
o segredo das coisa
12
151 GABRIELE GRAMELSBERGER SIMULATION AND SYSTEM U NDERST ANDING ABSTRACT Systems biology is based on a mathematized understanding of molecular biologi- cal processes. Because genetic networks are so complex, a system understandin g is required that allows for the appropriate modelling of these complex networks and its products up to the whole-cell scale. Since 2000 standardizatio ns in model- ling and simulation techniques have been established to support the communi- ty-wide endeavors for whole-cell simulations. The development of the Systems Biology Markup Language (SBML), in particular, has helped systems biologists achieve their goal. This paper explores the current developments of modelling and simulation in systems biology. It discusses the question as to whether an appropri- ate system understanding has been developed yet, or whether advanced software machineries of whole-cell simulations can compensate for the lack of system un- derstanding. 1. TOWARDS A SIMULATION-ORIENTATED BIOLOGY In a 2002 Natur e paper systems biology was dened as the “mathematical con - cepts […] to illuminate the principles underlying biology at a genetic, molecular, cellular and even organismal level.” 1 During the past years these mathematical concepts have become ‘whole-cell simulations’ in order to observe, and hopefully understand, the complex dynamic behavior of cells. Alread y in 1997 the very rst minimal cell was created in-silico, called the ‘virtual self-surviving cell (SSC)’, consisting of 120 in-silico synthesized genes of the 480 genes of M. genitalium and 7 genes from other species. 2 The virtual self-surviving cell absorbs up and metabo- lizes glucose, and generates ATP as an energy source for protein and membrane synthesis. As degradation is programmed into the SSC, it has to produce proteins and lipids constantly to survive. All the activities result from 495 reaction rules for enzymatic reactions, complex formations, transportations, and stochastic proc- esses, which are executed in parallel telling the SSC what to do at each millisecond time step. The aims of this whole-cell simulation are to observe the changes in the 1 Christopher Surridge (Ed.), “Nature inside: Computation al Biology”, in: Nature 420, 2002, 205-250, here p. 205. 2 Cf. Masaru T omita, “Whole-ce ll Simulation: A Grand Challenge of the 21 st Century”, in: TRENDS in Biotechnology 19, 6, 2001, pp. 205-210. H. Andersen et al. (eds.), New Challenges to Philosophy o f Science, The Philosophy of Science in a European Perspectiv e 4,
-
Upload
pablo-dumont -
Category
Documents
-
view
214 -
download
0
Transcript of o segredo das coisa

7/26/2019 o segredo das coisa
http://slidepdf.com/reader/full/o-segredo-das-coisa 1/11
151
GABRIELE GRAMELSBERGER
SIMULATION AND SYSTEM U NDERSTANDING
ABSTRACT
Systems biology is based on a mathematized understanding of molecular biologi-
cal processes. Because genetic networks are so complex, a system understanding
is required that allows for the appropriate modelling of these complex networks
and its products up to the whole-cell scale. Since 2000 standardizations in model-
ling and simulation techniques have been established to support the communi-
ty-wide endeavors for whole-cell simulations. The development of the Systems
Biology Markup Language (SBML), in particular, has helped systems biologists
achieve their goal. This paper explores the current developments of modelling and
simulation in systems biology. It discusses the question as to whether an appropri-
ate system understanding has been developed yet, or whether advanced software
machineries of whole-cell simulations can compensate for the lack of system un-
derstanding.
1. TOWARDS A SIMULATION-ORIENTATED BIOLOGY
In a 2002 Nature paper systems biology was dened as the “mathematical con-
cepts […] to illuminate the principles underlying biology at a genetic, molecular,
cellular and even organismal level.”1 During the past years these mathematical
concepts have become ‘whole-cell simulations’ in order to observe, and hopefully
understand, the complex dynamic behavior of cells. Already in 1997 the very rst
minimal cell was created in-silico, called the ‘virtual self-surviving cell (SSC)’,consisting of 120 in-silico synthesized genes of the 480 genes of M. genitalium and
7 genes from other species.2 The virtual self-surviving cell absorbs up and metabo-
lizes glucose, and generates ATP as an energy source for protein and membrane
synthesis. As degradation is programmed into the SSC, it has to produce proteins
and lipids constantly to survive. All the activities result from 495 reaction rules
for enzymatic reactions, complex formations, transportations, and stochastic proc-
esses, which are executed in parallel telling the SSC what to do at each millisecond
time step. The aims of this whole-cell simulation are to observe the changes in the
1 Christopher Surridge (Ed.), “Nature inside: Computational Biology”, in: Nature 420,
2002, 205-250, here p. 205.
2 Cf. Masaru Tomita, “Whole-cell Simulation: A Grand Challenge of the 21st Century”,
in: TRENDS in Biotechnology 19, 6, 2001, pp. 205-210.
H. Andersen et al. (eds.), New Challenges to Philosophy of Science,
The Philosophy of Science in a European Perspective 4,
DOI 10.1007/978-94-007-5845-2_13, © Springer Science+Business Media Dordrecht 2013

7/26/2019 o segredo das coisa
http://slidepdf.com/reader/full/o-segredo-das-coisa 2/11
152 Gabriele Gramelsberger
amount of substances inside the cell as well as the gene expression resulting from
these changes, to study the temporal patterns of change, and, nally, to conduct
experiments with the SSC, e.g. real-time gene knock-out experiments. Thus,
[t]his simple cell model sometimes shows unpredictable behavior and has delivered bio-
logically interesting surprises. When the extracellular glucose is drained and set to be zero,
intracellular ATP momentarily increases and then decreases […]. At rst, this nding was
confusing. Because ATP is synthesized only by the glycolysis pathway, it was assumed
that ATP would decrease when the glucose, the only source of energy, becomes zero. After
months of checking the simulation program and the cell model for errors, the conclusion is
that this observation is correct and a rapid deprivation of glucose supplement can lead to the
same phenomenon in living cells.3
The motivation of making use of simulation in biology is the desire to predict ef-fects of changes in cell behavior and guide further research in the lab. As metabolic
networks are extremely complex systems involving dozens and hundreds of genes,
enzymes, and other species, it is difcult to study these networks experimentally.
Therefore, in-silico studies are increasingly expanding the wet lab studies, but this
requires the full range of strategies necessary to establish a simulation-orientated
biology. These strategies are standardization, acquisition of sufcient spatiotem-
poral information about processes and parameters, creation of advanced software
machineries, and, last but not least, a coherent system understanding.
2. STANDARDIZATION
The situation of modeling and simulation in cell biology is characterized by a wide
variety of modeling practices and methods. There are thousands of simple mod-
els around, an increasing amount of simulations for more complex models, and
various computational tools to ease modeling. Each institute, each researcher cre-
ates his or her own model with slightly different concepts and meanings. Most of
these models are not comparable with each other, because “each author may use a
different modeling environment (and model representation language), [therefore]
such model denitions are often not straightforward to examine, test and reuse.”4
However, in 2000 this situation led to an effort to create an open and standardized
framework for modeling – the Systems Biology Markup Language (SBML). The
collaborative work on SBML was motivated by the goal to overcome “the cur -
rent inability to exchange models between different simulation and analysis tools
[which] has its roots in the lack of a common format for describing models.” 5
3 Tomita 2001, loc. cit ., here p. 208.4 Michael Hucka et al., “The Systems Biology Markup Language (SBML): A Medium
for Representation and Exchange of Biochemical Network Models”, in: Bioinformat-
ics 19, 2003, pp. 524–531, here p. 525.
5 Hucka et al. 2003, loc. cit ., here p. 524. SBML is organized as a community-wide open

7/26/2019 o segredo das coisa
http://slidepdf.com/reader/full/o-segredo-das-coisa 3/11
153Simulation and System Understanding
Therefore, SBML is based on the Extensible Markup Language (XML), a set of
rules for encoding documents in machine-readable form, developed in 1996 by the
World Wide Web Consortium. After three years of work with many contributions
from the community, in particular from teams developing simulation and analysis
packages for systems biology, SBML level 1 was released in 2003. As is the na-
ture of software development, level 2 was tackled immediately after the release of
level 1 containing more features; since 2010, with level 3, SBML has practically
become a lingua franca of model description in biology – according to Nature’s
website: “There are now hundreds of software systems supporting SBML; further,
many journals now suggest it as a format for submitting supplementary data.”6
The advantage of SBML is the standardization of modeling by dening con-
ceptual elements of chemical reactions. These elements are compartments, spe-
cies, reactions, parameters, unit denitions, and rules. For instance, a hypotheti-cal single-gene oscillatory circuit can be modeled with SBML as a simple, two
compartment model: one compartment for the nucleus, one for the surrounding
cell cytoplasm.7 This circuit can produce nine species and each species is dened
by its ‘name’, by its ‘initialAmount’, and optionally by ‘boundaryCondition’ and
‘charge’. The species produced result from the eight reactions which the hypo-
thetical single-gene circuit is capable of. “In SBML, reactions are dened using
lists of reactant species and products, their stoichiometric coefcients, and kinetic
rate laws.”8 This means that every reaction has to be specied by its ‘name’, by
the ‘species’ involved as reactants or products, by the attribute ‘reversible’, whichcan have the values false or true, and by the optional attribute ‘fast’. If the attribute
‘fast’ has the value true, “simulation/analysis packages may choose to use this in-
formation to reduce the number of [ordinary differential equations] ODEs required
and thereby optimize such computations.”9 Finally, ‘rules’ can be set to constrain
variables and parameters. However, SBML is solely a format to describe a model.
For simulation it has to be transferred to simulation and analysis packages that
support SBML. In 2003 nine simulation tools have supported SBML (Cellera-
tor , DBsolve, E-CELL, Gepasi, Jarnac, NetBuilder , ProMoT/DIVA, StochSim, and
Virtual Cell ), while today more than two hundred packages do. Based on the suc-cess of SBML, Systems Biology Graphical Notation (SBGN) has recently been
developed – and released in 2009 – as a community-wide open graphical standard
that allows three different views of biological systems: process descriptions, en-
standard based on open workshops and an editorial team for updates, which is elected
to a 3-year non-renewable term. (Cf. http://sbml.org/).
6 Nature Precedings: The Systems Biology Markup Language (SBML) Collection, (ac-
cessed on 6 January 2012). URL: http://precedings.nature.com/collections/sbml. An-
other description language for modeling is CellML.7 The example, and its notations, is taken from the initial SBML paper. Cf. Hucka et al.,
2003, loc. cit ., p. 526 ff.
8 Ibid ., p. 528.
9 Ibid ., p. 528.

7/26/2019 o segredo das coisa
http://slidepdf.com/reader/full/o-segredo-das-coisa 4/11
154 Gabriele Gramelsberger
tity relationships and activity ows, creating three different types of diagrams.
Furthermore, new software applications, like CellDesigner and CellPublisher , are
now built on both SBML and SBGN.
3. QUANTITATIVE DATA
Another important basis of preparing the stage for a simulation-orientated biology
is the acquisition of quantitative data as most simulation methods in biology are
based on differential equations, which describe the temporal development of a sys-
tem’s behavior. However, most data available in biology are qualitative data rep-
resenting the functions of genes, pathway maps, protein interaction, etc. “But for
simulation quantitative data (such as concentrations of metabolites and enzymes,ux rates, kinetic parameters and dissociation constants) are needed. A major chal-
lenge is to develop high-throughput technologies for measurement of inner-cellu-
lar metabolites.”10 Furthermore, these quantitative data have to be ne-grained.
“[T]raditional biological experiments tend to measure only the change before and
after a certain event. For computational analysis, data measured at a constant time
interval are essential in addition to traditional sampling points.”11 However, quan-
titative measurements of inner-cellular metabolites, protein synthesis, gene ex-
pression, etc. in ne-grained time series experiments are challenging experimental
biology. For instance, it is assumed that eukaryotic organisms contain between4,000 and 20,000 metabolites. Unlike proteins or RNA, the physical and chemical
properties of these metabolites are highly divergent and there is a “high proportion
of unknown analytes that is measured in metabolite proling. Typically, in current
[Gas-chromatography–mass-spectrometry] GC–MS-based metabolite proling, a
chemical structure has been unambiguously assigned in only 20–30% of the ana-
lytes detected.”12 Nevertheless, a typical GC-MS prole of one sample contains
300 to 500 analytes and generates a 20-megabyte data le. Quantitative data for
metabolite proles as well as for transcript and protein proling are usually ex-
pressed as ratios of a control sample. “In addition, absolute quantication is im- portant for understanding metabolic networks, as it is necessary for the calculation
of atomic balances or for using kinetic properties of enzymes to develop predictive
models.”13 For both types of quantitative data the changes in the samples, even for
tiny inuences, can be huge and it is difcult to achieve meaningful and reliable
10 Tomita 2001, loc. cit ., here p. 210. Cf. Jörg Stelling, et al., “Towards a Virtual Biologi-
cal Laboratory”, in: Hiroaki Kitano (Ed.), Foundations of Systems Biology. Cambridge
(Mass.): The MIT Press 2001, pp. 189-212.
11 Hiroaki Kitano, “Systems Biology: Toward System-level Understanding of BiologicalSystems”, in: Hiroaki Kitano, 2001, op. cit ., pp. 1-38, here p. 6.
12 Alisdair R. Fernie, et al., “Metabolite Proling: From Diagnostics to Systems Biol-
ogy”, in: Nature Reviews Molecular Cell Biology 5, 2004, pp. 763-769, here p. 764.
13 Fernie et al., 2004, here p. 765.

7/26/2019 o segredo das coisa
http://slidepdf.com/reader/full/o-segredo-das-coisa 5/11
155Simulation and System Understanding
results. Furthermore, these results do not come from ne-grained time series, let
alone cross-category measurements of “metabolites, proteins and/or mRNA from
the same sample […] to assess connectivity across different molecular entities.”14
However, ne-grained quantitative information is required for simulation in
biology. The answers to this challenge are manifold. One approach is to address
the measurement problem by creating new facilities, methods, and institutes. For
instance, in Japan a new institute has been set up “for this new type of simulation-
orientated biology, [… which] consists of three centers for metabolome research,
bioinformatics, and genome engineering, respectively.”15 Or alternatively, other
simulation methods for specic purposes have to be used “to deal with the lack of
kinetic information, […] for instance ux balance analysis (FBA).” 16 FBA does
not require dynamic data as it analyzes the capabilities of a reconstructed meta-
bolic network on basis of systemic mass-balance and reaction capacity constraints.Yet ux balance analysis does not give a unique solution for the ux distribution
– only an optimal distribution can be inferred.17 Another alternative could be the
estimation of unmeasurable parameters and variables by models. So-called non-
linear state-space models have been developed recently for the indirect determina-
tion of unknown parameters from measurements of other quantities. These models
take advantage of knowledge that is hidden in the system, by training the models
to learn more about themselves.
4. WHOLE-CELL SIMULATIONS
Whatever option is chosen to tackle the problems that come along with a simu-
lation-orientated biology, whole-cell simulations show great promise. On the one
hand they are needed for data integration,18 on the other hand the “ultimate goal
14 Ibid ., p. 768.
15 Tomita 2001, loc. cit ., here p. 210.
16 J. S. Edwards, R. U. Ibarra, B. O. Palsson, “In Silico Predictions of Escherichia coliMetabolic Capabilities are Consistent with Experimental Data”, in: Nature Biotechnol-
ogy 19, 2001, pp. 125–130, here p. 125.
17 “As a result of the incomplete set of constraints on the metabolic network (that is,
kinetic constant constraints and gene expression constraints are not considered), FBA
does not yield a unique solution for the ux distribution. Rather, FBA provides a solu-
tion space that contains all the possible steady-state ux distributions that satisfy the
applied constraints. Subject to the imposed constraints, optimal metabolic ux distri-
butions can be determined from the set of all allowable ux distributions using linear
programming (LP).” (Edwards, Ibarra, Palsson, 2001, loc cit ., here p. 125).
18 “[…] a crucial and obvious challenge is to determine how these, often disparate andcomplex, details can explain the cellular process under investigation. The ideal way
to meet this challenge is to integrate and organize the data into a predictive model.”
(Boris M. Slepchenko et al., “Quantitative Cell Biology with the Virtual Cell”, in:
TRENDS in Cell Biology 13, 11, 2003, pp. 570-576, here p. 570). Olaf Wolkenhauer

7/26/2019 o segredo das coisa
http://slidepdf.com/reader/full/o-segredo-das-coisa 6/11
156 Gabriele Gramelsberger
[…] is to construct a whole-cell model in silico […] and then to design a novel
genome based on the computer simulation and create real cells with the novel ge-
nome by means of genome engineering;”19 in brief: to enable the “computer aided
design (CAD) of useful microorganisms.”20 Projects like E-Cell or Virtual Cell –
just to mention two – aim “to develop the theories, techniques, and software plat-
forms necessary for whole-cell-scale modeling, simulation, and analysis.”21 E-Cell
and Virtual Cell differ in their conception as well as organization. Development of
the E-Cell software in C++ started in 1996 at the Laboratory for Bioinformatics at
Keio University, initiated by Masaru Tomita. In 1997 the 1.0beta version was used
to program the ‘virtual self-surviving cell’, which was accepted as an OpenSource
project by the Bioinformatics.org in 2000. The software development led to the
establishment of the Institute for Advanced Biosciences for metabolome research,
bioinformatics, and genome engineering in 2001 and by 2005 the Molecular Sci-ences Institute, Berkeley, and the Mitsubishi Space Co. Ltd, Amagasaki Japan, had
joined the project. E-Cell is used internationally by various research groups to re-
alize in-silico projects, e.g. on the dynamics of mitochondrial metabolism, on the
energy metabolism of E. coli, on glycolysis, etc.22 The software as well as the al-
ready programmed models can be downloaded from the web page. Unlike E-Cell ,
Virtual Cell is a freely accessible software platform by the Center for Cell Analysis
& Modeling of the University of Connecticut for building complex models with a
web-based Java interface. Thus, the “mathematic-savy user may directly specify
the complete mathematical description of the model, bypassing the schematicinterface.”23 Virtual Cell is conceived as an open community platform, providing
software releases and programmed models. Thus, the increasing availability of an
open community cyberinfrastructure for a simulation-orientated biology can be
observed as is already common for other disciplines like meteorology.24
and Ursula Klingmüller have expanded the denition of systems biology given in the
rst paragraph of this paper by adding “the integration of data, obtained from experi-
ments at various levels and associated with the ‘omics family’ of technologies.” (Olaf
Wolkenhauer, Ursula Klingmüller, “Systems Biology: From a Buzzword to a Life Sci-ence Approach”, in: BIOforum Europe 4, 2004, pp. 22-23, here p. 22).
19 Tomita 2001, loc. cit ., here p. 210.
20 Masaru Tomita, “Towards Computer Aided Design (CAD) of Useful Microorgan-
isms”, in: Bioinformatics 17, 12, 2001a, pp. 1091-1092.
21 Kouichi Takahashi et al., “Computational Challenges in Cell Simulation: A Software
Engineering Approach”, in: IEEE Intelligent Systems 5, 2002, pp. 64-71, here p. 64.
22 Cf. E-Cell: Homepage, (accessed on 6 January 2012). URL: http://www.e-cell.org/
ecell.
23 Virtual Cell: Homepage at the Center for Cell Analysis & Modeling, (accessed on 6
January 2012). URL: http://www.nrcam.uchc.edu/.24 Cf. Gabriele Gramelsberger, Johann Feichter, “Modeling the Climate System”, in: Ga-
briele Gramelsberger, Johann Feichter (Eds.), Climate Change and Policy. The Cal-
culability of Climate Change and the Challenge of Uncertainty. Heidelberg: Springer
2011, p. 44 ff.

7/26/2019 o segredo das coisa
http://slidepdf.com/reader/full/o-segredo-das-coisa 7/11
157Simulation and System Understanding
However, the aim of this simulation approach is the creation and distribu-
tion of complex models. These collaboratively advanced software machineries are
‘synthesizers’ for interconnecting all kinds of computational schemes and strat-
egies. Thus, complex systems are built in a bottom-up process by innumerous
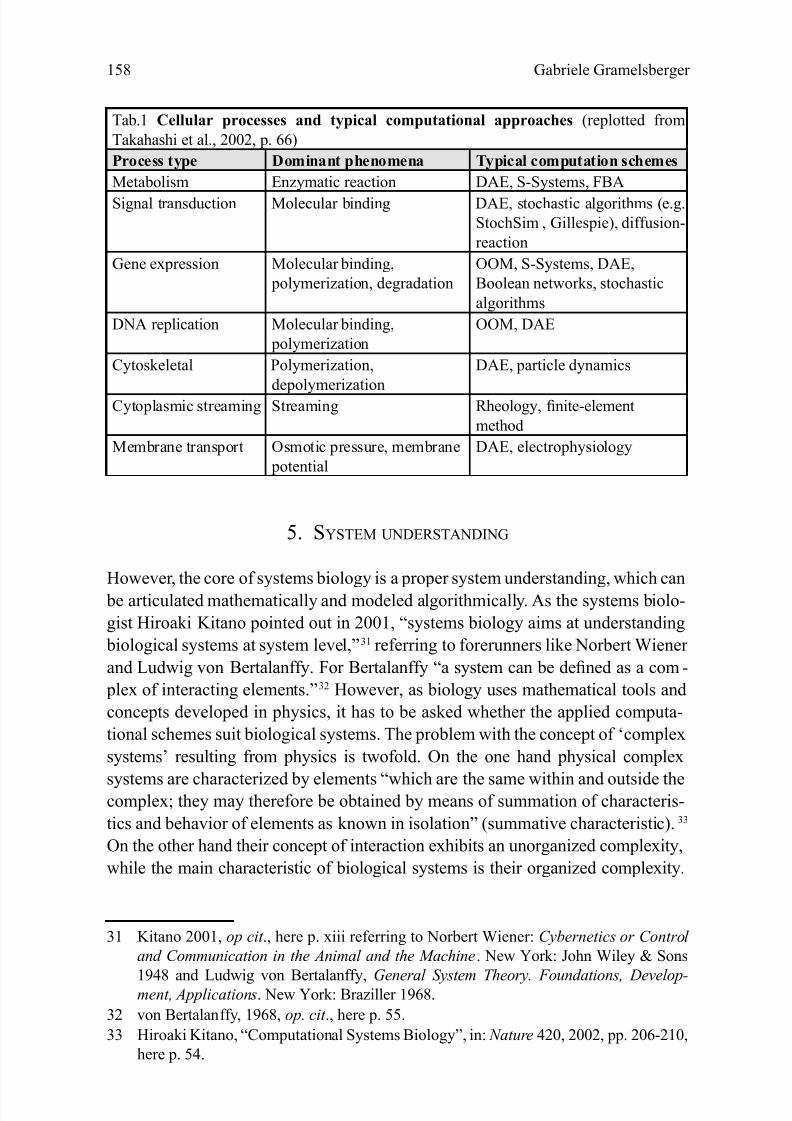
computational schemes. E-Cell , for example, combines object-oriented modeling
for DNA replication, Boolean networks and stochastic algorithms for gene expres-
sion, differential-algebraic equations (DAEs)25 and FBA for metabolic pathways,
SDEs and ODEs for other cellular processes (see Tab. 1).26 These advanced inte-
grative cell simulations provide in-silico experimental devices for hypothesis test-
ing and predictions, but also for data integration and the engineering of de-novo
cells. In such an in-silico experimental device genes can be turned on and off, sub-
stance concentrations and metabolites can be changed, etc. Observing the behavior
of the in-silico cell yields comparative insights and can lead to the discovery ofcausalities and interdependencies. Thus, “computers have proven to be invaluable
in analyzing these systems, and many biologists are turning to the keyboard.”27
Researchers involved in the development of the whole-cell simulator E-Cell have
outlined a ‘computational cell biology research cycle’ from wet experiments form-
ing cellular data and hypotheses, to qualitative and quantitative modeling, to pro-
gramming and simulation runs, to analysis and interpretation of the results, and
back to wet experiments for evaluation purposes.28 However, this is still a vision as
“biologists rarely have sufcient training in the mathematics and physics required
to build quantitative models, [therefore] modeling has been largely the purview oftheoreticians who have the appropriate training but little experience in the labora-
tory. This disconnection to the laboratory has limited the impact of mathematical
modeling in cell biology and, in some quarters, has even given modeling a poor
reputation.”29 In particular Virtual Cell tries to overcome this by offering an intui-
tive modeling workspace that is “abstracting and automating the mathematical and
physical operations involved in constructing models and generating simulations
from them.”30
25 A DAE combines one ordinary differential equation (ODE) for each enzyme reaction,
a stochimetric matrix, and algebraic equations for constraining the system.
26 Cf. Takahashi et al., 2002, loc. cit ., p. 66 ff.
27 Ibid ., p. 64.28 Cf. Ibid ., p. 64 ff.
29 Leslie M. Loew, et al., “The Virtual Cell Project”, in: Systems Biomedicine, 2010, pp.
273-288, here p. 274.
30 Loew, et al., 2010, loc. cit ., here p. 274.
7/26/2019 o segredo das coisa
http://slidepdf.com/reader/full/o-segredo-das-coisa 8/11
158 Gabriele Gramelsberger
Tab.1 Cellular processes and typical computational approaches (replotted from
Takahashi et al., 2002, p. 66)
Process type Dominant phenomena Typical computation schemes
Metabolism Enzymatic reaction DAE, S-Systems, FBA
Signal transduction Molecular binding DAE, stochastic algorithms (e.g.
StochSim , Gillespie), diffusion-
reaction
Gene expression Molecular binding,
polymerization, degradation
OOM, S-Systems, DAE,
Boolean networks, stochastic
algorithms
DNA replication Molecular binding,
polymerization
OOM, DAE
Cytoskeletal Polymerization,
depolymerization
DAE, particle dynamics
Cytoplasmic streaming Streaming Rheology, nite-element
method
Membrane transport Osmotic pressure, membrane
potential
DAE, electrophysiology
5. SYSTEM UNDERSTANDING
However, the core of systems biology is a proper system understanding, which can be articulated mathematically and modeled algorithmically. As the systems biolo-
gist Hiroaki Kitano pointed out in 2001, “systems biology aims at understanding
biological systems at system level,”31 referring to forerunners like Norbert Wiener
and Ludwig von Bertalanffy. For Bertalanffy “a system can be dened as a com-
plex of interacting elements.”32 However, as biology uses mathematical tools and
concepts developed in physics, it has to be asked whether the applied computa-
tional schemes suit biological systems. The problem with the concept of ‘complex
systems’ resulting from physics is twofold. On the one hand physical complex
systems are characterized by elements “which are the same within and outside thecomplex; they may therefore be obtained by means of summation of characteris-
tics and behavior of elements as known in isolation” (summative characteristic).33
On the other hand their concept of interaction exhibits an unorganized complexity,
while the main characteristic of biological systems is their organized complexity.
31 Kitano 2001, op cit ., here p. xiii referring to Norbert Wiener: Cybernetics or Control
and Communication in the Animal and the Machine. New York: John Wiley & Sons
1948 and Ludwig von Bertalanffy, General System Theory. Foundations, Develop-ment, Applications. New York: Braziller 1968.
32 von Bertalanffy, 1968, op. cit ., here p. 55.
33 Hiroaki Kitano, “Computational Systems Biology”, in: Nature 420, 2002, pp. 206-210,
here p. 54.

7/26/2019 o segredo das coisa
http://slidepdf.com/reader/full/o-segredo-das-coisa 9/11
159Simulation and System Understanding
Therefore, Kitano calls biological systems ‘symbiotic systems’ exhibiting coher -
ent rather than complex behavior:
It is often said that biological systems, such as cells, are ‘complex systems’. A popular
notion of complex systems is of very large numbers of simple and identical elements in-teracting to produce ‘complex’ behaviours. The reality of biological systems is somewhat
different. Here large numbers of functionally diverse, and frequently multifunctional, sets
of elements interact selectively and nonlinearly to produce coherent rather than complex
behaviours.
Unlike complex systems of simple elements, in which functions emerge from the properties
of the networks they form rather than from any specic element, functions in biological
systems rely on a combination of the network and the specic elements involved. […] In
this way, biological systems might be better characterized as symbiotic systems.34
In contrast to physics, biological elements are innumerous, functionally diverse,
and interact selectively in coupled and feedbacked sub-networks, thus character-
izing the properties of a biological system. The challenge for systems biology is:
how to conceive biological complexity? In retrospect, the basic question of sys-
tem understanding was already discussed in the 19th century’s mechanism-vitalism
debate.35 In the 20th century the idea of self-regulating systems emerged and two
different concepts have been developed against the complex system approach of
physics: the steady-state (Fliessgleichgewicht = ux equilibrium) or open sys-
tem concept by Bertalanffy and the feedback regulation concept of Wiener refer -ring to Walter B. Cannon’s biological concept of homeostasis.36 Bertalanffy aptly
describes the differences between both concepts. For him Cannon’s homeostatic
control and Wiener’s feedback systems are special classes of self-regulating sys-
tems. Both are
‘open’ with respect to incoming information, but ‘closed’ with respect to matter. The con-
cepts of information theory–particularly in the equivalence of information and negative
entropy–correspond therefore to ‘closed’ thermodynamics (thermostatics) rather than irre-
versible thermodynamics of open systems. However, the latter is presupposed if the system(like the living organism) is to be ‘self-organizing’. […]
Thus dynamics in open systems and feedback mechanisms are two different model con-
cepts, each right in its proper sphere. The open-system model is basically nonmechanistic,
and transcends not only conventional thermodynamics, but also one-way causality as is ba-
sic in conventional physical theory. The cybernetic approach retains the Cartesian machine
34 Kitano, 2002, loc cit ., here p. 206.
35 Cf. Ulrich Krohs, Georg Toepfer (Eds.), Philosophie der Biologie. Frankfurt: Suhr -
kamp 2005.
36 Cf. Ludwig von Bertalanffy, Theoretische Biologie. Berlin: Bornträger 1932; Ludwigvon Bertalanffy, Biophysik des Fließgleichgewichts. Berlin: Akademie Verlag 1952;
Wiener, 1948, op. cit .; Walter B. Cannon, “Organization for Physiological Homeosta-
sis”, in: Physiological Review 9, 1929, p. 397; Walter B. Cannon, The Wisdom of the
Body. New York: Norton 1932.

7/26/2019 o segredo das coisa
http://slidepdf.com/reader/full/o-segredo-das-coisa 10/11
160 Gabriele Gramelsberger
model of the organism, unidirectional causality and closed systems; its novelty lies in the
introduction of concepts transcending conventional physics, especially those of information
theory. Ultimately, the pair is a modern expression of the ancient antithesis of ‘process’ and
‘structure’; it will eventually have to be solved dialectically in some new synthesis.37
Following Bertalanffy’s ideas, “the living cell and organism is not a static pattern
or machine like structure consisting of more or less permanent ‘building materi-
als’ […, but] an ‘open system’.”38 While in physics systems are usually conceived
as closed ones, sometimes expanded by additional terms for energy exchange
with their environment, biological systems are open in regard to energy and mat-
ter. Therefore biological systems show characteristic principles of behavior like
steady state, equinality, and hierarchical organization. “In the steady state, the
composition of the system remains constant in spite of continuous exchange of
components. Steady states or Fliessgleichgewicht are equinal […]; i.e., the same
time-independent state may be reached from different initial conditions and in dif-
ferent ways – much in contrast to conventional physical systems where the equi-
librium state is determined by initial conditions.”39 This can lead to ‘overshoots’
and ‘false starts’ as a response to unstable states and external stimuli (adaptation),
because biological systems tend towards a steady state. Based on this concept the
overshoot of intercellular ATP as exhibited by the virtual self-surviving cell (see
Sect. 1) can be easily explained, while for a physicist it sounds mysteriously.
However, the basic question is: Does simulation support this new system un-
derstanding beyond the physical concept of complex systems? Can biological sys-
tems modeled based on non-summative characteristics, meaning that a complex
is not built up “step by step, by putting together the rst separate elements; […
but by using] constitutive characteristics […] which are dependent on the specic
relations within the complex; for understanding such characteristics we therefore
must know not only the parts, but also their relations.”40 The brief overview of
modeling practices with SBML has shown that each part (species, reaction, etc.)
has to be described explicitly. However, the important aspect is the interaction
between these parts (ux). Advanced software machineries allow complex inter -actions and feedbacks to be organized, e.g. feedbacks, loops, alternatives (jumps),
etc. Thus, the software machineries of whole-cell simulations function as ‘synthe-
sizers’ and can be seen as media for organizing complex relations and uxes. As
already dened by Herman Goldstine and John von Neumann in 1946, “coding
begins with the drawing of the ow diagrams.”41 These ow diagrams specify the
37 von Bertalanffy 1986, op. cit ., here p. 163.
38 Ibid ., p. 158.
39 Ibid ., p. 159.40 Ibid ., pp. 67 and 55.
41 Herman H. Goldstine, John von Neumann, “Planning and Coding Problems for an
Electronic Computing Instrument” (1947), Part II, vol. 1, in: John von Neumann, Col-
lected Work , vol. V: Design of Computers, Theory of Automata and Numerical Analy-

7/26/2019 o segredo das coisa
http://slidepdf.com/reader/full/o-segredo-das-coisa 11/11
161Simulation and System Understanding
various sequences of calculations as well as routines that have to be repeated. The
result is a complex choreography of command courses and loops; and “the relation
of the coded instructions to the mathematically conceived procedures of (numeri-
cal) solutions is not a statical one, that of a translation, but highly dynamical.”42
The dynamics of the actual path of computing through the instructions (simulation
run), which Goldstine and Neumann called the ‘modus procedendi’, requires an
ambitious choreography of possibilities and alternatives expressed by ‘if, then,
else, end if’-decisions, loops, and calls of other subroutines within each singular
le of a program. Moreover, object-oriented programming allows for ‘simulating’
complex, organizational structures by dening objects and classes with specic
properties for varying contexts, imitating selectivity and functional diversity.43
While the underlying mathematical tools and computational schemes, resulting
from physics, haven’t changed, advanced software machineries allow their ‘organ-ized complexity’. Thus, the simulation approach not just supports, but enables the
new system understanding for biology. One can ask, if it only bypasses the core
challenge of biological complexity by mimicking organized complexity. However,
if this is the case, biology has to inspire a new mathematics, just as physics did
four centuries ago by developing the calculus for describing the kinetics of spatio-
temporal systems.
FU Institute of Philosophy
Freie Universität Berlin
Habelschwerdter Allee 30
D-14195, Berlin
Germany
sis, Oxford: Pergamon Press 1963, pp. 80-151, here p. 100. Cf. Gabriele Gramels-
berger, “From Computation with Experiments to Experiments with Computation”, in:
Gabriele Gramelsberger (Ed.), From Science to Computational Sciences. Studies in the
History of Computing and its Inuence on Today’s Sciences. Zurich: Diaphanes, pp.
131-142.
42 Goldstine, Neumann, 1947, loc. cit ., here pp. 81-82.
43 Interestingly, the object-oriented programming paradigm – introduced in 1967 with
Simula 67 for physical simulations – was advanced for the programming language
C++, which originated in the telecommunication industry (Bell labs) in response to thedemand for more complex structures for organizing network trafc. Cf. Terry Shinn,
“When is Simulation a Research-Technology? Practices, Markets and Lingua Franca”,
in: Johannes Lenhard, Günter Küppers, Terry Shinn (Eds.), Simulation: Pragmatic
Construction of Reality. Dordrecht: Springer, 2006, pp. 187-203.


















