
Nanostructured Ca-based sorbents with high CO2 uptake efficiency
8
Chemical Engineering Science 64 (2009) 1936--1943 Contents lists available at ScienceDirect Chemical Engineering Science journal homepage: www.elsevier.com/locate/ces Nanostructured Ca-based sorbents with high CO 2 uptake efficiency Hong Lu a , Panagiotis G. Smirniotis a, ∗ , Frank O. Ernst b , Sotiris E. Pratsinis b a Chemical and Materials Engineering Department, University of Cincinnati, OH 45221-0012, USA b Particle Technology Laboratory, Department of Mechanical and Process Engineering, ETH Zurich, CH-8092 Z ¨ urich, Switzerland ARTICLE INFO ABSTRACT Article history: Received 21 September 2008 Received in revised form 16 December 2008 Accepted 31 December 2008 Available online 17 January 2009 Keywords: Adsorption Aerosol Calcium oxide Carbon dioxide Carbonation Nanostructure Separations Reaction-controlled Flame synthesis Nanosized materials Calcium-based carbon dioxide sorbents were made in the gas phase by scalable flame spray pyrolysis (FSP) and compared to the ones made by calcination (CAL) of selected calcium precursors. Such flame- made sorbents consisted of nanostructured CaO and CaCO 3 with twice as much specific surface area (40–60 m 2 /g) as the CAL-made sorbents. All FSP-made sorbents exhibited faster and higher CO 2 uptake capacity than all CAL-made sorbents at intermediate temperatures. CAL of calcium acetate monohydrate resulted in sorbents with the best CO 2 uptake among all CAL-made ones. At higher temperatures both FSP- and CAL-made sorbents (esp. from CaAc 2 · H 2 O) exhibited very high initial molar conversions (95%) but sintering contributed to grain growth that reduced the molar conversion down to 50%. In multiple carbonation/decarbonation cycles, the nanostructured FSP-made sorbents demonstrated stable, reversible and high CO 2 uptake capacity sustaining maximum molar conversion at about 50% even after 60 such cycles, indicating high potential for CO 2 uptake. The top performance of flame-made sorbents is best attributed to their nanostructure (30–50 nm grain size) that allows operation in the reaction-controlled carbonation regime rather than in the diffusion-controlled one when sorbents made with larger particles are employed. © 2009 Elsevier Ltd. All rights reserved. 1. Introduction Carbon dioxide from fossil fuel-fired power plants accounts for the largest anthropogenic CO 2 emissions (Herzog and Gollomb, 2004). Using metal oxide sorbents, such as calcium oxide, is one of the most potent ways to trap CO 2 and prevent its emission into the atmosphere (Silaban and Harrison, 1995; Reddy and Smirniotis, 2004; Alvarez and Abanades, 2005; Li et al., 2006; Lu et al., 2006, 2008; Manovic and Anthony, 2007; Martavaltzi and Lemonidou, 2008) in contrast to methylamine used today for CO 2 capture (Gupta and Fan, 2002; Aaron, 2005). Currently, amine-based absorption technology is the only available commercial technology, which captures CO 2 and regenerates solvent at low temperature range (313–433 K). This technology may be retrofitted or applied to post- or pre-combustion captures. Efforts were made to develop new sorbents such as amine-grafted SBA-15 to boost sorbent capacity (Khatri et al., 2006). Other emerging technologies include absorption with soluble carbonate, adsorption with activated carbon, molec- ular sieve, metal oxide, or ionic liquid (IL), membrane separation, and cryogenic distillation (Bates et al., 2002; Aaron, 2005; Figueroa et al., 2008). None of these technologies, however, can be applied ∗ Corresponding author. Tel.: +1 513 556 1474; fax: +1 513 556 3473. E-mail address: [email protected] (P.G. Smirniotis). 0009-2509/$ - see front matter © 2009 Elsevier Ltd. All rights reserved. doi:10.1016/j.ces.2008.12.038 easily above 823 K except for CaO-based sorbents. The CaO-based technology may be still a concept but has great potential for its large capacity and operation at a much higher temperature range (823–1223 K). Promising CaO sorbents could be applied in a carbona- tor in which sorbent captures CO 2 from gas streams before the gases are cooled down. As early as in 1967 (Curran et al., 1967), the CaO- based separation principle was proved in pilot tests involving flu- idized carbonation bed and calciner operated at 1088 and 1033 K in a gas-fired process. In gas-fired power plants, the exhaust gas temper- atures are 858–885 K even after heat recovery, which is suitable for high temperature CO 2 capture. Biomass-fired power plants, which are still not commercialized though, are highly possible to be applied for in situ CO 2 capture at around 973 K (Florin and Harris, 2008). If CaO-based capture is technology capable and cost competitive, then a gas stream could be required to be heated up/cooled down or pres- surized/vacuumed to meet the capture requirement before it enters a capture process. There are four large CO 2 point sources which are possible for future application of CO 2 capture: post-combustion systems, pre-combustion systems, oxy-fuel combustion systems, and industrial process streams. The first three sources are power generation related and account about one-third of all anthropogenic CO 2 emissions (more than 8.2 Gt ton CO 2 /year), while the industrial process streams account for about 4.3 Gt ton CO 2 /year. In a post- combustion system, the CO 2 concentrations are typical between 3% for a natural gas combined cycle and less than 15% for a coal-fired
Transcript of Nanostructured Ca-based sorbents with high CO2 uptake efficiency

Chemical Engineering Science 64 (2009) 1936 -- 1943
Contents lists available at ScienceDirect
Chemical Engineering Science
journal homepage: www.e lsev ier .com/ locate /ces
Nanostructured Ca-based sorbentswith high CO2 uptake efficiency
Hong Lua, Panagiotis G. Smirniotisa,∗, Frank O. Ernstb, Sotiris E. Pratsinisb
aChemical and Materials Engineering Department, University of Cincinnati, OH 45221-0012, USAbParticle Technology Laboratory, Department of Mechanical and Process Engineering, ETH Zurich, CH-8092 Zurich, Switzerland
A R T I C L E I N F O A B S T R A C T
Article history:Received 21 September 2008Received in revised form 16 December 2008Accepted 31 December 2008Available online 17 January 2009
Keywords:AdsorptionAerosolCalcium oxideCarbon dioxideCarbonationNanostructureSeparationsReaction-controlledFlame synthesisNanosized materials
Calcium-based carbon dioxide sorbents were made in the gas phase by scalable flame spray pyrolysis(FSP) and compared to the ones made by calcination (CAL) of selected calcium precursors. Such flame-made sorbents consisted of nanostructured CaO and CaCO3 with twice as much specific surface area(40–60m2/g) as the CAL-made sorbents. All FSP-made sorbents exhibited faster and higher CO2 uptakecapacity than all CAL-made sorbents at intermediate temperatures. CAL of calcium acetate monohydrateresulted in sorbents with the best CO2 uptake among all CAL-made ones. At higher temperatures bothFSP- and CAL-made sorbents (esp. from CaAc2 · H2O) exhibited very high initial molar conversions (95%)but sintering contributed to grain growth that reduced the molar conversion down to 50%. In multiplecarbonation/decarbonation cycles, the nanostructured FSP-made sorbents demonstrated stable, reversibleand high CO2 uptake capacity sustaining maximum molar conversion at about 50% even after 60 suchcycles, indicating high potential for CO2 uptake. The top performance of flame-made sorbents is bestattributed to their nanostructure (30–50nm grain size) that allows operation in the reaction-controlledcarbonation regime rather than in the diffusion-controlled one when sorbents made with larger particlesare employed.
© 2009 Elsevier Ltd. All rights reserved.
1. Introduction
Carbon dioxide from fossil fuel-fired power plants accounts forthe largest anthropogenic CO2 emissions (Herzog and Gollomb,2004). Using metal oxide sorbents, such as calcium oxide, is oneof the most potent ways to trap CO2 and prevent its emission intothe atmosphere (Silaban and Harrison, 1995; Reddy and Smirniotis,2004; Alvarez and Abanades, 2005; Li et al., 2006; Lu et al., 2006,2008; Manovic and Anthony, 2007; Martavaltzi and Lemonidou,2008) in contrast to methylamine used today for CO2 capture (Guptaand Fan, 2002; Aaron, 2005). Currently, amine-based absorptiontechnology is the only available commercial technology, whichcaptures CO2 and regenerates solvent at low temperature range(313–433K). This technology may be retrofitted or applied to post-or pre-combustion captures. Efforts were made to develop newsorbents such as amine-grafted SBA-15 to boost sorbent capacity(Khatri et al., 2006). Other emerging technologies include absorptionwith soluble carbonate, adsorption with activated carbon, molec-ular sieve, metal oxide, or ionic liquid (IL), membrane separation,and cryogenic distillation (Bates et al., 2002; Aaron, 2005; Figueroaet al., 2008). None of these technologies, however, can be applied
∗ Corresponding author. Tel.: +15135561474; fax: +15135563473.E-mail address: [email protected] (P.G. Smirniotis).
0009-2509/$ - see front matter © 2009 Elsevier Ltd. All rights reserved.doi:10.1016/j.ces.2008.12.038
easily above 823K except for CaO-based sorbents. The CaO-basedtechnology may be still a concept but has great potential for itslarge capacity and operation at a much higher temperature range(823–1223K). Promising CaO sorbents could be applied in a carbona-tor in which sorbent captures CO2 from gas streams before the gasesare cooled down. As early as in 1967 (Curran et al., 1967), the CaO-based separation principle was proved in pilot tests involving flu-idized carbonation bed and calciner operated at 1088 and 1033K in agas-fired process. In gas-fired power plants, the exhaust gas temper-atures are 858–885K even after heat recovery, which is suitable forhigh temperature CO2 capture. Biomass-fired power plants, whichare still not commercialized though, are highly possible to be appliedfor in situ CO2 capture at around 973K (Florin and Harris, 2008). IfCaO-based capture is technology capable and cost competitive, thena gas stream could be required to be heated up/cooled down or pres-surized/vacuumed to meet the capture requirement before it entersa capture process. There are four large CO2 point sources whichare possible for future application of CO2 capture: post-combustionsystems, pre-combustion systems, oxy-fuel combustion systems,and industrial process streams. The first three sources are powergeneration related and account about one-third of all anthropogenicCO2 emissions (more than 8.2Gt ton CO2/year), while the industrialprocess streams account for about 4.3Gt ton CO2/year. In a post-combustion system, the CO2 concentrations are typical between 3%for a natural gas combined cycle and less than 15% for a coal-fired

H. Lu et al. / Chemical Engineering Science 64 (2009) 1936 -- 1943 1937
combustion plant. However, in a pre-combustion system, the con-centration of CO2 in a shift reactor varies from 15% to 60% and thetotal pressure could be up to 70 bar. In an oxy-fuel combustion sys-tem, the concentrations of CO2 are even higher than 80%. In thosecases, a direct pressurization and sequestration would be a betteroption for CO2 separation and sequestration. In industrial processstreams (steel production, cement production, or ammonia produc-tion, etc), the CO2 content varies from 14% to 33%. All of these differ-ences in stream temperature and pressure, and CO2 concentrationindicate that there will not be a simple capture technology whichcan be applied unanimously to all capture systems.
The major challenge for CaO-based technology capturing CO2 isthe fast decay in sorbent performance during cyclic operations. CaOsorbents obtained from porous calcium carbonate (Gupta and Fan,2002) have been shown to reach 90% molar conversion at 973K.Salvador et al. (2003) used NaCl to enhance CaO sorbents perfor-mance. Reddy and Smirniotis (2004) enhanced the performance ofCaO sorbents by doping them with alkali metals that had zero affin-ity to N2 and O2 and rather low to H2O. Li et al. (2006) integrated CaOsorbents with Ca12Al14O33 and effectively enhanced the cyclic per-formance. Other inert materials were also investigated to enhancedurability (Lu et al., 2006; Albrecht et al., 2008). Steam reactivation(Kuramoto et al., 2003; Manovic and Anthony, 2007) was appliedfor promotion of performance on CaO sorbents. The reversible car-bonation (adsorption) and calcinations (CAL) of CaO with CO2 pro-vide great potential for separation of CO2 from high temperature gasstreams such as flue gases, coal gasification, fuel cell applications orchemical heat pumps (Aihara et al., 2001; Comas et al., 2004; Corellaet al., 2006, 2008; Florin and Harris, 2008). Promising future appli-cations for CaO-based sorbents are cost competitive. Recent studiesshow that carbon capture by using CaO-based sorbents would gen-erate costs 16–44US$/tCO2 avoided, which is competitive with thecost 13–74US$/tCO2 avoided by currentMEA technologies (Abanadeset al., 2007; MacKenzie et al., 2007).
While CaO sorbents are made largely by precipitation and/or CALof liquid or solid Ca-precursors, an alternative route is their aerosolsynthesis in flames. The latter technology is a fast, cost-effective andversatile process for large scale production (several tons per hour) ofcarbon black, fumed silica, alumina and pigmentary titania rangingfrom $1 to 6/kg, depending on specifications, with no liquid byprod-ucts (Strobel and Pratsinis, 2007). In particular, flame spray pyrolysis(FSP) allows synthesis of such particles with high specific surfacearea (SSA) and well defined chemical composition, as recentlydemonstrated both on a lab (Madler et al., 2002a) and pilot scale upto 1kg/h (Mueller et al., 2003). Furthermore, Li2ZrO3 particles weresynthesized by spray pyrolysis for CO2 capture and showed highercapacity than those by solid-state reaction-made sorbents (Choiet al., 2003). Huber et al. (2005) produced mixed CaO/CaCO3 parti-cles with SSA of 31–103m2/g for orthopedic applications by FSP ofCa-2-ethylhexanoate solutions.
Here the potential of such direct flame synthesis of calcium-basedsorbents for CO2 uptake from inexpensive precursors is explored andcompared to sorbents made conventionally by CAL at intermediateand high temperatures that can be encountered at various stagesin a process generating CO2. Sorbent carbonation capacity and cyclestability are investigated at high temperatures by thermogravimet-ric analysis while sorbent characteristics are determined by X-raydiffraction (XRD), nitrogen adsorption and microscopy.
2. Experimental
2.1. Sorbent synthesis by FSP
The experimental setup for synthesis of nanoscale powdersby FSP is described in detail elsewhere (Madler et al., 2002b). ACa-naphthenate precursor (∼35% in mineral spirits, Strem Chem.,
Inc.) was dissolved in xylene (Riedel-de-Haen, > 96%) and fed(5–9mL/min) by a syringe pump (Inotech R232) through the spraynozzle and dispersed by 3–5L/min oxygen (Pan Gas, > 99.95%) intofine droplets (pressure drop at the nozzle tip 1.5 bar). The resultingspray was ignited and maintained by a premixed (1.13 L/min CH4and 2.40 L/min O2) flame ring surrounding the spray capillary at aradius of 6mm (spacing was 0.15mm) (Madler et al., 2002b). Watercooling of nozzle and manifold prevents any precursor evaporationwithin the liquid feed lines or overheating of the nozzle (Madleret al., 2002a). An additional oxygen sheath flow of 5 L/min was fedthrough a sinter metal ring (9/17mm inner/outer diameter) sur-rounding the supporting flame to assure complete conversion of thereactants. The gas flows were monitored by calibrated mass flowcontrollers (Bronkhorst). With the aid of a vacuum pump, prod-uct particles were collected on a glass fiber filter (GF/D Whatman,25.7 cm in diameter) placed in a water-cooled holder 40 cm abovethe nozzle, keeping the off-gas temperature below 423K. The liquidfeed rate and the dispersion oxygen flow rate were varied in orderto select the product particle characteristics (Madler et al., 2002a).
2.2. Sorbent synthesis by CAL
Calcium oxide (Aldrich), calcium carbonate (Fisher) and calciumacetate monohydrate (Fisher) were used as CaO precursors. Sor-bents prepared from these three precursors by CAL are denoted asCaO–CaO, CaCO3–CaO, and CaAc2–CaO. The sorbents were calcinedas follows: the precursors were heated up from 323 to 1023K at10K/min in helium and kept at 1023K for 30min for full CAL fol-lowed by cooling down to room temperature at 15K/min (Reddy andSmirniotis, 2004).
2.3. Materials characterization
The Brunauer–Emmett–Teller (BET) equivalent SSA of the sor-bents was determined by five-point nitrogen adsorption isothermat 77K in the relative pressure range of p/p0 = 0.05–0.25 (Tristar,Micromeritics Instruments Corp.). All samples for BET measurementwere degassed at 423K for 1h. Assuming spherical primary particles,a BET equivalent particle diameter was calculated accounting for theweight fraction of CaO and CaCO3 in the sorbent, as extracted fromTGA measurements in Table 1 while only Ca compounds were con-sidered. Pore size distributions (PSD) were obtained by nitrogen ad-sorption and desorption isotherms at 77K on a Micromeritics ASAP2010 volumetric adsorption analyzer. Sorbents for pore distributionanalysis were degassed at 573K for 3h. The crystallite sizes, dXRD,were determined from XRD patterns recorded with a Bruker AXS D8Advance (Cu-K�, 40 kV, 40mA, scanning step 0.03◦, scanning time3 s/step) by the Rietveld method with TOPAS 2 software. The powderwas also analyzed by transmission electron microscopy (TEM) witha Zeiss microscope 912 Omega with ProScan and slow scan charge-coupled device (CCD) camera at 100kV.
2.4. CO2 chemisorption characterization
All carbonation/decarbonation experiments were conductedwith a Perkin-Elmer PyrisTM-1 thermogravimetric analyzer (TGA).This TGA was assisted by Perkin-Elmer thermal analysis gas station(TAGS) and PyrisTM v3.8 software. The TGA microbalance resolutionis 0.1�g. To maintain the TGA balance accurately, 45mL/min ofhelium (prepurified, Wright Bros, Inc.) flowed over both balanceand sample. The four gas channels of the TFGS can be controlledautomatically to introduce reaction gas or balance inert gas into thesystem.
The carbonation and decarbonation experiments, includingheating, cooling, and switching gases between CO2 (99.5%, Wright

1938 H. Lu et al. / Chemical Engineering Science 64 (2009) 1936 -- 1943
Table 1Synthesis conditions, BET specific surface areas, particle diameters, and weight percentages of CaO and CaCO3 of FSP-made sorbents.
Sorbent Ca precursor solutionfeeding rate (mL/min)
Dispersion O2 flowrate (L/min)
BET SSA (m2/g) Particle diameter(nm)a
CaOwt%b CaCO3 wt%b
F1 9 3 41 48 36 (37) 56 (55)F2 7 3 47 41 43 (43) 48 (48)F3 5 5 59 34 22 (22) 73 (73)
aDiameters are calculated from BET surface area.bWeight percentages are derived from TGA pretreatment curves (and XRD values).
Bros, Inc.) and helium were programmed and operated batchwise.Sorbents (ranging from 2 to 10mg) were placed in a platinum sam-ple holder and heated to the desired carbonation temperature at10K/min under 65mL/min flow of helium (45mL/min purge gasand 20mL/min balance gas). All samples were subjected to pre-treatment at 1023K for 30min before its first carbonation. Oncethe sample reached the carbonation temperature, 20mL/min he-lium was replaced by 20mL/min CO2 initiating the carbonation.Therefore, carbonation was conducted under 30vol% CO2 streambalanced by helium. The carbonation temperatures were variedhere while decarbonation temperature was fixed at 973K. All de-carbonation were done under helium atmosphere. Since CO2 partialpressure is zero and below its equilibrium pressure 0.03bar, CaCO3will decompose completely at 973K. The carbonation time was 5,60 or 300min while the decarbonation time was 30min, excepts asspecified. Sorbent weight and temperature were recorded continu-ously throughout the experiment. Rather long reaction times wereselected to test the stability of these sorbents which is a crucialcharacteristic for their large scale application.
3. Results and discussion
3.1. FSP-made sorbent characterization
Nanostructured sorbents with SSAs ranging from 40 to 60m2/gand various CaO and CaCO3 contents were made by FSP (Table 1).The BET equivalent primary particle diameters of all sorbents are30–50nm (Table 1). High flame temperatures (Camenzind et al.,2005; Madler et al., 2002b) cause the Ca-naphthenate precursors tobe initially converted into CaO particles that react (partially) withcombustion off-gas CO2 in the flame tail converting them into CaCO3.Huber et al. (2005) reported that amorphous CaCO3 made by FSPat low fuel/oxygen ratios from costly 2-ethylhexanoic acid solutionswhile high such ratios and slower cooling rates facilitated parti-cle crystallization. Crystalline CaCO3 was identified by XRD (Fig. 1)though limited, if any, amorphous material cannot exceed more than10% by weight given the flat XRD baselines here. This is consis-tent with Huber et al. (2005) who reported decreasing amorphousCaCO3 content when increasing high temperature particle residencetime by increasing the fuel to oxygen ratio (mL precursor/min overL O2/min) fed to the burner. High fuel/oxygen ratios increase theflame enthalpy density and maximum flame temperature and thusparticle crystallinity (Camenzind et al., 2005).
Before the first carbonation, all sorbents were pretreated in he-lium atmosphere following the temperature profile in Fig. 2. Physi-cally adsorbed gases and water were released first above 373K. At873K, CaCO3 begins to decompose into CaO. According to XRD, allcalcium is in the form of CaO or CaCO3. Therefore, the compositionsof the sorbents were extracted by mass balances of CaO and CaCO3on the sample weight before and after CaCO3 decomposition, e.g. atabout 30 and 60min, respectively (Fig. 2). There is more CaCO3 thanCaO in the as-prepared FSP-made sorbents (Table 1). These com-positions are in excellent agreement (within 2% error) to the onesdetermined by Rietveld XRD analysis.
30
**
#
# #
##
* **
*
**
#
*
Calcium carbonate
Calcium oxide
Inte
nsity
(a.
u.)
F1
2 Theta (degree)
*******##
#
#
#
*
F2
*****
** ##
#
#
#
*F3
40 50 60 70
Fig. 1. XRD patterns of as-prepared FSP-made sorbents (Table 1). The sharp spectraand rather flat baseline indicate that these are crystalline materials. CaO PDF#:00-004-0777 and CaCO3 PDF#: 00-005-0586.
3.2. Carbonation of FSP-made sorbents
Fig. 3 shows typical CO2 uptake performance of the FSP sorbentsin the form of molar conversion as a function of time at 973K. Thesamples were pretreated at 1023K under helium for 30min and thenexposed to CO2 for 5h at 973K. All the curves show monotonic in-crease of the sorbent conversion versus time during this carbonationperiod. A fast and complete decomposition of the carbonated CaCO3back to CaO was observed during the 30min decarbonation periodindicating reversible carbonation/decarbonation. The carbonation ofall sorbents exhibit two stages: in the initial stage of each carbon-ation period, conversion increased steeply followed by a plateau inthe second stage. The first stage is chemically controlled (Barker,1973; Bhatia and Perlmutter, 1983; Borgwardt, 1985) and starts af-ter about 2min induction period, which corresponds closely withtime taken for gas changeover from inert helium to CO2 contained

H. Lu et al. / Chemical Engineering Science 64 (2009) 1936 -- 1943 1939
040
60
80
100
F1
F2
F3
Temperature
Time (min)
Wei
ght %
dec
reas
ed
200
400
600
800
1000
CaO
CaCO3
Tem
pera
ture
(K
)
25 50 75 100
Fig. 2. Thermogravimetric analysis of the pretreatment of FSP-made sorbents(Table 1) showing the release of adsorbed H2O (∼600K) and CaCO3 (∼800K)conversion.
00
70
80
90
100
00
25
50
75
100
70% conversion means1 kg sorbent captured550 g CO2
F1
Mol
ar C
onve
rsio
n/%
Time (min)
F1
F2
F3
T = 973 K
100 200 300
1 2 3
Fig. 3. Carbonation of FSP-made sorbents (Table 1) at 973K showing high capacity.Inset image: initial carbonation performance of F1. Curve was drawn from the initialdata of the F1 in Fig. 3. There was 2min delay time for CO2 to reach reactor.
stream. The abrupt change is remarkable from the fast surface reac-tion controlled by chemical to much slower reaction controlled bydiffusion of CO2 gas through a product layer of CaCO3. The later theonset of the slow reaction, the higher the carbonation can progressor the higher the sorbent's capacity can reach. This change of thetwo-stage reaction is atypical for gas–solid reactions, but commonfor CaO carbonation in the literature (Barker, 1973). All sorbents ex-hibited high capacity of more than 90% molar conversion. SamplesF1 and F2 behaved similarly while F3 with the highest SSA (Table 1)showed the highest CO2 capacity and uptake rate as extracted fromthe steep slope in the first stage. The high performance is attributedto the nanostructured characteristics of these particles (large SSAand crystallite size). To elucidate the sorbents' fast kinetics, the in-set of Fig. 3 highlights the initial kinetics of F1 powder there. Within1–3min, the conversion of the sorbent has reached as high as 70%.This means that 550g CO2 can be captured by 1kg of sorbent in asingle cycle.
Following pretreatment, the Ca of all sorbents existed onlyin the form of CaO regardless of the initial CaCO3 content. It is
00
25
50
75
100
CaAc2-CaO
CaCO3-CaO
CaO-CaO
973K
Mol
ar c
onve
rsio
n (%
)
Time (min)100 200 300
Fig. 4. Conversion of three CAL-made sorbents at 973K indicating the high SSAand porous structure of the CaAc2–CaO sorbents (Table 2) related to their highperformance.
Table 2BET specific surface areas and pore volumes of CAL-made sorbents.
CaO precursor BET SSA (m2/g) Pore volume (cm3/g)
CaAc2–CaO 20 0.23CaCO3–CaO 5 0.06CaO–CaO 4 0.02
worth noting that the flame-made sorbents demonstrated similarperformance although they contained initially quite different per-centages of CaO and CaCO3 (Table 1). Therefore, the original crys-talline composition of the FSP-made sorbents did not play significantrole in their carbonation performance.
3.3. Carbonation of CAL-made sorbents
Fig. 4 shows the CO2 uptake of conventional calcined sorbents at973K. CaAc2–CaO exhibited much higher CO2 uptake than the othertwo sorbents. Table 2 and Fig. 5 suggest that the high performanceof sorbents made from CaAc2 is related to their large BET surfacearea and pore volume. The pore volume of the CaAc2-made CaO is aslarge as 0.23 cm3/g, which is 11 times larger than that of CaO-madesorbents and nearly four times larger than CaCO3-made sorbents.The CaAc2-made sorbents have not only much larger pore volumes(Table 2) but also narrower PSD than the other CAL-made sorbents.Thermogravimetric analysis revealed multiple precursor decompo-sitions till the final CaO sorbent porous structure obtained as below:
Ca(CH3COO)2H2O>∼400 K−→ Ca(CH3COO)2
>∼600 K−→ CaCO3>∼900 K−→ CaO
3.4. Comparison of FSP- and CAL-made sorbents
FSP-made sorbents contained more CaCO3 than CaO. Thus, car-bonation of FSP-made sorbents was compared to CAL-made sorbentsfrom commercial CaCO3. Fig. 6 shows the molar conversion as afunction of time for FSP-made and CaCO3-made sorbents at 973K.Both sorbents show similar kinetic behavior during the first stage ofcarbonation. However, the FSP-made sorbent exhibited much highercapacity during the 5-h-carbonation with CO2. Comparing Tables 1and 2, and Fig. 7a and b shows that the particle sizes and SSAs of
1940 H. Lu et al. / Chemical Engineering Science 64 (2009) 1936 -- 1943
10
0.0
0.1
0.2
0.3
0.4
dV/d
logD
por
e vo
lum
e (c
m-3
/g-A
)
Pore Diameter (A)
CaAc2-CaO
CaCO3-CaO
CaO-CaO
100 1000
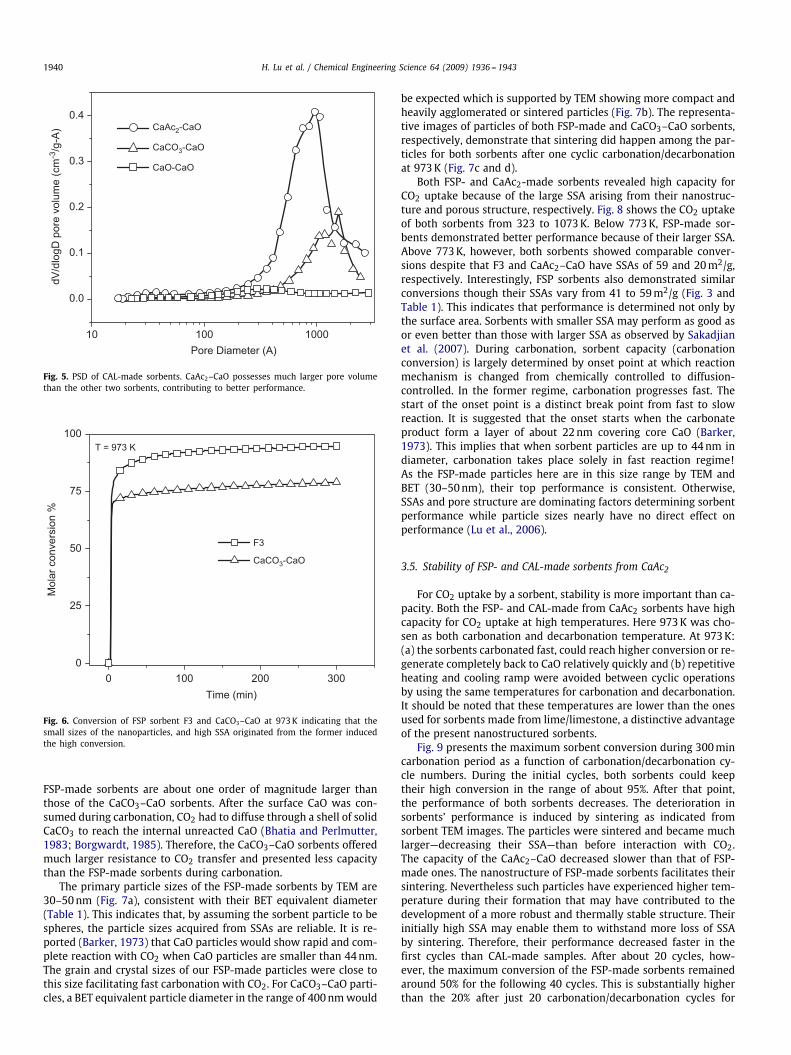
Fig. 5. PSD of CAL-made sorbents. CaAc2–CaO possesses much larger pore volumethan the other two sorbents, contributing to better performance.
00
25
50
75
100
F3
CaCO3-CaO
T = 973 K
Mol
ar c
onve
rsio
n %
Time (min)100 200 300
Fig. 6. Conversion of FSP sorbent F3 and CaCO3–CaO at 973K indicating that thesmall sizes of the nanoparticles, and high SSA originated from the former inducedthe high conversion.
FSP-made sorbents are about one order of magnitude larger thanthose of the CaCO3–CaO sorbents. After the surface CaO was con-sumed during carbonation, CO2 had to diffuse through a shell of solidCaCO3 to reach the internal unreacted CaO (Bhatia and Perlmutter,1983; Borgwardt, 1985). Therefore, the CaCO3–CaO sorbents offeredmuch larger resistance to CO2 transfer and presented less capacitythan the FSP-made sorbents during carbonation.
The primary particle sizes of the FSP-made sorbents by TEM are30–50nm (Fig. 7a), consistent with their BET equivalent diameter(Table 1). This indicates that, by assuming the sorbent particle to bespheres, the particle sizes acquired from SSAs are reliable. It is re-ported (Barker, 1973) that CaO particles would show rapid and com-plete reaction with CO2 when CaO particles are smaller than 44nm.The grain and crystal sizes of our FSP-made particles were close tothis size facilitating fast carbonation with CO2. For CaCO3–CaO parti-cles, a BET equivalent particle diameter in the range of 400nmwould
be expected which is supported by TEM showing more compact andheavily agglomerated or sintered particles (Fig. 7b). The representa-tive images of particles of both FSP-made and CaCO3–CaO sorbents,respectively, demonstrate that sintering did happen among the par-ticles for both sorbents after one cyclic carbonation/decarbonationat 973K (Fig. 7c and d).
Both FSP- and CaAc2-made sorbents revealed high capacity forCO2 uptake because of the large SSA arising from their nanostruc-ture and porous structure, respectively. Fig. 8 shows the CO2 uptakeof both sorbents from 323 to 1073K. Below 773K, FSP-made sor-bents demonstrated better performance because of their larger SSA.Above 773K, however, both sorbents showed comparable conver-sions despite that F3 and CaAc2–CaO have SSAs of 59 and 20m2/g,respectively. Interestingly, FSP sorbents also demonstrated similarconversions though their SSAs vary from 41 to 59m2/g (Fig. 3 andTable 1). This indicates that performance is determined not only bythe surface area. Sorbents with smaller SSA may perform as good asor even better than those with larger SSA as observed by Sakadjianet al. (2007). During carbonation, sorbent capacity (carbonationconversion) is largely determined by onset point at which reactionmechanism is changed from chemically controlled to diffusion-controlled. In the former regime, carbonation progresses fast. Thestart of the onset point is a distinct break point from fast to slowreaction. It is suggested that the onset starts when the carbonateproduct form a layer of about 22nm covering core CaO (Barker,1973). This implies that when sorbent particles are up to 44nm indiameter, carbonation takes place solely in fast reaction regime!As the FSP-made particles here are in this size range by TEM andBET (30–50nm), their top performance is consistent. Otherwise,SSAs and pore structure are dominating factors determining sorbentperformance while particle sizes nearly have no direct effect onperformance (Lu et al., 2006).
3.5. Stability of FSP- and CAL-made sorbents from CaAc2
For CO2 uptake by a sorbent, stability is more important than ca-pacity. Both the FSP- and CAL-made from CaAc2 sorbents have highcapacity for CO2 uptake at high temperatures. Here 973K was cho-sen as both carbonation and decarbonation temperature. At 973K:(a) the sorbents carbonated fast, could reach higher conversion or re-generate completely back to CaO relatively quickly and (b) repetitiveheating and cooling ramp were avoided between cyclic operationsby using the same temperatures for carbonation and decarbonation.It should be noted that these temperatures are lower than the onesused for sorbents made from lime/limestone, a distinctive advantageof the present nanostructured sorbents.
Fig. 9 presents the maximum sorbent conversion during 300mincarbonation period as a function of carbonation/decarbonation cy-cle numbers. During the initial cycles, both sorbents could keeptheir high conversion in the range of about 95%. After that point,the performance of both sorbents decreases. The deterioration insorbents' performance is induced by sintering as indicated fromsorbent TEM images. The particles were sintered and became muchlarger—decreasing their SSA—than before interaction with CO2.The capacity of the CaAc2–CaO decreased slower than that of FSP-made ones. The nanostructure of FSP-made sorbents facilitates theirsintering. Nevertheless such particles have experienced higher tem-perature during their formation that may have contributed to thedevelopment of a more robust and thermally stable structure. Theirinitially high SSA may enable them to withstand more loss of SSAby sintering. Therefore, their performance decreased faster in thefirst cycles than CAL-made samples. After about 20 cycles, how-ever, the maximum conversion of the FSP-made sorbents remainedaround 50% for the following 40 cycles. This is substantially higherthan the 20% after just 20 carbonation/decarbonation cycles for

H. Lu et al. / Chemical Engineering Science 64 (2009) 1936 -- 1943 1941
Fig. 7. TEM Images of (a) FSP sorbent F3; (b) CaCO3–CaO; (c) F3 after one carbonation/decarbonation cycle at 973K and (d) CaCO3–CaO after one carbonation/decarbonationcycle at 973K.
0
0
25
50
75
100
F3-CaO
CaAc2-CaO
973 K and 1073 K
973 K and 1073 K
773 K 773 K
573 K
323 K
Mol
ar c
onve
rsio
n %
Time (min)100 200 300
Fig. 8. Uptake of CO2 by FSP sorbent F3 and CaAc2–CaO at various temperatures.
CAL-made sorbents reviewed in the literature (Abanades, 2002).Note, however, that sorbents and experimental conditions em-ployed among these literature data are different. The sorbents hereunderwent decarbonation at relatively lower temperature (973K)while those in Abanades (2002) were between 1023 and 1333K.The capability of the nanostructured sorbents for fast CAL at 973K isadvantageous as sintering effects that dominate at higher tempera-tures are minimal at 973K while fast carbonation is maintained.
To explore further the high CO2 uptake of the FSP-made sorbents,the carbonation cycling time is reduced to 5-min-carbonation/5-min-decarbonation at (a) 973K, (b) 1073K, and under dynamicconditions (c) 5-min-carbonation at 973K and ramped decarbon-ation at 10K/min from 973K–1173K–973K for the first 20 cyclesof FSP sorbent F3 (Fig. 10). Comparing to Fig. 9, shorter carbon-ation/decarbonation cycles lead to slightly lower initial values ofthe molar conversion; however, similar overall values are observed.This is probably due to the fast reaction rate at initial stage. Thesorbents reached conversions of up to 70% within 3min. Decreasingcarbonation/decarbonation time would actually make the sorbentperformance even more “stable” as sintering effects are mitigatedduring the shorter thermal periods. Extending carbonation timeafter fast reaction period does not enhance (if not hurt) the overall

1942 H. Lu et al. / Chemical Engineering Science 64 (2009) 1936 -- 1943
0
40
60
80
100
F3
CaAc2-CaO
T = 973 K
Max
imum
car
bona
tion/
%
Carbonation/decarbonation cycle #20 40 60
Fig. 9. Carbonation/decarbonation (300min/30min) cycles on FSP sorbent F3 andCaAc2–CaO at 973K showing that the former sustained carbonation around 50%after 60 cycles.
0 5 10 15 200
20
40
60
80
a: 973 K
b: 1073 K
c: 973 K - 1173 K - 973 K
F3 (5 min carbonation)
Max
imum
con
vers
ion
(%)
Carbonation/decarbonation cycle #
Fig. 10. Carbonation/decarbonation (5min/5min) cycles on FSP sorbent F3 at: (a)973K; (b) 1073K and (c) carbonation for 5min at 973K and decarbonation for40min from 973 to 1173K and from 1173 back to 973K both at 10K/min. Thesorbent could reach high CO2 capture capacity, be completely regenerated in shorttime and be quite stable even at higher temperatures.
sorbent performance. Increasing decarbonation temperature slightlydecreases the molar conversion which may be attributed to sorbentsintering as discussed above. Decarbonation at low temperatures(e.g. 973K) is of advantage in view of industrial application wherethe sorbent has to withstand many more cycles.
4. Conclusions
Flame-made nanostructured sorbents demonstrated good per-formance for CO2 uptake at high temperatures (773–1073K) andprolonged carbonation/decarbonation cycles. This was attributed totheir high specific surface area (40–60m2/g) and corresponding fineparticle size (30–50nm) that allowed carbonation to take place at therapid reaction-controlled regime in contrast to diffusion-controlled
regime with larger sorbent particles in the literature. When cal-cium acetate monohydrate was used as sorbent precursor forCAL-made sorbent, fast CO2 uptake was observed at the initialcarbonation/decarbonation cycles. After about 10 cycles the per-formance of these sorbents, however, decreased steadily with in-creasing cycles. In contrast, flame-made sorbents presented stable,reversible maximum molar conversion around 50% after 60 cycles ofoperation indicating their high potential for CO2 uptake. Moreover,carbonation of flame-made sorbents can progress rapidly above823K, enabling less particle sintering and thus improved longevityfor such sorbents making them promising candidates for industrialscale CO2 capture applications.
Acknowledgments
We acknowledge the financial support of the US Departmentof Energy for awarding this Innovative Concepts Phase-II Program(Grant number DE-FG26-03-NT41810) and the Swiss Commissionfor Technology and Innovation (KTI) under Grant number 5773.2.
References
Aaron, D., 2005. Separation of CO2 from flue gas: a review. Separation Science andTechnology 40, 321–348.
Abanades, J.C., 2002. The maximum capture efficiency of CO2 using acarbonation/calcination cycle of CaO/CaCO3. Chemical Engineering Journal 90,303–306.
Abanades, J.C., Grasa, G., Alonso, M., Rodriguez, N., Anthony, E.J., Romeo, L.M., 2007.Cost structure of a postcombustion CO2 capture system using CaO. EnvironmentalScience & Technology 41, 5523–5527.
Aihara, M., Nagai, T., Matsushita, J., Negishi, Y., Ohya, H., 2001. Development ofporous solid reactant for thermal-energy storage and temperature upgrade usingcarbonation/decarbonation reaction. Applied Energy 69, 225–238.
Albrecht, K.O., Wagenbach, K.S., Satrio, J.A., Shanks, B.H., Wheelock, T.D., 2008.Development of a CaO-based CO2 sorbent with improved cyclic stability.Industrial & Engineering Chemistry Research 47, 7841–7848.
Alvarez, D., Abanades, J.C., 2005. Pore-size and shape effects on the recarbonationperformance of calcium oxide submitted to repeated calcination/recarbonationcycles. Energy & Fuels 19, 270–278.
Barker, R., 1973. The reversibility of the reaction CaCO3 = CaO+CO2. Journal ofApplied Chemistry and Biotechnology 23, 733–742.
Bates, E.D., Mayton, R.D., Ntai, I., Davis, J.H., 2002. CO2 capture by a task-specificionic liquid. Journal of the American Chemical Society 124, 926–927.
Bhatia, S.K., Perlmutter, D.D., 1983. Effect of the product layer on the kinetics of theCO2-lime reaction. A.I.Ch.E. Journal 29, 79–86.
Borgwardt, R.H., 1985. Calcination kinetics and surface area of dispersed limestoneparticles. A.I.Ch.E. Journal 31, 103–111.
Camenzind, A., Strobel, R., Pratsinis, S.E., 2005. Cubic or monoclinic Y2O3:Eu3+nanoparticles by one step flame spray pyrolysis. Chemical Physics Letters 416,193–197.
Choi, K.-H., Korai, Y., Mochida, I., 2003. Preparation of CO2 absorbent by spraypyrolysis. Chemistry Letters 32, 924–925.
Comas, J., Laborde, M., Amadeo, N., 2004. Thermodynamic analysis of hydrogenproduction from ethanol using CaO as a CO2 sorbent. Journal of Power Sources138, 61.
Corella, J., Toledo, J.M., Molina, G., 2006. Steam gasification of coal at low-medium(600–800 ◦C) temperature with simultaneous CO2 capture in fluidized bed atatmospheric pressure: the effect of inorganic species. 1. Literature review andcomments. Industrial & Engineering Chemistry Research 45, 6137–6146.
Corella, J., Toledo, J.M., Molina, G., 2008. Steam gasification of coal at low-medium(600–800 ◦C) temperature with simultaneous CO2 capture in a bubbling fluidizedbed at atmospheric pressure. 2. Results and recommendations for scaling up.Industrial & Engineering Chemistry Research 47, 1798–1811.
Curran, G.P., Fink, C.E., Gorin, E., 1967. Carbon dioxide-acceptor (coal) gasificationprocess. Studies of acceptor properties. Advances in Chemistry Series 69,141–165.
Figueroa, J.D., Fout, T., Plasynski, S., McIlvried, H., Srivastava, R.D., 2008. Advances inCO2 capture technology—the US Department of Energy's carbon sequestrationprogram. International Journal of Greenhouse Gas Control 2, 9.
Florin, N.H., Harris, A.T., 2008. Enhanced hydrogen production from biomass with insitu carbon dioxide capture using calcium oxide sorbents. Chemical EngineeringScience 63, 287.
Gupta, H., Fan, L.-S., 2002. Carbonation/calcination cycle using high reactivity calciumoxide for carbon dioxide separation from flue gas. Industrial & EngineeringChemistry Research 41, 4035–4042.
Herzog, H., Gollomb, D., 2004. Encyclopedia of Energy. Elsevier Science Inc., NewYork. pp. 277–287.

H. Lu et al. / Chemical Engineering Science 64 (2009) 1936 -- 1943 1943
Huber, M., Stark, W.J., Loher, S., Maciejewski, M., Krumeich, F., Baiker, A., 2005.Flame synthesis of calcium carbonate nanoparticles. Chemical Communications2005, 648–650.
Khatri, R.A., Chuang, S.S.C., Soong, Y., Gray, M., 2006. Thermal and chemicalstability of regenerable solid amine sorbent for CO2 capture. Energy & Fuels 20,1514–1520.
Kuramoto, K., Fujimoto, S., Morita, A., Shibano, S., Suzuki, Y., Hatano, H., Lin, S.Y.,Harada, M., Takarada, T., 2003. Repetitive carbonation–calcination reactionsof Ca-based sorbents for efficient CO2 sorption at elevated temperatures andpressures. Industrial & Engineering Chemistry Research 42, 975–981.
Li, Z.S., Cai, N.S., Huang, Y.Y., 2006. Effect of preparation temperature on cyclic CO2
capture and multiple carbonation–calcination cycles for a new Ca-based CO2
sorbent. Industrial & Engineering Chemistry Research 45, 1911–1917.Lu, H., Khan, A., Smirniotis, P.G., 2008. Relationship between structural properties
and CO2 capture performance of CaO-based sorbents obtained from differentorganometallic precursors. Industrial & Engineering Chemistry Research 47,6216–6220.
Lu, H., Reddy, E.P., Smirniotis, P.G., 2006. Calcium oxide-based sorbents for captureof carbon dioxide at high temperatures. Industrial & Engineering ChemistryResearch 45, 3944–3949.
MacKenzie, A., Granatstein, D.L., Anthony, E.J., Abanades, J.C., 2007. Economics ofCO2 capture using the calcium cycle with a pressurized fluidized bed combustor.Energy & Fuels 21, 920–926.
Madler, L., Kammler, H.K., Mueller, R., Pratsinis, S.E., 2002a. Controlled synthesis ofnanostructured particles by flame spray pyrolysis. Journal of Aerosol Science 33,369–389.
Madler, L., Stark, W.J., Pratsinis, S.E., 2002b. Flame-made ceria nanoparticles. Journalof Materials Research 17, 1356–1362.
Manovic, V., Anthony, E.J., 2007. Steam reactivation of spent CaO-based sorbentfor multiple CO2 capture cycles. Environmental Science & Technology 41,1420–1425.
Martavaltzi, C.S., Lemonidou, A.A., 2008. Development of new CaO-based sorbentmaterials for CO2 removal at high temperature. Microporous and MesoporousMaterials 110, 119–127.
Mueller, R., Madler, L., Pratsinis, S.E., 2003. Nanoparticle synthesis at high productionrates by flame spray pyrolysis. Chemical Engineering Science 58, 1969–1976.
Reddy, E.P., Smirniotis, P.G., 2004. High-temperature sorbents for CO2 made of alkalimetals doped on CaO supports. Journal of Physical Chemistry B 108, 7794–7800.
Sakadjian, B.B., Iyer, M.V., Gupta, H., Fan, L.-S., 2007. Kinetics and structuralcharacterization of calcium-based sorbents calcined under subatmosphericconditions for the high-temperature CO2 capture process. Industrial &Engineering Chemistry Research 46, 35–42.
Salvador, C., Lu, D., Anthony, E.J., Abanades, J.C., 2003. Enhancement of CaO for CO2
capture in an FBC environment. Chemical Engineering Journal 96, 187–195.Silaban, A., Harrison, D.P., 1995. High temperature capture of carbon dioxide:
characteristics of the reversible reaction between CaO(s) and CO2(g). ChemicalEngineering Communications 137, 177–190.
Strobel, R., Pratsinis, S.E., 2007. Flame aerosol synthesis of smart nanostructuredmaterials. Journal of Materials Chemistry 17, 4743–4756.


















