
Now what happens after inflammation? Some pathogens are very mild and are easily eradicated by...
33
Now what happens after inflammation? • Some pathogens are very mild and are easily eradicated by neutrophils and macrophages. But others can be deadly. • Bacteria, toxic molecules, virus may all cause direct cellular damage, initially at epithelial cells, that lead first to inflammation, including degranulation of mast cells, release of chemokines like histamine and… • vasodilation (swelling), redness, pain (prostaglandins), possibly pus (phagocytizing neutrophils). Herpes simplex is one example. Others include a sore throat, acne, a spider bite, a splinter, inflamed eyes etc. • For self-limiting pathogenic infections, following neutrophil migration out of vascular system, macrophages usually digest cell debris, and chemokine activation of fibroblasts starts re- epithelialization and healing. • But, some pathogens invade further than epithelial tissue and can cause inflammation and cell death that is life threatening, for example in all encephalitis infections — “-itis” means inflammation — or in rabies or gastritis. • When dendritic cells bind to the pathogen antigens, either from virus, the surface of microbial cells or the molecular debris of their digestion products, they can launch an ADAPTIVE IMMUNE RESPONSE, as happens with Herpes and many other infections (e.g. chicken pox [a Herpes virus], measles, small pox.
-
Upload
tyrone-lamb -
Category
Documents
-
view
215 -
download
0
Transcript of Now what happens after inflammation? Some pathogens are very mild and are easily eradicated by...

Now what happens after inflammation?• Some pathogens are very mild and are easily eradicated by neutrophils and
macrophages. But others can be deadly.• Bacteria, toxic molecules, virus may all cause direct cellular damage, initially at
epithelial cells, that lead first to inflammation, including degranulation of mast cells, release of chemokines like histamine and…
• vasodilation (swelling), redness, pain (prostaglandins), possibly pus (phagocytizing neutrophils). Herpes simplex is one example. Others include a sore throat, acne, a spider bite, a splinter, inflamed eyes etc.
• For self-limiting pathogenic infections, following neutrophil migration out of vascular system, macrophages usually digest cell debris, and chemokine activation of fibroblasts starts re-epithelialization and healing.
• But, some pathogens invade further than epithelial tissue and can cause inflammation and cell death that is life threatening, for example in all encephalitis infections — “-itis” means inflammation — or in rabies or gastritis.
• When dendritic cells bind to the pathogen antigens, either from virus, the surface of microbial cells or the molecular debris of their digestion products, they can launch an ADAPTIVE IMMUNE RESPONSE, as happens with Herpes and many other infections (e.g. chicken pox [a Herpes virus], measles, small pox.

INNATE IMMUNITY relies on detection of a microbial infection by key receptors (first discovered in insects) that have evolved to recognize and bind evolutionarily conserved molecules unique to microbes. Toll protein receptors bind these microbial molecules and activate inflammatory and innate immune responses. Binding to Toll-like receptors on dendritic cells — major surveyors of epithelial areas like skin and gut — triggers their maturation and can launch steps of the adaptive immune system.
After phagocytic cells, primarily dendritic cells, recognize common cell-surface antigens on microbes, phagocytosis leads to individual pathogen cell digestion and release of pathogen molecular debris, all of which may be antigenic.
Pathogen debris

Toll-like receptors recognize a variety of common microbial molecules. Several different TLRs bind different Pathogen-associated Molecular Patterns (PAMPs). Key PAMPs include lipopolysaccharides, peptidoglycan (found in bacterial membranes), dsRNA (found in many virus), flagellin (a protein found in bacterial flagella), lipoteichoic acid (found in Gram-positive bacteria). See Fig 35.4 in your book, p 713. Binding of PAMPs by TLRs on dendritic cells upregulates co-stimulatory molecules CD80 and CD86 as well as MHC-II molecules and dendritic cell maturation. Dendritic cells are ANTIGEN PRESENTING CELLS that will lead to the activation of T-lymphocytes.
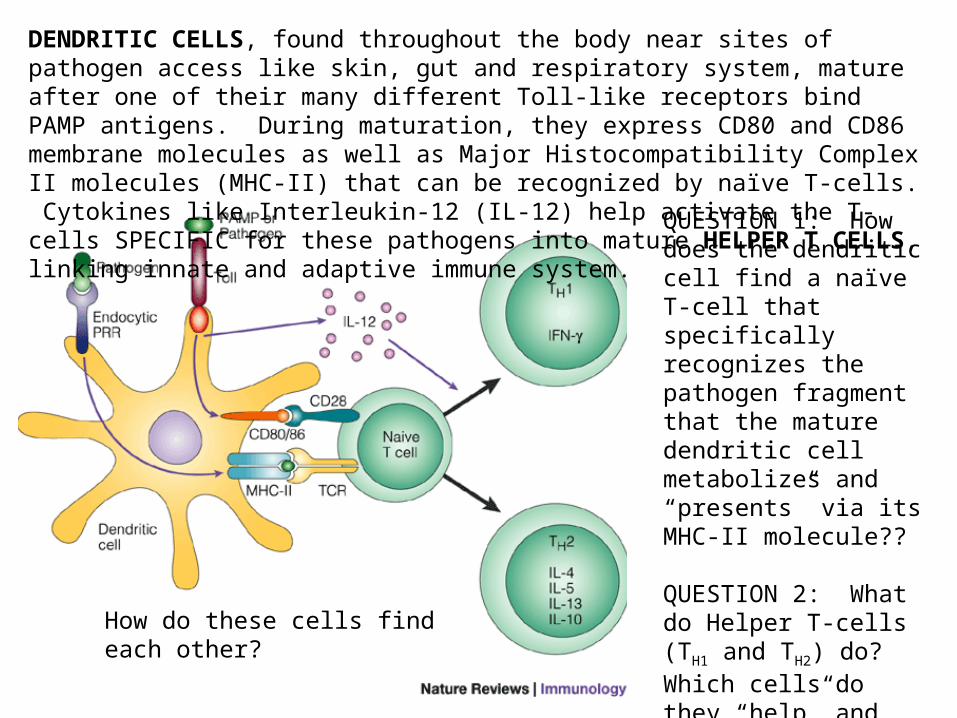
DENDRITIC CELLS, found throughout the body near sites of pathogen access like skin, gut and respiratory system, mature after one of their many different Toll-like receptors bind PAMP antigens. During maturation, they express CD80 and CD86 membrane molecules as well as Major Histocompatibility Complex II molecules (MHC-II) that can be recognized by naïve T-cells. Cytokines like Interleukin-12 (IL-12) help activate the T-cells SPECIFIC for these pathogens into mature HELPER T CELLS, linking innate and adaptive immune system.
QUESTION 1: How does the dendritic cell find a naïve T-cell that specifically recognizes the pathogen fragment that the mature dendritic cell metabolizes and “presents” via its MHC-II molecule??
QUESTION 2: What do Helper T-cells (TH1 and TH2) do? Which cells do they “help” and how do they do it?
ANSWERS TO BE SEEN!
How do these cells find each other?

A simplified image of antigen processing by an APC [antigen presenting cell] – dendritic and macrophage cells. Notice that the APC presents an antigen “fragment” following digestion of an entire microbe or endocytosis of microbial debris. Naïve Helper T cells that recognize these specific fragment conformations will bind to the APC cell through its MHC-II molecule, along with the CD-80/86 co-stimulators that have been up-regulated.
interleukins

A more elaborate picture of APC antigen presentation with MHC-II. Either entire microorganisms or microbial debris can be processed into presentation fragments through the APC endolysosome. The MHC-II molecules will not be upregulated until an antigen fragment binds to it within the APC. The Helper T-cell receptor [TCR] recognizes the MHC-II
molecule with its antigen fragment via the T-cell CD-4 co-receptor. The APC will also release IL-12 among other cytokines that launch maturation of the naïve T-cell into a mature Helper T-cell that is SPECIFIC for the particular antigen presented.

Dendritic APC
From previous slide where APC presents antigen to T-cell receptor with CD4 coreceptor (a helper T cell)
Two things are happening simultaneously: 1) dendritic cells have digested an invading microbe, traveled to a nearby lymph node, encountered a naïve T-4 cell and activated it through antigen presentation, and 2) free antigen from microbial debris has traveled to the same lymph node where it encounters a B-cell receptor
Within a nearby lymph node!

Where does this all occur?
Recall how fluids and small proteins escape capillaries into interstitial spaces. During an infection with capillary swelling, inflammation and cell destruction, the lymphatics also pick up microbe antigens and cell debris. Importantly, AP cells can migrate into lymphatics and move toward a nearby lymph node into which microbes and microbial antigens will flow.
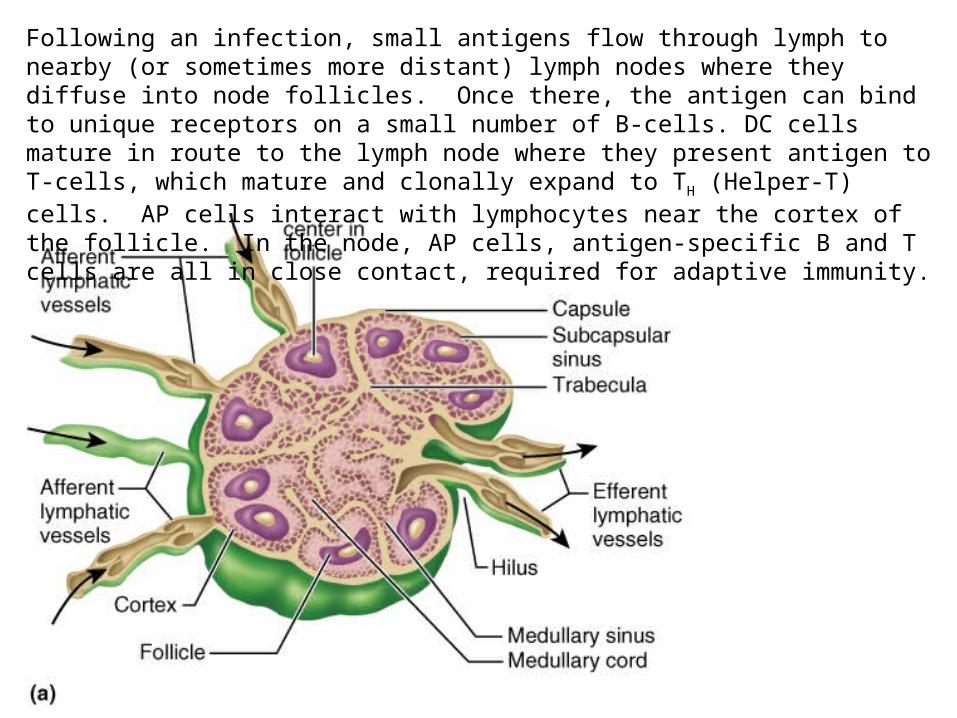
Following an infection, small antigens flow through lymph to nearby (or sometimes more distant) lymph nodes where they diffuse into node follicles. Once there, the antigen can bind to unique receptors on a small number of B-cells. DC cells mature in route to the lymph node where they present antigen to T-cells, which mature and clonally expand to TH (Helper-T) cells. AP cells interact with lymphocytes near the cortex of the follicle. In the node, AP cells, antigen-specific B and T cells are all in close contact, required for adaptive immunity.

What might happen in the lymph node?After — or even during — APC presentation of antigen to a naïve T-cell, a B-cell with the right B-cell receptor (BCR) can bind free antigen. The Ag will be processed via the IgD receptor and presented on the B-cells MHC-II protein. This is virtually identical to what the APC does. In fact, B-cells are also antigen-presenting cells. Ag presentation to the T-cell, remember, leads to its maturation as a TH1 or TH2 cell. This is a TH2 cell that will in turn activate the B-cell that also presents it with Ag by releasing B-cell specific interleukins.
This activated B-cell will now undergo maturation, eventually developing into a circulating PLASMA CELL which synthesizes and releases antibodies.
Dendritic cell presenting Ag to naïve T-cell

The B-cell receptor is an IMMUNOGLOBULIN, one of 5 classes of Ig molecules – IgA, IgD, IgE, IgG and IgM. The receptor is itself IgD, a membrane-bound immunoglobulin.
Clonal expansion and maturation to Plasma and Memory B cells.
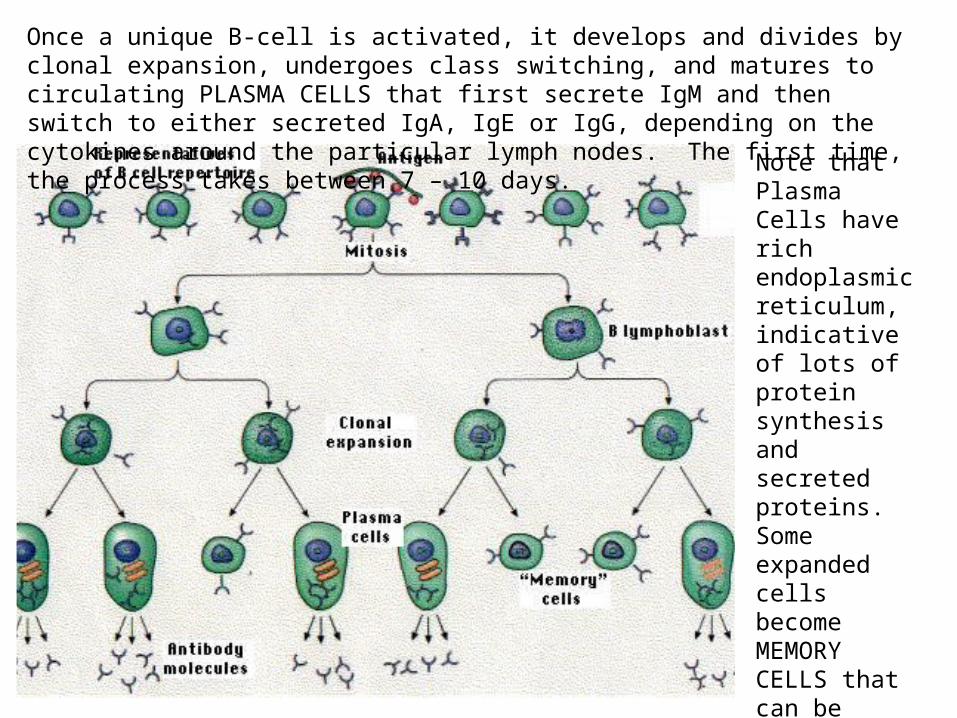
Once a unique B-cell is activated, it develops and divides by clonal expansion, undergoes class switching, and matures to circulating PLASMA CELLS that first secrete IgM and then switch to either secreted IgA, IgE or IgG, depending on the cytokines around the particular lymph nodes. The first time, the process takes between 7 – 10 days.
Note that Plasma Cells have rich endoplasmic reticulum, indicative of lots of protein synthesis and secreted proteins. Some expanded cells become MEMORY CELLS that can be activated to plasma cells very quickly after a 2nd infection.

• B-cell receptors that can bind free antigen (Ag) entering lymph nodes are all IMMUNOGLOBINS
• All Igs have 2 light chains and 2 heavy chains, and all 4 chains have a “constant” region and a “variable” region.
• The constant region may be one of 5 types, but the variable region may be one of more than 300,000,000 conformations!!!!
• The combinations emerge from random genetic shuffling, generated by recombinase enzymes
Notice that Igs are BIVALENT, meaning that two identical binding sites are generated on each Ig molecule. Any given B-cell makes only one type of Ig at a time. Also, any given T-cell makes only one type of T-cell receptor, out of the hundreds of millions of possibilities.

We’ve seen how RNA can be cut and spliced last semester. DNA recombination occurs normally in B and T cells to generate large numbers of unique sequences that are transcribed to generate far more individual proteins than there are genes! This diagram is of light-chain rearrangement. Heavy chains can generate even more combinations. When light and heavy chains combine, up to 3,500,000 different antibody receptors can be made within a single organism. V and J regions also mutate during B-cell maturation, generating even more combinations.
A naïve B-cell, upon stimulation by an antigen and activation from Helper T cells, matures to a PLASMA CELL, and clonally expands while switching its Ig class, first to IgM and later to IgA, IgE or IgG, all of which are secreted in large quantity by plasma B cells. Some cells mature to MEMORY B-CELLS

• An individual B-cell not only makes one variation of an Ig, with a binding site identical to the B-cell receptor.
• The cell SWITCHES the constant region as it develops after first encountering an Ag in the lymph node – called CLASS SWITCHING.
• But the binding site undergoes only small mutational variations, allowing its binding affinity to get stronger but not change significantly
1. The B-cell receptor itself is IgD2. The next antibody produced is IgM3. In lymph nodes around secretory
ducts, B-cells next produce IgA (found in tears, saliva, mucus)
4. IgE is the primary antibody elicited by parasitic antigens and is associated with hypersensitive reactions.
5. IgG is the most abundant (75%) and activates COMPLEMENT CASCADE, crosses the placenta and increases with bacterial and viral infections
After a B-cell first encounters a free antigen on its IgD receptor, the receptor is internalized, and the B-cell starts maturing and undergoing CLASS SWITCHING and variable-site mutations to secrete Igs with tighter binding affinities to the same antigen.

Bivalent antibodies can bind to microbe surfaces or to free antigens, a process called OPSONIZATION, thereby targeting them for destruction by MACROPHAGES, neutrophils or COMPLEMENT – a plasma protein cascade that enhances macrophage binding to opsonized microbes.
Membrane-attack complex

A simplified view of the connections between innate immunity and adaptive immunity. Unlike the innate system, adaptive immunity is specific, highly diverse, recognizes differences between self and non-self and is long-lasting.
The adaptive immune system operates as a clonal-selection system whereby scavenging and phagocytizing cells [APCs], dendritic cells and macrophages present non-self peptide antigens to HELPER T-cells in a highly specific way.
Helper T-cells clonally expand and link with B-cells that have bound soluble antigen, produced by microbe or cell digestion, causing development & expansion of the B-cells to produce antigen-specific soluble antibodies (HUMORAL IMMUNITY). At the same time, helper T-cells bind to other T-cells that develop to cytotoxic cells (CELLULAR IMMUNITY).

• Here we are at THURSDAY!

To review: activation of B-cells. Free antigen or microbial fragments bind to inactive B-cells in lymph nodes. Direct activation of B-cells is possible after B-cells process antigen and present via MHC-II to TH-cell receptor. Action is much more robust is TH-cell has been activated by dendritic APC leading to TH-cell clonal expansion. In either case B-cell clonally expands and differentiates to Plasma cell clones that express IgG, IgA or IgE
and to memory B cells. See Fig 35.12 for specificity of immunological memory. Note below IgM initial response followed quickly by IgG class switch. Response to second antigen challenge reflects memory cells which typically secrete IgG.
APC

• B-cell response is typical of adaptive immune reaction to extracellular pathogens (e.g. bacteria and parasites).
• Response to intracellular parasites typically involves activation of cytotoxic T-cells.
• In addition to MHC-II found on APCs, all cells except RBCs (including APCs) have another Major Histocompatibility Complex, MHC-I.
• Intracellular pathogens, for example, virus can result in inflammation and macrophage activity.
• As with dendritic cells, macrophages have MHC-II complexes that present antigen fragments — for example viral fragments, or fragments of cells killed by virus — to Helper T cells in lymph nodes.

Macrophages, like dendritic cells, are highly mobile and enter lymphatics after action at the site of a viral infection and inflammation. Notice macrophages don’t rely as extensively on Toll-like receptors. They phagocytize all cell debris around an infection. After migrating to lymph nodes and presenting Ag to the Helper T-cell that recognizes the Ag, secreted IL-2 promotes differentiation of the Helper T-cell into a TH1 cell that, in turn helps activate
cytotoxic T-cells in the lymph nodes. TC cells then migrate back to site of inflammation and infection. TC cells recognize infected cells — or other abnormal cells that macrophages have processed like some cancer cells — by their display of antigen on the MHC-I protein. ALL cells, except RBCs, can display antigens via MHC-I proteins.
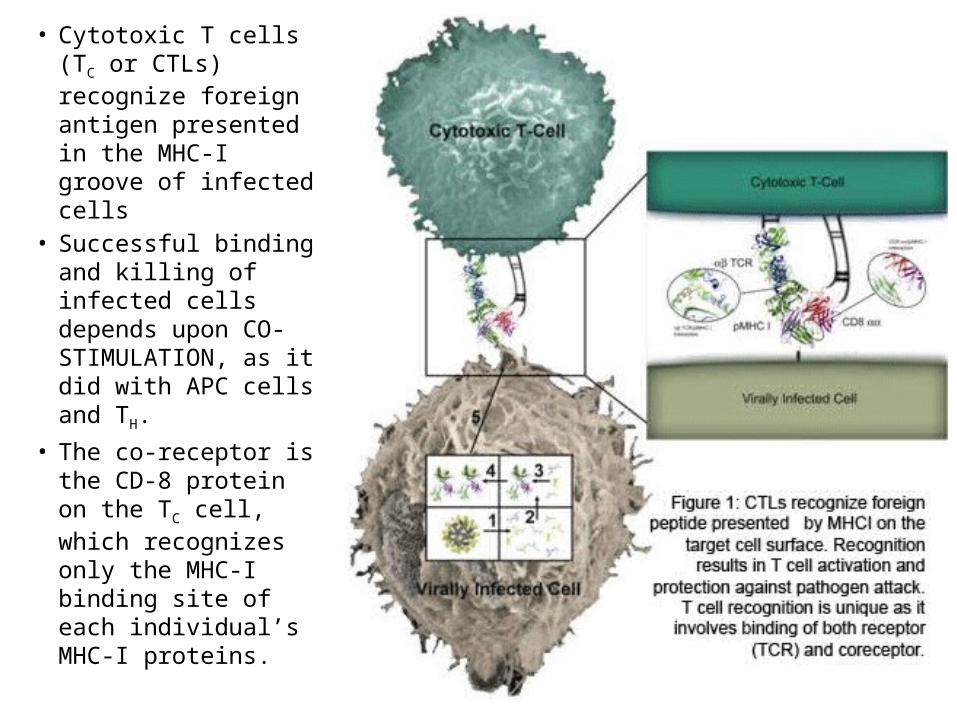
• Cytotoxic T cells (TC or CTLs) recognize foreign antigen presented in the MHC-I groove of infected cells
• Successful binding and killing of infected cells depends upon CO-STIMULATION, as it did with APC cells and TH.
• The co-receptor is the CD-8 protein on the TC cell, which recognizes only the MHC-I binding site of each individual’s MHC-I proteins.

CD8 acts as a coreceptor for regular MHC-I binding TC cells. Interactions with CD8 is too weak independent of the TCR, but it potentiates the TCR binding and secreting cytokines and development of cytotoxicity against a cell that has both the correct MHC-I protein with foreign antigen AND binding site for CD8.
A similar coreceptor activity occurs with TH cells, except that their coreceptor is the CD4 molecule. Frequently the cells are identified as T-4 or T-8 cells.
Once a TC cell recognizes an antigen fragment bound to the MCH-I of an infected cell, costimulation by CD8 results in cytotoxic responses, including release of PERFORIN, which can lead to osmotic cell death, and cytokines which launch apoptosis.

MHC-I molecules exhibit enormous polymorphic variability in a population. Most variability is at the a1 and a2 sites, which generate the antigen-binding groove. The groove accommodates peptides between 8-10 amino acids. There are 6 genes that encode Class I MHC in humans (remember, called HLA in humans). Each of us has only 6 of the many MHC-1 alleles, but it’s highly unlikely any of us have the same 6. With very rare exception, mature lymphocytes recognize only the MHC of the same individual (or identical twin).
Cytokines like IL-12 increase expression of MHC-1. NK cells and cytotoxic T-cells (TC or T8 cell) recognize either foreign MHC-1 molecules or foreign peptides in the groove of self-MHC-I complexes, but not empty MHC-I molecules.
9 amino acid peptide in MHC-I groove

Antigen presentation via MHC-I and MHC-II. LEFT PANEL. An invading virus can be digested via proteasomes to generate peptides 12-20 amino acids long. These are VIRAL ANTIGENS. Cells begin synthesizing MHC-I proteins upon infection, and the viral antigens are targeted to the ER, where the MHC-I binding pocket will bind them. The whole complex of MHC-I—antigen is then processed like a new membrane protein and inserted into the cell membrane. It will be recognized by Tcytotoxic lymphocyte cells of the adaptive immune system.
DC cells (dendritic) and macrophages process antigens via lysosomes and then display them in their MHC-II complexes on the cell surface…for a different function.
MHC-I processing MHC-II processing
APC cellsAll
cells

Infected cells can also secrete a-interferon, a cytokine that helps inhibit virus replication in neighboring cells that might become infected after the virus starts reproducing in abundance. Cells that secrete a-interferon may not survive a viral infection, but will slow the infection process significantly in other cells. Interferons also work to up-regulate proteasome digestion of foreign antigens and lysosome activity for digesting infectious microbes and also MHC-I synthesis in preparation for antigen display.
After AP cells are activated and have displayed antigen in their MHC-II cell-surface complexes, they then “present” the antigen to a key T-lymphocyte, the “naïve” T-cell. Naïve T-cells are T-lymphocytes that have been process in the thymus gland but have not undergone any further maturation via an infection.

Fig 35.13, p 721. A simplified graphic that diagrams the generation of TH1 and TH2 cells to promote cell-mediated TC cytotoxicity or B-cell production of antibodies that target extracellular pathogens, respectively. What’s missing in the diagram, however, are the different Helper T cells!!!!
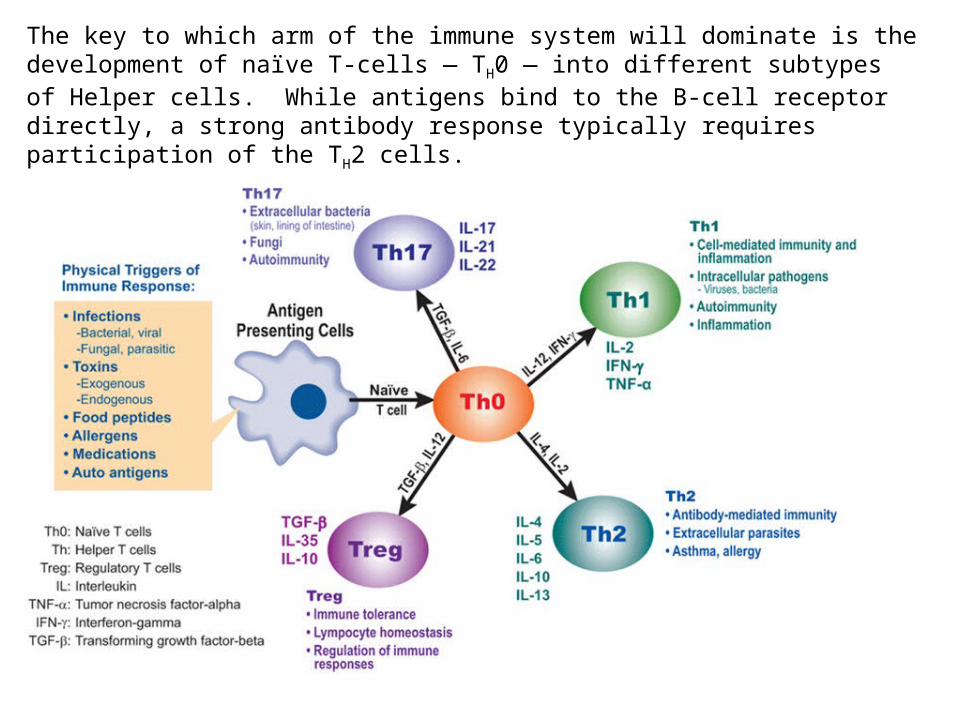
The key to which arm of the immune system will dominate is the development of naïve T-cells — TH0 — into different subtypes of Helper cells. While antigens bind to the B-cell receptor directly, a strong antibody response typically requires participation of the TH2 cells.


Intestinal immune reactions include constant surveillance of gut microbes by the processes of dendritic cells, movement of dendritic cells to gut lymphoid tissue like GALT (gut-associated lymphoid tissue) and Peyer’s Patches.
Ongoing local inflammatory reactions and antibody IgA production generally keeps gut microbe activity in check — but not always!!
Gut microbes
Intestinal lymph node

Goblet cells normally secrete mucus that limits exposure to bacteria. Paneth cells secrete antimicrobial a-defensins. Dendritic cells survey the intestinal mucosa by inserting dendritic processes through epithelial cell-cell junctions. Commensal and pathogenic bacterial PAMPs are differentiated by intestinal dendritic cells (usually), after which IgA-secreting B-cells are activated in intestinal lymph nodes, leading to an IgA environment that readily opsonizes the occasional pathogenic intestinal microbe that breaches the intestinal epithelial wall. Microbial recognition is integrated by epithelial cells and promotes intestinal cell health and survival by activating synthesis of cell-cell tight junction and antimicrobial proteins.
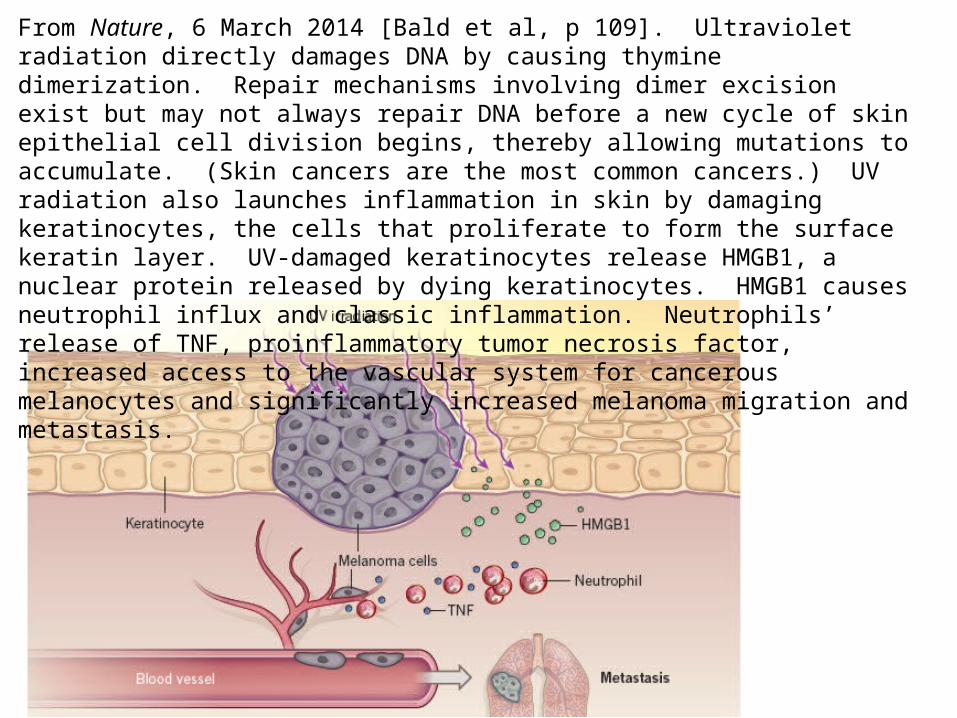
From Nature, 6 March 2014 [Bald et al, p 109]. Ultraviolet radiation directly damages DNA by causing thymine dimerization. Repair mechanisms involving dimer excision exist but may not always repair DNA before a new cycle of skin epithelial cell division begins, thereby allowing mutations to accumulate. (Skin cancers are the most common cancers.) UV radiation also launches inflammation in skin by damaging keratinocytes, the cells that proliferate to form the surface keratin layer. UV-damaged keratinocytes release HMGB1, a nuclear protein released by dying keratinocytes. HMGB1 causes neutrophil influx and classic inflammation. Neutrophils’ release of TNF, proinflammatory tumor necrosis factor, increased access to the vascular system for cancerous melanocytes and significantly increased melanoma migration and metastasis.

• Topics not covered in lecture that are important:
• Evasion of immune system• Origin of self-tolerance• IgE and allergies


















