
MSA- multiple sequence alignment Aligning many sequences is often preferable to pairwise...
98
MSA- multiple sequence alignment • Aligning many sequences is often preferable to pairwise comparisons. • Problem- Computational complexity of multiple alignments grows rapidly with the number of sequences being aligned.
-
date post
22-Dec-2015 -
Category
Documents
-
view
220 -
download
0
Transcript of MSA- multiple sequence alignment Aligning many sequences is often preferable to pairwise...

MSA- multiple sequence alignment
• Aligning many sequences is often preferable to pairwise comparisons.
• Problem- Computational complexity of multiple alignments grows rapidly with the number of sequences being aligned.

“Even using supercomputers or networks of workstations,
multiple sequence alignment is an intractable problem for more
than 20 or so sequences of average length and complexity.”

As a result, alignment methods using heuristics have been
developed. These methods, (including ClustalW) cannot
guarantee an optimal alignment, but can find near-optimal
alignments for larger number of sequences.

CLUSTALW
• Developed in 1988
• Begins by aligning closely related sequences and then adds increasingly divergent sequences to produce a complete msa.

• http://www.ncbi.nlm.nih.gov/
• http://www.ebi.ac.uk/clustalw/

Introduction to Molecular Phylogeny*
*Phylogeny- the evolutionary history of a group

Mutations Happen!
3 types possible:
• Deleterious
• Advantageous
• ???

Important Point:
• Much of variation that is observed among individuals must have little beneficial or detrimental effect and be essentially selectively neutral.
• Deleterious mutations are screened out. Advantageous mutations are rare.

Functional Constraints?
• Portions of genes that especially important are said to be under functional constraint and tend to accumulate changes very slowly.
• Ex. = histone proteins- practically every
amino acid is important. A yeast histone can replace a human histone.

Relative Rate of Change within -globin gene (4 mammals)
0
0.5
1
1.5
2
2.5
3
3.5
4
5'flank 5' UT intron 1 coding 3' UT 3' flank
# of substitutions / site / 100 my

Basis of Molecular Phylogenetics
• The evolution of species can be modeled as a bifurcating process- speciation is initiated when two populations become reproductively isolated.

Basis of Molecular Phylogenetics
• Once these two populations cease to interbreed, it is inevitable that they diverge due to random mutational processes.

Basis of Molecular Phylogenetics
• Over time, this branching process may repeat itself.
• A species is said to be related to some other species with which it shares a direct common ancestor.


Basis of Molecular Phylogenetics
• The amount of DNA sequence difference between a pair of organisms should indicate how recently those two organisms shared a common ancestor.


Basis of Molecular Phylogenetics
• The longer two populations remain reproductively isolated, the more DNA divergence will occur.
• The longer two populations remain reproductively isolated, the more protein divergence will occur.

Molecular Phylogeny is relatively new.
• Evolution by Natural Selection- Darwin/Wallace 1858
• Molecular Phylogeny 1960s ??

How it started . . ..
• In 1959, scientists determined the three-dimensional structures of two proteins that are found in almost every animal: hemoglobin and myoglobin.
• During the next two decades, myoglobin and hemoglobin sequences were determined for dozens of mammals, birds, reptiles, amphibians, fish, etc.

What they found . . .
• “This tree agreed completely with observations derived from paleontology and anatomy about the common descent of the corresponding organisms.”*
• *from Science and Creationism: A View from the National Academy of Sciences, 2nd Ed., 1999.

Organisms with high degrees of molecular similarity are expected
to be more closely related than those that are dissimilar.

Advantages of Molecular Phylogeny
• Can be used to decipher relationships between all living things
• Relying on anatomy can be misleading- Similar traits can evolve in organisms that are not closely related (i.e. convergent evolution lead to eyes in vertebrates, insects, and molluscs).

Word of Caution
Phylogenetic analysis is controversial. There are a wide variety of different methods for analyzing the data, and even the experts often disagree on the best method for analyzing the data.

Why so controversial??
2 Reasons:

#1 - Molecular vs.
Classical• How much weight is given to
molecular phylogenetic data, when it contrasts the findings of the traditional taxonomist??

. . . • The
phylogeny of whales :

• The
phylogeny of whales:

How many cars changed spaces during this 2 hour interval?
• Parking lot “A” at 2:00
• Parking lot “A” at 4:00

#2- Molecular Phylogeny requires statistical estimations.
• Parking lot “A” at 2:00
• Parking lot “A” at 4:00

Phylogenetic Data Analysis requires 4 steps
• 1) Alignment
• 2) Determine the substitution model
• 3) Tree Building
• 4) Tree Evaluation

STEP 1- Alignment
• Molecular phylogenetic analysis is dependent on a good alignment. An evolutionary tree based on an improper alignment is an erroneous tree.


Homology
It is critical to phylogenetic analysis that homologous characters be compared across species.
Webster’s New Collegiate- Fundamental similarity of structure due to descent from a common ancestral form.



Compare homologous genes and homologous characters:
• For DNA and proteins, this means that gaps must be placed correctly in multiple alignments to ensure that the same position is being compared for each species.

Homologous Genes? When could you accidentally compare
nonhomologous genes?
• Be careful if you comparing genes that are members of a gene family.
• Comparing a tubulin-3 from one species with a tubulin-6 from another will not generate accurate results.

What to align?
• Phylogenetic trees are generated by comparing DNA or protein. The molecule of choice depends on the question you are attempting to answer.

DNA
• contains more evolutionary information than protein :
• ATT GCG AAA CAC•
* * * *
• ATA GCC AAG CTC

Protein(same region analyzed only 1
difference)
• Ile-Ala-Lys- His
• Ile-Ala-Lys- Leu

DNA• high rate of base substitution
makes DNA best for very short term studies, e.g. closely-related species


*Homoplasy• Return of a character to its
original state, thus masking intervening mutational events. Every fourth mutation should result in a homoplasy.

Protein
• more reliable alignment than DNA:
fewer homoplasies than DNA • lower rate of substitution than
DNA; better for wide species comparisons


rRNA= ribosomal RNA• Best for very long term evolutionary
studies spanning biological kingdoms
• Selective processes constraining sequence evolution should be roughly the same across species boundaries

STEP 2- Determine the substitution model.
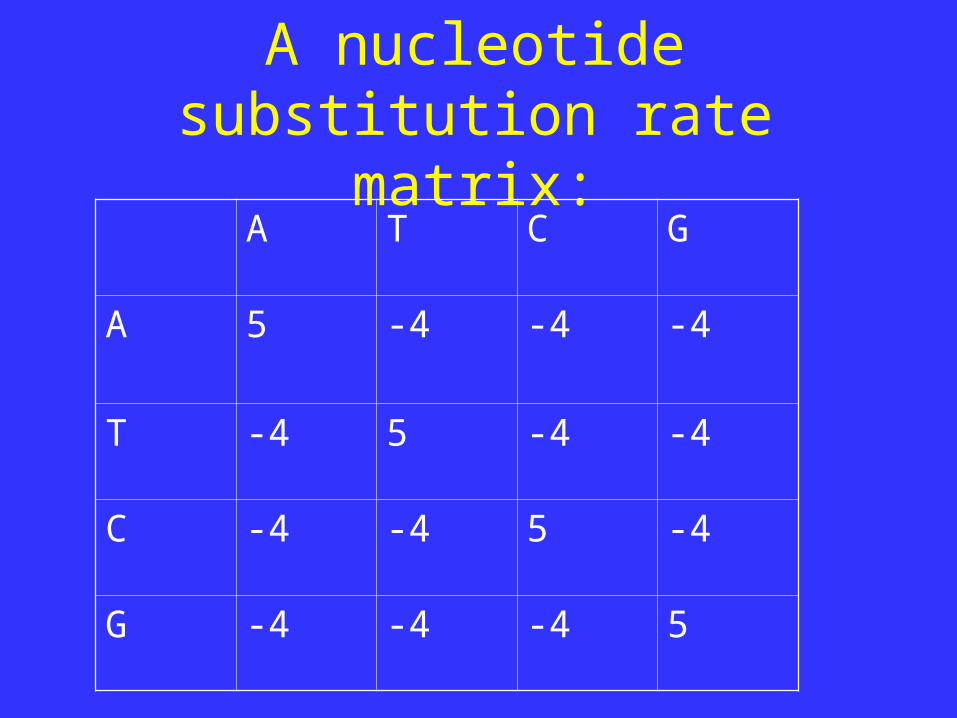
A nucleotide substitution rate matrix:
A T C G
A 5 -4 -4 -4
T -4 5 -4 -4
C -4 -4 5 -4
G -4 -4 -4 5

Step 3- Tree Building

Step 3- Tree Building
Tree terminology:
Nodes: branching points Branches: linesTopology: branching pattern

Branches can be rotated at a node, without changing the relationships.


Unrooted trees explain phylogenetic relationships; they say nothing about the directions
of evolution- the order of descent



There are two main tree drawing methods.
• - Character Methods• - Distance Methods
Both approaches are widely used and work well with most data sets.

Distance methods
Distance- a measure of the overall pairwise difference between two data sets.
The raw material for tree reconstruction is tabular summaries of the pairwise differences between all data sets to be analyzed

In distance methods, the first step is to calculate a matrix of all pairwise
differences between a set of sequences. Species A B C D
B 9 ----- ----- -----
C 8 11 ----- -----
D 12 15 10 -----
E 15 18 13 5

Distance methods
• Identify the sequence pairs that have the smallest number of sequence changes between them and are identified as ‘neighbors’. On a tree, these sequences share a common ancestor and are joined by a short branch.

UPGMA, pairwise distance and neighbor joining are distance
methods.
• They progressively group sequences, starting with those that are most alike.
• UPGMA = unweighted-pair-group method with arithmetic mean

Phylogenetic trees based on distance methods.
1) The two sequences that are closest together are connected at a node.
2) The process is repeated until all sequences are joined.
3) Addition of the last sequence defines the root of the tree.

The branch lengths may reflect the degree of similarity (and theoretically
reflect evolutionary time).• Scaled trees- when branch length are
proportional to the differences between base pairs.
• In the best of cases, scaled trees are additive (the physical length of branches connecting any two nodes is an accurate representation of their accumulated differences).


Phylogenetic trees based on distance methods.
• Relatively simple.
• Problem:
–May not be accurate!!

Character Methods
“There is no denying that distance-based methods “look at the big picture” and pointedly ignore much potentially valuable information.”

Character Methods
Analysis of individual characters are translated into evolutionary trees.
Character- a well-defined feature that can exist in a limited number of different states. (Ex. DNA and protein sequences)

The concept of parsimony is at the heart of all character-based
methods of phylogenetic reconstruction.
• The process of attaching preference to one evolutionary pathway over another on the basis of which pathway requires the invocation of the smallest number of mutational events.

Character-based methods of phylogenetic reconstruction.
• “The relationship that requires the fewest number of mutations to explain the current state of affairs is most likely to be correct”

First Step in Character Methods: Identify all of the informative
sites:

2nd step: Calculate the minimum number of substitutions at each informative site:

Final step:
After sequences are aligned, algorithms model each tree.

Maximum parsimony is a character method
• Character methods require a multiple sequence align. Analysis of informative ‘characters’ is used to construct an evolutionary tree.

Maximum Parsimony: General scientific criterion for
choosing among competing hypotheses states that we should
accept the hypothesis that explains the data most simply and efficiently.
• The tree requiring the _______ number of nucleic acid or amino acid substitutions is selected.

Maximum Parsimony:
• The algorithm searches for a tree that requires the smallest number of changes to explain the differences observed among the groups under study.

Character methods are best suited for . . .
• Sequences that are quite similar.
• Small number of sequences
The method is computationally time consuming as all possible trees are examined.

Phylogenetic trees based on maximum likelihood:
The aim is to find the tree (among all possible trees)
that has the highest likelihood of producing the observed data (statistical methods).

Phylogenetic trees based on maximum likelihood
are similar to maximum parsimony methods but also take into account the likelihood of specific mutations (ex. A G).

Mutation Rates Vary:
• Transitions (purine to purine or pyrimidine to pyrimidine) occur more frequently than transversions (purine to pyrimidine or pyrimidine to purine).

Many of the methods described require
significant amounts of computer time.
Why?

Number of possible rooted and unrooted trees
# of Data Sets # of Rooted Trees # of Unrooted Trees
2 1 1
3 3 1
4 15 3
5 105 15
10 34,459,425 2,027,025
15 213,458,046,767,875 7,905,853,580,625
20 8,200,794,532,637,891,559,375 221,643,095,476,699,771,875


Programs take shortcuts.• When a large number of tree is being
compared, it is impossible to score each tree. A shortcut algorithm establishes an upper limit. As it evaluates other trees, it throws out any tree exceeding the upper bound before the calculation is completed.

• Here are some 194 of the phylogeny packages, and 16 free servers, that I know about. Updates to these pages are made about twice a year.

Tree EvaluationEvery ‘tree drawing program’ will
generate a tree. The important question is whether or not the tree drawn is the right one.
• In some cases, there are many trees of similar probabilities.

Vertebrate -globins:


Bootstrap method of assessing tree reliability:
Inferred tree is constructed from data set.
Re-run the calculation on subsets of the data (resampling).
Resampling is repeated several (100-1000) times.


Bootstrap method
Bootstrap trees are constructed from the resampled data sets.
Bootstrap tree is compared to original inferred tree.
% of bootstrap trees supporting a node are determined for each node in the tree.

Molecular ClockAddition of time to phylogenetic
tree. Units of time are often in millions of years.
Assumption- substitution rates are constant over millions of years.

Molecular ClockRates of molecular evolution
for genes with similar functional constraints can be quite uniform. (Clock may run at different rates in different proteins.)

The End

•Evolutionary biology also has benefited greatly from genome-sequencing projects. The wealth of new genome data is helping to better resolve the tree of life, particularly its major branches. This has been especially true for prokaryotes, where more than 80 genomes have been sequenced so far and the results have greatly improved our view of the early history of life.

Problem- As the # of sequences increases, the # of possible trees increases dramatically
# of sequences # of trees
3 1
4 3
5 15
6 105
7 945
8 10,395
9 135,135
10 1,027,025
50 2.8 x 1074

Phylogenetic trees based on neighbor joining.
• Also utilizes a ‘distance matrix’
• Neighbor joining algorithm searches for sets of neighbors that minimize the total length of the tree.
• Can produce reasonable trees, especially when evolutionary distances are short.

• For vertebrates, many thorny issues remain to be resolved, such as the phylogeny of families and other major groups in the tree of life. For example, it is not yet known whether humans are closer to mice or to cattle because different results have been obtained with different gene analyses. On the other hand, there is no guarantee that complete genome sequences will immediately solve all phylogenetic questions, as evidenced by the continuing debate over the relationships among humans, flies, and nematodes. We will need to develop new statistical methods and bioinformatics tools to handle the greater volume of data and to unravel the complexities of molecular evolution.

• Choice of individual genes or proteins.

Distance matrices:
• Scoring matrices include values for all possible substitutions. Each mismatch between two sequences adds to the distance, and each identity subtracts from the distance.


















