Polymorphic haplotypes of CRELD1 differentially predispose Down syndrome and euploids individuals to...
6
RESEARCH ARTICLE Polymorphic Haplotypes of CRELD1 Differentially Predispose Down Syndrome and Euploids Individuals to Atrioventricular Septal Defect Priyanka Ghosh, 1 Pranami Bhaumik, 1 Sujoy Ghosh, 1,2 Umut Ozbek, 3 Eleanor Feingold, 3,4 Cheryl Maslen, 5 Biswanath Sarkar, 6 Vishmadeb Pramanik, 6 Priyanka Biswas, 1 Biswajit Bandyopadhyay, 7 and Subrata Kumar Dey 1 * 1 Human Genetics Research Unit, School of Biotechnology and Biological Sciences, West Bengal University of Technology, Kolkata, West Bengal, India 2 Department of Zoology, Sundarban Hazi Desarat College, Pathankhali, West Bengal, India 3 Department of Biostatistics, Graduate School of Public Health, University of Pittsburgh, Pittsburgh. Pennsylvania 4 Department of Human Genetics, Graduate School of Public Health, University of Pittsburgh, Pittsburgh, Pennsylvania 5 Division of Cardiovascular Medicine and OHSU Heart Research Center, Oregon Health & Science University, Portland, Oregon 6 DNA laboratory, Anthropological Survey of India, Kolkata, West Bengal, India 7 Rabindranath.Tagore International Institute of Cardiac Sciences, Kolkata, West Bengal, India Manuscript Received: 17 February 2012; Manuscript Accepted: 26 July 2012 To explore the role of CRELD1 variants on congenital heart defects, we sequenced the entire reading frame of CRELD1 in the samples from Kolkata and adjoining areas. Nearly, 400 par- ticipants were included in the genetic association study and they were stratified as Down syndrome (DS) with atrioventricular septal defect (AVSD), DS without AVSD, euploid with AVSD, and euploid without AVSD. A significant association was found between AVSD and three polymorphisms, namely rs9878047 (c.1049-129T > C), rs3774207 (c.1119C > T), and rs73118372 (c.1136T > C) among the Down syndrome and euploid individ- uals. The polymorphism rs73118372, involves a transition (c.1136T > C) that leads to change in amino acid methionine to threonine which alters protein secondary structure as confirmed by the bioinformatics software SOPMA. In addition, two haplo- types, C-T-C and C-T-T, in the order of loci rs9878047-rs3774207- rs73118372 were associated with incidence of AVSD among euploid and Down syndrome, with a slightly higher odds ratio in the later group. We hypothesize that these haplotypes increase the risk of AVSD, and the susceptibility is exacerbated in DS, possibly due to the trisomy 21 genetic background. Moreover, we report for the first time on an interaction between the mutant alleles of rs3774207 and rs73118372 which could disrupt the delicate balance between different CRELD1 isoforms. Ó 2012 Wiley Periodicals, Inc. Key words: Down syndrome; atrioventricular septal defect; CRELD1; single nucleotide polymorphism; splicing INTRODUCTION Down syndrome (DS), caused by trisomy of chromosome 21 (Ch21) is the most commonly identified genetic form of intellectual disability characterized by multiple congenital abnormalities of variable severity. Nearly, 50% of DS individuals suffer from a congenital heart defect (CHD) of which approximately 40–60% have an atrioventricular septal defect (AVSD) [Freeman et al., 1998]. Beside DS, AVSD occurs as a nonsyndromic sporadic CHD [Digilio et al., 1999], and also part of several other syndromes [Lin et al., 2006]. Grant sponsor: Council of Scientific and Industrial Research (CSIR), New Delhi, India; Grant number: 27(0225)/10/EMRII. *Correspondence to: Subrata Kumar Dey, Human Genetics Research Unit, School of Biotechnology and Biological Sciences. West Bengal University of Technology, BF—142, Salt Lake City, Sector I, Kolkata, West Bengal 700064, India. E-mail: [email protected] Article first published online in Wiley Online Library (wileyonlinelibrary.com): 00 Month 2012 DOI 10.1002/ajmg.a.35626 How to Cite this Article: Ghosh P, Bhaumik P, Ghosh S, Ozbek U, Feingold E, Maslen C, Sarkar B, Pramanik V, Biswas P, Bandyopadhyay B, Dey SK. 2012. Polymorphic haplotypes of CRELD1 differentially predispose Down syndrome and euploids individuals to atrioventricular septal defect. Am J Med Genet Part A. Ó 2012 Wiley Periodicals, Inc. 1
-
Upload
priyanka-ghosh -
Category
Documents
-
view
212 -
download
0
Transcript of Polymorphic haplotypes of CRELD1 differentially predispose Down syndrome and euploids individuals to...

RESEARCH ARTICLE
Polymorphic Haplotypes of CRELD1 DifferentiallyPredispose Down Syndrome and Euploids Individualsto Atrioventricular Septal DefectPriyanka Ghosh,1 Pranami Bhaumik,1 Sujoy Ghosh,1,2 Umut Ozbek,3 Eleanor Feingold,3,4
Cheryl Maslen,5 Biswanath Sarkar,6 Vishmadeb Pramanik,6 Priyanka Biswas,1
Biswajit Bandyopadhyay,7 and Subrata Kumar Dey1*1Human Genetics Research Unit, School of Biotechnology and Biological Sciences, West Bengal University of Technology, Kolkata,
West Bengal, India2Department of Zoology, Sundarban Hazi Desarat College, Pathankhali, West Bengal, India3Department of Biostatistics, Graduate School of Public Health, University of Pittsburgh, Pittsburgh. Pennsylvania4Department of Human Genetics, Graduate School of Public Health, University of Pittsburgh, Pittsburgh, Pennsylvania5Division of Cardiovascular Medicine and OHSU Heart Research Center, Oregon Health & Science University, Portland, Oregon6DNA laboratory, Anthropological Survey of India, Kolkata, West Bengal, India7Rabindranath.Tagore International Institute of Cardiac Sciences, Kolkata, West Bengal, India
Manuscript Received: 17 February 2012; Manuscript Accepted: 26 July 2012
To explore the role of CRELD1 variants on congenital heart
defects, we sequenced the entire reading frame of CRELD1 in
the samples from Kolkata and adjoining areas. Nearly, 400 par-
ticipants were included in the genetic association study and they
were stratified as Down syndrome (DS) with atrioventricular
septal defect (AVSD), DS without AVSD, euploid with AVSD,
and euploid without AVSD. A significant association was found
between AVSD and three polymorphisms, namely rs9878047
(c.1049-129T>C), rs3774207 (c.1119C>T), and rs73118372
(c.1136T>C) among the Down syndrome and euploid individ-
uals. The polymorphism rs73118372, involves a transition
(c.1136T>C) that leads to change in amino acid methionine to
threonine which alters protein secondary structure as confirmed
by the bioinformatics software SOPMA. In addition, two haplo-
types, C-T-C andC-T-T, in the order of loci rs9878047-rs3774207-
rs73118372 were associated with incidence of AVSD among
euploid and Down syndrome, with a slightly higher odds ratio
in the later group. We hypothesize that these haplotypes increase
the risk of AVSD, and the susceptibility is exacerbated in DS,
possibly due to the trisomy 21 genetic background. Moreover, we
report for the first time on an interaction between the mutant
alleles of rs3774207 and rs73118372 which could disrupt the
delicate balance between different CRELD1 isoforms.
� 2012 Wiley Periodicals, Inc.
Key words: Down syndrome; atrioventricular septal defect;
CRELD1; single nucleotide polymorphism; splicing
INTRODUCTION
Down syndrome (DS), caused by trisomy of chromosome 21
(Ch21) is themost commonly identified genetic formof intellectual
disability characterized by multiple congenital abnormalities of
variable severity. Nearly, 50% of DS individuals suffer from a
congenital heart defect (CHD) of which approximately 40–60%have an atrioventricular septal defect (AVSD) [Freeman et al.,
1998]. Beside DS, AVSD occurs as a nonsyndromic sporadic
CHD [Digilio et al., 1999], and also part of several other syndromes
[Lin et al., 2006].
Grant sponsor: Council of Scientific and Industrial Research (CSIR), New
Delhi, India; Grant number: 27(0225)/10/EMRII.
*Correspondence to:
Subrata Kumar Dey, Human Genetics Research Unit, School of
Biotechnology and Biological Sciences. West Bengal University of
Technology, BF—142, Salt Lake City, Sector I, Kolkata, West Bengal
700064, India. E-mail: [email protected]
Article first published online in Wiley Online Library
(wileyonlinelibrary.com): 00 Month 2012
DOI 10.1002/ajmg.a.35626
How to Cite this Article:Ghosh P, Bhaumik P, Ghosh S, Ozbek U,
Feingold E, Maslen C, Sarkar B, Pramanik V,
Biswas P, Bandyopadhyay B, Dey SK. 2012.
Polymorphic haplotypes of CRELD1
differentially predisposeDown syndrome and
euploids individuals to atrioventricular septal
defect.
Am J Med Genet Part A.
� 2012 Wiley Periodicals, Inc. 1

The developmental basis of AVSD is complex. Formerly known
as an endocardial cushiondefect, basedon thepremise that it occurs
due to a failure of the atrioventricular (AV) endocardial cushions to
fuse during embryonic cardiogenesis [Eisenberg and Markwald,
1995], recent studies indicate a role for the dorsal mesenchymal
protrusion (DMP) in AV septation, and suggest that abnormalities
of the DMP could also result in AVSD. Misalignment of the atria
over the ventricles or looping abnormalities can also result in an
AVSD.
The increased prevalence of AVSD among individuals with
trisomy 21 compared to euploid individuals suggests that causative
genes for AVSD may be located on chromosome 21 including
COL6A1, COL6A2 [Davies et al., 1994, 1995; Klewer et al., 1998],
DSCAM [Barlow et al., 2001], and SLC19A1 [Locke et al., 2010].
This is further supported by a recent study showing that over-
expression of DSCAM and COL6A2 cooperatively cause atrial
defects in mice [Grossman et al., 2011]. However, regardless of
genetic background AVSD is a complex trait with genetic
heterogeneity.
CRELD1 (cysteine-rich with EGF-like domains 1; MIM 607170)
is located at the locus 3p25.3 and was originally identified as a
candidate for theAVSD2 locus [Rupp et al., 2002]. Subsequently, it
was shown that missense mutations in CRELD1 are a dominant
susceptibility factor for AVSD both in euploid [Robinson et al.,
2003; Zatyka et al., 2005] and DS [Maslen et al., 2006; Ying et al.,
2010; Kusuma et al., 2011] individuals. The gene is expressed in
embryonic cardiac progenitor cells. To gain insight into the asso-
ciation of CRELD1 polymorphisms with AVSD in both DS and
euploid individuals, we sequenced the entire reading frame of the
gene in samples obtained from subjects in Kolkata (previously
known as Calcutta) and adjoining areas.
MATERIALS AND METHODS
The studywas designed and performed following the declaration of
Helsinki and approved by the institutional ethics committee.
SubjectsFor all the case–control analyses that we performed, the participat-
ing volunteers were stratified into four categories, namely DS with
AVSD (n¼ 121), euploid with AVSD (n¼ 96), DS without any
detectable CHD(n¼ 92), and euploidwithout any detectable CHD
(n¼ 102), and for simplicity, referring to these last two classes as
‘‘groups without AVSD.’’ All subjects. mostly Bengali, were
reported randomly to our laboratory from the hospitals within
the Kolkata municipal area. The volunteers in all four categories
were chosen from hospitals of same locality to maintain the
maximum socio-demographic similarity. The cardiac phenotype
status of all subjects was ascertained by the physicians through
echocardiography; all of the case positive volunteers exhibited
complete AVSD. The average age of the DS and euploid subjects
was 7 and 9 years, respectively. The subjects were enrolled in the
study after confirmation of karyotype profile and only complete
trisomy 21 cases were included as ‘‘DS’’ for the analyses. Informed
consent was obtained for tissue sample collection and subsequent
analyses.
Genotyping of the CRELD1 GeneAll sequencing was done by individuals blinded to the case/control
and phenotype status of enrolled subjects. The sequencing was
performed in gene-walking manner, using overlapping sets of
primers covering entire length of reading frame of CRELD1. The
purified PCR amplicons were used as a template for bi-directional
DNA sequencing using the ABI PRISM 3700 DNA Analyzer plat-
form. The sequence obtained was thoroughly scanned and com-
pared against the published sequence (NCBI accession number
NM_001031717.3) of CRELD1.
Functional Prediction of Detected VariantsWe employed an in-silico approach to predict the consequences of
missense variants on protein structure and function by using the
Bioinformatics programsPolyPhen [Sunyaev et al., 2000, 2001] and
MutPred [Li et al., 2009]. Anticipation of change in secondary
structure of protein of interest was done through the SOPMA
[Combet et al., 2000] program from Network Protein Sequencing
Analysis, Pole Bioinformatics Lyonnais, France. The software
Human Splicing Finder (Version 2.4.1) was employed to predict
the effect of polymorphism on transcript splicing [Desmet et al.,
2009].
Statistical AnalysesWe tested the association between genotypes and phenotypes using
Fisher’s exact tests on2� 2or2� 3 tables, as appropriate. For all the
analyses, odds ratio was reported along with a 95% confidence
interval (CI). Difference between the SNP effects in DS and euploid
categories was tested using a logistic regression model with geno-
type, DS/euploid status and interaction as predictors; the test of
interaction terms serves as a test for the difference between DS and
Euploid Individuals.
RESULTS
Sequencing ofCRELD1 in our study cohort identified three known
SNPs, namely rs9878047, rs3774207, and rs73118372, as deter-
mined through reference sequences from NCBI and HapMap
databases. The SNP rs9878047 is located within the intron that
joins exons 8 and 9b. The second one (rs3774207) is a synonymous
variant (c.1119C>T) located in exon 9b and the third one,
rs73118372 is a nonsynonymous variant (c.1136T>C) located
in exon 9b that results in the replacement of the codon ATG
with the codon ACG, leading to change of amino acid from
methionine to threonine at the amino acid position 379 of the
CRELD1 protein (p.M379T; Table I, Fig. 1). This amino acid
substitution is predicted to be potentially deleterious to the protein
by both the MutPred and PolyPhen-2 algorithms. The SNPs
rs9878047 and rs3774207 exhibit complete linkage disequilibrium
(LD) in our results and this association is likely to be universal as we
confirmed it through HapMap published data (D’¼ 1). These
SNPs also show relatively high LD with rs73118372.
In case–control analyses for the SNP rs73118372 in the DS
group, carriers of the minor allele (non wild ‘‘C’’ allele) exhibited
more than a fourfold increased odds of AVSD (OR¼ 4.88, 95% CI
2 AMERICAN JOURNAL OF MEDICAL GENETICS PART A

1.60–14.89, P¼ 0.0036) and in the euploid groups the odds ratio
was 3.64 (95% CI 0.95–13.92; Tables II and III). As expected given
the high LD among the SNPs, carriers of the minor allele for
rs9878047 also showed an elevated risk: OR¼ 3.44 (95% CI
1.29–9.14, P¼ 0.013) in the euploid group (Table II) and
OR¼ 4.27, (95% CI 1.87–9.74, P¼ 0.0002) in the DS group
(Table III). The degree of risk association remained identical for
the SNP locus rs3774207C>T (Tables II and III) with the geno-
types C/T and T/T being more susceptible for AVSD. We did not
observe a significant statistical difference between the odds ratios of
the DS and euploid groups (P¼ 0.46 for the interaction term in the
logistic regression model) for intronic polymorphism rs9878047.
In the second phase of analyses, we looked to see if any specific
haplotype was associated with AVSD risk, combining the above
mentioned three loci. We identified two haplotypes namely
C-T-C and C-T-T in the order of the loci rs9878047-rs3774207-
rs73118372 that are associated with risk among both the DS and
euploid samples (Table IV). The association is stronger inDS group
(P¼ 0.001) and within this category, the risk of AVSD was more
than fourfold with each copy of C-T-C haplotype compared to
T-C-T halotype (95% CI 1.60–14.89, P¼ 0.0035) and more than
three fold with each copy of C-T-T haplotype (95% CI 1.16–11.48,
P¼ 0.027; Table IV). This is highly consistent with the single-SNP
results, and suggests that the real association is likely to be with the
rs9878047-rs3774207 haplotype rather thanwith rs73118372.Hap-
lotype T-C-C (rs9878047-rs3774207-rs73118372) was not found
among both the euploid and DS samples.
Our final analysis step was in-silico functional/risk prediction.
The result of in-silico analysis as predicted byHuman Splice Finder
for the polymorphism rs3774207 (c.1119C>T) suggests a possible
deleterious effect on transcript splicing, as this synonymous change
is predicted to disrupt an exon splicing enhancer. The polymor-
phism rs7311872 (c.1136T>C) is predicted to disrupt an exon
splice silencer (ESS) and creates a new ESE (Table I, Fig. 2).
Moreover, both the programs PolyPhen and MutPred predict
an adverse effect of the non-synoymous variant c.1136T>C
(rs73118372) on CRELD1 secondary structure, which was con-
firmed subsequently in the SOPMA program.
DISCUSSION
CRELD1 is a putative cell adhesion molecule and is involved in
embryonic cardiogenesis. Previous studies have identifiedCRELD1
as a candidate gene for AVSD [Rupp et al., 2002] and mutations in
TABLE I. In Silico Inferred Effect of CRELD1 Polymorphisms Associated With AVSD
SNP locusNucleotidechange
Aminoacid change
Nature ofcodon change
Outcome of various software programs
Polyphen MutPred prediction Human splicing finderrs73118372 c.1136T> C p.M379T Non-synonymous
codingPossiblydamaging
Actionable*hypothesis
Disruption of ESS;creation of new ESE
rs3774207 c.1119C> T p.H373H Synonymous coding — — Disruption ofpotential ESE
rs9878047 c.1049-129T> C — Intronic — — —
ESE, exon splice enhancer; ESS, exon splice silencer.*Actionable hypothesis—variants with a MutPred general score (g)> 0.5 indicating the probability that the amino acid substitution is deleterious coupled with a property score P-value of P< 0.05predicting that specified structural or functional properties of the protein are impacted by the amino acid substitution.
FIG. 1. The chromatogramsshownucleotide changeat various SNP loci of CRELD1. A: Thepolymorphismat rs9878047 in the intron between the exons8
and 9b; B: Polymorphism at rs3774207 in exon 9b causes synonymous codon change; C: Polymorphism at rs73118372 of exon 9b causes
replacement of amino acid methionine to threonine.
GHOSH ET AL. 3

TABLE III. CRELD1 SNP Association With Risk of AVSD in DS Population
Polymorphic Genotype
Frequency
OR (95% CI) P valueDowns without AVSD (N¼ 92) Downs with AVSD (N¼ 121)rs9878047T/T 0.91 0.71 1.00T/C 0.07 0.24 4.186 (1.74–10.05) 0.0008C/C 0.01 0.04 4.88 (0.55–42.68) 0.21rs3774207C/C 0.91 0.71 1.00C/T 0.07 0.24 4.18 (1.74–10.05) 0.0008T/T 0.01 0.04 4.88 (0.55–42.68) 0.21rs73118372T/T 0.91 0.71 1.00T/C 0.04 0.16 4.88 (1.60–14.89) 0.0036
AVSD, atrioventricular septal defect; OR, odds ratio; CI, confidence interval.
TABLE II. CRELD1 SNP Association With Risk of AVSD in the Euploid Population
Polymorphic genotype
Frequency
OR (95% CI) P valueEuploid without AVSD (N¼ 102) Euploid with AVSD (N¼ 96)rs9878047
T/T 0.94 0.82 1.00T/C 0.04 0.12 3.64 (1.13–11.74) 0.03C/C 0.02 0.05 3.03 (0.57–16.08) 0.25
rs3774207C/C 0.94 0.82 1.00C/T 0.04 0.12 3.64 (1.13–11.74) 0.03T/T 0.02 0.05 3.03 (0.57–16.08) 0.25
rs73118372T/T 0.94 0.82 1.00T/C 0.02 0.09 3.64 (0.95–13.92) 0.07
AVSD, atrioventricular septal defect; OR, odds ratio; CI, confidence interval.
TABLE IV. Association of CRELD1 Haplotypes With AVSD Among Euploids and DS Individuals
Haplotypes
Polymorphic loci Phenotype
OR (95% CI) P valuers9878047 rs3774207 rs73118372 With AVSD Without AVSDEuploid (N¼ 198)1 T C T 0.82 0.94 1.002 C T C 0.09 0.03 3.64 (0.95–13.92) 0.073 C T T 0.08 0.03 3.24 (0.83–12.62) 0.11Down syndrome (N¼ 213)1 T C T 0.71 0.92 1.002 C T C 0.16 0.04 4.88 (1.60–14.89) 0.0033 C T T 0.11 0.04 3.566 (1.16–11.48) 0.027
OR, odds ratio; CI, confidence interval.
4 AMERICAN JOURNAL OF MEDICAL GENETICS PART A

this gene cause AVSD in euploid [Robinson et al., 2003; Zatyka
et al., 2005] as well as in DS [Maslen et al., 2006; Ying et al., 2010;
Kusuma et al., 2011] individuals. Considering these observations
together and to gain further insights into the role of CRELD1 in
AVSDpathogenesis,we sequenced theORFs and introns of the gene
to identify any probable risk haplotypes in CRELD1.
The genetic association analyses for individual SNP loci yields a
significant association between AVSD and three polymorphisms
namely, rs9878047 (c.1049-129T>C), rs3774207 (c.1119C>T),
rs73118372 (c.1136T>C) both in euploid and DS groups (Fig. 1).
Among these, the rs73118372 (c.1136T>C) is a nonsynonymous
coding variant that causes an amino acid replacement (p.M379T;
Table I) and its deleterious effect is predicted by the Polyphen and
MutPred bioinformatics programs. The variation in the protein is
predicted to be destabilizing. The SOPMAprogram indicates that it
may result in incorporation of a secondary alpha-helical structure,
whereasMutPred predicts that it will cause a gain ofmethylation at
K378 (P¼ 0.0274) and a loss of stability (P¼ 0.0487). Therefore,
this may be the risk allele that is tagged by the other SNPs in the
haplotype block.
The haplotype study identified C-T-C and C-T-T (rs9878047-
rs3774207-rs73118372) as predisposing to AVSD both in euploid
and DS groups. There was a statistically significant association of
these two haplotypes with AVSD, with much higher penetrance
among DS (Fig. 1; Table IV). The overall risk for developing an
AVSD is 2,000 times greater in DS than euploid individuals,
indicating that trisomy 21 significantly increases susceptibility
although it is not alone sufficient to cause the defect [Maslen
et al., 2006]. The higher degree of association between susceptible
haplotypes and AVSD among DS than in euploids indicates that
there is increased penetrance of a risk allele in DS. This is consistent
with a threshold model of disease where fewer risk factors are
required to breach the disease threshold on the trisomy 21 back-
ground since it already confers substantial risk. By contrast, the
euploid genetic background presents a generally low risk for AVSD
and therefore requires a large numberof risk factors, or risk factorof
larger effect size. In other words, the effect size of this risk variant is
sufficient to push individuals with trisomy 21 past the disease
threshold, whereas it is not sufficient to do the same in lower
risk euploid individuals.
The results of the analyses involving the polymorphic site
rs3774207 are also intriguing. Although the polymorphism
(c.1119C>T) involves a synonymous codon change, it is predicted
to disrupt an exon splicing enhancer (ESE), which could result in
altered pre-mRNA splicing, excluding exon 9B from the final
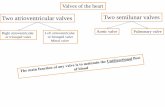
transcript. The CRELD1 has multiple isoforms resulting from
alternate splicing [Rupp et al., 2002]. The SNP rs3774207 is located
in exon 9B, which is an alternately spliced ORF. The 9B isoform
eliminates the transmembranedomain and is thereforenot tethered
to the cell surface (Fig. 2). Since the protein is otherwise identical to
the cell membrane-bound version of CRELD1, it may sequester
ligand thereby modulating CRELD1 activity.
Our results suggest the existence of a potential genetic inter-
action between the SNP loci rs3774207 and rs73118372. Theminor
allele ‘‘T’’ of rs3774207disrupts anESE, so therewouldbe adecrease
in inclusion of exon 9B. On other hand, the minor allele ‘‘C’’ of
rs73118372 creates a new ESE disrupting existing exon splicing
suppressor (ESS), thereby increases the exon 9B inclusion in final
transcript (Fig. 2). However, this allele also alters the protein
configuration, causing it to be unstable. Therefore, if the minor
alleles for both of these SNPs are present together, then there is a
cumulative effect that reduces the amount of expressed CRELD1-
9B isoform. This effect could disrupt a delicate balance between 9B
and non-9B isoforms expression. These are the first potentially
pathogenic alleles in this alternatively spliced exon to be associated
with AVSD, and the first example of an association with a dis-
ruption of an ESE and ESS in CRELD1.
FIG. 2. Alternative splicing of CRELD1 nascent transcript to produce
9b and non-9b isoform proteins. The 9b isoform does not carry the
transmembrane domain and hence is not tethered to cell
membrane unlike to non-9b isoforms that carries the
transmembrane domain. In normal pre-mRNA, exon 9b harbors
both exon splicing enhancer (ESE) and exon splicing silencer
(ESS) elements rs3774207 (T> C) disrupts ESE, results in the
skipping of exon 9b left panel rs73118372 (C> T) a
nonsynonymous variant (p.M379T) disrupts the ESS and creation
of new ESE, results in inclusion of exon 9b right panel.
GHOSH ET AL. 5

In summary, we have successfully demonstrated association
between three SNPs of CRELD1 and AVSD. For the first time,
we identified two CRELD1 haplotypes associated with AVSD
phenotype among DS and euploid individuals. The apparent larger
absolute risk of the haplotype association with DS compared to
euploid individuals supports a threshold model of disease for
AVSD. Previous studies have only identified rare variants in
CRELD1 that may play a role in the etiology of AVSD. It remains
to be seen if this association also occurs in other race/ethnic groups
or is specific to the Bengali population studied here.
ACKNOWLEDGMENTS
We would like to thank the families participated in the study and
professionals who helped us in collection of tissue samples. We are
thankful to the Director, Anthropological Survey of India, Kolkata,
for providing laboratory facilities for experimental work.
REFERENCES
BarlowGM, Chen XN, Shi ZY, Lyons GE, Kurnit DM, Celle L, Spinner NB,Zackai E, Pettenati MJ, Van Riper AJ, Vekemans MJ, Mjaatvedt CH,Korenberg JR. 2001. Down syndrome congenital heart disease: Anarrowed region and a candidate gene. Genet Med 3:91–101.
Combet C, Blanchet C, Geourjon C, Del�eage G. 2000. NPS@: Networkprotein sequence analysis. Trends Biochem Sci 25:147–150.
DaviesGE,HowardCM, FarrerMJ,ColemanMM,Cullen LM,WilliamsonR, Wyse RKH, Kessling AM. 1994. Unusual genotypes in the COL6A1gene in parents of children with trisomy-21 and major congenital heartdefects. Hum Genet 93:443–446.
DaviesGE,HowardCM, FarrerMJ,ColemanMM,Bennett LB,Cullen LM,Wyse RKH, Burn J, Williamson R, Kessling AM. 1995. Genetic variationin the COL6A1 region is associated with congenital heart defects intrisomy 21 (Down’s syndrome). Ann Hum Genet 59:253–269.
Desmet FO, Hamroun D, Lalande M, Collod-Beroud G, Claustres M,Beroud C. 2009. Human Splicing Finder: An online bioinformatics toolto predict splicing signals. Nucleic Acid Res 37:e67.
Digilio MC, Marino B, Toscano A, Giannotti A, Dallapiccola B. 1999.Atrioventricular canal defect without Down syndrome: A heterogeneousmalformation. Am J Med Genet 85:140–146.
Eisenberg LM, Markwald RR. 1995. Molecular regulation of atrioventric-ular valvuloseptal morphogenesis. Circ Res 77:1–6.
FreemanSB, Taft LF,DooleyKJ, AllranK, Sherman SL,HassoldTJ, KhouryMJ, Saker DM. 1998. Population-based study of congenital heart defectsin Down syndrome. Am J Med Genet 80:213–217.
GrossmanTR,Gamliel A,Wessells RJ, Taghli-LamallemO, JepsenK,OcorrK, Korenberg JR, Peterson KL, Rosenfeld MG, Bodmer R, Bier E. 2011.Over-expression of DSCAM and COL6A2 cooperatively generates con-genital heart defects. PLoS Genet 7:e1002344.
Klewer SE, Krob SL, Kolker SJ, Kitten GT. 1998. Expression of type VIcollagen in the developing mouse heart. Dev Dyn 211:248–255.
Kusuma L, Dinesh SM, Savitha MR, Krishnamurthy B, Narayanappa D,Ramachandra NB. 2011. Amaiden report on CRELD1 single-nucleotidepolymorphismassociation in congenital heart diseasepatients ofMysore,South India. Genet Test Mol Biomarkers 15:483–487.
Li B, Krishnan VG, Mort ME, Xin F, Kamati KK, Cooper DN, MooneySD, Radivojac P. 2009. Automated inference of molecular mechanismsof disease from amino acid substitutions. Bioinformatics 25:2744–2750.
LinAE, Belmont J,Malik S. 2006. Heart. In: StevensonRE,Hall JG, editors.Human malformations and related anomalies 2e. Oxford, UK: OxfordUniversity Press. p. 102.
Locke AE, Dooley KJ, Tinker SW, Cheong SY, Feingold E, Allen EG,Freeman SB, Torfs CP, Cua CL, Epstein MP, Wu MC, Lin X, CaponeG, Sherman SL, Bean LJ. 2010. Variation in folate pathway genescontributes to risk of congenital heart defects among individuals withDown syndrome. Genet Epidemiol 34:613–623.
Maslen CL, Babcock D, Robinson SW, Bean LJ, Dooley KJ, Willour VL,Sherman SL. 2006. CRELD1 mutations contribute to the occurrence ofcardiac atrioventricular septal defects in Down syndrome. Am J MedGenet Part A 140A:2501–2505.
Robinson SW, Morris CD, Goldmuntz E, Reller MD, Jones MA, SteinerRD, Maslen CL. 2003. Missense mutations in CRELD1 are associatedwith cardiac atrioventricular septal defects. Am J Hum Genet 72:1047–1052.
Rupp PA, Fouad GT, Egelston CA, Reifsteck CA, Olson SB, Knosp WM,Glanville RW, Thornburg KL, Robinson SW, Maslen CL. 2002. Identi-fication, genomic organization and mRNA expression of CRELD1, thefounding member of a unique family of matricellular proteins. Gene293:47–57.
Sunyaev S, Ramensky V, Bork P. 2000. Towards a structural basis ofhuman nonsynonymous single nucleotide polymorphisms. TrendsGenet 16:198–200.
Sunyaev S, RamenskyV,Koch I, LatheWIII, KondrashovAS, Bork P. 2001.Prediction of deleterious human alleles. Hum Mol Genet 10:591–597.
Guo Y, Shen J, Yuan L, Li F, Wang J, Sun K. 2010. Novel CRELD1 genemutations in patients with atrioventricular septal defect.World J Pediatr6:348–352.
ZatykaM, Priestley M, Ladusans EJ, Fryer AE, Mason J, Latif F, Maher ER.2005. Analysis of CRELD1 as a candidate 3p25 atrioventicular septaldefect locus (AVSD2). Clin Genet 67:526–528.
6 AMERICAN JOURNAL OF MEDICAL GENETICS PART A