
Probing Dense Packed Limits of a Hyperbranched Polymer through Ligand Binding and Size Selective...
20
Probing Dense Packed Limits of a Hyperbranched Polymer through Ligand Binding and Size Selective Catalysis Adam Ellis and Lance J. Twyman* Department of Chemistry, University of Sheffield, Brook Hill, Sheffield, South Yorkshire S3 7HF, U.K. * S Supporting Information ABSTRACT: In the area of dendritic chemistry (hyper- branched polymers and dendrimers) it is often generalized that dendrimers are the molecule of choice for smart, selective, or technical applications involving encapsulation or controlled/ selective environments. This is despite the fact that hyper- branched polymers (HBP)s are generally easier and cheaper to synthesize, making them more amenable to large-scale applications. Dendrimers have been successful in these applications by virtue of a dense packed or dense shell limit. This paper describes the synthesis of a series of narrowly dispersed HBPs possessing binding and catalytic cores with a high and uniform loading. Subsequent binding experiments clearly demonstrated the existence of a dense packed limit with respect to polymer molecular weight and ligand size. A series of catalytic experiments were also performed in an attempt to exploit these molecules and their dense packed limit to the area of shape/size selective catalysisan area where dendrimers have previously been used with celebrated success. However, although we were able to show the existence of a dense packed limit, we were initially unable to demonstrate any selectivity based on substrate size or shape. Nevertheless, further studies into core branching motif and multiplicity eventually enabled us to obtain a series of HBPs capable of perturbing the shape/size selectivity of a simple oxidation reaction involving two alkenes. Specifically, we were able to demonstrate a 3.5-fold shift in chemoselectivity toward a smaller alkene of lower reactivity. These results compare favorably with those obtained using dendrimers and allow us to conclude that, with careful thought regarding core design, HBPs are indeed capable of being applied to technical/smart applications involving controlled and selective environments. ■ INTRODUCTION Although hyperbranched polymers (HBPs) can be applied to a range of applications, 1 it is generally perceived that the most high-end or technical applications are reserved for dendrimers. 2 We wanted to challenge this idea and determine whether or not this generalization was valid. We were particularly interested in investigating applications that exploit the use of dense shell/ packing 3 properties when applied to selective encapsulation, release, or catalysis. To do this, we need to identify the specific structural and design requirements necessary for HBPs to operate at a similar level of performance when applied to the same high-end or technical applications normally reserved for dendrimers. These structural and design properties are controlled by the synthetic methods used to construct the relevant macromolecule (dendrimer or HBP). Dendrimers are synthesized using stepwise methods. 4 Although this is often tedious and time-consuming, it does lead to a series of well-defined structures known as generations. These molecules are monodisperse with regards to molecular weight, structure, and degree of branching (100% by virtue of complete and controlled reactionsi.e., all branching points have reacted). Furthermore, as each dendrimer possesses a unique molecular weight and therefore size, a dendrimer series can be considered quantized with respect to its generations. 5 As such, the internal environment, including any packing and closed shell properties, are switched on or off as we move from one particular generation to the next. This level of precision makes it relatively easy to control local environment (electronic and steric) and results in high selectivity and control in a range of applications. 6 A particularly good exemplifier of how this controlled structure can be exploited was described by Suslick and Moore. 7 Suslick and Moore showed how porphyrin cored dendrimers could be used to mimic the reactivity of cytochrome P450 8 and catalyze the oxidation of various alkenes in a shape/size selective way. The key experiment involved taking a 1:1 mixture of the two substrates and catalyzing the reaction using the porphyrin cored dendrimers. The two substrates used were a large/fat substrate with a high relative reactivity and the second smaller/thinner substrate possessing much lower relative reactivity. When catalyzed by a dendrimer, the results showed that selectivity shifted toward the smaller substrate reactivity, despite it possessing a lower inherent reactivity (compared to the larger substrate). This change in selectivity was by virtue of the dendrimer’s structure, which created a steric barrier around the porphyrin and through which the smaller substrate could pass through more easily. Received: June 27, 2013 Revised: August 1, 2013 Article pubs.acs.org/Macromolecules © XXXX American Chemical Society A dx.doi.org/10.1021/ma401344d | Macromolecules XXXX, XXX, XXX−XXX
Transcript of Probing Dense Packed Limits of a Hyperbranched Polymer through Ligand Binding and Size Selective...

Probing Dense Packed Limits of a Hyperbranched Polymer throughLigand Binding and Size Selective CatalysisAdam Ellis and Lance J. Twyman*
Department of Chemistry, University of Sheffield, Brook Hill, Sheffield, South Yorkshire S3 7HF, U.K.
*S Supporting Information
ABSTRACT: In the area of dendritic chemistry (hyper-branched polymers and dendrimers) it is often generalized thatdendrimers are the molecule of choice for smart, selective, ortechnical applications involving encapsulation or controlled/selective environments. This is despite the fact that hyper-branched polymers (HBP)s are generally easier and cheaper tosynthesize, making them more amenable to large-scaleapplications. Dendrimers have been successful in theseapplications by virtue of a dense packed or dense shell limit.This paper describes the synthesis of a series of narrowly dispersed HBPs possessing binding and catalytic cores with a high anduniform loading. Subsequent binding experiments clearly demonstrated the existence of a dense packed limit with respect topolymer molecular weight and ligand size. A series of catalytic experiments were also performed in an attempt to exploit thesemolecules and their dense packed limit to the area of shape/size selective catalysisan area where dendrimers have previouslybeen used with celebrated success. However, although we were able to show the existence of a dense packed limit, we wereinitially unable to demonstrate any selectivity based on substrate size or shape. Nevertheless, further studies into core branchingmotif and multiplicity eventually enabled us to obtain a series of HBPs capable of perturbing the shape/size selectivity of a simpleoxidation reaction involving two alkenes. Specifically, we were able to demonstrate a 3.5-fold shift in chemoselectivity toward asmaller alkene of lower reactivity. These results compare favorably with those obtained using dendrimers and allow us toconclude that, with careful thought regarding core design, HBPs are indeed capable of being applied to technical/smartapplications involving controlled and selective environments.
■ INTRODUCTION
Although hyperbranched polymers (HBPs) can be applied to arange of applications,1 it is generally perceived that the mosthigh-end or technical applications are reserved for dendrimers.2
We wanted to challenge this idea and determine whether or notthis generalization was valid. We were particularly interested ininvestigating applications that exploit the use of dense shell/packing3 properties when applied to selective encapsulation,release, or catalysis. To do this, we need to identify the specificstructural and design requirements necessary for HBPs tooperate at a similar level of performance when applied to thesame high-end or technical applications normally reserved fordendrimers. These structural and design properties arecontrolled by the synthetic methods used to construct therelevant macromolecule (dendrimer or HBP).Dendrimers are synthesized using stepwise methods.4
Although this is often tedious and time-consuming, it doeslead to a series of well-defined structures known as generations.These molecules are monodisperse with regards to molecularweight, structure, and degree of branching (100% by virtue ofcomplete and controlled reactionsi.e., all branching pointshave reacted). Furthermore, as each dendrimer possesses aunique molecular weight and therefore size, a dendrimer seriescan be considered quantized with respect to its generations.5 Assuch, the internal environment, including any packing and
closed shell properties, are switched on or off as we move fromone particular generation to the next. This level of precisionmakes it relatively easy to control local environment (electronicand steric) and results in high selectivity and control in a rangeof applications.6 A particularly good exemplifier of how thiscontrolled structure can be exploited was described by Suslickand Moore.7 Suslick and Moore showed how porphyrin coreddendrimers could be used to mimic the reactivity ofcytochrome P4508 and catalyze the oxidation of various alkenesin a shape/size selective way. The key experiment involvedtaking a 1:1 mixture of the two substrates and catalyzing thereaction using the porphyrin cored dendrimers. The twosubstrates used were a large/fat substrate with a high relativereactivity and the second smaller/thinner substrate possessingmuch lower relative reactivity. When catalyzed by a dendrimer,the results showed that selectivity shifted toward the smallersubstrate reactivity, despite it possessing a lower inherentreactivity (compared to the larger substrate). This change inselectivity was by virtue of the dendrimer’s structure, whichcreated a steric barrier around the porphyrin and through whichthe smaller substrate could pass through more easily.
Received: June 27, 2013Revised: August 1, 2013
Article
pubs.acs.org/Macromolecules
© XXXX American Chemical Society A dx.doi.org/10.1021/ma401344d | Macromolecules XXXX, XXX, XXX−XXX

Conversely, and despite being inherently more reactive, thelarger substrate had a more difficult journey to the centralporphyrin catalyst. These result demonstrated the ability of acore functionalized dendrimer and, more specifically, howpacking around a catalytic core could be exploited to influenceselectivity. If HBPs were to act in a same way to the dendrimersdescribed by Suslick and Moore, they would require a similarlevel of control with respect to their structure. In particular, wewould need a polymer that possessed a steric or dense packedlimit with respect to molecular weight. However, HBPs aresynthesized using a much simpler procedure involving abranching monomer and a single-step random polymerizationprocedure, generating a mixture of molecules differing inmolecular weight, branching architecture, and 3D structure.9
The resulting structures are poorly defined, and any change ininternal steric or electronic environment (i.e., dense shell/packing properties) is gradual and poorly defined with respectto size or molecular weight. It is this lack of control regardingthe internal environment that ultimately leads to poorperformance in selective encapsulation, release, or catalyticapplications. Nevertheless, we proposed to investigate whetheror not a well-defined hyperbranched polymer could be appliedas enzyme mimetics6,10 capable of replicating the same levels ofshape and size selectivity demonstrated by the dendrimersreported by Suslik and Moore.The specific aims of the work described in this paper are
threefold. Our initial aim was to obtain a narrowly dispersed,pseudo-generational series of HBPs possessing a catalytic/binding core in each and every polymer molecule. Havingachieved this, we would then determine whether or not (and atwhat point) an observable and well-def ined steric or densepacked limit existed. The steric properties of the HBP serieswould be assessed by studying ligand binding to the centralcore. Finally, catalytic experiments would be carried out eitherside of any steric limit to test for possible shape/size selectivity.If these aims could be achieved, then we would havedemonstrated that, in principle, HBPs can be applied to themore technical and high-end applications usually reserved fordendrimers.
■ RESULTS AND DISCUSSIONSynthesis of Tetra(acetoxyphenyl)porphyrin Cored
Hyperbranched Polymers. Because of the lack of controlregarding the synthesis of HBPs, dense packing will bedependent on a number of different and variable properties.These include molecular weight, which is effectively a measureof size, which in turn controls the packing density. However,this is not necessarily going to affect dense packing in the sameway observed for dendrimers and is due to the reduced degreeof branching that occurs with HBPs.11 This reduction inbranching results in a more flexible and open structure with lessgeometric constraints. As such, any dense packing is likely tooccur at a higher molecular weight for HBPs in comparison todendrimers. Another important parameter to be considered ispolydispersity. In a polydisperse solution there will be a mix ofpolymers with a range of molecular weights and sizes;inevitably, some of these will be below the dense packinglimit. This is not an issue for dendrimers, whose controlledstepwise synthesis ensures a monodisperse structure. A finalproblem relates to the level of core or catalyst incorporation.This is probably the most significant consideration whencomparing the properties of a hyperbranched polymer and adendrimer, particularly when trying to mimic the generational
effects observed with dendrimers. As a consequence of thecontrolled synthetic procedure used to construct dendrimers, itis trivial to place a catalytic group at the core.12 Specifically, thecore can be used to start or end the synthetic sequence,ensuring total incorporation (after purification). This is not thecase for hyperbranched polymers, where the core is oftendistributed unevenly across the full molecular weight range or isconcentrated in a particular molecular weight fraction.13 Forany study involving core incorporation and isolation, it isessential that the core distribution is controlled. Although it isdesirable for each and every polymer molecule to possess acore, it is not essential. However, it is paramount that the coreis distributed evenly across the polymers molecular weightdistribution, as this allows us to compare a series of HBPs thatdiffer in molecular weight (pseudo-generations).14 In aconventional one-pot hyperbranched polymer synthesis carriedout under kinetic conditions, an even distribution of coremolecule cannot be guaranteed, with core units beingdistributed randomly across the molecular weight range (ornot incorporated at all). Despite this difficulty there are anumber of methods that have been reported for coreincorporation.15 In our case we have used reversible reactionconditions to control core incorporation with respect todistribution and final molecular weight. A reversible reactionis dominated by thermodynamics and will result in a statisticaldistribution of core units across all molecular weights. If theconditions are such that the equilibrium is on the side of theproducts, then in principle a core molecule will be incorporatedin each and every polymer molecule. We approached thisproblem by applying a reversible/equilibrium procedure using atransesterification procedure.We elected to use 3,5-diacetoxybenzoic acid 116 as the AB2
monomer as it generates the same repeat unit used by Suslickand Moore when synthesizing their shape/size selectivecatalytic dendrimer.7 As such, the electronic environmentcreated within our HBP will be very similar to that generated inSuslick’s dendrimer. The AB2 monomer 1 was synthesized from3,5-dihydroxybenzoic acid by reaction with acetic anhydride(Scheme 1).15,16 Tetraacetoxyphenylporphyrin 3 was selected
as the central porphyrin unit as it contains four acetoxy groups,each of which can react with the carboxylic acid group on theAB2 repeat units (via a reversible transesterification process).The porphyrin 3 was synthesized using 4-acetoxybenzaldehyde2, which was obtained after acetylation of 4-hydroxybenzalde-hyde using acetyl chloride and triethylamine (Scheme 2).17
Scheme 1. Synthesis of 3,5-Diacetoxybenzoic Acid
Scheme 2. Synthesis of 4-Acetoxybenzaldehyde
Macromolecules Article
dx.doi.org/10.1021/ma401344d | Macromolecules XXXX, XXX, XXX−XXXB
The porphyrin was then synthesized as shown in Scheme 3,using the methods developed by Rothemund18 and Adler andLongo.19
Having obtained the building blocks, the next step was tosynthesize the porphyrin cored hyperbranched polymer. Aspreviously mentioned, a reversible strategy was used to ensurethe porphyrin core was evenly distributed across all molecularweights. We have previously used this strategy to synthesize a
HBP that possessed a p-nitrophenol core with a 100% level ofincorporation.3 Specifically, 3,5-diacetoxybenzoic acid 1 waspolymerized with porphyrin 3 in a 20:1 molar ratio usingdiphenyl ether as solvent (Scheme 4). The mixture was placedin a round-bottom flask and heated to 225 °C for 45 min. Thetemperature was then lowered to 180 °C and the pressurereduced to 5 mmHg for 4 h. Placing the flask under reducedpressure allows the removal of the acetic acid byproduct,driving the equilibrium in favor of the product. Once thereaction time was complete the crude mixture was dissolved inrefluxing tetrahydrofuran and precipitated into a large excess ofcold methanol to give the porphyrin cored polymer, TAPP-HBP 6 in 65% yield by mass. The presence of porphyrin in thepolymer sample was immediately apparent from the reddish-brown color of the product and was substantiated by 1H NMRresults, which showed small coincident sharp and broadresonances at chemical shifts corresponding to the porphyrinsaromatic protons at 8.89 ppm. This suggested a mixture of“f ree” and incorporated porphyrins with similar evidenceprovided by GPC. In addition to a broad peak for the polymer,a second peak was seen at the low molecular weight end of thechromatogram. This peak was small accounting for 6% of thetotal peak area. GPC analysis using a fixed wavelength UVdetector at 418 nm indicated that this peak was indeed fromfree porphyrin and at the same time demonstrated the presence
Scheme 3. Synthesis of TAPP 3, Zn-TAPP 4, and Fe-TAPP 5
Scheme 4. Synthesis of Porphyrin Cored Hyperbranched TAPP-HBP 6 and Metalated Derivatives
Macromolecules Article
dx.doi.org/10.1021/ma401344d | Macromolecules XXXX, XXX, XXX−XXXC

of porphyrin groups in the polymer itself. Unfortunately, due toinsolubility of the porphyrin in methanol, it was not possible toseparate “free” porphyrin from the polymer throughpurification of the polymer using the established triturationprocedure. Thus, chromatography using an alumina columnwas used to separate the free porphyrin from the rest of thepolymer mixture. Subsequent analysis confirmed removal offree porphyrin. GPC now showed a single broad trace coveringmolecular weights greater than that of the free porphyrin, andthe sharp peaks corresponding to unincorporated porphyrinwere no longer present in the 1H NMR. As well as theporphyrin resonances in the aromatic region, a stronglydeshielded peak at minus 2.03 ppm was also seen for the NHprotons. Along with a visible Soret band in the UV spectra,20
this is a diagnostic characteristic associated with all porphyr-ins.21 The resulting polymer sample possessed a molecularweight of 11 700 and polydispersity of 1.90. Mass spectrometrywas carried out on the bulk polymer sample, although problemswith disproportionate ionization meant that peaks were notobserved above ∼2000 mass units. The resulting spectrum didhowever demonstrate full incorporation of the porphyrin corefor the lower molecular weight fractions. Peaks were observedat 178 mass unit intervals, with each peak corresponding topolymers consisting of n monomer residues and a singleporphyrin core (674 + n178). Peaks from cyclized and“coreless” polymers were not observed. Although massspectrometry provided direct evidence for 100% coreincorporation, it was only true for the low molecular weightfractions (mass spectrometry on the higher molecular weightfractions was inconclusivesee above). To prove 100%incorporation across all molecular weights, the polymer neededto be fractionated into a series of different sizes and the level ofcore incorporation determined for each (using a non-massspectrometric method). Fractionation was also important withrespect to achieving the main aims of this work, specificallyensuring a series of narrowly dispersed “pseudo” dendrimerswhere potential dense packing properties could be observed.The polymer was therefore fractionated using preparative GPCin the form of a column packed with SX-1 Biobeads.22
The extent of core incorporation was quantified bycomparing the polymer’s molecular weight determined usingthe polymer’s bulk property (i.e., GPC) with the molecularweight obtained from a core group analysis (i.e., UV of theporphyrin core). Because of the way these values are calculated,it is unlikely that these two values will be identical. For example,it is well-known23 that GPC calibrated using linear standardsunderestimates molecular weight of a globular hyperbranchedpolymer. As such, the molecular weight cannot be lower thanthat obtained using GPC and can be referred to as Mn
min.15 Onthe other hand, it may be possible that molecular weightsdetermined using a core unit analysis may be overestimated.This is due to the assumption that ALL polymer molecules willpossess a core unit. If this is not the case, then polymerswithout core units will effectively contaminate the mixture andincrease the relative proportion of repeat units relative to coreunits. Therefore, in the case of core analysis the molecularweights obtained represent a maximum possible value and arereferred to as Mn
max.15 The data obtained for all polymerfractions are shown in Table 1. The apparent level ofincorporation for each fraction can be obtained by dividingMn
min by Mnmax; the results are also shown in Table 1. The ratio
of the two molecular weights indicates a level of incorporationaround 60% and is consistent across all fractions, demonstrating
the desired even distribution for the core molecule.Furthermore, having already proven 100% incorporation forthe low molecular weight fractions (via mass spectrometry), itfollows that the bulk polymer sample must also possess a coreincorporation approaching 100%.The two protons meta to the carboxyl function exist in a
number of different environments and resonate as a series ofsignals between 8.03 and 7.83 ppm in the 1H NMR spectrum.There are three well-defined peaks at 7.50, 7.35, and 7.20 ppmcorresponding to the para protons, which are present in threedistinctive environments. These are the dendritic, linear, andterminal repeat units; integrating these peaks and applyingFrechet’s equation returned a value of 50% for the degree ofbranching. After polymerization the porphyrin’s pyrrolicresonance could be seen as a singlet at 8.89 ppm. Althoughonly slightly shifted from the position of the same proton peakin the starting material, a split or unsymmetrical peak patternwas not observed. A similar situation was also observed for theporphyrin’s aromatic peak at 8.24 ppm. As such, there is noclear evidence for unreacted acetoxy groups (on the porphyrin)and confirms that all four of the porphyrin’s acetoxy groupshave reacted with monomer. However, the degree ofpolymerization from each site will vary dramatically, and avariety of structures will exist. If we consider two extremes, theyare: (a) A polymer with four large and roughly equal HBPsections (one from each of the pophyrin’s acetoxy groups).This structure is similar to that of a core functionalizeddendrimer. (b) A polymer with one very large and dominantHBP section (with a degree of polymerization equivalent to 4times that observed for each HBP section in (a) above). Thisstructure is more like that observed for dendrons (Figure 1).
Table 1. Minimum Core Incorporation Calculated fromGPC and UV Analysisa
fraction Mnmin (GPC) Mn
max (UV) minimum core incorporation (%)
bulk 8500 14250 591 3000 5000 602 5500 9500 583 11000 18500 614 15100 24500 62
aMnmin by GPC is a function of the minimum number of polymer
molecules, and Mnmax by UV is a function of the minimum number
porphyrin units. Thus, dividing Mnmin by Mn
max gives the minimumlevel of core incorporation. These are expressed as percentages.
Figure 1. For a single molecular weight there are a number of possiblestructures with respect to polymerization. Two extreme structures areshown above: (left) a tetrafunctionalized polymer possessing fourroughly equivalent HBP sections; (right) a monofunctionalizedpolymers with a large HBP section (di- and trisubstituted structuresas well as a number of other nonsymmetrical structures are alsopossible).
Macromolecules Article
dx.doi.org/10.1021/ma401344d | Macromolecules XXXX, XXX, XXX−XXXD

Nevertheless, due to the same back folding properties observedfor dendrons,24 the porphyrin units will remain encapsulated bythe polymer matrix, and a very similar environment will existfor all possible structures (of similar molecular weight).Although it was not possible to control the molecular weight
precisely via the core/monomer ratio, it was possible toinfluence the molecular weight by altering this ratio in favor ofwhatever product was desired. As described above, when usinga 20:1 ratio of AB2 monomer relative to the porphyrin core apolymer with an Mn of approximately 8500 was obtained. If theratio was doubled (to 40:1), then a polymer with roughlydouble the molecular weight was obtained (Mn of around 18500). While this ratio could be used to favor a certainapproximate molecular weight, the temperature, reaction time,and the quality of vacuum were paramount. For example, if thevacuum was poor, a relatively small polymer would beproduced regardless of the monomer/core ratio. This wasexemplified when a polymer with a very low Mn value of 2000(and a PD of around 4.0) was obtained when using a 20:1 ratio.This was much smaller than polymers obtained from similarexperiments (using the same 20:1 ratio) and was probably dueto a poor vacuum resulting in inefficient removal of the aceticacid byproduct. However, the product was returned to areaction flask containing solvent (diphenyl ether), and themixture was heated back up to 180 °C and a vacuum of 5mmHg applied. When the product was analyzed, an Mn valuesimilar to that recorded from previous experiments wasobtained (around 9000). These experiments confirm thereversible nature of the reaction and how this can be used togenerate hyperbranched polymers that possess specific andfunctional units with very high levels of incorporation. Asdescribed above, this is a key requirement when comparing theproperties of hyperbranched polymers with those ofdendrimers.Probing Dense Packing of the Zn-TAPP-HBP through
Ligand Binding Studies. After confirming the reversibilityand high/even levels of core incorporation of the hyper-branched polymer system, investigations were conducted todetermine any site isolation or dense packing properties of theresulting polymers. Specifically we wanted to determinewhether there was a sudden break in binding properties,similar to that observed for dendrimers at their dense packed ordense shell limit,3 or a linear or nonspecific relationshipbetween binding and molecular weight, as is the case for simplelinear polymers.25 The presence of a porphyrin at the centerallows for direct monitoring of access to the core through UVbinding experiments. This can be achieved using zincmetalloporphyrin cored polymers and studying the binding ofvarious nitrogen ligands, enabling any trends to be identifiedrelative to ligand size. In addition, analysis of binding affinity toa particular polymer fraction with respect to the different sizedligands would indicate whether the hyperbranched system wascapable of excluding larger ligands while allowing access tosmaller substratesa requirement for future size selectivecatalysis experiments. However, the first requirement was toobtain a pseudo-generational series of hyperbranched polymerspossessing a zinc metalloporphyrin core. One approach wouldbe to carry out a number of polymerizations using an increasingmonomer to core ratio. Alternatively, a bulk metalloporphyrinpolymer could be synthesized and then fractionated using thechromatographic method described above and separate thepolymer into the required pseudo-generational series ofhyperbranched polymers. We opted for this second method
as the first was not completely reliable or reproducible(extremely dependent on vacuum and temperature); fractiona-tion would also reduce the polydispersity. Therefore, the bulkhyperbranched polymer was resynthesized as described aboveusing a 40:1 molar ratio of AB2 monomer 1 and the porphyrinunit 3. Zinc was inserted into the porphryin core using excesszinc acetate at room temperature in DCM. Purification wasachieved by filtration to remove unreacted zinc acetate followedby reducing the volume of solvent by rotary evaporation beforetrituration into excess methanol to yield the zinc insertedhyperbranched polymer Zn-TAPP-HBP 7 in an overall yield of62% by mass (Scheme 4). Confirmation of successful insertionwas provided by 1H NMR, in which the highly shielded peak atminus 2.03 ppm was absent; this corresponds to the innerprotons that are removed upon metal insertion. Furtherconfirmation of successful functionalization was provided byUV/vis spectrometry in which the four Q-bands of the startingmaterial at 515, 548, 592, and 648 nm were replaced by tworesonances at 548 and 589 nm. For control experiments zincwas also inserted into the free base porphyrin 3 yielding thezinc porphyrin Zn-TAPP 4, using a similar procedure (Scheme3).Prior to performing the UV/vis titrations, it was necessary to
fractionate and purify the zinc functionalized polymer.Preparative size exclusion chromatography was employed inthe form of a glass column (3 cm diameter) using SX-1Biobeads and DCM as the eluent. This process allowed (500mg of) the polydisperse bulk hyperbranched polymer to beseparated into more discrete molecular weights with lowerpolydispersities, and the data are shown in Table 2.
It is clear from the table that early generations are very closein molecular weight; this placed a limit on the fractionationprocedure. The narrow molecular weight divergence at lowgenerations is irrelevant for quantized dendrimers but doescreate a difficulty for nonquantized poly dispersed hyper-branched polymers. By taking the molecular weight andestimating the number of repeat units for each fraction, wewere able to obtain a series of pseudo-generations. Thesevalues, along with those corresponding to the equivalentdendrimers, are shown in Table 2. Inevitably, overlap exists forsome of the lower molecular weight fractions due to the similarmolecular weight values moving from one generation toanother (this is less of a problem for higher generations as
Table 2. Pseudo-Generational Series of the Zn-TAPP-HBP7a
theoretical values forequivalent dendrimer
experimentally determined values forfractionated Zn-TAPP-HBP-5
MWrepeatunits generation fraction
MW(GPC-Mn)
repeatunits
pseudo-generation
1450 4 1.0 1 1650 5 1.02874 12 2.0 2 2150 8 1.55722 28 3.0 3 5900 29 3.011418 60 4.0 4 8750 45 3.523522 128 5.0 5 11500 62 4.0
6 13900 75 4.57 16800 91 5.0
aHalf-generations were assigned to hyperbranched polymers when thenumber of terminal groups and the molecular weights fell betweenthose for the full generations. Values for the equivalent dendrimer arealso shown for comparison.
Macromolecules Article
dx.doi.org/10.1021/ma401344d | Macromolecules XXXX, XXX, XXX−XXXE
the divergence of molecular weights becomes more markedbetween each generation). Consequently, a judgment was madeas to which pseudo-generation each fraction belonged. Thisdictated that some fractions are not included in the data set,and fractions with ambiguous molecular weights were not used;only fractions with molecular weights corresponding closely totheir pseudo-generation are included. Half-generations wereassigned to hyperbranched polymers when the number ofrepeat units and the molecular weights were between thetheoretical full generations (G1.5, G3.5, and G4.5).Once the zinc inserted polymer had been fractionated and
characterized, the next step was to probe whether or not thispolymeric system was capable of dense packing, core isolation,and size selectivity. Therefore, binding experiments werecarried out in the form of UV/vis titrations using pyridineand other pyridine derivatives.26 Binding experiment would becarried out using the fractionated hyperbranched polymers anda series of ligands with different molecular volumes or size. Ifdense packing was observed at different molecular weights forthis series of ligands, this could indicate a selective capability ofthese hyperbranched polymers with respect to size. The specificpyridyl ligands used must differ in size and must be free ofgroups in the ortho positions which may block the interaction.Based on these criteria, the following ligands were chosen:pyridine, 3,5-lutidine, and 3-phenylpyridine. These are shownalong with their space-filling models in Figure 2. The titrations
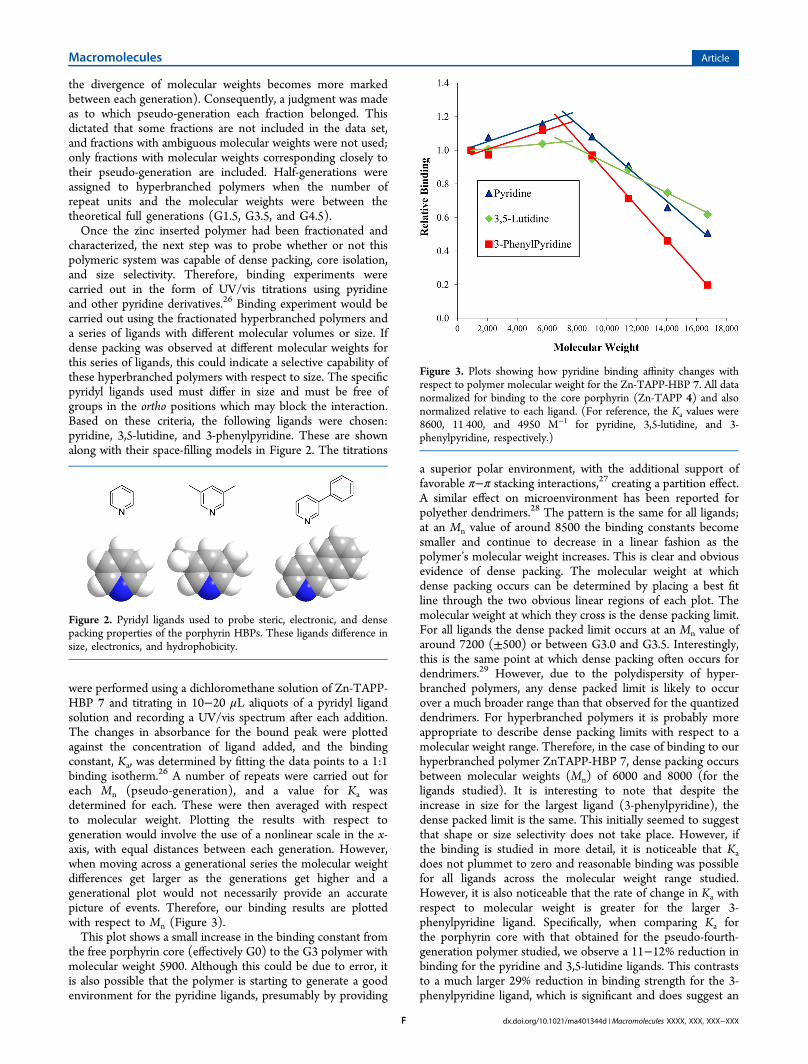
were performed using a dichloromethane solution of Zn-TAPP-HBP 7 and titrating in 10−20 μL aliquots of a pyridyl ligandsolution and recording a UV/vis spectrum after each addition.The changes in absorbance for the bound peak were plottedagainst the concentration of ligand added, and the bindingconstant, Ka, was determined by fitting the data points to a 1:1binding isotherm.26 A number of repeats were carried out foreach Mn (pseudo-generation), and a value for Ka wasdetermined for each. These were then averaged with respectto molecular weight. Plotting the results with respect togeneration would involve the use of a nonlinear scale in the x-axis, with equal distances between each generation. However,when moving across a generational series the molecular weightdifferences get larger as the generations get higher and agenerational plot would not necessarily provide an accuratepicture of events. Therefore, our binding results are plottedwith respect to Mn (Figure 3).This plot shows a small increase in the binding constant from
the free porphyrin core (effectively G0) to the G3 polymer withmolecular weight 5900. Although this could be due to error, itis also possible that the polymer is starting to generate a goodenvironment for the pyridine ligands, presumably by providing
a superior polar environment, with the additional support offavorable π−π stacking interactions,27 creating a partition effect.A similar effect on microenvironment has been reported forpolyether dendrimers.28 The pattern is the same for all ligands;at an Mn value of around 8500 the binding constants becomesmaller and continue to decrease in a linear fashion as thepolymer’s molecular weight increases. This is clear and obviousevidence of dense packing. The molecular weight at whichdense packing occurs can be determined by placing a best fitline through the two obvious linear regions of each plot. Themolecular weight at which they cross is the dense packing limit.For all ligands the dense packed limit occurs at an Mn value ofaround 7200 (±500) or between G3.0 and G3.5. Interestingly,this is the same point at which dense packing often occurs fordendrimers.29 However, due to the polydispersity of hyper-branched polymers, any dense packed limit is likely to occurover a much broader range than that observed for the quantizeddendrimers. For hyperbranched polymers it is probably moreappropriate to describe dense packing limits with respect to amolecular weight range. Therefore, in the case of binding to ourhyperbranched polymer ZnTAPP-HBP 7, dense packing occursbetween molecular weights (Mn) of 6000 and 8000 (for theligands studied). It is interesting to note that despite theincrease in size for the largest ligand (3-phenylpyridine), thedense packed limit is the same. This initially seemed to suggestthat shape or size selectivity does not take place. However, ifthe binding is studied in more detail, it is noticeable that Kadoes not plummet to zero and reasonable binding was possiblefor all ligands across the molecular weight range studied.However, it is also noticeable that the rate of change in Ka withrespect to molecular weight is greater for the larger 3-phenylpyridine ligand. Specifically, when comparing Ka forthe porphyrin core with that obtained for the pseudo-fourth-generation polymer studied, we observe a 11−12% reduction inbinding for the pyridine and 3,5-lutidine ligands. This contraststo a much larger 29% reduction in binding strength for the 3-phenylpyridine ligand, which is significant and does suggest an
Figure 2. Pyridyl ligands used to probe steric, electronic, and densepacking properties of the porphyrin HBPs. These ligands difference insize, electronics, and hydrophobicity.
Figure 3. Plots showing how pyridine binding affinity changes withrespect to polymer molecular weight for the Zn-TAPP-HBP 7. All datanormalized for binding to the core porphyrin (Zn-TAPP 4) and alsonormalized relative to each ligand. (For reference, the Ka values were8600, 11 400, and 4950 M−1 for pyridine, 3,5-lutidine, and 3-phenylpyridine, respectively.)
Macromolecules Article
dx.doi.org/10.1021/ma401344d | Macromolecules XXXX, XXX, XXX−XXXF

element of shape and/or size selectivity with respect to ligandsize and the polymer’s molecular weight; the data aresummarized in Table 3.Probing Shape and Size Selectivity of the Fe-TAPP-
HBP via Catalysis. The binding data presented aboveconfirmed the porphyrin cored hyperbranched polymerspossess a dense packed limit at a molecular weight around8000. The data also suggest the polymers were capable of sizeselective binding. That is, at a molecular weight below 8000 arelatively open structure persists, and this allows unhinderedaccess to the core, whereas at higher molecular weights (above8000) the polymer possesses a tighter more compact structureand access to the core is restricted. This effect is morepronounced for the bigger polymers and larger ligands.Therefore, assuming all other things are equal, it should bepossible for smaller substrates to access the core in preferenceto larger substrates. We now wish to exploit this effect andstudy the possibility of size selective catalysis. Suslick andMoore demonstrated the selective capability of similardendrimers using a mixture of linear and cyclic octenes.19 Inan attempt to make a valid comparison between dendrimersand hyperbranched polymers, we decided to study the samecatalytic reaction (Scheme 5), that is, the catalytic epoxidationof a linear alkene 9 and a cyclic alkene 11, in isolation andtogether in equal ratios.
The specific substrates selected were cyclooctene, which is arelatively large substrate, and 1-octene, which is a small (thin)linear alkene. The linear substrate has a terminal alkene, whichis relatively electron poor and therefore much less reactive thanthe cyclic alkene, which is more electrophilic. This increase isdue to the higher level of substitution and increased sigmadonation, leading to a more electron-rich alkene compared tothe equivalent linear system. This is an ideal scenario whentrying to study the effect of size on reactivity, specifically whentrying to encourage the reaction between a catalyst and a smallbut unreactive substrate, in the presence of a more reactive butlarger substrate. To test this, reactions were carried out on bothsubstrates using catalytically functionalized hyperbranchedpolymers below and above their dense packed limit. If a size/shape selective reaction is possible, we would expect anyreaction using the smaller (but unreactive) linear alkene
substrate 9 to occur equally well with a catalyticallyfunctionalized polymer above or below its dense packed limit.Conversely, we might predict that the larger cyclic alkenesubstrate 11 would struggle to reach the catalytic core of ahyperbranched polymer whose molecular weight was above thedense packed limit, resulting in a slower reaction and a loweryield. We proposed to study these reactions using the samemethod reported by Suslick and Moore,7 specifically usingiodosylbenzene as the oxygen source and a metalloporphyrin asthe catalyst. Although zinc porphyrins were used to study andprobe binding, they are not suitable as oxidation catalysts.There are a number of suitable metals that can be inserted intoa porphyrin;30 in this case iron (Fe(III)) was used as theproducts are robust and make good oxidation catalysts.31 Wetherefore set about synthesizing Fe(III) metalloporphyrin32
cored hyperbranched polymers above and below the densepacked limit as well as aiming for a polymer with a molecularweight close to the dense packed limit. For control andreference experiments we would also required the simpleFe(III) metalloporphyrin 5, which was synthesized as shown inScheme 3.Insertion of iron into free base porphyrin 3 was achieved by
reaction with a large excess of iron(II) chloride in the presenceof 2,6-lutidine.33 After refluxing for 4 h under nitrogen, themixture was exposed to air as it cooled, oxidizing the Fe(II) toFe(III). After work-up and column chromatography, the massspectra showed a peak corresponding to the target molecule,but also a series of peaks above the molecular ion. This reactionwas repeated a number of times, but on every occasion anidentical mass spectrum was obtained. It was assumed the extrapeaks were due to an unknown species strongly ligated to themetal center. Therefore, the porphyrin was dissolved indichloromethane and washed with 1 M hydrochloric acid.The organic phase was collected, the solvent removed and theproduct reanalyzed. On this occasion the EI mass spectrumcontained a single dominant peak of the molecular ion and nopeaks of a higher value. Further authentication for the productFe-TAPP 5 was provided by UV/vis analysis, which showed areduction in the number of Q-bands from four to two, adiagnostic change for a metal inserted porphyrin. Because ofthe paramagnetic iron, substantial broadening of the peaksoccurred in the 1H NMR spectrum.34 With the porphyrin itselfmetalated, it was also necessary to insert iron into theporphyrin cored hyperbranched polymer. The polymerizationwas repeated using the unmetalated porphyrin and metalinsertion performed after purification (Scheme 4). Thesynthesis was performed in a similar manner to that describedabove for the free porphyrin, using iron(II) chloride and 2,6-lutidine; however, after refluxing TAPP-HBP 6 for 4 h thesolution was filtered through a short plug of silica to removeunreacted iron chloride, washed with dilute HCl, andprecipitated into excess methanol to give Fe-TAPP-HBP 8 ingood yield. GPC analysis carried out after the acid washreturned an Mn value of 12 100, which was almost identical to
Table 3. Binding Data of Pyridyl Ligands to Zn-TAPP-HBP 7
ligand Ka porphyrin core (M−1) Ka of HBP G4 (Mn ∼ 11 500) ( M−1) onset of dense packing (Mn) reduction in bindinga (%)
pyridine 8600 (±400) 4700 (±200) 7200 (±500) 11 (±5)3,5-lutidine 11400 (±850) 6900 (±500) 7100 (±500) 12 (±7.5)3-phenylpyridine 4950 (±350) 1000 (±75) 7200 (±500) 29 (±3)
aCalculated by comparing the Ka values obtained from the porphyrin core unit (Zn-TAPP 4) and the pseudo-fourth-generation Zn-TAPP-HBP 7(Mn 11 500).
Scheme 5. Iron Porphyrin Catalysed Oxidation of Small/Thin Alkene 9 and a Larger/Fat Alkene 11
Macromolecules Article
dx.doi.org/10.1021/ma401344d | Macromolecules XXXX, XXX, XXX−XXXG

the Mn of TAPP-HBP 6 (11 700), confirming that the structureremained intact during metalation. As before, UV/vis analysisshowed that the number of Q-bands had been reduced fromfour to two, confirming iron insertion. Because of the size of thepolymer, which effectively shields the central iron, linebroadening in the 1H NMR was significantly reduced, andsome structural information remainedthe most important ofwhich was the disappearance of the free base porphyrin NHpeak at −2.03 ppm, confirming metal insertion.The bulk Fe-TAPP-HBP 8 was fractionated using a Biobead
column, and the molecular weights of each fraction wereanalyzed by GPC. Fractions were required with Mn valuesabove and below the dense packed limit, as well as one with anMn reasonably close to the dense packed limit (previouslyshown to be somewhere between the G3.0 and G3.5 pseudogeneration). Table 4 below shows the fractions selected for the
catalytic experiments along with data regarding the number ofrepeat units and the corresponding pseudo generation. Thecore unit would be used as reference and control catalyst duringthe future epoxidation and catalytic experiments and is alsoincluded in the table. Iodosylbenzene 13, was used as theoxygen source for the catalytic reactions and was synthesized asshown in Scheme 6. The synthesis is trivial and iodosylbenzene
was prepared by the stirring iodobenzene diacetate in 3 Msodium hydroxide for 45 min.35 The solid obtained wascollected and thoroughly washed with water followed bychloroform, if left to stand for a few days the product candecompose (and is particularly sensitive to prolonged exposureto light). However, as the product was easy to synthesis andcharacterize, a fresh batch was prepared each time a series ofcatalytic and epoxidation reactions were performed (a controlreaction was always carried out to test the activity of the oxygensource).
Now in a position to study the catalytic properties of thehyperbranched polymers, the individual oxidation of eachalkene with each polymer fraction (or porphyrin core) wasstudied. The reaction for the linear alkene 9 is shown inScheme 7. It is important to ensure that all catalyst/porphyrinconcentrations are equivalent when studying a series ofpolymers. Therefore, the concentration of all solutions withrespect to the amount of porphyrin was checked by UVspectroscopy and adjusted if necessary, to ensure equivalentporphyrin concentrations for all polymer fractions (seeExperimental section in the Supporting Information). As wellas the dominant epoxide products, a number of other oxidationproducts can also form, either directly or after initial formationof the epoxide. Therefore, the reactions were monitored byfollowing the reduction product, that is, the conversion ofiodosyl benzene to iodobenzene (which is a measure of alloxidation reactions catalyzed by the porphyrin).36 A secondseries of experiments would study the product yields when eachpolymer (and core) was reacted with an equimolar mixture ofboth alkenes. This set of experiments would indicate whether ornot the hyperbranched polymers could influence a shape/sizeselective reaction. Gas chromatography was used to quantifythe outcome of the catalyzed oxidation reactions by monitoringthe epoxide and the iodobenzene; however, it was firstnecessary to calibrate the instrument using the alkene startingmaterials, the epoxide products, the iodobenzene side product,and an internal standard (n-dodecane). An initial blankexperiment indicated that iodosylbenzene reacted with theporphyrin in the absence of alkene (producing iodobenzene).However, it has been shown that if a large excess of alkene isused, then this side reaction is suppressed (effectively saturatingthe rate of alkene oxidation).37 A second blank experiment wascarried out to ascertain if the alkenes could react with theoxygen source in the absence of porphyrin catalyst. The GCresults showed that although oxidation took place, the yieldswere extremely low and not at a level that would effect theresults from the catalytic experiments.With the instrument calibrated and the blank experiments
performed, investigation into the activity of the porphyrincatalyst could begin. The yield of oxidation products whenusing just the core porphyrin unit (i.e., pseudo-generation G =0) were 74% and 96% for the linear and cyclic alkenes,respectively.38 This indicates that the cyclic alkene is morereactive, which is consistent with previous results and theinherent differences in reactivity between a mono- anddisubstituted alkene.39 To make comparisons easier, the yieldsof the linear and cyclic species are normalized relative to thevalue obtained using the reference porphyrin (Fe-TAPP 5, i.e.,the G = 0 pseudo-generation) and plotted against pseudo-generation (Figure 4).40
The results for the linear alkene show that the activity of G2,G3, and G4 remained similar to that of the free porphyrin G0(within the 10% error). However, for the cyclic alkene, thereappears to be a steady decrease in yield and therefore activity.
Table 4. Theoretical Values for Pseudo-Generations vs theValues for Obtained Fractions of Fe-TAPP-HBP 8
molecular weight repeat unitsa
pseudo-generation theoretical obtained theoretical obtained
0 (Fe-TAPP) 901 901 0 02 3038 3200 12 133 5888 5400 28 254 11588 12100 60 63
aRounded to the nearest integer.
Scheme 6. Synthesis of Iodosylbenzene 13
Scheme 7. Oxidation of Alkene 9 to the Epoxide 10 and Other Oxidation Products
Macromolecules Article
dx.doi.org/10.1021/ma401344d | Macromolecules XXXX, XXX, XXX−XXXH
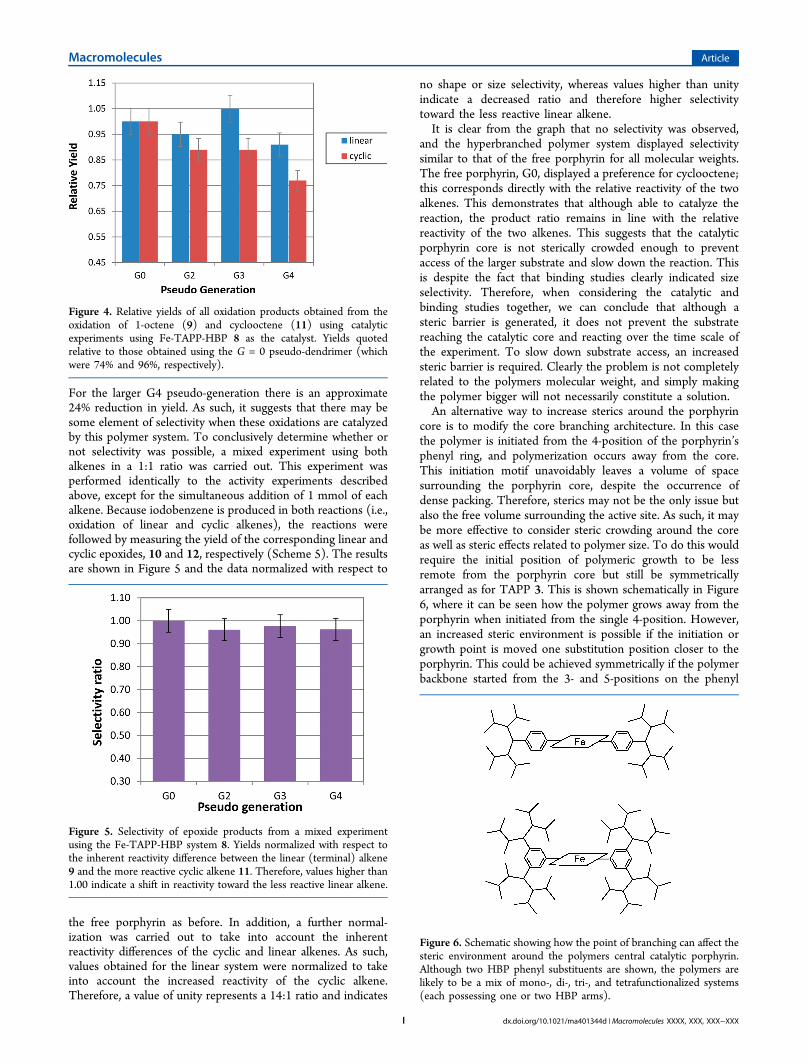
For the larger G4 pseudo-generation there is an approximate24% reduction in yield. As such, it suggests that there may besome element of selectivity when these oxidations are catalyzedby this polymer system. To conclusively determine whether ornot selectivity was possible, a mixed experiment using bothalkenes in a 1:1 ratio was carried out. This experiment wasperformed identically to the activity experiments describedabove, except for the simultaneous addition of 1 mmol of eachalkene. Because iodobenzene is produced in both reactions (i.e.,oxidation of linear and cyclic alkenes), the reactions werefollowed by measuring the yield of the corresponding linear andcyclic epoxides, 10 and 12, respectively (Scheme 5). The resultsare shown in Figure 5 and the data normalized with respect to
the free porphyrin as before. In addition, a further normal-ization was carried out to take into account the inherentreactivity differences of the cyclic and linear alkenes. As such,values obtained for the linear system were normalized to takeinto account the increased reactivity of the cyclic alkene.Therefore, a value of unity represents a 14:1 ratio and indicates
no shape or size selectivity, whereas values higher than unityindicate a decreased ratio and therefore higher selectivitytoward the less reactive linear alkene.It is clear from the graph that no selectivity was observed,
and the hyperbranched polymer system displayed selectivitysimilar to that of the free porphyrin for all molecular weights.The free porphyrin, G0, displayed a preference for cyclooctene;this corresponds directly with the relative reactivity of the twoalkenes. This demonstrates that although able to catalyze thereaction, the product ratio remains in line with the relativereactivity of the two alkenes. This suggests that the catalyticporphyrin core is not sterically crowded enough to preventaccess of the larger substrate and slow down the reaction. Thisis despite the fact that binding studies clearly indicated sizeselectivity. Therefore, when considering the catalytic andbinding studies together, we can conclude that although asteric barrier is generated, it does not prevent the substratereaching the catalytic core and reacting over the time scale ofthe experiment. To slow down substrate access, an increasedsteric barrier is required. Clearly the problem is not completelyrelated to the polymers molecular weight, and simply makingthe polymer bigger will not necessarily constitute a solution.An alternative way to increase sterics around the porphyrin
core is to modify the core branching architecture. In this casethe polymer is initiated from the 4-position of the porphyrin’sphenyl ring, and polymerization occurs away from the core.This initiation motif unavoidably leaves a volume of spacesurrounding the porphyrin core, despite the occurrence ofdense packing. Therefore, sterics may not be the only issue butalso the free volume surrounding the active site. As such, it maybe more effective to consider steric crowding around the coreas well as steric effects related to polymer size. To do this wouldrequire the initial position of polymeric growth to be lessremote from the porphyrin core but still be symmetricallyarranged as for TAPP 3. This is shown schematically in Figure6, where it can be seen how the polymer grows away from theporphyrin when initiated from the single 4-position. However,an increased steric environment is possible if the initiation orgrowth point is moved one substitution position closer to theporphyrin. This could be achieved symmetrically if the polymerbackbone started from the 3- and 5-positions on the phenyl
Figure 4. Relative yields of all oxidation products obtained from theoxidation of 1-octene (9) and cyclooctene (11) using catalyticexperiments using Fe-TAPP-HBP 8 as the catalyst. Yields quotedrelative to those obtained using the G = 0 pseudo-dendrimer (whichwere 74% and 96%, respectively).
Figure 5. Selectivity of epoxide products from a mixed experimentusing the Fe-TAPP-HBP system 8. Yields normalized with respect tothe inherent reactivity difference between the linear (terminal) alkene9 and the more reactive cyclic alkene 11. Therefore, values higher than1.00 indicate a shift in reactivity toward the less reactive linear alkene.
Figure 6. Schematic showing how the point of branching can affect thesteric environment around the polymers central catalytic porphyrin.Although two HBP phenyl substituents are shown, the polymers arelikely to be a mix of mono-, di-, tri-, and tetrafunctionalized systems(each possessing one or two HBP arms).
Macromolecules Article
dx.doi.org/10.1021/ma401344d | Macromolecules XXXX, XXX, XXX−XXXI

ring. In fact, this is exactly the same substitution pattern used bySuslick and Moore on their work on porphyrin coreddendrimers.7 Thus, it was postulated that this alternativeporphyrin core may be capable of providing the correct stericenvironment required for selectivity.Synthesis of Tetra(3,5-diacetoxyphenyl)porphyrin
Cored Hyperbranched Polymers. The more substitutedtarget porphyrin 16 could in principle be synthesized in amanner analogous to that used to construct the simple TAPPcore 3. Unfortunately the required 3,5-diacetoxybenzaldehydewas not easily available, and its acetylated precursor wasprohibitively expensive. However, 3,5-dimethoxybenzaldehydecould be used to assemble tetra-3,5-dimethoxyphenylporphyrin14, which could in turn be demethylated and finally acetylatedto give the target porphyrin, tetra-3,5-diacetoxyphenylporphyr-in, tetra-(3,5-DAPP) 16 (Scheme 8). Tetra(3,5-dimethoxyphenyl)porphyrin 14 was synthesized via the sameRothemund and Adler−Longo procedure used previously;18
equal molar quantities of 3,5-dimethoxybenzaldehyde andpyrrole were refluxed for 30 min and after work-up andpurification gave 3,5-dimethoxyphenylporphyrin 14 in 13%yield. The UV/vis absorption spectrum confirmed the presenceof the porphyrin displaying the characteristic Soret band at420.5 nm and four Q-bands at 514, 548, 588, and 648.5 nm.The 1H NMR spectrum contained peaks corresponding to theβ-pyrrolic protons at 8.96 ppm. The ortho and para aromaticprotons resonated at 7.43 and 6.93 ppm, respectively, and alarge singlet at 3.99 ppm was assigned to the methoxy protons.Finally, a peak corresponding to the highly shielded inner NHhydrogens could be seen at minus 2.81 ppm. The second stagewas to demethylate the octamethoxy-functionalized porphyrin;
this was achieved using a standard demethylation procedure.39
Specifically, porphyrin 14 was reacted with an excess of borontribromide, followed by water. The product obtained afterpurification showed that the methoxy peak at 55.6 ppm in 13CNMR was no longer present. Removal of the methoxy groupwas confirmed by 1H NMR, which showed that thecorresponding methoxy signal was absent. In addition, a largeOH peak was observed at 3425 cm−1 in the IR spectrum. Thisprocedure produced the desired tetra(3,5-dihydroxyphenyl)-porphyrin 15 in high purity and a yield approaching 90%. Thefinal synthetic step was acetylation; this was carried by refluxingporphyrin 15 in acetic anhydride for 6 h. Excess aceticanhydride and acetic acid byproduct were removed via vacuumdistillation, and the target tetra(3,5-diacetoxyphenyl)porphyrin16 was obtained in 64% yield after purification. 1H NMRanalysis showed a large singlet corresponding to threehydrogens from on the newly introduced acetoxy group at2.42 ppm. Mass spectrometry gave a molecular ion peak at1079 which corresponds to that required for the targetporphyrin.The next step was to repeat the polymerization using this
alternate porphyrin core. The reaction is shown in Scheme 9and was carried out using the same synthetic and purificationmethods described for TAPP-HBP 6. After precipitation fromtetrahydrofuran and methanol, followed by purification usingan alumina column, tetra(3,5-diacetoxyphenyl)porphyrin coredhyperbranched polymer 19 (tetra-(3,5-DAPP-HBP)) wasrecovered in good yield as a red/brown solid. SEC analysiswas carried out using RI and UV detection, with each traceshowing a single peak that was directly super imposable on theother. This confirms that the porphyrin was incorporated
Scheme 8. Synthesis of Tetra(3,5-diacetoxyphenyl)porphyrin 16 and Its Metalated Derivatives 17 and 18
Macromolecules Article
dx.doi.org/10.1021/ma401344d | Macromolecules XXXX, XXX, XXX−XXXJ

evenly across all molecular weights.15 Analysis returned an Mn
value of 8100 and a polydispersity of 2.64. 1H NMR confirmedporphyrin incorporation, showing the characteristic deshieldedpeak from the NH protons at minus 2.03 ppm. As with the 4-substituted HBP system, both SEC and NMR indicated verysmall peaks corresponding to unincorporated porphyrin(equivalent to just 2% of the total product and is completelygone after the fractionation step). Mass spectrometry showed aseries of peaks separated by 178 mass units, each correspondingto n monomer residues and a single porphyrin core (744 +n178). Peaks from cyclized and “coreless” polymers were notobserved.Probing Dense Packing of Zn-3,5-DAPP-HBP through
Ligand Binding Studies. To probe binding first required zincinsertion into the synthesized porphryin 16. This was carriedout using zinc acetate dihydrate as shown in Scheme 8. The
reaction was complete after 30 min, after which unreacted zincacetate dihydrate was removed by filtration, followed by flashchromatography to give Zn-tetra-(3,5-DAPP) 17 in 68% yield.Structural confirmation of Zn-tetra-(3,5-DAPP) 17 was
provided by 1H NMR, in which the highly shielded peak atminus 2.94 ppm was no longer present and UV/vis analysis,which showed the number of Q-bands had been reduced fromfour to two. Mass spectrometry showed only a single peak ofthe molecular ion with M/z of 1141. Metalation of tetra-(3,5-DAPP)-HBP 19 to give Zn-tetra-(3,5-DAPP)-HBP 20 is shownin Scheme 9 and was performed in an identical manner to thatdescribed above for the porphyrin core 16, with the exceptionthat filtration and trituration (from THF into methanol) wasused in place of chromatography for purification. UV/visspectroscopy showed a strong Soret band at 421.5 nm and thatthe number of Q-bands had been reduced from four to two.
Scheme 9. Synthesis of 3,5-(Diacetoxyphenyl)porphyrin Cored Hyperbranched Polymer Tetra-(3,5-DAPP)-HBP 19 (left) and aRepresentative Hyperbranched Wedge (right)
Table 5. Mn, Polydispersity, and Number of Repeat Units for Hyperbranched Polymer Zn-3,5-DAPP-HBP 20
Mn polydispersity no. of repeat units Mn polydispersity no. of repeat units
1142 1.00 0 (core unit) 8000 2.46 403100 1.81 13 9000 2.33 463700 1.82 16 10500 2.14 555000 1.72 24 11800 1.99 616100 3.12 30 12500 1.87 666800 2.74 34
Macromolecules Article
dx.doi.org/10.1021/ma401344d | Macromolecules XXXX, XXX, XXX−XXXK
SEC analysis confirmed the hyperbranched polymer remainedintact after the metalation reaction. The polymer was thenfractionated using size exclusion chromatography in the form ofa Biobead column; the results are shown in Table 5. Asdescribed above for the TAPP-HBP system, UV analysis on thefractions confirmed that the core was evenly distributed acrossall molecular weights.15 On this occasion, smaller fractions wereobtained, enabling more data points to be collected. This alsomeant that molecular weight differences between fractionswould be smaller. This allowed a more accurate analysis ofbinding data and effects, particularly the onset of dense packing.As the differences between fractions was smaller, there was alarge number of fractions between each pseudo-generation, andthere was no longer any value in assigning pseudo-generationsto the fractions (i.e., between pseudo-generation 3.0 and 4.0there are five fractions).As before, dense packing was probed by studying the binding
of various pyridyl ligands to the fractions of Zn-tetra-(3,5-DAPP)-HBP 20 (as well as to the core unit Zn-tetra-(3,5,DAPP) 17). The same three ligands were used, and toaid comparison, the data for all ligands are plotted on a singlegraph. The data have been normalized relative to the bindingconstants obtained for the porphyrin core as well as beingnormalized relative to each ligand (Ka values for each ligand aredifferent); the plots are shown in Figure 7.
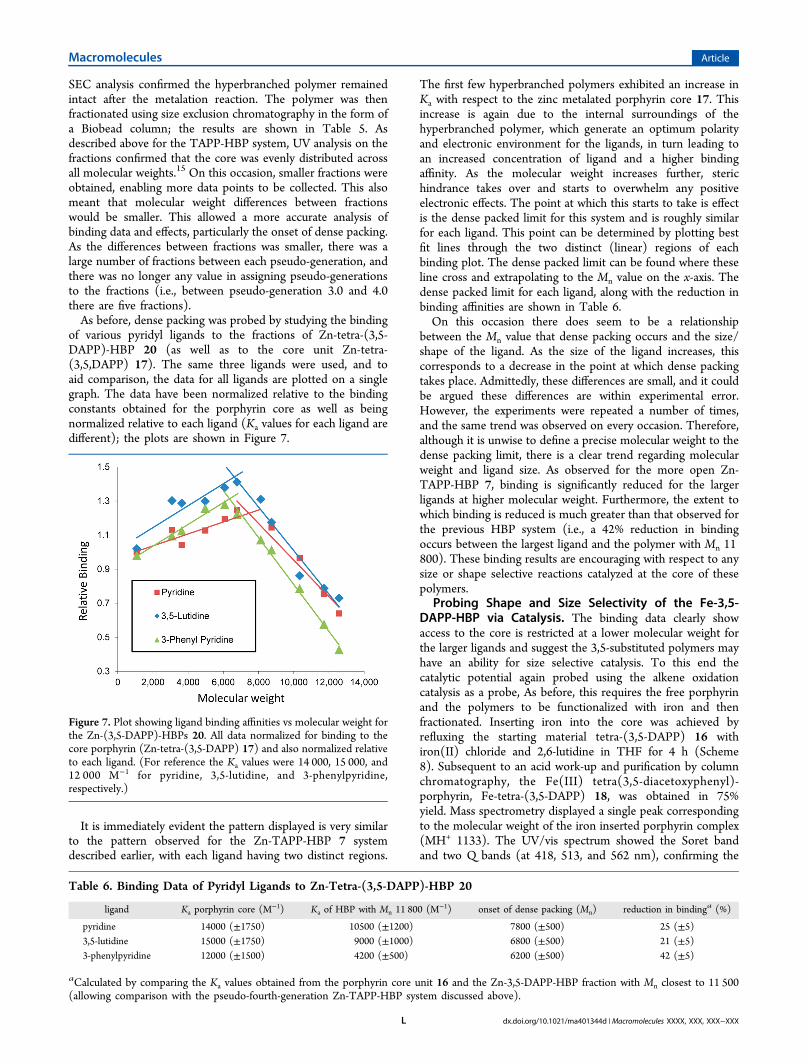
It is immediately evident the pattern displayed is very similarto the pattern observed for the Zn-TAPP-HBP 7 systemdescribed earlier, with each ligand having two distinct regions.
The first few hyperbranched polymers exhibited an increase inKa with respect to the zinc metalated porphyrin core 17. Thisincrease is again due to the internal surroundings of thehyperbranched polymer, which generate an optimum polarityand electronic environment for the ligands, in turn leading toan increased concentration of ligand and a higher bindingaffinity. As the molecular weight increases further, sterichindrance takes over and starts to overwhelm any positiveelectronic effects. The point at which this starts to take is effectis the dense packed limit for this system and is roughly similarfor each ligand. This point can be determined by plotting bestfit lines through the two distinct (linear) regions of eachbinding plot. The dense packed limit can be found where theseline cross and extrapolating to the Mn value on the x-axis. Thedense packed limit for each ligand, along with the reduction inbinding affinities are shown in Table 6.On this occasion there does seem to be a relationship
between the Mn value that dense packing occurs and the size/shape of the ligand. As the size of the ligand increases, thiscorresponds to a decrease in the point at which dense packingtakes place. Admittedly, these differences are small, and it couldbe argued these differences are within experimental error.However, the experiments were repeated a number of times,and the same trend was observed on every occasion. Therefore,although it is unwise to define a precise molecular weight to thedense packing limit, there is a clear trend regarding molecularweight and ligand size. As observed for the more open Zn-TAPP-HBP 7, binding is significantly reduced for the largerligands at higher molecular weight. Furthermore, the extent towhich binding is reduced is much greater than that observed forthe previous HBP system (i.e., a 42% reduction in bindingoccurs between the largest ligand and the polymer with Mn 11800). These binding results are encouraging with respect to anysize or shape selective reactions catalyzed at the core of thesepolymers.
Probing Shape and Size Selectivity of the Fe-3,5-DAPP-HBP via Catalysis. The binding data clearly showaccess to the core is restricted at a lower molecular weight forthe larger ligands and suggest the 3,5-substituted polymers mayhave an ability for size selective catalysis. To this end thecatalytic potential again probed using the alkene oxidationcatalysis as a probe, As before, this requires the free porphyrinand the polymers to be functionalized with iron and thenfractionated. Inserting iron into the core was achieved byrefluxing the starting material tetra-(3,5-DAPP) 16 withiron(II) chloride and 2,6-lutidine in THF for 4 h (Scheme8). Subsequent to an acid work-up and purification by columnchromatography, the Fe(III) tetra(3,5-diacetoxyphenyl)-porphyrin, Fe-tetra-(3,5-DAPP) 18, was obtained in 75%yield. Mass spectrometry displayed a single peak correspondingto the molecular weight of the iron inserted porphyrin complex(MH+ 1133). The UV/vis spectrum showed the Soret bandand two Q bands (at 418, 513, and 562 nm), confirming the
Figure 7. Plot showing ligand binding affinities vs molecular weight forthe Zn-(3,5-DAPP)-HBPs 20. All data normalized for binding to thecore porphyrin (Zn-tetra-(3,5-DAPP) 17) and also normalized relativeto each ligand. (For reference the Ka values were 14 000, 15 000, and12 000 M−1 for pyridine, 3,5-lutidine, and 3-phenylpyridine,respectively.)
Table 6. Binding Data of Pyridyl Ligands to Zn-Tetra-(3,5-DAPP)-HBP 20
ligand Ka porphyrin core (M−1) Ka of HBP with Mn 11 800 (M−1) onset of dense packing (Mn) reduction in bindinga (%)
pyridine 14000 (±1750) 10500 (±1200) 7800 (±500) 25 (±5)3,5-lutidine 15000 (±1750) 9000 (±1000) 6800 (±500) 21 (±5)3-phenylpyridine 12000 (±1500) 4200 (±500) 6200 (±500) 42 (±5)
aCalculated by comparing the Ka values obtained from the porphyrin core unit 16 and the Zn-3,5-DAPP-HBP fraction with Mn closest to 11 500(allowing comparison with the pseudo-fourth-generation Zn-TAPP-HBP system discussed above).
Macromolecules Article
dx.doi.org/10.1021/ma401344d | Macromolecules XXXX, XXX, XXX−XXXL

iron insertion. Insertion of iron into the bulk hyperbranchedpolymer 19 was carried out in analogous fashion, withpurification achieved by trituration from THF into coldmethanol, giving Fe(III) tetra(3,5-diacetoxyphenyl)porphyrincored hyperbranched polymer 21 (Fe-(3,5-DAPP)-HBP) asshown in Scheme 9. Once more, as for Fe-TAPP-HBP 8, linebroadening in the 1H NMR was reduced relative to that of thefree porphyrin alone. Crucially, the spectrum showed thedisappearance of the highly shielded inner porphyrin protons,confirming the metal insertion was a success. Furtherconfirmation was provided by UV/vis spectrometry whichshowed a reduction from four Q-bands at 514.5, 548.5, 587.5,and 645 nm to two at 511 and 569.5 nm. SEC analysis of thefunctionalized polymer confirmed that it had remained intactduring the procedure (Mn remained at 8100). The bulkpolymer 21 was then fractionated as previously described, andthe molecular weights were determined by GPC analysis.Although sufficient material for the catalysis study (plusrepeats) was required, it was attempted to maximize thenumber of data points by collecting relatively small fractions.The molecular weight obtained for each fraction along with thetheoretical values calculated for the equivalent dendrimers areshown in Table 7. The data show good agreement between the
values obtained for the fractionated polymer and the theoreticalvalues. The obtained molecular weights are all close enough toensure valid comparison using pseudo-generations.The catalytic reactions were carried out using the same
method and concentrations described for Fe-TAPP-HBP 8,again using UV spectroscopy to ensure that the porphyrinconcentrations were constant for all polymer fractions tested.The experiments independently studied the effect of catalystsize on the oxidation reactions of the linear alkene 9 and thecyclic alkene 11 (Figure 8). The yields of all oxidation productsfor each polymer catalyst are normalized with respect to theporphyrin core, Fe-tetra-(3,5-DAPP) 18. As expected, theporphyrin cored hyperbranched system was capable ofcatalyzing the oxidation of both alkenes. For the linear alkene9 the activity of G1 to G3.5 never drops below 90% of the valueobtained for the core unit. This is within the 5% error limits,and we can conclude that the reaction remains fairly constantfor these polymeric catalysts. However, there is a decrease inactivity for the larger G4.0 polymer, where the recorded yieldhas dropped to around 82% of the value obtained for the coreunit. Therefore, it can be concluded that the catalytic reactioninvolving the smaller linear alkene 9 occurs without hindranceuntil the largest polymer is used (Mn > 21 000). The oxidation
of the larger cyclic alkene 11 shows that there is initially a smalldecrease in activity for each polymer as size is increased.However, there is a notable decrease in yield for the G3.5 andG4 polymers, where the activity drops to 76% and 72% of thevalue obtained for the core unit. Therefore, in both cases apoint is reached when access to the catalytic core becomesmore hindered, either through dense packing of the polymerbackbone/shell or dense packing specifically around the centralporphyrin core. Significantly, this point occurs a generationearlier when the larger alkene substrate 11 is used. Overall, thecatalytic data concur well with the binding evidence thatshowed access to the porphyrin core is being restricted as thepseudo-generation increases, and this restriction is morepronounced for larger species. This was once again encouragingwith regard to possible shape and size selectivity reactions.To probe the possibility of a selective reaction, a mixed
experiment was carried out using a 1:1 mixture of the linearalkene 9 and the cyclic alkene 11. This experiment wasperformed as described for the Fe-TAPP-HBP systemdescribed above, and once again the reactions were followedby measuring the yield of epoxides, 10 and 12 (Scheme 5). Thedata are again normalized with respect to core porphyrin (Fe-tetra-(3,5-DAPP) 18) and the reactivity difference between thelinear and cyclic alkenes taken into account as before (Figure9). Although the data appear encouraging and the graph doesindicate a very small shift in selectivity (toward the less reactivelinear species), the change is less than 10%; therefore, onceagain it may be argued the data are within the error limits of theexperiment. It had been hoped that moving the growth/initiation points to the 3,5-position on the phenyl rings on theporphryin core would reduce the space around the porphyrincore sufficiently to achieve selectivity, and with respect to thezinc metalated polymers and ligand binding this was indeed thecase. However, no definitive shape or size selectivity could beobserved when using the iron metalated polymers tosimultaneously catalyze the epoxidation of the two alkenes.Although only a modest change in selectivity was observed
when the initiation/growth points were moved to the 3,5-positions, the results are an improvement over those obtainedusing the 4-substituted system studied earlier. In a final effort toimprove selectivity, we proposed to move the initiation/growth
Table 7. Theoretical Values for Pseudo-Generations vs theValues Obtained for Fractions of Fe(III) 3,5-DAPP-HBP21a
molecular weight repeat unitsa
pseudo-generation theoretical obtained theoretical obtained
0 (Fe-HBP-17) 1133 1133 0 01.5 3983 3500 15 132.5 8271 7750 37 373.0 11134 11500 56 593.5 16862 16500 83 864.0 22590 21500 120 117
aHalf-generations were assigned to hyperbranched polymers when thenumber of terminal groups and the molecular weights fell betweenthose for the full generations.
Figure 8. Relative yields of all oxidation products obtained from theoxidation of 1-octene 9 and cyclooctene 11 using catalytic experimentsusing Fe-3,5-DAPP-HBP 21 as the catalyst. Yields quoted relative tothose obtained using the G = 0 pseudo-dendrimer (which were 76%and 90%, respectively).
Macromolecules Article
dx.doi.org/10.1021/ma401344d | Macromolecules XXXX, XXX, XXX−XXXM

points one position further toward the porphyrin ring to the 2-and 6-positions. This substitution motif places the acetoxyfunctionalities directly above and below the plane of theporphyrin. As such, a steric environment around the catalyticcore would develop as soon as the polymer started to grow.This is shown schematically in Figure 10.Synthesis of Metalated Tetra(2,6-diacetoxyphenyl)-
porphyrin. The 2,6-substituted porphyrin 25 was synthesized
using an analogous method to that of the substituted 3,5-substituted diacetoxyphenylporphyrin, tetra(3,5-DAPP) 16.This dictated that the synthesis must begin with 2,6-dimethoxyporphyrin, which was synthesized from the corre-sponding 2,6-dimethoxybenzaldehyde, which, in turn wasobtained by reacting 1,3-dimethoxybenzene with butyllithiumand dimethylformamide (Scheme 10).40 After purification, 2,6-
dimethoxybenzaldehyde 22 was isolated as a pale yellow solidin a 90% yield. The 1H NMR spectrum shows a highlydeshielded peak corresponding to the aldehyde proton at 10.46ppm. The three aromatic protons are also easily distinguishedin the spectrum in the 6.55−7.44 ppm range as well as a largesinglet at 3.88 ppm corresponding to the six protons from themethoxy hydrogens.Although it was possible to synthesize the 2,6-substituted
porphyrin using the procedure described previously, the yieldwas very low (<1%). In an effort to improve the yield, thesynthesis was repeated using the low concentration approachdeveloped by Lindsey.40 This involved the use of the Lewis acidboron trifluoride diethyl etherate to synthesize a porphyri-nogen, followed by oxidation with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) as shown in Scheme 11. After columnchromatography the desired porphyrin was isolated in 9% as apurple crystalline solid (Scheme 11). The 1H NMR spectrumof 23 contained a singlet at 8.65 ppm, from the β-pyrrolicprotons, a triplet at 7.66 ppm from the para-aromatic hydrogen,a doublet at 6.94 ppm from the meta-aromatic protons, a largesinglet at 3.47 ppm from the methoxy protons, and a shieldedpeak at minus 2.52 ppm from the inner NH protons. Thiscombined with the UV/vis spectrum showing four Q-bands at513, 543.5, 589, and 650.5 nm and a Soret band at 417.5 nmconfirmed the synthesis was a success. The methyl groups of 23were removed using boron tribromide (as previously describedfor 15) to give the 2,6-dihydroxy derivative 24.41 Successfuldeprotection was confirmed by 13C NMR, which showed lossof the methoxy peak at 56.1 ppm, and by mass spectroscopy,which indicated a molecular ion at 855 Da.The introduction of the required acetoxy groups was carried
out using the same method described above for the 3,5-functionalized system. Purification using column chromatog-raphy gave the porphyrin 25 in 65% yield. Mass spectrometryindicated a molecular ion at 1079 Da, providing strong evidencethat the product had been formed. The product’s structure wasfinally confirmed by 1H NMR which displayed the characteristicpyrrolic and shielded inner protons of the porphyrin at 8.85ppm and minus 2.94 ppm, respectively, and a large singletcorresponding to the 24 acetoxy methyl protons at 1.11 ppm.Insertion of zinc to generate the metalated porphyrin 26 wascarried out using by refluxing the free base porphyrin with zincacetate dihydrate in chloroform for 30 min. (Compared to the4- and 3,5-substituted porphyrins, higher temperatures arerequired to insert zinc into the the 2,6-porphyrin system.) Afterwork-up and purification, the UV/vis spectrum of the crudereaction mixture now displayed only two Q-bands, indicating
Figure 9. Selectivity of epoxide products from a mixed experimentusing the Fe-tetra-(3,5-DAPP)-HBP 21. Yields normalized withrespect to the inherent reactivity difference between the linear(terminal) alkene 9 and the more reactive cyclic alkene 11. Therefore,values higher than 1.00 indicate a shift in reactivity toward the lessreactive linear alkene.
Figure 10. Schematic showing how steric crowding around theporphyrin core is more acute for the 2,6-substituted system (below)compared to the 3,5-substituted system (top). Moving the initiation/branching point one position closer to the porphyrin ring immediatelyeffects the steric environment around the catalytic porphyrin. Althoughtwo HBP phenyl substituents are shown, the polymers are likely to bea mix of mono-, di-, tri-, and tetrafunctionalized systems (eachpossessing 1 or 2 HBP arms).
Scheme 10. Synthesis of 2,6-Dimethoxybenzaldehyde 22
Macromolecules Article
dx.doi.org/10.1021/ma401344d | Macromolecules XXXX, XXX, XXX−XXXN

successful insertion. Further confirmation of zinc insertion wasconfirmed by mass spectrometry, which showed only a singlepeak corresponding to the functionalized porphyrin at 1141 Daand disappearance of the highly shielded inner porphyrinprotons in the 1H NMR (previously seen at minus 2.94 ppm).Insertion of iron proved to be more problematic. Following
the same method as for 5 and 18 and simply refluxing theporphyrin precursor with Fe(II) chloride and 2,6-lutidine didnot yield any of the metalated product. This is due to theincreased steric hindrance imposed by the eight acetatesubstituents. In an effort to circumvent this difficulty, thereaction was refluxed for 24 h; disappointingly, this did notsolve the problem. The use of Fe(CO)5 and iodine haspreviously been used to metalate “twin coronet” porphyrinsbearing four binaphthalene bridges.42 Unfortunately, afterseveral attempts, we were still unable to insert iron into the2,6-functionalized porphyrin. Therefore, the only option was toreduce the steric hindrance; consequently, the iron must beinserted into the porphyrin prior to the acetylation.Accordingly, the less hindered 5,10,15,20-tetrakis(2,6-dihydrox-yphenyl)-21H,23H-porphyrin 24 was metalated using iron(II)chloride in methanol at 50 °C for 3 h, as shown in Scheme12.31 Purification using column chromatography gave the
desired metalated porphyrin 27 in an excellent 98% yield. Thefirst indication the reaction had been a success was provided byUV/vis absorption spectroscopy, which showed a significantlybroadened peak when compared with the unfunctionalizedporphyrin 24. Final confirmation was provided by the massspectrum which displayed only a single peak corresponding tothe successfully functionalized porphyrin at 796 Da. The nextsynthetic step was to introduce acetoxy functionality; this wasperformed as before using acetic anhydride under reflux for 6 h.The acetic acid side product and excess solvent were thenremoved under vacuum and the crude product purified via flashchromatography giving the acetylated porphyrin 28 (Fe-tetra-
(2,6-DAPP)) in a yield of 40%. Although the spectrum wasbroadened (due to the paramagnetic iron), 1H NMRspectroscopy showed a large singlet at 1.55 ppm correspondingto the acetoxy protons on the product. Furthermore, the massspectrum displayed only one peak at 1133 Da corresponding tothe molecular ion.
Syn thes i s o f Z in c Me ta l a ted Te t r a ( 2 , 6 -diacetoxyphenyl)porphyrin Cored Hyperbranched Poly-mers. This polymer was synthesized using exactly the sameprocedure as that previously used to construct the two polymersystems discussed above. Specifically, 2,6-diacetoxyphenylpor-phyrin was reacted with a 20 M excess of the AB2 monomer,3,5-diacetoxybenzoic acid, as shown in Scheme 13. Afterpurification the polymer was isolated in good yield andpossessed an Mn of 11 306. UV and 1H NMR confirmedporphyrin incorporation and the successful synthesis of tetra-(2,6-DAPP)-HBP 29. The UV/vis spectrum showed an intenseband at 417 nm and Q-bands at 511, 539, 586, and 653 nm.Zinc was inserted into the corresponding polymer by refluxingthe polymer and excess zinc acetate in CH2Cl2. The crudereaction mixture was then filtered to remove unreacted zincacetate, the solvent was then removed, and the productprecipitated into methanol from refluxing chloroform. GPCanalysis showed the zinc metalated polymer 30 had a verysimilar molecular weight (11 710) to the free base polymer 29(11 306) subsequent to metal insertion. This confirms that zincinsertion does not damage the polymer.Successful insertion of zinc was confirmed by UV/vis
spectroscopy, which showed two Q-bands at 546.5 and 590.5nm. Furthermore, the 1H NMR spectrum showed that thehighly shielded peak at minus 2.94 ppm was absent. The bulkpolymer was fractionated using size exclusion chromatographyand analyzed by GPC; the data are shown in Table 8 (duringfractionation the very small amounts of residual unincorporatedporphyrin were removed; equivalent to around 5% of the totalproduct). Once again, monitoring UV absorption with respectto Mn/concentration confirmed that the core was evenlydistributed across all molecular weights.15 Dense packing wasonce again probed by studying the binding strengths of variouspyridyl ligands to the fractions of Zn-tetra-(2,6-DAPP)-HBP30. The same three ligands were used and to aid comparisons,and the data normalized relative to the binding constantsobtained for the porphyrin core unit Zn-tetra-(2,6-DAPP) 26,as well as being normalized relative to each ligand; the plots areshown in Figure 11. Although data were not available for a lowmolecular weight polymer, the plots obtained for each ligandare very different to those observed for Zn-TAPP-HBP 7 andZn-(3,5-DAPP)-HBP 20. For the Zn-(2,6-DAPP)-HBP 30,steric hindrance appears to be immediate (over the molecular
Scheme 11. Synthesis of the 2,6-Functionalized Porphyrins 23, 24, 24, and 26
Scheme 12. Synthesis of 2,6-Diacetoxyphenylporphyrin IronComplex 28
Macromolecules Article
dx.doi.org/10.1021/ma401344d | Macromolecules XXXX, XXX, XXX−XXXO

weights studied) and increase with polymer molecular weight.Interestingly, the binding strength decreases in an almost linearmanner up to an Mn value of 11 300, after which bindingappears to decrease at a slower rate.The binding data for each ligand are shown in Table 9.
Overall, there is a very large reduction in binding affinities forall ligands, with values between 72 and 79% being recorded forall three. Within error the values obtained are equivalent, withno ligand being hindered any more or less than the any other.Furthermore, with respect to core binding, we notice thatbinding affinity for each ligand was similar to the valuesobtained for the Zn-tetra-(3,5-DAPP)-HBP 20. This suggests asimilar steric environment around the central metal, at leastwith respect to the ligands studied. However, when comparingthe binding for the Zn-tetra-(3,5-DAPP)-HBP 20 and Zn-tetra-(2,6-DAPP)-HBP 30 polymer systems, it is clear that aconsiderable steric barrier is imposed on the latter. Conversely,a significantly improved binding environment is generated bythe Zn-tetra-(3,5-DAPP)-HBP 20. Specifically, when compar-ing the two polymer systems we observe a 35−50% drop inbinding for the Zn-tetra-(2,6-DAPP)-HBP 30 with Mn of 5900
(with respect to the simple porphyrin core). However, for theequivalent Zn-tetra-(3,5-DAPP)-HBP 20 with an Mn of 6100,we observe a 20−40% increase in binding affinity whencompared to its simple porphyrin core. This implies thatalthough dense packing is not a factor for the Zn-tetra-(2,6-DAPP) porphyrin core 26, it must be occurring very early onwithin the polymer series. For the polymer with molecularweight 11 300 (i.e., the HBP with a molecular weight closest tothe analysis used for the original TAPP polymer system), the
Scheme 13. Synthesis of Tetra-(2,6-DAPP)-HBP 29, Zn-Tetra-(2,6-DAPP)-HBP 30, and Fe-Tetra-(2,6-DAPP)-HBP 31
Table 8. Mn, Polydispersity, and Number of Repeat Units forHyperbranched Polymer Zn-2,6-DAPP-HBP 30
Mn polydispersityno. of repeat
unitspseudo-
generation
1142 (Zn-2,6-DAPP) 05900 3.74 27 2.08800 2.93 43 2.511300 2.53 57 3.013225 2.55 68 3.5
Figure 11. Plot showing ligand binding affinities vs molecular weightfor the Zn-2,6-DAPP-HBP 30. All data normalized for binding to thecore porphyrin (Zn-2,6-DAPP 26) and also normalized relative toeach ligand. (For reference, the Ka values were 14 900, 13 900, and 12100 M−1 for pyridine, 3,5-lutidine, and 3-phenylpyridine, respectively.)
Macromolecules Article
dx.doi.org/10.1021/ma401344d | Macromolecules XXXX, XXX, XXX−XXXP
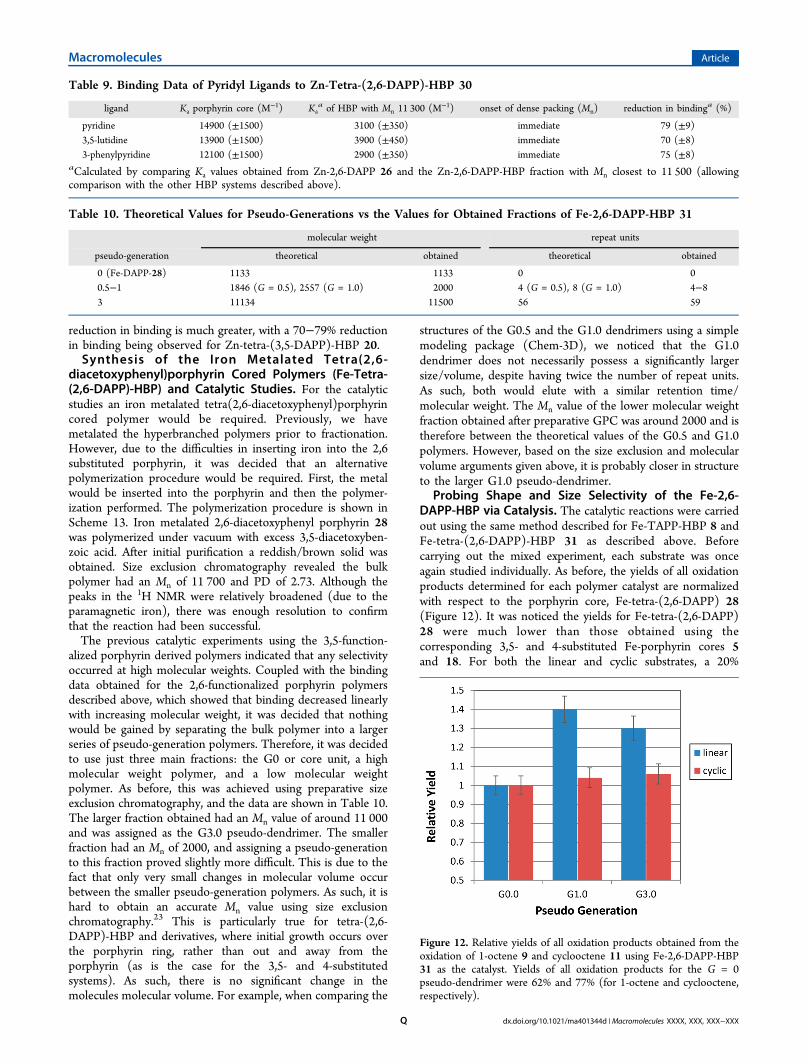
reduction in binding is much greater, with a 70−79% reductionin binding being observed for Zn-tetra-(3,5-DAPP)-HBP 20.Synthesis of the Iron Metalated Tetra(2,6-
diacetoxyphenyl)porphyrin Cored Polymers (Fe-Tetra-(2,6-DAPP)-HBP) and Catalytic Studies. For the catalyticstudies an iron metalated tetra(2,6-diacetoxyphenyl)porphyrincored polymer would be required. Previously, we havemetalated the hyperbranched polymers prior to fractionation.However, due to the difficulties in inserting iron into the 2,6substituted porphyrin, it was decided that an alternativepolymerization procedure would be required. First, the metalwould be inserted into the porphyrin and then the polymer-ization performed. The polymerization procedure is shown inScheme 13. Iron metalated 2,6-diacetoxyphenyl porphyrin 28was polymerized under vacuum with excess 3,5-diacetoxyben-zoic acid. After initial purification a reddish/brown solid wasobtained. Size exclusion chromatography revealed the bulkpolymer had an Mn of 11 700 and PD of 2.73. Although thepeaks in the 1H NMR were relatively broadened (due to theparamagnetic iron), there was enough resolution to confirmthat the reaction had been successful.The previous catalytic experiments using the 3,5-function-
alized porphyrin derived polymers indicated that any selectivityoccurred at high molecular weights. Coupled with the bindingdata obtained for the 2,6-functionalized porphyrin polymersdescribed above, which showed that binding decreased linearlywith increasing molecular weight, it was decided that nothingwould be gained by separating the bulk polymer into a largerseries of pseudo-generation polymers. Therefore, it was decidedto use just three main fractions: the G0 or core unit, a highmolecular weight polymer, and a low molecular weightpolymer. As before, this was achieved using preparative sizeexclusion chromatography, and the data are shown in Table 10.The larger fraction obtained had an Mn value of around 11 000and was assigned as the G3.0 pseudo-dendrimer. The smallerfraction had an Mn of 2000, and assigning a pseudo-generationto this fraction proved slightly more difficult. This is due to thefact that only very small changes in molecular volume occurbetween the smaller pseudo-generation polymers. As such, it ishard to obtain an accurate Mn value using size exclusionchromatography.23 This is particularly true for tetra-(2,6-DAPP)-HBP and derivatives, where initial growth occurs overthe porphyrin ring, rather than out and away from theporphyrin (as is the case for the 3,5- and 4-substitutedsystems). As such, there is no significant change in themolecules molecular volume. For example, when comparing the
structures of the G0.5 and the G1.0 dendrimers using a simplemodeling package (Chem-3D), we noticed that the G1.0dendrimer does not necessarily possess a significantly largersize/volume, despite having twice the number of repeat units.As such, both would elute with a similar retention time/molecular weight. The Mn value of the lower molecular weightfraction obtained after preparative GPC was around 2000 and istherefore between the theoretical values of the G0.5 and G1.0polymers. However, based on the size exclusion and molecularvolume arguments given above, it is probably closer in structureto the larger G1.0 pseudo-dendrimer.
Probing Shape and Size Selectivity of the Fe-2,6-DAPP-HBP via Catalysis. The catalytic reactions were carriedout using the same method described for Fe-TAPP-HBP 8 andFe-tetra-(2,6-DAPP)-HBP 31 as described above. Beforecarrying out the mixed experiment, each substrate was onceagain studied individually. As before, the yields of all oxidationproducts determined for each polymer catalyst are normalizedwith respect to the porphyrin core, Fe-tetra-(2,6-DAPP) 28(Figure 12). It was noticed the yields for Fe-tetra-(2,6-DAPP)28 were much lower than those obtained using thecorresponding 3,5- and 4-substituted Fe-porphyrin cores 5and 18. For both the linear and cyclic substrates, a 20%
Table 9. Binding Data of Pyridyl Ligands to Zn-Tetra-(2,6-DAPP)-HBP 30
ligand Ka porphyrin core (M−1) Kaa of HBP with Mn 11 300 (M−1) onset of dense packing (Mn) reduction in bindinga (%)
pyridine 14900 (±1500) 3100 (±350) immediate 79 (±9)3,5-lutidine 13900 (±1500) 3900 (±450) immediate 70 (±8)3-phenylpyridine 12100 (±1500) 2900 (±350) immediate 75 (±8)
aCalculated by comparing Ka values obtained from Zn-2,6-DAPP 26 and the Zn-2,6-DAPP-HBP fraction with Mn closest to 11 500 (allowingcomparison with the other HBP systems described above).
Table 10. Theoretical Values for Pseudo-Generations vs the Values for Obtained Fractions of Fe-2,6-DAPP-HBP 31
molecular weight repeat units
pseudo-generation theoretical obtained theoretical obtained
0 (Fe-DAPP-28) 1133 1133 0 00.5−1 1846 (G = 0.5), 2557 (G = 1.0) 2000 4 (G = 0.5), 8 (G = 1.0) 4−83 11134 11500 56 59
Figure 12. Relative yields of all oxidation products obtained from theoxidation of 1-octene 9 and cyclooctene 11 using Fe-2,6-DAPP-HBP31 as the catalyst. Yields of all oxidation products for the G = 0pseudo-dendrimer were 62% and 77% (for 1-octene and cyclooctene,respectively).
Macromolecules Article
dx.doi.org/10.1021/ma401344d | Macromolecules XXXX, XXX, XXX−XXXQ
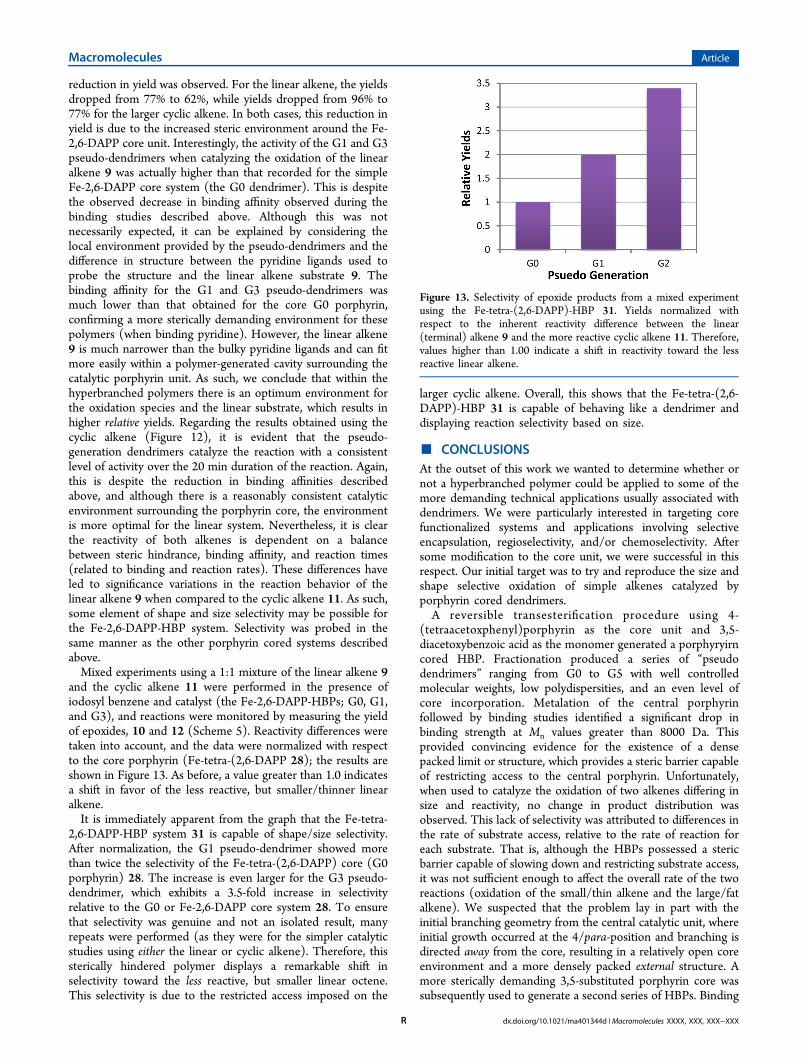
reduction in yield was observed. For the linear alkene, the yieldsdropped from 77% to 62%, while yields dropped from 96% to77% for the larger cyclic alkene. In both cases, this reduction inyield is due to the increased steric environment around the Fe-2,6-DAPP core unit. Interestingly, the activity of the G1 and G3pseudo-dendrimers when catalyzing the oxidation of the linearalkene 9 was actually higher than that recorded for the simpleFe-2,6-DAPP core system (the G0 dendrimer). This is despitethe observed decrease in binding affinity observed during thebinding studies described above. Although this was notnecessarily expected, it can be explained by considering thelocal environment provided by the pseudo-dendrimers and thedifference in structure between the pyridine ligands used toprobe the structure and the linear alkene substrate 9. Thebinding affinity for the G1 and G3 pseudo-dendrimers wasmuch lower than that obtained for the core G0 porphyrin,confirming a more sterically demanding environment for thesepolymers (when binding pyridine). However, the linear alkene9 is much narrower than the bulky pyridine ligands and can fitmore easily within a polymer-generated cavity surrounding thecatalytic porphyrin unit. As such, we conclude that within thehyperbranched polymers there is an optimum environment forthe oxidation species and the linear substrate, which results inhigher relative yields. Regarding the results obtained using thecyclic alkene (Figure 12), it is evident that the pseudo-generation dendrimers catalyze the reaction with a consistentlevel of activity over the 20 min duration of the reaction. Again,this is despite the reduction in binding affinities describedabove, and although there is a reasonably consistent catalyticenvironment surrounding the porphyrin core, the environmentis more optimal for the linear system. Nevertheless, it is clearthe reactivity of both alkenes is dependent on a balancebetween steric hindrance, binding affinity, and reaction times(related to binding and reaction rates). These differences haveled to significance variations in the reaction behavior of thelinear alkene 9 when compared to the cyclic alkene 11. As such,some element of shape and size selectivity may be possible forthe Fe-2,6-DAPP-HBP system. Selectivity was probed in thesame manner as the other porphyrin cored systems describedabove.Mixed experiments using a 1:1 mixture of the linear alkene 9
and the cyclic alkene 11 were performed in the presence ofiodosyl benzene and catalyst (the Fe-2,6-DAPP-HBPs; G0, G1,and G3), and reactions were monitored by measuring the yieldof epoxides, 10 and 12 (Scheme 5). Reactivity differences weretaken into account, and the data were normalized with respectto the core porphyrin (Fe-tetra-(2,6-DAPP 28); the results areshown in Figure 13. As before, a value greater than 1.0 indicatesa shift in favor of the less reactive, but smaller/thinner linearalkene.It is immediately apparent from the graph that the Fe-tetra-
2,6-DAPP-HBP system 31 is capable of shape/size selectivity.After normalization, the G1 pseudo-dendrimer showed morethan twice the selectivity of the Fe-tetra-(2,6-DAPP) core (G0porphyrin) 28. The increase is even larger for the G3 pseudo-dendrimer, which exhibits a 3.5-fold increase in selectivityrelative to the G0 or Fe-2,6-DAPP core system 28. To ensurethat selectivity was genuine and not an isolated result, manyrepeats were performed (as they were for the simpler catalyticstudies using either the linear or cyclic alkene). Therefore, thissterically hindered polymer displays a remarkable shift inselectivity toward the less reactive, but smaller linear octene.This selectivity is due to the restricted access imposed on the
larger cyclic alkene. Overall, this shows that the Fe-tetra-(2,6-DAPP)-HBP 31 is capable of behaving like a dendrimer anddisplaying reaction selectivity based on size.
■ CONCLUSIONSAt the outset of this work we wanted to determine whether ornot a hyperbranched polymer could be applied to some of themore demanding technical applications usually associated withdendrimers. We were particularly interested in targeting corefunctionalized systems and applications involving selectiveencapsulation, regioselectivity, and/or chemoselectivity. Aftersome modification to the core unit, we were successful in thisrespect. Our initial target was to try and reproduce the size andshape selective oxidation of simple alkenes catalyzed byporphyrin cored dendrimers.A reversible transesterification procedure using 4-
(tetraacetoxphenyl)porphyrin as the core unit and 3,5-diacetoxybenzoic acid as the monomer generated a porphyryirncored HBP. Fractionation produced a series of “pseudodendrimers” ranging from G0 to G5 with well controlledmolecular weights, low polydispersities, and an even level ofcore incorporation. Metalation of the central porphyrinfollowed by binding studies identified a significant drop inbinding strength at Mn values greater than 8000 Da. Thisprovided convincing evidence for the existence of a densepacked limit or structure, which provides a steric barrier capableof restricting access to the central porphyrin. Unfortunately,when used to catalyze the oxidation of two alkenes differing insize and reactivity, no change in product distribution wasobserved. This lack of selectivity was attributed to differences inthe rate of substrate access, relative to the rate of reaction foreach substrate. That is, although the HBPs possessed a stericbarrier capable of slowing down and restricting substrate access,it was not sufficient enough to affect the overall rate of the tworeactions (oxidation of the small/thin alkene and the large/fatalkene). We suspected that the problem lay in part with theinitial branching geometry from the central catalytic unit, whereinitial growth occurred at the 4/para-position and branching isdirected away from the core, resulting in a relatively open coreenvironment and a more densely packed external structure. Amore sterically demanding 3,5-substituted porphyrin core wassubsequently used to generate a second series of HBPs. Binding
Figure 13. Selectivity of epoxide products from a mixed experimentusing the Fe-tetra-(2,6-DAPP)-HBP 31. Yields normalized withrespect to the inherent reactivity difference between the linear(terminal) alkene 9 and the more reactive cyclic alkene 11. Therefore,values higher than 1.00 indicate a shift in reactivity toward the lessreactive linear alkene.
Macromolecules Article
dx.doi.org/10.1021/ma401344d | Macromolecules XXXX, XXX, XXX−XXXR

studies again identified a dense packed limit at a molecularweight around 8000 Da. Furthermore, the same data indicatedthat the larger polymer fractions possessed a more stericallycrowded environment that was more sensitive to ligand sizeand geometry (when compared to the 4-substituted system).This was encouraging and was backed up by initial catalyticstudies showing how the relative yields of oxidation productobtained from the larger alkene were smaller than thoseobtained using the smaller alkene. This suggested that shapeand size selectivity might be possible when oxidizing bothalkenes at the same time, and although the results did show animprovement in selectivity, it was very small. In a final attemptto impose steric demand at the core, a 2,6-disubstitutedporphyrin unit was used. The branching motif would force theresulting polymer structure to grow directly over and aroundthe catalytic porphyrin core. Binding studies on this moresterically demaning HBP system revealed a very differentbinding profile to that obtained using the other porphyrincored systems. In the case of the 2,6 system, a linear decrease inbinding was observed as molecular weight increased, whichindicates a severe steric environment with any dense packedlimit being immediate and strong. Catalytic studies carried outindependently on the two alkenes revealed higher thanexpected yields in both cases, which suggests that the polymerenvironment (around the core) might be helping the catalyticprocess. This effect seemed to be particularly strong for thelinear alkene, where increased relative yields were observedwith respect to molecular weight. When the two alkenes werereacted together with the catalysts, a marked switch in relativeselectivity was observed. When yields were compared to thoseobtained using the nonpolymeric catalysts, the inherently lessreactive linear substrate was 3−4 times more reactive relative tothe larger bulkier cyclic alkene (after normalization with respectto the inherent background reactivities). We suggest that thischange in selectivity is due to the core’s branching motif, whichencourages growth over and around the reactive porphyrin,rather than away from the core, as was the case with the othercore units.If we compare these results to those obtained by Suslick and
Moore using analogous dendrimers, we can conclude thatHBPs can indeed be applied as genuine alternatives todendrimers in a number of advanced and high-techapplicationsparticularly those requiring site isolation, encap-sulation, and release. Notwithstanding this generic claim, it isimportant to appreciate that simply “bulking up” the polymer isnot enough and that it is equally important to consider how thepolymer’s architecture and core branching motif will affect thespecific environment directly around any active site/core unit.
■ ASSOCIATED CONTENT
*S Supporting InformationSynthesis and characterization of all compounds. This materialis available free of charge via the Internet at http://pubs.acs.org.
■ AUTHOR INFORMATION
Corresponding Author*E-mail [email protected] (L.J.T.).
NotesThe authors declare no competing financial interest.
■ ACKNOWLEDGMENTS
We thank the EPSRC for providing funds through the DTAscheme to support a studentship.
■ REFERENCES(1) (a) Liu, J.; Zhong, Y.; Lam, J. W. Y.; Lu, P.; Hong, Y.; Yu, Y.; Yue,Y.; Faisal, M.; Sung, H. H. Y.; Williams, I. D.; Wong, K. S.; Tang, B. Z.Macromolecules 2010, 43, 4921. (b) Zhu, S.-W.; Shi, W. F. Polym.Degrad. Stab. 2002, 75, 543. (c) Kirkorian, K.; Ellis, A.; Twyman, L. J.Chem. Soc. Res. 2012, 41, 6138.(2) (a) Klajnert, B.; Peng, L.; Cena, V. Dendrimers in BiomedicalApplications; Royal Society of Chemistry: Cambridge, 2013.(b) Reymond, J. L.; Darbre, T. Org. Biomol. Chem. 2012, 10, 1483.(c) Wen, S.; Li, K.; Cai, H.; Chen, Q.; Shen, M.; Huang, Y.; Peng, C.;Houa, W.; Zhuc, M.; Zhangb, G.; Shi, X. Biomaterials 2013, 34, 1570.(d) Isea1, R.; Hoebeke1, J.; Mayo-García, R. Am. J. Bioinf. Comput.Biol. 2013, 1, 1.(3) (a) de Gennes, P. G.; Hervet, H. J. Phys. Lett. 1983, 44, 351.(b) Lescanec, R.; Muthukumar, M. Macromolecules 199.0, 23, 2280.(c) Pandita, D.; Santos, J. L.; Rodrigues, J. O.; Pego, A. P.; Granja, P.L.; Tomas, H. Biomacromolecules 2011, 12, 472.(4) Newkome, G. R.; Moorefield, C. N.; Vogtle, F. DendriticMolecules; VCH: Weinheim, 1996.(5) Tomalia, D. A. New J. Chem. 2012, 36, 264.(6) Wallace, M.; Ellis, A.; Twyman, L. J. Chem. Commun. 2013, 49,8063.(7) (a) Bhyrappa, P.; Young, J. K.; Moore, J. S.; Suslick, K. S. J. Am.Chem. Soc. 1996, 118, 5708. (b) Bhyrappa, P.; Young, J. K.; Moore, J.S.; Suslick, K. S. J. Mol. Catal. A: Chem. 1996, 113, 109.(8) (a) Pulkkinen, J.; Zachar, P.; Borek, V. A.; Laatikainen, R.; Kral,V. J. Mol. Catal. A: Chem. 2004, 219, 21. (b) Schenning, A. P. H. J.;Hubert, D. H. W.; Feiters, M. C.; Nolte, R. J. M. Langmuir 1996, 12,1572. (c) Niedercorn, F.; Ledon, H.; Tkatchenko, I. New J. Chem.1988, 12, 897. (d) Ostovic, D.; Bruice, T. C. J. Am. Chem. Soc. 1989,111, 6511. (e) Mashiko, T.; Dolphin, D.; Nakano, T.; Traylor, T. G. J.Am. Chem. Soc. 1985, 107, 3735. (f) Schenning, A. P. H. J.; Spelberg, J.H. L.; Hubert, D. H. W.; Feiters, M. C.; Molte, R. J. M. Chem.Eur. J.1998, 4, 871.(9) (a) Yates, C. R.; Hayes, W. Eur. Polym. J. 2004, 40, 1257.(b) Gao, C.; Yan, D. Prog. Polym. Sci. 2004, 29, 183. (c) Kim, Y. H. J.;Polym. Sci., Part A: Polym. Chem. 1998, 36, 1685. (d) Grayson, S. M.;Frechet, J. M. J. Chem. Rev. 2001, 101, 3819.(10) (a) Liu, L.; Breslow, R. J. Am. Chem. Soc. 2002, 124 (18), 4978.(b) Liu, L.; Rozenman, M.; Breslow, R. J. Am. Chem. Soc. 2002, 124(43), 12660. (c) Kirby, A. J. Angew. Chem., Int. Ed. 1996, 35, 706.(11) Holter, D.; Burgath, A.; Frey, H. Acta Polym. 1997, 48, 30.(12) King, A. S. H. K.; Martin, I.; Twyman, L. J. Chem. Soc. Res. 2002,31, 69.(13) Bharathi, P.; Moore, J. S. Macromolecules 2000, 33, 3212.(14) Zheng, X.; Oviedo, I. R.; Twyman, L. J. Macromolecules 2008,41, 7776.(15) (a) Gittins, P. J.; Alston, J.; Ge, Y.; Twyman, L. J.Macromolecules 2004, 37, 7428. (b) Twyman, L. J.; Ge, Y. Chem.Commun. 2006, 1658. (c) Gittins, P. J.; Twyman, L. J. J. Am. Chem.Soc. 2005, 127 (6), 1646.(16) Turner, S. R.; Voit, B. I.; Mourey, T. H. Macromolecules 1993,26, 4617.(17) (a) Hulsman, N.; Medema, J. P.; Bos, C.; Jongejan, A.; Leurs, R.;Smit, M. J.; de, E. I. J. P.; Richel, D.; Wijtmans, M. J. Med. Chem. 2007,50, 2424. (b) Liao, C.-C.; Wang, C.-S.; Sheu, H.-S.; Lai, C. K.Tetrahedron 2008, 64, 7977.(18) Rothemund, P. J. Am. Chem. Soc. 1935, 57, 2010.(b) Rothemund, P. J. Am. Chem. Soc. 1936, 58, 625.(19) Adler, A. D.; Longo, F. R.; Finarelli, J. D.; Goldmacher, J.;Assour, J.; Korsakoff, L. J. Org. Chem. 1967, 32, 476.(20) Soret, J. L. C. R. Chim. 1883, 97, 1267.(21) Caughey, W. S.; Kosk, W. S. Biochemistry 1962, 1 (5), 923.
Macromolecules Article
dx.doi.org/10.1021/ma401344d | Macromolecules XXXX, XXX, XXX−XXXS

(22) Friley, B. K.; Phelps, J. B.; Kincaid, J. R. J. Chromatogr. 1983,258, 310. (b) Gilbert, J.; Shepherd, M. J.; Wallwork, M. A. J.Chromatogr. 1985, 320, 361.(23) (a) Matthews, O. A.; Shipway, A. N.; Stoddart, J. F. Prog. Polym.Sci. 1998, 23, 1. (b) Frechet, J. M. J.; Hawker, C. J. Compr. Polym. Sci.,2nd Suppl. 1996, 71. (c) Twyman, L. J.; Beezer, A. E.; Mitchell, J. C. J.Chem. Soc., Perkin Trans. 1 1994, 4, 407.(24) (a) Chow, H.-F.; Mak, C. C. J. Org. Chem. 1997, I62, 5116.(b) Mak, C. C.; Chow, H.-F. Macromolecules 1997, 30, 1228.(25) (a) Xiao, F.; Sun, X.; Wu, X.; Zhao, J.; Luo, Y. J. Organomet.Chem. 2012, 713, 96. (b) Etrych, T.; Subr, V.; Jir Strohalm, J.; Sírova,M.; Ríhova, B.; Ulbrich, K. J. Controlled Release 2012, 164, 346.(c) Huang, M.; Khor, E.; Lim, L. Y. Pharm. Res. 2004, 21, 344.(26) UV titrations were performed by monitoring changes in theporphyrin’s Soret band, which appeared at 418 (±0.5 nm) for allfractions studied. For information on binding analysis and techniques,see : Schneider, , H. J.; Yatsimirsky, , A. Principles and Methods inSupramolecular Chemistry; Wiley: Chichester, 2000,.(27) Hunter, C. A.; Sanders, J. K. M. J. Am. Chem. Soc. 1990, 112,5525.(28) (a) Hawker, C. J.; Wooley, K. L.; Frechet, J. M. J. J. Am. Chem.Soc. 1993, 115, 4375. (b) Piotti, M. E.; Rivera, F., Jr.; Bond, R.; F.Hawker, C. J.; Frechet, J. M. J. J. Am. Chem. Soc. 1999, 121, 9471.(29) (a) Boris, D.; Rubinstein, M. Macromolecules 1996, 29 (22),7251. (b) Jansen, J. F. G. A.; Meijer, E. W.; de Brabander-van denBerg, E. M. M. J. Am. Chem. Soc. 1995, 117, 4417. (c) Burnett, J. L.;King, A. S. H.; Martin, I. K.; Twyman, L. J. Tetrahedron Lett. 2002, 43,2431.(30) Sanders, J. K. M. S; Bampos, N.; Clyde-Watson, Z.; Darling, S.L.; Hawley, J. C.; Kim, H. J.; Mak, C. C.; Webb, S. J.; AxialCordination Chemistry of Metalporphyrins. In The PorphyrinHandbook; Kadish, K. M., Smith, K. M., Guilard, R., Eds.; AcademicPress: San Diego, 2000; Vol. 3, Chapter 15.(31) We have also investigated to use manganese porphyrins butfound the polymeric products to be unstable and therefore unreliable.(32) Adler, A. D.; Longo, F. R.; Kampas, F.; Kim, J. J. Inorg. Nucl.Chem. 1970, 32, 2443.(33) Twyman, L. J.; Ellis, A.; Gittins, P. J. Macromolecules 2011, 44,6365.(34) Otting, G. J. Biomol. NMR 2008, 42, 1.(35) Dauban, P.; Saniere, L.; Tarrade, A.; Dodd, R. H. J. Am. Chem.Soc. 2001, 123, 7707.(36) Lindsay, S. J. R.; Sleath, P. R. J. Chem. Soc., Perkin Trans. 2 1982,1009.(37) Collman, J. P.; Brauman, J. I.; Hampton, P. D.; Tanaka, H.;Bohle, D. S.; Hembre, R. T. J. Am. Chem. Soc. 1990, 112, 7980.(38) The yields of just the epoxide 10 were 12% and 40% for thelinear and cyclic alkene, respectively.(39) Clayden, J.; Greeves, N.; Warren, S. Organic Chemistry; OxfordUniversity Press: Oxford, 2012; Chapter 19.(40) Experiments were also performed on the bulk polymer and theyields and selectivities were similar to those obtained using the coreporphyrins. See: Zheng, X.; Oviedo, I. R.; Twyman, L. J. Macro-molecules 2008, 41, 7776.(41) Huang, L.; Chen, Y.; Gao, G.-Y.; Zhang, X. P. J. Org. Chem.2003, 68, 8179.(42) (a) Wagner, R. W.; Lindsey, J. S. J. Am. Chem. Soc. 1994, 116,9759. (b) Wagner, R. W.; Johnson, T. E.; Lindsey, J. S. J. Am. Chem.Soc. 1996, 118, 11166. (c) Prathapan, S.; Johnson, T. E.; Lindsey, J. S.J. Am. Chem. Soc. 1993, 115, 7519.(43) (a) Adams, R.; Bockstahler, T. E. J. Am. Chem. Soc. 1952, 74,5346. (b) Haight, A. R.; Bailey, A. E.; Baker, W. S.; Cain, M. H.; Copp,R. R.; DeMattei, J. A.; Ford, K. L.; Henry, R. F.; Hsu, M. C.; Keyes, R.F.; King, S. A.; McLaughlin, M. A.; Melcher, L. M.; Nadler, W. R.;Oliver, P. A.; Parekh, S. I.; Patel, H. H.; Seif, L. S.; Staeger, M. A.;Wayne, G. S.; Wittenberger, S. J.; Zhang, W. Org. Process Res. Dev.2004, 8, 897.(44) (a) Tani, F.; Matsu-ura, M.; Ariyama, K.; Setoyama, T.;Shimada, T.; Kobayashi, S.; Hayashi, T.; Matsuo, T.; Hisaeda, Y.;
Naruta, Y. Chem.Eur. J. 2003, 9, 862. (b) Matsu-ura, M.; Tani, F.;Naruta, Y. J. Am. Chem. Soc. 2002, 124, 1941.
Macromolecules Article
dx.doi.org/10.1021/ma401344d | Macromolecules XXXX, XXX, XXX−XXXT


















