
Radical Cations•+: Generation, Reactivity, Stability 5Me 5H –6.5 26.1 e H H H H H H–B+ B...
65
Radical Cations •+ : Generation, Reactivity, Stability MacMillan Group Meeting 4-27-11 by Anthony Casarez R A R A
-
Upload
nguyendieu -
Category
Documents
-
view
216 -
download
1
Transcript of Radical Cations•+: Generation, Reactivity, Stability 5Me 5H –6.5 26.1 e H H H H H H–B+ B...

Radical Cations•+: Generation, Reactivity, Stability
MacMillan Group Meeting 4-27-11
by Anthony Casarez
R A R A

Three Main Modes to Generate Radical Cations
Electrochemical oxidation (anodic oxidation)
Chemical oxidation
Photoinduced electron transfer (PET)
D A D A
DAnode
D
D A D A* D Ah!
D A D* A D Ah!
1)
2)
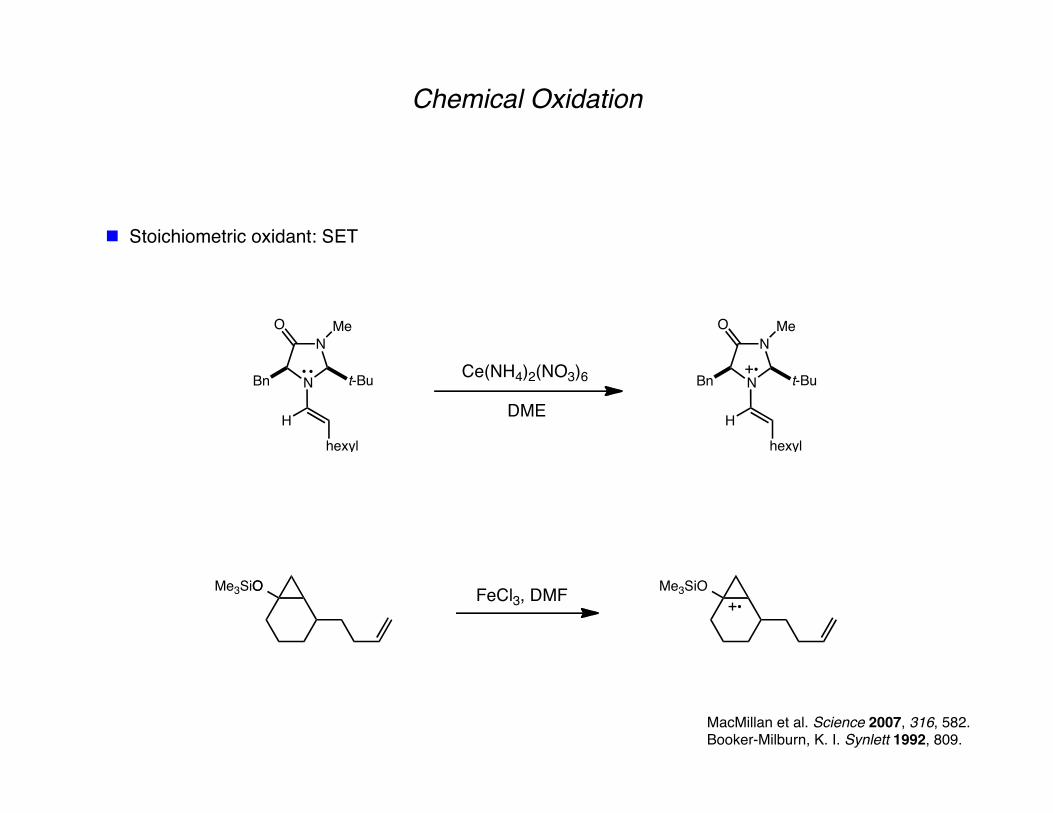
Chemical Oxidation
Stoichiometric oxidant: SET
MacMillan et al. Science 2007, 316, 582. Booker-Milburn, K. I. Synlett 1992, 809.
H
hexyl
DME
N
NBn
O Me
t-Bu
H
hexyl
N
NBn
O Me
t-BuCe(NH4)2(NO3)6
OMe3SiOFeCl3, DMF
Me3SiO

Photoinduced Electron Transfer
PET: Organic arene
OMe
NC CN
h!, MeCN, MeOH
OMe
CN
CN
PET: Metal mediated
Arnold, D. R.; Maroulis, A. J. J. Am. Chem. Soc. 1976, 98, 5931. Ischay, M. A.; Lu, Z.; Yoon, T. P. J. Am. Chem. Soc. 2010, 132, 8572.
R
MeOh!, MgSO4,
MeNO2, Ru(bpy)32+
MeN NMeR
MeO
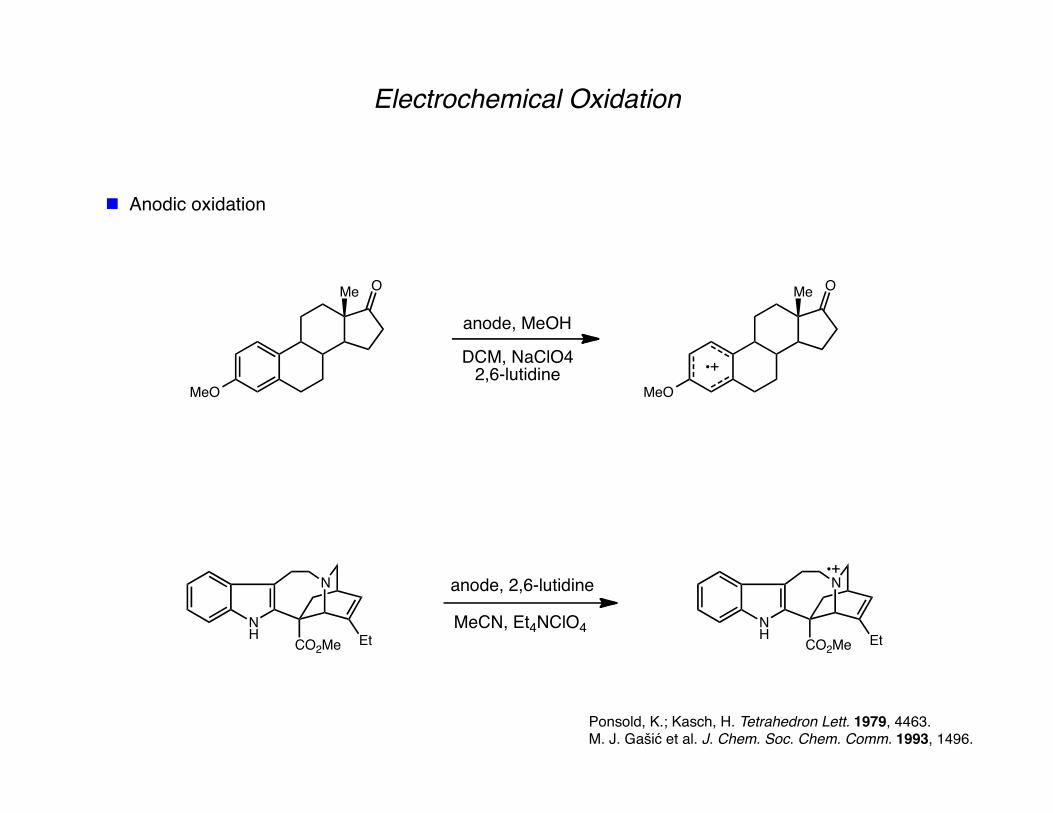
Electrochemical Oxidation
Anodic oxidation
Ponsold, K.; Kasch, H. Tetrahedron Lett. 1979, 4463. M. J. Gašić et al. J. Chem. Soc. Chem. Comm. 1993, 1496.
MeO
MeO
anode, MeOH
DCM, NaClO42,6-lutidine
MeO
MeO
NH
N
EtCO2Me
anode, 2,6-lutidine
MeCN, Et4NClO4 NH
N
EtCO2Me

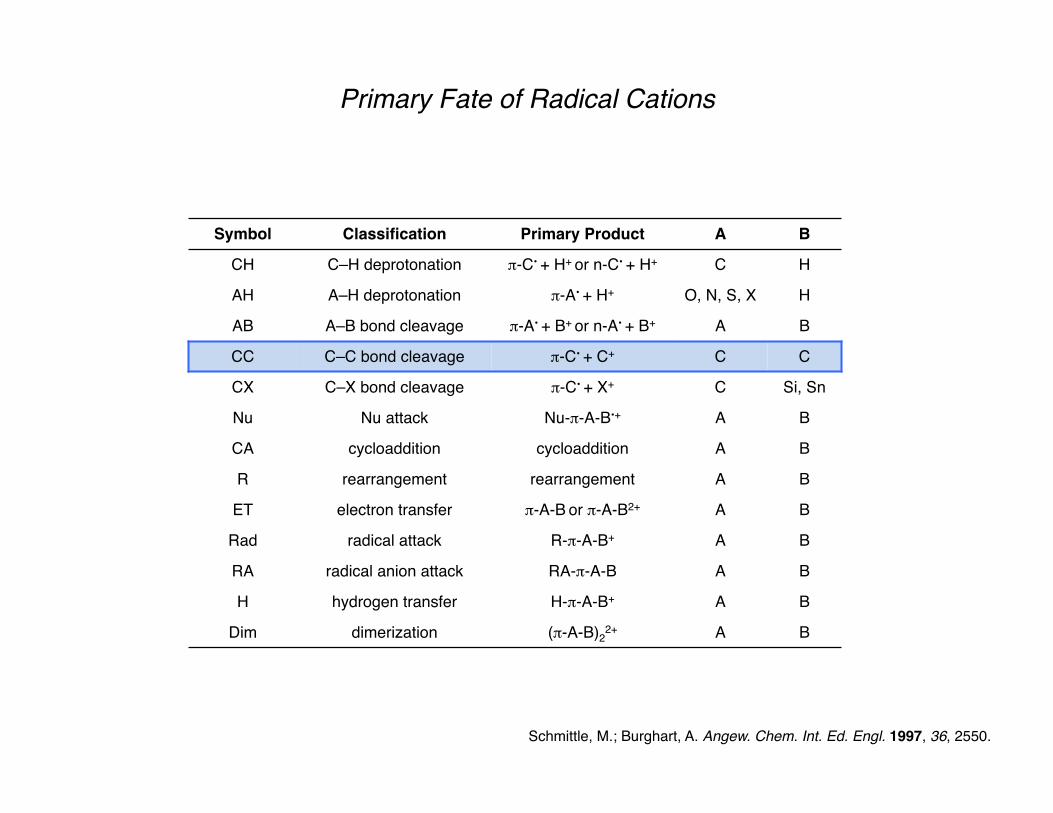
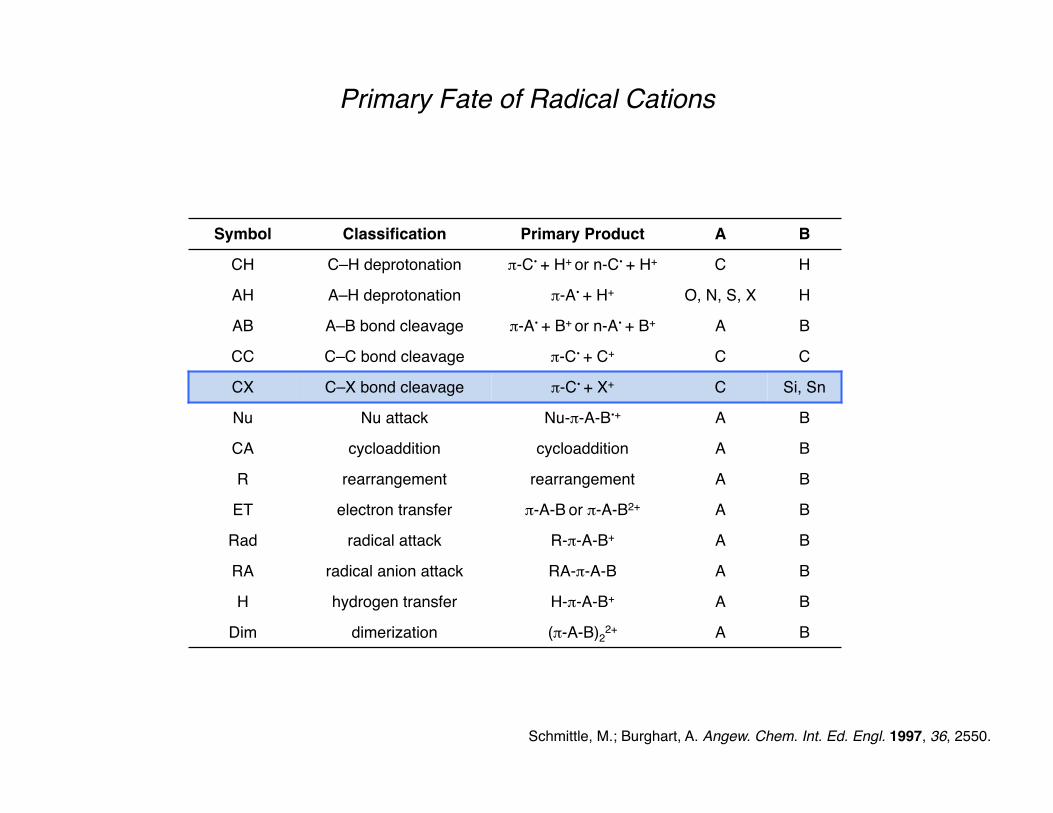
Primary Fate of Radical Cations
A radical cation will be generated from the electrophore on the molecule with the lowest oxidation potential (usually π or n, where n = nonbonding electrons).
The chemistry of the resultant radical cation is determined from the functionality around its periphery.
Deprotonation of the radical cation is a major pathway, resulting in a radical which adheres to typical radical reactivity patterns.
Secondary reactions play a major role in our generation and use of radical cations.
Schmittle, M.; Burghart, A. Angew. Chem. Int. Ed. Engl. 1997, 36, 2550.
Key Points
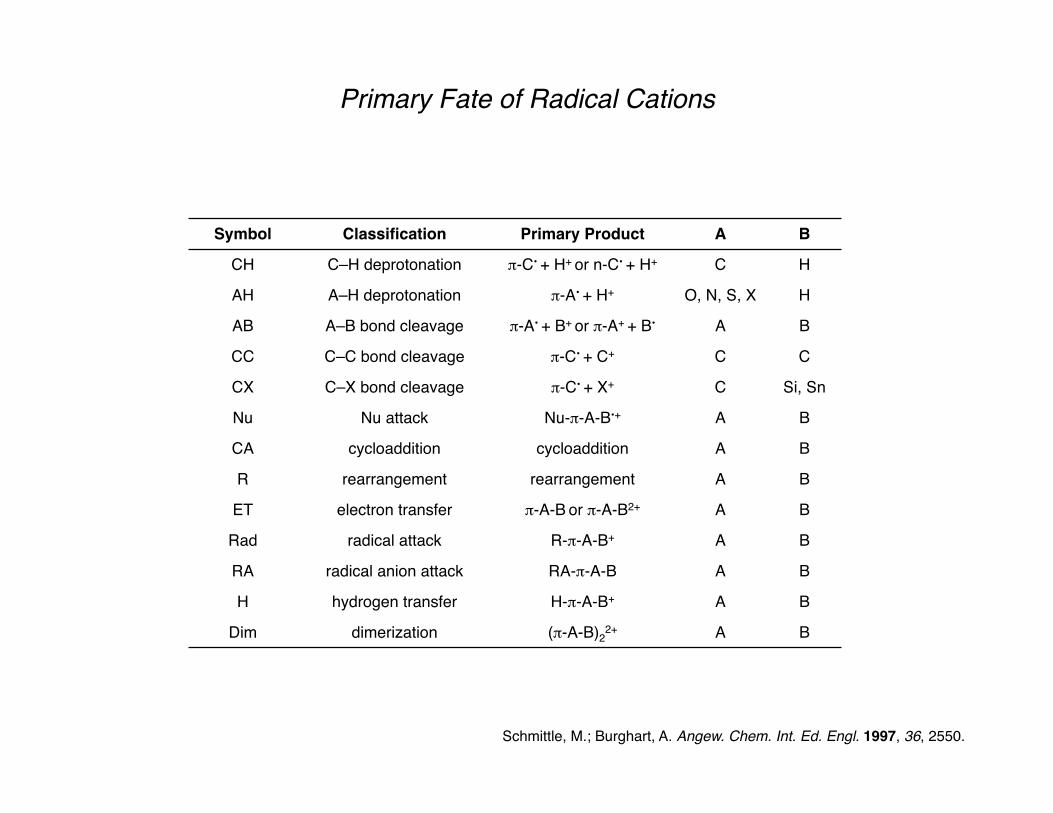
Primary Fate of Radical Cations
Schmittle, M.; Burghart, A. Angew. Chem. Int. Ed. Engl. 1997, 36, 2550.
Symbol Classification Primary Product A B
CH C–H deprotonation π-C• + H+ or n-C• + H+ C H
AH A–H deprotonation π-A• + H+ O, N, S, X H
AB A–B bond cleavage π-A• + B+ or π-A+ + B• A B
CC C–C bond cleavage π-C• + C+ C C
CX C–X bond cleavage π-C• + X+ C Si, Sn
Nu Nu attack Nu-π-A-B•+ A B
CA cycloaddition cycloaddition A B
R rearrangement rearrangement A B
ET electron transfer π-A-B or π-A-B2+ A B
Rad radical attack R-π-A-B+ A B
RA radical anion attack RA-π-A-B A B
H hydrogen transfer H-π-A-B+ A B
Dim dimerization (π-A-B)22+ A B

Primary Fate of Radical Cations
Schmittle, M.; Burghart, A. Angew. Chem. Int. Ed. Engl. 1997, 36, 2550.
Symbol Classification Primary Product A B
CH C–H deprotonation π-C• + H+ or n-C• + H+ C H
AH A–H deprotonation π-A• + H+ O, N, S, X H
AB A–B bond cleavage π-A• + B+ or π-A+ + B• A B
CC C–C bond cleavage π-C• + C+ C C
CX C–X bond cleavage π-C• + X+ C Si, Sn
Nu Nu attack Nu-π-A-B•+ A B
CA cycloaddition cycloaddition A B
R rearrangement rearrangement A B
ET electron transfer π-A-B or π-A-B2+ A B
Rad radical attack R-π-A-B+ A B
RA radical anion attack RA-π-A-B A B
H hydrogen transfer H-π-A-B+ A B
Dim dimerization (π-A-B)22+ A B
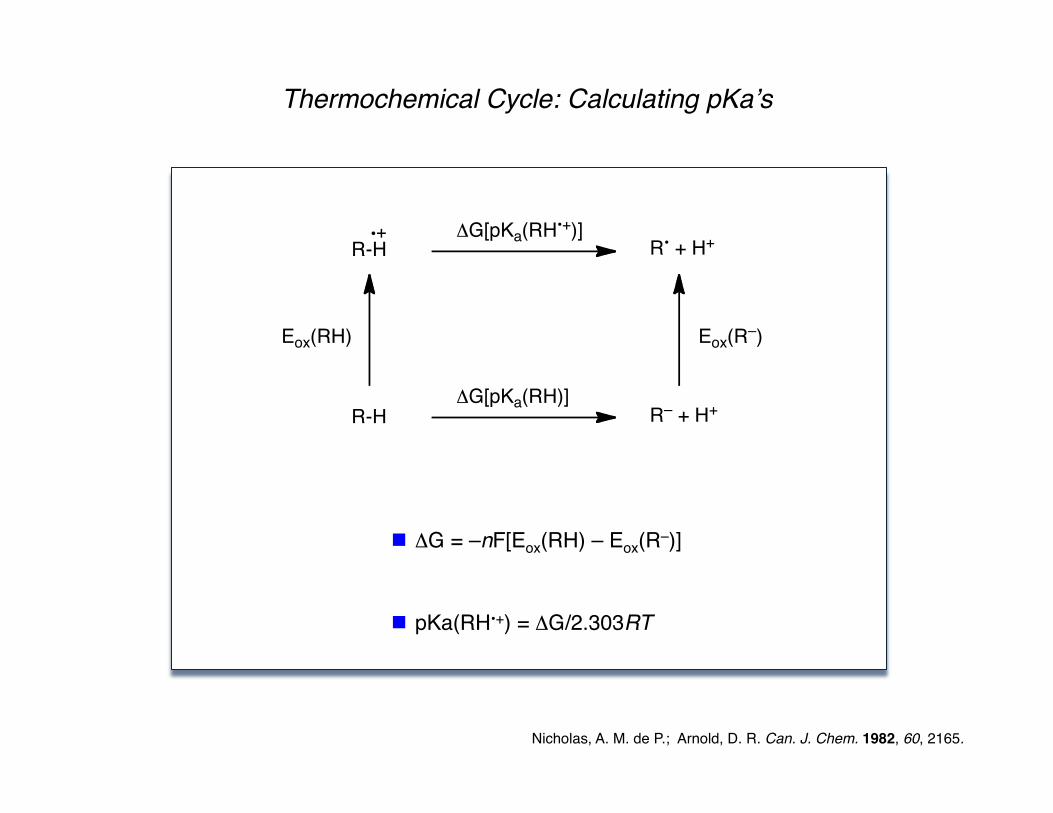
Nicholas, A. M. de P.; Arnold, D. R. Can. J. Chem. 1982, 60, 2165.
Thermochemical Cycle: Calculating pKaʼs
ΔG = –nF[Eox(RH) – Eox(R–)]
pKa(RH•+) = ΔG/2.303RT
R-H
Eox(RH)
R-H!G[pKa(RH•+)]
R• + H+
!G[pKa(RH)]
Eox(R–)
R– + H+

Deprotonation
a) Bordwell et al. J. Phys. Org. Chem. 1988, 1, 209. b) Bordwell et al. J. Phys. Org. Chem.1988, 1, 225. c) Bordwell, F. G.; Cheng, J.-P. J. Am. Chem. Soc. 1989, 111, 1792. d) Bordwell, F. G.; Satish, A. V. J. Am. Chem. Soc. 1992, 114, 10173. e) Bordwell, F. G.; Cheng, J.-P. J. Am. Chem. Soc. 1988, 110, 2872. f) Bordwell, F. G.; Cheng, J.-P.; Bausch, M. J. J. Am. Chem. Soc. 1988, 110, 2867.
Acidity of a radical cation is severely increased compared to the neutral counterpart.
R-H pKa (π-RH•+) pKa (π-RH) reference
PhCH2CN –32 21.9 a
PHCH2SO2Ph –25 23.4 b
Ph2CH2 –25 32.2 c
PhCH3 –20 43 c
indene (3-H) –18 20.1 d
CpH –17 18.0 c,e
fluorene (9-H) –17 22.6 f
C5Me5H –6.5 26.1 e
H
H
H
H
HH–B
+
B
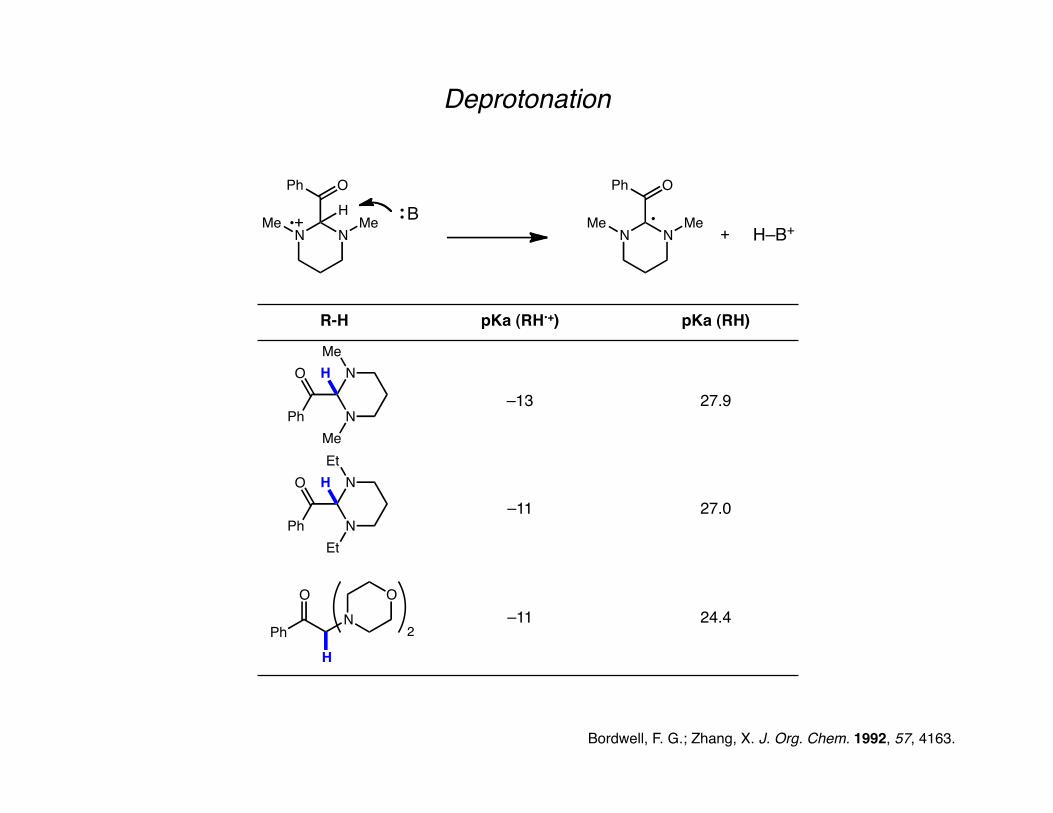
Deprotonation
Bordwell, F. G.; Zhang, X. J. Org. Chem. 1992, 57, 4163.
R-H pKa (RH•+) pKa (RH)
–13 27.9
–11 27.0
–11 24.4
N
NO
Ph
H
Me
Me
N
NO
Ph
H
Et
Et
Ph
O
N
O
2
H
N N
OPh
H B
H–B+MeMe
N N
OPh
MeMe
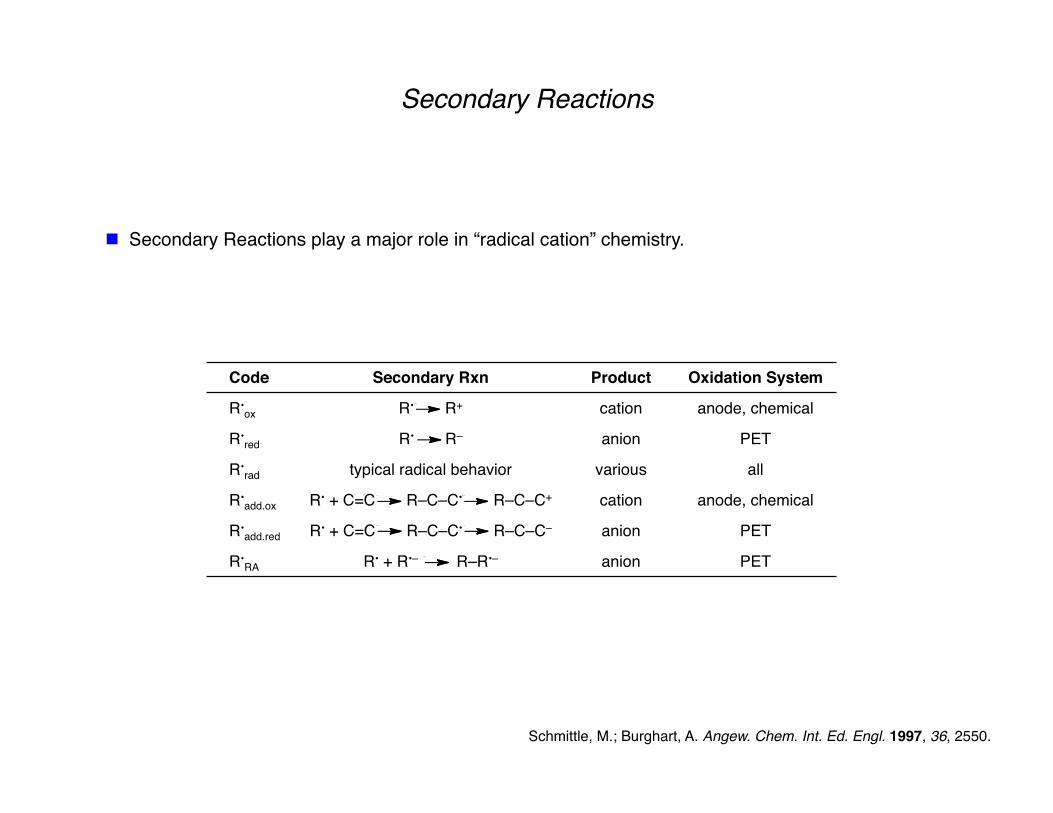
Secondary Reactions
Secondary Reactions play a major role in “radical cation” chemistry.
Code Secondary Rxn Product Oxidation System
R•ox R• R+ cation anode, chemical
R•red R• R– anion PET
R•rad typical radical behavior various all
R•add.ox R• + C=C R–C–C• R–C–C+ cation anode, chemical
R•add.red R• + C=C R–C–C• R–C–C– anion PET
R•RA R• + R•– R–R•– anion PET
Schmittle, M.; Burghart, A. Angew. Chem. Int. Ed. Engl. 1997, 36, 2550.

Ponsold, K.; Kasch, H. Tetrahedron Lett. 1979, 4463.
π-RH•+ Deprotonation
π-RH•+ deprotonation followed by a secondary reactivity pathway (benzylic radical)
MeO
MeO
anode, MeOH
DCM, NaClO4
2,6-lutidine95%
MeO
MeO
HB
MeO
MeO
anode
MeO
MeO
MeOH
B
MeO
MeO
MeO
HMe
O
MeO
MeO
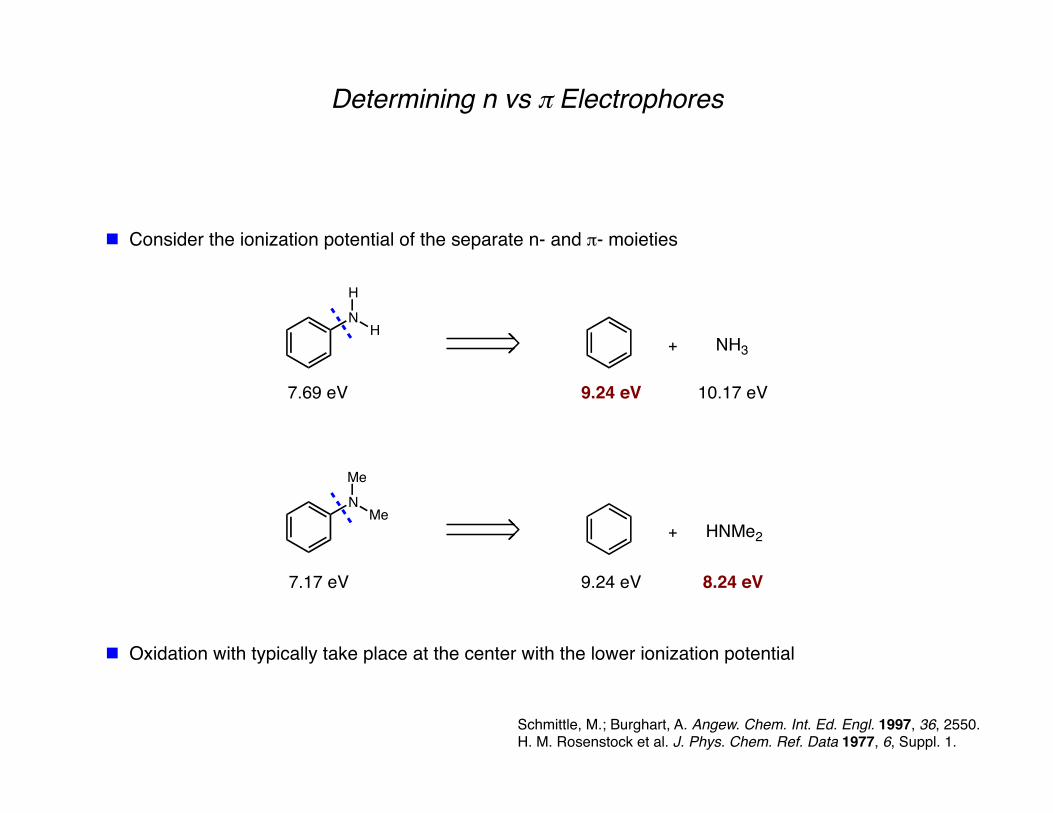
Determining n vs π Electrophores
Oxidation with typically take place at the center with the lower ionization potential
Consider the ionization potential of the separate n- and π- moieties
NH
H
NMe
Me
NH3
9.24 eV 10.17 eV
HNMe2
9.24 eV 8.24 eV
7.69 eV
7.17 eV
Schmittle, M.; Burghart, A. Angew. Chem. Int. Ed. Engl. 1997, 36, 2550. H. M. Rosenstock et al. J. Phys. Chem. Ref. Data 1977, 6, Suppl. 1.
T. Shono et al. J. Am. Chem. Soc. 1982, 104, 5753.
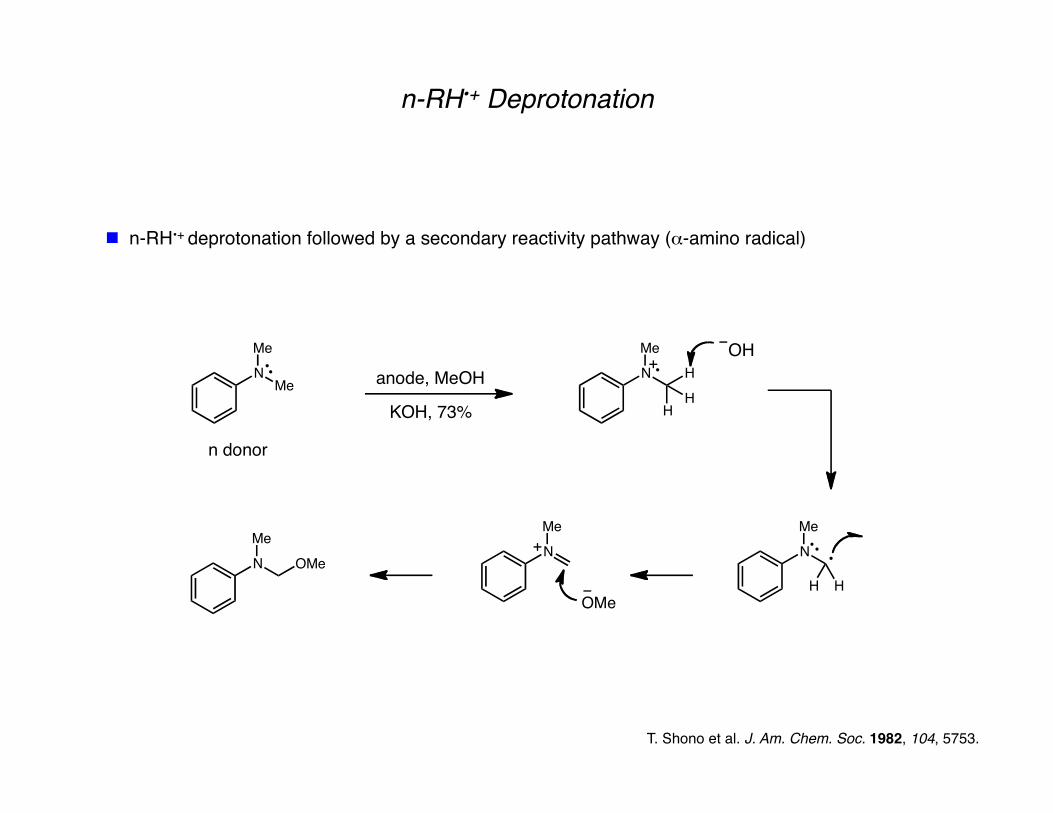
n-RH•+ Deprotonation
n-RH•+ deprotonation followed by a secondary reactivity pathway (α-amino radical)
NMe
Me
anode, MeOH
KOH, 73%
N
Me
H
H
H
OH
N
Me
H H
N
Me
OMe
N
Me
OMe
n donor

T. Shono et al. J. Am. Chem. Soc. 1982, 104, 5753.
n-RH•+ Deprotonation
NH
H
! donor
anodeN
H
H
H2N NH2
HN
NH2
N
N
Typical arene oxidation products are observed
Observation of these products indicates that the arene is the site of oxiation, not N-lone pairs

Albini et al. J. Chem. Soc. Chem. Commun. 1995, 41.
σ-CH•+ Deprotonation
Deprotonation of the radical ion pair happens by the solvent
NC CN
CNNC
h!, 81%
MeCN
NC CN
CNNCH
MeCN

Albini et al. J. Chem. Soc. Chem. Commun. 1995, 41.
σ-CH•+ Deprotonation
Deprotonation of the radical ion pair happens by the solvent
NC CN
CNNC
h!, 81%
MeCN
NC CN
CNNCH
MeCN
NC CN
CNNC
NCCN
CNNC
Adm
Adm CN
CNNC
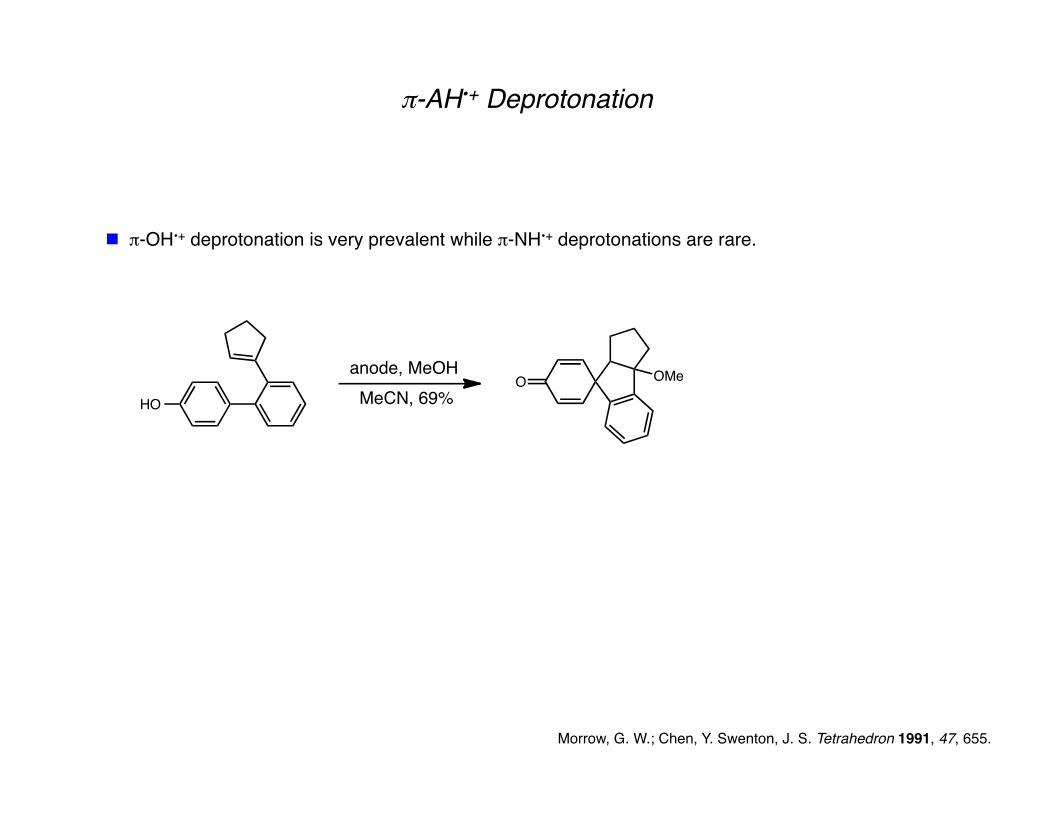
Morrow, G. W.; Chen, Y. Swenton, J. S. Tetrahedron 1991, 47, 655.
π-AH•+ Deprotonation
π-OH•+ deprotonation is very prevalent while π-NH•+ deprotonations are rare.
OOMe
HO
anode, MeOH
MeCN, 69%

Morrow, G. W.; Chen, Y. Swenton, J. S. Tetrahedron 1991, 47, 655.
π-AH•+ Deprotonation
π-OH•+ deprotonation is very prevalent while π-NH•+ deprotonations are rare.
O
HO
MeCN
O
OOOMe
HOMe
–H+
HO
anode, MeOH
MeCN, 69%
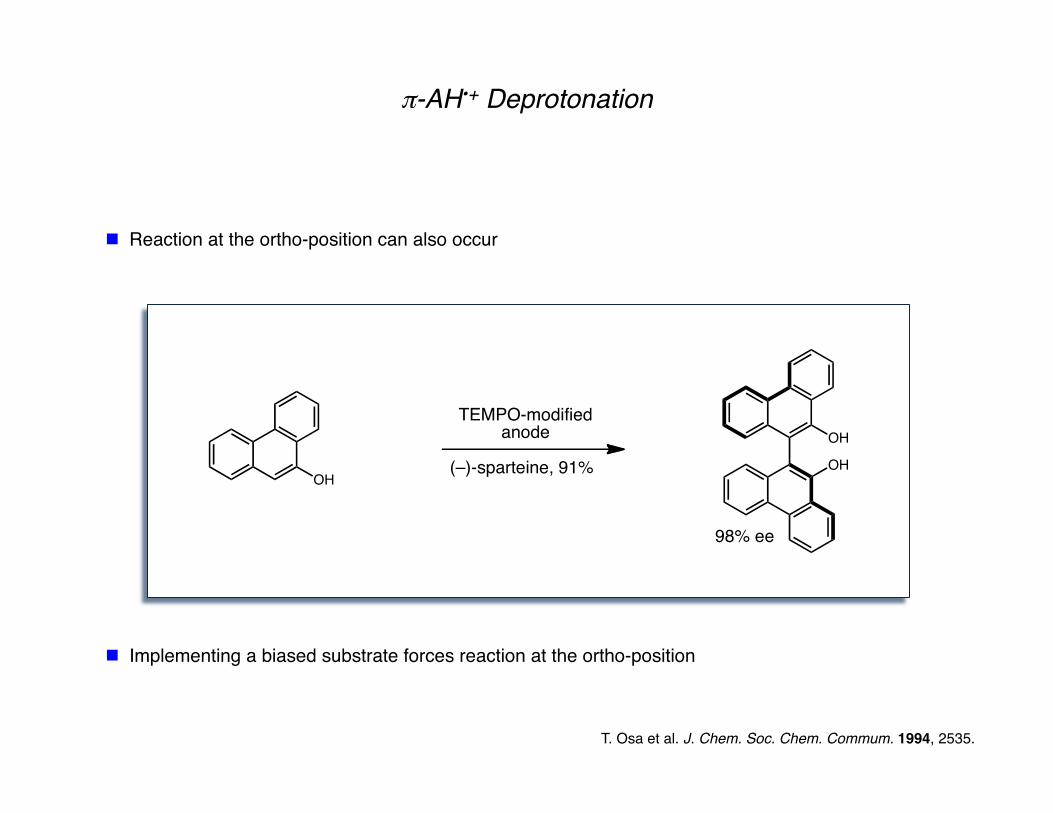
T. Osa et al. J. Chem. Soc. Chem. Commum. 1994, 2535.
π-AH•+ Deprotonation
Reaction at the ortho-position can also occur
OH
TEMPO-modifiedanode
(–)-sparteine, 91%
OH
OH
98% ee
Implementing a biased substrate forces reaction at the ortho-position

Shine, H. J.; Hoque, A. K. M. M. J. Org. Chem. 1988, 53, 4349.
n-AH•+ Deprotonation: Very Limited
The details surrounding the cycloaddition below are speculative at best.
NNHPh
Ph
H
Th•+ ClO4–
MeCN
NNHPh
Ph
H
N Me
–e–
–2H+ N
NPh
N
Me
Ph
The limited examples show how rare these deprotonations are
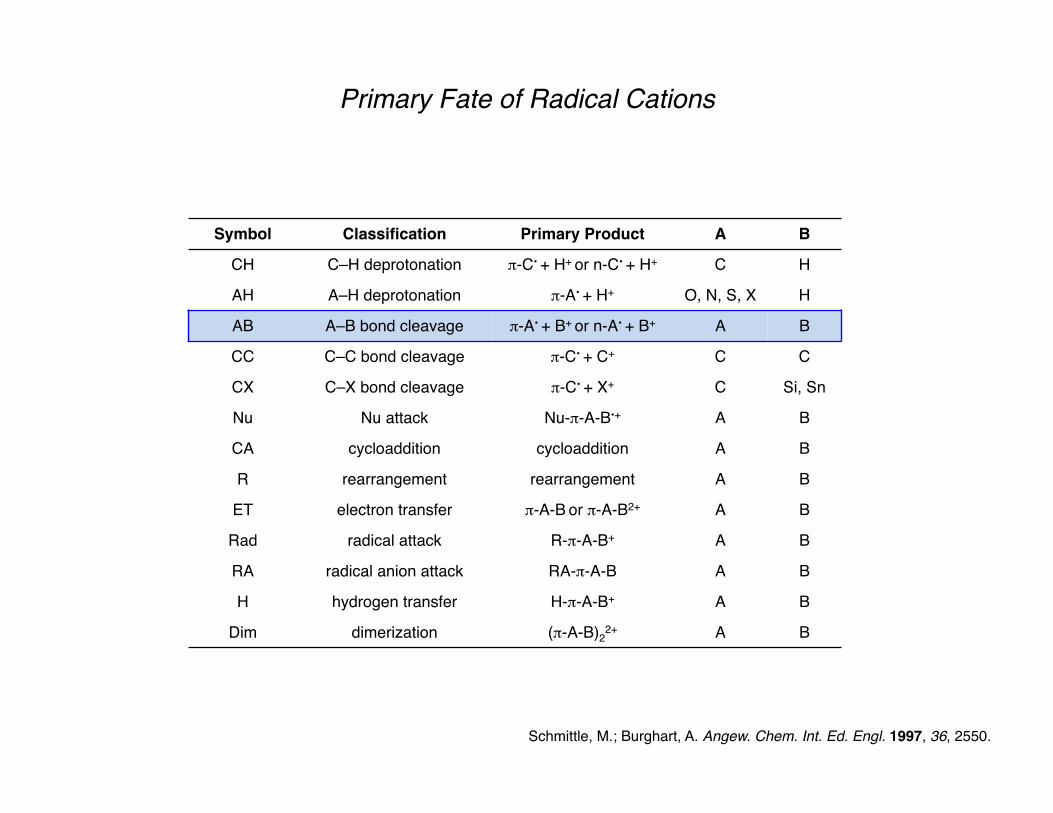
Primary Fate of Radical Cations
Schmittle, M.; Burghart, A. Angew. Chem. Int. Ed. Engl. 1997, 36, 2550.
Symbol Classification Primary Product A B
CH C–H deprotonation π-C• + H+ or n-C• + H+ C H
AH A–H deprotonation π-A• + H+ O, N, S, X H
AB A–B bond cleavage π-A• + B+ or n-A• + B+ A B
CC C–C bond cleavage π-C• + C+ C C
CX C–X bond cleavage π-C• + X+ C Si, Sn
Nu Nu attack Nu-π-A-B•+ A B
CA cycloaddition cycloaddition A B
R rearrangement rearrangement A B
ET electron transfer π-A-B or π-A-B2+ A B
Rad radical attack R-π-A-B+ A B
RA radical anion attack RA-π-A-B A B
H hydrogen transfer H-π-A-B+ A B
Dim dimerization (π-A-B)22+ A B
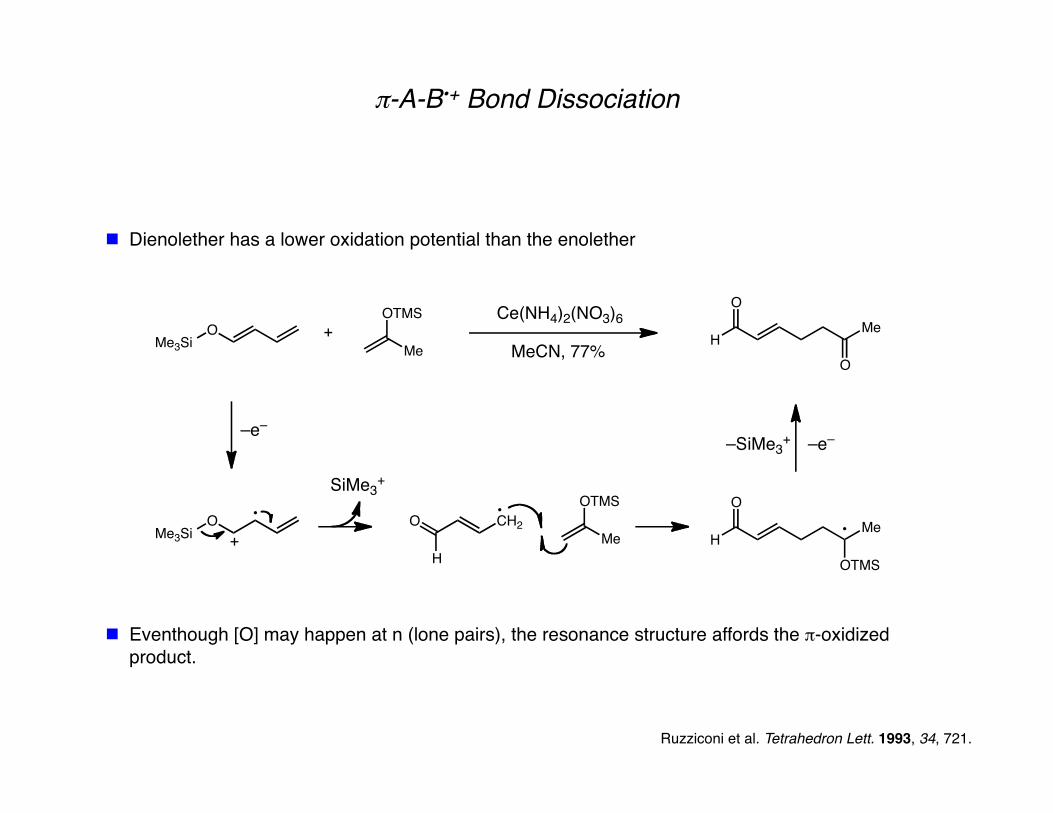
π-A-B•+ Bond Dissociation
O
Me
OTMS
MeCN, 77%
Ce(NH4)2(NO3)6
H
O
Me
O
Me3Si
Dienolether has a lower oxidation potential than the enolether
OMe3Si
–e–
O
SiMe3+
H
CH2
Me
OTMS
H
O
Me
OTMS
–e––SiMe3+
Eventhough [O] may happen at n (lone pairs), the resonance structure affords the π-oxidized product.
Ruzziconi et al. Tetrahedron Lett. 1993, 34, 721.

π-A-B•+ Bond Dissociation
Certain Mukaiyama–Michael reactions are postulated to operate via a radical cation initiated fragmentation
OEt
OTES
DCM , –78 °C
LA
Ph
O
Ph
O
Me
Me
OEt
OTES
Me
Me
Me Me
Me Me
OEt
O
Lewis Acid 4°–4° product 4°–2° product Product ratio
Bu2Sn(OTf)2 85 0 100:0
SnCl4 95 0 100:0
Et3SiClO4 93 0 100:0
TiCl4 97 2.4 97:3
J. Otera et al. J. Am. Chem. Soc. 1991, 113, 4028.

Mukaiyama–Michael Proposed Catalytic Cycle
J. Otera et al. J. Am. Chem. Soc. 1991, 113, 4028.
SnCl4Ph
O
Me
Me
OEt
OTES
Me
MeOEt
OTES
Me
Me
Ph
O
Me
Me
SnCl4
Ph
O
Me
Me
SnCl3
Cl–Et3SiCl
Ph
O
Me
Me
OEt
O
Me
Me
Cl3Sn
Et3SiCl
Ph
Et3SiO Me Me
Me Me
OEt
O
e– transfer
R•–R• coupling complexation
catalyst regeneration

n-A-B•+ Bond Dissociation
Adam, W.; Sendelbach, J. J. Org. Chem. 1993, 58, 5316.
N
N
Me
Me
OHh!, DCA
Ph2
O Me
Me
N
N
Me
Me
OHN2
OH
Me
Me
Me
OH
Me
BET
Limited mostly to azoalkanes and triazines
Primary Fate of Radical Cations
Schmittle, M.; Burghart, A. Angew. Chem. Int. Ed. Engl. 1997, 36, 2550.
Symbol Classification Primary Product A B
CH C–H deprotonation π-C• + H+ or n-C• + H+ C H
AH A–H deprotonation π-A• + H+ O, N, S, X H
AB A–B bond cleavage π-A• + B+ or n-A• + B+ A B
CC C–C bond cleavage π-C• + C+ C C
CX C–X bond cleavage π-C• + X+ C Si, Sn
Nu Nu attack Nu-π-A-B•+ A B
CA cycloaddition cycloaddition A B
R rearrangement rearrangement A B
ET electron transfer π-A-B or π-A-B2+ A B
Rad radical attack R-π-A-B+ A B
RA radical anion attack RA-π-A-B A B
H hydrogen transfer H-π-A-B+ A B
Dim dimerization (π-A-B)22+ A B

π-C-C•+ Bond Dissociation
Fragmentation of a β-ether generates an oxocarbenium ion
Arnold, D. R.; Maroulis, A. J. J. Am. Chem. Soc. 1976, 98, 5931.
MeO
NC CN
h!, MeCN, MeOH
MeOMeO
HOMe
OMeMeO
DCB
–H+
H+
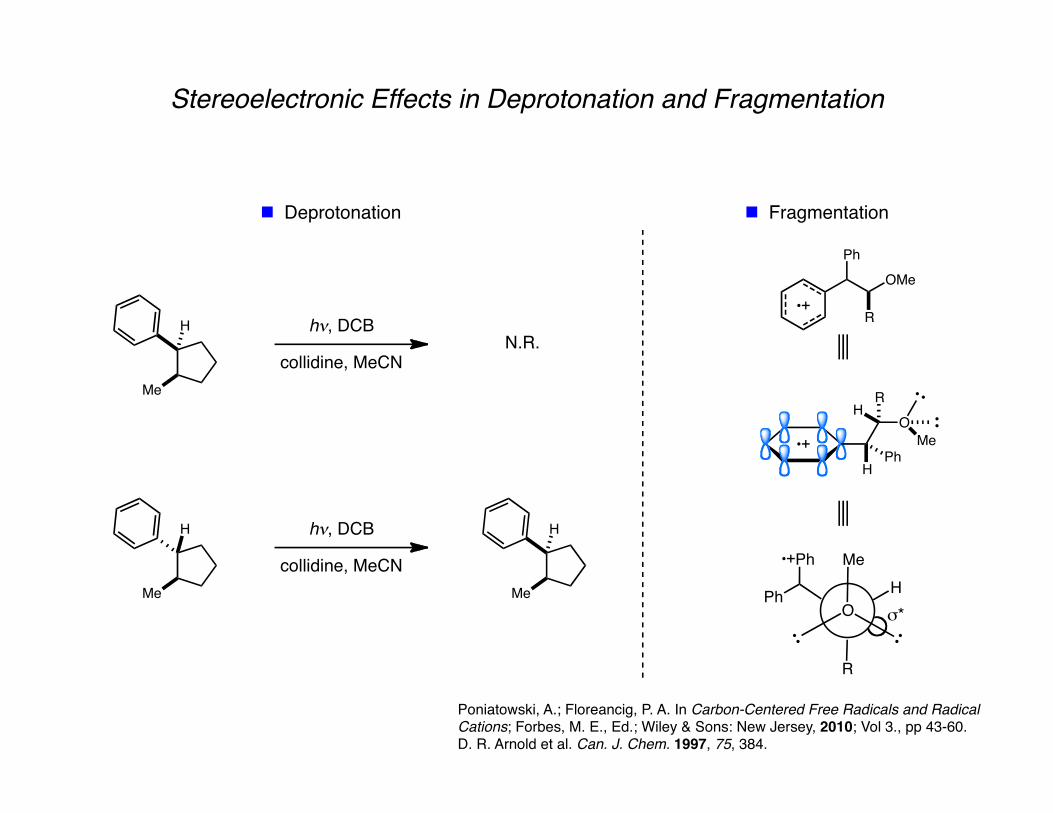
Stereoelectronic Effects in Deprotonation and Fragmentation
Poniatowski, A.; Floreancig, P. A. In Carbon-Centered Free Radicals and Radical Cations; Forbes, M. E., Ed.; Wiley & Sons: New Jersey, 2010; Vol 3., pp 43-60. D. R. Arnold et al. Can. J. Chem. 1997, 75, 384.
Deprotonation Fragmentation
Me
h!, DCB
collidine, MeCN
N.R.
Me
h!, DCB
collidine, MeCN
Me
H
HH
O
Ph
RH
H
Me
OMe
R
Ph
H
R
Me
"*Ph
Ph
O
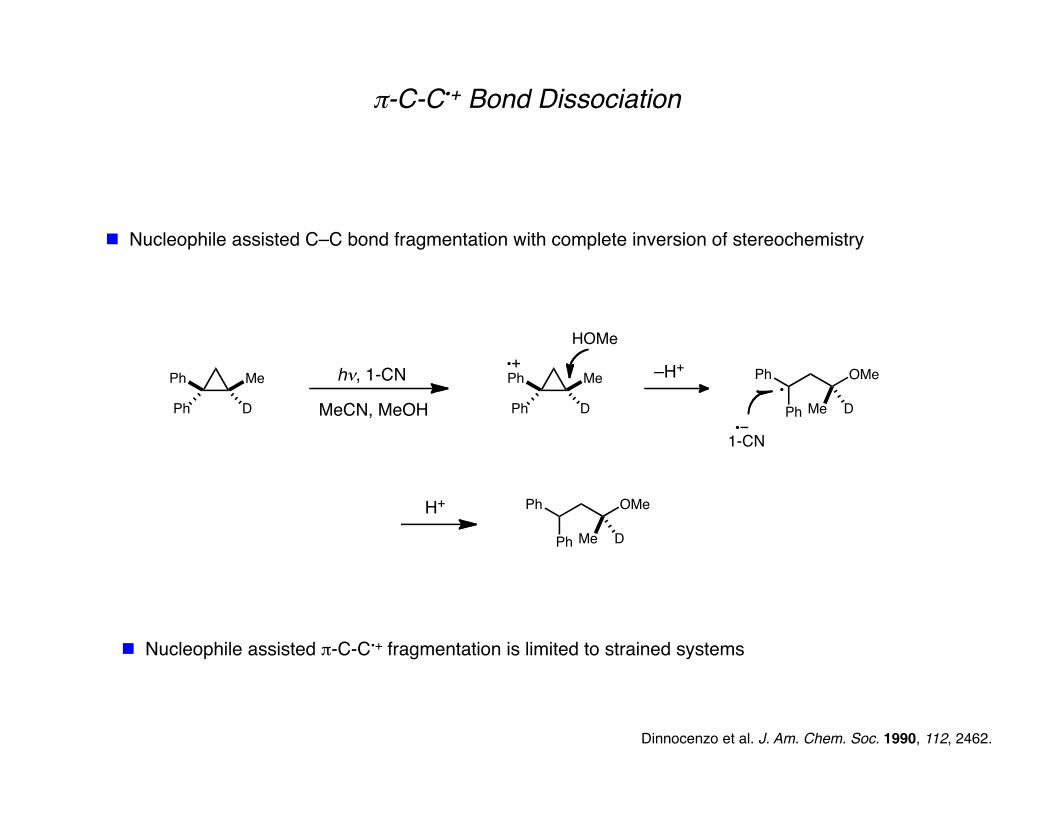
π-C-C•+ Bond Dissociation
Nucleophile assisted C–C bond fragmentation with complete inversion of stereochemistry
Dinnocenzo et al. J. Am. Chem. Soc. 1990, 112, 2462.
Ph
Ph
Me
D
h!, 1-CN
MeCN, MeOH
Ph
Ph
Me
D
HOMe
Ph
Ph
OMe
Me D
–H+
1-CN
H+ Ph
Ph
OMe
Me D
Nucleophile assisted π-C-C•+ fragmentation is limited to strained systems

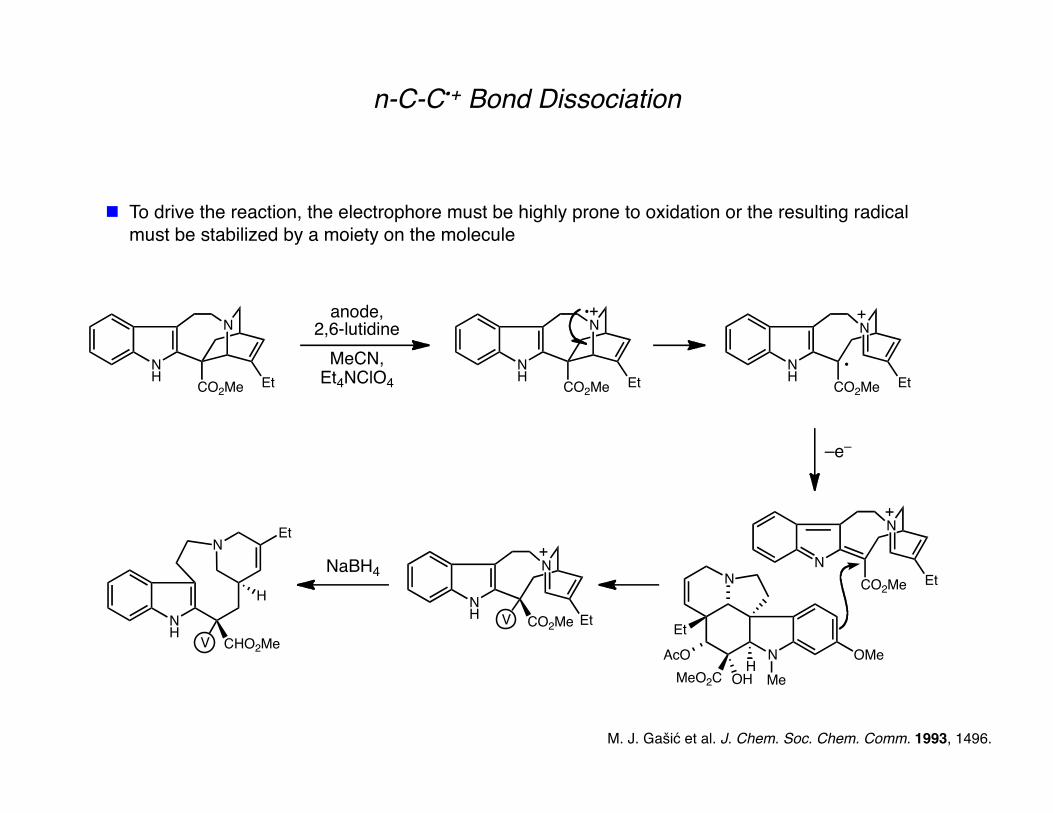
n-C-C•+ Bond Dissociation
To drive the reaction, the electrophore must be highly prone to oxidation or the resulting radical must be stabilized by a moiety on the molecule
M. J. Gašić et al. J. Chem. Soc. Chem. Comm. 1993, 1496.
NH
N
EtCO2Me
anode, 2,6-lutidine
MeCN, Et4NClO4
NH
N
EtCO2MeNH
N
EtCO2Me
–e–
N
N
EtCO2Me
N
N
H
EtOMe
OHMeO2CAcO
NH
N
EtCO2MeV
NaBH4
NH
N
H
CHO2MeV
Et
Me
n-C-C•+ Bond Dissociation
To drive the reaction, the electrophore must be highly prone to oxidation or the resulting radical must be stabilized by a moiety on the molecule
M. J. Gašić et al. J. Chem. Soc. Chem. Comm. 1993, 1496.
NH
N
EtCO2Me
anode, 2,6-lutidine
MeCN, Et4NClO4
NH
N
EtCO2MeNH
N
EtCO2Me
–e–
N
N
EtCO2Me
N
N
H
EtOMe
OHMeO2CAcO
NH
N
EtCO2MeV
NaBH4
NH
N
H
CHO2MeV
Et
Me

Me
AcO
O
O
TCB, h!
RSH, H2O, 90%
AcO
Me
O
O
OHTCB
Me
AcO
O
O
AcO
Me
O
O
TCB
H2O
n-C-C•+ Bond Dissociation
A. Albini et al. Tetrahedron Lett. 1994, 50, 575.
Easily oxidized acetal

σ-C-C•+ Bond Dissociation
Booker-Milburn, K. I. Synlett 1992, 809.
Me3SiOFeCl3, DMF
OH
H
Cl
H
Cl3Fe
–SiMe3
64%
Me3SiO
Trapping of the pendant olefin and Cl• abstraction form the bicycle
ESR studies on strained systems show that the inner C-C bond is selectively weakened
Primary Fate of Radical Cations
Schmittle, M.; Burghart, A. Angew. Chem. Int. Ed. Engl. 1997, 36, 2550.
Symbol Classification Primary Product A B
CH C–H deprotonation π-C• + H+ or n-C• + H+ C H
AH A–H deprotonation π-A• + H+ O, N, S, X H
AB A–B bond cleavage π-A• + B+ or n-A• + B+ A B
CC C–C bond cleavage π-C• + C+ C C
CX C–X bond cleavage π-C• + X+ C Si, Sn
Nu Nu attack Nu-π-A-B•+ A B
CA cycloaddition cycloaddition A B
R rearrangement rearrangement A B
ET electron transfer π-A-B or π-A-B2+ A B
Rad radical attack R-π-A-B+ A B
RA radical anion attack RA-π-A-B A B
H hydrogen transfer H-π-A-B+ A B
Dim dimerization (π-A-B)22+ A B
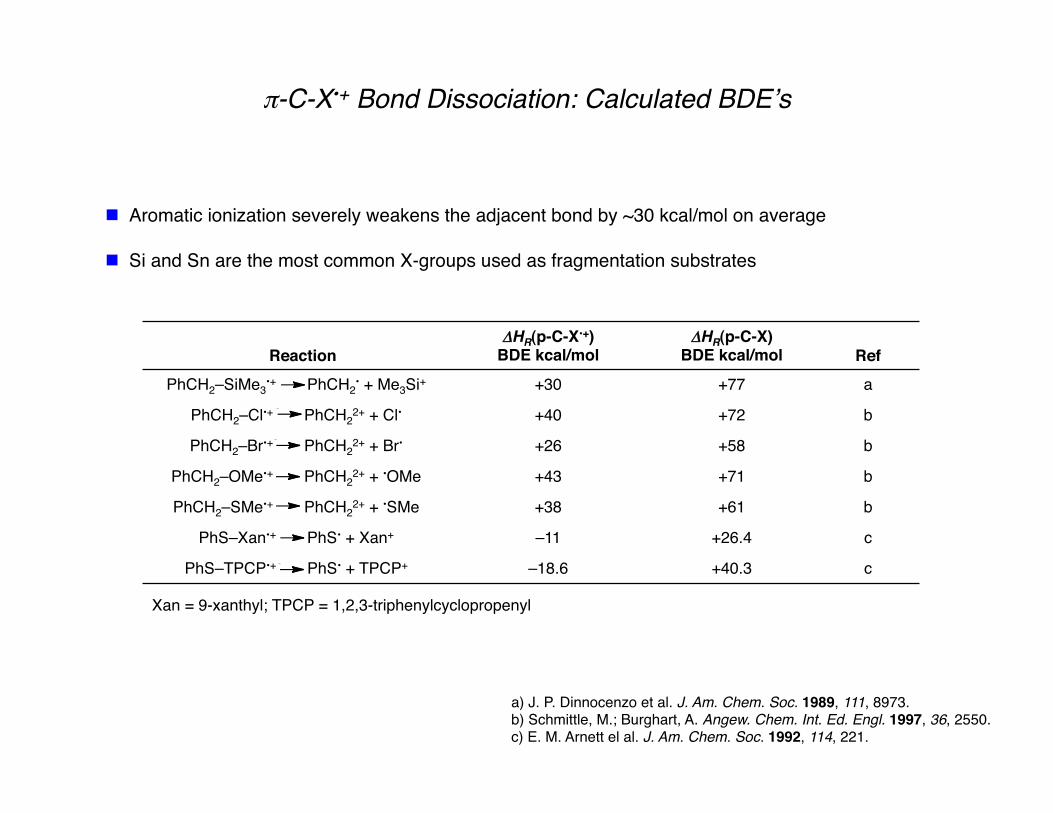
π-C-X•+ Bond Dissociation: Calculated BDEʼs
Aromatic ionization severely weakens the adjacent bond by ~30 kcal/mol on average
Si and Sn are the most common X-groups used as fragmentation substrates
Reaction ΔHR(p-C-X•+)
BDE kcal/mol ΔHR(p-C-X)
BDE kcal/mol Ref PhCH2–SiMe3
•+ PhCH2• + Me3Si+ +30 +77 a
PhCH2–Cl•+ PhCH22+ + Cl• +40 +72 b
PhCH2–Br•+ PhCH22+ + Br• +26 +58 b
PhCH2–OMe•+ PhCH22+ + •OMe +43 +71 b
PhCH2–SMe•+ PhCH22+ + •SMe +38 +61 b
PhS–Xan•+ PhS• + Xan+ –11 +26.4 c
PhS–TPCP•+ PhS• + TPCP+ –18.6 +40.3 c
a) J. P. Dinnocenzo et al. J. Am. Chem. Soc. 1989, 111, 8973. b) Schmittle, M.; Burghart, A. Angew. Chem. Int. Ed. Engl. 1997, 36, 2550. c) E. M. Arnett el al. J. Am. Chem. Soc. 1992, 114, 221.
Xan = 9-xanthyl; TPCP = 1,2,3-triphenylcyclopropenyl
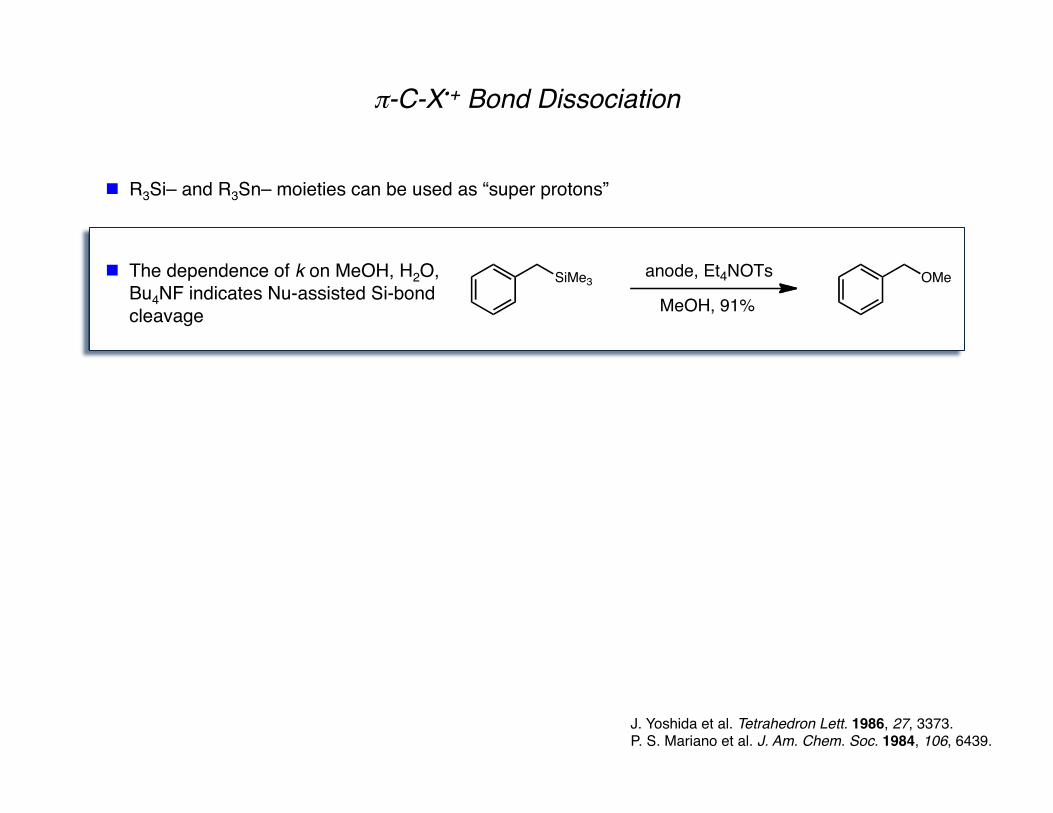
π-C-X•+ Bond Dissociation
J. Yoshida et al. Tetrahedron Lett. 1986, 27, 3373. P. S. Mariano et al. J. Am. Chem. Soc. 1984, 106, 6439.
R3Si– and R3Sn– moieties can be used as “super protons”
The dependence of k on MeOH, H2O, Bu4NF indicates Nu-assisted Si-bond cleavage
SiMe3 OMeanode, Et4NOTs
MeOH, 91%
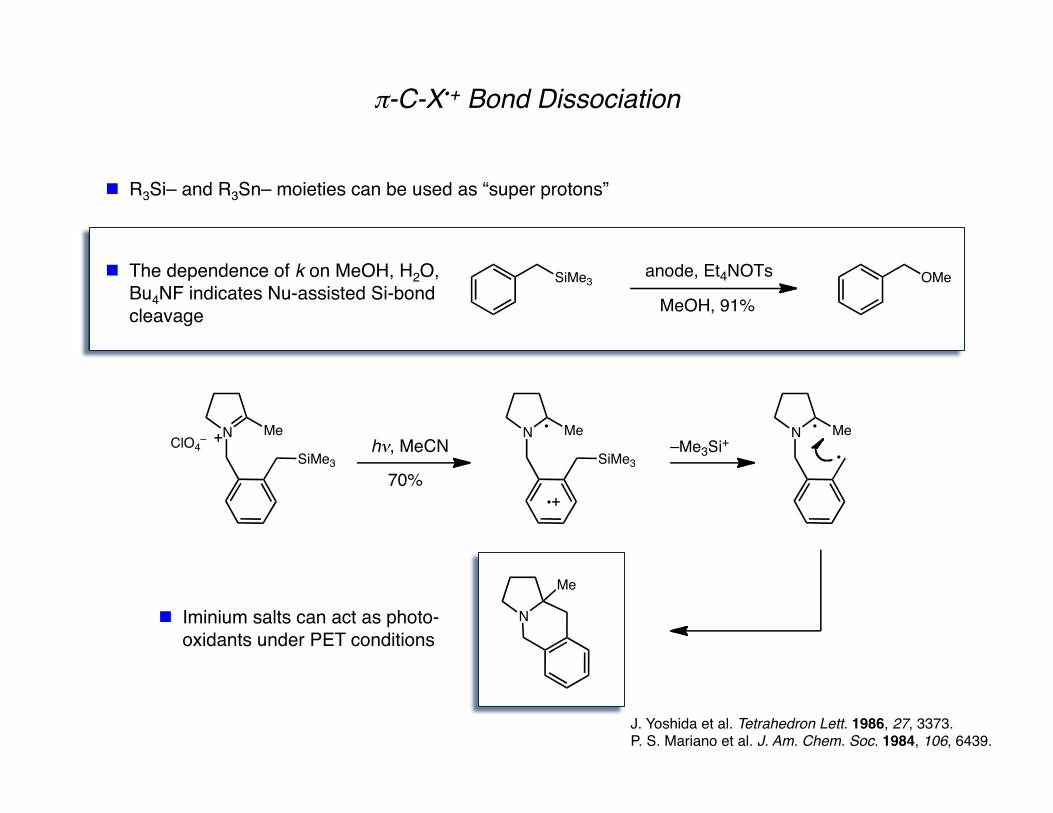
π-C-X•+ Bond Dissociation
J. Yoshida et al. Tetrahedron Lett. 1986, 27, 3373. P. S. Mariano et al. J. Am. Chem. Soc. 1984, 106, 6439.
R3Si– and R3Sn– moieties can be used as “super protons”
The dependence of k on MeOH, H2O, Bu4NF indicates Nu-assisted Si-bond cleavage
SiMe3 OMeanode, Et4NOTs
MeOH, 91%
Iminium salts can act as photo-oxidants under PET conditions
N
SiMe3
MeClO4
–h!, MeCN
70%
N
SiMe3
Me N Me
–Me3Si+
N
Me
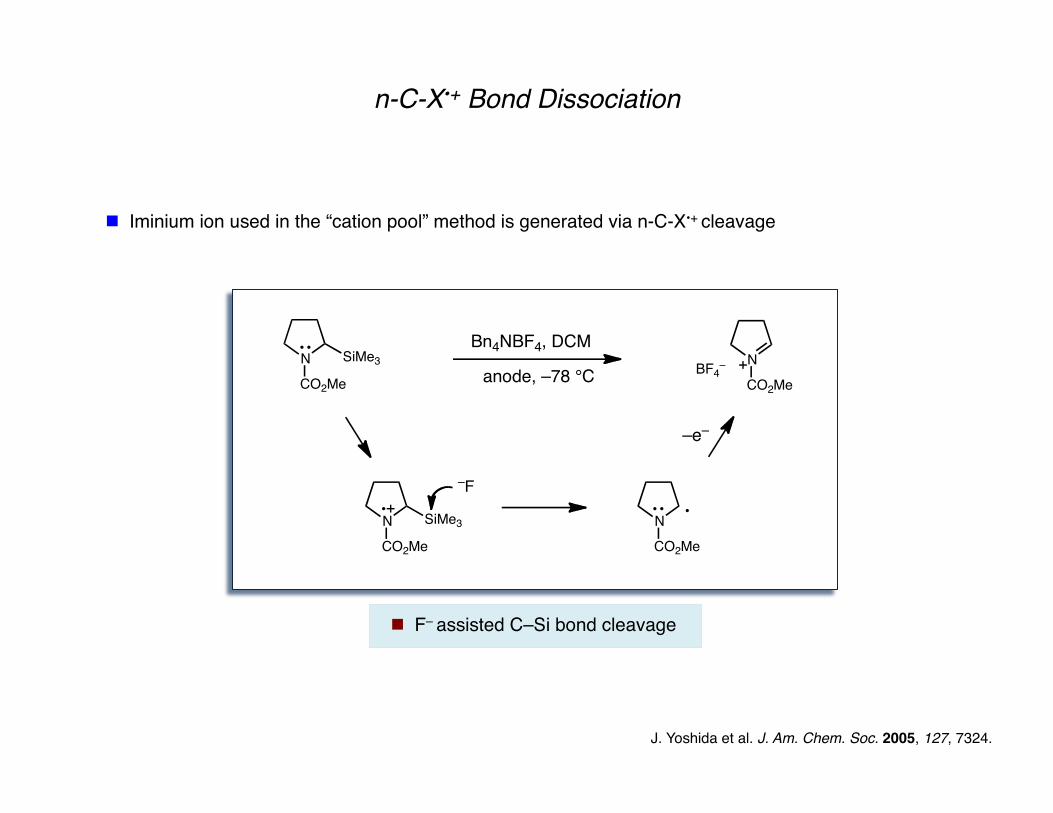
n-C-X•+ Bond Dissociation
J. Yoshida et al. J. Am. Chem. Soc. 2005, 127, 7324.
N
CO2Me
BF4–
N
CO2Me
SiMe3
Bn4NBF4, DCM
anode, –78 °C
N
CO2Me
SiMe3
–F
N
CO2Me
–e–
Iminium ion used in the “cation pool” method is generated via n-C-X•+ cleavage
F– assisted C–Si bond cleavage
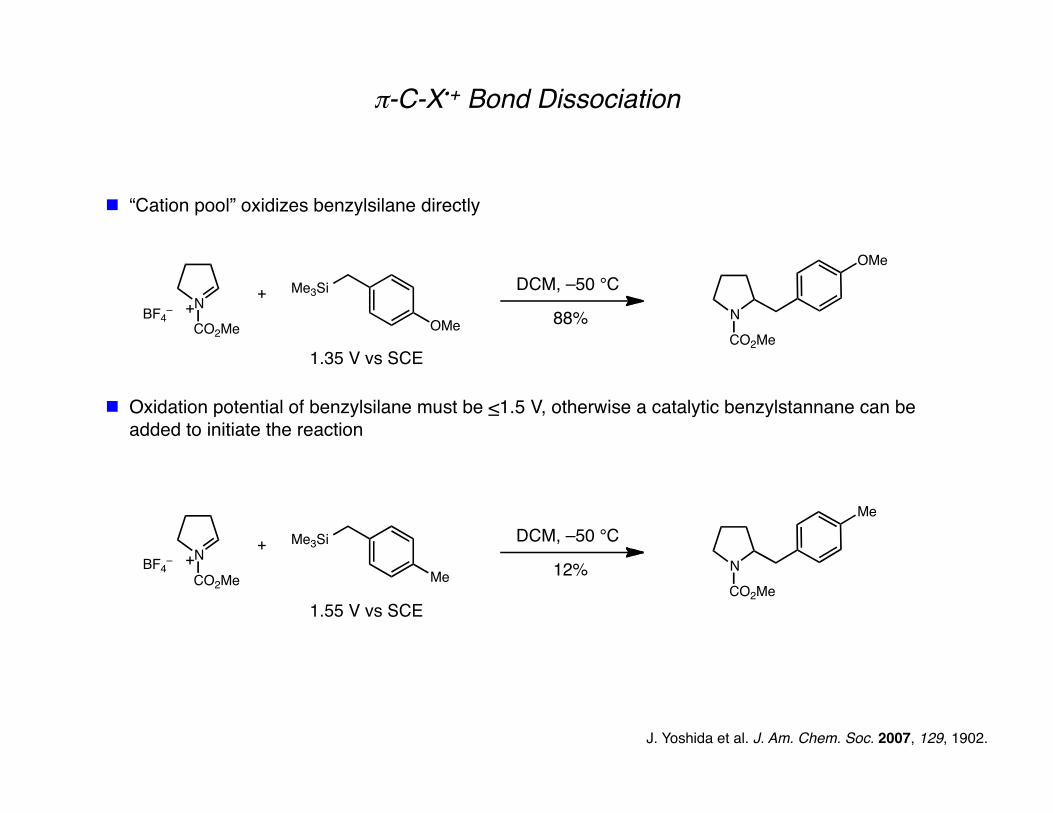
π-C-X•+ Bond Dissociation
“Cation pool” oxidizes benzylsilane directly
Oxidation potential of benzylsilane must be <1.5 V, otherwise a catalytic benzylstannane can be added to initiate the reaction
NBF4
–
CO2Me OMe
Me3Si DCM, –50 °C
N
CO2Me
OMe
88%
1.35 V vs SCE
N
CO2Me Me
Me3Si DCM, –50 °C
N
CO2Me
Me
12%
1.55 V vs SCE
BF4–
J. Yoshida et al. J. Am. Chem. Soc. 2007, 129, 1902.

π-C-X•+ Bond Dissociation
“Cation pool” oxidizes benzylsilane directly
Oxidation potential of benzylsilane must be <1.5 V, otherwise a catalytic benzylstannane can be added to initiate the reaction
NBF4
–
CO2Me OMe
Me3Si DCM, –50 °C
N
CO2Me
OMe
88%
1.35 V vs SCE
Me
Bu3Sn
10 mol%
J. Yoshida et al. J. Am. Chem. Soc. 2007, 129, 1902.
N
CO2Me Me
Me3Si DCM, –50 °C
N
CO2Me
Me
97%
1.55 V vs SCE
BF4–
OMe
NCO2Me
BF4–
NCO2Me
BF4–
OMe
Me3Si
OMe
Me3Si
Me3Si+
NCO2Me
NCO2Me
OMe
OMe
Me3Si
NCO2Me
OMe
Propagation
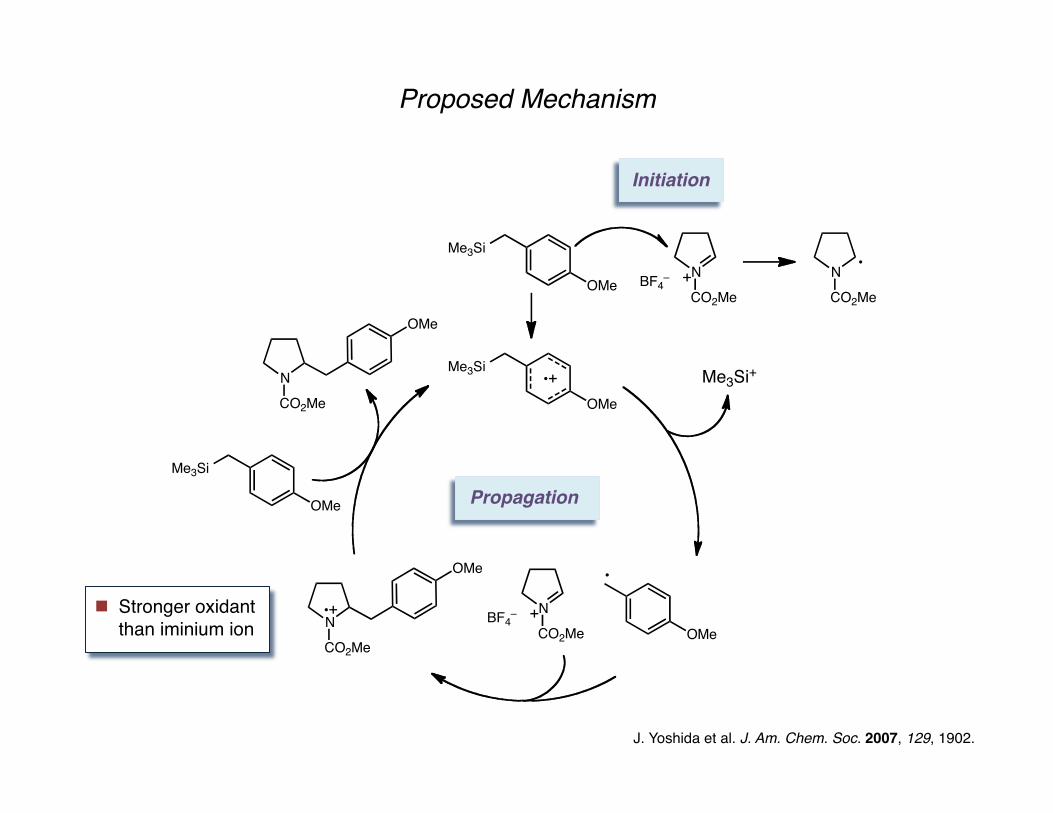
Proposed Mechanism
J. Yoshida et al. J. Am. Chem. Soc. 2007, 129, 1902.
Propagation
Initiation
Stronger oxidant than iminium ion

n-C-X•+ Bond Dissociation
Pandy, G.; Reddy, G. D. Tetrahedron Lett. 1992, 33, 6533. Steckhan et al. Angew. Chem. Int. Ed. Engl. 1995, 34, 2137. Mariano et al. J. Org. Chem. 1992, 57, 6037.
N
SiMe3 DCN, h!
i-PrOH, 90% N
HMe
97:3 dr
O
O
NBn
SiMe3
DCA, h!
MeCN, 59%
Nhexyl
O
OMe
SiMe3
DCA, h!, 67%
MeCn/MeOH
NO O
Me
Nhexyl
O
OMe
NO O
Me
N O
N
H
N O
O
O
Bn
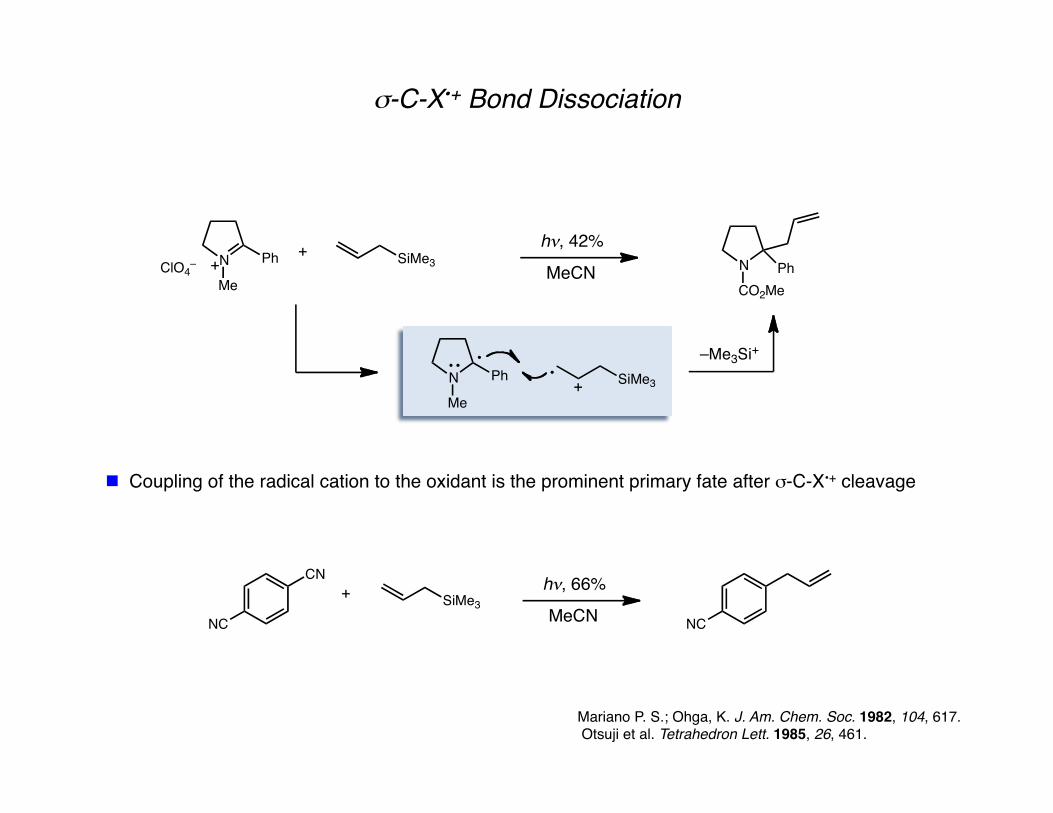
σ-C-X•+ Bond Dissociation
Mariano P. S.; Ohga, K. J. Am. Chem. Soc. 1982, 104, 617. Otsuji et al. Tetrahedron Lett. 1985, 26, 461.
CN
NC
h!, 66%
MeCNSiMe3
NC
NClO4
–
Me
SiMe3
h!, 42%
N
CO2Me
PhPh
N
Me
Ph SiMe3
–Me3Si+
MeCN
Coupling of the radical cation to the oxidant is the prominent primary fate after σ-C-X•+ cleavage

Primary Fate of Radical Cations
Schmittle, M.; Burghart, A. Angew. Chem. Int. Ed. Engl. 1997, 36, 2550.
Symbol Classification Primary Product A B
CH C–H deprotonation π-C• + H+ or n-C• + H+ C H
AH A–H deprotonation π-A• + H+ O, N, S, X H
AB A–B bond cleavage π-A• + B+ or n-A• + B+ A B
CC C–C bond cleavage π-C• + C+ C C
CX C–X bond cleavage π-C• + X+ C Si, Sn
Nu Nu attack Nu-π-A-B•+ A B
CA cycloaddition cycloaddition A B
R rearrangement rearrangement A B
ET electron transfer π-A-B or π-A-B2+ A B
Rad radical attack R-π-A-B+ A B
RA radical anion attack RA-π-A-B A B
H hydrogen transfer H-π-A-B+ A B
Dim dimerization (π-A-B)22+ A B

Nu-π-A-B•+
Mihelcic, J.; Moeller, K D. J. Am. Chem. Soc. 2004, 126, 9106. Trauner et al. J. Am. Chem. Soc. 2006, 128, 17057.
OTBS
TBSOO
Me
Me
Meanode, LiClO4
2,6-lutidineMeOH/DCM, 88%
O
TBSOO
Me
Me
Me
H
Moeller en route to (–)-alliacol A
Trauner en route to (–)-guanacastepene E
O
TBDPSO
MeMe
Me
Me
OBnTBSO
anode, LiClO42,6-lutidine
MeOH/DCM, 81% O
TBDPSO
MeMe
Me
Me
OBnOMeO
H
H
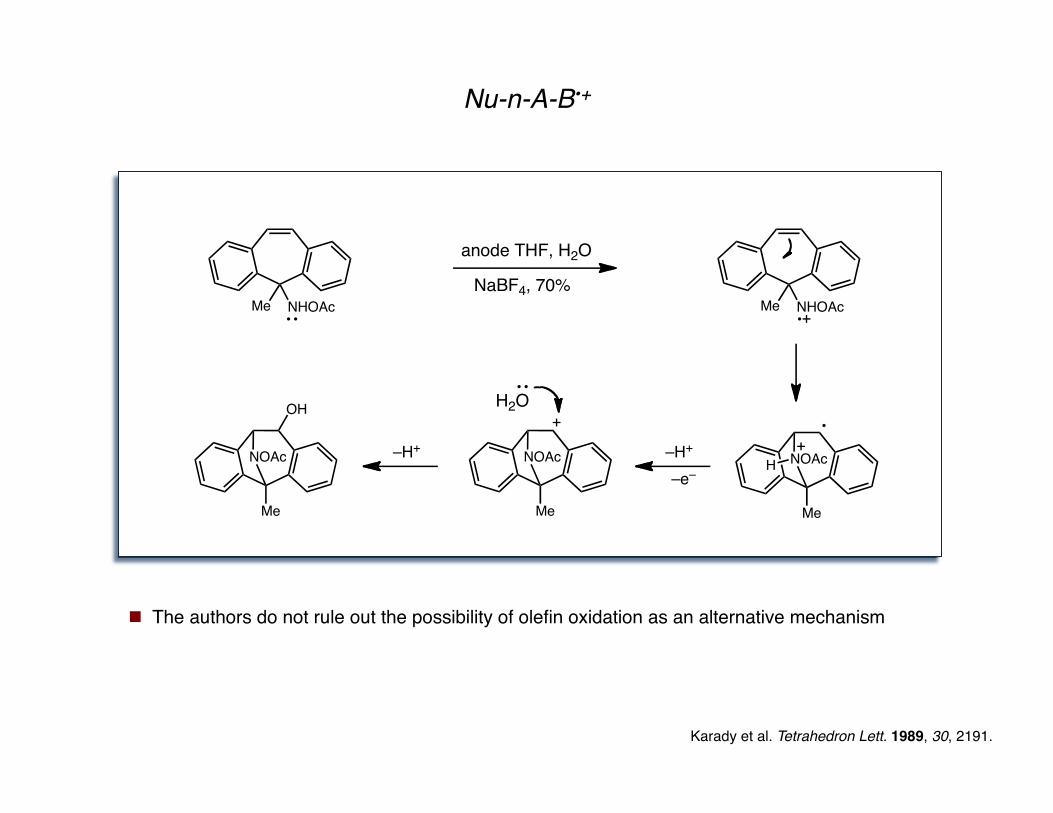
Nu-n-A-B•+
Karady et al. Tetrahedron Lett. 1989, 30, 2191.
Me NHOAc
anode THF, H2O
NaBF4, 70%
Me
NOAc
Me NHOAc
Me
NOAcH
–H+
–e–
Me
NOAc
OHH2O
–H+
The authors do not rule out the possibility of olefin oxidation as an alternative mechanism

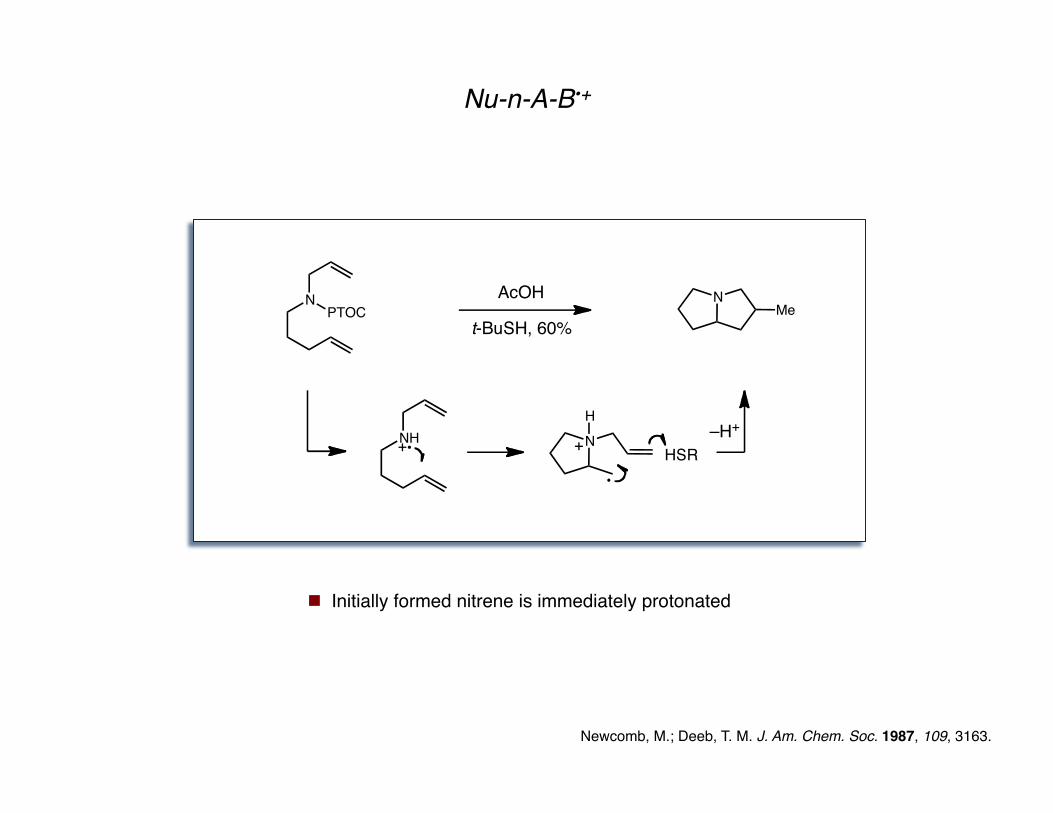
Nu-n-A-B•+
Newcomb, M.; Deeb, T. M. J. Am. Chem. Soc. 1987, 109, 3163.
NPTOC
t-BuSH, 60%
AcOH NMe
N
S
O
O
PTOC =
Nu-n-A-B•+
Newcomb, M.; Deeb, T. M. J. Am. Chem. Soc. 1987, 109, 3163.
NPTOC
t-BuSH, 60%
AcOH NMe
NH N
H
HSR
–H+
Initially formed nitrene is immediately protonated
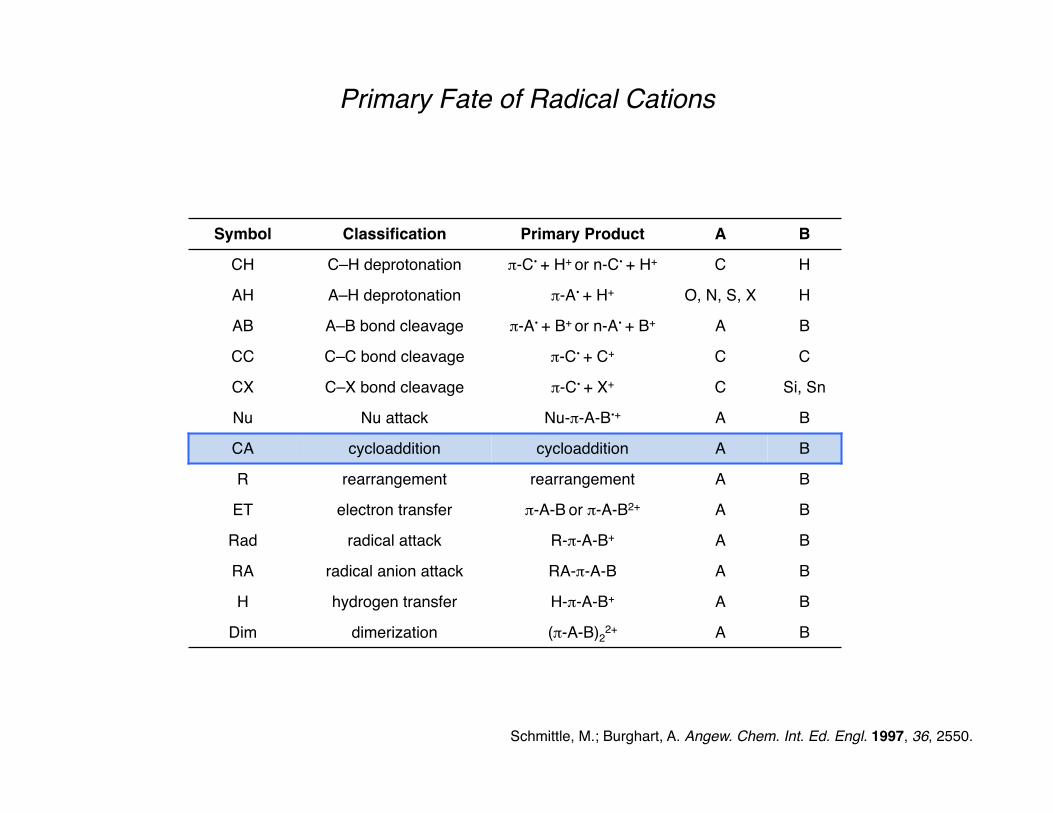
Primary Fate of Radical Cations
Schmittle, M.; Burghart, A. Angew. Chem. Int. Ed. Engl. 1997, 36, 2550.
Symbol Classification Primary Product A B
CH C–H deprotonation π-C• + H+ or n-C• + H+ C H
AH A–H deprotonation π-A• + H+ O, N, S, X H
AB A–B bond cleavage π-A• + B+ or n-A• + B+ A B
CC C–C bond cleavage π-C• + C+ C C
CX C–X bond cleavage π-C• + X+ C Si, Sn
Nu Nu attack Nu-π-A-B•+ A B
CA cycloaddition cycloaddition A B
R rearrangement rearrangement A B
ET electron transfer π-A-B or π-A-B2+ A B
Rad radical attack R-π-A-B+ A B
RA radical anion attack RA-π-A-B A B
H hydrogen transfer H-π-A-B+ A B
Dim dimerization (π-A-B)22+ A B

Cycloaddition
a) Calhoun, G. C.; Schuster, G. B. J. Am. Chem. Soc. 1984, 106, 6870. b) Lorenz, K. T.; Bauld, N. L. J. Am. Chem. Soc. 1987, 109, 1157. c) Takamuku et al. J. Org. Chem. 1991, 56, 6240. d) Schepp, N. P; Johnston, L. J J. Am. Chem. Soc. 1994, 116, 6895. e) Schepp, N. P; Johnston, L. J J. Am. Chem. Soc. 1994, 116, 10330.
Reaction k [M–1s–1] comments reference
3 x 108 Mostly [4 + 2] endo adduct; ratio of/to [2 + 2] varied by concentration a, b
<6.7 x 107 1) [2 + 2] terminated by C-H•– deprotonation 2) concerted reaction b
1) 1.4 x 109 2) 1.4 x 109
3) 1 x 1010
1) concerted cycloaddition assumed 2) distonic 1,4-radical cation intermediate c,d
7 x 108 21% Diels–Alder adduct e
1.5 x 106 80% Diels–Alder adduct e
p-An
p-AnMe
p-AnMe
p-AnMe
p-An
p-An
Experimental kinetic data for radical cation cycloadditions (rt)

Diels–Alder [4 + 2] Cycloaddition
Bauld et al. J. Am. Chem. Soc. 1981, 109, 3163. Lorenz, K. T.; Bauld, N. L. J. Am. Chem. Soc. 1987, 109, 1157
Suprafacial selectivity observed
Shown to operate via a cation radical chain mechanism
DP + Ar3N•+ Ar3N + DP•+
DP•+ + D A•+
A•+ + DP A + DP•+
DCM, 70%
H
H
5:1 endo/exo
Ar3N –SbCl5
2DP•+ dication DP•+ H+ + DP•
Propagation
Termination
k = 3 x 108 [M–1s–1]

Diels–Alder [4 + 2] Cycloaddition
Shown to operate via a cation radical chain mechanism
DCM, 70%
H
H
5:1 endo/exo
Ar3N –SbCl5
Bauld et al. J. Am. Chem. Soc. 1981, 109, 3163. Lorenz, K. T.; Bauld, N. L. J. Am. Chem. Soc. 1987, 109, 1157
Propagator H
H
H
H
Ar3N
Initiator
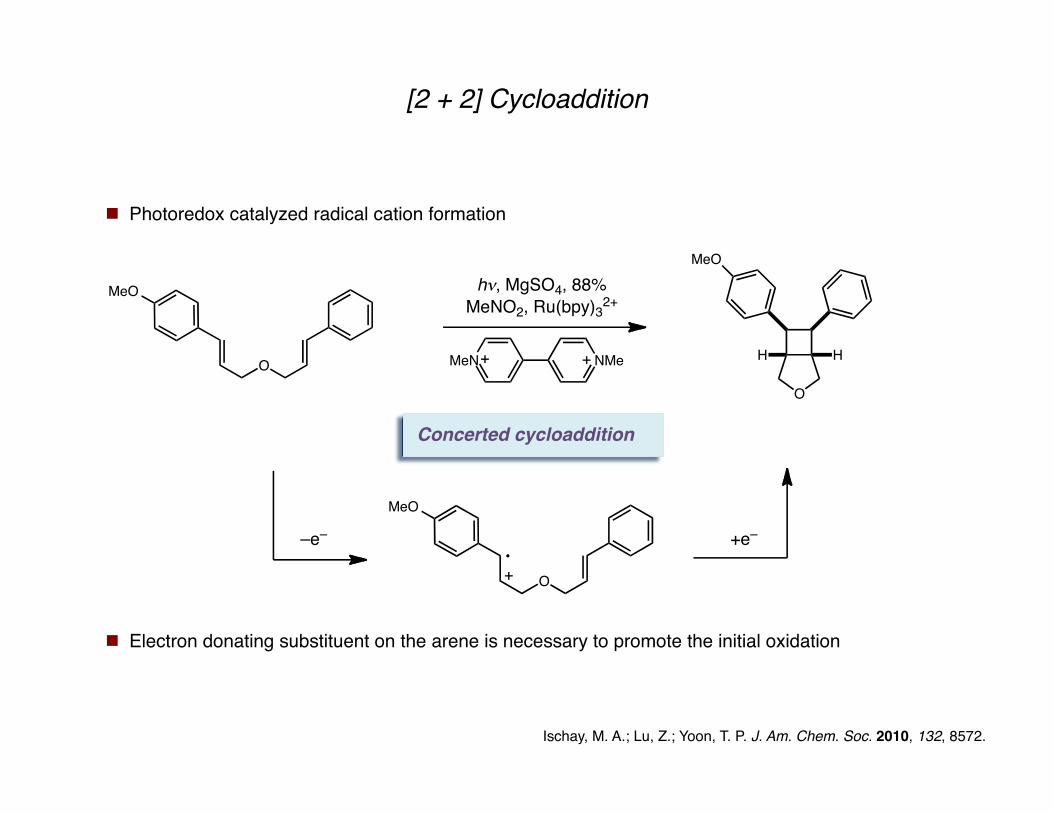
[2 + 2] Cycloaddition
Photoredox catalyzed radical cation formation
Ischay, M. A.; Lu, Z.; Yoon, T. P. J. Am. Chem. Soc. 2010, 132, 8572.
Concerted cycloaddition
–e– +e–
MeO
O
Electron donating substituent on the arene is necessary to promote the initial oxidation
MeO
O
O
HH
MeO
h!, MgSO4, 88%
MeNO2, Ru(bpy)32+
MeN NMe
MeO
O
O
HH
MeO
h!, MgSO4, 82%
MeNO2, Ru(bpy)32+
MeN NMe
O
HH
MeO
h!, MgSO4, 71%
MeNO2, Ru(bpy)32+
MeN NMeO
OMe
6:1 dr
5:1 dr
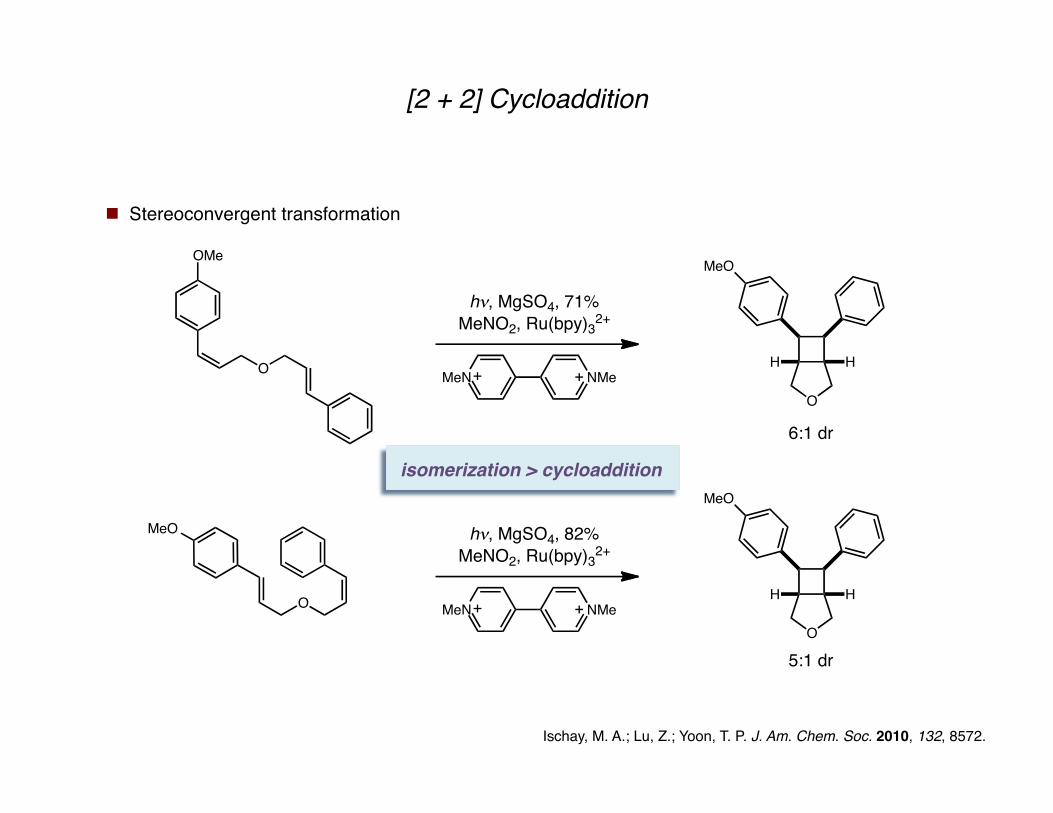
[2 + 2] Cycloaddition
Stereoconvergent transformation
Ischay, M. A.; Lu, Z.; Yoon, T. P. J. Am. Chem. Soc. 2010, 132, 8572.
isomerization > cycloaddition
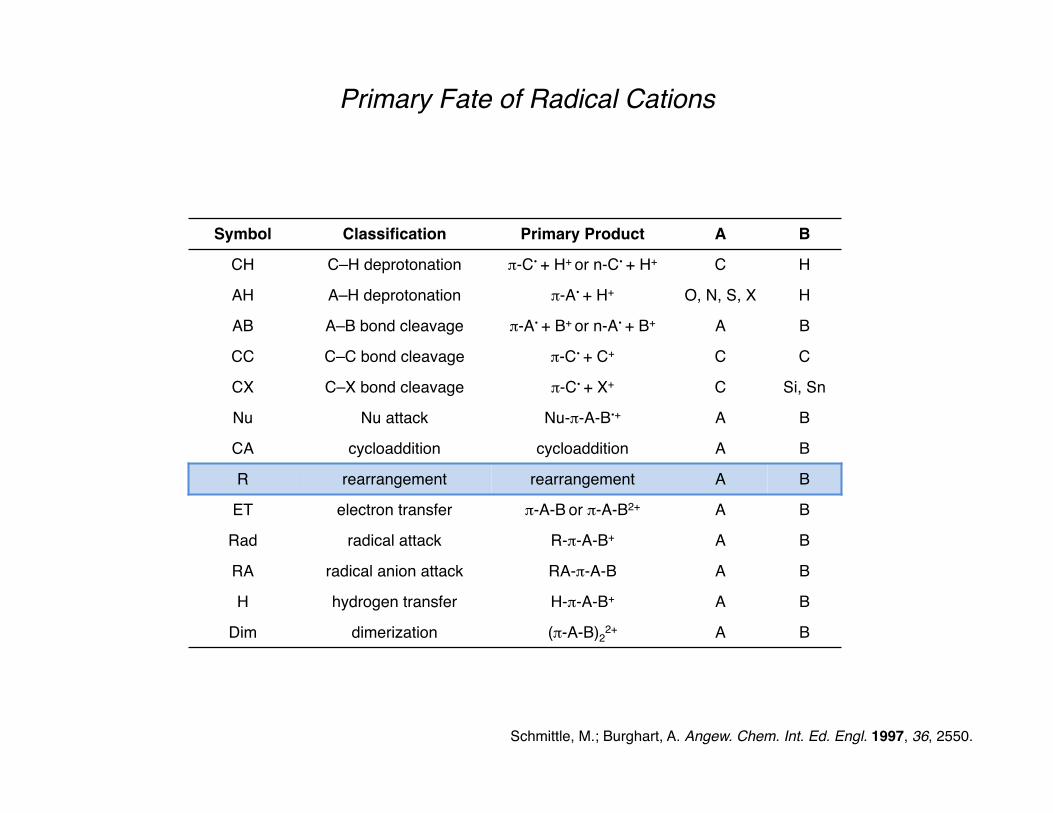
Primary Fate of Radical Cations
Schmittle, M.; Burghart, A. Angew. Chem. Int. Ed. Engl. 1997, 36, 2550.
Symbol Classification Primary Product A B
CH C–H deprotonation π-C• + H+ or n-C• + H+ C H
AH A–H deprotonation π-A• + H+ O, N, S, X H
AB A–B bond cleavage π-A• + B+ or n-A• + B+ A B
CC C–C bond cleavage π-C• + C+ C C
CX C–X bond cleavage π-C• + X+ C Si, Sn
Nu Nu attack Nu-π-A-B•+ A B
CA cycloaddition cycloaddition A B
R rearrangement rearrangement A B
ET electron transfer π-A-B or π-A-B2+ A B
Rad radical attack R-π-A-B+ A B
RA radical anion attack RA-π-A-B A B
H hydrogen transfer H-π-A-B+ A B
Dim dimerization (π-A-B)22+ A B
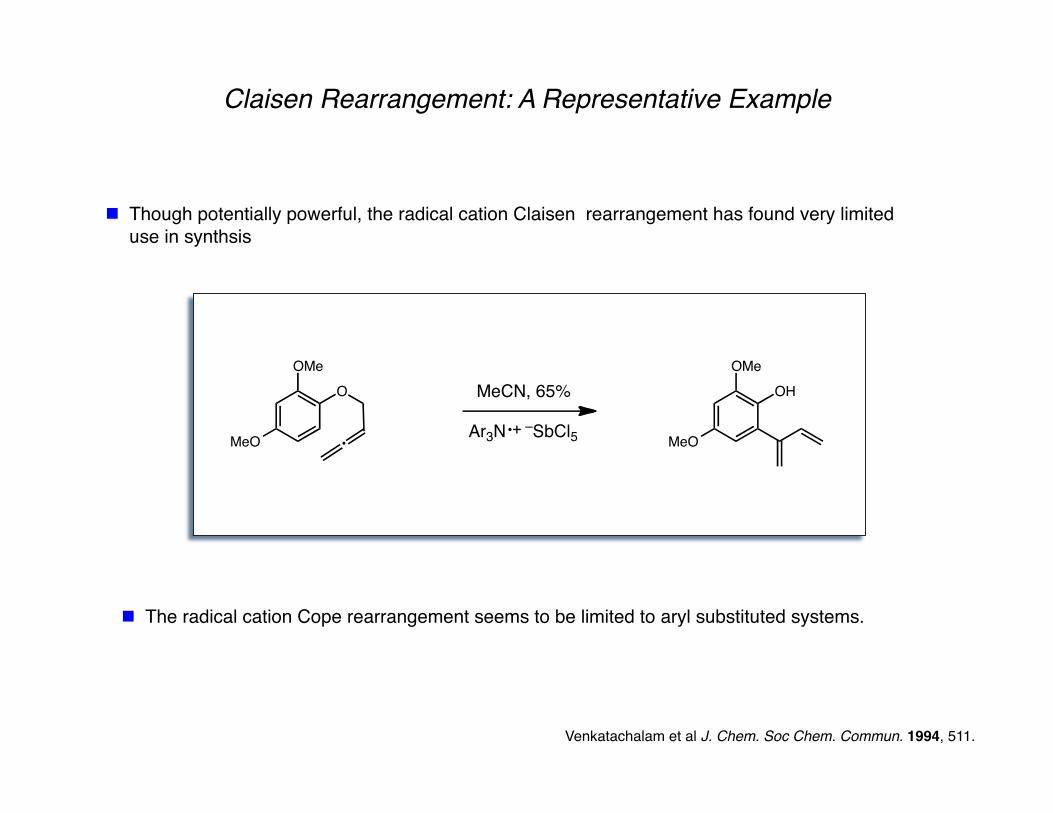
Claisen Rearrangement: A Representative Example
Though potentially powerful, the radical cation Claisen rearrangement has found very limited use in synthsis
Venkatachalam et al J. Chem. Soc Chem. Commun. 1994, 511.
OMe
MeO
O
•
MeCN, 65%
Ar3N –SbCl5
OMe
MeO
OH
The radical cation Cope rearrangement seems to be limited to aryl substituted systems.

Primary Fate of Radical Cations
Schmittle, M.; Burghart, A. Angew. Chem. Int. Ed. Engl. 1997, 36, 2550.
Symbol Classification Primary Product A B
CH C–H deprotonation π-C• + H+ or n-C• + H+ C H
AH A–H deprotonation π-A• + H+ O, N, S, X H
AB A–B bond cleavage π-A• + B+ or n-A• + B+ A B
CC C–C bond cleavage π-C• + C+ C C
CX C–X bond cleavage π-C• + X+ C Si, Sn
Nu Nu attack Nu-π-A-B•+ A B
CA cycloaddition cycloaddition A B
R rearrangement rearrangement A B
ET electron transfer π-A-B or π-A-B2+ A B
Rad radical attack R-π-A-B+ A B
RA radical anion attack RA-π-A-B A B
H hydrogen transfer H-π-A-B+ A B
Dim dimerization (π-A-B)22+ A B
Both radical (Rad) and radical anion (RA) have been previously discussed
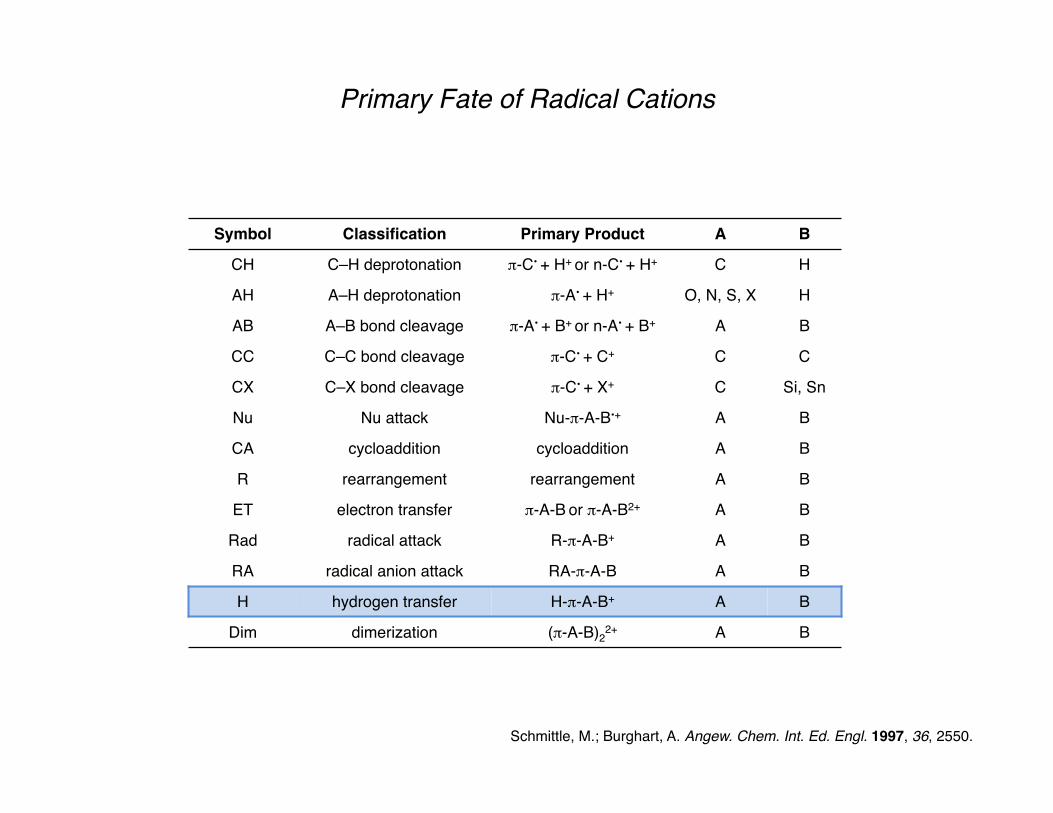
Primary Fate of Radical Cations
Schmittle, M.; Burghart, A. Angew. Chem. Int. Ed. Engl. 1997, 36, 2550.
Symbol Classification Primary Product A B
CH C–H deprotonation π-C• + H+ or n-C• + H+ C H
AH A–H deprotonation π-A• + H+ O, N, S, X H
AB A–B bond cleavage π-A• + B+ or n-A• + B+ A B
CC C–C bond cleavage π-C• + C+ C C
CX C–X bond cleavage π-C• + X+ C Si, Sn
Nu Nu attack Nu-π-A-B•+ A B
CA cycloaddition cycloaddition A B
R rearrangement rearrangement A B
ET electron transfer π-A-B or π-A-B2+ A B
Rad radical attack R-π-A-B+ A B
RA radical anion attack RA-π-A-B A B
H hydrogen transfer H-π-A-B+ A B
Dim dimerization (π-A-B)22+ A B
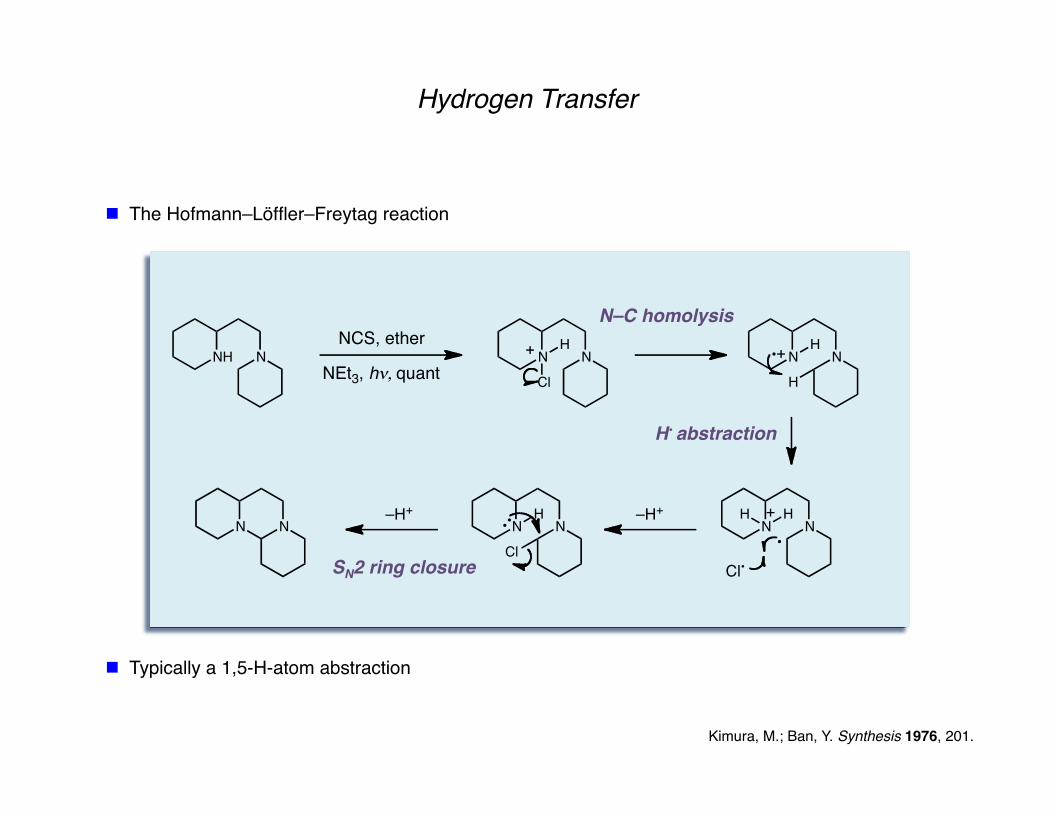
Hydrogen Transfer
The Hofmann–Löffler–Freytag reaction
Kimura, M.; Ban, Y. Synthesis 1976, 201.
NH N
NCS, ether
NEt3, h!, quantN N
Cl
HN N
H
H
N NH
Cl•
HN N
H –H+
Cl
N N–H+
Typically a 1,5-H-atom abstraction
N–C homolysis
H• abstraction
SN2 ring closure
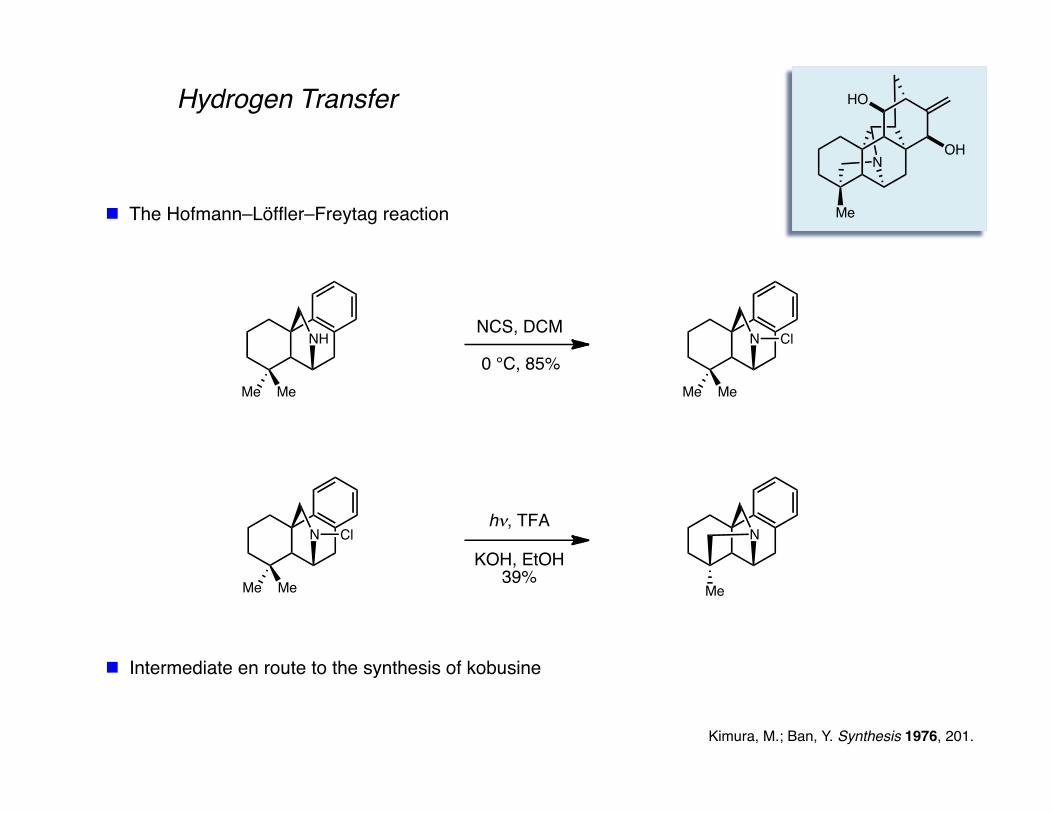
Hydrogen Transfer
The Hofmann–Löffler–Freytag reaction
Kimura, M.; Ban, Y. Synthesis 1976, 201.
Intermediate en route to the synthesis of kobusine
Me
OH
HO
N
NH
MeMe
NCS, DCM
0 °C, 85%N
MeMe
Cl
hν, TFA
KOH, EtOH39%
NN
MeMe
Cl
Me
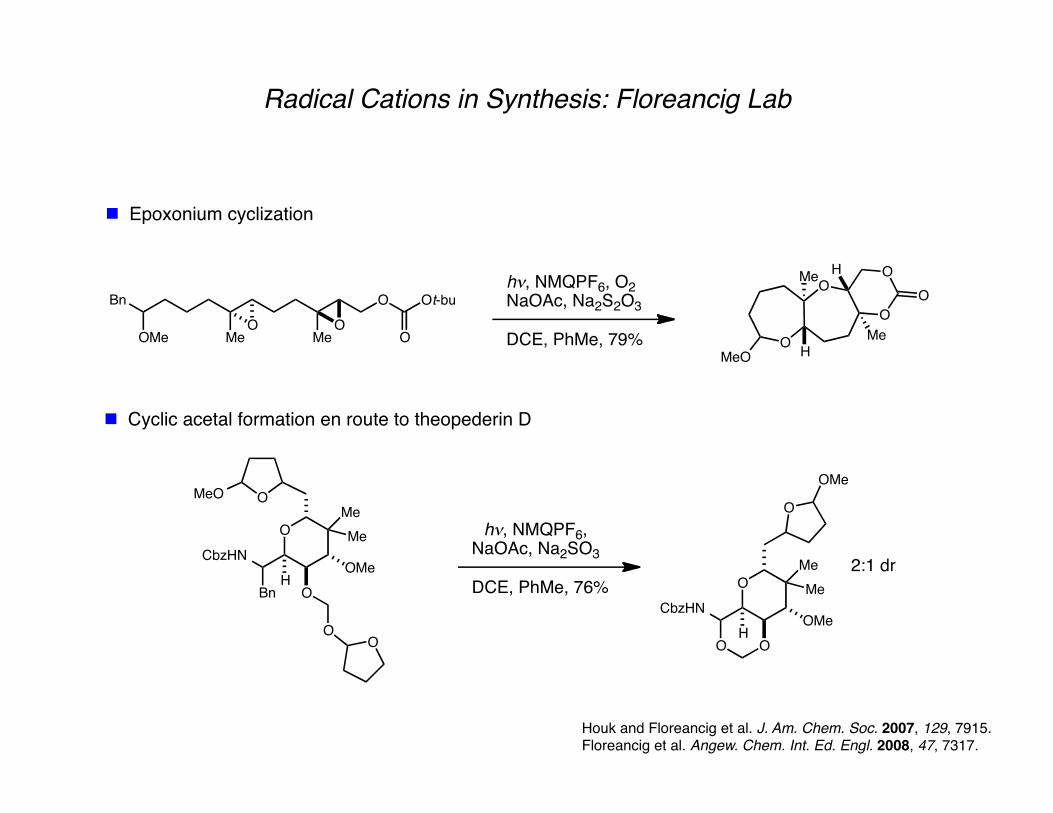
Radical Cations in Synthesis: Floreancig Lab
Epoxonium cyclization
Houk and Floreancig et al. J. Am. Chem. Soc. 2007, 129, 7915. Floreancig et al. Angew. Chem. Int. Ed. Engl. 2008, 47, 7317.
Cyclic acetal formation en route to theopederin D
Bn O
OMe MeO
MeO
Ot-bu
O
h!, NMQPF6, O2
NaOAc, Na2S2O3
DCE, PhMe, 79% O
O
O
OMe
Me
H
H
O
MeO
O
O
OMeH
OMeO
CbzHN
Bn
OO
Me
Me h!, NMQPF6,NaOAc, Na2SO3
DCE, PhMe, 76% O
O
OMeH
O
OMe
CbzHN
O
Me
Me
2:1 dr

Radical Cations in Synthesis
Used in Moellerʼs synthesis of nemorensic acid
Moeller et al J. Am. Chem. Soc. 2002, 124, 10101. Boger et al J. Am. Chem. Soc. 2008, 130, 420.
Bogerʼs synthesis of vinblastine
S
OHMe S
Me
O
Me
MeMe
Me
S
S
MeO
anode, 71%
Et4NOTsMeOH/THF
NH
N
EtCO2Me
ii. Fe2(ox)3–NaBH4air, 0 °C, lutidine
43%N
N
H
EtMeO
HO CO2MeOAc
i. FeCl3, 2hNH
N
H
MeO2C
Et
N
N
H
EtMeO
HO CO2MeOAc
OH
Me
Me

Radical Cations in Synthesis: MacMillan Lab
N
NBn
O Me
t-Bu
R
SOMO-activated
H
R
O
H
R
O
!-enolation
!-allylation
!-vinylation
H
O
R
!-nitroalkylation
HR
O
!-arylation
intramolecular!-arylation
R'
O
H
H
O
R'
R'
R'
R'
H
R
O NO2
R'
















![Novel Triphenylantimony(V) and Triphenylbismuth(V ... · carboxylic acid hydrogen (at 10.77 ppm and 11.14 ppm for HL1 and HL2, respectively) [26,34,35], indicating the deprotonation](https://static.fdocuments.us/doc/165x107/5e08a1757e14ab0ea37c3808/novel-triphenylantimonyv-and-triphenylbismuthv-carboxylic-acid-hydrogen-at.jpg)

