
Natural Gas Conversion Into Value Added Chemicals Using ...
195
Louisiana State University LSU Digital Commons LSU Doctoral Dissertations Graduate School March 2019 Natural Gas Conversion Into Value Added Chemicals Using Solid Acid Catalysts Swarom Ravindra Kanitkar Louisiana State University and Agricultural and Mechanical College, [email protected] Follow this and additional works at: hps://digitalcommons.lsu.edu/gradschool_dissertations Part of the Catalysis and Reaction Engineering Commons is Dissertation is brought to you for free and open access by the Graduate School at LSU Digital Commons. It has been accepted for inclusion in LSU Doctoral Dissertations by an authorized graduate school editor of LSU Digital Commons. For more information, please contact[email protected]. Recommended Citation Kanitkar, Swarom Ravindra, "Natural Gas Conversion Into Value Added Chemicals Using Solid Acid Catalysts" (2019). LSU Doctoral Dissertations. 4865. hps://digitalcommons.lsu.edu/gradschool_dissertations/4865
Transcript of Natural Gas Conversion Into Value Added Chemicals Using ...

Louisiana State UniversityLSU Digital Commons
LSU Doctoral Dissertations Graduate School
March 2019
Natural Gas Conversion Into Value AddedChemicals Using Solid Acid CatalystsSwarom Ravindra KanitkarLouisiana State University and Agricultural and Mechanical College, [email protected]
Follow this and additional works at: https://digitalcommons.lsu.edu/gradschool_dissertations
Part of the Catalysis and Reaction Engineering Commons
This Dissertation is brought to you for free and open access by the Graduate School at LSU Digital Commons. It has been accepted for inclusion inLSU Doctoral Dissertations by an authorized graduate school editor of LSU Digital Commons. For more information, please [email protected].
Recommended CitationKanitkar, Swarom Ravindra, "Natural Gas Conversion Into Value Added Chemicals Using Solid Acid Catalysts" (2019). LSU DoctoralDissertations. 4865.https://digitalcommons.lsu.edu/gradschool_dissertations/4865

i
NATURAL GAS CONVERSION INTO VALUE ADDED CHEMICALS
USING SOLID ACID CATALYSTS
A Dissertation
Submitted to the Graduate Faculty of the Louisiana State University and
Agricultural and Mechanical College in partial fulfillment of the
requirements for the degree of Doctor of Philosophy
in
The Department of Chemical Engineering
by
Swarom R. Kanitkar
B.Tech., Visvesvaraya National Institute of Technology, 2010
M.S., University of Louisiana at Lafayette, 2014
May 2019

ii
Acknowledgements
First of all, I would like to thank my advisor Dr. James J. Spivey for allowing me to
join his group. He has been guiding me kindly through my five years of studying and
research. He has always been supportive, and he provided great facilities and
opportunities for the students to build a strong academic & research background.
I also want to thank my committee members (Dr. Kerry Dooley, Dr. George
Stanley, Dr. Kunlun Ding, and Dr. Huiming Bao) for their support, guidance, and valuable
advice. I would like to thank Chevron for Chevron Innovation Research Fund (LSU
foundation fund), and National Science Foundation (NSF) for EAGER grant (# 1644895)
for the financial support for superacid work. I also thank LSU’s Office of Research and
Economic Development (ORED) for the funding of superacid as well as aromatization
work. I really appreciate the help and encouragement of my labmates (Nitin Kumar, Zi
Wang, Ashraf Abedin, and Srikar Bhattar) who helped me complete this research, and I
am glad they all have become good friends of mine. I would like to thank Dr. Amitava Roy
from Center for Advanced Microstructures & Devices, LSU for setting up the XAS
experiments. I am thankful to Dr. Clayton Loehn and Dr. Dongmei Cao from LSU SIF
(Shared Instrumentation Facility) for help with various characterizations. I thank Ms.
Wanda Leblanc from LSU Geology and Mr. Tommy Blanchard from LSU Oceanography
for ICP-OES sample preparation and measurement. I also would like to thank the
collaborators of my research, Dr. James Carter and Dr. Graham Hutchings from Cardiff
University, UK for their valuable advice and support.
I also want to thank my fellow graduate students in this department, including
Pragathi Darapaneni, Xun Cheng, Daniel Guedes de Oliveira, Vidyadhar Manee, and

iii
Abhijit Rao for helping me out in studying and in life. I would also like to thank my sister
Gouravee Athavale and my brother-in-law Amit Athavale for their constant support and
for keeping me motivated throughout my PhD. I would also like to thank my wife (Ketaki)
and my extended family for their support and encouragement through the final stages of
my PhD. Last but not least, I am really thankful to my parents (Mrs. Dhanashree and Dr.
Ravindra Kanitkar) for their love, support and encouragement throughout my 30 years of
life.

iv
Contents
Acknowledgements ..........................................................................................................ii
Abstract ...........................................................................................................................vi
Chapter 1 . Introduction ................................................................................................... 1 1.1 Research objective ................................................................................................ 1
1.2 Justification of the research ................................................................................... 1
1.3 Rationale for studying solid acids ........................................................................... 4
1.4 Rationale for studying metals (Mo) supported on solid acids ................................. 5
1.5 Outline of the dissertation ...................................................................................... 5
1.6 References ............................................................................................................. 6
Chapter 2 . Literature Review .......................................................................................... 9 2.1 Conversion using a gas-phase superacid ............................................................ 11
2.2 Methane dehydroaromatization ............................................................................ 16
2.3 References ........................................................................................................... 26
Chapter 3 . Light Alkane Aromatization: Efficient Use of Natural Gas ........................... 39 3.1 Introduction .......................................................................................................... 39
3.2 Aromatization of light alkanes .............................................................................. 43
3.3 Future perspective ............................................................................................... 67
3.4 References ........................................................................................................... 72
Chapter 4 . Low Temperature Direct Conversion of Methane using a Solid Superacid . 80 4.1 Introduction .......................................................................................................... 80
4.2 Experimental section ............................................................................................ 82
4.3 Results and discussion ........................................................................................ 87
4.4 Conclusions ......................................................................................................... 95
4.5 References ........................................................................................................... 96
Chapter 5 . Methane Dehydroaromatization over Molybdenum supported on Sulfated Zirconia Catalysts ........................................................................................................ 100
5.1 Introduction ........................................................................................................ 100
5.2 Experimental ...................................................................................................... 103
5.3 Results and discussion ...................................................................................... 109
5.4 Conclusions ....................................................................................................... 135
5.5 References ......................................................................................................... 136
Chapter 6 . Future Work .............................................................................................. 143 6.1 Continuation of superacid work .......................................................................... 143
6.2 Continuation of Mo/SZ work ............................................................................... 143
Appendix A. Permission to Use Copyrighted Materials ............................................... 144

v
Appendix B. Proof of Principal Authorship ................................................................... 155
Appendix C. Supplemental information for chapter 4 .................................................. 158
Appendix D. Supplemental information for chapter 5 .................................................. 172
Vita .............................................................................................................................. 187

vi
Abstract
Direct, non-oxidative conversion of natural gas to value-added chemicals has been
identified as one of the grand challenges of the 21st century. Circumventing indirect and
costly reforming steps is highly sought after and solid acids can play an important role in
that.
The direct conversion of methane to higher hydrocarbons and hydrogen can be
catalyzed using “superacids” at < 450 °C. Reported superacid catalysts in solid, liquid,
and gas phase included sulfated zirconia (SZ), HF-SbF5, FSO3H-SbF5, and HBr-AlBr3.
Liquid and gas phase superacids presented difficulties in separation while the solid ones
provided low yields. Here, we report a new class of Br-based solid superacids, AlBrx/H-
ZSM-5 (“ABZ-5”, x = 1 or 2). ABZ-5 is based on gas-phase HBr/AlBr3. This solid catalyst
is synthesized using a vapor-phase process in which AlBr3 vapor is grafted on to solid H-
ZSM-5. This catalyst is characterized using NH3-TPD, XRD, and DRIFTS. The results
show that ABZ-5 is significantly more active than SZ and showed methane conversions
of ~1% at 300 °C using ABZ-5. Hydrocarbon products observed in the temperature range
of 200-400 °C include both C2-C6 hydrocarbons and aromatics.
In another approach to methane activation, Mo is doped on solid SZ to create a
catalyst similar to Mo/H-ZSM-5, but with a different solid acid for methane
dehydroaromatization (DHA). These catalysts were characterized using Raman, XPS,
DRIFTS, SEM-EDS, HRTEM, XRD, XANES and other temperature programmed
techniques. Raman spectra confirmed the formation of Mo = O and O-Mo-O bonds on the
surface of SZ support. DRIFTS confirmed that there was little difference in acid sites when
Mo was doped on SZ, except at higher Mo loadings. XPS, XANES, and HRTEM analyses

vii
showed that MoO3 is converted to MoOxCy and is further converted to Mo2C as the DHA
reaction progresses. Further, these catalysts were evaluated for methane DHA reaction.
All of these catalysts showed methane conversions of 5-20 % at temperatures of 600-
700 °C. In each case, the catalysts deactivated steadily, attributable to strong coking on
the surface, as confirmed with TPO. A comparison with literature showed that Mo/SZ has
comparable activity to Mo/H-ZSM-5 at around 650-675 °C temperature range.

1
Chapter 1 . Introduction
1.1 Research objective
The purpose of this research is to synthesize, characterize, and evaluate the catalysts
for converting methane (CH4) into value-added chemicals including ethylene, benzene,
hydrogen, and other hydrocarbons. The objective of the research includes:
Synthesize a solid acid catalyst based on AlBr3 supported on inorganic support
in order to oligomerize methane into value-added chemicals (C2+).
Synthesize a bifunctional solid acid catalyst based on Mo and sulfated zirconia
(SZ) in order to dehydro-aromatize methane into ethylene, benzene, and H2.
Utilize various characterization methods to characterize these both type of
catalysts under ex-situ conditions.
1.2 Justification of the research
The shale gas revolution has presented the US with a unique opportunity. These
immense, previously unattainable reserves of natural gas have already had major impacts
on the energy and chemical industries and have helped the US reduce CO2 emissions [1-
4]. The resources of shale gas have allowed the price of natural gas to remain low in the
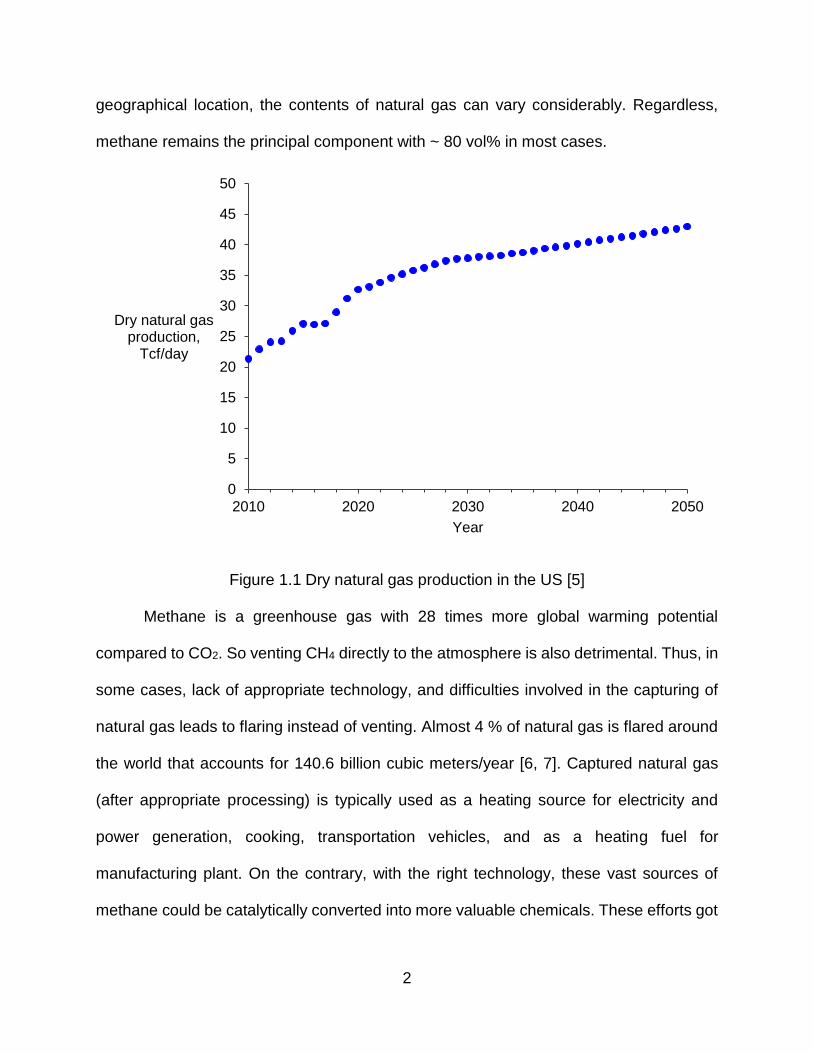
US and it is projected to remain low (below $ 5.1 per million BTU through 2023 [2]). Figure
1.1 shows the dry natural gas production over the last decade and its projections until
2050 as forecasted by US Energy Information Administration (EIA). It is projected to grow
significantly at a rate of 8 – 10 % every year for the next couple of years.
Natural gas is comprised of light alkanes such as methane, ethane, propane,
butane, isobutane as well as other gases such as CO2, He, N2. Depending on the
2
geographical location, the contents of natural gas can vary considerably. Regardless,
methane remains the principal component with ~ 80 vol% in most cases.
Figure 1.1 Dry natural gas production in the US [5]
Methane is a greenhouse gas with 28 times more global warming potential
compared to CO2. So venting CH4 directly to the atmosphere is also detrimental. Thus, in
some cases, lack of appropriate technology, and difficulties involved in the capturing of
natural gas leads to flaring instead of venting. Almost 4 % of natural gas is flared around
the world that accounts for 140.6 billion cubic meters/year [6, 7]. Captured natural gas
(after appropriate processing) is typically used as a heating source for electricity and
power generation, cooking, transportation vehicles, and as a heating fuel for
manufacturing plant. On the contrary, with the right technology, these vast sources of
methane could be catalytically converted into more valuable chemicals. These efforts got
0
5
10
15
20
25
30
35
40
45
50
2010 2020 2030 2040 2050
Dry natural gas production,
Tcf/day
Year

3
a recent boost by the shale gas revolution forcing the prices of natural gas to remain low
and thus, making it an attractive feedstock for the production of high value chemicals.
Value-added chemicals from natural gas include C2+ hydrocarbons and aromatics.
These include high-volume intermediates such as ethylene and propylene, which are
widely used in the production of polyethylene and polypropylene. Other products include
C4+ hydrocarbons in the gasoline range. Another class of high-value chemicals is
aromatics, including benzene, toluene, naphthalene, and related intermediates. These
aromatics are often used as precursors for the production of heavier aromatics. Benzene,
however, is the most valuable product because it serves as a feedstock for valuable
polymeric chains (styrenics, nylons, polycarbonates, phenol-formaldehyde resins, and
polyurethanes) [8, 9]. One specific example is the conversion of benzene to
ethylbenzene, which is then reacted to styrene. Benzene is also used as a feedstock for
the production of cumene, a starting point for the production of phenol and acetone. These
two are further converted to bisphenol-A and it leads to polycarbonate and epoxy resins.
Other uses of benzene include its use as a feedstock for the production of cyclohexane,
a precursor for Nylon-6, and Nylon-6,6 polymers. Conventionally all these value-added
chemicals have been produced through processing of crude oil, which has limited
reserves around the world. However, if the feedstock for these value-added chemicals
could be switched to natural gas, it will result in cleaner energy with a lower carbon
footprint. This has been highlighted in a recent report by NAP (National Academic Press)
[10], which says as a result of shale gas revolution, “the U.S. chemical industry is in the
process of switching from naphtha, derived from crude oil, as its major feedstock to
natural gas and natural gas liquids” and further the report identified ‘catalytic conversion

4
of natural gas to higher value chemicals’ as one of the significant challenges facing
researchers today.
1.3 Rationale for studying solid acids
Solid acids have been an integral part of modern day catalysis. This class of
materials have a range of applications and in particular, in the area of heterogeneous
catalysis. As the demand for green and sustainable processes has been growing, solid
acids play a more and more vital role in the development of new processes [11]. Their
primary purpose is the replacement of corrosive liquid and gas phase acid catalysts,
which are often difficult to separate and generate a lot of waste.
One class of solid acids involve what is known as solid superacids. Superacids are
materials that have very high acidity, stronger than 100% H2SO4 (Ho = -12). These
materials have the ability to protonate even methane, which has one of the strongest C-
H bonds. These catalysts in liquid form have been demonstrated previously, as shown by
George Olah in his Nobel Prize using FSO3H-SbF5 [12]. Recent work [13] at LSU has
shown that a gas-phase superacid using HBr-AlBr3 is active, but neither a liquid- nor a
gas-phase superacid is feasible in practice. This is because the separation of the
products from the catalyst is prohibitively complex. Having demonstrated that a gas-
phase HBr-AlBr3 superacid is active, the question is whether the same superacidity in the
gas-phase can be demonstrated as a solid. This is not straightforward, as has been
shown by a number of attempts to heterogenize homogeneous catalysts [14, 15]. To the
best of our knowledge, no study has shown oligomerization of methane using solid HBr-
AlBr3 based catalysts.

5
1.4 Rationale for studying metals (Mo) supported on solid acids
Sulfated Zirconia (SZ) is a well-known solid superacid containing both strong
Brønsted and Lewis acid sites. SZ is known to have stronger acidity than many
conventional zeolites such as HY, H-ZSM-5, H-MOR. Methane dehydroaromatization
(DHA) is a widely studied reaction using Mo supported on H-ZSM-5 or H-MCM-22,
containing both Mo sites as well as zeolitic Brønsted acid sites [16, 17]. Although widely
studied, incremental improvements and limited success have hampered the
commercialization of this process. In principle, Mo supported on SZ can function as a bi-
functional catalyst similar to Mo/H-ZSM-5 for the DHA reaction. This catalyst can also be
used in acid-catalyzed reactions such as dehydrogenation, hydroconversion, or
aromatization. Although Mo/SZ has been studied for heptane hydroconversion [18], we
are aware of no reports of the application of similar catalyst for methane DHA.
1.5 Outline of the dissertation
Chapter 2 gives a review of the literature dealing with the conversion of natural gas
into value added chemicals. Primarily, it discusses the approaches based on solid acid
catalysts that include superacids, and a metal supported on solid acids.
Chapter 3 is a book chapter published in Natural Gas Processing: from Midstream
to Downstream [19]. It reviews recent progress in light alkane activation, especially in
ethane aromatization area.
Chapters 4 and 5 are the peer-reviewed journal articles that the author has
published. Chapter 4 is published in ChemCatChem journal [20] and it discusses the low
temperature direct conversion of methane using a solid superacid. Solid acid catalysts
based on AlBr3 supported on inorganic supports are synthesized and further

6
characterized using NH3-TPD, DRIFTS, XRD, and other techniques. Finally, these
catalysts are evaluated for CH4 oligomerization. Chapter 5 is published in Applied
Catalysis A: General [21]. The paper is based on methane dehydroaromatization using
Mo supported on sulfated zirconia catalyst. These catalysts were characterized using
various techniques including Raman, DRIFTS, NH3-TPD, XANES, HRTEM, and others.
Further, these catalysts were tested for CH4 dehydroaromatization. Effect of physical
properties such as Mo content, temperature, and space velocity was also studied.
Chapter 6 deals with possible future work that could be carried out as a
continuation of this thesis.
1.6 References
[1] S. Jenner, A.J. Lamadrid, Shale gas vs. coal: Policy implications from environmental impact comparisons of shale gas, conventional gas, and coal on air, water, and land in the United States, Energy Policy, 53 (2013) 442-453. [2] Q. Wang, X. Chen, A.N. Jha, H. Rogers, Natural gas from shale formation – The evolution, evidences and challenges of shale gas revolution in United States, Renewable and Sustainable Energy Reviews, 30 (2014) 1-28. [3] T. Boersma, C. Johnson, The Shale Gas Revolution: U.S. and EU Policy and Research Agendas, Review of Policy Research, 29 (2012) 570-576. [4] K. Aruga, The U.S. shale gas revolution and its effect on international gas markets, Journal of Unconventional Oil and Gas Resources, 14 (2016) 1-5. [5] U. EIA, Annual Energy Outlook 2018, in: U.D.o. Energy (Ed.), US DOE, 2018. [6] Global Gas Flaring Reduction Partnership (GGFR), in, World Bank, 2018. [7] Natural gas production. Available from: https://yearbook.enerdata.net/natural-gas/world-natural-gas-production-statistics.html [8] Benzene, in: Chemical Economics Handbook, IHS Markit, 2018. Available from: https://ihsmarkit.com/products/benzene-chemical-economics-handbook.html [9] J.S. Plotkin, Benzene’s Unusual Supply–Demand Dilemma, in: INDUSTRIAL CHEMISTRY AND ENGINEERING, American Chemical Society, 2015. Available from:

7
https://www.acs.org/content/acs/en/pressroom/cutting-edge-chemistry/benzenes-unusual-supply-demand-dilemma.html. [10] National Academies of Sciences, Engineering, Medicine. The Changing Landscape of Hydrocarbon Feedstocks for Chemical Production: Implications for Catalysis: Proceedings of a Workshop, The National Academies Press, Washington, DC, 2016. [11] P. Gupta, S. Paul, Solid acids: Green alternatives for acid catalysis, Catalysis Today, 236, Part B (2014) 153-170. [12] G.A. Olah, R.H. Schlosberg, Chemistry in super acids. I. Hydrogen exchange and polycondensation of methane and alkanes in FSO3H-SbF5 ("magic acid") solution. Protonation of alkanes and the intermediacy of CH5+ and related hydrocarbon ions. The high chemical reactivity of "paraffins" in ionic solution reactions, Journal of the American Chemical Society, 90 (1968) 2726-2727. [13] S. Vasireddy, S. Ganguly, J. Sauer, W. Cook, J.J. Spivey, Direct conversion of methane to higher hydrocarbons using AlBr3-HBr superacid catalyst, Chemical Communications, 47 (2011) 785-787. [14] A.E.C. Collis, I.T. Horváth, Heterogenization of homogeneous catalytic systems, Catalysis Science & Technology, 1 (2011) 912-919. [15] D.T. Genna, A.G. Wong-Foy, A.J. Matzger, M.S. Sanford, Heterogenization of Homogeneous Catalysts in Metal–Organic Frameworks via Cation Exchange, Journal of the American Chemical Society, 135 (2013) 10586-10589. [16] J.J. Spivey, G. Hutchings, Catalytic aromatization of methane, Chemical Society Reviews, 43 (2014) 792-803. [17] Z.R. Ismagilov, E.V. Matus, L.T. Tsikoza, Direct conversion of methane on Mo/ZSM-5 catalysts to produce benzene and hydrogen: achievements and perspectives, Energy & Environmental Science, 1 (2008) 526-541. [18] F.F. Oloye, A.J. McCue, J.A. Anderson, n-Heptane hydroconversion over sulfated-zirconia-supported molybdenum carbide catalysts, Applied Petrochemical Research, 6 (2016) 341-352. [19] S.R. Kanitkar, J.J. Spivey, Light alkane aromatization : efficient use of natural gas, in: N. Elbashir, M. El-Halwagi; I. Economou; K. Hall (Ed.) Natural Gas Processing from Midstream to Downstream, John Wiley and Sons, Hoboken, NJ, 2019. [20] S. Kanitkar, J.H. Carter, G.J. Hutchings, K. Ding, J.J. Spivey, Low Temperature Direct Conversion of Methane using a Solid Superacid, ChemCatChem, 10 (2018) 5019-5024.

8
[21] S. Kanitkar, M.A. Abedin, S. Bhattar, J.J. Spivey, Methane Dehydroaromatization over Molybdenum supported on Sulfated Zirconia Catalysts, Applied Catalysis A: General, 575 (2019) 25-37.

9
Chapter 2 . Literature Review
CH4 is a thermodynamically stable, non-polar molecule with one of the strongest
C-H bonds (First C-H bond breaking strength: 434 kJ/mol) [1]. This makes it very difficult
to activate methane often requiring strong oxidative and corrosive environments, and high
temperatures [2]. CH4 conversion to syngas using reforming reactions and subsequent
conversion of syngas to higher hydrocarbons, alcohols, oxygenates through Fisher-
Tropsch synthesis is a widely practiced approach. These processes often involve multiple
steps and are very energy intensive. The first step is reforming, which accounts for 70 %
of the cost of the overall process [3, 4]. This was one of the reasons why Shell recently
cancelled a GTL plant in Louisiana [5]. Other approaches include OCM (Oxidative
Coupling of Methane) [6], Reforming (Dry/Bi/Oxy/Steam) [7-10], Partial Oxidation (PO)
[11], Halogenation [12] or even Sulfidation [2]. A summary of most of these oxidative
routes for methane conversion could be found in Figure 2.1.
On the other hand, a direct, non-oxidative conversion of methane would
circumvent the costly reforming steps and would produce value added products like
hydrocarbons directly in one step, reducing the overall cost. Both heterogeneous and
homogeneous conversion has been reported. Heterogeneous catalysts involved the use
of solid catalysts like zeolites (H-ZSM-5) [13], lattice confined single Fe sites embedded
in a SiO2 matrix [14], metal sulfides [2], sulfated zirconia [15], etc. Another heterogeneous
type was explored by Olah in 1969 using a liquid phase superacid, FSO3H-SbF5 or
commonly known as Magic Acid [16]. One homogeneous attempt involved the use of gas
phase superacid HBr-AlBr3 [17] that will be discussed in detail in section 2.1 Conversion
using a gas-phase superacid below. Other attempts involve the use of microwaves [18],

10
plasma [19, 20], UV induced [21] etc. A summary of some of the direct catalytic
conversion attempts is shown in Table 2.1.
Figure 2.1 Summary of oxidative conversion routes for CH4
These direct conversion attempts can primarily be divided into two types: moderate
temperature ones, and high temperature ones. Moderate temperature conversion of
methane involves the use of superacids in all phases: gas, liquid, and solid. These were
used around 150-450 °C. High temperature conversion (> 500 °C) of methane involves
dehydroaromatization, and gas phase reactions that won't occur on the catalyst surface
[14].
Despite the use of higher temperatures, various oxidants, or use of superacids,
product yield values reported have been very low and often hampered by strong coking
at least on solid catalysts. Homogeneous superacids showed high conversion levels but

11
the product distribution was difficult to control, and the products obtained were hard to
separate due to the homogeneous nature of the catalyst. This requires a solid catalyst.
Table 2.1 Summary of some of the direct conversion attempts from literature
Catalyst Type Catalyst
Phase
Methane
Conversion (%)
Temperature
(K)
Pressure
(bar) Reference
Fe©-SiO2 Solid 48.0 % 1370 K 1 bar [14]
PdS/ZrO2 Solid 16.0 % 1325 K 1 bar [2]
SO42-/ZrO2 Solid 4.6 % 673 K 1 bar [15]
Zeolite Solid 20 % 983 K 1 bar [13]
FSO3H-SbF5 Liquid 100 % 423 K 1.5 bar [16]
HBr-AlBr3 Gas 99.9 % 673 K 1 bar [17]
2.1 Conversion using a gas-phase superacid
AlBr3 is a stronger Lewis acid than AlCl3 based on Fluoride Ion Affinity [22]. Also
when it is mixed with HBr, forms a binary L/B superacid which has been shown to be one
of the strongest superacids in the gas phase [23]. Recently, this superacid (in the
presence of benzene) identified the first structural determination of a well-ordered
benzenium cation ([C6H7]+) formed by the protonation of benzene at about 0 °C [24],
highlighting the importance of this superacid. Being such a strong superacid, it was a
probable candidate for the study of methane oligomerization. Vasireddy et al. [17] carried
out direct catalytic oligomerization of methane over gas phase superacid [H+ - AlBr4-].
This reaction was carried out in a single step and at atmospheric pressure to yield C2+
hydrocarbons and hydrogen. Gas-phase products up to C8 were observed whereas,
liquid-phase products ranging from C6 to C26 were observed upon analysis of the red oil,
an accumulation of higher hydrocarbons and superacid obtained from a number of runs.

12
This sample showed that higher hydrocarbons were produced, but could not be
quantified. The highest level of conversion of 99.9 % was observed at 400 °C and the
mechanism proposed was similar to the protolytic oligo-condensation as suggested by
Olah [16].
Despite high levels of conversion of methane, hydrocarbon % C yields were very
low (e.g. for gas phase C2-C8 hydrocarbons, the yields were in the range of 0.01-0.15 %).
Additionally, a large portion of the converted carbon could not be accounted for. Liquid
phase products in the red oil were hard to detect and quantify and also were hard to
separate from the dissolved HBr-AlBr3 superacid. Other than that, some of the liquid
phase C5+ hydrocarbons were partially retained in the scrubber [17]. Overall, a full system
carbon balance was not possible because of the difficulty in separating the gas-phase
superacid from the hydrocarbons.
These problems could be solved if a solid superacid with an acidic strength high
enough to protonate methane, can be synthesized and employed that would make the
process heterogeneous. A solid catalyst is necessary to develop a viable process for the
conversion of methane using this type of catalysis.
2.1.1 Solid superacids
Solid superacids are a class of well-known materials that have been studied over
the past 3-4 decades. One of the first definitions include solids that have acid strength
higher than that of 100 % sulfuric acid i.e. (H0 < -11.9) [25]. One of the few solid
superacids reported include zeolites (ZSM-5) [26], Nafion [27], SbF5 and AlCl3 supported
on oxide supports like (SiO2 [28-30], Al2O3 [31], SiO2-Al2O3 [32], TiO2 [25, 33] , ZrO2 [34]).
In the 1980s, Hino and Arata [35] developed another strong solid superacid namely

13
Sulfated Zirconia (SZ) that had (Ho ~ -16) as determined by the titration with Hammett
indicators. Later on Olah and coworkers [23] classified solid superacids as zeolitic acids,
polymeric resin sulfonic acids, enhanced acidity solids, and immobilized superacids. Out
of these catalysts, SZ catalyst went on to become one of the most studied solid acid
catalyst due to its high acidity, relatively high thermal stability, and ease of preparation.
Attempts have been reported in the literature for direct conversion of methane over
solid superacids, in particular using SZ catalysts. Rezgui et al. [36], Hua et al. [37], Martin
and Schmal [38] observed primarily C2 hydrocarbons (mostly ethane). Fraenkel [15] on
the other hand observed primarily ethylene, iso-butane, and small amounts of propane.
Hydrogen was observed indicating oligocondensation mechanism (shown in Figure 2.2
below) similar to the one proposed by Olah for liquid superacid [16] and others did not
observe any due to scavenging of oxygen by the dihydrogen [15] or due to incorporation
into the carbonaceous deposits [36].
Figure 2.2 Oligo-condensation mechanism for methane proposed by Olah [16]
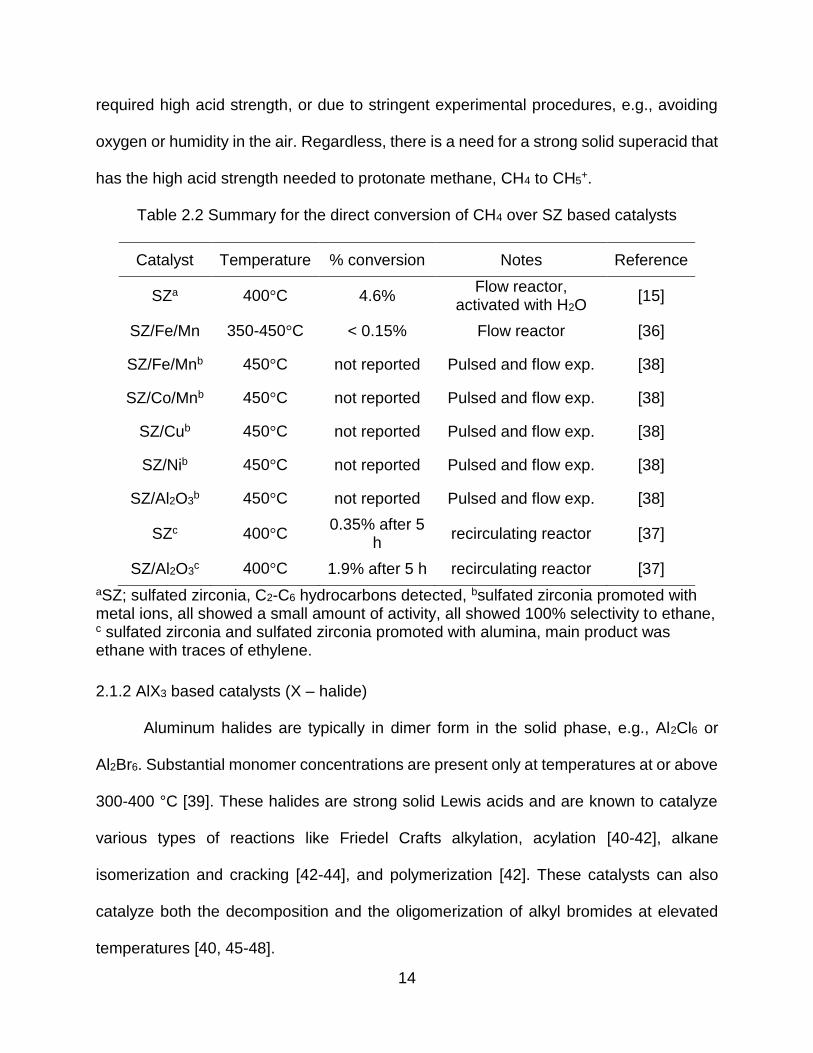
A summary of the performance of SZ based catalysts for methane conversion is
presented in Table 2.2 below. Despite the advances in solid superacid research, results
on methane oligomerization are very limited (Table 2.2), typically either due to lack of the
14
required high acid strength, or due to stringent experimental procedures, e.g., avoiding
oxygen or humidity in the air. Regardless, there is a need for a strong solid superacid that
has the high acid strength needed to protonate methane, CH4 to CH5+.
Table 2.2 Summary for the direct conversion of CH4 over SZ based catalysts
Catalyst Temperature % conversion Notes Reference
SZa 400°C 4.6% Flow reactor,
activated with H2O [15]
SZ/Fe/Mn 350-450°C < 0.15% Flow reactor [36]
SZ/Fe/Mnb 450°C not reported Pulsed and flow exp. [38]
SZ/Co/Mnb 450°C not reported Pulsed and flow exp. [38]
SZ/Cub 450°C not reported Pulsed and flow exp. [38]
SZ/Nib 450°C not reported Pulsed and flow exp. [38]
SZ/Al2O3b 450°C not reported Pulsed and flow exp. [38]
SZc 400°C 0.35% after 5
h recirculating reactor [37]
SZ/Al2O3c 400°C 1.9% after 5 h recirculating reactor [37]
aSZ; sulfated zirconia, C2-C6 hydrocarbons detected, bsulfated zirconia promoted with metal ions, all showed a small amount of activity, all showed 100% selectivity to ethane, c sulfated zirconia and sulfated zirconia promoted with alumina, main product was ethane with traces of ethylene.
2.1.2 AlX3 based catalysts (X – halide)
Aluminum halides are typically in dimer form in the solid phase, e.g., Al2Cl6 or
Al2Br6. Substantial monomer concentrations are present only at temperatures at or above
300-400 °C [39]. These halides are strong solid Lewis acids and are known to catalyze
various types of reactions like Friedel Crafts alkylation, acylation [40-42], alkane
isomerization and cracking [42-44], and polymerization [42]. These catalysts can also
catalyze both the decomposition and the oligomerization of alkyl bromides at elevated
temperatures [40, 45-48].

15
Aluminum chloride, Al2Cl6 is a widely used acid catalyst in industry. However, the
generation of inorganic and acidic waste through leaching remains a problem, making
separation of products difficult. This has led several researchers to investigate the
possibility of supporting Al2Cl6 on various types of solid supports like SiO2 [30, 49-57],
Al2O3 [58], mesoporous silica like MCM-41 [59, 60], or polystyrene [61-63]. These
supported aluminum chloride catalysts showed high activity in various acid-catalyzed
processes including alkylation [64], polymerization [55, 58], isomerization [50], and
Mannich synthesis [54].
These supported Al2Cl6 catalysts were characterized by using various techniques
by various groups. It included a range of different analytical techniques like Fourier
Transform Infra-Red (FTIR) spectroscopy with pyridine as a probe molecule [28, 50, 54,
55, 60], NH3-Temperature Programmed Desorption (TPD) [60], solid-state Magic Angle
Spinning Nuclear Magnetic Resonance (MAS NMR) spectroscopy (27Al [28, 55, 65], 1H
[54], 29Si [65]), titration with various Hammett indicators (m-nitrotoluene, p-nitrotoluene
etc.) [57], isomerization of n-butane to isobutane as a test reaction [29]. Based on these
results silica supported Al2Cl6 was claimed to be a superacid by Drago et al. [28].
However, these catalysts have not been investigated for methane oligomerization,
despite that they are clear candidates for methane oligomerization. Both AlCl3 and AlBr3
can catalyze the decomposition and oligomerization of the alkyl bromides at elevated
temperatures [40, 45-47] and form superelectrophiles in the presence of
polyhalomethanes [66, 67].
In summary, AlCl3 or AlBr3 can be supported onto solid supports to create solid
acid catalysts. These acid catalysts can further be treated with HBr to create an analogue

16
of the gas phase superacid [H+/AlBr4-]. This catalyst can then be tested for converting
methane into higher hydrocarbons and possibly H2. To the best of our knowledge, there
is no study focusing on the development of HBr treated supported AlBr3 catalysts to create
solid acid catalysts for the oligomerization of methane.
2.2 Methane dehydroaromatization
Dehydroaromatization (DHA) of methane is also currently one of the most widely
researched routes. This direct (non-oxidative) route converts methane into benzene and
hydrogen at relatively high temperatures (~ 600-700 °C).
6 CH4 C6H6 + 9 H2 (ΔG298 K = +433 kJ/mol of Benzene,
ΔH298K = +530 kJ/mol of Benzene) (2.1)
Aromatization of methane was first reported over silica catalyst in 1966 in a
Science paper [68]. However, it was not until the early ‘90s when this research picked up
pace after the first report of aromatization of methane over metal supported on zeolite
catalysts by Wang et al. in 1993 [69]. After this, a series of literature ranging from different
metals to zeolites, with different promoters got published. In addition, an emphasis was
given on the pretreatment and the reaction conditions that have been well studied. This
research received recent impetus from the shale gas revolution that made immense
resources of natural gas accessible. Till date, a vast amount of literature has been
presented and it has been well summarized in recent reviews [70-73].
It is generally accepted that this reaction requires a bifunctional catalyst [70-73]
although, there is one recent report [74] that has opened a new debate of monofunctional
vs. bifunctional mechanism. In the typical bifunctional mechanism, the activation of
methane occurs on a metal site and it undergoes dimerization into C2 species. These C2
species subsequently undergo cyclization and aromatization over Brønsted acid sites

17
(BAS) provided by the support. Thus the metal site and an acid site create a bifunctionality
and this mechanism can be shown as below in equation 2.2:
CH4Mo site→ C2Hy
BAS→ C6H6 + Other Aromatics + H2 ↑ (2.2)
It is believed that there exists an analogy between methane aromatization (DHA)
and methanol to hydrocarbons (MTH) reaction over the same H-ZSM-5 zeolite. MTH
reaction is thought to proceed through hydrocarbon pool mechanism [75] in which,
carbonaceous deposits inside the zeolite channels act as an active site over which the
methanol is converted to ethylene [71]. Because of the nature of this type of reaction,
various parameters that can be physical and/or chemical are of high importance. Physical
parameters can include temperature, pressure, space velocity, pore size, and pore
structure of the support. Chemical parameters can include metal content, acidity of the
support, and nature of the active site (that is often debated). There are several other
smaller factors that can also play a key role. For example, the proximity between the
metal site and the acid site as has been highlighted by many researchers [73, 74, 76].
Apart from these two functionalities, another aspect that is of utmost importance is the
shape selectivity of the support [74].
Mo has been found to be the most active out of the various metals studied for this
reaction that includes Mo [69-71, 77-80], Re [81, 82], Zn [83-88], Ag [89, 90], In [91] etc.
Among various studied supports, H-ZSM-5 and H-MCM-22 have been the most active,
and shape selective catalysts [70, 71].
2.2.1 Metal site
The active metal site is generated in-situ or sometimes can be generated ex-situ
and then the reaction is carried out on these catalysts. Various spectroscopic techniques

18
have been used to investigate this that include XAS [92-94], FTIR [77, 95, 96], XPS [78,
79, 97, 98], Raman [99-101], XRD [80], etc. Because of the complex transformations for
the active metal, in-situ characterizations and spectroscopic investigations carry great
importance. Although, relatively severe reaction conditions (high temperatures and
reducing environments) present significant challenges. Some of these challenges have
been well documented in a recent review [73].
There are various hypotheses about the exact nature of the metal site which,
haven’t been investigated much for metals other than Mo [73]. The consensus suggests
Mo to be in the oxidized form (MoO3) for fresh calcined catalyst [77, 102, 103]. This oxide
needs to be reduced to a lower oxidation state (between +6 and +4) in order to activate
C-H bond [73, 79, 93] mostly in the carbidic form. Further, it has been found that the
carbidic form of Mo behaves similarly to the noble metals [104-107] that is due to the
changes in the electronic properties of Mo due to C [73]. When this fresh oxide catalyst
is kept under reducing conditions such as CH4, or CH4 + H2, it undergoes reduction and
simultaneous carburization which is said to proceed through following reactions [108]:
4 MoO3 + CH4 = 4 MoO2 + CO2 + 2 H2O (2.3)
4 MoO2 + 4 CH4 = 2 Mo2C + CO2 + 5 H2O + CO + 3 H2 (2.4)
Many recent reports [76, 94] suggest this transformation might occur through some
intermediate steps such as MoO3 MoOxCy Mo2C. In this system, either of Mo
phases: MoOxCy (oxy-carbidic) [109] or Mo2C (carbidic) [110] are said to be active.
Although, a very recent study observed only fully carbided species (Mo1.6C3) to be active
as compared to MoOxCy based on operando EXAFS [94]. Among molybdenum carbides,
four different phases have been reported in the literature [107]: α, β, η, and γ. For DHA

19
reaction, only α-MoCx-1 [111], and β-Mo2C [80, 110, 112] have been found to be active
and are the only ones reported.
It also has been observed in the literature that the amount of metal decides the
location (outside/inside pore) and the state of the metal (oxidic/carbidic). Some reports
have reported with the use of XRD [113], HRTEM [80], XAS [77], FTIR [114] that below
5-6 wt% Mo content, Mo is in a finely dispersed state. These studies further showed that
these lower Mo concentrations showed the highest activity per metal atom. If the metal
content goes above 5-6 wt%, it forms aggregates of metal oxides. Larger the clump,
higher is the reduction temperature required for reducing MoO3 [70]. This formation of
aggregate metal oxides has been attributed to metal/Al ratio, which for higher metal
content is > 1. This leads to insufficient sites for anchoring metal as Al is believed to be
the anchoring site apart from providing acidity to the zeolite. In this, metal is linked to the
framework Al through oxygen while replacing proton of the BAS [73].
Ismagilov et al. [70] summarized some of the factors that affect the location of
molybdenum. These were: concentration [102, 114], temperature [78, 114], calcination
time as well as composition of gas phase during calcination [78, 115]. These studies have
indicated that higher Mo concentrations lead to Mo oxides on the external zeolite surface.
2.2.2 Acid site
Brønsted acid sites (BAS) from the support carry out the cyclization and
oligomerization of olefinic intermediates such as ethylene into benzene, and other higher
aromatic compounds. For the most widely studied Mo/H-ZSM-5 catalyst, base zeolite
provides BAS originating through charge compensation due to Al substitution. These sites
can also act as anchors for supporting Mo [70, 73]. Vollmer et al. [73] have summarized

20
the functions of BAS that include: dispersion of metal in the pores of the support, and the
Brønsted acidity of the support. A recent study [74] also suggested that BAS in H-ZSM-5
can dictate the distribution of the Mo oxide phase between the external zeolite surface
and the micropores in the zeolite.
The concentration of acid sites and the distribution and location of those sites in
the zeolite framework are crucial to determine the final performance of the catalyst. For
ex. if the acid sites are on the external surface of the zeolite, they contribute significantly
to the coking and the type of coke that forms is often characterized as refractory [116,
117]. On the other hand, acid sites inside the zeolite channels mainly contribute to the
formation of benzene and other aromatics and also to the stabilization of Mo oxide species
[74].
2.2.3 Other supports
Martinez et al. [118] studied Mo supported on ITQ-2 material. This material is
derived from MCM-22. It possesses 12 MR (membered ring) supercages and 10 MR
sinusoidal channel system of 0.40 nm X 0.59 nm both similar to MCM-22. Additionally, it
consists of thin sheets of 2.5 nm in height presenting a hexagonal array of 0.7 nm X 0.7
nm ‘cups’. This material presents a higher surface area than MCM-22 [119]. This material
when treated with acid, showed higher benzene selectivity as compared with MCM-22.
Structure of MCM-22 could be represented as shown in Figure 2.3 and structure of ITQ-
2 can be represented as shown in Figure 2.4.

21
Figure 2.3 Structure of MCM-22 [120]
Figure 2.4 Possible structure of ITQ-2. Reproduced from [119] with permission from Elsevier

22
Xu et al. [121] studied DHA over Mo supported on ITQ-13 zeolite having high silica
content. This catalyst showed better selectivity towards benzene as compared to H-ZSM-
5 supported catalyst, but lower activity and stability as compared to H-ZSM-5 supported
catalyst. ITQ-13 structure consists 9 MR with 0.4 nm X 0.49 nm, straight 10 MR with 0.47
nm X 0.51 nm, and sinusoidal 10 MR with 0.48 nm X 0.57 nm dimensions. Structure of
ITQ-13 could be as shown in Figure 2.5. 10 MR could be seen when viewed along [010]
plane and 9 MR could be seen when viewed along [100].
Figure 2.5 Structure of ITQ-13 zeolite viewed along two different planes [120]
Liu and coworkers [122, 123] synthesized and studied Mo supported on IM-5
catalysts for DHA. These catalysts achieved better benzene yields and better stability
than that of Mo/H-ZSM-5 at same reaction conditions. Performance of these catalytic

23
materials (Mo/IM-5) was recently improved further in terms of benzene yields and stability
by Liu and Kan [124] using mesoporous SBA-15, and MCM-48 as the silica sources. This
improvement was attributed to the generation of mesoporous systems within the zeolite
crystals. It was further speculated that this led to easier access to the active sites and
favored diffusion of products.
In another approach, Heng and Kan along with co-workers reported TNU-9 as
support for Mo in DHA [125]. This catalyst also showed improved benzene yields and
stability as compared to Mo/H-ZSM-5. Secondary type of mesoporosity was speculated
to be the reason for this performance.
Kan et al. [126] also studied MCM-49, which is a three dimensional analogue of
MCM-22. This catalyst also showed high benzene yield and longer catalyst life.
Performance of these materials was attributed to the enhanced diffusion of aromatic
products as an outcome of the shorter nano-sized channels of MCM-41.
Other supports that have been studied most commonly include acidic supports
such as zeolites H-ZSM-8 [127], H-ZSM-11 [127], H-SSZ-13 [74], H-Mordenite [74] or
some inorganic ones like SiO2 [74], Al2O3 [74]. Zhang et al. [127] did a comprehensive
study on a variety of supports to support Mo for methane DHA reaction. The study
included H-ZSM-8, H-ZSM-11, H-SAPO-5, H-SAPO-11, H-SAPO-34, H-X, H-Y, H-MOR,
H-Beta, H-MCM-41. Activities of these catalysts was found to be in the following
decreasing order: Mo/H-ZSM-11 > Mo/H-ZSM-5 > Mo/H-ZSM-8 > Mo/H-Beta > Mo/H-
MCM-41 > Mo/H-SAPO-34 > Mo/H-MOR ≈ Mo/H-X ≈ Mo/H-Y > Mo/H-SAPO-5 ≈ Mo/H-
SAPO-11. This difference was primarily ascribed to the difference in the pore structure of
the various zeolites. Overall, zeolites with two-dimensional pore structure and with pore

24
diameters closer to that of benzene showed high activity in the aromatization reaction.
Although, authors [127] did point out that the difference could not exclusively be attributed
to the shape or pore structure and they further speculated acidity to be also a key factor.
2.2.4 Sulfated zirconia
Sulfated zirconia (SZ) as discussed previously in section 2.1.1 Solid superacids, is
a well-known and widely studied solid superacid. A large amount of literature is present
on these materials ever since it was first discovered in the 80s. This literature has been
well summarized in some books [128], and review articles [129-131]. Sulfated zirconia
has been tested in a variety of reactions involving butane isomerization, reduction of NO
with methane [132, 133], Friedel crafts alkylation/acylation, fuel cell electrodes [134-138],
etc. A more comprehensive summary of the type of reactions catalyzed by sulfated
zirconia could be obtained in the books and review articles aforementioned.
Different types of SZ have been reported depending on the final application such
as mesoporous/nano-sized. The preparation of SZ can occur through a variety of
procedures such as single step/two step, sol-gel route, co-precipitation route, or using
various precursors: ZrCl4/ZrOCl2/ZrO(NO3)2 etc. Properties of the end product (SZ) are
significantly influenced by various synthesis parameters that can be both physical as well
as chemical. Physical parameters involve drying temperatures, calcination temperatures.
Chemical parameters, on the other hand, involve type of precursor, precipitating agent,
pH, sulfating agent. All these parameters contribute to the final phase of the material
which can be amorphous, tetragonal, or monoclinic. Effects of these individual
parameters have been well documented in books and review articles mentioned
previously.

25
Acidity in SZ originates from sulfate anions, which when modifies zirconium oxide
forms strong acid sites. There have been many theories in the literature that discuss these
effects and thus, many structures have been proposed for sulfated zirconia. Some of them
can be summarized as shown in Figure 2.6 below.
Figure 2.6 Different structures of sulfated zirconia as proposed by (a) Hino and Arata [35, 139], (b) Ward and Ko [140], (c) Bolis et al. [141]
SZ is known to have stronger acidity as compared to some of the other zeolites
such as H-ZSM-5, H-Mordenite etc. This has been proved in the recent literature using
isobutane conversion reaction [142]. Additionally, the same authors calculated acidity to
be higher and stronger for SZ on the Hammett scale. The values are tabulated in Table
2.3:
Table 2.3 Comparison of acidity of SZ with other solid acids based on two references
Solid Acid Ho value [128] Ho value [142]
SZ -16.1 -18.0
H-ZSM-5 -13.2 -10.0
H-Mordenite -14.5 -14.0
Considering the bifunctional mechanism of DHA, a catalyst similar to Mo/H-ZSM-
5 can be created using Mo supported on SZ. Preparation of Mo-SZ has been studied
before albeit for a different reaction. This system has been studied for n-heptane
hydroconversion by Oloye et al. [143] In this work, authors used extensive

26
characterization techniques such as XRD, TGA, FTIR, Raman, TPR, SEM-EDX, CO
chemisorption, and N2 physisorption. Using this system, the same authors could obtain
Mo2C particles finely dispersed over tetragonal sulfated zirconia. Further, it was observed
that with the help of this catalyst RON (Research octane number) could be increased from
0 to 50. Based on this work from Oloye et al. [143] it could be seen that a stable Mo2C
supported on SZ system could be obtained.
To the best of our knowledge, this strong acidity in SZ has not been tested for
methane DHA. SZ being a strong solid superacid and with apparently higher acid strength
than conventional zeolites like H-ZSM-5 or H-Mordenite, it can catalyze the oligomeric
species in the bifunctional mechanism faster. So, it might be worth investigating Mo
supported on SZ (Mo/SZ) system for DHA. Thus, it is proposed to investigate a
bifunctional catalyst Mo/SZ for the first time for DHA of methane.
2.3 References
[1] S.J. Blanksby, G.B. Ellison, Bond Dissociation Energies of Organic Molecules, Accounts of Chemical Research, 36 (2003) 255-263. [2] Q. Zhu, S.L. Wegener, C. Xie, O. Uche, M. Neurock, T.J. Marks, Sulfur as a selective ‘soft’ oxidant for catalytic methane conversion probed by experiment and theory, Nat Chem, 5 (2013) 104-109. [3] B. Patel, LNG vs. GTL (F-T): An economic and technical comparison, in: Gastech Conference, 2005, pp. 28. [4] Y. Larry Song, B.F. Burke, The GTL Industry, Hydrocarbon Engineering, 11 (2006) 12-16. [5] B. Olson, Shell Halts $20 Billion Louisiana Gas-to-Liquids Project, in: S. Warren (Ed.), www.bloomberg.com. [6] G.E. Keller, M.M. Bhasin, Synthesis of ethylene via oxidative coupling of methane, Journal of Catalysis, 73 (1982) 9-19.

27
[7] N. Kumar, Z. Wang, S. Kanitkar, J.J. Spivey, Methane reforming over Ni-based pyrochlore catalyst: deactivation studies for different reactions, Applied Petrochemical Research, (2016) 1-7. [8] D. Pakhare, J. Spivey, A review of dry (CO2) reforming of methane over noble metal catalysts, Chemical Society Reviews, 43 (2014) 7813-7837. [9] G.A. Olah, A. Goeppert, M. Czaun, G.K.S. Prakash, Bi-reforming of Methane from Any Source with Steam and Carbon Dioxide Exclusively to Metgas (CO–2H2) for Methanol and Hydrocarbon Synthesis, Journal of the American Chemical Society, 135 (2013) 648-650. [10] V.R. Choudhary, K.C. Mondal, T.V. Choudhary, Oxy-methane reforming over high temperature stable NiCoMgCeOx and NiCoMgOx supported on zirconia–haffnia catalysts: Accelerated sulfur deactivation and regeneration, Catalysis Communications, 8 (2007) 561-564. [11] B. Christian Enger, R. Lødeng, A. Holmen, A review of catalytic partial oxidation of methane to synthesis gas with emphasis on reaction mechanisms over transition metal catalysts, Applied Catalysis A: General, 346 (2008) 1-27. [12] A. Zhang, S. Sun, Z.J.A. Komon, N. Osterwalder, S. Gadewar, P. Stoimenov, D.J. Auerbach, G.D. Stucky, E.W. McFarland, Improved light olefin yield from methyl bromide coupling over modified SAPO-34 molecular sieves, Physical Chemistry Chemical Physics, 13 (2011) 2550-2555. [13] Y. Wu, L. Emdadi, Z. Wang, W. Fan, D. Liu, Textural and catalytic properties of Mo loaded hierarchical meso-/microporous lamellar MFI and MWW zeolites for direct methane conversion, Applied Catalysis A: General, 470 (2014) 344-354. [14] X. Guo, G. Fang, G. Li, H. Ma, H. Fan, L. Yu, C. Ma, X. Wu, D. Deng, M. Wei, D. Tan, R. Si, S. Zhang, J. Li, L. Sun, Z. Tang, X. Pan, X. Bao, Direct, Nonoxidative Conversion of Methane to Ethylene, Aromatics, and Hydrogen, Science, 344 (2014) 616-619. [15] D. Fraenkel, Methane conversion over sulfated zirconia, Catalysis Letters, 58 (1999) 123-125. [16] G.A. Olah, G. Klopman, R.H. Schlosberg, Super acids. III. Protonation of alkanes and intermediacy of alkanonium ions, pentacoordinated carbon cations of CH5+ type. Hydrogen exchange, protolytic cleavage, hydrogen abstraction; polycondensation of methane, ethane, 2,2-dimethylpropane and 2,2,3,3-tetramethylbutane in FSO3H-SbF5, Journal of the American Chemical Society, 91 (1969) 3261-3268.

28
[17] S. Vasireddy, S. Ganguly, J. Sauer, W. Cook, J.J. Spivey, Direct conversion of methane to higher hydrocarbons using AlBr3-HBr superacid catalyst, Chemical Communications, 47 (2011) 785-787. [18] A. Domínguez, B. Fidalgo, Y. Fernández, J.J. Pis, J.A. Menéndez, Microwave-assisted catalytic decomposition of methane over activated carbon for -free hydrogen production, International Journal of Hydrogen Energy, 32 (2007) 4792-4799. [19] C. Xu, X. Tu, Plasma-assisted methane conversion in an atmospheric pressure dielectric barrier discharge reactor, Journal of Energy Chemistry, 22 (2013) 420-425. [20] B. Wang, W. Yan, W. Ge, X. Duan, Methane conversion into higher hydrocarbons with dielectric barrier discharge micro-plasma reactor, Journal of Energy Chemistry, 22 (2013) 876-882. [21] L. Li, S. Fan, X. Mu, Z. Mi, C.-J. Li, Photoinduced Conversion of Methane into Benzene over GaN Nanowires, Journal of the American Chemical Society, 136 (2014) 7793-7796. [22] I. Krossing, Fluoride Ion Affinity. Available from: https://portal.uni-freiburg.de/molchem/research/lewis-acids/fia. [23] G.A. Olah, G.K. Surya Prakash, Á. Molnár, J. Sommer, Superacid Systems, in: Superacid Chemistry, John Wiley & Sons, Inc., 2009, pp. 35-82. [24] F. Scholz, D. Himmel, L. Eisele, W. Unkrig, I. Krossing, The Superacid HBr/AlBr3: Protonation of Benzene and Ordered Crystal Structure of [C6H7]+[Al2Br7]−, Angewandte Chemie International Edition, 53 (2014) 1689-1692. [25] K. Tanabe, New solid acids and bases : their catalytic properties, Tokyo : Kodansha ; Amsterdam ; New York : Elsevier ; New York, N.Y., U.S.A. : [Distributors] for the U.S.A. and Canada, Elsevier Science Pub. Co., 1989., 1989. [26] L.E. Kitaev, E.E. Kolesnikova, E.N. Biryukova, N.V. Kolesnichenko, S.N. Khadzhiev, Formation of superacid centers in the structure of zeolite ZSM-5, Russian Journal of Physical Chemistry A, 87 (2013) 668-673. [27] E. Ponomareva, M.-A. López-Martínez, D. Wigger, M.V. Morales, I. Melián-Cabrera, Novel reactivation allows effective reuse of Nafion® super-acid nano-catalyst, Applied Catalysis A: General, 569 (2019) 134-140. [28] R.S. Drago, S.C. Petrosius, C.W. Chronister, Characterization and improvements in the synthesis of the novel solid superacid AlCl2(SG)n, Inorganic Chemistry, 33 (1994) 367-372.

29
[29] R.S. Drago, S.C. Petrosius, P.B. Kaufman, Alkylation, isomerization and cracking activity of a novel solid acid, AlCl2(SG)n, Journal of Molecular Catalysis, 89 (1994) 317-327. [30] T. Xu, N. Kob, R.S. Drago, J.B. Nicholas, J.F. Haw, A Solid Acid Catalyst at the Threshold of Superacid Strength: NMR, Calorimetry, and Density Functional Theory Studies of Silica-Supported Aluminum Chloride, Journal of the American Chemical Society, 119 (1997) 12231-12239. [31] A. Krzywicki, M. Marczewski, S. Malinowski, “Superacid” alumina catalysts. Al2O3−P2O5 and Al2O3−AlCl3 catalysts for the isomerization and cracking of n-hexane, Reaction Kinetics and Catalysis Letters, 8 (1978) 25-28. [32] H. Hattori, O. Takahashi, M. Takagi, K. Tanabe, Solid super acids: Preparation and their catalytic activities for reaction of alkanes, Journal of Catalysis, 68 (1981) 132-143. [33] J.R. Sohn, Recent advances in solid superacids, J. Ind. Eng. Chem., 10 (2004) 1-15. [34] T. Kozo, H. Hideshi, THE SYNTHESIS OF SOLID SUPER ACIDS AND THE ACTIVITY FOR THE REACTIONS OF BUTANE AND ISOBUTANE, Chemistry Letters, 5 (1976) 625-626. [35] M. Hino, K. Arata, Synthesis of solid superacid catalyst with acid strength of H0[less-than-or-eq]-16.04, Journal of the Chemical Society, Chemical Communications, (1980) 851-852. [36] S. Rezgui, A. Liang, T.K. Cheung, B.C. Gates, Methane conversion to ethane in the
presence of iron‐ and manganese‐promoted sulfated zirconia, Catalysis Letters, 53 (1998) 1-2. [37] W. Hua, A. Goeppert, J. Sommer, Methane activation in the presence of Al2O3-promoted sulfated zirconia, Applied Catalysis A: General, 219 (2001) 201-207. [38] R.L. Martins, M. Schmal, Methane activation on superacidic catalysts based on oxoanion modified zirconium oxide, Applied Catalysis A: General, 308 (2006) 143-152. [39] K. Aarset, Q. Shen, H. Thomassen, A.D. Richardson, K. Hedberg, Molecular Structure of the Aluminum Halides, Al2Cl6, AlCl3, Al2Br6, AlBr3, and AlI3, Obtained by Gas-Phase Electron-Diffraction and ab Initio Molecular Orbital Calculations, The Journal of Physical Chemistry A, 103 (1999) 1644-1652. [40] J.D. Heldman, Aliphatic Friedel—Crafts Reactions. I. The Alkylation of Butanes by Methyl and Ethyl Bromide, Journal of the American Chemical Society, 66 (1944) 1791-1793.

30
[41] J.M. Crafts, C. Friedel, Organic chemistry, Journal of the Chemical Society, 32 (1877) 725-791. [42] A. Corma, H. García, Lewis Acids: From Conventional Homogeneous to Green Homogeneous and Heterogeneous Catalysis, Chemical Reviews, 103 (2003) 4307-4366. [43] A.L. Glasebrook, N.E. Phillips, W.G. Lovell, The Action of Aluminum Halides on n-Pentane, Journal of the American Chemical Society, 58 (1936) 1944-1948. [44] O. Grummitt, E.E. Sensel, W.R. Smith, R.E. Burk, H.P. Lankelma, The Action of Aluminum Bromide on Paraffin Hydrocarbons. I. n-Hexane and n-Heptane, Journal of the American Chemical Society, 67 (1945) 910-914. [45] J.J. Burbage, A.B. Garrett, Phase Rule Studies in Anhydrous Aluminum Bromide, The Journal of Physical Chemistry, 56 (1952) 730-733. [46] D.G. Walker, THE ASSOCIATION EQUILIBRIUM IN THE METHYL BROMIDE—ALUMINUM BROMIDE SYSTEM. ESTIMATED BONDING STRENGTHS OF ALUMINUM BROMIDE-ADDITION MOLECULES WITH METHYL BROMIDE, PENTENE AND BENZENE, The Journal of Physical Chemistry, 64 (1960) 939-941. [47] H.C. Brown, W.J. Wallace, Addition Compounds of Aluminum Halides with Alkyl Halides1, Journal of the American Chemical Society, 75 (1953) 6279-6285. [48] N. Osterwalder, W.J. Stark, Direct Coupling of Bromine-Mediated Methane Activation and Carbon-Deposit Gasification, ChemPhysChem, 8 (2007) 297-303. [49] J. Liao, L. Bao, W. Wang, Y. Xie, J. Chang, W. Bao, L. Chang, Preparation of AlCl3/silica gel catalyst for simultaneously removing thiophene and olefins from coking benzene by inclosed grafting method, Fuel Processing Technology, 117 (2014) 38-43. [50] G.A. Fuentes, J.V. Boegel, B.C. Gates, n-butane isomerization catalyzed by supported aluminum chloride, Journal of Catalysis, 78 (1982) 436-444. [51] A. El Maatougui, J. Azuaje, E. Sotelo, O. Caamano, A. Coelho, Silica-Supported Aluminum Chloride-Assisted Solution Phase Synthesis of Pyridazinone-Based Antiplatelet Agents, ACS Combinatorial Science, 13 (2010) 7-12. [52] K.P. Boroujeni, K. Parvanak, Efficient and solvent-free synthesis of bis-indolylmethanes using silica gel supported aluminium chloride as a reusable catalyst, Chinese Chemical Letters, 22 (2011) 939-942. [53] R. Cámera, R. Rimada, G. Romanelli, J.C. Autino, P. Vázquez, Silica-supported aluminum chloride as catalyst for the tetrahydropyranylation of thymol, Catalysis Today, 133–135 (2008) 822-827.

31
[54] Z. Li, X. Ma, J. Liu, X. Feng, G. Tian, A. Zhu, Silica-supported aluminum chloride: A recyclable and reusable catalyst for one-pot three-component Mannich-type reactions, Journal of Molecular Catalysis A: Chemical, 272 (2007) 132-135. [55] V. Sage, J.H. Clark, D.J. Macquarrie, Cationic polymerization of styrene using mesoporous silica supported aluminum chloride, Journal of Molecular Catalysis A: Chemical, 198 (2003) 349-358. [56] L.R. Zupp, V.L. Campanella, D.M. Rudzinski, F. Beland, R. Priefer, Microwave-assisted silica-supported aluminum chloride-catalyzed Friedel-Crafts alkylation, Tetrahedron Letters, 53 (2012) 5343-5346. [57] R. Kumar, A. Kumar, A. Khanna, Synthesis, characterization and kinetics of AlCl3 supported on silica superacid catalysts for the formation of linear alkylbenzenes, Reaction Kinetics, Mechanisms and Catalysis, 106 (2012) 141-155. [58] T. Cai, M. He, X. Shi, X. Wang, D. Han, L. Lü, A study on structural suitability of immobilized aluminum chloride catalyst for isobutene polymerization, Catalysis Today, 69 (2001) 291-296. [59] M. Xu, A. Arnold, A. Buchholz, W. Wang, M. Hunger, Low-Temperature Modification of Mesoporous MCM-41 Material with Sublimated Aluminum Chloride in Vacuum, The Journal of Physical Chemistry B, 106 (2002) 12140-12143. [60] X.S. Zhao, M.G.Q. Lu, C. Song, Immobilization of aluminum chloride on MCM-41 as a new catalyst system for liquid-phase isopropylation of naphthalene, Journal of Molecular Catalysis A: Chemical, 191 (2003) 67-74. [61] K.P. Borujeni, A.R. Massah, Synthesis and application of polystyrene supported aluminium triflate as a new polymeric Lewis acid catalyst, Reactive and Functional Polymers, 66 (2006) 1126-1131. [62] L. Dai, Y. Zhang, Q. Dou, X. Wang, Y. Chen, Chemo/regioselective Aza-Michael additions of amines to conjugate alkenes catalyzed by polystyrene-supported AlCl3, Tetrahedron, 69 (2013) 1712-1716. [63] B. Tamami, K. Parvanak Borujeny, Chemoselective tetrahydropyranylation of alcohols and phenols using polystyrene supported aluminium chloride as a catalyst, Tetrahedron Letters, 45 (2004) 715-718. [64] D. Dubé, S. Royer, D. Trong On, F. Béland, S. Kaliaguine, Aluminum chloride grafted mesoporous molecular sieves as alkylation catalysts, Microporous and Mesoporous Materials, 79 (2005) 137-144. [65] S. Sato, G.E. Maciel, Structures of aluminum chloride grafted on silica surface, Journal of Molecular Catalysis A: Chemical, 101 (1995) 153-161.

32
[66] I. Akhrem, A. Orlinkov, Polyhalomethanes Combined with Lewis Acids in Alkane Chemistry, Chemical Reviews, 107 (2007) 2037-2079. [67] I. Akhrem, Selective and effective functionalization of alkanes and cycloalkanes with CO by polyhalomethanes combined with aluminum halides in organic media, Topics in Catalysis, 6 (1998) 27-39. [68] J. Oró, J. Han, High-Temperature Synthesis of Aromatic Hydrocarbons from Methane, Science, 153 (1966) 1393. [69] L. Wang, L. Tao, M. Xie, G. Xu, J. Huang, Y. Xu, Dehydrogenation and aromatization of methane under non-oxidizing conditions, Catalysis Letters, 21 (1993) 35-41. [70] Z.R. Ismagilov, E.V. Matus, L.T. Tsikoza, Direct conversion of methane on Mo/ZSM-5 catalysts to produce benzene and hydrogen: achievements and perspectives, Energy & Environmental Science, 1 (2008) 526-541. [71] J.J. Spivey, G. Hutchings, Catalytic aromatization of methane, Chemical Society Reviews, 43 (2014) 792-803. [72] K. Sun, D.M. Ginosar, T. He, Y. Zhang, M. Fan, R. Chen, Progress in Nonoxidative Dehydroaromatization of Methane in the Last 6 Years, Industrial & Engineering Chemistry Research, 57 (2018) 1768-1789. [73] I. Vollmer, I. Yarulina, F. Kapteijn, J. Gascon, Progress in Developing a Structure-Activity Relationship for the Direct Aromatization of Methane, ChemCatChem, 11 (2019) 39-52. [74] N. Kosinov, F.J.A.G. Coumans, E.A. Uslamin, A.S.G. Wijpkema, B. Mezari, E.J.M. Hensen, Methane Dehydroaromatization by Mo/HZSM-5: Mono- or Bifunctional Catalysis?, ACS Catalysis, 7 (2017) 520-529. [75] S. Ilias, A. Bhan, Mechanism of the Catalytic Conversion of Methanol to Hydrocarbons, ACS Catalysis, 3 (2013) 18-31. [76] C.H.L. Tempelman, E.J.M. Hensen, On the deactivation of Mo/HZSM-5 in the methane dehydroaromatization reaction, Applied Catalysis B: Environmental, 176-177 (2015) 731-739. [77] S. Liu, L. Wang, R. Ohnishi, M. Ichikawa, Bifunctional Catalysis of Mo/HZSM-5 in the Dehydroaromatization of Methane to Benzene and Naphthalene XAFS/TG/DTA/MASS/FTIR Characterization and Supporting Effects, Journal of Catalysis, 181 (1999) 175-188. [78] D. Wang, J.H. Lunsford, M.P. Rosynek, Characterization of a Mo/ZSM-5 Catalyst for the Conversion of Methane to Benzene, Journal of Catalysis, 169 (1997) 347-358.

33
[79] D. Wang, J.H. Lunsford, M.P. Rosynek, Catalytic conversion of methane to benzene over Mo/ZSM-5, Topics in Catalysis, 3 (1996) 289-297. [80] E.V. Matus, I.Z. Ismagilov, O.B. Sukhova, V.I. Zaikovskii, L.T. Tsikoza, Z.R. Ismagilov, J.A. Moulijn, Study of Methane Dehydroaromatization on Impregnated Mo/ZSM-5 Catalysts and Characterization of Nanostructured Molybdenum Phases and Carbonaceous Deposits, Industrial & Engineering Chemistry Research, 46 (2007) 4063-4074. [81] L. Wang, R. Ohnishi, M. Ichikawa, Selective Dehydroaromatization of Methane toward Benzene on Re/HZSM-5 Catalysts and Effects of CO/CO2 Addition, Journal of Catalysis, 190 (2000) 276-283. [82] R. Ohnishi, M. Ichikawa, Effect of CO2/CO Addition in Dehydroaromatization of Methane on Zeolite-Supported Mo and Re Catalysts Towards Hydrogen, Benzene and Naphthalene, Catalysis Surveys from Japan, 5 (2002) 103-110. [83] B.S. Liu, Y. Zhang, J.F. Liu, M. Tian, F.M. Zhang, C.T. Au, A.S.C. Cheung, Characteristic and Mechanism of Methane Dehydroaromatization over Zn-Based/HZSM-5 Catalysts under Conditions of Atmospheric Pressure and Supersonic Jet Expansion, The Journal of Physical Chemistry C, 115 (2011) 16954-16962. [84] V. Abdelsayed, M.W. Smith, D. Shekhawat, Investigation of the stability of Zn-based HZSM-5 catalysts for methane dehydroaromatization, Applied Catalysis A: General, 505 (2015) 365-374. [85] V.B. Kazansky, A.I. Serykh, E.A. Pidko, DRIFT study of molecular and dissociative adsorption of light paraffins by HZSM-5 zeolite modified with zinc ions: methane adsorption, Journal of Catalysis, 225 (2004) 369-373. [86] G. Qi, Q. Wang, J. Xu, J. Trébosc, O. Lafon, C. Wang, J.-P. Amoureux, F. Deng, Synergic Effect of Active Sites in Zinc-Modified ZSM-5 Zeolites as Revealed by High-Field Solid-State NMR Spectroscopy, Angewandte Chemie International Edition, 55 (2016) 15826-15830. [87] A.A. Gabrienko, S.S. Arzumanov, A.V. Toktarev, I.G. Danilova, I.P. Prosvirin, V.V. Kriventsov, V.I. Zaikovskii, D. Freude, A.G. Stepanov, Different Efficiency of Zn2+ and ZnO Species for Methane Activation on Zn-Modified Zeolite, ACS Catalysis, 7 (2017) 1818-1830. [88] A.A. Gabrienko, S.S. Arzumanov, M.V. Luzgin, A.G. Stepanov, V.N. Parmon, Methane Activation on Zn2+-Exchanged ZSM-5 Zeolites. The Effect of Molecular Oxygen Addition, The Journal of Physical Chemistry C, 119 (2015) 24910-24918.

34
[89] T. Baba, H. Sawada, Conversion of methane into higher hydrocarbons in the presence of ethylene over H-ZSM-5 loaded with silver cations, Physical Chemistry Chemical Physics, 4 (2002) 3919-3923. [90] A.A. Gabrienko, S.S. Arzumanov, I.B. Moroz, A.V. Toktarev, W. Wang, A.G. Stepanov, Methane Activation and Transformation on Ag/H-ZSM-5 Zeolite Studied with Solid-State NMR, The Journal of Physical Chemistry C, 117 (2013) 7690-7702. [91] A.A. Gabrienko, S.S. Arzumanov, I.B. Moroz, I.P. Prosvirin, A.V. Toktarev, W. Wang, A.G. Stepanov, Methane Activation on In-Modified ZSM-5: The State of Indium in the Zeolite and Pathways of Methane Transformation to Surface Species, The Journal of Physical Chemistry C, 118 (2014) 8034-8043. [92] W. Ding, S. Li, G. D Meitzner, E. Iglesia, Methane Conversion to Aromatics on Mo/H-ZSM5: Structure of Molybdenum Species in Working Catalysts, The Journal of Physical Chemistry B, 105 (2001) 506-513. [93] L.G. Inés, O. Ramon, R. Mauro, G. Pieter, B.S. W., W.B. M., B.A. M., Molybdenum Speciation and its Impact on Catalytic Activity during Methane Dehydroaromatization in
Zeolite ZSM‐5 as Revealed by Operando X‐Ray Methods, Angewandte Chemie, 128 (2016) 5301-5305. [94] M. Agote-Arán, A.B. Kroner, H.U. Islam, W.A. Sławiński, D.S. Wragg, I. Lezcano-González, A.M. Beale, Determination of Molybdenum Species Evolution during Non-Oxidative Dehydroaromatization of Methane and its Implications for Catalytic Performance, ChemCatChem, 11 (2019) 473-480. [95] W. Liu, Y. Xu, Methane Dehydrogenation and Aromatization over Mo/HZSM-5: In Situ FT–IR Characterization of Its Acidity and the Interaction between Mo Species and HZSM-5, Journal of Catalysis, 185 (1999) 386-392. [96] B. Rhimi, M. Mhamdi, V.N. Kalevaru, A. Martin, Synergy between vanadium and molybdenum in bimetallic ZSM-5 supported catalysts for ethylene ammoxidation, RSC Advances, 6 (2016) 65866-65878. [97] B.S. Liu, L. Jiang, H. Sun, C.T. Au, XPS, XAES, and TG/DTA characterization of deposited carbon in methane dehydroaromatization over Ga–Mo/ZSM-5 catalyst, Applied Surface Science, 253 (2007) 5092-5100. [98] N. Kosinov, F.J.A.G. Coumans, G. Li, E. Uslamin, B. Mezari, A.S.G. Wijpkema, E.A. Pidko, E.J.M. Hensen, Stable Mo/HZSM-5 methane dehydroaromatization catalysts optimized for high-temperature calcination-regeneration, Journal of Catalysis, 346 (2017) 125-133.

35
[99] W. Li, G.D. Meitzner, R.W. Borry, E. Iglesia, Raman and X-Ray Absorption Studies of Mo Species in Mo/H-ZSM5 Catalysts for Non-Oxidative CH4 Reactions, Journal of Catalysis, 191 (2000) 373-383. [100] R.W. Borry, Y.H. Kim, A. Huffsmith, J.A. Reimer, E. Iglesia, Structure and Density of Mo and Acid Sites in Mo-Exchanged H-ZSM5 Catalysts for Nonoxidative Methane Conversion, The Journal of Physical Chemistry B, 103 (1999) 5787-5796. [101] J. Gao, Y. Zheng, J.-M. Jehng, Y. Tang, I.E. Wachs, S.G. Podkolzin, Identification of molybdenum oxide nanostructures on zeolites for natural gas conversion, Science, 348 (2015) 686-690. [102] Y. Xu, Y. Shu, S. Liu, J. Huang, X. Guo, Interaction between ammonium heptamolybdate and NH4ZSM-5 zeolite: the location of Mo species and the acidity of Mo/HZSM-5, Catalysis Letters, 35 (1995) 233-243. [103] Y. Xu, S. Liu, X. Guo, L. Wang, M. Xie, Methane activation without using oxidants over Mo/HZSM-5 zeolite catalysts, Catalysis Letters, 30 (1994) 135-149. [104] J.-S. Choi, J.-M. Krafft, A. Krzton, G. Djéga-Mariadassou, Study of Residual Oxygen Species over Molybdenum Carbide Prepared During In Situ DRIFTS Experiments, Catalysis Letters, 81 (2002) 175-180. [105] W. Wu, Z. Wu, C. Liang, P. Ying, Z. Feng, C. Li, An IR study on the surface passivation of Mo2C/Al2O3 catalyst with O2, H2O and CO2, Physical Chemistry Chemical Physics, 6 (2004) 5603-5608. [106] C. Li, M. Zheng, A. Wang, T. Zhang, One-pot catalytic hydrocracking of raw woody biomass into chemicals over supported carbide catalysts: simultaneous conversion of cellulose, hemicellulose and lignin, Energy & Environmental Science, 5 (2012) 6383-6390. [107] C. Wan, Y.N. Regmi, B.M. Leonard, Multiple Phases of Molybdenum Carbide as Electrocatalysts for the Hydrogen Evolution Reaction, Angewandte Chemie, 126 (2014) 6525-6528. [108] D. Ma, Y. Shu, M. Cheng, Y. Xu, X. Bao, On the Induction Period of Methane Aromatization over Mo-Based Catalysts, Journal of Catalysis, 194 (2000) 105-114. [109] B. Li, S. Li, N. Li, H. Chen, W. Zhang, X. Bao, B. Lin, Structure and acidity of Mo/ZSM-5 synthesized by solid state reaction for methane dehydrogenation and aromatization, Microporous and Mesoporous Materials, 88 (2006) 244-253. [110] L. Chen, J. Lin, H.C. Zeng, K.L. Tan, Non-oxidative methane conversion into aromatics on mechanically mixed Mo/HZSM-5 catalysts, Catalysis Communications, 2 (2001) 201-206.

36
[111] C. Bouchy, I. Schmidt, J.R. Anderson, C.J.H. Jacobsen, E.G. Derouane, S.B. Derouane-Abd Hamid, Metastable fcc α-MoC1−x supported on HZSM5: preparation and catalytic performance for the non-oxidative conversion of methane to aromatic compounds, Journal of Molecular Catalysis A: Chemical, 163 (2000) 283-296. [112] S.B. Derouane-Abd Hamid, J.R. Anderson, I. Schmidt, C. Bouchy, C.J.H. Jacobsen, E.G. Derouane, Effect of the activation procedure on the performance of Mo/H-MFI catalysts for the non-oxidative conversion of methane to aromatics, Catalysis Today, 63 (2000) 461-469. [113] M. Rahman, A. Sridhar, S.J. Khatib, Impact of the presence of Mo carbide species prepared ex situ in Mo/HZSM-5 on the catalytic properties in methane aromatization, Applied Catalysis A: General, 558 (2018) 67-80. [114] L.Y. Chen, L.W. Lin, Z.S. Xu, X.S. Li, T. Zhang, Dehydro-oligomerization of Methane to Ethylene and Aromatics over Molybdenum/HZSM-5 Catalyst, Journal of Catalysis, 157 (1995) 190-200. [115] H. Liu, W. Shen, X. Bao, Y. Xu, Methane dehydroaromatization over Mo/HZSM-5 catalysts: The reactivity of MoCx species formed from MoOx associated and non-associated with Brönsted acid sites, Applied Catalysis A: General, 295 (2005) 79-88. [116] T. Behrsing, H. Jaeger, J.V. Sanders, Coke deposits on H-ZSM-5 zeolite, Applied Catalysis, 54 (1989) 289-302. [117] C.H.L. Tempelman, V.O. de Rodrigues, E.R.H. van Eck, P.C.M.M. Magusin, E.J.M. Hensen, Desilication and silylation of Mo/HZSM-5 for methane dehydroaromatization, Microporous and Mesoporous Materials, 203 (2015) 259-273. [118] A. Martínez, E. Peris, G. Sastre, Dehydroaromatization of methane under non-oxidative conditions over bifunctional Mo/ITQ-2 catalysts, Catalysis Today, 107-108 (2005) 676-684. [119] A. Corma, V. Fornés, J.M. Guil, S. Pergher, T.L.M. Maesen, J.G. Buglass, Preparation, characterisation and catalytic activity of ITQ-2, a delaminated zeolite, Microporous and Mesoporous Materials, 38 (2000) 301-309. [120] Ch. Baerlocher and L.B. McCusker, Database of Zeolite Structures. Available from: http://www.iza-structure.org/databases/ [121] C. Xu, J. Guan, S. Wu, M. Jia, T. Wu, Q. Kan, Catalytic performance of zeolite ITQ-13 with 9- and 10-member rings for methane dehydroaromation, Reaction Kinetics, Mechanisms and Catalysis, 99 (2010) 193-199.

37
[122] H. Liu, S. Wu, Y. Guo, F. Shang, X. Yu, Y. Ma, C. Xu, J. Guan, Q. Kan, Synthesis of Mo/IM-5 catalyst and its catalytic behavior in methane non-oxidative aromatization, Fuel, 90 (2011) 1515-1521. [123] H. Liu, J. Hu, Z. Li, S. Wu, L. Liu, J. Guan, Q. Kan, Synthesis of zeolite IM-5 under rotating and static conditions and the catalytic performance of Mo/H-IM-5 catalyst in methane non-oxidative aromatization, Kinetics and Catalysis, 54 (2013) 443-450. [124] H. Liu, Q. Kan, Improved performance of hierarchical porous Mo/H-IM-5 catalyst in methane non-oxidative aromatization, Applied Petrochemical Research, 7 (2017) 97-105. [125] H. Liu, S. Yang, S. Wu, F. Shang, X. Yu, C. Xu, J. Guan, Q. Kan, Synthesis of Mo/TNU-9 (TNU-9 Taejon National University No. 9) catalyst and its catalytic performance in methane non-oxidative aromatization, Energy, 36 (2011) 1582-1589. [126] Q. Kan, D. Wang, N. Xu, P. Wu, X. Yang, S. Wu, T. Wu, Nonoxidative aromatization of methane over Mo/MCM-49 catalyst, in: X. Bao, Y. Xu (Eds.) Studies in Surface Science and Catalysis, Elsevier, 2004, pp. 631-636. [127] C.-L. Zhang, S. Li, Y. Yuan, W.-X. Zhang, T.-H. Wu, L.-W. LIn, Aromatization of methane in the absence of oxygen over Mo-based catalysts supported on different types of zeolites, Catalysis Letters, 56 (1998) 207-213. [128] K.M. Arata, H., Solid Superacids, Nova Science Publishers, New York, 2011. [129] X. Song, A. Sayari, Sulfated Zirconia-Based Strong Solid-Acid Catalysts: Recent Progress, Catalysis Reviews, 38 (1996) 329-412. [130] G.D. Yadav, J.J. Nair, Sulfated zirconia and its modified versions as promising catalysts for industrial processes Microporous Mesoporous Mater., 33 (1999) 1-48. [131] B.M. Reddy, M.K. Patil, Organic Syntheses and Transformations Catalyzed by Sulfated Zirconia, Chemical Reviews, 109 (2009) 2185-2208. [132] L.F. Córdoba, W.M.H. Sachtler, C. Montes de Correa, NO reduction by CH4 over Pd/Co-sulfated zirconia catalysts, Applied Catalysis B: Environmental, 56 (2005) 269-277. [133] E.M. Holmgreen, M.M. Yung, U.S. Ozkan, Pd-supported on sulfated monoclinic zirconia for the reduction of NO2 with methane under lean conditions, Catalysis Letters, 111 (2006) 19-26. [134] S. Ren, G. Sun, C. Li, S. Song, Q. Xin, X. Yang, Sulfated zirconia–Nafion composite membranes for higher temperature direct methanol fuel cells, Journal of Power Sources, 157 (2006) 724-726.

38
[135] C. Bi, H. Zhang, Y. Zhang, X. Zhu, Y. Ma, H. Dai, S. Xiao, Fabrication and investigation of SiO2 supported sulfated zirconia/Nafion® self-humidifying membrane for proton exchange membrane fuel cell applications, Journal of Power Sources, 184 (2008) 197-203. [136] S. Tominaka, N. Akiyama, F. Croce, T. Momma, B. Scrosati, T. Osaka, Sulfated zirconia nanoparticles as a proton conductor for fuel cell electrodes, Journal of Power Sources, 185 (2008) 656-663. [137] Y. Zhang, H. Zhang, X. Zhu, C. Bi, Promotion of PEM Self-Humidifying Effect by Nanometer-Sized Sulfated Zirconia-Supported Pt Catalyst Hybrid with Sulfonated Poly(Ether Ether Ketone), The Journal of Physical Chemistry B, 111 (2007) 6391-6399. [138] Y. Zhai, H. Zhang, J. Hu, B. Yi, Preparation and characterization of sulfated zirconia (SO42−/ZrO2)/Nafion composite membranes for PEMFC operation at high temperature/low humidity, Journal of Membrane Science, 280 (2006) 148-155. [139] K. Arata, M. Hino, Preparation of superacids by metal oxides and their catalytic action, Materials Chemistry and Physics, 26 (1990) 213-237. [140] D.A. Ward, E.I. Ko, One-Step Synthesis and Characterization of Zirconia-Sulfate Aerogels as Solid Superacids, Journal of Catalysis, 150 (1994) 18-33. [141] V. Bolis, G. Magnacca, G. Cerrato, C. Morterra, Microcalorimetric Characterization of Structural and Chemical Heterogeneity of Superacid SO4/ZrO2 Systems†, Langmuir, 13 (1997) 888-894. [142] D. Fraenkel, N.R. Jentzsch, C.A. Starr, P.V. Nikrad, Acid strength of solids probed by catalytic isobutane conversion, Journal of Catalysis, 274 (2010) 29-51. [143] F.F. Oloye, A.J. McCue, J.A. Anderson, n-Heptane hydroconversion over sulfated-zirconia-supported molybdenum carbide catalysts, Applied Petrochemical Research, 6 (2016) 341-352.

39
Chapter 3 . Light Alkane Aromatization: Efficient Use of Natural Gas1
3.1 Introduction
Energy and environment have been two major focuses of research in the twenty‐
first century that are closely related. As the population grows and cities expand, the
demand for energy inevitably increases, as does the incentive for clean energy. Despite
significant research efforts for biofuel development, and for alternative energy sources
(wind, solar, hydrothermal, etc.), fossil fuels still dominate world energy demands. Coal
and oil are widely used, but natural gas is generally considered to be cleaner and more
efficient in many applications. The shale gas revolution has presented a unique
opportunity to address these challenges by expanding the use of natural gas. The
immense, previously unattainable, reserves of natural gas have already had major
impacts on the energy and chemical industries and have helped countries like the United
States, reduce CO2 emissions [1-4].
3.1.1 Shale gas revolution
Advances in horizontal drilling and hydrofracking have led to an increase in shale
gas production. In the United States alone, it rose from 1% to 20% from 2000 to 2010
causing a decrease in the prices of natural gas. It is this dramatic effect that is often
termed the “Shale gas revolution” [4].
This has significantly lowered natural gas prices, and projections show that they
will stay at current low levels through 2023 [3]. Recently, a 5% drop in prices of natural
1 Reprinted from Kanitkar, S., & Spivey, J. J. (2019). “Light Alkane Aromatization: Efficient Use of Natural Gas”. In N. Elbashir, M. M. El-Halwagi, K. Hall, I. Economou (Eds.) Natural Gas Processing from Midstream to Downstream (pp. 379-402). Hoboken, NJ: John Wiley & Sons. With permission from John Wiley and Sons.

40
gas was reported just for the change from 2015 to 2016, whereas European and Asian
markets saw a 20-25% drop in natural gas prices to compensate for the supply of natural
gas [5]. The shale gas revolution has also provided an alternative to coal‐fired power
plants, thereby helping reduce CO2 emissions [6]. This makes natural gas an economic
alternative feedstock over conventional fuels. This has resulted in providing a significant
incentive for new research in the area of natural gas conversion: in particular, methane
and light alkanes.
3.1.2 Composition of natural gas
Natural gas composition can vary significantly based on the location of the reserve
and also depending on whether it is associated or non‐associated with oil. In either case,
primary components of natural gas include C1, C2, C3, CO2, N2, H2S, and trace levels of
higher hydrocarbons. Table 3.1 shows the composition of natural gas from some of the
fields around the world. Regardless of the variety in composition of various wells, CH4
remains the principal component, ∼49-84%.
Table 3.1 Composition of natural gas from various wells around the world [7]
Composition
(mol %) ↓
Field Name
Xifeng,
China
Tengiz,
Kazakhastan
Ekofisk,
Norway
Salt Creek,
USA
Boscan,
Venezuela
N2 2 2 0 1 2
CO2 2 4 3 1 6
C1 49 60 79 81 84
C2 13 10 10 9 4
C3 19 3 4 4 2
C4+ 15 6 3 3 1
H2S 0 15 1 0 1
Methane conversion to higher-value products is one of the grand challenges for
the scientific community working in the area of catalysis and electrocatalysis. Despite the

41
strong impetus, and as indicated by recent reviews [8-11], direct non-oxidative conversion
of methane is yet to be widely commercialized.
Secondary important components of natural gas are ethane and propane that
typically constitute 2-5 % (combined) of natural gas. Although in some wells, up to 8-10
% of ethane along with 1-3 % propane could also be seen. N-butane on the other hand,
could be observed around ~ 0.1 % of natural gas in composition. These light alkanes are
relatively easier to activate than methane and are principal feedstocks for the production
of olefins and plastics, through dehydrogenation or oxidative coupling processes. Ethane
is the principal feedstock for the production of ethylene (C2=), which is subsequently
converted to a variety of valuable chemicals such as polyethylene, ethylene oxide,
ethylene dichloride etc. The production of ethylene from ethane is carried out via steam
cracking and is a well-established process.
Production of ethane has increased significantly over the past few years as a result
of the shale gas revolution. This has caused costs of ethane to drop to a low level, and
this downward trend in price is expected to continue over the next few years (Figure 3.1).
This makes ethane an attractive feedstock for aromatization into benzene, toluene, and
higher‐value hydrocarbons, along with pure H2. Therefore, the chemical industry
recognizes that an economically desirable goal will be to replace naphtha with light
alkanes as feedstock, which would also reduce the environmental footprint of aromatics
synthesis.
The aromatization of methane is one key potential process of interest to the
chemical industry:
6 CH4 → C6H6 + 9H2, ∆𝐺298 𝐾 = +102.4 𝑘𝑐𝑎𝑙
𝑚𝑜𝑙⁄ , ∆𝐻298 𝐾 = +116.3𝑘𝑐𝑎𝑙
𝑚𝑜𝑙⁄ (3.1)

42
Current research has demonstrated only net yields of 5-10% of benzene, and rapid
deactivation of coke has limited commercialization of aromatization [8-11]. Hydrogen is a
valuable byproduct of the aromatization process, and the appeal of a reaction that
produces both benzene and hydrogen suggests that industrial laboratories are developing
a viable process.
Figure 3.1 Ethane production and prices over the last few years [12]
In addition to aromatization of methane, aromatization of light alkanes is a potential
process. In general, acid catalysts have been effective in the aromatization of light
alkanes, in particular the pentasil types: ZSM-5 etc. Various types of catalysts based on
bi-functionality in which metals like Mo, Ga, Pt, Re, Zn were doped on the ZSM-5 are
studied. Despite being widely investigated, ethane aromatization is not yet
commercialized due to poor catalyst stability, high temperature requirements, and coking
issues with the catalysts. Although several reviews [13-16] exist on light alkane
conversion to aromatics, no recent review show progress in the last decade. The chapter
0
2
4
6
8
10
12
14
0
200
400
600
800
1000
1200
1400
2009 2010 2011 2012 2013 2014 2015 2016 2017
Avg
. a
nn
ua
l p
rices (
$/m
mB
tu)
Th
ou
san
d b
arr
els
per
day
Year

43
here reviews recent progress in the area of acid catalyzed light alkane aromatization (in
particular, ethane). This chapter focuses on the effect of acidity, pore structure, and metal
functions on the aromatization of ethane to aromatics.
3.2 Aromatization of light alkanes
3.2.1 Thermodynamics and short history
Alkanes are a class of hydrocarbons having only sigma bonds. Although singly
bonded, these are more difficult to activate than the stronger double bonded olefins, thus
requiring more severe conditions, e.g., high temperatures. Alkanes with a high number of
carbon atoms are easier to activate than their lower counterparts[17] because C-C bond
is weaker than C-H bond and a surrounding carbon influences the C-H bond dissociation
energy. Thus, the more the number of ‘C’ atoms in an alkane, the easier it is to break the
C-H bond. Often, this breaking of the first C-H bond is the activation step for alkane
activation. This exemplifies the reactivity for the first 4 alkanes (light alkanes):
CH4 < C2H6 < C3H8 < C4H10
This is clearly reflected by the thermodynamics. Figure 3.2 shows aspects of
thermodynamics for light alkane conversion into aromatics. For simplicity, reactions and
products shown in eq. 3.2) were considered for the calculations:
Alkane (C1 − C4) → Benzene, Toluene, Ethylbenzene, Xylene (BTEX) + H2 ↑ (3.2)
As seen from Figure 3.2, the reactivities for all the four basic alkanes differ quite
considerably. Appreciable levels of conversion (20–30%) could thermodynamically be
possible only at ∼700 °C (for methane), ∼550 °C (for ethane), ∼450 °C (for propane), and
∼250 °C (for n‐butane). Because high activation temperatures are required for methane

44
and ethane, efforts are focused on aromatization of C3 and C4, as will be discussed in
section 3.2.2 Existing technologies.
21C2H6 → 2C6H6 + 2C7H8 + 2C8H10 + 39H2 (3.3)
∆G298 K = + 336.6 kcal
mol⁄ , ∆H298 K = + 458.7 kcal
mol⁄
Figure 3.2 Thermodynamics for light alkane aromatization (Plotted using HSC
Chemistry 8)
For the reaction depicted in equation 3.3, a plot of ΔG vs. temperature is shown in
Figure 3.3. High levels of conversion for the forward reaction are possible for ΔG ≤ 0 after
which reaction becomes spontaneous. By observing Figure 3.3, for ethane aromatization
from equation 3.3, this is spontaneous only at temperatures ≥ 700 °C.
One way to overcome thermodynamic limitations is to add oxidant along with
ethane on the catalyst. This reduces the energy barrier, making it relatively easier to
activate ethane. Common oxidants involve CO2, NOx, and O2. However, the presence of
0
10
20
30
40
50
60
70
80
90
100
0 200 400 600 800 1000 1200 1400
Co
nve
rsio
n (
%)
Temperature (oC)
Ethane
Propane
n-Butane
Methane

45
an additional reactant changes the reaction pathway considerably and dehydrogenation
reactions (oxidative dehydrogenation) might be favored over aromatization reactions [18].
Figure 3.3 Gibbs free energy plot for ethane aromatization (plotted using HSC
Chemistry 8)
Aromatization of olefins was first reported around 1946 by Russian workers [14]
and it was not until the 1970s when aromatization of light alkanes (C3-C5) was first
reported by Csicsery [17, 19-22]. He described aromatization on Pt/Al2O3 and transition
metal oxides (MoO3, Cr2O3, V2O5, WO3, MnO) doped on Al2O3 at around 550-600 °C.
Primary products observed were xylenes and toluene but the product distribution was
found to be influenced by the strength of the acid sites over the catalyst. Coking was
evident and the rate of coke formation was influenced by temperature, residence time,
and catalyst on-stream time. Although, transition metal oxides were found to be better
-200
-150
-100
-50
0
50
100
150
200
250
300
350
400
0 200 400 600 800 1000
ΔG
(kca
l/m
ol)
Temperature (°C)
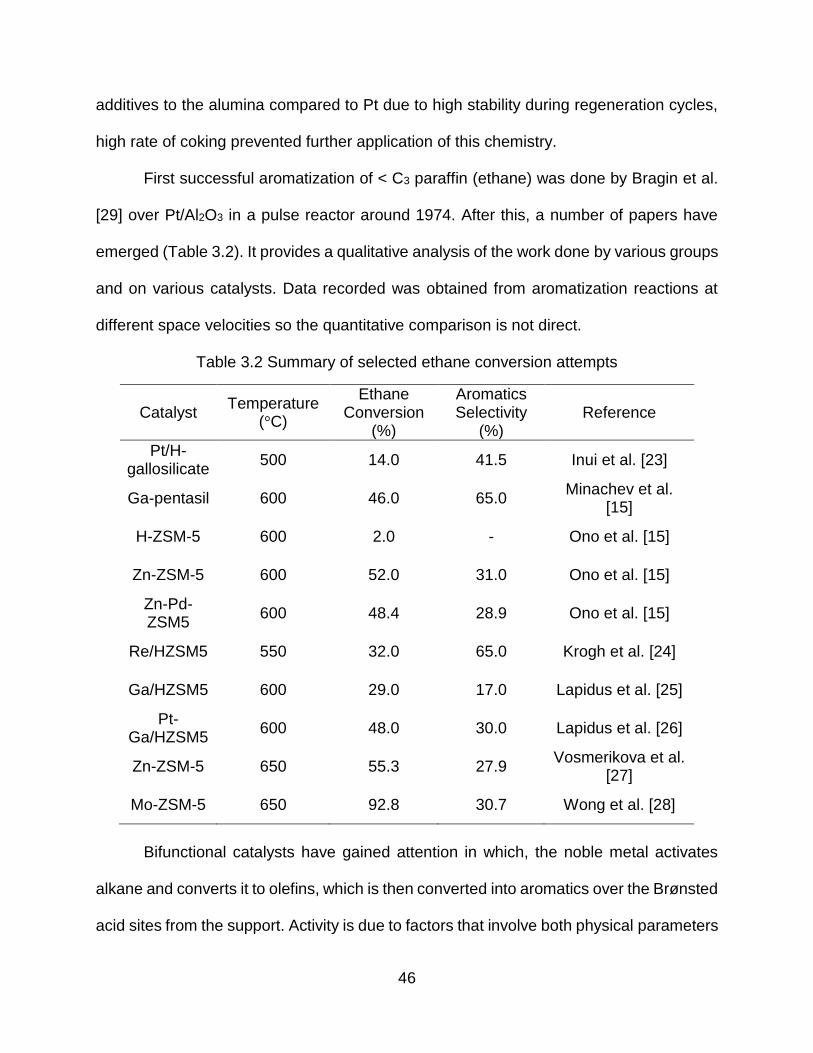
46
additives to the alumina compared to Pt due to high stability during regeneration cycles,
high rate of coking prevented further application of this chemistry.
First successful aromatization of < C3 paraffin (ethane) was done by Bragin et al.
[29] over Pt/Al2O3 in a pulse reactor around 1974. After this, a number of papers have
emerged (Table 3.2). It provides a qualitative analysis of the work done by various groups
and on various catalysts. Data recorded was obtained from aromatization reactions at
different space velocities so the quantitative comparison is not direct.
Table 3.2 Summary of selected ethane conversion attempts
Catalyst Temperature
(°C)
Ethane Conversion
(%)
Aromatics Selectivity
(%) Reference
Pt/H-gallosilicate
500 14.0 41.5 Inui et al. [23]
Ga-pentasil 600 46.0 65.0 Minachev et al.
[15]
H-ZSM-5 600 2.0 - Ono et al. [15]
Zn-ZSM-5 600 52.0 31.0 Ono et al. [15]
Zn-Pd-ZSM5
600 48.4 28.9 Ono et al. [15]
Re/HZSM5 550 32.0 65.0 Krogh et al. [24]
Ga/HZSM5 600 29.0 17.0 Lapidus et al. [25]
Pt-Ga/HZSM5
600 48.0 30.0 Lapidus et al. [26]
Zn-ZSM-5 650 55.3 27.9 Vosmerikova et al.
[27]
Mo-ZSM-5 650 92.8 30.7 Wong et al. [28]
Bifunctional catalysts have gained attention in which, the noble metal activates
alkane and converts it to olefins, which is then converted into aromatics over the Brønsted
acid sites from the support. Activity is due to factors that involve both physical parameters

47
(temperature, pressure, space velocity, or even the pretreatment conditions etc.) and
chemical parameters (type and amount of metal exchanged, type of support,
concentration of acid sites, Si/Al ratio). Some of the key chemical parameters will be
discussed here.
3.2.2 Existing technologies
Aromatization processes began to appear in commercial practice around the
1970s after the discovery of ZSM-5 zeolite. This zeolite found widespread applications
ranging from FCC additive for propylene production to octane number improvement [30].
Many notable aromatization processes were developed, including those of M-2 forming
developed by Mobil, Aroforming by IFP and Salutec, Z-forming by Mitsubishi and
Chiyoda, Alpha forming by Toyo and Sanyo, Cyclar process by BP-UOP, GTA technology
by Sinopec Luoyang. Most of them have similar feedstocks (light naphthas, lower olefins,
LPGs, or light alkanes), similar aromatic yields albeit with different unit operations or
reactor setups. Catalysts used are primarily ZSM-5 or modified ZSM-5.
Mobil developed M-2 forming process in 1986 that involved aromatization of olefins
as well as C5, C6 alkanes over a HZSM5 catalyst [31]. However, the feedstock can be as
different as LPG or light olefinic cuts, or paraffinic or unsaturated gasolines [32]. One
limitation with the M-2 process is that the side reaction of dehydrogenation shifts the
equilibrium giving less aromatics and less hydrogen yield making it less economical [33].
Aroforming process that was similar to Cyclar, was developed around the 1980s
as a joint venture between France’s IFP and Salutec from Australia. Similar to M-2
forming, the feedstock for this process also involved LPGs and light naphthas that were
converted into BTX and hydrogen [32]. This process uses a shape selective zeolite

48
catalyst doped with a metal oxide but has a product distribution similar to that of Cyclar
Process [34].
Mitsubishi and Chiyoda had developed ‘Z forming’ process around 1983 that
converted light naphtha or LPG into aromatic hydrocarbons over an acidity adjusted
catalyst. The process was used by Nippon Oil till 2008 but was abandoned due to low
yield. This process is based on a bifunctional catalyst consisting of a metal (most likely
zinc) modified zeolite (ZSM-5) base material. The reforming reactors are of an adiabatic,
fixed-bed type. Operating conditions are 500-600 °C at < 100 psia. BTX yields of up to
55-58 wt% are observed [35].
Chevron Phillips’ Aromax process that converts light naphtha into aromatic
hydrocarbons was developed around the 1980s. First commercial demonstration of the
Aromax technology was in 1992 which afterward continued. Feedstock for this process
primarily consists of C6-C8 hydrocarbons with high selectivity to benzene, toluene, and
hydrogen. The process also involves a highly efficient sulfur control system to eliminate
catalyst poisoning. Currently, four plants around the world (US, Spain, Japan, and Saudi
Arabia) make use of this technology [36]. Process flow diagram for the Aromax process
could be depicted in Figure 3.4.
Sinopec Luoyang’s GTA technology makes use of Olefinic C4 streams to convert
into aromatics at around 500 °C over a bifunctional metal modified ZSM5 catalyst. This
process adopts a fixed-bed reaction-regeneration system and it is feasible with short
flows. This process is highly selective to aromatics (55-60 wt%) yield and it does not
generate non aromatic components eliminating the need for aromatic extraction unit [35].

49
Figure 3.4 Chevron Philips' Aromax Technology Process [36]
BP-UOP’s successful ‘CYCLAR’ process that involves aromatization of propane
and butanes, was first demonstrated at BP’s refinery at Grange-Mouth, UK in 1991. This
commercial set up with a capacity of 1000 barrels per day was active for almost 8 years.
A second commercial unit was set up in the Middle East around 1999 with a capacity of
45,000 barrels per day and was in operation as of 2013 [37]. Cyclar process has aromatic
(BTX) yields of around 58 – 60 % on a metal doped (mostly Ga) H-ZSM5 catalyst that is
easily coked and requires continuous regeneration. This regeneration is carried out using
UOP’s trademarked CCR (continuous catalyst regeneration) technology. Schematic for a
typical CYCLAR process unit is shown in Figure 3.5.
3.2.3 Role of metals (Mo, Pt, Ga, Re, Zn)
It is well understood that the MFI zeolite by itself is not active for the aromatization
reactions. In order to enhance its activity, doping of metals is necessary into the zeolite

50
Figure 3.5 Schematic for CYCLAR process [37]
framework. These metal dopants lead to distinct changes in the properties of the undoped
zeolite and often forms a bifunctional catalyst. It is commonly believed that ethane is first
activated on the metal species and is then dehydrogenated into ethylene. This ethylene
forms olefinic intermediates and undergoes various oligomerization reactions, and
cyclization reactions take place on the Brønsted acid site from the zeolite itself.
Bifunctional mechanism could be depicted as shown in Figure 3.6.
Since the first step of ethane activation is crucial, it often determines the rate of
formation of olefinic species and subsequently the rate of aromatization. Thus, it becomes
important that the metal is well dispersed in the zeolite structure [38, 39]. Different metals
have different activation energies for activation of ethane and introduction of these metal
ions in the zeolite structure changes the concentration of both types of acid sites: Lewis
and Brønsted. Various noble and base metals have been tested so far including Cr, Fe,
Mn, Pd, Pt, Re, Ru, W, Zn, Mo, Ga etc. However, the most active and very widely studied
metal dopants are Pt, Ga, Zn, and Mo.

51
Figure 3.6 Bifunctional mechanism over metal doped HZSM5 catalysts
One relatively new metal dopant is Re. It has also been seen that when a small
metal dopant (as a promoter) is added to an already metal doped zeolite, it can enhance
the activity that bifunctional catalyst. Some examples include the addition of Pt into
Ga/ZSM-5 [26, 40, 41], the addition of W into Mo/ZSM-5 [28] etc.
3.2.3.1 Mo/ZSM-5
Molybdenum is an active metal dopant for the dehydroaromatization of light
alkanes. Most noticeable activity for Mo based zeolite catalyst has been in the methane
dehydroaromatization at around 700-800 °C [42, 43]. It is hypothesized that methane gets
activated into CHx that subsequently forms a dimer on the Mo2C species and the
dimerized form is oligomerized, then cyclized on the Brønsted acid sites of the zeolite[8].
H-ZSM-5 and H-MCM-22 have been the two prominent zeolitic/molecular sieve supports
for incorporating Mo, demonstrating high activity in the aromatization of methane/ethane.
Molybdenum can typically be doped on the zeolite using incipient wetness
impregnation (IWI), often with the ammonium salt of Mo [43-45]. Other procedures
involved could be solid state ion exchange [46, 47] that involves the physical mixing of
MoO3 and Zeolite and heating the mixture. However, for aromatization reactions, at least

52
on methane, IWI prepared Mo catalyst showed greater selectivity to aromatics than the
ion exchanged catalysts [47]. The low selectivity of ion exchanged catalysts was
attributed to the formation of large MoOx clusters that were unable to diffuse/migrate
inside the ZSM5 pores.
In terms of ethane or propane aromatization on Mo based catalysts, there have
been few reports. Wong et al. [28] studied Mo/HZSM5 and W/HZSM5 for ethane
aromatization that showed very high levels of ethane conversion (93 %) and (25 %)
respectively. However, selectivity for aromatics differed significantly with Mo doped
catalyst, which favored cracking to CH4 more than aromatization. This was attributed to
the strong activity of Mo in both the dehydrogenation reaction (first step in ethane
activation) and strong activity in the reaction between H2 from first step combining with
ethane to get cracked into methane. A sequence of reactions [28] could be depicted as
shown in eqns. 3.4 and 3.5.
𝐶2𝐻6 → 𝐶2𝐻4 + 𝐻2 (3.4)
𝐻2 + 𝐶2𝐻6 → 2 𝐶𝐻4 (3.5)
Samanta et al. [40] reported reduction in a number of strong Brønsted acid sites
when Mo was doped into H-ZSM-5 despite the increase in the overall acidity that most
likely occurs due to the exchange of zeolitic proton with the metal itself. However,
generated extra acidity was attributed to the MoO3 Lewis acid sites. Around 40 %
conversion of ethane with 35 % selectivity to aromatics was reported at 650 °C over these
catalysts. Compared to Ga or GaPt doped catalysts, Mo doped catalysts had lower
activity. Mo doped catalysts favored ethylene more than Ga or GaPt doped catalysts. This
likely would have been due to the generation of extra Lewis acidity that would have
favored dehydrogenation over aromatization. Since dehydrogenation is thought to be the

53
initial activation step, reduction in strong Brønsted acid sites would have caused less
aromatization of the olefinic species generated from the dehydrogenation.
3.2.3.2 Pt/ZSM-5
Platinum can be incorporated onto the ZSM5 support typically by IWI of H2PtCl6
(chloroplatinic acid) salt[40] or by ion exchange [48] of NH4-ZSM5 using ammonium salts
of Pt like Pt(NH3)4Cl2. Another method that could be used for depositing Pt into the pores
of zeolite is the vapor phase impregnation using platinum acetyl acetonate, Pt(acac)2 as
a precursor.
Platinum (Pt) based catalysts have been effective in dehydrogenation of
propane[49, 50] and also for dehydrogenolysis activity[51] most likely due to H2
abstraction. Despite this, Pt by itself shows very low but stable activity (10% conversion)
with very high (97%) selectivity to ethylene and almost negligible amounts of aromatics
[40]. Lapidus et al. [41] however found around 27% ethane conversion with aromatic
selectivity of about 51.5 % on Pt loaded ZSM-5 catalyst. These differences could be due
to different Pt loading (0.3 wt%)[41] in one case whereas (0.5 wt%)[40] in other. Also, the
parent H-ZSM5 zeolite material had different SiO2/Al2O3 ratios, SiO2/Al2O3 = 30:1 [41] and
SiO2/Al2O3 = 50:1 for the other material [40] that would have more of an influence than
the Pt loading itself.
Despite a lack of strong activity in ethane aromatization by itself, Pt can be a very
effective promoter [40] and can form bimetallic nanoclusters as depicted in Figure 3.7
along with Ga thereby enhancing the activity of the overall catalyst multiple times and at
lower deactivation rates.

54
Figure 3.7 Possible representation of Ga-Pt nano-clusters
3.2.3.3 Ga/H-ZSM-5
Gallium can be incorporated into the zeolite support by numerous methods
including IWI of [Ga(NO3)3.xH2O] salt [40, 41], Ion exchange with Gallium salts [52], vapor
phase deposition using volatile GaX3 (X = halide) precursors [53, 54], physical mixing of
Ga2O3 with ZSM-5 [55-57] or directly adding gallium salt during the synthesis of ZSM-5
to form gallosilicate or galloaluminosilicates.
Gallium has been an effective and well-known dopant in the zeolites for the
aromatization of propane [13]. Gallium is favored over platinum because of stronger
aromatization activity. Ono et al. [15, 58] reported ethane aromatization over Ga/ZSM-5
based catalysts. After this, numerous reports [25, 26, 40, 41, 51, 59] have emerged.
Gallium activates ethane by abstracting hydrogen from ethane thereby forming ethylene.
The mechanism for hydrogen removal from ethane is presented by Keipert et al. [51] as
shown in Figure 3.8. However, like Mo, Ga also has a tendency to exchange with the

55
protons on the zeolite surface thereby causing the reduction in the strong acid sites on
the surface of the zeolite [40].
Figure 3.8 Elementary steps in ethane dehydrogenation over Ga based catalysts [51]
There have been various reports about the nature of active Ga species for ethane
aromatization. Some reports [59] speculate it to be multinuclear gallium clusters rather
than mononuclear ones. On the other hand, Yakerson et al. [60], based on the extensive
IR study and electron microscopy, had pointed out that Ga2O3 on the external surface is
the active species for this reaction. It is widely accepted that Ga2O3 favors
dehydrogenation through H2 recombinative desorption. However, Dooley et al. [61]
correctly pointed out about transformations in the catalyst that can occur while reduction
under H2 or during pretreatment with lower hydrocarbons or even under reaction
conditions. Sometimes, these treatments or transformations can result in the formation of
Ga+ that can replace H+ while migrating into the zeolite channels. In a more recent study
[62], ethane dehydrogenation was studied over Ga/ZSM5 specifically to probe the role of
various Ga species including: Ga2O3, GaO+, Ga+, and GaH2+. A noticeable drop in the
acidity was observed when Ga/ZSM-5 was reduced under H2 and it was attributed to the

56
formation of reduced Ga species (Ga+ and GaH2+). These reduced species can undergo
exchange with the protons and replace the acid sites, which was confirmed using NH3-
TPD and H2-TPR. Further, these reduced species showed better dehydrogenation activity
than the oxide species Ga2O3/GaO+ for ethane aromatization. Authors[62] ranked the
dehydrogenation activity for different species as follows:
GaO+ ~ Ga2O3 < Ga+ < GaH2+
Because dehydrogenation is the initiation step in the aromatization of light alkanes,
it becomes important to reduce Ga species (Ga+, GaH2+). However, these reduced
species can cause reduction of strong acid sites, and this may hamper the oligomerization
step that forms aromatics. Therefore, a balance between the number of reduced Ga
species and Brønsted acid sites might be necessary for optimal activity of Ga/ZSM-5
catalyst. Lapidus et al. [41] found the optimal loading of Ga to be around 2.0% for the
pentasil zeolites.
3.2.3.4 Re/H-ZSM-5
Rhenium can typically be introduced into the zeolite (H-ZSM-5) using IWI of
ammonium perrheneate salt [(NH4)2ReO4.4H2O]. Like most other metallic species, Re in
metallic state is often the active species for alkane activation [63]. Encapsulated Re
clusters have also been reported to be active for CH4 dehydroaromatization and in fact,
can be 30% faster than the MoCx catalyst [64]. Also, these clusters have been shown to
be active for propane activation irrespective of Re content.
Two studies have reported doping of rhenium into the ZSM5 zeolite [24, 38] for
ethane aromatization. Both studies doped Re onto ZSM-5 with SiO2/Al2O3 ratio of 30:1.
Although, the loading of Re was different. Krogh et al. [24] compared Re/ZSM-5 vs.
Zn/ZSM-5 and found out that although Re exhibited a lower activity initially, it deactivated

57
less rapidly than the Zn/ZSM-5 and thus showed higher and stable ethane conversion
values over long runs. Performance of Re/ZSM-5 against Zn/ZSM-5 could be observed
in Figure 3.9. The difference was attributed to the sublimation of Zn species that did not
occur in the case of Re/ZSM-5 which is more stable at those temperatures.
Figure 3.9 Ethane conversion on Re/ZSM5 and Zn/ZSM5 (1 atm, 650 °C, 400 mg
catalyst, WHSV = 1.81 h-1). Reprinted from “Re/HZSM‐5: a new catalyst for ethane
aromatization with improved stability”, Catalysis Communications, 4(12), 627–630,
2003, with permission from Elsevier
Solymosi et al. [38] reported that Re was promoting the activation of the C-H bond
that resulted in more ethane conversion and higher aromatics selectivity. However, higher
loadings of Re resulted in catalysts with lower activity. It could have been that higher
loading led to poor dispersion of Re metal, making the catalyst less active.
3.2.3.5 Zn/H-ZSM-5
One of the most widely studied dopant in the zeolite framework for aromatization
of ethane is zinc. Zinc can be introduced onto/into the ZSM5 support using various

58
techniques: IWI using precursors Zn(NO3)2.6H2O or ZnCl2, ZnO, ZnS, etc., ion exchange
with a corresponding salt, or mechanical mixing of Zn salt with the zeolite. Heemsoth et
al. [65] found that the catalyst prepared by a solid-state reaction of Zn dust and HZSM5
gave a similar activity for ethane aromatization as of the one where Zn was impregnated
onto HZSM5. Zn salt can also be added to the gel during the synthesis of the zeolite to
incorporate Zn into the zeolite framework [66]. Newer techniques of deposition are also
coming into practice such as CVD using dimethyl zinc, Zn(CH3)2 that allows for almost
stoichiometric substitution of Brønsted acid sites (BAS) with Zn2+ ions [67]. This leads to
a heterogeneous distribution of extra-framework Zn2+ species that is more active than
isolated Zn2+ species obtained during Ion Exchange (IE) or incipient wetness
impregnation (IWI). However, the catalysts prepared by the CVD technique were less
active in propane aromatization than the catalysts prepared with IE or IWI due to the
lesser abundance of active Zn2+ and multinuclear zinc oxide species [67].
Zn2+ species have proven to be an effective dehydrogenation site for light alkanes
including methane. Apart from Zn2+, other divalent Zn species that can activate light
alkanes include [Zn-O-Zn]2+ or small clusters of ZnO [68]. Arzumanov et al. [66] found a
similar mechanism of ethane activation using 13C MAS NMR in which dissociative
adsorption of ethane occurs on ZnO species to form Zn-ethyl species. A similar result
was obtained by Gabrienko et al. [69, 70] and Xu et al. [71] in which, the catalyst prepared
by partial substitution of Brønsted acid sites with Zn2+, could activate methane by forming
Zn-methyl species even at room temperature. Some of the proposed Zn sites as
suggested by Mehdad and Lobo [68] are depicted in Figure 3.10.

59
Figure 3.10 Various Zn2+ sites in ZSM5 and Silicalite. (a) Zn(II) stabilized by two
framework Al atoms, (b) Zn(OH)+ stabilized by one framework Al, (c) oxygen‐bridged Zn dimer, (d) (ZnO)n cluster. Reproduced from “Ethane and ethylene aromatization on zinc‐containing zeolites”, Catalysis Science and Technology, 7(16), 3562–3572, 2017, with permission of The Royal Society of Chemistry
The amount of zinc onto the zeolite support thus has a substantial influence on the
activity of the catalyst. This is due to the generation of Lewis acid sites when Zn is
introduced, and at the same time reduction in Brønsted acid sites of the zeolite. Lapidus
et al. [72] studied the effect of Zn content into the HZSM5 zeolite and found the optimal
loading of the Zn to be in between 3-5 wt%. Effect of Zn content on the ethane conversion
and aromatics selectivity as found by the same authors is shown in Table 3.3.
Table 3.3 Effect of Zn content on the ethane conversion and selectivity to aromatics [72] (T = 600 °C, Volume hourly space velocity = 450 h-1). Also, conversions and yields of
aromatics are per pass of ethane. Reproduced from “Zinc‐containing zeolite catalysts for ethane aromatization prepared by solid‐state modification”, Russian Chemical Bulletin International Edition, 52(5), 1094–1099, 2003, with permission of Springer
Zn content (wt %)
C2H6 conversion
(%)
Aromatics Yield (%)
Products composition (wt %)
C6 C7 C8 C9 C10-12
0 8.4 2.3 37.2 39.0 11.5 1.4 10.9
0.75 30.7 15.5 33.2 27.0 6.0 1.9 31.9
1.5 41.9 20.1 29.4 27.0 6.4 1.1 41.8
3.0 49.5 21.9 25.3 25.2 6.6 1.1 41.8
5.0 51.5 23.7 26.7 24.2 6.0 1.3 41.8
7.5 50.9 21.4 25.6 25.0 5.9 0.8 42.7

60
Similar results were observed by Mehdad and Lobo [68] who found out the optimal
concentration to be around 5 wt%. Above 5 wt% loading of Zn, blockage of pores due to
ZnO deposition was observed. They found that at low Zn content, ethylene was favored
instead of aromatics which was attributed to the presence of high density of Brønsted
acid sites. At high Zn content, concentration of Brønsted acid sites had decreased, and
the presence of generated Lewis acid sites would have favored aromatization instead of
ethylene. However, higher zinc content also favored CH4 formation. This is in contrast
with Anunziata et al. [66] observations with the ion exchanged Zn-ZSM-11, where an
increase in the Zn content decreased CH4 formation.
This might have been due to the difference in the support materials: ZSM-5 vs.
ZSM-11. Although they are quite similar, the channel system differs in that the ZSM5 has
crosslinked pores as compared to the later in which the pores are straight linked as could
be seen in Figure 3.11. Also, the shape selectivity and acidity on these two supports are
different leading to differences in activity.
Figure 3.11 Pore channel system for ZSM-5 (a) and ZSM-11 (b) materials. Reprinted from “Hierarchical ZSM-11 with intergrowth structures: Synthesis, characterization and catalytic properties”, Journal of Energy Chemistry, 22(5), 761-768, 2013, with permission from Elsevier
Although Zn doped ZSM5 catalyst is an active catalyst, one of the main challenges
with its use is the volatilization of Zn, which occurs under reducing reaction conditions

61
most likely due to low melting point of 420 °C (high vapor pressure). For stable and long
runs of ethane aromatization over Zn-ZSM-5, a constant source of Zn is required to
replenish its loss.
3.2.3.6 Promoters
Pb, Mo, and Fe have been used as promoters in Zn doped ZSM-11 catalysts [66].
It could be done through IE. Most of these promoters generate Lewis acid sites that
increase the ethane conversion as well as the selectivity to aromatics. Effect of promoters
on the concentration of Lewis and Brønsted acid sites as observed by Anunziata et al.
[66] is shown in Table 3.4. It could be seen that except for Fe, Mo and Pb doping
increased the concentration of Lewis acid sites.
Table 3.4 Effect of promoters on the concentration of acid sites [66] (pyridine based
FTIR, after desorption at 400 °C for 4 h), site concentration is in absorbance units
Zn-ZSM-11 (Si/Al = 17)
Zn-Pb-ZSM-11 (Si/Al = 17)
Zn-Mo-ZSM-11 (Si/Al = 17)
Zn-Fe-ZSM-11 (Si/Al = 17)
Brønsted sites 0.009 0.004 0.009 0.020
Lewis sites 0.030 0.033 0.044 0.029
W was also used as a promoter for Mo based ZSM5 catalysts[28] that improved
the overall stability of the catalyst and increased selectivity to ethane aromatization, and
was attributed to the formation Mo-W oxide, either bimetallic or closely interacted. Authors
further concluded that W might be providing an ensemble effect thus reducing the rate of
formation of coke and preventing the sintering of Mo active species.
Pt has also been used as a very effective promoter for Ga based ZSM-5 catalysts
to enhance its activity in the aromatization of lower alkanes. Also, the introduction of Pt
into Ga based zeolites considerably reduces the formation of byproducts methane and

62
ethylene [25]. It is believed that the dehydrogenation step gets accelerated in the
presence of Pt in Ga doped zeolites [59] through the reduction in the activation energy
for dehydrogenation and reduction in the hydrogen desorption energy too [26]. Further, it
is speculated that Pt forms bimetallic alloy particles with the Ga and yields active sites in
the zeolite framework [26, 59]. Although the presence of Pt does not help prevent coke
formation, it partially removes coke that occurs due to hydrogenolysis through hydrogen
spillover [40]. This results in less refractive coke as compared to Ga based ZSM-5 without
Pt.
Pd has also been tested as a promoter for Ga based ZSM-5 catalysts by Zaikovskii
et al. [73]. However, Pd did not promote the aromatizing activity of Ga-ZSM-5 significantly
because of poor dispersion of Pd over the catalyst. In contrast, the same study showed
higher activity for Pt promoted Ga-ZSM-5 catalyst due to uniform dispersion of Pt over
the catalyst. Similar results were obtained for Pt and Pd promoted Zn-ZSM-5 catalysts
[74] in which, Pt promoted Zn-ZSM-5 catalyst showed higher activity in ethane
aromatization as compared to Pd promoted Zn-ZSM-5 catalyst. Pd though can have an
effect on stabilization of Zn by alloying with it to suppress the volatilization of Zn [75].
3.2.4 Effect of pore structure (ZSM-5, ZSM-8, ZSM-11, ZSM-12)
Pore structure can be important for the aromatization reaction as it involves
complex mechanisms, thus nature of the support can have considerable effects on
reaction rates of different stages of the process. This results in considerably different
activities for ethane aromatization over these catalysts.
MFI or pentasil type zeolite, ZSM-5 is arguably the most active and the most
extensively studied material for creating bifunctional catalysts. A pentasil unit typically

63
consists of 8 five-membered rings. The ZSM-5 has interconnected 10-membered straight
and sinusoidal pores that result in three dimensional pore network. It is a medium pore
zeolite with pore dimensions of around 0.53 nm x 0.56 nm (straight pores) and 0.51 nm x
0.56 nm (sinusoidal pores) [13]. One advantage with the medium pore zeolites such as
ZSM-5 is that it can selectively perform conversions of light hydrocarbons into mono-
cyclic aromatics (formation of higher hydrocarbons is restricted) while minimizing coke
formation. This is not possible with the small pore zeolites (requires severe conditions) or
large pore zeolites (heavy aromatics production, faster deactivation) [76]. Some of the
zeolite structures that have been studied in ethane aromatization are depicted in Figure
3.12.
Figure 3.12 Various zeolite structures [77] (a) ZSM-5, (b) ZSM-11, (c) ZSM-12

64
Pore structure can be more important of a factor as compared to the acidity of the
catalysts as observed by Vosmerikova et al. [27] who studied ethane aromatization on Zn
modified zeolites of the type (ZSM-5, ZSM-8, ZSM-11, ZSM-12). Zn modified ZSM-12 had
the highest concentration of acid sites, both strong and weak but still, Zn modified ZSM-
5 catalyst showed highest levels of conversion and selectivity to aromatics. Catalysts with
the ZSM-8 and ZSM-11 pore structure showed lower activity overall in terms of ethane
conversion as well as selectivity to aromatics. ZSM-8 based catalyst differs from ZSM-5
only in terms of available surface area (lower than for ZSM-5) and pore volume (lower
than for ZSM-5) but these two factors seem to have a high effect on the overall
performance of the catalyst [27].
ZSM-8 based catalyst showed lower ethane conversions and low aromatic yields.
Similar results in terms of ethane conversion and aromatics selectivity were obtained on
ZSM-11 based catalyst. However, ZSM-11 based catalyst showed high selectivity
towards naphthalene that might have been due to slightly larger pore volume that allows
condensation of carbenium ion type intermediates and olefinic species that eventually
lead to higher hydrocarbons [78].
Despite stronger and higher distribution of acid sites, ZSM-12 based catalysts had
lowest aromatization activity as compared to dehydrogenation (~ 80 % selectivity).
Authors [27] observed C2-C4 alkenes on the ZSM-12 based catalyst and despite low
ethane conversions, it formed large amounts of coke, possibly due to polymerization of
intermediate compounds and slightly larger pore channels. It is possible that high acidity
might have favored side reactions such as cracking leading principally to ethylene. Zn
doped SSZ-13 or Chabazite type zeolite having very small pores has also been tried for

65
ethane aromatization [68] although it did not show any apparent activity. This inactivity
was not clearly attributed but was thought to be due to geometric differences. In the same
study, authors also studied Zn doped on Beta (BEA) zeolite but this catalyst was 2.5 times
slower than the Zn-ZSM-5 for the aromatization of ethane. The exact values of conversion
and selectivity to aromatics were not reported. Structures for Chabazite (SSZ-13) and
Beta type zeolites are shown in Figure 3.13.
Figure 3.13 Structures of zeolites [77]: (a) CHA (SSZ-13) and (b) BEA (Beta)
3.2.5 Effect of acidity (Si/Al ratio etc.)
Mehdad and Lobo [68] pointed out the importance of balance between the
concentration of Lewis acid sites (LAS) and Brønsted acid sites (BAS). Metal doping
introduces or increases Lewis acid sites depending on the type of dopant (for e.g. Zn)
that are useful in activation of ethane through dehydrogenation, whereas Brønsted acid
sites are useful for aromatization of olefinic intermediates. BAS can also play a role as an

66
ion exchange site for metal dopants. Based on their studies over ZSM-11 supports and
different metal dopants, Anunziata et al. [66] pointed out that high LAS produced high
concentrations of hydrogen acceptor species thus favoring dehydrogenation and
ultimately ethane conversion and aromatic hydrocarbon production. On the contrary, BAS
promoted cracking activity and C1 production.
Si/Al ratio also influences the conversion of ethane and selectivity to aromatic
hydrocarbons [38]. As the Si/Al ratio increases, conversion drops down as well as the
selectivity to aromatics. However, the dehydrogenation route is favored resulting in more
ethylene yield. This effect was observed in the case of Ga-Pt systems [41]. Aluminum
content in the catalyst is directly related to the strong Brønsted acid sites that are in turn
necessary for the oligomerization or cyclization of the olefinic species yielding aromatics.
Lapidus et al. [41] recommended optimal Si/Al ratio to be ≤ 30 for Ga-Pt based system
based on the ethane conversion data as shown in Table 3.5.
Table 3.5 Ethane aromatization on Pt-Ga pentasils having different SiO2 /Al 2O3 molar ratios [41] (T = 600 °C, volume hourly space velocity = 450 h–1). Reprinted from “Ethane aromatization on Ga-Pt pentasil zeolites”, Petroleum Chemistry, 48(2), 83-86, 2008, with permission from Springer
SiO2/Al2O3 mol.
Metal, wt% XEthane
(%) YAr (%) SAr(%)
Smethane (%)
Sethylene (%)
SC3-C4
(%) Ga Pt
30 0.5 0.3 47.1 30.3 64.3 19.5 7.0 -
56 0.5 0.3 39.9 24.0 60.2 23.3 6.5 -
90 0.5 0.3 27.2 14.8 54.5 11.1 19.5 4.1
30 2.0 0.3 47.6 30.1 63.2 20.1 7.8 -
56 2.0 0.3 24.7 12.7 51.4 7.7 21.5 8.9
90 2.0 0.3 17.0 4.2 24.8 3.5 53.9 7.6

67
In another study, Lapidus et al. [72] studied 5% Zn loaded onto ZSM-5 with
different Si/Al ratio only to obtain similar results in which, highest conversion of ethane
and the highest yield of aromatics was obtained on the catalyst with Si/Al ratio equal to
30. Mehdad and Lobo [68] on the other hand, studied 5% Zn on ZSM-5 catalyst with Si/Al
ratio below 30. They found that as the ratio increased from 11.5 to 25, the rate of ethane
conversion as well as the rate of aromatization, dropped down. However, rate of
deactivation also had decreased (less aromatization).
Acidity or the Si/Al ratio, in some cases, can also influence the bifunctionality of
the catalyst. Ausavasukhi and Sooknoi [62] observed that Si/Al ratio influenced the
dispersion of Ga species in the zeolite structure thereby affecting the overall
dehydrogenation activity. This was due to less availability of acid sites for exchange with
Ga species in the case of higher Si/Al ratio zeolites.
3.3 Future perspective
The main challenges associated with the conversion of natural gas are rapid
catalyst deactivation through coking, loss in acid sites, and also in some catalysts loss of
active dopant metal such as Zn through volatilization. All these factors demand constant
regeneration of the catalysts as well as a constant source of replenishment of the active
dopant metal. These processes can be expensive but are often employed on industrial
scales for other light alkanes (e.g., propane based Cyclar process). Approaches to reduce
the formation of coke include the addition of a promoter that lowers the rate of formation
of coke or by addition of an oxidant (CO, CO2, O2, NOx). Other options to improve process
performance include shifting of the thermodynamic equilibrium with the use of
permselective membrane that can selectively remove produced H2 to drive the reaction

68
forward. H2 absorbing materials such as Zr2Fe can also be used as pointed out in the
earlier reviews [14].
Palladium membrane has been tested for propane aromatization by some
research groups [79] and it was found that the activity increased several times by
extracting hydrogen from the reaction products. Other membranes reported in the
literature include ceramic materials (La5.5W0.6Mo0.4O11.25-δ)[80]. However, in these
hydrogen selective membranes, along with an increase in the rate of aromatization, the
rate of coke formation also increases [81], causing a rapid deactivation of the catalyst.
Introduction of an oxidant like oxygen has been shown to inhibit coke production without
affecting the selectivity to benzene. Thus, the membranes that can be employed should
be of multifunctional nature i.e. it should selectively be able to remove H2 from the reaction
and at the same time, provide O2 to remove coke formation from the system. These
membranes can be multifunctional or two separate membranes for O2 and H2 can be
used (from inside and outside of the reaction zone). A typical schematic for a
multifunctional membrane (separate membranes for O2 and H2) system is depicted in
Figure 3.14.
Very recently, a similar multifunctional membrane reactor model was proposed and
optimized by Fouty et al. [82]. Variety of factors determine the overall performance of this
multifunctional membrane reactor system. It was determined that for high performance of
this system: membrane should have a high permeation rate (0.01 mol/s·m2·atm1/4) and
high selectivity to H2 (> 105). Other factors that will also affect the performance included
selectivity to O2 (slow permeation), reaction temperatures, feed conditions, reactor
dimensions, and membrane thicknesses.

69
Figure 3.14 Schematic for a multifunctional membrane reactor
Practical application of a multifunctional BaZrO3 membrane in the methane
aromatization reaction was recently shown by Morejudo et al. [81] This membrane was
electrochemically able to remove H2 while, at the same time, provide O2 to remove carbon
deposits. This resulted in high aromatics yield as well as high stability of the catalysts.
Membranes like these could also be employed for the conversion of ethane or even for
the conversion of a mixed feed: methane and ethane or directly natural gas. Ethane being
easier to activate, a high yield, low temperature process might be feasible.
Addition of oxidants such as CO2 has led to an increase in the aromatization
activity in terms of both the conversion as well as the aromatics yield [83] while at the
same time suppressing ethylene yield. In the same study, authors also observed that the
addition of steam enhanced the dehydrogenation activity of Ga promoted catalysts. This
positive effect was attributed to the additional reaction of steam and CO2 with the coke
on the catalyst surface. CO2 reacts with desorbed H2 to undergo reverse water gas shift

70
reaction as CO and H2O were observed in the reaction products along with aromatics.
(eq. 3.6):
𝐶𝑂2 + 𝐻2 ↔ 𝐶𝑂 +𝐻2𝑂 (3.6)
The other reason is the Boudouard reaction (eq. 3.7), which removes deposited
coke.
𝐶𝑂2 + 𝐶 ↔ 2 𝐶𝑂 (3.7)
Although some activity is observed due to CO2 addition, it could also be due to
steam generated from the reverse water gas shift reaction as pointed out by Nakagawa
et al. [83]. On the contrary, Nishi et al. [84] had proposed that the improvement in the
activity of the catalyst under the presence of CO2 was due to the kinetic effect instead of
the thermodynamic effect. Also, the authors thought that the increase in activity was due
to an increase in acid sites in the presence of CO2 that can interact with zeolite lattice and
form Brønsted acid sites.
As pointed out in an earlier review [14], the addition of an oxidant leads to
additional side reactions such as the formation of CO and H2 like a syngas mixture along
with methane and other cracking products. Controlling these reactions then becomes
important. The ideal catalyst should have high activity in the aromatization of ethane with
low selectivity for side reactions. This will ensure high yield to desired aromatics under
the presence of an oxidant. Steam, when added as a co-reactant, enhances the
conversion of ethane in the dehydrogenation reaction as observed by Nakagawa et al.
[83] but at the same time, selectivity to CH4 also had increased.
Zn doped ZSM-5 catalysts have shown high activity in ethane aromatization over
the years. As discussed before, the primary issue with these catalysts is the volatilization

71
of Zn because of the high vapor pressure of Zn at reaction temperatures. There have
been numerous attempts to minimize Zn volatilization. Earlier attempts focused on
reducing Zn vapor pressure by alloying it with expensive metals such as Pd [75], Au [75],
Ag [75] or some attempts focused on Ga [85]. These attempts resulted in the reduction
of the volatilization rate of Zn, but improvements were not significant. In another
subsequent patent, authors [86] proclaimed a novel method of addition of either
nonmetallic oxides such as CO2, CO, NOx, Steam, etc. or of nonmetallic sulfur compound
such as H2S. These seem to have improved the rate of loss of Zn considerably, if not
completely.
H2S or some of the oxidants like CO2 are already found as constituents of natural
gas. Therefore, if an alloyed Zn on HZSM5 catalyst is used for directly converting natural
gas to aromatics, this may provide a viable pathway ahead. This may accomplish a couple
of things:
1. Promoted or alloyed Zn on HZSM5 has high activity in the aromatization reaction
and at the same time, volatilization of the Zn would be suppressed.
2. If natural gas is directly used or a combined feed of lower alkanes along with
CO2 and H2S, volatilization of Zn also may get suppressed.
3. Presence of an oxidant will reduce coke formation and also will enhance the
overall activity of the catalyst.
Other approaches for minimizing zinc volatilization could include a regeneration unit
wherein Zn or ZnO could be re-supplied to the spent catalyst through solid state ion
exchange [14]. Catalysts prepared by these methods are effective and this type of

72
synthesis could be done in-situ by adding the appropriate amounts of Zn or ZnO along
with heat treatment.
Although the scientific advancements in terms of membrane technologies, other
separation technologies and even for catalyst development have been impressive,
significant progress is needed before the ethane aromatization process is put into
commercial practice. Ethane is an important commodity for the production of ethylene
and other valuable chemicals. The range of products that can be produced from ethane
can further be exploited if aromatization of ethane becomes commercially feasible. This
would allow efficient use of natural gas, much of which is flared for lack of a practical,
competitive process. A strong research emphasis from the scientific community should
be given to the ethane aromatization. Methane aromatization is the next grand challenge,
but ethane should be given equal attention because it is much easier to activate than
methane.
3.4 References
[1] S. Jenner, A.J. Lamadrid, Shale gas vs. coal: Policy implications from environmental impact comparisons of shale gas, conventional gas, and coal on air, water, and land in the United States, Energy Policy, 53 (2013) 442-453. [2] Q. Wang, X. Chen, A.N. Jha, H. Rogers, Natural gas from shale formation – The evolution, evidences and challenges of shale gas revolution in United States, Renewable and Sustainable Energy Reviews, 30 (2014) 1-28. [3] T. Boersma, C. Johnson, The Shale Gas Revolution: U.S. and EU Policy and Research Agendas, Review of Policy Research, 29 (2012) 570-576. [4] K. Aruga, The U.S. shale gas revolution and its effect on international gas markets, Journal of Unconventional Oil and Gas Resources, 14 (2016) 1-5. [5] BP, Natural gas prices, Available from: https://www.bp.com/en/global/corporate/energyeconomics/ statistical-review-of-world-energy/natural-gas/natural-gas-prices.html.

73
[6] R.S. Middleton, R. Gupta, J.D. Hyman, H.S. Viswanathan, The shale gas revolution: Barriers, sustainability, and emerging opportunities, Applied Energy, 199 (2017) 88-95. [7] PLOS ONE, Available from: http://journals.plos.org/plosone/article/asset?unique&id=info:doi/10.1371/journal.pone .0144141.s001. [8] J.J. Spivey, G. Hutchings, Catalytic aromatization of methane, Chemical Society Reviews, 43 (2014) 792-803. [9] W. Taifan, J. Baltrusaitis, CH4 conversion to value added products: Potential, limitations and extensions of a single step heterogeneous catalysis, Applied Catalysis B: Environmental, 198 (2016) 525-547. [10] C. Karakaya, R.J. Kee, Progress in the direct catalytic conversion of methane to fuels and chemicals, Progress in Energy and Combustion Science, 55 (2016) 60-97. [11] T.V. Choudhary, E. Aksoylu, D.W. Goodman, Nonoxidative activation of methane, Catalysis Reviews-Science and Engineering, 45 (2003) 151-203. [12] US EIA, 2015. Available from: https://www.eia.gov/energyexplained/index.cfm?page=hgls_prices#tab1.
[13] A. Bhan, W. Nicholas Delgass, Propane Aromatization over HZSM‐5 and Ga/HZSM‐5 Catalysts, Catalysis Reviews, 50 (2008) 19-151. [14] A. Hagen, F. Roessner, Ethane to Aromatic Hydrocarbons: Past, Present, Future, Catalysis Reviews, 42 (2000) 403-437. [15] Y. Ono, Transformation of Lower Alkanes into Aromatic Hydrocarbons over ZSM-5 Zeolites, Catalysis Reviews, 34 (1992) 179-226. [16] M.A. Bañares, Supported metal oxide and other catalysts for ethane conversion: a review, Catalysis Today, 51 (1999) 319-348. [17] S.M. Csicsery, Dehydrocyclodimerization: III. Dehydrocyclodimerization of butanes over transition metal oxide catalysts, Journal of Catalysis, 17 (1970) 315-322. [18] Y. Cheng, H. Gong, C. Miao, W. Hua, Y. Yue, Z. Gao, Ga2O3/HSSZ-13 for dehydrogenation of ethane: Effect of pore geometry of support, Catalysis Communications, 71 (2015) 42-45. [19] S.M. Csicsery, Dehydrocyclodimerization: II. Dehydrocyclodimerization of propane and pentane over supported platinum catalyst, Journal of Catalysis, 17 (1970) 216-218.

74
[20] S.M. Csicsery, Dehydrocyclodimerization: I. Dehydrocyclodimerization of butanes over supported platinum catalysts, Journal of Catalysis, 17 (1970) 207-215. [21] S.M. Csicsery, Dehydrocyclodimerization: V. The mechanism of the reaction, Journal of Catalysis, 18 (1970) 30-32. [22] S.M. Csicsery, Dehydrocyclodimerization: IV. The reactions of butenes, Journal of Catalysis, 17 (1970) 323-330. [23] T.M. Inui, Akihiko, Selective aromatization of light hydrocarbons on polyfunctional metallosilicate catalysts, in: ACS Fall National Meeting, American Chemical Society, New York, NY, 1991. [24] A. Krogh, A. Hagen, T.W. Hansen, C. Hviid Christensen, I. Schmidt, Re/HZSM-5: a new catalyst for ethane aromatization with improved stability, Catal. Commun., 4 (2003) 627-630. [25] A.L. Lapidus, A.L. Lapidus, M.N. Mikhailov, M.N. Mikhailov, A.A. Dergachev, A.A. Dergachev, G.M. Zhidomirov, G.M. Zhidomirov, I.V. Mishin, I.V. Mishin, The nature of active sites in Pt promoted GaZSM-5 catalysts, React. Kinet. Catal. Lett., 87 (2006) 249-254. [26] A.L. Lapidus, M.N. Mikhailov, A.A. Dergachev, I.V. Mishin, Structure of active sites of Ga-Pt zeolite catalysts of alkane aromatization, Dokl. Phys. Chem., 408 (2006) 175-177. [27] L. Vosmerikova, Y. Barbashin, A. Vosmerikov, Catalytic aromatization of ethane on zinc-modified zeolites of various framework types, Petroleum Chemistry, 54 (2014) 420-425. [28] S.-T. Wong, Y. Xu, L. Wang, S. Liu, G. Li, M. Xie, X. Guo, Methane and ethane activation without adding oxygen: promotional effect of W in Mo-W/HZSM-5, Catalysis Letters, 38 (1996) 39-43. [29] O.V. Bragin, T.V. Vasina, A.V. Preobrazhenskii, Catalytic aromatization of ethylene and ethane, Bulletin of the Academy of Sciences of the USSR, Division of chemical science, 33 (1984) 56-63. [30] T.F. Degnan, G.K. Chitnis, P.H. Schipper, History of ZSM-5 fluid catalytic cracking additive development at Mobil, Microporous and Mesoporous Materials, 35 (2000) 245-252. [31] N.Y. Chen, T.Y. Yan, M2 forming - a process for aromatization of light hydrocarbons, Industrial & Engineering Chemistry Process Design and Development, 25 (1986) 151-155.

75
[32] C. Marcilly, Acido-Basic Catalysis. Volume 2. Application to Refining and Petrochemistry. Editions Technip, 2013. [33] G.A. Olah, Á. Molnár, Hydrocarbons from Petroleum and Natural Gas, in: Hydrocarbon Chemistry, John Wiley & Sons, Inc., 2003, pp. 30-84. [34] F. Yuzo, T. Ken, W. Jun, U. Yutaka, T. Hiroshi, Thermal Activation of Alkane C–H Bonds by Palladium Catalysts. Carbonylation of Alkanes with Carbon Monoxide, Chemistry Letters, 18 (1989) 1687-1688. [35] N. Syed, Process Economics Program - Aromatics from Light Hydrocarbons in, IHS, 2014. [36] Chevron Phillips Process. Available from: http://www.cpchem.com/en-us/rnt/licensing/aromaxtech/pages/process.aspx. [37] T. Foley, CYCLAR™ Process Produces High-Quality Aromatic Products, Available from: https://www.uop.com/cyclar-process-produces-high-quality-aromatic-products/. [38] F. Solymosi, P. Tolmacsov, Conversion of Ethane into Benzene on Re/ZSM-5, Catal. Lett., 93 (2004) 7-11. [39] O.V. Chetina, T.V. Vasina, V.V. Lunin, Aromatization of ethane over Pt,Ga/HZSM-5 catalyst and the effect of intermetallic hydrogen acceptor on the reaction, Applied Catalysis A: General, 131 (1995) 7-14. [40] A. Samanta, X. Bai, B. Robinson, H. Chen, J. Hu, Conversion of Light Alkane to Value-Added Chemicals over ZSM-5/Metal Promoted Catalysts, Industrial & Engineering Chemistry Research, (2017). [41] A.L. Lapidus, A.A. Dergachev, V.A. Kostina, A.A. Silakova, Ethane aromatization on Ga-Pt pentasil zeolites, Neftekhimiya, 48 (2008) 83-86. [42] Y. Wu, L. Emdadi, Z. Wang, W. Fan, D. Liu, Textural and catalytic properties of Mo loaded hierarchical meso-/microporous lamellar MFI and MWW zeolites for direct methane conversion, Applied Catalysis A: General, 470 (2014) 344-354. [43] E.V. Matus, O.B. Sukhova, I.Z. Ismagilov, L.T. Tsikoza, Z.R. Ismagilov, Peculiarities of dehydroaromatization of CH4-C2H6 and CH4 over Mo/ZSM-5 catalysts, React. Kinet. Catal. Lett., 98 (2009) 59-67. [44] F. Solymosi, A. Szo˝ke, Conversion of ethane into benzene on Mo2C/ZSM-5 catalyst, Applied Catalysis A: General, 166 (1998) 225-235. [45] T. Osawa, I. Nakano, O. Takayasu, Dehydrogenation of Methane over Mo/ZSM-5. Effects of Additives in the Methane Stream, Catal. Lett., 86 (2003) 57-62.

76
[46] W. Ding, S. Li, G. D Meitzner, E. Iglesia, Methane Conversion to Aromatics on Mo/H-ZSM5: Structure of Molybdenum Species in Working Catalysts, The Journal of Physical Chemistry B, 105 (2001) 506-513. [47] L.M. Petkovic, D.M. Ginosar, Comparison of Two Preparation Methods on Catalytic Activity and Selectivity of Ru-Mo/HZSM5 for Methane Dehydroaromatization, Journal of Fuels, 2014 (2014) 7. [48] G.N. Folefoc, J. Dwyer, Dispersion of platinum in Pt/ZSM-5 zeolites, Journal of Catalysis, 136 (1992) 43-49. [49] C. Yu, Q. Ge, H. Xu, W. Li, Propane dehydrogenation to propylene over Pt-based catalysts, Catalysis Letters, 112 (2006) 197-201. [50] G. Liu, L. Zeng, Z.-J. Zhao, H. Tian, T. Wu, J. Gong, Platinum-Modified ZnO/Al2O3 for Propane Dehydrogenation: Minimized Platinum Usage and Improved Catalytic Stability, ACS Catalysis, 6 (2016) 2158-2162. [51] O.P. Keipert, D. Wolf, P. Schulz, M. Baerns, Kinetics of ethane aromatization over a gallium-doped H-ZSM-5 catalyst, Applied Catalysis A: General, 131 (1995) 347-365. [52] Z. Li, A.W. Lepore, M.F. Salazar, G.S. Foo, B.H. Davison, Z. Wu, C.K. Narula, Selective conversion of bio-derived ethanol to renewable BTX over Ga-ZSM-5, Green Chemistry, 19 (2017) 4344-4352. [53] B.S. Kwak, W.M.H. Sachtler, Characterization and Testing of Ga/HZSM-5 Prepared by Sublimation of GaCl3 into HZSM-5, Journal of Catalysis, 141 (1993) 729-732. [54] B.S. Kwak, W.M.H. Sachtler, W.O. Haag, Catalytic Conversion of Propane to Aromatics: Effects of Adding Ga and/or Pt to HZSM-5, Journal of Catalysis, 149 (1994) 465-473. [55] G.L. Price, V. Kanazirev, Ga2O3/HZSM-5 propane aromatization catalysts: Formation of active centers via solid-state reaction, Journal of Catalysis, 126 (1990) 267-278. [56] N.S. Gnep, J.Y. Doyemet, A.M. Seco, F.R. Ribeiro, M. Guisnet, Conversion of light alkanes to aromatic hydrocarbons: II. Role of gallium species in propane transformation on GaZSM5 catalysts, Applied Catalysis, 43 (1988) 155-166. [57] N.S. Gnep, J.Y. Doyemet, M. Guisnet, Conversion of Light Alkanes Into Aromatic Hydrocarbons. 3. Aromatization of Propane and Propene on Mixtures of HZSM5 and of Ga2O3, in: H.G. Karge, J. Weitkamp (Eds.) Studies in Surface Science and Catalysis, Elsevier, 1989, pp. 153-162.

77
[58] Y. Ono, H. Nakatani, H. Kitagawa, E. Suzuki, The Role of Metal Cations in the Transformation of Lower Alkanes into Aromatic Hydrocarbons, in: T. Inui (Ed.) Studies in Surface Science and Catalysis, Elsevier, 1989, pp. 279-290. [59] M.N. Mikhailov, I.V. Mishin, L.M. Kustov, A.L. Lapidus, Structure and reactivity of Pt/GaZSM-5 aromatization catalyst, Microporous Mesoporous Mater., 104 (2007) 145-150. [60] V.I. Yakerson, T.V. Vasina, L.I. Lafer, V.P. Sytnyk, G.L. Dykh, A.V. Mokhov, O.V. Bragin, K.M. Minachev, The properties of zinc and gallium containing pentasils — The catalysts for the aromatization of lower alkanes, Catalysis Letters, 3 (1989) 339-345. [61] K.M. Dooley, G.L. Price, V.I. Kanazirev, V.I. Hart, Gallium-loaded zeolites for light paraffin aromatization: evidence for exchanged gallium cation active centers, Catalysis Today, 31 (1996) 305-315. [62] A. Ausavasukhi, T. Sooknoi, Tunable activity of [Ga]HZSM-5 with H2 treatment: Ethane dehydrogenation, Catalysis Communications, 45 (2014) 63-68.
[63] L. Wang, R. Ohnishi, M. Ichikawa, Novel rhenium‐based catalysts for dehydrocondensation of methane with CO/CO2 towards ethylene and benzene, Catalysis Letters, 62 (1999) 29-33. [64] H.S. Lacheen, P.J. Cordeiro, E. Iglesia, Isolation of Rhenium and ReOx Species within ZSM5 Channels and their Catalytic Function in the Activation of Alkanes and Alkanols, Chemistry – A European Journal, 13 (2007) 3048-3057. [65] J. Heemsoth, E. Tegeler, F. Roessner, A. Hagen, Generation of active sites for ethane aromatization in ZSM-5 zeolites by a solid-state reaction of zinc metal with Brønsted acid sites of the zeolite, Microporous and Mesoporous Materials, 46 (2001) 185-190. [66] O.A. Anunziata, G.A. Eimer, L.B. Pierella, Catalytic Activity of ZSM-11 Zeolites Modified with Metal Cations for the Ethane Conversion, Catalysis Letters, 75 (2001) 93-97. [67] S.M.T. Almutairi, B. Mezari, P.C.M.M. Magusin, E.A. Pidko, E.J.M. Hensen, Structure and Reactivity of Zn-Modified ZSM-5 Zeolites: The Importance of Clustered Cationic Zn Complexes, ACS Catalysis, 2 (2012) 71-83. [68] A. Mehdad, R.F. Lobo, Ethane and ethylene aromatization on zinc-containing zeolites, Catalysis Science & Technology, 7 (2017) 3562-3572. [69] A.A. Gabrienko, S.S. Arzumanov, A.V. Toktarev, I.G. Danilova, I.P. Prosvirin, V.V. Kriventsov, V.I. Zaikovskii, D. Freude, A.G. Stepanov, Different Efficiency of Zn2+ and

78
ZnO Species for Methane Activation on Zn-Modified Zeolite, ACS Catalysis, 7 (2017) 1818-1830. [70] A.A. Gabrienko, S.S. Arzumanov, M.V. Luzgin, A.G. Stepanov, V.N. Parmon, Methane Activation on Zn2+-Exchanged ZSM-5 Zeolites. The Effect of Molecular Oxygen Addition, The Journal of Physical Chemistry C, 119 (2015) 24910-24918. [71] J. Xu, A. Zheng, X. Wang, G. Qi, J. Su, J. Du, Z. Gan, J. Wu, W. Wang, F. Deng, Room temperature activation of methane over Zn modified H-ZSM-5 zeolites: Insight from solid-state NMR and theoretical calculations, Chemical Science, 3 (2012) 2932-2940. [72] A.L. Lapidus, A.A. Dergachev, V.A. Kostina, I.V. Mishin, Zinc-containing zeolite catalysts for ethane aromatization prepared by solid-state modification, Russ. Chem. Bull., 52 (2003) 1094-1099. [73] V.I. Zaikovskii, L.N. Vosmerikova, A.V. Vosmerikov, Ethane aromatization on galloaluminosilicate modified with platinum and palladium, Kinet. Catal., 53 (2012) 731-736. [74] U. Mroczek, W. Reschetilowski, K. Pietzsch, K.H. Steinberg, Aromatization of ethane on Pt and Pd supported Zn- and Mn-ZSM-5 zeolites, Reaction Kinetics and Catalysis Letters, 43 (1991) 539-544. [75] W.O. Haag, T.J. Huang, Conversion of olefinic naphtha, in, Google Patents, 1978. https://patents.google.com/patent/US4097367A/ [76] C.H. Bartholomew, R.J. Farrauto, Fundamentals of industrial catalytic processes. [electronic resource], Hoboken, N.J. : Wiley, c2006. 2nd ed., 2006. [77] Ch. Baerlocher and L.B. McCusker, Database of Zeolite Structures:, in, 2017.
Available from: http://www.izastructure.org/databases/. [78] E.G. Derouane, P. Dejaifve, Z. Gabelica, J.C. Vedrine, Molecular shape selectivity of ZSM-5, modified ZSM-5 and ZSM-11 type zeolites, Faraday Discussions of the Chemical Society, 72 (1981) 331-344. [79] S. Natesakhawat, N.C. Means, B.H. Howard, M. Smith, V. Abdelsayed, J.P. Baltrus, Y. Cheng, J.W. Lekse, D. Link, B.D. Morreale, Improved benzene production from methane dehydroaromatization over Mo/HZSM-5 catalysts via hydrogen-permselective palladium membrane reactors, Catalysis Science & Technology, 5 (2015) 5023-5036. [80] J. Xue, Y. Chen, Y. Wei, A. Feldhoff, H. Wang, J. Caro, Gas to Liquids: Natural Gas Conversion to Aromatic Fuels and Chemicals in a Hydrogen-Permeable Ceramic Hollow Fiber Membrane Reactor, ACS Catalysis, 6 (2016) 2448-2451.

79
[81] S.H. Morejudo, R. Zanón, S. Escolástico, I. Yuste-Tirados, H. Malerød-Fjeld, P.K. Vestre, W.G. Coors, A. Martínez, T. Norby, J.M. Serra, C. Kjølseth, Direct conversion of methane to aromatics in a catalytic co-ionic membrane reactor, Science, 353 (2016) 563-566. [82] N. Fouty, J. Carrasco, F. Lima, Modeling and Design Optimization of Multifunctional Membrane Reactors for Direct Methane Aromatization, Membranes, 7 (2017) 48. [83] K. Nakagawa, C. Kajita, Y. Ide, M. Okamura, S. Kato, H. Kasuya, N.o. Ikenaga, T. Kobayashi, T. Suzuki, Promoting effect of carbon dioxide on the dehydrogenation and
aromatization of ethane over gallium‐loaded catalysts, Catalysis Letters, 64 (2000) 215-221. [84] K.E. Nishi, Masato; Satsuma, Atsushi; Hattori, Tadashi; Murakami, Yuichi:, Effect of Carbon Dioxide on Aromatization of Ethane over Metal-loaded HZSM-5 Catalysts, Sekiyu Gakkaishi, 39 (1996) 260-266. [85] Y.F. Chu, A.W. Chester, Process for converting propane to aromatics over zinc-gallium zeolite, in, Google Patents, 1984. Available from: https://www.google.com/patents/US4490569. [86] S.B. McCullen, P.G. Rodewald, Stabilization of zinc on catalysts, in, Google Patents, 1989. Available from: https:/www.google.com/patents/US4849568.

80
Chapter 4 . Low Temperature Direct Conversion of Methane using a Solid Superacid2
4.1 Introduction
Methane conversion to higher-value products has been a topic of research for
decades, but the “shale gas revolution” has made low-cost natural gas readily available.
This has presented a unique opportunity to take advantage of the immense, previously
inaccessible reserves of natural gas. This has already had major impacts on the energy
and chemical industries [1-3].
Methane is a thermodynamically stable, non-polar molecule containing strong,
equivalent (C-H) bonds (434 kJ/mol) [4]. Activation of methane typically requires very high
temperatures and oxidants like O2 (partial oxidation/oxidative coupling) [5], CO2/H2O
(reforming) [6], S2 (sulfidation) [7], or Br2 (bromination) [8]. These reactions produce
intermediates like syngas or CH3OH/CH3SH/CH3Br, which are subsequently converted
into hydrocarbons or oxygenates. In these reactions, methane activation at elevated
temperatures leads to another challenge: complete dehydrogenation of methane into
elemental carbon or coke.
Direct (non-oxidative) conversion of methane is an alternative to processes that
require intermediates such as syngas. Catalysts involving direct conversion include
lattice-confined single Fe sites [9] or modified-zeolites [10-13] at very high temperatures
(700-1100 °C). One particularly promising approach for the direct conversion of methane
at lower temperatures is based on superacids (Figure 4.1).
2 Reprinted from S. Kanitkar, J. Carter, G. Hutchings, K. Ding, & J. J. Spivey (2018) Low temperature direct conversion of methane using a solid superacid. ChemCatChem, 10, 5019-5024. With permission from John Wiley & Sons.

81
Figure 4.1 Current and proposed process for converting methane into olefins and fuels. At present, methane is reformed into syngas and then transformed into higher hydrocarbons through the Fischer-Tropsch synthesis. This indirect process requires high temperatures and pressures. Methane oligomerization using solid superacids offers a direct route to converting methane into higher hydrocarbons under mild conditions
These catalysts were first demonstrated by Olah [14], using liquid “magic acid”
(FSO3H-SbF5). Other superacids have been reported for the oligomerization of methane:
(SbF5-HF) [15], (HBr-AlBr3) [16, 17], and sulfated zirconia [18, 19].
HBr-AlBr3 is a particularly promising gas-phase superacid catalyst. This is a proven
strong superacid, and has been compared to the strongest known superacid, “magic acid”
[20]. Recently [17], this gas-phase superacid has been shown to convert CH4 into higher
hydrocarbons (C2 – C26) and H2 at temperatures as low as 200-400 °C. Although CH4
conversions were relatively high, separating the catalyst from the gaseous products was
difficult, making it practically impossible to carry out a carbon balance, or to envision a

82
practical process based on a gas-phase superacid. This could be addressed by
incorporating the proven gaseous HBr-AlBr3 superacid onto a solid support.
Any process based on direct conversion of methane using superacids at the
temperatures reported here (200-400 °C) must address the equilibria limitations. At the
conditions of interest here, equilibrium methane conversion is ~ 0.5 to 12 % (with coke
formation allowed) depending on the product composition, including hydrogen,
alkanes/alkenes, polynuclear aromatics, among others. It is outside the scope of our
study here to determine if a feasible process based on superacid catalysis of methane
oligomerization is practical. However, we recognize that a solid superacid could be an
essential element of a practical process.
We are aware of no report of a solid superacid based on bromine for methane
oligomerization, despite the potential significance of an active solid superacid based on
the gas-phase HBr/AlBr3 catalyst sites. Our study here describes the synthesis,
characterization and direct conversion of methane to higher hydrocarbons using a solid
superacid, AlBrx/H-ZSM-5 (“ABZ-5”, x = 1 or 2). Though the oligomerization of methane
is the focus here, there are clear significant opportunities in related acid-catalyzed
processes, e.g. alkylations and acylations [21-23]; alkane isomerization and cracking [23-
25]; and polymerization [23].
4.2 Experimental section
4.2.1 Materials
AlBr3 (anhydrous, 98%) and NH4-ZSM-5 (SiO2/Al2O3 = 50:1) were purchased from
Alfa Aesar. SiO2 gel was purchased from PQ Corporation. Zr(OH)4 (97%) was purchased
from Sigma Aldrich Inc. and H2SO4 (95–98%) was purchased from Malinckrodt Chemicals

83
Inc. Silver Nitrate (AgNO3), 0.0141 N solution was purchased as is from Macron Fine
Chemicals. Pyridine (50 ppm) in gas phase (balance Argon) was purchased from Praxair
Inc. 10% NH3/He (ultra-high purity), 10% CH4/Ar (ultra-high purity) were purchased from
Airgas Inc.
4.2.2 Catalyst preparation
ABZ-5 was prepared as follows: H-ZSM-5 was prepared by calcining NH4-ZSM-5
at 500 °C for 3 h in a muffle furnace. In order to optimize the -OH concentration on the
surface of the as-prepared H-ZSM-5, it was washed in three alternate cycles of HBr (1 M)
and deionized water and then dried in a vacuum at 80 °C for 72 h before being exposed
to the room atmosphere [25]. Then, in separate vials, AlBr3 (1 g) and the washed H-ZSM-
5 (1 g) were loaded into a Teflon lined SS autoclave (Parr Instruments Inc., Moline, IL)
inside a glovebox. The autoclave was then sealed and put inside an isothermal oven at
180 °C for 72 h in order for AlBr3 to vaporize and react with the hydroxyl groups on the
surface of the zeolite. The excess pressure generated during the synthesis (HBr
formation) was released by opening the autoclave in a fume hood. The autoclave was
then returned to the glovebox where the catalyst was recovered. Masses of all the
materials were recorded in order to check for the weight gain by the H-ZSM-5 sample.
ABSi catalyst was also prepared using the same methodology but substituting the H-
ZSM-5 for the SiO2 gel.
Sulfated zirconia (SZ) was prepared using a previously reported literature
procedure [26]. Briefly, Zr(OH)4 gel was washed with H2SO4 (0.5 M) solution and the
washed product was dried at 110 °C overnight in an isothermal oven. Then the material
was subsequently heated to 550 °C at a ramp rate of 5 °C/min and calcined at this

84
temperature for 4 h under static air. The solid sulfated zirconia powder obtained was then
stored for further use. Formation of sulfated zirconia was confirmed by XRD on the
prepared sample, which showed a diffraction pattern similar to the one previously
reported [26].
4.2.3 Ammonia-TPD
Ammonia-TPD was carried out using AMI-200 reactor system (Altamira
Instruments Inc., Pittsburgh, PA) in conjunction with an Ametek LC-D Mass Spectrometer.
Typically, 25 mg of catalyst was weighed and loaded in a quartz tube reactor. The catalyst
was pretreated at 100 °C for 30 min under He flow to clean the catalyst surface. After
pretreatment, the sample was cooled down to 50 °C, and ammonia was adsorbed by
flowing it through the catalyst bed for 1 h. After ammonia adsorption, 25 ml min-1 of He
was flowed for 40 min to remove any physisorbed/residual ammonia. Then, the Mass
Spectrometer and TCD detectors were turned on and the temperature was ramped up at
10 °C min-1 from 50 to 500 °C. Masses that were analyzed included 16 (NH3), 17 (NH3),
18 (H2O), 81 (HBr).
4.2.4 Pyridine-DRIFTS
DRIFTS experiments using pyridine as a probe molecule were carried out using
Thermo Scientific Nicolet 6700 FTIR equipped with Harrick Praying Mantis reaction cell
fitted with KBr windows. Spectra of all the samples were recorded with a spectral
resolution of 4 cm-1 in region going from 4000-650 cm-1. In a typical experiment, the IR
cell was loaded with the catalyst sample inside the glovebox. The sample was then
transferred to the spectrometer and pretreated at 100 °C for 30 min. under He flow (30
sccm) to clean surface of the catalyst. After pretreatment, the sample was cooled down

85
to 25 °C and a background spectrum was recorded. At this temperature, catalyst was
saturated with pyridine vapors for 45 min. The saturated sample was then treated under
He flow for 30 min. Sample was then treated at 100 °C for 10 min and cooled to room
temperature and the actual spectrum was recorded. Similar spectra were recorded at
room temperature after 10 min. treatments were carried out at 200 °C, 300 °C, and 400
°C to check the thermal stability of the acid sites on catalysts.
FTIR of hydroxyl region was carried out in the same aforementioned DRIFTS cell
under Helium flow and at various temperatures. Some IR catalyst samples were also
recorded using Bruker Alpha IR spectrometer using Diamond ATR crystal in the range of
4000-400 cm-1.
4.2.5 TPO
Temperature programmed oxidation (TPO) experiments were carried out in an
Altamira AMI-200 catalyst characterization system in conjunction with Ametek Dycor
Quadlink residual gas analyzer. Typically, 30 mg of spent catalyst was loaded in the
reactor tube and the temperature was ramped up from 30 °C to 900 °C at a rate of 10 K
min-1 under a flow of 10%O2/He. Masses (m/z) that were analyzed for this procedure
included 44 (CO2), 28 (CO), 18 (H2O), 32 (O2), 4 (Helium).
4.2.6 XRD
X-ray diffraction studies were conducted at Shared Instrument Facility (SIF) at LSU
using PAN Analytical EMPYREAN diffractometer with a generator voltage of 45 kV and a
tube current of 40 mA. The scan range chosen was from 5 to 90° at a step size of
0.02626°.

86
4.2.7 XPS
XPS analysis of all the samples was carried out using ScientaOmicron ESCA 2SR
XPS/Auger instrument at Shared Instrument Facility (SIF) at LSU. All the samples were
ran at a characteristic energy of 1486.7 eV and with an acquisition time of 2 s.
4.2.8 Reaction studies
CH4 oligomerization was carried out in a custom built reactor equipped with a
Hamilton ¼” OD glass lined SS tube. Typically, the supported catalyst (0.10 g) was loaded
into the reactor tube and was heated to the desired reaction temperature at which time
methane was passed over the catalyst. Downstream of the reactor, a caustic wash tank
was fitted (aq. KOH, 0.1 N) to neutralize HBr (when used) during the treatment of the
catalyst. Products were analyzed using Shimadzu GC2014 (FID) equipped with Restek
RT-Q-Bond column (30 m x 0.53 mm x 20 μm). A schematic could be found in Figure C.1,
Appendix C. Downstream of the reactor the outlet line going to the GC (for analysis), was
not heated because we didn’t observe any condensation as analytes of interest were few
ppm.
4.2.9 Silver nitrate (AgNO3) test
AgNO3 is a common test for identifying halide ions and can also be used to quantify
the amount of halides present in a sample. This test was used in the estimation of bromine
(Br) content as well as to confirm the presence of bromide ions in the HBr dissolved
aqueous sample. In a typical test, measured amount of catalyst (0.2 g) was dissolved in
water (although its moisture sensitive, here only dissolution of bromide ions in the
aqueous phase was desired), then using 1 M potassium chromate (K2CrO4) indicator
solution, it was titrated against 0.0141 N silver nitrate to the reddish-brown endpoint. For

87
the HBr dissolved water samples, it was directly titrated with silver nitrate solution using
potassium chromate indicator solution.
4.3 Results and discussion
4.3.1 Comparison with previous research
Table 4.1 compares the literature on direct conversion of methane using solid
superacids with a representative result on ABZ-5 reported here (300 oC).
Table 4.1 Comparison of performance of ABZ-5 with other solid superacids
Catalyst Temp[a]
(°C)
Space velocity (L gcat
-1 h-1) Conv. (%) Notes Ref.
SZ[b]/Fe/Mn 350-450 0.76 < 0.15 Flow reactor [19]
SZ/Fe/Mn[c] 450 4.5 NR* Pulsed and flow exp.
[26]
SZ/Co/Mn[c] 450 4.5 NR Pulsed and flow exp.
[26]
SZ/Cu[c] 450 4.5 NR Pulsed and flow exp.
[26]
SZ/Ni[c] 450 4.5 NR Pulsed and flow exp.
[26]
SZ/Al2O3[c] 450 4.5 NR
Pulsed and flow exp.
[26]
SZ[d] 400 0.6 0.35 after
5 h recirculating
reactor [27]
SZ/Al2O3[d] 400 0.6 1.9 after 5 h
recirculating reactor
[27]
ABZ-5 300 18 ~ 1 Fixed bed
reactor Current Work
[a] Temperature, [b] Sulfated Zirconia, [c] sulfated zirconia promoted with metal ions, all showed small activities, all showed 100 % selectivity to ethane, [d] sulfated zirconia and sulfated zirconia promoted with alumina, main product was ethane with traces of ethylene. *NR – Not reported
Table 4.1 shows that methane conversions in the literature are not typically given.
For studies in which conversions are reported, values vary from 0.15-1.9%. However, a
direct comparison of ~1% conversion for the ABZ-5 catalyst with the literature is not
possible because of the different reaction conditions. For example, Table 4.1 shows that

88
results for ABZ-5 at the temperature of 300 °C, while reported studies were carried out at
higher temperatures, 350-450 °C. SV values in the literature were 0.44-4.5 L gcat-1 h-1,
while here the space velocity was 18 L gcat-1 h-1, a factor of 4-40 times greater than those
reported here. Table 4.1 suggests that ABZ-5 is more active than SZ. In order to test this,
a direct comparison was carried out.
4.3.2 Direct comparison of ABZ-5 with other solid superacids
Table 4.2 directly compares the activity of four solid superacids in CH4
oligomerization at identical conditions: H-ZSM-5 (as-received), and three catalysts
synthesized here: sulfated zirconia (SZ), AlBr3 supported on SiO2 (ABSi) and AlBr3
supported on ZSM-5 (ABZ-5). An intermediate temperature of 300 °C was selected to
compare solid superacid product selectivities directly.
Table 4.2 shows that only ABZ-5 is active, while there are no measurable products
from any of the other catalysts at these conditions. A decrease in TOF (turnover
frequency) with TOS (time on stream) for ABZ-5 indicates deactivation. With time, there
is a general increase in ethylene and propane selectivity. Selectivity to aromatics is
significant, even when methane conversion is ~1%. The aromatic selectivity also changes
on-stream: after 1 h, ethylbenzene and toluene are the major products, but after 3 h, no
ethyl benzene was observed and the selectivity to xylene and toluene increased
significantly.
Despite low methane conversions, these results provide clear evidence of
methane activation at temperatures as low as 300 °C and demonstrates that ABZ-5 is a
strong solid superacid.

89
Table 4.2 Time on stream (TOS) activity of ABZ-5 catalyst and its comparison against various catalysts for CH4 oligomerization (300 °C, 1 atm, 9 Lgcat-1h-1, CH4 conv. ~ 1% for ABZ-5)
[a] Benzene, [b] Toluene, [c] Ethyl Benzene, [d] Ortho-, meta-, para- Xylene, [e] Selectivity is
calculated based on total number of observed products. (-) = not detected
4.3.3 Acidity measurements
The acidity of these superacids was characterized using pyridine-DRIFTS, and
NH3-TPD (Figure 4.2[b]) and the crystallinity was characterized using XRD (Figure C.3,
Appendix C). Pyridine-DRIFTS uses pyridine as a probe molecule to identify Brønsted
and Lewis acid sites. Figure 4.2[a] compares the IR spectra of H-ZSM-5 and ABZ-5
catalysts, which shows the effect on the IR spectra when AlBr3 is added to H-ZSM-5.
DRIFTS spectra of both catalysts at 300 °C show the presence of strong Brønsted
acid sites (~ 1545-1550 cm-1). Although there are very few differences between fresh
ABZ-5 and H-ZSM-5, one noticeable difference is a new shoulder that corresponds to a
Brønsted acid site at 1540 cm-1 in the case of ABZ-5. This shoulder is apart from the main
peak at 1547 cm-1, which must have been generated due to the grafting of AlBr3 on H-
ZSM-5.
Cat. TOS (h)
TOF (h-1)
Product Selectivity (%)[e]
C2= C3 C4 B[a] T[b] EB[c] X
(o,m,p)[d]
ABZ-5 1 0.1 3.0 7.9 2.3 0.5 18.5 59.6 8.1
ABZ-5 2 0.015 8.2 14.9 - - 26.1 41.3 9.5
ABZ-5 3 0.004 13.5 17.8 - - 38 - 30.6
Blank (Quartz Wool)
1 0 - - - - - - -
ABSi 1 0 - - - - - - -
H-ZSM-5 1 0 - - - - - - -
SZ 1 0 - - - - - - -

90
Figure 4.2 [a] Comparison of DRIFTS spectra at 300 °C for (a) Fresh ABZ-5, (b) H-ZSM-5, and (c) Spent ABZ-5. Region corresponding to (L) = Lewis acid sites, (B) = Brønsted acid sites; [b] NH3-TPD for (a) Fresh ABZ-5, (b) H-ZSM-5, and (c) Spent* ABZ-5. *Spent catalyst refers to ABZ-5 recovered after CH4 oligomerization reaction ran at 300 °C, 1 atm, and 9 L gcat-1 h-1 for 16 h

91
Also, the Brønsted (“B”) acid sites appeared to be stable at high temperatures,
although the concentration of Lewis acid sites decreased with increasing temperatures
(Figure C.5, Appendix C). Acid catalysts having peaks in the same range of wavenumbers
are reported to be solid superacids [28-32]. The spent ABZ-5 catalyst shows significantly
lower intense peaks, indicating either a loss of acid sites or lack of access to the acid
sites. Figure C.7 (Appendix C) shows that H-ZSM-5 maintains acid sites up to at least
400 °C, and it is unlikely that there is a loss of Brønsted acid sites from this zeolite. Table
C.1 (Appendix C) shows that there is some bromine loss during the reaction, but at least
the Brønsted sites due to zeolite should remain because they are stable. The presence
of coke on the spent catalyst, as indicated by the decrease in TOF with time (Table 4.2),
is likely responsible for blocking access to the acid sites.
Acidity of both ABZ-5 and H-ZSM-5 was also characterized using NH3-TPD. Figure
4.2 [b] shows the results. Both catalysts show two principal peaks, one at ~ 150-200 °C
that can be characterized as a low temperature (LT) or weakly bound ammonia site [33,
34], and the second one, at ~ 370-400 °C, attributable to a high temperature (HT) or
strongly bound ammonia site [33, 34], which probably is able to protonate NH3 to form
NH4+. Similar TPD peaks were also observed when AlCl3 was grafted on mesoporous
MCM-41 zeolites[35]. The low temperature peaks are often identified as ammonia
adsorbed through hydrogen co-ordinate bond [34]. The literature suggests that this weak
acidity has little catalytic significance [36, 37]. In the case of ABZ-5, both the LT and HT
peaks are present but shifted to temperatures higher than H-ZSM-5. The LT, weak acid
peak for ABZ-5 is centered at ~ 210 °C compared to ~165 °C for H-ZSM-5. The HT, strong
acid peaks are very similar: ~400 °C for ABZ-5 compared to ~390 °C for H-ZSM-5. Both

92
peak shifts, and the total acidity, as measured by the areas under the NH3-TPD curves,
clearly indicate an increase in acidic strength due to the presence of AlBr3 [38]. Note that
the difference in total acidity between ABZ-5 and H-ZSM-5 is almost entirely due to that
of the strong, HT acidity, even though the acid strength, as measured by the shift in the
peak temperature, is comparable (~ 10 °C shift for HT peak). For spent catalyst, NH3-
TPD also showed significantly fewer acid sites and the loss was seen in both the LT and
HT regions. Table 4.3 shows that ABZ-5 clearly has more acid sites, which can be
attributed to the AlBr3 grafting, which generated new acid sites in the H-ZSM-5. The spent
ABZ-5 catalyst on the other hand, did show a loss in the number of acid sites (Figure
4.2[b] and Table 4.3). This could either be due to the loss of Br from ABZ-5 during reaction
or due to lack of access to the acid sites from coke.
Table 4.3 Amount of acid sites on zeolite catalysts using NH3-TPD
Catalyst Conc. of acid sites
(mmol/g)
H-ZSM-5 1.19
ABZ-5 (fresh) 1.55
ABZ-5 (spent) 0.85
4.3.4 Effect of temperature
Reactions were run on ABZ-5 at three temperatures from 200 to 400 °C. The effect
of temperature on CH4 oligomerization is shown in Table 4.4. ABZ-5 showed activity at
each temperature. Higher temperatures (400 °C) resulted in higher rate, as measured by
TOF. There is a general increase in total selectivity to aromatics with temperature, with
toluene selectivity being the only clear trend with temperature. These aromatic products
are thought to be precursors for polynuclear aromatic compounds and coke. Formation

93
of these products at a faster rate at higher temperatures likely leads to faster deactivation.
Similar deactivation is also typically observed in methane aromatization [39].
Table 4.4 Effect of temperature on methane oligomerization over ABZ-5 catalyst (1 atm, 9 L gcat-1 h-1). (-) = not detected; measured at 1 h
Temp. (°C)
TOF (h-1)
Product selectivity (%)
C2= C3 C4 B T EB X (o,m,p)
M[a]
200 0.01 17.6 30.4 10.7 31.6 9.7 - - -
300 0.1 3 7.9 2.3 0.49 18.6 59.6 8.1 -
400 0.08 7.8 14.4 1.4 9.0 35.6 24.7 7.1 -
[a] Mesitylene
4.3.5 Effect of space velocity
Space velocity (SV) significantly affects hydrocarbon selectivity. ABZ-5 was
studied at three different space velocities (3.6, 9 and 18 L gcat-1 h-1). The corresponding
activity results are presented in Table 4.5.
Table 4.5 Effect of space velocity on methane oligomerization over ABZ-5 catalyst (1 atm, 300 °C). Samples are taken at 1 h time on stream
Space Velocity
(L gcat-1 h-1)
TOF (h-1)
Product selectivity (%)
C2= C3 C4 B T EB X
(o,m,p) M
3.6 0.16 4.2 0.75 2.1 1.97 8.4 20.1 5.6 56.9
9 0.1 3 7.9 2.3 0.49 18.6 59.6 8.1 -
18 0.02 17 25.6 5.5 4.6 17.1 30.2 - -
The total aromatic selectivity is relatively high at all conditions tested here,
consistent with coke formation. TOF increases at lower SV, as expected. However, there
is no clear trend of total aromatic selectivity with SV, nor with any specific aromatic
compounds. The only clear trend in selectivity is that of propane selectivity, which

94
increases with increasing SV, indicating that propane is an intermediate. However, there
is no obvious relationship between propane and any measurable aromatics.
4.3.6 TPO
Results in Table 4.2, Table 4.4, and Table 4.5 clearly show the relatively high total
selectivity to aromatics. Figure 4.3 compares the amount and reactivity of coke analyzed
in the four catalysts of interest here: fresh catalyst and spent catalysts after being run at
three reaction temperatures (200 °C, 300 °C, and 400 °C). Peak TPO positions were
similar for all runs, ~ 420 °C, indicating amorphous carbon on the surface of all three
spent catalysts [40]. TPO results (Table 4.6) show slightly different levels of coke
deposition depending on reaction temperature.
Table 4.6 Carbon deposition from oligomerization reactions
Catalyst Carbon deposited
(mmol/g)
Fresh ABZ-5 2.53
Spent ABZ-5 (after 16 h reaction at 200 oC)
3.00
Spent ABZ-5 (after 16 h reaction at 300 oC)
3.60
Spent ABZ-5 (after 16 h reaction at 400 oC)
3.47
TPO results for the 300 °C catalyst show a small peak at ~120 °C that is not shown in the
others. In addition, there is more coke at the 420 °C peak than the other two runs. This is
perhaps consistent with greater aromatic selectivity and TOF for the 300 °C catalyst
(Table 4.4). [The anomalous TPO peak at ~120 °C for the fresh catalyst may be due to
trace amounts of CO2 adsorbed in handling].

95
Figure 4.3 TPO comparison for ABZ-5 after CH4 oligomerization runs (1 atm, 9 Lgcat-1h-
1) at various temperatures on ABZ-5 catalyst (a) fresh catalyst, (b) 200 °C, (c) 300 °C, (d) 400 °C (after running reaction at respective temperatures for ~ 1000 min)
4.4 Conclusions
Vapor grafted AlBr3 on to H-ZSM-5 has been used to synthesize AlBrx/H-ZSM-5
catalyst (“ABZ-5”). Grafting of AlBr3 creates new Brønsted acid sites in the H-ZSM-5
framework, as observed from the NH3-TPD and pyridine-DRIFTS. These acid sites, in
synergy with the existing acid sites from the H-ZSM-5 framework, are likely the cause for
the protonation of methane via an oligo-condensation mechanism [14], producing higher

96
hydrocarbons. This catalyst has been shown to be active in the oligomerization of
methane at temperatures as low as 200 °C.
4.5 References
[1] S. Jenner, A.J. Lamadrid, Shale gas vs. coal: Policy implications from environmental impact comparisons of shale gas, conventional gas, and coal on air, water, and land in the United States, Energy Policy, 53 (2013) 442-453. [2] Q. Wang, X. Chen, A.N. Jha, H. Rogers, Natural gas from shale formation – The evolution, evidences and challenges of shale gas revolution in United States, Renewable and Sustainable Energy Reviews, 30 (2014) 1-28. [3] T. Boersma, C. Johnson, The Shale Gas Revolution: U.S. and EU Policy and Research Agendas, Review of Policy Research, 29 (2012) 570-576. [4] S.J. Blanksby, G.B. Ellison, Bond Dissociation Energies of Organic Molecules, Accounts of Chemical Research, 36 (2003) 255-263. [5] G.E. Keller, M.M. Bhasin, Synthesis of ethylene via oxidative coupling of methane, Journal of Catalysis, 73 (1982) 9-19. [6] N. Kumar, Z. Wang, S. Kanitkar, J.J. Spivey, Methane reforming over Ni-based pyrochlore catalyst: deactivation studies for different reactions, Applied Petrochemical Research, (2016) 1-7. [7] Q. Zhu, S.L. Wegener, C. Xie, O. Uche, M. Neurock, T.J. Marks, Sulfur as a selective ‘soft’ oxidant for catalytic methane conversion probed by experiment and theory, Nat Chem, 5 (2013) 104-109. [8] A. Zhang, S. Sun, Z.J.A. Komon, N. Osterwalder, S. Gadewar, P. Stoimenov, D.J. Auerbach, G.D. Stucky, E.W. McFarland, Improved light olefin yield from methyl bromide coupling over modified SAPO-34 molecular sieves, Physical Chemistry Chemical Physics, 13 (2011) 2550-2555. [9] X. Guo, G. Fang, G. Li, H. Ma, H. Fan, L. Yu, C. Ma, X. Wu, D. Deng, M. Wei, D. Tan, R. Si, S. Zhang, J. Li, L. Sun, Z. Tang, X. Pan, X. Bao, Direct, Nonoxidative Conversion of Methane to Ethylene, Aromatics, and Hydrogen, Science, 344 (2014) 616-619. [10] Y. Wu, L. Emdadi, Z. Wang, W. Fan, D. Liu, Textural and catalytic properties of Mo loaded hierarchical meso-/microporous lamellar MFI and MWW zeolites for direct methane conversion, Applied Catalysis A: General, 470 (2014) 344-354.

97
[11] S. Ma, X. Guo, L. Zhao, S. Scott, X. Bao, Recent progress in methane dehydroaromatization: From laboratory curiosities to promising technology, Journal of Energy Chemistry, 22 (2013) 1-20. [12] P. Schwach, X. Pan, X. Bao, Direct Conversion of Methane to Value-Added Chemicals over Heterogeneous Catalysts: Challenges and Prospects, Chemical Reviews, 117 (2017) 8497-8520. [13] K. Sun, D.M. Ginosar, T. He, Y. Zhang, M. Fan, R. Chen, Progress in Nonoxidative Dehydroaromatization of Methane in the Last 6 Years, Industrial & Engineering Chemistry Research, 57 (2018) 1768-1789. [14] G.A. Olah, G. Klopman, R.H. Schlosberg, Super acids. III. Protonation of alkanes and intermediacy of alkanonium ions, pentacoordinated carbon cations of CH5+ type. Hydrogen exchange, protolytic cleavage, hydrogen abstraction; polycondensation of methane, ethane, 2,2-dimethylpropane and 2,2,3,3-tetramethylbutane in FSO3H-SbF5, Journal of the American Chemical Society, 91 (1969) 3261-3268. [15] G.A. Olah, Y. Halpern, J. Shen, Y.K. Mo, Electrophilic reactions at single bonds. XII. Hydrogen-deuterium exchange, protolysis (deuterolysis), and oligocondensation of alkanes with superacids, Journal of the American Chemical Society, 95 (1973) 4960-4970. [16] G.A. Olah, R.H. Schlosberg, Chemistry in super acids. I. Hydrogen exchange and polycondensation of methane and alkanes in FSO3H-SbF5 ("magic acid") solution. Protonation of alkanes and the intermediacy of CH5+ and related hydrocarbon ions. The high chemical reactivity of "paraffins" in ionic solution reactions, Journal of the American Chemical Society, 90 (1968) 2726-2727. [17] S. Vasireddy, S. Ganguly, J. Sauer, W. Cook, J.J. Spivey, Direct conversion of methane to higher hydrocarbons using AlBr3-HBr superacid catalyst, Chemical Communications, 47 (2011) 785-787. [18] W. Hua, A. Goeppert, J. Sommer, Methane activation in the presence of Al2O3-promoted sulfated zirconia, Applied Catalysis A: General, 219 (2001) 201-207. [19] S. Rezgui, A. Liang, T.K. Cheung, B.C. Gates, Methane conversion to ethane in the
presence of iron‐ and manganese‐promoted sulfated zirconia, Catalysis Letters, 53 (1998) 1-2. [20] D. Farcasiu, S.L. Fisk, M.T. Melchior, K.D. Rose, Evaluation of super acid strength from the protonation of benzene. Comparison of HF-SbF5, HF-TaF5, and HBr-AlBr3 systems, The Journal of Organic Chemistry, 47 (1982) 453-457.

98
[21] J.D. Heldman, Aliphatic Friedel—Crafts Reactions. I. The Alkylation of Butanes by Methyl and Ethyl Bromide, Journal of the American Chemical Society, 66 (1944) 1791-1793. [22] J.M. Crafts, C. Friedel, Organic chemistry, Journal of the Chemical Society, 32 (1877) 725-791. [23] A. Corma, H. García, Lewis Acids: From Conventional Homogeneous to Green Homogeneous and Heterogeneous Catalysis, Chemical Reviews, 103 (2003) 4307-4366. [24] A.L. Glasebrook, N.E. Phillips, W.G. Lovell, The Action of Aluminum Halides on n-Pentane, Journal of the American Chemical Society, 58 (1936) 1944-1948. [25] O. Grummitt, E.E. Sensel, W.R. Smith, R.E. Burk, H.P. Lankelma, The Action of Aluminum Bromide on Paraffin Hydrocarbons. I. n-Hexane and n-Heptane, Journal of the American Chemical Society, 67 (1945) 910-914. [26] R.L. Martins, M. Schmal, Methane activation on superacidic catalysts based on oxoanion modified zirconium oxide, Applied Catalysis A: General, 308 (2006) 143-152. [27] W. Hua, A. Goeppert, J. Sommer, Methane activation in the presence of Al2O3-promoted sulfated zirconia, Applied Catalysis A: General, 219 (2001) 201-207. [28] W.-H. Chen, H.-H. Ko, A. Sakthivel, S.-J. Huang, S.-H. Liu, A.-Y. Lo, T.-C. Tsai, S.-B. Liu, A solid-state NMR, FT-IR and TPD study on acid properties of sulfated and metal-promoted zirconia: Influence of promoter and sulfation treatment, Catalysis Today, 116 (2006) 111-120. [29] R. Buzzoni, S. Bordiga, G. Ricchiardi, C. Lamberti, A. Zecchina, G. Bellussi, Interaction of Pyridine with Acidic (H-ZSM5, H-β, H-MORD Zeolites) and Superacidic (H-Nafion Membrane) Systems: An IR Investigation, Langmuir, 12 (1996) 930-940. [30] Y. Wu, L. Qin, G. Zhang, L. Chen, X. Guo, M. Liu, Porous Solid Superacid SO42–/Fe2–xZrxO3 Fenton Catalyst for Highly Effective Oxidation of X-3B under Visible Light, Industrial & Engineering Chemistry Research, 52 (2013) 16698-16708. [31] M. Marczewski, H. Marczewska, K. Witosławski, Superacid properties of
Molecular Catalysis A: Chemical, 97 (1995) 101-110. [32] H. Hattori, O. Takahashi, M. Takagi, K. Tanabe, Solid super acids: Preparation and their catalytic activities for reaction of alkanes, Journal of Catalysis, 68 (1981) 132-143. [33] L. Rodríguez-González, F. Hermes, M. Bertmer, E. Rodríguez-Castellón, A. Jiménez-López, U. Simon, The acid properties of H-ZSM-5 as studied by NH3-TPD and 27Al-MAS-NMR spectroscopy, Applied Catalysis A: General, 328 (2007) 174-182.

99
[34] A.S. Al-Dughaither, H. de Lasa, HZSM-5 Zeolites with Different SiO2/Al2O3 Ratios. Characterization and NH3 Desorption Kinetics, Industrial & Engineering Chemistry Research, 53 (2014) 15303-15316. [35] D. Dubé, S. Royer, D. Trong On, F. Béland, S. Kaliaguine, Aluminum chloride grafted mesoporous molecular sieves as alkylation catalysts, Microporous and Mesoporous Materials, 79 (2005) 137-144. [36] N. Katada, H. Igi, J.H. Kim, M. Niwa, Determination of the Acidic Properties of Zeolite by Theoretical Analysis of Temperature-Programmed Desorption of Ammonia Based on Adsorption Equilibrium, J. Phys. Chem. B, 5647 (1997) 5969. [37] N. Katada, M. Niwa, Analysis of Acidic Properties of Zeolitic and Non-Zeolitic Solid Acid Catalysts Using Temperature-Programmed Desorption of Ammonia, Catal. Surv. Asia, 8 (2004) 161. [38] H. Xiao, J. Zhang, X. Wang, Q. Zhang, H. Xie, Y. Han, Y. Tan, A highly efficient Ga/ZSM-5 catalyst prepared by formic acid impregnation and in situ treatment for propane aromatization, Catalysis Science & Technology, 5 (2015) 4081-4090. [39] Z.R. Ismagilov, E.V. Matus, L.T. Tsikoza, Direct conversion of methane on Mo/ZSM-5 catalysts to produce benzene and hydrogen: achievements and perspectives, Energy & Environmental Science, 1 (2008) 526-541. [40] C.A. Querini, Coke characterization, in: J.J. Spivey, G.W. Roberts (Eds.) Catalysis: Volume 17, The Royal Society of Chemistry, 2004, pp. 166-209.

100
Chapter 5 . Methane Dehydroaromatization over Molybdenum supported on Sulfated Zirconia Catalysts3
5.1 Introduction
The conversion of methane into higher value chemicals is one of the grand
challenges of 21st century. The shale gas revolution has made these immense,
previously unattainable resources of natural gas accessible and economically
competitive. Natural gas is considered to be a cleaner source than other conventional
fuels such as coal and oil. This has helped some countries such as USA to reduce its
CO2 emissions by direct application of natural gas in the power plants [1, 2]. Conventional
end-uses of natural gas include power generation or flaring, but it can also be used as a
feedstock for the production of higher-value chemicals. Conversion of methane into
higher-value chemicals has been long sought by researchers, but with limited success
and generally with only incremental improvements.
The principal processes for conversion of methane can be categorized into non-
oxidative or oxidative. Examples of oxidative route are as shown in Figure 5.1. These
include OCM (Oxidative Coupling of Methane) [3], Reforming (Dry/Bi/Oxy/Steam) [4-7],
(PO) Partial Oxidation [8], Halogenation [9] or even Sulfidation [10]. Except for Steam
Reforming of Methane (SRM) and Fisher Tropsch (FT), none of the other oxidative
processes are commercialized and are often limited by the net yields of desired products.
Though commercially practiced, SRM produces a syngas (CO+H2) mixture, which is
subsequently converted into higher hydrocarbons, via processes such as Fisher Tropsch
3 Reprinted from S. Kanitkar, Md. A. Abedin, S. Bhattar, J. J. Spivey. (2019) Methane dehydroaromatization over Mo supported on sulfated zirconia catalysts. Applied Catalysis A: General. 575, 25-37. With permission from Elsevier B.V.

101
synthesis. Reforming processes produce commercially practical yields only at
temperatures of 700-800 °C, which often results into high energy requirements and thus
high costs [11]. On the contrary, non-oxidative conversion routes are based on a single
step, thus requiring lesser energy and less cost. Some of the attempts involve use of what
is known as superacids: HF-FSO3H [12, 13], HF-SbF5 [12, 13] or HBr-AlBr3 [14] or sulfated
zirconia (SZ) [15-17]. Other significant recent attempts involved use of lattice confined
single Fe sites[18], use of metal modified zeolites (Mo [19, 20], Re [21], Ag [22], Zn [23]),
or even recently using GaN nanowires under UV irradiation [24].
Figure 5.1 Oxidative conversion routes of methane to higher hydrocarbons
One of the potentially most important non-oxidative methane conversion
processes is dehydroaromatization (DHA), the oligomerization of methane to benzene
and hydrogen as shown in equation 5.1 below:
6 CH4 ↔ C6H6(g) + 9 H2 ↑ (∆𝐺298𝐾 = +433𝑘𝐽
𝑚𝑜𝑙, ∆𝐻298𝐾 = +530
𝑘𝐽
𝑚𝑜𝑙) (5.1)

102
DHA was first reported around 1966 in which methane was passed over silica gel
at temperatures of around 1000 °C, with aromatic yields of 4-6 % [25]. It was not until
1990s that DHA attracted attention [19, 20]. Molecular sieves such as H-ZSM-5 zeolite
and H-MCM-22 have been shown to be the most active supports for this reaction [26-28].
Many reports based on doping of noble metals such as Mo, Re, or W into these supports
for the aromatization of methane have also been reported [29]. Nevertheless, the
literature shows that Mo-doped catalysts have been the most widely-studied catalysts for
DHA. Despite a great deal of research, and significant improvements to the process, e.g.,
using membranes to separate hydrogen, DHA has not yet been commercialized [30-32].
It is widely accepted that active molybdenum oxycarbide or carbide species are
generated in-situ under reducing reaction conditions and under the flow of methane [26,
27]. These species can also be intentionally generated using other carbon sources
(CO2/CO) under H2 flow [26, 27]. Even though the nature of active species
oxycarbidic/carbidic is not fully understood [29], it is commonly believed that exchanged
molybdenum [33] species activate methane by activating one of the C-H bonds that lead
to the formation of CHx species, forming a dimer: C2Hy. This dimer is then oligomerized
and cyclized into higher hydrocarbons and aromatics on the Brønsted acid sites (BAS) of
the molecular sieve. This mechanism has been opened for debate as to whether it is
bifunctional or monofunctional, based on one study [34]. Regardless, this typical
bifunctional mechanism can be depicted as shown in equation 5.2 (BAS=Brønsted acid
site) [26]:
CH4Mo site↔ C2Hy
BAS↔ C6H6 + C7H8 + C8H10 + H2 (5.2)

103
There are a number of solid acids that could, in principle, catalyze the C2Hy
species. For example, sulfated zirconia (SZ) is a well-known solid superacid that has been
studied and a number of variations have been produced (mesoporous, nanosized, e.g.)
[35-37]. SZ possesses strong BAS that can play a key role in the oligomerization of
dimeric species. In principle, a similar bifunctional catalyst could be synthesized if Mo is
doped onto a solid acid. Although doping of Mo on SZ is not novel [38]; to the best of our
knowledge, no study has shown DHA of methane on these catalysts.
The present work tests these Mo/SZ catalysts for DHA of methane and also the
effect of Mo loading at reaction conditions of interest. For this purpose, 3 different
catalysts were synthesized: SZ (w/o Mo), and two Mo loaded SZ catalysts with two
different loadings of Mo (1 %, 5 %). These catalysts were further evaluated for DHA and
were characterized using spectroscopic techniques including Raman, DRIFTS, XRD,
SEM-EDS, XANES, HRTEM, XPS. The hypothesis is that an active DHA catalyst can be
prepared based on Mo/SZ, incorporating the requisite metal and acid sites for this
reaction.
5.2 Experimental
5.2.1 Materials
Zirconium hydroxide, Zr(OH)4 (97%) and ammonium molybdate tetrahydrate
(NH4)6Mo7O24.4H2O were both purchased from Sigma Aldrich Inc. H2SO4 (95–98.0 %)
was purchased from Malinckrodt Chemicals Inc. Ultra-high purity grade H2, CH4, Ar, and
10% O2/He were all purchased from Airgas Inc. NH4-ZSM-5 (Si/Al=50:1) was purchased
from Alfa Aesar Inc. All Mo standards (Mo powder, MoS2, MoO3, MoO2, β-Mo2C) were
purchased from Alfa Aesar Inc. with a purity of 99+%.

104
5.2.2 Catalyst preparation
Sulfated zirconia was prepared based on literature methods [36] . In a typical
preparation, about 35 g of Zr(OH)4 was mixed with 500 ml of 0.5M H2SO4 solution and
the mixture was stirred for 2 h, followed by vacuum filtration and subsequently dried
overnight at 110 °C. The dried catalyst powder was then calcined at 550 °C for 4 h under
static air to get the sulfated zirconia catalyst. Different loadings of molybdenum were
incorporated onto sulfated zirconia using standard impregnation method. In a typical
preparation, known quantity of ammonium molybdate tetrahydrate was dissolved in 50 ml
of water and to this solution, appropriate quantity of sulfated zirconia was added, and the
solution was stirred for 2 h after which a similar procedure of vacuum filtration, overnight
drying (110 °C) and calcination at 550 °C for 4 h in static air was followed. This yielded
the final Mo supported on sulfated zirconia catalysts. Similar procedure was also used for
preparing Mo supported on H-ZSM-5 catalysts. For H-ZSM-5 preparation, as purchased
NH4-ZSM-5 was calcined at 550 °C for 4 h under static air.
5.2.3 ICP-OES
Inductively coupled plasma-optical emission spectroscopy (ICP-OES) was carried
out using Varian-MPX spectrophotometer in Wetland Biogeochemical Lab at LSU
Oceanography. The digestions of samples were carried out using a mixture of borate flux:
Lithium Tetraborate (49.75 wt%), Lithium Metaborate (49.75 wt%), and Lithium Iodide
(0.5 wt%). In a typical procedure, 0.2 g of catalyst sample was mixed with 2 g of borate
flux mixture. This mixture was fused in a furnace at 1000 °C and subsequently dissolved
in a warm 10% HNO3 solution. This solution was diluted subsequently and used for ICP-
OES analysis.

105
5.2.4 BET
Breauner Emmett Teller (BET) surface area analysis was done using Altamira
AMI-200 catalyst characterization system using N2 monolayer adsorption. Three-point
BET with 10%, 20% and 30% N2 concentrations in Helium was used to estimate the
surface areas of these catalysts.
5.2.5 Raman spectroscopy
Analysis by Raman spectroscopy for all catalyst samples was carried out using
Renishaw inVia Raman microscope equipped with a 532 nm (green) laser. Spectra
measurements were carried out at 10mW power and with 50 μm slit aperture.
5.2.6 DRIFTS
Diffuse Reflectance Infrared Fourier Transform Spectroscopy (DRIFTS)
experiment using pyridine as a probe molecule was carried out in a Thermo Scientific
Nicolet 6700 FTIR equipped with Harrick Praying Mantis reaction cell fitted with KBr
windows. Spectra of all the samples were recorded with a spectral resolution of 4 cm−1 in
region going from 4000-650 cm−1. In a typical experiment, IR cell was loaded with the
catalyst sample inside the glovebox. Sample was then brought on stream and pretreated
at 100 °C for 30 min under He flow to clean the surface of the catalyst from adsorbed
impurities. After pretreatment, sample was cooled down to 25 °C and a background
spectrum was recorded. At this temperature, catalyst was saturated with pyridine vapors
for 3 h. Saturated sample was then treated under He flow for 30 min to remove
physisorbed pyridine from the catalyst surface and from the cell chamber. Sample was
then treated at 100 °C for 10 min under He flow and cooled back to room temperature
and the actual spectrum was recorded. Similar spectra were recorded at room

106
temperature after 10 min. treatments at 200 °C, 300 °C, and 400 °C to check the thermal
stability of the acid sites on catalysts.
5.2.7 Ammonia-TPD
Ammonia-TPD was carried out using Altamira AMI-200 reactor system in
conjunction with Ametek Mass Spectrometer (MS). Typically, 50 mg of catalyst was
weighed and loaded in a quartz tube reactor. Catalyst was pretreated at 100 °C for 30
min under He flow to clean the catalyst surface. After pretreatment, sample was cooled
down to 50 °C, and ammonia was adsorbed by flowing it through the catalyst bed for 1 h.
Post ammonia adsorption, 25 sccm of He was flown for 40 min to remove any
physisorbed/residual ammonia. At this time, Mass Spectrometer and TCD detector were
turned on and the temperature was ramped up at 10 °C/min. from 50 to 500 °C. The TCD
signal is usually a combination of signals from various gases that are generated or are
being flowed in the system. In order to clearly distinguish various gases coming out, MS
was used to track signals for following masses: 4, 16, 17, 18, 27, 32, and 82. Based on
ammonia (m/e=16) signals from MS with respect to temperature, amounts of ammonia
desorbed and peak positions were calculated.
5.2.8 SEM-EDS
SEM analysis of the samples was done at LSU Shared Instrument Facilities (SIF)
using FEI quanta 3D FIB/SEM equipped with Ametek EDAX accessory. Samples were
analyzed at a voltage of 5 kV and at a resolution of 3 μm.
5.2.9 HRTEM
High Resolution Transmission Electron Microscopy (HRTEM) was used to study
these type of catalysts. This was particularly useful in confirming the loading of Mo onto

107
the sulfated zirconia sample and also to confirm the formation of Mo2C after carburization
of the fresh catalysts was done. HRTEM scans of these catalysts were obtained using
JEOL JEM-2011 Scanning TEM at an accelerating voltage of 200 kV.
5.2.10 XRD measurements
X-ray diffraction (XRD) measurements for all the samples was carried out using
PANalytical EMPYREAN diffractometer with Cu Kα radiation. Sample was collected by
scanning the data from 5° to 90° with a step size of 0.026°. Data analysis of XRD of all
samples was done using PANalytical X’Pert software.
5.2.11 XPS
X-ray Photoelectron Spectroscopy (XPS) for the samples was performed at
Shared Instruments Facility (SIF) in LSU. All the analyses were performed on Scienta
Omicron ESCA 2SR instrument with Mg as an X-ray source at 15 kV and at a pass energy
of 40. Post processing of XPS data was performed using CasaXPS software.
5.2.12 XANES
X-ray Absorption Near Edge Spectroscopy (XANES) studies of the catalyst were
performed at the electron storage ring of J. Bennett Johnston, Sr., Center for Advanced
Microstructures and Devices (CAMD) of Louisiana State University, Baton Rouge,
Louisiana. CAMD operates the ring operating at 1.3 GeV with current between 100 mA
to 50 mA. The Mo K edge measurements were made at the High Energy Xray
Spectroscopy (HEXAS) beamline located on an 11-pole wiggler operating at 5.5 T. The
LIII edge measurements were made at the Low Energy X-ray Absorption Spectroscopy
(LEXAS) beamline, a windowless beamline on a bending magnet with a 13 μm Kapton
tape separating the ring from the beamline. Both beamlines use Lemonnier double crystal

108
monochromator with design modifications made at the University of Bonn. At lower
energy InSb 111 crystals (resolution ca. 0.5 eV) were used while at higher energy water-
cooled Ge 422 crystals (resolution ca. 2 eV) were used. For Mo K edge measurement,
the Hexas beamline was calibrated with a Mo Foil at 20 keV. The beamline has three ion
chambers with a Mo foil in between the second and third chambers for monitoring the
calibration. Mixtures of argon, and xenon gases were used to achieve 20%, 30% and
30% absorption in the first, second and third chamber, respectively.
Mo standards used in the analysis were: MoO3 (99.9% metals basis, Alfa Aesar),
Mo foil (200 mesh, 99.9% metals basis, Alfa Aesar), and Mo2C (99.9% metals basis, Alfa
Aesar). The standards were measured in transmission while the catalyst samples were
placed on Kapton tape and scanned in fluorescence using a seven-element Ketek silicon
drift detector with a total area of 560 mm2 and a resolution of ca. 135 eV.
The low energy beamline was calibrated with the zinc sulfate white being at
2481.44 eV. The ion chambers used air as ionizing gas, the chamber being evacuated to
50 Torr. A single-element Ketek silicon drift detector with an active area of 150 mm2 was
used for fluorescence signal detection.
5.2.13 Carburization and dehydroaromatization (DHA)
MoOxCy or Mo2C, which are the active species for DHA reaction were generated
in-situ. For this, a procedure from the literature [38] was followed. First, Mo catalyst
(oxidized form) was reduced under H2 at 10 K/min ramp rate from room temperature to
650 °C. After 650 °C temperature was reached, CH4 gas was introduced into the reaction
system for 4 h at a ratio of 1:4 (CH4:H2). This treatment led to carburization of the MoOx
species. After 4 h, hydrogen and methane was turned off and the system was purged with

109
argon. Subsequently pure CH4 was introduced at the desired reaction temperature to
carry out DHA reaction. All of these sequences were carried out in an Altamira AMI 200HP
reactor system equipped with a SGE glass lined SS tube. In a typical experiment, reactor
tube would be loaded with (0.3-1) g of carburized catalyst and small quantities of CH4 (10
sccm) and Argon (5 sccm, internal standard) would be passed over the catalyst bed.
Reaction products were analyzed downstream using Shimadzu GC2014 (FID, 2 TCD’s)
equipped with Restek RT-Q-Bond column (30m × 0.53 mm × 20 μm) in conjunction with
Shimadzu QP2010 GC-MS system. We would like to point out that we were not able to
measure Naphthalene in our GC system. Also, estimating coke content at any point
during reaction is a challenge. So, based on the products observed and the amount of
methane converted, we estimated combined naphthalene and coke content at any time
on stream.
Following formulas were used for the activity calculation:
% CH4 conversion = conc. CH4in− conc. CH4out
conc. CH4in x 100 (5.3)
Selectivities were calculated based on observable products and naphthalene and
coke content was estimated based on the amount of methane converted.
%C selectivity = carbon number in the product × conc. Product
conc. CH4 reacted× 100 (5.4)
5.3 Results and discussion
5.3.1 Physico-chemical properties of the catalyst
Physical and chemical properties of the catalysts are listed in Table 5.1. Base SZ
catalysts in the literature are reported to have surface areas of 50-100 m2/g [39].The
present SZ catalyst has a surface area of 84 m2/g, well within the expected value. As the
Mo was doped onto SZ, it led to slight decrease in the surface area, as expected, due to

110
blockage of pores as expected from the loading of Mo [38, 40]. However, in the case of
1% doping of Mo, there is no statistical difference between the BET surface areas.
Table 5.1 shows the elemental Mo loading to be 0.84 and 3.92 wt% as compared
to intended 1 and 5 wt% loading. We believe that it is quite possible that some of the Mo
was lost during vacuum filtration while synthesis of the catalysts.
Table 5.1 Physico-chemical properties of Mo/SZ
Catalyst Mo
loading, % (intended)
Mo loading, wt% (ICP)
Mo loading, wt%
(EDS)
S loading, wt% (ICP)
BET Surface Area (m2/g)
SZ 0 0 0 3.5 84
1% Mo/SZ 1 0.84 1.6 2.99 87
5% Mo/SZ 5 3.92 4.5 1.95 76
S (sulfur) loading from ICP also is shown in Table 5.1. It shows that the base SZ
had sulfur content of 3.5 wt%, which was reduced as Mo was doped into this catalyst.
This decrease was proportional to the amount of Mo loaded. When 1wt% Mo was loaded,
it decreased slightly from 3.5 to 2.99 and further to 1.95 wt% when, 5 wt% Mo was loaded.
This is quite possible because when Mo is impregnated onto this catalyst, it interacts with
or replaces the SO42- species on the surface. This could be seen subsequently in Raman
spectroscopy as well.
5.3.2 Raman spectroscopy
Raman spectroscopy is used to detect non-IR specific bond vibrations and can be
useful to analyze inorganic compounds which are not IR-active. Figure 5.2 shows Raman
spectra for a sulfated zirconia as a base sample (a) and a transition of 5% Mo/SZ catalyst
from oxidized form (b) to a carburized one (c), and to a spent one (d).

111
Figure 5.2 Raman spectra for samples at various treatment (a) base SZ, (b) fresh 5%Mo/SZ, (c) carburized 5%Mo/SZ, (d) spent 5%Mo/SZ
Common bands observed for sulfated zirconia include ~ 270 cm-1, ~ 320 cm-1, ~
460 cm-1, ~ 650 cm-1 and 1025 cm-1. These bands below 700 cm-1 are attributed to
tetragonal ZrO2 vibrations [36], while the band at 1025 cm-1 is attributed to vibrations from
sulfate groups on ZrO2 surface. When Mo was introduced into this catalyst, two distinct
new bands appear around ~ 820 cm-1 and ~ 970 cm-1 Raman shifts (shown in the box in
Figure 5.2). These bands are attributed to the Mo-O-Mo, and Mo=O vibrations [41]
respectively. This confirms doping of Mo on the surface of sulfated zirconia.
An attempt to measure the Raman spectra for spent catalyst samples, or even for
a carburized fresh catalyst, was difficult because these samples were grey/dark, causing
a strong absorption of the energy from the excitation source [42]. Similar difficulties were

112
also experienced by the same authors [42] in obtaining FTIR spectra for carburized MoO3
samples. Despite of this, we made an attempt to focus on a different spot location and
this provided meaningful spectra for these samples. Moving from (b) to (c) and to (d), i.e.
from an oxidized catalyst to a spent catalyst, most of the bands corresponding to t-ZrO2
show very little difference. Also, band corresponding to Mo-O-Mo vibration (~ 820 cm-1)
appears to be normal but the band corresponding to Mo=O vibration (~ 970 cm-1) appears
to have shifted higher (~ 995 cm-1). This shift might indicate the presence of a carbidic
form of molybdenum [38]. In addition to these differences, there are two new bands at
1340 cm-1 and 1600 cm-1 in the case of carburized and spent samples, which may be
attributed to the D and G bands of graphitic carbon [43, 44]. As expected, these bands
were intense for the spent sample than the carburized one, indicative of more carbon
deposited on the surface.
Another noticeable change between these samples is the loss of intensity for the
band at ~ 1025 cm-1 corresponding to the sulfate groups, which may be due to the
interaction between Mo and SO42- groups during the impregnation process and also
probably due to volatility of SO42- species during the carburization and the DHA reaction
[38].
5.3.3 DRIFTS
DRIFTS in conjunction with pyridine as a probe molecule was used to characterize
the acid sites of the catalyst. Base SZ catalyst is known to have both Brønsted and Lewis
strong acid sites [16, 17, 36]. This was confirmed by observing the shift in vibrations from
adsorbed pyridine at ~ 1445, 1610 cm-1 (co-ordinated pyridine), and ~ 1540 cm-1, 1640
cm-1 (protonated pyridine) that are present in all the samples [45]. Bands at ~ 1490 cm-1

113
correspond to pyridine adsorbed on both Lewis and Brønsted sites [45, 46]. A comparison
of DRIFTS spectra for catalysts with different loadings of Mo is presented in Figure 5.3.
Figure 5.3 Comparison of SZ and Mo doped SZ catalysts after pyridine desorption at 100 °C (a) SZ, (b) 1% Mo/SZ, (c) 5% Mo/SZ, “B” = Brønsted, “L” = Lewis
When Mo was doped in different amounts on the base SZ catalyst, the type of acid
sites did not change. Both Brønsted and Lewis acid sites were still present and there was
little qualitative difference in the concentration of acid sites. However, as the loading of
the Mo increased, bands corresponding to both types of acid sites shifted slightly towards
lower wavenumbers. This indicates a slight decrease in the strength of the acidic sites,
possibly due to interaction of Mo oxides with the hydroxyl groups and oxygens on the
surface of SZ [47]. Also, a qualitative slight increase in the concentration of Lewis acid

114
sites could be seen. This could have been due to generation of MoO3 species that are
Lewis acidic in nature [47, 48].
The thermal stability of acid sites up to 400 °C was tested [shown in Appendix D,
D.2.2 DRIFTS]. Despite higher temperatures, all the catalysts maintained both types of
acid sites and the pattern was quite similar for all.
5.3.4 NH3-TPD
Ammonia TPD of all the catalysts showed strong acid sites. TPD results for Mo-
doped catalysts followed a trend similar to that of base SZ catalyst. Several reports [36,
37, 46] suggest that beyond 600-700 °C, sulfates on the surface start to decompose
(considering that the calcination temperature was 550 °C). This often generates a false
signal in the TCD, but this signal can be correctly distinguished from the ammonia signal
using a mass spectrometer as was done in the present case. A signal corresponding to
(m/e = 32) started to appear beyond 630 °C, confirming the decomposition of sulfate
species [46].
Figure 5.4 shows a comparison of NH3-TPD curves for all four different catalysts:
base SZ, and three Mo doped SZ catalysts (1%, and 5% doping). Sulfated zirconia
typically has a desorption peak at around 160 °C and a long shoulder that decreases
slowly down to ~ 550 °C. As Mo was doped in increasing concentrations onto SZ, the low-
temperature peaks (~160 °C) are essentially the same as that of SZ alone. The shapes
of the TPD are remarkably similar for lower Mo content, suggesting that the acidity is due
to SZ alone. At higher loading, shapes of the curve slightly change indicating a reduction
in acid sites. The total acidity, as measured by TPD, also decreases with loading. There
are several explanations: one is that the high loading may cover the catalyst surface
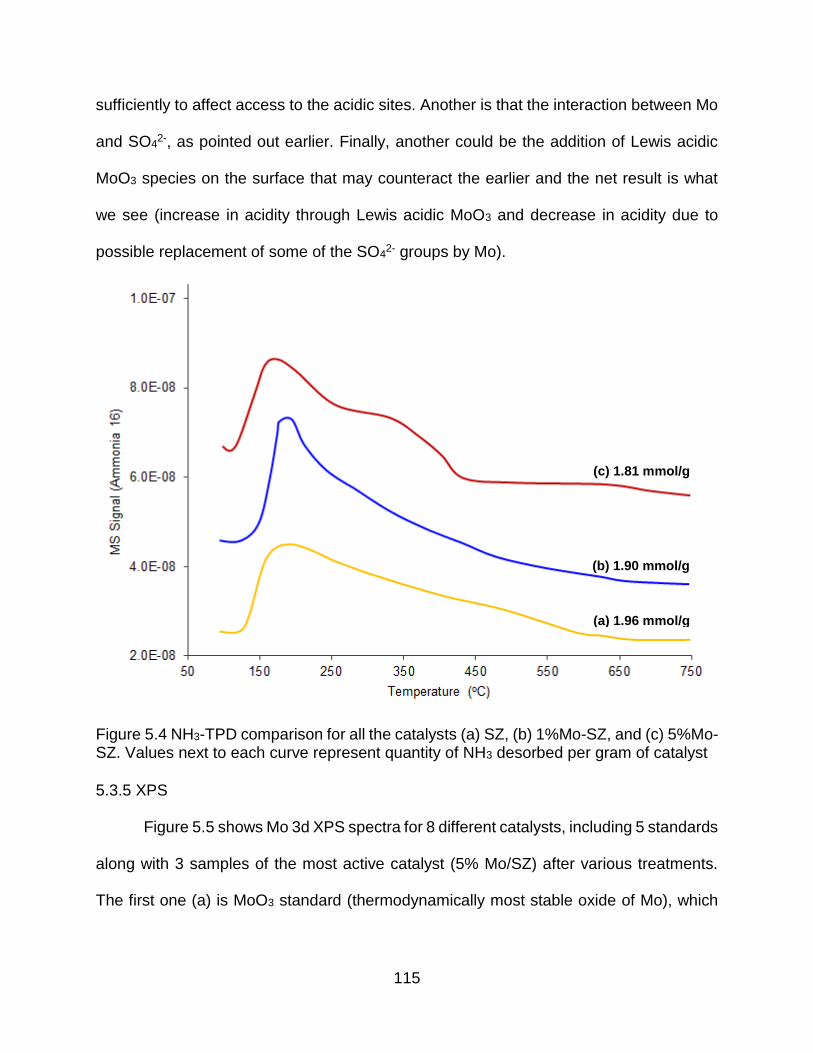
115
sufficiently to affect access to the acidic sites. Another is that the interaction between Mo
and SO42-, as pointed out earlier. Finally, another could be the addition of Lewis acidic
MoO3 species on the surface that may counteract the earlier and the net result is what
we see (increase in acidity through Lewis acidic MoO3 and decrease in acidity due to
possible replacement of some of the SO42- groups by Mo).
Figure 5.4 NH3-TPD comparison for all the catalysts (a) SZ, (b) 1%Mo-SZ, and (c) 5%Mo-SZ. Values next to each curve represent quantity of NH3 desorbed per gram of catalyst
5.3.5 XPS
Figure 5.5 shows Mo 3d XPS spectra for 8 different catalysts, including 5 standards
along with 3 samples of the most active catalyst (5% Mo/SZ) after various treatments.
The first one (a) is MoO3 standard (thermodynamically most stable oxide of Mo), which
(a) 1.96 mmol/g
(b) 1.90 mmol/g
(c) 1.81 mmol/g

116
shows standard 3d spin orbit coupling of 3d5/2 (BE = 233 eV) and 3d3/2,corresponding to
a +6 oxidation state [49].
Figure 5.5 XPS comparison of Mo 3d for (a) MoO3 std., (b) Fresh 5%Mo/SZ (oxide form), (c) Carburized 5% Mo/SZ, (d) Spent 5%Mo/SZ (after reaction for 1000 min at 650 °C), (e) β-Mo2C std., (f) MoO2 std., (g) MoS2 std., (h) Mo metal powder std

117
Sample (b) shows the fresh catalyst (oxidized form) without any treatment. This
sample very closely resembles with that of (a), indicating the presence of MoO3 on the
fresh sample. The third sample (c) is the fresh (oxidized form) carburized for 4 h under a
flow of CH4:H2 at a ratio of (1:4) at 650 °C after reduction under H2. This sample shows
peaks corresponding to MoO3 and another third peak at BE ~ 229 eV. This peak likely
corresponds to oxy-carbide phase of Mo (MoOxCy). The fourth sample (d) is the spent
catalyst sample after running the carburized sample in the CH4 aromatization reaction at
650 °C for 1000 min on stream. This sample also shows three peaks with two resembling
MoO3 and the third one slightly shifted towards right (BE ~ 228.6 eV) as compared to
carburized form. This more closely resembles the Mo2C phase. In order to verify this,
standards directly purchased from the manufacturer were tested: β-Mo2C (e), MoO2 (f),
MoS2 (g), Mo powder (h). Mo2C standard shows peaks corresponding to oxide phase and
a small third peak at BE ~ 228.4 eV. MoO2 is another form of Mo oxide which clearly
shows three peaks: two corresponding to MoO3, however, the third one corresponds to
+4 oxidation state (3d5/2) [50]. Since the catalysts of interest here involve Mo and S and
especially Mo ‘3d’ and S ‘2s’ regions overlap, so to remove any doubt about formation of
MoS2 or overlapping issues, the MoS2 sample (g) was tested. It shows two large peaks
that correspond to 3d5/2 and 3d3/2 of Mo corresponding to +4 oxidation state [51] and a
small peak at ~226 eV, corresponding to S ‘2s’. Clearly, MoS2 contains substantial
amount of sulfur and still S ‘2s’ peak is weak. Thus, in the samples of interest (Mo/SZ), S
content being very small compared to the ‘S’ content from MoS2, we expect negligible
influence due to S ‘2s’ peak and the peaks we observed correspond only to Mo 3d. Finally,
the Mo powder standard (h) would be expected to be in reduced form, Mo (0). For this

118
standard, we saw three peaks, two corresponding to Mo (+6), one shoulder that might
correspond to some intermediate oxide phase (+5), and a small peak at ~ 228.2 eV, which
most likely corresponds to the reduced Mo (0) [50]. On the basis of these observations,
the carburized sample is initially in the oxycarbide phase, then subsequently transformed
into Mo2C under reaction conditions.
Note that the samples were not run in-situ, so it is possible that part of the samples
was oxidized due to exposure to room conditions, forming peaks corresponding to these
oxidized parts of the catalyst, even in standards such as Mo2C, Mo powder, or MoO2,
which show peaks corresponding to MoO3.
5.3.6 XANES
Mo K-edge (Figure 5.6) and LIII-edge XANES (D.2.5 XANES, Appendix D) spectra
provided the evidence for the formation of Mo2C or MoOxCy phases, which are generally
believed to be the active sites for methane conversion. Figure 5.6 shows K-edge XANES
spectra for various Mo catalyst samples along with two reference samples of MoO3 and
Mo2C. Spectra for MoO3, fresh catalyst (oxidized form), and carburized show a pre-edge
feature that indicates the oxidation state of +6 and is attributable to the quadrupole/dipole
transition from 1s to 4d orbital [33]. After the pre-edge region, the edge region indicates
a sharp jump in the energy of absorption edge corresponding to 1s to 5p dipole transition.
Spectra for the fresh catalyst sample resembles MoO3 closely with a similar pre-
edge feature that is commonly observed in the case of Mo/H-ZSM-5 catalysts [33, 52,
53]. For the carburized, and the spent sample, there is a reduction in the energy of
absorption edge, indicating the reduced state of Mo in both these samples (carburized
and spent). This reduction in edge energy from fresh (oxidized form) to spent (after

119
reaction) was around ~ 7 eV that corresponds well with the literature [33, 52]. Mo2C
reference and the spent catalyst sample have similar absorption edge energies.
Figure 5.6 K-edge XANES spectra for catalyst samples: (a) MoO3 – std., (b) Fresh 5%Mo/SZ (oxidized form), (c) Carburized 5%Mo/SZ, (d) Spent 5%Mo/SZ, (e) Mo2C-std
Also, for the carburized and the spent sample, the pre-edge peak disappears. This
disappearance is apparent as pointed out by the arrow in Figure 5.6. Since the absorption
edge energies for carburized and spent catalysts are higher than Mo2C, it provides
(a)
(b)
(c)
(d)
(e)

120
evidence for the existence of more oxidized species than Mo2C (Mo, +2), such as MoOxCy
that disappear as the reaction progresses and spent catalyst closely resembles Mo2C [52,
53].
Figure 5.6 shows that the reference samples: MoO3 and Mo2C have stronger
oscillations in the EXAFS region farther from the edge. However, 5% Mo/SZ samples with
different treatments (fresh, carburization, and reaction) do not have these oscillations in
the EXAFS region. This might be an indication that the well dispersed Mo species in the
SZ matrix are without any long range order [33].
5.3.7 HRTEM
HRTEM was performed on all of the catalysts but the results for the 5% Mo/SZ are
shown here since it has the highest activity (section 5.3.8 Reaction data). Four different
images are shown for four different phases: SZ, 5% Mo/SZ (fresh - oxide form), 5% Mo/SZ
(carburized form), and 5% Mo/SZ (spent form). All these images also have SADP
(selected area diffraction pattern) images in the inset.
There are two phases of ZrO2 in XRD (D.2.4 XRD, Appendix D), monoclinic (m)
and tetragonal (t), and tetragonal being the primary phase. In HRTEM analysis, primarily
monoclinic (m-ZrO2) planes (111) were observed with few t-ZrO2 (011). Figure 5.7 shows
an image for the base SZ. Planes corresponding to m-ZrO2 and t-ZrO2 are seen.
However, a bulk analysis of the sample shows rings corresponding to t-ZrO2 (011, 002,
020, 121, etc.) as shown in the inset of Figure 5.7. This illustrates the SADP for a zoomed-
out sample. This confirms the observations from XRD (D.2.4 XRD, Appendix D) that the
bulk phase of ZrO2 was tetragonal.

121
Figure 5.7 HRTEM for base sulfated zirconia (SZ)
When Mo was impregnated on SZ, new planes appear, corresponding to MoO3
species, as seen in Figure 5.8. Further, Mo appears finely dispersed in the SZ matrix [54,
55] as seen in Figure 5.8. Tetragonal zirconia (t-ZrO2) was also detected that often is the
crystalline part of sulfated zirconia as detected in XRD [36].
For the carburized catalyst, in addition to ZrO2 planes, other planes were observed.
Some of these planes correspond to MoO3. There are also planes that correspond to
Mo2C and there are some that do not belong to either MoO3 or Mo2C, along with ZrO2.
This could be due to the formation of an intermediate phase of MoOxCy [56, 57]. Formation
of Mo2C was also confirmed using HRTEM of carburized sample of 5% Mo loaded on SZ
catalyst, as observed in Figure 5.9. A small portion on the carburized sample showed d
spacing corresponding to (111, 201) plane of Mo2C. When this carburized sample is run
further under DHA conditions for 16 h, there are changes in the lattice planes.

122
Figure 5.8 HRTEM for fresh 5% Mo/SZ catalyst
Figure 5.10 shows HRTEM of a spent 5% Mo/SZ catalyst sample. Greater
amounts of carbon were observed, and this carbon can be divided into two types: graphitic
(primarily around the edges) and amorphous (over the entire sample). There are also 2-
3 nm particles around the edges, which most likely correspond to Mo2C nanoparticles
[54]. Surrounding these particles, there are few layers of graphitic carbon. Similar results
can be observed in the literature [54, 55, 58, 59], which confirmed the presence of Mo2C
in H-ZSM-5.
These results show that Mo is finely dispersed in SZ matrix and it also shows the
state of Molybdenum through different phases of reaction and treatments: Fresh
Carburized Spent. TEM analysis confirmed the conclusions drawn from other
characterizations and explains this Mo transition from oxidized form into the oxycarbide

123
Figure 5.9 HRTEM for carburized 5%Mo/SZ catalyst
Figure 5.10 HRTEM for spent 5% Mo/SZ catalyst

124
form and finally into the carbidic form. TEM also supported the observations from XRD
(D.2.4 XRD, Appendix D) about the bulk phases of the sample.
5.3.8 Reaction data
5.3.8.1 Effect of Mo content
Results of reaction runs from the Mo-doped SZ catalysts varied considerably
(Figure 5.11 and Figure 5.12) albeit majority of the runs had naphthalene and coke as
primary products. Among other products included ethylene, ethane, propylene, propane,
and aromatics including benzene, toluene and xylenes along with hydrogen.
Figure 5.11 Reaction data for methane DHA over 1% Mo/SZ catalyst (650 °C, 1 atm, 0.6 L gcat-1 h-1)
There are several general trends of conversion and selectivity as a function of Mo
loading and time. First, methane conversion decreases with time at all Mo loadings. This
can be attributed to coke formation, which likely forms almost initially and accumulates

125
with time-on-stream. Second, selectivity to aromatics is generally ~ 10-15 % initially, but
decreases rapidly with time. These compounds are believed to be the precursors for
polynuclear aromatic compounds, which comprise coke.
The rate at which methane conversion decreased varied slightly from catalyst to
catalyst, depending on Mo loading. The 1% Mo/SZ showed a rapid drop in conversion of
methane as compared to 5% Mo/SZ. For the 1% Mo/SZ catalyst, the conversion dropped
from ~ 14 to ~ 5% in 16 hours. However, 5% Mo loaded catalyst showed more gradual
decrease in the conversion of methane, from ~ 20 to 6.5%.
Figure 5.12 Reaction data for methane DHA over over 5% Mo/SZ catalyst (650 °C, 1 atm, 600 ml/gcat-h)
For the 5% Mo/SZ catalyst, even the selectivity to non-coke products for this
catalyst did not show a rapid drop as compared to the 1% Mo/SZ. Overall, 5% Mo loaded
catalyst showed higher overall conversion compared to the other catalyst.

126
Ethylene selectivity is a clear anomaly. For both catalysts, ethylene selectivity
generally increased with time up to ~ 10-12%. For the 1% Mo/SZ catalyst, ethylene
selectivity does not significantly decrease with time after 600-700 min, even when
methane conversion decreases. For the 5% Mo/SZ catalyst, there is some decrease in
ethylene selectivity after 600-700 min. It is clear that the dimerization of methane is a
primary product of methane conversion on both catalysts, and subsequent conversion to
measurable products is relatively less selective. This supports the general observation
that active Mo sites are responsible for the production of C2 dimers. Higher loadings of
Mo contain more active sites, but these sites are of comparable selectivity.
Over time, coke deposition starts to block the BAS, decreasing the rate of
formation of aromatics and blocking access to the Mo active sites, though selectivity to
ethylene is relatively steady beyond ~700 min on stream. These results indicate that the
coke deactivates BAS first and subsequently deactivates Mo active sites. Similar results
have been observed in the literature [60] in which it was found that the rate of formation
of C2-C3 hydrocarbons increased with decreasing benzene and naphthalene selectivity
rates. The large amount of naphthalene and coke is likely due to high rates of cyclization
and dimerization of olefinic C2 intermediates over strong acid sites from sulfated zirconia.
Selectivity to higher aromatics such as ethylbenzene or xylenes (o, m, p) increased
over time (not shown) but was negligible. During an initial period of several hours, there
was no measurable formation of these compounds. Only benzene and toluene could be
observed during these initial hours. As the methane conversion decreased, the formation
of these higher aromatics increased. This trend was true for both the catalysts.

127
Coke deposition is the most probable reason for deactivation of these catalysts,
although loss of SO42- groups (SI, 5.6.3.1) from the surface of sulfated zirconia at high
temperatures could also be responsible for deactivation [46]. This loss could not be ruled
out because during NH3-TPD, starting at around 620 °C, the oxygen signal (m/z = 32)
was detected, indicative of an oxide compound decomposition. ZrO2 being very stable
compound (m.p. = 2715 °C), it is unlikely that Zr-O bonds would break down. Neither
would any molybdenum oxide-based compound because MoO3 sublimes at ~ 1150 °C.
The only remaining source of oxygen then could be from SO42- groups, which are co-
ordinated on the surface of ZrO2. Similar decomposition of sulfated ZrO2 has been
observed in the literature [61], indicating that O2 from the ZrO2 structure is highly unlikely,
so the oxygen from SO4-2 could be responsible for that signal.
5.3.8.2 Effect of temperature
Since 5% Mo doped SZ activity/selectivity was representative of the other
catalysts, the effect of reaction temperature was studied using this catalyst. Methane DHA
has been typically studied in the temperature range of around 700 °C [27]. Sulfated
zirconia is not stable beyond 700 °C, and starts to decompose at ~ 620 °C [62]. Thus, the
effect of temperature on 5% Mo doped SZ was studied at 600, 650, and 700 °C, spanning
this temperature range of interest here.
Table 5.2 shows that methane conversion increased with temperature, reaching ~
20 % at 700 °C. The aliphatic and aromatic selectivities show no clear trend with
temperature. The results in Figure 5.13 and Figure 5.14 are the net result of competing
rates of reaction, deactivation, and the rate of sulfate decomposition from surface. The
reaction rate increases with temperature while deactivation due to sulfate decomposition

128
is more rapid as well. This has been observed on Mo/H-ZSM-5 catalysts as well [27, 63],
in which higher temperatures led to higher activity but faster deactivation because rate of
naphthalene and coke formation also increases. This is apparent for Mo/SZ catalysts as
seen in Table 5.2 as well.
Table 5.2 Effect of temperature on CH4 DHA over 5% Mo/SZ catalyst (1 atm, 0.6 L gcat-1 h-1). Results are at 2 hrs into the reaction
Temperature (°C)
CH4 conversion
(%)
Aromatics selectivity (%)
Aliphatics selectivity
(%)
Naphthalene + coke selectivity
(%)
600 5.43 16.0 16.3 67.6
650 17.0 14.0 9.4 76.6
700 20.2 11.0 10.7 78.2
Figure 5.13 shows that ethylene increases monotonically with time, reaching ~8-
9 % selectivity at higher temperatures, then decreases. Selectivity goes through a
Figure 5.13 Effect of temperature on ethylene selectivity over 5% Mo/SZ catalyst (0.6 L gcat-1 h-1, 1 atm)

129
maximum. In the case of higher temperatures, selectivity drops quickly. Activity decreases
more rapidly at higher temperatures. This might be due to a faster rate of deactivation at
higher temperatures. Except for lower temperature of 600 °C, selectivity continues to
increase with time on stream.
Benzene selectivity also goes through a maximum and then decreases constantly
with time at each temperature (Figure 5.14).
Figure 5.14 Effect of temperature on benzene selectivity over 5%Mo/SZ catalyst (1 atm, 0.6 L gcat-1 h-1)
A rise in ethylene selectivity and a drop in benzene selectivity suggests that the
sites for conversion of the dimers to benzene are deactivated by coke deposition with
time, and this trend is seen at all three temperatures tested here. But this does not mean
that there is no deposition of coke on methane activation sites. Note that methane
conversion is decreasing as well, so that the yield of ethylene is decreasing despite the
fact that the selectivity is increasing. This indicates that coke is deposited on Mo sites as

130
well, as shown by HRTEM that graphitic carbon layers around Mo particles (see Figure
5.10).
The main difference for the lower benzene selectivity at 650 °C and 700 °C, as
compared to 600 °C, might be due to loss of BAS through loss of volatile SO42-. Similar
conclusions can also be drawn from TPO (Figure D.11, Appendix D) ran for these three
spent catalyst samples. Catalyst ran at 700 °C showed highest amount of coke deposited
only to be followed by catalyst ran at 650 °C and the least amount of coke was observed
on the catalyst ran at 600 °C.
5.3.8.3 Effect of space velocity
Figure 5.15, Figure 5.16, and Figure 5.17 show that higher space velocities over
5% Mo/SZ lead to both lower CH4 conversion (Figure 5.15), and to lower benzene
selectivity (Figure 5.17). This is expected because often this reaction is not considered
as mass transfer limited [63]. Figure 5.16 shows that ethylene selectivity initially increases
slowly, corresponding to high initial benzene production (Figure 5.17). However, this
changes very quickly with time-on-stream as ethylene selectivity goes through a
maximum whereas, benzene selectivity decreases rapidly. This can be attributed to coke
formation, which blocks BAS and limiting the aromatization of ethylene.
Ethylene selectivity reaches a similar steady state in all three SVs (Figure 5.16),
but the maximum is reached at earlier times-on-stream as the space velocity is increased.
The maxima in ethylene selectivity represents the result of two rates: ethylene formation
from methane, and ethylene reaction leading to benzene. With time, benzene selectivities
decrease to near-zero values, while ethylene selectivities reach a steady state. This
means that although very little benzene is formed after ~1000 min, active sites capable
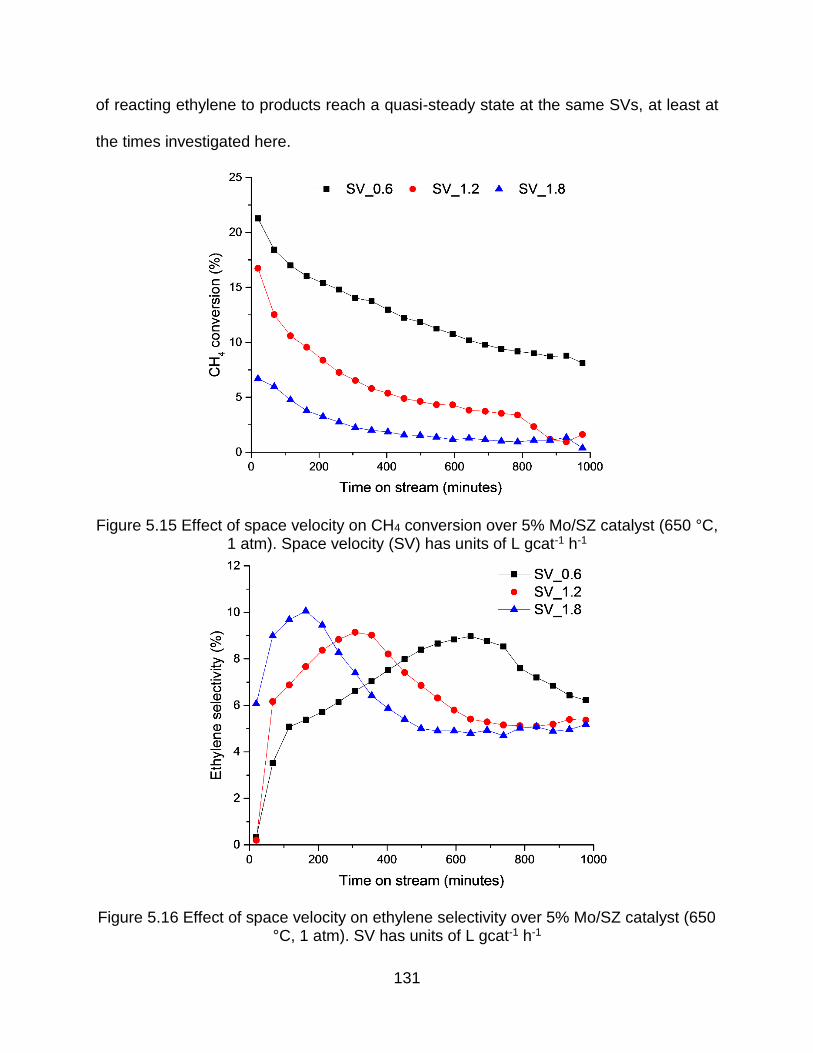
131
of reacting ethylene to products reach a quasi-steady state at the same SVs, at least at
the times investigated here.
Figure 5.15 Effect of space velocity on CH4 conversion over 5% Mo/SZ catalyst (650 °C, 1 atm). Space velocity (SV) has units of L gcat-1 h-1
Figure 5.16 Effect of space velocity on ethylene selectivity over 5% Mo/SZ catalyst (650 °C, 1 atm). SV has units of L gcat-1 h-1

132
Coking leads to blockage of BAS that in turn affects the production of benzene. Mo
sites are also blocked but possibly at a slower rate than BAS. Thus, selectivity to ethylene
reaches a steady state value although the methane conversion is still decreasing.
TPO (Figure D.12, Appendix D) results on the spent catalysts showed that the
greatest coke deposition was observed on the lowest space velocity of 0.6 Lgcat-1h-1,
while the lowest coke deposition was found on the catalyst ran at highest space velocity
of 1.8 Lgcat-1h-1. This was surprising because the rate of decay in benzene formation
(Figure 5.17) and time to reach maximum ethylene selectivity (Figure 5.16) was faster in
the case of higher space velocity, whereas the amounts of coke at those space velocities
were lower. This can be attributed to the preferential nature of coke formation near outer
surface of catalyst particles [63, 64].
Figure 5.17 Effect of space velocity on benzene selectivity over 5% Mo/SZ catalyst (650 °C, 1 atm). SV has units of L gcat-1 h-1
Additionally, in these types of reactions benzene production is often associated
with H2 generated in this reaction. Thus, in the case of lower space velocity, more H2 is

133
generated than at higher space velocity. This additional hydrogen might be the answer to
the disparity between amount of coke and corresponding activity for Mo/SZ catalysts
similar to Mo/H-ZSM-5 [53, 64-66].
5.3.6.4 Comparison with literature
A comparison of Mo/SZ with Mo supported on different supports from the literature
is shown in Table 5.3. This shows that benzene yield on Mo/SZ at 650 °C is comparable
to Mo/H-ZSM-5 at 675 °C. At 700 °C, benzene and the overall aromatics yield for Mo/H-
ZSM-5 are far greater compared to Mo/SZ. Compared to other acidic supports such as
H-Mordenite or H-SSZ-13 that possess strong acidity, sulfated zirconia shows greater
benzene yields (Table 5.3). Although, this is not a direct comparison because
physical/chemical factors such as pretreatment, space velocity, Mo content can contribute
to this difference. For example, the sulfate may not be stable at temperatures 700 °C, and
the lack of shape selectivity [34] in sulfated zirconia, may affect the comparison of Mo/SZ
and the literature.
Table 5.3 Comparison of reaction performance with literature
Catalyst Mo
content (wt%)
Temperature (°C)
Space velocity
(Lgcat-1h-1)
Max. Benzene yield (%)
Reference
Mo/H-ZSM-5 6 675 1.5 3 [63]
Mo/H-ZSM-5 6 700 1.5 6 [63]
Mo/H-ZSM-5 5 700 1.5 6 [60]
Mo/H-ZSM-5 5 700 0.8a 11 [31]
Mo/H-Mordenite 5 700 2.0a 0.5 [34]
Mo/H-SSZ-13 5 700 2.0a negligible [34]
Mo/SZ 5 650 1.2 1.8 Current work
Mo/SZ 5 700 0.6 3 Current work

134
Although Mo/SZ has no shape selectivity, the strong acidity in SZ is sufficient to
catalyze the aromatization reaction. The literature shows that benzene selectivities on
Mo/H-ZSM-5 are 50-90% and thus, is more active for aromatization of methane to
benzene than Mo/SZ. Mo/SZ is more selective for non-oxidative conversion of methane
to heavier aromatics such as naphthalene and coke.
5.3.7 Temperature programmed oxidation (TPO)
Figure 5.18 shows TPO results for 1 % and 5 % Mo/SZ catalysts.
Figure 5.18 TPO profiles for the two spent catalysts (after DHA reaction for 16 h at 650 °C, 0.6 L gcat-1h-1, 1 atm)
There are distinct differences among the two spent catalysts, as expected due to
different activities of methane DHA. Based on the TPO peak temperatures, the coke on
both catalysts can be described as either amorphous or soft coke, corresponding to peak

135
temperatures of ~ 400 °C [67] or polymeric, and hard coke corresponding to peak
temperatures of ~ 500-600 °C [68]. This hard coke is often associated with deactivation
of strong BAS. The TPO results for the 1% and 5% Mo/SZ catalysts showed similar
pattern of peak TPO positions.
For 1% Mo/SZ, there is one large peak at ~ 590 °C while one at ~510 °C for 5%
Mo/SZ. Both can be attributed to polymeric coke. This shift in peak positions is attributable
to the amount of acidity of the catalyst (NH3-TPD results). More acidity might correspond
to higher peak oxidation temperature [68]. For 1% Mo/SZ, an identifiable small peak can
be seen at ~ 440 °C, which can be designated as amorphous coke [67]. Another small
peak at ~ 685 °C for the 5% Mo/SZ can be designated as graphitic coke. Overall, 5%
Mo/SZ catalyst that showed greater activity in DHA but also greater amounts of coke
(Table 5.4).
Table 5.4 Quantification of coke using TPO (after ~ 16 h on stream)
Catalyst Amount of coke deposited
(mmol/gcat)
1% Mo/SZ 5.80
5% Mo/SZ 10.5
5.4 Conclusions
Supported Mo/SZ catalyst is active for the DHA reaction. SEM-EDS and ICP-OES
confirmed the actual loading of 1%, and 5% Mo on SZ. Pyridine-based DRIFTS and NH3-
TPD analysis shows that both Brønsted and Lewis sites are present on these catalysts.
Further analysis confirmed that Mo loading did not significantly affect acidity. At the higher
5% Mo loading, more Lewis acid sites were observed than SZ, however BAS were
weakened by the addition of Mo, possibly due to interaction between sulfate and Mo.

136
Higher loadings of Mo resulted in lower total acidity. The presence of the apparent active
sites, MoOxCy were confirmed through XANES, XPS, and HRTEM. Mo loading strongly
influenced the dispersion and number of available sites for methane activation. It appears
that the loss of sulfate content and significant coking leads to deactivation of the catalysts.
A literature comparison of benzene selectivities shows that Mo/H-ZSM-5 is far more
active than Mo/SZ. Mo/SZ was found to be more selective for non-oxidative conversion
of methane to heavier aromatics such as naphthalene and coke.
5.5 References
[1] R.S. Middleton, R. Gupta, J.D. Hyman, H.S. Viswanathan, The shale gas revolution: Barriers, sustainability, and emerging opportunities, Applied Energy, 199 (2017) 88-95. [2] A. Press, CO2 emissions in U.S. drop to 20-year low, in, Politico, 2012. Available from: https://www.politico.com/story/2012/08/co2-emissions-in-us-drop-to-20-year-low-079802 [3] G.E. Keller, M.M. Bhasin, Synthesis of ethylene via oxidative coupling of methane, Journal of Catalysis, 73 (1982) 9-19. [4] N. Kumar, Z. Wang, S. Kanitkar, J.J. Spivey, Methane reforming over Ni-based pyrochlore catalyst: deactivation studies for different reactions, Applied Petrochemical Research, (2016) 1-7. [5] D. Pakhare, J. Spivey, A review of dry (CO2) reforming of methane over noble metal catalysts, Chemical Society Reviews, 43 (2014) 7813-7837. [6] G.A. Olah, A. Goeppert, M. Czaun, G.K.S. Prakash, Bi-reforming of Methane from Any Source with Steam and Carbon Dioxide Exclusively to Metgas (CO–2H2) for Methanol and Hydrocarbon Synthesis, Journal of the American Chemical Society, 135 (2013) 648-650. [7] V.R. Choudhary, K.C. Mondal, T.V. Choudhary, Oxy-methane reforming over high temperature stable NiCoMgCeOx and NiCoMgOx supported on zirconia–haffnia catalysts: Accelerated sulfur deactivation and regeneration, Catalysis Communications, 8 (2007) 561-564. [8] B. Christian Enger, R. Lødeng, A. Holmen, A review of catalytic partial oxidation of methane to synthesis gas with emphasis on reaction mechanisms over transition metal catalysts, Applied Catalysis A: General, 346 (2008) 1-27.

137
[9] A. Zhang, S. Sun, Z.J.A. Komon, N. Osterwalder, S. Gadewar, P. Stoimenov, D.J. Auerbach, G.D. Stucky, E.W. McFarland, Improved light olefin yield from methyl bromide coupling over modified SAPO-34 molecular sieves, Physical Chemistry Chemical Physics, 13 (2011) 2550-2555. [10] Q. Zhu, S.L. Wegener, C. Xie, O. Uche, M. Neurock, T.J. Marks, Sulfur as a selective ‘soft’ oxidant for catalytic methane conversion probed by experiment and theory, Nat Chem, 5 (2013) 104-109. [11] A. Iulianelli, S. Liguori, J. Wilcox, A. Basile, Advances on methane steam reforming to produce hydrogen through membrane reactors technology: A review, Catalysis Reviews, 58 (2016) 1-35. [12] G.A. Olah, G. Klopman, R.H. Schlosberg, Super acids. III. Protonation of alkanes and intermediacy of alkanonium ions, pentacoordinated carbon cations of CH5+ type. Hydrogen exchange, protolytic cleavage, hydrogen abstraction; polycondensation of methane, ethane, 2,2-dimethylpropane and 2,2,3,3-tetramethylbutane in FSO3H-SbF5, Journal of the American Chemical Society, 91 (1969) 3261-3268. [13] G.A. Olah, R.H. Schlosberg, Chemistry in super acids. I. Hydrogen exchange and polycondensation of methane and alkanes in FSO3H-SbF5 ("magic acid") solution. Protonation of alkanes and the intermediacy of CH5+ and related hydrocarbon ions. The high chemical reactivity of "paraffins" in ionic solution reactions, Journal of the American Chemical Society, 90 (1968) 2726-2727. [14] S. Vasireddy, S. Ganguly, J. Sauer, W. Cook, J.J. Spivey, Direct conversion of methane to higher hydrocarbons using AlBr3-HBr superacid catalyst, Chemical Communications, 47 (2011) 785-787. [15] W. Hua, A. Goeppert, J. Sommer, Methane activation in the presence of Al2O3-promoted sulfated zirconia, Applied Catalysis A: General, 219 (2001) 201-207. [16] D. Fraenkel, Methane conversion over sulfated zirconia, Catalysis Letters, 58 (1999) 123-125. [17] S. Rezgui, A. Liang, T.K. Cheung, B.C. Gates, Methane conversion to ethane in the
presence of iron‐ and manganese‐promoted sulfated zirconia, Catalysis Letters, 53 (1998) 1-2. [18] X. Guo, G. Fang, G. Li, H. Ma, H. Fan, L. Yu, C. Ma, X. Wu, D. Deng, M. Wei, D. Tan, R. Si, S. Zhang, J. Li, L. Sun, Z. Tang, X. Pan, X. Bao, Direct, Nonoxidative Conversion of Methane to Ethylene, Aromatics, and Hydrogen, Science, 344 (2014) 616-619. [19] D. Wang, J.H. Lunsford, M.P. Rosynek, Catalytic conversion of methane to benzene over Mo/ZSM-5, Topics in Catalysis, 3 (1996) 289-297.

138
[20] L. Wang, L. Tao, M. Xie, G. Xu, J. Huang, Y. Xu, Dehydrogenation and aromatization of methane under non-oxidizing conditions, Catalysis Letters, 21 (1993) 35-41. [21] L. Wang, R. Ohnishi, M. Ichikawa, Selective Dehydroaromatization of Methane toward Benzene on Re/HZSM-5 Catalysts and Effects of CO/CO2 Addition, Journal of Catalysis, 190 (2000) 276-283. [22] A.A. Gabrienko, S.S. Arzumanov, A.V. Toktarev, I.G. Danilova, I.P. Prosvirin, V.V. Kriventsov, V.I. Zaikovskii, D. Freude, A.G. Stepanov, Different Efficiency of Zn2+ and ZnO Species for Methane Activation on Zn-Modified Zeolite, ACS Catalysis, 7 (2017) 1818-1830. [23] A.A. Gabrienko, S.S. Arzumanov, M.V. Luzgin, A.G. Stepanov, V.N. Parmon, Methane Activation on Zn2+-Exchanged ZSM-5 Zeolites. The Effect of Molecular Oxygen Addition, The Journal of Physical Chemistry C, 119 (2015) 24910-24918. [24] L. Li, S. Fan, X. Mu, Z. Mi, C.-J. Li, Photoinduced Conversion of Methane into Benzene over GaN Nanowires, Journal of the American Chemical Society, 136 (2014) 7793-7796. [25] J. Oró, J. Han, High-Temperature Synthesis of Aromatic Hydrocarbons from Methane, Science, 153 (1966) 1393. [26] J.J. Spivey, G. Hutchings, Catalytic aromatization of methane, Chemical Society Reviews, 43 (2014) 792-803. [27] Z.R. Ismagilov, E.V. Matus, L.T. Tsikoza, Direct conversion of methane on Mo/ZSM-5 catalysts to produce benzene and hydrogen: achievements and perspectives, Energy & Environmental Science, 1 (2008) 526-541. [28] K. Sun, D.M. Ginosar, T. He, Y. Zhang, M. Fan, R. Chen, Progress in Nonoxidative Dehydroaromatization of Methane in the Last 6 Years, Industrial & Engineering Chemistry Research, 57 (2018) 1768-1789. [29] I. Vollmer, I. Yarulina, F. Kapteijn, J. Gascon, Progress in Developing a Structure-Activity Relationship for the Direct Aromatization of Methane, ChemCatChem, 11 (2019) 39-52. [30] A. Kumar, K. Song, L. Liu, Y. Han, A. Bhan, Absorptive Hydrogen Scavenging for Enhanced Aromatics Yield During Non-oxidative Methane Dehydroaromatization on Mo/H-ZSM-5 Catalysts, Angewandte Chemie International Edition, 57 (2018) 15577-15582. [31] N. Kosinov, F.J.A.G. Coumans, G. Li, E. Uslamin, B. Mezari, A.S.G. Wijpkema, E.A. Pidko, E.J.M. Hensen, Stable Mo/HZSM-5 methane dehydroaromatization catalysts

139
optimized for high-temperature calcination-regeneration, Journal of Catalysis, 346 (2017) 125-133. [32] M. Agote-Arán, A.B. Kroner, H.U. Islam, W.A. Sławiński, D.S. Wragg, I. Lezcano-González, A.M. Beale, Determination of Molybdenum Species Evolution during Non-Oxidative Dehydroaromatization of Methane and its Implications for Catalytic Performance, ChemCatChem, 11 (2019) 473-480. [33] L.G. Inés, O. Ramon, R. Mauro, G. Pieter, B.S. W., W.B. M., B.A. M., Molybdenum Speciation and its Impact on Catalytic Activity during Methane Dehydroaromatization in
Zeolite ZSM‐5 as Revealed by Operando X‐Ray Methods, Angewandte Chemie, 128 (2016) 5301-5305. [34] N. Kosinov, F.J.A.G. Coumans, E.A. Uslamin, A.S.G. Wijpkema, B. Mezari, E.J.M. Hensen, Methane Dehydroaromatization by Mo/HZSM-5: Mono- or Bifunctional Catalysis?, ACS Catalysis, 7 (2017) 520-529. [35] Y. Luo, Z. Mei, N. Liu, H. Wang, C. Han, S. He, Synthesis of mesoporous sulfated zirconia nanoparticles with high surface area and their applies for biodiesel production as effective catalysts, Catalysis Today, 298 (2017) 99-108. [36] K.M. Arata, H., Solid Superacids, Nova Science Publishers, New York, 2011. [37] B.M. Reddy, M.K. Patil, Organic Syntheses and Transformations Catalyzed by Sulfated Zirconia, Chemical Reviews, 109 (2009) 2185-2208. [38] F.F. Oloye, A.J. McCue, J.A. Anderson, n-Heptane hydroconversion over sulfated-zirconia-supported molybdenum carbide catalysts, Applied Petrochemical Research, 6 (2016) 341-352. [39] N. Katada, J.-i. Endo, K.-i. Notsu, N. Yasunobu, N. Naito, M. Niwa, Superacidity and Catalytic Activity of Sulfated Zirconia, The Journal of Physical Chemistry B, 104 (2000) 10321-10328. [40] E.A. El-Sharkawy, A.S. Khder, A.I. Ahmed, Structural characterization and catalytic activity of molybdenum oxide supported zirconia catalysts, Microporous and Mesoporous Materials, 102 (2007) 128-137. [41] A.S.C. Brown, J.S.J. Hargreaves, S.H. Taylor, A study of “superacidic” MoO3/ZrO2 catalysts for methane oxidation, Catalysis Letters, 57 (1999) 109-113. [42] T.-c. Xiao, A.P.E. York, V.C. Williams, H. Al-Megren, A. Hanif, X.-y. Zhou, M.L.H. Green, Preparation of Molybdenum Carbides Using Butane and Their Catalytic Performance, Chemistry of Materials, 12 (2000) 3896-3905.

140
[43] A.C. Ferrari, Raman spectroscopy of graphene and graphite: Disorder, electron–phonon coupling, doping and nonadiabatic effects, Solid State Communications, 143 (2007) 47-57. [44] J. Schwan, S. Ulrich, V. Batori, H. Ehrhardt, S.R.P. Silva, Raman spectroscopy on amorphous carbon films, Journal of Applied Physics, 80 (1996) 440-447. [45] E.P. Parry, An infrared study of pyridine adsorbed on acidic solids. Characterization of surface acidity, Journal of Catalysis, 2 (1963) 371-379. [46] W.-H. Chen, H.-H. Ko, A. Sakthivel, S.-J. Huang, S.-H. Liu, A.-Y. Lo, T.-C. Tsai, S.-B. Liu, A solid-state NMR, FT-IR and TPD study on acid properties of sulfated and metal-promoted zirconia: Influence of promoter and sulfation treatment, Catalysis Today, 116 (2006) 111-120. [47] B. Li, S. Li, N. Li, H. Chen, W. Zhang, X. Bao, B. Lin, Structure and acidity of Mo/ZSM-5 synthesized by solid state reaction for methane dehydrogenation and aromatization, Microporous and Mesoporous Materials, 88 (2006) 244-253. [48] W. Liu, Y. Xu, Methane Dehydrogenation and Aromatization over Mo/HZSM-5: In Situ FT–IR Characterization of Its Acidity and the Interaction between Mo Species and HZSM-5, Journal of Catalysis, 185 (1999) 386-392. [49] T.A. Patterson, J.C. Carver, D.E. Leyden, D.M. Hercules, A surface study of cobalt-molybdena-alumina catalysts using x-ray photoelectron spectroscopy, The Journal of Physical Chemistry, 80 (1976) 1700-1708. [50] T.H. Fleisch, G.J. Mains, An XPS study of the UV reduction and photochromism of MoO3 and WO3, The Journal of chemical physics, 76 (1982) 780-786. [51] K. Suzuki, M. Soma, T. Onishi, K. Tamaru, Reactivity of molybdenum disulfide surfaces studied by XPS, Journal of Electron Spectroscopy and Related Phenomena, 24 (1981) 283-287. [52] W. Ding, S. Li, G. D Meitzner, E. Iglesia, Methane Conversion to Aromatics on Mo/H-ZSM5: Structure of Molybdenum Species in Working Catalysts, The Journal of Physical Chemistry B, 105 (2001) 506-513. [53] H. Aritani, H. Shibasaki, H. Orihara, A. Nakahira, Methane dehydroaromatization over Mo-modified H-MFI for gas to liquid catalysts, Journal of Environmental Sciences, 21 (2009) 736-740. [54] E.V. Matus, I.Z. Ismagilov, O.B. Sukhova, V.I. Zaikovskii, L.T. Tsikoza, Z.R. Ismagilov, J.A. Moulijn, Study of Methane Dehydroaromatization on Impregnated Mo/ZSM-5 Catalysts and Characterization of Nanostructured Molybdenum Phases and

141
Carbonaceous Deposits, Industrial & Engineering Chemistry Research, 46 (2007) 4063-4074. [55] Z.R.M. Ismagilov, E. V.; Ismagilova, I. Z.; Kerzhentseva, I. Z.; Zailovskiia, V. I.; Dosumovb, K. D.; Mustafinc, A. G. , Structural Changes of Mo/ZSM-5 Catalysts During the Methane Dehydroaromatization, Eurasian Chemico Technological Journal, 12 (2010) 9-16. [56] S.R.J. Likith, C.A. Farberow, S. Manna, A. Abdulslam, V. Stevanović, D.A. Ruddy, J.A. Schaidle, D.J. Robichaud, C.V. Ciobanu, Thermodynamic Stability of Molybdenum Oxycarbides Formed from Orthorhombic Mo2C in Oxygen-Rich Environments, The Journal of Physical Chemistry C, 122 (2018) 1223-1233. [57] P. Delporte, F.d.r. Meunier, C. Pham-Huu, P. Vennegues, M.J. Ledoux, J. Guille, Physical characterization of molybdenum oxycarbide catalyst; TEM, XRD and XPS, Catalysis Today, 23 (1995) 251-267. [58] E.V. Matus, O.B. Sukhova, I.Z. Ismagilov, L.T. Tsikoza, Z.R. Ismagilov, Peculiarities of dehydroaromatization of CH4-C2H6 and CH4 over Mo/ZSM-5 catalysts, React. Kinet. Catal. Lett., 98 (2009) 59-67. [59] C.H.L. Tempelman, E.J.M. Hensen, On the deactivation of Mo/HZSM-5 in the methane dehydroaromatization reaction, Applied Catalysis B: Environmental, 176-177 (2015) 731-739. [60] S.J. Han, S.K. Kim, A. Hwang, S. Kim, D.-Y. Hong, G. Kwak, K.-W. Jun, Y.T. Kim, Non-oxidative dehydroaromatization of methane over Mo/H-ZSM-5 catalysts: A detailed analysis of the reaction-regeneration cycle, Applied Catalysis B: Environmental, 241 (2019) 305-318. [61] R. Srinivasan, R.A. Keogh, D.R. Milburn, B.H. Davis, Sulfated Zirconia Catalysts: Characterization by TGA/DTA Mass Spectrometry, Journal of Catalysis, 153 (1995) 123-130. [62] M. Bi, H. Li, W.-P. Pan, W.G. Lloyd, Thermal studies of metal promoted sulfated zirconia, (1996). [63] C. Karakaya, S.H. Morejudo, H. Zhu, R.J. Kee, Catalytic Chemistry for Methane Dehydroaromatization (MDA) on a Bifunctional Mo/HZSM-5 Catalyst in a Packed Bed, Industrial & Engineering Chemistry Research, 55 (2016) 9895-9906. [64] Y. Xu, Y. Song, Y. Suzuki, Z.-G. Zhang, Effect of superficial velocity on the coking behavior of a nanozeolite-based Mo/HZSM-5 catalyst in the non-oxidative CH4 dehydroaromatization at 1073 K, Catalysis Science & Technology, 3 (2013) 2769-2777.

142
[65] K. Skutil, M. Taniewski, Some technological aspects of methane aromatization (direct and via oxidative coupling), Fuel Processing Technology, 87 (2006) 511-521. [66] H. Ma, R. Ohnishi, M. Ichikawa, Highly Stable Performance of Methane Dehydroaromatization on Mo/HZSM-5 Catalyst with a Small Amount of H2 Addition into Methane Feed, Catalysis Letters, 89 (2003) 143-146. [67] C.A. Querini, Coke characterization, in: J.J. Spivey, G.W. Roberts (Eds.) Catalysis: Volume 17, The Royal Society of Chemistry, 2004, pp. 166-209. [68] F. Denardin, O.W. Perez-Lopez, Tuning the acidity and reducibility of Fe/ZSM-5 catalysts for methane dehydroaromatization, Fuel, 236 (2019) 1293-1300.

143
Chapter 6 . Future Work
Research is an ever learning, adapting and evolving process. Following are some
ideas that could be explored in the future as a continuation of the work presented here.
6.1 Continuation of superacid work
Zeolites range widely and extensively and among them, hundreds of different types
of zeolites exist. The superacid work presented in this thesis was based upon supporting
AlBr3 on H-ZSM-5. On similar lines, the following work could be assessed for its feasibility:
Supporting AlBr3 on other zeolites such as H-Mordenite (highest acidity among
zeolites), H-SSZ-13 (used in MTO reaction), H-Beta.
Gas phase superacid catalysis: Flowing a mixture of HBr, AlBr3, and CH4 over a
zeolite (H-ZSM-5/H-MOR) and checking what reaction products could be obtained.
Development of high temperature superacids: Ceramic Acids, Heteropoly Acids
(Keggin type) – Controlling coke formation in these structures would be important.
6.2 Continuation of Mo/SZ work
Many other metals can be supported on SZ (sulfated zirconia) and this catalyst
can find applications in other reactions such as light alkane aromatization.
Supporting Zn on SZ and evaluating this catalyst for ethane aromatization.
Supporting Ga on SZ and evaluating this catalyst for propane aromatization.
Evaluating the effect of promoters such as Pt, Pd, Cr on these metals in reactions
aforementioned.
Stabilization of sulfate on SZ surface, and introducing mesoporosity in the SZ
matrix. Using this as a support for Mo doping and subsequent evaluation in
methane DHA.
144
Appendix A. Permission to Use Copyrighted Materials
A.1. Permission to use Figure 2.4

145
A.2. Permission to use Chapter 3 text

146
A.3. Permission to use Chapter 3 (All figures and tables)

147
A.4. Permission to use Figure 3.9

148
A.5. Permission to use Figure 3.10

149
A.5. Permission to use Figure 3.10 (contd.)

150
A.6. Permission to use Figure 3.11

151
A.7. Permission to use Table 3.3

152
A.8. Permission to use Table 3.5

153
A.9. Permission to use Chapter 4

154
A.10. Permission to use Chapter 5

155
Appendix B. Proof of Principal Authorship
B.1. Chapter 3

156
B.2. Chapter 4

157
B.3. Chapter 5

158
Appendix C. Supplemental information for chapter 4
C.1 Materials and methods
A schematic for a reaction system is shown in Figure C.1.
Figure C.1 Schematic for CH4 reaction system
From the NH3-TPD calculations (Table 4.3, chapter 4) and the calculations for gain
in weight after grafting, it was observed that roughly 80 % of the Al sites formed through
grafting were active for NH3 adsorption and thus were used in the TOF calculation (shown
below). Another assumption that was made was the species on the surface of H-ZSM-5
are –AlBr2 with one Br losing through the reaction with hydroxyl group as shown below:
AlBr3 + -OH -O-AlBr2 + HBr (C.1)
An example calculation is as shown below:
Weight of H-ZSM-5 (before grafting) = 1.40 g,
Weight of ABZ-5 (AlBr3 grafted H-ZSM-5) = 1.53 g.

159
Difference in weight = 0.13 g
Molecular weight of –AlBr2 = 189 g/mol moles of AlBr2 grafted = 0.13 g/189
g.mol-1 = 0.000688 mol AlBr2
Moles of Al = 0.000688 mol/1.53 gcat = 0.000449 mol.gcat-1
Difference in acidity from NH3-TPD = 1556 μmol.gcat-1 – 1190 μmol.gcat-1 = 366
μmol.gcat-1
So 0.000449 mol Al adsorbed 366 μmol NH3 1 mol Al adsorbed ~ 0.82 mol of
NH3
This calculation was used in calculating TOF and thus 80% of Al sites were assumed to
be active.
C.2 Results and discussion
The grafting of AlBr3 on H-ZSM-5 was confirmed by several means: (a) gain in
weight of the zeolite after grafting, (b) the apparent color change from white to skin (also
confirmed using UV-Vis), (c) capturing the HBr gas generated during grafting in deionized
water and checking for the change in pH before and after grafting. In the (c) test, the water
pH became acidic side and the specific gravity of the water increased, consistent with the
capture of HBr. Finally, a silver nitrate (AgNO3) test was also carried out on the water
sample on the captured HBr. The test was positive, indicating that bromine was captured.
Collectively, these tests confirm the reaction between AlBr3 and surface hydroxyls of H-
ZSM-5, forming the grafted AlBrx/H-ZSM-5 complex.
It was also attempted to characterize the grafting using FTIR, Raman
spectroscopy. However, analytical capabilities of FTIR limited the inorganic bonds with
vibrations in the far-IR region that are not within the standard range available on all IR

160
spectrometers. With the Raman, excessive fluorescence from sample prevented from
obtaining any meaningful spectra of these catalysts.
There were other limitations in using X-ray edge techniques. For example,
overlapping of ‘Br’ L edge peaks with ‘Al’ K edge peaks that prevented quantification of
the elements using EDX (Energy Dispersive X-ray) spectroscopic technique. Further, 27Al
MAS NMR could not distinguish between the Al from the H-ZSM-5 framework and the Al
from AlBr3 grafting, because the contribution from Al in H-ZSM-5 was too large.
Hammett indicators are often employed to measure the superacidity of solid
catalysts. However, the Hammett indicator technique is typically based on a color change
from basic to acidic upon addition of the solid catalyst in the indicator solution. This
technique works well with the white powders that do not change the color of the indicator
solution on its own[1]. However, in the present case, the catalysts were already colored
so the Hammett indicator technique could not be employed for assessing the
acidity/superacidity of the present catalysts.
Another test of superacidity of catalysts acidity in the literature is the isomerization
of butane to isobutane at room temperature [2]. However, in the case of H-ZSM-5, butane
appears to be trapped inside the zeolite pores at low concentrations at temperatures <
100 °C. Only if the catalyst is heated to temperatures > 100 °C, butane appears to be
desorbed and could be analyzed using FID. However, carrying out the reaction of butane
to isobutane at elevated temperature is often claimed not to be a true measure of
superacidity [2, 3].

161
C.2.1 Catalyst preparation
Table C.1 describes the physico-chemical properties of the main catalyst of
interest here. Base H-ZSM-5 had a measured surface area of 435 m2/g that decreased
to 380 m2/g after the AlBr3 grafting (ABZ-5). This indicates that AlBr3 was grafted on to H-
ZSM-5. Spent ABZ-5 after the reaction at 300 oC had a surface area of 422 m2/g that
shows a slight drop from the H-ZSM-5 but a higher surface area than ABZ-5. This could
have been due to two opposite mechanisms: bromine loss would result in increase in
surface area than ABZ-5, while deposition of coke that would decrease the surface by
blocking the pores. If so, the net effect is a slight decrease of surface area compared to
the H-ZSM-5.
Table C.1 Physico-chemical properties of the catalysts
Catalyst BET
surface area (m2/g)
Al content through
XPS (at%)
Br content through
XPS (at%)
Br content through AgNO3 test (wt%)
H-ZSM-5 435 1.15 - -
Fresh ABZ-5 380 0.98 0.06 2.29
Spent ABZ-5* 422 0.94 0.04 1.43
* Spent Catalyst – ABZ-5 recovered after CH4 oligomerization at 300 oC, 1 atm, 9 L gcat-1hr-1 for 16 hrs
C.2.2 Thermodynamics
In the case of halide-based catalysts, often the possibility of formation of methyl
halide (CH3X) is considered. However, based on the thermodynamic calculations (using
HSC Chemistry 8.6) under the presence of AlBrx species, the thermodynamic extent of
CH3Br formation from CH4 is negligible. Typically formation of these species (CH3Br)
requires the presence of a stronger oxidant such as Br2 that are able to generate
significant quantities [4].

162
Figure C.2 Thermodynamic CH4 conversion against temperature (a) with coke formation allowed, (b) without coke formation allowed (calculated using HSC Chemistry 8.6)
Table C.2 Thermodynamic CH4 conversion versus temperature with and without coke
formation allowed
Temperature (oC)
CH4 conversion (%)
w/o coke w/coke
25 0 0.002
100 0.002 0.04
200 0.023 0.577
300 0.118 3.463
400 0.558 12.57
Methane conversions at the temperatures of interest here, range from 0.5 – 12 %
(Table C.2) depending on whether coke formation is allowed/not allowed. Figure C.2
shows a comparison of CH4 conversion at various temperatures from 25 oC to 1000 oC
with and without coke formation is allowed.

163
C.2.3 XRD
Figure C.3 shows a comparison between H-ZSM-5 sample, fresh AlBr3 grafted H-
ZSM-5 (ABZ-5) sample, and spent ABZ-5 sample. All three spectra show the
characteristic peaks of H-ZSM-5. When compared based H-ZSM-5 (a) and fresh ABZ-5
(b), XRD shows no extra features arising from the grafting of AlBr3.
Figure C.3 XRD comparison of (a) H-ZSM-5, (b) Fresh ABZ-5, (c) Spent ABZ-5
This indicates that AlBrx species are well dispersed and no long-range order exists.
Further no measurable difference between fresh and spent catalyst (b, c; respectively)
could be seen indicating no structural change in the base zeolite H-ZSM-5 during the
reaction, as expected due to low reaction temperatures (200- 400 oC) of interest here.
C.2.4 DRIFTS
It is known that three type of hydroxyls [5] are present on H-ZSM-5: one that is
attached to both Al, Si (Al-OH-Si), one being attached only to Si- commonly known as

164
silanols (Si-OH), and the third one being non-framework hydroxyls. Al-OH-Si hydroxyl
groups are the acidic ones having IR vibration band ~ 3610 cm-1, silanols are the terminal
–OH groups with IR vibration ~ 3740 cm-1, while the non-framework hydroxyls show
bands around 3670 – 3690 cm-1. Figure C.4 shows the IR spectrum for H-ZSM-5 and
ABZ-5 catalyst in the –OH group region from 3600-3700 cm-1. It can be observed for both
H-ZSM-5 and ABZ-5, that there exist three IR bands in the –OH region (3580 cm-1, 3650
cm-1, 3740 cm-1) that can be attributed to acidic hydroxyl, non-framework hydroxyls, and
silanols. When AlBr3 was grafted on the H-ZSM-5, we observed a decrease in the
intensity of the band around 3740 cm-1 corresponding to the silanol group. This is
consistent with the hypothesis that Br atoms from AlBr3 react with –OH groups on the
surface of H-ZSM-5. Similar decrease in the intensity of peaks corresponding to silanol
has been observed when AlCl3 was grafted on siliceous MCM-41 [6]. This decrease in
the intensity is most likely due to occurrence of the following reaction:
AlBr3 + -OH -O-AlBr2 + HBr
Thermal stability of the acid sites on the most active catalyst, ABZ-5 was also
tested using pyridine-DRIFTS. Spectra for ABZ-5 catalyst after pyridine desorption at
temperatures up to 400 °C are shown in Figure C.5. Both Lewis and Brønsted acid sites
are present on this catalyst at room temperature.
As the temperature is increased, as expected, bands corresponding to hydrogen
bound pyridine start decreasing, as do the bands corresponding to the Lewis acid site.
On the contrary, Brønsted acid sites appear to be stable at all temperatures.

165
Figure C.4 IR spectra for hydroxyl group region for H-ZSM-5 (dotted) and ABZ-5 (undotted), (a) at 25 oC, (b) at 100 oC, (c) at 200 oC, (d) at 300 oC, (e) at 400 oC

166
Figure C.5 DRIFTS spectra for ABZ-5 catalyst after pyridine desorption at (a) 25 oC, (b) 100 oC, (c) 200 oC, (d) 300 oC, (e) 400 oC
Figure C.6 DRIFTS spectra for SZ catalysts after pyridine desorption at (a) 25 oC, (b) 100 oC, (c) 200 oC, (d) 300 oC, (e) 400 oC

167
Figure C.6 and Figure C.7 also show stable acid sites of both type (L and B) up to
high temperatures ~ 400 °C for catalysts: Sulfated Zirconia as well as base H-ZSM-5
respectively. Although strength of the acid sites must be stronger in the case ABZ-5
compared to these two catalysts.
Figure C.7 DRIFTS spectra for H-ZSM-5 catalysts after pyridine desorption at (a) 25 oC,
(b) 100 oC, (c) 200 oC, (d) 300 oC, (e) 400 oC
A comparison at 300 oC for H-ZSM-5 versus ABZ-5 (Figure 4.2[a], chapter 4)
shows that both the spectra are very similar, except for a small shoulder at 1540 cm-1 that
is present in the case of ABZ-5 and not in H-ZSM-5. We believe that this could have been
due to grafting of AlBr3 on H-ZSM-5 creating a new Brønsted acid site. It is known that
DRIFTS is not a quantitative technique [7] and often transmission is the most commonly
used technique for quantification. We thus did not attempt to quantify acid sites based on
these results. However, based on just the area under curve (Table C.3), that more pyridine

168
adsorbed on the fresh ABZ-5 catalyst than H-ZSM-5, supporting the hypothesis that there
are more acid sites on fresh ABZ-5 than base H-ZSM-5.
Table C.3 Area under the curve for pyridine DRIFTS
Catalyst
Area under curve (acid sites)
L (1450 cm-1)
L+B (1490 cm-1)
B (1540 cm-1)
L (1620 cm-1)
B (1640 cm-1)
H-ZSM-5 - 3.07 4.53 2.57 3.23
Fresh ABZ-5 - 3.69 5.63 3.06 4.03
Spent ABZ-5 - 1.17 1.37 - 0.50
When spent ABZ-5 was also tested using pyridine DRIFTS, area under the curve
was clearly very less, indicating less access to the acid sites possibly due to blocking of
the pores by coke formation and due to loss of ‘Br’.
C.2.5 Reaction runs
To avoid the ambiguity of whether the activity originated from H-ZSM-5 or purely
due to AlBr3 doping, H-ZSM-5 was subjected to further analysis. At 300 °C, no significant
hydrocarbon peaks were observed for H-ZSM-5, indicating no activity. However, when
the same H-ZSM-5 is tested at 400 °C, low levels of oligomerization products are
detected, as shown in Table C.4, but the concentration is much lower compared to those
formed on the ABZ-5 catalyst. Additionally, after 2 h on-stream, only trace amounts of C2
– C5 hydrocarbons were detected.
To calculate the TOF in these (and subsequent) runs, both the methane
conversion rate and a valid value for the number of superacid sites are needed. In the
experiments discussed here, the conversion of methane is measured by the difference
between the inlet concentration (5% from a high-purity tank) and the outlet concentration,
both measured using a GC/MSD (4.2.8 Reaction studies, chapter 4).

169
Table C.4 Time on stream (TOS) of CH4 oligomerization on H-ZSM-5 at 400 oC, 9 L gact-1h-1, 1 atm. (-) = not detected
Time (hr)
TOF (h-1)
Product selectivity (%)
C2= C3 C4 C5 B T EB X (o,m,p)
1 0.002 49.0 - - - 11.2 31.9 - 7.9
2 - 99.9 - - - - - - -
3 - 99.9 - - - - - - -
Blank - - - - - - - - -
Detection limits for the hydrocarbon analytes of interest here are 0.1 ppm in these
experiments, which makes it difficult to accurately estimate the methane conversion
values (e.g. 5 % vs. 4.99 %), considering the uncertainties associated with even slight
fluctuations in flow controllers, catalyst weights, temperature of the reaction. In addition,
the superacid sites may differ at different temperatures. Thus, here in the present results,
we do not report methane conversion values. This, and because methane conversions
are typically closer to 1%, a single value for the TOF at all reaction conditions is not
possible, even if reaction conditions change slightly. We find that in the literature of
superacid methane oligomerization, TOF is often not reported, presumably due to the
uncertainties associated with these experiments.
In terms of detecting hydrogen, TCD is most often used. However, detecting low
concentrations of hydrogen is extremely difficult as TCD relies heavily on the difference
in thermal conductivities, which should be large for a detectable signal. In the present
case, the differences between the inlet and outlet concentration would be ~ 50 – 100 ppm,
which was clearly far below the detection limits on the analytical instruments used here,
and thus although hydrogen is produced, it could not be quantified.
Attempts for direct conversion of methane have been reported in the literature over
to over solid superacids, in particular using sulfated zirconia catalysts. Rezgui et al. [8],

170
Hua et al. [9], Martin and Schmal [10] observed primarily C2 hydrocarbons (mostly
ethane). Hydrogen was observed, indicating an oligocondensation mechanism similar to
the one proposed by Olah for liquid superacids [11], although others did not report any
hydrogen, possibly due to scavenging of oxygen by the dihydrogen [12] or due to
incorporation into the carbonaceous deposits [8].
C.3 References
[1] G.A. Olah, G.K. Surya Prakash, Á. Molnár, J. Sommer, General Aspects, in: Superacid Chemistry, John Wiley & Sons, Inc., 2009, pp. 1-34. [2] V. Adeeva, H.-Y. Liu, B.-Q. Xu, W.H. Sachtler, Alkane isomerization over sulfated zirconia and other solid acids, Topics in Catalysis, 6 (1998) 61-76. [3] J.R. Sohn, Recent advances in solid superacids, J. Ind. Eng. Chem., 10 (2004) 1-15. [4] K. Ding, H. Metiu, G.D. Stucky, The Selective High-Yield Conversion of Methane Using Iodine-Catalyzed Methane Bromination, ACS Catalysis, 3 (2013) 474-477. [5] H. Berndt, A. Martin, H. Kosslick, B. Lücke, Comparison of the acidic properties of ZSM-5 zeolites isomorphously substituted by Ga, In, B and Fe, Microporous Materials, 2 (1994) 197-204. [6] V.R. Choudhary, K. Mantri, AlCl3-Grafted Si–MCM-41: Influence of Thermal Treatment Conditions on Surface Properties and Incorporation of Al in the Structure of MCM-41, Journal of Catalysis, 205 (2002) 221-225. [7] T. Armaroli, T. Bécue, S. Gautier, Diffuse Reflection Infrared Spectroscopy (Drifts): Application to the in Situ Analysis of Catalysts, Oil & Gas Science and Technology - Rev. IFP, 59 (2004) 215-237. [8] S. Rezgui, A. Liang, T.K. Cheung, B.C. Gates, Methane conversion to ethane in the
presence of iron‐ and manganese‐promoted sulfated zirconia, Catalysis Letters, 53 (1998) 1-2. [9] W. Hua, A. Goeppert, J. Sommer, Methane activation in the presence of Al2O3-promoted sulfated zirconia, Applied Catalysis A: General, 219 (2001) 201-207. [10] R.L. Martins, M. Schmal, Methane activation on superacidic catalysts based on oxoanion modified zirconium oxide, Applied Catalysis A: General, 308 (2006) 143-152.

171
[11] G.A. Olah, Y. Halpern, J. Shen, Y.K. Mo, Electrophilic reactions at single bonds. XII. Hydrogen-deuterium exchange, protolysis (deuterolysis), and oligocondensation of alkanes with superacids, Journal of the American Chemical Society, 95 (1973) 4960-4970. [12] D. Fraenkel, Methane conversion over sulfated zirconia, Catalysis Letters, 58 (1999) 123-125.

172
Appendix D. Supplemental information for chapter 5
D.1 Thermodynamics
Thermodynamics for methane dehydroaromatization is quite complex and can
involve lot of possibilities. Often, the question comes whether the formation of coke should
be included or not in the thermodynamic calculations because the conversion values,
selectivity, and all other parameters change considerably. Coke is one of the most stable
entity in the overall system so given chance, all methane can get converted into is coke
and H2 unless some kinetic limitations drive the selectivity to value added products such
as aromatics, low C hydrocarbons through catalysis. For the purpose of thermodynamic
calculations, commercial software HSC Chemistry 8.1 is used. Figure D.1 shows CH4
conversion values for the DHA reaction with and without the formation of coke and the
conversion values differ considerably.
When coke formation is allowed, following reaction was used for the calculation:
𝐶𝐻4 → 𝐶𝑥𝐻𝑦(𝑥 = 1 − 8) + 𝐶10𝐻8 + 𝐶14𝐻10 + 𝐶(𝑠) + 𝐻2 (D.1)
When coke formation is not allowed, following reaction equation was used
𝐶𝐻4 → 𝐶𝑥𝐻𝑦(𝑥 = 1 − 8) + 𝐶10𝐻8 + 𝐶14𝐻10 + 𝐻2 (D.2)
When coke formation is allowed, methane conversion gets meaningful numbers
beyond 200 oC, however, when coke formation is not allowed, methane conversion gets
meaningful numbers beyond 400 oC. For the temperatures of interest in the current study
(600 – 700 oC), conversions without coke, range from (6% - 13%), whereas with coke,
they range from (60 - 80%).

173
Figure D.1 Thermodynamic CH4 conversion for dehydroaromatization (Press. = 1 bar) (a) with coke, (b) without coke. (Calculated using HSC Chemistry 8.1)
D.2 Characterizations
D.2.1 Raman spectroscopy
Raman spectra for base sulfated zirconia (SZ) and 2 other fresh catalysts (oxidized
form) is shown in Figure D.2. This spectroscopy was carried out only to confirm loading
of Mo on the sulfated zirconia and to see if this leads to any changes in the type and
concentration of surface species.When Mo was loaded onto base SZ, two key changes
could be observed. One was the reduction in the intensity of the peak corresponding to
sulfate (S=O) vibrations (~ 1025 cm-1), and the other one being formation of Mo-O-Mo (~
830 cm-1) and Mo=O species (~ 970 cm-1) [1]. At all the loadings of Mo, similar two types
of species were observed. Other vibrations below 800 cm-1 Raman shift, correspond
primarily to tetragonal ZrO2 species [2, 3] that has been discussed in the main manuscript.
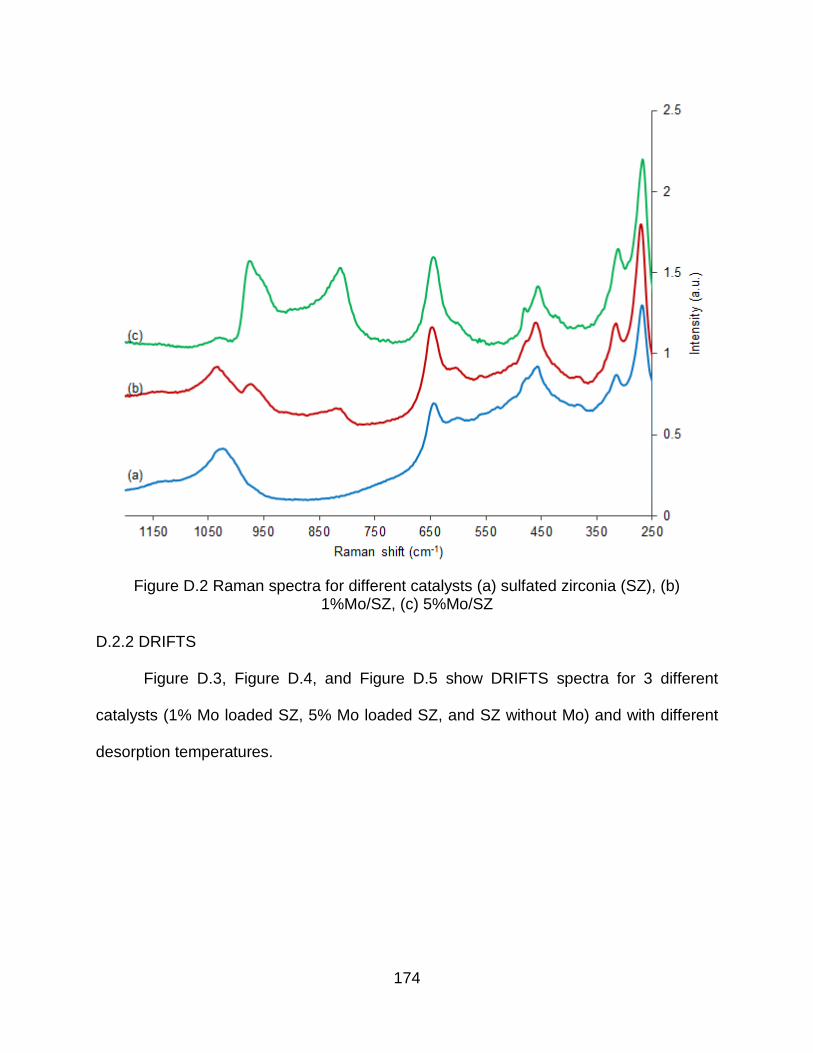
174
Figure D.2 Raman spectra for different catalysts (a) sulfated zirconia (SZ), (b)
1%Mo/SZ, (c) 5%Mo/SZ
D.2.2 DRIFTS
Figure D.3, Figure D.4, and Figure D.5 show DRIFTS spectra for 3 different
catalysts (1% Mo loaded SZ, 5% Mo loaded SZ, and SZ without Mo) and with different
desorption temperatures.

175
Figure D.3 DRIFTS spectra for 1%Mo/SZ after pyridine desorption at various
temperatures: a) 25 oC, (b) 100 oC, (c) 200 oC, (d) 300 oC, (e) 400 oC
Figure D.4 DRIFTS spectra for 5%Mo/SZ after desorption at various temperatures: (a)
25 oC, (b) 100 oC, (c) 200 oC, (d) 300 oC, (e) 400 oC

176
Figure D.5 DRIFTS spectra for sulfated zirconia (SZ) after pyridine desorption at various
temperatures: (a) 25 oC, (b) 100 oC, (c) 200 oC, (d) 300 oC, (e) 400 oC
All the catalysts showed similar type of acid sites and of both types: Lewis (~1450
cm-1, 1620 cm-1) and Brønsted (~1540 cm-1, 1640 cm-1) and acid sites at 1490 cm-1
correspond to Lewis + Brønsted type [3]. All the catalysts showed stable acid sites even
when the temperatures for desorption were raised to 400 oC. It is expected that the
Brønsted acid sites must be stable even at reaction temperatures (600 – 700 oC) although
this stability was not tested in-situ due to DRIFTS cell limitations.
D.2.3 SEM
SEM images of some of the catalysts tested are shown in Figure D.6. When a SZ
catalyst is doped with Molybdenum, we could see formation of some rod like structures
(Figure D.6-b) that most probably correspond to the formation of molybdenum oxides.
Carburized samples and spent catalyst samples show structures similar to sulfated
zirconia (SZ) samples and without any rod like structures confirming that Mo oxides were

177
converted into Mo2C or MoOxCy species as interpreted by various researchers [4, 5].
Spent catalysts (Figure D.6-d) also show some agglomeration of particles as is expected
to occur during coking of the catalysts. These results were further confirmed by TEM.
Figure D.6 SEM spectra of various catalysts (a) Sulfated Zirconia (SZ), (b) fresh 5 % Mo/SZ, (c) carburized 5%Mo/SZ, (d) spent 5%Mo/SZ
D.2.4 XRD
Sulfated zirconia has a clear and distinct XRD pattern that has been widely
reported in the literature [3, 6]. It is also known that, depending on the calcination

178
conditions, different phases of sulfated zirconia can be present. Since the present
catalysts were prepared by calcining at 550 oC, primarily tetragonal phases are expected
to be present [3]. Although, some impurities can exist due to calcination conditions and
can lead to small amount of other phases such as monoclinic. Figure D.7 shows XRD
spectra for 8 different catalyst samples that includes sulfated zirconia (SZ), fresh and
spent 1% Mo doped on sulfated zirconia, fresh and spent 5% Mo doped on sulfated
zirconia, and fresh and spent 10% Mo doped on sulfated zirconia, and also a carburized
5% Mo doped on sulfated zirconia (SZ). SZ catalyst sample clearly showed all the
characteristic peaks primarily corresponding to the tetragonal phase (ICDD PDF #
811544) although some small features at 28o and 31o were observed to be present,
attributable to monoclinic ZrO2 (ICDD PDF # 37-1484). Mo doped catalysts (all samples:
fresh, carburized, spent) did not show any difference in the XRD spectra when compared
with SZ. This suggest that Mo is well dispersed in the sulfated zirconia and most likely in
the amorphous form although possibility of crystallinity on small scale cannot be
neglected that can be confirmed using other techniques like TEM. This also suggests that
the expected MoOxCy and Mo2C are not visible by XRD for carburized and spent samples
and most likely even after the change in phase from MoO3 (fresh) to Mo2C (spent), the
form appeared amorphous and without long range order. Also, no significant difference
in the XRD indicates that the base structure of SZ did not change during the reaction of
DHA and was stable. Similar results have been reported in the literature [2] for 5%Mo
loaded on sulfated zirconia.

179
Figure D.7 XRD spectra for various catalysts: (a) SZ, (b) Fresh 1%Mo/SZ, (c) Spent 1%Mo/SZ, (d) Fresh 5%Mo/SZ, (e) Spent 5%Mo/SZ, (f) Carburized 5%Mo/SZ
D.2.5 XANES
Figure D.8 shows LIII edge spectra for Mo catalyst samples along with 2 reference
samples of MoO3 and Mo2C. Fresh catalyst resembles very well with MoO3 reference
sample with the feature of split peaks. These split peaks correspond to t2g and eg splitting
of 4d orbitals [7] and indicates tetrahedral co-ordination. Similar peaks could also be
observed in the case of carburized catalysts meaning some of the oxide features of these
catalysts are still preserved. Spent catalysts on the contrary, showed absorption edge at
low energy indicating a reduction of Mo and closely resembling Mo2C with absorption
edge around similar energy [7, 8].

180
Figure D.8 LIII edge XANES spectra for 5% Mo/SZ catalysts with different treatments: (a) MoO3-std., (b) fresh 5%Mo/SZ, (c) carburized 5%Mo/SZ, (d) spent 5%Mo/SZ, and
(e) Mo2C-std.
D.2.6 XPS
In order to support claims of Mo2C formation, C ‘1s’ XPS was also carried out.
Figure D.9 shows C ‘1s’ XPS spectra for four samples: fresh 5%Mo/SZ, carburized
5%Mo/SZ, spent 5%Mo/SZ, and Mo2C (as purchased).
Fresh catalyst shows peak corresponding to adventitious carbon at 284.6 eV.
Carburized catalyst show slightly more carbon counts than that of fresh catalyst and the
peak is slighted shifted towards lower Binding Energy. Mo2C-std shows similar feature
but the principal peak is also shifted towards lower Binding Energy. Spent 5% Mo/SZ
catalyst showed the highest amount of carbon deposited as expected and the peak was

181
asymmetric with a long tail indicative of sp2 type of carbon [9, 10]. The peak position for
principal peak more closely matches that of Mo2C. This is indicative of Mo transition from
MoO3 MoOxCy Mo2C.
Figure D.9 'C-1s' XPS spectra for various stages of catalysts; (a) fresh 5%Mo/SZ, (b)
carburized 5%Mo/SZ, (c) Mo2C-std., (d) spent 5%Mo/SZ
D.2.7 H2-TPR
In order to test the possibility of formation of H2S under H2 rich environments, H2-
TPR was carried out using a 10% H2/Ar stream. Depending on the type of sample, we

182
observed signals corresponding to H2S starting at different temperature range, as could
be observed in Figure D.10.
Figure D.10 H2S evolution through H2-TPR (10 K/min)
For 1% Mo loaded SZ catalyst, a sharp peak at around ~ 590 oC could be
observed. This sample had highest sulfur content amongst all the samples. 5%Mo loaded
SZ catalyst showed a much earlier H2S evolution starting at ~ 485 oC but a much smaller
peak. It also showed another peak at 540 oC. But considering the area, it showed much
less sulfur formation. When a 5%Mo/SZ was calcined at 650 oC, it showed no sulfur
formation, indicating that only thermally stable sulfate was present. This procedure can
be used to make more stable catalysts. Further, ICP-OES results discussed in Table D.1
indicate that not all sulfur reacted with hydrogen to form H2S, and there was still
measurable sulfur present.

183
D.3 Additional data
D.3.1 Deactivation of catalysts
We postulated two deactivation mechanisms for our catalysts: one being loss of
sulfur, and the other one being coke deposition. The coke deposition part is already
discussed in the main text. Regarding the loss of sulfur, we observed it through NH3-TPD
as well as through ICP. Table D.1 shows the elemental composition for the representative
sample (5% Mo/SZ) for fresh and spent conditions.
Table D.1 Elemental composition for fresh and spent catalysts
Catalyst Mo loading,
wt% (ICP-OES)
S loading, wt%
(ICP-OES)
SZ 0 3.5
Fresh 5%Mo/SZ
3.92 1.95
Spent* 5%Mo/SZ
3.95 1.25
* Spent catalyst refers to the sample recovered after 4 hrs pretreatment + 16 hrs of reaction at
650 oC over a fresh (oxidized) catalyst.
It shows that the Mo did not undergo any change in elemental composition
indicating that Mo did not come off during the carburization as well as during the reaction.
On the contrary, a loss in the content of sulfur is observed for the fresh and spent catalyst.
This can primarily be attributed to the volatility of sulfur from SO42- at these temperatures.
Temperature programmed oxidation (TPO) profiles for spent catalysts from other
runs such as for samples ran at different temperatures (Figure D.11) and for samples ran
at different space velocities (Figure D.12) are also shown below. They correlate well with
what was seen in terms of activity of these catalysts at different temperatures and different
space velocities.

184
Figure D.11 TPO curves for spent catalyst samples ran at various temperatures
(5%Mo/SZ, 1 atm, 0.6 Lgcat-1hr-1)
Figure D.12 TPO profile for spent catalysts ran at different space velocities (5%Mo/SZ, 1 atm, 650 oC). Units for space velocity are: Lgcat-1hr-1

185
D.3.2 Regeneration of catalysts
Regeneration of catalysts was also considered. One attempt to regenerate the
catalyst involved oxidizing the coke using the TPO procedure, then reducing the catalyst
and subsequently carburizing it as mentioned in the chapter 5 (section 5.2.13
Carburization and dehydroaromatization (DHA)). This regenerated catalyst was re-run
following the carburization sequence, however, the activity was not restored and the
catalyst showed significantly lower CH4 conversions and lower aromatic selectivities. This
might have oxidized coke from the catalyst surface, but temperatures higher than 650 oC
and longer times are required. This would have caused a significant loss of sulfur and a
loss in the Brønsted acidity. A systematic study of the regeneration of these catalysts, or
stabilization of sulfate, is beyond the scope of this paper.
D.4 References
[1] A.S.C. Brown, J.S.J. Hargreaves, S.H. Taylor, A study of “superacidic” MoO3/ZrO2 catalysts for methane oxidation, Catalysis Letters, 57 (1999) 109-113. [2] F.F. Oloye, A.J. McCue, J.A. Anderson, n-Heptane hydroconversion over sulfated-zirconia-supported molybdenum carbide catalysts, Applied Petrochemical Research, 6 (2016) 341-352. [3] K.M. Arata, H., Solid Superacids, Nova Science Publishers, New York, 2011. [4] Z.R. Ismagilov, E.V. Matus, L.T. Tsikoza, Direct conversion of methane on Mo/ZSM-5 catalysts to produce benzene and hydrogen: achievements and perspectives, Energy & Environmental Science, 1 (2008) 526-541. [5] E.V. Matus, I.Z. Ismagilov, O.B. Sukhova, V.I. Zaikovskii, L.T. Tsikoza, Z.R. Ismagilov, J.A. Moulijn, Study of Methane Dehydroaromatization on Impregnated Mo/ZSM-5 Catalysts and Characterization of Nanostructured Molybdenum Phases and Carbonaceous Deposits, Industrial & Engineering Chemistry Research, 46 (2007) 4063-4074. [6] B.M. Reddy, M.K. Patil, Organic Syntheses and Transformations Catalyzed by Sulfated Zirconia, Chemical Reviews, 109 (2009) 2185-2208.

186
[7] E.J. Lede, F.G. Requejo, B. Pawelec, J.L.G. Fierro, XANES Mo L-Edges and XPS Study of Mo Loaded in HY Zeolite, The Journal of Physical Chemistry B, 106 (2002) 7824-7831. [8] H. Aritani, H. Shibasaki, H. Orihara, A. Nakahira, Methane dehydroaromatization over Mo-modified H-MFI for gas to liquid catalysts, Journal of Environmental Sciences, 21 (2009) 736-740. [9] B.S. Liu, L. Jiang, H. Sun, C.T. Au, XPS, XAES, and TG/DTA characterization of deposited carbon in methane dehydroaromatization over Ga–Mo/ZSM-5 catalyst, Applied Surface Science, 253 (2007) 5092-5100. [10] N. Kosinov, F.J.A.G. Coumans, G. Li, E. Uslamin, B. Mezari, A.S.G. Wijpkema, E.A. Pidko, E.J.M. Hensen, Stable Mo/HZSM-5 methane dehydroaromatization catalysts optimized for high-temperature calcination-regeneration, Journal of Catalysis, 346 (2017) 125-133.

187
Vita
Swarom Ravindra Kanitkar was born in Aurangabad, Maharashtra, India in 1988.
He earned his Bachelor of Technology degree in Chemical Engineering at Visvesvaraya
National Institute of Technology in Nagpur, Maharashtra, India. He then attended the
University of Louisiana at Lafayette to earn a Master’s degree in Engineering with a
Chemical Engineering option where he worked on ‘Modeling and analysis of production
of propylene glycol using copper chromite catalysts.’ As his interest grew in catalysis, he
decided to join the Department of Chemical Engineering at LSU to pursue his Ph.D. study
and worked on the catalytic conversion of natural gas. He expects to graduate in May
2019.


















