
Polymorphonuclear Neutrophils Improve Replication of Chlamydia
10
of January 11, 2019. This information is current as Vivo upon MyD88-Dependent Attraction In Chlamydia pneumoniae Replication of Polymorphonuclear Neutrophils Improve Thomas Miethke Susanne Dürr, Ulrich Heinzmann, Hermann Wagner and Schiemann, Nina Wantia, Clarissa U. Prazeres da Costa, Nuria Rodriguez, Falko Fend, Luise Jennen, Matthias http://www.jimmunol.org/content/174/8/4836 doi: 10.4049/jimmunol.174.8.4836 2005; 174:4836-4844; ; J Immunol References http://www.jimmunol.org/content/174/8/4836.full#ref-list-1 , 12 of which you can access for free at: cites 34 articles This article average * 4 weeks from acceptance to publication Fast Publication! • Every submission reviewed by practicing scientists No Triage! • from submission to initial decision Rapid Reviews! 30 days* • Submit online. ? The JI Why Subscription http://jimmunol.org/subscription is online at: The Journal of Immunology Information about subscribing to Permissions http://www.aai.org/About/Publications/JI/copyright.html Submit copyright permission requests at: Email Alerts http://jimmunol.org/alerts Receive free email-alerts when new articles cite this article. Sign up at: Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists All rights reserved. Copyright © 2005 by The American Association of 1451 Rockville Pike, Suite 650, Rockville, MD 20852 The American Association of Immunologists, Inc., is published twice each month by The Journal of Immunology by guest on January 11, 2019 http://www.jimmunol.org/ Downloaded from by guest on January 11, 2019 http://www.jimmunol.org/ Downloaded from
Transcript of Polymorphonuclear Neutrophils Improve Replication of Chlamydia

of January 11, 2019.This information is current as
Vivo upon MyD88-Dependent Attraction InChlamydia pneumoniaeReplication of
Polymorphonuclear Neutrophils Improve
Thomas MiethkeSusanne Dürr, Ulrich Heinzmann, Hermann Wagner andSchiemann, Nina Wantia, Clarissa U. Prazeres da Costa, Nuria Rodriguez, Falko Fend, Luise Jennen, Matthias
http://www.jimmunol.org/content/174/8/4836doi: 10.4049/jimmunol.174.8.4836
2005; 174:4836-4844; ;J Immunol
Referenceshttp://www.jimmunol.org/content/174/8/4836.full#ref-list-1
, 12 of which you can access for free at: cites 34 articlesThis article
average*
4 weeks from acceptance to publicationFast Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
Submit online. ?The JIWhy
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists All rights reserved.Copyright © 2005 by The American Association of1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on January 11, 2019http://w
ww
.jimm
unol.org/D
ownloaded from
by guest on January 11, 2019
http://ww
w.jim
munol.org/
Dow
nloaded from

Polymorphonuclear Neutrophils Improve Replication ofChlamydia pneumoniae In Vivo upon MyD88-DependentAttraction1
Nuria Rodriguez,* Falko Fend,† Luise Jennen,‡ Matthias Schiemann,* Nina Wantia,*Clarissa U. Prazeres da Costa,* Susanne Durr,* Ulrich Heinzmann,‡ Hermann Wagner,* andThomas Miethke2*
Chlamydia pneumoniae, an obligate intracellular bacterium, causes pneumonia in humans and mice. In this study, we show thatGR1�/CD45� polymorphonuclear neutrophils (PMN) surprisingly increase the bacterial load of C. pneumoniae in vivo. Uponintranasal infection of wild-type mice, the lung weight is increased; the cytokines TNF, IL-12p40, and IFN-�, as well as thechemokines keratinocyte-derived chemokine, MCP-1, and MIP-2 are secreted; and GR1�/CD45� PMN are recruited into lungs3 days postinfection. In contrast, in infected MyD88-deficient mice, which lack a key adaptor molecule in the signaling cascade ofTLRs and IL-1R family members, the increase of the lung weight is attenuated, and from the analyzed cyto- and chemokines, onlyIL-12p40 is detectable. Upon infection, almost no influx of inflammatory cells into lungs of MyD88-deficient mice can be observed.Six days postinfection, however, MyD88-deficient mice were able to produce TNF, IFN-�, keratinocyte-derived chemokine, andMCP-1 in amounts similar to wild-type mice, but failed to secrete IL-12p40 and MIP-2. At this time point, the infection increasedthe lung weight to a level similar to wild-type mice. Curiously, the chlamydial burden in MyD88-deficient mice 3 days postinfectionis lower than in wild-type mice, a finding that can be reproduced in wild-type mice by depletion of GR1� cells. In analyzing howPMN influence the chlamydial burden in vivo, we find that PMN are infected and enhance the replication of C. pneumoniae inepithelial cells. Thus, the lower chlamydial burden in MyD88-deficient mice can be explained by the failure to recruit PMN. TheJournal of Immunology, 2005, 174: 4836–4844.
P olymorphonuclear neutrophils (PMN)3 are crucial for in-nate host defense against bacteria and fungi. Upon che-mokine-mediated attraction, they rapidly ingest and kill
extracellular microbial pathogens via release of reactive oxygenspecies, proteolytic enzymes, and antimicrobial peptides (1, 2).During pneumonia, the ELR� CXC chemokines KC and MIP-2were reported to control recruitment of PMN (1). Several obligateor facultative intracellular microbial pathogens such as Yersiniaenterocolitica or Leishmania major developed mechanisms toevade destruction upon ingestion by PMN (3, 4). Among thisgroup of pathogens, microorganisms such as Ehrlichia (3) or Chla-mydia pneumoniae (5) even replicate in PMN, indicating that PMNmight not participate in the control of these microorganismsin vivo.
C. pneumoniae is responsible for up to 10% of all cases ofcommunity-acquired pneumonia in humans (6), and also causes
pneumonia in mice (7). The primary cell type of the lung infectedby the microorganism are bronchial epithelial cells (8). Withinthese cells, C. pneumoniae enters a Chlamydia-specific cycle ofreplication. The cycle is initiated by metabolically inert, but infec-tious elementary bodies (EB) that subsequently develop into retic-ulate bodies (RB) and divide by binary fission. After 48–72 h, RBrevert back to EB that leave the cell and infect neighboring cells.Within the first 2–3 days postinfection, infected pulmonary areasare characterized by a cellular infiltrate consisting among other celltypes mainly of PMN (8).
The Gram-negative, obligate intracellular bacterium C. pneu-moniae is recognized by the innate immune system via TLRs thatdetect microbes via recognition of pathogen-associated microbialpatterns, such as endotoxin, peptidoglycan, bacterial DNA, andothers (9). We have shown in a previous study that murine bonemarrow-derived dendritic cells sense C. pneumoniae via TLR2 andTLR4 (10). The relative importance of the two TLRs to triggercellular reactions varies with the cellular response analyzed. Forinstance, TNF release by bone marrow-derived dendritic cellsstimulated with C. pneumoniae is clearly dependent on TLR2, butindependent from TLR4, whereas IL-12p40 secretion depends onTLR2 and TLR4 (10). Because TLR2- and TLR4-induced signal-ing cascades result in different cellular reactions, more than oneadaptor is probably recruited to TLRs upon recognition of thisbacterium.
Upon interaction with their ligand, TLRs recruit up to threedifferent adaptor molecules. Among these, MyD88 appears to in-teract with all TLRs (11, 12) and also with IL-1R family members,whereas Toll-IL-1R domain-containing adaptor protein (TIRAP,also known as Mal) associates with TLR1, 2, 4, and 6 (13, 14), andToll-IL-1R domain-containing adaptor molecule-1 (TICAM-1,
Institutes of *Medical Microbiology, Immunology, and Hygiene and †Pathology,Technical University of Munich, Munich, Germany; and ‡GSF National ResearchCenter for Environment and Health, Institute of Pathology, Neuherberg, Germany
Received for publication July 21, 2004. Accepted for publication January 3, 2005.
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby marked advertisement in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.1 This work was supported by Deutsche Forschungsgemeinschaft MI 471/1-1.2 Address correspondence and reprint requests to Dr. Thomas Miethke, Institute ofMedical Microbiology, Immunology and Hygiene, Technical University of Munich,Trogerstr. 9, 81675 Munich, Germany. E-mail address: [email protected] Abbreviations used in this paper: PMN, polymorphonuclear neutrophil; EB, ele-mentary body; IFU, inclusion-forming unit; RB, reticulate body; RT, room temper-ature; TICAM-1, Toll-IL-1R domain-containing adaptor molecule-1; TIRAP, Toll-IL-1R domain-containing adaptor protein; KC, keratinocyte-derived chemokine.
The Journal of Immunology
Copyright © 2005 by The American Association of Immunologists, Inc. 0022-1767/05/$02.00
by guest on January 11, 2019http://w
ww
.jimm
unol.org/D
ownloaded from

also known as Toll-IL-7R domain-containing adaptor inducingIFN-�) with TLR3 and TLR4 (15). The majority of inflammatoryresponses, however, depend on MyD88. Yet, macrophages fromMyD88-deficient mice are still able to activate the MAPKs JNK1,ERK1/2, and p38, as well as NF-�B, albeit with a delayed kinetic(16). It was initially assumed that TIRAP represents the MyD88-independent adaptor (17), but later studies using gene-deficientmice indicated that MyD88 and TIRAP cooperate (13, 14). How-ever, a recently defined third adaptor, TICAM-1, was shown toactivate the IFN-� promoter in a MyD88-independent manner (15,18). Upon interaction of TLRs with MyD88, IL-1R-associated ki-nase 1 and 4 are recruited, which in turn interact with TNFR-associated factor 6 (12). This signaling cascade leads to the acti-vation of NF-�B, and MAPKs such as ERK1/2, p38, and JNK1/2(16). As a final consequence, TLR signaling contributes to thecontrol of microbial infections by the host in vivo. Thus, TLR4-mutant mice were shown to be highly susceptible to infection withSalmonella typhimurium (19). Furthermore, TLR2-deficient micedisplayed a higher sensitivity to infection with Staphylococcus au-reus (20) and were more susceptible to meningitis caused by Strep-tococcus pneumoniae (21). MyD88-deficient mice were also un-able to control infections by Staphylococcus aureus (20). Incontrast, in a model of polymicrobial peritonitis, MyD88 defi-ciency improved resistance against sepsis presumably via a reduc-tion of hyperinflammation (22).
In this study, we infected mice intranasally with C. pneumoniaeand analyzed the relevance of MyD88 for the initiation of inflam-matory responses by the host as well as its influence on the rep-lication of this obligate intracellular microorganism. The compar-ison of wild-type and MyD88-deficient mice revealed a new roleof PMN in the replication of C. pneumoniae.
Materials and MethodsStrains of mice
C57BL/6 mice were purchased from Harlan Winkelmann. Breeding pairsof MyD88-deficient mice were kindly provided by Dr. S. Akira (Depart-ment of Host Defense, Research Institute for Microbial Diseases, OsakaUniversity, Osaka, Japan) and bred in our own animal facility. Mice were8� backcrossed to C57BL/6 mice.
Reagents and mAbs
The allophycocyanin-labeled mAb specific for CD11b (553312), PE-la-beled anti-CD45 mAb (553081), FITC-labeled anti-GR1 mAb (553126),and CD16/32-specific mAb (553142) to block Fc receptors were purchasedfrom BD Pharmingen. For depletion of GR1� cells in vivo, purified GR1mAb (RB6-8C5, 150 �g/mouse), kindly provided by Dr. J. Zerrahn (MaxPlanck Institute for Infection Biology, Berlin, Germany), was used.
Infection protocol
Mice were anesthesized with an i.p. injection of Ketamin (150 �l/mouse).Subsequently, mice were infected intranasally with C. pneumoniae by ap-plying 30 �l of PBS containing 2.5 � 106 inclusion-forming units (IFU) ofthe microorganism or PBS alone.
Isolation of pulmonary cells
To isolate pulmonary cells, mice were sacrificed by CO2 inhalation 3 dayspostinfection with C. pneumoniae. The lungs were flushed with 10 ml ofPBS applied through the right atrium of the heart to remove blood. There-after, the organ was cut into small pieces in a 60-mm plate and digested for10 min with collagenase VIII (400 U/100 �l, room temperature (RT),C-2139; Sigma-Aldrich) and subsequently for another 30 min (400 U ofcollagenase/100 �l in 2 ml of RPMI 1640, 0% FCS, 37°C). The digestedmaterial was filtered through a cell strainer of 100 �m pore size (BDDiscovery Labware Europe) to remove debris. The cells were incubatedwith ammonium chloride (0.15 M, 3 min, RT) to lyse erythrocytes. Theremaining cells were washed in PBS containing 3% FCS and analyzedby FACS.
Detection of chemokines and cytokines
The chemokines keratinocyte-derived chemokine (KC), MCP-1, andMIP-2 as well as the cytokines TNF, IFN-�, and IL-12p40 were deter-mined in lungs of infected mice by commercially available ELISAs (duoset for KC, MIP-2, TNF, IFN-�, and IL-12p40, R&D Systems; MCP-1from BD Biosciences). The assays were performed, as described by themanufacturer. Lungs were isolated from mice and minced to homogeneityin 500 �l of PBS. After centrifugation (2000 � g, 5 min), the supernatantwas analyzed for its cyto- or chemokine content in duplicates.
FACS analysis and cell sorting
The cellular composition in lungs postinfection was determined by FACS.The isolated cells were preincubated with ethydium monacide (2 �g/ml, 20min, 4°C; Molecular Probes) to exclude dead cells and anti-CD16/32 (100�g/ml, 10 min, 4°C) to block Fc receptors. Subsequently, cells were doublestained (30 min, 4°C) with PE-labeled mAb directed against CD45 (1 �g/ml) and FITC-labeled GR1 mAb (1 �g/ml). After three wash steps, thecells were fixed with 2% paraformaldehyde and flow cytometric analysiswas performed (BD Biosiences). All FACS data were analyzed usingFlowJo (Tree Star).
Pulmonary cells were stained with APC-labeled anti-CD11b mAb, PE-labeled anti-CD45 mAb, and FITC-labeled anti-GR1 mAb. Cells weregated on CD45, and CD11b�/GR1� and CD11b�/GR1� subsets weresorted with a MoFlow instrument (DakoCytomation).
Giemsa stain
For Giemsa staining, sorted GR1�/CD45� cells isolated from lungs ofcontrol and infected mice were cytospinned (72.3 � g, 5 min). Thereafter,cells were fixed with methanol (100%, 5 min, RT) and stained with Giemsa(20 min, RT).
Disruption of PMN
Sorted GR1�/CD45� cells were vortexed with glass beads in 2.5 ml of cellculture medium (RPMI 1640, 0% FCS, 10 min). Supernatants were trans-ferred to another tube to discard glass beads and again centrifuged (400 �g, 5 min, RT) to remove cellular debris. The supernatant containing theequivalent of 8.25 � 105 cells/well was added to HEp2 cells (3 � 105
cells/well). After 48 h of culture, chlamydial inclusions were visualized byconfocal microscopy.
Histology and immunohistochemistry
Infected wild-type (C57BL/6) and MyD88-deficient mice were sacrificedby CO2 inhalation, and lungs were perfusion fixed with buffered 10% For-malin and in total embedded in paraffin. Sections were stained with H&E.
Immunohistochemistry with a mAb specific for heat shock protein 60 ofC. pneumoniae (Affinity BioReagents) in a dilution of 1/1000 was per-formed on an automated immunostainer (Benchmark; Ventana MedicalSystems) using established protocols. In brief, slides were dewaxed, andthe subsequent procedure including heat-induced Ag retrieval as well asblocking of nonspecific binding was performed in the immunostainer. De-tection of bound Ab was performed with a biotinylated secondary Ab andalkaline phosphatase-conjugated streptavidin (Ventana Medical Systems),using Fast Red as substrate. The same procedure with omission of theprimary Ab was performed as negative control.
For quantitative evaluation, immunoreactive inclusion bodies werecounted over 50 randomly selected high power fields (�400). Inclusionbodies in the bronchial epithelium and in alveolar lining cells were eval-uated separately.
Confocal microscopy
To quantify the numbers of C. pneumoniae in lungs of infected mice, thesupernatant of homogenized lung specimens was used to infect HEp2 cells.After 48 h, HEp2 cells were fixed and permeabilized with methanol/ace-tone (1:1 v/v, 5 min), stained with FITC-labeled mAb specific for chla-mydial endotoxin, and counterstained with Evans blue (ACI-FITC, Progen;Biotechnik). Cells were washed and embedded in glycerin/PBS (50%) andanalyzed by confocal microscopy (LSM510; Carl Zeiss Jena).
To detect C. pneumoniae in GR1� cells, CD45� cells were purified byMACS (Miltenyi Biotec) from lungs taken from mice 3 days postinfectionor from mock-infected mice. FcRs were blocked, as described above, andcells were stained with a FITC-labeled GR1 mAb (1 �g/ml, 30 min, 4°C).After washing (PBS containing 3% FCS), cells were fixed with parafor-maldehyde (2%, 30 min), washed again, and permeabilized with saponin(1%, 30 min). Simultaneously, cells were stained with an Alexa546-labeledmAb specific for chlamydial endotoxin. Cells were washed three times (1%
4837The Journal of Immunology
by guest on January 11, 2019http://w
ww
.jimm
unol.org/D
ownloaded from

saponin/PBS containing 3% FCS), followed by three wash steps with PBSand 3% FCS.
Electron microscopy
GR1�/CD45� cells were purified by cell sorting on day 3 postinfectionwith C. pneumoniae from collagenase-treated lungs. The cells were washedwith PBS, fixed with paraformaldehyde (2%) plus glutaraldehyde (0.1%)for 20 min, 37°C, and postfixed in osmium tetroxide. Subsequently, sam-ples were dehydrated with a graded ethanol series and embedded in Epon,and ultrathin sections were prepared. Sections were washed carefully withdeionized H2O and stained with uranyl acetate (0.5%) plus lead citrate(3%). Sections were examined at 80 kV with a Zeiss EM 10 GR transmis-sion electron microscope.
Statistics
Multiple comparisons were analyzed by ANOVA, and as post hoc test theHolm-Sidak method was used. The t test was used to compare two groups.
ResultsInflammatory responses of MyD88-deficient mice upon infectionwith C. pneumoniae are severely impaired on day 3postinfection
To analyze the ability of MyD88-deficient mice to recognize theobligate intracellular bacterium C. pneumoniae, gene-deficient andwild-type mice were infected intranasally with 2.5 � 106 IFU ofthe microorganism. Three days postinfection, lungs of wild-typemice contained elevated levels of TNF, IL-12p40, and IFN-�, aswell as the chemokines KC, MCP-1, and MIP-2 (Fig. 1A). In con-trast, the cyto- and chemokine response of MyD88-deficient micewas much weaker, with the notable exception of IL-12p40, indi-cating that this cytokine was secreted in vivo during C. pneu-moniae infection in a MyD88-independent fashion (Fig. 1A). Totalcellular numbers of the lungs of wild-type mice increased 2- to3-fold 3 days after infection, but did not change in lungs ofMyD88-deficient mice (data not shown). As a consequence of theinflammatory response, the lung weight of wild-type mice rosesubstantially (Fig. 1B). The increment of lung weight was attenu-ated in the case of MyD88-deficient mice and significantly lowerthan in wild-type mice (Fig. 1B). In line with these findings, ahistologic analysis of lungs 3 days postinfection with C. pneu-moniae revealed that lungs of wild-type mice showed an extensiveinfiltration of pulmonary tissue with leukocytes, which was almostnot detectable in MyD88-deficient mice (Fig. 2). However, 6 dayspostinfection, MyD88-deficient mice were able to produce TNF,IFN-�, KC, and MCP-1 in amounts similar to wild-type mice, butfailed to secrete IL-12p40 and MIP-2 (Fig. 1A). In addition, theinfection increased the lung weight in both strains of mice to thesame level 6 days postinfection (Fig. 1B). Thus, it appears thatMyD88 is of critical importance to trigger inflammatory responsesinduced by C. pneumoniae during the early period of infection.
FIGURE 1. Impaired inflammatory response in MyD88-deficient miceupon infection with C. pneumoniae. A, C57BL/6 (wild type (WT), n � 6,u) or MyD88-deficient mice (n � 6, f) were intranasally infected with
2.5 � 106 IFU/mouse and sacrificed 3 days later. Of each strain, twouninfected mice served as negative controls (�). In addition, C57BL/6(WT, n � 3, u) or MyD88-deficient mice (n � 3, f) were infected for 6days with 2.5 � 106 IFU/mouse. One mouse of each strain served asnegative control (�). Cyto- and chemokines were determined in the su-pernatant of lung homogenates via ELISA, as described in Materials andMethods. Error bars represent SD. �, p � 0.001; #, p � 0.05; ANOVA posthoc Holm-Sidak. B, Lung weight of C57BL/6 (WT, n � 6, u) or MyD88-deficient mice (n � 6, f) 3 days upon infection with 2.5 � 106 IFU/mouse.Of each mouse strain, two uninfected mice were used as negative controls(�). The lung weight was also determined 6 days after infection with 2.5 �106 IFU/mouse (C57BL/6, n � 5, u; MyD88-deficient mice, n � 5, f).Two uninfected mice of each strain served as negative controls (�). �, p �0.001; ANOVA post hoc Holm-Sidak.
4838 NEUTROPHILS IMPROVE REPLICATION OF C. pneumoniae
by guest on January 11, 2019http://w
ww
.jimm
unol.org/D
ownloaded from

On day 3 postinfection, �30% of the CD45� pulmonary infil-trate was also GR1 positive in wild-type mice (Fig. 3, A and B).This cellular subset did not increase in MyD88-deficient mice (Fig.3, A and B). Purification of GR1�/CD45� cells by cell sorting andsubsequent Giemsa staining revealed that the vast majority of cellsdisplayed the morphology of PMN (Fig. 3B).
Chlamydial burden in MyD88-deficient mice is lower than inwild-type mice
Because the majority of inflammatory responses analyzed in thisstudy were not induced in MyD88-deficient mice, we expected thechlamydial burden in these mice to be higher than in wild-typemice. In contrast to our expectation, we found that the microor-ganism was detected in even lower amounts in MyD88-deficientthan wild-type mice (Fig. 4). A semiquantitative real-time PCRapproach confirmed these results (data not shown). When chla-mydial burden was analyzed 6 days postinfection, the number ofmicroorganisms found in lungs of MyD88-deficient mice wasabout the same as on day 3 postinfection (Fig. 4). However, wild-type mice decreased chlamydial burden to the level of MyD88-
deficient mice 6 days postinfection (Fig. 4). Thus, althoughMyD88-deficient mice are severely impaired to mount an inflam-matory response against C. pneumoniae, the replication of the mi-croorganism during the early phase of infection appears to be lesseffective in these mice.
Depletion of GR1� cells in vivo reduces chlamydial burden
Because GR1�/CD45� cells were not attracted into infected lungsof MyD88-deficient mice, we wondered whether these cells mayinfluence the chlamydial burden in wild-type mice. Therefore, wedepleted wild-type mice from GR1� cells and infected them 1 daylater with C. pneumoniae. Fig. 5A shows that GR1�/CD45� cellswere completely absent in depleted mice 3 days postinfection. Fur-thermore, the chlamydial burden was lower in GR1-depleted micecompared with untreated wild-type mice at this time point (Fig.5B). Thus, GR1� cells appear to support the replication of C.pneumoniae in vivo.
PMN are infected with and support the replication of C.pneumoniae in epithelial cells
In an attempt to identify the mechanism in which PMN increasethe chlamydial burden in wild-type mice in vivo, we examinedwhether PMN are infected with C. pneumoniae in vivo. Confocalmicroscopy of CD45� cells purified from infected lungs demon-strated that a substantial fraction of the GR1� subset contained C.pneumoniae (Fig. 6a). Next, we analyzed the possibility that themicroorganism replicates in GR1� PMN by electron microscopy,as claimed recently for human PMN (5). Electron microscopy ofex vivo purified GR1�/CD45� cells confirmed the results obtainedwith confocal microscopy, and EB could clearly be visualized inPMN (Fig. 6, b and c, arrow). Also, a few RB were found, indi-cating that C. pneumoniae replicated to some extent in these cells(Fig. 6c, arrowhead). Some of the EB isolated from PMN at day 3postinfection were indeed living, because coincubation of HEp2cells with a lysate of purified PMN gave rise to chlamydial inclu-sions (Fig. 6d).
The total number of chlamydial inclusions found in PMN perinfected lung is much smaller than the difference in chlamydialburden between untreated and GR1-depleted mice. Therefore, wespeculated that PMN in addition stimulate the replication of C.pneumoniae in bronchial epithelial cells. We addressed this as-sumption by sorting PMN from lungs of untreated or infectedwild-type mice and cocultured the cells with HEp2 cells infectedwith C. pneumoniae just before the addition of PMN. Confocalmicroscopy revealed that PMN isolated from mock or infectedmice increased the number of chlamydial inclusions more than2.5-fold, while GR1�/CD11b� cells were less effective (Fig. 7).
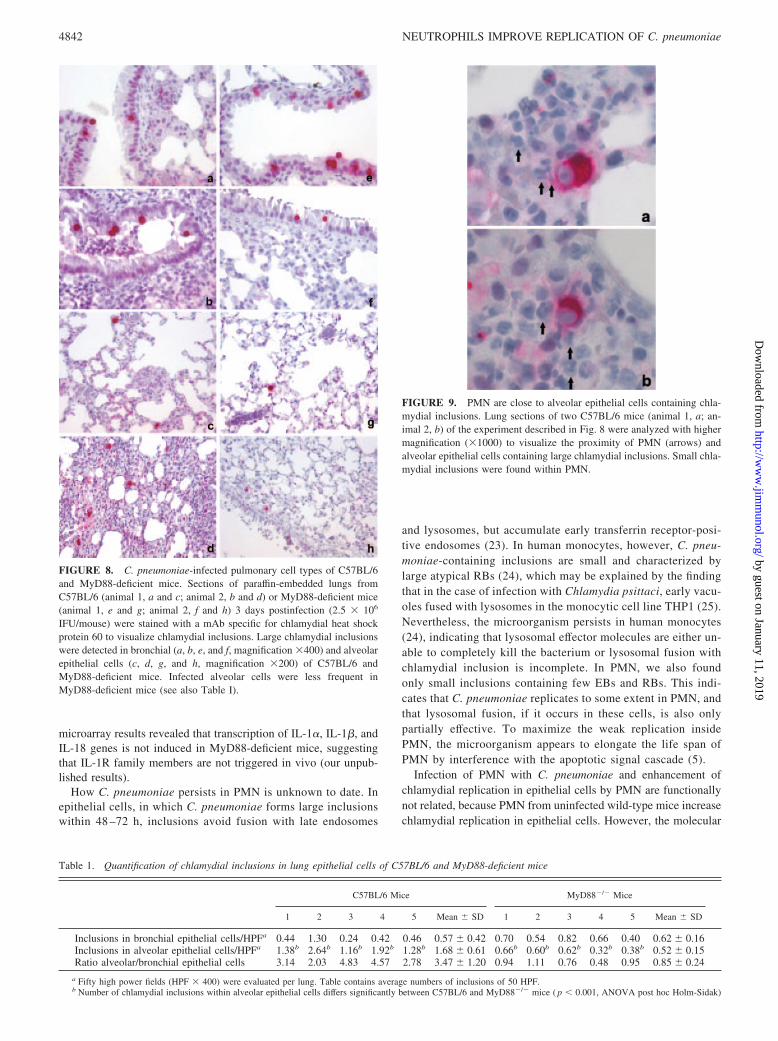
We also determined the anatomical localization of chlamydialinclusions in infected lungs of wild-type and MyD88-deficientmice and their spatial relationship to PMN using immunohisto-chemistry. Although similar numbers of large chlamydial inclu-sions were found within bronchial epithelial cells in both strains ofmice, which indicates that MyD88-deficient epithelial cells are inprinciple able to replicate the microorganism as efficiently as ep-ithelial cells from wild-type mice, alveolar epithelial cells wererarely infected in MyD88-deficient mice compared with wild-typemice (Fig. 8 and Table I). Of note, infected alveolar epithelial cellswere regularly surrounded by PMN (Figs. 8 and 9), suggesting thatPMN might support chlamydial replication in alveolar epithelialcells in vivo. In agreement with data detailed above, PMN of wild-type mice contained small chlamydial inclusions (Fig. 9).
We conclude from these findings that PMN increased chlamyd-ial burden in two ways: they act as cellular site of infection by C.
FIGURE 2. Histologic analysis of the cellular infiltrate in wild-type vsMyD88-deficient mice. Lung histology of wild-type (a–d) and MyD88-deficient mice (e–h) 3 days after infection with C. pneumoniae (2.5 � 106
IFU/mouse). H&E stain, �250 (a–c and e–g); �40 (d and h). Mock-infected animals (a and e) show normal bronchi and alveoli. Infected wild-type animals (b and c) show massive infiltrates of PMN in a peribronchio-lar and perivascular distribution. The low power view (d) demonstrates thewide extent of inflammatory changes, accompanied by focal hemorrhage.MyD88-deficient mice show essentially no (f) or only mild and focal (g)inflammatory changes with occasional PMN (arrow). The low power view(h) demonstrates the preservation of the lung architecture and the absenceof major inflammatory changes in MyD88-deficient mice.
4839The Journal of Immunology
by guest on January 11, 2019http://w
ww
.jimm
unol.org/D
ownloaded from

pneumoniae and, in addition, amplify the replication of the micro-organism in epithelial cells.
DiscussionMyD88 is the central adaptor of the TLR-signaling cascade inter-acting with all known TLRs, but also with IL-1R family members.In this study, we analyzed the importance of this adaptor moleculefor host defense against the obligate intracellular bacterium C.pneumoniae. In the early phase of infection, i.e., 3 days postinfec-tion, most of the inflammatory responses analyzed were switchedoff, as expected. In addition, gene microarray analysis of lungsrevealed that 312 genes were induced �2- and up to 27-fold by C.pneumoniae infection in wild-type mice. Of these genes, 197 werebelow a 2-fold induction in MyD88-deficient mice, indicating thatMyD88 is crucial for the initiation of host responses against thisintracellular pathogen (data not shown). Surprisingly, chlamydialreplication in MyD88-deficient was less efficient during the earlyphase of infection, a conclusion based on the finding that bacterialburden in the lung was lower in comparison with wild-type mice.This unexpected result is explained by the observation that PMNsupport the replication of C. pneumoniae in wild-type mice.MyD88-deficient mice, however, are unable to recruit this popu-lation of cells to sites of inflammation.
At present, it is unclear whether the inflammatory defects ob-served in MyD88-deficient mice upon infection with C. pneu-moniae are due to impaired TLR signaling and/or failure of sig-naling through IL-1R family members. Our preliminary results,
FIGURE 4. Lower chlamydial burden in MyD88-deficient mice. Threeand 6 days postinfection (2.5 � 106 IFU/mouse), chlamydial burden wasdetermined in the supernatant of lung homogenates of C57BL/6 (wild type(WT), day 3, n � 6; day 6, n � 5, f) and MyD88�/� mice (day 3, n � 6;day 6, n � 5, �). Chlamydial burden was quantified by transfer of lungsupernatants to HEp2 cells. Forty-eight hours later, IFU in HEp2 cells werecounted. The graph represents the pooled data from two independent ex-periments. �, p � 0.001; ANOVA post hoc Holm-Sidak.
FIGURE 3. Recruitment of PMN into infected lungsof wild-type, but not MyD88-deficient mice. A, Lungsof C. pneumoniae-infected (2.5 � 106 IFU/mouse, day3) C57BL/6 mice (n � 2) and MyD88-deficient mice(n � 2) as well as mock-infected controls (n � 1) weredigested with collagenase. Single cell suspensions werestained with a FITC-labeled mAb specific for GR1 anda PE-labeled mAb specific for CD45. Top graphs, Showthe FACS analysis from mock infected; middle and bot-tom graphs, from C. pneumoniae-infected animals. B,Shows the percentage of GR1�/CD45� cells from in-fected C57BL/6 (wild type (WT), n � 4, u) andMyD88-deficient mice (n � 4, f). Two uninfected miceof each strain served as controls. �, p � 0.001; ANOVApost hoc Holm-Sidak. C, The experiment was per-formed, as described in A, with the exception thatGR1�/CD45� cells of C57BL/6 mice were sorted andtheir morphology was analyzed by Giemsa staining.
4840 NEUTROPHILS IMPROVE REPLICATION OF C. pneumoniae
by guest on January 11, 2019http://w
ww
.jimm
unol.org/D
ownloaded from

however, suggest that TLR signaling is important. Thus, mice dou-ble deficient for TLR2 and TLR4 display a similar phenotype uponinfection with C. pneumoniae, i.e., the numbers of microorganismsin the lung are lower and pulmonary recruitment of PMN is highlyattenuated. In addition, murine dendritic cells sense C. pneumoniaein a TLR2- and TLR4-dependent fashion (10). Furthermore, gene
FIGURE 7. PMN support the replication of C. pneumoniae in epithelialHEp2 cells. GR1�/CD11b� or GR1�/CD11b� cells were sorted frommock-infected (f) or day 3 infected C57BL/6 mice (u). Both cell typeswere added at a density of 2 � 105 cells/well to HEp2 cell cultures (2 �105 cells/well), which were infected with C. pneumoniae at an multiplicityof infection of 1 just before the addition of lung cells. �, Represents C.pneumoniae-infected HEp2 cell cultures without addition of inflammatorycells. After 48 h of culture, cells were lysed, and the supernatant of thelysate was transferred to a new well seeded with uninfected HEp2 cells(2.5 � 105 cells/well). After another culture period of 30 h, chlamydialinclusions were counted using confocal microscopy. Error bars representSD of results obtained with five to eight individual cultures. The graphrepresents data from three pooled experiments. �, p � 0.001; ANOVA posthoc Holm-Sidak.
FIGURE 5. Depletion of GR1� cells in vivo decreases chlamydial burden.A, C57BL/6 mice (n � 2) were depleted from GR1� cells by injecting i.p. 150�g/mouse anti-GR1 mAb 1 day before infection with C. pneumoniae (2.5 �106 IFU/mouse, right graphs). Control C57BL/6 mice (n � 2) were pretreatedi.p. with PBS and subsequently infected (2.5 � 106 IFU/mouse, left graphs).One mouse was neither depleted nor infected (top graph). Three days postin-fection, lungs were removed and digested with collagenase. Single cell sus-pensions were stained with a FITC-labeled mAb specific for GR1 and a PE-labeled mAb specific for CD45. B, C57BL/6 mice (n � 4) were depleted fromGR1� cells and infected, as described in A (�). PBS-pretreated and infectedC57BL/6 mice (n � 5) served as controls (f). Chlamydial burden was deter-mined 3 days after infection. The graph represents the pooled data from twoindependent experiments. �, t test, p � 0.012.
FIGURE 6. C. pneumoniae infects and generates small inclusions inPMN in vivo. a, CD45� cells were purified by magnetic separation fromcollagenase-treated lungs 3 days after infection of C57BL/6 mice with C.pneumoniae (2.5 � 106 IFU/mouse). Cells were stained with FITC-labeledanti-GR1 mAb. C. pneumoniae inclusions were visualized by an Alexa546-labeled mAb specific for chlamydial endotoxin. Cells were analyzed byconfocal microscopy. b, GR1�/CD45� cells were selected by cell sortingfrom collagenase-treated lungs 3 days after infection of C57BL/6 micewith C. pneumoniae (2.5 � 106 IFU/mouse) and analyzed by electronmicroscopy (magnification �22,000). Arrow points to a chlamydial inclu-sion. c, Further electronic close-up. Arrow points to an EB, and arrowheadto an RB. d, FACS-purified GR1�/CD45� cells were minced by vortexingwith glass beads. After centrifugation of the lysate, the supernatant (equiv-alent of 8.25 � 105 cells/well) was transferred to HEp2 cells (3 � 105
cells/well). After 48 h of culture, chlamydial inclusions were visualized byconfocal microscopy. (arrows).
4841The Journal of Immunology
by guest on January 11, 2019http://w
ww
.jimm
unol.org/D
ownloaded from
microarray results revealed that transcription of IL-1�, IL-1�, andIL-18 genes is not induced in MyD88-deficient mice, suggestingthat IL-1R family members are not triggered in vivo (our unpub-lished results).
How C. pneumoniae persists in PMN is unknown to date. Inepithelial cells, in which C. pneumoniae forms large inclusionswithin 48 –72 h, inclusions avoid fusion with late endosomes
and lysosomes, but accumulate early transferrin receptor-posi-tive endosomes (23). In human monocytes, however, C. pneu-moniae-containing inclusions are small and characterized bylarge atypical RBs (24), which may be explained by the findingthat in the case of infection with Chlamydia psittaci, early vacu-oles fused with lysosomes in the monocytic cell line THP1 (25).Nevertheless, the microorganism persists in human monocytes(24), indicating that lysosomal effector molecules are either un-able to completely kill the bacterium or lysosomal fusion withchlamydial inclusion is incomplete. In PMN, we also foundonly small inclusions containing few EBs and RBs. This indi-cates that C. pneumoniae replicates to some extent in PMN, andthat lysosomal fusion, if it occurs in these cells, is also onlypartially effective. To maximize the weak replication insidePMN, the microorganism appears to elongate the life span ofPMN by interference with the apoptotic signal cascade (5).
Infection of PMN with C. pneumoniae and enhancement ofchlamydial replication in epithelial cells by PMN are functionallynot related, because PMN from uninfected wild-type mice increasechlamydial replication in epithelial cells. However, the molecular
Table 1. Quantification of chlamydial inclusions in lung epithelial cells of C57BL/6 and MyD88-deficient mice
C57BL/6 Mice MyD88�/� Mice
1 2 3 4 5 Mean � SD 1 2 3 4 5 Mean � SD
Inclusions in bronchial epithelial cells/HPFa 0.44 1.30 0.24 0.42 0.46 0.57 � 0.42 0.70 0.54 0.82 0.66 0.40 0.62 � 0.16Inclusions in alveolar epithelial cells/HPFa 1.38b 2.64b 1.16b 1.92b 1.28b 1.68 � 0.61 0.66b 0.60b 0.62b 0.32b 0.38b 0.52 � 0.15Ratio alveolar/bronchial epithelial cells 3.14 2.03 4.83 4.57 2.78 3.47 � 1.20 0.94 1.11 0.76 0.48 0.95 0.85 � 0.24
a Fifty high power fields (HPF � 400) were evaluated per lung. Table contains average numbers of inclusions of 50 HPF.b Number of chlamydial inclusions within alveolar epithelial cells differs significantly between C57BL/6 and MyD88�/� mice ( p � 0.001, ANOVA post hoc Holm-Sidak)
FIGURE 8. C. pneumoniae-infected pulmonary cell types of C57BL/6and MyD88-deficient mice. Sections of paraffin-embedded lungs fromC57BL/6 (animal 1, a and c; animal 2, b and d) or MyD88-deficient mice(animal 1, e and g; animal 2, f and h) 3 days postinfection (2.5 � 106
IFU/mouse) were stained with a mAb specific for chlamydial heat shockprotein 60 to visualize chlamydial inclusions. Large chlamydial inclusionswere detected in bronchial (a, b, e, and f, magnification �400) and alveolarepithelial cells (c, d, g, and h, magnification �200) of C57BL/6 andMyD88-deficient mice. Infected alveolar cells were less frequent inMyD88-deficient mice (see also Table I).
FIGURE 9. PMN are close to alveolar epithelial cells containing chla-mydial inclusions. Lung sections of two C57BL/6 mice (animal 1, a; an-imal 2, b) of the experiment described in Fig. 8 were analyzed with highermagnification (�1000) to visualize the proximity of PMN (arrows) andalveolar epithelial cells containing large chlamydial inclusions. Small chla-mydial inclusions were found within PMN.
4842 NEUTROPHILS IMPROVE REPLICATION OF C. pneumoniae
by guest on January 11, 2019http://w
ww
.jimm
unol.org/D
ownloaded from

mechanism(s) involved in the latter process is unclear. A similarfunction has been reported for the monocyte-like cell U937. Thus,coculture of U937 cells with human endothelial cells enhanced theinfection of the latter cells by C. pneumoniae (26). Subsequently,it was suggested that the infectivity-enhancing factor is identicalwith human insulin-like growth factor 2 (27). Using a differentcellular system, a recent study demonstrated that human mono-cytes also enhance the growth of C. pneumoniae in arterial smoothmuscle cells (28). In this case, cell to cell contact was required, andit was suggested that mannose 6-phosphate receptor expressed onmonocytes is implicated in this process. Other possible mecha-nisms include the regulation of intracellular tryptophan levels inepithelial cells by PMN because C. pneumoniae lacks tryptophanbiosynthesis genes (29) and thus is completely dependent on host-derived tryptophan (30). Increasing the supply of this amino acidvia TGF-�-mediated down-regulation of the tryptophan-degradingenzyme indoleamine 2,3-dioxygenase or the tryptophanyl-tRNAsynthetase could represent potential mechanisms (31). TGF-�, inturn, might be produced by PMN because it was reported thatTGF-� mRNA was induced after stimulation of human PMN withMycobacterium bovis bacillus Calmette-Guerin (32).
In comparison with wild-type mice, the chlamydial load appearsto be more reduced in MyD88-deficient mice than in GR1-depletedmice, indicating that in addition to PMN, another cell type mightsupport the replication of C. pneumoniae. As discussed above, thehuman monocyte-like cell U937 was able to amplify chlamydialreplication in smooth muscle and epithelial cells (26, 28). In ourexperiments, the GR1�/CD11b� cell population, which may in-clude monocytes/macrophages, also weakly amplified chlamydialreplication, although the differences were not statistically signifi-cant. Thus, further experimentation is needed to clarify this issue.
It remains to be seen whether other adaptors aside from MyD88are relevant for host defense against C. pneumoniae. However,release of IL-12p40 at day 3 postinfection appears to be MyD88independent. Also, our experiments show that on day 6 postinfec-tion, TNF, IFN-�, KC, and MCP-1 are secreted MyD88 indepen-dently. IL-12 participates in the control of C. pneumoniae repli-cation in vivo, because anti IL-12p40 treatment results in higherchlamydial titers, but less severe pathological changes within thelung of infected mice (33). From the adaptors identified to date,only TICAM-1 (Toll-IL-7R domain-containing adaptor-inducingIFN-�) appears to operate MyD88 independently (15, 18). TI-CAM-1 is crucially involved in the induction of IFN-� (34), butalso in endotoxin-mediated secretion of IL-12p40 (15). In vitro,IL-12p40 secretion induced by C. pneumoniae depends partiallyon TLR4 (10), and TICAM-1 not only interacts with TLR3 (34),but also with TLR4 (18). On the one hand, these findings argue fora role of TICAM-1 during recognition of C. pneumoniae. In con-trast, our preliminary data show that TLR4 appears not to partic-ipate in release of IL-12p40 in vivo.
In conclusion, the results of this study describe a new role forPMN in the replication of the intracellular pathogen C. pneu-moniae in vivo: attracted MyD88 dependently to the site of in-flammation, they increase chlamydial burden during the first daysof infection.
AcknowledgmentsWe thank Dr. S. Akira for providing MyD88-deficient mice. We are alsograteful to Dr. J. Zerrahn for the generous supply with anti-GR1 mAb. Wethank Carmen Hartmann for expert technical assistance with immunohis-
tochemistry. Many thanks to Dr. B. Holzmann for critical reading of themanuscript.
DisclosuresThe authors have no financial conflict of interest.
References1. Mizgerd, J. P. 2002. Molecular mechanisms of neutrophil recruitment elicited by
bacteria in the lungs. Semin. Immunol. 14:123.
2. Faurschou, M., and N. Borregaard. 2003. Neutrophil granules and secretory ves-icles in inflammation. Microbes Infect. 5:1317.
3. Allen, L. A. 2003. Mechanisms of pathogenesis: evasion of killing by polymor-phonuclear leukocytes. Microbes Infect. 5:1329.
4. Laskay, T., G. van Zandbergen, and W. Solbach. 2003. Neutrophil granulocytes:Trojan horses for Leishmania major and other intracellular microbes? TrendsMicrobiol. 11:210.
5. Van Zandbergen, G., J. Gieffers, H. Kothe, J. Rupp, A. Bollinger, E. Aga,M. Klinger, H. Brade, K. Dalhoff, M. Maass, et al. 2004. Chlamydia pneumoniaemultiply in neutrophil granulocytes and delay their spontaneous apoptosis. J. Im-munol. 172:1768.
6. Kuo, C. C., L. A. Jackson, L. A. Campbell, and J. T. Grayston. 1995. Chlamydiapneumoniae (TWAR). Clin. Microbiol. Rev. 8:451.
7. Yang, Z. P., C. C. Kuo, and J. T. Grayston. 1993. A mouse model of Chlamydiapneumoniae strain TWAR pneumonitis. Infect. Immun. 61:2037.
8. Yang, Z. P., P. K. Cummings, D. L. Patton, and C. C. Kuo. 1994. Ultrastructurallung pathology of experimental Chlamydia pneumoniae pneumonitis in mice.J. Infect. Dis. 170:464.
9. Medzhitov, R. 2001. Toll-like receptors and innate immunity. Nat. Rev. Immunol.1:135.
10. Prebeck, S., C. Kirschning, S. Durr, C. da Costa, B. Donath, K. Brand,V. Redecke, H. Wagner, and T. Miethke. 2001. Predominant role of Toll-likereceptor 2 versus 4 in Chlamydia pneumoniae-induced activation of dendriticcells. J. Immunol. 167:3316.
11. Medzhitov, R., P. Preston-Hurlburt, E. Kopp, A. Stadlen, C. Chen, S. Ghosh, andC. A. J. Janeway. 1998. MyD88 is an adaptor protein in the hToll/IL-1 receptorfamily signaling pathways. Mol. Cell 2:253.
12. Barton, G. M., and R. Medzhitov. 2003. Toll-like receptor signaling pathways.Science 300:1524.
13. Horng, T., G. M. Barton, R. A. Flavell, and R. Medzhitov. 2002. The adaptormolecule TIRAP provides signalling specificity for Toll-like receptors. Nature420:329.
14. Yamamoto, M., S. Sato, H. Hemmi, H. Sanjo, S. Uematsu, T. Kaisho,K. Hoshino, O. Takeuchi, M. Kobayashi, T. Fujita, et al. 2002. Essential role forTIRAP in activation of the signalling cascade shared by TLR2 and TLR4. Nature420:324.
15. Yamamoto, M., S. Sato, H. Hemmi, K. Hoshino, T. Kaisho, H. Sanjo,O. Takeuchi, M. Sugiyama, M. Okabe, K. Takeda, and S. Akira. 2003. Role ofadaptor TRIF in the MyD88-independent Toll-like receptor signaling pathway.Science 301:640.
16. Kawai, T., O. Adachi, T. Ogawa, K. Takeda, and S. Akira. 1999. Unresponsive-ness of MyD88-deficient mice to endotoxin. Immunity 11:115.
17. Horng, T., G. M. Barton, and R. Medzhitov. 2001. TIRAP: an adapter moleculein the Toll signaling pathway. Nat. Immunol. 2:835.
18. Hoebe, K., X. Du, P. Georgel, E. Janssen, K. Tabeta, S. O. Kim, J. Goode, P. Lin,N. Mann, S. Mudd, et al. 2003. Identification of Lps2 as a key transducer ofMyD88-independent TIR signalling. Nature 424:743.
19. O‘Brien, A. D., D. L. Rosenstreich, I. Scher, G. H. Campbell, R. P. MacDermott,and S. B. Formal. 1980. Genetic control of susceptibility to Salmonella typhi-murium in mice: role of the LPS gene. J. Immunol. 124:20.
20. Takeuchi, O., K. Hoshino, and S. Akira. 2000. Cutting edge: TLR2-deficient andMyD88-deficient mice are highly susceptible to Staphylococcus aureus infection.J. Immunol. 165:5392.
21. Echchannaoui, H., K. Frei, C. Schnell, S. L. Leib, W. Zimmerli, andR. Landmann. 2002. Toll-like receptor 2-deficient mice are highly susceptible toStreptococcus pneumoniae meningitis because of reduced bacterial clearing andenhanced inflammation. J. Infect. Dis. 186:798.
22. Weighardt, H., S. Kaiser-Moore, R. M. Vabulas, C. J. Kirschning, H. Wagner,and B. Holzmann. 2002. Cutting edge: myeloid differentiation factor 88 defi-ciency improves resistance against sepsis caused by polymicrobial infection.J. Immunol. 169:2823.
23. Al Younes, H. M., T. Rudel, and T. F. Meyer. 1999. Characterization and intra-cellular trafficking pattern of vacuoles containing Chlamydia pneumoniae in hu-man epithelial cells. Cell Microbiol. 1:237.
24. Airenne, S., H. M. Surcel, H. Alakarppa, K. Laitinen, J. Paavonen, P. Saikku, andA. Laurila. 1999. Chlamydia pneumoniae infection in human monocytes. Infect.Immun. 67:1445.
4843The Journal of Immunology
by guest on January 11, 2019http://w
ww
.jimm
unol.org/D
ownloaded from

25. Ojcius, D. M., R. Hellio, and A. Dautry-Varsat. 1997. Distribution of endosomal,lysosomal, and major histocompatibility complex markers in a monocytic cellline infected with Chlamydia psittaci. Infect. Immun. 65:2437.
26. Lin, T. M., L. A. Campbell, M. E. Rosenfeld, and C. C. Kuo. 2000. Monocyte-Endothelial cell coculture enhances infection of endothelial cells with Chlamydiapneumoniae. J. Infect. Dis. 181:1096.
27. Lin, T. M., L. A. Campbell, M. E. Rosenfeld, and C. C. Kuo. 2001. Humanmonocyte-derived insulin-like growth factor-2 enhances the infection of humanarterial endothelial cells by Chlamydia pneumoniae. J. Infect. Dis. 183:1368.
28. Puolakkainen, M., L. A. Campbell, T. M. Lin, T. Richards, D. L. Patton, and C. C. Kuo.2003. Cell-to-cell contact of human monocytes with infected arterial smooth-muscle cellsenhances growth of Chlamydia pneumoniae. J. Infect. Dis. 187:435.
29. Kalman, S., W. Mitchell, R. Marathe, C. Lammel, J. Fan, R. W. Hyman,L. Olinger, J. Grimwood, R. W. Davis, and R. S. Stephens. 1999. Comparativegenomes of Chlamydia pneumoniae and C. trachomatis. Nat. Genet. 21:385.
30. Mehta, S. J., R. D. Miller, J. A. Ramirez, and J. T. Summersgill. 1998. Inhibitionof Chlamydia pneumoniae replication in HEp-2 cells by interferon-�: role oftryptophan catabolism. J. Infect. Dis. 177:1326.
31. Yuan, W., A. Collado-Hidalgo, T. Yufit, M. Taylor, and J. Varga. 1998. Modu-lation of cellular tryptophan metabolism in human fibroblasts by transforminggrowth factor-�: selective inhibition of indoleamine 2,3-dioxygenase and tryp-tophanyl-tRNA synthetase gene expression. J. Cell. Physiol. 177:174.
32. Suttmann, H., N. Lehan, A. Bohle, and S. Brandau. 2003. Stimulation of neu-trophil granulocytes with Mycobacterium bovis bacillus Calmette-Guerin induceschanges in phenotype and gene expression and inhibits spontaneous apoptosis.Infect. Immun. 71:4647.
33. Geng, Y., K. Berencsi, Z. Gyulai, T. Valyi-Nagy, E. Gonczol, and G. Trinchieri.2000. Roles of interleukin-12 and � interferon in murine Chlamydia pneumoniaeinfection. Infect. Immun. 68:2245.
34. Oshiumi, H., M. Matsumoto, K. Funami, T. Akazawa, and T. Seya. 2003. TI-CAM-1, an adaptor molecule that participates in Toll-like receptor 3-mediatedinterferon-� induction. Nat. Immunol. 4:161.
4844 NEUTROPHILS IMPROVE REPLICATION OF C. pneumoniae
by guest on January 11, 2019http://w
ww
.jimm
unol.org/D
ownloaded from


















