
p53-Dependent DNA damage response sensitive to editing ...
6
p53-Dependent DNA damage response sensitive to editing-defective tRNA synthetase in zebrafish Youngzee Song a,b,c,1 , Yi Shi a,b,c,1 , Tristan M. Carland d,e,f , Shanshan Lian g , Tomoyuki Sasaki g , Nicholas J. Schork d,e,f , Steven R. Head h , Shuji Kishi g,2 , and Paul Schimmel a,b,c,g,2 a The Scripps Laboratories for tRNA Synthetase Research, The Scripps Research Institute, La Jolla, CA 92037; b Department of Cell and Molecular Biology, The Skaggs Institute for Chemical Biology, The Scripps Research Institute, La Jolla, CA 92037; c Department of Chemistry, The Skaggs Institute for Chemical Biology, The Scripps Research Institute, La Jolla, CA 92037; d The Scripps Translational Science Institute, The Scripps Research Institute, La Jolla, CA 92037; e Department of Molecular and Experimental Medicine, The Scripps Research Institute, La Jolla, CA 92037; f Department of Human Biology, The J. Craig Venter Institute, La Jolla, CA 92037; g Department of Metabolism and Aging, The Scripps Research Institute, Jupiter, FL 33458; and h Next Generation Sequencing and Microarray Core Facility, The Scripps Research Institute, La Jolla, CA 92037 Contributed by Paul Schimmel, May 25, 2016 (sent for review February 27, 2016; reviewed by Doron Betel, Feng Cui, and Shuo Lin) Brain and heart pathologies are caused by editing defects of transfer RNA (tRNA) synthetases, which preserve genetic code fidelity by re- moving incorrect amino acids misattached to tRNAs. To extend under- standing of the broader impact of synthetase editing reactions on organismal homeostasis, and based on effects in bacteria ostensibly from small amounts of mistranslation of components of the replication apparatus, we investigated the sensitivity to editing of the vertebrate genome. We show here that in zebrafish embryos, transient overex- pression of editing-defective valyl-tRNA synthetase (ValRS ED ) activated DNA break-responsive H2AX and p53-responsive downstream pro- teins, such as cyclin-dependent kinase (CDK) inhibitor p21, which pro- motes cell-cycle arrest at DNA damage checkpoints, and Gadd45 and p53R2, with pivotal roles in DNA repair. In contrast, the response of these proteins to expression of ValRS ED was abolished in p53-deficient fish. The p53-activated downstream signaling events correlated with suppression of abnormal morphological changes caused by the editing defect and, in adults, reversed a shortened life span (followed for 2 y). Conversely, with normal editing activities, p53-deficient fish have a normal life span and few morphological changes. Whole-fish deep sequencing showed genomic mutations associated with the editing defect. We suggest that the sensitivity of p53 to expression of an editing-defective tRNA synthetase has a critical role in promot- ing genome integrity and organismal homeostasis. mistranslation | genomic fidelity | shortened lifespan | morphological changes | genomic mutations I n the first step of protein synthesis, amino acids are attached to their cognate tRNAs in reactions catalyzed by aminoacyl-tRNA synthetases (1, 2). Mistakes of aminoacylation occur with a frequency of less than 1% (3, 4), and if not corrected, will result in the insertion of a mischarged amino acid into a growing polypeptide chain at the wrong codon (5–7). This sort of error is usually corrected by a uni- versal editing mechanism in tRNA synthetases, which hydrolyti- cally removes the mischarged amino acid from the tRNA before the wrong amino acid is inserted into a nascent protein (8–14). Serious pathologies and cell death result from rare errors in protein synthesis (mistranslation). For example, a mild editing defect of an alanyl-tRNA synthetase (AlaRS) in the mouse was shown to cause the death of Purkinje cells and result in neurodegeneration (5). More recently, cardioproteinopathy was also shown to have connections to mistranslation (15). Although the cellular and physiological impor- tance of editing has been established, the extent of how editing defects impact major cellular mechanisms is only beginning to be clarified. Because assaults on the genome by environmental factors lead to somatic cell mutations that cause diseases in humans, we were especially interested in the role editing might play in protecting ver- tebrates against DNA damage. Earlier work in bacteria was sup- portive of such a connection (16– 18), and later work established that the occasional misincorporation of the wrong amino acids into en- zymes of the DNA replication and repair apparatus was mutagenic by virtue of error-prone repair of DNA damage (19) (Fig. 1A). Here, we investigated whether this phenomenon could extend to vertebrates. Because coupling of mistranslation and genome fidelity would have obvious implications for organismal homeostasis and disease, we chose the zebrafish, a popular model organism (with many of the same genes and disorders as mammals) (20, 21) as a system to explore the linkage between translation and ge- nome fidelity. Using editing-defective (ED) valyl-tRNA synthetase (ValRS), we demonstrated that, in response to mistranslation, the DNA damage response (DDR) is activated, as shown by phos- phorylation of H2AX and up-regulation of p53 downstream genes. In addition, ED ValRS shortened the lifespan of p53-deficient zebrafish, and whole-fish genomic DNA sequencing showed ge- nomic mutations were associated with the editing defect. Thus, p53 surveillance of mistranslation links translational and genomic fidelity to sustain vertebrate homeostasis. ED ValRS Causes Cell Death in Zebrafish For a mistranslational system, we used an ED tRNA synthetase, which has impaired ability to correct an error of aminoacylation of its cognate tRNA. For this purpose, ValRS, which by editing elim- inates the occasional production of Thr-tRNA Val (22), was chosen. Replacement of a universally conserved T535 (which is T539 in zebrafish ValRS) with a T535P substitution yields an enzyme that is completely deficient in editing and retains full aminoacylation ac- tivity (6). This mutant was designated as ValRS ED , where ED de- notes “editing-defective. ” As shown by direct analysis of mammalian Significance Although DNA damage is a well-known cause of disease, its connection, if any, to mistranslation is unclear. Here, we used an editing-defective tRNA synthetase to show how an editing defect in zebrafish leads to severe consequences during development and to a shortened overall life span of adult fish. These effects were p53-dependent and reflect a strong connection of mis- translation to the DNA damage response. In addition, deep se- quencing of genomic DNA revealed that expression of the editing-defective tRNA synthetase was mutagenic. We suggest that the p53-dependent DNA damage response links translational and genomic fidelity to promote vertebrate homeostasis. Author contributions: Y. Song, Y. Shi, S.R.H., S.K., and P.S. designed research; Y. Song, Y. Shi, S.L., and T.S. performed research; Y. Song, Y. Shi, T.M.C., N.J.S., S.K., and P.S. analyzed data; and Y. Song and P.S. wrote the paper. Reviewers: D.B., Weill Cornell Medical College; F.C., Rochester Institute of Technology; and S.L., University of California, Los Angeles. The authors declare no conflict of interest. 1 Y. Song and Y. Shi contributed equally to this work. 2 To whom correspondence may be addressed. Email: [email protected] or schimmel@ scripps.edu. This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10. 1073/pnas.1608139113/-/DCSupplemental. 8460–8465 | PNAS | July 26, 2016 | vol. 113 | no. 30 www.pnas.org/cgi/doi/10.1073/pnas.1608139113 Downloaded by guest on November 27, 2021
Transcript of p53-Dependent DNA damage response sensitive to editing ...

p53-Dependent DNA damage response sensitive toediting-defective tRNA synthetase in zebrafishYoungzee Songa,b,c,1, Yi Shia,b,c,1, Tristan M. Carlandd,e,f, Shanshan Liang, Tomoyuki Sasakig,Nicholas J. Schorkd,e,f, Steven R. Headh, Shuji Kishig,2, and Paul Schimmela,b,c,g,2
aThe Scripps Laboratories for tRNA Synthetase Research, The Scripps Research Institute, La Jolla, CA 92037; bDepartment of Cell and Molecular Biology, TheSkaggs Institute for Chemical Biology, The Scripps Research Institute, La Jolla, CA 92037; cDepartment of Chemistry, The Skaggs Institute for ChemicalBiology, The Scripps Research Institute, La Jolla, CA 92037; dThe Scripps Translational Science Institute, The Scripps Research Institute, La Jolla, CA 92037;eDepartment of Molecular and Experimental Medicine, The Scripps Research Institute, La Jolla, CA 92037; fDepartment of Human Biology, The J. CraigVenter Institute, La Jolla, CA 92037; gDepartment of Metabolism and Aging, The Scripps Research Institute, Jupiter, FL 33458; and hNext GenerationSequencing and Microarray Core Facility, The Scripps Research Institute, La Jolla, CA 92037
Contributed by Paul Schimmel, May 25, 2016 (sent for review February 27, 2016; reviewed by Doron Betel, Feng Cui, and Shuo Lin)
Brain and heart pathologies are caused by editing defects of transferRNA (tRNA) synthetases, which preserve genetic code fidelity by re-moving incorrect amino acids misattached to tRNAs. To extend under-standing of the broader impact of synthetase editing reactions onorganismal homeostasis, and based on effects in bacteria ostensiblyfrom small amounts ofmistranslation of components of the replicationapparatus, we investigated the sensitivity to editing of the vertebrategenome. We show here that in zebrafish embryos, transient overex-pression of editing-defective valyl-tRNA synthetase (ValRSED) activatedDNA break-responsive H2AX and p53-responsive downstream pro-teins, such as cyclin-dependent kinase (CDK) inhibitor p21, which pro-motes cell-cycle arrest at DNA damage checkpoints, and Gadd45 andp53R2, with pivotal roles in DNA repair. In contrast, the response ofthese proteins to expression of ValRSED was abolished in p53-deficientfish. The p53-activated downstream signaling events correlated withsuppression of abnormal morphological changes caused by the editingdefect and, in adults, reversed a shortened life span (followed for2 y). Conversely, with normal editing activities, p53-deficient fishhave a normal life span and few morphological changes. Whole-fishdeep sequencing showed genomic mutations associated with theediting defect. We suggest that the sensitivity of p53 to expressionof an editing-defective tRNA synthetase has a critical role in promot-ing genome integrity and organismal homeostasis.
mistranslation | genomic fidelity | shortened lifespan | morphologicalchanges | genomic mutations
In the first step of protein synthesis, amino acids are attached totheir cognate tRNAs in reactions catalyzed by aminoacyl-tRNA
synthetases (1, 2). Mistakes of aminoacylation occur with a frequencyof less than 1% (3, 4), and if not corrected, will result in the insertionof a mischarged amino acid into a growing polypeptide chain at thewrong codon (5–7). This sort of error is usually corrected by a uni-versal editing mechanism in tRNA synthetases, which hydrolyti-cally removes the mischarged amino acid from the tRNA before thewrong amino acid is inserted into a nascent protein (8–14).Serious pathologies and cell death result from rare errors in protein
synthesis (mistranslation). For example, a mild editing defect of analanyl-tRNA synthetase (AlaRS) in the mouse was shown to causethe death of Purkinje cells and result in neurodegeneration (5). Morerecently, cardioproteinopathy was also shown to have connections tomistranslation (15). Although the cellular and physiological impor-tance of editing has been established, the extent of how editingdefects impact major cellular mechanisms is only beginning to beclarified. Because assaults on the genome by environmental factorslead to somatic cell mutations that cause diseases in humans, we wereespecially interested in the role editing might play in protecting ver-tebrates against DNA damage. Earlier work in bacteria was sup-portive of such a connection (16–18), and later work established thatthe occasional misincorporation of the wrong amino acids into en-zymes of the DNA replication and repair apparatus was mutagenic byvirtue of error-prone repair of DNA damage (19) (Fig. 1A).
Here, we investigated whether this phenomenon could extendto vertebrates. Because coupling of mistranslation and genomefidelity would have obvious implications for organismal homeostasisand disease, we chose the zebrafish, a popular model organism(with many of the same genes and disorders as mammals) (20, 21)as a system to explore the linkage between translation and ge-nome fidelity. Using editing-defective (ED) valyl-tRNA synthetase(ValRS), we demonstrated that, in response to mistranslation, theDNA damage response (DDR) is activated, as shown by phos-phorylation of H2AX and up-regulation of p53 downstream genes.In addition, ED ValRS shortened the lifespan of p53-deficientzebrafish, and whole-fish genomic DNA sequencing showed ge-nomic mutations were associated with the editing defect. Thus,p53 surveillance of mistranslation links translational and genomicfidelity to sustain vertebrate homeostasis.
ED ValRS Causes Cell Death in ZebrafishFor a mistranslational system, we used an ED tRNA synthetase,which has impaired ability to correct an error of aminoacylation ofits cognate tRNA. For this purpose, ValRS, which by editing elim-inates the occasional production of Thr-tRNAVal (22), was chosen.Replacement of a universally conserved T535 (which is T539 inzebrafish ValRS) with a T535P substitution yields an enzyme that iscompletely deficient in editing and retains full aminoacylation ac-tivity (6). This mutant was designated as ValRSED, where ED de-notes “editing-defective.” As shown by direct analysis of mammalian
Significance
Although DNA damage is a well-known cause of disease, itsconnection, if any, to mistranslation is unclear. Here, we used anediting-defective tRNA synthetase to show howan editing defectin zebrafish leads to severe consequences during developmentand to a shortened overall life span of adult fish. These effectswere p53-dependent and reflect a strong connection of mis-translation to the DNA damage response. In addition, deep se-quencing of genomic DNA revealed that expression of theediting-defective tRNA synthetase was mutagenic. We suggestthat the p53-dependent DNA damage response links translationaland genomic fidelity to promote vertebrate homeostasis.
Author contributions: Y. Song, Y. Shi, S.R.H., S.K., and P.S. designed research; Y. Song, Y. Shi,S.L., and T.S. performed research; Y. Song, Y. Shi, T.M.C., N.J.S., S.K., and P.S. analyzed data;and Y. Song and P.S. wrote the paper.
Reviewers: D.B., Weill Cornell Medical College; F.C., Rochester Institute of Technology;and S.L., University of California, Los Angeles.
The authors declare no conflict of interest.1Y. Song and Y. Shi contributed equally to this work.2To whom correspondence may be addressed. Email: [email protected] or [email protected].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1608139113/-/DCSupplemental.
8460–8465 | PNAS | July 26, 2016 | vol. 113 | no. 30 www.pnas.org/cgi/doi/10.1073/pnas.1608139113
Dow
nloa
ded
by g
uest
on
Nov
embe
r 27
, 202
1
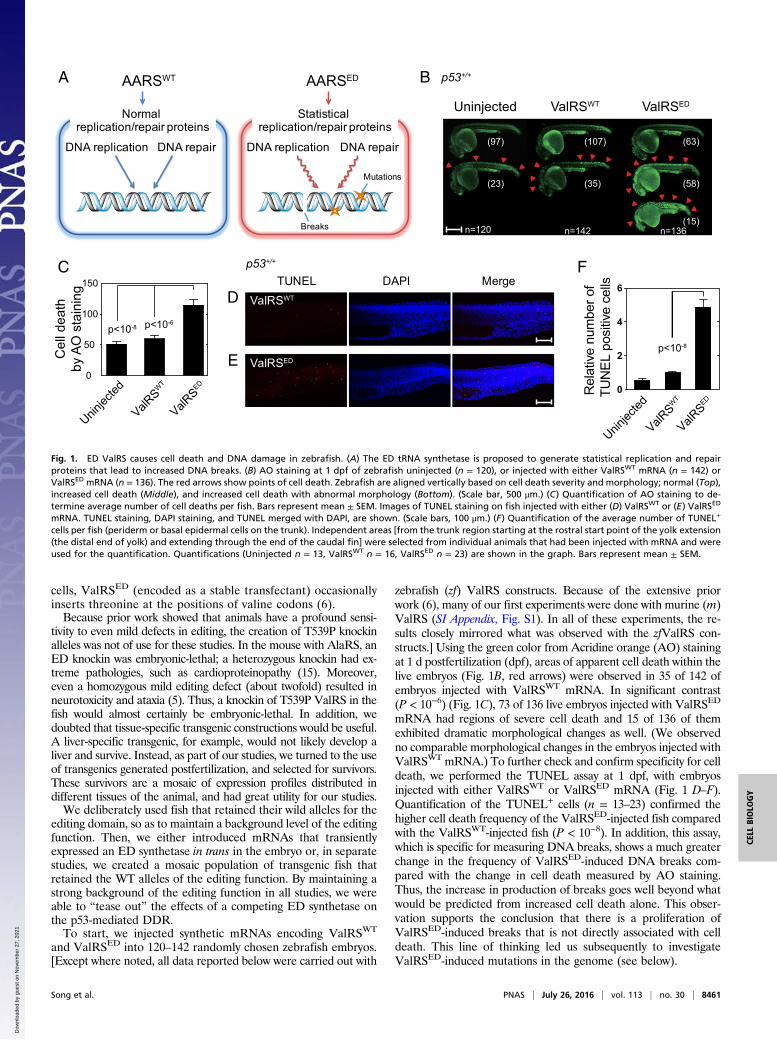
cells, ValRSED (encoded as a stable transfectant) occasionallyinserts threonine at the positions of valine codons (6).Because prior work showed that animals have a profound sensi-
tivity to even mild defects in editing, the creation of T539P knockinalleles was not of use for these studies. In the mouse with AlaRS, anED knockin was embryonic-lethal; a heterozygous knockin had ex-treme pathologies, such as cardioproteinopathy (15). Moreover,even a homozygous mild editing defect (about twofold) resulted inneurotoxicity and ataxia (5). Thus, a knockin of T539P ValRS in thefish would almost certainly be embryonic-lethal. In addition, wedoubted that tissue-specific transgenic constructions would be useful.A liver-specific transgenic, for example, would not likely develop aliver and survive. Instead, as part of our studies, we turned to the useof transgenics generated postfertilization, and selected for survivors.These survivors are a mosaic of expression profiles distributed indifferent tissues of the animal, and had great utility for our studies.We deliberately used fish that retained their wild alleles for the
editing domain, so as to maintain a background level of the editingfunction. Then, we either introduced mRNAs that transientlyexpressed an ED synthetase in trans in the embryo or, in separatestudies, we created a mosaic population of transgenic fish thatretained the WT alleles of the editing function. By maintaining astrong background of the editing function in all studies, we wereable to “tease out” the effects of a competing ED synthetase onthe p53-mediated DDR.To start, we injected synthetic mRNAs encoding ValRSWT
and ValRSED into 120–142 randomly chosen zebrafish embryos.[Except where noted, all data reported below were carried out with
zebrafish (zf) ValRS constructs. Because of the extensive priorwork (6), many of our first experiments were done with murine (m)ValRS (SI Appendix, Fig. S1). In all of these experiments, the re-sults closely mirrored what was observed with the zfValRS con-structs.] Using the green color from Acridine orange (AO) stainingat 1 d postfertilization (dpf), areas of apparent cell death within thelive embryos (Fig. 1B, red arrows) were observed in 35 of 142 ofembryos injected with ValRSWT mRNA. In significant contrast(P < 10−6) (Fig. 1C), 73 of 136 live embryos injected with ValRSED
mRNA had regions of severe cell death and 15 of 136 of themexhibited dramatic morphological changes as well. (We observedno comparable morphological changes in the embryos injected withValRSWT mRNA.) To further check and confirm specificity for celldeath, we performed the TUNEL assay at 1 dpf, with embryosinjected with either ValRSWT or ValRSED mRNA (Fig. 1 D–F).Quantification of the TUNEL+ cells (n = 13–23) confirmed thehigher cell death frequency of the ValRSED-injected fish comparedwith the ValRSWT-injected fish (P < 10−8). In addition, this assay,which is specific for measuring DNA breaks, shows a much greaterchange in the frequency of ValRSED-induced DNA breaks com-pared with the change in cell death measured by AO staining.Thus, the increase in production of breaks goes well beyond whatwould be predicted from increased cell death alone. This obser-vation supports the conclusion that there is a proliferation ofValRSED-induced breaks that is not directly associated with celldeath. This line of thinking led us subsequently to investigateValRSED-induced mutations in the genome (see below).
DNA replication DNA repairDNA replication DNA repair
Breaks
Mutations
Normalreplication/repair proteins
AARSWT AARSED
Statisticalreplication/repair proteins
A B
Uninjected ValRSWT ValRSED
(63)
(58)
n=136(15)
(107)
(35)
(97)
n=120 n=142
(23)
p53+/+
C
0
50
100
150
ValRSW
T
ValRSED
p<10-8 p<10-6
ValRSWT
ValRSED
D
E
F
Rel
ativ
enu
mbe
rof
TUN
ELpo
sitiv
ece
lls
0
2
4
6
p<10-8
TUNEL DAPI Mergep53+/+
Cel
l dea
th
by A
O s
tain
ing
Uninjec
ted
ValRSW
T
ValRSED
Uninjec
ted
Fig. 1. ED ValRS causes cell death and DNA damage in zebrafish. (A) The ED tRNA synthetase is proposed to generate statistical replication and repairproteins that lead to increased DNA breaks. (B) AO staining at 1 dpf of zebrafish uninjected (n = 120), or injected with either ValRSWT mRNA (n = 142) orValRSED mRNA (n = 136). The red arrows show points of cell death. Zebrafish are aligned vertically based on cell death severity and morphology; normal (Top),increased cell death (Middle), and increased cell death with abnormal morphology (Bottom). (Scale bar, 500 μm.) (C) Quantification of AO staining to de-termine average number of cell deaths per fish. Bars represent mean ± SEM. Images of TUNEL staining on fish injected with either (D) ValRSWT or (E) ValRSED
mRNA. TUNEL staining, DAPI staining, and TUNEL merged with DAPI, are shown. (Scale bars, 100 μm.) (F) Quantification of the average number of TUNEL+
cells per fish (periderm or basal epidermal cells on the trunk). Independent areas [from the trunk region starting at the rostral start point of the yolk extension(the distal end of yolk) and extending through the end of the caudal fin] were selected from individual animals that had been injected with mRNA and wereused for the quantification. Quantifications (Uninjected n = 13, ValRSWT n = 16, ValRSED n = 23) are shown in the graph. Bars represent mean ± SEM.
Song et al. PNAS | July 26, 2016 | vol. 113 | no. 30 | 8461
CELL
BIOLO
GY
Dow
nloa
ded
by g
uest
on
Nov
embe
r 27
, 202
1

Pursuant to more rigorously addressing the question of artifactsfrom injection or overexpression variability, we investigated levelsof both ValRSWT and ValRSED in normal p53+/+ fish (SI Appendix,Fig. S2). These data show that expression of ValRSED is less thanor equal to that of ValRSWT. However, with three different dosesof the two ValRS’s, ValRSED consistently gave much more celldeath (SI Appendix, Fig. S3). This difference was even seen at thelowest dose, whereas the uninjected and ValRSWT mRNA injectedzebrafish were the same (SI Appendix, Fig. S3). (In experimentsdescribed below, unless otherwise noted, we then used this “lowestdose.”) These results came from three independent experiments,using about 59–136 fish in each of seven groups. These additionaldata further support that the effects of ValRSED are not a result ofmicroinjection/overexpression artifacts.
ED ValRS Activates DNA Damage-Responsive H2AXTo more sharply focus on DNA breaks per se, we looked at theH2AX protein that, in response to DNA damage, is rapidly phos-phorylated at S139 by ATM (Ataxia telangiectasia mutated), ATR(Ataxia telangiectasia and Rad3 related), and DNA-PK (DNA-dependent protein kinase), among others (Fig. 2A) (23, 24). For thisanalysis, we used UV-treated zebrafish embryos as a positive con-trol. The activation of H2AX to give γ-H2AX was determined bymicroscopy and quantitation of staining with a specific antibody(α-γ-H2AX). With fish injected with ValRSED mRNA, even in theabsence of UV-irradiation, the amount of γ-H2AX was increased(Fig. 2B). With an enlarged view of γ-H2AX staining, strong greensignals were found in the UV control and in ValRSED mRNAinjected fish, but not in the uninjected or ValRSWT mRNA-injected fish (Fig. 2C). The γ-H2AX staining was quantified and isplotted in Fig. 2D. Whereas the uninjected and ValRSWT mRNA-injected samples showed background levels of γ-H2AX stain-ing, the ValRSED mRNA-injected fish showed significantly ele-vated γ-H2AX levels (P < 10−5). Thus, the editing defect by itselfleads to DNA breaks. These results further support the idea thatsuppression of miscoding is protective against DNA damage.
ED ValRS Activates the p53 PathwayTo verify the expected DDR in the subject animals (Fig. 2), wemonitored downstream markers of p53 activation for our analysis.These markers included cyclin-dependent kinase (CDK) inhibitorp21, which promotes cell-cycle arrest at DNA damage checkpoints(25), and Gadd45 (26) and p53R2 (27), which have pivotal roles inDNA repair. Strikingly, although embryos injected with mRNAencoding ValRSWT showed little change in expression of thesep53-downstream target genes, p21, gadd45, and p53R2 were allsignificantly up-regulated in embryos that harbored ValRSED (allP < 10−2) (Fig. 3A). (Because of the inevitable presence of cellsnot harboring ValRSED in the samples, we speculate that the levelof activation of the downstream markers may be underestimated.)We also checked Mdm2, an E3 ubiquitin ligase, which negativelyregulates the presence of p53 proteins to suppress p53-inducedapoptosis (28). Neither injected ValRSWT nor ValRSED mRNAaffected mdm2 expression (SI Appendix, Fig. S4A). As expectedfrom the cell-death–associated AO staining seen in Fig. 1 B and C,we observed that ValRSED mRNA injection caused increased ex-pression of the apoptosis-inducing bax (29) (SI Appendix, Fig. S4A).These results demonstrated a strong sensitivity of the induction ofthe DDR to miscoding in zebrafish.In addition to using ValRSWT as a control, we created an
ALGA substitution of the highly conserved HLGH motif impor-tant for ValRS aminoacylation activity. This mutant (ValRSAD) isaminoacylation-defective, but retains the editing activity encodedby a separate site for editing (30). [Here the mouse ValRS con-struct was used. In a separate analysis, we found that cell death insurviving fish, as monitored by AO staining, was similar for fer-tilized eggs injected with WT and ED constructs of either zfValRSmRNA or mValRS mRNA (SI Appendix, Fig. S1 A–D). In addi-tion, the mRNA levels of the p53 downstream target genes werealso similar for both the zfValRS mRNA- and mValRS mRNA-injected fish (SI Appendix, Fig. S1 E and F). This similarity isconsistent with the high conservation of the two ValRS se-quences.] We found that 5 of 24 embryos injected withmValRSAD
mRNA had localized regions of cell death (SI Appendix, Fig. S5 Aand B), similar to those injected with ValRSWT mRNA (10 of 56)(SI Appendix, Fig. S1 A and B). However, the quantification of theAO staining suggested that there was a statistically significant dif-ference in cell death between the mValRSWT and the mValRSAD
mRNA-injected zebrafish (P < 10−2) (SI Appendix, Fig. S5B). Thisobservation could be a manifestation of prior results showing thatactive-site mutations can impede bacterial cell growth because theinactive synthetase retains its tRNA binding site and soaks up thecognate tRNA in abortive complexes (31).Next, we examined expression of the three p53 DDR down-
stream target genes: p21, gadd45, and p53R2. Expression of thesegenes did not differ from those injected with ValRSWT mRNA(SI Appendix, Fig. S5C). [Although expression of mdm2 was notaltered significantly by injection of ValRSAD mRNA, the apo-ptosis marker bax was elevated (P < 10−2), and thus consistent withthe somewhat higher cell death associated with ValRSAD-expressingfish (SI Appendix, Fig. S5 A and B).] These results show that theDDR is not directly sensitive to the site for aminoacylation.
In p53-Deficient Fish, ED ValRS Leads to Increased AbnormalMorphologyTo further investigate the connection of miscoding to thep53-directed DDR, we performed mRNA injections into embryosthat were p53-deficient (p53zdf1/zdf1). Here again, the expressionlevels of ValRSWT and ValRSED were similar (SI Appendix, Fig.S6). Additionally, as in the p53+/+ background, with three differentdoses of the two ValRS’s, ValRSED consistently gave much more celldeath (SI Appendix, Fig. S7). In these experiments, the p53-deficiencyhad little or no morphology effect on either uninjected embryosor embryos injected with mRNA for ValRSWT (Fig. 3B). With
ValRSWT
Uninjected
ValRSED
UV
UV
05
10152060
70
80
H2A
X s
tain
ing
(Inte
nsity
/ nu
cleu
s, x
1000
)γ
-
p<10-5
No UV
A B
C D
p53+/+
Uninjected
ValRSWT ValRSED
H2AX
H2AXP
ATM/ATRDNA-PK
UV
Uninjec
ted
ValRSW
T
ValRSED
Fig. 2. Response of H2AX to an ED ValRS. (A) Cartoon of H2AX phos-phorylation. (B) γ-H2AX staining in uninjected (n = 60), ValRSWT mRNA in-jected (n = 65), ValRSED mRNA injected (n = 79), or UV treated zebrafish(n = 30) at 1 dpf. (Scale bar, 250 μm.) (C) Enlarged pictures of γ-H2AXstaining in the nuclei of different samples of zebrafish. (Scale bar, 40 μm.) (D)Quantification of γ-H2AX staining. Bars represent mean ± SEM.
8462 | www.pnas.org/cgi/doi/10.1073/pnas.1608139113 Song et al.
Dow
nloa
ded
by g
uest
on
Nov
embe
r 27
, 202
1

ValRSED mRNA injections into p53+/+ fish, we observed cell deathin zebrafish with both normal and abnormal morphology (Fig. 1 Band C). However, in the p53zdf1/zdf1 fish injected with ValRSED
mRNA, cell death was only observed in zebrafish with abnormalmorphology (Fig. 3 B and C). Thus, 36 of 93 of the surviving embryosinjected with ValRSEDmRNA showedmany localized regions of AOstaining and had simultaneously undergone morphological changes,whereas the remaining 57 fish had neither. (The limited cell deathwithout morphological changes in the ValRSED-expressing p53+/+
fish of Fig. 1 B and C is consistent with the protective role of p53against more extensive damage to the whole organism.) As expected,because of the absence of p53 activity, there was not a statisti-cally significant difference between ValRSWT and ValRSED mRNA-injected embryos in the expression of any of the p53 downstreammarkers p21, gadd45, p53R2, mdm2, and bax (Fig. 3D and SI Ap-pendix, Fig. S4B). It is worth noting that, with both the p53+/+ andp53zdf1/zdf1 fish, the protein levels of WT and mutant p53 were un-affected by the expression of ValRSWT or ValRSED (SI Appendix,Figs. S8 and S9). [The p53 +/zdf1 heterozygote behaves like the p53+/+
fish for the activation of p53 downstream targets and tumor pre-disposition after γ-irradiation (32). For that reason, we did not feeljustified to pursue confirmatory work showing that p53 heterozygoteswould behave the same as the p53 WT fish in our system.]
ED ValRS Shortens the Short- and Long-Term Survival ofZebrafishAccumulation of DNA damage can lead to mutations throughthe error-prone DNA repair machinery and ostensibly shortenthe lifespan (33, 34). To investigate this question, we redirectedour efforts to p53zdf1/zdf1 transgenic fish that expressed ValRSED.We inserted the cDNA encoding ValRSWT and ValRSED intothe pT2-mCherry vector, which fuses the coding sequence of themCherry vector to the coding sequence for the C terminus of theValRS’s. (We designate these constructs as pT2-ValRSWT andpT2-ValRSED, respectively.) The resulting vector constructionswere then injected into one-cell stage p53zdf1/zdf1 embryos. After2 dpf (when the fish appear developed), we sorted for mCherry+
fish under a fluorescent microscope and harvested samples for
Western blot analysis to confirm the fusion protein of the expectedsize was indeed produced (SI Appendix, Fig. S10).Next, we investigated both the short- and long-term survival of
fish harboring WT and ED alleles of ValRS. For the short-termanalysis, ValRSWT or ValRSED mRNAs were injected into sep-arate fertilized p53+/+ or p53zdf1/zdf1 eggs. Deaths of embryoswere monitored during 6–24 h postfertilization (hpf). Both p53+/+
and p53zdf1/zdf1 fish injected with ValRSED mRNA showed in-creased embryo death (approximately 13–35% increase comparedwith WT) compared with the uninjected and ValRSWT mRNA-injected fish (which were similar) (SI Appendix, Fig. S11).For the analysis of long-term survival, we injected p53+/+ and
p53zdf1/zdf1 fertilized eggs, separately, with either the pT2-vectoralone or the pT2-ValRSWT or pT2-ValRSED construct. For thiswork we injected pT2-ValRS constructs into the embryos that werecreated from siblings’ group mating (a group mating of a few malesand females in the same generation). This protocol established sixseparate groups of fish for long-term survival analysis. After de-velopment, and depending on the experiment, mCherry+ fish wereselected and maintained over a period of almost 2 y. The long-termsurvival rates of these fish were then compared. We observed that,in the p53zdf1/zdf1 background, the long-term survival of fish har-boring the pT2-vector alone and the pT2-ValRSWT construct werenot significantly different. In contrast, fish harboring the pT2-ValRSED construct had sharply reduced survival rates (P < 10−3)(Fig. 4). Consistent with the analysis shown above of the p53-dependent effects of expression of ValRSED on the DDR, the long-term survival of fish harboring the pT2-vector alone, and of thoseharboring either the pT2-ValRSWT or pT2-ValRSED construct,were not different in a statistically significant way in the p53+/+
background (SI Appendix, Fig. S12). Thus, the p53-dependent re-sponse to DNA insults caused by mistranslation preserves the life-span in the fish.
Increased Genomic Variants Associate with the Presence ofan ED tRNA synthetaseTo confirm that fish harboring ValRSED had accumulated mutations,we selected from the siblings’ cohort, one zebrafish at 2 dpf expressing
A
Rel
ativ
e m
RN
A le
vel
ValRSEDp<10-2
Uninjected
p<10-4 p<10-3
p53+/+
p21 gadd45 p53R2
2.5
2.0
1.5
1.0
0.5
0.0
ValRSWT
D
0.0
0.2
0.4
0.6
0.8
1.0
1.2Uninjected
p53zdf1/zdf1
p21 gadd45 p53R2
ValRSWT
Rel
ativ
e m
RN
A le
vel
ValRSED
Uninjected ValRSWT ValRSED
n=118 n=90 n=93
(105) (80)
(10)
(57)
(36)(13)
B p53zdf1/zdf1
C
0
50
100
150
200
250
p<10-3
Cel
l dea
th
by A
O s
tain
ing
ValRSW
T
ValRSED
Uninjec
ted
p<10-3
Fig. 3. ED ValRS activates p53 in zebrafish. (A) RT-PCR results of p53 downstream markers p21, gadd45,and p53R2 in ValRSWT or ValRSED mRNA-injectedzebrafish. The bar graphs show the mean values ±SEM after normalization to the β-actin level. (B)Uninjected (n = 118), ValRSWT mRNA-injected (n =90), or ValRSED mRNA-injected (n = 93) p53zdf1/zdf1
zebrafish were stained with AO at 1 dpf. The redarrows show points of cell death. Zebrafish arealigned based on cell death severity. (Scale bar,500 μm.) (C) Quantification of AO staining. Barsrepresent mean ± SEM. (D) RT-PCR results of p53downstream markers p21, gadd45, and p53R2 inp53zdf1/zdf1 zebrafish injected with either ValRSWT orValRSED mRNA. The bar graphs show the meanvalues ± SEM after normalization to the β-actin level.
Song et al. PNAS | July 26, 2016 | vol. 113 | no. 30 | 8463
CELL
BIOLO
GY
Dow
nloa
ded
by g
uest
on
Nov
embe
r 27
, 202
1
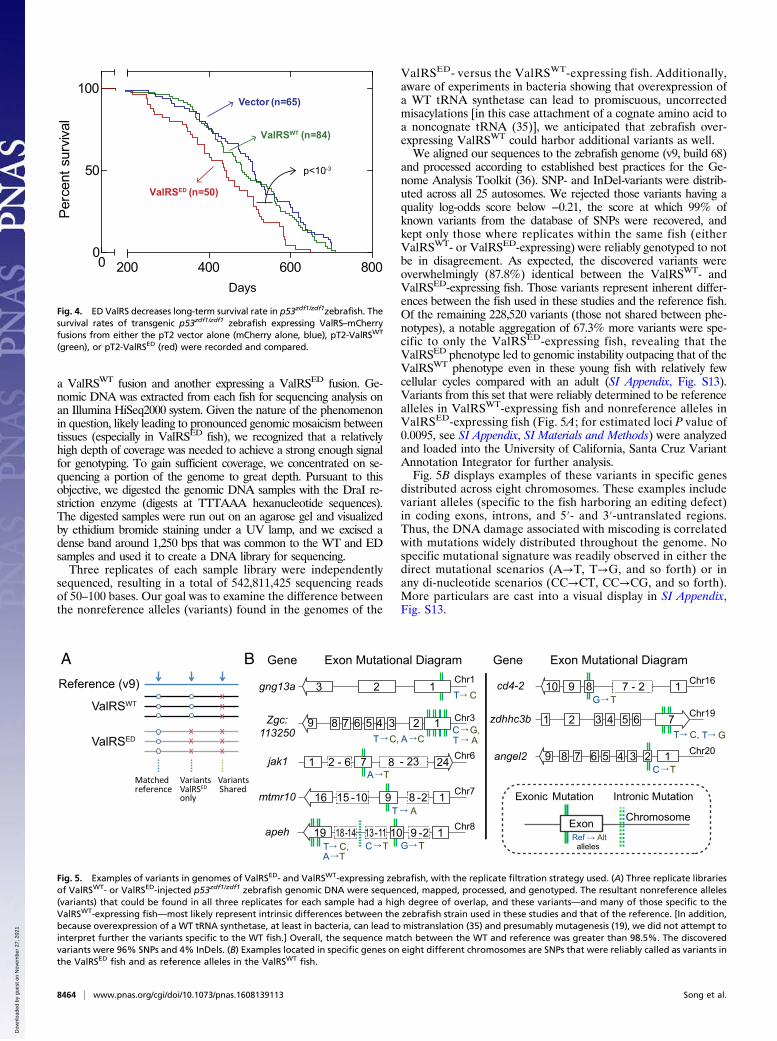
a ValRSWT fusion and another expressing a ValRSED fusion. Ge-nomic DNA was extracted from each fish for sequencing analysis onan Illumina HiSeq2000 system. Given the nature of the phenomenonin question, likely leading to pronounced genomic mosaicism betweentissues (especially in ValRSED fish), we recognized that a relativelyhigh depth of coverage was needed to achieve a strong enough signalfor genotyping. To gain sufficient coverage, we concentrated on se-quencing a portion of the genome to great depth. Pursuant to thisobjective, we digested the genomic DNA samples with the DraI re-striction enzyme (digests at TTTAAA hexanucleotide sequences).The digested samples were run out on an agarose gel and visualizedby ethidium bromide staining under a UV lamp, and we excised adense band around 1,250 bps that was common to the WT and EDsamples and used it to create a DNA library for sequencing.Three replicates of each sample library were independently
sequenced, resulting in a total of 542,811,425 sequencing readsof 50–100 bases. Our goal was to examine the difference betweenthe nonreference alleles (variants) found in the genomes of the
ValRSED- versus the ValRSWT-expressing fish. Additionally,aware of experiments in bacteria showing that overexpression ofa WT tRNA synthetase can lead to promiscuous, uncorrectedmisacylations [in this case attachment of a cognate amino acid toa noncognate tRNA (35)], we anticipated that zebrafish over-expressing ValRSWT could harbor additional variants as well.We aligned our sequences to the zebrafish genome (v9, build 68)
and processed according to established best practices for the Ge-nome Analysis Toolkit (36). SNP- and InDel-variants were distrib-uted across all 25 autosomes. We rejected those variants having aquality log-odds score below −0.21, the score at which 99% ofknown variants from the database of SNPs were recovered, andkept only those where replicates within the same fish (eitherValRSWT- or ValRSED-expressing) were reliably genotyped to notbe in disagreement. As expected, the discovered variants wereoverwhelmingly (87.8%) identical between the ValRSWT- andValRSED-expressing fish. Those variants represent inherent differ-ences between the fish used in these studies and the reference fish.Of the remaining 228,520 variants (those not shared between phe-notypes), a notable aggregation of 67.3% more variants were spe-cific to only the ValRSED-expressing fish, revealing that theValRSED phenotype led to genomic instability outpacing that of theValRSWT phenotype even in these young fish with relatively fewcellular cycles compared with an adult (SI Appendix, Fig. S13).Variants from this set that were reliably determined to be referencealleles in ValRSWT-expressing fish and nonreference alleles inValRSED-expressing fish (Fig. 5A; for estimated loci P value of0.0095, see SI Appendix, SI Materials and Methods) were analyzedand loaded into the University of California, Santa Cruz VariantAnnotation Integrator for further analysis.Fig. 5B displays examples of these variants in specific genes
distributed across eight chromosomes. These examples includevariant alleles (specific to the fish harboring an editing defect)in coding exons, introns, and 5′- and 3′-untranslated regions.Thus, the DNA damage associated with miscoding is correlatedwith mutations widely distributed throughout the genome. Nospecific mutational signature was readily observed in either thedirect mutational scenarios (A→T, T→G, and so forth) or inany di-nucleotide scenarios (CC→CT, CC→CG, and so forth).More particulars are cast into a visual display in SI Appendix,Fig. S13.
Days
Perc
ents
urvi
val
00
50
100
200 400 600 800
p<10-3
Vector (n=65)
ValRSWT (n=84)
ValRSED (n=50)
Fig. 4. ED ValRS decreases long-term survival rate in p53zdf1/zdf1zebrafish. Thesurvival rates of transgenic p53zdf1/zdf1 zebrafish expressing ValRS–mCherryfusions from either the pT2 vector alone (mCherry alone, blue), pT2-ValRSWT
(green), or pT2-ValRSED (red) were recorded and compared.
A B
xx
x
Matchedreference
xx
x
xx
x
VariantsShared
VariantsValRSEDonly
Reference (v9)
ValRSWT
ValRSED
Gene Exon Mutational DiagramChr1
Chr6
Chr7
Chr8
Chr16
Chr19
Chr20
Gene Exon Mutational Diagram
T C, A CC G,T A
T C
Chr3
A T
T A
T C,A T
C T G T
G T
T GT C,
C T
gng13a
Zgc:113250
jak1
mtmr10
apeh
cd4-2
zdhhc3b
angel2
Fig. 5. Examples of variants in genomes of ValRSED- and ValRSWT-expressing zebrafish, with the replicate filtration strategy used. (A) Three replicate librariesof ValRSWT- or ValRSED-injected p53zdf1/zdf1 zebrafish genomic DNA were sequenced, mapped, processed, and genotyped. The resultant nonreference alleles(variants) that could be found in all three replicates for each sample had a high degree of overlap, and these variants—and many of those specific to theValRSWT-expressing fish—most likely represent intrinsic differences between the zebrafish strain used in these studies and that of the reference. [In addition,because overexpression of a WT tRNA synthetase, at least in bacteria, can lead to mistranslation (35) and presumably mutagenesis (19), we did not attempt tointerpret further the variants specific to the WT fish.] Overall, the sequence match between the WT and reference was greater than 98.5%. The discoveredvariants were 96% SNPs and 4% InDels. (B) Examples located in specific genes on eight different chromosomes are SNPs that were reliably called as variants inthe ValRSED fish and as reference alleles in the ValRSWT fish.
8464 | www.pnas.org/cgi/doi/10.1073/pnas.1608139113 Song et al.
Dow
nloa
ded
by g
uest
on
Nov
embe
r 27
, 202
1

DiscussionThe zebrafish system used here seems to be especially robust fordetection of p53-sensitive DNA damage and therefore could beuseful as a “tester system” analogous to the Ames test for carcin-ogens (37). With this system, a powerful genome-protective effectof an editing mechanism that suppresses miscoding was demon-strated. Even in the p53zdf1/zdf1 background, the WT editing ac-tivity was sufficiently robust to suppress miscoding so that fishembryo morphology and a normal life span were maintained.However, in that same p53zdf1/zdf1 background, the miscoding-defectwe introduced was transdominant, because all fish still harbored thediploid WT alleles for the native tRNA synthetase. This observa-tion highlights the sensitivity of the genome to miscoding.The results also independently reveal a critical protective effect of
the p53-directed surveillance of miscoding. Despite miscoding, themorphological changes in fish embryos and lifespan decrease ofadult fish were much less altered in the p53+/+ background. (Becausethese changes were marginal, we did not attempt to investigate ge-nome sequence alterations.) Thus, in circumstances where p53 sur-veillance of miscoding is compromised, mechanisms like editing thatsuppress miscoding are particularly important, because even mildmiscoding could, for example, contribute to the “mutator pheno-type” seen in disease connected to p53 defects (38–40).
MethodsAO Staining for Detecting Cell Death in Zebrafish Embryos. The zebrafish(Danio rerio, strain: AB/Tu; sex, N/D; age, 0–2.5 y old) used in our study weremaintained at 28.5 °C under continuous water flow and filtration with au-tomatic control for a 14-h/10-h light/dark cycle. The night before injection,male and female fish were placed in a 1-L tank containing a fish mating cagewith an inner mesh and divider. Zebrafish embryos were obtained from
natural spawning by removing the divider and stimulating with light. Theembryos were kept at 28.5 °C before and after microinjection.
Synthetic mRNAs (made with mMESSAGE mMACHINE kit, Ambion) wereinjected into the one-cell stage embryos at a dosage of 250–1,000 pg perembryo (sample size was determined by standards of the field of three in-dependent experiments of 30–50 embryos each). For the injection of pT2vector constructs, the dosage of DNA was ∼1 ng per embryo together with∼250 pg transposase-encoding mRNA. At 1 dpf, dead embryos were removedand all live embryos were used for experiments. For AO staining, live zebrafishembryos were dechorionated in pronase (2.0 mg/mL in egg water, 5 mM NaCl,0.17 mMKCl, 0.33 mM CaCl2, 0.33 mMmagnesium sulfate) for 3–5min and rinsedfive times in egg water at 1 dpf. Embryos were then incubated in 10 μg/mLAO (Sigma A-6014) in egg water for 15 min at 28.5 °C, followed by five quickrinses. Embryos were anesthetized in 160 μg/mL tricaine (3-aminobenzoicacid ethyl ester; Sigma A-5040), mounted laterally on the glass slide, andphotographed with a Nikon fluorescent microscope (AZ100) equipped witha Nikon CCD camera (Qimaging Retiga 2000R). All of the experiments in-volving zebrafish had been conducted according to the guidelines estab-lished by the Institutional Animal Care and Use Committee (IACUC) at TheScripps Research Institute, IACUC approval number 09–0009. Results shownare from three independent experiments.
Other Procedures. Additional procedures are described in SI Appendix, SIMaterials and Methods and Table S1.
ACKNOWLEDGMENTS. We thank Profs. Susan Ackerman (Jackson Labora-tories, Bar Harbor), Arnold Levine (Institute for Advance Studies, PrincetonUniversity), Xiaohua Wu (The Scripps Research Institute), and Leonard Zon(Children’s Hospital, Harvard Medical School, and Harvard University) forhelpful comments on this work. This work was supported by NationalCancer Institute Grant CA92577; NIH NIAAA Grants AA007456 andAA013525; aTyr Pharma; and by a fellowship from the National Foundationfor Cancer Research.
1. Carter CW, Jr (1993) Cognition, mechanism, and evolutionary relationships in ami-noacyl-tRNA synthetases. Annu Rev Biochem 62:715–748.
2. Giegé R (2006) The early history of tRNA recognition by aminoacyl-tRNA synthetases.J Biosci 31(4):477–488.
3. Loftfield RB (1963) The frequency of errors in protein biosynthesis. Biochem J 89:82–92.4. Loftfield RB, Vanderjagt D (1972) The frequency of errors in protein biosynthesis.
Biochem J 128(5):1353–1356.5. Lee JW, et al. (2006) Editing-defective tRNA synthetase causes protein misfolding and
neurodegeneration. Nature 443(7107):50–55.6. Nangle LA, Motta CM, Schimmel P (2006) Global effects of mistranslation from an
editing defect in mammalian cells. Chem Biol 13(10):1091–1100.7. Netzer N, et al. (2009) Innate immune and chemically triggered oxidative stress
modifies translational fidelity. Nature 462(7272):522–526.8. Baldwin AN, Berg P (1966) Transfer ribonucleic acid-induced hydrolysis of valylade-
nylate bound to isoleucyl ribonucleic acid synthetase. J Biol Chem 241(4):839–845.9. Eldred EW, Schimmel PR (1972) Rapid deacylation by isoleucyl transfer ribonucleic
acid synthetase of isoleucine-specific transfer ribonucleic acid aminoacylated withvaline. J Biol Chem 247(9):2961–2964.
10. Martinis SA, Boniecki MT (2010) The balance between pre- and post-transfer editingin tRNA synthetases. FEBS Lett 584(2):455–459.
11. Yadavalli SS, Ibba M (2012) Quality control in aminoacyl-tRNA synthesis its role intranslational fidelity. Adv Protein Chem Struct Biol 86:1–43.
12. Kumar S, Das M, Hadad CM, Musier-Forsyth K (2012) Substrate and enzyme functionalgroups contribute to translational quality control by bacterial prolyl-tRNA synthetase.J Phys Chem B 116(23):6991–6999.
13. Schreier AA, Schimmel PR (1972) Transfer ribonucleic acid synthetase catalyzed de-acylation of aminoacyl transfer ribonucleic acid in the absence of adenosine mono-phosphate and pyrophosphate. Biochemistry 11(9):1582–1589.
14. Beuning PJ, Musier-Forsyth K (2000) Hydrolytic editing by a class II aminoacyl-tRNAsynthetase. Proc Natl Acad Sci USA 97(16):8916–8920.
15. Liu Y, et al. (2014) Deficiencies in tRNA synthetase editing activity cause car-dioproteinopathy. Proc Natl Acad Sci USA 111(49):17570–17575.
16. Murphy HS, Humayun MZ (1997) Escherichia coli cells expressing a mutant glyV(glycine tRNA) gene have a UVM-constitutive phenotype: Implications for mecha-nisms underlying the mutA or mutC mutator effect. J Bacteriol 179(23):7507–7514.
17. Dorazi R, Lingutla JJ, Humayun MZ (2002) Expression of mutant alanine tRNAs in-creases spontaneous mutagenesis in Escherichia coli. Mol Microbiol 44(1):131–141.
18. Balashov S, Humayun MZ (2002) Mistranslation induced by streptomycin provokes aRecABC/RuvABC-dependent mutator phenotype in Escherichia coli cells. J Mol Biol315(4):513–527.
19. Bacher JM, Schimmel P (2007) An editing-defective aminoacyl-tRNA synthetase is mu-tagenic in aging bacteria via the SOS response. Proc Natl Acad Sci USA 104(6):1907–1912.
20. Dooley K, Zon LI (2000) Zebrafish: A model system for the study of human disease.Curr Opin Genet Dev 10(3):252–256.
21. Amsterdam A, Hopkins N (2006) Mutagenesis strategies in zebrafish for identifyinggenes involved in development and disease. Trends Genet 22(9):473–478.
22. Fersht AR, Kaethner MM (1976) Enzyme hyperspecificity. Rejection of threonine bythe valyl-tRNA synthetase by misacylation and hydrolytic editing. Biochemistry 15(15):3342–3346.
23. Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM (1998) DNA double-strandedbreaks induce histone H2AX phosphorylation on serine 139. J Biol Chem 273(10):5858–5868.
24. Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ (2001) ATM phosphorylates histoneH2AX in response to DNA double-strand breaks. J Biol Chem 276(45):42462–42467.
25. Bunz F, et al. (1998) Requirement for p53 and p21 to sustain G2 arrest after DNAdamage. Science 282(5393):1497–1501.
26. Maeda T, et al. (2002) GADD45 regulates G2/M arrest, DNA repair, and cell death inkeratinocytes following ultraviolet exposure. J Invest Dermatol 119(1):22–26.
27. Tanaka H, et al. (2000) A ribonucleotide reductase gene involved in a p53-dependentcell-cycle checkpoint for DNA damage. Nature 404(6773):42–49.
28. Buschmann T, Fuchs SY, Lee CG, Pan ZQ, Ronai Z (2000) SUMO-1 modification ofMdm2 prevents its self-ubiquitination and increases Mdm2 ability to ubiquitinatep53. Cell 101(7):753–762.
29. Yin C, Knudson CM, Korsmeyer SJ, Van Dyke T (1997) Bax suppresses tumorigenesisand stimulates apoptosis in vivo. Nature 385(6617):637–640.
30. Lin L, Schimmel P (1996) Mutational analysis suggests the same design for editingactivities of two tRNA synthetases. Biochemistry 35(17):5596–5601.
31. Schmidt E, Schimmel P (1993) Dominant lethality by expression of a catalytically in-active class I tRNA synthetase. Proc Natl Acad Sci USA 90(15):6919–6923.
32. Berghmans S, et al. (2005) tp53 mutant zebrafish develop malignant peripheral nervesheath tumors. Proc Natl Acad Sci USA 102(2):407–412.
33. Vogel H, Lim DS, Karsenty G, Finegold M, Hasty P (1999) Deletion of Ku86 causes earlyonset of senescence in mice. Proc Natl Acad Sci USA 96(19):10770–10775.
34. Vijg J, Suh Y (2013) Genome instability and aging. Annu Rev Physiol 75:645–668.35. Swanson R, et al. (1988) Accuracy of in vivo aminoacylation requires proper balance of
tRNA and aminoacyl-tRNA synthetase. Science 242(4885):1548–1551.36. Van der Auwera GA, et al. (2013) From FastQ data to high-confidence variant calls:
The Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics 43:11.10.1–11.10.33.
37. Ames BN, Lee FD, DurstonWE (1973) An improved bacterial test system for the detectionand classification of mutagens and carcinogens. Proc Natl Acad Sci USA 70(3):782–786.
38. Havre PA, Yuan J, Hedrick L, Cho KR, Glazer PM (1995) p53 inactivation by HPV16 E6results in increased mutagenesis in human cells. Cancer Res 55(19):4420–4424.
39. Bishop AJ, et al. (2003) Atm-, p53-, and Gadd45a-deficient mice show an increasedfrequency of homologous recombination at different stages during development.Cancer Res 63(17):5335–5343.
40. Feng Z, Hu W, Rajagopal G, Levine AJ (2008) The tumor suppressor p53: Cancer andaging. Cell Cycle 7(7):842–847.
Song et al. PNAS | July 26, 2016 | vol. 113 | no. 30 | 8465
CELL
BIOLO
GY
Dow
nloa
ded
by g
uest
on
Nov
embe
r 27
, 202
1













![Research Paper p53 increase mitochondrial copy number via ... · p53 was announced to be a regulator of metabolism recently [16], and some report has been published, describing p53](https://static.fdocuments.us/doc/165x107/5f1224eac6aba03d512fda6f/research-paper-p53-increase-mitochondrial-copy-number-via-p53-was-announced.jpg)




