
Post-Translational Processing Volume 2 * Molecular Genetic Analysis of Populations (2nd edition)...
30
Transcript of Post-Translational Processing Volume 2 * Molecular Genetic Analysis of Populations (2nd edition)...


Post-Translational Processing

The Practical Approach Series
SERIES EDITOR
B. D. HAMESSchool of Biochemistry and Molecular Biology
University of Leeds, Leeds LS2 9JT, UK
/
See also the Practical Approach web site at http://www.oup.co.uk/PAS* indicates new and forthcoming titles
Affinity ChromatographyAffinity SeparationsAnaerobic MicrobiologyAnimal Cell Culture
(2nd edition)Animal Virus PathogenesisAntibodies IAntibodies IIAntibody Engineering
* Antisense TechnologyApplied Microbial PhysiologyBasic Cell CultureBehavioural NeuroscienceBioenergeticsBiological Data AnalysisBiomechanics—MaterialsBiomechanics—Structures and
SystemsBiosensorsCarbohydrate Analysis
(2nd edition)Cell-Cell InteractionsThe Cell CycleCell Growth and Apoptosis
* Cell SeparationCellular CalciumCellular Interactions in
DevelopmentCellular Neurobiology
* Chromatin* Chromosome Structural
AnalysisClinical ImmunologyComplement
* Crystallization of Nucleic Acidsand Proteins (2nd edition)
Cytokines (2nd edition)The CytoskeletonDiagnostic Molecular
Pathology IDiagnostic Molecular
Pathology IIDNA and Protein Sequence
AnalysisDNA Cloning 1: Core
Techniques (2nd edition)DNA Cloning 2: Expression
Systems (2nd edition)DNA Cloning 3: Complex
Genomes (2nd edition)

DNA Cloning 4: MammalianSystems (2nd edition)
* Drosophila (2nd edition)Electron Microscopy in BiologyElectron Microscopy in
Molecular BiologyElectrophysiologyEnzyme AssaysEpithelial Cell CultureEssential Developmental
BiologyEssential Molecular Biology IEssential Molecular Biology II
* Eukaryotic DNA ReplicationExperimental NeuroanatomyExtracellular MatrixFlow Cytometry (2nd edition)Free RadicalsGas ChromatographyGel Electrophoresis of Nucleic
Acids (2nd edition)if Gel Electrophoresis of Proteins
(3rd edition)Gene Probes 1Gene Probes 2Gene TargetingGene Transcription
if Genome MappingGlycobiology
if Growth Factors and ReceptorsHaemopoiesis
if High ResolutionChromotography
Histocompatibility TestingHIV Volume 1HIV Volume 2
* HPLC of Macromolecules(2nd edition)
Human Cytogenetics I(2nd edition)
Human Cytogenetics II(2nd edition)
Human Genetic DiseaseAnalysis
* Immobilized Biomolecules inAnalysis
Immunochemistry 1Immunochemistry 2Immunocytochemistry
if In Situ Hybridization(2nd edition)
lodinated Density GradientMedia
Ion Channelsif Light Microscopy (2nd edition)
Lipid Modification of ProteinsLipoprotein AnalysisLiposomesMammalian Cell BiotechnologyMedical ParasitologyMedical VirologyMHC Volume 1MHC Volume 2
* Molecular Genetic Analysis ofPopulations (2nd edition)
Molecular Genetics of YeastMolecular Imaging in
NeuroscienceMolecular NeurobiologyMolecular Plant Pathology IMolecular Plant Pathology IIMolecular VirologyMonitoring Neuronal Activity

Mutagenicity Testing* Mutation Detection
Neural Cell CultureNeural TransplantationNeurochemistry (2nd edition)Neuronal Cell LinesNMR of Biological
MacromoleculesNon-isotopic Methods in
Molecular BiologyNucleic Acid HybridisationOligonucleotides and
AnaloguesOligonucleotide SynthesisPCR1PCR2
*PCR3:PCR In SituHybridization
Peptide AntigensPhotosynthesis: Energy
TransductionPlant Cell BiologyPlant Cell Culture (2nd edition)Plant Molecular BiologyPlasmids (2nd edition)PlateletsPostimplantation Mammalian
Embryos
Preparative CentrifugationProtein Blotting
* Protein Expression Vol 1* Protein Expression Vol 2
Protein EngineeringProtein Function (2nd editionProtein PhosphorylationProtein Purification
ApplicationsProtein Purification MethodsProtein SequencingProtein Structure
(2nd edition)Protein Structure PredictionProtein TargetingProteolytic EnzymesPulsed Field Gel
ElectrophoresisRNA Processing IRNA Processing II
* RNA-Protein InteractionsSignalling by InositidesSubcellular FractionationSignal Transduction
if Transcription Factors(2nd edition)
Tumour Immunobiology

Post-TranslationalProcessing
A Practical Approach
Edited by
S. J. HIGGINSSchool of Biochemistry and Molecular Biology,
University of Leeds, Leeds
and
B. D. HAMESSchool of Biochemistry and Molecular Biology,
University of Leeds, Leeds
OXTORDUNIVERSITY PRESS

OXTORDUNIVERSITY PRESS
Great Clarendon Street, Oxford OX2 6DPOxford University Press is a department of the University of Oxford
and furthers the University's aim of excellence in research, scholarship,and education by publishing worldwide in
Oxford New YorkAthens Auckland Bangkok Bogotd Buenos Aires Calcutta
Cape Town Chennai Dar es Salaam Delhi Florence Hong Kong IstanbulKarachi Kuala Lumpur Madrid Melbourne Mexico City Murnbai
Nairobi Paris Sao Paulo Singapore Taipei Tokyo Toronto Warsawand associated companies in Berlin Ibadan
Oxford is a registered trade mark of Oxford University Press
Published in the United Statesby Oxford University Press Inc., New York
© Oxford University Press 1999
All rights reserved. No part of this publication may be reproduced,stored in a retrieval system, or transmitted, in any form or by any means,
without the prior permission in writing of Oxford University Press.Within the UK, exceptions are allowed in respect of any fair dealing for the
purpose of research or private study, or criticism or review, as permittedunder the Copyright, Designs and Patents Act, 1988, or in the case ofreprographic reproduction in accordance with the terms of licenses
issued by the Copyright Licensing Agency. Enquiries concerningreproduction outside those terms and in other countries should be
sent to the Rights Department, Oxford University Press,at the address above.
This book is sold subject to the condition that it shall not, by wayof trade or otherwise, be lent, re-sold, hired out, or otherwise circulatedwithout the publisher's prior consent in any form of binding or cover
other than that in which it is published and without a similar conditionincluding this condition being imposed on the subsequent purchaser
Users of books in the Practical Approach Series are advised that prudentlaboratory safety procedures should be followed at all times. Oxford
University Press makes no representation, express or implied, in respect ofthe accuracy of the material set forth in books in this series and cannotaccept any legal responsibility or liability for any errors or omissions
that may be made.
A catalogue record for this book is available from the British Library
Library of Congress Cataloging in Publication Data(Data available)
ISBN 0-19-963794-6 (Hbk)0-19-963795-4 (Pbk)
Typeset by Footnote Graphics,Warminster, Wilts
Printed in Great Britain by Information Press, Ltd,Eynsham, Oxon.

Preface
Some years ago we edited a book for The Practical Approach series entitledTranscription and translation: a practical approach. When the time came toconsider organizing a second edition, it rapidly became clear that no one bookof the desired size could include in sufficient detail the myriad of importantnew techniques. As a result, a decision was taken to produce a collection ofbooks to cover this important area. Gene transcription: a practical approachand two volumes of RNA processing: a practical approach have since beenpublished. Now, this book, Post-translational processing: a practical approach,and its companion volume, Protein expression: a practical approach, completethe 'mini-series' by providing a comprehensive and up-to-date coverage of thesynthesis and subsequent processing of proteins.
Post-translational processing: a practical approach begins with a chapter onprotein sequence analysis by Jeff Keen and Alison Ashcroft. Joachim Rassowthen covers essential methods for the study of protein folding and import intoorganelles. Next follow three chapters which describe the major covalent mod-ification events of phosphorylation (by Ivar Walaas and Anne 0stvold), glyco-sylation (by David Ashford and Fran Platt), and lipid modification (by NigelHooper and Jeff Mcllhinney). Another key area, proteolytic processing, is thesubject of a chapter by John Hutton et al. The final two chapters focus on pro-tein turnover in mammalian cells (by Aaron Ciechanover and BerndWiederanders) and in yeast (by Wolfgang Hilt and Dieter Wolf).
Those researchers who require practical guidance on the synthesis of pro-teins in vitro or in vivo for study are advised to consult the companion volume,Protein expression: a practical approach, which describes the expression ofcloned DNA or RNA templates in all the major in vitro and in vivo systems,both prokaryotic and eukaryotic, as well as methods for monitoring expres-sion.
The overriding goals of Post-translational processing: a practical approachare to describe, in precise detail, tried and tested versions of key protocols forthe active researcher, and to provide all the support required to make the tech-niques work optimally, including hints and tips for success, advice on potentialpitfalls, and guidance on data interpretation. We thank the authors for theirdiligence in writing such strong chapters and for accepting the editorialchanges we suggested. The end-result is a comprehensive compendium of thebest of current methodology in this subject area. It is a book designed both tobe used at the laboratory bench and to be read at leisure to gain insight intofuture experimental approaches.
LeedsAugust 1998
S.J.H.B.D.H.

This page intentionally left blank

Contents
List of ContributorsAbbreviations
1. Sequence analysis of expressed proteinsJeffN. Keen and Alison E. Ashcroft
1. Introduction
2. N-terminal sequence analysisAutomated sequencing
3. Sample preparationSample preparation by SDS-PAGESample preparation by HPLCOther procedures for sample preparation
4. N-terminal blockingProtein fragmentation
5. C-terminal sequencingChemical analysis of the C-terminusEnzymic analysis of the C-terminus
6. Mass spectrometric analysis of proteinsA brief guide to mass spectrometersMolecular mass determinationSequencing by mass spectrometry
References
2. Protein folding and import into organellesJoachim Rassow
1. Introduction
2. Preparation of preprotein substrates for in vitro import intoorganelles
Preparation of mRNA for in vitro translationSynthesis of preproteins in the reticulocyte lysateSynthesis of preproteins in wheat germ lysateSynthesis of preproteins in yeast cytosolSynthesis of preproteins in Escherichia coliDihydrofolate reductase as a model protein for import studies
xv
xvii
33
9101212
1417
212122
25252934
40
43
43
44444446474747
1
1

Contents
3. Import of proteins into mitochondriaIsolation of mitochondria for import studiesImport of preproteins into isolated mitochondriaGeneration of translocation intermediatesLocalization of imported proteins
4. Import of proteins into microsomes (endoplasmic reticulum)Isolation of microsomesImport of preproteins into microsomes
5. Import of proteins into other organelles
6. Analysis of protein import into organellesMonitoring the association of proteins with membranesAnalysis of protein complexes involved in organelle importAnalysis of protein folding after import into organelles
Acknowledgements
References
3. Analysis of protein phosphorylation5. Ivor Walaas and Anne Carine 0stvold
1. Introduction
2. Investigating protein phosphorylation systems
3. Phosphorylation of proteins in intact preparationsGeneral considerationsIntact animalsTissue slicesIsolated cells
4. Phosphorylation of proteins in cell-free preparationsGeneral considerationsLabelling and stimulation of cell-free preparations
5. Analysis of phosphorylated proteinsGeneral considerationsQuantification of phosphoproteinsPhosphoprotein separationProtein isolation by immunomethodsAnalysis of multisite phosphorylationPhosphoamino acid analysisAnalysis of the state of phosphorylation
6. Analysis of protein kinasesGeneral considerationsAnalysis of protein kinase activity in vitroAnalysis of specific protein kinases in vitro
x
4950566368
707073
74
75757790
91
92
95
95
96
9696969798
9999
100
101101101102103103107108
110110110113

Contents
Analysis of protein kinase activity in intact cellsPurification of protein kinases
7. Phosphoprotein phosphatasesGeneral considerationsP-Ser/P-Thr phosphoprotein phosphatasesTyrosine-specific phosphoprotein phosphatases
References
4. Protein glycosylationDavid A. Ashford and Fran Platt
1. IntroductionProtein glycosylationOligosaccharide structuresGlycosylation pathwaysCharacterization of protein glycosylation
2. Is my protein glycosylated?Colorimetric methodsProprietary detection methodsLectin binding
3. Study of whole protein glycosylationDetection of Af-glycosylationMonosaccharide compositionLectin binding analysisSusceptibility to endoglycosidase HOther methods
4. Analysis of glycosylation sitesProtease mapping of glycopeptidesGlycopeptide identification and analysis
5. Analysis of glycan structureGlycan releaseGlycan labellingGlycan separationStructural analysis of glycans
6. Manipulation of protein glycosylation
References
5. Lipid modification of proteinsNigel M. Hooper and R. A. Jeffrey Mcllhinney
1. Introduction
122123
123123123127
128
135
135135136137140
140140141142
144144146149150151
152152154
155155159161167
170
172
175
175
xi

Contents
2. Protein acylationGeneral considerationsEnzymology
3. Identification of acylated proteinsGeneral pointsLabelling cultured cells with fatty acidsAnalysis of acylated proteinsMyristoylated proteins
4. Glycosyl-phosphatidylinositol membrane anchorage ofproteins
Structure of GPI anchorsSignals for attachment of a GPI anchor to a protein
5. Identification of GPI anchorageGeneral pointsRelease of GPI-anchored proteins by bacterial
phosphatidylinositol-specific phospholipase CDifferential detergent solubilizationDetection of the cross-reacting determinantMetabolic labelling
6. Prenylation
References
6. Proteolytic processingJohn M. W. Creemers, Elaine M. Bailyes, Iris Lindberg,and John C. Hutton
1. Introduction
2. Immunoadsorbent assay of PCI and PC2Calcium-dependence of prohormone convertases
3. Expression of prohormone convertases in DG44 CHO cells
4. Fluorogenic assay for PCI and PC2PCI assayPC2 assay
5. Transient expression with recombinant vaccinia virus V.V.:T7Introduction
References
7. Protein degradation in mammalian cellsAaron Ciechanover and Bernd Wiederanders
1. The ubiquitin-proteasome pathway in mammalian cells
175175176
177177179180184
188188188
190190
191194195200
200
201
205
205
208212
213
216217218
220220
222
225
225
xii

Contents
IntroductionPreparation of cell extracts for monitoring conjugation and degradationFractionation of cell extracts for monitoring conjugation and
degradationLabelling of proteolytic substratesConjugation of proteolytic substratesDegradation of proteolytic substratesUse of inhibitors to study proteasome function
2. Proteolysis in mammalian lysosomesIntroductionIsolation of mammalian lysosomesLysosomal peptidasesMeasurement of lysosomal protein degradation
Acknowledgements
References
8. Protein degradation and proteinases in yeastWolfgang Hilt and Dieter H. Wolf
1. IntroductionAdvantages of yeast for studying eukaryotic cell biology
2. Growth of yeast cells and preparation of cell extracts
3. Analysis of protein turnoverProtein degradation in vivoDegradation of individual proteins
4. The proteasome and protein degradation in yeast
5. Proteinase yscD, a major cytoplasmic peptidaseIntroductionAssay of proteinase yscD
6. Protein degradation in the vacuoleIntroductionAssay of vacuolar peptidasesPurification of yeast vacuolar proteinasesIsolation of yeast vacuoles
AcknowledgementsReferences
Appendix
Index
225229
230233236241241
244244245251257
261
261
265
265266
268
270270272
275
283283284
285285286294299
300
301
303
309
xiii

This page intentionally left blank

Contributors
ALISON E. ASHCROFTSchool of Biochemistry and Molecular Biology, University of Leeds, LeedsLS2 9JT, UK.
DAVID A. ASHFORDGlycobiology: Research and Analytical, Department of Biology, University ofYork, PO Box 373, York YO1 5YW, UK.
ELAINE M. BAILYESDepartment of Clinical Biochemistry, University of Cambridge, Adden-brooke's Hospital, Hills Road, Cambridge CB2 2QR, UK.
AARON CIECHANOVERDepartment of Biochemistry, The Bruce Rappaport Faculty of Medicine andthe Rappaport Institute for Research in the Medical Sciences, Technion-IsraelInstitute of Technology, PO Box 9649, Haifa 31096, Israel.
JOHN M. W. CREEMERSCentre for Human Genetics, University of Leuven, Herestraat 49, B-3000Leuven, Belgium.
WOLFGANG HILTInstitut fur Biochemie, Universitat Stuttgart, Pfaffenwaldring 55, D-70569Stuttgart, Germany.
NIGEL M. HOOPERSchool of Biochemistry and Molecular Biology, University of Leeds, LeedsLS2 9JT, UK.
JOHN C. HUTTONBarbara Davis Center for Childhood Diabetes, University of Colorado HealthSciences, 4200 East 9th Avenue, Box B140, Denver, CO 80262, USA.
JEFF N. KEENSchool of Biochemistry and Molecular Biology, University of Leeds, LeedsLS2 9JT, UK.
IRIS LINDBERGBarbara Davis Center for Childhood Diabetes, University of Colorado HealthSciences, 4200 East 9th Avenue, Box B140, Denver, CO 80262, USA.
R. A. JEFFREY MCILHINNEYMRC Anatomical Neuropharmacology Unit, Mansfield Road, Oxford OX13TH, UK.

Contributors
ANNE CARINE 0STVOLDNeurochemical Laboratory, University of Oslo, PO Box 1115, Blindern,N-0317 Oslo, Norway.
FRAN PLATTOxford Glycobiology Institute, Department of Biochemistry, University ofOxford, South, Parks Road, Oxford OX1 3QU, UK.
JOACHIM RASSOWInstitut fur Biochemie und Molekularbiologie, Universitat Freiburg, HermannHerder Strasse 7, D-79104 Freiburg, Germany.
S. IV AR WALAASNeurochemical Laboratory, University of Oslo, PO Box 1115, Blindern,N-0317 Oslo, Norway.
BERND WIEDERANDERSInstitut fur Biochemie, Klinikum der Friedrich-Schiller-Universitat Jena,Nonnenplan 2, D-07740 Jena, Germany.
DIETER H. WOLFInstitut fur Biochemie, Universitat Stuttgart, Pfaffenwaldring 55, D-70569Stuttgart, Germany.
xvi

Abbreviations
1,10-PA275
AAAACAcamcAMCAPSATPATP--/-SATZBNABSABzCA-074Me®
CaMcAMPCAPSCbzCdkCECF
CHAPS
CONCRDCTPDCIDEDEAEDGG-U-GEEDHFRDITCDMPDMSODSSDTT€405
E-64®
1,10-orf/iophenanthrolineabsorbance (at 275 nm)arylamineATP/ADP carrieracetylamino methylcoumarin7-amido-4-methylcoumarineammonium persulfateadenosine triphosphateadenosine 5'-0-(3-thiotriphosphate)anilinothiazolinoneB-naphthylamidebovine serum albuminbenzoylN-(L-3-frww-propylcarbamoyloxirane-2-carbonyl)-L-isoleucyl-L-prolyl-methyl estercalcium calmodulinadenosine 3',5'-cyclic monophosphate3-(cyclohexylamino)-l-propanesulfonicacidcarboxybenzoylcyclin-dependent kinasecapillary electrophoresisconcentration of fluorophore3-[(3-cholamidopropyl)dimethylammonio]-l-propanesulfonatecontrolcross-reacting determinantcytidine 5'-triphosphatedichloroisocoumarindelayed extractiondiethylaminoethyldes-Gly-Gly-ubiquitin-Gly-ethyl esterdihydrofolate reductasephenyl diisothiocyanatedimethyl pimelimidatedimethyl sulfoxidedisuccinimidyl suberatedithiothreitolmolar extinction coefficient (at 405 nm)A^L-S-fraws-carboxirane-Z-carbony^-L-leucyl-agmatine

Abbreviations
ECL enhanced chemiluminescenceEDC l-ethyl-3-(3-dimethylamino propyl)carbodiimideEDTA ethylenediaminetetraacetic acidEGTA ethylene glycol-0,O'-bis(2-aminoethyl)-N,N,N',N'-
tetraacetic acidELISA enzyme-linked immunosorbent assayERK extracellular-regulated kinasesES electrosprayFPLC fast performance liquid chromatographyFT-ICR Fourier transform-ion cyclotron resonanceGEE glycine-ethyl esterGPI glycosyl phosphatidylinositolHepes N-(2-hydroxyethyl)piperazine-N'-(2-ethanesulfonic acid)HIV human immunodeficiency virusHMG high mobility groupHPLC high performance liquid chromatographyHPV human papillomavirusIgG immunoglobulin class GkDa kilo DaltonKRB Krebs Ringer bicarbonate (medium)
nex excitation wavelengthLC liquid chromatographyLDH lactate dehydrogenaseLM lysosomal fractionMALDI matrix-assisted laser desorption ionizationMALDITOF-MS matrix-assisted laser desorption ionization time-of-flight
mass spectrometryMAPK mitogen-activated protein kinaseMARKS myristoylated alanine-rich C-kinase substrateMCS multiple cloning siteMeUb methylated ubiquitinMES 2-(morpholino)ethanesulfonic acidMHC maj or histocompatibility complex(M + H)+ protonated molecular ion(M - H)~ deprotonated molecular ionMo methoxyMoBNA 4-methoxy-p-naphthylamideMOPS 3-(Af-morpholino)propanesulfonic acidMS mass spectrometryMS-MS tandem mass spectrometryMTX methotrexateMV mineral-vitamin
xviii
KRP Krebs Ringer phosphate (buffer)
aem emission wavelength

Abbreviations
m/zNEMNMTOD578
PAGEPBSPCPCRPDGFPEGPGPHPipesPI-PLCPITCPKAPKCPMSFpNAPP2APPOp.s.i.P-SerP-ThrPTHFTPP-TyrPVDFQ-TOFSDSSDS-PAGESulfo-MBSSTITBSTCATFATONTHTLCTMPDTOPTPCKTrypsin-TPCK
UbAl
mass-to-charge ratioAf-ethylmaleimidemyristoyl-CoA:protein N-myristoyl transferaseoptical density (at 578 nm)polyacrylamide gel electrophoresisphosphate-buffered salineprohormone convertasepolymerase chain reactionplatelet-derived growth factorpolyethylene glycolpeptidylglutamyl-peptide hydrolysing activitypiperazine-N,N'-bis(2-ethanesulfonicacid)phosphatidylinositol-specific phospholipase Cphenyl isothiocyanateprotein kinase A (cyclic AMP-dependent protein kinase)phospholipid-dependent protein kinasephenylmethylsulfonyl fluoridep-nitroanilideprotein phosphatase 2A2,5-diphenyloxazolepounds per square inchphosphoserinephosphothreoninephenylthiohydantoinphosphotyrosine-specific phosphatasephosphotyrosinepolyvinylidene difluoridequadrupole-time-of-flightsodium dodecyl sulfateSDS-polyacrylamide gel electrophoresism-maleimidobenzoyl-N-hydroxy-sulfosuccinimide estersoybean trypsin inhibitorTris-buffered salinetrichloroacetic acidtrifluoroacetic acidtrans Golgi networkthiohydantointhin-layer chromatographytetramethyl phenylene diaminetime-of-flightAf-tosyl-L-phenylalanine chloromethyl ketonetrypsin treated with AT-tosyl-L-phenylalanine chloromethylketoneubiquitin aldehyde
xix

Abbreviations
UBCUVVmax
volVSGYPDZZ-F-F-CHN2
ubiquitin-carrier protein or ubiquitin-conjugating enzymeultravioletmaximal velocityvolumevariant surface glycoproteinyeast extract, peptone, dextrosebenzyloxycarbonyl-Z-L-phenylalanyl-L-phenylalanyl-diazomethylketone
xx

Sequence analysis of expressedproteins
JEFF N. KEEN and ALISON E. ASHCROFT
1. IntroductionThe generation of recombinant proteins may be considered a relatively facileprocess nowadays. The wide range of expression systems available, and themarketing of specialized molecular biological kits by a number of manu-facturers, virtually ensure that reasonable quantities of recombinant proteinscan be produced in most biochemistry laboratories, although there are notableexceptions such as some integral membrane proteins. The engineering ofappropriate 'tags' (e.g. hexa-His, StrepTag) into the DNA sequence encodingany particular protein frequently enables single-step affinity purification of theexpressed polypeptide. Similarly, the creation of a fusion protein, with thedesired polypeptide linked to glutathione 5-transferase for example, allowsready expression and purification of the protein. The incorporation of a uniqueprotease-sensitive site (e.g. for Factor Xa) into the linking region between thetwo polypeptide domains enables the desired product to be removed from thefusion protein. However, unequivocal proof of the identity of the expressedprotein and detailed analysis of the synthetic fidelity are essential prerequisitesfor its use in experimental systems, particularly in structural and functionalanalysis, and are absolutely critical prior to release of the protein into theenvironment in numerous biotechnological applications. The confirmation ofN- and C-termini is of paramount importance, and amino acid sequence analysisremains the most unambiguous technique for this. This is complemented bymass spectrometric methods, particularly utilizing electrospray ionization, foraccurate mass measurement of the expressed protein. This in turn enables acomparison to be made with the theoretical value, which may indicate thepresence of post-translational modifications and lead to their identification.
Although biochemical research has encountered major changes over theyears, the basics of protein sequencing methodology have barely changed sincePehr Edman introduced his degradative chemistry for N-terminal sequencingin the 1950s (1, 2). However, relatively minor improvements to the overallchemistry, coupled with extensive automation, have led to greatly enhanced
1

Jeff N. Keen and Alison E. Ashcroft
sensitivity and consequently the routine determination of long sequences.Only the relatively recent introduction of biological mass spectrometry hasoffered a better alternative strategy in certain cases.
It has been suggested that the extremely rapid determination of DNAsequences will eliminate the requirement for routine protein sequencing, butin fact the two approaches have become increasingly complementary. Proteinsequencing has remained central to modern molecular biological research,although the emphasis of the work has necessarily changed. Today, ratherthan sequencing entire proteins, the requirements are much more as pre-requisites for DNA cloning work, providing the information required for thedesign of oligonucleotide probes and PCR primers. Sequencing also providesdata for the manufacture of synthetic peptides for antibody production, forprotein identification, and for the study of post-translational modifications. Inthe quality control of recombinant proteins, a few residues of N-terminalsequence are sufficient to confirm identity and the correctness of the readingframe. The rapid rise in the manufacture of recombinant proteins has also ledto the perfection of C-terminal sequencing chemistry (3, 4) and the develop-ment of automated C-terminal analysis instrumentation to complement N-terminal sequencers and thus assist in the routine characterization of expressedproteins.
The introduction of fully-automated instrumentation to perform multiplecycles of the Edman chemistry reproducibly has eliminated much labour-intensive and inefficient manipulation of samples. Manual methods are virtuallyunused nowadays, as most researchers can obtain access to core facilitiesoperating automated sequencers. Procedures for the removal of contaminantsfrom samples prior to sequencing, the means by which the sample is exposedto the chemistry, the optimization of that chemistry, the reduction of side-product formation, and identification methods for the product have all beenimproved to enable the routine determination of sequences from ever-smalleramounts of material. Thus, the sensitivity of the technique has been reducedfrom nanomolar to sub-picomolar levels. In many cases sensitivity is notcritical, as expressed proteins are not limited in supply, but in others this is anissue and minimization of usage is then important, e.g. for integral membraneproteins expressed from baculovirus vectors in insect cells.
In contrast to N-terminal sequencing, the determination of the C-terminalsequence of a protein has proved to be extremely difficult. Traditional enzymicapproaches have been both laborious and relatively ineffective, sometimesproducing only a few ambiguous residues. Recently, however, successful auto-mated chemical approaches have been developed, allowing several residuesto be identified reliably from a few hundred picomoles of material. The imple-mentation of these approaches has been driven by the need to fully characterizeexpressed proteins.
The introduction of mass spectrometry (MS) into the field of proteinsequence analysis has been a significant change. The technique has become an
2

1: Sequence analysis of expressed proteins
almost essential complementary approach to chemical protein sequencing.Accurate mass information aids the interpretation of sequence data, par-ticularly with respect to post-translational modifications. MS analysis of eitherN-terminal or C-terminal sequencing 'ladders' generated by incomplete re-actions can provide sequence information. MS approaches are also used forthe direct determination of amino acid sequences. Spontaneous or directedfragmentation products of peptides from protein digests can be analysedto generate sequence information, which is particularly useful for theidentification and characterization of known sequences.
In this chapter, we will describe the essential details of modern chemical N-and C-terminal protein sequencing. The importance of sample preparationand some recommended methods to produce good samples will be provided.The use of MS in sequence analysis and its prospects for the future will also bediscussed.
2. N-terminal sequence analysisModern automated protein sequencers utilize the degradative chemistry de-veloped in the 1950s by Pehr Edman (1, 2) for the determination of the N-terminal sequences of proteins (Figure 1). The protein sample is first madealkaline by exposure to a volatile amine, then exposed to the Edman reagent,phenyl isothiocyanate (PITC), which reacts with the N-terminal amino group(and some side chains). Excess reagents are then washed away from the sampleusing a variety of organic solvents and the modified N-terminal residue iscyclized and cleaved from the polypeptide chain using anhydrous trifluoro-acetic acid (TFA). The released N-terminal residue is washed into a secondreaction chamber for conversion from the relatively unstable anilinothiazo-linone (ATZ)-derivative to a more stable phenylthiohydantoin (PTH)-aminoacid which can be identified subsequently by reverse-phase HPLC. The trun-cated protein remains in the original reaction chamber where it can undergofurther rounds of Edman degradation, leading eventually to the generation ofa sequence of residues.
2.1 Automated sequencingCommercial automated sequencers utilize various approaches for subjectingthe sample to the Edman chemistry, and differ slightly in the details of thechemistry. In solid phase instruments (e.g. the MilliGen ProSequencer) thesample is attached covalently to a support membrane, whereas in gas phase orliquid pulse equipment (e.g. PE Applied Biosystems; Hewlett-Packard) thesample is simply adsorbed non-covalently onto the support. The differencebetween covalent and non-covalent attachment can govern the exact cycleprogramme used and the efficiency of the washing steps; covalent attachmentallows much more stringent washing. The vast majority of work, however, is
3
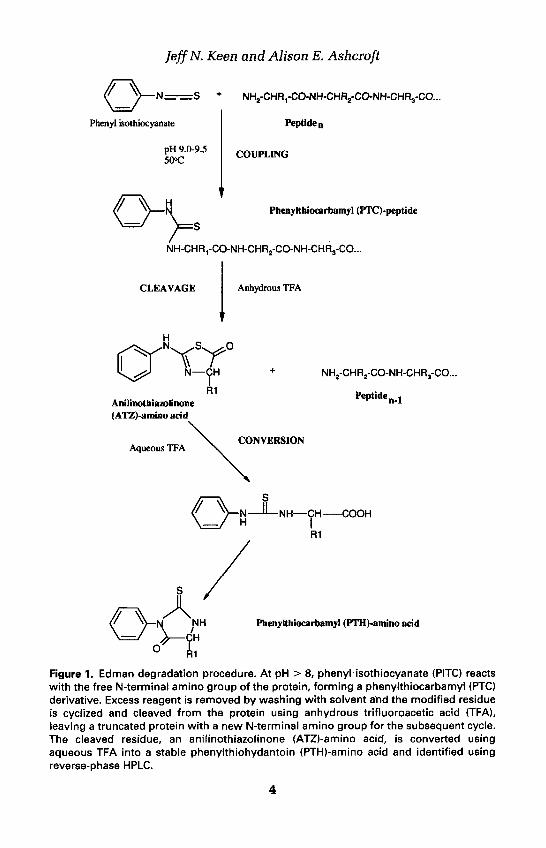
Jeff N. Keen and Alison E. Ashcroft
Figure 1. Edman degradation procedure. At pH > 8, phenyl isothiocyanate (PITC) reactswith the free N-terminal amino group of the protein, forming a phenylthiocarbamyl (PTC)derivative. Excess reagent is removed by washing with solvent and the modified residueis cyclized and cleaved from the protein using anhydrous trifluoroacetic acid (TFA),leaving a truncated protein with a new N-terminal amino group for the subsequent cycle.The cleaved residue, an anilinothiazolinone (ATZ)-amino acid, is converted usingaqueous TFA into a stable phenylthiohydantoin (PTH)-amino acid and identified usingreverse-phase HPLC.
4

1: Sequence analysis of expressed proteins
Table 1. Comparison of absorptive and solid phase sequencinga
Adsorptive sequencing
Sample adsorbed to PVDF membraneor glass fibre disc
Tolerant of small amounts of contaminants
Sample washout problem
Loss of charged residues in reactionchamberLong cycle timesSDS a problem in line blockage and samplewashout
Solid phase sequencing
Sample attached covalently to DITCmembrane (via Lys), AA membrane (viaAsp/Glu), or polyamino polymer (via Lys)on PVDFCoupling intolerant of primary amines/thiolgroups (polyamino polymer, DITC) oracidic groups (AA)Low yield of attached residuesNo washout; stringent washing providesclean background and allows long runsCharged residues (e.g. phosphorylatedamino acids) recoveredReduced cycle times, due to high flow ratesSDS solubilization of samples possible
'Abbreviations: PVDF polyvinylidene difluoride; DITC diisothiocyanate; AA arylamine.
carried out using adsorptive technology, which is convenient for mostsamples. Table 1 compares and contrasts these two approaches.
2.1.1 Adsorptive sequencingThe sample may be applied to a glass fibre disc, usually pre-treated with apolycationic carrier (polybrene) to aid in the entrapment of the protein usingionic and H bond interactions (5). Alternatively, and preferably, the samplemay be dried onto a polyvinylidene difluoride (PVDF) membrane, either bydirect spotting, by electrophoretic transfer following SDS-PAGE, or by usingProSpin or ProSorb cartridges (see Section 3.3). As the protein is only adsorbedand not bound covalently to the support, care must be taken to ensure thatthe sample is not washed too quickly out of the system during the varioussequencing reactions. Thus, adsorptive sequencers use gaseous delivery and/or small pulses of liquid to supply the solubilizing reagents (PITC and TFA)and to reduce the exposure of the sample to liquid. Solvent washes are alsokept low to minimize washout whilst still removing excess reagents and by-products.
2.1.2 Biphasic column technologyHewlett-Packard developed an alternative strategy to subject the sample tothe Edman chemistry. The sample is applied to a biphasic column, comprisingreverse-phase material in one-half and ion exchange material in the other.The sample, which may be several millilitres of protein solution contaminatedwith high levels of buffer salts and/or detergents, is applied to the reverse-phase material and washed thoroughly with a polar solvent (e.g. 2% (v/v)
5

Jeff N. Keen and Alison E. Ashcroft
aqueous TFA), causing inorganic ions to elute. The reverse-phase segment ofthe column (now containing the protein) is then attached to the ion exchangepart and placed in the sequencer. Small organic impurities are washed to wasteduring the initial stages of sequencing, but the proteinaceous material becomestrapped at the interface between the two resins as the organic solvent washesencounter the sample and leach it from the reverse-phase resin.
2.1.3 Solid phase sequencingSolid phase sequence analysis, pioneered by Richard Laursen (6), hasgenerally not been implemented in core sequencing facilities. However, it hasproven to be a particularly effective approach for the sequencing of extremelyhydrophobic samples, such as integral membrane proteins, which tend toelute rapidly from instruments utilizing adsorptive procedures. In solid phaseinstruments the sample is attached covalently to the support matrix, usually achemically modified PVDF membrane, activated with either phenyl diisothio-cyanate (DITC) (Protocol 1) or arylamine (AA) (Protocol 2). Once attachedcovalently to the support membrane, the delivery of reagents and solventwashes can be greatly increased for efficient reaction and for thorough removalof excess reagents and by-products with minimal sample washout. The samplemust be chemically clean prior to covalent attachment to the membrane,otherwise attachment may be severely compromised. Alternatively, followingelectroblotting onto a PVDF membrane after SDS-PAGE, the protein canbe immobilized by cross-linking it to an overlying polymeric matrix, themembrane entrapment procedure (Protocol 3), which limits washout (7).
For methods of preparation for the protein samples used in Protocols 1-3,see Section 3.
Protocol 1. Solid phase attachment of protein to Sequelon-DITCmembrane discs
Equipment and reagents
• Lyophilized protein sample for analysis* • Sequelon-DITC membrane disc (PerSeptive. 0.2 M 4-methylmorpholine, 0.1% (w/v) SDSb Biosystems). Sonicator bath (optional, Jencons) • 0-2 M 4-methylmorpholine, 50% (v/v). Heating block at 56°C propan-2-ol
Method1. Dissolve 10-1000 pmol of the protein sample in 35 ul 0.2 M 4-methyl-
morpholine, 0.1% SDS. Warm the solution to 56°C and sonicate it ifnecessary to aid solubilization.
2. Wet the membrane in 0.2 M 4-methylmorpholine, 50% propan-2-oland place it in the cap of a microcentrifuge tube.
3. Apply the solubilized protein sample to the wetted membrane andallow it to dry at 56°C.C
6

1: Sequence analysis of expressed proteins
4. Wet the membrane with 5 ul 0.2 M 4-methylmorpholine, 50% propan-2-ol and allow it to redry.d
"The sample must be free of contaminating primary amines and thiols which will seriouslycompromise coupling (see Section 3 for purification methods).bThe SDS concentration can be increased to 2% (w/v) for very hydrophobic proteins.cAttachment to the membrane occurs via the N-terminus and lysine side chains.dFollowing this coupling step, the membrane may be washed in 0.2 M 4-methylmorpholine,50% propan-2-ol to remove excess salts and detergent.
Protocol 2. Solid phase attachment of protein to Sequelon-AAmembrane discs
Equipment and reagents• Lyophilized protein sample for analysis" • Sequelon-AA membrane disc (PerSeptive. 50% (vM aqueous acetonitrile Biosystems)'• Coupling buffer: 10 mg/ml 1-ethyl-3-(3- • Heating block at 56°C
dimethylaminopropyl) carbodiimide (EDOin 2-(morpholino)ethanesulfonic acid (MES)buffer pH 5b
Method
1. Dissolve 10-1000 pmol of the protein sample in 30 ul 50% acetonitrile.
2. Apply the solution to the Sequelon-AA membrane disc in 10 ul aliquotsand allow it to dry at 56°C between additions.
3. Transfer the disc to room temperature and add 10 u.1 coupling buffer.
4. Allow it to dry at room temperature (about 30 min).d
"The sample must be free of contaminating organic acids (e.g. acetate), phosphate, anddetergents (e.g. SDS) which will prevent attachment of the protein to the membrane (seeSection 3 for purification methods).b Supplied as part of the kit; the concentration of the MES buffer is unspecified.c Sequelon-AA discs are supplied in a kit containing solid EDC and MES buffer.dThe protein attaches to the membrane covalently via the C-terminus and acidic side chains.
Protocol 3. Membrane entrapment procedurea
Equipment and reagents• PVDF membrane onto which the protein
sample has been deposited by spotting orby electroblotting
• Fine forceps« 0.1% (v/v) PITC in ethyl acetate• Heating block at 55°C• 2% (v/v) triethylamine in 50% (v/v) aqueous
methanol
• 0.1% (w/v) DITC in ethyl acetatei Poly(allylamine) hydrochloride solution:
0.1% (w/v) poly(allylamine) hydrochloride(Aldrich) in 2% (v/v) triethylamine, 50% (v/v)aqueous methanol
7

Jeff N. Keen and Alison E. Ashcroft
Protocol 3. Continued
Method1. Hold the PVDF membrane with fine forceps and pipette 5 ul 0.1% PITC
in ethyl acetate onto each side. Allow it to air dry (15-20 sec).b
2. Place the PVDF membrane on the heating block at 55°C and add 30 ul2% triethylamine in 50% methanol. Allow it to dry (7-8 min).
3. Return the PVDF membrane to room temperature and add 5 ul 0.1%DITC in ethyl acetate to each side. Allow it to air dry (15-20 sec).c
4. Place the PVDF membrane at 55°C and add 30 ul poly(allylamine)hydrochloride solution. Allow it to dry (5-6 min).d
5. Add 20 ul 2% triethylamine in 50% methanol and allow it to dry (10min).
6. Wash the membrane thoroughly with methanol, then with water, andfinally with methanol again. Allow it to dry.
aFrom ref. 7.bA proportion of the primary amino groups are modified, allowing their later identification.cThe remaining amino groups are modified for cross-linking.dThe modified amino groups cross-link to the polymer, immobilizing the protein on themembrane.
2.1.4 Amino acid identificationMost automated sequencers use on-line HPLC to identify the modified aminoacids recovered at each cycle of sequencing. In N-terminal sequencers thereleased amino acid is first converted to the stable PTH-derivative and theninjected onto a reverse-phase C18 resin equilibrated with acetate buffer andeluted with an increasing gradient of acetonitrile. The exact buffer compo-nents and compositions vary between instruments, but most use essentiallythe same approach. The PTH-amino acids are generally detected at 269 nm,the absorbance maximum of the PTH moiety. Sensitivity is around 1 pmolusing UV detection, but attempts have been made by various manufacturersto enhance this, e.g. by the use of diode array detectors (ratio of 269/293 nm;Beckman/Porton) or previous chromatogram subtraction (MilliGen; PEApplied Biosystems). The recent introduction of a capillary LC system (PEApplied Biosystems Precise cLC) improves sensitivity to 50-100 fmol. Theuse of radiolabelled samples or fluorescent derivatives may also enhancesensitivity, but these approaches have not been incorporated successfully intoroutine analysis. However, in the case of recombinant proteins which gener-ally are available in relatively large quantities, sensitivity is rarely an issue andthus identification of the amino acids is facile.
Amino acid identification may be a problem in cases where the protein hasbeen modified post-translationally. The modified residue may not be re-covered from the reaction cartridge (e.g. in the case of highly charged phos-
8

1: Sequence analysis of expressed proteins
phoamino acids, which are insoluble in the organic transfer solvent) or maydisplay modified behaviour during HPLC and not be recognized (e.g. fasteluting glycosylated residues), leading to a 'blank' cycle in the sequencer run.Alternatively, an amino acid may be particularly prone to degradation duringthe Edman reaction and its presence not detected (e.g. cysteine, tryptophan).Cysteine can be positively identified if it is first modified to protect the labileside chain (e.g. by S-pyridylethylation; Protocol 4). Tryptophan can usually beidentified at very low yield, but at times can be overlooked quite easily.Phosphoserine can be identified if it is converted to 5-ethylcysteine prior toanalysis (8), although alternative means of identification (e.g. utilizing MS)may be preferable.
In the case of recombinant proteins, a 'blank' cycle may not be a problem,since the flanking sequences can be used to confirm the identity of the ex-pressed polypeptide. If absolute identification of the 'missing' residue isrequired, then analysis using MS may be more appropriate. An accurate massdetermined by MS may be sufficient to identify a modification of the protein,by comparison to the mass predicted from the DNA sequence, or tandem MSsequencing of a peptide containing the modified amino acid may be requiredfor absolute confirmation (see Section 6.1.3).
Protocol 4. Modification of cysteine using 4-vinyl-pyridinea
Equipment and reagents• Lyophilized protein sample for analysis• 0.2 M Tris-HCI pH 8.5, 6 M guanidinium chloride
. 0.5 M DTT• 4-Vinyl-pyridine (Aldrich)b
Method
1. Dissolve 10-1000 pmol of the protein sample in 50 ul 0.2 M Tris-HCIpH 8.5, 6 M guanidinium chloride.
2. Add 0.5 M DTT to a final concentration of 10 mM. Incubate the mixturefor 1 h at 37°C.
3. Add 2 ul 4-vinyl-pyridine. Incubate for 1 h at 37°C.
4. Desalt the sample by appropriate means.c
aFrom ref. 9.b 4-Vinyl-pyridine is unstable. Use recently purchased reagent that is stored under argon at -20°C.cGel filtration or solvent precipitation are suitable methods. Alternatively the sample may betransferred to a PVDF membrane using ProSpin or ProSorb cartridges (see Section 3.3).
3. Sample preparationFor efficient sequence analysis, protein samples must be extremely clean.Substances which compromise the Edman chemistry (e.g. primary amines and
9


















