
MSU Nanotechnology Education and Research Center...
76
ATOMISTIC SIMULATIONS ON ATOMISTIC SIMULATIONS ON SUPERCOMPUTERS USING SUPERCOMPUTERS USING OPEN SOURCE CODES OPEN SOURCE CODES Joint Institute for High Temperatures Joint Institute for High Temperatures of Russian Academy of Sciences of Russian Academy of Sciences Igor Morozov Igor Morozov MSU Nanotechnology Education and Research Center International School “Computer simulation of advanced materials”, 16–21 July 2012 July 19, 2012
Transcript of MSU Nanotechnology Education and Research Center...

ATOMISTIC SIMULATIONS ON ATOMISTIC SIMULATIONS ON SUPERCOMPUTERS USINGSUPERCOMPUTERS USING
OPEN SOURCE CODESOPEN SOURCE CODES
Joint Institute for High TemperaturesJoint Institute for High Temperaturesof Russian Academy of Sciencesof Russian Academy of Sciences
Igor MorozovIgor Morozov
MSU Nanotechnology Education and Research CenterInternational School “Computer simulation of advanced materials”, 16–21 July 2012
July 19, 2012

Contents1. Atomistic simulations in material science:
- Molecular Dynamics (MD)- Monte-Carlo (MC)
2. Choice of the simulation software:open source vs. commercial
3. Challenges for atomistic simulation codes:- Interaction models- Parallel scaling- Using GPUs- Atomistic simulations on the Grid

First papers on MD simulations

The record numbers of particles taken for MD simulations in different years
4 nm
1964 1.000 Rahman1984 200.000 Abraham1990 1.000.000 Swope, Anderson1994 100.000.000 Beazley, Lomdahl1997 1.213.857.792 Stadler1997 1.399.440.000 Müller1999 5.180.116.000 Roth2000 8.500.000.000 Vashishta2003 19.000.416.964 Kadau, Germann, Lomdahl2005 160.000.000.000 Kadau, Germann, Lomdahl2006 320.000.000.000 Kadau, Germann, Lomdahl2008 1.000.000.000.000 Kadau, Germann
4 m

Mflops 1964 . CDC 6600 10 MHz 1 CPUsGflops 1985 . Cray 2 125 MHz 8 CPUsTflops 1997 . ASCI Red 200 MHz 9152 CPUsPflops 2008 . Roadrunner 3,2 GHz 122400 CoresEflops 2018 . 108 – 109
Years, Flop/s and the numbers of coresYears, Flop/s and the numbers of cores
Voevodin V.V., XII International supercomputing conference «Scientific Service in the Internet: Supercomputing Centers and Challenges», September 21, 2010.

4 nm
1964 1.000 Rahman1984 200.000 Abraham1990 1.000.000 Swope, Anderson1994 100.000.000 Beazley, Lomdahl1997 1.213.857.792 Stadler1997 1.399.440.000 Müller1999 5.180.116.000 Roth2000 8.500.000.000 Vashishta2003 19.000.416.964 Kadau, Germann, Lomdahl2005 160.000.000.000 Kadau, Germann, Lomdahl2006 320.000.000.000 Kadau, Germann, Lomdahl2008 1.000.000.000.000 Kadau, Germann
4 m
The record numbers of particles taken for MD simulations in different years

Multiscale approach in material sciences
Original at Berkeley Lab We site: http://www.lbl.gov/CS/html/exascale4energy/nuclear.html

Choice of the simulation code
Open sourceFree of charge Allows modificationsImplements cutting edge technologiesTested by many users over the internetSupported by a community of developers
ProprietaryReliableEasier to install and configureProfessional supportUsually has better support for different platforms
Creating new code on the basis of an open source project

Basic simulation algorithmsMD, MC, energy minimization, thermostats, boundary conditions, neighbor lists, optimized electrostatics
Particle interaction modelsForce fields for pair and many-body potentials, customization
Trajectory analysis toolsBinary trajectory output, plausible modules for calculation of thermodynamics, correlation functions, etc. including averaging
Parameter input and variationParameter input languages, batch runs, workflows
Parallel executionParallel efficiency on modern supercomputing clusters
Optimization for a special hardware, GPUVisualization
Key features of atomistic simulation codes

Basic simulation algorithmsMD, MC, energy minimization, thermostats, boundary conditions, neighbor lists, optimized electrostatics
Particle interaction modelsForce fields for pair and many-body potentials, customization
Trajectory analysis toolsBinary trajectory output, plausible modules for calculation of thermodynamics, correlation functions, etc. including averaging
Parameter input and variationParameter input languages, batch runs, workflows
Parallel executionParallel efficiency on modern supercomputing clusters
Optimization for a special hardware, GPUVisualization
Key features of atomistic simulation codes

Open source packages for atomistic simulations
-
+
+
++
++
+
GPU
MD for metals and ceramics+++pthreadsCXMDSimple MD, QM/MM, molecular design+++OpenMPFortranTINKER
Coarse-grained models of proteins and nucleic acids
+++C/C++RedMD
High parallel scalability, designed for biomolecular, visualization (VMD)
MPIC++NAMD/VMD
Basic algorithms and force fields, Replica exchange, coarse-grained models, Tcl GUI
++MPIFortran, TclMOILParallel MD for AMBER force field+MPIFortranMDynaMix
High parallel scalability, wide range of potentials and analysis tools
+++MPIC++LAMMPS
General-purpose MD highly optimized for GPUs
++C++, PythonHOOMD-blue
High-precision MD for the large-scale simulation of simple and complex liquids, HDF5 output
+C++HALMD
High performance MD, designed for biological systems and polymers
+MPICGROMACS
General purpose MD, HDF5 output, Java GUI
+Fortran, C++, Java
DL_POLY*
User specified force field (FFML), QM/MM calculations with Empirical Valence Bond (EVB)
++CAdun
CommentMCMDMinParallelLanguagePackage name

SIMULATIONSIMULATIONALGORITHMSALGORITHMS

Molecular Dynamics: Trajectory CalculationSet initial coordinates and positions
Calculate forces between particles
Perform MD step solving the equations of motion
Calculate thermodynamical parameters.Check energy conservation.Output to the trajectory file.
0 0 0 0, , 1,...i i i ir t r v t v i N
i
Ni r
rrUF ),..,( 1
1 1,i n i n i n i nr t r t v t v t
?1 MAXn tt Exitno yes

Monte-Carlo: Metropolis algorithmSet initial coordinates and positions
Randomly displace particles
Calculate the potential energy change
Accept with the given probability:
0 0 0 0, , 1,...i i i ir t r v t v i N
1 1 1 1( , , ),n n n n ni NU U r r U U U
MAXn n Exitno yes
1max max, [0, ], ( ) 1n n
i i i ir r
0 10 exp
UU U
the move is accepted
Return to the previous state
Next step

Equations of motion in Classical MD
1
0 0
1''( ) ( , ,..., ), 1, ,
(0) , (0) .
k k Nk
r t F t r r k Nm
R R V V( )( )kk
U RF Rr
( ) ( ) ( , ) ( , ) .., , , .extk i j i j k
k i j i j kkU R U r U r r rt v r r
1{ ,..., }NR r r 1{ ,..., }NV v vConsider a system of N particles with positions
and velocities .
The Newton’s equations of motion:
Force acting of k-th particle:
General form of the interaction potential:
( )( )
k
kj kj kjpairkj
j jkj kj kj
r U r rF f r
r r r( , ) ( ) ( )i j i j ijU r r U r r U r
Particular case of the pairwise potential:

INTERACTION MODELSINTERACTION MODELS

Interaction models and simulation techniquesParticle-in-cell, hydrodynamic codes
Coarse-grained, discontinuous molecular dynamics
Simple pairwise potentials for atoms ormolecules with fixed atomic bonds
Complicated many-body potentials for atoms (EAM, MEAM, ReaxFF, Tersoff, etc.)
Classical MD for electrons and ions
Wave Packet MD, Electron Force Field
QM/MM hybrid models
Quantum MD (Car-Parinello, DFT, TD-DFT)
Numerical solution of the Schrödinger equation
System size
< 1nm
> 1 mA
tomistic
simulations

Particle-in-cell, hydrodynamic codes
Coarse-grained, discontinuous molecular dynamics
Simple pairwise potentials for atoms ormolecules with fixed atomic bonds
Complicated many-body potentials for atoms (EAM, MEAM, ReaxFF, Tersoff, etc.)
Classical MD for electrons and ions
Wave Packet MD, Electron Force Field
QM/MM hybrid models
Quantum MD (Car-Parinello, DFT, TD-DFT)
Numerical solution of the Schrödinger equation
System size
< 1nm
> 1 m
Interaction models and simulation techniquesA
tomistic
simulations

Force field for interaction of atoms in a molecule
))cos(1(2
)(2
~)(
2
44),...,(
20,
20,
1 1 0
612
1
nVkllk
rqq
rrrrU
torsions
n
bonds anglesii
iii
i
N
i
N
ij ij
ji
ij
ij
ij
ijijN
Non-bonded interactions(van der Waals)
+
+
-
Non-bonded interactions(electrostatic)
Bond stretching
Angle bendingBond rotation
(torsion)
Source: Pingwen Zhang, Molecular Dynamics Simulations (http://math.xtu.edu.cn/myphp/math/image/MD.ppt)

Andrey G. Kalinichev, Seminar talk,JIHT RAS, Moscow, Russia, October 1, 2009
Guillot B (2002) What we have learnt during three decades of computer simulations on water. Journal of Molecular Liquids 101, 219-260.
Finney JL (2004) Water? What's so special about it? Phil. Trans. R. Soc. Lond. B 359, 1145-1165.
Classical Molecular Models of Water

Andrey G. Kalinichev, Seminar talk,JIHT RAS, Moscow, Russia, October 1, 2009
The Structure of H2O Molecule and Classical Intermolecular Potentials
Ab-initio quantum mechanicalEmpirical and semi-empiricalRigid vs flexiblePoint polarizability“Charges on springs” modelsCore-shell models
U Aij/rij12 - Bij/rij
6 + qiqj / rij) +Short-range repulsion Van der Waals Coulombic
+ ½ kb(rij - r0)2 + ½ k ( ij - 0)2
bond stretching bond bending
SPCSPC/ETIP3P
MCYTIPS2TIP4P
BNSST2TIP5P

Particle-in-cell, hydrodynamic codes
Coarse-grained, discontinuous molecular dynamics
Simple pairwise potentials for atoms ormolecules with fixed atomic bonds
Complicated many-body potentials for atoms (Tersoff, EAM, MEAM, ReaxFF, etc.)
Classical MD for electrons and ions
Wave Packet MD, Electron Force Field
QM/MM hybrid models
Quantum MD (Car-Parinello, DFT, TD-DFT)
Numerical solution of the Schrödinger equation
System size
< 1nm
> 1 m
Interaction models and simulation techniquesA
tomistic
simulations

N
iji j
E U
1 2e e ( )ij ijr rij ij c ijU A B f r
/0 ,ijz b
ijB B e
cos
,
( ) ,( )
ijk
ndik
ijk i j ij
w rz c ew r
2( ) ( ) .rcw r f r e
1.5 2 2.5 3 3.5 4
0
5
10
15 E, eV
r12, Å
r12
Many-body Tersoff potential
Parameters for Si:(J. Tersoff, Phys. Rev. B, 37 (1988) 6991)

1.5 2 2.5 3 3.5 4
0
5
10
15N
iji j
E U
1 2e e ( )ij ijr rij ij c ijU A B f r
/0 ,ijz b
ijB B e
cos
,
( ) ,( )
ijk
ndik
ijk i j ij
w rz c ew r
2( ) ( ) .rcw r f r e
E, eV
r12, Å
r12
r13
Many-body Tersoff potential
Parameters for Si:(J. Tersoff, Phys. Rev. B, 37 (1988) 6991)

1.5 2 2.5 3 3.5 4
0
5
10
15N
iji j
E U
1 2e e ( )ij ijr rij ij c ijU A B f r
/0 ,ijz b
ijB B e
cos
,
( ) ,( )
ijk
ndik
ijk i j ij
w rz c ew r
2( ) ( ) .rcw r f r e
E, eV
r12, Å
r12
r13
Many-body Tersoff potential
Parameters for Si:(J. Tersoff, Phys. Rev. B, 37 (1988) 6991)

i j ij ij iji j i i j
U F r r
Embedded Atom Method - EAM
Pair potential
Electron density (transfer function)
Embedding function
r
(r)
(rij)
( )iji j i
U r?

(r)
i j ij ij iji j i i j
U F r r
Pair potential
Electron density (transfer function)
Embedding function
(r)F( )
Embedded Atom Method - EAM

Atomistic model of spallation processat high rate shock*
Vimp Vsur
impactor target
Model parameters:1. = 300 2. Number of Cu atoms 112 0003. Impactor / target mass ratio 1/64. Initial defect number density 0.05
* A. Yu. Kuksin, G.E.Norman, V. V. Stegailov, and A. V. Yanilkin // Journal of Engineering Thermophysics, 2009, Vol. 18, No. 3, pp. 197–226.

Shock wave in Al crystal with Cu precipitates
Vp = 1800 m/sN = 5 million atoms T=300K
Cu precipitatespiston
Vp
atomsof defects
thin slice
200x80x80
* A. Yu. Kuksin, G.E.Norman, V. V. Stegailov, and A. V. Yanilkin // Journal of Engineering Thermophysics, 2009, Vol. 18, No. 3, pp. 197–226.

Source: http://lammps.sandia.gov/workshops/Feb10/Aidan_Thompson/ReaxFF_LAMMPS_2010.pdf


Particle-in-cell, hydrodynamic codes
Coarse-grained, discontinuous molecular dynamics
Simple pairwise potentials for atoms ormolecules with fixed atomic bonds
Complicated many-body potentials for atoms (EAM, MEAM, ReaxFF, Tersoff, etc.)
Classical MD for electrons and ions
Wave Packet MD, Electron Force Field
QM/MM hybrid models
Quantum MD (Car-Parinello, DFT, TD-DFT)
Numerical solution of the Schrödinger equation
System size
< 1nm
> 1 m
Interaction models and simulation techniquesA
tomistic
simulations

Density-Temperature Diagram
100 101 102 103 104 105 106 107 108108
1010
10121014
1016
1018
1020
10221024
1026
1028100 101 102 103 104 105 106 107 108
1081010101210141016101810201022102410261028
Classical gas
Quantum gas
Classical nonideal
plasma
Te, K
ne, cm-3
Degenerate nonideal
plasma
Fae /2
110DN
1
/F kT
1/3 243
en ekT
Nonideality parameter for
electrons
343
DD e
rN n
Number of electrons in the Debye sphere
Degeneracy parameter
MDMD
ionization

Breaking into the atom
Source: Alexey Shaitan, International School “Computer simulation of advanced materials”, Moscow, Russia, July 16–21, 2012
Approximations, restrictions and gains
1. BO approx. (adiabatic approx.)No nonadiabatic processes (charge-transfer reactions,
photochemistry)2. Ground state
No electronic excitations, no electron-phonon coupling, etc
3. Classical approx. for nuclei movementNo proton transfer, atom tunneling, etc
On the good side – big systems, long evolution times, nice parallelization and scaling
TD Schrödinger eq. – 3 atomsMD – 10^9 atoms, up to milliseconds

Ionization and cluster expansion «Coulomb explosion»
In the initial stateIn the initial state the ions are located the ions are located in the nodes of Na55 in the nodes of Na55 icosahedralicosahedral crystal. crystal. Electrons are on top of ions.Electrons are on top of ions.
IonIon--electron mass ratio iselectron mass ratio is 4191041910
ErfErf--like electron ion potentiallike electron ion potential is usedis usedwith with UUminmin = = -- 5.1eV 5.1eV
Typical times of:Typical times of:cluster expansioncluster expansion >> 100100fsfselectron oscillationselectron oscillations << 11fsfs
MD simulations of ionized sodium clusters
0 100 200 3000
0.4
0.8
1.2
1.6
t, fs
rrms, nm
Laser heating
Frozen ionst0 = 100fs
t0 = 200fs
2
1
53
iN
rms ii
r r
Cluster expansion
laser pulse: I = 5·1012 W/cm2, = 50 fs
trajectories of ions
electrons

Ionization and cluster expansion «Coulomb explosion»
In the initial stateIn the initial state the ions are located the ions are located in the nodes of Na55 in the nodes of Na55 icosahedralicosahedral crystal. crystal. Electrons are on top of ions.Electrons are on top of ions.
IonIon--electron mass ratio iselectron mass ratio is 4191041910
ErfErf--like electron ion potentiallike electron ion potential is usedis usedwith with UUminmin = = -- 5.1eV 5.1eV
Typical times of:Typical times of:cluster expansioncluster expansion >> 100100fsfselectron oscillationselectron oscillations << 11fsfs
MD simulations of ionized sodium clusters
0 100 200 3000
0.4
0.8
1.2
1.6
t, fs
rrms, nm
Laser heating
Frozen ionst0 = 100fs
t0 = 200fs
2
1
53
iN
rms ii
r r
Cluster expansion
laser pulse: I = 5·1012 W/cm2, = 50 fs
trajectories of ions
electrons
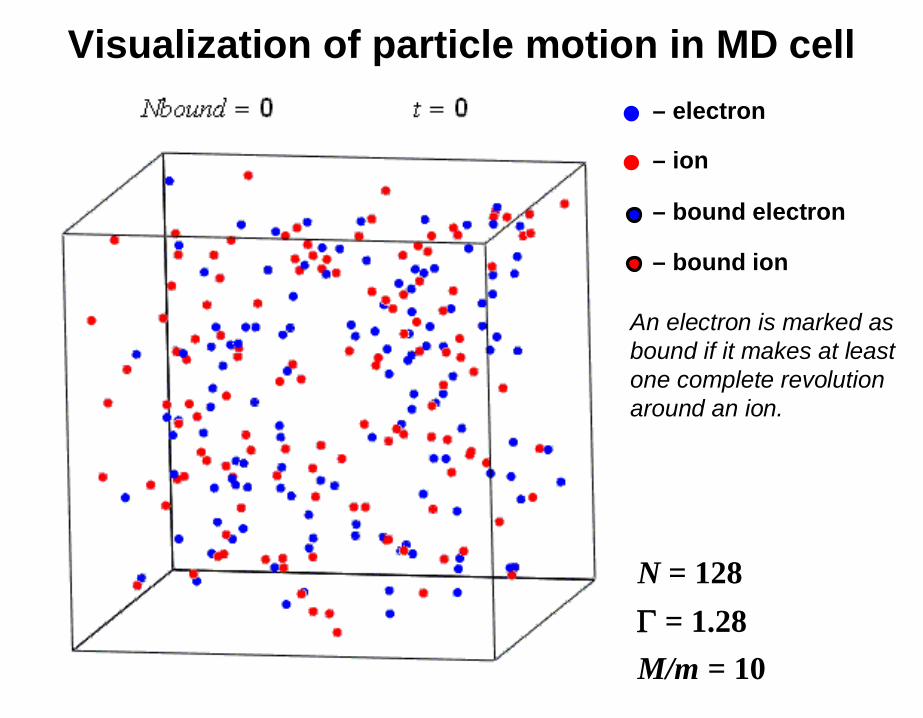
N = 128= 1.28
M/m = 10
An electron is marked as bound if it makes at least one complete revolution around an ion.
– ion
– electron
– bound ion
– bound electron
Visualization of particle motion in MD cell

0.1 0.2 0.3 0.4 0.5 0.6
8
6
4
2
0Coulomb
Deutsch
Cut-off
Error function
Kelbg
r1
kTV
2ekTr
mkTthie
ei
rr
exp11
ei
rr
erf1
ei
rFr1
))erf(1()exp(1)( 2 xxxxF mkTie 2
1,;1,1 rrr
Models for electron-ion interaction potentials

0.1 0.2 0.3 0.4 0.5 0.6
8
6
4
2
0Coulomb
Deutsch
Cut-off
Error function
Corrected Kelbg
r1
kTV
2ekTr
mkTthie
ei
rr
exp11
ei
rr
erf1
))erf(1()exp(1)( 2 xxxxF
22
2 exp~1
eieiei
ei
rkT
eAekTrrF
r
mkTie 2
1,;1,1 rrr
Models for electron-ion interaction potentials

Particle-in-cell, hydrodynamic codes
Coarse-grained, discontinuous molecular dynamics
Simple pairwise potentials for atoms ormolecules with fixed atomic bonds
Complicated many-body potentials for atoms (EAM, MEAM, ReaxFF, Tersoff, etc.)
Classical MD for electrons and ions
Wave Packet MD, Electron Force Field
QM/MM hybrid models
Quantum MD (Car-Parinello, DFT, TD-DFT)
Numerical solution of the Schrödinger equation
System size
< 1nm
> 1 m
Interaction models and simulation techniquesA
tomistic
simulations

)()(24
3exp2
3),( 22
4/3
2 rxprxx iipt
“Physical” parameters (8 real numbers per particle): (r, p) particle coordinate and momentum (3D vectors)
width of the packet ( > 0)p “momentum” of the width
Gaussian wave packet (WP) for a single particle:
r
p
Wave Packet Molecular Dynamics

),()},({ tt kk
k xx
22 2
2 2 2
9ˆ erf2 2 8 2( ) / 3
kk k l klext
k k k lk kl k l
e e rH H Hm m m r
pp
( ) , ( )) , ,( ( )k
k
k kk k
kk
H H HHt t tt pp
r pp r
,dq HNdt q
0 00 0
0 00
0 11 0
0 11 00
N
Wave Packet Molecular DynamicsHamiltonian
Norm-matrix
Equations of motion
2 2
,
ˆ ˆ ˆ ˆ ˆ ˆˆ ˆ ˆ2
ie ei ee ext k ext
k k i k mk i k m
eq eH K V V H Hm x R x x
Many-electron wave function
Total energy
** ln ( ) ( )N
q qq q
(Hartree approximation)

0 1 2 3 4 5 6 7
-80
-70
-60
-50
Eortho, exp = –59.4eV
Epara, exp = –78.98eV
Epara, HF = –77.87eV
Nwp
1 2 3 4 5 6 70.0001
0.001
0.01
0.1
1
Nwp
Ground state of simple atoms represented by multiple Gaussian wave packets
-2 -1 0 1 20
1
2
1
x
2
3
Hydrogen Helium

Excited states of Hydrogen represented by multiple Gaussian wave packets
-12 -8 -4 0 4
I(E)
E, eV
E0 E1
0.35 0.36 0.37 0.38 0.39 0.4 0.410
0.01
0.02
0.03
0.04Eabs, a.u.
, a.u.
Lyman-
Absorbed energy depending on the laser frequency in the area
of 1s 2s transitionNwp = 5, T = 200a.u., E0 = 0.005a.u.
Spectrum of the time correlation function after
excitation

Ionization of H(1s) atom by a laser pulse*
* Comparison with J.P. Hansen, J. Lu, L.B. Madsen, H.M. Nilsen // Phys. Rev. A, 2001, V. 64, P. 033418.
bound free
Classical Trajectory Motne-Carlo (CTMC):
0partE
tot
NP
N
WPMD(1 electron, 3 WPs):
free freeP
0.1 1 100.001
0.01
0.1
1 w

Electron force field approach*
* J.T. Su, W.A. Goddard III, Excited Electron Dynamics Modeling ofWarm Dense Matter, Phys. Rev. Lett., 2007, v. 99, p. 185003.

PARALLEL EXECUTIONPARALLEL EXECUTION
LLNL SequoiaSupercomputer

Top-500: System Architecture


Domain decompositionDomain decomposition
rcut
Lr
iLr
cutr
skin
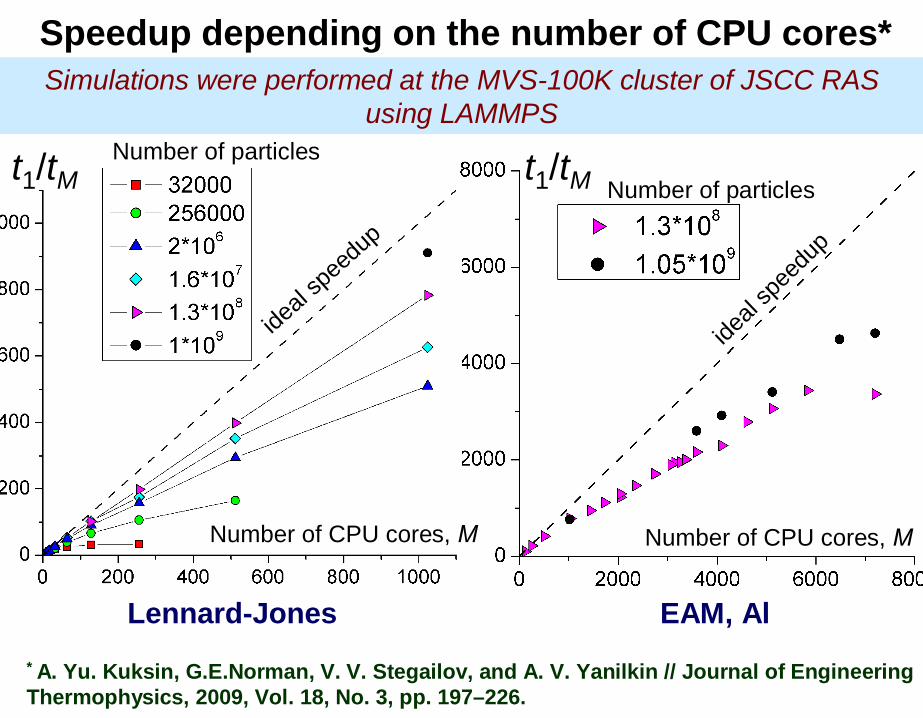
Speedup depending on the number of CPU cores*
ideal
spee
dup
ideal
spee
dup
Number of CPU cores,
t1/tM t1/tMNumber of particles
Lennard-Jones EAM, Al
Simulations were performed at the MVS-100K cluster of JSCC RASusing LAMMPS
* A. Yu. Kuksin, G.E.Norman, V. V. Stegailov, and A. V. Yanilkin // Journal of Engineering Thermophysics, 2009, Vol. 18, No. 3, pp. 197–226.
Number of CPU cores,
Number of particles

The number of particles is fixed toN = 32 000
Lennard-Jones interaction potential
with large cut-off lengthRcut = 7.0
Computing clusterMIPT–60
ApplicationLAMMPS
CPU number limit due tointerprocess communications
* A. Yu. Kuksin, G.E.Norman, V. V. Stegailov, and A. V. Yanilkin // Journal of Engineering Thermophysics, 2009, Vol. 18, No. 3, pp. 197–226.
1 10 100 10000.1
1.0
10.0
100.0
1000.0 Computational time, s
Number of CPU cores
Total timeComputation of forcesCommunications

NAMD: weak scaling for different clusters
Source: http://www.ks.uiuc.edu/Research/namd/performance.html

ACCELERATION USING ACCELERATION USING GPUsGPUs

Top 500 Supercomputers (June 2012)*
* http://www.top500.org

IBM
2011 IBM Corporation60
Accelerators in supercomputers
: Top500.org., XIII
: », 14-24 2011 .
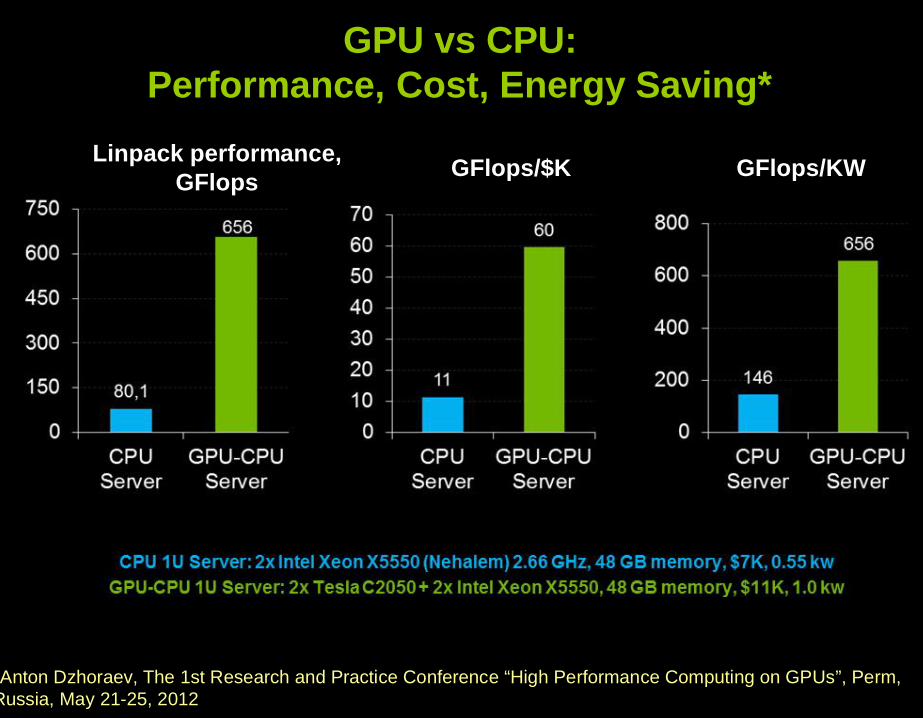
*Anton Dzhoraev, The 1st Research and Practice Conference “High Performance Computing on GPUs”, Perm, Russia, May 21-25, 2012
GPU vs CPU:Performance, Cost, Energy Saving*
GFlops/$K GFlops/KWLinpack performance, GFlops

*Original location: http://www.chepr.ru/index.php?rasd=info&id=75
Comparison of CPU and GPU architectures*

Highly Optimized Object Oriented Molecular DynamicsDevelopers: J.A. Anderson, A. TravessetIowa State University, Ames, IA 50011, USAhttp://codeblue.umich.edu/hoomd-blue/, Free.
Large-scale Atomic/Molecular Massively Parallel SimulatorSandia National Lab, USAhttp://lammps.sandia.gov, Free.
NAMD/VMDUniversity of Illinois at Urbana-Champaign (UIUC), USAhttp://www.ks.uiuc.edu/Research/namd, Free.
MDGPUMDGPU
LAMMPSLAMMPS
ACEACE--MD MD AMBERAMBER
GPU-enabled MD simulation packages
HALMDHALMD+
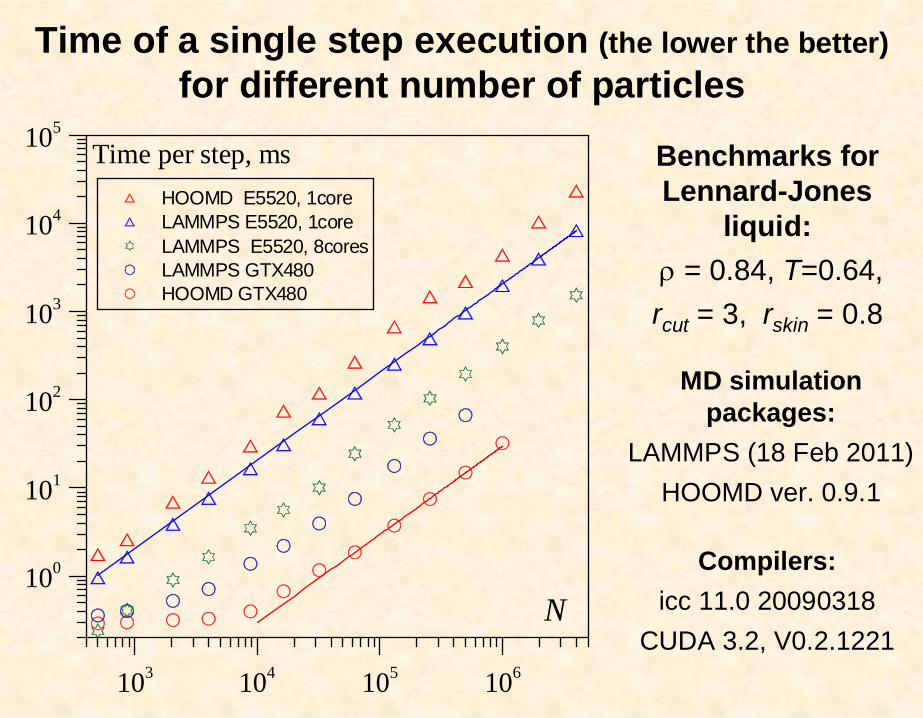
Benchmarks for Lennard-Jones
liquid:= 0.84, T=0.64,
rcut = 3, rskin = 0.8
103 104 105 106
100
101
102
103
104
105
HOOMD E5520, 1coreLAMMPS E5520, 1coreLAMMPS E5520, 8coresLAMMPS GTX480HOOMD GTX480
Time per step, ms
NCompilers:
icc 11.0 20090318CUDA 3.2, V0.2.1221
MD simulation packages:
LAMMPS (18 Feb 2011)HOOMD ver. 0.9.1
Time of a single step execution (the lower the better)for different number of particles

6060 XX
Benchmarks for Lennard-Jones
liquid:= 0.19, T=1.0,
rcut = 3, rskin = 0.8
Compilers:icc 11.0 20090318
CUDA 3.2, V0.2.1221
MD simulation packages:
LAMMPS (20 May 2010)HOOMD ver. 0.9.0
103 104 105 106
0
20
40
60
GPU GTX480/CPU E5520(1core)GPU GTX480/CPU E5520 - LAMMPSGPU C2050/CPU E5520(1core)GPU C1060/CPU E5520(1core)
PGPU /PCPU
N
Ratio between GPU and CPU performancesdepending on the number of particles
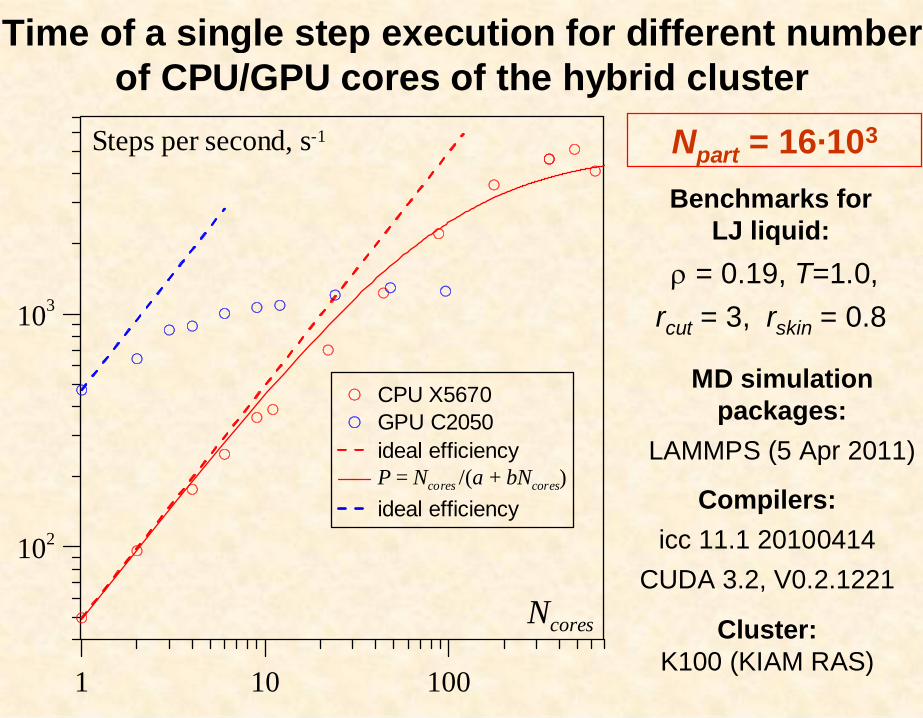
Benchmarks forLJ liquid:
= 0.19, T=1.0,rcut = 3, rskin = 0.8
Compilers:icc 11.1 20100414
CUDA 3.2, V0.2.1221
MD simulation packages:
LAMMPS (5 Apr 2011)
1 10 100
102
103
CPU X5670GPU C2050ideal efficiencyP = Ncores /(a + bNcores)ideal efficiency
Steps per second, s-1
Ncores
Npart = 16·103
Time of a single step execution for different number of CPU/GPU cores of the hybrid cluster
Cluster:K100 (KIAM RAS)
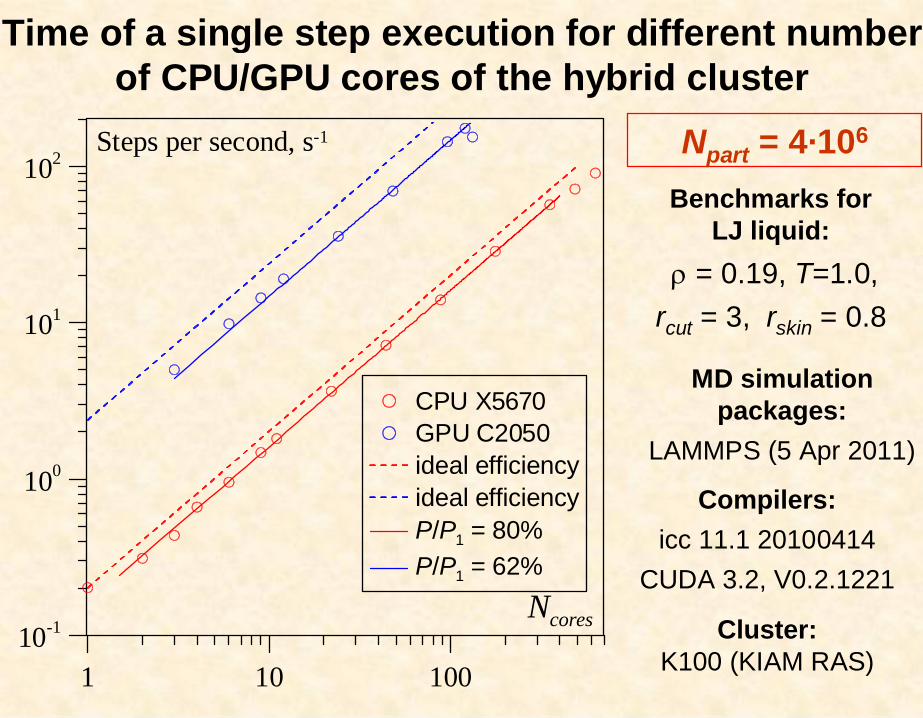
Npart = 4·106
1 10 10010-1
100
101
102
CPU X5670GPU C2050ideal efficiencyideal efficiencyP/P1 = 80%P/P1 = 62%
Steps per second, s-1
Ncores
Time of a single step execution for different number of CPU/GPU cores of the hybrid cluster
Benchmarks forLJ liquid:
= 0.19, T=1.0,rcut = 3, rskin = 0.8
Compilers:icc 11.1 20100414
CUDA 3.2, V0.2.1221
MD simulation packages:
LAMMPS (5 Apr 2011)
Cluster:K100 (KIAM RAS)

11000 X0 XGPU:
NVIDIA Tesla M2050CUDA 4.0, V0.2.1221
CPU:Xeon E5520 (Nehalem),
2.27GHz, 8Mb L3Intel compilerver. 11.0.083
Interactions:Coulomb
(long-range)100 1000 10000 100000
1
10
100PGPU /PCPU
N
Ratio between GPU and CPU performancesdepending on the number of particles

RIKEN RIKEN ““Protein ExplorerProtein Explorer””: : special purpose system for molecular dynamicsspecial purpose system for molecular dynamics
1 unit = Dual-core Intel Xeon CPU+ 24 MD-GRAPE chips
201 units achieve 1 PFlops for MD

ATOMISTIC SIMULATIONSATOMISTIC SIMULATIONSON THE GRIDON THE GRID

MD (or MC) numerical experiment usually goes through a number of stages:
Low level: • prepare system in the initial state• propagate it through the chain of other states• obtain system properties from the MD trajectory
High level: • average over a set of initial states• perform parameter sweep• perform optimal parameter searchetc
need for predefined workflow scenarios
need for workflow
Requirements to Grid middleware

production
Init
Systemcycle
Finalize
logical
Scenarios and workflow graphs
s0
s1
s2
s3
s4
A2 B2
A2 B1
A1 B2
A1 B1
select parameter A
select parameter B
Graph elements Scenario examples

• Automatic workflow generation from GridMD function calls inside the main application• Component architecture, easy interfacing to other MD packages.
• All required components for MD experiment: boundary conditions, thermodynamic ensembles, etc.)
• Support for various job managers• Cross-platform design, compiled for Linux and Windows
• Open source code (available at http://gridmd.sourceforge.net)
Open source GridMD library
GridMD is a C++ class library intended for constructing simulation applications and running them in distributed environments• Single application for serial (debug) and distributed execution

Batch system (PBS, ...)
Workingnode
Cluster
local shell, ssh, Globus, etc
Workingnode
Workingnode
Workingnode
User codeUser code
MPI
Submission of the tasks to parallel and distributed systems
Simulation program

Grid Middleware
Cluster
local shell, ssh,Globus, etc
User codeUser code
MPI
User codeUser code
MPI
User codeUser code
MPI
i = 1 i = 2 i = N
Workingnode
PBS Cluster
Workingnode
PBS Cluster
Workingnode
PBS
Submission of the tasks to parallel and distributed systems
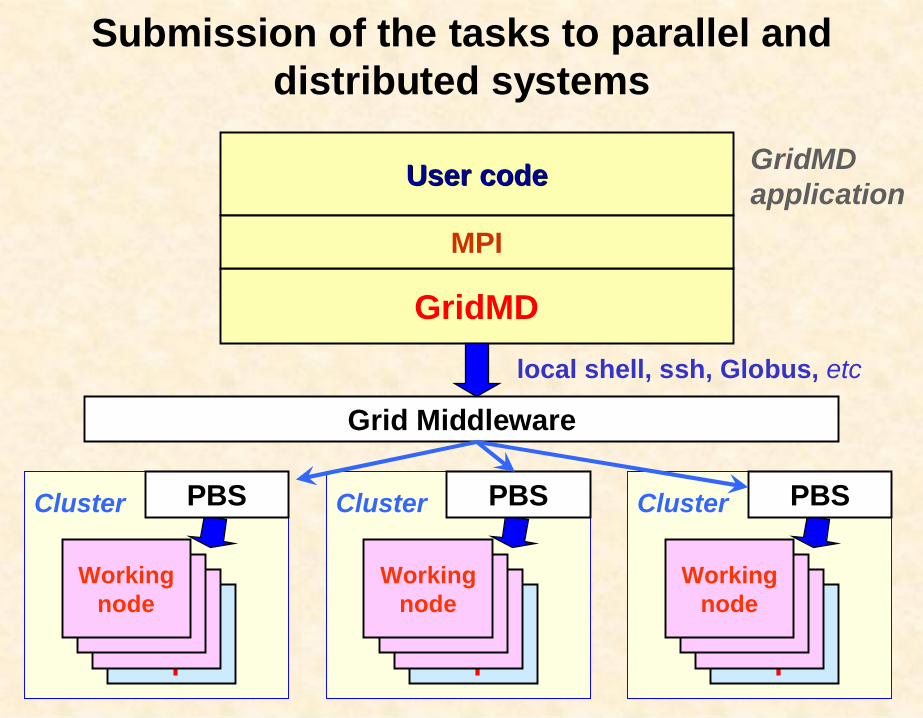
Grid Middleware
Cluster
User codeUser code
GridMD
Workingnode
PBS Cluster
Workingnode
PBS Cluster
Workingnode
PBS
local shell, ssh, Globus, etc
MPI
GridMDapplication
Submission of the tasks to parallel and distributed systems

User codeUser codeparameter sweep, optimization, workflows
GridMDworkflow scenarios, internal job manager
MPI
Nimrod Nimrod workflowsworkflows
PBSPBS
DEISA ShellDEISA Shell JSCC RASJSCC RAS
local shell,ssh,
Globus, etc
Submission of the tasks to parallel and distributed systems
GridMDapplication

Open source packages for atomistic simulations
-
+
+
++
++
+
GPU
MD for metals and ceramics+++pthreadsCXMDSimple MD, QM/MM, molecular design+++OpenMPFortranTINKER
Coarse-grained models of proteins and nucleic acids
+++C/C++RedMD
High parallel scalability, designed for biomolecular, visualization (VMD)
MPIC++NAMD/VMD
Basic algorithms and force fields, Replica exchange, coarse-grained models, Tcl GUI
++MPIFortran, TclMOILParallel MD for AMBER force field+MPIFortranMDynaMix
High parallel scalability, wide range of potentials and analysis tools
+++MPIC++LAMMPS
General-purpose MD highly optimized for GPUs
++C++, PythonHOOMD-blue
High-precision MD for the large-scale simulation of simple and complex liquids, HDF5 output
+C++HALMD
High performance MD, designed for biological systems and polymers
+MPICGROMACS
General purpose MD, HDF5 output, Java GUI
+Fortran, C++, Java
DL_POLY*
User specified force field (FFML), QM/MM calculations with Empirical Valence Bond (EVB)
++CAdun
CommentMCMDMinParallelLanguagePackage name

Source: http://www.stfc.ac.uk/cse/resources/pdf/biomolbenchiii.pdf
Benchmarks for bimolecular simulations
H.H. Loeffler, M.D. Winn, Large biomolecularsimulations on HPC platforms: III. AMBER, CHARMM, GROMACS, LAMMPS and NAMD, 15 March 2012

CONCLUSIONSOpen source projects are particularly suitable for scientific research because they are customizable, extensible, implement a variety of ionization models and optimization techniques.
Quasiclassical atomistic simulations of chemical reactions, ionization/recombination processes and electron-ion relaxation are still a challenge.
GPUs can highly accelerate MD simulations in particular for long-ranged interactions.
Averaging and parameter sweep lead allow to run simulations on Grid and Cloud environments.
Our web site: http://www.ihed.ras.ru/norman


















