
Probing the onset of dense shell packing by measuring the aminolysis rates for a series amine...
8
Reactive & Functional Polymers 66 (2006) 187–194 www.elsevier.com/locate/react 1381-5148/$ - see front matter 2005 Elsevier B.V. All rights reserved. doi:10.1016/j.reactfunctpolym.2005.07.022 Probing the onset of dense shell packing by measuring the aminolysis rates for a series amine terminated dendrimers John Burnett, Amy S.H. King, Lance J. Twyman ¤ Chemistry Department, Dainton Building, University of SheYeld, Brook Hill, SheYeld S3 7HF, UK Available online 30 September 2005 Abstract In an eVort to probe dendritic structure, the initial rate of an aminolysis reaction was measured for a series of dendri- mers with 4, 8, 16, 32 and 64 terminal amine groups (1st–5th generation dendrimers, respectively). From these experi- ments, a relationship between size and rate was observed. In all cases, the rate was faster than the control experiment using a simple amine (N-acetylethylene diamine), with the 4th generation dendrimer displaying the maximum rate enhancement (28-fold with respect to the control experiment). The observed increase in rates was due to: (i) hydropho- bic binding and (ii) transition state stabilization, (dependent on dendrimer generation). These experimental results sup- ported a preliminary surface density analysis, which predicted the onset of a closed shell/globular structure at or around the 4th generation PAMAM dendrimer. 2005 Elsevier B.V. All rights reserved. Keywords: Dendrimers; Catalysis; Dense shell; Dense packing 1. Introduction Enzymes act by binding their substrates in close proximity to speciWc functional groups that are capable of either reacting with the substrate or sta- bilising a particular transition state [1]. Synthetic, supramolecular and biomimetic chemists have adopted these principles and over many years con- structed a variety of model compounds capable of mimicking the action of certain enzymes [2,3]. Of particular note, with respect to a macromolecular approach are the surfactants or micellar based sys- tems [4–6]. It is well known that carrying out cer- tain reactions in aqueous micellar media can alter the rates and pathways of many reactions, such as hydrolysis [7,8], nucleophilic addition [9–11], epox- idation [12], halogenation [13], and pericyclic reac- tions [14,15], which can all be eVected in the presence of micelles. For example, the rate con- stants for the 1,3-dipolar cycloaddition of benzo- nitrile oxide with various dipolarophiles were determined in aqueous media and it was found * Corresponding author. Tel.: +44 0114 222 9560; fax: +44 0114 273 8673. E-mail address: l.j.twyman@sheYeld.ac.uk (L.J. Twyman).
-
Upload
john-burnett -
Category
Documents
-
view
215 -
download
1
Transcript of Probing the onset of dense shell packing by measuring the aminolysis rates for a series amine...

Reactive & Functional Polymers 66 (2006) 187–194
www.elsevier.com/locate/react
Probing the onset of dense shell packing by measuring the aminolysis rates for a series amine terminated dendrimers
John Burnett, Amy S.H. King, Lance J. Twyman ¤
Chemistry Department, Dainton Building, University of SheYeld, Brook Hill, SheYeld S3 7HF, UK
Available online 30 September 2005
Abstract
In an eVort to probe dendritic structure, the initial rate of an aminolysis reaction was measured for a series of dendri-mers with 4, 8, 16, 32 and 64 terminal amine groups (1st–5th generation dendrimers, respectively). From these experi-ments, a relationship between size and rate was observed. In all cases, the rate was faster than the control experimentusing a simple amine (N-acetylethylene diamine), with the 4th generation dendrimer displaying the maximum rateenhancement (28-fold with respect to the control experiment). The observed increase in rates was due to: (i) hydropho-bic binding and (ii) transition state stabilization, (dependent on dendrimer generation). These experimental results sup-ported a preliminary surface density analysis, which predicted the onset of a closed shell/globular structure at or aroundthe 4th generation PAMAM dendrimer. 2005 Elsevier B.V. All rights reserved.
Keywords: Dendrimers; Catalysis; Dense shell; Dense packing
1. Introduction
Enzymes act by binding their substrates in closeproximity to speciWc functional groups that arecapable of either reacting with the substrate or sta-bilising a particular transition state [1]. Synthetic,supramolecular and biomimetic chemists haveadopted these principles and over many years con-structed a variety of model compounds capable of
* Corresponding author. Tel.: +44 0114 222 9560; fax: +440114 273 8673.
E-mail address: [email protected] (L.J. Twyman).
1381-5148/$ - see front matter 2005 Elsevier B.V. All rights reservedoi:10.1016/j.reactfunctpolym.2005.07.022
mimicking the action of certain enzymes [2,3]. Ofparticular note, with respect to a macromolecularapproach are the surfactants or micellar based sys-tems [4–6]. It is well known that carrying out cer-tain reactions in aqueous micellar media can alterthe rates and pathways of many reactions, such ashydrolysis [7,8], nucleophilic addition [9–11], epox-idation [12], halogenation [13], and pericyclic reac-tions [14,15], which can all be eVected in thepresence of micelles. For example, the rate con-stants for the 1,3-dipolar cycloaddition of benzo-nitrile oxide with various dipolarophiles weredetermined in aqueous media and it was found
d.

188 J. Burnett et al. / Reactive & Functional Polymers 66 (2006) 187–194
that these pericyclic reactions (with both electronrich and electron poor dipolarophiles) are acceler-ated by the presence of sodium dodecyl sulphate(SDS) micelles [14,15]. The observed increase inrate (which only occurs above the CMC) has beenattributed to the fact that the hydrophobic dipol-arophile is located at the anionic micellar surface,which increases the eVective molarity of the sub-strate. The reaction rate continues to increase asthe concentration of micelles increases which indi-cates that the binding of the substrates to themicelles is relatively weak and that a high concen-tration of micelles is necessary for complete bind-ing of the reagents. Despite the weak substratebinding in this case, there is at least a 4-foldincrease in reaction rate above the CMC of SDS.
It is generally observed that the mechanism ofcatalysis in micellar systems involves a combinationof factors, including hydrophobic binding, transi-tion state stabilisation and substrate concentration[16]. Despite the considerable success of micelles,some major problems associated with their use stillremain. Micelles are created under equilibrium con-ditions, so as each micelle forms it will immediatelybegin to fall apart. Another problematic area is thatof critical micelle concentration (CMC); these canvery dramatically depending on the nature of thesurfactant. Micelle formation is also temperaturedependent. In order for these systems to be moreeYcient, a static or covalent micelle is required.Dendrimers are monodisperse, highly symmetricalmacromolecules with a tree-like complexity and ahigh density of terminal groups [17]. The surfacefunctionality can be tailored, for example, adaptingthe terminal groups to make them more or lesshydrophilic, which changes their solubility proper-ties. When these terminal groups are either chargedor polar, the resulting dendrimers have similarstructural and solubility properties to micelles, andcan be considered as static covalent micelles.
2. Experimental
2.1. PAMAM dendrimers (1–5)
The amine terminated PAMAM dendrimers(generations one to four with 4–32 terminal NH2
groups respectively, are shown in Fig. 1; the largerWfth generation dendrimer, with 64 terminalgroups, is not shown for clarity) were purchasedfrom Aldrich Chemicals as solutions in methanol(20% wt vol.). The purity of the dendrimers wasconWrmed using 1H, 13C NMR spectroscopy andGC analysis before use in the aminolysis rateenhancement studies.
2.2. Rate measurements
Solutions of N-acetyl ethylenediamine, ethy-lenediamine, and dendrimer generations 1–5 weremade up in a Tris buVer solution so that an abso-lute total amine concentration of 6.4 mM wasachieved in each case, i.e. concentrations of 6.4(NAEDA), 3.2 (EDA), 1.6 (G1), 0.8 (G2), 0.4 (G3),0.2 (G4), 0.1 (G5) mM were used. For the rateenhancement studies, a dendrimer or NAEDAsolution (1.9 ml, 1.2 £ 10¡5 mol amine) in pH 8.5buVer was added to a UV cuvette with stirring.About 0.1 ml (1.4 £ 10¡8 mol) of a 1.4 mM solutionof p-nitrophenol acetate (8) (in acetonitrile) wasadded to the cuvette quickly and the UV absor-bance at 410 nm was measured every 10 s for aperiod of 20 min (using an Hitachi U-2010 spectro-photometer). The background hydrolysis of PNPAin aqueous solution was measured in the same way.The concentration of phenolate by product wasdetermined from a standard Beer–Lambert plotobtained using 10 solutions of p-nitrophenol in0.1 M Tris buVer solution, pH 8.5 (solution concen-trations ranged from 1 £ 10¡5 to 1 £ 10¡4).
3. Results and discussion
Recent reports have described the use of watersoluble dendrimers as solubilising agents, whichencapsulate hydrophobic guests in their interiors[18,19]. Although these molecules are more diYcultto construct (relative to simple surfactants), theyare static, exist at all concentrations and persistacross a wide range of temperatures. It should,therefore, be possible for these molecules to cata-lyse or accelerate the rates of various reactions. Todemonstrate the feasibility of this proposal the ini-tial rate of an aminolysis reaction using a large
J. Burnett et al. / Reactive & Functional Polymers 66 (2006) 187–194 189
PAMAM dendrimer with 64 terminal amine coloured and its formation can be followed using
groups 1 was measured. This initial rate was com-pared to that obtained using 64 equivalents of thesimple amine N-acetyl ethylenediamine (NAEDA),2, Scheme 1. NAEDA (2) was chosen as our con-trol/test reaction because it resembles the outerdomain of a PAMAM dendrimer and thereforeacts as an isolated and reactive unit independent ofthe macroscopic dendrimer backbone. The selectedprocess is shown in Scheme 1, this involves thereaction between the ester substrate, p-nitrophenolacetate (3), and the amine (dendrimer 1 or amine2). p-Nitrophenol acetate (3) was selected becausethe leaving group, p-nitrophenolate (5), is highlyUV spectroscopy (�max 410 nm in aqueous buVer).All reactions were performed in buVered water atpH 8.5 (Tris buVer, 0.1 M). In all cases, an acylatedamine (4 and 6) was produced along with the p-nitrophenolate (3), as shown in Scheme 1. SpeciW-
cally, a small volume of a concentrated solution ofp-nitrophenyl acetate was added directly to a UVcuvette containing a solution of amines 1 or 2 inaqueous buVer (pH 8.5) with stirring. The tworeactions were performed so that the starting con-centration of amine groups were the same,enabling a direct comparison of rates to be made.The absorbance at 410 nm was measured every 10 s
Fig. 1. 1st– 4th generation amine terminated PAMAM dendrimers.
N
NHON
NHO
NHO HN
O
NH2
N
HN
OHN
O NH2
NHO
N
NH2
H2N
NHON
NHO
NHO HN
O
NH2
N
NH
O NH
O NH2
NHO
N
NH2
H2N
N
HN ON
HN O
HNONH
O
H2N
N
NH
ONH
OH2N
HN
O
N
H2N
NH2
HN ON
HN O
HNONH
O
H2N
N
HNO
HN
OH2N
HNO
N
H2N
NH2
N
NHON
NHO
NH2
HN
ONH2
NHON
NHO
NH2
NH
ONH2
N
HN ON
HN O
H2N
NH
OH2N
HN ON
HN O
H2N
HNO
H2N
N
NHONH2
NHONH2
N
HN OH2N
HN OH2N
N
NHON
NHO
NHO HN
O
NH
N
HN
N
HNOHN
O
H2N
N
NH2
NHOHN
O
NH2
N
H2N
NH
O
NHO
HN
O NH
N
HN
HNO
NHO
NH2
NNH2
NH
O
HN
O NH2
N
NH2
NHON
NHO
NHO HN
O
NH
N
HN
HNONH
O
H2N
N
NH2
NHOHN
O
NH2
N
H2N
HN
O HN
O
NO
HN
N
HN
HNO
NHO
NH2
N NH2
NHO
NH
O NH2
N
NH2
HN ON
HN O
HN ONHO
HN
N
NH
NHO HN
O
NH2
N
H2N
HN ONH
O
H2N
N
NH2
NH
OHN
O
HNON
H
N
NH
NHO
HN O
H2N
NH2N
NH
O
HN
OH2N
N
H2N
HN ON
HN O
HN ONHO
HN
N
NH
NHO HN
O
NH2
N
H2N
HN ONH
O
H2N
N
NH2
HN
OHNO
NH
OHN
N
NH
NHO
HN O
H2N
NH2N
HN
O
NH
OH2N
N
H2N
190 J. Burnett et al. / Reactive & Functional Polymers 66 (2006) 187–194
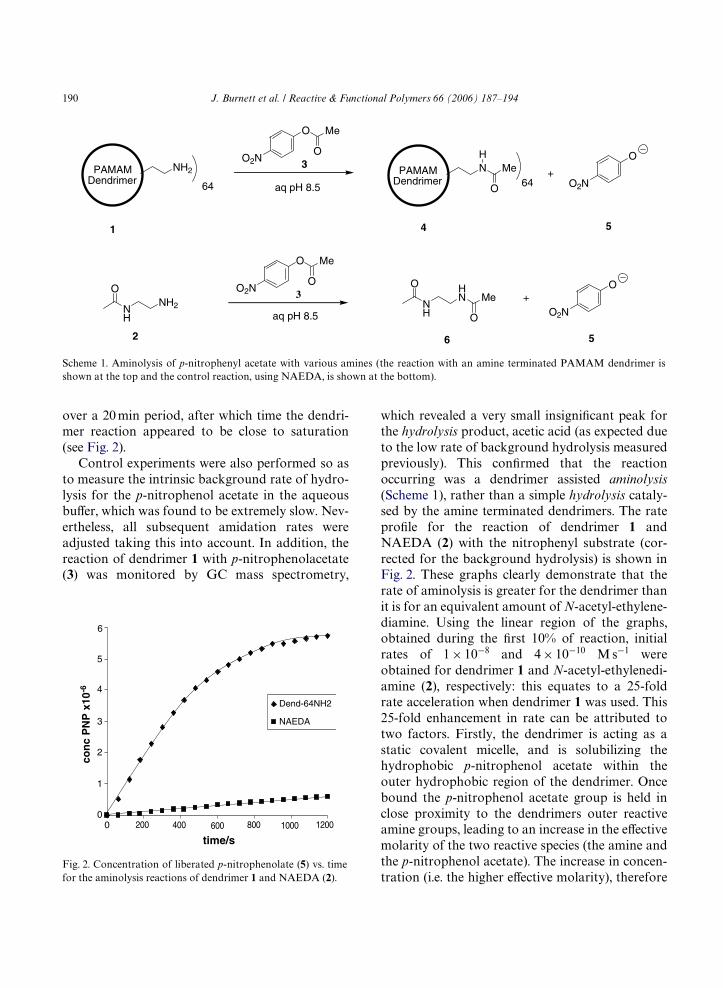
over a 20 min period, after which time the dendri-mer reaction appeared to be close to saturation(see Fig. 2).
Control experiments were also performed so asto measure the intrinsic background rate of hydro-lysis for the p-nitrophenol acetate in the aqueousbuVer, which was found to be extremely slow. Nev-ertheless, all subsequent amidation rates wereadjusted taking this into account. In addition, thereaction of dendrimer 1 with p-nitrophenolacetate(3) was monitored by GC mass spectrometry,
Fig. 2. Concentration of liberated p-nitrophenolate (5) vs. timefor the aminolysis reactions of dendrimer 1 and NAEDA (2).
0
1
2
3
4
5
6
0 200 400 600 800 1000 1200
time/s
con
c P
NP
x10
-6
Dend-64NH2
NAEDA
which revealed a very small insigniWcant peak forthe hydrolysis product, acetic acid (as expected dueto the low rate of background hydrolysis measuredpreviously). This conWrmed that the reactionoccurring was a dendrimer assisted aminolysis(Scheme 1), rather than a simple hydrolysis cataly-sed by the amine terminated dendrimers. The rateproWle for the reaction of dendrimer 1 andNAEDA (2) with the nitrophenyl substrate (cor-rected for the background hydrolysis) is shown inFig. 2. These graphs clearly demonstrate that therate of aminolysis is greater for the dendrimer thanit is for an equivalent amount of N-acetyl-ethylene-diamine. Using the linear region of the graphs,obtained during the Wrst 10% of reaction, initialrates of 1 £ 10¡8 and 4 £ 10¡10 M s¡1 wereobtained for dendrimer 1 and N-acetyl-ethylenedi-amine (2), respectively: this equates to a 25-foldrate acceleration when dendrimer 1 was used. This25-fold enhancement in rate can be attributed totwo factors. Firstly, the dendrimer is acting as astatic covalent micelle, and is solubilizing thehydrophobic p-nitrophenol acetate within theouter hydrophobic region of the dendrimer. Oncebound the p-nitrophenol acetate group is held inclose proximity to the dendrimers outer reactiveamine groups, leading to an increase in the eVectivemolarity of the two reactive species (the amine andthe p-nitrophenol acetate). The increase in concen-tration (i.e. the higher eVective molarity), therefore
Scheme 1. Aminolysis of p-nitrophenyl acetate with various amines (the reaction with an amine terminated PAMAM dendrimer isshown at the top and the control reaction, using NAEDA, is shown at the bottom).
O
O2N O
O2N
Me
O
NH2 +aq pH 8.5
3
4
PAMAMDendrimer 64
1
2
NH
NH2
O
NH
HN
O+
N PAMAMDendrimer
H
O
Me64
O
O2N
5
O
O2N
Me
O
aq pH 8.5
3
O
Me
5
6
J. Burnett et al. / Reactive & Functional Polymers 66 (2006) 187–194 191
contributes signiWcantly to the observed rateenhancement. A second possible factor involvesthe dendrimers internal amide groups [20], thesemay be able to stabilise the transition state as itforms during the aminolysis reaction, lowering theactivation energy and, therefore, increasing therate of reaction.
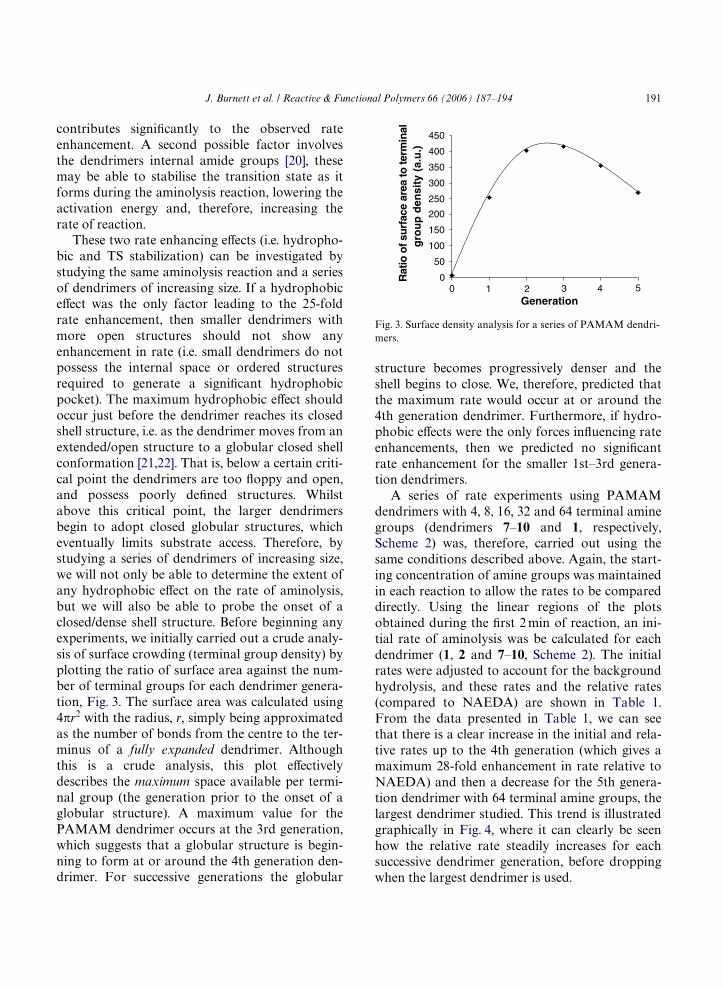
These two rate enhancing eVects (i.e. hydropho-bic and TS stabilization) can be investigated bystudying the same aminolysis reaction and a seriesof dendrimers of increasing size. If a hydrophobiceVect was the only factor leading to the 25-foldrate enhancement, then smaller dendrimers withmore open structures should not show anyenhancement in rate (i.e. small dendrimers do notpossess the internal space or ordered structuresrequired to generate a signiWcant hydrophobicpocket). The maximum hydrophobic eVect shouldoccur just before the dendrimer reaches its closedshell structure, i.e. as the dendrimer moves from anextended/open structure to a globular closed shellconformation [21,22]. That is, below a certain criti-cal point the dendrimers are too Xoppy and open,and possess poorly deWned structures. Whilstabove this critical point, the larger dendrimersbegin to adopt closed globular structures, whicheventually limits substrate access. Therefore, bystudying a series of dendrimers of increasing size,we will not only be able to determine the extent ofany hydrophobic eVect on the rate of aminolysis,but we will also be able to probe the onset of aclosed/dense shell structure. Before beginning anyexperiments, we initially carried out a crude analy-sis of surface crowding (terminal group density) byplotting the ratio of surface area against the num-ber of terminal groups for each dendrimer genera-tion, Fig. 3. The surface area was calculated using4�r2 with the radius, r, simply being approximatedas the number of bonds from the centre to the ter-minus of a fully expanded dendrimer. Althoughthis is a crude analysis, this plot eVectivelydescribes the maximum space available per termi-nal group (the generation prior to the onset of aglobular structure). A maximum value for thePAMAM dendrimer occurs at the 3rd generation,which suggests that a globular structure is begin-ning to form at or around the 4th generation den-drimer. For successive generations the globular
structure becomes progressively denser and theshell begins to close. We, therefore, predicted thatthe maximum rate would occur at or around the4th generation dendrimer. Furthermore, if hydro-phobic eVects were the only forces inXuencing rateenhancements, then we predicted no signiWcantrate enhancement for the smaller 1st–3rd genera-tion dendrimers.
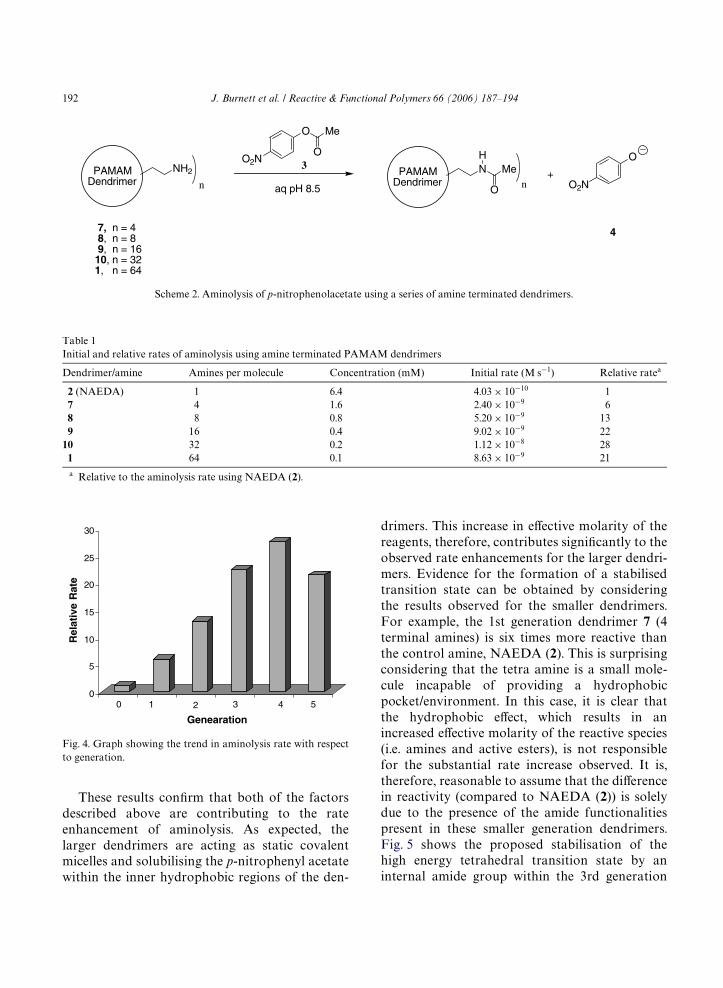
A series of rate experiments using PAMAMdendrimers with 4, 8, 16, 32 and 64 terminal aminegroups (dendrimers 7–10 and 1, respectively,Scheme 2) was, therefore, carried out using thesame conditions described above. Again, the start-ing concentration of amine groups was maintainedin each reaction to allow the rates to be compareddirectly. Using the linear regions of the plotsobtained during the Wrst 2 min of reaction, an ini-tial rate of aminolysis was be calculated for eachdendrimer (1, 2 and 7–10, Scheme 2). The initialrates were adjusted to account for the backgroundhydrolysis, and these rates and the relative rates(compared to NAEDA) are shown in Table 1.From the data presented in Table 1, we can seethat there is a clear increase in the initial and rela-tive rates up to the 4th generation (which gives amaximum 28-fold enhancement in rate relative toNAEDA) and then a decrease for the 5th genera-tion dendrimer with 64 terminal amine groups, thelargest dendrimer studied. This trend is illustratedgraphically in Fig. 4, where it can clearly be seenhow the relative rate steadily increases for eachsuccessive dendrimer generation, before droppingwhen the largest dendrimer is used.
Fig. 3. Surface density analysis for a series of PAMAM dendri-mers.
0
50
100
150
200
250
300
350
400
450
0Generation
Rat
io o
f sur
face
are
a to
term
inal
gro
up
den
sity
(a.
u.)
1 2 3 4 5
192 J. Burnett et al. / Reactive & Functional Polymers 66 (2006) 187–194
These results conWrm that both of the factorsdescribed above are contributing to the rateenhancement of aminolysis. As expected, thelarger dendrimers are acting as static covalentmicelles and solubilising the p-nitrophenyl acetatewithin the inner hydrophobic regions of the den-
Fig. 4. Graph showing the trend in aminolysis rate with respectto generation.
0
5
10
15
20
25
30
Rel
ativ
e R
ate
0 1 2 3 4 5
Genearation
drimers. This increase in eVective molarity of thereagents, therefore, contributes signiWcantly to theobserved rate enhancements for the larger dendri-mers. Evidence for the formation of a stabilisedtransition state can be obtained by consideringthe results observed for the smaller dendrimers.For example, the 1st generation dendrimer 7 (4terminal amines) is six times more reactive thanthe control amine, NAEDA (2). This is surprisingconsidering that the tetra amine is a small mole-cule incapable of providing a hydrophobicpocket/environment. In this case, it is clear thatthe hydrophobic eVect, which results in anincreased eVective molarity of the reactive species(i.e. amines and active esters), is not responsiblefor the substantial rate increase observed. It is,therefore, reasonable to assume that the diVerencein reactivity (compared to NAEDA (2)) is solelydue to the presence of the amide functionalitiespresent in these smaller generation dendrimers.Fig. 5 shows the proposed stabilisation of thehigh energy tetrahedral transition state by aninternal amide group within the 3rd generation
Scheme 2. Aminolysis of p-nitrophenolacetate using a series of amine terminated dendrimers.
O
O2N O
O2N
Me
O
NH2 +aq pH 8.5
3
4
PAMAMDendrimer n
7, n = 4 8, n = 8 9, n = 1610, n = 321, n = 64
N PAMAMDendrimer
H
O
Men
Table 1Initial and relative rates of aminolysis using amine terminated PAMAM dendrimers
a Relative to the aminolysis rate using NAEDA (2).
Dendrimer/amine Amines per molecule Concentration (mM) Initial rate (M s¡1) Relative ratea
2 (NAEDA) 1 6.4 4.03 £ 10¡10 17 4 1.6 2.40 £ 10¡9 68 8 0.8 5.20 £ 10¡9 139 16 0.4 9.02 £ 10¡9 22
10 32 0.2 1.12 £ 10¡8 281 64 0.1 8.63 £ 10¡9 21
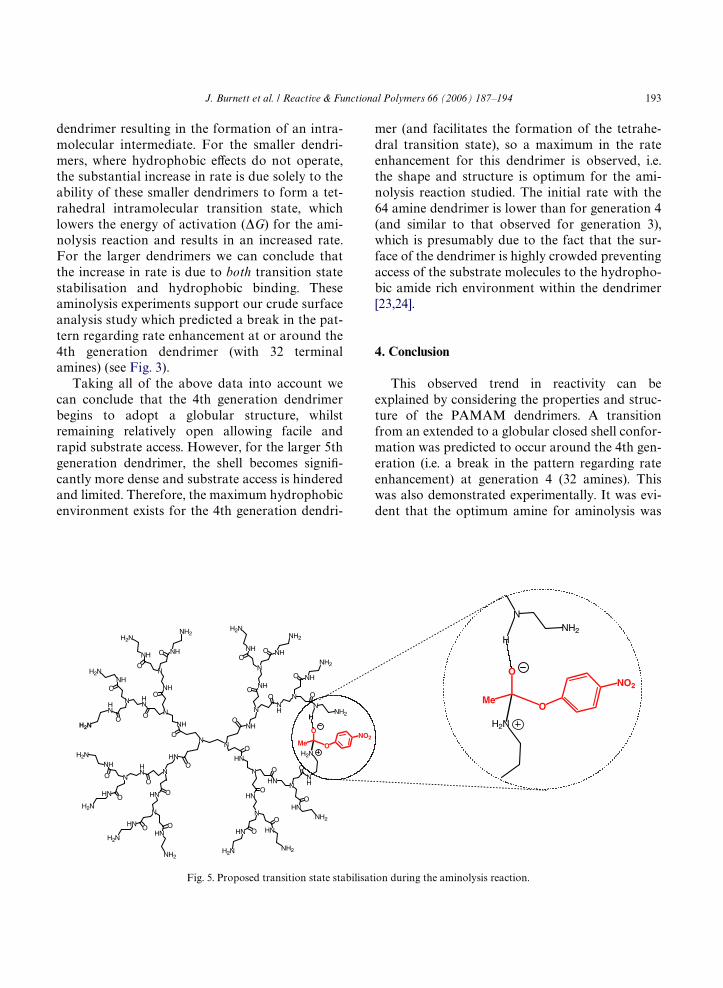
J. Burnett et al. / Reactive & Functional Polymers 66 (2006) 187–194 193
dendrimer resulting in the formation of an intra-molecular intermediate. For the smaller dendri-mers, where hydrophobic eVects do not operate,the substantial increase in rate is due solely to theability of these smaller dendrimers to form a tet-rahedral intramolecular transition state, whichlowers the energy of activation (�G) for the ami-nolysis reaction and results in an increased rate.For the larger dendrimers we can conclude thatthe increase in rate is due to both transition statestabilisation and hydrophobic binding. Theseaminolysis experiments support our crude surfaceanalysis study which predicted a break in the pat-tern regarding rate enhancement at or around the4th generation dendrimer (with 32 terminalamines) (see Fig. 3).
Taking all of the above data into account wecan conclude that the 4th generation dendrimerbegins to adopt a globular structure, whilstremaining relatively open allowing facile andrapid substrate access. However, for the larger 5thgeneration dendrimer, the shell becomes signiW-
cantly more dense and substrate access is hinderedand limited. Therefore, the maximum hydrophobicenvironment exists for the 4th generation dendri-
mer (and facilitates the formation of the tetrahe-dral transition state), so a maximum in the rateenhancement for this dendrimer is observed, i.e.the shape and structure is optimum for the ami-nolysis reaction studied. The initial rate with the64 amine dendrimer is lower than for generation 4(and similar to that observed for generation 3),which is presumably due to the fact that the sur-face of the dendrimer is highly crowded preventingaccess of the substrate molecules to the hydropho-bic amide rich environment within the dendrimer[23,24].
4. Conclusion
This observed trend in reactivity can beexplained by considering the properties and struc-ture of the PAMAM dendrimers. A transitionfrom an extended to a globular closed shell confor-mation was predicted to occur around the 4th gen-eration (i.e. a break in the pattern regarding rateenhancement) at generation 4 (32 amines). Thiswas also demonstrated experimentally. It was evi-dent that the optimum amine for aminolysis was
Fig. 5. Proposed transition state stabilisation during the aminolysis reaction.
NNH
O NN
O
NHO
NH
HN
O
HN
O
N
HNO
NH
ON
O
N
NH2
NH2
NH2
O
Me O
NO2
H2N
NHO
N
NHONHO
H2NNH2
HNO
N
HN O HN
O
NH2H2N
H
NHN
ON
NH
O
HN O
HN
NH
O
HN
O
N
NHO
HN
ON
O
N
H2N
H2N
H2N
H2N
HN O
N
HN OHN
O
NH2
H2N
NHO
N
NHONH
O
H2NNH2
NNH2
O
MeO
NO2
H2N
H

194 J. Burnett et al. / Reactive & Functional Polymers 66 (2006) 187–194
the dendrimer possessing 32 terminal aminegroups (the 4th generation). Two factors wereresponsible for the observed rate accelerations: (i)transition state stabilization and (ii) provision of ahydrophobic pocket that positions the substrate inclose proximity to the reactive amine units. Whichof these factors dominated depends on the dendri-mer’s size/generation. Below a certain point, thedendrimers do not possess signiWcant hydrophobicdomains, whilst above this critical point dendri-mers have an overly crowded surface hinderingsubstrate assess. We, therefore, conclude that the4th generation dendrimer maintains a suYcientlyopen structure allowing the p-nitrophenyl acetatesubstrate to enter its hydrophobic environmentunhindered. Furthermore, once inside, there issuYcient room and Xexibility to allow any confor-mation changes required for transition state stabil-ization. Further work is going on in our laboratoryto study similar systems capable of acting as staticcovalent micelles for application in solubilizationdrug.
References
[1] H. Dugas, Bioorganic Chemistry. A Chemical Approach toEnzyme Action, Springer-Verlag, New York, 1996.
[2] M. Kamieth, F.G. Klarner, Chem. Unserer Zeit 31 (1997) 97.[3] A. Wiseman, H. Dalton, Trends Biotechnol. 5 (1987) 241.[4] X. Zeng, Y. Chen, J. Dispers. Sci. Technol. 21 (2000) 449.
[5] M. Ruasse, I.B. Blagoeva, R. Ciri, L. Garcia-Rio, J.R. Leis,A. Marques, J. Mejuto, E. Monnier, Pure Appl. Chem. 69(1997) 1923.
[6] J.F. Rathman, Curr. Opin. Colloid Interf. Sci. 1 (1996) 514.[7] S. Cheng, X. Zeng, J. Dispers. Sci. Technol. 21 (2000) 655.[8] E.H. Cordes, Reaction Kinetics in Micelles, Plenum Press,
New York, 1973.[9] C.A. Bunton, R. Bacaloglu, J. Colloid Interf. Sci. 115 (1987) 288.
[10] J. Baumucker, M. Calzadilla, M. Centeno, G. Lehrmann,M. Urdanela, P. Lindquist, D. Dunham, M. Price, D. Sears,E.H. Cordes, J. Am. Chem. Soc. 94 (1972) 8164.
[11] C. Swiswanto, T. Battal, O.E. Schuss, J.F. Rathman, Lang-muir 13 (1997) 6047.
[12] D. Monti, P. Tagliatesta, G. Mancini, T. Boschi, Angew.Chem., Int. Ed. 37 (1998) 1131.
[13] D.A. Jaeger, R.E. Robertson, J. Org. Chem. 32 (1977) 3298.[14] D. Van Mersbergen, J.W. Wijnen, J.B.F.N. Engberts, J. Org.
Chem. 63 (1998) 8801.[15] D.A. Jaeger, D. Wang, Tetrahedron Lett. 33 (1992) 6415.[16] G.R. Newkome, C.N. MoorWeld, F. Vögtle, Dendritic Mol-
ecules; Concepts, Synthesis and Perspectives, VCH-Wein-heim, 1996.
[17] X. Zeng, Y. Cheng, J. Dispers. Sci. Technol. 21 (2000) 449.[18] L.J. Twyman, A.E. Beezer, R. Esfand, M.J. Hardy, J.C.
Mitchell, Tetrahedron Lett. 40 (1999) 1743.[19] C.J. Hawker, K.L. Wooley, J.M.J. Fréchet, J. Chem. Soc.,
Perkin Trans. 1 (1993) 1287.[20] D.J. Evans, A. Kanagasooriam, A. Williams, J. Mol. Catal.
85 (1993) 21.[21] J.F.J.A. Jansen, E.W. Meijer, J. Am. Chem. Soc. 117 (1995)
4417.[22] M.E. Piotti, F. Rivera, R. Bond, C.J. Hawker, J.M.J. Fré-
chet, J. Am. Chem. Soc. 121 (1999) 9471.[23] I.K. Martin, L.J. Twyman, Tetrahedron Lett. 42 (2001) 1123.[24] J. Burnett, A.S. King, I.K. Martin, L.J. Twyman, Tetrahe-
dron Lett. 42 (2002) 2431.


















