
Pcr & gel
15
0 Gene cloning Polymerase Chain Reaction (PCR) & Gel Electrophoresis Submitted to: Dr. Ambreen Ahmed Submitted by: Jannat Iftikhar B11-16 Semester: 6 th Department of botany University of the Punjab, LHR.
-
Upload
jannat-iftikhar -
Category
Science
-
view
158 -
download
3
Transcript of Pcr & gel

0
Gene
cloning
Polymerase Chain
Reaction (PCR) &
Gel Electrophoresis
Submitted to: Dr. Ambreen Ahmed
Submitted by: Jannat Iftikhar
B11-16 Semester: 6th
Department of botany University of the Punjab,
LHR.

1
Contents
1. Polymerase chain reaction (PCR) 1) Introduction
2) History
3) Polymerase chain reaction a. Requirements
b. Procedure
c. Post amplification detection
4) Modification and different types of PCR
5) Applications of PCR
2. Gel electrophoresis
1) Introduction
2) History
3) Preparation
4) Procedure
a. Making and preparing gel
b. Loading and running gel
c. Post electrophoresis staining
5) Applications of gel electrophoresis

2
1.Polymerase chain reaction
1) Introduction:
The polymerase chain reaction (PCR) is arguably the most powerful laboratory technique ever invented.
The ease with which it can be done, the relatively low cost, and it’s unique combination of specificity
and sensitivity coupled with great flexibility has led to a true revolution in genetics. PCR has opened
doors to areas hidden to all but a few for most of the history of genetics.
2) History:
The polymerase chain reaction was conceptualized and operationalized by Kary Mullis and colleagues at
Cetus Corporation in the early 1980’s. The method was first formally presented at the American Society
of Human Genetics Conference in October of 1985 and the first clinical application for PCR, an analysis
of sickle cell anemia, was published the same year. In its initial form, PCR was tedious and labor
intensive. However, the advent of a method by which a specific DNA sequence could be isolated from its
genomic context and amplified virtually without limit. The breakthrough came with the isolation and
purification of thermostable DNA polymerases. This allowed for PCR to be automated and soon the first
programmable PCR thermal cyclers appeared on the market. With this technique it is possible to make
virtually unlimited copies of a single DNA molecule even though it is initially present in a mixture
containing many different DNA molecules. It is used to amplify a specific DNA (target) sequence lying
between known positions (flanks) on a double-stranded (ds) DNA molecule. The polymerase chain
reaction can be used to amplify both double and single stranded DNA. Kary Mullis was awarded the
1993 Nobel Prize in Chemistry, not even ten years after its introduction.
3) POLYMERASE CHAIN REACTION (PCR):
PCR stands for the Polymerase Chain Reaction. In order to perform PCR, one must know at least a
portion of the sequence of the target DNA molecule that has to be copied. Generally, PCR amplifies
small DNA targets 100-1000 base pairs (bp) long. It is technically difficult to amplify targets >5000 bp
long. A pair of single stranded oligonucleotide primers, which have DNA sequences complementary to
the flanking regions of the target sequence, must be synthesized. The primers are complementary to
either end of the target sequence but lie on opposite strands. The primers are usually 20-30 nucleotides
long and bind to complementary flanking region at 3' end.
a. Requirements: ƒ
i. Thermal cycler (thermocycler)
ii. PCR amplification mix typically containing
iii. Sample dsDNA with a target sequence
iv. Thermostable DNA polymerase
v. Two oligonucleotide primers

3
vi. Deoxynucleotide triphosphates (dNTPs)
vii. Reaction buffer containing magnesium ions and other components
b. Procedure:
1. The DNA molecule carrying a target sequence is denatured by heat at 90-95oC for 20 seconds.
The two strands separate due to breakage of the hydrogen bonds holding them together.
Oligonucleotide primers are added.
2. A reaction mixture containing all four deoxynucleotide triphosphates (dATP, dCTP, dGTP, dTTP)
and a thermostable DNA polymerase is added. A DNA polymerase (Taq) that is not denatured by the
high temperature needed to separate the DNA strands is used. It is usually sourced from Thermus
aquaticus, a bacterium isolated from hot springs. 3. The mixture is allowed to cool to a lower
temperature (50-65oC). Each strand of DNA molecule becomes annealed with an oligonucleotide
primer complementary to either end of the target sequence. Primer anne aling takes 20 seconds.
4. The temperature is raised to 60-75oC and primers are extended by the action of DNA polymerase
for 30 seconds. The polymerase synthesizes complementary sequence the 5' to 3' direction away
from each of the primers. If the template contains an A nucleotide, the enzyme adds on a T
nucleotide to the primer. If the template contains a G, it adds a C to the new chain. Polymerization
continues until each newly synthesized strand has proceeded far enough to contain the site
recognized by the other primer. At this point there would be exactly two copies of the target DNA
sequence.
5. The mixture is heated again at 90-95oC to denature the molecules and separate the strands and
the cycle repeated. Each new strand then acts as a template for the next cycle of synthesis. Thus
amplification proceeds at an exponential (logarithmic) rate, i.e. amount of DNA produced doubles at
each cycle. The amplified product at the end of PCR is called amplicon. (fig.1)
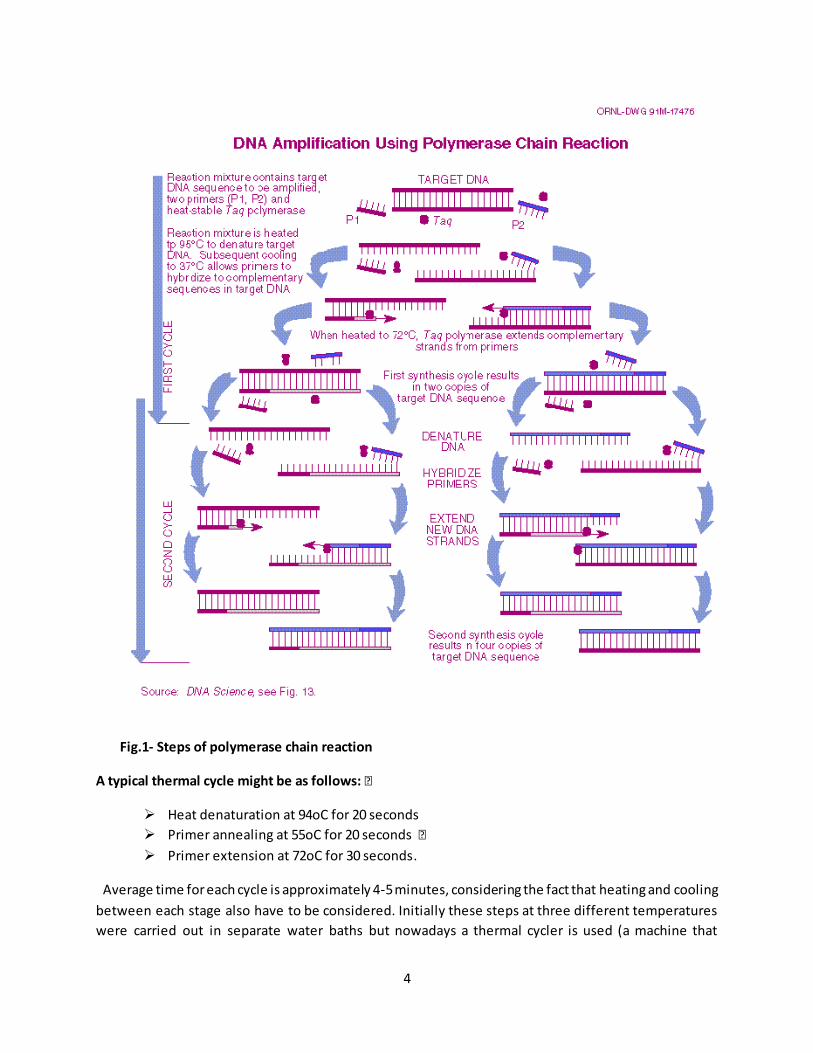
4
Fig.1- Steps of polymerase chain reaction
A typical thermal cycle might be as follows: ƒ
Heat denaturation at 94oC for 20 seconds
Primer annealing at 55oC for 20 seconds ƒ
Primer extension at 72oC for 30 seconds.
Average time for each cycle is approximately 4-5 minutes, considering the fact that heating and cooling
between each stage also have to be considered. Initially these steps at three different temperatures
were carried out in separate water baths but nowadays a thermal cycler is used (a machine that

5
automatically changes the temperature at the correct time for each of the stages and can be
programmed to carry out a set number of cycles). After the introduction of thermocycler, each cycle of
replication can be completed in less than 5 minutes. After 30 cycles, a single molecu le of DNA is
amplified into more than a billion copies (230 = 1.02 x 109).
c. Post amplification detection:
Following PCR, the amplification product can be detected using gel electrophoresis followed by ethidium
bromide staining and visualization with uv transillumination. Visualization of a band containing DNA
fragments of a particular size can indicate the presence of the target sequence in the original DNA
sample. Absence of a band may indicate that the target sequence was not present in the original DNA
sample. Confirmation of the amplicons can be made by southern blotting using specific probes.
4) Modifications and different types of PCR are:
Nested PCR, Multiplex PCR, RT-PCR, Touchdown PCR, Arbitrarily Primed PCR, Inverse PCR, Allele Specific
PCR, Asymmetric PCR, "Hot Start" PCR, Core Sample PCR, Degenerate PCR and PCR-Elisa.
5) Applications of PCR:
Amplification of small amounts of DNA for further analysis by DNA fingerprinting.
The analysis of ancient DNA from fossils.
Mapping the human (and other species) genome.
The isolation of a particular gene of interest from a tissue sample.
Generation of probes: large amount of probes can be synthesized by this technique.
Production of DNA for sequencing: Target DNA in clone is amplified using appropriate primers
and then its sequence determined. Helpful in conditions where amount of DNA is small.
Analysis of mutations: Deletions and insertions in a gene can be detected by differences in size
of amplified product.
Diagnosis of monogenic diseases (single gene disorders): For pre-natal diagnosis, PCR is used to
amplify DNA from foetal cells obtained from amniotic fluid. PCR has also proved very important
in carrier testing.
Detection of microorganisms: Especially of organisms and viruses that are difficult to culture or
take long time to culture or dangerous to culture.
The PCR has even made it possible to analyze DNA from microscope slides of tissue preserved
years before.
Detection of microbial genes responsible for some aspect of pathogenesis or antibiotic
resistance. Crucial forensic evidence may often be present in very small quantities, e.g. one
human hair, body fluid stain (blood, saliva, semen). PCR can generate sufficient DNA from a
single cell.

6
6) Limitations of PCR:
PCR is an extremely sensitive technique but is prone to contamination from extraneous DNA, leading to
false positive results. Another potential problem is due to cross-contamination between samples. It is
for this reason that sample preparation, running PCR and post-amplification detection must be carried
out in separate rooms. Concentration of Mg is very crucial as low Mg2+ leads to low yields (or no yield)
and high Mg2+ leads to accumulation of nonspecific products. Non-specific binding of primers and
primer-primer dimmer formation are other possible reasons for unexpected results. Reagents and
equipments are costly, hence can’t be afforded by small laboratories.
2. Gel electrophoresis
1. Introduction:
Agarose gel electrophoresis is a quick and easy molecular technique used to analyze and separate
nucleic acids based on their size (i.e. how many base pairs a molecule is composed of). Electrophoresis
takes advantage of the fact that DNA’s phosphate backbone is negatively charged. Thus when DNA is
placed in an electric field, it will migrate toward the positive electrode. The differential ability of DNA to
move through a gel based on its size doesn’t really depend on the electric field or the charged properties
of DNA, but more importantly on the composition of the gel. Agarose is a complex polymer that forms a
matrix through which DNA travels when subjected to an electric field.
Smaller DNA runs FARTHER down the gel than bigger DNA
In general, the rate of travel of linear DNA in an electric field varies inversely with its molecular
weightand depends on 4 things:
• The strength of the E field, or voltage applied – DNA will run “faster” in a stronger field. You may think
that larger DNA runs faster than smaller DNA as more voltage is applied because larger DNA is more
negatively charged simply because it is larger. But this is not necessarily the case. As explained above,
DNA of different sizes move at different rates because of their relative ease at which they can get
through the agarose matrix, not because a big DNA is more charged than a small DNA. The only time the
charge of large DNA molecules makes a difference is at very high voltages. Therefore, gels are usually
run at voltages less than 125V.
• Concentration of agarose – DNA runs “slower” in a more tightly woven net (i.e more concentrated
agarose).
• Buffer used – the same DNA will run slightly different in different types and concentrations of buffer.
• Conformation of DNA – Circular and supercoiled DNA run faster than linear DNA. Thus if you have a
1000 bp linear fragment and a 1000 bp bacterial plasmid which is circular, the plasmid will migrate
farther down the gel than the linear molecule even though they are the same molecular weight .

7
2. History:
The history of electrophoresis begins in earnest with the work of Arne Tiselius in the 1930s, and
new separation processes and chemical analysis techniques based on electrophoresis continue to be
developed into the 21st century. Tiselius, with support from the Rockefeller Foundation, developed the
"Tiselius apparatus" for moving boundary electrophoresis, which was described in 1937 in the well-
known paper "A New Apparatus for Electrophoretic Analysis of Colloidal Mixtures". The method spread
slowly until the advent of effective zone electrophoresis methods in the 1940s and 1950s, which
used filter paper or gels as supporting media. By the 1960s, increasingly sophisticated gel
electrophoresis methods made it possible to separate biological molecules based on minute physical
and chemical differences, helping to drive the rise of molecular biology. Gel electrophoresis and related
techniques became the basis for a wide range of biochemical methods, such as protein
fingerprinting, Southern blot and similar blotting procedures,DNA sequencing, and many more. Some
important events are given here:
1930s – first reports of the use of sucrose for gel electrophoresis
1955 – introduction of starch gels, mediocre separation
1959 – introduction of acrylamide gels; disc electrophoresis (Ornstein and Davis); accurate
control of parameters such as pore size and stability; and (Raymond and Weintraub)
1966 – agar gels
1969 – introduction of denaturing agents especially SDS separation of protein subunit (Weber
and Osborn)
1970 – Laemmli separated 28 components of T4 phage using a stacking gel and SDS
1972 – agarose gels with ethidium bromide stain.
1975 – 2-dimensional gels (O’Farrell); isoelectric focusing then SDS gel electrophoresis
1977 – sequencing gels
1983 – pulsed field gel electrophoresis enables separation of large DNA molecules
1983 – introduction of capillary electrophoresis
A 1959 book on electrophoresis by Milan Bier cites references from the 1800s. However, Oliver
Smithies made significant contributions. Bier states: "The method of Smithies ... is finding wide
application because of its unique separatory power." Taken in context, Bier clearly implies that
Smithies' method is an improvement.
3. Preparation: a. Materials:
Gel box/tank, gel tray, gel comb(s), power supply, UV viewing table, camera (or more sophisticated gel
viewing and image producing equipment), pipettes, pipette tips, KimWipes, gloves, goggles, 250 ml
Erlenmeyer flask, graduated cylinder

8
Ethidium Bromide (10mg/ml)
Agarose, Gene Pure GQA (all purpose gels)
Agarose, Gene Pure 3:1 (high resolution applications, such as <200 bp DNA fragments or RFLP)
DNA ladder/marker,
DNA ladder/marker,
DNA ladder/marker, quantitative,
Gel Loading Dye, 6X, Blue-Orange
10X TBE (stock)
in a 1L beaker: 850 ml of dH2O, 40 ml of 0.5M EDTA (pH8), 108 g Tris base stir add 55g Boric Acid, stir
pour into graduated cylinder and fill up to 1L with dH2O autoclave if it precipitates over time, re-
autoclave Storage: RT 5X TBE (stock) in a 1L beaker: 850 ml of dH2O, 20 ml of 0.5M EDTA (pH 8), 54g
Tris base stir add 27.5 g Boric Acid, stir pour into graduated cylinder and fill up to 1L with dH2O
autoclave Storage: RT
1X TBE (working)
dilute 2 L of above solution to 10 L with dH2O or 200 ml of above to 1L. Note: you can use sterile water
and autoclave, but it’s not necessary Storage: RT 0.5X TBE (working) **This is the concentration of
buffer most commonly used for gels in the Treseder Lab. If your gels are not turning out well, you can
try 1X buffer. ***For best results, use the same concentration and batch of buffer in both the gel and
the gel box dilute 1 L of above to 10 L with dH2O or 100 ml of above to 1 L. Note: you can use sterile
water and autoclave, but it’s not necessary Storage: RT
0.5M EDTA (pH 8.0)
46.5 EDTA 200ml water 5 g of NaOH pellets to adjust pH to 8.0 (EDTA won’t dissolve until pH is 8.0)
b. Samples, Combs, and Loading Volume:
First, get an idea of what you want to view in the gel and how many lanes you will need (i.e. how many
samples you have). Don’t forget to leave at least one well for DNA ladder/marker. If you plan to purify
the DNA bands out of this gel via gel extraction, see below. We have 2 gel apparatuses: large and small. I
only use the large one when I have more than 22-24 lanes to load, when I need to skip lanes, or when I
want to load more than 15-20 uL of sample. The small gel is easier and faster to deal with, so use that
one if you can. The gels have several combs - choose the ones appropriate for how many lanes you
want your gel to have. You can do a double-decker gel if you need that many lanes or you can do just
one row of lanes. A single row is sometimes helpful if you need to run the gel for a long time in order to
get good separation of DNA fragments that are very close in size. Otherwise there is no difference

9
between a double row gel and a single row gel except that in a double-row gel you just have to be sure
that your top samples don’t run down into the bottom section. The other important thing to note about
the combs is that the teeth on the different sides of the comb have different widths. With the small gel,
use the fat side of the comb if you want to load up to 15-20 uL of sample. The skinnier side creates wells
that can only reliably hold up to 10 or 12 uL. But if you want to have deeper wells to accommodate more
sample volume, you can also increase the gel height by using more buffer to create the gel. Or just use
the fat side of the comb, or use the large gel which creates larger wells.
4. Procedure: Making and Preparing the Gel :
1. Figure out the best concentration of agarose to use based on what size DNA you want to visualize.
The DNA will show up pretty much on any gel, but the resolution will be better if you follow the table
below. On that note, however, don’t worry about measuring exactly when making the gel, because a
small difference in concentration doesn’t matter that much. Only worry about exact measurements
when you need to be super consistent between gels (e.g. RFLP). Also, agarose is a little expensive, so
don’t make a 2 or 3 % gel unless you really need to separate fragments that only differ in size by a few
base pairs. Gels less than 1% are flimsy, however, so handle them carefully. The more concentrated a gel
is, the stiffer and easier to handle it is. Good rules of thumb are: to look at PCR products make a 1.5 %
gel and to look at genomic DNA make a 1% gel.
2. Make the melted agarose solution. The small gel is made with 35-40 mL of buffer and the large gel is
made with 80-100 mL of buffer. The percent agarose is calculated in g/mL as in the following: To make a
1% gel in the small gel tray, use 0.35 grams of agarose in 35 mL of buffer. 0.35g / 35ml = .01 or 1%
Weigh out the appropriate amount of agarose using the designated agarose weigh boat that is on the
first shelf above the electrophoresis bench. Put the agarose in a 250 ml erlenmyer flask. In a graduated
cylinder, measure out the appropriate volume of 0.5 X TBE Buffer for the appropriate size gel tray and
pour it into the flask. Swirl a little bit. Stuff 2 crumpled kimwipes into the opening of t he flask.
Microwave for 30 sec then swirl, and then microwave another 30 sec, or until it boils but before it tries
to bubble out of the flask. Use a paper towel or flask holder to grab the flask (watch out, it’s hot!) and
swirl it around well. Make sure there are no little undissolved agarose floaties… if there are, microwave
again. Remove the kimwipes and let cool on bench for a few minutes.
3. While the melted agarose is cooling, tape up the open ends of the gel tray with lab labeling tape.
Leave the rubber gaskets on the ends of the tray, just make sure to press down on the tape so there is
no way for the agarose to ooze out when you pour it into the tray. Also, choose the combs you want to
use and put them into the slots on the tray.
4. If there is still visible steam coming out of the flask, let it cool a few more minutes on the bench, run
it under cool tap water for a bit, or hold it in a cool water bath for a bit. If it gets too cool it will harden
up, which is not a problem – just microwave it again. The melted gel must be a little cooled off before
you add Ethidium Bromide (EtBr is toxic, so avoid creating toxic vapor by adding EtBr to hot liquid) and

10
before you pour it into the tray (it can crack the tray if it’s too hot). You can be pretty certain the gel is
cool enough if you can hold the flask at the bottom comfortably.
5. Add Ethidium Bromide. EtBr is necessary to stain the DNA so we can see it under UV light. Because it
works be intercalating the base pairs in the major groove of double-stranded DNA, Ethidium Bromide is
also a mutagen. Therefore, PLEASE USE CAUTION WHEN USING ETHIDIUM BROMIDE. Use gloves, and
proper disposal procedures. Ethidium Bromide can also be used as a stain after the gel has already been
run. We do this occasionally in the Treseder lab, but not that often because it tacks on another hour to
the procedure. Gels tend to look nicer and clearer if stained afterwards, but just including it inside the
gel is easier and gets the job done. If you need to stain the gel af terwards, refer to the gel staining
protocol.
The final concentration of EtBr in the gel should be about 0.3 - 0.5 ug/mL. The stock solution is 10
mg/mL. So for the small gel consisting of 35 mL, I add 1.1 – 1.25 uL of EtBr. Pipette up the amount of
EtBr you need, with any pipettor directly out of the stock solution. Pipette it out into the E-flask, and
pipette up and down and submerge the pipette tip to get all the EtBr off the tip. If you successfully got
off all the EtBr go ahead and put the tip in the regular tip waste container. If not, put the tip in the EtBr
waste. Swirl the melted gel slurry very well.
6. Cast the gel. Pour the slurry slowly into the taped gel tray. Add combs if you haven’t already. Tip the
tray slightly to get the slurry into every crevice and pop any bubbles with the corner of a kimwipe. Be
sure to check for bubbles around the teeth of the comb(s). Let the tray stand on a level surface for about
20 minutes or until the gel has that plastic-y sheen to it. (Before the gel is hard, it is perfectly
transparent, but becomes less clear as it hardens.)
7. While you are waiting for the gel to set, get out all the things you need to thaw, and let them thaw on
the bench. (If you are going to store the gel overnight, don’t do this step until you are actually going to
run the gel.) Things that will need to thaw include:
6 X loading dye/buffer DNA ladder/molecular marker DNA samples you want to run
8. Prepare the gel box. Carefully lift up the combs from the hardened gel. If you don’t have time to run
the gel today, you can wrap it up with plastic wrap and store it overnight in the refrigerator. Otherwise,
go ahead and remove the tape from the sides and place the entire tray into the gel box/tank. Place the
gel tray containing the hardened gel into the box/tank so that the top of the gel (top = the end with the
wells nearest to the edge) is at the negative (black) electrode-end of the tank. Pour enough 0.5 X TBE
(use the same batch of buffer as used to create the gel) into the tank such that the gel is completely
submerged. It only needs to be covered in about a mm of buffer. Try to place the tank in a location that
will be convenient for loading and running – it is not a good idea to move the entire tank once you’ve
already loaded the wells.
Loading and Running the Gel :

11
Please use the appropriate set of pipettors based on what type of DNA you are dealing with. If you are
looking at PCR products, use the “PCR products only” pipettors and barrier tips. Never use the “PCR
products only” pipettors when working with non-amplified DNA.
1. Mix the DNA with the loading dye and load the wells. This can be done either on a piece of parafilm
(good for running small volumes of sample quickly) or in separate eppendorf tubes. Decide what volume
of DNA you need to load – this is usually dependent on the concentration of the DNA, but one often
does not know what concentration the sample is in before it is run on a gel. (It is probably a good idea to
measure your DNA concentration at least roughly in the spec beforehand.) Usually a couple of uLs is
enough to see on a gel. For PCR products and total genomic DNA, 3-4 uL is sufficient. If you are doing a
gel extraction, you will probably want to use 15 or 20 ul. Pipette out the appropriate volume of 6 X
loading buffer in little beads on the parafilm. Because it is 6X concentration, use about 1 ul of dye for
every 5 ul of DNA that you will be loading. But beware: the dye is mostly glycerol and does not like to
pipette very accurately. Also, it is better to use too much dye than too little. If you don’t have enough
dye, the DNA won’t sink into the well. So, considering all these things, I like to set the pipettor for about
1 ul when pipetting the dye, even if I am only going to load 3 or 4 ul of DNA. Work quickly or only pipette
a few dye aliquots at a time because the dye likes to evaporate off the parafilm. Next pipette out the
volume of DNA you want to use (if you are using PCR products, please use the PCR pipettors) and
pipette it up and down with a bead of dye on the parafilm to mix. If you don’t want to waste any DNA,
then change your pipettor setting up one ul and then suck up all the DNA/dye mixture into the tip and
carefully dip the pipette into one of the wells in the gel and pipette it out slowly. You should see the
colored solution literally look as though it is sinking. If you change the pipette setting in order to get up
all the DNA, just be sure to stay consistent between all the samples you will be loading. Otherwise, you
can leave the pipette’s setting as is, and just leave a little unused DNA/dye on the parafilm. Continue
this procedure with all your samples. Try to keep your loading technique consistent between samples
and always have some mechanism to remember what sample each well contains. And always change
your pipette tip between samples. Just take note if some stray DNA gets into a neighboring well – you
can even load every other well if this is a concern. Try standing/sitting in diff erent positions, or sliding
the gel tray slightly within the tank – sometimes there is a glare that can make loading difficult. Also,
make sure not to stick the tip down into the well too far or you may puncture the bottom of the well.
And don’t forget that you can use your non-pipetting hand to rest or stabilize the barrel of the pipette
while pipetting. Another trick that sometimes makes loading easier, is to just pipette up and down the
buffer inside each well before loading to “rinse” out the well.
Don’t forget to load a DNA ladder into at least one of your wells. DNA ladders, also called molecular
weight markers, are used as a standard so that you know how far DNA of a known size migrates in your
gel.
We have 3 different ladders:
1. 100 bp ladder – good for fragments that are 100 bp – 1 or 2 kilobases (kb)
2. 1 kb ladder – good for fragments larger than 500 bp

12
3. Quant-it Ladder – quantitative ladder for determining approximate DNA concentration. This is the one
with the purple top, and already contains dye. So do NOT add dye to this one.
For Ladders 1 and 2 (both have a pale green top), use 5 ul of ladder + 1 ul dye. For the Quant-it ladder,
do not add anything, just pipette out 10 ul of ladder and load directly.
2. After loading the wells, I usually add Ethidium Bromide to the positive end (red electrode, or end
opposite the wells in the gel) of the gel tank. Because EtBr runs toward the negative electrode, the EtBr
inside the gel will move towards the top of the gel, leaving the bottom half of the gel a different color.
To correct for this, add about 1.25uL of EtBr to the buffer in the positive end of the gel tank. Swish it
around with the pipette tip to mix. Remember to dispose of EtBr waste appropriately.
3. Place the lid on the gel tank and be sure the electrode connections are secure and tight.
4. Turn on the power supply by flipping the switch. Set the voltage appropriately. Quick diagnostic gels
are usually run at 90-105 V. But for clearer bands and a prettier picture, you can use as low as 60 V. The
lower the voltage, the longer you will have to run the gel. Also, if you need to resolve very small DNA
fragments or fragments that are close in length, the voltage should be lower.
5. Set your timer. For quick diagnostic gels in the small gel box. Run longer to resolve bands better. Do
not run too long or at too high of voltage that you DNA runs off the gel!
6. When timer is done, turn off the power supply. Take off the gel box lid and remove the gel tray.
Careful here: remember to wear gloves, and do not let the gel slip out of the tray! Transfer the gel in it’s
tray to the UV-viewing device. Slide the gel out of the tray and directly onto the UV table. Cover with the
plexi-glass cover attached to the UV-table. Turn on the UV. Remember to wear goggles! Note: there are
2 UV settings. One makes the bands look brighter – use this one to take picture. The lower intensity
setting is useful for gel extractions, such that the DNA is not exposed to high UV while you are cutting
out the gel.
7. Take a polaroid picture of your gel. Wearing goggles, place the tube-like end of the camera so it is
standing upright on the UV table, directly over the gel, with the handle end of the camera facing up.
Being as still as possible, pull the trigger on the handle to take the picture. Pull the strip of paper out of
the camera to release the exposed Polaroid. Do NOT peel off the paper yet. Let the picture set for 5
minutes before opening. When peeling the paper off the photo, try not to touch the li quidy stuff on the
sides – the exposure chemicals are skin irritants.
8. If you aren’t doing a gel extraction, dispose of the gel in the EtBr hazardous waste. You can leave the
used buffer in the gel tank for several more runs, just be sure to add to the buffer to completely
submerge the gel. For best gel results, use completely fresh buffer. To clean: wash gel tray, lid, and tank
with mild soap and warm water (not hot!) and rinse with DI.
9. To do a gel extraction: Label appropriate number of 1.5 ml tubes for each band you will be extracting.
Put UV setting at the lower intensity. Using a clean razor blade or scalpel, carefully cut out the piece of
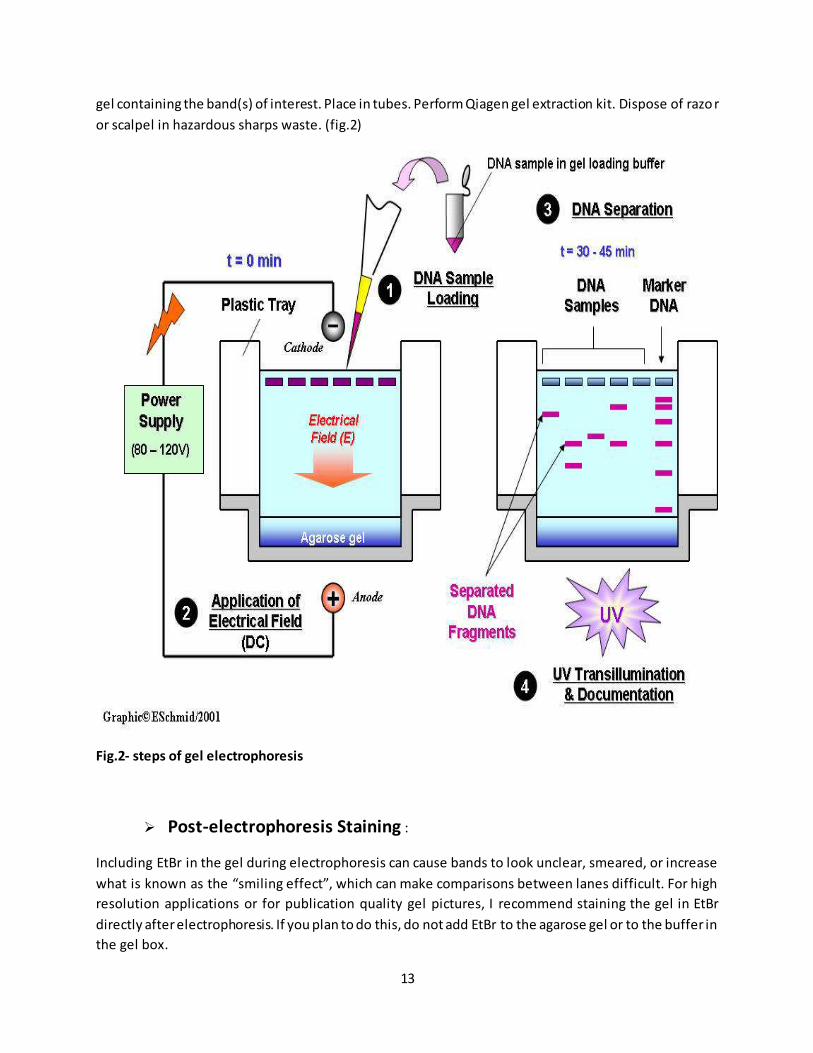
13
gel containing the band(s) of interest. Place in tubes. Perform Qiagen gel extraction kit. Dispose of razo r
or scalpel in hazardous sharps waste. (fig.2)
Fig.2- steps of gel electrophoresis
Post-electrophoresis Staining :
Including EtBr in the gel during electrophoresis can cause bands to look unclear, smeared, or increase
what is known as the “smiling effect”, which can make comparisons between lanes difficult. For high
resolution applications or for publication quality gel pictures, I recommend staining the gel in EtBr
directly after electrophoresis. If you plan to do this, do not add EtBr to the agarose gel or to the buffer in
the gel box.

14
1. Prepare a shallow plastic container (with lid) to contain enough solution to reach a depth of 2-4 mm
(high enough to cover the gel). Solution = DI water containing 0.5-1 ug/ml of EtBr.
2. When gel is done running, turn off power supply and remove gel tray containing the gel.
3. Slide the gel out of the tray into the container with the EtBr solution. Put lid on.
4. Put container on shaker. Turn shaker on, medium setting. Cover container with foil to preserve EtBr
potency, especially if you plan to re-use the staining solution.
5. Let shake for at least 20 minutes.
6. Transfer the gel to a similar container with only DI water. Shake gel in DI for 5-10 minutes.
7. View/Image gel with UV. If gel is over-stained, repeat step 6 for up to 1 hour. 8. Staining solution can
be re-used several times. Dispose of all solutions in EtBr liquid waste receptacles.
Applications of gel electrophoresis:
Gel electrophoresis allows scientists to separate strands of DNA--allowing for easier identification and examination of its components. As the DNA moves through the gel, it helps separate the larger and smaller sections from each other. This process offers several beneficial applications of providing genetic information. Illness
Gel electrophoresis can help scientists identify damaged genes, including oncogenes. It can also help identify certain genetic diseases such as sickle cell anemia, Huntington’s disease and Duchenne muscular dystrophy, according to the State University of New York. Gel electrophoresis can also identify viruses.
Family
Gel electrophoresis can determine paternity as well as establish genetic links between larger groups of people, according to Colorado University.
Animals
Gel electrophoresis offers many benefits in fields dealing with animals. Zoos can use this technique to reduce the negative impact of inbreeding. Taxonomists and evolutionary biologists can learn more about evolutionary relationships, identify species and document differences between them. Thos e studying animal behavior can use DNA to glean information about kinship of animals and how it impacts interaction with other animals.
Crime
Scientists can also use this procedure to examine tissue samples found at crime scenes.


















