
Pairwise Sequence Alignment and Scoring Matrices Xiaole Shirley Liu And Jun Liu Stat 115 Lecture 3.
46
Pairwise Sequence Alignment and Scoring Matrices Xiaole Shirley Liu And Jun Liu Stat 115 Lecture 3
-
Upload
griffin-fowler -
Category
Documents
-
view
230 -
download
0
Transcript of Pairwise Sequence Alignment and Scoring Matrices Xiaole Shirley Liu And Jun Liu Stat 115 Lecture 3.

Pairwise Sequence Alignmentand Scoring Matrices
Xiaole Shirley Liu
And
Jun Liu
Stat 115 Lecture 3

STAT 1152
Mol Bio Quick Facts
• Building block of DNA is deoxyribonucleic acid
• Building block of protein is amino acid– Protein is a peptide, a long peptide
• Only exons can code for functional proteins or RNAs– Introns are spliced out

STAT 1153
Outline
• Motivation and introduction• Dynamic programming
– Global sequence alignment• Needleman-Wunsch• 3 steps: Initialize, fill matrix, trace back• Gap penalties
– Local sequence alignment, Smith-Waterman
• Scoring matrices– PAM– BLOSUM

STAT 1154
Pairwise Sequence Alignment
• Given: two sequences, scoring for match/mismatch/gap
• Goal: find pairing of letters in the two sequences that optimize the total scoreThis is a hard example.
That is another easy example.
This is a --hard---- example.
|| ||||| | | |||||||||
That is another easy example.
gap
match
mismatch

STAT 1155
Align Biological Sequences
• DNA (4 nt + gap)TTGATCAC
TTTA-CAC
• Protein (20 aa + gap)RKVA--GMAKPNM
RKIAVAAASKPAV
• Sometimes > 4 nt for DNA and > 20 aa for proteins– A word on IUPAC

STAT 1156
IUPAC for DNA
A adenosine
C cytidine
G guanine
T thymidine
U uridine
R G A (purine)
Y T C (pyrimidine)
K G T (keto)
M A C (amino)
S G C (strong)
W A T (weak)
B C G T (not A)
D A G T (not C)
H A C T (not G)
V A C G (not T)
N A C G T (any)
– gap
STAT 1157
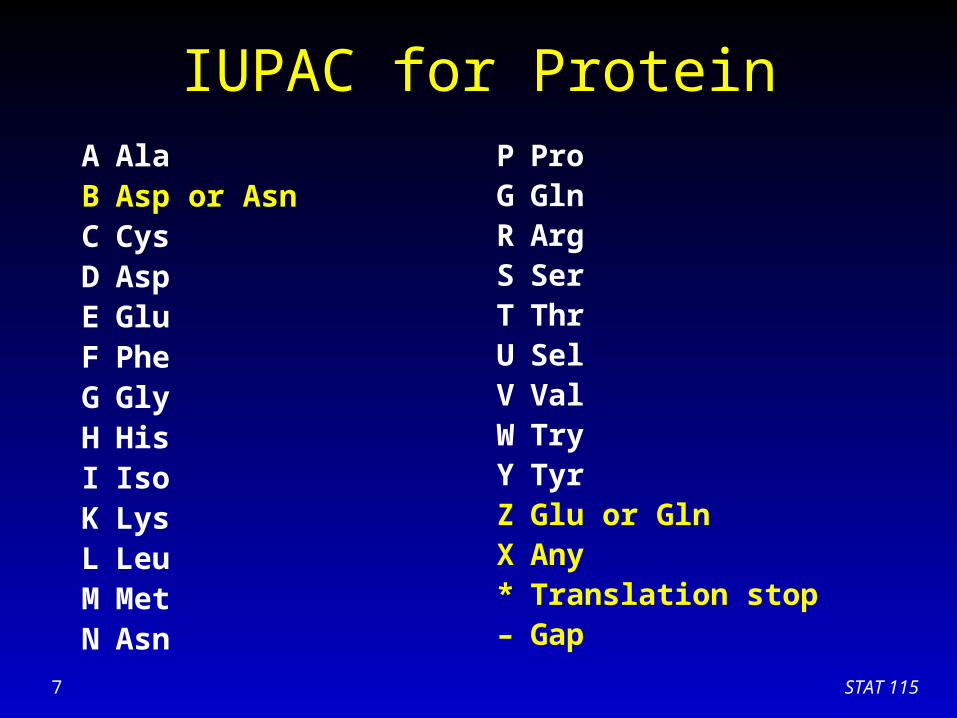
IUPAC for ProteinA AlaB Asp or AsnC CysD AspE GluF PheG GlyH HisI IsoK LysL LeuM MetN Asn
P ProG GlnR ArgS SerT ThrU SelV ValW TryY TyrZ Glu or GlnX Any* Translation stop– Gap

STAT 1158
Why Align Two Sequences
• If two sequences are similar, they might share the same ancestor
• If two sequences are similar, they may share the same structure, therefore similar function
• In genome sequencing assembly, if two sequences have overlapping similar regions, they might be connected to represent longer sequenced region.

STAT 1159
Scoring Schemes• Match, mismatch, gap score determines the final
alignmentThis is a --hard---- example.
|| ||||| | | |||||||||
That is another easy example.
• Match OK, mismatch costly, gap cheap.This is a-- h-ard---- example.
Th--at is anothe-r easy example.
• Match cheap, mismatch cheap, gap costly.This is a hard example.------
That is another easy example.

STAT 11510
Dot Matrix Approach
• Naïve algorithm
• Dot – match, find diagonal lines
• Can’t afford more complex scoring
• Visual analysis,
hard to find
optimal alignments

STAT 11511
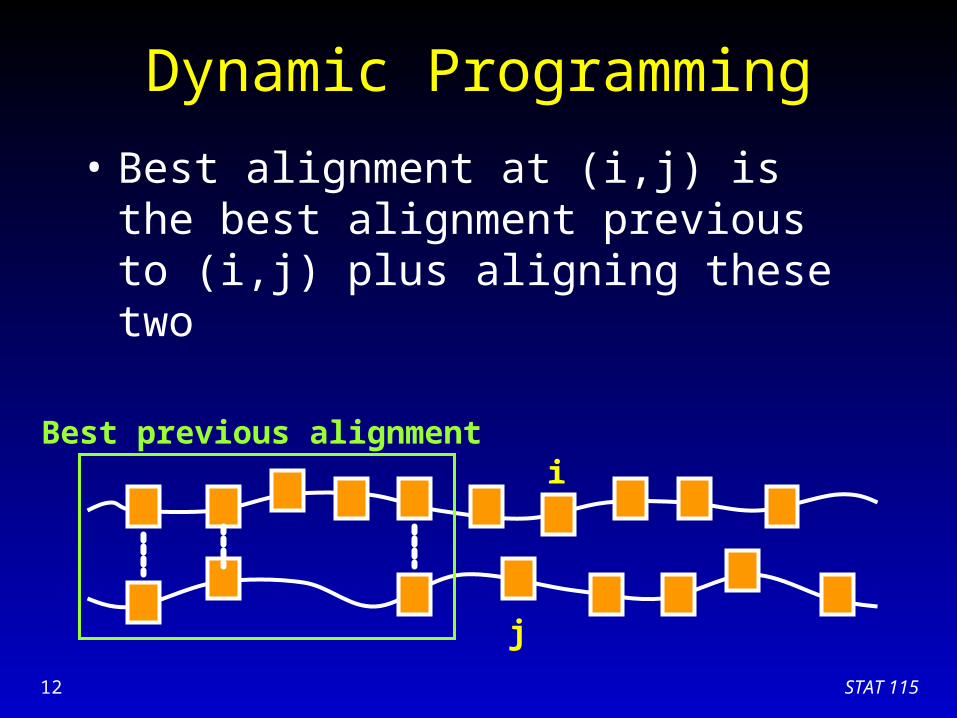
Dynamic Programming
• Essence of dynamic programming:– Store the sub-problem solutions for later use– Best alignment at (i,j) is the best alignment
previous to (i,j) plus aligning these two
• Earliest method, Needleman & Wunsch 1969
• Still the best (sensitive and optimal) algorithm for pair-wise alignment
STAT 11512
Dynamic Programming
• Best alignment at (i,j) is the best alignment previous to (i,j) plus aligning these two
i
j
Best previous alignment
STAT 11513
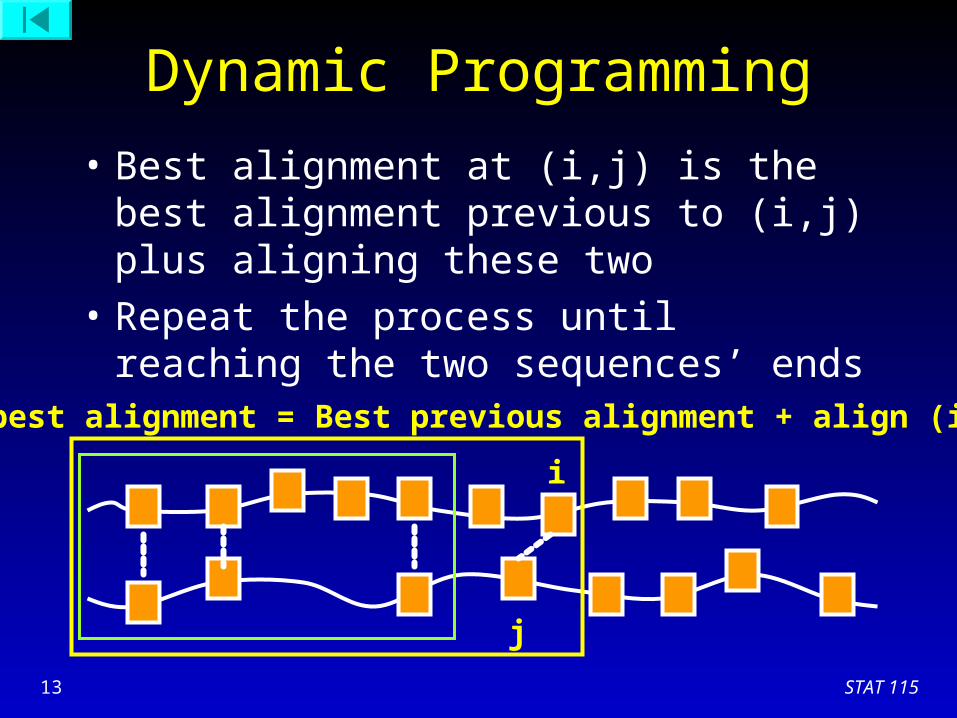
Dynamic Programming
• Best alignment at (i,j) is the best alignment previous to (i,j) plus aligning these two
• Repeat the process until reaching the two sequences’ ends
i
j
New best alignment = Best previous alignment + align (i,j)

STAT 11514
Dynamic Programming Steps
• Initialize NxM matrix– N, M are the length of the two sequences
• Fill the matrix– Each element record the current best score,
and pointer to the previous best alignment– Always search the previous column and row
for the best previous alignment
• Trace back to obtain optimal alignment
STAT 11515
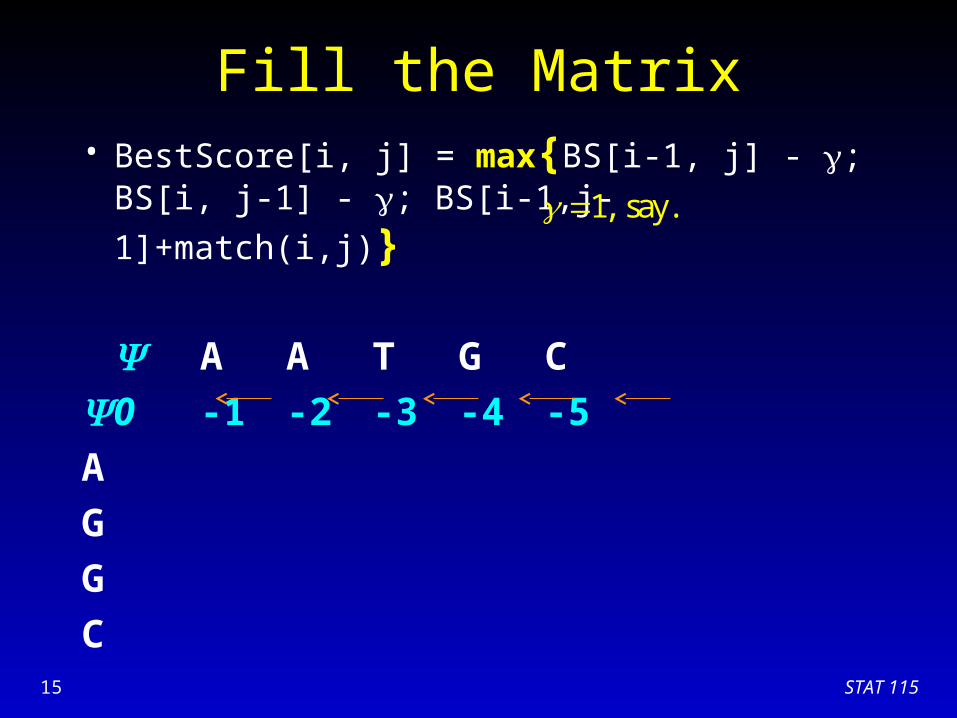
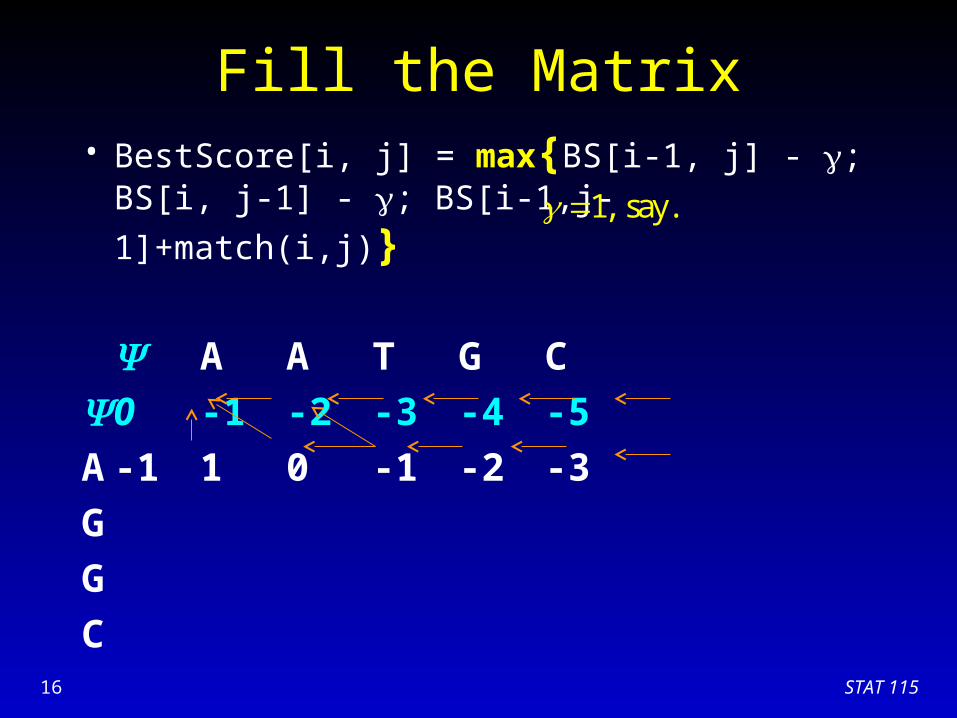
Fill the Matrix• BestScore[i, j] = max{BS[i-1, j] - ; BS[i, j-1] - ;
BS[i-1,j-1]+match(i,j)}
A A T G C
0 -1 -2 -3 -4 -5
A
G
G
C
1, say.
STAT 11516
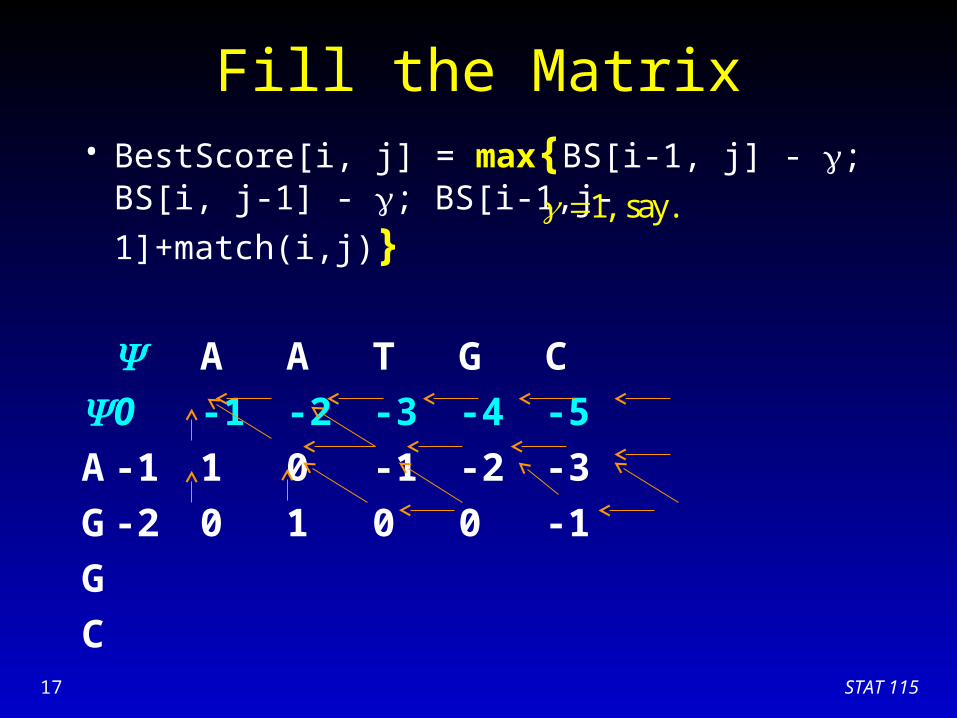
Fill the Matrix• BestScore[i, j] = max{BS[i-1, j] - ; BS[i, j-1] - ;
BS[i-1,j-1]+match(i,j)}
A A T G C
0 -1 -2 -3 -4 -5
A -1 1 0 -1 -2 -3
G
G
C
1, say.
STAT 11517
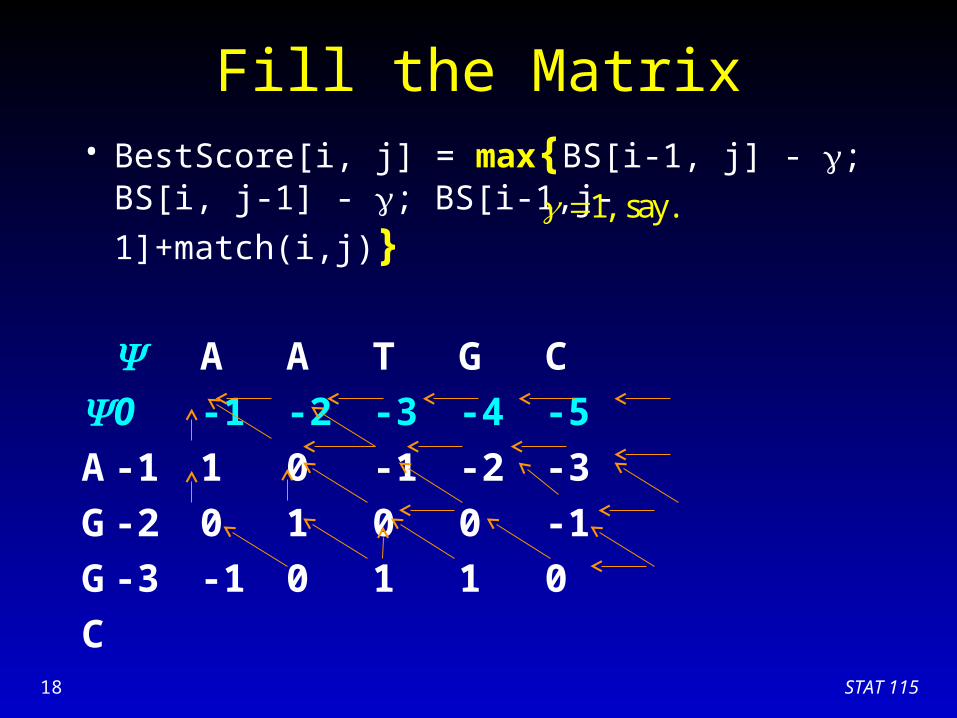
Fill the Matrix• BestScore[i, j] = max{BS[i-1, j] - ; BS[i, j-1] - ;
BS[i-1,j-1]+match(i,j)}
A A T G C
0 -1 -2 -3 -4 -5
A -1 1 0 -1 -2 -3
G -2 0 1 0 0 -1
G
C
1, say.
STAT 11518
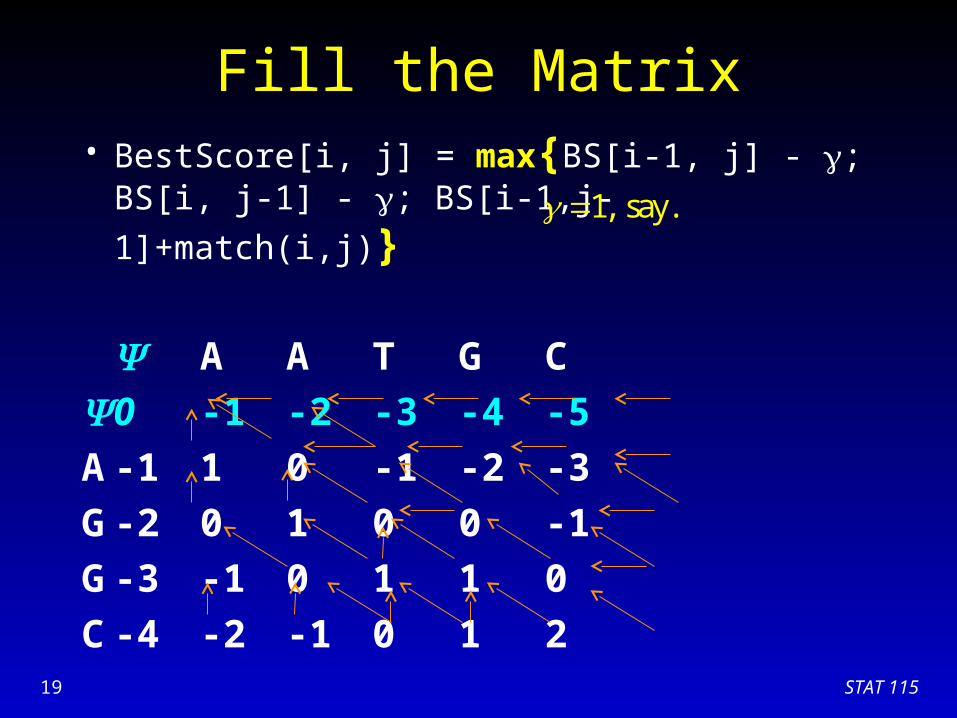
Fill the Matrix• BestScore[i, j] = max{BS[i-1, j] - ; BS[i, j-1] - ;
BS[i-1,j-1]+match(i,j)}
A A T G C
0 -1 -2 -3 -4 -5
A -1 1 0 -1 -2 -3
G -2 0 1 0 0 -1
G -3 -1 0 1 1 0
C
1, say.
STAT 11519
Fill the Matrix• BestScore[i, j] = max{BS[i-1, j] - ; BS[i, j-1] - ;
BS[i-1,j-1]+match(i,j)}
A A T G C
0 -1 -2 -3 -4 -5
A -1 1 0 -1 -2 -3
G -2 0 1 0 0 -1
G -3 -1 0 1 1 0
C -4 -2 -1 0 1 2
1, say.

STAT 11520
Fill the Matrix• BestScore[i, j] = max{BS[i-1, j] - ; BS[i, j-1] - ;
BS[i-1,j-1]+match(i,j)}
A A T G C
0 -1 -2 -3 -4 -5
A -1 1 0 -1 -2 -3
G -2 0 1 0 0 -1
G -3 -1 0 1 1 0
C -4 -2 -1 0 1 2
1, say.
Trace back
AATGC-AGGC
AATGCAG-GC

STAT 11521
Alignment Recursion(linear gap penalty)
F(i,j) F(i-1,j)
F(i,j-1)F(i-1,j-1)
( 1, 1) ( , )
( , ) max ( 1, )
( , 1)
i jF i j s x y
F i j F i j
F i j
C A T T G
TCATG
0 -1 -2 -3 -4 -5-1
1-2-3-4-5
E.g., { }( , ) 2 , 1x ys x y I 0-1-2
0 -10332
554
0-12 1
46

STAT 11522
Affine Gap Penalty
• Gap penalty function: – Typically a>b (e.g., a=12; b=2)
• An order O(nm) algorithm.
( ) ( 1)g a g b
F(i,j)
( , ) max{ ( 1, 1) ( , ); ( , ); ( , )}i j h vF i j F i j s x y F i j F i j
( , ) max{ ( , 1) , ( , 1) }h hF i j F i j a F i j b
( , ) max{ ( 1, ) , ( 1, ) }v vF i j F i j a F i j b
Key: keep 3 functions, each recording a directional optimum.

STAT 11523
Gap Penalty
• Gap penalty = g + l e
A A T G C
A 1 1 0 0 0
G 0 1 1 1.4 0.3
G
C
g – gap startl – gap lengthe – gap extend
e.g. g = -0.5 e = -0.1
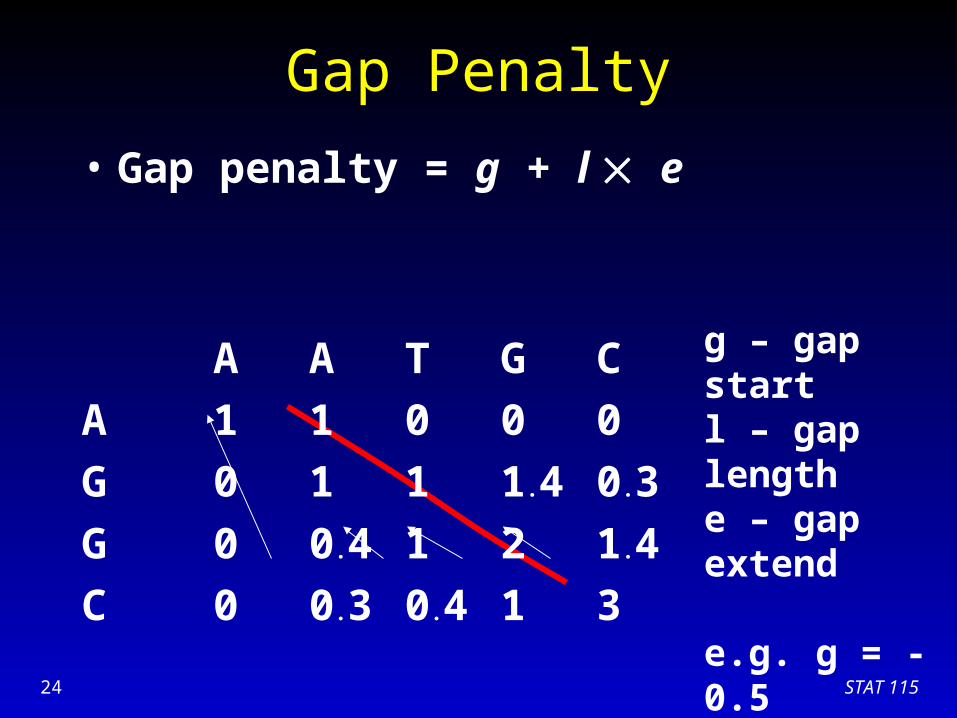
STAT 11524
Gap Penalty
• Gap penalty = g + l e
A A T G C
A 1 1 0 0 0
G 0 1 1 1.4 0.3
G 0 0.4 1 2 1.4
C 0 0.3 0.4 1 3
g – gap startl – gap lengthe – gap extend
e.g. g = -0.5 e = -0.1

STAT 11525
End Gaps
• Should we penalize gaps at the ends?
ATCCGCATACCGGA
--CCGCATAC----• If two sequences similar length and entire
sequences are supposed to be similar, penalize.
• If two sequences have very different length, do not penalize (most of the time, ignore end gap penalties)

STAT 11526
Global vs. Local Alignment
• Global: Needleman-Wunsch– Find best alignment for the whole 2 sequences– Could have no penalty for mismatches/gaps– Trace back from lower right corner to upper left
corner
• Local: Smith-Waterman– Find high scoring subsequences– E.g. Two proteins only share one similar
functional domain– Can be achieved by modifying global
alignment method

STAT 11527
Local Alignment Modifications
1. Use negative mismatch and gap penalties2. The minimum score for [i, j] is 0
– If S[i,j] < 0, rewrite it to 0, point to self– If previous col/row is all 0, S[i,j] point to self
3. The best score is sought anywhere in the matrix
– Not just last column and last row (should keep a global pointer to the best score)
– Trace back until a cell pointing to itself (not necessary to the beginning of the two sequences)
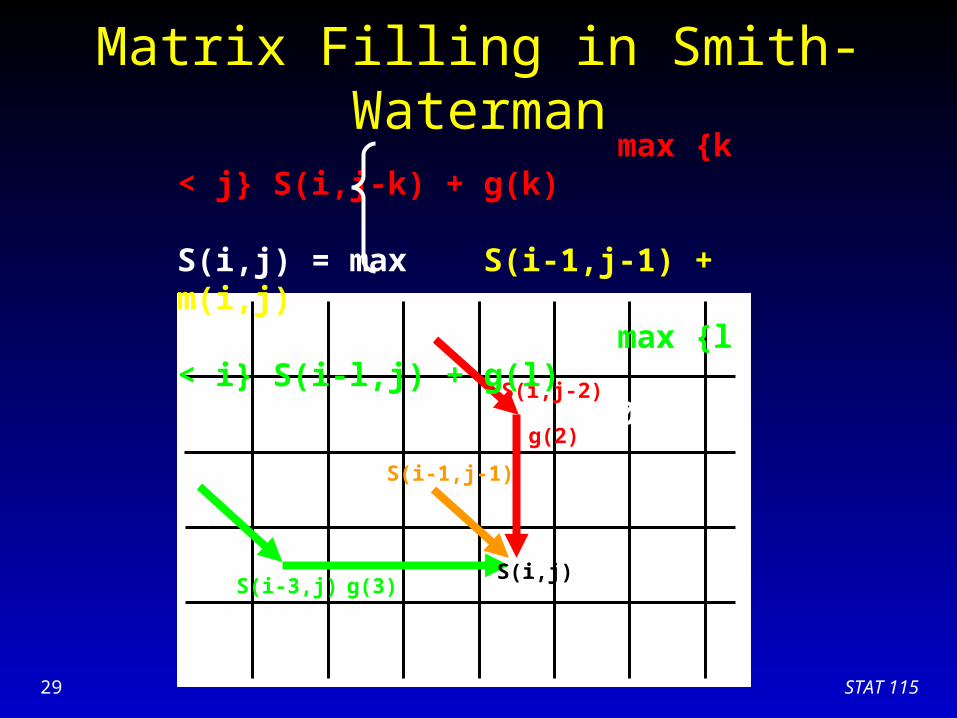
STAT 11529
Matrix Filling in Smith-Waterman
S(i,j)S(i-3,j)
S(i,j-2)
g(3)
g(2)
S(i-1,j-1)
max {k < j} S(i,j-k) + g(k) S(i,j) = max S(i-1,j-1) + m(i,j) max {l < i} S(i-l,j) + g(l) 0

Example
STAT 11530

Finding suboptimal alignments
STAT 11531

STAT 11532
Smith-Waterman
• Negative mis-match & negative gaps• Scoring matrix >= 0• Trace from maximum
Seq. T j j+1 … … … … nM A T C H E S
Seq. S 0 0 0 0 0 0 0 0i T 0 0 0 5 0 0 0 2i+1 H 0 0 0 0 2 10 2 0… A 0 0 5 0 0 2 9 3… T 0 0 0 10 2 0 9 3… C 0 0 0 2 23 15 7 3… H 0 0 0 0 15 33 25 17… E 0 0 0 0 7 25 39 31m R 0 0 0 0 0 17 31 38
A T C H EA T C H E

STAT 11533
Scoring Matrices
• For DNA, match + 5, mismatch – 4
• For proteins, different amino acid pairs receive different scores– Consideration: size, shape, electric charge, van
der Waals interaction, ability to form salt, hydrophobic, and hydrogen bonds
– Substitution matrices • Often symmetrical
• + / – scores:
functional similarity

STAT 11534
PAM Matrices
• MO Dayhoff 1978
• PAM: percent accepted mutations– Database of 1572 changes in 71 groups of
closely related proteins (> 85% similar)– Construct phylogenetic tree of each group,
tabulate probability of amino acid changes between every pairs of aa
– For statistician: Markov chain transition b/w aa pairs

STAT 11535
PAM Matrix Family
• PAM-N– PAM-0: 1 on diagonal and 0 all the rest– PAM-N: what would happen if N out of 100 aa
mutate – For statisticians: matrix multiplication N times– Bigger N, more diverged substitution matrice
• Final matrix
102/]})(
)([log]
)(
)([{log
1
1210
2
2110
aaFreq
aatoaaPb
aaFreq
aatoaaPb
STAT 11536
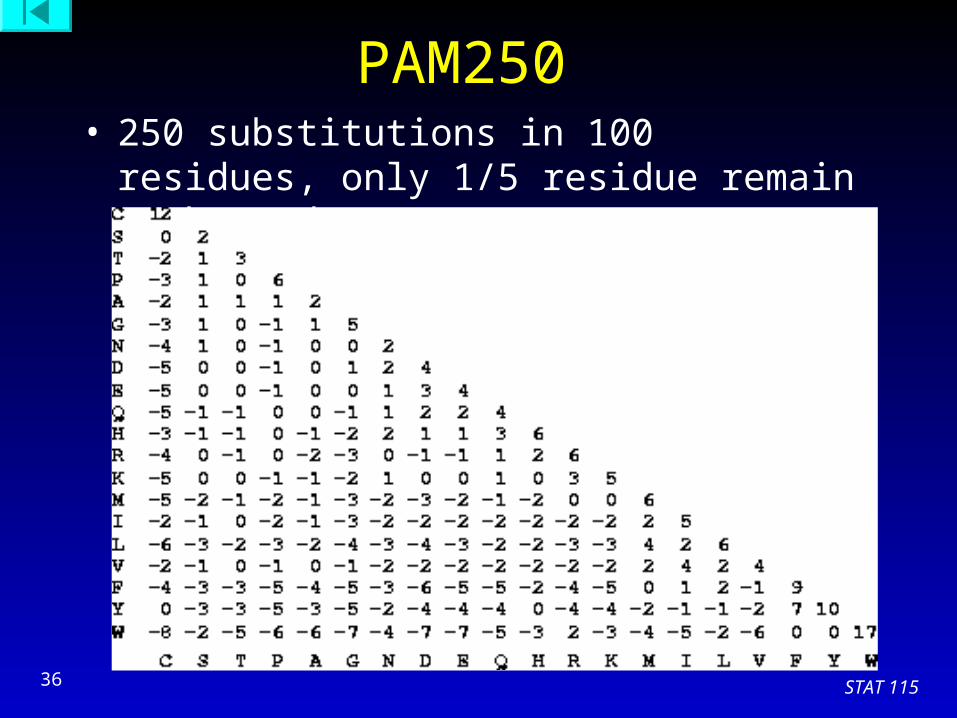
PAM250 • 250 substitutions in 100 residues, only 1/5 residue
remain unchanged

STAT 11537
BLOSUM Matrices
• BLOcks amino acid SUbstitution Matrices
• Henikoff and Henikoff, PNAS. 1992, 89:10915-9– Check >500 protein families in the Prosite
database (Bairoch 1991)– Find ~2000 blocks of aligned segments
• BLOCKS database
• Characterize ungapped patterns 3-60 aa long
• Check aligned columns for observed substitutions

STAT 11538
BLOSUM Matrix Entry
• How frequently do aa appear
• How often do we expect to see i, j together
• How often do we actually see them together in all the alignments
• BLOSUM entry
ji ff ,
jiij ffe 2
ijq
)/(log2 2 ijijij eqs

STAT 11539
BLOSUM62 Matrix

STAT 11540
BLOSUM Matrices
• Blocks are grouped before looking at aa substitutions– BLOSUMN: if sequences > N% identical, their
contributions are weighted to sum to 1
• Most widely used: BLOSUM62 and PAM250

STAT 11541
More About Dynamic Programming• Example 1: Suppose I have x0 amount of savings at retirement,
and also receive st amount of social security payment every year. Annual interest rate is t, what is an optimal spending plan if I want to leave zero dollars at the end (say, year 5)?
Year 0 Year 1 Year 2 Year 3 Year 4
x0 x1 x2 x3 x4
s0 s1 s2 s3 s4
u0 u1 u2 u3 u4
1 0
0 0 0
(1 )( )
xx s u
2 1
1 1 1
(1 )( )xx s u
5 4
4 4 4
(1 )( )xx s u
4 3
3 3 3
(1 )( )xx s u
3 2
2 2 2
(1 )( )xx s u
4 4 4u s x
Maximizing total spending? 0 1 4u u u
3 4 3 4
3 3 3 3(1 )( )
u u u s
x s u
3 0u
40 1 4u u u

STAT 11542
Example 2: Secretary Problem
• We get to observe the “qualities” of m secretaries: X1,…, Xm sequentially according to a random order. Our goal is to maximize the probability of finding the “best” candidate with no looking back!
• Heuristic: We start our reasoning backwards. Suppose we have seen X1,…, Xj, should we stop or go on?

STAT 11543
• What if we wait till the last person?
• What if we wait till having two people left?– Strategy: if m-1st person is better than previous
ones, take her; otherwise wait till the last one.
• Get a recursion? If we let go j-1 people, and take the best-person-so-far starting from jth person ...
1
1 11
1m mP Pm m
1
1 11j jP P
m j
Let’s start reasoning ...1
mPm
1 1 1
1 1
j
m j m
Pj maximized at
2.718...
m mj
e

STAT 11544
Final “answer”
• We should reject the first 37% of the candidates and start to look: recruit the first person who is the best among all that have been interviewed.
• The chance of getting the best one is ~37% !

STAT 11545
Summary
• Dynamic programming finds optimal alignment between two sequences– Keep subproblem solution for later use– Needleman & Wunsch, global– Smith & Waterman, local
• Scoring sensitive to gap penalty and substitution matrices used– Substitution matrices capture aa similarity– PAM and BLOSUM matrices

STAT 11546
Question for Thoughts
• Given a string of integers (both positive and negative)– E.g. 3, -1, -5, 2, 4, -3, 6, -4, 2, 5, -8, 3, 1, -7, 6
• Can you read each number only ONCE, and tell from which number to which number you will get the largest sum?– 3, -1, -5, 2, 4, -3, 6, -4, 2, 5, -8, 3, 1, -7, 6, -2– Largest sum = 12
• Hint: dynamic programming

STAT 11547
Acknowledgement
• Russ Altman
• Theodor Hanekamp
• Eric Rouchka


















