
Payers’Guide toNew FDA Approvals
52
Payers’ Guide to New FDA Approvals ©2011 Engage Healthcare Communications, LLC www.AHDBonline.com ™ Special Feature THE PEER-REVIEWED FORUM FOR EVIDENCE IN BENEFIT DESIGN ™ FOR PAYERS, PURCHASERS, POLICYMAKERS, AND OTHER HEALTHCARE STAKEHOLDERS MAY/JUNE 2011 VOLUME 4 I NUMBER 2 I SPECIAL FEATURE
-
Upload
dalia-buffery -
Category
Documents
-
view
219 -
download
1
description
American Health & Drug Benefits
Transcript of Payers’Guide toNew FDA Approvals

Payers’ Guide to New FDA Approvals
©2011 Engage Healthcare Communications, LLCwww.AHDBonline.com
™ Special Feature
THE PEER-REVIEWED FORUM FOR EVIDENCE IN BENEFIT DESIGN™
FOR PAYERS, PURCHASERS, POLICYMAKERS, AND OTHER HEALTHCARE STAKEHOLDERS
MAY/JUNE 2011 VOLUME 4 I NUMBER 2 I SPECIAL FEATURE

i
INDICATION®
WARNING: POTENTIAL FOR ABUSE, IMPORTANCE OF PROPER PATIENT SELECTION AND LIMITATIONS OF USE
Potential for AbuseEXALGO contains hydromorphone, an opioid agonist and a Schedule II controlled substance with an abuse liability similar to other opioid analgesics. EXALGO can be abused in a manner similar to other opioid agonists, legal or illicit. These risks should be considered when administering, prescribing, or dispensing EXALGO in situations where the healthcare professional is concerned about increased risk of misuse, abuse, or diversion. Schedule II opioid substances which include hydromorphone, morphine, oxycodone, fentanyl, oxymorphone and methadone have the highest potential for abuse and risk of fatal overdose due to respiratory depression.Proper Patient SelectionEXALGO is an extended-release formulation of hydromorphone hydrochloride indicated for the management of moderate to severe pain in opioid tolerant patients when a continuous around-the-clock opioid analgesic is needed for an extended period of time.
Patients considered opioid tolerant are those who are taking at least 60 mg oral morphine per day, 25 mcg transdermal fentanyl/hour, 30 mg oral oxycodone/day, 8 mg oral hydromorphone/day, 25 mg oral oxymorphone/day or an equianalgesic dose of another opioid, for a week or longer.EXALGO is for use in opioid tolerant patients only.Fatal respiratory depression could occur in patients who are not opioid tolerant.Accidental consumption of EXALGO, especially in children, can result in a fatal overdose of hydromorphone.Limitations of UseEXALGO is not indicated for the management of acute or postoperative pain.EXALGO is not intended for use as an as-needed analgesic. EXALGO tablets are to be swallowed whole and are not to be broken, chewed, dissolved, crushed or injected. Taking broken, chewed, dissolved or crushed EXALGO or its contents leads to rapid release and absorption of a potentially fatal dose of hydromorphone.
IMPORTANT RISK INFORMATION
W
s

®
Please see brief summary of Full Prescribing Information, including boxed warning, on following pages.
I
2 a
n
c
t o
WHERE IS HER DAY HEADED WITHOUT A 24-HOUR PAIN MEDICATION?
EXALGO® puts the power of hydromorphone into a once-daily dose, so your patients can worry less about their medicine wearing off.
To find out more, visit www.EXALGO.com.
11:11:23 AM

EXALGO® (hydromorphone HCl)Extended-Release Tablets
WARNING: POTENTIAL FOR ABUSE, IMPORTANCE OF PROPER PATIENTSELECTION AND LIMITATIONS OF USE
Potential for AbuseEXALGO contains hydromorphone, an opioid agonist and a Schedule II controlled substance with an abuse liability similar to other opioid analgesics. EXALGO can be abused in a manner similar to other opioid agonists, legal or illicit. These risks should be considered when administering, prescribing, or dispensing EXALGO in situations where the healthcare professional is concerned about increased risk of misuse, abuse, or diversion. Schedule II opioid substances which include hydromorphone, morphine, oxycodone, fentanyl, oxymorphone and methadone have the highest potential for abuse and risk of fatal overdose due to respiratory depression [see Drug Abuse and Dependence (9)].
Proper Patient SelectionEXALGO is an extended-release formulation of hydromorphone hydrochloride indicated for the management of moderate to severe pain in opioid tolerant patients when a continuous around-the-clock opioid analgesic is needed for an extended period of time. Patients considered opioid tolerant are those who are taking at least 60 mg oral morphine per day, 25 mcg transdermal fentanyl/hour, 30 mg of oral oxycodone/day, 8 mg oral hydromorphone/day, 25 mg of oral oxymorphone/day or an equianalgesic dose of another opioid, for a week or longer [see Indications and Usage (1) and Dosage and Administration (2)].
EXALGO is for use in opioid tolerant patients only [see Indications and Usage (1) and Dosage and Administration (2)].
Fatal respiratory depression could occur in patients who are not opioid tolerant.
Accidental consumption of EXALGO, especially in children, can result in a fatal overdose of hydromorphone [see Warnings and Precautions (5.1)].
Limitations of UseEXALGO is not indicated for the management of acute or postoperative pain [see Indications and Usage (1)].
EXALGO is not intended for use as an as-needed analgesic [see Indications and Usage (1)].
EXALGO tablets are to be swallowed whole and are not to be broken, chewed, dissolved, crushed or injected. Taking broken, chewed, dissolved or crushed EXALGO or its contents leads to rapid release and absorption of a potentially fatal dose of hydromorphone [see Warnings and Precautions (5)].
CONTRAINDICATIONSOpioid Non-Tolerant PatientsEXALGO is contraindicated in opioid non-tolerant patients. Fatal respiratory depression could occur in patients who are not opioid tolerant. Impaired Pulmonary FunctionEXALGO is contraindicated in patients with significant respiratory depression, especially in the absence of resuscitative equipment or in unmonitored settings and in patients with acute or severe bronchial asthma or hypercarbia.Paralytic IleusEXALGO is contraindicated in patients who have or are suspected of having a paralytic ileus. Preexisting Gastrointestinal (GI) Surgery or Narrowing of GI TractEXALGO is contraindicated in patients who have had surgical procedures and/or underlying disease that would result in narrowing of the gastrointestinal tract, or have “blind loops” of the gastrointestinal tract or gastrointestinal obstruction. Allergy or HypersensitivityEXALGO is contraindicated in patients with known hypersensitivity to any of its components including the active agent, hydromorphone hydrochloride or known allergy to sulfite-containing medications [see Warnings and Precautions (5.8)].
WARNINGS AND PRECAUTIONSInformation Essential for Safe AdministrationEXALGO tablets are to be swallowed whole, and are not to be broken, chewed, crushed, dissolved or injected. Taking broken, chewed, crushed, dissolved EXALGO or its contents leads to the rapid release and absorption of a potentially fatal dose of hydromorphone [see Boxed Warning]. EXALGO is for use only in opioid tolerant patients. Ingestion of EXALGO may cause fatal respiratory depression when administered to patients who are not opioid tolerant [see Boxed Warning].EXALGO tablets must be kept in a secure place out of the reach of children. Accidental consumption of EXALGO, especially in children, can result in a fatal overdose of hydromorphone.Misuse and AbuseEXALGO contains hydromorphone, an opioid agonist, and is a Schedule II controlled substance. Opioid agonists have the potential for being abused and are sought by drug abusers and people with addiction disorders and are subject to criminal diversion. EXALGO can be abused in a manner similar to other opioid agonists, legal or illicit. This should be considered when prescribing or dispensing EXALGO in situations where the healthcare professional is concerned about an increased risk of misuse, abuse, or diversion. Breaking, crushing, chewing, or dissolving the contents of an EXALGO tablet results in the uncontrolled delivery of the opioid and poses a significant risk of overdose and death [see Drug Abuse and Dependence (9)]. If attempts are made to extract the drug from the hard outer shell for purposes of parenteral abuse, the injection of tablet excipients may be toxic and may result in lethal complications. Concerns about abuse, addiction, and diversion should not prevent the proper management of pain. However, all patients treated with opioids, including EXALGO, require careful monitoring for signs of abuse and addiction, since use of opioid analgesic products carries the risk of addiction even under appropriate medical use. Healthcare professionals should contact their State Professional Licensing Board or State Controlled Substances Authority for information on how to prevent and detect abuse or diversion of this product.
Respiratory Depression Respiratory depression is the chief hazard of EXALGO. Respiratory depression occurs more frequently in elderly or debilitated patients as well as those suffering from conditions accompanied by hypoxia or hypercapnia when even moderate therapeutic doses may dangerously decrease pulmonary ventilation, and when opioids are given in conjunction with other agents that depress respiration.Use EXALGO with extreme caution in patients with conditions accompanied by hypoxia, hypercapnia, or decreased respiratory reserve such as asthma, chronic obstructive pulmonary disease or cor pulmonale, severe obesity, sleep apnea, myxedema, kyphoscoliosis or CNS depression. In these patients, even moderate therapeutic doses of hydromorphone may decrease respiratory drive while simultaneously increasing airway resistance to the point of apnea. In these patients, consider alternative non-opioid analgesics, and use EXALGO only under careful medical supervision at the lowest effective dose.
Interactions with Alcohol and Other CNS DepressantsThe concurrent use of EXALGO with other central nervous system (CNS) depressants, including but not limited to other opioids, illicit drugs, sedatives, hypnotics, general anesthetics, phenothiazines, muscle relaxants, other tranquilizers, and alcohol, increases the risk of respiratory depression, hypotension, and profound sedation, potentially resulting in coma or death. Use with caution and in reduced dosages in patients taking CNS depressants. Avoid concurrent use of alcohol and EXALGO [see Clinical Pharmacology (12.3)].
Head Injury and Increased Intracranial PressureIn the presence of head injury, intracranial lesions or a preexisting increase in intracranial pressure, the respiratory depressant effects of EXALGO and its potential to elevate cerebrospinal fluid pressure (resulting from vasodilation following CO2 retention) may be markedly exaggerated. Furthermore, EXALGO can produce effects on pupillary response and consciousness, which may obscure neurologic signs of further increases in intracranial pressure in patients with head injuries.
Hypotensive EffectEXALGO may cause severe hypotension. There is added risk to individuals whose ability to maintain blood pressure has been compromised by a depleted blood volume, or after concurrent administration with drugs such as phenothiazines, general anesthetics, or other agents that compromise vasomotor tone. Administer EXALGO with caution to patients in circulatory shock, since vasodilation produced by the drug may further reduce cardiac output and blood pressure.
Gastrointestinal EffectsBecause the EXALGO tablet is nondeformable and does not appreciably change in shape in the GI tract, do not administer EXALGO to patients with preexisting severe gastrointestinal narrowing (pathologic or iatrogenic, for example: esophageal motility disorders small bowel inflammatory disease, “short gut” syndrome due to adhesions or decreased transit time, past history of peritonitis, cystic fibrosis, chronic intestinal pseudoobstruction, or Meckel’s diverticulum). There have been reports of obstructive symptoms in patients with known strictures or risk of strictures, such as previous GI surgery, in association with the ingestion of drugs in nondeformable extended-release formulations. The administration of EXALGO may obscure the diagnosis or clinical course in patients with acute abdominal condition.It is possible that EXALGO tablets may be visible on abdominal x-rays under certain circumstances, especially when digital enhancing techniques are utilized.
SulfitesEXALGO contains sodium metabisulfite, a sulfite that may cause allergic-type reactions including anaphylactic symptoms and life-threatening or less severe asthmatic episodes in certain susceptible people. The overall prevalence of sulfite sensitivity in the general population is unknown and probably low. Sulfite sensitivity is seen more frequently in asthmatic than in nonasthmatic people.
MAO InhibitorsEXALGO is not recommended for use in patients who have received MAO inhibitors within 14 days, because severe and unpredictable potentiation by MAO inhibitors has been reported with opioid analgesics.
Special Risk GroupsEXALGO should be administered with caution in elderly (≥ 65 years) and debilitated patients and in patients who are known to be sensitive to central nervous system depressants, such as those with cardiovascular, pulmonary, renal, or hepatic disease [see Use in Specific Populations (8)].EXALGO should also be used with caution in the following conditions: adrenocortical insufficiency (e.g., Addison’s disease); delirium tremens; myxedema or hypothyroidism; prostatic hypertrophy or urethral stricture; and, toxic psychosis. EXALGO may aggravate convulsions in patients with convulsive disorders, and all opioids may induce or aggravate seizures in some clinical settings.
Use in Pancreatic/Biliary Tract DiseaseEXALGO can cause an increase in biliary tract pressure as a result of spasm in the sphincter of Oddi. Caution should be exercised in the administration of EXALGO to patients with inflammatory or obstructive bowel disorders, acute pancreatitis secondary to biliary tract disease and in patients about to undergo biliary surgery.
Driving and Operating MachineryEXALGO may impair the mental and/or physical abilities needed to perform potentially hazardous activities such as driving a car or operating machinery. Caution patients accordingly. Also warn patients about the potential combined effects of EXALGO with other CNS depressants, including other opioids, phenothiazines, sedative/hypnotics, and alcohol [see Drug Interactions (7)].
Precipitation of Withdrawal Mixed agonist/antagonist analgesics (i.e., pentazocine, nalbuphine, and butorphanol) should not be administered to patients who have received or are receiving a course of therapy with a pure opioid agonist analgesic, including EXALGO. In these patients, mixed agonists/antagonists analgesics may reduce the analgesic effect and/or may precipitate withdrawal symptoms. Do not abruptly discontinue EXALGO.Clinical conditions or medicinal products that cause a sudden and significant shortening of gastrointestinal transit time may result in decreased hydromorphone absorption with EXALGO and may potentially lead to withdrawal symptoms in patients with a physical dependence on opioids.
ADVERSE REACTIONSThe following serious adverse reactions are discussed elsewhere in the labeling:
[see Warnings and Precautions (5.3)]Head Injury and Increased Intracranial Pressure [see Warnings and Precautions (5.5)]
[see Warnings and Precautions (5.6)] [see Warnings and Precautions (5.7)]
[see Overdosage (10)][see Warnings and Precautions (5.13)]
Clinical Studies ExperienceBecause clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.EXALGO was administered to a total of 2,524 patients in 15 controlled and uncontrolled clinical studies. Of these, 423 patients were exposed to EXALGO for greater than 6 months and 141 exposed for greater than one year. The overall incidence of adverse reactions in patients greater than 65 years of age was higher, with a greater than 5% difference in rates for constipation and nausea when compared with younger patients. The overall incidence of adverse reactions in female patients was higher, with a greater than 5% difference in rates for nausea, vomiting, constipation and somnolence when compared with male patients.A 12-week double-blind, placebo-controlled, randomized withdrawal study was conducted in opioid tolerant patients with moderate to severe low back pain [see Clinical Studies (14)]. A total of 447 patients were enrolled into the open-label titration phase with 268 patients randomized into the double-blind treatment phase. The adverse reactions that were reported in at least 2% of the patients are contained in Table 1.
Table 1. Number (%) of Patients with Adverse Reactions Reported in ≥2% of
Patients with Moderate to Severe Low Back Pain During the Open-Label Titration Phase or Double-Blind Treatment Phase by Preferred Term
Preferred Term Open-Label Double-Blind Treatment Phase Titration Phase EXALGO (N=447) EXALGO (N=134) Placebo (N=134)Constipation 69 (15) 10 (7) 5 (4)Nausea 53 (12) 12 (9) 10 (7)Somnolence 39 (9) 1 (1) 0 (0)Headache 35 (8) 7 (5) 10 (7)Vomiting 29 (6) 8 (6) 6 (4)Drug Withdrawal Syndrome 22 (5) 13 (10) 16 (12)Pruritus 21 (5) 1 (1) 0 (0)Dizziness 17 (4) 3 (2) 2 (1)Asthenia a 16 (4) 2 (1) 6 (4)Insomnia 13 (3) 7 (5) 5 (4)Diarrhea 13 (3) 5 (4) 9 (7)Back Pain 13 (3) 6 (4) 8 (6)Dry Mouth 13 (3) 2 (1) 0 (0)Edema Peripheral 13 (3) 3 (2) 1 (1)Hyperhidrosis 13 (3) 2 (1) 2 (1)Anorexia b 10 (2) 2 (1) 0 (0)Arthralgia 9 (2) 8 (6) 3 (2)Anxiety 9 (2) 0 (0) 4 (3)Abdominal Pain c 9 (2) 4 (3) 3 (2)Muscle Spasms 5 (1) 3 (2) 1 (1)Weight Decreased 3 (1) 4 (3) 3 (2)
a Fatigue was grouped and reported with asthenia b Decreased appetite was grouped and reported with anorexia c Abdominal pain upper was grouped and reported with abdominal pain
The adverse reactions that were reported in at least 2% of the total treated patients (N=2,474) in the 14 chronic clinical trials are contained in Table 2.
Table 2.Number (%) of Patients with Adverse Reactions Reported in ≥2% of Patientswith Chronic Pain Receiving EXALGO in 14 Clinical Studies by Preferred TermPreferred Term All Patients (N=2,474)Constipation 765 (31)Nausea 684 (28)Vomiting 337 (14)Somnolence 367 (15) Headache 308 (12)Asthenia a 272 (11)Dizziness 262 (11)Diarrhea 201 (8) Pruritus 193 (8) Insomnia 161 (7) Hyperhidrosis 143 (6) Edema Peripheral 135 (5)Anorexia b 139 (6) Dry Mouth 121 (5) Abdominal Pain c 115 (5) Anxiety 95 (4) Back Pain 95 (4) Dyspepsia d 88 (4) Depression 81 (3) Dyspnea e 76 (3) Muscle Spasms 74 (3) Arthralgia 72 (3) Rash 64 (3) Pain in Extremity 63 (3) Pain 58 (2) Drug Withdrawal Syndrome 55 (2)Pyrexia 52 (2) Fall 51 (2) Chest Discomfort f 51 (2)
a Fatigue was grouped and reported with asthenia b Decreased appetite was grouped and reported with anorexia c Abdominal pain upper was grouped and reported with abdominal pain d Reflux esophagitis, gastroesophageal reflux disease and Barrett’s esophagus were grouped and
reported with dyspepsia e Dyspnea exacerbated and dyspnea exertional were grouped and reported with dyspnea f Chest pain and non-cardiac chest pain were grouped and reported with chest discomfort
BRIEF SUMMARY - Consult fullprescribing information before use.

The following Adverse Reactions occurred in patients with an overall frequency of <2% and are listed in descending order within each System Organ Class:Cardiac disorders: palpitations, tachycardia, bradycardia, extrasystolesEar and labyrinth disorders: vertigo, tinnitus Endocrine disorders: hypogonadismEye disorders: vision blurred, diplopia, dry eye, miosis Gastrointestinal disorders: �atulence, dysphagia, hematochezia, abdominal distension, hemorrhoids, abnormal feces, intestinal obstruction, eructation, diverticulum, gastrointestinal motility disorder, large intestine perforation, anal �ssure, bezoar, duodenitis, ileus, impaired gastric emptying, painful defecation General disorders and administration site conditions: chills, malaise, feeling abnormal, feeling hot and cold, feeling jittery, hangover, di�culty in walking, feeling drunk, hypothermiaInfections and infestations: gastroenteritis, diverticulitisInjury, poisoning and procedural complications: contusion, overdoseInvestigations: weight decreased, hepatic enzyme increased, blood potassium decreased, blood amylase increased, blood testosterone decreased, oxygen saturation decreasedMetabolism and nutrition disorders: dehydration, �uid retention, increased appetite, hyperuricemiaMusculoskeletal and connective tissue disorders: myalgia Nervous system disorders: tremor, sedation, hypoesthesia, paraesthesia, disturbance in attention, memory impairment, dysarthria, syncope, balance disorder, dysgeusia, depressed level of consciousness, coordination abnormal, hyperesthesia, myoclonus, dyskinesia, hyperre�exia, encephalopathy, cognitive disorder, convulsion, psychomotor hyperactivityPsychiatric disorders: confusional state, nervousness, restlessness, abnormal dreams, mood altered, hallucination, panic attack, euphoric mood, paranoia, dysphoria, listless, crying, suicide ideation, libido decreased, aggressionRenal and urinary disorders: dysuria, urinary retention, urinary frequency, urinary hesitation, micturition disorderReproductive system and breast disorders: erectile dysfunction, sexual dysfunction Respiratory, thoracic and mediastinal disorders: rhinorrhoea, respiratory distress, hypoxia, bronchospasm, sneezing, hyperventilation, respiratory depressionSkin and subcutaneous tissue disorders: erythema Vascular disorders: �ushing, hypertension, hypotension
DRUG INTERACTIONSCNS DepressantsThe concomitant use of EXALGO with central nervous system depressants such as hypnotics, sedatives, general anesthetics, antipsychotics and alcohol may cause additive depressant e�ects and respiratory depression. Additionally, hypotension and profound sedation or coma could occur. When this combination is indicated, the dose of one or both agents should be reduced. The concomitant use of alcohol should be avoided [see Clinical Pharmacology (12.3)]. Monoamine Oxidase (MAO) Inhibitors MAO inhibitors may cause CNS excitation or depression, hypotension or hypertension if co-administered with opioids including EXALGO. EXALGO is not intended for patients taking MAO inhibitors or within 14 days of stopping such treatment.Mixed Agonist/Antagonist Opioid AnalgesicsThe concomitant use of EXALGO with morphine agonist/antagonists (buprenor-phone, nalbuphine, pentazocine) could lead to a reduction of the analgesic e�ect by competitive blocking of receptors, thus leading to risk of withdrawal symptoms. Therefore, this combination is not recommended.AnticholinergicsAnticholinergics or other medications with anticholinergic activity when used concurrently with EXALGO may result in increased risk of urinary retention and/or severe constipation, which may lead to paralytic ileus.Cytochrome P450 EnzymesIn vitro data suggest that hydromorphone in clinically relevant concentrations has minimal potential to inhibit the activity of human hepatic CYP450 enzymes including CYP1A2, 2C9, 2C19, 2D6, 3A4, and 4A11.
USE IN SPECIFIC POPULATIONSPregnancy Teratogenic E�ects Pregnancy Category C: There are no adequate and well-controlled studies in pregnant women. Hydromorphone crosses the placenta. EXALGO should be used during pregnancy only if the potential bene�t justi�es the potential risk to the fetus [see Use in Speci�c Populations (8.2)].Hydromorphone was not teratogenic in pregnant rats given oral doses up to 6.25 mg/kg/day or in pregnant rabbits administered oral doses up to 25 mg/kg/day during the period of organogenesis (~1.2 times the human exposure following 32 mg/day). Hydromorphone administration to pregnant Syrian hamsters and CF-1 mice during major organ development revealed teratogenicity likely the result of maternal toxicity associated with sedation and hypoxia. In Syrian hamsters given single subcutaneous doses from 14 to 258 mg/kg during organogenesis (gestation days 8 to 10), doses ≥ 19 mg/kg hydromorphone produced skull
fo noisufni suounitnoC .)sisihcsoinarc dna ylahpecnexe( snoitamrof lamhydromorphone (5 mg/kg, s.c.) via implanted osmotic mini pumps during organogenesis (gestation days 7 to 10) produced soft tissue malformations (cryptorchidism, cleft palate, malformed ventricals and retina), and skeletal variations (supraoccipital, checkerboard and split sternebrae, delayed ossi�cation of the paws and ectopic ossi�cation sites). The malformations and variations observed in the hamsters and mice were at doses approximately three-fold higher and <one-fold lower, respectively, than a 32 mg human daily oral dose on a body surface area basis.Nonteratogenic E�ectsIn the pre- and post-natal e�ects study in rats, neonatal viability was reduced at 6.25 mg/kg/day (~1.2 times the human exposure following 32 mg/day).Neonates born to mothers who have been taking opioids regularly prior to delivery will be physically dependent. The withdrawal signs include irritability and excessive crying, tremors, hyperactive re�exes, increased respiratory rate, increased stools, sneezing, yawning, vomiting, and fever. The intensity of the syndrome does not always correlate with the duration of maternal opioid use or dose. There is no consensus on the best method of managing withdrawal. Approaches to the treatment of the syndrome have included supportive care and, if indicated, drugs such as paregoric or phenobarbital.
Labor and DeliveryEXALGO is not recommended for use in women during and immediately prior to labor and delivery. Administration of EXALGO to the mother shortly before delivery may result in some degree of respiratory depression in the neonate. However, neonates whose mothers received opioid analgesics during labor should be observed closely for signs of respiratory depression. Nursing MothersLow concentrations of hydromorphone have been detected in human milk in clinical trials. Withdrawal symptoms can occur in breastfeeding infants when maternal administration of an opioid analgesic is stopped. Nursing should not be undertaken while a patient is receiving EXALGO since hydromorphone is excreted in the milk.Pediatric UseThe safety and e�ectiveness of EXALGO in pediatric patients 17 years of age and younger have not been established.Geriatric UseElderly patients have been shown to be more sensitive to the adverse e�ects of EXALGO compared to the younger population. Therefore, use extra caution when prescribing EXALGO in elderly patients and reduce the initial dose. Neonatal Withdrawal SyndromeChronic maternal use of opiates or opioids during pregnancy coexposes the fetus. The newborn may experience subsequent neonatal withdrawal syndrome (NWS). Manifestations of NWS include irritability, hyperactivity, abnormal sleep pattern, high-pitched cry, tremor, vomiting, diarrhea, weight loss, and failure to gain weight. The onset, duration, and severity of the disorder di�er based on such factors as the addictive drug used, time and amount of mother’s last dose, and rate of elimination of the drug from the newborn. Approaches to the treatment of this syndrome have included supportive care and, when indicated, drugs such as paregoric or phenobarbital.Hepatic ImpairmentIn a study that used a single 4 mg oral dose of immediate-release hydromor-phone tablets, four-fold increases in plasma levels of hydromorphone (Cmax and AUC0- ) were observed in patients with moderate hepatic impairment (Child-Pugh Group B). Start patients with moderate hepatic impairment on a reduced dose and closely monitored during dose titration. The pharmacokinetics of hydromorphone in severe hepatic impairment patients have not been studied. Further increase in Cmax and AUC0- of hydromorphone in this group is expected, therefore, use an even more conservative starting dose [see Dosage and Administration (2.4)].Renal ImpairmentRenal impairment a�ected the pharmacokinetics of hydromorphone and its metabolites following administration of a single 4 mg dose of immediate-release tablets. The e�ects of renal impairment on hydromorphone pharmacokinetics were two-fold and four-fold increases in plasma levels of hydromorphone (Cmax and AUC0-48h) in moderate (CLcr = 40 to 60 mL/min) and severe (CLcr < 30 mL/min) impairment, respectively. In addition, in patients with severe renal impairment hydromorphone appeared to be more slowly eliminated with longer terminal elimination half-life (40 hours) compared to subjects with normal renal function (15 hours). Start patients with moderate renal impairment on a reduced dose and closely monitored during dose titration. As EXALGO is only intended for once daily administration, consider use of an alternate analgesic that may permit more �exibility with the dosing interval in patients with severe renal impairment [see Dosage and Administration (2.4)].
DRUG ABUSE AND DEPENDENCEControlled SubstanceEXALGO contains hydromorphone, a Schedule II controlled substance with a high potential for abuse similar to fentanyl, methadone, morphine, oxycodone, and oxymorphone. EXALGO can be abused and is subject to misuse, abuse, addiction, and criminal diversion [see Warnings and Precautions (5.2)]. The high drug content in the extended release formulation adds to the risk of adverse outcomes from abuse.Abuse All patients treated with opioids, including EXALGO, require careful monitoring for signs of abuse and addiction, because use of opioid analgesic products carries the risk of addiction even under appropriate medical use.Addiction is a primary, chronic, neurobiologic disease, with genetic, psychosocial, and environmental factors in�uencing its development and manifestations. It is characterized by behaviors that include one or more of the following: impaired control over drug use, compulsive use, continued use despite harm, and craving. “Drug-seeking” behavior is very common to addicts and drug abusers. Drug-seeking tactics include emergency calls or visits near the end of o�ce hours, refusal to undergo appropriate examination, testing or referral, repeated claims of loss of prescriptions, tampering with prescriptions and reluctance to provide prior medical records or contact information for other treating physician(s). “Doctor shopping” (visiting multiple prescribers) to obtain additional prescriptions is common among drug abusers, people su�ering from untreated addiction and criminals seeking drugs to sell. Abuse and addiction are separate and distinct from physical dependence and tolerance. Physicians should be aware that addiction may not be accompanied by concurrent tolerance and symptoms of physical dependence in all addicts. In addition, abuse of opioids can occur in the absence of true addiction and is characterized by misuse for non-medical purposes, often in combination with other psychoactive substances. Since EXALGO may be diverted for non-medical use, careful record-keeping of prescribing information, including quantity, frequency, and renewal requests is strongly advised. Proper assessment of the patient, proper prescribing practices, periodic re-evaluation of therapy, and proper dispensing and storage are appropriate measures that help to limit abuse of opioid drugs. EXALGO is intended for oral use only. Misuse or abuse by breaking, crushing, chewing, or dissolving EXALGO poses a hazard of overdose and death. This risk is increased with concurrent abuse of EXALGO with alcohol and other substances. With intravenous abuse, the tablet excipients, especially polyethylene oxide, can be expected to result in necrosis and in�ammation of cardiac tissues. In addition, parenteral drug abuse is commonly associated with transmission of infectious disease such as hepatitis and HIV.Healthcare professionals should contact their State Professional Licensing Board or State Controlled Substances Authority for information on how to prevent and detect abuse or diversion of this product.DependenceTolerance is a state of adaptation in which exposure to a drug induces changes that result in a diminution of one or more of the drug’s e�ects over time.
Tolerance could occur to both the desired and undesired e�ects of drugs, and may develop at di�erent rates for di�erent e�ects.Physical dependence is a state of adaptation that is manifested by an opioid speci�c withdrawal syndrome that can be produced by abrupt cessation, rapid dose reduction, decreasing blood level of the drug, and/or administration of an antagonist. The opioid abstinence or withdrawal syndrome is characterized by some or all of the following: restlessness, lacrimation, rhinorrhea, yawning, perspiration, chills, piloerection, myalgia, mydriasis, irritability, anxiety, backache, joint pain, weakness, abdominal cramps, insomnia, nausea, anorexia, vomiting, diarrhea, increased blood pressure, respiratory rate, or heart rate.Infants born to mothers physically dependent on opioids will also be physically dependent and may exhibit respiratory di�culties and withdrawal symptoms [see Use in Speci�c Populations (8.1, 8.2)].
OVERDOSAGESymptomsAcute overdosage with opioids can be manifested by respiratory depression, somnolence progressing to stupor or coma, skeletal muscle �accidity, cold and clammy skin, constricted pupils, and sometimes bradycardia, hypotension and death. The extended release characteristics of EXALGO should also be taken into account when treating the overdose. Even in the face of improvement, continued medical monitoring is required because of the possibility of extended e�ects. Deaths due to overdose could occur with abuse and misuse of EXALGO.Due to the delayed mean apparent peak plasma level of EXALGO occurring at 16 hours following administration as well as the 11 hour mean elimination half-life of EXALGO, patients who receive an overdose will require an extended period of monitoring and treatment that may go beyond 24 to 48 hours. TreatmentGive primary attention to the re-establishment of a patent airway and institution of assisted or controlled ventilation. Employ supportive measures (including oxygen and vasopressors) in the management of circulatory shock and pulmonary edema accompanying overdose as indicated. Cardiac arrest or arrhythmias will require advanced life support techniques.The pure opioid antagonists, such as naloxone and naltrexone are speci�c antidotes to respiratory depression from opioid overdose. Since the duration of reversal would be expected to be less than the duration of action of hydromorphone in EXALGO, the patient must be carefully monitored until spontaneous respiration is reliably re-established. EXALGO will continue to release and add to the hydromorphone load for up to 24 hours after administration and the management of an overdose should be monitored accordingly, at least 24 to 48 hours beyond the overdose.Only administer opioid antagonists in the presence of clinically signi�cant respiratory or circulatory depression secondary to hydromorphone overdose. In patients who are physically dependent on any opioid agonist including EXALGO, an abrupt or complete reversal of opioid e�ects may precipitate an acute abstinence syndrome. The severity of the withdrawal syndrome produced will depend on the degree of physical dependence and the dose of the antagonist administered. Please see the prescribing information for the speci�c opioid antagonist for details of their proper use.
OROS is a registered trademark of ALZA Corporation.EXALGO is a registered trademark of Mallinckrodt Inc.COVIDIEN, COVIDIEN with logo and Covidien logo are U.S. and/or internationally registered trademarks of Covidien AG.
© 2010 Mallinckrodt Inc., a Covidien company
Distributed by:Mallinckrodt Brand Pharmaceuticals, Inc.Hazelwood, MO 63042 USA
Issued 11/2010
Mallinckrodt
3-4 5 11:11:24 AM

6 I AMERICAN HEALTH & DRUG BENEFITS I May/June 2011
AHDB3911
The individual drug updates have been sponsored by theirmanufacturers. The drug manufacturers had editorial controlover these articles.
7 INTRODUCTIONNew Drug Approvals in 2010: Another Difficult YearGary M. Owens, MD
12 EXALGO® (hydromorphone HCl) Extended-Release Tablets, CII: A Once-DailyExtended-Release Option for the Management of Chronic Pain
Stakeholder Perspective by Gary Rice, RPh, MS, MBA
22 FDA Approvals of Brand-Name Prescription Drugs in 2010
32 KAPVAY™: Clonidine Hydrochloride Extended-Release Tablets for the Treatment of Attention-Deficit/Hyperactivity DisorderTodd Parker, PhD, and Jillian Gee, PhD
Stakeholder Perspectives Floyd R. Sallee, MD, PhD
41 The Current Drug Pipeline and New Approvals in 2011: A Managed Care PerspectiveDiana Papshev, PharmD; Chantell M. Reagan, PharmD
48 FDA Approvals of Brand-Name Prescription Drugs, January-May 2011
TABLE OF CONTENTS
MAY/JUNE 2011 VOLUME 4, NUMBER 2, SPECIAL FEATURE
™ ™
THE PEER-REVIEWED FORUM FOR EVIDENCE IN BENEFIT DESIGN™
FOR PAYERS, PURCHASERS, POLICYMAKERS, AND OTHER HEALTHCARE STAKEHOLDERS
PublisherNicholas Englezos
Associate PublisherMaurice Nogueira
Editorial DirectorDalia Buffery
Associate EditorLara J. Reiman732-992-1892
Editorial AssistantJessica A. Smith
Senior Production ManagerLynn Hamilton
Quality Control DirectorBarbara Marino
Business ManagerBlanche Marchitto
Founding Editor-in-ChiefRobert E. Henry
[email protected]’s note: The material presented in this supplement is intended to be a thorough, objective, balanced pres-entation of clinical information. The opinions expressed in this professional educational supplement are those ofthe authors, presenters, and/or panelists and do not necessarily reflect those of the Publisher, Editors, or EditorialBoard of American Health & Drug Benefits, or the sponsor. Dosages, indications, and methods of use for productsreferred to in this professional educational supplement may reflect the clinical experience of the authors, presen-ters, and/or panelists or may be derived from the professional literature or other clinical sources and are not neces-sarily the same as indicated in the package insert for the product. This publication may contain or discuss off-labeluses of commercial products or investigational uses not cleared for marketing. Readers are advised to consult the fullprescribing information before administering any product. Neither the Editors nor the Publisher assume any respon-sibility for any injury and/or damage to persons or property arising out of or related to any use of the material men-tioned in this publication.
American Health & Drug Benefits, ISSN 1942-2962 (print); ISSN 1942-2970 (online), is published 6 times a year byEngage Healthcare Communications, LLC, 241 Forsgate Drive, Suite 205A, Monroe Twp, NJ 08831. Copyright ©2011 by Engage Healthcare Communications, LLC. All rights reserved. American Health & Drug Benefits and ThePeer-Reviewed Forum for Evidence in Benefit Design are trademarks of Engage Healthcare Communications, LLC. Nopart of this publication may be reproduced or transmitted in any form or by any means now or hereafter known, elec-tronic or mechanical, including photocopy, recording, or any informational storage and retrieval system, withoutwritten permission from the Publisher. Printed in the United States of America.
For permission to reuse material from American Health & Drug Benefits (ISSN 1942-2962), please access www.copy-right.com <http:// www.copyright.com/> or contact the Copyright Clearance Center, Inc. (CCC), 222 RosewoodDrive, Danvers, MA 01923, 978-750-8400.
POSTMASTER: CORRESPONDENCE REGARDING SUBSCRIPTIONS OR CHANGE OF ADDRESSshould be directed to CIRCULATION DIRECTOR, American Health & Drug Benefits, 241 Forsgate Drive,Suite 205A, Monroe Twp, NJ 08831. Fax: 732-992-1881.
Mission StatementAmerican Health & Drug Benefits is foundedon the concept that health and drug benefitshave undergone a transformation: the econo -metric value of a drug is of equal importanceto clinical outcomes as it is to serving as thebasis for securing coverage in formularies andbenefit designs. Because benefit designs aregreatly affected by clinical, business, and pol-icy conditions, this journal offers a forum forstakeholder integration and collaborationtoward the improvement of healthcare.
This publication further provides benefitdesign de cision makers the integrated industryinformation they require to devise formulariesand benefit designs that stand up to today’sspecial healthcare delivery and business needs.
Contact Information:For reprints, subscription information, andeditorial queries, please contact:[email protected]
T: 732-992-1892F: 732-992-1881

INTRODUCTION
7May/June 2011 www.AHDBonline.com
The year 2010 represented anotherdifficult period for the pharmaceuti-cal industry. In a similar fashion to
the 2 previous years, a relatively small num-ber of new drugs were approved by the USFood and Drug Administration’s (FDA’s)Center for Drug Evaluation and Research(CDER). In total, there were 15 or 16 newmolecular entities (NMEs) and between 6and 13 biologic license applications (BLAs)approved, depending on whether one counts all entities,including vaccines, for example. (Different lists have dif-ferent numbers, and it is not easy to reach a unanimousagreement on this, based on the FDA website.) Overall,these numbers represent less than the number of NMEsapproved in either 2008 or 2009, but the same or moreBLAs approved.
So far, FDA approvals in the past decade have beenrelatively modest, with only 2004 seeing more than 30NMEs approved (Figure).1 It appears that the concept oflooking to manufacture new drugs for use in large pri-mary care markets is a thing of the past. We now infre-quently see the launch of “blockbuster” drugs seen in thepast, such as Pfizer’s Lipitor or Schering’s Claritin.Rather, the focus is clearly turning toward specialtyproducts and orphan drugs.
To this end, the approval of biologic entities repre-sents close to half of all new agents approved in 2010. Inthe biologic space, 2010 saw entries into relativelycrowded fields—including rheumatoid arthritis, with thelaunch of Roche’s Actemra (tocilizumab), and Novartis’Gilenya (fingolimod), which is indicated for multiplesclerosis. However, there were also 6 new orphan drugslaunched that targeted diverse conditions (Table 1).
New Drugs: The P&T Committee PerspectiveAlthough 2010 saw a small number of new medica-
tions reaching the market, Pharmacy & Therapeutics(P&T) Committees clearly had the basis for some veryinteresting discussions. In addition to the 6 orphan drugsin areas where previous therapies were either nonexist-ent or nonmedical (eg, surgery for Dupuytren’s contrac-tures), there were 11 new drugs launched with require-ments for the REMS (risk evaluation and mitigation
strategy) programs, as well as many newindications for previously approved drugsthat require a REMS program.
Table 2 lists the total number of newdrugs—NMEs and BLAs—approved in2010.1 Of note, only 1 manufacturer hadmore than 1 drug on this list. That soleexception is Amgen’s denosumab, whichwas first approved as Prolia for osteoporosis,and was later approved under the brand
name Xgeva for skeletal metastases.As noted, several drugs have and will continue to
generate considerable discussion with various P&TCommittees as these new entities—molecular or bio-logic—come up for a P&T Committee review. Forexample, 2010 saw the approval of fingolimod for thetreatment of multiple sclerosis. This approval repre-sents the first oral therapy for multiple sclerosis, andthis medication has a unique mechanism of action as asphingosine 1-phosphate receptor modulator. P&TCommittee discussions centered on the relative patientpreference for an oral agent versus the injectable ther-apies, balanced by the known long-term data and out-comes for the injectables. Similarly, dalfampridine(Ampyra) became the first agent to be considered “add-on” therapy to selected patients with multiple sclerosisto improve walking time. Concern about appropriateuse of this drug, and how to measure its effects, domi-nated the discussions.
Probably some of the liveliest P&T Committee dis-cussions centered on the launch of dabigatran (Pradaxa).Dabigatran, which is indicated for stroke prevention inpatients with atrial fibrillation, represents the first directthrombin inhibitor to reach the market. This is the firstnew oral anticoagulant therapy to come to the market inmore than 60 years. Warfarin (Coumadin) has been thegold standard for patients who need anticoagulation, butthe narrow therapeutic window and significant potentialfor drug–drug interactions make warfarin a difficult drugto manage for patients and for physicians.
Discussions in this category attempted to balancethe low cost of warfarin therapy versus the more expen-sive new anticoagulant agent. Considerations for labo-ratory monitoring of warfarin, bleeding complications,
New Drug Approvals in 2010: AnotherDifficult YearGary M. Owens, MDPresident, Gary Owens Associates

INTRODUCTION
8 AMERICAN HEALTH & DRUG BENEFITS May/June 2011
and drug interactions had to be balanced with the costof the new agent. In addition, the current drug pipelinecontains at least 2 factor Xa inhibitors that are likely tobe direct competitors for dabigatran and ultimately towarfarin. This caused many P&T Committee membersto take a wait-and-see approach to this category.
Expect the oral anticoagulation category to be the sub-ject of P&T Committee discussions well into 2011 andeven 2012.
Amgen’s denosumab (Prolia), a first-in-class mono-clonal antibody that is specific for receptor activator ofnuclear factor kappa-B ligand (RANKL), was approvedin June 2010 for the treatment of postmenopausal osteo-porosis. A twice-yearly injectable medication for osteo-porosis, this agent offers an attractive alternative to bis-phosphonate therapy, which has long been the mainstayof osteoporosis therapy. Among the benefits of deno-sumab is that it is thought to act earlier to prevent bonedestruction than the bisphosphonates. Again, P&TCommittee discussions centered on the relative low costof generic bisphosphonates such as alendronate(Fosamax), which is partially offset by the known lowcompliance rates for this category of drugs, balancedagainst the higher cost of denosumab, which has thepotential to offer better compliance. To make thingseven more complex, later in the year denosumab alsoreceived approval under the brand name Xgeva for theprevention of skeletal-related events in patients withbone metastases from solid tumors.
1996 1997 1998 1999 2000 2001 2002 2003 2004 2005 2006 2007 2008 2009 2010
60
50
40
30
20
10
0
FDA indicates US Food and Drug Administration.Reprinted with permission from Mullard A. Nat Rev Drug Discov. 2011;10:82-85.
Figure FDA Drug Approvals Since 1996
Numbe
r of d
r ug s
ap p
r ov e
d
New molecular entities
Biologic license applications
Table 1 2010 Orphan Drugs
Agent Manufacturer Condition
Ampyra (dalfampridine) Acorda Multiple sclerosis
Carbaglu (carglumic acid)
Orphan Europe Hyperammonemiaresulting fromNAGS deficiency
Krystexxa (pegloticase) Savient Gout
Lumizyme (alglucosidase alfa)
Genzyme Noninfantile Pompe disease
Xiaflex (collagenaseclostridium histolyticum)
Auxilium Dupuytren’s contracture
NAGS indicates N-acetylglutamate synthase.
53
39
30
35
2724
17
21
31
18 1816
2119
15
42 3
6 6
2
5675
23
76
3

INTRODUCTION
9May/June 2011 www.AHDBonline.com
Continued
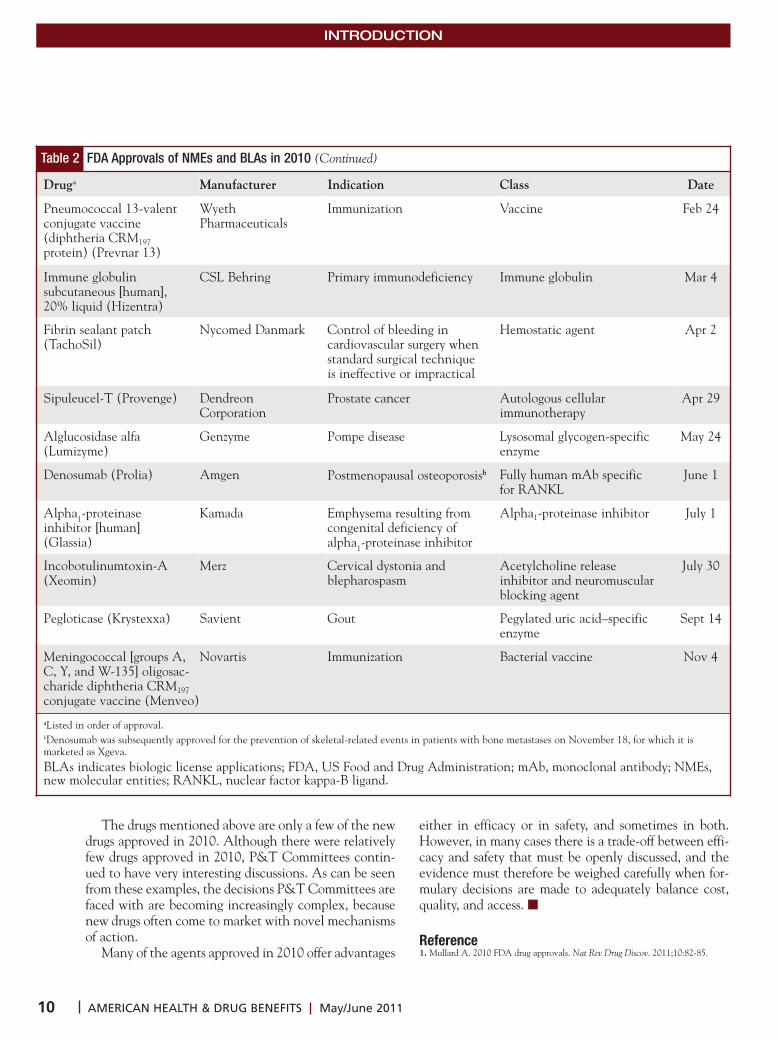
Table 2 FDA Approvals of NMEs and BLAs in 2010
Drug Manufacturer Indication Class Date
NMEs
Dalfampridine (Ampyra) Acorda Therapeutics Improving walking in patientswith multiple sclerosis
Potassium channel blocker Jan 22
Liraglutide (Victoza) Novo Nordisk Type 2 diabetes Glucagon-like peptide 1 receptor agonist
Jan 25
Velaglucerase alfa (VPriv) Shire Gaucher disease Recombinant human beta-glucocerebrosidase
Feb 26
Carglumic acid (Carbaglu) Orphan Europe Acute hyperammonemia Carbamoyl phosphate synthetase 1 activator
Mar 18
Polidocanol (Asclera) Chemische FabrikKreussler
Uncomplicated spider veinsand uncomplicated reticularveins
Sclerosing agent Mar 30
Everolimus (Zortress) Novartis Prophylaxis of organ rejectionin adults at low-to-moderateimmunologic risk receiving a kidney transplant
Immunosuppressant(macrolide)
Apr 22
Estradiol valerate anddienogest (Natazia)
Bayer Contraception Estrogen and progestin combi-nation oral contraceptive
May 6
Cabazitaxel (Jevtana) sanofi-aventis Prostate cancer Microtubule inhibitor June 17
Alcaftadine (Lastacaft) VistakonPharmaceuticals
Allergic conjunctivitis H1 histamine receptor agonist July 28
Ulipristal (Ella) Lab HRA Pharma Contraception Progesterone receptor modulator
Aug 13
Fingolimod (Gilenya) Novartis Multiple sclerosis Sphingosine 1-phosphatereceptor modulator
Sept 21
Dabigatran (Pradaxa) Boehringer Ingelheim Stroke prevention in atrial fibrillation
Direct thrombin inhibitor Oct 10
Lurasidone (Latuda) Sunovion Schizophrenia Atypical antipsychotic agent Oct 28
Ceftaroline fosamil (Teflaro)
Cerexa Skin and skin-structure infections, community-acquired pneumonia
Broad-spectrum cephalosporinantibiotic
Oct 29
Tesamorelin (Egrifta) Theratechnologies HIV lipodystrophy Growth hormone–releasing factor analog
Nov 10
Eribulin (Halaven) Eisai Breast cancer Microtubule inhibitor Nov 15
BLAs
Tocilizumab (Actemra) Genentech Rheumatoid arthritis Humanized mAb specific forthe interleukin-6 receptor
Jan 8
Collagenase clostridium histolyticum (Xiaflex)
Auxilium Dupuytren’s contracture Purified collagenase clostridiumhistolyticum
Feb 2
INTRODUCTION
10 AMERICAN HEALTH & DRUG BENEFITS May/June 2011
The drugs mentioned above are only a few of the newdrugs approved in 2010. Although there were relativelyfew drugs approved in 2010, P&T Committees contin-ued to have very interesting discussions. As can be seenfrom these examples, the decisions P&T Committees arefaced with are becoming increasingly complex, becausenew drugs often come to market with novel mechanismsof action.
Many of the agents approved in 2010 offer advantages
either in efficacy or in safety, and sometimes in both.However, in many cases there is a trade-off between effi-cacy and safety that must be openly discussed, and theevidence must therefore be weighed carefully when for-mulary decisions are made to adequately balance cost,quality, and access. ■
Reference1. Mullard A. 2010 FDA drug approvals. Nat Rev Drug Discov. 2011;10:82-85.
Table 2 FDA Approvals of NMEs and BLAs in 2010 (Continued)
Druga Manufacturer Indication Class Date
Pneumococcal 13-valentconjugate vaccine (diphtheria CRM197protein) (Prevnar 13)
WyethPharmaceuticals
Immunization Vaccine Feb 24
Immune globulin subcutaneous [human], 20% liquid (Hizentra)
CSL Behring Primary immunodeficiency Immune globulin Mar 4
Fibrin sealant patch(TachoSil)
Nycomed Danmark Control of bleeding in cardiovascular surgery whenstandard surgical technique is ineffective or impractical
Hemostatic agent Apr 2
Sipuleucel-T (Provenge) Dendreon Corporation
Prostate cancer Autologous cellularimmunotherapy
Apr 29
Alglucosidase alfa(Lumizyme)
Genzyme Pompe disease Lysosomal glycogen-specificenzyme
May 24
Denosumab (Prolia) Amgen Postmenopausal osteoporosisb Fully human mAb specific for RANKL
June 1
Alpha1-proteinase inhibitor [human] (Glassia)
Kamada Emphysema resulting from congenital deficiency of alpha1-proteinase inhibitor
Alpha1-proteinase inhibitor July 1
Incobotulinumtoxin-A(Xeomin)
Merz Cervical dystonia and blepharospasm
Acetylcholine release inhibitor and neuromuscularblocking agent
July 30
Pegloticase (Krystexxa) Savient Gout Pegylated uric acid–specificenzyme
Sept 14
Meningococcal [groups A,C, Y, and W-135] oligosac-charide diphtheria CRM197conjugate vaccine (Menveo)
Novartis Immunization Bacterial vaccine Nov 4
aListed in order of approval. bDenosumab was subsequently approved for the prevention of skeletal-related events in patients with bone metastases on November 18, for which it is marketed as Xgeva. BLAs indicates biologic license applications; FDA, US Food and Drug Administration; mAb, monoclonal antibody; NMEs,new molecular entities; RANKL, nuclear factor kappa-B ligand.

References: 1. Plantinga LC, Crews DC, Coresh J, et al; for the CDC CKD Surveillance Team. Prevalence of chronic kidney disease in US adults with undiagnosed diabetes or prediabetes. Clin J Am Soc Nephrol. 2010;5(4):673-682. 2. Parving H-H, Lewis JB, Ravid M, Remuzzi G, Hunsicker LG; for the DEMAND Investigators. Prevalence and risk factors for microalbuminuria in a referred cohort of type II diabetic patients: a global perspective. Kidney Int. 2006;69:2057-2063. 3. Zhang X, Saaddine JB, Chou C-F, et al. Prevalence of diabetic retinopathy in the United States, 2005-2008. JAMA. 2010;304(6):649-656. 4. Gregg EW, Gu Q, Williams D, et al. Prevalence of lower extremity diseases associated with normal glucose levels, impaired fasting glucose, and diabetes among U.S. adults aged 40 or older. Diabetes Res Clin Pract. 2007;77(3):485-488. 5. American Diabetes Association. Nephropathy in diabetes. Diabetes Care. 2004;27(suppl 1):S79-S83. 6. American Diabetes Asso-ciation. Standards of medical care in diabetes—2011. Diabetes Care. 2011;34(suppl 1):S11-S61. 7. Fong DS,Ferris FL, Aiello LP, Klein R. Diabetic retinopathy. Diabetes Care. 2004;27(10):2540-2553. 8. Boulton AJM, Arezzo JC, Malik RZ, et al. Diabetic somatic neuropathies. Diabetes Care. 2004;27(6):1458-1486.
Renal impairment is the leading microvascular complication associated with type 2 diabetes (over 40%), followed by retinopathy (28.5%) and neuropathy (19.4%)— it is important to recognize these complications as soon as possible1-4
Microalbuminuria (albumin in the urine 30 mg/day or 20 µg/min) is the earliest clinical evidence of renal disease6
Regular dilated eye examinations can be effective in detecting vision-threatening diabetic retinopathy6,7
Because diabetic neuropathy may be asymptomatic in about 50% of patients, it is important to conduct a physical examination of lower extremities and feet annually6,8
THERE COULD BE DANGER BELOW
Copyright ©2011, Boehringer Ingelheim Pharmaceuticals, Inc. All rights reserved. (5/11) DI93803MHC
ExploringDiabetes.com
It’s important to recognize and screen for microvascular complications in patients with type 2 diabetes as early as possible.7 Effective management of diabetes can help prevent or slow the progression of microvascular complications.
ADVERTISEMENT
1:25 PM

Burden and Impact of Chronic PainIn the United States, the estimated mean preva-
lence of chronic pain is a staggering 35.5%, with anestimated 105 million Americans reporting chronicpain.1,2 Untreated or inadequately controlled pain cansignificantly impact a person’s quality of life, includingthe ability to concentrate, perform a job, socialize,exercise, sleep restfully, perform chores, or participatein leisure activities.3 Findings from a recent survey of27,035 adults (18 years of age or older) in the UnitedStates suggest that chronic pain imposes a substantialburden on our society.4 In fact, chronic pain was report-ed to occur in approximately one-third of the popula-tion, with lower back pain cited as the primary cause ofpain, followed by osteoarthritis pain. The prevalence ofchronic pain, defined as chronic, recurrent, or long-lasting pain (lasting for at least 6 months) was higherfor women (34.3%) than men (26.7%) and increasedwith age.4
According to the National Institute of NeurologicalDisorders and Stroke, low back pain is the most com-mon cause of job-related disability and lost productivi-ty, costing an estimated $50 billion per year in theUnited States.5 One study showed that employees whoreported back pain or arthritis lost productive time of5.2 hours per week.6 Back pain is the leading cause ofdisability in Americans under 45 years of age.7Furthermore, as the prevalence of pain increases withage, pain in the elderly will rise over the next 2 decadesas the baby boom generation (people born between1946 and 1964) reaches the age of 65.8,9
Opioid Therapy for Chronic PainOpioids are one of the most widely prescribed drug
classes for the treatment of pain in the United States.An estimated 4.3 million adults in this country take opi-oids regularly.10 However, many of these patients do notachieve acceptable pain relief. A survey of patients withchronic pain who were receiving opioids (N = 303)showed that only 23% reported their medications were“very effective.”11
One of the challenges of opioid therapy is considera-tion of the individual patient’s needs because no 2 pa -tients are alike. Regimens must be carefully planned andtitrated to an effective dose while opioid risks are man-aged; yet even with dose titration, adequate pain reliefmay not be achieved.12 In a study of 86 chronic pain suf-ferers, it took as many as 5 different extended-release opi-oids in succession to effectively treat 81% of the patients.13
Another Treatment Option for Chronic PainManagement
Another therapeutic option for the management ofchronic pain became available in March 2010, whenEXALGO® (hydromorphone HCl) Extended-ReleaseTablets, CII, was approved by the Food and Drug Admin -istration (FDA).14 EXALGO, an extended-release oralformulation of the opioid agonist hydromorphonehydrochloride, is indicated for the management of mod-erate to severe pain in opioid-tolerant patients requiringcontinuous around-the-clock opioid analgesia for anextended period of time.
EXALGO is not intended for use as an as-neededanalgesic and is not indicated for the management ofacute or postoperative pain.
EXALGO is the only extended-release formulation ofhydromorphone available in the United States. Theimmediate-release formulation of hydromorphone hasbeen used in this country for at least 80 years.15,16Hydromorphone is classified as a Schedule II prescrip-tion opioid.17
Clinical PharmacologyUtilizing the OROS® Push-PullTM osmotic pump
delivery system, EXALGO releases hydromorphone at acontrolled rate over an extended period to achieve once-daily dosing.17 With EXALGO, steady-state plasma con-centrations are reached after 3 or 4 days and sustainedthroughout the 24-hour dosing interval.18 This OROS®
Push-PullTM delivery system is a well-established tech-nology that has been used in several marketed prescrip-tion and nonprescription products since 1989.17
12 AMERICAN HEALTH & DRUG BENEFITS May/June 2011
EXALGO® (hydromorphone HCl) Extended-Release Tablets, CII: A Once-DailyExtended-Release Option for the Management ofChronic Pain
See pages 20-21 for brief summary of Full Prescribing Information.

Hour 6 Hour 12 Hour 18 Hour 24
4
3
2
1
0
Figure Mean Plasma Hydromorphone Concentrations Over Time after Administration of EXALGO q 24 h or Hydromorphone IR q 6 h
Day 5 postdose
Plas
ma hy
drom
orph
one (ng/mL)
HMIR 4 mg q 6 h
EXALGO 16 mg q 24 h
EXALGO® (hydromorphone HCl) Extended-ReleaseTablets, CII, has a bioavailability comparable to imme-diate-release hydromorphone given 4 times daily, andproduces steady-state concentrations of hydromorphonethat comparably reduce the drug’s peaks and troughs (seeFigure).18
Several studies conducted in healthy volunteers showthat EXALGO continually releases hydromorphoneresulting in consistent plasma levels over a 24-hour peri-od.19-21 The once-daily formulation of EXALGO providesthis 24-hour continual drug delivery with diminishedpeak-trough fluctuation compared with immediate-release hydromorphone.22 Peak plasma concentrationsare reached approximately 12-16 hours after administra-tion.18 EXALGO has an apparent elimination half-life of8-15 hours, and steady-state plasma concentrations arereached after 3-4 days of dosing.18 Its pharmacokineticsare not affected by food as indicated by bioequivalencewhen administered under fed and fasting conditions.18Therefore, EXALGO may be administered withoutregard to meals.18
Based on a randomized, open-label, crossover study inhealthy volunteers, EXALGO was shown to maintainsteady-state plasma drug concentrations within the samerange as the immediate-release formulation of hydro-morphone, at the same total daily dose, with less peak-to-trough fluctuation.23 The study participants (29 com-
pleted treatment) were randomly assigned to receiveEXALGO 16 mg once daily and immediate-releasehydromorphone 4 mg 4 times daily for 5 consecutivedays.23 A washout period of 7-14 days separated the 2treatments. The maximum measured plasma concentra-tion during steady state (Cmaxss) of EXALGO was lower,and the time of Cmaxss (tmaxss) was longer, compared withthe immediate-release formulation. Overall, systemicexposure to hydromorphone as assessed by the AUC0-24was similar between both treatment formulations, withsteady-state conditions attained by day 4. In addition,peak-to-trough fluctuations were significantly smallerwith EXALGO compared with immediate-releasehydromorphone (61% vs 172%).23
In a study measuring drug liking, single oral doseswere compared with immediate-release hydromorphonein a double-blind, randomized, placebo-controlled, 2-part crossover study (N = 30).24 When twice the dose ofEXALGO (16 mg) was administered intact vs immedi-ate-release hydromorphone (8 mg), the maximum drugliking (Emax) occurred later with EXALGO and was sig-nificantly lower, compared with immediate-releasehydromorphone.24
Clinical Study DataThe analgesic effect of EXALGO was demonstrated
in multiple clinical studies. A double-blind, placebo-
13May/June 2011 www.AHDBonline.com
Source: Reference 18.
Time
See pages 20-21 for brief summary of Full Prescribing Information.

controlled, randomized, withdrawal study of 268 opioid-tolerant patients with moderate to severe low back painfor at least 6 months was conducted to evaluate the effi-cacy of EXALGO® (hydromorphone HCl) Extended-Release Tablets, CII.15 The study consisted of a screeningvisit, an open-label conversion/titration phase, and adouble-blind treatment phase. All patients were on astable dose of a different opioid before entering the study.A total of 447 patients were enrolled into the open-labeltitration phase, with 268 patients randomized into thedouble-blind treatment phase; of the 110 patients whocompleted the double-blind phase, 49% of patientstreated with EXALGO and 33% of those treated withplacebo completed the 12-week treatment period.18Significantly more patients in the placebo group werediscontinued from the double-blind phase, comparedwith those in the EXALGO group (67% vs 50%). In theEXALGO and placebo groups, the most common rea-sons for discontinuation were insufficient analgesic effi-cacy (11.9% and 29.9%), noncompliance (8.2% and8.2%), unacceptable rescue medication usage (6.0% and9.0%), and adverse events (6.7% and 3.0%).25
During the conversion and titration phase of thestudy, all patients were titrated and converted toEXALGO. Patients in the EXALGO group and theplacebo group had a 50% reduction in mean pain inten-sity score, from 6.4 at the screening visit to 3.2 in theweek prior to randomization.25 In the double-blind treat-ment phase of the study, limited immediate-releasehydromorphone was permitted as rescue medication.Patients taking EXALGO had a significant reductionin pain intensity (measured on an 11-point numericalrating scale ranging from 0 for no pain to 10 for worstpossible pain) at the 12-week study endpoint andthroughout the study (P <0.001). The median changefrom baseline was 0.2 units for patients treated withEXALGO compared with 1.6 units for the placebo group.25
The analgesic efficacy of EXALGO was maintainedin the long-term, based on an open-label extension trialof 388 patients (106 completed at least 1 year of thera-py; 38 withdrew because of lack of efficacy; and 56 with-drew because of adverse events) with chronic cancer andnon-cancer pain followed for 1 year.26 During this 12-month study, analgesic efficacy was maintained withonly moderate increases in daily dose.26 The most com-mon adverse events included nausea (24%), constipa-tion (19.3%), and vomiting (16.8%).
Safety Profile and Metabolic PropertiesThe most common adverse reactions associated with
EXALGO (>10%) include constipation (31%), nausea(28%), vomiting, somnolence, headache, and dizziness.18
EXALGO was administered to a total of 2,524 patientsin 15 controlled and uncontrolled clinical studies. Ofthese, 423 patients were exposed to EXALGO for greaterthan 6 months and 151 were exposed for greater than 1year. The overall incidence of adverse reactions inpatients greater than 65 years of age was higher, with agreater than 5% difference in rates for constipation andnausea when compared with younger patients. The over-all incidence of adverse reactions in female patients washigher, with a greater than 5% difference in rates fornausea, vomiting, constipation, and somnolence whencompared with male patients.18
EXALGO has the potential to induce severe respira-tory depression. Other serious adverse events includeincreased intracranial pressure in the presence of a headinjury, hypotensive effect, gastrointestinal effects, andcardiac arrest.18 For detailed Important Risk Informationsee below.
EXALGO has a reduced potential for certain druginteractions because it minimally interacts with thecytochrome P450 pathway and produces no active drugmetabolites.18 Given this consideration, hydromorphonemay be suitable for patients taking multiple medications.
The pharmacokinetic profile of EXALGO is not sig-nificantly affected by alcohol, and exhibits no evidenceof “dose dumping” of hydromorphone.27 Therefore,consuming alcohol with EXALGO does not acceleratethe release of hydromorphone into the bloodstream.However, due to a central nervous system (CNS) depres-sant effect, patients should not consume alcohol whiletaking EXALGO.18
Once-Daily DosingAvailable in several strengths for once-daily dosing,
EXALGO is provided in 8 mg, 12 mg, and 16 mg tabletsto facilitate conversion. For conversion to EXALGO inopioid-tolerant patients, the dose range of EXALGOstudied in clinical trials was 8 mg to 64 mg. EXALGOtablets may be administered once every 24 hours with orwithout food. EXALGO is for use in opioid-tolerantpatients only; no patient should receive EXALGO as thefirst opioid.18
Appropriate management of conversion and titrationis crucial. The first time a patient takes EXALGO, hydro-morphone levels gradually increase over 6-8 hours.28Particularly during the initial transition period, otheranalgesics may be needed to treat breakthrough pain.
Conversion in Opioid-Tolerant PatientsNo fixed conversion ratio is likely to be satisfactory in
all patients, especially in patients receiving large opioiddoses.18 In opioid-tolerant patients, patients should be
14 AMERICAN HEALTH & DRUG BENEFITS May/June 2011
See pages 20-21 for brief summary of Full Prescribing Information.

converted from prior opioid therapy to EXALGO®
(hydromorphone HCl) Extended-Release Tablets, CII,using a simple conversion ratio of 5 to 1 (morphineequivalent to hydromorphone), because hydromor-phone is approximately 5 times more potent than mor-phine.29 EXALGO treatment can be initiated 18 hoursafter removal of a fentanyl patch.18 For each 25 mcg/hourfentanyl transdermal dose, the equianalgesic dose ofEXALGO is 12 mg every 24 hours.
Conversion ratios between other various opioids andEXALGO are shown in the Table. All other extended-release opioids must be discontinued when beginningEXALGO therapy. A stepwise manner is recommend-ed when converting, starting in general, with an initial25%-50% reduction of the calculated equianalgesicdose (see Table).18,25,30 Other key points regarding con-version include:• Increasing or decreasing the dose in the second step
depends on the patient reassessment,30 which shouldbe conducted every 3-4 days18 until a stable dose isreached.
• If reassessment indicates that the pain is severe,
dose increase is recommended; increase the dosegradually by 25%-50% every 3-4 days until a stabledose is reached.18
• If reassessment reveals moderate pain, mild confu-sion, and the use of multiple other drugs, it is judi-cious to reduce the dose by an additional 15%.30In selecting the initial dose of EXALGO, attention
should be given to the following18:• The daily dose, potency, and specific characteristics
of the opioid the patient has been taking previously• The reliability of the relative potency estimate used
to calculate the equivalent hydromorphone doseneeded
• The patient’s degree of opioid tolerance• The age, general condition, and medical status of
the patient• The concurrent non-opioid analgesics and other
medications, such as those with CNS activity• The type and severity of the patient’s pain• The balance between pain control and adverse effects• Risk factors for abuse, addiction, or diversion, includ-
ing a history of abuse, addiction, or diversion.
15May/June 2011 www.AHDBonline.com
Table Conversion Ratios from Various Opioids to EXALGOa
Previous Opioid Hydromorphone Codeine Hydrocodone Methadoneb Morphine Oxycodone Oxymorphone
Multiply the:
Approximate dailyequivalent oral dose
12 mg 200 mg 30 mg 20 mg 60 mg 30 mg 20 mg
By the:
Oral conversionratioc
1 0.06 0.4 0.6 0.2 0.4 0.6
• Select opioid• Sum the total daily dose• Multiply the dose by the conversion ratio to calculate the approximate oral hydromorphone equivalent• In general, start EXALGO therapy by administering 25%-50% of the calculated total daily dose of EXALGO every 24 hours
• Dosing titration should occur gradually (25%-50%) every 3 to 4 days If EXALGO is discontinued, doses should also be tapered gradually (25%-50%) every 2 or 3 days
aThe conversion ratios and approximate doses in this conversion table are to be used only for the conversion from current opioid therapy to EXALGO.bIt is extremely important to monitor all patients closely when converting from methadone to other opioid agonists. The ratio betweenmethadone and other opioid agonists may vary widely as a function of previous dose exposure. Methadone has a long half-life and tends to accumulate in the plasma.cThe ratio for conversion of oral opioid dose to approximate hydromorphone equivalent dose.Source: References 18, 25, 30.
See pages 20-21 for brief summary of Full Prescribing Information.

16 AMERICAN HEALTH & DRUG BENEFITS May/June 2011
See pages 20-21 for brief summary of Full Prescribing Information.
WARNING: POTENTIAL FOR ABUSE, IMPORTANCE OF PROPER PATIENT SELECTION AND LIMITATIONS OF USE
Potential for AbuseEXALGO contains hydromorphone, an opioid agonist anda Schedule II controlled substance with an abuse liabilitysimilar to other opioid analgesics. EXALGO can beabused in a manner similar to other opioid agonists,legal or illicit. These risks should be considered whenadministering, prescribing, or dispensing EXALGO insituations where the healthcare professional isconcerned about increased risk of misuse, abuse, ordiversion. Schedule II opioid substances which includehydromorphone, morphine, oxycodone, fentanyl,oxymorphone and methadone have the highest potentialfor abuse and risk of fatal overdose due to respiratorydepression.
Proper Patient SelectionEXALGO is an extended-release formulation ofhydromorphone hydrochloride indicated for themanagement of moderate to severe pain in opioidtolerant patients when a continuous around-the-clockopioid analgesic is needed for an extended period of
time. Patients considered opioid tolerant are those whoare taking at least 60 mg oral morphine per day, 25 mcgtransdermal fentanyl/hour, 30 mg oral oxycodone/day, 8mg oral hydromorphone/day, 25 mg oral oxymorphone/day or an equianalgesic dose of another opioid, for aweek or longer.EXALGO is for use in opioid tolerant patients only.Fatal respiratory depression could occur in patients whoare not opioid tolerant.Accidental consumption of EXALGO, especially inchildren, can result in a fatal overdose ofhydromorphone.
Limitations of UseEXALGO is not indicated for the management of acute orpostoperative pain.EXALGO is not intended for use as an as-neededanalgesic.EXALGO tablets are to be swallowed whole and are notto be broken, chewed, dissolved, crushed or injected.Taking broken, chewed, dissolved or crushed EXALGO orits contents leads to rapid release and absorption of apotentially fatal dose of hydromorphone.
IMPORTANT RISK INFORMATION
• EXALGO® is also contraindicated in patients who:
- need management of mild pain or pain not expectedto persist
- have significant impaired respiratory function includingthose with acute or severe bronchial asthma orhypercarbia
- have or are suspected to have paralytic ileus
- have narrowed or obstructed gastrointestinal tractincluding those from previous surgery or “blind loops” inthe GI tract
- have known hypersensitivity to any componentsincluding hydromorphone hydrochloride and sulfites.
• Avoid concurrent use of alcohol and EXALGO. Concurrentuse of EXALGO with CNS depressants, including alcohol,increases risk of respiratory depression, hypotension, andprofound sedation, potentially resulting in coma or death.EXALGO may impair the ability to drive a car or operatemachinery.
• Not intended in patients who have received MAOinhibitors within 14 days of starting EXALGO.
• Use with caution in reduced doses in older or debilitatedpatients, as well as patients with renal or hepaticinsufficiency, Addison’s disease, delirium tremens,myxedema or hypothyroidism, prostatic hypertrophy orurethral stricture, toxic psychosis. May aggravateconvulsions in patients with convulsive disorders; mayinduce or aggravate seizures in some clinical settings.Consider use of an alternate analgesic in patients withsevere renal impairment.
• Respiratory depression, which occurs more frequentlyin elderly or debilitated patients, is the chief hazardwith EXALGO.
• Serious adverse events could also include hypotensiveeffects, GI effects, cardiac arrest from overdose andprecipitation of withdrawal. Most common adverseevents (>10%) seen in clinical studies (N = 2474) were:constipation (31%), nausea (28%), vomiting, somnolence,headache, asthenia and dizziness.
• Use EXALGO with extreme caution in patients susceptibleto intracranial effects of CO2 retention.
• Do not abruptly discontinue EXALGO.

17May/June 2011 www.AHDBonline.com
Risk Evaluation and Mitigation StrategyIn February 2009, the FDA notified manufacturers of
certain opioid drug products that a Risk Evaluation andMitigation Strategy (REMS) for these drugs would berequired to ensure that the benefits of the drug outweighthe risk to the patient.31 Effective March 25, 2008, aspart of the Food and Drug Administration AmendmentsAct of 2007 (FDAAA), the FDA was given the author-ity to require certain drugs or biologics to have a REMSin place. This REMS requirement was enacted for thosedrugs that provide an important benefit to patients butmay be especially dangerous if not used properly.31 In July2010, the FDA presented a proposal for a class REMS forlong-acting and extended-release opioid agents.32
An EXALGO REMS has been implemented inresponse to the FDA requirement. The REMS programprovides education and training for patients andhealthcare professionals designed to inform patientsand healthcare professionals about the potential forabuse, misuse, overdose, and addiction of EXALGO,and about the safe use of EXALGO. It includes severalelements to assure safe use, including a “dear health-care professional letter,” full prescribing information, amedication guide, a prescribing brochure, and an essen-tial information form. These REMS elements are avail-able online at www.exalgorems.com.
ConclusionEXALGO® (hydromorphone HCl) Extended-Release
Tablets, CII, a long-acting option for the management ofmoderate to severe pain in opioid-tolerant patients, pro-vides consistent, prolonged analgesia in a convenient,once-daily formulation. Using the OROS® Push-Pull™osmotic pump delivery system, EXALGO continuallyreleases hydromorphone resulting in consistent plasmalevels over a 24-hour period at steady state with dimin-ished peak-trough fluctuation18,22 compared with immedi-ate-release hydromorphone.
EXALGO is the only extended-release formulation ofhydromorphone currently available in the United States.EXALGO is provided in several strengths for once-dailydosing, including 8 mg, 12 mg, and 16 mg tablets, tofacilitate conversion. The pharmacokinetic characteris-tics of EXALGO suggest that it may be well suited forthe long-term management of chronic pain.22 As withother agents in the opioid class of drugs, appropriatepatient selection and use are im portant considerations.
Funding Source Funding for this article was provided by Covidien.
References1. Turk DC. Pain hurts—individuals, significant others, and society! Am Pain Soc Bull.2006;16. www.ampainsoc.org/pub/bulletin/win06/pres1.htm. Accessed February 3, 2011.
2. Harstall C, Ospina M. How prevalent is chronic pain? Pain Clin Updates. 2003;11:1-4. www.iasp-pain.org/AM/AMTemplate.cfm?Section=HOME,HOME&TEMPLATE=/CM/ContentDisplay.cfm&CONTENTID=7594&SECTION=HOME,HOME.Accessed February 3, 2011.3.American Pain Society. Resources, chronic pain in America: roadblocks to relief, conclu-sions. www.ampainsoc.org/links/roadblocks/conclude_road.htm. Accessed February 4, 2011.4. Johannes CB, Le TK, Zhou X, et al. The prevalence of chronic pain in UnitedStates adults: results of an internet-based survey. J Pain. 2010;11:1230-1239.5. National Institute of Neurological Disorders and Stroke, National Institutes ofHealth. Low back pain—fact sheet. January 31, 2011. www.ninds.nih.gov/disorders/backpain/detail_backpain.htm. Accessed January 31, 2011.6. Stewart WF, Ricci JA, Chee E, et al. Lost productive time and cost due to com-mon pain conditions in the US workforce. JAMA. 2003;290:2443-2454. 7. American Pain Society. APS press room—media backgrounder. www.ampainsoc.org/press/backgrounder.htm. Accessed February 3, 2011.8. Vincent GK, Velkoff VA. The Next Four Decades, the Older Population in theUnited States: 2010 to 2050. Current Population Reports: 25-1138. US CensusBureau, Washington, DC: May 2010.9.US Department of Health and Human Services, Administration on Aging. A profileof older Americans: 2007. www.agingcarefl.org/aging/AOA-2007profile.pdf. AccessedFebruary 3, 2011.10. Parsells Kelly J, Cook SF, Kaufman DW, et al. Prevalence and characteristics ofopioid use in the US adult population. Pain. 2008;138:507-513.11. David Michaelson & Company. Voices of chronic pain, a national study con-ducted for the American Pain Foundation. May 2006. www.painfoundation.org/media/resources/voices-survey-report.pdf. Accessed February 3, 2011. 12. Chou R, Fanciullo GJ, Fine PG, et al. Clinical guidelines for the use of chronicopioid therapy in chronic noncancer pain. J Pain. 2009;10:113-130.13. Quang-Cantagrel ND, Wallace MS, Magnuson SK. Opioid substitution toimprove the effectiveness of chronic noncancer pain control: a chart review. AnesthAnalg. 2000;90:933-937. 14. FDA approves Exalgo™ extended-release tablets. Medical News Today. Press release.March 4, 2010. www.medicalnewstoday.com/articles/181115.php. Accessed January 31, 2011.15. Murray A, Hagen NA. Hydromorphone. J Pain Symptom Manage. 2005;29(suppl 1):57-66.16.Quigley C, Wiffen P. A systematic review of hydromorphone in acute and chron-ic pain. J Pain Symptom Manage. 2003;25:169-178.17. Neuromed Pharmaceuticals. Exalgo™ (hydromorphone HCl) extended-releasetablets CII. NDA 21-217. September 23, 2009.18. Exalgo (hydromorphone HCl) prescribing information. Hazelwood, MO:Mallinckrodt, a Covidien company; 2010. www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/AnestheticAndLifeSupportDrugsAdvisory Committee/UCM183035.pdf. Accessed February 3, 2011.19. Drover DR, Angst MS, Valle M, et al. Input characteristics and bioavailabilityafter administration of immediate and a new extended-release formulation of hydro-morphone in healthy volunteers. Anesthesiology. 2002;97:827-836.20.Angst MS, Drover DR, Lötsch J, et al. Pharmacodynamics of orally administeredsustained-release hydromorphone in humans. Anesthesiology. 2001;94:67-73. 21. Sathyan G, Xu E, Thipphawong J, Gupta SK. Pharmacokinetic profile of a 24-hour controlled-release OROS formulation of hydromorphone in the presence andabsence of food. BC Clin Pharmacol. 2007;7:2.22. Gupta S, Sathyan G. Advances in the long-term management of chronic pain:recent evidence with OROS® hydromorphone, a novel, once-daily, long-acting opi-oid analgesic, providing constant analgesia with OROS® hydromorphone. J PainSymptom Manage. 2007;33(suppl 1):S19-S24. 23. Moore KT, St-Fleur D, Marricco NC, et al. Steady-state pharmacokinetics ofextended-release hydromorphone (OROS hydromorphone): a randomized study inhealthy volunteers. J Opioid Manag. 2007;6:351-358.24. Shram MJ, Sathyan G, Khanna S, et al. Evaluation of the abuse potential ofextended release hydromorphone versus immediate release hydromorphone. J ClinPsychopharmacol. 2010;30:25-33.25.Hale M, Khan A, Kutch M, Li S. Once-daily OROS hydromorphone ER comparedwith placebo in opioid-tolerant patients with chronic low back pain. Curr Med ResOpin. 2010;26(6):1505-1518. Erratum in: Curr Med Res Opin. 2010;26:1505-1518.26.Wallace M, Moulin DE, Rauck RL, et al. Long-term safety, tolerability, and efficacyof OROS hydromorphone in patients with chronic pain. J Opioid Manag. 2009;5:97-105.27. Sathyan G, Sivakumar K, Thipphawong J. Pharmacokinetic profile of a 24-hourcontrolled-release OROS formulation of hydromorphone in the presence of alcohol.Curr Med Res Opin. 2008;24:297-305.28. Sathyan G, Xu E, Thipphawong J, Gupta SK. Pharmacokinetic investigation ofdose proportionality with a 24-hour controlled-release formulation of hydromor-phone. BMC Clin Pharmacol. 2007;7:3.29. Palangio M, Northfelt DW, Portenoy RK, et al. Dose conversion and titrationwith a novel, once-daily, OROS osmotic technology, extended-release hydromor-phone formulation in the treatment of chronic malignant or nonmalignant pain.J Pain Symptom Manage. 2002;23:355-368.30. Fine PG, Portenoy RK; Ad Hoc Expert Panel on Evidence Review andGuidelines for Opioid Rotation. Establishing “best practices” for opioid rotation:conclusions of an expert panel. J Pain Symptom Manage. 2009;38(3):418-425. 31. US Department of Health & Human Services, US Food and Drug Administra -tion. Background on opioid REMS. November 20, 2009. www.fda.gov/Drugs/DrugSafety/InformationbyDrugClass/ucm187975.htm. Accessed February 4, 2011. 32. US Department of Health & Human Services, US Food and Drug Administra -tion. Opioid drugs and risk evaluation and mitigation strategies (REMS). September23, 2010. www.fda.gov/Drugs/DrugSafety/InformationbyDrugClass/ucm163647.htm.Accessed February 4, 2011.
See pages 20-21 for brief summary of Full Prescribing Information.

At least 76 million individuals in the United Statessuffer from pain.1 The annual cost associatedwith chronic pain, including healthcare expens-
es, lost income, and lost productivity, in the UnitedStates is estimated to be $100 billion.2 When chronicpain is not adequately treated, it can have significantdetrimental effects on one’s mood and social and occupa-tional functions, and it can also increase the rate ofcomorbidities.2,3 Chronic pain also impairs quality of lifeand emotional well-being and is strongly correlated withpsychiatric comorbidities, such as depression.3,4
Managed Care Organizations Although chronic pain is a common complaint among
patients within managed care, pain management has notbeen a major focus of most health plans, in contrast toother chronic disorders, such as diabetes, asthma, cardio-vascular disease, or hypertension.5,6 This is partly becauseof the inability of managed care organizations (MCOs) toquantify or classify chronic pain.7 This condition is diffi-cult to track, because providers typically omit a pain diag-nosis, although pain may be the patient’s primary reasonfor the office visit.7
In addition, it is difficult to code the severity of painby standard International Classification of Diseases coding,even if a pain-related diagnosis is used as the primarycode. Therefore, assessing the economic costs and thera-pies used for treating chronic pain has been challenging.8
The escalating cost of pain medications also poses amajor challenge to pain treatment in managed care,because pain management medication options havechanged from primarily lower-cost generic formulationsto branded, more expensive formulations, includingextended release, patches, and buccal dosage forms. MostMCOs restrict access to pain medications through theuse of formularies, prior authorization (PA), and tieredcopayment. Formulary restriction is the most commoncost-control mechanism used in managed care.
Although tiered copayment systems may control thecost of more expensive new pain medications, there is noevidence that the total cost of pain care is controlled.Considerable debate surrounds the usefulness of PA anddrug utilization management in reducing the costs of pain
medications. Some MCOs indicate that these processesalienate physicians and are a source of dissatisfaction forpatients.8 These processes can also be used by MCOs toeducate physicians about appropriate utilization of painmedications, and often result in significant cost-savings.
Advocates of PA contend that by asking physiciansto document the reasons for prescribing a particularpain medication, the physician learns appropriate clin-ical practice, while saving the cost of inappropriate pre-scribing.9 Formularies should include opioids that havebeen shown to manage pain most effectively, whichwill minimize long-term costs related to inadequatepain management.
New Drug Formulation OptionsThe new drug formulation EXALGO® (hydromor-
phone HCl) Extended-Release Tablets, CII, is a oncedaily extended-release oral formulation indicated foronce daily administration for the management of moder-ate to severe pain in opioid-tolerant patients who requirecontinuous, around-the-clock opioid analgesia for anextended period of time.
The use of around-the-clock medication has beenshown to be an effective approach for treating chronicpain. Guidelines issued by the American Pain Society asfar back as 1999 state that, in most cases, the preferredroute of opioid administration is oral, as a result of con-venience, flexibility, and relative steadiness of the opioidconcentrations in the blood. The advantages of around-the-clock therapy and an oral medication are some of thereasons for the development of controlled-release, oralformulations of opioids (eg, morphine, oxycodone,hydromorphone, and hydrocodone). The reduced dosingfrequency makes the oral medication more convenientfor patients, making it easier for them to comply with thedosing regimen.
Around-the-clock therapeutic coverage, and the pos-sible increased compliance afforded by controlled-releaseformulations, can make opioids even more effective inthe management of chronic pain.10 High-tech oral deliv-ery systems such as the OROS® Push-PullTM osmoticpump delivery system produces steady-state concentra-tions of hydromorphone that comparably reduce the
STAKEHOLDER PERSPECTIVE
18 AMERICAN HEALTH & DRUG BENEFITS May/June 2011
Extended-Release Formulation ofHydromorphone Available in the United StatesBy Gary Rice, RPh, MS, MBAVice President of Operations, ITSRx, Houston, TX
See pages 20-21 for brief summary of Full Prescribing Information.

drug’s peaks and troughs; once steady state is achieved,this should significantly reduce the need for additionalanalgesics for breakthrough pain.11
ConclusionThe directive to “first, do not harm” is anything but
simple in today’s pain therapy strategies, and nowhere isits complexity more evident than in treating chronic painwith opioids. Patients in pain who rely on opioids fortheir pain management and for improved functiondeserve access to safe and effective medications.Depriving them of optimal pain relief does them harm.These same life-restoring medications cause harm topatients who may be at risk for addiction and abuse.
Managed care navigates in a complex triad of clinicalpractice standards, population-based medicine, andpatient variability to clinical responses. Chronic pain hasno definitive solution, just a collection of clinical optionsfor many patients. Extended-release forms of the opioidpain medication offer another option in the armamentar-ium of chronic pain therapy considerations. ■
Disclosure StatementMr Rice reported no conflicts of interest in relation to thisarticle.
References1. Moskowitz MA. Advances in understanding chronic pain: mechanisms of painmodulation and relationship to treatment. Neurology. 2002;59(suppl 20):S1.2. Gureje O, Von Korff M, Simon GE, Gater R. Persistent pain and well-being: aWorld Health Organization study in primary care. JAMA. 1998;280:147-151. 3. Stewart WF, Ricci JA, Chee E, et al. Lost productive time and cost due to com-mon pain conditions in the US workforce. JAMA. 2003;290:2443-2454.4. National Institutes of Health. NIH guide: new directions in pain research: I.Bethesda, MD: National Institutes of Health. September 4, 1998. http://grants.nih.gov/grants/guide/pa-files/PA-98-102.html. Accessed February 20, 2011.5. Polatin PB, Mayer TG. Occupational disorders and the management of chronicpain. Orthop Clin North Am. 1996;27:881-890.6. Liu AC, Byrne E. Cost of care for ambulatory patients with low back pain. J FamPract. 1995;40:449-455.7. American Pain Society. Chronic pain in America: roadblocks to relief. www.ampainsoc.org/links/roadblocks. Accessed February 24, 2011.8. Lande SD. The Problem of Pain in Managed Care. In: Lande SD, Kulich RJ, eds.Managed Care and Pain. Glenview, IL: American Pain Society; 2000:15-36.9. Zagari MJ, Mazonson PD, Longton WC. Pharmacoeconomics of chronic nonma-lignant pain. Pharmacoeconomics. 1996;10:356-377.10. Reder RF. Opioid formulations: tailoring to the needs in chronic pain. Eur J Pain.2001;5(suppl A):109-111.11. EXALGO (hydromorphone HCl) prescribing information. Hazelwood, MO:Mallinckrodt, a Covidien company; 2010.
STAKEHOLDER PERSPECTIVE
19May/June 2011 www.AHDBonline.com
See pages 20-21 for brief summary of Full Prescribing Information.

EXALGO® (hydromorphone HCl)Extended-Release Tablets
WARNING: POTENTIAL FOR ABUSE, IMPORTANCE OF PROPER PATIENTSELECTION AND LIMITATIONS OF USE
Potential for AbuseEXALGO contains hydromorphone, an opioid agonist and a Schedule II controlled substance with an abuse liability similar to other opioid analgesics. EXALGO can be abused in a manner similar to other opioid agonists, legal or illicit. These risks should be considered when administering, prescribing, or dispensing EXALGO in situations where the healthcare professional is concerned about increased risk of misuse, abuse, or diversion. Schedule II opioid substances which include hydromorphone, morphine, oxycodone, fentanyl, oxymorphone and methadone have the highest potential for abuse and risk of fatal overdose due to respiratory depression [see Drug Abuse and Dependence (9)].
Proper Patient SelectionEXALGO is an extended-release formulation of hydromorphone hydrochloride indicated for the management of moderate to severe pain in opioid tolerant patients when a continuous around-the-clock opioid analgesic is needed for an extended period of time. Patients considered opioid tolerant are those who are taking at least 60 mg oral morphine per day, 25 mcg transdermal fentanyl/hour, 30 mg of oral oxycodone/day, 8 mg oral hydromorphone/day, 25 mg of oral oxymorphone/day or an equianalgesic dose of another opioid, for a week or longer [see Indications and Usage (1) and Dosage and Administration (2)].
EXALGO is for use in opioid tolerant patients only [see Indications and Usage (1) and Dosage and Administration (2)].
Fatal respiratory depression could occur in patients who are not opioid tolerant.
Accidental consumption of EXALGO, especially in children, can result in a fatal overdose of hydromorphone [see Warnings and Precautions (5.1)].
Limitations of UseEXALGO is not indicated for the management of acute or postoperative pain [see Indications and Usage (1)].
EXALGO is not intended for use as an as-needed analgesic [see Indications and Usage (1)].
EXALGO tablets are to be swallowed whole and are not to be broken, chewed, dissolved, crushed or injected. Taking broken, chewed, dissolved or crushed EXALGO or its contents leads to rapid release and absorption of a potentially fatal dose of hydromorphone [see Warnings and Precautions (5)].
CONTRAINDICATIONSOpioid Non-Tolerant PatientsEXALGO is contraindicated in opioid non-tolerant patients. Fatal respiratory depression could occur in patients who are not opioid tolerant. Impaired Pulmonary FunctionEXALGO is contraindicated in patients with significant respiratory depression, especially in the absence of resuscitative equipment or in unmonitored settings and in patients with acute or severe bronchial asthma or hypercarbia.Paralytic IleusEXALGO is contraindicated in patients who have or are suspected of having a paralytic ileus. Preexisting Gastrointestinal (GI) Surgery or Narrowing of GI TractEXALGO is contraindicated in patients who have had surgical procedures and/or underlying disease that would result in narrowing of the gastrointestinal tract, or have “blind loops” of the gastrointestinal tract or gastrointestinal obstruction. Allergy or HypersensitivityEXALGO is contraindicated in patients with known hypersensitivity to any of its components including the active agent, hydromorphone hydrochloride or known allergy to sulfite-containing medications [see Warnings and Precautions (5.8)].
WARNINGS AND PRECAUTIONSInformation Essential for Safe AdministrationEXALGO tablets are to be swallowed whole, and are not to be broken, chewed, crushed, dissolved or injected. Taking broken, chewed, crushed, dissolved EXALGO or its contents leads to the rapid release and absorption of a potentially fatal dose of hydromorphone [see Boxed Warning]. EXALGO is for use only in opioid tolerant patients. Ingestion of EXALGO may cause fatal respiratory depression when administered to patients who are not opioid tolerant [see Boxed Warning].EXALGO tablets must be kept in a secure place out of the reach of children. Accidental consumption of EXALGO, especially in children, can result in a fatal overdose of hydromorphone.Misuse and AbuseEXALGO contains hydromorphone, an opioid agonist, and is a Schedule II controlled substance. Opioid agonists have the potential for being abused and are sought by drug abusers and people with addiction disorders and are subject to criminal diversion. EXALGO can be abused in a manner similar to other opioid agonists, legal or illicit. This should be considered when prescribing or dispensing EXALGO in situations where the healthcare professional is concerned about an increased risk of misuse, abuse, or diversion. Breaking, crushing, chewing, or dissolving the contents of an EXALGO tablet results in the uncontrolled delivery of the opioid and poses a significant risk of overdose and death [see Drug Abuse and Dependence (9)]. If attempts are made to extract the drug from the hard outer shell for purposes of parenteral abuse, the injection of tablet excipients may be toxic and may result in lethal complications. Concerns about abuse, addiction, and diversion should not prevent the proper management of pain. However, all patients treated with opioids, including EXALGO, require careful monitoring for signs of abuse and addiction, since use of opioid analgesic products carries the risk of addiction even under appropriate medical use. Healthcare professionals should contact their State Professional Licensing Board or State Controlled Substances Authority for information on how to prevent and detect abuse or diversion of this product.
Respiratory Depression Respiratory depression is the chief hazard of EXALGO. Respiratory depression occurs more frequently in elderly or debilitated patients as well as those suffering from conditions accompanied by hypoxia or hypercapnia when even moderate therapeutic doses may dangerously decrease pulmonary ventilation, and when opioids are given in conjunction with other agents that depress respiration.Use EXALGO with extreme caution in patients with conditions accompanied by hypoxia, hypercapnia, or decreased respiratory reserve such as asthma, chronic obstructive pulmonary disease or cor pulmonale, severe obesity, sleep apnea, myxedema, kyphoscoliosis or CNS depression. In these patients, even moderate therapeutic doses of hydromorphone may decrease respiratory drive while simultaneously increasing airway resistance to the point of apnea. In these patients, consider alternative non-opioid analgesics, and use EXALGO only under careful medical supervision at the lowest effective dose.
Interactions with Alcohol and Other CNS DepressantsThe concurrent use of EXALGO with other central nervous system (CNS) depressants, including but not limited to other opioids, illicit drugs, sedatives, hypnotics, general anesthetics, phenothiazines, muscle relaxants, other tranquilizers, and alcohol, increases the risk of respiratory depression, hypotension, and profound sedation, potentially resulting in coma or death. Use with caution and in reduced dosages in patients taking CNS depressants. Avoid concurrent use of alcohol and EXALGO [see Clinical Pharmacology (12.3)].
Head Injury and Increased Intracranial PressureIn the presence of head injury, intracranial lesions or a preexisting increase in intracranial pressure, the respiratory depressant effects of EXALGO and its potential to elevate cerebrospinal fluid pressure (resulting from vasodilation following CO2 retention) may be markedly exaggerated. Furthermore, EXALGO can produce effects on pupillary response and consciousness, which may obscure neurologic signs of further increases in intracranial pressure in patients with head injuries.
Hypotensive EffectEXALGO may cause severe hypotension. There is added risk to individuals whose ability to maintain blood pressure has been compromised by a depleted blood volume, or after concurrent administration with drugs such as phenothiazines, general anesthetics, or other agents that compromise vasomotor tone. Administer EXALGO with caution to patients in circulatory shock, since vasodilation produced by the drug may further reduce cardiac output and blood pressure.
Gastrointestinal EffectsBecause the EXALGO tablet is nondeformable and does not appreciably change in shape in the GI tract, do not administer EXALGO to patients with preexisting severe gastrointestinal narrowing (pathologic or iatrogenic, for example: esophageal motility disorders small bowel inflammatory disease, “short gut” syndrome due to adhesions or decreased transit time, past history of peritonitis, cystic fibrosis, chronic intestinal pseudoobstruction, or Meckel’s diverticulum). There have been reports of obstructive symptoms in patients with known strictures or risk of strictures, such as previous GI surgery, in association with the ingestion of drugs in nondeformable extended-release formulations. The administration of EXALGO may obscure the diagnosis or clinical course in patients with acute abdominal condition.It is possible that EXALGO tablets may be visible on abdominal x-rays under certain circumstances, especially when digital enhancing techniques are utilized.
SulfitesEXALGO contains sodium metabisulfite, a sulfite that may cause allergic-type reactions including anaphylactic symptoms and life-threatening or less severe asthmatic episodes in certain susceptible people. The overall prevalence of sulfite sensitivity in the general population is unknown and probably low. Sulfite sensitivity is seen more frequently in asthmatic than in nonasthmatic people.
MAO InhibitorsEXALGO is not recommended for use in patients who have received MAO inhibitors within 14 days, because severe and unpredictable potentiation by MAO inhibitors has been reported with opioid analgesics.
Special Risk GroupsEXALGO should be administered with caution in elderly (≥ 65 years) and debilitated patients and in patients who are known to be sensitive to central nervous system depressants, such as those with cardiovascular, pulmonary, renal, or hepatic disease [see Use in Specific Populations (8)].EXALGO should also be used with caution in the following conditions: adrenocortical insufficiency (e.g., Addison’s disease); delirium tremens; myxedema or hypothyroidism; prostatic hypertrophy or urethral stricture; and, toxic psychosis. EXALGO may aggravate convulsions in patients with convulsive disorders, and all opioids may induce or aggravate seizures in some clinical settings.
Use in Pancreatic/Biliary Tract DiseaseEXALGO can cause an increase in biliary tract pressure as a result of spasm in the sphincter of Oddi. Caution should be exercised in the administration of EXALGO to patients with inflammatory or obstructive bowel disorders, acute pancreatitis secondary to biliary tract disease and in patients about to undergo biliary surgery.
Driving and Operating MachineryEXALGO may impair the mental and/or physical abilities needed to perform potentially hazardous activities such as driving a car or operating machinery. Caution patients accordingly. Also warn patients about the potential combined effects of EXALGO with other CNS depressants, including other opioids, phenothiazines, sedative/hypnotics, and alcohol [see Drug Interactions (7)].
Precipitation of Withdrawal Mixed agonist/antagonist analgesics (i.e., pentazocine, nalbuphine, and butorphanol) should not be administered to patients who have received or are receiving a course of therapy with a pure opioid agonist analgesic, including EXALGO. In these patients, mixed agonists/antagonists analgesics may reduce the analgesic effect and/or may precipitate withdrawal symptoms. Do not abruptly discontinue EXALGO.Clinical conditions or medicinal products that cause a sudden and significant shortening of gastrointestinal transit time may result in decreased hydromorphone absorption with EXALGO and may potentially lead to withdrawal symptoms in patients with a physical dependence on opioids.
ADVERSE REACTIONSThe following serious adverse reactions are discussed elsewhere in the labeling:
[see Warnings and Precautions (5.3)]Head Injury and Increased Intracranial Pressure [see Warnings and Precautions (5.5)]
[see Warnings and Precautions (5.6)] [see Warnings and Precautions (5.7)]
[see Overdosage (10)][see Warnings and Precautions (5.13)]
Clinical Studies ExperienceBecause clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.EXALGO was administered to a total of 2,524 patients in 15 controlled and uncontrolled clinical studies. Of these, 423 patients were exposed to EXALGO for greater than 6 months and 141 exposed for greater than one year. The overall incidence of adverse reactions in patients greater than 65 years of age was higher, with a greater than 5% difference in rates for constipation and nausea when compared with younger patients. The overall incidence of adverse reactions in female patients was higher, with a greater than 5% difference in rates for nausea, vomiting, constipation and somnolence when compared with male patients.A 12-week double-blind, placebo-controlled, randomized withdrawal study was conducted in opioid tolerant patients with moderate to severe low back pain [see Clinical Studies (14)]. A total of 447 patients were enrolled into the open-label titration phase with 268 patients randomized into the double-blind treatment phase. The adverse reactions that were reported in at least 2% of the patients are contained in Table 1.
Table 1. Number (%) of Patients with Adverse Reactions Reported in ≥2% of
Patients with Moderate to Severe Low Back Pain During the Open-Label Titration Phase or Double-Blind Treatment Phase by Preferred Term
Preferred Term Open-Label Double-Blind Treatment Phase Titration Phase EXALGO (N=447) EXALGO (N=134) Placebo (N=134)Constipation 69 (15) 10 (7) 5 (4)Nausea 53 (12) 12 (9) 10 (7)Somnolence 39 (9) 1 (1) 0 (0)Headache 35 (8) 7 (5) 10 (7)Vomiting 29 (6) 8 (6) 6 (4)Drug Withdrawal Syndrome 22 (5) 13 (10) 16 (12)Pruritus 21 (5) 1 (1) 0 (0)Dizziness 17 (4) 3 (2) 2 (1)Asthenia a 16 (4) 2 (1) 6 (4)Insomnia 13 (3) 7 (5) 5 (4)Diarrhea 13 (3) 5 (4) 9 (7)Back Pain 13 (3) 6 (4) 8 (6)Dry Mouth 13 (3) 2 (1) 0 (0)Edema Peripheral 13 (3) 3 (2) 1 (1)Hyperhidrosis 13 (3) 2 (1) 2 (1)Anorexia b 10 (2) 2 (1) 0 (0)Arthralgia 9 (2) 8 (6) 3 (2)Anxiety 9 (2) 0 (0) 4 (3)Abdominal Pain c 9 (2) 4 (3) 3 (2)Muscle Spasms 5 (1) 3 (2) 1 (1)Weight Decreased 3 (1) 4 (3) 3 (2)
a Fatigue was grouped and reported with asthenia b Decreased appetite was grouped and reported with anorexia c Abdominal pain upper was grouped and reported with abdominal pain
The adverse reactions that were reported in at least 2% of the total treated patients (N=2,474) in the 14 chronic clinical trials are contained in Table 2.
Table 2.Number (%) of Patients with Adverse Reactions Reported in ≥2% of Patientswith Chronic Pain Receiving EXALGO in 14 Clinical Studies by Preferred TermPreferred Term All Patients (N=2,474)Constipation 765 (31)Nausea 684 (28)Vomiting 337 (14)Somnolence 367 (15) Headache 308 (12)Asthenia a 272 (11)Dizziness 262 (11)Diarrhea 201 (8) Pruritus 193 (8) Insomnia 161 (7) Hyperhidrosis 143 (6) Edema Peripheral 135 (5)Anorexia b 139 (6) Dry Mouth 121 (5) Abdominal Pain c 115 (5) Anxiety 95 (4) Back Pain 95 (4) Dyspepsia d 88 (4) Depression 81 (3) Dyspnea e 76 (3) Muscle Spasms 74 (3) Arthralgia 72 (3) Rash 64 (3) Pain in Extremity 63 (3) Pain 58 (2) Drug Withdrawal Syndrome 55 (2)Pyrexia 52 (2) Fall 51 (2) Chest Discomfort f 51 (2)
a Fatigue was grouped and reported with asthenia b Decreased appetite was grouped and reported with anorexia c Abdominal pain upper was grouped and reported with abdominal pain d Reflux esophagitis, gastroesophageal reflux disease and Barrett’s esophagus were grouped and
reported with dyspepsia e Dyspnea exacerbated and dyspnea exertional were grouped and reported with dyspnea f Chest pain and non-cardiac chest pain were grouped and reported with chest discomfort
BRIEF SUMMARY - Consult fullprescribing information before use.
20

The following Adverse Reactions occurred in patients with an overall frequency of <2% and are listed in descending order within each System Organ Class:Cardiac disorders: palpitations, tachycardia, bradycardia, extrasystolesEar and labyrinth disorders: vertigo, tinnitus Endocrine disorders: hypogonadismEye disorders: vision blurred, diplopia, dry eye, miosis Gastrointestinal disorders: �atulence, dysphagia, hematochezia, abdominal distension, hemorrhoids, abnormal feces, intestinal obstruction, eructation, diverticulum, gastrointestinal motility disorder, large intestine perforation, anal �ssure, bezoar, duodenitis, ileus, impaired gastric emptying, painful defecation General disorders and administration site conditions: chills, malaise, feeling abnormal, feeling hot and cold, feeling jittery, hangover, di�culty in walking, feeling drunk, hypothermiaInfections and infestations: gastroenteritis, diverticulitisInjury, poisoning and procedural complications: contusion, overdoseInvestigations: weight decreased, hepatic enzyme increased, blood potassium decreased, blood amylase increased, blood testosterone decreased, oxygen saturation decreasedMetabolism and nutrition disorders: dehydration, �uid retention, increased appetite, hyperuricemiaMusculoskeletal and connective tissue disorders: myalgia Nervous system disorders: tremor, sedation, hypoesthesia, paraesthesia, disturbance in attention, memory impairment, dysarthria, syncope, balance disorder, dysgeusia, depressed level of consciousness, coordination abnormal, hyperesthesia, myoclonus, dyskinesia, hyperre�exia, encephalopathy, cognitive disorder, convulsion, psychomotor hyperactivityPsychiatric disorders: confusional state, nervousness, restlessness, abnormal dreams, mood altered, hallucination, panic attack, euphoric mood, paranoia, dysphoria, listless, crying, suicide ideation, libido decreased, aggressionRenal and urinary disorders: dysuria, urinary retention, urinary frequency, urinary hesitation, micturition disorderReproductive system and breast disorders: erectile dysfunction, sexual dysfunction Respiratory, thoracic and mediastinal disorders: rhinorrhoea, respiratory distress, hypoxia, bronchospasm, sneezing, hyperventilation, respiratory depressionSkin and subcutaneous tissue disorders: erythema Vascular disorders: �ushing, hypertension, hypotension
DRUG INTERACTIONSCNS DepressantsThe concomitant use of EXALGO with central nervous system depressants such as hypnotics, sedatives, general anesthetics, antipsychotics and alcohol may cause additive depressant e�ects and respiratory depression. Additionally, hypotension and profound sedation or coma could occur. When this combination is indicated, the dose of one or both agents should be reduced. The concomitant use of alcohol should be avoided [see Clinical Pharmacology (12.3)]. Monoamine Oxidase (MAO) Inhibitors MAO inhibitors may cause CNS excitation or depression, hypotension or hypertension if co-administered with opioids including EXALGO. EXALGO is not intended for patients taking MAO inhibitors or within 14 days of stopping such treatment.Mixed Agonist/Antagonist Opioid AnalgesicsThe concomitant use of EXALGO with morphine agonist/antagonists (buprenor-phone, nalbuphine, pentazocine) could lead to a reduction of the analgesic e�ect by competitive blocking of receptors, thus leading to risk of withdrawal symptoms. Therefore, this combination is not recommended.AnticholinergicsAnticholinergics or other medications with anticholinergic activity when used concurrently with EXALGO may result in increased risk of urinary retention and/or severe constipation, which may lead to paralytic ileus.Cytochrome P450 EnzymesIn vitro data suggest that hydromorphone in clinically relevant concentrations has minimal potential to inhibit the activity of human hepatic CYP450 enzymes including CYP1A2, 2C9, 2C19, 2D6, 3A4, and 4A11.
USE IN SPECIFIC POPULATIONSPregnancy Teratogenic E�ects Pregnancy Category C: There are no adequate and well-controlled studies in pregnant women. Hydromorphone crosses the placenta. EXALGO should be used during pregnancy only if the potential bene�t justi�es the potential risk to the fetus [see Use in Speci�c Populations (8.2)].Hydromorphone was not teratogenic in pregnant rats given oral doses up to 6.25 mg/kg/day or in pregnant rabbits administered oral doses up to 25 mg/kg/day during the period of organogenesis (~1.2 times the human exposure following 32 mg/day). Hydromorphone administration to pregnant Syrian hamsters and CF-1 mice during major organ development revealed teratogenicity likely the result of maternal toxicity associated with sedation and hypoxia. In Syrian hamsters given single subcutaneous doses from 14 to 258 mg/kg during organogenesis (gestation days 8 to 10), doses ≥ 19 mg/kg hydromorphone produced skull
fo noisufni suounitnoC .)sisihcsoinarc dna ylahpecnexe( snoitamrof lamhydromorphone (5 mg/kg, s.c.) via implanted osmotic mini pumps during organogenesis (gestation days 7 to 10) produced soft tissue malformations (cryptorchidism, cleft palate, malformed ventricals and retina), and skeletal variations (supraoccipital, checkerboard and split sternebrae, delayed ossi�cation of the paws and ectopic ossi�cation sites). The malformations and variations observed in the hamsters and mice were at doses approximately three-fold higher and <one-fold lower, respectively, than a 32 mg human daily oral dose on a body surface area basis.Nonteratogenic E�ectsIn the pre- and post-natal e�ects study in rats, neonatal viability was reduced at 6.25 mg/kg/day (~1.2 times the human exposure following 32 mg/day).Neonates born to mothers who have been taking opioids regularly prior to delivery will be physically dependent. The withdrawal signs include irritability and excessive crying, tremors, hyperactive re�exes, increased respiratory rate, increased stools, sneezing, yawning, vomiting, and fever. The intensity of the syndrome does not always correlate with the duration of maternal opioid use or dose. There is no consensus on the best method of managing withdrawal. Approaches to the treatment of the syndrome have included supportive care and, if indicated, drugs such as paregoric or phenobarbital.
Labor and DeliveryEXALGO is not recommended for use in women during and immediately prior to labor and delivery. Administration of EXALGO to the mother shortly before delivery may result in some degree of respiratory depression in the neonate. However, neonates whose mothers received opioid analgesics during labor should be observed closely for signs of respiratory depression. Nursing MothersLow concentrations of hydromorphone have been detected in human milk in clinical trials. Withdrawal symptoms can occur in breastfeeding infants when maternal administration of an opioid analgesic is stopped. Nursing should not be undertaken while a patient is receiving EXALGO since hydromorphone is excreted in the milk.Pediatric UseThe safety and e�ectiveness of EXALGO in pediatric patients 17 years of age and younger have not been established.Geriatric UseElderly patients have been shown to be more sensitive to the adverse e�ects of EXALGO compared to the younger population. Therefore, use extra caution when prescribing EXALGO in elderly patients and reduce the initial dose. Neonatal Withdrawal SyndromeChronic maternal use of opiates or opioids during pregnancy coexposes the fetus. The newborn may experience subsequent neonatal withdrawal syndrome (NWS). Manifestations of NWS include irritability, hyperactivity, abnormal sleep pattern, high-pitched cry, tremor, vomiting, diarrhea, weight loss, and failure to gain weight. The onset, duration, and severity of the disorder di�er based on such factors as the addictive drug used, time and amount of mother’s last dose, and rate of elimination of the drug from the newborn. Approaches to the treatment of this syndrome have included supportive care and, when indicated, drugs such as paregoric or phenobarbital.Hepatic ImpairmentIn a study that used a single 4 mg oral dose of immediate-release hydromor-phone tablets, four-fold increases in plasma levels of hydromorphone (Cmax and AUC0- ) were observed in patients with moderate hepatic impairment (Child-Pugh Group B). Start patients with moderate hepatic impairment on a reduced dose and closely monitored during dose titration. The pharmacokinetics of hydromorphone in severe hepatic impairment patients have not been studied. Further increase in Cmax and AUC0- of hydromorphone in this group is expected, therefore, use an even more conservative starting dose [see Dosage and Administration (2.4)].Renal ImpairmentRenal impairment a�ected the pharmacokinetics of hydromorphone and its metabolites following administration of a single 4 mg dose of immediate-release tablets. The e�ects of renal impairment on hydromorphone pharmacokinetics were two-fold and four-fold increases in plasma levels of hydromorphone (Cmax and AUC0-48h) in moderate (CLcr = 40 to 60 mL/min) and severe (CLcr < 30 mL/min) impairment, respectively. In addition, in patients with severe renal impairment hydromorphone appeared to be more slowly eliminated with longer terminal elimination half-life (40 hours) compared to subjects with normal renal function (15 hours). Start patients with moderate renal impairment on a reduced dose and closely monitored during dose titration. As EXALGO is only intended for once daily administration, consider use of an alternate analgesic that may permit more �exibility with the dosing interval in patients with severe renal impairment [see Dosage and Administration (2.4)].
DRUG ABUSE AND DEPENDENCEControlled SubstanceEXALGO contains hydromorphone, a Schedule II controlled substance with a high potential for abuse similar to fentanyl, methadone, morphine, oxycodone, and oxymorphone. EXALGO can be abused and is subject to misuse, abuse, addiction, and criminal diversion [see Warnings and Precautions (5.2)]. The high drug content in the extended release formulation adds to the risk of adverse outcomes from abuse.Abuse All patients treated with opioids, including EXALGO, require careful monitoring for signs of abuse and addiction, because use of opioid analgesic products carries the risk of addiction even under appropriate medical use.Addiction is a primary, chronic, neurobiologic disease, with genetic, psychosocial, and environmental factors in�uencing its development and manifestations. It is characterized by behaviors that include one or more of the following: impaired control over drug use, compulsive use, continued use despite harm, and craving. “Drug-seeking” behavior is very common to addicts and drug abusers. Drug-seeking tactics include emergency calls or visits near the end of o�ce hours, refusal to undergo appropriate examination, testing or referral, repeated claims of loss of prescriptions, tampering with prescriptions and reluctance to provide prior medical records or contact information for other treating physician(s). “Doctor shopping” (visiting multiple prescribers) to obtain additional prescriptions is common among drug abusers, people su�ering from untreated addiction and criminals seeking drugs to sell. Abuse and addiction are separate and distinct from physical dependence and tolerance. Physicians should be aware that addiction may not be accompanied by concurrent tolerance and symptoms of physical dependence in all addicts. In addition, abuse of opioids can occur in the absence of true addiction and is characterized by misuse for non-medical purposes, often in combination with other psychoactive substances. Since EXALGO may be diverted for non-medical use, careful record-keeping of prescribing information, including quantity, frequency, and renewal requests is strongly advised. Proper assessment of the patient, proper prescribing practices, periodic re-evaluation of therapy, and proper dispensing and storage are appropriate measures that help to limit abuse of opioid drugs. EXALGO is intended for oral use only. Misuse or abuse by breaking, crushing, chewing, or dissolving EXALGO poses a hazard of overdose and death. This risk is increased with concurrent abuse of EXALGO with alcohol and other substances. With intravenous abuse, the tablet excipients, especially polyethylene oxide, can be expected to result in necrosis and in�ammation of cardiac tissues. In addition, parenteral drug abuse is commonly associated with transmission of infectious disease such as hepatitis and HIV.Healthcare professionals should contact their State Professional Licensing Board or State Controlled Substances Authority for information on how to prevent and detect abuse or diversion of this product.DependenceTolerance is a state of adaptation in which exposure to a drug induces changes that result in a diminution of one or more of the drug’s e�ects over time.
Tolerance could occur to both the desired and undesired e�ects of drugs, and may develop at di�erent rates for di�erent e�ects.Physical dependence is a state of adaptation that is manifested by an opioid speci�c withdrawal syndrome that can be produced by abrupt cessation, rapid dose reduction, decreasing blood level of the drug, and/or administration of an antagonist. The opioid abstinence or withdrawal syndrome is characterized by some or all of the following: restlessness, lacrimation, rhinorrhea, yawning, perspiration, chills, piloerection, myalgia, mydriasis, irritability, anxiety, backache, joint pain, weakness, abdominal cramps, insomnia, nausea, anorexia, vomiting, diarrhea, increased blood pressure, respiratory rate, or heart rate.Infants born to mothers physically dependent on opioids will also be physically dependent and may exhibit respiratory di�culties and withdrawal symptoms [see Use in Speci�c Populations (8.1, 8.2)].
OVERDOSAGESymptomsAcute overdosage with opioids can be manifested by respiratory depression, somnolence progressing to stupor or coma, skeletal muscle �accidity, cold and clammy skin, constricted pupils, and sometimes bradycardia, hypotension and death. The extended release characteristics of EXALGO should also be taken into account when treating the overdose. Even in the face of improvement, continued medical monitoring is required because of the possibility of extended e�ects. Deaths due to overdose could occur with abuse and misuse of EXALGO.Due to the delayed mean apparent peak plasma level of EXALGO occurring at 16 hours following administration as well as the 11 hour mean elimination half-life of EXALGO, patients who receive an overdose will require an extended period of monitoring and treatment that may go beyond 24 to 48 hours. TreatmentGive primary attention to the re-establishment of a patent airway and institution of assisted or controlled ventilation. Employ supportive measures (including oxygen and vasopressors) in the management of circulatory shock and pulmonary edema accompanying overdose as indicated. Cardiac arrest or arrhythmias will require advanced life support techniques.The pure opioid antagonists, such as naloxone and naltrexone are speci�c antidotes to respiratory depression from opioid overdose. Since the duration of reversal would be expected to be less than the duration of action of hydromorphone in EXALGO, the patient must be carefully monitored until spontaneous respiration is reliably re-established. EXALGO will continue to release and add to the hydromorphone load for up to 24 hours after administration and the management of an overdose should be monitored accordingly, at least 24 to 48 hours beyond the overdose.Only administer opioid antagonists in the presence of clinically signi�cant respiratory or circulatory depression secondary to hydromorphone overdose. In patients who are physically dependent on any opioid agonist including EXALGO, an abrupt or complete reversal of opioid e�ects may precipitate an acute abstinence syndrome. The severity of the withdrawal syndrome produced will depend on the degree of physical dependence and the dose of the antagonist administered. Please see the prescribing information for the speci�c opioid antagonist for details of their proper use.
OROS is a registered trademark of ALZA Corporation.EXALGO is a registered trademark of Mallinckrodt Inc.COVIDIEN, COVIDIEN with logo and Covidien logo are U.S. and/or internationally registered trademarks of Covidien AG.
© 2010 Mallinckrodt Inc., a Covidien company
Distributed by:Mallinckrodt Brand Pharmaceuticals, Inc.Hazelwood, MO 63042 USA
Issued 11/2010
Mallinckrodt
3-4 5 11:11:24 AM
21

22 I AMERICAN HEALTH & DRUG BENEFITS I May/June 2011
Ampyra (NME)(Dalfampridine; Acorda)Class: Potassium channel blockerIndication: To improve walking in patients with multiple sclerosisApproval considerations: Priority review, orphan drug, REMS
Asclera (NME)(Polidocanol; Chemische Fabrik Kreussler)Class: Sclerosing agentIndication: Treatment of uncomplicated spider veins(varicose veins ≤1 mm in diameter) and uncompli -cated reticular veins (varicose veins 1-3 mm in diameter) in the lower extremity
Axiron(Testosterone; Eli Lilly)Class: Androgen, steroid (topical)Indication: Androgen replacement therapy in males forconditions associated with a deficiency or absence ofendogenous testosteroneApproval consideration: REMS
Butrans (new formulation)(Buprenorphine; Purdue Pharma)Class: Opioid agonistIndication: Management of moderate-to-severe chronicpain in patients requiring a continuous, around-the-clock opioid analgesic for an extended period of timeApproval consideration: REMS
Carbaglu (NME)(Carglumic acid; Orphan Europe)Class: Carbamoyl phosphate synthetase 1 activatorIndication: Adjunctive therapy for the treatment of acutehyperammonemia resulting from the deficiency of thehepatic enzyme N-acetylglutamate synthase (NAGS)and maintenance therapy for the treatment of chronichyperammonemia resulting from NAGS deficiencyApproval considerations: Priority review, orphan drug
Cysview(Hexaminolevulinate hydrochloride; Photocure ASA)Class: Optical imaging agentIndication: For cystoscopic detection of nonmuscle inva-sive papillary cancer of the bladder in patients suspectedor known to have lesion(s) on the basis of a prior cysto -
scopy; used with the Karl Storz D-Light C PhotodynamicDiagnostic system to perform cystoscopy with the bluelight setting (mode 2) as an adjunct to the white lightsetting (mode 1)Approval consideration: Priority review
Egrifta (NME)(Tesamorelin acetate; Theratechnologies)Class: Growth hormone–releasing factor analog Indication: Reduction of excess abdominal fat in HIV-infected patients with lipodystrophy
Ella (NME)(Ulipristal acetate; Lab HRA Pharma)Class: Progesterone agonist/antagonist emergency contraceptionIndication: Prevention of pregnancy after unprotectedintercourse or a known or suspected contraceptive failure;not intended for routine use as a contraceptive
Exalgo(Hydromorphone hydrochloride; Covidien/Mallinckrodt)Class: Opioid agonistIndication:Management of moderate-to-severe pain inopioid-tolerant patients requiring continuous, around-the-clock opioid analgesia for an extended period of timeApproval considerations: Priority review, REMS
Gilenya (NME)(Fingolimod hydrochloride; Novartis)Class: Sphingosine 1-phosphate receptor modulatorIndication: Treatment of patients with relapsing formsof multiple sclerosis to reduce the frequency of clinicalexacerbations and to delay the accumulation of physi-cal disabilityApproval considerations: Priority review, REMS
Glassia (BLA)(Alpha1-proteinase inhibitor [human]; Kamada)Class: Alpha1-proteinase inhibitorIndication: Treatment of chronic augmentation andmaintenance therapy in individuals with emphysemaresulting from congenital deficiency of alpha1-proteinaseinhibitor, also known as alpha1-antitrypsin
Halaven (NME)(Eribulin mesylate; Eisai)Class: Microtubule inhibitorIndication: Treatment of patients with metastatic breast
FDA Approvals of Brand-NamePrescription Drugs in 2010I. New Pharmaceuticals, NMEs, Biologics

23
2010 FDA Approvals of Brand-Name Drugs
cancer who have previously received at least 2chemotherapeutic regimens for the treatment ofmetastatic disease; prior therapy should have includedan anthracycline and a taxane in either the adjuvant ormetastatic settingApproval consideration: Priority review
Hizentra (BLA)(Immune globulin subcutaneous [human], 20% liquid;CSL Behring)Class: Immune globulinIndication: Treatment of primary immunodeficiency disease
Jevtana (NME)(Cabazitaxel; sanofi-aventis)Class: Taxane antimicrotubuleIndication: Treatment of patients with hormone-refractory metastatic prostate cancer previously treatedwith a docetaxel-containing treatment regimenApproval consideration: Priority review
Kapvay(Clonidine hydrochloride; Shionogi)Class: Alpha2-adrenergic receptor agonistIndication: Treatment of attention-deficit/hyperactivitydisorder in children and adolescents aged 6 to 17 years
Krystexxa (BLA)(Pegloticase; Savient Pharmaceuticals)Class: Pegylated uric acid–specific enzyme Indication: Treatment of gout in adult patients whofailed conventional gout therapy to control hyper-uricemia and manage the signs and symptoms of goutApproval considerations: Priority review, orphan drug,REMS
Lastacaft (NME)(Alcaftadine ophthalmic solution; VistakonPharmaceuticals)Class: H1 histamine receptor antagonist + mast-cellstabilizerIndication: Prevention of itching associated with allergic conjunctivitis
Latuda (NME)(Lurasidone hydrochloride; Sunovion Pharmaceuticals)Class: Atypical antipsychoticIndication: Treatment of schizophrenia in adults
Lumizyme (BLA)(Alglucosidase alfa; Genzyme Corporation)Class: Lysosomal glycogen-specific enzyme Indication: Treatment of patients aged ≥8 years withnoninfantile Pompe disease who do not have evidenceof cardiac hypertrophyApproval considerations: Priority review, orphan drug, REMS
Menveo (BLA)(Meningococcal [groups A, C, Y, and W-135] oligosaccharide diphtheria CRM197 conjugate vaccine; Novartis)Class: Bacterial vaccineIndication: For active immunization to prevent invasivemeningococcal disease caused by Neisseria meningitidisserogroups A, C, Y, and W-135 when administered toindividuals aged 2 through 55 years
Moxeza(Moxifloxacin hydrochloride ophthalmic solution;Alcon Pharmaceuticals)Class: Topical fluoroquinoloneIndication: Treatment of bacterial conjunctivitis causedby susceptible strains of the following organisms:Aerococcus viridans, Corynebacterium macginleyi,Enterococcus faecalis, Micrococcus luteus, Staphylococcusarlettae, S aureus, S capitis, S epidermidis, S haemolyticus, S hominis, S saprophyticus, S warneri, Streptococcus mitis,Streptococcus pneumoniae, Streptococcus parasanguinis,Escherichia coli, Haemophilus influenzae, Klebsiella pneu -moniae, Propionibacterium acnes, and Chlamydia trachomatis
Natazia (NME)(Estradiol valerate/dienogest; Bayer HealthCare)Class: Estrogen + progestinIndication: For the prevention of pregnancy
Nuedexta(Dextromethorphan hydrobromide + quinidine sulfate;Avanir Pharmaceuticals)Class: Uncompetitive N-methyl-D-aspartate receptorantagonist/sigma-1 agonist plus a CYP450 2D6 inhibitorIndication: Treatment of pseudobulbar affectApproval consideration: Priority review
Pradaxa (NME)(Dabigatran etexilate mesylate; Boehringer Ingelheim)Class: Direct thrombin inhibitorIndication: To reduce the risk of stroke and systemicembolism in patients with nonvalvular atrial fibrillationApproval consideration: Priority review
Prevnar-13 (BLA)(Pneumococcal 13-valent conjugate vaccine [diphtheria CRM197 protein]; Wyeth)Class: VaccineIndication: Active immunization for the prevention ofinvasive disease caused by Streptococcus pneumoniaeserotypes 1, 3, 4, 5, 6A, 6B, 7F, 9V, 14, 18C, 19A, 19F,and 23F, for use in children aged 6 weeks through 5years; for the prevention of otitis media caused byStreptococcus pneumoniae serotypes 4, 6B, 9V, 14, 18C,19F, and 23F
May/June 2011 I www.AHDBonline.com I

II. New Combinations, Formulations, Indications Actemra (new indication)(Tocilizumab; Genentech)Class: Interleukin-6 receptor inhibitorIndication: Treatment for reducing signs and symptomsin adult patients with moderately to severely activerheumatoid arthritis who have had an inadequateresponse to 1 or more tumor necrosis factor–antagonisttherapiesApproval consideration: REMS
Alsuma (new formulation) (Sumatriptan succinate; King Pharmaceuticals)Class: 5-HT1B/1D receptor agonist Indication:Acute treatment of migraine attacks, with orwithout aura; acute treatment of cluster headache episodes
Amturnide (new combination)(Aliskiren, amlodipine + hydrochlorothiazide; Novartis)
24 I AMERICAN HEALTH & DRUG BENEFITS I May/June 2011
Prolia (BLA)(Denosumab; Amgen)Class: RANKL inhibitorIndication: Treatment of postmenopausal women withosteoporosis at high risk for fractureApproval consideration: REMSSee also Xgeva
Provenge (BLA)(Sipuleucel-T; Dendreon Corporation)Class: Autologous cellular immunotherapyIndication: Treatment of men with asymptomatic orminimally symptomatic metastatic castrate-resistant(hormone refractory) prostate cancer
TachoSil (BLA)(Fibrin sealant patch; Nycomed Danmark)Class: Hemostatic agent (patch)Indication: Adjunct to hemostasis in cardiovascular surgery when control of bleeding by standard surgicaltechniques—such as suture, ligature, or cautery—is ineffective or impractical
Teflaro (NME)(Ceftaroline fosamil; Cerexa)Class: Cephalosporin antibacterialIndication: Treatment of acute bacterial skin and skin-structure infections and community-acquired bacterial pneumonia
Victoza (NME)(Liraglutide recombinant; Novo Nordisk)Class: Glucagon-like peptide-1 receptor agonistIndication: Adjunct to diet and exercise to improveglycemic control in adults with type 2 diabetes melli-tus Approval consideration: REMS
VPriv (NME)(Velaglucerase alfa; Shire Human Genetic Therapies)
Class: Hydrolytic lysosomal glucocerebroside-specificenzymeIndication: Long-term enzyme replacement therapy forpediatric and adult patients with type 1 Gaucher diseaseApproval considerations: Priority review, orphan drug
Xeomin (BLA)(Incobotulinumtoxin-A; Merz Pharmaceuticals)Class: Acetylcholine release inhibitor + neuromuscularblockerIndication: Treatment of adults with cervical dystonia, todecrease the severity of abnormal head position andneck pain in both botulinum toxin–naïve and previous-ly treated patients; treatment of blepharospasm in adultspreviously treated with onabotulinumtoxin-AApproval consideration: REMS
Xgeva (BLA)(Denosumab; Amgen)Class: RANKL inhibitorIndication: To help prevent skeletal-related events inpatients with bone metastases from solid tumorsApproval consideration: REMSSee also Prolia
Xiaflex (BLA)(Collagenase clostridium histolyticum; AuxiliumPharmaceuticals)Class: Miscellaneous uncategorized agentsIndication: Treatment of advanced Dupuytren’s contrac-ture with a palpable cordApproval considerations: Priority review, orphan drug,REMS
Zortress (NME)(Everolimus; Novartis)Class: Immunosuppressant (macrolide)Indication: Prophylaxis of organ rejection in adultpatients at low-to-moderate immunologic risk receiv-ing a kidney transplantApproval consideration: REMS

25
2010 FDA Approvals of Brand-Name Drugs
May/June 2011 I www.AHDBonline.com I
Class: Direct renin inhibitor + calcium channel blocker+ diureticIndication: Treatment of hypertension
Aricept (new formulation)(Donepezil hydrochloride; Eisai)Class: Reversible acetylcholinesterase inhibitor (piperidine derivative)Indication: Treatment of dementia of the Alzheimer’s type
Aridol/Aridol Kit (new formulation)(Mannitol; Pharmaxis)Class: Sugar alcoholIndication: Assessment of bronchial hyperresponsivenessin patients aged ≥6 years who do not have clinicallyapparent asthma
Atelvia (new formulation)(Risedronate sodium; Warner Chilcott)Class: BisphosphonateIndication: Treatment of osteoporosis in postmenopausalwomen
Benicar (new indication)(Olmesartan medoxomil; Daiichi Sankyo)Class: Angiotensin II receptor blockerIndication: Treatment of hypertension, alone or withother antihypertensive agents, in adults and childrenaged 6 to 16 years
Beyaz (new combination)(Drospirenone + ethinyl estradiol + levomefolate calcium; Bayer HealthCare)Class: Progestin + estrogen + folate combination oralcontraceptiveIndication: For prevention of pregnancy; treatment ofsymptoms of premenstrual dysphoric disorder for womenwho choose to use an oral contraceptive for contracep-tion; treatment of moderate acne for women at least 14 years old only if the patient desires an oral contra-ceptive for birth control; to raise folate levels in womenwho choose to use an oral contraceptive
Botox (new indication)(Onabotulinumtoxin-A; Allergan)Class: Acetylcholine release inhibitor + neuromuscularblocking agentIndication: Treatment of upper limb spasticity in adults,cervical dystonia in adults to reduce the severity ofabnormal head position and neck pain, severe axillaryhyperhidrosis that is inadequately managed with topicalagents in adults, blepharospasm associated with dystoniain patients aged ≥12 years, strabismus in patients aged≥12 years, chronic migraine
Bromday (new formulation)(Bromfenac ophthalmic solution; ISTAPharmaceuticals)Class: Nonsteroidal anti-inflammatory drugIndication: Treatment of postoperative inflammationand reduction of ocular pain in patients who haveundergone cataract extraction
Carpine (new formulation)(Pilocarpine hydrochloride; Alcon)Class: Muscarinic cholinergic agonistIndication: Reduction of elevated intraocular pressure inpatients with open-angle glaucoma or ocular hyperten-sion; management of acute angle-closure glaucoma; pre-vention of postoperative elevated intraocular pressureassociated with laser surgery; induction of miosisApproval consideration: Priority review
Cayston (new formulation)(Aztreonam; Gilead)Class: Monobactam antibacterialIndication: Treatment of cystic fibrosis in patientsknown to have Pseudomonas aeruginosa in the lungs
Cuvposa (new formulation)(Glycopyrrolate; Shionogi)Class: Acetylcholine receptor inhibitor Indication: Treatment of chronic severe drooling inpatients aged 3 to 16 years with neurologic conditionsassociated with problem drooling (eg, cerebral palsy)Approval consideration: Priority review
Cymbalta (new indication)(Duloxetine hydrochloride; Eli Lilly)Class: Serotonin + norepinephrine reuptake inhibitorIndication: Treatment of major depressive disorder, gen-eralized anxiety disorder, diabetic peripheral neuropath-ic pain, fibromyalgia, chronic musculoskeletal pain
Differin (new formulation)(Adapalene; Galderma Laboratories)Class: RetinoidIndication: Topical treatment of acne vulgaris in patientsaged ≥12 years
Dulera (new combination)(Formoterol fumarate dihydrate + mometasone furoate;Schering)Class: Corticosteroid + long-acting beta2-agonistIndication: Treatment of asthma in patients aged ≥12 yearsApproval consideration: REMS
Fortesta (new formulation)(Testosterone; Endo Pharmaceuticals)Class: Steroid androgenIndication: Testosterone replacement therapy in males

for primary hypogonadism (congenital or acquired) and hypogonadotrophic hypogonadism (congenital or acquired) associated with a deficiency or absence of endogenous testosteroneApproval consideration: REMS
Herceptin (new indication)(Trastuzumab; Genentech)Class: HER2/neu receptor antagonistIndication: Treatment of HER2-overexpressing cancerand treatment of HER2-overexpressing metastatic gas-tric or gastroesophageal junction adenocarcinoma
H.P. Acthar Gel (new indication)(Repository corticotropin; Questcor Pharmaceuticals)Class: Adrenocorticotropic hormone analogIndication: Monotherapy for infantile spasms in personsaged <2 years; treatment of exacerbations of multiplesclerosis in adultsApproval consideration: REMS
Jalyn (new combination)(Dutasteride + tamsulosin hydrochloride;GlaxoSmithKline)Class: Type I and II 5-alpha-reductase inhibitor +alpha1A-blockerIndication: Treatment of symptomatic benign prostatichyperplasia in men with an enlarged prostate
Kombiglyze XR (new combination)(Metformin hydrochloride + saxagliptin; Bristol-MyersSquibb)Class: Dipeptidyl peptidase-4 inhibitor + biguanidecombinationIndication: Adjunct to diet and exercise to improveglycemic control in adults with type 2 diabetes mellituswhen treatment with both saxagliptin and metformin is appropriate
Lamictal XR (new formulation)(Lamotrigine; GlaxoSmithKline)Class: Phenyltriazine (antiepileptic drug)Indication: Adjunctive therapy for primary generalizedtonic-clonic seizures and partial-onset seizures with orwithout secondary generalization in patients aged ≥13 yearsApproval consideration: REMS
Lamivudine, Nevirapine, and Stavudine (new combination)(Lamivudine + stavudine; Hetero Drugs)Class: Nucleoside analog (reverse transcriptase inhibitor)Indication: HIV-1 infection
Lo Loestrin FE (new formulation)(Ethinyl estradiol + norethindrone acetate, ferrousfumarate; Warner Chilcott)
Class: Progestin + estrogen combination oral contraceptiveIndication: Use by women to prevent pregnancyApproval consideration: REMS
Lumigan (new indication)(Bimatoprost; Allergan)Class: Prostaglandin analogIndication: Reduction of elevated intraocular pressurein patients with open-angle glaucoma or ocular hypertension
Lyrica (new formulation)(Pregabalin; Pfizer)Class: Alpha2-delta ligandIndication: Neuropathic pain associated with diabeticperipheral neuropathy, postherpetic neuralgia, adjunc-tive therapy for adult patients with partial-onsetseizures, fibromyalgiaApproval consideration: REMS
Namenda XR (new formulation)(Memantine hydrochloride; Forest Laboratories)Class: N-methyl-D-aspartate receptor antagonistIndication: Treatment of moderate-to-severe dementia ofthe Alzheimer’s type
Norvir (new formulation)(Ritonavir; Abbott Laboratories)Class: HIV protease inhibitorIndication: Treatment of HIV-1 infection in combina-tion with other antiretroviral agents
Oleptro (new formulation)(Trazodone hydrochloride; Labopharm)Class: TriazolopyridineIndication: Major depressive disorderApproval consideration: REMS
Orabloc (new combination)(Articaine hydrochloride + epinephrine bitartrate;Pierrel)Class: Amide local anesthetic Indication: Local, infiltrative, or conductive anesthesiain simple and complex dental procedures
Oravig (new formulation)(Miconazole; Strativa Pharmaceuticals)Class: Azole antifungalIndication: Local treatment of oropharyngeal candidia-sis in adults
Ozurdex (new formulation)(Dexamethasone intravitreal implant; Allergan)Class: CorticosteroidIndication: Treatment of macular edema after branch
26 I AMERICAN HEALTH & DRUG BENEFITS I May/June 2011

27
2010 FDA Approvals of Brand-Name Drugs
May/June 2011 I www.AHDBonline.com I
retinal vein occlusion or central retinal vein occlusionand noninfectious uveitis affecting the posterior seg-ment of the eyeApproval consideration: Priority review
Pennsaid (new formulation)(Diclofenac sodium; Covidien)Class: Nonsteroidal anti-inflammatory drugIndication: Treatment of signs and symptoms ofosteoarthritis of the knee(s)
Silenor (new formulation)(Doxepin hydrochloride; Somaxon Pharmaceuticals)Class: H1 receptor antagonistIndication: Treatment of insomnia characterized by difficulties with sleep maintenanceApproval consideration: REMS
Sprix (new formulation)(Ketorolac tromethamine; Roxro Pharmaceuticals)Class: Nonsteroidal anti-inflammatory drugIndication: Short-term management (up to 5 days) of acute moderate-to-moderately severe pain
Staxyn (new formulation)(Vardenafil hydrochloride; Bayer HealthCare)Class: Phosphodiesterase type 5 inhibitor (cGMP-specific)Indication: Treatment of erectile dysfunction
Suboxone (new combination)(Buprenorphine + naloxone; Reckitt Benckiser)Class: Opioid (partial agonist-antagonist) + opioidantagonistIndication:Maintenance treatment of opioid dependenceApproval consideration: REMS
Suprep Bowel Prep Kit (new combination)(Magnesium sulfate anhydrous + potassium sulfate +sodium sulfate; Braintree Laboratories)Class: Osmotic bowel cleanserIndication: Cleansing of the colon in preparation forcolonoscopy in adultsApproval consideration: REMS
Tekamlo (new combination)(Aliskiren hemifumarate + amlodipine besylate;Novartis)Class: Renin inhibitor + dihydropyridine calcium channel blockerIndication: Hypertension
Tramadol hydrochloride (new formulation)(Tramadol hydrochloride; Cipher Pharmaceuticals)Class: Opioid agonist
Indication: Management of moderate-to-moderatelysevere chronic pain in adults who require around-the-clock treatment of their pain for an extended period of time
Trelstar (new formulation)(Triptorelin pamoate; Watson Laboratories)Class: Gonadotropin-releasing hormone agonist Indication: Palliative treatment of advanced prostatecancer
Tribenzor (new combination)(Amlodipine besylate + hydrochlorothiazide + olmesartan medoxomil; Daiichi Sankyo)Class: Angiotensin II receptor blocker + dihydropyridinecalcium channel blocker + thiazide diureticIndication: Treatment of hypertension; not indicated forinitial therapy
Vimovo (new combination)(Esomeprazole magnesium + naproxen; AstraZeneca)Class: Nonsteroidal anti-inflammatory drug + protonpump inhibitorIndication: Relief of signs and symptoms of osteoarthri-tis, rheumatoid arthritis, and ankylosing spondylitis andto decrease the risk of developing gastric ulcers inpatients at risk of developing nonsteroidal anti-inflam-matory drug–associated gastric ulcers
Vimpat (new formulation)(Lacosamide; UCB Pharma)Class: Sodium channel inactivatorIndication: Adjunctive therapy in the treatment of partial-onset seizures in patients with epilepsy aged ≥17 yearsApproval consideration: REMS
Vivitrol (new formulation)(Naltrexone; Alkermes)Class: Opioid antagonistIndication: Treatment of alcohol dependence in patientsable to abstain from alcohol in an outpatient settingbefore initiation of treatment and for the prevention ifrelapse to opioid dependence after opioid detoxificationApproval considerations: Priority review, REMS
Xifaxan (new indication)(Rifaximin; Salix Pharmaceuticals)Class: Rifamycin antibacterialIndication: Treatment of patients (aged ≥12 years) withtravelers’ diarrhea caused by noninvasive strains ofEscherichia coli and reduction in risk of overt hepaticencephalopathy recurrence in patients aged ≥18 yearsApproval consideration: Priority review

III. Newly Approved Old Drugs Previously Marketed without FDA Approval(Drugs were available before the establishment of the FDA)
Zuplenz (new formulation)(Ondansetron; Strativa Pharmaceuticals/Par Pharmaceutical Companies)Class: Selective 5-HT3 receptor antagonistIndication: Prevention of nausea and vomiting associat-ed with highly emetogenic cancer chemotherapy; pre-vention of nausea and vomiting associated with initialand repeat courses of moderately emetogenic cancerchemotherapy; prevention of nausea and vomitingassociated with radiotherapy in patients receiving totalbody irradiation, single high-dose fraction to abdomen,or daily fractions to the abdomen; prevention of postoperative nausea and/or vomiting
Zymaxid (new indication)(Gatifloxacin ophthalmic solution; Allergan)Class: Fluoroquinolone anti-infectiveIndication: Treatment of bacterial conjunctivitis caused by susceptible strains of Haemophilus influenzae,Staphylococcus aureus, S epidermidis, Streptococcus mitis group, Streptococcus oralis, and Streptococcus pneumoniae
BLA indicates biologic license application; NME, new molecular entity; REMS, risk evaluation and mitigation strategy.
I AMERICAN HEALTH & DRUG BENEFITS I May/June 201128
Oxycodone Hydrochloride(Oxycodone hydrochloride; Lehigh Valley Technologies)Class: Opioid agonistIndication: Management of moderate-to-severe acuteand chronic pain where the use of an opioid analgesic is appropriateApproval consideration: REMS
Pancreaze(Pancrelipase; Ortho-McNeil Janssen)Class: Digestive enzymesIndication: Treatment of exocrine pancreatic insuffi ciency resulting from cystic fibrosis or other conditions

© 2011 Novo Nordisk 0311-00001944-1 April 2011
Victoza® is a registered trademark and VictozaCare™ is a trademark of Novo Nordisk A/S.
To learn how Victoza® can help your patients manage their type 2 diabetes, visit VictozaPro.com/Benefi t.
For patients who need more than metformin
Help grab type 2 diabetes by the roots.Once-daily Victoza® provides powerful and sustained* reductions in A1C and:
Impacts beta-cell function to help regulate insulin secretion
May reduce weight – Victoza® is not indicated for the management of obesity – Weight change was a secondary end point in clinical trials
* Victoza® was evaluated in a 52-week monotherapy trial and in fi ve 26-week, add-on combination trials.
Indications and usageVictoza® is indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus. Because of the uncertain relevance of the rodent thyroid C-cell tumor fi ndings to humans, prescribe Victoza® only to patients for whom the potential benefi ts are considered to outweigh the potential risk. Victoza® is not recommended as fi rst-line therapy for patients who have inadequate glycemic control on diet and exercise. In clinical trials of Victoza®, there were more cases of pancreatitis with Victoza® than with comparators. Victoza® has not been studied suffi ciently in patients with a history of pancreatitis to determine whether these patients are at increased risk for pancreatitis while using Victoza®. Use with caution in patients with a history of pancreatitis. Victoza® is not a substitute for insulin. Victoza® should not be used in patients with type 1 diabetes mellitus or for the treatment of diabetic ketoacidosis, as it would not be effective in these settings. The concurrent use of Victoza® and insulin has not been studied.
Important safety informationLiraglutide causes dose-dependent and treatment-duration-dependent thyroid C-cell tumors at clinically relevant exposures in both genders of rats and mice. It is unknown whether Victoza® causes thyroid C-cell tumors, including medullary thyroid carcinoma (MTC), in humans, as human relevance could not be ruled out by clinical or nonclinical studies. Victoza® is contraindicated in patients with a personal or family history of MTC and in patients with Multiple Endocrine Neoplasia syndrome type 2 (MEN 2). Based on the fi ndings in rodents, monitoring with serum calcitonin or thyroid ultrasound was performed during clinical trials, but this may have increased the number of unnecessary thyroid surgeries. It is unknown whether monitoring with serum calcitonin or thyroid ultrasound will mitigate human risk of thyroid C-cell tumors. Patients should be counseled regarding the risk and symptoms of thyroid tumors.If pancreatitis is suspected, Victoza® should be discontinued. Victoza® should not be re-initiated if pancreatitis is confi rmed.When Victoza® is used with an insulin secretagogue (e.g. a sulfonylurea) serious hypoglycemia can occur. Consider lowering the dose of the insulin secretagogue to reduce the risk of hypoglycemia.There have been no studies establishing conclusive evidence of macrovascular risk reduction with Victoza® or any other antidiabetic drug. The most common adverse reactions, reported in ≥5% of patients treated with Victoza® and more commonly than in patients treated with placebo, are headache, nausea, diarrhea, and anti-liraglutide antibody formation. Immunogenicity-related events, including urticaria, were more common among Victoza®-treated patients (0.8%) than among comparator-treated patients (0.4%) in clinical trials. Victoza® has not been studied in type 2 diabetes patients below 18 years of age and is not recommended for use in pediatric patients. Victoza® should be used with caution in patients with renal impairment and in patients with hepatic impairment.
Please see brief summary of Prescribing Information on adjacent page.

Victoza® (liraglutide [rDNA origin] injection)Rx OnlyBRIEF SUMMARY. Please consult package insert for full prescribing information.
WARNING: RISK OF THYROID C-CELL TUMORS: Liraglutide causes dose-dependent and treatment-duration-dependent thyroid C-cell tumors at clinically relevant exposures in both genders of rats and mice. It is unknown whether Victoza® causes thyroid C-cell tumors, including medullary thyroid carcinoma (MTC), in humans, as human relevance could not be ruled out by clinical or nonclinical studies. Victoza® is contraindicated in patients with a per-sonal or family history of MTC and in patients with Multiple Endocrine Neoplasia syndrome type 2 (MEN 2). Based on the findings in rodents, monitoring with serum calcitonin or thyroid ultrasound was performed during clinical trials, but this may have increased the number of unnecessary thyroid surgeries. It is unknown whether monitoring with serum calcitonin or thyroid ultrasound will mitigate human risk of thyroid C-cell tumors. Patients should be coun-seled regarding the risk and symptoms of thyroid tumors [see Contraindications and Warnings and Precautions].
INDICATIONS AND USAGE: Victoza® is indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus. Important Limitations of Use: Because of the uncertain relevance of the rodent thyroid C-cell tumor findings to humans, pre-scribe Victoza® only to patients for whom the potential benefits are considered to outweigh the potential risk. Victoza® is not recommended as first-line therapy for patients who have inadequate glycemic control on diet and exercise. In clinical trials of Victoza®, there were more cases of pancreatitis with Victoza® than with comparators. Victoza® has not been studied sufficiently in patients with a history of pancreatitis to determine whether these patients are at increased risk for pancreatitis while using Victoza®. Use with caution in patients with a history of pancreatitis. Victoza® is not a substitute for insulin. Victoza® should not be used in patients with type 1 dia-betes mellitus or for the treatment of diabetic ketoacidosis, as it would not be effective in these settings. The concurrent use of Victoza® and insulin has not been studied.CONTRAINDICATIONS: Victoza® is contraindicated in patients with a personal or family history of medullary thyroid carcinoma (MTC) or in patients with Multiple Endocrine Neoplasia syn-drome type 2 (MEN 2).WARNINGS AND PRECAUTIONS: Risk of Thyroid C-cell Tumors: Liraglutide causes dose-dependent and treatment-duration-dependent thyroid C-cell tumors (adenomas and/or carcinomas) at clinically relevant exposures in both genders of rats and mice. Malignant thyroid C-cell carcinomas were detected in rats and mice. A statistically significant increase in cancer was observed in rats receiving liraglutide at 8-times clinical exposure compared to controls. It is unknown whether Victoza® will cause thyroid C-cell tumors, including medullary thyroid car-cinoma (MTC), in humans, as the human relevance of liraglutide-induced rodent thyroid C-cell tumors could not be determined by clinical or nonclinical studies [see Boxed Warning, Contra-indications]. In the clinical trials, there have been 4 reported cases of thyroid C-cell hyperplasia among Victoza®-treated patients and 1 case in a comparator-treated patient (1.3 vs. 0.6 cases per 1000 patient-years). One additional case of thyroid C-cell hyperplasia in a Victoza®-treated patient and 1 case of MTC in a comparator-treated patient have subsequently been reported. This comparator-treated patient with MTC had pre-treatment serum calcitonin concentrations >1000 ng/L suggesting pre-existing disease. All of these cases were diagnosed after thyroidectomy, which was prompted by abnormal results on routine, protocol-specified measurements of serum calcitonin. Four of the five liraglutide-treated patients had elevated calcitonin concentrations at baseline and throughout the trial. One liraglutide and one non-liraglutide-treated patient devel-oped elevated calcitonin concentrations while on treatment. Calcitonin, a biological marker of MTC, was measured throughout the clinical development program. The serum calcitonin assay used in the Victoza® clinical trials had a lower limit of quantification (LLOQ) of 0.7 ng/L and the upper limit of the reference range was 5.0 ng/L for women and 8.4 ng/L for men. At Weeks 26 and 52 in the clinical trials, adjusted mean serum calcitonin concentrations were higher in Victoza®-treated patients compared to placebo-treated patients but not compared to patients receiving active comparator. At these timepoints, the adjusted mean serum calcitonin values (~ 1.0 ng/L) were just above the LLOQ with between-group differences in adjusted mean serum calcitonin values of approximately 0.1 ng/L or less. Among patients with pre-treatment serum calcitonin below the upper limit of the reference range, shifts to above the upper limit of the reference range which persisted in subsequent measurements occurred most frequently among patients treated with Victoza® 1.8 mg/day. In trials with on-treatment serum calcitonin measurements out to 5-6 months, 1.9% of patients treated with Victoza® 1.8 mg/day developed new and persistent calci-tonin elevations above the upper limit of the reference range compared to 0.8-1.1% of patients treated with control medication or the 0.6 and 1.2 mg doses of Victoza®. In trials with on-treat-ment serum calcitonin measurements out to 12 months, 1.3% of patients treated with Victoza® 1.8 mg/day had new and persistent elevations of calcitonin from below or within the reference range to above the upper limit of the reference range, compared to 0.6%, 0% and 1.0% of patients treated with Victoza® 1.2 mg, placebo and active control, respectively. Otherwise, Victoza® did not produce consistent dose-dependent or time-dependent increases in serum calcitonin. Patients with MTC usually have calcitonin values >50 ng/L. In Victoza® clinical trials, among patients with pre-treatment serum calcitonin <50 ng/L, one Victoza®-treated patient and no comparator-treated patients developed serum calcitonin >50 ng/L. The Victoza®-treated patient who developed serum calcitonin >50 ng/L had an elevated pre-treatment serum calcitonin of 10.7 ng/L that increased to 30.7 ng/L at Week 12 and 53.5 ng/L at the end of the 6-month trial. Follow-up serum calci-tonin was 22.3 ng/L more than 2.5 years after the last dose of Victoza®. The largest increase in serum calcitonin in a comparator-treated patient was seen with glimepiride in a patient whose serum calcitonin increased from 19.3 ng/L at baseline to 44.8 ng/L at Week 65 and 38.1 ng/L at Week 104. Among patients who began with serum calcitonin <20 ng/L, calcitonin elevations to >20 ng/L occurred in 0.7% of Victoza®-treated patients, 0.3% of placebo-treated patients, and 0.5% of active-comparator-treated patients, with an incidence of 1.1% among patients treated with 1.8 mg/day of Victoza®. The clinical significance of these findings is unknown. Counsel patients regarding the risk for MTC and the symptoms of thyroid tumors (e.g. a mass in the neck, dysphagia, dyspnea or persistent hoarseness). It is unknown whether monitoring with serum calcitonin or thyroid ultrasound will mitigate the potential risk of MTC, and such monitoring may increase the risk of unnecessary procedures, due to low test specificity for serum calcitonin and a high background incidence of thyroid disease. Patients with thyroid nodules noted on physical examination or neck imaging obtained for other reasons should be referred to an endocrinolo-gist for further evaluation. Although routine monitoring of serum calcitonin is of uncertain value in patients treated with Victoza®, if serum calcitonin is measured and found to be elevated, the
patient should be referred to an endocrinologist for further evaluation. Pancreatitis: In clinical trials of Victoza®, there were 7 cases of pancreatitis among Victoza®-treated patients and 1 case among comparator-treated patients (2.2 vs. 0.6 cases per 1000 patient-years). Five cases with Victoza® were reported as acute pancreatitis and two cases with Victoza® were reported as chronic pancreatitis. In one case in a Victoza®-treated patient, pancreatitis, with necrosis, was observed and led to death; however clinical causality could not be established. One additional case of pan-creatitis has subsequently been reported in a Victoza®-treated patient. Some patients had other risk factors for pancreatitis, such as a history of cholelithiasis or alcohol abuse. There are no con-clusive data establishing a risk of pancreatitis with Victoza® treatment. After initiation of Victoza®, and after dose increases, observe patients carefully for signs and symptoms of pancreatitis (including persistent severe abdominal pain, sometimes radiating to the back and which may or may not be accompanied by vomiting). If pancreatitis is suspected, Victoza® and other potentially suspect medications should be discontinued promptly, confirmatory tests should be performed and appropriate management should be initiated. If pancreatitis is confirmed, Victoza® should not be restarted. Use with caution in patients with a history of pancreatitis. Use with Medications Known to Cause Hypoglycemia: Patients receiving Victoza® in combination with an insulin secretagogue (e.g., sulfonylurea) may have an increased risk of hypoglycemia. In the clinical trials of at least 26 weeks duration, hypoglycemia requiring the assistance of another person for treat-ment occurred in 7 Victoza®-treated patients and in two comparator-treated patients. Six of these 7 patients treated with Victoza® were also taking a sulfonylurea. The risk of hypoglycemia may be lowered by a reduction in the dose of sulfonylurea or other insulin secretagogues [see Adverse Reactions]. Macrovascular Outcomes: There have been no clinical studies establishing con-clusive evidence of macrovascular risk reduction with Victoza® or any other antidiabetic drug.ADVERSE REACTIONS: Clinical Trials Experience: Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice. The safety of Victoza® was evaluated in a 52-week monotherapy trial and in five 26-week, add-on combination therapy trials. In the monotherapy trial, patients were treated with Victoza® 1.2 mg daily, Victoza® 1.8 mg daily, or glimepiride 8 mg daily. In the add-on to metformin trial, patients were treated with Victoza® 0.6 mg, Victoza® 1.2 mg, Victoza® 1.8 mg, placebo, or glimepiride 4 mg. In the add-on to glimepiride trial, patients were treated with Victoza® 0.6 mg, Victoza® 1.2 mg, Victoza® 1.8 mg, placebo, or rosiglitazone 4 mg. In the add-on to metformin + glimepiride trial, patients were treated with Victoza® 1.8 mg, placebo, or insulin glargine. In the add-on to metformin + rosiglitazone trial, patients were treated with Victoza® 1.2 mg, Victoza® 1.8 mg or placebo. Withdrawals: The incidence of withdrawal due to adverse events was 7.8% for Victoza®-treated patients and 3.4% for comparator-treated patients in the five controlled trials of 26 weeks duration or longer. This difference was driven by withdrawals due to gastrointestinal adverse reactions, which occurred in 5.0% of Victoza®-treated patients and 0.5% of comparator-treated patients. The most common adverse reactions leading to withdrawal for Victoza®-treated patients were nausea (2.8% versus 0% for comparator) and vomiting (1.5% versus 0.1% for comparator). Withdrawal due to gastrointestinal adverse events mainly occurred during the first 2-3 months of the trials. Tables 1, 2 and 3 summarize the adverse events reported in 5% of Victoza®-treated patients in the six controlled trials of 26 weeks duration or longer.Table 1: Adverse events reported in 5% of Victoza®-treated patients or 5% of glimepiride-treated patients: 52-week monotherapy trial
All Victoza® N = 497 Glimepiride N = 248Adverse Event Term (%) (%)Nausea 28.4 8.5Diarrhea 17.1 8.9Vomiting 10.9 3.6Constipation 9.9 4.8Upper Respiratory Tract Infection 9.5 5.6Headache 9.1 9.3Influenza 7.4 3.6Urinary Tract Infection 6.0 4.0Dizziness 5.8 5.2Sinusitis 5.6 6.0Nasopharyngitis 5.2 5.2Back Pain 5.0 4.4Hypertension 3.0 6.0
Table 2: Adverse events reported in 5% of Victoza®-treated patients and occur-ring more frequently with Victoza® compared to placebo: 26-week combination therapy trials
Add-on to Metformin TrialAll Victoza® +
Metformin N = 724Placebo +
Metformin N = 121Glimepiride +
Metformin N = 242Adverse Event Term (%) (%) (%)Nausea 15.2 4.1 3.3Diarrhea 10.9 4.1 3.7Headache 9.0 6.6 9.5Vomiting 6.5 0.8 0.4
Add-on to Glimepiride TrialAll Victoza® +
Glimepiride N = 695Placebo +
Glimepiride N = 114Rosiglitazone +
Glimepiride N = 231Adverse Event Term (%) (%) (%)Nausea 7.5 1.8 2.6Diarrhea 7.2 1.8 2.2Constipation 5.3 0.9 1.7Dyspepsia 5.2 0.9 2.6

Add-on to Metformin + GlimepirideVictoza® 1.8 + Metformin +
Glimepiride N = 230
Placebo + Metformin + Glimepiride
N = 114
Glargine + Metformin + Glimepiride
N = 232Adverse Event Term (%) (%) (%)Nausea 13.9 3.5 1.3Diarrhea 10.0 5.3 1.3Headache 9.6 7.9 5.6Dyspepsia 6.5 0.9 1.7Vomiting 6.5 3.5 0.4
Add-on to Metformin + RosiglitazoneAll Victoza® + Metformin +
Rosiglitazone N = 355Placebo + Metformin
+ Rosiglitazone N = 175Adverse Event Term (%) (%)Nausea 34.6 8.6Diarrhea 14.1 6.3Vomiting 12.4 2.9Decreased Appetite 9.3 1.1Anorexia 9.0 0.0Headache 8.2 4.6Constipation 5.1 1.1Fatigue 5.1 1.7
Table 3: Treatment-Emergent Adverse Events in 26 Week Open-Label Trial versus Exenatide (Adverse events with frequency 5% and occurring more frequently with Victoza® compared to exenatide are listed)
Victoza® 1.8 mg once daily + metformin and/or
sulfonylurea N = 235
Exenatide 10 mcg twice daily + metformin and/or
sulfonylurea N = 232Preferred Term (%) (%)Diarrhea 12.3 12.1Dyspepsia 8.9 4.7Constipation 5.1 2.6
Gastrointestinal adverse events: In the five clinical trials of 26 weeks duration or longer, gastroin-testinal adverse events were reported in 41% of Victoza®-treated patients and were dose-related. Gastrointestinal adverse events occurred in 17% of comparator-treated patients. Events that occurred more commonly among Victoza®-treated patients included nausea, vomiting, diarrhea, dyspepsia and constipation. In a 26-week study of Victoza® versus exenatide, both in combination with metformin and/or sulfonylurea overall gastrointestinal adverse event incidence rates, includ-ing nausea, were similar in patients treated with Victoza® and exenatide. In five clinical trials of 26 weeks duration or longer, the percentage of patients who reported nausea declined over time. Approximately 13% of Victoza®-treated patients and 2% of comparator-treated patients reported nausea during the first 2 weeks of treatment. In a 26 week study of Victoza® versus exenatide, both in combination with metformin and/or sulfonylurea, the proportion of patients with nausea also declined over time. Immunogenicity: Consistent with the potentially immunogenic properties of protein and peptide pharmaceuticals, patients treated with Victoza® may develop anti-liraglutide antibodies. Approximately 50-70% of Victoza®-treated patients in the five clinical trials of 26 weeks duration or longer were tested for the presence of anti-liraglutide antibodies at the end of treatment. Low titers (concentrations not requiring dilution of serum) of anti-liraglutide antibodies were detected in 8.6% of these Victoza®-treated patients. Sampling was not performed uniformly across all patients in the clinical trials, and this may have resulted in an underestimate of the actual percentage of patients who developed antibodies. Cross-reacting anti-liraglutide antibod-ies to native glucagon-like peptide-1 (GLP-1) occurred in 6.9% of the Victoza®-treated patients in the 52-week monotherapy trial and in 4.8% of the Victoza®-treated patients in the 26-week add-on combination therapy trials. These cross-reacting antibodies were not tested for neutral-izing effect against native GLP-1, and thus the potential for clinically significant neutralization of native GLP-1 was not assessed. Antibodies that had a neutralizing effect on liraglutide in an in vitro assay occurred in 2.3% of the Victoza®-treated patients in the 52-week monotherapy trial and in 1.0% of the Victoza®-treated patients in the 26-week add-on combination therapy trials. Among Victoza®-treated patients who developed anti-liraglutide antibodies, the most common category of adverse events was that of infections, which occurred among 40% of these patients compared to 36%, 34% and 35% of antibody-negative Victoza®-treated, placebo-treated and active-control-treated patients, respectively. The specific infections which occurred with greater frequency among Victoza®-treated antibody-positive patients were primarily nonserious upper respiratory tract infections, which occurred among 11% of Victoza®-treated antibody-positive patients; and among 7%, 7% and 5% of antibody-negative Victoza®-treated, placebo-treated and active-control-treated patients, respectively. Among Victoza®-treated antibody-negative patients, the most common category of adverse events was that of gastrointestinal events, which occurred in 43%, 18% and 19% of antibody-negative Victoza®-treated, placebo-treated and active-con-trol-treated patients, respectively. Antibody formation was not associated with reduced efficacy of Victoza® when comparing mean HbA1c of all antibody-positive and all antibody-negative patients. However, the 3 patients with the highest titers of anti-liraglutide antibodies had no reduction in HbA1c with Victoza® treatment. In clinical trials of Victoza®, events from a composite of adverse events potentially related to immunogenicity (e.g. urticaria, angioedema) occurred among 0.8% of Victoza®-treated patients and among 0.4% of comparator-treated patients. Urticaria accounted for approximately one-half of the events in this composite for Victoza®-treated patients. Patients who developed anti-liraglutide antibodies were not more likely to develop events from the immu-nogenicity events composite than were patients who did not develop anti-liraglutide antibodies. Injection site reactions: Injection site reactions (e.g., injection site rash, erythema) were reported in approximately 2% of Victoza®-treated patients in the five clinical trials of at least 26 weeks duration. Less than 0.2% of Victoza®-treated patients discontinued due to injection site reactions.
Papillary thyroid carcinoma: In clinical trials of Victoza®, there were 6 reported cases of papillary thyroid carcinoma in patients treated with Victoza® and 1 case in a comparator-treated patient (1.9 vs. 0.6 cases per 1000 patient-years). Most of these papillary thyroid carcinomas were <1 cm in greatest diameter and were diagnosed in surgical pathology specimens after thyroidectomy prompted by findings on protocol-specified screening with serum calcitonin or thyroid ultra-sound. Hypoglycemia: In the clinical trials of at least 26 weeks duration, hypoglycemia requiring the assistance of another person for treatment occurred in 7 Victoza®-treated patients (2.6 cases per 1000 patient-years) and in two comparator-treated patients. Six of these 7 patients treated with Victoza® were also taking a sulfonylurea. One other patient was taking Victoza® in combina-tion with metformin but had another likely explanation for the hypoglycemia (this event occurred during hospitalization and after insulin infusion) (Table 4). Two additional cases of hypoglyce-mia requiring the assistance of another person for treatment have subsequently been reported in patients who were not taking a concomitant sulfonylurea. Both patients were receiving Victoza®, one as monotherapy and the other in combination with metformin. Both patients had another likely explanation for the hypoglycemia (one received insulin during a frequently-sampled intra-venous glucose tolerance test, and the other had intracranial hemorrhage and uncertain food intake).Table 4: Incidence (%) and Rate (episodes/patient year) of Hypoglycemia in the 52-Week Monotherapy Trial and in the 26-Week Combination Therapy Trials
Victoza® Treatment
Active Comparator
Placebo Comparator
Monotherapy Victoza®
(N = 497)Glimepiride
(N = 248)None
Patient not able to self−treat 0 0 —Patient able to self−treat 9.7 (0.24) 25.0 (1.66) —Not classified 1.2 (0.03) 2.4 (0.04) —Add-on to Metformin
Victoza® + Metformin (N = 724)
Glimepiride + Metformin (N = 242)
Placebo + Metformin (N = 121)
Patient not able to self−treat 0.1 (0.001) 0 0Patient able to self−treat 3.6 (0.05) 22.3 (0.87) 2.5 (0.06)Add-on to Glimepiride Victoza® +
Glimepiride (N = 695)
Rosiglitazone + Glimepiride
(N = 231)
Placebo + Glimepiride
(N = 114)Patient not able to self−treat 0.1 (0.003) 0 0Patient able to self−treat 7.5 (0.38) 4.3 (0.12) 2.6 (0.17)Not classified 0.9 (0.05) 0.9 (0.02) 0Add-on to Metformin + Rosiglitazone
Victoza® + Metformin +
Rosiglitazone (N = 355)
None
Placebo + Metformin +
Rosiglitazone (N = 175)
Patient not able to self−treat 0 — 0Patient able to self−treat 7.9 (0.49) — 4.6 (0.15)Not classified 0.6 (0.01) — 1.1 (0.03)Add-on to Metformin + Glimepiride
Victoza® + Metformin + Glimepiride
(N = 230)
Insulin glargine + Metformin + Glimepiride
(N = 232)
Placebo + Metformin + Glimepiride
(N = 114)Patient not able to self−treat 2.2 (0.06) 0 0Patient able to self−treat 27.4 (1.16) 28.9 (1.29) 16.7 (0.95)Not classified 0 1.7 (0.04) 0
In a pooled analysis of clinical trials, the incidence rate (per 1,000 patient-years) for malig-nant neoplasms (based on investigator-reported events, medical history, pathology reports, and surgical reports from both blinded and open-label study periods) was 10.9 for Victoza®, 6.3 for placebo, and 7.2 for active comparator. After excluding papillary thyroid carcinoma events [see Adverse Reactions], no particular cancer cell type predominated. Seven malignant neoplasm events were reported beyond 1 year of exposure to study medication, six events among Victoza®-treated patients (4 colon, 1 prostate and 1 nasopharyngeal), no events with placebo and one event with active comparator (colon). Causality has not been established. Laboratory Tests: In the five clinical trials of at least 26 weeks duration, mildly elevated serum bilirubin concentra-tions (elevations to no more than twice the upper limit of the reference range) occurred in 4.0% of Victoza®-treated patients, 2.1% of placebo-treated patients and 3.5% of active-comparator-treated patients. This finding was not accompanied by abnormalities in other liver tests. The significance of this isolated finding is unknown.OVERDOSAGE: In a clinical trial, one patient with type 2 diabetes experienced a single overdose of Victoza® 17.4 mg subcutaneous (10 times the maximum recommended dose). Effects of the overdose included severe nausea and vomiting requiring hospitalization. No hypoglycemia was reported. The patient recovered without complications. In the event of overdosage, appropriate supportive treatment should be initiated according to the patient’s clinical signs and symptoms.More detailed information is available upon request. For information about Victoza® contact: Novo Nordisk Inc., 100 College Road West, Princeton, New Jersey 08540, 1−877-484-2869Date of Issue: December 22, 2010 Version: 2Manufactured by: Novo Nordisk A/S, DK-2880 Bagsvaerd, DenmarkVictoza® is a registered trademark of Novo Nordisk A/S. Victoza® is covered by US Patent Nos. 6,268,343; 6,458,924; and 7,235,627 and other patents pending. Victoza® Pen is covered by US Patent Nos. 6,004,297; 6,235,004; 6,582,404 and other patents pending.© 2011 Novo Nordisk 140586-R2 2/2011

32 AMERICAN HEALTH & DRUG BENEFITS May/June 2011
Attention-deficit/hyperactivity disorder (ADHD)is characterized by a persistent pattern of inat-tention, impulsivity, and hyperactivity that is
more frequently displayed and severe than is typicallyobserved in peers. This disorder causes impairment ofsocial, work, and academic functioning (eg, poor aca-demic performance and increased use of school-basedresources)1,2 and increases healthcare utilization, withannual costs associated with ADHD in children andadolescents in the United States estimated at more than$42 billion.3 The worldwide prevalence of ADHD inchildren and adolescents is estimated to be 5.3%.4 In theUnited States, approximately 2.4 million children(8.7%) aged 8 to 15 years are believed to have ADHD.5,6The standard of care for patients with uncomplicatedADHD is psychostimulant monotherapy.7-9 However,some patients (up to 35%) have an inadequate clinicalresponse to stimulants due to intolerable adverse eventsor suboptimal symptom relief.8,10 Thus, there is an unmetneed for effective, well-tolerated therapies for the treat-ment of ADHD.
Clonidine is an alpha2-adrenergic agonist, and imme-diate-release (IR) clonidine has been used off-label forthe treatment of patients with ADHD either as amonotherapy or as part of a multimodal regimen withpsychostimulants.11-14 However, IR clonidine is associat-ed with widely fluctuating plasma drug concentrationsand requires frequent dosing.15 These rapid fluctuationsin the plasma concentration of clonidine may be associ-ated with peak-and-trough–related side effects (eg, seda-tion, dry mouth),16 which can limit the therapeutic use-fulness of IR clonidine. Furthermore, because IRclonidine is not indicated for the treatment of ADHD, itdoes not have labeling to provide dosing, safety, or otherinstructions and precautions for its use for ADHD.
KAPVAY™ (clonidine hydrochloride extended-releasetablets; Shionogi Inc, Florham Park, NJ) was approvedby the US Food and Drug Administration in September2010 for the treatment of ADHD as monotherapy or asadjunctive therapy to stimulant medications.17 KAPVAYis an extended-release formulation of clonidine that hasa different pharmacokinetic profile than IR clonidine.
The KAPVAY clinical development program includedthree phase 3 trials consisting of 2 randomized, multi-center, double-blind, placebo-controlled trials and anopen-label, 12-month safety trial.
Clinical PharmacologyThe novel formulation of KAPVAY provides a dis-
tinctly different pharmacokinetic profile compared withIR clonidine. A direct comparison of the pharmacoki-netic profiles of KAPVAY and a marketed IR clonidineformulation (Catapres®; Boehringer Ingelheim Phar ma -ceuticals, Inc, Ridgefield, CT) in healthy adult volun-teers (N = 15) demonstrated that KAPVAY providedextended release of clonidine.18 In this single-dosecrossover study, peak plasma concentration (Cmax) withKAPVAY was reduced by approximately 50% (235pg/mL vs 443 pg/mL for IR clonidine), and the meantime to reach the Cmax (Tmax) was extended by ~5 hourscompared with IR clonidine (mean peak absorptionwithin 7 hours for KAPVAY vs 2 hours for IR clonidine).Thus, KAPVAY minimizes the peak-and-trough plasmaconcentrations observed with IR clonidine. The half-lifeof KAPVAY was similar to that of IR clonidine, and nofood effects were observed. This pharmacokinetic profilesuggested that this extended-release formulation of cloni-dine might overcome some of the limitations of IR cloni-dine. Three randomized-controlled phase 3 trials weresubsequently initiated to evaluate the efficacy and safetyof KAPVAY as a monotherapy for children and adoles-cents with ADHD and as adjunctive therapy for patientswith an inadequate response to a stimulant regimen.
Monotherapy with KAPVAYThe efficacy and safety of KAPVAY as a fixed-dose
monotherapy for the treatment of ADHD was evaluatedin a phase 3, multicenter, 8-week, randomized, double-blind, placebo-controlled trial (NCT00556959; Table).Patients aged 6 to 17 years who were diagnosed witheither hyperactive- or combined-subtype ADHD accord-ing to DSM-IV-TR criteria (N = 236) were randomizedto receive monotherapy with KAPVAY 0.1 mg twicedaily (0.2 mg/d), 0.2 mg twice daily (0.4 mg/d), or a
KAPVAY™: Clonidine Hydrochloride Extended-Release Tablets for the Treatmentof Attention-Deficit/Hyperactivity DisorderTodd Parker, PhD, and Jillian Gee, PhD, MedThink SciCom, Inc

33May/June 2011 www.AHDBonline.com
Table Phase 3 Clinical Trials Evaluating KAPVAY for the Treatment of Attention-Deficit/Hyperactivity Disorder
KAPVAY Fixed-Dose Monotherapy for ADHD15,18-21
Study design Population Groups End points Key results
Phase 3, randomized, 8-week, multicenter,double-blindescalation study
Children and adolescents (aged6-17 years) withhyperactive- or combined inattentive/hyperactive–subtype ADHD
Placebo (n = 76)
KAPVAY• 0.2 mg/d (n = 74) • 0.4 mg/d (n = 78)
Primary• Reduction inADHD-RS-IVtotal score withKAPVAY vsplacebo
Secondary• ADHD-RS-IVsubscales
• CPRS • CGI • PGA • HADS • Safety
Efficacy at week 5 (KAPVAY vs placebo)• Significant improvement inADHD-RS-IV scores (P <0.0001 for both groups)
• Significant improvement inADHD-RS-IV inattentionand hyperactivity/impulsivitysubscale scores (P <0.002 forboth groups)
• Significant improvement inCPRS, CGI, HADS, and PGA
Safety• Most common TEAEs weremild-to-moderate somno-lence and fatigue
• No discernible significant cardiac concerns
KAPVAY Flexible-Dose Combination Therapy for ADHD15,20-22
Study design Population Groups End points Key results
Phase 3, randomized, 8-week, multicenter,double-blind,dose-escalationstudy
Children and adolescents (aged6-17 years) withhyperactive- orcombined inattentive/hyperactive–subtype ADHDwith inadequatecontrol of symp-toms with a sta-ble stimulantregimen
Placebo + stimulants(n = 96)a
KAPVAY + stimu-lants (n = 102)
Primary• Reduction inADHD-RS-IVtotal score withKAPVAY + stimulants vsplacebo + stimulants
Secondary• ADHD-RS-IVsubscales
• CPRS • CGI • PGA • HADS • Safety
Efficacy at week 5 (KAPVAY +stimulants vs placebo + stimulants)• Significant improvement inADHD-RS-IV scores (P <0.0091)
• Significant improvement inADHD-RS-IV subscalescores, CPRS, CGI, andPGA
Safety• Most common TEAEs wereheadache, somnolence, andfatigue
• Parameters were not affectedby psychostimulant class used
• No discernible significant cardiac concerns
aOne patient in this group for whom there were no postbaseline assessments was not included in the intention-to-treat population.ADHD indicates attention-deficit/hyperactivity disorder; ADHD-RS-IV, ADHD Rating Scale-IV; CGI, ClinicalGlobal Impression; CPRS, Conners’ Parent Rating Scale; HADS, Horacek Adrenergic Dysregulation Scale; PGA,Parent Global Assessment; TEAEs, treatment-emergent adverse events.

34 AMERICAN HEALTH & DRUG BENEFITS May/June 2011
placebo.19 Doses of KAPVAY were initiated at 0.1 mg/dduring the first week and escalated by 0.1 mg/d each weekuntil patients reached their assigned dose during the ini-tial 3 weeks. The target dose was maintained for at least 2weeks through week 5, and during the last 3 weeks (ie,between weeks 5 and 8), the dose was tapered by 0.1mg/d/week to 0.1 mg/d by week 8. Patients who could nottolerate their assigned doses were discontinued from thestudy. The primary efficacy end point was mean change inADHD Rating Scale-IV (ADHD-RS-IV) total scorefrom baseline to week 5; the last observation carried for-ward method was used for patients who discontinuedearlier. Safety data collected from the safety population(ie, patients who received at least 1 dose; n = 230)included adverse events, vital signs, laboratory values,and electrocardiograms (ECGs).18
Baseline demographic characteristics were balancedbetween treatment groups.18 The mean improvement inADHD-RS-IV total score from baseline to week 5 (ie,primary end point) was significantly greater in theKAPVAY 0.2-mg/d (–15.6) and 0.4-mg/d (–16.5)groups than in the placebo group (–7.5; P <0.0001 forboth active treatment groups).19 Moreover, a statistical-ly significant improvement in ADHD-RS-IV total scorewas observed by week 1 in the KAPVAY 0.2-mg/d groupand week 2 in the KAPVAY 0.4-mg/d group comparedwith the placebo group. A post hoc analysis determinedthat the effect sizes were 0.713 (95% confidence inter-val [CI], 0.38 to 1.04) and 0.766 (95% CI, 0.44 to 1.09)for the KAPVAY 0.2-mg/d and 0.4-mg/d groups, respec-tively. In addition to improvement in ADHD-RS-IVtotal score, significant improvements in the KAPVAYgroups were observed for secondary end points, such asthe ADHD-RS-IV hyperactivity- and inattention-subscale scores, Conners’ Parent Rating Scale (CPRS),Clinical Global Impression of Improvement (CGI-I),Clinical Global Impression of Severity (CGI-S), andParent Global Assessment (PGA).
The most common treatment-emergent adverseevents (TEAEs) associated with KAPVAY were somno-lence and fatigue, which were also the most commonTEAEs that led to discontinuation.19 Fourteen of 230patients (6%) experienced a severe TEAE. No seriousadverse events were reported. There were no clinicallymeaningful changes in chemistry or laboratory tests,and mean decreases in blood pressure and pulse ratewere not clinically significant. Mean systolic and dias-tolic blood pressure were increased in the placebo group(1.1 and 0.3 mm Hg, respectively), unchanged orincreased in the KAPVAY 0.2-mg/d group (0 and 0.6mm Hg), and decreased in the KAPVAY 0.4-mg/d group(–5.8 and –4.6 mm Hg).20 Heart rate was increased in
the placebo group (0.9 bpm), unchanged in the KAP-VAY 0.2-mg/d group, and decreased in the KAPVAY0.4-mg/d group (–4.3 bpm). Furthermore, there wereno discontinuations due to bradycardia, and no patientshad a QTc ≥500 msec.18-20 This study demonstrated thatKAPVAY monotherapy significantly reduced symptomsof ADHD and was generally well tolerated.
Combination Therapy with KAPVAYIn addition to the fixed-dose monotherapy trial, the
efficacy and safety of KAPVAY as an adjunct to stimu-lant therapy was examined in a phase 3, 8-week, multi-center, randomized, double-blind, placebo-controlledtrial (NCT00641329; Table). Patients aged 6-17 yearswith ADHD (N = 198) who had an inadequateresponse to stimulants after at least 4 weeks on a stablestimulant regimen were randomized to receive eitherKAPVAY (0.1-0.4 mg/d; doses >0.1 mg/d administeredas 2 doses) added on to baseline stimulant medicationor placebo plus baseline stimulant treatment.15,21,22 BothKAPVAY and stimulant doses were allowed to beadjusted to achieve an optimal efficacy and safety pro-file.22 As in the monotherapy trial, the primary efficacyend point was mean change in ADHD-RS-IV totalscore from baseline to week 5. Secondary end pointsincluded ADHD-RS-IV subscale scores, PGA, CGI-I,CGI-S, CPRS, and safety. In addition, a post hocanalysis of effect size was performed.
A total of 198 patients were randomized to receiveeither KAPVAY plus stimulants (n = 102) or placeboplus stimulants (n = 96).22 Demographics were similarbetween groups, and there were no differences in theuse of concomitant medications (ie, stimulants or otherapproved non-ADHD medications). Statistically sig-nificant improvements in ADHD-RS-IV total score forthe KAPVAY plus stimulants group were observedstarting at week 2 and continuing through week 7. Atweek 5, the mean improvement in ADHD-RS-IV totalscore (ie, primary end point) was significantly greaterin the KAPVAY plus stimulants group (–15.7) than inthe placebo plus stimulants group (–11.5; P = 0.009).15Significant improvements in ADHD subscale scores(ie, inattention and hyperactivity), CPRS, CGI-I,CGI-S, and PGA were also observed.22 Furthermore,when effect size with respect to ADHD-RS-IV totalscore was examined post hoc, the overall effect size forKAPVAY plus stimulants versus placebo plus stimu-lants was 0.34. These data demonstrate that KAPVAYprovides an incremental benefit as an add-on therapyfor those patients who have an inadequate response tostimulants alone.
The rates of TEAEs were similar between groups

(45% for KAPVAY plus stimulants and 41% for place-bo plus stimulants).22 Common TEAEs occurring in≥5% of patients in the KAPVAY plus stimulants groupwere headache, somnolence, fatigue, upper abdominalpain, nasal congestion, cough, irritability, pharyngola -r yngeal pain, insomnia, increased body temperature,and dizziness. A total of 5 patients discontinuedbecause of a TEAE (4 in the placebo plus stimulantsgroup and 1 in the KAPVAY plus stimulants group).Changes in ECGs were not considered clinically signif-icant. Mean ECG changes were generally mild, with nosignificant changes to suggest a drug-related effect, andthere were no incidents of symptomatic bradycardia ordifferences between groups in numbers of patients withan increase in QTc interval. These findings show thatthe addition of KAPVAY for 8 weeks to a stimulantregimen that did not achieve an adequate response wasgenerally well tolerated and resulted in statistically sig-nificant ADHD symptom improvements.
12-Month Safety StudyAs part of the KAPVAY clinical development pro-
gram, a prospectively designed open-label, 12-monthextension study was conducted to examine the safety ofa flexible dosing regimen of twice-daily KAPVAY (0.1-0.4 mg/d) when administered chronically under regularclinical conditions either as monotherapy or in combi-nation with other ADHD medication(s). In this trial,patients who completed the monotherapy or combina-tion-therapy trial or who discontinued for a reasonother than safety were eligible to participate(NCT00723190).15 Patients (N = 303) received flexi-ble dosing of KAPVAY (0.1-0.4 mg/d; doses >0.1 mg/dadministered twice daily) as monotherapy or in combi-nation with other ADHD medication(s), and doses ofKAPVAY and other medications could have beentitrated to optimize safety while maximizing efficacy.15,20
In addition to adverse events, data on vital signs andECGs were obtained.20 At the time of a planned inter-im analysis (June 2, 2009), 14 of 215 patients (7%) hadTEAEs leading to discontinuation.15 Somnolence(27%) and fatigue (11%) were among the most com-mon TEAEs reported; the majority (96%) of TEAEswere mild to moderate. An in-depth analysis of cardiacdata at the conclusion of the study indicated that,although 1 patient discontinued because of dizziness,there were no discontinuations due to bradycardia, andfew patients had a prolonged QTc interval (≥450msec).20 This study demonstrated that administrationof KAPVAY (up to 0.4 mg/d) with or without otherADHD medications was well tolerated for up to 1 yearwith no significant cardiac TEAEs observed.
ConclusionsIn much the same way that the extended-release for-
mulations of stimulants evolved, alpha2-adrenergic ago-nist formulations have evolved to address the limitationsof currently marketed IR formulations. KAPVAY is anovel, extended-release oral formulation of clonidinethat, when administered as directed, results in reducedpeak plasma concentrations and extends the time neces-sary to reach the maximum plasma concentration com-pared with marketed IR clonidine. Because of differencesin its pharmacokinetic profile, KAPVAY cannot be usedinterchangeably with the IR formulations of clonidine(ie, patients should be tapered off of IR clonidine beforereceiving KAPVAY).
Results from three phase 3 trials demonstrated thattwice-daily treatment with KAPVAY (up to 0.4 mg/d)provides significant improvements in the core symptomsof ADHD for patients aged 6 to 17 years with ADHDwhen administered as monotherapy or as adjunctivetherapy to those patients with an inadequate responseto a stimulant regimen. The efficacy observed withKAPVAY was both statistically significant and clinical-ly meaningful. Furthermore, the effect size of KAPVAYwhen administered as a monotherapy (ie, 0.713 and0.766 for the 0.2-mg/d and 0.4-mg/d groups, respective-ly) is comparable to that observed with other nonstim-ulants for ADHD (eg, atomoxetine, guanfacine extend-ed release).19 In addition, KAPVAY (up to 0.4 mg/d) wasgenerally safe and well tolerated for up to 12 months ofchronic use. As such, KAPVAY is an important newtherapeutic option for the treatment of children andadolescents with ADHD alone or as part of a multi-modal regimen in patients experiencing an inadequateresponse to stimulants. ■
Disclosure StatementMedical writing and editorial services were provided by
MedThink SciCom, Inc, Raleigh, NC, with funding fromShionogi Inc, Florham Park, NJ.
References1. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Dis -orders, 4th ed. Text Revision. Washington, DC: American Psychiatric Association; 2000.2. Loe IM, Feldman HM. Academic and educational outcomes of children with ADHD.J Pediatr Psychol. 2007;32(6):643-654.3. Pelham WE, Foster EM, Robb JA. The economic impact of attention-deficit/hyperactivity disorder in children and adolescents. J Pediatr Psychol. 2007;32(6):711-727.4. Polanczyk G, de Lima MS, Horta BL, et al. The worldwide prevalence of ADHD: asystematic review and metaregression analysis. Am J Psychiatry. 2007;164(6):942-948.5. Froehlich TE, Lanphear BP, Epstein JN, et al. Prevalence, recognition, and treat-ment of attention-deficit/hyperactivity disorder in a national sample of US children.Arch Pediatr Adolesc Med. 2007;161(9):857-864.6. Merikangas KR, He JP, Brody D, et al. Prevalence and treatment of mental disor-ders among US children in the 2001-2004 NHANES. Pediatrics. 2010;125(1):75-81.7. Findling RL. Evolution of the treatment of attention-deficit/hyperactivity disorder
35May/June 2011 www.AHDBonline.com

36 AMERICAN HEALTH & DRUG BENEFITS May/June 2011
in children: a review. Clin Ther. 2008;30(5):942-957.8. Pliszka S, and the AACAP Work Group on Quality Issues. Practice parameter forthe assessment and treatment of children and adolescents with attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2007;46(7):894-921.9. Clinical practice guideline: treatment of the school-aged child with attention-deficit/hyperactivity disorder. Pediatrics. 2001;108(4):1033-1044.10. Barbaresi WJ, Katusic SK, Colligan RC, et al. Long-term stimulant medicationtreatment of attention-deficit/hyperactivity disorder: results from a population-basedstudy. J Dev Behav Pediatr. 2006;27(1):1-10.11. Hazell PL, Stuart JE. A randomized controlled trial of clonidine added to psy-chostimulant medication for hyperactive and aggressive children. J Am Acad ChildAdolesc Psychiatry. 2003;42(8):886-894.12. Hunt RD, Minderaa RB, Cohen DJ. Clonidine benefits children with attentiondeficit disorder and hyperactivity: report of a double-blind placebo-crossover thera-peutic trial. J Am Acad Child Psychiatry. 1985;24(5):617-629.13. Schvehla TJ, Mandoki MW, Sumner GS. Clonidine therapy for comorbid atten-tion deficit hyperactivity disorder and conduct disorder: preliminary findings in achildren’s inpatient unit. South Med J. 1994;87(7):692-695.14. Efron D, Hiscock H, Sewell JR, et al. Prescribing of psychotropic medications forchildren by Australian pediatricians and child psychiatrists. Pediatrics. 2003;111(2):372-375.15. Kollins S, Findling RL, Wigal SB, et al. Modified-release clonidine for the treat-ment of children/adolescents with ADHD. Poster presented at 56th Annual Meetingof the American Academy of Child and Adolescent Psychiatry. October 27-November 1, 2009; Honolulu, HI.
16. Keränen A, Nykänen S, Taskinen J. Pharmacokinetics and side-effects of cloni-dine. Eur J Clin Pharmacol. 1978;13(2):97-101.17. KAPVAY [package insert]. Florham Park, NJ: Shionogi Inc; 2010.18. Jain R, Kollins S, Bailey C, et al. Developing a sustained release formulation ofclonidine for the treatment of children and adolescents with attention deficit hyper-activity disorder (ADHD). Poster presented at 47th Annual Meeting of the AmericanCollege of Neuropsychopharmacology; December 7-11, 2008; Scottsdale, AZ.19. Jain R, Segal S, Kollins SH, Khayrallah M. Clonidine extended-release tabletsfor pediatric patients with attention-deficit/hyperactivity disorder. J Am Acad ChildAdolesc Psychiatry. 2011;50(2):171-179.20. Jain R, Findling R, Kollins SH, Khayrallah M. Cardiac data from the use of cloni-dine hydrochloride extended-release tablets in children and adolescents with atten-tion-deficit/hyperactivity disorder. Poster presented at 57th Annual Meeting of theAmerican Academy of Child and Adolescent Psychiatry. October 26-31, 2010; NewYork, NY.21. Findling R, Jain R, Kollins SH, Khayrallah M. Safety and efficacy of clonidinehydrochloride extended-release tablets by weight-adjusted dose in children and ado-lescents with attention-deficit/hyperactivity disorder. Poster presented at 57thAnnual Meeting of the American Academy of Child and Adolescent Psychiatry.October 26-31, 2010; New York, NY.22. Forman N, Yoon M, Wang C, Cavanaugh P. An extended-release formulation ofclonidine for the treatment of ADHD in children and adolescents with suboptimalresponse to stimulants. Poster presented at 22nd Annual International Conferenceon ADHD. November 11-13, 2010; Atlanta, GA.
Alpha2-Adrenergic Agonists in Combinationwith Psychostimulants a Potential NewApproach to ADHD TreatmentBy Floyd R. Sallee, MD, PhDProfessor of Psychiatry, University of Cincinnati, OH
Attention-deficit/hyperactivity disorder (ADHD) isa common psychiatric disorder that is recognizedworldwide.1 The prevalence of ADHD in chil-
dren has been estimated to be as high as 12%, makingit one of the most common psychiatric dis orders, afflict-ing 4.5 million children in the United States alone.2The consequences of ADHD can be devastating overthe lifespan of the individual, impacting academic- andwork-related performance, as well as family and peerrelations, and is also associated with high healthcareutilization.3-5 The presentation of ADHD can involveaspects of temperament (ie, impulsivity), motivation(ie, reward), behavior (ie, hyperactivity), and cognition(ie, inattention, working memory).6-8 The serious andimpairing nature of ADHD coupled with its range ofmultidimensional symptoms, presents a therapeuticchallenge for pharmacologic management.
The psychostimulant drug class has long been con-sidered first-line pharmacotherapy for ADHD,9 offeringsubstantial efficacy for most patients; however, weknow that at least 25% to 35% of those afflicted withADHD will not respond adequately to psychostimulanttherapy.10,11 Hence, the need for an expansion of thekinds of treatments, both pharmaceutical and behav-ioral, that address the various symptom domains inher-ent in the disorder.
Lack of benefit from psychostimulants can encompassinadequate symptom relief, intolerable side effects, ornonadherence.4,12 This coupled with the fact that psy-chostimulants are contraindicated in many patients,including those with a history of drug abuse or sympto-matic cardiovascular disease, makes the case for nonpsy-chostimulant options. Lingering concerns regarding theabuse, misuse, and diversion of the psychostimulant drug
STAKEHOLDER PERSPECTIVE

class have also contributed to the search for therapeuticalternatives such as the alpha2-adrenergic agonists.13,14
Prior to the systematic evaluation of alpha2-adrenergicagonists as possible treatments for ADHD, only atomox-etine was available as a nonpsychostimulant alternativefor pharmacologic management. Although atomoxetinewas a definite advance, offering long-duration symptomcoverage and documented im provement in health-relatedquality of life, its drawbacks include significant lag timefor therapeutic onset measured in weeks, and failure tomeet or beat the efficacy associated with osmoticallyreleased methylphenidate or extended-release mixedampheta mine salts in direct comparator trials.15 Still, thesearch for nonstimulant ADHD treatment continuesdespite the lack of success demonstrated by some thera -peutic drug class candidates, such as the novel neuronalnicotinic receptor partial agonist ABT-089.16
It is all the more remarkable that alpha2-adrenergicagonists, which had been used off-label to treat ADHDfor more than two decades, should now be reformulat-ed to take advantage of modern drug delivery technol-ogy, and thereby overcome limitations of rapid absorp-tion and clearance as well as tolerability problemsassociated with their pharmacometric properties.17 Newcontrolled release alpha2-adrenergic agonists havemore gradual absorption profiles over older immediate-release formulations, which has served to improve tol-erability and safety for children and adolescents.18 Atthe same time, the extended-release aspect of thesenewer agents has overcome the need for multiple dailydosing when treating ADHD with immediate-releaseclonidine19 or guanfacine.
Recent large-scale randomized controlled trials havefound clonidine extended-release, as well as guanfacineextended-release to be safe, tolerable, and effective forthe treatment of ADHD in children and adolescents.17Combination psychostimulant add-on studies have alsobeen completed with these new reformulations (ie,guanfacine extended-release and clonidine extended-release) to demonstrate that the addition of alpha2-adrenergic agonist to psychostimulant therapy can signif-icantly enhance efficacy, without compromising safety.
Alpha2-adrenergic agonists offer the possibility of anew approach to ADHD treatment inherent in combi-nation therapy with psychostimulants, to either reducethe side effect burden of the latter (ie, heart rate andblood pressure elevations) or to potentially work in anadditive fashion to enhance therapeutic effectiveness.17
This combination therapy approach comes at a time
when the field is recognizing the importance of maxi-mal ADHD symptom reduction, if not remission ofsymptoms, as a goal in ADHD treatment.20,21 What con-stitutes remission is controversial, but the preponder-ance of evidence would suggest that decreasing symp-tom burden is associated with improved functioning.20Arguably, combination therapy of alpha2-adrenergicagonist added to a psychostimulant could be an effec-tive strategy for many partial responders and mayachieve the optimal functioning implicit in the con-cept of “remission” for ADHD patients generally.20 ■
References1. Faraone SV, Sergeant J, Gillberg C, Biederman J. The worldwide prevalence ofADHD: is it an American condition? World Psychiatry. 2003;2:104-113.2. Bloom B, Cohen RA, Freeman G. Summary health statistics for U.S. children:National Health Interview Survey, 2008. Vital Health Stat 10. 2009;244:1-81.3. Barbaresi WJ, Katusic SK, Colligan RC, et al. Long-term school outcomes forchildren with attention-deficit/hyperactivity disorder: a population-based perspec-tive. J Dev Behav Pediatr. 2007;28:265-273.4. Biederman J, Faraone SV. Attention-deficit hyperactivity disorder. Lancet.2005;366:237-248.5. Cuffe SP, Moore CG, McKeown R. ADHD and health services utilization in thenational health interview survey. J Atten Disord. 2009;12:330-340. 6. Castellanos FX, Tannock R. Neuroscience of attention-deficit/hyperactivity dis-order. Nat Rev Neurosci. 2002;3:617-628.7. Nigg JT. Neuropsychologic theory and findings in attention-deficit/hyperactivitydisorder: the state of the field and salient challenges for the coming decade. BiolPsychiatry. 2005;57:1424-1435.8. Sonuga-Barke EJ. Psychological heterogeneity in AD/HD—a dual pathway modelof behaviour and cognition. Behav Brain Res. 2002;10:29-36.9. Pliszka SR, Crismon ML, Hughes CW, et al., and the Texas Consensus ConferencePanel on Pharmacotherapy of Childhood Attention-Deficit/ Hyperactivity Disorder.The Texas Children’s Medication Algorithm Project: revision of the algorithm forpharmacotherapy of attention-deficit/hyperactivity disorder. J Am Acad Child AdolescPsychiatry. 2006;45:642-657.10. Elia J, Ambrosini PJ, Rapoport JL. Treatment of attention-deficit-hyperactivitydisorder. N Engl J Med. 1999;340:780-788.11. Wood JG, Crager JL, Delap CM, Heiskell KD. Beyond methylphenidate: non-stimulant medications for youth with ADHD. J Atten Disord. 2007;11:341-350.12. Wilens TE, Spencer TJ. The stimulants revisited. Child Adolesc Psychiatr Clin NAm. 2000;9:573-603.13. Faraone SV, Wilens TE. Effect of stimulant medications for attention-deficit/hyperactivity disorder on later substance use and the potential for stimulantmisuse, abuse, and diversion. J Clin Psychiatry. 2007;68:15-22.14. Wilens TE. A sobering fact: ADHD leads to substance abuse. J Am Acad ChildAdolesc Psychiatry. 2011;50:6-8.15. Garnock-Jones KP, Keating GM. Atomoxetine: a review of its use in attention-deficit hyperactivity disorder in children and adolescents. Paediatr Drugs. 2009;11:203-226. 16. Wilens TE, Gault LM, Childress A, et al. Safety and efficacy of ABT-089 inpediatric attention-deficit/hyperactivity disorder: results from two randomizedplacebo-controlled clinical trials. J Am Acad Child Adolesc Psychiatry. 2011; 50:73-84.17. Sallee FR. The role of alpha2-adrenergic agonists in attention-deficit /hyperac-tivity disorder. Postgrad Med. 2010;122:78-87.18. Daviss WB, Patel NC, Robb AS, et al. Clonidine for attention-deficit/hyper-activity disorder: II. ECG changes and adverse events analysis. J Am Acad ChildAdolesc Psychiatry. 2008;47:189-198.19. Palumbo DR, Sallee FR, Pelham WE Jr, et al., the CAT Study team. Clo nidinefor attention-deficit/hyperactivity disorder: I. Efficacy and tolerabi lity outcomes.J Am Acad Child Adolesc Psychiatry. 2008;47:180-188.20. Ramos-Quiroga JA, Casas M. Achieving remission as a routine goal of pharma-cotherapy in attention-deficit hyperactivity disorder. CNS Drugs. 2011;25:17-36.21. Steele M, Jensen PS, Quinn DM. Remission versus response as the goal of ther-apy in ADHD: a new standard for the field? Clin Ther. 2006;28:1892-1908.
STAKEHOLDER PERSPECTIVE
37May/June 2011 www.AHDBonline.com

moving millimeters
NEW EDARBI: Head-to-head efficacy trials versus the highest marketed doses of DIOVAN® (320 mg) and BENICAR® (40 mg)
REDUCTIONS IN 24-HR MEAN AMBULATORY SBP AT WEEK 61,2
Mean ambulatory baseline: Study 1=144.9 mm Hg
EDARBI 80 mg was statistically superior to DIOVAN® 320 mg and BENICAR® 40 mg in reducing 24-hr mean ambulatory and clinic SBP1
▼ Similar results were observed across two other comparator studies: Study 2 vs BENICAR 40 mg and Study 3 vs DIOVAN 320 mg
▼ Clinic SBP differences between EDARBI and active comparators were consistent with mean ambulatory results
IMPORTANT SAFETY INFORMATION
WARNING: AVOID USE IN PREGNANCYWhen pregnancy is detected, discontinue EDARBI as soon as possible. Drugs that act directly on the renin-angiotensin system can cause injury and death to the developing fetus.
▼ Avoid fetal or neonatal exposure. ▼ Correct volume or salt depletion prior to administration of EDARBI, or start
treatment at 40 mg.▼ Monitor for worsening renal function in patients with renal impairment.▼ In patients with an activated renin-angiotensin system, as by volume or salt
depletion, renin-angiotensin-aldosterone system (RAAS) blockers such as azilsartan medoxomil can cause excessive hypotension. In patients whose renal function may depend on the activity of the renin-angiotensin system, e.g., with renal artery stenosis, treatment with RAAS blockers has been associated with oliguria or progressive azotemia and rarely with acute renal failure and death.
▼ Monitor renal function periodically in patients receiving EDARBI and NSAIDs who are also elderly, volume-depleted, or who have compromised renal function.
▼ The most common adverse reaction in adults was diarrhea (2%).For further information, please see adjacent Brief Summary of Prescribing Information.
INDICATIONEDARBI is an angiotensin II receptor blocker (ARB) indicated for the treatment of hypertension, either alone or in combination with other antihypertensive agents.
Study 1 Design: A 6-week, randomized, double-blind, placebo-controlled, forced-titration study in patients (N = 1,291) with clinic SBP ≥150 mm Hg and ≤180 mm Hg and 24-hr mean SBP ≥130 mm Hg and ≤170 mm Hg. The primary endpoint was change in 24-hr mean ambulatory SBP. Placebo lowered 24-hr mean ambulatory SBP by 0.3 mm Hg. Data shown are placebo corrected.
References: 1. EDARBI Prescribing Information. 2. White WB, Weber MA, Sica D, et al. Effects of the angiotensin receptor blocker azilsartan medoxomil versus olmesartan and valsartan on ambulatory and clinic blood pressure in patients with stages 1 and 2 hypertension. Hypertension. 2011;57:413-420.
P<0.001 vs DIOVAN 320 mgP=0.009 vs BENICAR 40 mg
EDARBI 80 mg
-14.3 mm Hg
BENICAR 40 mg
-11.7 mm Hg
DIOVAN 320 mg
-10.0 mm Hg
STUDY 1
EDARBI is a trademark of Takeda Pharmaceutical Company Limited registered with the U.S. Patent and Trademark Office and used under license by Takeda Pharmaceuticals America, Inc. Trademarks are the property of their respective owners. ©2011 Takeda Pharmaceuticals North America, Inc. All rights reserved. LXA-00331 04/11

Generally, adverse reactions were mild, not dose related and similar regardless of age, gender and race.In placebo controlled monotherapy trials, diarrhea was reported up to 2% in patients treated with Edarbi 80 mg daily compared with 0.5% of patients on placebo.Other adverse reactions with a plausible relationship to treatment that have been reported with an incidence of ≥0.3% and greater than placebo in more than 3300 patients treated with Edarbi in controlled trials are listed below:Gastrointestinal Disorders: nauseaGeneral Disorders and Administration Site Conditions: asthenia, fatigueMusculoskeletal and Connective Tissue Disorders: muscle spasmNervous System Disorders: dizziness, dizziness postural Respiratory, Thoracic and Mediastinal Disorders: coughClinical Laboratory FindingsIn controlled clinical trials, clinically relevant changes in standard laboratory parameters were uncommon with administration of Edarbi.Serum creatinine: Small reversible increases in serum creatinine are seen in patients receiving 80 mg of Edarbi. The increase may be larger when coadministered with chlorthalidone or hydrochlorothiazide.In addition, patients taking Edarbi who had moderate to severe renal impairment at baseline or who were >75 years of age were more likely to report serum creatinine increases.Hemoglobin/Hematocrit: Low hemoglobin, hematocrit, and RBC counts were observed in 0.2%, 0.4%, and 0.3% of Edarbi-treated subjects, respectively. None of these abnormalities were reported in the placebo group. Low and high markedly abnormal platelet and WBC counts were observed in <0.1% of subjects.DRUG INTERACTIONSNo clinically significant drug interactions have been observed in studies of azilsartan medoxomil or azilsartan given with amlodipine, antacids, chlorthalidone, digoxin, fluconazole, glyburide, ketoconazole, metformin, pioglitazone, and warfarin. Therefore, Edarbi may be used concomitantly with these medications.Non-Steroidal Anti-Inflammatory Agents including Selective Cyclooxygenase-2 Inhibitors(COX-2 Inhibitors)In patients who are elderly, volume-depleted (including those on diuretic therapy), or who have compromised renal function, co-administration of NSAIDs, including selective COX-2 inhibitors, with angiotensin II receptor antagonists, including azilsartan, may result in deterioration of renal function, including possible acute renal failure. These effects are usually reversible. Monitor renal function periodically in patients receiving azilsartan and NSAID therapy.The antihypertensive effect of angiotensin II receptor antagonists, including azilsartan, may be attenuated by NSAIDs, including selective COX-2 inhibitors.USE IN SPECIFIC POPULATIONSPregnancyPregnancy Category C (first trimester) and D (second and third trimesters). There is no clinical experience with the use of Edarbi in pregnant women.Nursing MothersIt is not known if azilsartan is excreted in human milk, but azilsartan is excreted at low concentrations in the milk of lactating rats. Because of the potential for adverse effects on the nursing infant, a decision should be made whether to discontinue nursing or discontinue the drug, taking into account the importance of the drug to the mother. Pediatric UseSafety and effectiveness in pediatric patients under 18 years of age have not been established.Geriatric UseNo dose adjustment with Edarbi is necessary in elderly patients. Of the total patients in clinical studies with Edarbi, 26% were elderly (65 years of age and older); 5% were75 years of age and older. Abnormally high serum creatinine values were more likely to be reported for patients age 75 or older. No other differences in safety or effectiveness were observed between elderly patients and younger patients, but greater sensitivity of some older individuals cannot be ruled out.Renal ImpairmentDose adjustment is not required in patients with mild-to-severe renal impairment or end-stage renal disease. Patients with moderate to severe renal impairment are more likely to report abnormally high serum creatinine values.Hepatic ImpairmentNo dose adjustment is necessary for subjects with mild or moderate hepatic impairment. Edarbi has not been studied in patients with severe hepatic impairment.OVERDOSAGELimited data are available related to overdosage in humans. During controlled clinical trials in healthy subjects, once daily doses up to 320 mg of Edarbi were administered for 7 days and were well tolerated. In the event of an overdose, supportive therapy should be instituted as dictated by the patient’s clinical status. Azilsartan is not dialyzable.CLINICAL PHARMACOLOGYMechanism of ActionAngiotensin II is formed from angiotensin I in a reaction catalyzed by angiotensin-converting enzymes (ACE, kinase II). Angiotensin II is the principal pressor agent of the renin-angiotensin system, with effects that include vasoconstriction, stimulation of synthesis and release of aldosterone, cardiac stimulation, and renal reabsorption of sodium. Azilsartan blocks the vasoconstrictor and aldosterone-secreting effects of angiotensin II by selectively blocking the binding of angiotensin II to the AT1 receptor in many tissues, such as vascular smooth muscle and the adrenal gland. Its action is, therefore, independent of the pathway for angiotensin II synthesis.
BRIEF SUMMARY OF FULL PRESCRIBING INFORMATION for Edarbi(azilsartan medoxomil) tablets
WARNING: AVOID USE IN PREGNANCYWhen pregnancy is detected, discontinue Edarbi as soon as possible. Drugs that act directly on the renin-angiotensin system can cause injury and death to the developing fetus.
INDICATIONS AND USAGEEdarbi is an angiotensin II receptor blocker (ARB) indicated for the treatment of hypertension. It may be used alone or in combination with other antihypertensive agents.CONTRAINDICATIONSNoneWARNINGS AND PRECAUTIONSFetal/Neonatal Morbidity and MortalityDrugs that act directly on the renin-angiotensin system can cause fetal and neonatal morbidity and death when administered to pregnant women during the second and third trimester. When pregnancy is detected, Edarbi should be discontinued as soon as possible.The use of drugs that act directly on the renin-angiotensin system during the second and third trimesters of pregnancy has been associated with fetal and neonatal injury, including hypotension, neonatal skull hypoplasia, anuria, reversible or irreversible renal failure, and death. Oligohydramnios has also been reported, presumably resulting from decreased fetal renal function; oligohydramnios in this setting has been associated with fetal limb contractures, craniofacial deformation, and hypoplastic lung development. Prematurity, intrauterine growth retardation, and patent ductus arteriosus have also been reported, although it is not clear whether these occurrences were due to exposure to the drug.These adverse effects do not appear to have resulted from intrauterine drug exposure that has been limited to the first trimester. Mothers whose embryos and fetuses are exposed to an angiotensin II receptor antagonist only during the first trimester should be so informed. Nonetheless, when patients become pregnant, physicians should have the patient discontinue the use of Edarbi as soon as possible.Rarely (probably less often than once in every thousand pregnancies), no alternative to a drug acting on the renin-angiotensin system is available. In these rare cases, the mother should be apprised of the potential hazards to the fetus and serial ultrasound examinations should be performed to assess the intra-amniotic environment.If oligohydramnios is observed, Edarbi should be discontinued unless it is considered life-saving for the mother. Contraction stress testing, a nonstress test or biophysical profiling may be appropriate, depending upon the week of pregnancy. Patients and physicians should be aware, however, that oligohydramnios may not appear until after the fetus has sustained irreversible injury.Infants with histories of in utero exposure to an angiotensin II receptor antagonist should be closely observed for hypotension, oliguria, and hyperkalemia. If oliguria occurs, attention should be directed toward support of blood pressure and renal perfusion. Exchange transfusion or dialysis may be required as a means of reversing hypotension and/or substituting for impaired renal function.Hypotension in Volume- or Salt-Depleted PatientsIn patients with an activated renin-angiotensin system, such as volume- and/or salt-depleted patients (eg, those being treated with high doses of diuretics), symptomatic hypotension may occur after initiation of treatment with Edarbi. Correct volume or salt depletion prior to administration of Edarbi, or start treatment at 40 mg. If hypotension does occur, the patient should be placed in the supine position and, if necessary, given an intravenous infusion of normal saline. A transient hypotensive response is not a contraindication to further treatment, which usually can be continued without difficulty once the blood pressure has stabilized.Impaired Renal FunctionAs a consequence of inhibiting the renin-angiotensin system, changes in renal function may be anticipated in susceptible individuals treated with Edarbi. In patients whose renal function may depend on the activity of the renin-angiotensin system (e.g., patients with severe congestive heart failure, renal artery stenosis, or volume depletion), treatment with angiotensin-converting enzyme inhibitors and angiotensin receptor blockers has been associated with oliguria or progressive azotemia and rarely with acute renal failure and death. Similar results may be anticipated in patients treated with Edarbi.In studies of ACE inhibitors in patients with unilateral or bilateral renal artery stenosis, increases in serum creatinine or blood urea nitrogen have been reported. There has been no long-term use of Edarbi in patients with unilateral or bilateral renal artery stenosis, but similar results may be expected.ADVERSE REACTIONSClinical Trials ExperienceBecause clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.A total of 4814 patients were evaluated for safety when treated with Edarbi at doses of 20, 40 or 80 mg in clinical trials. This includes 1704 patients treated for at least6 months; of these, 588 were treated for at least 1 year.Treatment with Edarbi was well-tolerated with an overall incidence of adverse reactions similar to placebo. The rate of withdrawals due to adverse events in placebo-controlled monotherapy and combination therapy trials was 2.4% (19/801) for placebo, 2.2% (24/1072) for Edarbi 40 mg, and 2.7% (29/1074) for Edarbi80 mg. The most common adverse event leading to discontinuation, hypotension/orthostatic hypotension, was reported by 0.4% (8/2146) patients randomized to Edarbi 40 mg or 80 mg compared to 0% (0/801) patients randomized to placebo.

An AT2 receptor is also found in many tissues, but this receptor is not known to be associated with cardiovascular homeostasis. Azilsartan has more than a 10,000-fold greater affinity for the AT1 receptor than for the AT2 receptor.Blockade of the renin-angiotensin system with ACE inhibitors, which inhibit the biosynthesis of angiotensin II from angiotensin I, is widely used in the treatment of hypertension. ACE inhibitors also inhibit the degradation of bradykinin, a reaction catalyzed by ACE. Because azilsartan does not inhibit ACE (kinase II), it should not affect bradykinin levels. Whether this difference has clinical relevance is not yet known. Azilsartan does not bind to or block other receptors or ion channels known to be important in cardiovascular regulation.Blockade of the angiotensin II receptor inhibits the negative regulatory feedback of angiotensin II on renin secretion, but the resulting increased plasma renin activity and angiotensin II circulating levels do not overcome the effect of azilsartan on blood pressure.PharmacodynamicsAzilsartan inhibits the pressor effects of an angiotensin II infusion in a dose-related manner. An azilsartan single dose equivalent to 32 mg azilsartan medoxomil inhibited the maximal pressor effect by approximately 90% at peak, and approximately 60% at 24 hours. Plasma angiotensin I and II concentrations and plasma renin activity increased while plasma aldosterone concentrations decreased after single and repeated administration of Edarbi to healthy subjects; no clinically significant effects on serum potassium or sodium were observed.Effect on Cardiac Repolarization: A thorough QT/QTc study was conducted to assess the potential of azilsartan to prolong the QT/QTc interval in healthy subjects. There was no evidence of QT/QTc prolongation at a dose of 320 mg of Edarbi.PharmacokineticsAbsorption: Azilsartan medoxomil is hydrolyzed to azilsartan, the active metabolite, in the gastrointestinal tract during absorption. Azilsartan medoxomil is not detected in plasma after oral administration. Dose proportionality in exposure was established for azilsartan in the azilsartan medoxomil dose range of 20 mg to 320 mg after single or multiple dosing.The estimated absolute bioavailability of azilsartan following administration of azilsartan medoxomil is approximately 60%. After oral administration of azilsartan medoxomil, peak plasma concentrations (Cmax) of azilsartan are reached within1.5 to 3 hours. Food does not affect the bioavailability of azilsartan.Distribution: The volume of distribution of azilsartan is approximately 16L. Azilsartan is highly bound to human plasma proteins (>99%), mainly serum albumin. Protein binding is constant at azilsartan plasma concentrations well above the range achieved with recommended doses.In rats, minimal azilsartan-associated radioactivity crossed the blood-brain barrier. Azilsartan passed across the placental barrier in pregnant rats and was distributed to the fetus.Metabolism and Elimination: Azilsartan is metabolized to two primary metabolites. The major metabolite in plasma is formed by O-dealkylation, referred to as metabolite M-II, and the minor metabolite is formed by decarboxylation, referred to as metabolite M-I. Systemic exposures to the major and minor metabolites in humans were approximately 50% and less than 1% of azilsartan, respectively. M-I and M-II do not contribute to the pharmacologic activity of Edarbi. The major enzyme responsible for azilsartan metabolism is CYP2C9.Following an oral dose of 14C-labeled azilsartan medoxomil, approximately55% of radioactivity was recovered in feces and approximately 42% in urine, with 15% of the dose excreted in urine as azilsartan. The elimination half-life of azilsartan is approximately 11 hours and renal clearance is approximately 2.3 mL/min. Steady-state levels of azilsartan are achieved within 5 days and no accumulation in plasma occurs with repeated once-daily dosing.Special PopulationsThe effect of demographic and functional factors on the pharmacokinetics of azilsartan was studied in single and multiple dose studies. Pharmacokinetic measures indicating the magnitude of the effect on azilsartan are presented in Figure 1 as change relative to reference (test/reference). Effects are modest and do not call for dosage adjustment.Figure 1 Impact of intrinsic factors on the pharmacokinetics of azilsartan
Population Description PK Fold Change and 90% CI Recommendation
No dose adjustment
No dose adjustment
No dose adjustment
No dose adjustment
No dose adjustment
No dose adjustment
No dose adjustment
No dose adjustment
No dose adjustment
NO EXPERIENCE
NO EXPERIENCE
0.5 1.0 1.5 2.0 2.5 3.0
Change relative to reference
CmaxAUC
CmaxAUC
CmaxAUC
CmaxAUC
CmaxAUC
CmaxAUC
CmaxAUC
CmaxAUC
CmaxAUC
AGE
>65y/18-45y
GENDER
Females/Males
RACE
Whites/Blacks
RENAL IMPAIRMENT
Mild/Normal
HEPATIC IMPAIRMENT
Mild/Normal
PEDIATRIC
Moderate/Normal
Severe/Normal
Moderate/Normal
Severe/Normal
ESRD/Normal
NONCLINICAL TOXICOLOGY Carcinogenesis, Mutagenesis, Impairment of Fertility Carcinogenesis: Azilsartan medoxomil was not carcinogenic when assessed in 26-week transgenic (Tg.rasH2) mouse and 2-year rat studies. The highest doses tested (450 mg azilsartan medoxomil/kg/day in the mouse and 600 mg azilsartan medoxomil/kg/day in the rat) produced exposures to azilsartan that are 12 (mice) and 27 (rats) times the average exposure to azilsartan in humans given the maximum recommended human dose (MRHD, 80 mg azilsartan medoxomil/day). M-II was not carcinogenic when assessed in 26-week Tg.rasH2 mouse and 2-year rat studies. The highest doses tested (approximately 8000 mg M-II/kg/day (males) and 11,000 mg M-II/kg/day (females) in the mouse and 1000 mg M-II/kg/day (males) and up to 3000 mg M-II/kg/day (females) in the rat) produced exposures that are, on average, about 30 (mice) and 7 (rats) times the average exposure to M-II in humans at the MRHD.Mutagenesis: Azilsartan medoxomil, azilsartan, and M-II were positive for structural aberrations in the Chinese Hamster Lung Cytogenetic Assay. In this assay, structural chromosomal aberrations were observed with the prodrug, azilsartan medoxomil, without metabolic activation. The active moiety, azilsartan was also positive in this assay both with and without metabolic activation. The major human metabolite, M-II was also positive in this assay during a 24 hr assay without metabolic activation.Azilsartan medoxomil, azilsartan, and M-II were devoid of genotoxic potential in the Ames reverse mutation assay with Salmonella typhimurium and Escherichia coli, the in vitro Chinese Hamster Ovary Cell forward mutation assay, the in vitro mouse lymphoma (tk) gene mutation test, the ex vivo unscheduled DNA synthesis test, and the in vivo mouse and/or rat bone marrow micronucleus assay.Impairment of Fertility: There was no effect of azilsartan medoxomil on the fertility of male or female rats at oral doses of up to 1000 mg azilsartan medoxomil/kg/day [6000 mg/m2 (approximately 122 times the MRHD of 80 mg azilsartan medoxomil/60 kg on a mg/m2 basis)]. Fertility of rats also was unaffected at doses of up to 3000 mg M-II/kg/day. Animal Toxicology and/or Pharmacology Reproductive Toxicology: In peri- and postnatal rat development studies, adverse effects on pup viability, delayed incisor eruption and dilatation of the renal pelvis along with hydronephrosis were seen when azilsartan medoxomil was administered to pregnant and nursing rats at 1.2 times the MRHD on a mg/m2 basis. Reproductive toxicity studies indicated that azilsartan medoxomil was not teratogenic when administered at oral doses up to 1000 mg azilsartan medoxomil/kg/day to pregnant rats (122 times the MRHD on a mg/m2 basis) or up to 50 mg azilsartan medoxomil/kg/day to pregnant rabbits (12 times the MRHD on a mg/m2 basis). M-II also was not teratogenic in rats or rabbits at doses up to 3000 mg M-II/kg/day. Azilsartan crossed the placenta and was found in the fetuses of pregnant rats and was excreted into the milk of lactating rats.PATIENT COUNSELING INFORMATIONSee FDA-approved patient labeling (Patient Information).General InformationPregnancy: Female patients of childbearing age should be told that use of drugs like Edarbi that act on the renin-angiotensin system during pregnancy can cause serious problems in the fetus and infant including low blood pressure, poor development of skull bones, kidney failure, and death. These consequences do not appear to have resulted from intrauterine drug exposure that has been limited to the first trimester. Discuss other treatment options with female patients planning to become pregnant. Women using Edarbi who become pregnant should notify their physicians as soon as possible.Distributed byTakeda Pharmaceuticals America, Inc.Deerfield, IL 60015Edarbi is a trademark of Takeda Pharmaceutical Company Limited registered with the U.S. Patent and Trademark Office and used under license by Takeda Pharmaceuticals America, Inc.©2011 Takeda Pharmaceuticals America, Inc.For more detailed information, see the full prescribing information for Edarbi at www.edarbi.com or contact Takeda Pharmaceuticals America, Inc. at 1-877-825-3327.AZL074 R1; February 2011 L-LXA-0211-2

41May/June 2011 www.AHDBonline.com
In 2010, the majority of the USFood and Drug Adminis tration(FDA) drug approvals were
focused on expanded indicationsand new dosing forms or formula-tions (Figure).1 Accord ing to theestimate reported by DrugManagementForum.com (which is ownedby InformaCeutica), only 23 newdrugs and biologics were approvedin 2010, including 21 new molecular entities (NMEs)approved by the FDA’s Center for Drug Evaluation andResearch and 2 biologics approved by the Center forBiologics Evaluation and Research.1,2
This is substantially lower than was projected by theseauthors in 2010,3 primarily because of this unexpectedlylow FDA approval rate. For example, of the 15 signifi-cant candidates in the pipeline that were highlightedlast year,3 only 3 were actually approved in 2010. TheFDA’s requests for new trials, additional data, or inclusionin the risk evaluation and mitigation strategy (REMS)program for the remaining 12 agents have resulted indrug development delays or program discontinuations.
New trials were requested for Bydureon (exenatide),Daliresp (roflumilast), Esbriet (pirfenidone), motaviz -umab, and Naproxcinod (nitronaproxen). Subsequent ly,roflumilast was approved in 2011 and development ofmotavizumab has been discontinued.
Additional information was also requested for belat-acept, Brilinta (ticagrelor), Lorqess (lorcaserin),Neutroval (XM02), Qnexa (phentermine and topira-
mate), Replagal (agalsidase alfa),and Uplyso (taliglucerase alfa). ForQnexa, the FDA has also requestedadjustments in the REMS program.
New FDA Approvals in 2011Considering the 2010-2011 ap -
proval rate, and the number ofnew drug applications submittedto the FDA in 2010,4 approxi-
mately 20 to 25 new drugs are expected to be approvedthis year. The greatest impact of these ap provals willconcentrate in several categories, including:• Cardiovascular disease• Immunology-related conditions • Oncology• Antiviral agents for infectious diseases.
The following 22 drugs, biologics, and related deviceshave already been approved by the FDA as of May 23,2011; 12 of these are classified as NMEs and 2 are newbiologics5:• Abstral sublingual tablets CII (fentanyl; ProStrakan)• Adenovirus type 4 and type 7 vaccine, live, oral (Teva)• Benlysta (belimumab; HGS/GlaxoSmithKline)• Corifact (factor XIII concentrate, human; CSL Behring)• Daliresp (roflumilast; Forest/Nycomed)• Duexis (ibuprofen and famotidine; Horizon)• Edarbi (azilsartan; Takeda)• Edurant (rilpivirine; Tibotec)• Epicyn HydroGel (Oculus)• Gel-One (development code: Gel-200; Seikagaku)• Gralise tablets (gabapentin; Depomed/Abbott)• Horizant (gabapentin enacarbil extended-release;
GlaxoSmithKline and Xenoport)• Incivek (telaprevir; Vertex)• Makena (hydroxyprogesterone caproate injection;
KV Pharm/Hologic)• Natroba topical suspension 0.9% (spinosad; ParaPRO)• Phoslyra (calcium acetate 667 mg/5 mL; Fresenius)• Tradjenta (linagliptin; Boehringer Ingelheim)• Vandetanib tablets (AstraZeneca)
The Current Drug Pipeline and NewApprovals in 2011: A Managed CarePerspectiveDi ana Papshev, Phar mD, and Chant el l M. Reagan, Phar mD
Dr Papshev is Partner and Dr Reagan is Managing Director, InformaCeutica, Yardley, PA
InformaCeutica is a drug management information companyfocused on developing interactive tools for managed care decision makers. InformaCeutica’s offerings include DrugManagementForum.com and DrugPipelineForecast.com.DrugManagementForum.com allows managed care profession-als to connect and share ideas on common issues and experi-ences in drug benefit management. DrugPipelineForecast.comoffers a comprehensive tool for tracking late-phase drug pipelinedevelopments from the managed care perspective.
Diana Papshev Chantell M. Reagan

42 AMERICAN HEALTH & DRUG BENEFITS May/June 2011
Adapted with permission from www.drugmanagementforum.com. Accessed February 10, 2011.
Figure Significant Brand-Name Drugs Approved by the FDA in 2010
Antiinfectives
Antineoplastics
Cardiovascular
Central nervous system
Dermatology
Endocrine/diabetes
Enzyme replacements and modifiers
Gastrointestinal drugs
Hematologic drugs
Immunologic drugs and vaccines
Obstetrics and gynecology
Ophthalmologic drugs
Orthopedic agents
Respiratory drugs
Transplant
Urologic agents
New molecular entity
New dosage/formulation
New biologics
Expanded indications
1 4
2 1 2 6
1 4 2
1 18 7
1 4
2 5 1
2 2 2
2
2
1 3 3 3
1 5
1 3 2
1
1
2
2
0 7.5 15 22.5 30
• Victrelis (boceprevir; Merck)• Viibryd (vilazodone; Trovis/Clinical Data)• Yervoy (ipilimumab injection for intravenous use;
Bristol-Myers Squibb)• Zytiga (abiraterone; Centocor).
In particular, 7 of these agents (Table 1) may be on theradar for health plans and pharmacy benefit managers.Most attention is focused on the 2 drugs for hepatitis Cvirus (HCV) infection—telaprevir and boceprevir. Thesenew HCV therapies are expected to change the standardof care for this condition and to have significant impacton drug utilization and cost. In clinical trials, both drugshave demonstrated an opportunity for shorter treatment
duration and high efficacy rates in treatment-naïvepatients with HCV and in those who were previously poorresponders to other HCV therapies.
Benlysta, Yervoy, and Zytiga each represent a signif-icant innovation in their respective therapeutic areas.All 3 are expected to reach blockbuster status.
Makena, although not a novel treatment, has gener-ated considerable controversy in the managed carecommunity and the general public. At the center of thiswas the substantial increase in price in comparison withpreviously custom-compounded therapy (17P, hydroxy -progesterone caproate) and the threat of Makena’s mar-ket exclusivity. In an unprecedented move, the FDApublicly announced that it was not planning to takeenforcement action against pharmacies compounding17P. Since then, KV Pharmaceuticals, the manufactur-er of Makena, lowered the price to $690 per injection(nearly 55%).
The 2011 Drug PipelineThe 10 other products that may be approved later in
Benlysta, Yervoy, and Zytiga each represent a significant innovation in their respectivetherapeutic areas. All 3 are expected to reach blockbuster status.

The Current Drug Pipeline
43May/June 2011 www.AHDBonline.com
Table 1 Significant FDA Approvals through May 23, 2011
Drug, formulationApproval
date IndicationEstimated peak annual sales, $ Implications for managed care
Viibryd (vilazodone), oral tablet
1/2011 Major depressivedisorder
1.7B SSRI and 5-HT1A receptor agonist,positioned as having less impact on sexual dysfunction than traditionalagents; will be entering crowded generic market
Makena (hydroxy -progesterone caproate), injection
2/2011 Pretermlabor/delivery
413M First drug approved for this indication;previously similar products were compounded
Benlysta (belimumab),IV infusion
3/2011 Immunology (SLE)
2.7B First new drug approved for lupus treatment in 50 years
Yervoy (ipilimumab), IV infusion
3/2011 Immunology/oncology(advancedmelanoma)
2B First-in-class CTL-4 inhibitor; firsttreatment that demonstrated prolongedsurvival; also being studied in prostateand lung cancers
Zytiga (abiraterone), oral tablet
4/2011 Oncology(advanced
prostate cancer[second-
/third-line])
1.5B One of few agents to demonstrateprolonged survival (in patients who werepreviously treated with chemotherapy);expected to be evaluated in earlier stages of prostate cancer
Victrelis (boceprevir), oral tablet
5/2011 Antiviral (hepatitis C)
>1B Novel mechanism of action—HCVprotease inhibitor; showed superiorefficacy in combination with pegylatedinterferon and ribavirin in treatment-naïve and experienced patients(excluding null responders); dependingon patient characteristics and treatmentresponse, overall treatment duration canbe reduced to 28 weeks (24 weeks forVictrelis); also being evaluated inpatients coinfected with HIV; expectedto compete with Incivek
Incivek (telaprevir) oral tablet
5/2011 Antiviral (hepatitis C)
3B-6B Novel mechanism of action—HCVprotease inhibitor; showed superiorefficacy in combination with pegylatedinterferon and ribavirin in treatment-naïve and experienced patients(including null responders); dependingon patient characteristics and treatmentresponse, overall treatment duration canbe reduced to 24 weeks (12 weeks forIncivek); also being evaluated inpatients coinfected with HIV; expectedto compete with Victrelis
5-HT1A indicates 5-hydroxytryptamine; B, billion; CTL-4, cytotoxic T-lymphocyte 4; HCV, hepatitis C virus; IV, intravenous; M, million; SLE, systemic lupus erythematosus; SSRI, selective serotonin reuptake inhibitor.Sources: www.DrugManagementForum.com; EvaluatePharma. World Preview 2016. www.evaluatepharma.com/EvaluatePharma_World_Preview_2016.aspx; EvaluatePharma. Weekly Market Movers, March 11, 2011. www.evaluatepharma.com.

44 AMERICAN HEALTH & DRUG BENEFITS May/June 2011
Table 2 Significant Nonspecialty Pipeline Candidates in 2011 (Starting 5/24/2011)
Drug
Estimated FDA
approvala
Therapeutic category
(investigated use)Estimated peakannual sales, $
Implications for managed care
Arcapta (indacaterol),inhalation
3Q 2011b Respiratory (COPD)
>1B Once-daily long-acting beta-agonistwhich showed superiority over twice-daily Serevent (salmeterol) and some clinical benefits overSpiriva (tiotropium); combinationagents with mometasone and glyco -pyrronium are being developed; newtrials were initiated after FDA con-cern regarding dosing regimens in10/2009 and highest dose in 3/2011;FDA advisory board endorses “low-dose” in 3/2011
Brilinta (ticagrelor),oral
7/20/2011c CV (ACS) 1.9B Third antiplatelet drug to competewith Plavix (clopidogrel) and Effient(prasugrel); showed superiority toclopidogrel in CV events, withoutincreased bleeding risk; unlikely tohave black box warning for issues withCYP2C19 metabolism; FDA requestedin 9/2010 additional analysis of thePLATO trial
Btripla (rilpivirine,emtricitabine, teno-fovir), oral fixed dose
8/10/2011c Antiviral (HIV) 1.1B Expected to be successor to Atripla(efavirenz, emtricitabine, tenofovir),with an improved safety profile
Dapagliflozin, oral 10/28/2011c Endocrine (type 2diabetes)
>1B First-in-class SGLT-2; showed addi-tive effect in combination withJanuvia (sitagliptin)
Xarelto (rivaroxaban),oral
4Q 2011b CV (AF, VTE) 1.6-3B Direct factor Xa inhibitor; showedsuperiority to Lovenox (enoxaparin)in VTE prevention and warfarin instroke prevention with no increase inintracranial bleeding; expected tocompete with LMWHs and oral anticoagulants
aEstimated approval as of May 2011, subject to change. bEstimated approval time is based on publicly available information. cEstimated approval time is based on announced Prescription Drug User Fee Act date.ACS indicates acute coronary syndrome; AF, atrial fibrillation; B, billion; COPD, chronic obstructive pulmonarydisease; CV, cardiovascular; LMWHs, low-molecular-weight heparins; Q, quarter; SGLT-2, sodium–glucose cotrans-porter type 2; VTE, venous thromboembolism. Sources: www.DrugPipelineForecast.com; www.DrugManagementForum.com; EP Vantage. EvaluatePharma. WorldPreview 2016. www.evaluatepharma.com/EvaluatePharma_World_Preview_2016.aspx.

The Current Drug Pipeline
45May/June 2011 www.AHDBonline.com
Table 3 Significant Specialty Candidates in Drug Pipeline in 2011 (after 5/23/2011)
Drug, formulation
Estimated FDA
approvala
Therapeutic category
(investigated use)Estimated peakannual sales, $ Implications for managed care
Nulogix (belatacept), IV
6/15/2011b Immunology (renal transplant)
500M First transplant drug approved in 13years; novel mechanism of action; firstselective co stimulation blocker for pro-phylaxis of acute renal graft rejection;concerns with PML and kidney rejec-tion; FDA requested additional data in4/2010 from an ongoing phase 3 trial
VEGF Trap-Eye,(aflibercept), intravitreal injection
8/20/2011b Ophthalmology (wet age-related
macular degeneration)
870M Fusion protein targeting VEGF-A andPIGF; expected to compete withLucentis (ranibizumab) on the basis ofnoninferiority data and less frequentdosing (every 2 months); also beingevaluated for central retinal veinocclusion, diabetic macular edema, andother eye diseases (IV form is also beingstudied in oncologic conditions)
Brentuximab, IV 8/30/2011b Oncology (refractory
lymphomas)
>250M First-in-class antibody drug conjugatewith anti-CD30 monoclonal antibodyattached to monomethyl auristatin E;also being evaluated as first-linetherapy in combination withchemotherapy (greater sales potential)
Crizotinib, oral 4Q 2011c Oncology (advanced NSCLC)
755M Oral agent, first-in-class ALK inhibitorfor patients with ALK-positiveadvanced NSCLC
Neutroval (XM02), SC/IV
4Q 2011c Hematology/oncology
(neutropenia)
N/A First biosimilar granulocyte colony-stimulating factor (with full BLAsubmission); likely to compete withfilgrastim based on price andnoninferiority data in head-to-headtrials; FDA requested additional data in10/2010, but no new trials were required
aEstimated approval time as of May 2011, subject to change. bExpected approval time is based on the announced Prescription Drug User Fee Act date. cExpected approval time is based on publicly available information. ALK indicates anaplastic lymphoma kinase; BLA, biologic license application; IV, intravenous; M, million; NSCLC,non–small-cell lung cancer; PML, progressive multifocal leukoencephalopathy; Q, quarter; SC, subcutaneous; VEGF, vascular endothelial growth factor. Sources: www.DrugPipelineForecast.com; www.DrugManagementForum.com; EP Vantage. EvaluatePharma WorldPreview 2016. www.evaluatepharma.com/EvaluatePharma_World_Preview_2016.aspx.

46 AMERICAN HEALTH & DRUG BENEFITS May/June 2011
2011 also have the potential to significantly impact drugutilization and trend:• Arcapta (indacaterol)• Brentuximab• Brilinta (ticagrelor)• Btripla (rilpivirine, emtricitabine, tenofovir)• Crizotinib• Dapagliflozin• Neutroval• Nulogix (belatacept)• VEGF Trap-Eye (aflibercept)• Xarelto (rivaroxaban).
Among 5 significant nonspecialty candidates in thedrug pipeline (Table 2), Xarelto and Brilinta are partic-ularly predicted to have moderate-to-significant impacton drug utilization.
Under the specialty category, pipeline agents with thegreatest potential in 2011 have already been approved.In addition, there are 5 other serious contenders (Table3) likely to receive approval this year.
Of these agents, VEGF Trap-Eye is most likely toreach blockbuster sales. This ophthalmic product is also
of interest to managed care organizations, because it isexpected to compete directly with Lucentis.
Unlike other specialty pipeline candidates, Neutrovalis a follow-on biologic expected to be positioned againstNeupogen (filgrastim). Neutroval has been available inEurope as a biosimilar, but in the United States it isbeing reviewed under a full biologic license applicationpathway. It will be interesting to see how the entry ofNeutroval may affect the dynamics of the granulocytecolony-stimulating factor drug class. ■
References1. Drug Management Forum. Significant brand approvals for 2010. www.drugmanagementforum.com/forum/new-brand-drug-approvals-month/24780-significant-brand-approvals-2010-a.html. Accessed March 4, 2011.2. US Food and Drug Administration. New molecular entities approved in 2010 byCDER. www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/DrugandBiologicApprovalReports/UCM242677.pdf.Accessed March 4, 2011.3. Papshev D, Reagan CM. The new drug pipeline: what is on the horizon for man-aged care in 2010? Am Health Drug Benefits. 2010;3(3 suppl 10):S258-S264.4. US Food and Drug Administration. Comparison of NMEs approved in 2010 toprevious years. www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/DrugandBiologicApprovalReports/UCM242695.pdf. Accessed March 4, 2011.5. Drug Management Forum. New brand drug approvals by month. www.drugmanagementforum.com/forum/new-brand-drug-approvals-month/. Accessed March 4, 2011.
Signature (Required) Specialty
Date (Required) Address
Name City/State/ZIP
Company E-mail
Title Phone
Request your SUBSCRIPTION to
AmeRicAn HeAltH & DRug Benefits®
Please provide all information indicated, including date and signature. INCOMPLETE CARDS WILL NOT BE PROCESSED.
❑ YES! I would like to receive American Health & Drug Benefits® as well as related educational supplements.❑ NO. Please discontinue my subscription.
Fax to: 732.992.1881

AVBCC is the fastest growing national specialty organization dedicated to improving the care of
cancer patients and their quality of life.
The organization was established to provide a network for payers and oncology healthcare professionals to interactand network in order to promote optimal care for patients
and their families.
2011 REGIONAL MEETINGS
Sponsorship Opportunities Available
www.AVBCConline.orgRegister online at
Past Sponsors:
September 24Chicago, IL
October 1San Francisco, CA
November 19Tampa, FL
2012 NATIONAL MEETINGMarch 29-31, Houston, TX
�� �� � �� �

48 I AMERICAN HEALTH & DRUG BENEFITS I May/June 2011
Abstral(Fentanyl citrate; ProStrakan)Class: Opioid analgesicIndication: Management of breakthrough pain in cancerpatients aged ≥18 years who are already receiving andare tolerant to opioid therapy for their underlying per-sistent cancer painApproval consideration: REMS
Benlysta (NME)(Belimumab; Human Genome Sciences/GlaxoSmithKline)Class: B-lymphocyte stimulator-specific inhibitorIndication: Treatment of adult patients with active,autoantibody-positive systemic lupus erythematosus
Corifact(Factor XIII concentrate, human; CSL Behring)Class: Fibrin-stabilizing factor concentrateIndication: Routine prophylactic treatment of congenitalfactor XIII (FXIII) deficiency
Daliresp (NME)(Roflumilast; Forest Research Institute)Class: Selective phosphodiesterase-4 inhibitorIndication: To reduce the risk of chronic obstructive pul-monary disease (COPD) exacerbations in patients withsevere COPD and a history of exacerbations
DaTscan (NME)(Ioflupane I123 injection; GE Healthcare)Class: RadiopharmaceuticalIndication: To assist in the evaluation of adult patientswith suspected Parkinsonian syndromesApproval consideration: Priority review
Dificid(Fidaxomicin; Optimer Pharma)Class: Macrolide antibacterialIndication: Treatment of Clostridium difficile–associateddiarrhea
Duexis(Ibuprofen + famotidine; Horizon Pharma)Class: Nonsteroidal anti-inflammatory drug + histamineH2-receptor antagonistIndication: Relief of signs and symptoms of rheumatoidarthritis and osteoarthritis and to decrease the risk ofdeveloping upper gastrointestinal ulcers
Edarbi (NME)(Azilsartan medoxomil; Takeda)Class: Angiotensin II receptor blockerIndication: Treatment of hypertension
Edurant (NME)(Rilpivirine; Tibotec)Class: HIV type 1 specific, non-nucleoside reverse transcriptase inhibitorIndication: Treatment of HIV-1 infection in treatment-naïve patients
Gadavist (NME)(Gadobutrol; Bayer HealthCare)Class: Macrocyclic gadolinium-based contrast agentIndication: For intravenous use in diagnostic magneticresonance imaging to visualize areas with disruptedblood-brain barrier and/or abnormal CNS vascularity
Horizant (NME)(Gabapentin enacarbil; GlaxoSmithKline)Class: Gamma-aminobutyric acid analogsIndication: Treatment of moderate-to-severe primaryrestless legs syndrome
Incivek (NME)(Telaprevir; Vertex Pharmaceuticals)Class: Hepatitis C virus NS3/4A protease inhibitorIndication: Treatment of genotype 1 chronic hepatitis Cin adult patients with compensated liver diseaseApproval consideration: Priority review
Makena(Hydroxyprogesterone caproate injection; Hologic)Class: ProgestinIndication: To reduce the risk of preterm birth in womenwith a singleton pregnancy who have a history of single-ton spontaneous preterm birth
Natroba (NME)(Spinosad; ParaPRO)Class: PediculicideIndication: Topical treatment of head lice infestations inpatients aged ≥4 years
Sylatron (NME)(Peginterferon alfa-2b; Merck)Class: Alpha interferon
FDA Approvals of Brand-NamePrescription Drugs, January-May 2011

49
2011 FDA Approvals of Brand-Name Drugs
Indication: Adjuvant treatment of melanoma withmicroscopic or gross nodal involvement within 84 daysof definitive surgical resection
Tradjenta (NME)(Linagliptin; Boehringer Ingelheim)Class: Dipeptidyl peptidase-4 inhibitorIndication: To improve glycemic control in adults withtype 2 diabetes mellitus
Vandetanib (NME)(Vandetanib; AstraZeneca)Class: Tyrosine kinase inhibitorIndication: Treatment of medullary thyroid cancer thatcannot be removed by surgery or that has spreadApproval considerations: Priority review, orphan drug, REMS
Victrelis (NME)(Boceprevir; Schering)Class: Hepatitis C virus NS3/4A protease inhibitorIndication: Treatment of chronic hepatitis C genotype 1 infectionApproval consideration: Priority review
Viibryd (NME)(Vilazodone hydrochloride; Forest Laboratories)Class: AntidepressantIndication: Treatment of major depressive disorderApproval consideration: REMS
Yervoy (NME)(Ipilimumab; Bristol-Myers Squibb)Class: Human cytotoxic T-lymphocyte antigen 4–blocking antibodyIndication: For unresectable or metastatic melanomaApproval consideration: REMS
Zytiga (NME)(Abiraterone acetate; Centocor Ortho Biotech)Class: CYP17 inhibitorIndication: Treatment of patients with metastatic castration-resistant prostate cancer who have receivedprevious chemotherapy containing docetaxelApproval consideration: Priority review
May/June 2011 I www.AHDBonline.com I
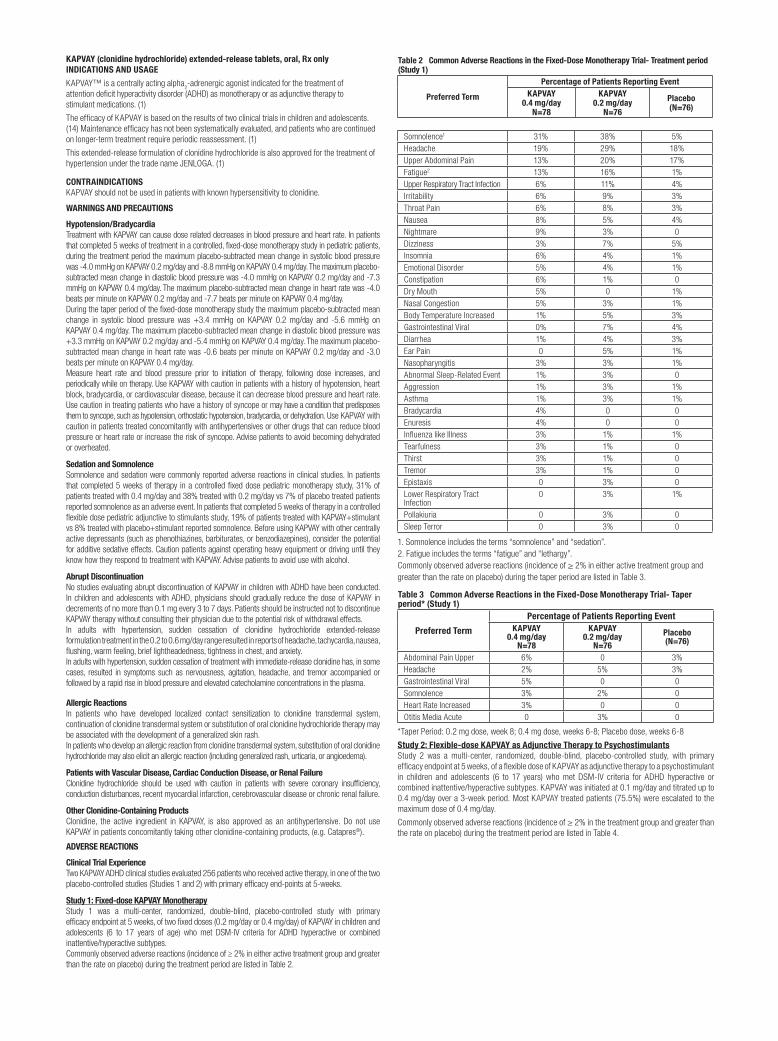
KAPVAY (clonidine hydrochloride) extended-release tablets, oral, Rx only INDICATIONS AND USAGE
KAPVAY™ is a centrally acting alpha2-adrenergic agonist indicated for the treatment of
attention deficit hyperactivity disorder (ADHD) as monotherapy or as adjunctive therapy to stimulant medications. (1)
The efficacy of KAPVAY is based on the results of two clinical trials in children and adolescents. (14) Maintenance efficacy has not been systematically evaluated, and patients who are continued on longer-term treatment require periodic reassessment. (1)
This extended-release formulation of clonidine hydrochloride is also approved for the treatment of hypertension under the trade name JENLOGA. (1)
CONTRAINDICATIONSKAPVAY should not be used in patients with known hypersensitivity to clonidine.
WARNINGS AND PRECAUTIONS
Hypotension/BradycardiaTreatment with KAPVAY can cause dose related decreases in blood pressure and heart rate. In patients that completed 5 weeks of treatment in a controlled, fixed-dose monotherapy study in pediatric patients, during the treatment period the maximum placebo-subtracted mean change in systolic blood pressure was -4.0 mmHg on KAPVAY 0.2 mg/day and -8.8 mmHg on KAPVAY 0.4 mg/day. The maximum placebo-subtracted mean change in diastolic blood pressure was -4.0 mmHg on KAPVAY 0.2 mg/day and -7.3 mmHg on KAPVAY 0.4 mg/day. The maximum placebo-subtracted mean change in heart rate was -4.0 beats per minute on KAPVAY 0.2 mg/day and -7.7 beats per minute on KAPVAY 0.4 mg/day.During the taper period of the fixed-dose monotherapy study the maximum placebo-subtracted mean change in systolic blood pressure was +3.4 mmHg on KAPVAY 0.2 mg/day and -5.6 mmHg on KAPVAY 0.4 mg/day. The maximum placebo-subtracted mean change in diastolic blood pressure was +3.3 mmHg on KAPVAY 0.2 mg/day and -5.4 mmHg on KAPVAY 0.4 mg/day. The maximum placebo-subtracted mean change in heart rate was -0.6 beats per minute on KAPVAY 0.2 mg/day and -3.0 beats per minute on KAPVAY 0.4 mg/day.Measure heart rate and blood pressure prior to initiation of therapy, following dose increases, and periodically while on therapy. Use KAPVAY with caution in patients with a history of hypotension, heart block, bradycardia, or cardiovascular disease, because it can decrease blood pressure and heart rate. Use caution in treating patients who have a history of syncope or may have a condition that predisposes them to syncope, such as hypotension, orthostatic hypotension, bradycardia, or dehydration. Use KAPVAY with caution in patients treated concomitantly with antihypertensives or other drugs that can reduce blood pressure or heart rate or increase the risk of syncope. Advise patients to avoid becoming dehydrated or overheated.
Sedation and SomnolenceSomnolence and sedation were commonly reported adverse reactions in clinical studies. In patients that completed 5 weeks of therapy in a controlled fixed dose pediatric monotherapy study, 31% of patients treated with 0.4 mg/day and 38% treated with 0.2 mg/day vs 7% of placebo treated patients reported somnolence as an adverse event. In patients that completed 5 weeks of therapy in a controlled flexible dose pediatric adjunctive to stimulants study, 19% of patients treated with KAPVAY+stimulant vs 8% treated with placebo+stimulant reported somnolence. Before using KAPVAY with other centrally active depressants (such as phenothiazines, barbiturates, or benzodiazepines), consider the potential for additive sedative effects. Caution patients against operating heavy equipment or driving until they know how they respond to treatment with KAPVAY. Advise patients to avoid use with alcohol.
Abrupt DiscontinuationNo studies evaluating abrupt discontinuation of KAPVAY in children with ADHD have been conducted. In children and adolescents with ADHD, physicians should gradually reduce the dose of KAPVAY in decrements of no more than 0.1 mg every 3 to 7 days. Patients should be instructed not to discontinue KAPVAY therapy without consulting their physician due to the potential risk of withdrawal effects.In adults with hypertension, sudden cessation of clonidine hydrochloride extended-release formulation treatment in the 0.2 to 0.6 mg/day range resulted in reports of headache, tachycardia, nausea, flushing, warm feeling, brief lightheadedness, tightness in chest, and anxiety.In adults with hypertension, sudden cessation of treatment with immediate-release clonidine has, in some cases, resulted in symptoms such as nervousness, agitation, headache, and tremor accompanied or followed by a rapid rise in blood pressure and elevated catecholamine concentrations in the plasma.
Allergic ReactionsIn patients who have developed localized contact sensitization to clonidine transdermal system, continuation of clonidine transdermal system or substitution of oral clonidine hydrochloride therapy may be associated with the development of a generalized skin rash.In patients who develop an allergic reaction from clonidine transdermal system, substitution of oral clonidine hydrochloride may also elicit an allergic reaction (including generalized rash, urticaria, or angioedema).
Patients with Vascular Disease, Cardiac Conduction Disease, or Renal FailureClonidine hydrochloride should be used with caution in patients with severe coronary insufficiency, conduction disturbances, recent myocardial infarction, cerebrovascular disease or chronic renal failure.
Other Clonidine-Containing ProductsClonidine, the active ingredient in KAPVAY, is also approved as an antihypertensive. Do not use KAPVAY in patients concomitantly taking other clonidine-containing products, (e.g. Catapres®).
ADVERSE REACTIONS
Clinical Trial ExperienceTwo KAPVAY ADHD clinical studies evaluated 256 patients who received active therapy, in one of the two placebo-controlled studies (Studies 1 and 2) with primary efficacy end-points at 5-weeks.
Study 1: Fixed-dose KAPVAY Monotherapy Study 1 was a multi-center, randomized, double-blind, placebo-controlled study with primary efficacy endpoint at 5 weeks, of two fixed doses (0.2 mg/day or 0.4 mg/day) of KAPVAY in children and adolescents (6 to 17 years of age) who met DSM-IV criteria for ADHD hyperactive or combined inattentive/hyperactive subtypes.Commonly observed adverse reactions (incidence of ≥ 2% in either active treatment group and greater than the rate on placebo) during the treatment period are listed in Table 2.
Table 2 Common Adverse Reactions in the Fixed-Dose Monotherapy Trial- Treatment period (Study 1)
Preferred Term
Percentage of Patients Reporting Event KAPVAY
0.4 mg/day N=78
KAPVAY 0.2 mg/day
N=76
Placebo (N=76)
Somnolence1 31% 38% 5% Headache 19% 29% 18% Upper Abdominal Pain 13% 20% 17% Fatigue2 13% 16% 1% Upper Respiratory Tract Infection 6% 11% 4% Irritability 6% 9% 3% Throat Pain 6% 8% 3% Nausea 8% 5% 4% Nightmare 9% 3% 0 Dizziness 3% 7% 5% Insomnia 6% 4% 1% Emotional Disorder 5% 4% 1% Constipation 6% 1% 0 Dry Mouth 5% 0 1% Nasal Congestion 5% 3% 1% Body Temperature Increased 1% 5% 3% Gastrointestinal Viral 0% 7% 4% Diarrhea 1% 4% 3% Ear Pain 0 5% 1% Nasopharyngitis 3% 3% 1% Abnormal Sleep-Related Event 1% 3% 0 Aggression 1% 3% 1% Asthma 1% 3% 1% Bradycardia 4% 0 0 Enuresis 4% 0 0 Influenza like Illness 3% 1% 1% Tearfulness 3% 1% 0 Thirst 3% 1% 0 Tremor 3% 1% 0 Epistaxis 0 3% 0 Lower Respiratory Tract Infection
0 3% 1%
Pollakiuria 0 3% 0 Sleep Terror 0 3% 0
1. Somnolence includes the terms “somnolence” and “sedation”.2. Fatigue includes the terms “fatigue” and “lethargy”.Commonly observed adverse reactions (incidence of ≥ 2% in either active treatment group and greater than the rate on placebo) during the taper period are listed in Table 3.
Table 3 Common Adverse Reactions in the Fixed-Dose Monotherapy Trial- Taper period* (Study 1)
Preferred Term
Percentage of Patients Reporting Event KAPVAY
0.4 mg/day N=78
KAPVAY 0.2 mg/day
N=76
Placebo (N=76)
Abdominal Pain Upper 6% 0 3% Headache 2% 5% 3% Gastrointestinal Viral 5% 0 0 Somnolence 3% 2% 0 Heart Rate Increased 3% 0 0 Otitis Media Acute 0 3% 0
*Taper Period: 0.2 mg dose, week 8; 0.4 mg dose, weeks 6-8; Placebo dose, weeks 6-8
Study 2: Flexible-dose KAPVAY as Adjunctive Therapy to PsychostimulantsStudy 2 was a multi-center, randomized, double-blind, placebo-controlled study, with primary efficacy endpoint at 5 weeks, of a flexible dose of KAPVAY as adjunctive therapy to a psychostimulant in children and adolescents (6 to 17 years) who met DSM-IV criteria for ADHD hyperactive or combined inattentive/hyperactive subtypes. KAPVAY was initiated at 0.1 mg/day and titrated up to 0.4 mg/day over a 3-week period. Most KAPVAY treated patients (75.5%) were escalated to the maximum dose of 0.4 mg/day.
Commonly observed adverse reactions (incidence of ≥ 2% in the treatment group and greater than the rate on placebo) during the treatment period are listed in Table 4.
11:31 AM

USE IN SPECIFIC POPULATIONSPregnancyPregnancy Category C: Oral administration of clonidine hydrochloride to pregnant rabbits during the period of embryo/fetal organogenesis at doses of up to 80 mcg/kg/day (approximately 3 times the oral maximum recommended daily dose [MRHD] of 0.4 mg/day on a mg/m2 basis) produced no evidence of teratogenic or embryotoxic potential. In pregnant rats, however, doses as low as 15 mcg/kg/day (1/3 the MRHD on a mg/m2 basis) were associated with increased resorptions in a study in which dams were treated continuously from 2 months prior to mating and throughout gestation. Increased resorptions were not associated with treatment at the same or at higher dose levels (up to 3 times the MRHD) when treatment of the dams was restricted to gestation days 6-15. Increases in resorptions were observed in both rats and mice at 500 mcg/kg/day (10 and 5 times the MRHD in rats and mice, respectively) or higher when the animals were treated on gestation days 1-14; 500 mcg/kg/day was the lowest dose employed in this study. No adequate and well-controlled studies have been conducted in pregnant women. Because animal reproduction studies are not always predictive of human response, this drug should not be used during pregnancy unless clearly needed.
Nursing MothersSince clonidine hydrochloride is excreted in human milk, caution should be exercised when KAPVAY is administered to a nursing woman.
Pediatric UseA study was conducted in which young rats were treated orally with clonidine hydrochloride from day 21 of age to adulthood at doses of up to 300 mcg/kg/day, which is approximately 3 times the maximum recommended human dose (MRHD) of 0.4 mg/day on a mg/m2 basis. A slight delay in onset of preputial separation was seen in males treated with the highest dose (with a no-effect dose of 100 mcg/kg/day, which is approximately equal to the MRHD), but there were no drug effects on fertility or on other measures of sexual or neurobehavioral development.
KAPVAY has not been studied in children with ADHD less than 6 years old.
Patients with Renal ImpairmentThe impact of renal impairment on the pharmacokinetics of clonidine in children has not been assessed. The initial dosage of KAPVAY should be based on degree of impairment. Monitor patients carefully for hypotension and bradycardia, and titrate to higher doses cautiously. Since only a minimal amount of clonidine is removed during routine hemodialysis, there is no need to give supplemental KAPVAY following dialysis.
Adult Use in ADHDKAPVAY has not been studied in adult patients with ADHD.
DRUG ABUSE AND DEPENDENCEControlled SubstanceKAPVAY is not a controlled substance and has no known potential for abuse or dependence.
OVERDOSAGESymptomsClonidine overdose: hypertension may develop early and may be followed by hypotension, bradycardia, respiratory depression, hypothermia, drowsiness, decreased or absent reflexes, weakness, irritability and miosis. The frequency of CNS depression may be higher in children than adults. Large overdoses may result in reversible cardiac conduction defects or dysrhythmias, apnea, coma and seizures. Signs and symptoms of overdose generally occur within 30 minutes to two hours after exposure.
TreatmentConsult with a Certified Poison Control Center for up-to-date guidance and advice.
© 2010 Shionogi Pharma, Inc. Florham Park, NJ 07932 Last modified 10/2010
Table 4 Common Adverse Reactions in the Flexible-Dose Adjunctive to Stimulant Therapy Trial- Treatment Period (Study 2)
Preferred Term Percentage of Patients Reporting Event
KAPVAY+STM (N=102)
PBO+STM (N=96)
Somnolence1 19% 8% Fatigue2 16% 4%
Abdominal Pain Upper 12% 7% Nasal Congestion 6% 5% Throat Pain 6% 3% Decreased Appetite 5% 4% Body Temperature Increased 4% 2% Dizziness 4% 2% Insomnia 4% 2% Epistaxis 3% 0 Rhinorrhea 3% 0 Abdominal Pain 2% 1% Anxiety 2% 0 Pain in Extremity 2% 0
1. Somnolence includes the terms: “somnolence” and “sedation”.
2. Fatigue includes the terms “fatigue” and “lethargy”.
Commonly observed adverse reactions (incidence of ≥ 2% in the treatment group and greater than the rate on placebo) during the taper period are listed in Table 5.
Table 5 Common Adverse Reactions in the Flexible-Dose Adjunctive to Stimulant Therapy Trial- Taper Period* (Study 2)
Preferred TermPercentage of Patients Reporting EventKAPVAY+STM
(N=102)PBO+STM
(N=96)Nasal Congestion 4% 2% Headache 3% 1% Irritability 3% 2% Throat Pain 3% 1% Gastroenteritis Viral 2% 0 Rash 2% 0
*Taper Period: weeks 6-8.
Most common adverse reactions, defined as events that were reported in at least 5% of drug-treated patients and at least twice the rate as in placebo patients, during the treatment period were somnolence, fatigue, upper respiratory tract infection, irritability, throat pain, insomnia, nightmares, emotional disorder, constipation, nasal congestion, increased body temperature, dry mouth, and ear pain. The most common adverse reactions that were reported during the taper phase were upper abdominal pain and gastrointestinal virus.Adverse Reactions Leading to DiscontinuationThirteen percent (13%) of patients receiving KAPVAY discontinued from the pediatric monotherapy study due to adverse events, compared to 1% in the placebo group. The most common adverse reactions leading to discontinuation of KAPVAY monotherapy treated patients were from somnolence/sedation (5%) and fatigue (4%). Less common adverse reactions leading to discontinuation (occurring in approximately 1% of patients) included: formication, vomiting, prolonged QT, increased heart rate, and rash. In the pediatric adjunctive treatment to stimulants study, one patient discontinued from KAPVAY + stimulant group because of bradyphrenia.
Effects on Laboratory Tests, Vital Signs, and ElectrocardiogramsKAPVAY treatment was not associated with any clinically important effects on any laboratory parameters in either of the placebo-controlled studies.
Mean decreases in blood pressure and heart rate were seen [see Warnings and Precautions (5.1)].
There were no changes on ECGs to suggest a drug-related effect.
DRUG INTERACTIONSNo drug interaction studies have been conducted with KAPVAY in children. The following have been reported with other oral immediate release formulations of clonidine.
Interactions with CNS-depressant DrugsClonidine may potentiate the CNS-depressive effects of alcohol, barbiturates or other sedating drugs.
Interactions with Tricyclic AntidepressantsIf a patient is receiving clonidine hydrochloride and also taking tricyclic antidepressants the hypotensive effects of clonidine may be reduced.
Interactions with Drugs Known to Affect Sinus Node Function or AV Nodal ConductionDue to a potential for additive effects such as bradycardia and AV block, caution is warranted in patients receiving clonidine concomitantly with agents known to affect sinus node function or AV nodal conduction (e.g., digitalis, calcium channel blockers and beta-blockers).
Use with other products containing clonidineDo not use KAPVAY concomitantly with other products containing clonidine (e.g. Catapres®).
Antihypertensive DrugsUse caution when KAPVAY is administered concomitantly with antihypertensive drugs, due to the potential for additive pharmacodynamic effects (e.g., hypotension, syncope) [see Warnings and Precautions (5.2)].
11:31 AM

• When added to a stimulant, extended-release Kapvay™ demonstrated statistically signifi cant improvement of ADHD symptoms compared with a stimulant alone at the end of 5 weeks of treatment, as measured by the ADHD RS-IV total score
IndicationKapvay™ (clonidine hydrochloride) extended-release tablets are indicated for the treatment of attention defi cit/hyperactivity disorder (ADHD) as monotherapy or as adjunctive therapy to stimulant medications in children and adolescents ages 6-17. The effi cacy of Kapvay™ is based on the results of 2 clinical trials in children and adolescents. Kapvay™ is indicated as an integral part of a total treatment program for ADHD that may include other measures (psychological, educational, and social) for patients with this syndrome.The effectiveness of Kapvay™ for longer-term use (more than 5 weeks) has not been systematically evaluated in controlled trials; therefore, the physician electing to use Kapvay™ for extended periods should periodically re-evaluate the long-term usefulness of the drug for the individual patient.
Important Safety Information• Kapvay™ should not be used in patients with known hypersensitivity to clonidine• Kapvay™ can cause dose-related decreases in blood pressure and heart rate. Use caution in treating patients who have a history of syncope
or may have a condition that predisposes them to syncope, such as hypotension, orthostatic hypotension, bradycardia, or dehydration. Use with caution in patients treated concomitantly with antihypertensives or other drugs that can reduce blood pressure or heart rate or increase the risk of syncope
• Somnolence/Sedation were commonly reported adverse reactions in clinical studies with Kapvay™. Potential for additive sedative effects with CNS-depressant drugs. Advise patients to avoid use with alcohol. Caution patients against operating heavy equipment or driving until they know how they respond to Kapvay™
• Patients should be instructed not to discontinue Kapvay™ therapy without consulting their physician due to the potential risk of withdrawal effects. Kapvay™ should be discontinued slowly in decrements of no more than 0.1 mg every 3 to 7 days
• In patients who have developed localized contact sensitization or other allergic reaction to clonidine in a transdermal system, substitution of oral clonidine hydrochloride therapy may be associated with the development of a generalized skin rash, urticaria, or angioedema. Use cautiously in patients with vascular disease, cardiac conduction disease, or chronic renal failure: Monitor carefully and uptitrate slowly
• Clonidine may potentiate the CNS-depressive effects of alcohol, barbiturates or other sedating drugs• Use caution when Kapvay™ is administered concomitantly with antihypertensive drugs, due to the additive pharmacodynamic effects
(e.g., hypotension, syncope)• Kapvay™ should not be used during pregnancy unless clearly needed. Since clonidine hydrochloride is excreted in human milk, caution should be
exercised when Kapvay™ is administered to a nursing woman• Caution is warranted in patients receiving clonidine concomitantly with agents known to affect sinus node function or AV nodal conduction
(e.g., digitalis, calcium channel blockers and beta-blockers) due to a potential for additive effects, such as bradycardia and AV block• Clonidine, the active ingredient in Kapvay™, is also approved as an antihypertensive. Do not use
Kapvay™ in patients concomitantly taking other clonidine-containing products, (e.g., Catapres® [clonidine hydrochloride], JENLOGA)
• Common adverse reactions (incidence at least 5% and twice the rate of placebo) include: somnolence, fatigue, upper respiratory tract infection, irritability, throat pain, insomnia, nightmares, emotional disorder, constipation, nasal congestion, increased body temperature, dry mouth, and ear pain
Extended-Release Formulation
© 2011 Shionogi Pharma, Inc. Atlanta, Georgia. All rights reserved. KAP10-PAD-002-00
* Kapvay™ was FDA approved on September 28, 2010.
Kapvay™ is a trademark of Shionogi Pharma, Inc.Catapres® is a registered trademark of Boehringer Ingelheim.
Please see Brief Summary of full Prescribing Information on the adjacent page.
When you treat Attention Defi cit/Hyperactivity Disorder (ADHD) with stimulants, for some patients, a question may be...
8:18 PM


















