polyamide nanofil-ration membrane (NF-200) are summarized in Table 8,nd the rejection of these compounds depends on a
ig. 33. Arsenite (©), arsenate (�), chloride (�), and sulfate (�) rejectionf a Nitto-Denko Co., Ltd. aromatic polyamide nanofiltration membraneES-10). Cross-flow experiments were performed at an unspecified tem-erature and an applied pressure difference of 2.4 bar (34.8 psi) with around water feed that was adjusted to contain 200 �g L−1 of arsenite,00 �g L−1 of arsenate, and 300 �g L−1 of chloride (the concentration ofulfate in the feed was not reported). Copyright 1998. Reproduced from287] with permission from Elsevier Ltd.
Hydrophobic–ionic 93.8
. The ionic strength of the feed solution was maintained at 0.01 mol L−1
e recovery was 8% [313].
number of factors that include the compound’s hydropho-bic/hydrophilic and neutral/charged characteristics, size,and tendency to sorb into the membrane [313]. Addi-tional fundamental structure/property relationships areneeded to guide the development of membrane poly-mers optimized for applications that involve these typesof molecules.
6. Summary
Many opportunities exist for studying fundamentalwater and salt transport structure–property relationshipsin membrane polymers. The design and optimizationof membrane polymers for a variety of water and salttransport-critical applications depend on development ofthese structure–property relationships.
Water content plays a significant role in determiningthe properties of a polymer. As water sorption increases,for both uncharged and charged polymers, diffusion andpermeability coefficients tend to increase and materialsbecome generally less selective. This trend is consistentwith expectations from free volume theory. For these rea-sons, water sorption and swelling must be controlled, andthis control can often be accomplished by varying theextent of charge functionality, cross-linking, or polymerarchitecture/morphology. It is not surprising that the activelayers of highly water/salt selective commercially availablepolyamide membranes sorb relatively little water.
Salt sorption tends to be sensitive to the chemical natureof the polymer, and several factors likely contribute to theability of a polymer to exclude salt. Polymer charge can beused to modify the salt sorption characteristics of a polymerby Donnan exclusion. However, incorporation of polymercharge often leads to increased water swelling, and thesecompeting characteristics must be managed.
Salt sorption and transport in both uncharged andcharged polymers is largely reported as sodium chloridesorption and transport. Studies on salts other than sodiumchloride are needed to further understand the influence ofion size and charge on fundamental salt transport prop-erties. Additionally, the effects of multicomponent salt
solutions on salt transport in both charged and unchargedpolymers have not been well explored to date. As manypractical applications involve multicomponent salt solu-tions, better understanding of multicomponent sorption
n Polymer Science 39 (2014) 1– 42
Fig. A.1. Water concentration, activity, and hydrostatic pressure profilesacross a non-porous polymer film. The water volume fraction, �m
w , massfraction, wm
w , and water activity, amw , are shown to be a linear function of
position in the film for simplicity, but in some cases, these profiles will
32 G.M. Geise et al. / Progress i
and diffusion phenomena is critical for optimizing polymerproperties. Fundamental structure/property relationshipsare needed to effectively design materials to controltransport of water and small molecules such as boron,arsenic, and organic compounds to optimize polymers fora variety of applications. In charged polymers, most ofthe fundamental studies have been performed on cationexchange materials, such as sulfonated polymers. In avariety of applications (e.g., electrodialysis, reverse elec-trodialysis, capacitive deionization, etc.), both cation andanion exchange membranes are required, and fundamen-tal structure/properties studies, such as those described inthis review, are virtually nonexistent in the open literaturefor anion exchange membrane materials.
Techniques for analyzing fundamental properties ofthin active layer polymer films of commercial membranesare important for connecting structure–property relation-ships developed for thick film polymers with those of thinfilms. Water sorption, ion transport, and morphology aresome of the properties that can be measured for thin filmactive layer membrane polymers.
Acknowledgements
This work was partially supported by Kraton Perfor-mance Polymers, Inc. (Houston, TX), the NSF’s Partnershipfor Innovation (PFI) Accelerating Innovative Research (AIR)Program (Grant no. IIP-1237857), the NSF Division ofChemical, Bioengineering, Environmental, and TransportSystems (Grants nos. 1160128 and 1160069), and the NSFCenter for Layered Polymeric Systems (CLiPS) (Grant no.DMR-0423914).
Appendix A. Relating hydraulic water permeabilityto diffusive water permeability and the averagewater diffusion coefficient
The hydraulic permeability, PHw , of a polymer film is
defined experimentally as [81,101]
PHw ≡ nwLH
�p − ��(A.1)
where nw is the steady-state volumetric water flux throughthe film, LH is the freely water swollen film thickness,and �p and �� are the hydrostatic and osmotic pressuredifferences across the film, respectively. The permeabilityis normalized by the freely water swollen film thickness,rather than the film thickness during the permeation mea-surement, because LH can be measured much more easilythan the film thickness during permeation, L, which couldbe slightly less than LH owing to some loss of liquid fromthe film due to the gradient in water concentration presentduring the permeation measurement. Fig. A.1 presents theprofiles of hydrostatic pressure, water concentration, andthermodynamic activity across a film undergoing waterpermeation. The figure introduces some of the nomencla-
ture used in this discussion.A constitutive equation for flux is required to relate thehydraulic water permeability to the water sorption, Kw, andeffective water diffusion, Dw, coefficients via the diffusive
not necessarily be linear [107]. The superscript “m” represents variablesevaluated inside the polymer film (i.e., membrane), and “s” representsvariables evaluated in the solution surrounding the polymer film.
water permeability, Pw, and the solution diffusion model[106,107]
Pw = Kw Dw (A.2)
The ternary (i.e., water, salt, and polymer)Maxwell–Stefan equations are the starting point toformulate this constitutive equation [107,314,315]. Whenfrictional coupling is negligible, as has been shown tobe reasonable for desalination systems [107], and watertransport is one-dimensional, in the x direction, the ternaryMaxwell–Stefan equation is [107,314]
nw = −DTwp
�mw
1 − �mw − �m
s
d ln amw
dx(A.3)
where �mw and �m
s are the volume fractions of water andsalt in the polymer, respectively, DT
wp is the water diffusioncoefficient in the polymer, and am
w is the activity of water inthe polymer. Typically, the volume fraction of salt sorbed inthe polymer is negligible compared to the volume fractionof water sorbed in the polymer (i.e., �m
s � �mw ), so the water
flux may be simplified as follows:
nw = −DTwp
�mw
1 − �mw
d ln amw
dx(A.4)
Since the driving force for mass transfer is oftenexpressed in terms of composition gradients ratherthan activity gradients, the diffusion coefficient is oftencombined with a thermodynamic factor to account fornon-ideal solution behavior. Thus, the thermodynamicallycorrected mutual diffusion coefficient, Dwp, is typicallydefined as follows [107,314,316]:
Dwp ≡ DT
(∂ ln am
w
)(A.5)
wp ∂ ln �mw T,p
Introducing this diffusion coefficient into Eq. (A.4) per-mits the driving force for water transport to be written in

n Polym
tm
n
tcc
n
w
D
n
wbutc⟨
n
nwowt
w
wxusopTaadeaae
G.M. Geise et al. / Progress i
erms of the concentration gradient of water in the polymerembrane:
w = −Dwp
1 − �mw
d�mw
dx(A.6)
Eq. (A.6) can be integrated across the thickness ofhe film during the permeation experiment, L, and theoncentration-averaged effective diffusion coefficient, Dw,an be introduced to obtain:
wL = Dw ln
[1 − �m
wL
1 − �mw0
](A.7)
here Dw is defined as:
w ≡ 1
ln[(1 − �m
wL)/(1 − �mw0)]
�mw0∫
�mwL
Dwp
(1 − �mw )
d�mw (A.8)
Eq. (A.7) can be re-written as follows:
w = Dw
L
(�m
w0 − �mwL
) ln[�m
pL/�mp0
]�m
pL − �mp0
(A.9)
here �mp is the volume fraction of polymer in the mem-
rane phase, and the relationship �mp = 1 − �m
w has beensed. This relationship is valid as long as the volume frac-ion of salt in the film is low. The log mean polymeroncentration in the film is defined as follows:
�mp
⟩=
�mpL − �m
p0
ln(
�mpL/�m
p0
) = �mw0 − �m
wL
ln[(1 − �mwL)/(1 − �m
w0)](A.10)
Eqs. (A.9) and (A.10) can be combined to obtain
w = Dw
L⟨
�mp
⟩ (�mw0 − �m
wL
)(A.11)
For practical purposes, it is useful to replace the thick-ess of the film during the permeation measurement, L,hich typically cannot be measured directly, with that
btained by swelling the polymer sample in pure water, LH,hich can be readily measured. A polymer mass balance in
he swollen film relates these two thicknesses [107]
mp0�HLHA = A
L∫0
�pwmp dx (A.12)
here wmp is the mass fraction of polymer at any position
in the film, wmp0 is the mass fraction of polymer at the
pstream face when the polymer is equilibrated in the feedolution, A is the film surface area normal to the directionf water flow, �p is the film density during steady stateermeation, and �H is the fully water swollen film density.he left hand side of Eq. (A.12) is the mass of polymer in
film that has been freely swollen in the feed solution,nd the right hand side of Eq. (A.12) is the mass of the filmuring a permeation experiment, so these two masses are
qual to one another. This would be the case if, for example,film, freely swollen in the feed solution, was loaded into permeation cell of fixed area, and a water permeationxperiment were then performed using that film.
er Science 39 (2014) 1– 42 33
For many polymers of interest for desalination, the massof polymer per unit volume of swollen film during the per-meability measurement, �p, is practically equal to the massof polymer per unit volume of a film freely swollen in thefeed solution, �H, so these terms cancel, and Eq. (A.12) issimplified as follows:
wmp0LH =
L∫0
wmp dx (A.13)
The steady-state concentration profile, when the diffu-sion coefficient is constant, is [317]
wmp = wm
p0
(wm
pL
wmp0
)x⁄L(A.14)
Inserting this concentration profile into Eq. (A.13) andintegrating yields:
wmp0LH =
⟨wm
p
⟩L (A.15)
where⟨
wmp
⟩is the log mean average weight fraction
of polymer across the film. Because the flux equation isexpressed in terms of volume fractions, it is convenient toexpress Eq. (A.15) in terms of volume fractions, which maybe done formally as shown below:
�mp0
(wm
p0
�mp0
)LH =
⟨�m
p
⟩(⟨wmp
⟩⟨�m
p
⟩)
L (A.16)
where the bracket notation (i.e., 〈 〉) refers to the logarithmicmean as discussed for Eq. (A.10).
Because the water content is not typically very differ-ent between the feed and permeate faces of a polymerfilm being used in a permeation experiment, the terms inparenthesis in Eq. (A.16) effectively cancel for polymersthat sorb less than approximately 75 volume percent waterand are exposed to applied pressure differences of less than1000 psi (69 bar). Thus, Eq. (A.16) can be simplified as
LH�mp0 = L
⟨�m
p
⟩(A.17)
which provides the desired relationship between LH and L.This result is then inserted into Eq. (A.11):
nw = Dw
LH(
1 − �mw0
) (�mw0 − �m
wL
)(A.18)
Typically, the volume fractions of water dissolved in thepolymer at its upstream face (i.e., feed or high pressureside), �m
w0, and downstream (i.e., permeate or low pres-sure side) face, �m
wL , are written in terms of the externallyimposed pressure and salt concentration differences acrossthe polymer film. The first step to relate the internal waterconcentration in the membrane to the external hydrostaticand osmotic pressure differences is to equate the chemicalpotential of water in the polymer to that in the surround-ing solutions at both the upstream and downstream faces of
the film. Following this approach, the ratio of water activityin the polymer film at the downstream and upstream facesof the film may be expressed in terms of the osmotic pres-sure difference and applied hydrostatic pressure difference
n Polym
34 G.M. Geise et al. / Progress iacross the film [107,132]
amwL
amw0
= exp
[− Vw
RT(�p − ��)
]∼= 1 − Vw
RT(�p − ��)
(A.19)
where the activity of water in the polymer is equal to that inthe external solution at the upstream face (i.e., am
w0 = asw0),
R is the ideal gas constant, T is the absolute temperature,and Vw is the partial molar volume of water, which at ambi-ent temperature may be approximated as the molar volumeof water, 18 cm3 mol−1. The linear approximation of theexponential term is valid for any practical scenario involv-ing liquid water permeation through a polymer film [107].To use Eq. (A.19) with Eq. (A.18), a connection must bemade between the volume fraction of water in the polymerand the thermodynamic activity of water in the polymer.Because the change in the water content from the feed tothe permeate side of the membrane is often small, the dif-ference in the volume fraction of water across the film canbe adequately described by the first term of the followingTaylor series expansion:
�mw0
∼= �mwL +
(am
w0 − amwL
) d�mw
damw
∣∣∣am
w0
(A.20)
Eq. (A.19) can be combined with Eq. (A.20) to yield
�mw0 − �m
wL = �mw0
Vw
RT(�p − ��)
d ln �mw
d ln amw
∣∣∣am
w0
(A.21)
Eq. (A.21) provides the desired relation between thewater concentration difference across the membrane as aresult of externally imposed hydrostatic and osmotic pres-sure differences. Combining Eqs. (A.18) and (A.21), the fluxequation becomes
nw = Dw�mw0
LH(
1 − �mw0
) Vw
RT(�p − ��)
d ln �mw
d ln amw
∣∣∣am
w0
(A.22)
The volume fraction of water in the polymer at theupstream face, �m
w0, is related to the water sorption coeffi-cient, Kw, by
Kw = �mw0Mw
CswVw
(A.23)
where Mw is the molar mass of water and Csw is the con-
centration of water in the contiguous solution. For saltsolutions that are sufficiently dilute, Cs
w is the density ofpure water, and the water sorption coefficient and volumefraction of water in the polymer at the upstream face areequal (i.e., Kw = �m
w0). In this case, the diffusive water per-meability can be introduced according to Eq. (A.2), and Eq.(A.22) becomes
nw = Pw
LH(
1 − �mw0
) Vw
RT(�p − ��)
d ln �mw
d ln amw
∣∣∣am
w0
(A.24)
Eq. (A.24) contains both a convective (frame of refer-ence) correction term, 1/
(1 − �m
w0
), and a thermodynamic( )∣
correction factor, d ln �mw /d ln am
w∣am
w0, which accounts for
non-ideal behavior of water sorbed in the polymer matrix.Two scenarios are considered below, one in which the con-centration of water in the polymer is low, which minimizes
er Science 39 (2014) 1– 42
the frame of reference and thermodynamic effects, andanother where the water content in the polymer is highenough that these effects measurably influence the waterflux.
A.1. Polymers that sorb relatively little water
When the volume fraction of water in the polymeris small (i.e., when �m
w � 1), the convective (frame ofreference) term in Eq. (A.24) is essentially equal to 1. Addi-tionally, the derivative in Eq. (A.24) also approaches 1 forpolymers that sorb relatively little water. The derivative inEq. (A.24) can be written as
d ln �mw
d ln amw
= 1
1 + d ln mw
d ln �mw
(A.25)
where mw is the activity coefficient of water in the polymer
(i.e., amw = m
w �mw ). If the relation between the water activity
in the polymer and the water concentration in the polymeris described by the Flory–Huggins equation, the derivativeon the right-hand side of Eq. (A.25) is [201,202]
d ln mw
d ln �mw
= −�mw − 2��m
w
(1 − �m
w
)(A.26)
As �mw becomes small relative to 1,
(d ln m
w /d ln �mw
)→
0, so(
d ln �mw /d ln am
w
)→ 1. Therefore, for polymers that
sorb relatively little water, Eq. (A.24) becomes
nw = Pw
LH
Vw
RT(�p − ��) (A.27)
which is the commonly cited expression for the water fluxthrough dense polymers [106,115,132]. In this limit, Eqs.(A.1) and (A.27) can be combined to relate the hydraulicwater permeability to the diffusive water permeability inthis limit:
Pw = PHw
RT
Vw(A.28)
A.2. Highly swollen polymers
Consider a highly swollen polymer (e.g., a hydrogel)where, for example, �m
w = 0.9. For such a material, theeffect of the convective (frame of reference) correctionfactor on water flux alone (not including the thermody-namic correction) could be approximately a factor of 10(i.e., 1/(1 − 0.9) = 10. Therefore, the full version of the fluxequation (i.e., Eq. (A.24)) should be used. Eq. (A.24) can bewritten as follows (assuming �m
w0 = Kw):
nw = DwKw
LH (1 − Kw)Vw
RT(�p − ��)
d ln �mw
d ln amw
∣∣∣am
w0
(A.29)
where, at equilibrium, the activity of water in the film atthe upstream face of the polymer film, am
w0, is equal to theactivity of water in the upstream (or feed) solution, as
w0,which, for salt solutions, can be determined experimentally
[318] or calculated using a model such as the Pitzer model[130,319]. Eq. (A.29) relates the water flux to the effectivediffusion and water sorption coefficients, but the deriva-tive must be evaluated using either experimental data or
G.M. Geise et al. / Progress in Polymer Science 39 (2014) 1– 42 35
Table A.1Equations for relating hydraulic water permeability to diffusive water permeability.
Physical situation Diffusive water permeability (Pw) Equation no.
Polymers with low water uptake (i.e., �mw � 1) Pw = KwDw = PH
wRTVw
(A.28)
Highly swollen polymers Experimental data Pw = KwDw = PHw
RTVw
(1−Kw )ı
(A.31)
Flory–Huggins Pw = KwDw = PHw
RTVw
(1 − Kw)2 (1 − 2�Kw) (A.34)
Note: PHw ≡ nwLH
.
avwe
moitpidotuo
t
P
t
a
(
n
p
P
A
hlads
limit �mw � 1 because Kw = 0.62, so, as mentioned earlier,
�mw = 0.62. Therefore, experimental water sorption data
were used to directly calculate the effective diffusion coef-
Table A.2Properties of an XLPEGDA hydrogel cross-linked in the presence of 60 wt%water.
�p − ��
thermodynamic model (e.g., Flory–Huggins) to relate theolume fraction of water in the polymer to the activity ofater in polymer and, in turn, to the activity of water in the
xternal solution at equilibrium.The derivative in Eq. (A.29) can be evaluated experi-
entally by measuring the equilibrium volume fractionf water in the polymer as a function of water activityn the solution surrounding the polymer. Experimentally,his measurement is typically performed by equilibratingolymer films in water vapor at specified relative humid-
ty values (see the example provided below) [320,321]. Theerivative in Eq. (A.29) can then be determined as the slopef the water volume fraction versus activity isotherm inhe limit where the water activity approaches that of thepstream solution (e.g., unity for a feed solution composedf pure water):
ı = d ln �mw
d ln amw
∣∣∣am
w0
(A.30)
Thus, the flux equation (i.e., Eq. (A.29)) can be writteno relate hydraulic permeability to diffusive permeability:
w = PHw
RT
Vw
(1 − Kw)ı
(A.31)
Alternatively, ı can be evaluated using a model, such ashe Flory–Huggins theory [201,202]
mw = �m
w exp[
1 − �mw + �
(1 − �m
w
)2]
(A.32)
Eq. (A.32) can be used to evaluate the derivative in Eq.A.29) to give the following:
w = Pw
LH(1 − Kw)2 (1 − 2�Kw)
Vw
RT(�p − ��) (A.33)
In this case, the relation between hydraulic and diffusiveermeability is as follows [81,131]
w = PHw
RT
Vw(1 − Kw)2 (1 − 2�Kw) (A.34)
.3. Summary
This formalism provides a framework for relatingydraulic permeability, which is readily measured in the
aboratory, to diffusive water permeability, which has fundamental dependence on the water sorption andiffusion coefficients in the polymer [106,107]. Table A.1ummarizes the three cases considered.
A.4. An example using a cross-linked hydrogel
A comparison can be made between the methodsdiscussed in the preceding section by considering across-linked poly(ethylene glycol) diacrylate hydrogel(XLPEGDA) [101]. This hydrogel was prepared via UV cross-linking using 0.1 wt% 1-hydroxycyclohexyl phenyl ketoneas the initiator [101]. When this hydrogel was cross-linkedin the presence of 60 wt% water, the resulting hydrogel hasthe properties recorded in Table A.2. The � parameter wasdetermined by forcing the pure water uptake in the poly-mer to be described by the Flory–Huggins equation. Sincethis material is cross-linked in the presence of a diluent, itmay be more appropriate to use the Flory–Rehner [196] orPeppas–Merrill [199] equations to model the water uptake.However, these equations require knowledge of param-eters, such as cross-link density, that can be difficult todetermine independently in many materials of interest, sowe use the Flory–Huggins model for illustrative purposesonly.
Based upon the parameters in Table A.2, the diffusivewater permeability, Pw, of the XLPEGDA hydrogel is cal-culated to be 3.8 × 10−5− cm2 s−1 using Eq. (A.28). Theeffective diffusion coefficient, Dw, calculated using Eq. (A.2)is 6.2 × 10−5 cm2 s−1. This value, however, exceeds the self-diffusion coefficient of water, which is 2.6×10−5 cm2 s−1 atthe temperature of the experiment (25 ◦C) because the con-vective frame of reference and thermodynamic correctionfactors have not been explicitly evaluated, so the effec-tive diffusion coefficient contains contributions from bothof these factors [155]. This result is not an indication ofconvective pore flow of water through the polymer film,but rather, it illustrates the potential importance of theframe of reference and thermodynamic correction terms[107,131,322,323].
According to Table A.2, this polymer does not satisfy the
Property Value References
PHw 10.0 L �m m−2 h−1 bar−1 [101]
Kw 0.62 [101]� 0.68
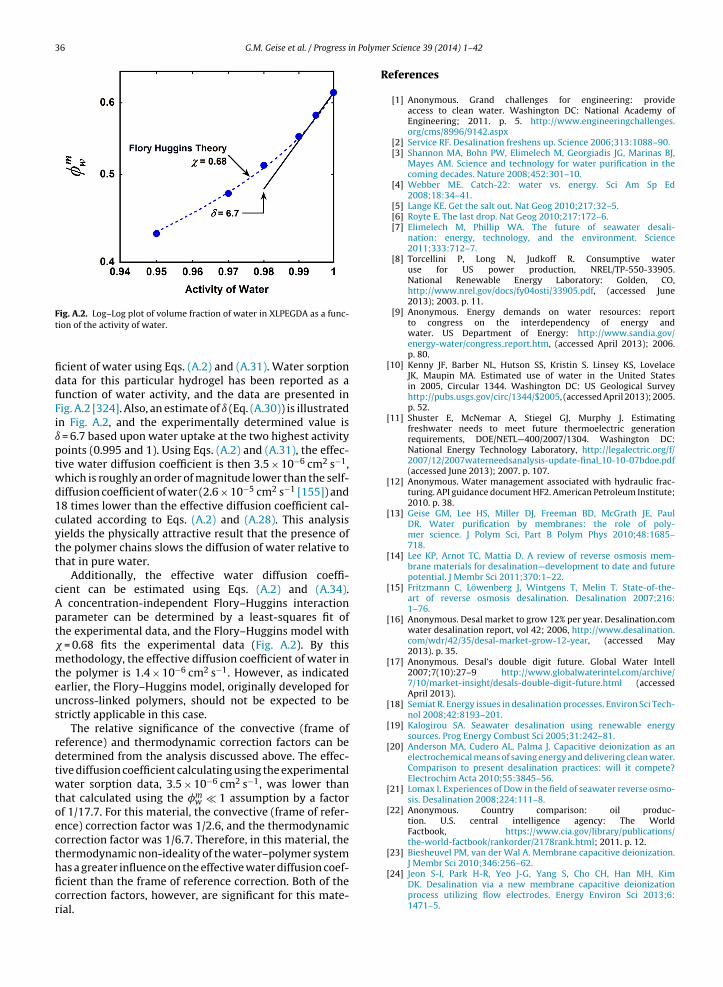
36 G.M. Geise et al. / Progress in Polym
J Membr Sci 2010;346:256–62.[24] Jeon S-I, Park H-R, Yeo J-G, Yang S, Cho CH, Han MH, Kim
Fig. A.2. Log–Log plot of volume fraction of water in XLPEGDA as a func-tion of the activity of water.
ficient of water using Eqs. (A.2) and (A.31). Water sorptiondata for this particular hydrogel has been reported as afunction of water activity, and the data are presented inFig. A.2 [324]. Also, an estimate of ı (Eq. (A.30)) is illustratedin Fig. A.2, and the experimentally determined value isı = 6.7 based upon water uptake at the two highest activitypoints (0.995 and 1). Using Eqs. (A.2) and (A.31), the effec-tive water diffusion coefficient is then 3.5 × 10−6 cm2 s−1,which is roughly an order of magnitude lower than the self-diffusion coefficient of water (2.6 × 10−5 cm2 s−1 [155]) and18 times lower than the effective diffusion coefficient cal-culated according to Eqs. (A.2) and (A.28). This analysisyields the physically attractive result that the presence ofthe polymer chains slows the diffusion of water relative tothat in pure water.
Additionally, the effective water diffusion coeffi-cient can be estimated using Eqs. (A.2) and (A.34).A concentration-independent Flory–Huggins interactionparameter can be determined by a least-squares fit ofthe experimental data, and the Flory–Huggins model with� = 0.68 fits the experimental data (Fig. A.2). By thismethodology, the effective diffusion coefficient of water inthe polymer is 1.4 × 10−6 cm2 s−1. However, as indicatedearlier, the Flory–Huggins model, originally developed foruncross-linked polymers, should not be expected to bestrictly applicable in this case.
The relative significance of the convective (frame ofreference) and thermodynamic correction factors can bedetermined from the analysis discussed above. The effec-tive diffusion coefficient calculating using the experimentalwater sorption data, 3.5 × 10−6 cm2 s−1, was lower thanthat calculated using the �m
w � 1 assumption by a factorof 1/17.7. For this material, the convective (frame of refer-ence) correction factor was 1/2.6, and the thermodynamiccorrection factor was 1/6.7. Therefore, in this material, thethermodynamic non-ideality of the water–polymer systemhas a greater influence on the effective water diffusion coef-
ficient than the frame of reference correction. Both of thecorrection factors, however, are significant for this mate-rial.er Science 39 (2014) 1– 42
References
[1] Anonymous. Grand challenges for engineering: provideaccess to clean water. Washington DC: National Academy ofEngineering; 2011. p. 5. http://www.engineeringchallenges.org/cms/8996/9142.aspx
[2] Service RF. Desalination freshens up. Science 2006;313:1088–90.[3] Shannon MA, Bohn PW, Elimelech M, Georgiadis JG, Marinas BJ,
Mayes AM. Science and technology for water purification in thecoming decades. Nature 2008;452:301–10.
[4] Webber ME. Catch-22: water vs. energy. Sci Am Sp Ed2008;18:34–41.
[5] Lange KE. Get the salt out. Nat Geog 2010;217:32–5.[6] Royte E. The last drop. Nat Geog 2010;217:172–6.[7] Elimelech M, Phillip WA. The future of seawater desali-
nation: energy, technology, and the environment. Science2011;333:712–7.
[8] Torcellini P, Long N, Judkoff R. Consumptive wateruse for US power production, NREL/TP-550-33905.National Renewable Energy Laboratory: Golden, CO,http://www.nrel.gov/docs/fy04osti/33905.pdf, (accessed June2013); 2003. p. 11.
[9] Anonymous. Energy demands on water resources: reportto congress on the interdependency of energy andwater. US Department of Energy: http://www.sandia.gov/energy-water/congress report.htm, (accessed April 2013); 2006.p. 80.
[10] Kenny JF, Barber NL, Hutson SS, Kristin S. Linsey KS, LovelaceJK, Maupin MA. Estimated use of water in the United Statesin 2005, Circular 1344. Washington DC: US Geological Surveyhttp://pubs.usgs.gov/circ/1344/$2005, (accessed April 2013); 2005.p. 52.
[11] Shuster E, McNemar A, Stiegel GJ, Murphy J. Estimatingfreshwater needs to meet future thermoelectric generationrequirements, DOE/NETL—400/2007/1304. Washington DC:National Energy Technology Laboratory, http://legalectric.org/f/2007/12/2007waterneedsanalysis-update-final 10-10-07bdoe.pdf(accessed June 2013); 2007. p. 107.
[12] Anonymous. Water management associated with hydraulic frac-turing. API guidance document HF2. American Petroleum Institute;2010. p. 38.
[13] Geise GM, Lee HS, Miller DJ, Freeman BD, McGrath JE, PaulDR. Water purification by membranes: the role of poly-mer science. J Polym Sci, Part B Polym Phys 2010;48:1685–718.
[14] Lee KP, Arnot TC, Mattia D. A review of reverse osmosis mem-brane materials for desalination—development to date and futurepotential. J Membr Sci 2011;370:1–22.
[15] Fritzmann C, Löwenberg J, Wintgens T, Melin T. State-of-the-art of reverse osmosis desalination. Desalination 2007;216:1–76.
[16] Anonymous. Desal market to grow 12% per year. Desalination.comwater desalination report, vol 42; 2006, http://www.desalination.com/wdr/42/35/desal-market-grow-12-year, (accessed May2013). p. 35.
[17] Anonymous. Desal’s double digit future. Global Water Intell2007;7(10):27–9 http://www.globalwaterintel.com/archive/7/10/market-insight/desals-double-digit-future.html (accessedApril 2013).
[18] Semiat R. Energy issues in desalination processes. Environ Sci Tech-nol 2008;42:8193–201.
[19] Kalogirou SA. Seawater desalination using renewable energysources. Prog Energy Combust Sci 2005;31:242–81.
[20] Anderson MA, Cudero AL, Palma J. Capacitive deionization as anelectrochemical means of saving energy and delivering clean water.Comparison to present desalination practices: will it compete?Electrochim Acta 2010;55:3845–56.
[21] Lomax I. Experiences of Dow in the field of seawater reverse osmo-sis. Desalination 2008;224:111–8.
[22] Anonymous. Country comparison: oil produc-tion. U.S. central intelligence agency: The WorldFactbook, https://www.cia.gov/library/publications/the-world-factbook/rankorder/2178rank.html; 2011. p. 12.
[23] Biesheuvel PM, van der Wal A. Membrane capacitive deionization.
DK. Desalination via a new membrane capacitive deionizationprocess utilizing flow electrodes. Energy Environ Sci 2013;6:1471–5.

n Polym
G.M. Geise et al. / Progress i[25] Zhao R, Satpradit O, Rijnaarts HHM, Biesheuvel PM, van der WalA. Optimization of salt adsorption rate in membrane capacitivedeionization. Water Res 2013;47:1941–52.
[26] Kim Y-J, Kim J-H, Choi J-H. Selective removal of nitrate ions by con-trolling the applied current in membrane capacitive deionization(MCDI). J Membr Sci 2013;429:52–7.
[27] Lee J-B, Park K-K, Eum H-M, Lee C-W. Desalination of a thermalpower plant wastewater by membrane capacitive deionization.Desalination 2006;196:125–34.
[28] Porada S, Sales BB, Hamelers HVM, Biesheuvel PM. Water desalina-tion with wires. J Phys Chem Lett 2012;3:1613–8.
[29] Zhao R, Biesheuvel PM, van der Wal A. Energy consumption andconstant current operation in membrane capacitive deionization.Energy Environ Sci 2012;5:9520–7.
[30] Liu F, Schaetzle O, Sales BB, Saakes M, Buisman CJN, Hamelers HVM.Effect of additional charging and current density on the perfor-mance of capacitive energy extraction based on Donnan potential.Energy Environ Sci 2012;5:8642–50.
[31] Długołecki P, van der Wal A. Energy recovery in membrane capac-itive deionization. Environ Sci Technol 2013;47:4904–10.
[32] Pilat B. Practice of water desalination by electrodialysis. Desalina-tion 2001;139:385–92.
[33] Nagarale RK, Gohil GS, Shahi VK. Recent developments onion-exchange membranes and electro-membrane processes. AdvColloid Interface Sci 2006;119:97–130.
[34] Xu TW. Ion exchange membranes: state of their development andperspective. J Membr Sci 2005;263:1–29.
[35] Van der Bruggen B, Vandecasteele C. Distillation vs. membrane fil-tration: overview of process evolutions in seawater desalination.Desalination 2002;143:207–18.
[36] Xu T, Huang C. Electrodialysis-based separation technologies: acritical review. AlChE J 2008;54:3147–59.
[37] Kim Y, Walker WS, Lawler DF. Competitive separation of di- vs.mono-valent cations in electrodialysis: effects of the boundarylayer properties. Water Res 2012;46:2042–56.
[38] Cath TY, Childress AE, Elimelech M. Forward osmosis: prin-ciples, applications, and recent developments. J Membr Sci2006;281:70–87.
[39] Gray GT, McCutcheon JR, Elimelech M. Internal concentrationpolarization in forward osmosis: role of membrane orientation.Desalination 2006;197:1–8.
[40] McCutcheon JR, Elimelech M. Influence of concentrative and dilu-tive internal concentration polarization on flux behavior in forwardosmosis. J Membr Sci 2006;284:237–47.
[41] McCutcheon JR, McGinnis RL, Elimelech M. A novelammonia–carbon dioxide forward (direct) osmosis desalinationprocess. Desalination 2005;174:1–11.
[42] McCutcheon JR, McGinnis RL, Elimelech M. Desalination byammonia–carbon dioxide forward osmosis: influence of draw andfeed solution concentrations on process performance. J Membr Sci2006;278:114–23.
[43] McGinnis RL, Elimelech M. Energy requirements ofammonia–carbon dioxide forward osmosis desalination.Desalination 2007;207:370–82.
[44] McCutcheon JR, Elimelech M. Modeling water flux in forwardosmosis: implications for improved membrane design. AlChE J2007;53:1736–44.
[45] Cath TY, Elimelech M, McCutcheon JR, McGinnis RL, Achilli A,Anastasio D, Brady AR, Childress AE, Farrg IV, Hancock NT, LampiJ, Nghiem LD, Xie M, Yip NY. Standard methodology for evalu-ating membrane performance in osmotically driven membraneprocesses. Desalination 2013;312:31–8.
[46] Chung T-S, Zhang S, Wang KY, Su J, Ling MM. Forward osmo-sis processes: yesterday, today and tomorrow. Desalination2010;257:78–81.
[47] Logan BE, Elimelech M. Membrane-based processes for sustainablepower generation using water. Nature 2012;488:313–9.
[48] Długołecki P, Nijmeijer K, Metz S, Wessling M. Current status of ionexchange membranes for power generation from salinity gradients.J Membr Sci 2008;319:214–22.
[49] Długołecki P, Gambier A, Nijmeijer K, Wessling M. Practical poten-tial of reverse electrodialysis as process for sustainable energygeneration. Environ Sci Technol 2009;43:6888–94.
[50] Post JW, Veerman J, Hamelers HVM, Euverink GJW, Metz SJ,
Nymeijer K, Buisman CJN. Salinity-gradient power: evaluation ofpressure-retarded osmosis and reverse electrodialysis. J Membr Sci2007;288:218–30.[51] Turek M, Bandura B. Renewable energy by reverse electrodialysis.Desalination 2007;205:67–74.
er Science 39 (2014) 1– 42 37
[52] Vermaas DA, Kunteng D, Saakes M, Nijmeijer K. Fouling inreverse electrodialysis under natural conditions. Water Res2013;47:1289–98.
[53] Luo X, Cao X, Mo Y, Xiao K, Zhang X, Liang P, Huang X. Powergeneration by coupling reverse electrodialysis and ammoniumbicarbonate: implication for recovery of waste heat. ElectrochemCommun 2012;19:25–8.
[54] Veerman J, Saakes M, Metz SJ, Harmsen GJ. Reverse electrodialysis:performance of a stack with 50 cells on the mixing of sea and riverwater. J Membr Sci 2009;327:136–44.
[55] Guler E, Zhang Y, Saakes M, Nijmeijer K. Tailor-made anion-exchange membranes for salinity gradient power generation usingreverse electrodialysis. ChemSusChem 2012;5:2262–70.
[56] Vermaas DA, Bajracharya S, Sales BB, Saakes M, Hamelers B, Nijmei-jer K. Clean energy generation using capacitive electrodes in reverseelectrodialysis. Energy Environ Sci 2013;6:643–51.
[57] Song X, Liu Z, Sun D. Energy recovery from concentrated sea-water brine by thin-film nanofiber composite pressure retardedosmosis membranes with high power density. Energy Environ Sci2013;6:1199–210.
[58] Kim YC, Elimelech M. Potential of osmotic power generation bypressure retarded osmosis using seawater as feed solution: analysisand experiments. J Membr Sci 2013;429:330–7.
[59] Thorsen T, Holt T. The potential for power production fromsalinity gradients by pressure retarded osmosis. J Membr Sci2009;335:103–10.
[60] Skilhagen SE, Dugstad JE, Aaberg RJ. Osmotic power–power pro-duction based on the osmotic pressure difference between waterswith varying salt gradients. Desalination 2008;220:476–82.
[61] Achilli A, Cath TY, Childress AE. Power generation with pressureretarded osmosis: an experimental and theoretical investigation. JMembr Sci 2009;343:42–52.
[62] Gerstandt K, Peinemann KV, Skilhagen SE, Thorsen T, Holt T. Mem-brane processes in energy supply for an osmotic power plant.Desalination 2008;224:64–70.
[63] Lonsdale HK. The growth of membrane technology. J Membr Sci1982;10:81–181.
[64] Lonsdale HK, Merten U, Riley RL. Transport properties of celluloseacetate osmotic membranes. J Appl Polym Sci 1965;9:1341–62.
[65] Anonymous. GE water & process technologies: pure watermembranes, http://www.gewater.com/products/consumables/pure water elements/index.jsp, (accessed April 2013); 2012. p. 1.
[66] Anonymous. Koch membrane systems: Fluid Systems® ROGA®
RO series, http://www.kochmembrane.com/Membrane-Products/Spiral/Reverse-Osmosis/Fluid-System-ROGA-RO-Series.aspx,(accessed April 2013); 2012. p. 1.
[67] Anonymous. Toyobo water treatment membranes: Hollosep®
http://www.-global.com/seihin/ro/tokucho.htm, (accessed June2013); 2006. p. 1.
[68] Anonymous. TriSep cellulose acetate RO membranes, http://membranes.trisep.com/viewitems/reverse-osmosis-membranes/sb-cellulose-acetate-blend-2?, (accessed June 2013); 2012. p. 1.
[69] Petersen RE, Cadotte JE. Thin film composite reverse osmosis mem-branes. In: Porter MC, editor. Handbook of industrial membranetechnology. Park Ridge, NJ: Noyes Publications; 1990. p. 307–48.
[70] Petersen RJ. Composite reverse osmosis and nanofiltration mem-branes. J Membr Sci 1993;83:81–150.
[71] Cadotte JE. Interfacially synthesized reverse osmosis mem-brane. US patent 4277344. FilmTec Corporation: Minetonka MN;1981.
[72] Cadotte JE. Reverse osmosis membrane. US patent 4259183. Mid-west Research Institute, Kansas City: MO; 1981.
[73] Cadotte JE. Evolution of composite reverse osmosis membranes. In:Lloyd DR, editor. Materials science of synthetic membranes—ACSsymposium series vol 269. Washington, DC: American ChemicalSociety; 1985. p. 273–94.
[74] Otero JA, Mazarrasa O, Villasante J, Silva V, Prádanos P, Calvo JI,Hernández A. Three independent ways to obtain information onpore size distributions of nanofiltration membranes. J Membr Sci2008;309:17–27.
[75] Lonsdale HK, Milstead CE, Cross BP, Graber FM. Study of rejec-tion of various solutes by reverse osmosis membranes, Officeof Saline Water Research and Development progress report no.
447. Washington, DC: US Department of the Interior; 1969.p. 115.[76] Lonsdale HK, Riley RL, Milstead CE, LaGrange LD, Douglas AS, SachsSB. US Department of Interior report no. 577. Washington, DC:Office of Saline Water; 1970. p. 143.

n Polym
38 G.M. Geise et al. / Progress i[77] Strathmann H, Michaels AS. Polymer–water interaction and itsrelation to reverse osmosis desalination efficiency. Desalination1977;21:195–202.
[78] Frommer MA, Murday JS, Messalem RM. Solubility and diffusivityof water and of salts in an aromatic polyamide film. Eur Polym J1973;9:367–73.
[79] McGrath JE, Park HB, Freeman BD. Chlorine resistant desalinationmembranes based on directly sulfonated poly(arylene ether sul-fone) copolymers. US patent 8,028,842 B2; 2011.
[80] Park HB, Freeman BD, Zhang Z-B, Sankir M, McGrath JE. Highlychlorine-tolerant polymers for desalination. Angew Chem Int Ed2008;120:6108–13.
[81] Xie W, Cook J, Park HB, Freeman BD, Lee CH, McGrath JE. Fundamen-tal salt and water transport properties in directly copolymerizeddisulfonated poly(arylene ether sulfone) random copolymers. Poly-mer 2011;52:2032–43.
[82] Xie W, Geise GM, Freeman BD, Lee CH, McGrath JE. Influenceof processing history on water and salt transport proper-ties of disulfonated polysulfone random copolymers. Polymer2012;53:1581–92.
[83] Parise PL, Allegrezza Jr AE, Parekh BS. Reverse osmosis: chlorine-resistant polysulfone reverse osmosis membrane and module.Ultrapure Water 1987;(October):54–65.
[84] Allegrezza Jr AE, Parekh BS, Parise PL, Swiniarski EJ, White JL. Chlo-rine resistant polysulfone reverse osmosis modules. Desalination1987;64:285–304.
[85] Kimura SG. Reverse osmosis performance of sulfonated poly(2,6-dimethylphenylene ether) ion exchange membranes. Ind Eng ChemRes 1971;10:335–9.
[86] Mauritz KA, Moore RB. State of understanding of Nafion. Chem Rev2004;104:4535–85.
[87] Bonner OD, Smith LL. A selectivity scale for some bivalent cationson Dowex-50. J Phys Chem 1957;61:326–9.
[88] Miyoshi H. Donnan dialysis with ion-exchange membranes. III. Dif-fusion coefficients using ions of different valence. Sep Sci Technol1999;34:231–41.
[89] Bauman WC, Skidmore JR, Osmun RH. Dowex 50. Ind Eng Chem1948;40:1350–5.
[90] Pushpa KK, Nandan D, Iyer RM. Thermodynamics of watersorption by perfluorosulphonate (Nafion-117) and polystyrene-divinylbenzene sulphonate (Dowex 50W) ion-exchangeresins at 298 ± 1 K. J Chem Soc, Faraday Trans 1988;1(84):2047–56.
[91] Freeman B, Yampol’skii Y. Membrane gas separation. West Sussex:John Wiley & Sons; 2010. p. 392.
[92] Yampol’skii Y, Freeman B. Membrane gas separation. West Sussex:John Wiley & Sons Ltd; 2010. p. 392.
[93] Yampol’skii Y, Pinnau I, Freeman BD. Materials science of mem-branes for gas and vapor separation. London: Wiley; 2006.p. 445.
[94] Koros WJ, Fleming GK. Membrane-based gas separation. J MembrSci 1993;83:1–80.
[95] Freeman BD. Basis of permeability/selectivity tradeoff rela-tions in polymeric gas separation membranes. Macromolecules1999;32:375–80.
[96] Robeson LM. Correlation of separation factor versus permeabilityfor polymeric membranes. J Membr Sci 1991;62:165–85.
[97] Robeson LM. The upper bound revisited. J Membr Sci2008;320:390–400.
[98] Robeson LM, Freeman BD, Paul DR, Rowe BW. An empir-ical correlation of gas permeability and permselectivity inpolymers and its theoretical basis. J Membr Sci 2009;341:178–85.
[99] Yasuda H, Lamaze CE, Ikenberry LD. Permeability of solutes throughhydrated polymer membranes. Part I. Diffusion of sodium chloride.Makromol Chem 1968;118:19–35.
[100] Ju H, McCloskey BD, Sagle AC, Wu Y-H, Kusuma VA, FreemanBD. Cross-linked poly(ethyleneoxide) fouling resistant coatingmaterials for oil/water separation. J Membr Sci 2008;307:260–7.
[101] Ju H, Sagle AC, Freeman BD, Mardel JI, Hill AJ. Characterizationof sodium chloride and water transport in poly(ethylene oxide)hydrogels. J Membr Sci 2010;358:131–41.
[102] Sagle AC, Ju H, Freeman BD, Sharma MM. PEG-based hydrogel mem-
brane coatings. Polymer 2009;50:756–66.[103] Gong X, Bandis A, Tao A, Meresi G, Wang Y, Inglefield PT, Jones AA,Wen W-Y. Self-diffusion of water, ethanol, and decafluropentane inperfluorosulfonate ionomer by pulse field gradient NMR. Polymer2001;42:6485–92.
er Science 39 (2014) 1– 42
[104] Helfferich F. Ion exchange. New York: Dover Publications; 1995. p.640.
[105] Sata T. Ion exchange membranes: preparation, characterization,modification and application. Cambridge: The Royal Society ofChemistry; 2004. p. 308.
[106] Wijmans JG, Baker RW. The solution–diffusion model: a review. JMembr Sci 1995;107:1–21.
[107] Paul DR. Reformulation of the solution–diffusion theory of reverseosmosis. J Membr Sci 2004;241:371–86.
[108] Stern SA. Polymers for gas separations: the next decade. J MembrSci 1994;94:1–65.
[109] Fried JR, Sadat-Akhavi M, Mark JE. Molecular simulation of gas per-meability: poly(2,6-dimethyl-1,4-phenylene oxide). J Membr Sci1998;149:115–26.
[110] Petropoulos JH. Mechanisms and theories for sorption and diffusionof gases in polymers. In: Paul DR, Yampolskii YP, editors. Polymericgas separation membranes. Boca Ration, FL: CRC Press; 1994. p.17–81.
[111] Merten U. Flow relationships in reverse osmosis. Ind Eng ChemFundam 1963;2:229–32.
[112] Reid CE, Breton EJ. Water and ion flow across cellulosic membranes.J Appl Polym Sci 1959;1:133–43.
[113] Reid CE, Kuppers JR. Physcial characteristics of osmotic membranesof organic polymers. J Appl Polym Sci 1959;2:264–72.
[114] Merten U. Desalination by reverse osmosis. Cambridge: MIT Press;1966. p. 289.
[115] Baker RW. Membrane technology and applications. 2nd ed. NewYork: John Wiley; 2004. p. 538.
[116] Lonsdale HK, Merten U, Tagami M. Phenol transport in celluloseacetate membranes. J Appl Polym Sci 1967;11:1807–20.
[117] Bason S, Oren Y, Freger V. Ion transport in the polyamide layer ofRO membranes: composite membranes and free-standing films. JMembr Sci 2011;367:119–26.
[118] Yaroshchuk AE, Boiko YP, Makovetskiv AL. Some properties ofelectrolyte solutions in nanoconfinement revealed by the mea-surement of transient filtration potential after pressure switch off.Langmuir 2005;21:7680–90.
[119] Yaroshchuk A, Karpenko L, Ribitsch V. Measurements of transientmembrane potential after current switch-off as a tool to study theelectrochemical properties of supported thin nanoporous layers. JPhys Chem B 2005;109:7834–42.
[120] Pusch W. Measurement techniques of transport through mem-branes. Desalination 1986;59:105–98.
[121] Demisch HU, Pusch W. Electrical and electroosmotic transportbehavior of asymmetric cellulose acetate membranes. I. Transportbehavior in dialysis–osmosis experiments. J Colloid Interface Sci1980;76:445–63.
[122] Yaroshchuk AE, Makovetskiv AL, Boiko YP, Galinker EW. Non-steady-state membrane potential: theory and measurements bya novel technique to determine the ion transport numbers inactive layers of nanofiltration membranes. J Membr Sci 2000;172:203–21.
[123] Yaroshchuk AE, Ribitsch V. The uses of non-steady-state membranecharacterization techniques for the study of transport properties ofactive layers of nanofiltration membranes: theory with experimen-tal examples. Chem Eng J 2000;80:203–14.
[124] Paul DR. The role of membrane pressure in reverse osmosis. J ApplPolym Sci 1972;16:771–82.
[125] Wijmans JG, Baker RW. A simple predictive treatment of the per-meation process in pervaporation. J Membr Sci 1993;79:101–13.
[126] Bhanushali D, Kloos S, Kurth C, Bhattacharyya D. Performance ofsolvent-resistant membranes for non-aqueous systems: solventpermeation results and modeling. J Membr Sci 2001;189:1–21.
[127] Luthra SS, Yang X, Freitas dos Santos LM, White LS, Livingston AG.Phase-transfer catalyst separation and re-use by solvent resistantnanofiltration membranes. Chem Commun 2001;16:1468–9.
[128] Lee CH. Theory of reverse osmosis and some other membrane per-meation operations. J Appl Polym Sci 1975;19:83–95.
[129] Smith JM, Van Ness HC, Abbott MM. Introduction to chemical engi-neering thermodynamics. 6th ed. New York: McGraw-Hill; 2001.p. 789.
[130] Pitzer KS. A thermodynamic model for aqueous solutions of liquid-like density. Rev Mineral Geochem 1987;17:97–142.
[131] Paul DR. Relation between hydraulic permeability and diffusion
in homogeneous swollen membranes. J Polym Sci, Polym Phys Ed1973;11:289–96.[132] Geise GM, Park HB, Sagle AC, Freeman BD, McGrath JE. Waterpermeability and water/salt selectivity tradeoff in polymers fordesalination. J Membr Sci 2011;369:130–8.

n Polym
G.M. Geise et al. / Progress i[133] Geise GM, Freeman BD, Paul DR. Sodium chloride diffusion insulfonated polymers for membrane applications. J Membr Sci2013;427:186–96.
[134] Rosenbaum S, Cotton O. Steady-state distribution of water in cel-lulose acetate membrane. J Polym Sci 1969;7:101–9.
[135] Paul DR. Diffusive transport in swollen polymer membranes. In:Hopfenberg HB, editor. Permeability of plastic films and coatings:to gases, vapors, and liquids. New York: Plenum Press; 1974. p.35–48.
[136] Puleo AC, Paul DR, Kelly SS. The effect of degree of acetylation ongas sorption and transport behavior in cellulose acetate. J MembrSci 1989;47:301–32.
[137] Ham JS, Bolen MC, Hughes JK. The use of high pressure to studypolymer–solvent interaction. J Polym Sci 1962;57:25–40.
[138] Anonymous. DOWTM FILMTECTM reverse osmosis andnanofiltration elements, http://www.dowwaterandprocess.com/products/ronf.htm, (accessed June 2913); 2012. p. 1.
[139] Anonymous. Hydranautics seawater composite (SWC) membranes,http://www.membranes.com/index.php?pagename=swc; 2012. p.1.
[140] Gehman SD. Molecular weight distribution of network chainsand swelling pressure of vulcanizates. Rubber Chem Technol1967;40:532–43.
[141] Xie W, Park HB, Cook J, Lee CH, Byun G, Freeman BD,McGrath JE. Advances in membrane materials: desalination mem-branes based on directly copolymerized disulfonated poly(aryleneether sulfone) random copolymers. Water Sci Technol 2010;61:619–24.
[142] Zaikov GE, Iordanskii AP, Markin VS. Diffusion of electrolytes inpolymers. Utrecht, The Netherlands: VSP; 1988. p. 321.
[143] Robeson LM, Hwu HH, McGrath JE. Upper bound relationship forproton exchange membranes: empirical relationship and relevanceof phase separated blends. J Membr Sci 2007;302:70–7.
[144] Pivovar BS, Wang Y, Cussler EL. Pervaporation membranes in directmethanol fuel cells. J Membr Sci 1999;154:155–62.
[145] Mehta A, Zydney AL. Permeability and selectivity analysis for ultra-filtration membranes. J Membr Sci 2005;249:245–9.
[146] Yip NY, Elimelech M. Performance limiting effects in power gener-ation from salinity gradients by pressure retarded osmosis. EnvironSci Technol 2011;45:10273–82.
[147] Freger V. Swelling and morphology of the skin layer of polyamidecomposite membranes: an atomic force microscopy study. EnvironSci Technol 2004;38:3168–75.
[148] Geise GM, Falcon LP, Freeman BD, Paul DR. Sodium chloride sorp-tion in sulfonated polymers for membrane applications. J MembrSci 2012;423-424:195–208.
[149] Yaroshchuk A. Dielectric exclusion of ions from membranes. AdvColloid Interface Sci 2000;85:193–230.
[150] Cohen MH, Turnbull D. Molecular transport in liquids and glasses.J Chem Phys 1959;31:1164–9.
[151] Xie W, Ju H, Geise GM, Freeman BD, Mardel JI, Hill AJ, McGrathJE. Effect of free volume on water and salt transport proper-ties in directly copolymerized disulfonated poly(arylene ethersulfone) random copolymers. Macromolecules 2011;44:4428–38.
[152] Fell CJD, Hutchison HP. Diffusion coefficients for sodium and potas-sium chlorides in water at elevated temperatures. J Chem Eng Data1971;16:427–9.
[153] Geise GM, Freeman BD, Paul DR. Characterization of a novelsulfonated pentablock copolymer for desalination applications.Polymer 2010;51:5815–22.
[154] Nightingale ER. Phenomenological theory of ion solvation, effectiveradii of hydrated ions. J Phys Chem 1959;63:1381–7.
[155] Wang JH, Robinson CV, Edelman IS. Self-diffusion and structureof liquid water. III. Measurement of the self-diffusion of liquidwater with H2, H3 and O18 as tracers. J Am Chem Soc 1953;75:466–70.
[156] Meares P. The mechanism of water transport in membranes. PhilosTrans R Soc London, Ser B 1977;278:113–50.
[157] Matteucci S, Yampolskii Y, Freeman BD, Pinnau I. Transport of gasesand vapors in glassy and rubbery polymers. In: Yampolskii Y, Pin-nau I, Freeman BD, editors. Materials science of membranes for gasand vapor separation. London: Wiley; 2006. p. 1–47.
[158] Barrer RM. Permeability of organic polymers. Trans Faraday Soc
1940;35:644–8.[159] Conway BE. Ionic hydration in chemistry and biophysics. Amster-dam: Elsevier; 1981. p. 774.
[160] Robinson RA, Stokes RH. Electrolyte solutions. 2nd ed. Mineola, NY:Dover; 2002. p. 590.
er Science 39 (2014) 1– 42 39
[161] Afanas’ev VN. Solvation of electrolytes and nonelectrolytes in aque-ous solutions. J Phys Chem B 2011;115:6541–63.
[162] Doyle DA, Cabral JM, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, ChaitBT, MacKinnon R. The structure of the potassium channel: molecu-lar basis of K+ conduction and selectivity. Science 1998;280:69–77.
[163] Marcus Y, Hefter G. Ion pairing. Chem Rev 2006;106:4585–621.[164] Pauling L. The sizes of ions and the structure of ionic crystals. J Am
Chem Soc 1927;49:765–90.[165] Brown TL, LeMay Jr HE, Bursten BE, Burdge JR. Chemistry: the cen-
tral science. 9th ed. Upper Saddle River, NJ: Pearson Education Inc;2003. p. 1152.
[166] Demisch HU, Pusch W. Ion exchange capacity of cellulose acetatemembranes. J Electrochem Soc 1976;123:370–4.
[167] Mafe S, Ramirez P, Pellicer J. Activity coefficients and Donnan coionexclusion in charged membranes with weak-acid fixed chargegroups. J Membr Sci 1998;138:269–77.
[168] Strathmann H. Ion exchange membranes. In: Winston Ho WS,Sirkar KK, editors. Membrane handbook. New York: Van NostrandReinhold; 1992. p. 230–45.
[169] Stumm W, Morgan JJ. Aquatic chemistry: chemical equilibria andrates in natural waters. 3rd ed. New York: John Wiley & Sons, Inc;1996. p. 1022.
[170] Greenlee LF, Lawler DF, Freeman BD, Marrot B, Moulin P. Reverseosmosis desalination: water sources, technology, and today’s chal-lenges. Water Res 2009;43:2317–48.
[171] van Wagner EM, Sagle AC, Sharma MM, Freeman BD. Effect of cross-flow testing conditions, including feed pH and continuous feedfiltration, on commercial reverse osmosis membrane performance.J Membr Sci 2009;345:97–109.
[172] Haynes WM. CRC handbook of chemistry and physics (Internet Ver-sion 2011). 91st ed Boca Raton, FL: CRC Press; 2011. p. 2610.
[173] Kvaratskheliya RK, Kvaratskheliya ER. Electrochemical behavior ofbenzenepolycarboxylic acids on solid electrodes in aqueous andmixed solutions. Russ J Electrochem 2006;42:978–81.
[174] Van Wagner EM. Polyamide desalination membrane characteriza-tion and surface modification to enhance fouling resistance. Austin,TX: The University of Texas at Austin; 2010. p. 175.
[175] Chechina ON, Sokolov SV, Berenblit VV, Soshin VA. Dissocia-tion constants of polyfluorocarboxylic acids. Russ J Appl Chem2007;80:1770–2.
[176] Twardowski Z, Yeager HL, O’Dell B. A comparison of perfluori-nated carboxylate and sulfonate exchange polymers. II. Sorptionand transport properties in concentrated solution. J ElectrochemSoc 1982;129:328–32.
[177] Cerfontain H, Schnitger BW. Solutes in sulfuric acid, 4. Ionization ofarenesulfonic acids; determination of pKa values by UV techniques.Recl Trav Chim Pays-Bas 1972;91:199–208.
[178] Dobrynin AV, Rubinstein M. Theory of polyelectrolytes in solutionsand at surfaces. Prog Polym Sci 2005;30:1049–118.
[179] Newman J, Thomas-Alyea KE. Electrochemical systems. 3rd edHoboken, NJ: Wiley-Interscience; 2004. p. 647.
[180] Paddison SJ, Reagor DW, Zawodzinski TA. High frequency dielec-tric studies of hydrated Nafion® . J Electroanal Chem 1998;459:91–7.
[181] Oberbroeckling KJ, Dunwoody DC, Minteer SD, Leddy J. Density ofNafion exchanged with transition metal complexes and tetram-ethyl ammonium, ferrous, and hydrogen ions: commercial andrecast films. Anal Chem 2002;74:4794–9.
[182] García-Fresnadillo D, Marazuela MD, Moreno-Bondi MC, Orel-lana G. Luminescent Nafion membranes dyed with ruthenium(II)complexes as sensing materials for dissolved oxygen. Langmuir1999;15:6451–9.
[183] Heyde ME, Peters CR, Anderson JE. Factors influencing reverseosmosis rejection of inorganic solutes from aqueous solution. J Col-loid Interface Sci 1975;50:467–87.
[184] Glueckauf E. A new approach to ion-exchange polymers. Proc R SocLondon, Ser A 1962;268:350–70.
[185] Glueckauf E, Watts RE. The Donnan law and its application toion-exchanger polymers. Proc R Soc London, Ser A 1962;268:339–49.
[186] Petropoulos JH. Membrane transport properties in relation tomicroscopic and macroscopic structural inhomogeneity. J MembrSci 1990;52:305–23.
[187] Donnan FG. The theory of membrane equilibria. Chem Rev
1924;1:73–90.[188] Pintauro PN, Bennion DN. Mass transport of electrolytes inmembranes. 2. Determination of sodium chloride equilibriumand transport parameters for Nafion. Ind Eng Chem Fundam1984;23:234–43.

n Polym
40 G.M. Geise et al. / Progress i[189] Goosen MFA, Sablani SS, Ai-Hinai H, Ai-Obeidani S, Al-Belushi R,Jackson D. Fouling of reverse osmosis and ultrafiltration mem-branes: a critical review. Sep Sci Technol 2004;39:2261–97.
[190] Contreras AE, Steiner Z, Miao J, Kasher R, Li Q. Studying the roleof common membrane surface functionalities on adsorption andcleaning of organic foulants using QCM-D. Environ Sci Technol2011;45:6309–15.
[191] Chen S, Jiang S. An new avenue to nonfouling materials. Adv Mater2008;20:335–8.
[192] Mo Y, Tiraferri A, Yip NY, Adout A, Huang X, Elimelech M. Improvedantifouling properties of polyamide nanofiltration membranes byreducing the density of surface carboxyl groups. Environ Sci Tech-nol 2012;46:13253–61.
[193] Passaniti LKR. Solubility measurements in partially disulfonatedpoly(arylene ether sulfone) for reverse osmosis water purificationapplications. MS thesis. Austin, TX: The University of Texas; 2010.p. 50.
[194] Rosenbaum S, Mahon HI, Cotton O. Permeation of water andsodium chloride through cellulose acetate. J Appl Polym Sci1967;11:2041–65.
[195] Bajpai SK, Singh S. Analysis of swelling behavior ofpoly(methacrylamide-co-methacrylic acid) hydrogels andeffect of synthesis conditions on water uptake. React Funct Polym2006;66:431–40.
[196] Flory PJ, Rehner JJ. Statistical mechanics of cross-linked polymernetworks. II. Swelling. J Chem Phys 1943;11:521–6.
[197] Hodge RM, Bastow TJ, Edward GH, Simon GP, Hill AJ. Free volumeand the mechanism of plasticization in water–swollen poly(vinylalcohol). Macromolecules 1996;29:8137–43.
[198] Hodge RM, Edward GH, Simon GP. Water absorption and statesof water in semicrystalline poly(vinyl alcohol) films. Polymer1996;37:1371–6.
[199] Peppas NA, Merril EW. Poly(vinyl alcohol) hydrogels: reinforce-ment of radiation-cross-linked networks by crystallization. J PolymSci Polym Chem Ed 1976;14:441–5.
[200] Burghoff H-G, Pusch W. The thermodynamic state of water in cel-lulose acetate membranes. Polym Eng Sci 1980;20:305–9.
[201] Flory PJ. Thermodynamics of high polymer solutions. J Chem Phys1942;10:51–61.
[202] Huggins ML. Thermodynamic properties of solutions of long-chaincompounds. Ann NY Acad Sci 1942;43:1–32.
[203] Khare AR, Peppas N. Swelling/deswelling of anionic copolymer gels.Biomaterials 1995;16:559–67.
[204] Iijima T, Obara T, Isshiki M, Seki T, Adachi K. Ionic transportof alkali chlorides in Nylon membrane. J Colloid Interface Sci1978;63:421–5.
[205] Jadwin TA, Hoffman AS, Vieth WR. Cross-linked poly(hydroxyethylmethacrylate) membranes for desalination by reverse osmosis. JAppl Polym Sci 1970;14:1339–59.
[206] Hickner MA. Ion-containing polymers: new energy & clean water.Mater Today 2010;13:34–41.
[207] Welgemoed TJ, Schutte CF. Capacitive deionizationTechnologyTM: an alternative desalination solution. Desalination2005;183:327–40.
[208] Gabelich CJ, Yun TI, Coffey BM, Suffet IHM. Effects of aluminumsulfate and ferric chloride coagulant residuals on polyamide mem-brane performance. Desalination 2002;150:15–30.
[209] Glater J, Hong S-k, Elimelech M. The search for a chlorine-resistant reverse osmosis membrane. Desalination 1994;95:325–45.
[210] Glater J, Zachariah MR. A mechanistic study of halogen interac-tion with polyamide reverse-osmosis membranes. ACS symposiumseries vol 281. Washington, DC: American Chemical Society; 1985.p. 345–58.
[211] Lowell Jr JR, Friesen DT, McCray SB, McDermott SD, Brose DJ,Ray RJ. Model compounds as predictors of chlorine sensitivityof interfacial-polymer reverse-osmosis membranes. Internationalcongress on membranes and membrane processes, Tokyo; 1987. p.354–5.
[212] Paul M, Park HB, Freeman BD, Roy A, McGrath JE, Riffle JS. Syn-thesis and crosslinking of partially disulfonated poly(arylene ethersulfone) random copolymers as candidates for chlorine resistantreverse osmosis membranes. Polymer 2008;49:2243–52.
[213] Guiver MD, Croteau S, Hazlett JD, Kutowy O. Synthesis and char-
acterization of carboxylated polysulfones. Br Polym J 1990;23:29–39.[214] Fox DW, Shenian P. Sulfonated polyphenylene ether cationexchange resin. US patent 3,259,592. General Electric Company;1966.
er Science 39 (2014) 1– 42
[215] Willis CL, Handlin DL, Trenor SR, Mather BD. Sulfonated blockcopolymers, method for making same, and various uses for suchblock copolymers. US patent 7,737,224 B2. Kraton Polymers US LLC:Houston, TX; 2010.
[216] Willis CL, Handlin DL, Trenor SR, Mather BD. Process for prepar-ing sulfonated block copolymers and various uses for such blockcopolymers. US patent application 2010/0203784 A1. Kraton Poly-mers US LLC: Houston TX; 2010.
[217] Noshay A, Robeson LM. Sulfonated polysulfone. J Appl Polym Sci1976;20:1885–903.
[218] Andrew M, Mukundan T, McGrath JE. Sulfonated poly(arylene ethersulfone) copolymers—acid and salt form: potential biofunctionalpolymers. J Bioact Compat Polym 2004;19:315–29.
[219] Connolly DJ, Gresham WF. Fluorocarbon vinyl ether polymers. USpatent 3,282,875. EI du Pont; 1966.
[220] Weiss RA, Sen A, Pottick LA, Willis CL. Block copolymerionomers: 2. Viscoelastic and mechanical properties ofsulphonated poly(styrene-ethylene/butylene-styrene). Polymer1991;32:2785–92.
[221] Weiss RA, Sen A, Willis CL, Pottick LA. Block copoly-mer ionomers: 1. Synthesis and physical properties ofsulphonated poly(styrene–ethylene/butylene–styrene). Polymer1991;32:1867–74.
[222] Geis GM, Willis CL, Doherty CM, Hill AJ, Bastow TJ, Ford J, Winey KI,Freeman BD, Paul DR. Characterization of aluminum-neutralizedsulfonated styrenic pentablock copolymer films. Ind Eng Chem Res2013;52:1056–68.
[223] Flood J, Dubois D, Willis CL, Bening R. Sulfonated styrenicpentablock copolymer membranes for high water transport rateapplications. In: ANTEC 2009—Proceedings of the 67th annual tech-nical conference & exhibition; 2009. p. 107–12.
[224] Willis CL. Metal-neutralized sulfonated block copolymers, pro-cess for making them and their use. US patent application2011/0086977 A1. Kraton Performance Polymers, Inc, Houston TX,1; 2011.
[225] Bonner OD, Payne WH. Equilibrium studies of some univalent ionson Dowex 50. J Phys Chem 1954;58:183–5.
[226] Argersinger Jr WJ, Davidson AW, Bonner OD. Thermodynamics andion exchange phenomena. Trans Kans Acad Sci 1950;53:404–10.
[227] Yeager HL, Twardowski Z, Clarke LM. A comparison of perfluori-nated carboxylate and sulfonate ion exchange polymers, I. Diffusionand water sorption. J Electrochem Soc 1982;129:324–7.
[228] Overbeek JT. The Donnan equilibrium. Prog Biophys Biophys Chem1956;6:57–84.
[229] Petropoulos JH, Tsimboukis DG, Kouzeli K. Non-equipotential vol-ume membrane models, relation between the Glueckauf andequipotential surface models. J Membr Sci 1983;16:379–89.
[230] McKelvey JG, Spiegler KS, Wyllie MRJ. Ultrafiltration of salt solu-tions through ion exchange membranes. Chem Eng Prog Symp Ser1959;55:199–208.
[231] Bason S, Freger V. Phenomenological analysis of transport of mono-and divalent ions in nanofiltration. J Membr Sci 2010;360:389–96.
[232] Berezina NP, Timofeev SV, Kononenko NA. Effect of condition-ing techniques of perfluorinated sulphocationic membranes ontheir hydrophylic and electrotransport properties. J Membr Sci2002;209:509–18.
[233] Lysova AA, Stenina IA, Gorbunova YG, Yaroslavtsev AB. Preparationof MF-4SC composite membranes with anisotropic distribution ofpolyaniline and ion-transport asymmetry. Polym Sci Ser B PolymChem 2011;53:35–41.
[234] Yaroshchuk AE, Ribitsch V. The use of trace ions for advanced char-acterisation of transport properties of NF membranes in electrolytesolutions: theoretical analysis. J Membr Sci 2002;201:85–94.
[235] Yaroshchuk A, Martínez-Lladó X, Llenas L, Rovira M, Pablo Jd.Solution–diffusion–film model for the description of pressure-driven trans-membrane transfer of electrolyte mixtures: onedominant salt and trace ions. J Membr Sci 2011;368:192–201.
[236] Bartels C, Franks R. Hydranautics, membrane desalination work-shop. International congress on membranes and membraneprocesses, Honolulu, HI; 2008.
[237] Geise GM, Freeman BD, Paul DR. Comparison of the perme-ation of MgCl2 versus NaCl in highly-charged sulfonated polymermembranes. In: Escobar IC, Van der Bruggen B, editors. Modernapplications in membrane science and technology. Washington DC:
American Chemical Society; 2011. p. 239–45.[238] Klein RJ, Welna DT, Weikel AL, Allcock HR, Runt J. Counter-ion effects on ion mobility and mobile ion concentration ofdoped polyphosphazene and polyphosphazene ionomers. Macro-molecules 2007;40:3990–5.

n Polym
G.M. Geise et al. / Progress i[239] Jardat M, Hribar-Lee B, Vlachy V. Self-diffusion coefficients ofions in the presence of charged obstacles. Phys Chem Chem Phys2008;10:449–57.
[240] Yaroshchuk A, Boiko Y, Makovetskiv A. Electrochemical perm-selectivity of active layers and diffusion permeability of sup-ports of an asymmetric and a composite NF membranestudied by concentration-step method. Desalination 2009;245:374–87.
[241] Sivashinsky N, Tanny GB. Ionic heterogeneities in sulfonated poly-sulfone films. J Appl Polym Sci 1983;28:3235–45.
[242] Elabd YA, Walker CW, Beyer FL. Triblock copolymer ionomermembranes: Part II. Structure characterization and its effects ontransport properties and direct methanol fuel cell performance. JMembr Sci 2004;231:181–8.
[243] Miyoshi H, Chubachi M, Yamagami M, Kataoka T. Characteris-tic coefficients for equilibrium between solution and Neoseptaor Selemion cation exchange membranes. J Chem Eng Data1992;37:120–4.
[244] Miyoshi H, Yamagami M, Chubachi M, Kataoka T. Characteristiccoefficients of cation-exchange membranes for bivalent cations inequilibrium between the membrane and solution. J Chem Eng Data1994;39:595–8.
[245] Bontha JR, Pintauro PN. Water orientation and ion solvationeffects during multicomponent salt partitioning in a Nafion cationexchange membrane. Chem Eng Sci 1994;49:3835–51.
[246] Weiss RA, Lundberg RD, Turner SR. Comparisons of styreneionomers prepared by sulfonating polystyrene and copolymeriz-ing styrene with styrene sulfonate. J Polym Sci Polym Chem Ed1985;23:549–68.
[247] Maruf SH, Ahn DU, Greenberg AR, Ding Y. Glass transition behav-iors of interfacially polymerized polyamide barrier layers on thinfilm composite membranes via nano-thermal analysis. Polymer2011;52:2643–9.
[248] Kim YS, Dong L, Hickner MA, Glass TE, Webb V, McGrath JE. Stateof water in disulfonated poly(arylene ether sulfone) copolymersand a perfluorosulfonic acid copolymer (Nafion) and its effect onphysical and electrochemical properties. Macromolecules 2003;36:6281–5.
[249] Rowe BW, Pas SJ, Hill AJ, Suzuki R, Freeman BD, Paul DR. A variableenergy positron annihilation lifetime spectroscopy study of physi-cal aging in thin glassy polymer films. Polymer 2009;50:6149–56.
[250] Struik LCE. Physical aging in amorphous polymers and other mate-rials. Amsterdam: Elsevier; 1978. p. 229.
[251] Willis CL, Handlin DL, Trenor SR, Mather BD. Composition contain-ing sulfonated block copolymers and articles made therefrom. USpatent 7,919,565 B2. Kraton Polymers US LLC, Houston TX; 2011.
[252] Willis CL, Handlin DL, Trenor SR, Mather BD. Method for varying thetransport properties of a film cast from a sulfonated copolymer. USpatent 8,084,546 B2. Kraton Polymers US LLC, Houston TX; 2011.
[253] McGrath JE, Harrison W, Wodzinski Jr TA. Multiblock copolymerscontaining hydrophilic hydrophobic segments for proton exchangemembrane. US patent application 10/595654; December 20, 2007.
[254] Choi J-H, Willis CL, Winey KI. Structure–property relation-ship in sulfonated pentablock copolymers. J Membr Sci2012;394–395:169–74.
[255] Coronell O, Marinas BJ, Zhang X, Cahill DG. Quantification of func-tional groups and modeling of their ionization behavior in theactive layer of FT30 reverse osmosis membrane. Environ Eng Sci2008;42:5260–6.
[256] Freger V. Nanoscale heterogeneity of polyamide membranesformed by interfacial polymerization. Langmuir 2003;19:4791–7.
[257] Tung K-L, Jean Y-C, Nanda D, Lee K-R, Hung W-S, Lo C-H, Lai J-Y. Characterization of multilayer nanofiltration membranes usingpositron annihilation spectroscopy. J Membr Sci 2009;343:147–56.
[258] Cahill DG, Freger V, Kwak S-Y. Microscopy and microanaly-sis of reverse-osmosis and nanofiltration membranes. MRS Bull2008;33:27–32.
[259] Chan EP, Young AP, Lee J-H, Chung JY, Stafford CM. Swelling of ultra-thin crosslinked polyamide water desalination membranes. J PolymSci, Part B Polym Phys 2013;51:385–91.
[260] Zhang X, Cahill DG, Coronell O, Marinas BJ. Absorption of waterin the active layer of reverse osmosis membranes. J Membr Sci2009;331:143–51.
[261] Bernstein R, Belfer S, Freger V. Toward improved boron removal in
RO by membrane modification: feasibility and challenges. EnvironSci Technol 2011;45:3613–20.[262] Hung PVX, Cho S-H, Moon S-H. Prediction of boron trans-port through seawater reverse osmosis membranes usingsolution–diffusion model. Desalination 2009;247:33–44.
er Science 39 (2014) 1– 42 41
[263] Mane PP, Park PK, Hyung H, Brown JC, Kim JH. Modeling boron rejec-tion in pilot- and full-scale reverse osmosis desalination processes.J Membr Sci 2009;338:119–27.
[264] Mnif A, Hamrouni B, Dhahbi M. Boron removal by membrane pro-cesses. Desalin Water Treat 2009;5:119–23.
[265] Hilal N, Kim GJ, Somerfield C. Boron removal from saline water: acomprehensive review. Desalination 2011;273:23–35.
[266] Redondo J, Busch M, De Witte JP. Boron removal from seawaterusing FILMTECTM high rejection SWRO membranes. Desalination2003;156:229–38.
[267] Pastor MR, Ruiz AF, Chillon MF, Rico DP. Influence of pH in theelimination of boron by means of reverse osmosis. Desalination2001;140:145–52.
[268] Taniguchi M, Fusaoka Y, Nishikawa T, Kurihara M. Boronremoval in RO seawater desalination. Desalination 2004;167:419–26.
[269] Cengeloglu Y, Arslan G, Tor A, Kocak I, Dursun N. Removal ofboron from water by using reverse osmosis. Sep Purif Technol2008;64:141–6.
[270] Koseoglu H, Kabay N, Yuksel M, Sarp S, Arar O, Kitis M.Boron removal from seawater using high rejection SWROmembranes—impact of pH, feed concentration, pressure, and cross-flow velocity. Desalination 2008;227:253–63.
[271] Ozurk N, Kavak D, Kose TE. Boron removal from aqueous solutionby reverse osmosis. Desalination 2008;223:1–9.
[272] Xu Y, Jiang JQ. Technologies for boron removal. Ind Eng Chem Res2008;47:16–24.
[273] La Y-H, Diep J, Al-Rasheed R, Miller D, Krupp L, Geise GM,Vora A, Davis B, Nassar M, Freeman BD, McNeil M, Dubois G.Enhanced desalination performance of polyamide bi-layer mem-branes prepared by sequential interfacial polymerization. J MembrSci 2013;437:33–9.
[274] Dydo P. Transport model for boric acid, monoborate and boratecomplexes across thin-film composite reverse osmosis membrane.Desalination 2013;311:69–79.
[275] Dydo P, Turek M. Boron transport and removal using ion-exchangemembranes: a critical review. Desalination 2013;310:2–8.
[276] Teychene B, Collet G, Gallard H, Croue JP. A comparative study ofboron and arsenic(III) rejection from brackish water by reverseosmosis membranes. Desalination 2013;310:109–14.
[277] Jin X, She QH, Ang XL, Tang CYY. Removal of boron and arsenic byforward osmosis membrane: influence of membrane orientationand organic fouling. J Membr Sci 2012;389:182–7.
[278] Zhai XF, Meng JQ, Li R, Ni L, Zhang YF. Hypochlorite treatmenton thin film composite RO membrane to improve boron removalperformance. Desalination 2011;274:136–43.
[279] Meng JQ, Yuan J, Kang YL, Zhang YF, Du QY. Surface glyco-sylation of polysulfone membrane towards a novel complexingmembrane for boron removal. J Colloid Interface Sci 2012;368:197–207.
[280] Anonymous. Guidelines for drinking-water quality, 3rdedition—incorporating first and second addenda; volume 1: rec-ommendations. Geneva Switzerland: World Health Organization;2008. p. 518.
[281] Anonymous. Guidelines for drinking-water quality, 4th ed. GenevaSwitzerland: World Health Organization; 2011. p. 541.
[282] Ning RY. Arsenic removal by reverse osmosis. Desalination2002;143:237–41.
[283] Fogarassy E, Galambos I, Bekassy-Molnar E, Vatai G. Treatment ofhigh arsenic content wastewater by membrane filtration. Desali-nation 2009;240:270–3.
[284] Smedley PL, Kinniburgh DG. A review of the source, behaviourand distribution of arsenic in natural waters. Appl Geochem2002;17:517–68.
[285] Sato Y, Kang M, Kamei T, Magara Y. Performance of nanofiltrationfor arsenic removal. Water Res 2002;36:3371–7.
[286] Kartinen EO, Martin CJ. An overview of arsenic removal processes.Desalination 1995;103:79–88.
[287] Urase T, Oh J-i, Yamamoto K. Effect of pH on rejection of differentspecies of arsenic by nanofiltration. Desalination 1998;117:11–8.
[288] Waypa JJ, Elimelech M, Hering JG. Arsenic removal by RO and NFmembranes. J AWWA 1997;89:102–14.
[289] Seidel A, Waypa JJ, Elimelech M. Role of charge (Donnan) exclusionin removal of arsenic from water by a negatively charged porous
nanofiltration membrane. Environ Eng Sci 2001;18:105–13.[290] Vrijenhoek EM, Waypa JJ. Arsenic removal from drinking water by a“loose” nanofiltration membrane. Desalination 2000;130:265–77.
[291] van derBruggen B, Vandecasteele C. Removal of pollutants fromsurface water and groundwater by nanofiltration: overview of

n Polym
42 G.M. Geise et al. / Progress ipossible applications in the drinking water industry. Environ Pollut2003;122:435–45.
[292] Kang M, Kawasaki M, Tamada S, Kamei T, Magara Y. Effect of pHon the removal of arsenic and antimony using reverse osmosismembranes. Desalination 2000;131:293–8.
[293] Shih MC. An overview of arsenic removal by pressure-driven mem-brane processes. Desalination 2005;172:85–97.
[294] Gholami MM, Mokhtari MA, Aameri A, Fard MRA. Application ofreverse osmosis technology for arsenic removal from drinkingwater. Desalination 2006;200:725–7.
[295] Chan BKC, Dudeney AWL. Reverse osmosis removal of arsenicresidues from bioleaching of refractory concentrates. Miner Eng2008;21:272–8.
[296] Walker M, Seiler RL, Meinert M. Effectiveness of household reverse-osmosis systems in a Western US region with high arsenic ingroundwater. Sci Total Environ 2008;389:245–52.
[297] Uddin MT, Mozumder MSI, Islam MA, Deowan SA, Hoinkis J.Nanofiltration membrane process for the removal of arsenic fromdrinking water. Chem Eng Technol 2007;30:1248–54.
[298] Lin TF, Hsiao HC, Wu JK, Hsiao HC, Yeh JC. Removal of arsenicfrom groundwater using point-of-use reverse osmosis and distill-ing devices. Environ Technol 2002;23:781–90.
[299] Gao Y, de Jubera AMS, Marinas BJ, Moore JS. Nanofiltration mem-branes with modified active layer using aromatic polyamidedendrimers. Adv Funct Mater 2013;23:598–607.
[300] Nghiem LD, Coleman PJ. NF/RO filtration of the hydrophobicionogenic compound triclosan: transport mechanisms and theinfluence of membrane fouling. Sep Purif Technol 2008;62:709–16.
[301] Nghiem LD, Schafer AI, Elimelech M. Removal of natural hormonesby nanofiltration membranes: measurement, modeling, and mech-anisms. Environ Sci Technol 2004;38:1888–96.
[302] Kimura K, Amy G, Drewes J, Watanabe Y. Adsorption of hydrophobiccompounds onto NF/RO membranes: an artifact leading to overes-timation of rejection. J Membr Sci 2003;221:89–101.
[303] Snyder SA, Adham S, Redding AM, Cannon FS, DeCarolis J, Oppen-heimer J, Wert EC, Yoon Y. Role of membranes and activatedcarbon in the removal of endocrine disruptors and pharmaceut-icals. Desalination 2007;202:156–81.
[304] Yoon Y, Westerhoff P, Snyder SA, Wert EC. Nanofiltration andultrafiltration of endocrine disrupting compounds, pharmaceut-icals and personal care products. J Membr Sci 2006;270:88–100.
[305] Yoon Y, Westerhoff P, Snyder SA, Wert EC, Yoon J. Removalof endocrine disrupting compounds and pharmaceuticals bynanofiltration and ultrafiltration membranes. Desalination2007;202:16–23.
[306] Verliefde ARD, Cornelissen ER, Heijman SGJ, Petrinic I, LuxbacherT, Amy GL, van der Bruggen B, van Dijk JC. Influence of membranefouling by (pretreated) surface water on rejection of pharmaceu-tically active compounds (PhACs) by nanofiltration membranes. J
Membr Sci 2009;330:90–103.[307] Verliefde ARD, Cornelissen ER, Heijman SGJ, Verberk J, Amy GL,van der Bruggen B, van Dijk JC. The role of electrostatic interac-tions on the rejection of organic solutes in aqueous solutions withnanofiltration. J Membr Sci 2008;322:52–66.
er Science 39 (2014) 1– 42
[308] Verliefde ARD, Heijman SG, Cornelissen ER, Amy G, van der BruggenB, van Dijk JC. Influence of electrostatic interactions on the rejectionwith NF and assessment of the removal efficiency during NF/GACtreatment of pharmaceutically active compounds in surface water.Water Res 2007;41:3227–40.
[309] Kosutic K, Dolar D, Asperger D, Kunst B. Removal of antibioticsfrom a model wastewater by RO/NF membranes. Sep Purif Technol2007;53:244–9.
[310] Radjenovic J, Petrovic M, Ventura F, Barcelo D. Rejection of pharma-ceuticals in nanofiltration and reverse osmosis membrane drinkingwater treatment. Water Res 2008;42:3601–10.
[311] Kim JH, Park PK, Lee CH, Kwon HH. Surface modification ofnanofiltration membranes to improve the removal of organicmicro-pollutants (EDCs and PhACs) in drinking water treatment:graft polymerization and cross-linking followed by functionalgroup substitution. J Membr Sci 2008;321:190–8.
[312] Zazouli MA, Susanto H, Nasseri S, Ulbricht M. Influences of solutionchemistry and polymeric natural organic matter on the removalof aquatic pharmaceutical residuals by nanofiltration. Water Res2009;43:3270–80.
[313] Yangali-Quintanilla V, Sadmani A, McConville M, Kennedy M, AmyG. Rejection of pharmaceutically active compounds and endocrinedisrupting compounds by clean and fouled nanofiltration mem-branes. Water Res 2009;43:2349–62.
[314] Ribeiro Jr CP, Freeman BD, Paul DR. Modeling of multicomponentmass transfer across polymer films using a thermodynamically con-sistent formulation of the Maxwell–Stefan equations in terms ofvolume fractions. Polymer 2011;52:3970–83.
[315] Bird RB, Stewart WE, Lightfoot EN. Transport phenomena. 2nd edNew York: Wiley; 2002. p. 895.
[316] Doghieri F, Biavati D, Sarti GC. Solubility and diffusivity of ethanolin PTMSP: effects of activity and of polymer aging. Ind Eng ChemRes 1996;35:2420–30.
[317] Paul DR, Ebra-Lima OM. Pressure-induced diffusion of organic liq-uids through highly swollen polymer membranes. J Appl Polym Sci1970;14:2201–24.
[318] Pitzer KS, Peiper JC, Busey RH. Thermodynamic properties ofaqueous sodium chloride solutions. J Phys Chem Ref Data1984;13:1–102.
[319] Pitzer KS, Mayorga G. Thermodynamics of electrolytes. II. Activityand osmotic coefficients for strong electrolytes with one or bothions univalent. J Phys Chem 1973;77:2300–8.
[320] Gregor HP, Sundheim BR, Held KM, Waxman MH. Ion-exchangeresins. V. Water–vapor sorption. J Colloid Sci 1952;7:511–34.
[321] Jeck S, Scharfer P, Kind M. Water sorption in physically crosslinkedpoly(vinyl alcohol) membranes: an experimental investigation ofSchroeder’s paradox. J Membr Sci 2009;337:291–6.
[322] Paul DR. Further comments on the relation between hydraulicpermeation and diffusion. J Polym Sci Polym Phys Ed1974;12:1221–30.
[323] Paul DR. The solution–diffusion model for swollen membranes. SepPurif Methods 1976;5:33–50.
[324] Sagle AC. PEG hydrogels as anti-fouling coatings for reverse osmosismembranes. PhD thesis. Austin, TX: The University of Texas; 2009.p. 168.


















