
PNA Directed Genome Rare Cutting: New Developments PDF/MFK and Demidov PNA book...
19
1 PAGE PROOFS PAGE PROOFS 9 PNA Directed Genome Rare Cutting: New Developments Maxim D. Frank-Kamenetskii and Vadim V. Demidov Abstract This chapter deals with the PNA- assisted rare cleavage (PARC) of duplex DNA. The technique is a variety of the general ‘Achilles’ heel´ cleavage strategy and uses PNA oligomers to protect very few sites on genomic DNA against enzymatic methylation. As a result, the PARC technique makes it possible to convert common restriction enzymes into a pool of infrequent genome cutters. These artificial genome cutting systems cover the range of recognition specificities, where very few, if any, cutters are now available. Here, we present the PARC- based method for robust purification of yeast artificial chromosomes (YACs) from host chromosomes with similar lengths and demonstrate the PARC potential for rare fragmentation of human DNA. Further progress in the PARC approach also includes the use of pseudocomplementary PNAs (pcPNAs) as sequence-unrestricted duplex DNA-binding ligands.
Transcript of PNA Directed Genome Rare Cutting: New Developments PDF/MFK and Demidov PNA book...

PNA Directed Genome Rare Cutting
1
PAGE PROOFS
PAGE PROOFS
9
PNA Directed Genome RareCutting: New Developments
Maxim D. Frank-Kamenetskiiand Vadim V. Demidov
Abstract
This chapter deals with the PNA-assisted rare cleavage (PARC) ofduplex DNA. The technique is a variety of the general ‘Achilles’ heel´cleavage strategy and uses PNA oligomers to protect very few siteson genomic DNA against enzymatic methylation. As a result, the PARCtechnique makes it possible to convert common restriction enzymesinto a pool of infrequent genome cutters. These artificial genomecutting systems cover the range of recognition specificities, wherevery few, if any, cutters are now available. Here, we present the PARC-based method for robust purification of yeast artificial chromosomes(YACs) from host chromosomes with similar lengths and demonstratethe PARC potential for rare fragmentation of human DNA. Furtherprogress in the PARC approach also includes the use ofpseudocomplementary PNAs (pcPNAs) as sequence-unrestrictedduplex DNA-binding ligands.

Frank-Kamenetskii and Demidov
2
PAGE PROOFS
Background
DNA rare fragmentation is used in genomics for genome analysis andmay find applications in DNA technology for handling with large DNAmolecules. However, a limited number of rare-cutting restrictionenzymes (1-4) and homing endonucleases have been identified (4-7).To extend the range of natural DNA rare cutters, their directedmodification or selection (8, 9), coupling with DNA methylases (10)and employment of specially inserted sites (11, 12) could be used.Also, artificial ‘restriction enzymes´ combining a variety of DNAbinding and cleavage functions have been created (13-15). Still, theprogress in this direction is limited, especially for creation of 8-12 bpcutters (16). Considering that this range of recognition specificitiescorresponds to the most useful sizes of large DNAs–from several dozenkilobases to few megabases, further expansion of approaches for rarecutting the genomes is highly desirable.
In 1996, we proposed a variety of the ‘Achilles’ heel´ rare cleavagestrategy (17, 18), the PARC approach (19-21; see refs. 16 and 20 forschematics), which is based on targeting the double-stranded DNA(dsDNA) with cationic pyrimidine bis-PNAs (22-27). A major ideaunderlying this method is that sequence-specific binding of bis-PNAto relatively short dsDNA sequences should block DNA recognition
Figure 1. Schematics of the PARC method involving pcPNAs. In contrast to triplex-forming pyrimidine bis-PNAs, sequence-unrestricted recognition of dsDNA bypcPNAs via the double-duplex invasion makes it now possible to completely coverthe desired methylation/restriction site. For the original similar PARC schematicswith bis-PNAs, see refs. 16 and 20.
genomic DNA
bind pcPNAsmethylate
inactivate methylasedisrupt PNA/DNA complex
digest byrestriction enzyme
Legend:
site cleavable byrestriction enzyme
methylated site, notcleavable
PNA binding site
pair of pcPNAs

PNA Directed Genome Rare Cutting
3
PAGE PROOFS
PAGE PROOFS
by the DNA methylase in case the PNA binding site overlaps with theenzyme binding site. At that time, only pyrimidine PNAs could beused for dsDNA targeting, and hence for PARC. Nevertheless, thisvariety of PNAs has potentially provided, despite its sequence-limitations, with a large pool of different nonnatural DNA rare cutters(16, 19-21).
Recently, a new kind of PNAs, pseudocomplementary PNAs(pcPNAs), has been designed that can bind to essentially any chosendsDNA site (28-30). In contrast to pyrimidine PNAs that form withdsDNA the invasion triplexes (23, 27, 31), pcPNAs sequence-specifically target the DNA duplex via the double-duplex invasionmode of DNA recognition (28-30). These novel PNAs, when employedin the framework of ‘Achilles’ heel´ strategy (Figure 1), make it alsopossible to infrequently cut genomic DNA (29), thus further extendingthe range of PARC-based DNA rare cutters.
Altogether, the use of pyrimidine bis-PNAs and pcPNAs can readilyyields a number of PARC cutters with 8-12 bp recognition specificities(see examples below). As a result, a difficult task of rare cutting thegenomes with unknown sequences using the diverse cutters becomespossible. This may significantly improve and simplify numeroustechniques in the entire field of DNA analysis, including genomemapping, cloning and sequencing.
Protocol
Materials
High-molecular-weight DNA samples with freely dissolved or agarose-embedded (for very large, sub-megabase or longer DNAs; see refs.32, 33) molecules; a pair of methylation/restriction enzymes withcoincident DNA recognition specificities; bis-PNA or pcPNAs having7-or 8-mer DNA target site overlapping with or embodying theenzymatic targets (bis-PNAs can be purchased from somebiotechnological companies (e.g., from Applied Biosystems) orobtained by manual (34) and automated (35; see also this book)

Frank-Kamenetskii and Demidov
4
PAGE PROOFS
syntheses in a lab from the commercially available protected PNAmonomers; pcPNA oligomers and corresponding protected monomersare presently not commercially available); buffer solutions: PNAbinding buffer (buffer A: any of suitable buffers (10-20 mM) with pH6.5-7.0; addition of 1-2 mM EDTA and 10 mM NaCl is recommended,in case they are not the buffer components); DNA methylase buffer(buffer B: use an appropriate commercially available or self-preparedbuffer for enzymatic methylation with pH close to 7.0 containing EDTAor some other chelating reagent); ‘stop´ buffer solution (buffer C: 1%SDS; 500 mM NaCl; 10 mM EDTA; 50 mM Tris-HCl; pH 9.0;preincubate buffer at 37oC before use to dissolve all components);restriction enzyme buffer (buffer D: use any appropriate commerciallyavailable buffer for restriction digest; to prevent precipitation, avoidK+ cations if possible; addition of SAM or BSA is recommended forsome restriction enzymes).
Step 1: PNA Targeting
For DNA solution, incubate the DNA sample (typically ≤1 µg) withthe requisite PNA(s) in 10-100 µl of buffer A for about 1 h at 37oC.The necessary PNA concentration is usually within 0.2-0.5 µM rangedepending on the PNA binding affinity and incubation time. Afterthat, remove unbound PNA by gel filtration of the sample throughSephadex G-50.
In case of agarose-embedded DNA, PNA-DNA complexes are formedat 37oC by shaking incubation of the 10-50 µl gel sample containing~1 µg of DNA for several hours with 50 µl of 0.2-0.5 µM PNA(s)dissolved in buffer A. Note that more concentrated PNA may berequired for the in-gel PNA targeting, as compared to the similartreatment in solution. The gel-filtration procedure is replaced here bywashing the gel sample twice for 30 min in 100 µl of buffer B.
To enhance the specificity of PNA binding (if necessary), prior washingfrom PNA in high-salt buffer solution (up to 1 M NaCl) and incubationof the sample at elevated temperature (~50oC) for a short time (≤10min) is recommended: incorrect PNA-DNA complexes are less stable

PNA Directed Genome Rare Cutting
5
PAGE PROOFS
PAGE PROOFS
than correct ones, therefore their differential dissociation may improvethe selectivity of DNA targeting (24, 27, 36).
Step 2: DNA Methylation
Next, the gel sample containing DNA targeted by PNA is saturated at4oC (a low temperature is used to prevent the contaminatingendonuclease activity) for about 10 h with the appropriate quantity ofDNA methylase (usually, 10-20 U of freshly diluted methylase arenecessary) in 50 µl of buffer B. To make the enzyme more stable, anaddition of BSA (up to100 µg/ml) to buffer B is advised at this stage.Periodical tipping of the sample is required. Then, SAM is added upto 300 µM and DNA methylation is performed for 2-4 h at 37oC withshaking. Incubating the sample in 100 µl of buffer C for 20-30 min at37oC stops the methylation. Thereafter the methylase is finallyinactivated and the PNA-DNA complex is dissociated by incubationof the sample for about 1 h at near 60oC (dependent on the meltingpoint of the agarose matrix) in the same buffer. Similar treatment isperformed with DNA solutions except less amount of methylase andmethylation time are necessary, and gel filtration is used here to changethe solution.
Step 3: DNA Cleavage
Afterwards, the ‘stop´ solution is removed, and DNA sample isequilibrated at least thrice in 100-200 µl of buffer D for 30 min at37oC with shaking. In case that buffer D contains K+ cations, washingthe DNA sample twice in 100 µl of 50 mM Tris-HCl (pH 7.5) for 30min at 37oC before its equilibration in buffer D is stronglyrecommended to avoid precipitation of dodecyl sulfate potassium saltfrom buffer C. DNA digestion is performed by 5-15 U of a desiredrestriction enzyme for about 5 h at 37oC (in some cases, othertemperatures may be required for restriction enzyme activity) withperiodical shaking by tipping. The digestion is stopped by incubatingthe sample for 20 min at 37oC in 50 mM EDTA (pH 8.0). Similartreatment is performed with DNA solutions except less restriction

Frank-Kamenetskii and Demidov
6
PAGE PROOFS
enzyme and digestion time are necessary, and gel filtration is usedhere to change the solution.
Step 4: PFGE Separation
If necessary, equilibrate the sample in TE buffer (pH 7.5) and checkthe PARC digest by pulsed-field gel electrophoresis (PFGE) or usethis procedure for preparative purposes. Additional materials andequipment: the pulsed-field certified or chromosomal grade agaroseand a PFGE apparatus; use the electrophoretic-pure reagents on allprevious stages and the low-melting agarose plugs compatible withPFGE.
Examples
Example 1: Purification of YACs
YACs are commonly used as convenient vectors to clone very longpieces of DNA and to transfer large genes or gene clusters intomammalian cells (37-39). One of the problems with the YACtechnology stems from the fact that YACs are maintained in yeastcells as single-copy objects and are very similar to yeast endogenouschromosomes in their general construction and size. As a result, theYAC isolation, which is usually based on the PFGE separation (37,40), is often complicated by the presence of comigrating or closelymigrating host chromosomes. Therefore, significant contamination ofPFGE-isolated YACs by endogenous yeast DNA often occurs, whichcan compromise their use for subsequent DNA transfer, subcloning,sequencing and amplification procedures (38, 40, 41). Here we showan application of the PARC method to PFGE purify YACs fromcomigrating endogenous yeast chromosomes with similar lengths (42).
The sequencing data for S. cerevisiae indicate that each yeastchromosome contains several unique 12-bp-long sites with genericsequence Pu7GCGCPy suitable for the PARC strategy. Specifically,the Pu7G octapurine part of these sites can be targeted by bis-PNAs

PNA Directed Genome Rare Cutting
7
PAGE PROOFS
PAGE PROOFS
with sequences Py7C-linker-JPy7 (here and below, J denotespseudoisocytosine and linker consists of three 8-amino-3,6-dioxaoctanoic acid units (22)). The PNA binding will mask the adjacentrecognition site of DNA methylase HhaI (GCGC) from methylation.Consequently, the restriction enzyme HaeII should cut, after the PNAremoval, only those designated yeast chromosome that overlaps witha specific YAC during PFGE and carries the unique unmethylated sitePuGCGCPy.
Note that any chosen 12 bp DNA sequence is met once per ~10 Mb,on average. It is therefore highly unlikely that a YAC, whose sequenceis usually unknown but normally does not exceed 2 Mb, will carry thesame target site for PARC. Hence, YAC should remain intact whereasthe comigrating chromosome will be quantitatively digested. In caseYAC will also be cut by the given bis-PNA/M.HhaI/R.HaeIIcombination (M and R stand here for methylation and restriction), thePARC method with another bis-PNA targeted to different site can beapplied to eliminate only the required endogenous yeast chromosome.It is virtually improbable that the same YAC will be cut again.
Figure 2 demonstrates the feasibility of this approach (42). Yeast strain731_G_2 (CEPH human YAC library) was chosen, which is derivedfrom the commonly used S. cerevisiae AB1380 and contains a YACvector with an insert from human chromosome 2. The size of thisYAC practically coincides with the size of yeast Chr II (see Figure 1,lane 3; here and below, Chr followed by a Roman numeral abbreviatesa specific yeast host chromosome). Chr II contains near its center theAG4A2GCGC2 site suitable for the PARC procedure. We accordinglysplit Chr II into two fragments by using cationic bis-PNA [TC4T2C-linker-JT2J4T](+4) (PNA 1) and a pair of M/R enzymes (Figure 2A).As a result, we segregated YAC from Chr II (Figure 2BC): quantitativedensitometry confirmed that at least 90% of Chr II was cut while ~90%of YAC remained unaltered.
To check the efficiency of the PARC-based YAC purification for DNAsubcloning, we sequenced DNA fragments recovered from the gelslices corresponding to YAC after PFGE separation and HindIIIdigestion and subcloned into pUC19. Before the PARC treatment, 5

Frank-Kamenetskii and Demidov
8
PAGE PROOFS
out of 10 randomly chosen subclones carried Chr II DNA. By contrast,none of 10 randomly chosen subclones obtained after the PARCtreatment contained yeast DNA: they all carried only human DNAderived from YAC.
One more example with another YAC not shown here demonstratesthe universality of the proposed approach (42). This YAC carries afragment of human chromosome 6 and significantly overlaps withboth Chr II and Chr XIV (second yeast chromosome from the top inFigure 2). Thus, now two yeast chromosomes have to be selectivelycut by the PARC method leaving YAC intact. To this end, PNA 2,
Figure 2. Segregation of YAC from Chr II using the PARC method with PNA 1/M.HhaI/R.HaeII combination. (A) The PFGE separation of host chromosomes (yeaststrain AB1380) before (lane 1) and after (lane 2) application of the PARC method.Fr I and Fr II designate two fragments of Chr II, which appear after the PARCmethod is applied. Fr II is very close to Chr III, which is also cut (19). (B) and (C)The PFGE separation of untreated (lanes 3, 5, 7, 9) and PARC-digested (lanes 4, 6,8, 10) DNA extracted from the yeast strain 731_G_2 carrying YAC. For details ofDNA extraction, PARC procedure and PFGE analysis, see ref. 42. Southern blothybridization (lanes 5, 6, 9, 10) was performed with PCR-generatedchemiluminescent probes (random-primer labeling) using Chr II (B) or human Alurepeats (C) as templates. All other lanes were visualized by staining with EtBr.Reproduced with minor modifications from ref. 42.

PNA Directed Genome Rare Cutting
9
PAGE PROOFS
PAGE PROOFS
[T2C2T3C-linker-JT3J2T2](+5), was additionally employed to target ChrXIV. This chromosome carries the PARC target site A2G2A3GCGC2
for PNA 2/M.HhaI/R.HaeII combination near the chromosomal end.Hence, the PARC digest is expected to result in a large fragment ofChr XIV coinciding with Chr X (third yeast chromosome from thetop in Figure 2), which migrates well ahead of YAC.
Using this pair of PNAs and the M.HhaI/R.HaeII pair of enzymes,successful segregation of the second YAC from two comigratingchromosomes has been achieved (42). Altogether, these data provethat the PARC procedure based on the M.HhaI/R.HaeII pair of enzymesand a pool of appropriate bis-PNAs may be an efficient tool forpurification of intact YACs by creating convenient PFGE ‘windows’in the corresponding range of DNA lengths. Due to very high selectivityof the PARC strategy, YACs must remain intact whereas the hostchromosomes will be completely degraded directly in the agarose gelplug carrying extracted YACs.
Example 2: Use of pcPNAs
We have found that pcPNAs as short as octamers form stable andsequence-specific complexes with duplex DNA and effectivelycompete with DNA methylases for common binding sites (29).Interference of pcPNAs with methylases protects selected DNA sitescontaining all four nucleobases from enzymatic methylation. ThepcPNA-assisted protection against enzymatic methylation is moreefficient when the PNA-binding site embodies the methylase-recognition site rather than overlaps it (see schematics in Figure 1).The experiment, whose results are presented in Figure 3, demonstratesthat this pcPNA ability can be used for sequence-unrestricted DNArare cleavage, similar to pyrimidine bis-PNAs.
In this study, the self-pseudocomplementary PNA 3,[sUsUGDsUCDD](+4), was employed that contains 2,6-diaminopurine(D) and 2-thiouracil (sU) as pseudocomplementary substitutes for Aand T, respectively (29). 35.9-kb-long adenovirus-2 DNA that carriesthe pcPNA-binding sites at positions 4.0 and 29.4 kb was subjected to

Frank-Kamenetskii and Demidov
10
PAGE PROOFS
the PARC treatment using the PNA 3/M.dam/R.DpnII combination.Note here that binding site for PNA 3 embodies the M.dam/R.DpnIIrecognition site, GATC. Also note that besides a pair of correct PNA-binding sites, two adenoviral DNA sites partially complementary toPNA 3 are located at 15.3 and 32.9 kb. They have the identicalsequence, [G]TGATCAA, which is end-mismatched relative to thepcPNA binding site (the mismatched nucleobase is in brackets). Theseare the only singly-mismatched PNA-binding sites on adenoviral DNAthat include the correct methylation/restriction site.
Figure 3, lane 2 shows that unprotected/unmethylated adenoviral DNAis digested by R.DpnII into many short fragments. When this restrictionenzyme is used after binding of 1 µM PNA 3 and subsequentmethylation with M.dam, only a few large fragments are observed(Figure 3, lane 5). The lengths of the resulting DNA fragments (4, 6.5,11, 15 and 25 kb) correspond to the PNA-assisted protection of three
Figure 3. Application of the pcPNA-based PARC method to rare cut adenovirus-2DNA with the PNA 3/M.dam/R.DpnII combination. Lane 1: control without anytreatment; lane 2: the R.DpnII digest; lane 3: the R.DpnII digest with prior methylation(no pcPNA); lanes 4 and 5: the R.DpnII digest with prior PNA incubation (3 and 1µM PNA, respectively) and subsequent methylation. Lanes M1 and M2 are 1 and 5kbp size ladders. For details of PARC procedure and PFGE analysis, see ref. 29,from which this figure is reproduced.

PNA Directed Genome Rare Cutting
11
PAGE PROOFS
PAGE PROOFS
sites in adenoviral DNA. Thus, as expected, full protection of the twocorrect PNA-binding sites plus partial protection of one of the end-mismatched sites (located at 15.3 kb) was observed. Although bothend-mismatched PNA-binding sites are sequence-identical to eachother, only one of them was evidently protected from methylation byPNA 3. The reason for this is presently unknown and requires furtherstudies.
Lower concentrations of PNA 3 resulted in incomplete digestion ofviral DNA due to incomplete binding of PNA and, as a result, lack offull protection of PNA-binding sites from enzymatic methylation (notshown). Interestingly, three-fold increase of the PNA concentrationalso yielded non-digested DNA along with a pair of longer, about 29and 32 kb, fragments again due to incomplete PNA binding (Figure 3,lane 4). Note that at a higher PNA concentration 11 and 15 kb fragmentscorresponding to mismatched PNA binding were observed with muchlower yield. We ascribe this effect to self-inhibition of the pcPNAinvasion into dsDNA due to the polycationic character of PNA 3 (30).At elevated PNA concentrations, positively charged PNA stabilizesthe DNA double helix against PNA invasion, as other cations do (29).
Based on these data, we therefore conclude that use of pcPNAs withinthe framework of the PARC strategy makes it possible to convert 4-bp frequent DNA cutter into 7-8 bp rare cutters. They both recognizeand cut the dsDNA sites with mixed sequence of all four nucleobases.Mention that thus obtained PARC-based cutters have recognitionspecificities, TGATCAA and TTGATCAA, previously beingunavailable.
Example 3: Rare Cut of Human DNA
Figure 4 demonstrates the results of the ‘blind´ PARC treatment ofthe YAC-cloned human DNA without prior knowledge of its sequence.In this example, 1.8-Mb-long YAC from yeast strain 680_C_10 (CEPHhuman YAC library) was chosen that carries a large insert from humanchromosome 16. Both bis-PNA (PNA 1) and pcPNA (PNA 3) wereindependently tested here.
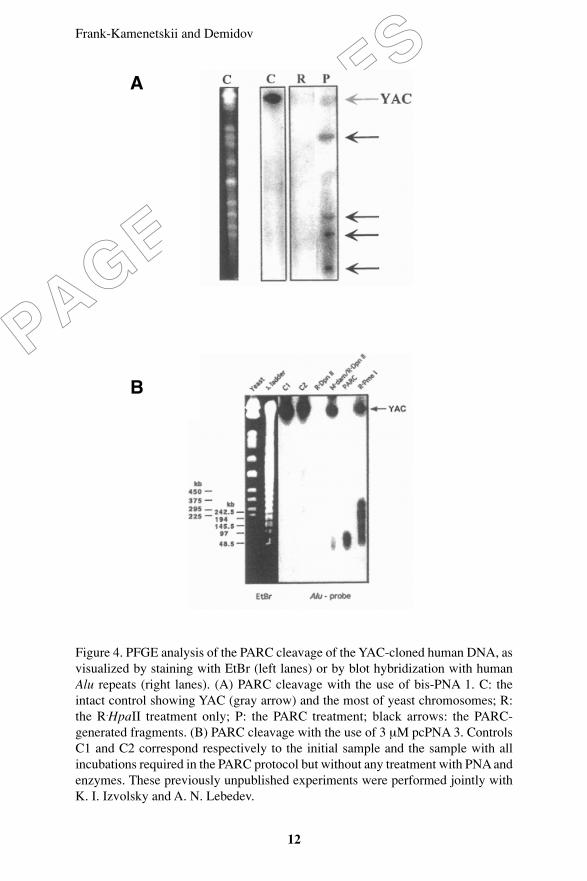
Frank-Kamenetskii and Demidov
12
PAGE PROOFS
Figure 4. PFGE analysis of the PARC cleavage of the YAC-cloned human DNA, asvisualized by staining with EtBr (left lanes) or by blot hybridization with humanAlu repeats (right lanes). (A) PARC cleavage with the use of bis-PNA 1. C: theintact control showing YAC (gray arrow) and the most of yeast chromosomes; R:the R.HpaII treatment only; P: the PARC treatment; black arrows: the PARC-generated fragments. (B) PARC cleavage with the use of 3 µM pcPNA 3. ControlsC1 and C2 correspond respectively to the initial sample and the sample with allincubations required in the PARC protocol but without any treatment with PNA andenzymes. These previously unpublished experiments were performed jointly withK. I. Izvolsky and A. N. Lebedev.
A
B

PNA Directed Genome Rare Cutting
13
PAGE PROOFS
PAGE PROOFS
Figure 4A shows that using the PNA 1/M.HpaII/R.HpaII combination,the YAC under study is rarely cut into four major fragments withlengths from ~100 to ~800 kb. Thus, the PARC combination used inthis experiment works as a 9-bp rare cutter. Figure 4B shows that useof the PNA 3/M.dam/R.DpnII combination yields dozens of unresolvedYAC fragments within the 50-90 kb size range, as expected for a 8-bprare cutter. This digest is compatible with that of the natural 8-bp cutter,R.PmeI, whose recognition site, GTTTAAAC, has the same basecomposition as the pcPNA-based PARC cutter. Note that R.DpnII alonecleaves YAC into a number of very short (≤1 kb), fast movingfragments that leave the gel during the PFGE analysis of largerfragments and hence are not detected in Figure 4B.
We may therefore conclude that by choosing an appropriatecombination of bis-PNA and/or pcPNA with some pair of methylation/restriction enzymes, the recognition specificity of PARC cutters canbe adjusted in a desired way to be used in a rare fragmentation ofgenomic DNA with unknown sequence.
Discussion
The data presented in this chapter clearly demonstrate the promisingPNA potential to provide the genomic researchers with essentiallyunlimited number of DNA rare cutters featuring 8-12 bp recognitionspecificities. Consequently, the PNA-assisted DNA rare fragmentationcould be done both in a directed way using the known DNA sequenceand absolutely ‘blindly´ without any prior sequence information, asour examples show. Note in this connection that another quite popularmethod of DNA rare cleavage with similar to PARC methodology,RARE (most recently reviewed in ref. 43), can cut DNA only at siteswith known sequences.
One more drawback of RARE, the non-specific background cleavage,is due to both the relatively low specificity of RecA-assistedhybridization (44, 45) and the presence of magnesium-activatedmethylase-contaminating nucleases (46). Since the PARC method doesnot require magnesium cations at the PNA-targeting/methylation steps

Frank-Kamenetskii and Demidov
14
PAGE PROOFS
and because the formation of PNA-DNA complexes is a highlysequence-specific process (23-30), PARC can become the method ofchoice in some applications.
Finally note that the PARC method, as any ‘Achilles’ heel´-basedapproach, leaves the resulting fragmented genome thoroughlymethylated, which may impose some limitations on the further use ofthe PARC products. As an option, another PNA-based, methylation-free method can be used for DNA rare cut in case non-methylatedproducts are required. This alternative method involves the use of PNAopeners (47; also see the next chapter of this book) and endonucleasesspecific to single-stranded DNA, e.g. S1 or mung bean nucleases,which site-specifically attack the DNA site locally exposed by PNAopeners (48, 49).
Acknowledgements
We thank Peter E. Nielsen, Michael Egholm and James M. Coull forthe continuous providing our research group with PNAs. We appreciateall colleagues contributed to these studies. We are also grateful toBoston University and the National Institutes of Health for financialsupport.
References
1. Kovall, R.A., and Matthews, B.W. 1999. Type II restrictionendonucleases: structural, functional and evolutionary relationships.Curr. Opin. Chem. Biol. 3: 578-583.
2. Pingoud, A., and Jeltsch, A. 2001. Structure and function of type IIrestriction endonucleases. Nucleic Acids Res. 29: 3705-3727.
3. Chandrasegaran, S. 2001. Restriction enzymes. In: Encyclopediaof Life Sciences. Nature Publishing Group. p.1-7.
4. Roberts, R.J., and Macelis, D. 2001. REBASE – restriction enzymesand methylases. Nucleic Acids Res. 29: 268-269.

PNA Directed Genome Rare Cutting
15
PAGE PROOFS
PAGE PROOFS
5. Chevalier, B.S., and Stoddard, B.L. 2001. Homing endonucleases:structural and functional insight into the catalysts of intron/inteinmobility. Nucleic Acids Res. 29: 3757-3774.
6. Thierry, A., Perrin, A., Boyer, J., Fairhead, C., Dujon, B., Frey, B.,and Schmitz, G. 1991. Cleavage of yeast and bacteriophage T7genomes at a single site using the rare cutter endonuclease I-Sce I.Nucleic Acids Res. 19: 189-190.
7. Bremer, M.C.D., Gimble, F.C., Thorne,r J., and Smith, C.L. 1992.VDE endonuclease cleaves Saccharomyces cerevisiae genomic DNAat a single site: physical mapping of the VMA1 gene. Nucleic AcidsRes. 20: 5484.
8 Lanio, T., Jeltsch, A., and Pingoud, A. 2000. On the possibilities andlimitations of rational protein design to expand the specificity ofrestriction enzymes: a case study employing EcoRV as the target.Protein Eng. 13: 275-281.
9. Gruen, M., Chang, K., Serbanescu, I., and Liu, D.R. 2002. An invivo selection system for homing endonuclease activity. NucleicAcids Res. 30:e29.
10. Nelson, M., and McClelland, M. 1992. Use of DNAmethyltransferase/endonuclease enzyme combinations for megabasemapping of chromosomes. Methods Enzymol. 216: 279-303.
11. Hanish, J., and McClelland, M. 1991. Enzymatic cleavage of abacterial chromosome at a transposon-inserted rare site. NucleicAcids Res. 19: 829-832.
12. Vernick, K.D., and McCutchan, T.F. 1998. A novel class ofsupercoil-independent nuclease hypersensitive site is comprised ofalternative DNA structures that flank eukaryotic genes. J. Mol. Biol.279: 737-751.
13. Dervan, P.B. 1992. Reagents for the site-specific cleavage ofmegabase DNA. Nature. 359: 87-88.
14. Helene, C. 1993. Sequence-selective recognition and cleavage ofdouble-helical DNA. Curr. Opin. Biotechnol. 4: 29-36.
15. Chen, C.B., Milne, L., Landgraf, R., Perrin, D.M., and Sigman,D.S. 2001. Artificial nucleases. ChemBioChem 2: 735-740.

Frank-Kamenetskii and Demidov
16
PAGE PROOFS
16. Demidov, V.V., and Frank-Kamenetskii, M.D. 1999. PNA directedgenome rare cutting. In: Peptide Nucleic Acids: Protocols andApplications. P.E. Nielsen and M. Egholm, eds. Horizon ScientificPress, Wymondham, UK, p. 163-174.
17. Koob, M. 1992. Conferring new cleavage specificities on restrictionendonucleases. Methods Enzymol. 216: 321-329.
18. Koob, M.D. 1995. Achilles’ cleavage. In: Molecular Biology andBiotechnology: A Comprehensive Desk Reference. R.A. Meyers,ed. VCH Publishers, New York, USA. p. 1-3.
19. Veselkov, A.G., Demidov, V.V., Frank-Kamenetskii, M.D., andNielsen, P.E. 1996. PNA as a rare genome-cutter. Nature. 379: 214.
20. Veselkov, A.G., Demidov, V.V., Nielsen, P.E., and Frank-Kamenetskii, M.D. 1996. A new class of genome rare cutters. NucleicAcids Res. 24: 2483-2488.
21. Frank-Kamenetskii, M.D., Veselkov, A.G., and Demidov, V.V.1999. Nucleic acid clamps. US patent No. 6,004,750.
22. Egholm, M., Christensen, L., Dueholm, K.L., Buchardt, O., Coull,J., and Nielsen, P.E. 1995. Efficient pH-independent sequence-specific DNA binding by pseudoisocytosine-containing bis-PNA.Nucleic Acids Res. 23: 217-222.
23. Demidov, V.V., Yavnilovich, M.V., Belotserkovskii, B.P., Frank-Kamenetskii, M.D., and Nielsen, P.E. 1995. Kinetics and mechanismof polyamide “peptide”. nucleic acid binding to duplex DNA. Proc.Natl. Acad. Sci. USA. 92: 2637-2641.
24. Demidov, V.V., Yavnilovich, M.V., and Frank-Kamenetskii, M.D.1997. Kinetic analysis of specificity of duplex DNA targeting byhomopyrimidine PNAs. Biophys. J. 72: 2763-2769.
25. Kuhn H., Demidov, V.V., Frank-Kamenetskii, M.D., and Nielsen,P.E. 1998. Kinetic sequence discrimination of cationic bis-PNAsupon targeting of double-stranded DNA. Nucleic Acids Res. 26: 582-587.
26. Kuhn H., Demidov, V.V., Nielsen, P.E., and Frank-Kamenetskii,M.D. 1999. An experimental study of mechanism and specificity ofpeptide nucleic acid PNA. binding to duplex DNA. J. Mol. Biol.

PNA Directed Genome Rare Cutting
17
PAGE PROOFS
PAGE PROOFS
286: 1337-1345.
27. Demidov, V.V., and Frank-Kamenetskii, M.D. 2001. Sequence-specific targeting of duplex DNA by peptide nucleic acids via triplexstrand invasion. Methods. 23: 108-122.
28. Lohse, J., Dahl, O., and Nielsen, P.E. 1999. Double duplex invasionby peptide nucleic acid: a general principle for sequence-specifictargeting of double-stranded DNA. Proc. Natl. Acad. Sci. USA. 96:11804-11808.
29. Izvolsky, K.I., Demidov, V.V., Nielsen, P.E., and Frank-Kamenetskii, M.D. 2000. Sequence-specific protection of duplexDNA against restriction and methylation enzymes bypseudocomplementary PNAs. Biochemistry. 39: 10908-10913.
30. Demidov, V.V., Protozanova, E., Izvolsky, K.I., Price, C., Nielsen,P.E., and Frank-Kamenetskii, M.D. 2002. Kinetics and mechanismof the DNA double helix invasion by pseudocomplementary peptidenucleic acids. Proc. Natl. Acad. Sci. USA. 99: 5953-5958.
31. Nielsen, P.E., Egholm, M., and Buchardt, O. 1994. Evidence for(PNA)2/DNA triplex structure upon binding of PNA to dsDNA bystrand displacement. J. Mol. Recog. 7: 165-170.
32. Sambrook, J., Fritsch, E.F., and Maniatis, T. 1989. MolecularCloning: A Laboratory Manual, Book 1, chapter 6.53. C. Nolan, ed.CSHL Press, Plainview, USA.
33. Finney, M. 1995. Preparation and analysis of DNA. Pulsed-fieldgel electrophoresis. In: Short Protocols in Molecular Biology. F.Ausubel et al., eds. Wiley, New York, USA. p. 2.15-2.19.
34. Norton, J.C., Waggenspack, J.H., Varnum, E., and Corey, D.R.1995. Targeting peptide nucleic acid-protein conjugates to structuralfeatures within duplex DNA. Bioorg. Med. Chem. 3: 437-445.
35. Mayfield, L.D., and Corey, D.R. 1999. Automated synthesis ofpeptide nucleic acids and peptide nucleic acid-peptide conjugates.Anal. Biochem. 268: 401-404.
36. Krasil’nikova, M.M., and Veselkov, A.G. 1996. Enhancing thespecificity of peptide-nucleic acid binding with DNA. Mol. Biol.Mosk.. 30: 226-230.

Frank-Kamenetskii and Demidov
18
PAGE PROOFS
37. Whittaker, P.A., and Anand, R. 1995. Cloning from PFGE-purifiedgenomic DNA fragments and YACs. In: Pulsed Field GelElectrophoresis. A Practical Approach. A.P. Monaco, ed. OxfordUniversity Press, New York, USA. p. 69-94.
38. Larin, Z. 1995. Functional analysis of mammalian genomes usingyeast artificial chromosomes. Ibidem. p. 139-157.
39. Mendez, M.J., Green, L.L., Corvalan, J.R., Jia, X.C., Maynard-Currie, C.E., Yang, X.D., Gallo, M.L., Louie, D.M., Lee, D.V.,Erickson, K.L., Luna, J., Roy, C.M., Abderrahim, H., Kirschenbaum,F., Noguchi, M., Smith, D.H., Fukushima, A., Hales, J.F., Klapholz,S., Finer, M.H., Davis, C.G., Zsebo, K.M., and Jakobovits, A. 1997.Functional transplant of megabase human immunoglobulin locirecapitulates human antibody response in mice. Nature Genet. 15:146-156.
40. Whittaker P.A., Mathrubutham M., and Wood L. 1993. Constructionof phage sublibraries from nanogram quantities of YAC DNA purifiedby preparative PFGE. Trends Genet. 9: 195-196.
41. Parimoo, S., Patanjali, S.R., Shukla, H., Chaplin, D.D., andWeissman, S.M. 1991. cDNA selection: efficient PCR approach forthe selection of cDNAs encoded in large chromosomal DNAfragments. Proc. Natl. Acad. Sci. USA. 88: 9623-9627.
42. Izvolsky, K.I., Demidov, V.V., Bukanov, N.O., and Frank-Kamenetskii, M.D. 1998. Yeast artificial chromosome segregationfrom host chromosomes with similar lengths. Nucleic Acids Res.26: 5011-5012.
43. Ferrin, L.J. 2001. Flexible genetic engineering using RecA protein.Mol. Biotechnol. 18:233-241.
44. Schoenfeld, T., Harper T.A., and Slater M.R. 1995. Simplifiedprotocols for using RecA cleavage and protection to target restrictionenzyme action. Promega Notes. 50: 14-17.
45. Malkov, V.A., Sastry, L., and Camerini-Otero, R.D. 1997. RecAprotein assisted selection reveals a low fidelity of recognition ofhomology in a duplex DNA by an oligonucleotide. J. Mol. Biol.271: 168-177.

PNA Directed Genome Rare Cutting
19
PAGE PROOFS
PAGE PROOFS
46. Gnirke, A., Iadonato, S., Kwok, P., and Olson, M. 1994. Physicalcalibration of yeast artificial chromosome contig maps by RecA-assisted restriction endonuclease (RARE) cleavage. Genomics. 24:199-210.
47. Demidov, V.V. 2001. PD-loop technology: PNA openers at work.Expert Rev. Mol. Diagn. 1: 343-351.
48. Demidov, V, Frank-Kamenetskii, M.D., Egholm, M., Buchardt,O., and Nielsen, P.E. 1993. Sequence selective double strand DNAcleavage by PNA targeting using nuclease S1. Nucleic Acids Res.21: 2103-2107.
49. Demidov, V.V., and Frank-Kamenetskii, M.D. 2002. PNA openersand their applications. In: Peptide Nucleic Acids: Methods andProtocols. P.E. Nielsen, ed. Humana Press, Totowa, USA. p. 119-130.


















