
Mpc(maltolyl p coumarate) 2007 shinky et al jnr (mpc dementia models)
12
A Novel Compound, Maltolyl p-coumarate, Attenuates Cognitive Deficits and Shows Neuroprotective Effects In Vitro and In Vivo Dementia Models Ki Young Shin, 1 Geon Ho Lee, 1 Cheol Hyoung Park, 1 Hee Jin Kim, 1 Soo-Hyun Park, 2 Seonghan Kim, 1 Hye-Sun Kim, 1 Kwan-Sun Lee, 3 Beom Young Won, 4 Hyung Gun Lee, 4 Jin-Ho Choi, 2 and Yoo-Hun Suh 1,4 * 1 Department of Pharmacology, College of Medicine, National Creative Research Initiative Center for Alzheimer’s Dementia and Neuroscience Research Institute, MRC, Seoul National University, Seoul, Korea 2 Faculty of Food Science and Biotechnology, Pukyong National University, Busan, Korea 3 Central Research Institute, Hanmi Pharmaceutical Company, Gyeonggi-do, Korea 4 Braintropia Company, Gyeonggi-do, Korea To develop a novel and effective drug that could enhance cognitive function and neuroprotection, we newly synthesized maltolyl p-coumarate by the esterifi- cation of maltol and p-coumaric acid. In the present study, we investigated whether maltolyl p-coumarate could improve cognitive decline in scopolamine- injected rats and in amyloid beta peptide 1–42 -infused rats. Maltolyl p-coumarate was found to attenuate cog- nitive deficits in both rat models using passive avoid- ance test and to reduce apoptotic cell death observed in the hippocampus of the amyloid beta peptide 1–42 - infused rats. We also examined the neuroprotective effects of maltolyl p-coumarate in vitro using SH-SY5Y cells. Cells were pretreated with maltolyl p-coumarate, before exposed to amyloid beta peptide 1–42 , glutamate or H 2 O 2 . We found that maltolyl p-coumarate signifi- cantly decreased apoptotic cell death and reduced re- active oxygen species, cytochrome c release, and cas- pase 3 activation. Taking these in vitro and in vivo results together, our study suggests that maltolyl p- coumarate is a potentially effective candidate against Alzheimer’s disease that is characterized by wide spread neuronal death and progressive decline of cog- nitive function. V V C 2007 Wiley-Liss, Inc. Key words: Alzheimer’s disease; apoptosis; cognitive impairment; maltolyl p-coumarate; oxidative stress Many neurodegenerative diseases including Alzhei- mer’s disease (AD), Parkinson’s disease, and amyotrophic lateral sclerosis are distinguished by the gradual loss of specific sets of neurons that results in deficits of move- ment and cognitive function (Thompson, 1995; Satry and Rao, 2000). AD is especially characterized by the presence of amyloid beta peptide (Ab deposition in neu- ritic plaques and by neuronal loss in brain regions involved in learning and memory processes (Hyman et al., 1984; Yankner, 1996; Mattson, 1997; Suh and Checler, 2002). Hippocampal neurons that are related to learning and memory (Karczmar, 1993) are known to be particularly vulnerable in AD (Whitehouse et al., 1982; Dutar et al., 1995; Winkler et al., 1995). Ab deposition may play a fatal role in the pathogenesis of AD by inducing neuronal cell death through the processes, such as glutamate-mediated excitotoxicity, increased produc- tion of reactive oxygen species (ROS) or hydrogen per- oxide-mediated apoptosis (Behl et al., 1994; Goodman and Mattson, 1994; Mark et al., 1997; Suh and Checler, 2002). Because oxidative stress is one of the major causes of neurodegeneration with aging (Leutner et al., 2001; Floyd and Hensley, 2002) and the subsequent cell death results in cognitive decline, an antioxidant may be an effective candidate that improves impairments of learning and memory processing (Petkov et al., 1993; Wo ¨rtwein et al., 1994; D’Hooge and De Deyn, 2001). Thus, a drug that could ameliorate neurotoxicity and cognitive dysfunction should be effective to treat various neurode- generative diseases including AD. We have reported previously the biologic activities of the root extract of the reed (Phragmites commnis) (Choi et al., 1993, 1997a,b, 1998). p-Coumaric acid, a compo- nent of reed root extract, is a phenolic acid that possesses K.Y. Shin and G.H. Lee contributed equally to this work. Contract grant sponsor: Ministry of Science and Technology, Korea; Contract grant sponsor: BK21 Life science. *Correspondence to: Dr. Yoo-Hun Suh, Department of Pharmacology, College of Medicine, Seoul National University, 28 Yeongeon-dong, Jongno-gu, Seoul, 110-799, Korea. E-mail: [email protected] Received 9 February 2007; Revised 6 April 2007; Accepted 7 April 2007 Published online 28 June 2007 in Wiley InterScience (www. interscience.wiley.com). DOI: 10.1002/jnr.21397 Journal of Neuroscience Research 85:2500–2511 (2007) ' 2007 Wiley-Liss, Inc.
-
date post
08-Apr-2016 -
Category
Documents
-
view
219 -
download
0
description
Â
Transcript of Mpc(maltolyl p coumarate) 2007 shinky et al jnr (mpc dementia models)

A Novel Compound, Maltolylp-coumarate, Attenuates Cognitive Deficitsand Shows Neuroprotective Effects InVitro and In Vivo Dementia Models
Ki Young Shin,1 Geon Ho Lee,1 Cheol Hyoung Park,1 Hee Jin Kim,1
Soo-Hyun Park,2 Seonghan Kim,1 Hye-Sun Kim,1 Kwan-Sun Lee,3 Beom YoungWon,4 Hyung Gun Lee,4 Jin-Ho Choi,2 and Yoo-Hun Suh1,4*1Department of Pharmacology, College of Medicine, National Creative Research Initiative Center forAlzheimer’s Dementia and Neuroscience Research Institute, MRC, Seoul National University, Seoul, Korea2Faculty of Food Science and Biotechnology, Pukyong National University, Busan, Korea3Central Research Institute, Hanmi Pharmaceutical Company, Gyeonggi-do, Korea4Braintropia Company, Gyeonggi-do, Korea
To develop a novel and effective drug that couldenhance cognitive function and neuroprotection, wenewly synthesized maltolyl p-coumarate by the esterifi-cation of maltol and p-coumaric acid. In the presentstudy, we investigated whether maltolyl p-coumaratecould improve cognitive decline in scopolamine-injected rats and in amyloid beta peptide1–42-infusedrats. Maltolyl p-coumarate was found to attenuate cog-nitive deficits in both rat models using passive avoid-ance test and to reduce apoptotic cell death observedin the hippocampus of the amyloid beta peptide1–42-infused rats. We also examined the neuroprotectiveeffects of maltolyl p-coumarate in vitro using SH-SY5Ycells. Cells were pretreated with maltolyl p-coumarate,before exposed to amyloid beta peptide1–42, glutamateor H2O2. We found that maltolyl p-coumarate signifi-cantly decreased apoptotic cell death and reduced re-active oxygen species, cytochrome c release, and cas-pase 3 activation. Taking these in vitro and in vivoresults together, our study suggests that maltolyl p-coumarate is a potentially effective candidate againstAlzheimer’s disease that is characterized by widespread neuronal death and progressive decline of cog-nitive function. VVC 2007 Wiley-Liss, Inc.
Key words: Alzheimer’s disease; apoptosis; cognitiveimpairment; maltolyl p-coumarate; oxidative stress
Many neurodegenerative diseases including Alzhei-mer’s disease (AD), Parkinson’s disease, and amyotrophiclateral sclerosis are distinguished by the gradual loss ofspecific sets of neurons that results in deficits of move-ment and cognitive function (Thompson, 1995; Satryand Rao, 2000). AD is especially characterized by thepresence of amyloid beta peptide (Ab deposition in neu-ritic plaques and by neuronal loss in brain regionsinvolved in learning and memory processes (Hyman
et al., 1984; Yankner, 1996; Mattson, 1997; Suh andChecler, 2002). Hippocampal neurons that are related tolearning and memory (Karczmar, 1993) are known to beparticularly vulnerable in AD (Whitehouse et al., 1982;Dutar et al., 1995; Winkler et al., 1995). Ab depositionmay play a fatal role in the pathogenesis of AD byinducing neuronal cell death through the processes, suchas glutamate-mediated excitotoxicity, increased produc-tion of reactive oxygen species (ROS) or hydrogen per-oxide-mediated apoptosis (Behl et al., 1994; Goodmanand Mattson, 1994; Mark et al., 1997; Suh and Checler,2002). Because oxidative stress is one of the major causesof neurodegeneration with aging (Leutner et al., 2001;Floyd and Hensley, 2002) and the subsequent cell deathresults in cognitive decline, an antioxidant may be aneffective candidate that improves impairments of learningand memory processing (Petkov et al., 1993; Wortweinet al., 1994; D’Hooge and De Deyn, 2001). Thus, adrug that could ameliorate neurotoxicity and cognitivedysfunction should be effective to treat various neurode-generative diseases including AD.
We have reported previously the biologic activitiesof the root extract of the reed (Phragmites commnis) (Choiet al., 1993, 1997a,b, 1998). p-Coumaric acid, a compo-nent of reed root extract, is a phenolic acid that possesses
K.Y. Shin and G.H. Lee contributed equally to this work.
Contract grant sponsor: Ministry of Science and Technology, Korea;
Contract grant sponsor: BK21 Life science.
*Correspondence to: Dr. Yoo-Hun Suh, Department of Pharmacology,
College of Medicine, Seoul National University, 28 Yeongeon-dong,
Jongno-gu, Seoul, 110-799, Korea. E-mail: [email protected]
Received 9 February 2007; Revised 6 April 2007; Accepted 7 April
2007
Published online 28 June 2007 in Wiley InterScience (www.
interscience.wiley.com). DOI: 10.1002/jnr.21397
Journal of Neuroscience Research 85:2500–2511 (2007)
' 2007 Wiley-Liss, Inc.

free radical scavenging and antioxidant properties(Hertog et al., 1995; Kato et al., 1997; Lodovici et al.,2001; Niwa et al., 2001; Abdel-Wahab et al., 2003).Additionally, maltol is suggested to be a functional agentthat prevents oxidative damage in mice brains (Kimet al., 2004), in horse blood plasma (Lee and Lee, 2005),and in human neuroblastoma cells (Yang et al., 2006).Because both parent compounds, maltol and p-couma-rate, are all known compounds, we want to developnew compound having more therapeutic efficacy againstAlzheimer’s disease. Based on these previous findings,we synthesized maltolyl p-coumarate (MPC) by theesterification of maltol isolated from Korean ginseng(Panax ginseng C.A. Meyer) and p-coumaric acid. Weconfirmed that MPC has been successfully synthesizedthrough NMR (nuclear magnetic resonance) and GC/MS (gas chromatography/mass spectroscopy) method.
In the present study, we investigated whetherMPC could improve cognitive decline in scopolamine-injected or Ab1–42-infused rats and reduce apoptotic celldeath in the brains of the Ab1–42-infused rats. We alsochecked the neuroprotective effects of MPC in vitrousing SH-SY5Y cells.
MATERIALS AND METHODS
Synthesis of MPC
The structure of MPC is shown in Figure 1A. Fourgrams of p-coumaric acid was added to a mixed solution of 16ml of pyridine and 8 ml of acetic anhydride. The mixture wasstirred overnight at room temperature and vacuum-filtered.The dried solid was added to 15 ml of chloroform, stirred for10 min, and vacuum-filtered. The product was washed withchloroform to obtain 3.0 g of p-acetoxycinnamic acid (Fig.1C). 2.8 g of p-acetoxycinnamic acid was dissolved in 40 mlof dimethylformamide. Thionyl chloride (1.05 ml) was addedto the solution. The reaction mixture was stirred for 1.5 hr at2158C to 2208C. Pyridine (2.3 ml) was added followed bythe addition of 1.62 g of maltol (Fig. 1D). The temperature
was allowed to rise to room temperature. After stirring over-night, the reaction mixture was vacuum-filtered. The productwas washed with water and was vacuum-dried to obtain3.92 g of maltolyl p-acetoxycinnamate (Fig. 1B). Maltolyl p-acetoxycinnamate (6 g) was dissolved in 60 ml of methanol,30 ml of triethylamine, and 20 ml of water and stirred for2.5 hr at 20–258C. The reaction mixture was evaporated todryness under reduced pressure. The residue was taken up in50 ml of methanol and the suspension was stirred for 10 minand then vacuum-filtered. The product was washed withmethanol and vacuum-dried to obtain 4.51 g of MPC.
Reagents and Antibodies
Ab1–42 peptide was purchased from US Peptide (Ran-cho Cucamonga, CA) and aged by incubation in 0.1 M PBS(pH 7.4) at 378C for 7 days. Anti-cytochrome c and anti-b-tubulin antibodies were obtained from Santa Cruz Biotechno-logy (Santa Cruz, CA) and anti-COX IV and anti-cleavedcaspase-3 antibodies were purchased from Cell Signaling(Danvers, MA). All other chemicals and reagents were pur-chased from Sigma (St. Louis, MO). MPC was dissolved ini-tially in PBS solution containing 2% dimethylsulfoxide(DMSO). All drugs were prepared just before use.
Animals
Seven-week-old Wistar rats were housed in a specificpathogen-free room that was automatically maintained on a12-hr light–dark cycle at 258C and proper humidity. Theanimals were also given food and water ad lib. All experimentswere carried out in accordance with the Guidelines forAnimal Experiments of Ethics Committee of Seoul NationalUniversity.
Animal Models
Scopolamine Injection. Some researchers have usedscopolamine-induced rats to screen the effects of drugs oncognitive enhancement (Park et al., 2000; van der Staay andBouger, 2005). Rats were given scopolamine (1 mg/kg, i.p.)1 hr before the acquisition trial. A single dose of MPC (100mg/kg, p.o.) was given to rats 30 min after scopolamine treat-ment and after another 30 min.
Ab1–42 Infusion. The surgical techniques used in thisstudy were described previously (Itoh et al., 1996; Nitta et al.,1997; Oka et al., 1999, 2000). Rats were anesthetized with so-dium pentobarbital (25 mg/kg, i.p.). Continuous infusion ofAb1–42 (600 pmol/day) was maintained for a week by attach-ment of an infusion kit connected to an osmotic mini-pump(Alzet 1007D; Alza, CA). The infusion kit was implanted intothe right ventricle (1.2 mm posterior to the bregma, 1.5 mmlateral to the midline, 4.0 mm ventral to the surface of theskull) according to the brain atlas of Paxinos and Watson(1986). The guide cannula was secured with dental cement andstainless steel skull screws. Rats infused by Ab1–42 for a weekwere injected with MPC (10 mg/kg, p.o.) or vitamin E (10mg/kg, p.o.) 2 weeks from the first day of Ab1–42 infusion.
Fig. 1. The structure and synthesis of MPC. (A) The structure ofMPC was shown. The molecular formula of MPC is as follows: 1H-NMR (d, DMSO-d6): 2.26 (s, 3H, CH3C 5 O), 6.42 (d, 1H, mal-tol, CH 5 CH), 6.58 (d, 1H, CH 5 CH), 6.82 (d, 2H, benzenering), 7.61 (d, 2H, benzene ring), 7.73 (d, 1H, CH 5 CH), 8.11 (d,1H, maltol CH 5 CH). Maltolyl p-acetoxycinnamate (B), the pre-cursor component of MPC, was synthesized by the esterificationbetween p-acetoxycinnamic acid (C) and maltol (D). Description onthe MPC synthesis method was presented in Materials and Methodsin detail.
Neuroprotective Effects of MPC 2501
Journal of Neuroscience Research DOI 10.1002/jnr

Passive Avoidance Test
A step-through type passive avoidance test apparatus(Model PACS-30, Columbus Instruments Int.) was used toevaluate the effects of MPC on learning and memory, asdescribed previously (Shen et al., 1990). The shuttle box is di-vided into two chambers of equal size (23.5 3 15.5 3 15.5cm) separated by a guillotine door (6.5 3 4.5 cm). The lightchamber is equipped and rats can enter the dark chamberthrough the guillotine door. Rats were placed initially in thelight chamber with the door open. On entering the darkcompartment, the door is closed automatically. Training wasrepeated until the rats entered the dark compartment within20 sec (training trial). Rats were placed in the illuminatedchamber 24 hr after the training trial. When rats entered thedark chamber, electrical foot shock (0.5 mA) was deliveredfor 3 sec through the grid floor and the door was closed auto-matically (acquisition trial). The rats were again placed in theilluminated chamber 24 hr after the acquisition trial and thelatency to enter the dark chamber was measured for 300 sec(retention trial). If a rat did not enter the dark chamber withinthe cut-off time (300 sec), it was assigned a latency value of300 sec. In scopolamine-injected group, rats were given sco-polamine (1 mg/kg, i.p.), 1 hr before the acquisition trial. Asingle dose of MPC (100 mg/kg, p.o.) was given to rats 30min after scopolamine treatment and after another 30 min,rats were placed in the illuminated chamber. In Ab1–42-infu-sion group, rats were injected with MPC (10 mg/kg, p.o.) orvitamin E (10 mg/kg, p.o.) during 2 weeks before the trainingtrial.
Evaluation of Apoptosis With TUNEL Method
Terminal deoxynucleotidyltransferase (TdT)-mediateddUTP nick-end labeling (TUNEL) staining was carried outaccording to the manufacturer’s protocol (In situ cell deathdetection kit; TMR Red; Roche Diagnostics GmbH, Ger-many). Apoptotic cell death was visualized by TUNELmethod (Gavrieli et al., 1992). For immunohistochemistryassay, brains were removed from animals and immersed in 4%paraformaldehyde. After fixation, the brains were embeddedin paraffin and cut 7 lm coronal sections. For immunocyto-chemistry assay, cells were immersed in 4% paraformaldehyde.TUNEL reaction was labeled with TMR red and analyzed byconfocal microscopy with appropriate filters (LSM510, CarlZeiss, Germany). DAPI (1 lM) staining was carried out fornucleus staining. TUNEL positive cells in three random fieldswere counted in two sets of experiments and expressed as per-centage of the number of TUNEL positive cells/the totalnumber of cells in each field of the hippocampus.
Cell Culture
SH-SY5Y cells were maintained in DMEM (Life Tech-nologies, Inc., Grand Island, NY) supplemented with 10%FBS (GIBCO BRL, Gaithersburg, MD) and 0.3% antibioticsat 378C in 5% CO2. SH-SY5Y cells were pretreated withMPC (25, 50 or 100 lM) or vitamin E (50 lM) for 4 hrbefore the treatment with Ab1–42, glutamate or H2O2.
Cell Viability Test
WST-1-metabolizing activity was determined accordingto the manufacturer’s instructions (Roche, Indianapolis, IN).SH-SY5Y cells were plated in a 96-well plate at a density of 83 103 cells/well. As reported previously (Park et al., 2002),cells were allowed to adhere to plates for 24 hr. MPC wasintroduced into the media of SH-SY5Y cells 4 hr beforetreatment with 25 lM Ab1–42, 1 mM glutamate, or 200 lMH2O2, which play vital roles in neurodegenerative diseases,such as AD (Behl et al., 1994; Goodman and Mattson, 1994;Mark et al., 1997; Suh and Checler, 2002). This colorimetricassay measures the metabolic activity of viable cells. Briefly,after incubating cells treated with various reagents, 10 llWST-1 was added to the culture media. The culture wasincubated at 378C in a humidified atmosphere of 95% air and5% CO2 for 1 hr. The absorbance of the reaction product wasmeasured with an ELISA reader (Bio-Rad, Germany) at awavelength of 450 nm.
Measurement of ROS Generation
Intracellular ROS in SH-SY5Y cells were assayed usingdye 20,70-dichlorofluoroscein diacetate (DCFH-DA; MolecularProbes, Eugene, OR). Cells were washed with HEPES-buf-fered saline (HBS) and incubated in the dark for 1 hr in HBScontaining 200 lM of DCFH-DA. On incubation, DCFH-DA is taken up by cells where intracellular esterase cleaves themolecule to DCFH, which is oxidized to DCF in the pres-ence of H2O2. The total fluorescence was measured using aspectrofluorometer (Molecular Devices, Sunnyvale, CA) at anemission wavelength of 488 nm and an excitation wavelengthof 524 nm (Wang and Zhu, 2003).
Western Blotting
Cells were lysed in a lysis buffer containing 50 mM Tris(pH 7.4), 150 mM NaCl, 1% Triton, 0.5% SDS, 0.5% DOC,and protease inhibitors. Protein was resolved in SDS poly-acrylamide gel, electrophoresed at 30–50 lg of protein/lane,and transferred onto a nitrocellulose membrane (AmershamPharmacia, Buckinghamshire, UK). The protein blot was con-firmed with appropriate antibodies and detected using horse-radish peroxidase-conjugated secondary antibody (AmershamPharmacia). Immunoreactive bands were visualized using anECL enhanced chemiluminescence system (ECL; AmershamPharmacia). For detection for cytochrome c, the mitochon-drial and cytosolic fractions were obtained according to theprevious method (Kang et al., 1995). The purity of thefractions was checked with subcellular markers, i.e., anti-cyto-chrome c oxidase IV for mitochondrial fraction and anti-b-tubulin for cytosol.
Statistical Analysis
Data are presented as means 6 SEM. These results wereanalyzed using one-way analysis of variance (ANOVA) fol-lowed by Turkey’s post-hoc test or two-way ANOVA, inwhich P < 0.05 was considered to be statistically significant.
2502 Shin et al.
Journal of Neuroscience Research DOI 10.1002/jnr
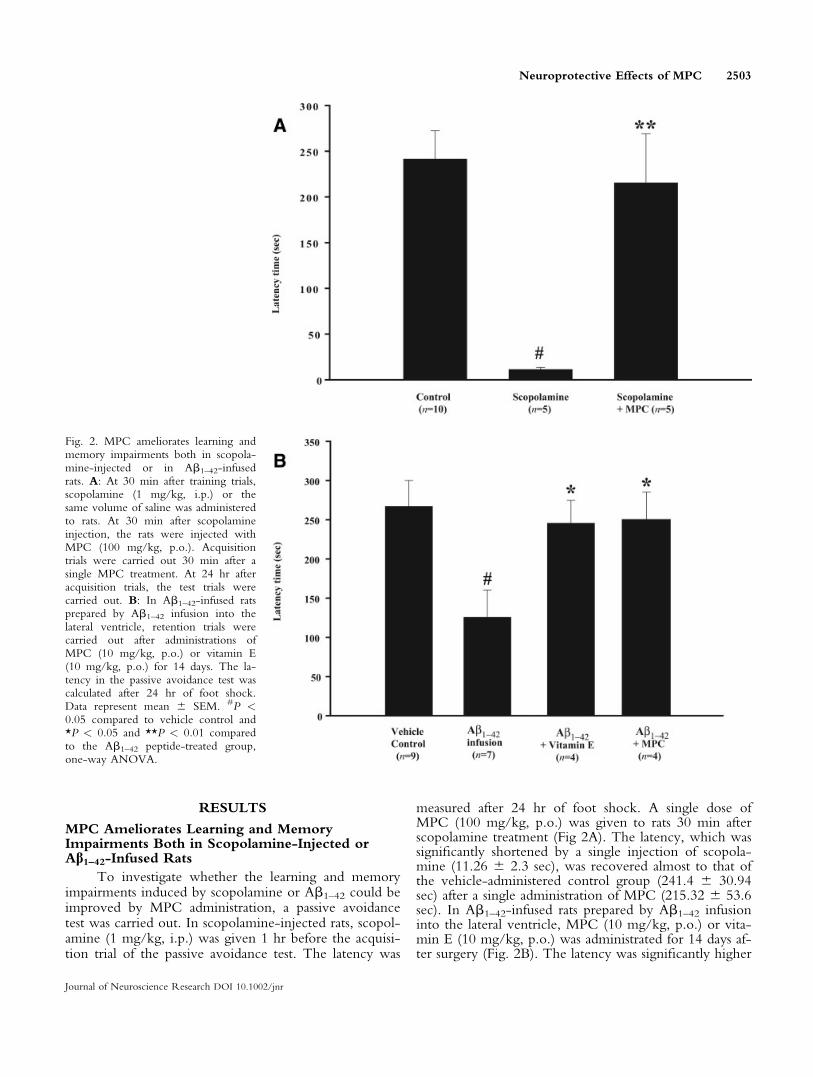
RESULTS
MPC Ameliorates Learning and MemoryImpairments Both in Scopolamine-Injected orAb1–42-Infused Rats
To investigate whether the learning and memoryimpairments induced by scopolamine or Ab1–42 could beimproved by MPC administration, a passive avoidancetest was carried out. In scopolamine-injected rats, scopol-amine (1 mg/kg, i.p.) was given 1 hr before the acquisi-tion trial of the passive avoidance test. The latency was
measured after 24 hr of foot shock. A single dose ofMPC (100 mg/kg, p.o.) was given to rats 30 min afterscopolamine treatment (Fig 2A). The latency, which wassignificantly shortened by a single injection of scopola-mine (11.26 6 2.3 sec), was recovered almost to that ofthe vehicle-administered control group (241.4 6 30.94sec) after a single administration of MPC (215.32 6 53.6sec). In Ab1–42-infused rats prepared by Ab1–42 infusioninto the lateral ventricle, MPC (10 mg/kg, p.o.) or vita-min E (10 mg/kg, p.o.) was administrated for 14 days af-ter surgery (Fig. 2B). The latency was significantly higher
Fig. 2. MPC ameliorates learning andmemory impairments both in scopola-mine-injected or in Ab1–42-infusedrats. A: At 30 min after training trials,scopolamine (1 mg/kg, i.p.) or thesame volume of saline was administeredto rats. At 30 min after scopolamineinjection, the rats were injected withMPC (100 mg/kg, p.o.). Acquisitiontrials were carried out 30 min after asingle MPC treatment. At 24 hr afteracquisition trials, the test trials werecarried out. B: In Ab1–42-infused ratsprepared by Ab1–42 infusion into thelateral ventricle, retention trials werecarried out after administrations ofMPC (10 mg/kg, p.o.) or vitamin E(10 mg/kg, p.o.) for 14 days. The la-tency in the passive avoidance test wascalculated after 24 hr of foot shock.Data represent mean 6 SEM. #P <0.05 compared to vehicle control and*P < 0.05 and **P < 0.01 comparedto the Ab1–42 peptide-treated group,one-way ANOVA.
Neuroprotective Effects of MPC 2503
Journal of Neuroscience Research DOI 10.1002/jnr

in MPC- (249.73 6 34.72 sec) or vitamin E-adminis-tered group (245.6 6 29.02 sec) than in vehicle-adminis-tered Ab1–42-infused group (125.69 6 34.64 sec).
MPC Reduces Apoptotic Cell Death in CA1Region of Ab1–42-Infused Rats
To examine whether the apoptotic cell death ofneurons could be attenuated by MPC, TUNEL stainingwas carried out using the brain slices of rats. Ab1–42-infused rats were administrated with MPC (10 mg/kg,p.o.) or vitamin E (10 mg/kg, p.o.) for 14 days afterinfusion of Ab1–42 for a week. Figure 3A,B showed thatapoptotic cell death of CA1 region was observed muchless in MPC- (14.81 6 9.89%) or vitamin E-adminis-tered group (13.08 6 5.42%) than in vehicle-adminis-tered Ab1–42-infused group (49.52 6 3.22%).
MPC Exerts Neuroprotective Effects AgainstAb1–42, Glutamate or H2O2 in SH-SY5Y Cells
We studied the effects of MPC on neuronal celldeath induced by Ab1–42, glutamate, or H2O2. As meas-ured by WST-1 assay, cell viability was significantlydecreased compared to vehicle-treated control in SH-SY5Y cells at 48 and 12 hr after treatment with Ab1–42
(64.61 6 2.92% vs. vehicle treated control cells) or glu-tamate only (46.14 6 8.11%), respectively (Fig. 4A,B).Pretreatment with MPC, however, significantly attenu-ated the decrease in cell viability in a dose-dependentmanner compared to the Ab1–42-treated (25 lM MPC,70.32 6 3.03; 50 lM MPC, 74.24 6 3.38; 100 lMMPC, 79.17 6 4.19%) or glutamate-treated cells (25lM MPC, 49.74 6 5.98; 50 lM MPC, 78.49 6 5.32;100 lM MPC, 91.56 6 5.78%). Pretreatment with 50 lMvitamin E also significantly attenuated the decrease incell viability induced by Ab1–42 (80.51 6 1.85%).
MPC Reduces the ROS Accumulation by H2O2
Treatment in SH-SY5Y Cells
We checked the effects of MPC on ROS at 24 hrafter treatment with H2O2 (200 lM) for 1hr (Fig.4C,D). Pretreatment with MPC (25, 50, or 100 lM) for4 hr before treatment with H2O2 ameliorated thedecreases in cell viability (25 lM MPC, 59.3 6 4.46;50 lM MPC, 72.45 6 3.55; 100 lM MPC, 75.76 67.74% vs. vehicle treated control) and significantlydecreased ROS production (25 lM MPC, 79.66 66.51; 50 lM MPC, 73.17 6 5.01; 100 lM MPC, 69.216 6.44% vs. vehicle-treated H2O2 treated cells) in adose-dependent manner, as compared to vehicle-treatedH2O2-group. Pretreatment with 50 lM vitamin E sig-nificantly recovered cell viability (71.49 6 3.75%) andROS production (69.67 6 4.27%).
Pretreatment of MPC Attenuates Apoptotic CellDeath by H2O2 in SH-SY5Y Cells
To examine whether apoptotic cell death by oxida-tive stress could be protected by MPC, TUNEL stainingwas carried out using the SH-SY5Y cells 24 hr after
being treated with H2O2 (200 lM) for 1hr (Fig. 5A,B).TUNEL stained cells were observed much less in the50 lM MPC-treated group (31.32 6 6.43% vs. total cellnumbers) or 50 lM vitamin E-treated group (39.98 63.33% vs. total cell numbers) for 4 hr before treatmentwith H2O2 than in H2O2-treated group (63.71 6 3.32%vs. total cell numbers).
Pretreatment With MPC Reduces the Release ofCytochrome c and the Activation of Caspase 3Induced by H2O2 in Neuronal Cells
Cytochrome c release into cytosol and caspase 3activation were measured at 24 hr after treatment withH2O2 (200 lM) in SH-SY5Y cells by Western blotting.Pretreatment with MPC (25, 50, or 100 lM) for 4 hrbefore treatment with H2O2 decreased the release ofcytochrome c into cytosolic fraction and the activationof caspase 3 in a dose-dependent manner, compared tovehicle treated H2O2-treated cells (Fig. 6A,C).
DISCUSSION
In the present study, we report that a newly syn-thesized compound, MPC, improves learning andmemory deficits in two dementia animal models, i.e.,scopolamine-injected or Ab1–42-infused rats. In addition,MPC protects SH-SY5Y cells against Ab1–42, glutamate,or H2O2. The experimental design used in this studywas prepared as previous reports on drug screening(Nitta et al., 1997; Yamada et al., 1999; Park et al.,2000, 2002; Nakamura et al., 2001; Jhoo et al., 2004;Kim et al., 2004). MPC was newly synthesized by theesterification of maltol isolated from Korean ginseng(Panax ginseng C.A. Meyer) and p-coumaric acidextracted from reed (Phragmites commnis). Because bothparent compounds, maltol and p-coumaric acid areknown compounds, we want to develop a new com-pound having better therapeutic efficacy against Alzhei-mer’s disease. Several scientists have already reported thatthe antioxidant effects of the parent compounds wereless active than vitamin E or other compounds (Lee andShibamoto, 2000; Wei et al., 2001; Etoh et al., 2004).We found, however, that MPC had similar antioxidanteffects to vitamin E, suggesting that MPC has strongerantioxidant effects than the parent compounds.
We reported previously that maltol protected hip-pocampal neurons against oxidative damage in the brainsof mice treated with kainic acid. Administration withmaltol (100 mg/kg) remarkably increased the total gluta-thione level and glutathione peroxidase activity, attenuat-ing kainic acid-induced neuronal loss in the hippocampus(Kim et al., 2004). Maltol of aroma extracts, which wasisolated from beans, also inhibited malonaldehyde forma-tion from horse blood plasma oxidized with Fenton’s rea-gent (Lee and Lee, 2005). Additionally, p-coumaric acidisolated from the root extract of reed is a phenolic acidthat is widely distributed in plants. It constitutes a part ofthe human diet (Scalbert and Williamson, 2000) and pos-sesses free radical scavenging and antioxidant properties
2504 Shin et al.
Journal of Neuroscience Research DOI 10.1002/jnr

Fig. 3. Apoptotic cells in CA1 region were reduced by MPC admin-istration in Ab1–42-infused rats. Rat brains from Ab1–42-infused ratsadministered with vehicle, MPC or vitamin E were embedded inparaffin and sections were cut at 7 lm thick. Then, brain slices wereimmunostained with TUNEL staining (A). The percentage of
TUNEL-positive neurons is shown in the graph (B). The resultswere normalized as percentage ratios compared to the total cell num-bers. #P < 0.05 compared to the vehicle and *P < compared toAb1–42 peptide-treated group, one-way ANOVA. DIC, differentialinterference contrast. Scale bar 5 50 lm.

(Hertog, 1995; Kato et al., 1997; Lodovici et al., 2001;Abdel-Wahab et al., 2003). Moreover, p-coumaric acidderivatives exhibited inhibitory activity stronger than thatof vitamin C or E on peroxynitrite-mediated lipoproteinnitration (Niwa et al., 2001).
To characterize a pharmacologic property of MPC,a newly synthesized compound, we investigated whetherMPC could improve learning and memory deficits inscopolamine-injected or Ab-infused rats.
First, we reported that MPC ameliorated scopola-mine-induced learning and memory impairments, using apassive avoidance test. Scopolamine, one of muscarinicreceptor antagonists, can induce amnesia in animals by
blocking cholinergic neurotransmission. The effects ofscopolamine are generally interpreted within the frame-work of the cholinergic hypothesis of cognitive dysfunc-tion (Bartus et al., 1982; Bartus, 2000). As a conse-quence, an animal model with scopolamine-inducedamnesia has widely been used as a pharmacological modelto test the effectiveness of new cognition-enhancingdrugs (Park et al., 2000; van der Staay and Bouger,2005). It must be sure that learning impaired by scopola-mine administration could be studied in a passive avoid-ance test (Stone et al., 1988). In the recent study, wefound that MPC increased activities of choline acetyl-transferase (ChAT) in the brains of SAMP8 mice (data
Fig. 4. MPC reduces neurotoxicity induced by Ab1–42, glutamate orH2O2 and the production of ROS induced by H2O2 in SH-SY5Ycells. SH-SY5Y cells were plated in a 96-well plate at a density of 8 3103 cells/well. MPC was introduced into the media of SH-SY5Ycells 4 hr before treatment with 25 lM Ab1–42 peptide (A), 1 mMglutamate (B), or 200 lM H2O2 (C) for 48, 12, and 1 hr, respec-tively. WST-1-metabolizing activity was determined according to the
manufacturer’s instructions (Roche). The intracellular level of ROSwas checked using DCFH-DA (D). MPC (25, 50, or 100 lM) waspretreated for 4 hr before treatment with H2O2. Data were expressedas the percent of vehicle control value 6 SEM. At least two experi-ments were carried out in triplicate. #P < 0.05 compared to each ve-hicle-treated control and *P < 0.05 and **P < 0.01 compared toAb1–42-, glutamate-, or H2O2-treated group, one-way ANOVA.
2506 Shin et al.
Journal of Neuroscience Research DOI 10.1002/jnr
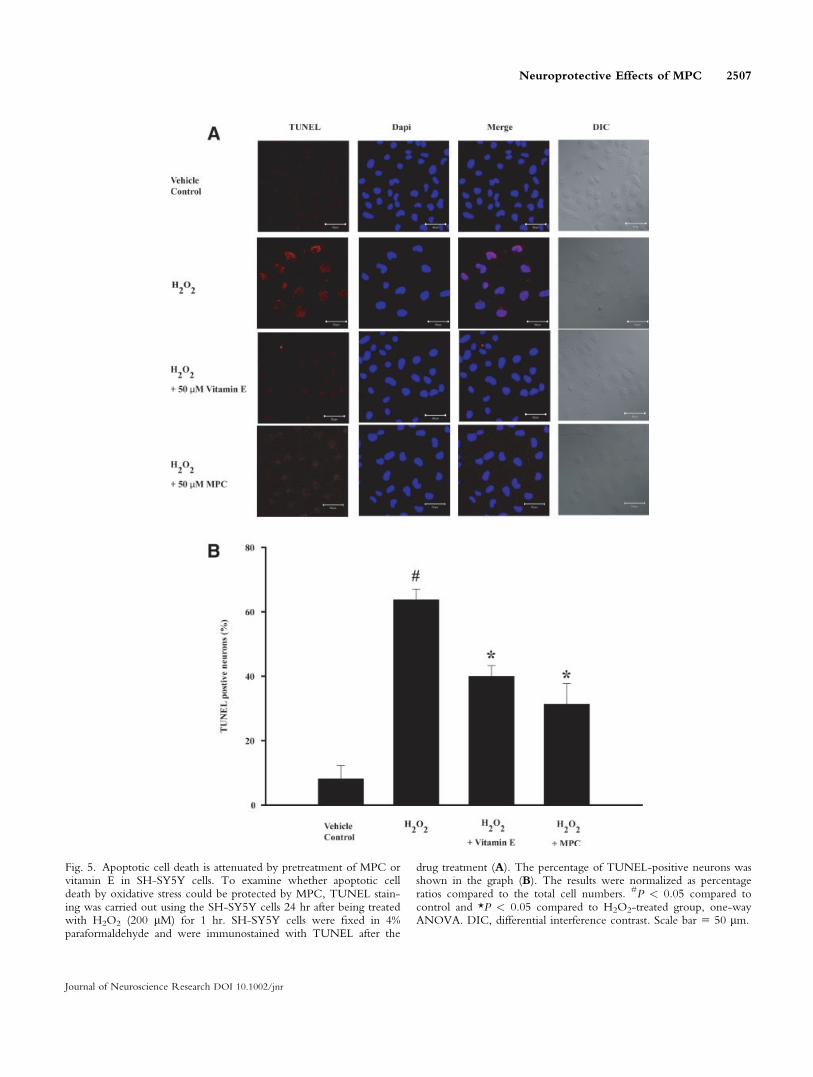
Fig. 5. Apoptotic cell death is attenuated by pretreatment of MPC orvitamin E in SH-SY5Y cells. To examine whether apoptotic celldeath by oxidative stress could be protected by MPC, TUNEL stain-ing was carried out using the SH-SY5Y cells 24 hr after being treatedwith H2O2 (200 lM) for 1 hr. SH-SY5Y cells were fixed in 4%paraformaldehyde and were immunostained with TUNEL after the
drug treatment (A). The percentage of TUNEL-positive neurons wasshown in the graph (B). The results were normalized as percentageratios compared to the total cell numbers. #P < 0.05 compared tocontrol and *P < 0.05 compared to H2O2-treated group, one-wayANOVA. DIC, differential interference contrast. Scale bar 5 50 lm.
Neuroprotective Effects of MPC 2507
Journal of Neuroscience Research DOI 10.1002/jnr
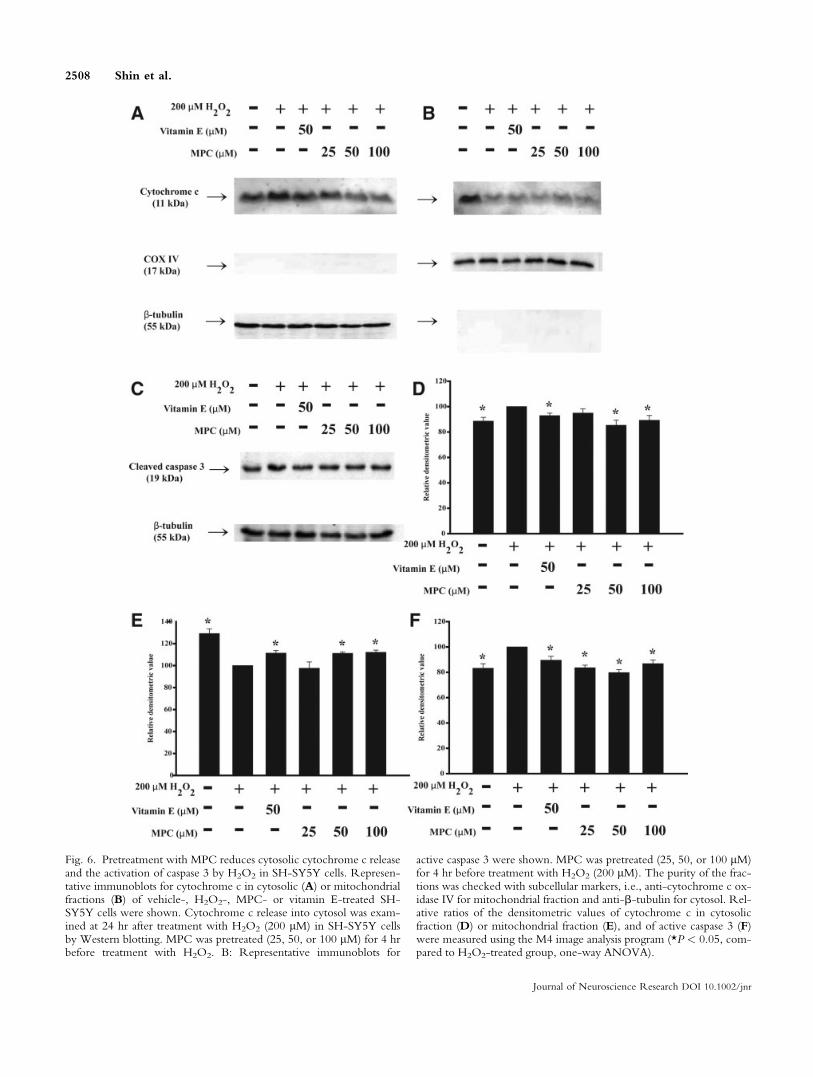
Fig. 6. Pretreatment with MPC reduces cytosolic cytochrome c releaseand the activation of caspase 3 by H2O2 in SH-SY5Y cells. Represen-tative immunoblots for cytochrome c in cytosolic (A) or mitochondrialfractions (B) of vehicle-, H2O2-, MPC- or vitamin E-treated SH-SY5Y cells were shown. Cytochrome c release into cytosol was exam-ined at 24 hr after treatment with H2O2 (200 lM) in SH-SY5Y cellsby Western blotting. MPC was pretreated (25, 50, or 100 lM) for 4 hrbefore treatment with H2O2. B: Representative immunoblots for
active caspase 3 were shown. MPC was pretreated (25, 50, or 100 lM)for 4 hr before treatment with H2O2 (200 lM). The purity of the frac-tions was checked with subcellular markers, i.e., anti-cytochrome c ox-idase IV for mitochondrial fraction and anti-b-tubulin for cytosol. Rel-ative ratios of the densitometric values of cytochrome c in cytosolicfraction (D) or mitochondrial fraction (E), and of active caspase 3 (F)were measured using the M4 image analysis program (*P < 0.05, com-pared to H2O2-treated group, one-way ANOVA).
2508 Shin et al.
Journal of Neuroscience Research DOI 10.1002/jnr

not shown). Accordingly, MPC can improve scopola-mine-induced cognitive impairment through the activa-tion of ChAT.
Second, we used Ab1–42 infused rats prepared byAb1–42 infusion into the cerebral ventricle for 1 week.In the passive avoidance test, we showed that MPCadministration (10 mg/kg, p.o.) for 2 weeks improvedcognitive deficits induced by Ab1–42 infusion. The Ab-infused animal model produces several pathologic hall-marks of AD including amyloid deposition, robustneuroinflammation, decrease in synaptic markers, neuro-nal death, and memory impairments (Nitta et al., 1997;Suh and Checler, 2002; Craft et al., 2004). This modelalso exhibits impairments of nicotine- and K1-stimulatedacetylcholine or dopamine release from the frontal cor-tex/hippocampus and striatum, respectively (Itoh et al.,1996). In conclusion, the Ab-infused model could beuseful as an animal model for evaluating developmentprocesses at the early or middle stages of Alzheimer-likedementia (Nakamura et al., 2001).
To examine neuroprotective effects of MPC invitro, we investigated the effects of MPC on the neuro-toxicity induced by Ab1–42, glutamate, or H2O2 in SH-SY5Y neuroblastoma cells. We show that MPC couldreduce Ab1–42-induced toxicity in the cells. Numeroussenile plaques have been found in the brains of ADpatients at autopsy (Roth et al., 1966; Suh and Checler,2002) and these senile plaques consist of fibrillar depositsof Ab peptides (Hardy, 1997; Suh and Checler, 2002).Ab peptide deposition precedes the development ofneurofibrillary change (Mann, 1985), and high micro-molar concentrations of Ab peptides are neurotoxic(Yankner et al., 1989; Suh and Checler, 2002). Mecha-nistic studies of Ab neurotoxicity in cell culture showeda dependence on glutamate-mediated excitotoxicity,mediation of ROS production, and hydrogen peroxide-mediated apoptosis (Behl et al., 1994; Goodman &Mattson, 1994; Mark et al., 1997). Several studies havesuggested a strategy that decreases Ab neurotoxicitymight be beneficial for AD therapeutics (Behl et al.,1992; Chyan et al., 1999; Luo et al., 2002).
We showed that MPC protected cultured neuronalcells against Ab1–42. The Ab1–42 treatment in the cellculture decreased cell death by about 30%. Ab1–42-induced cell death was inhibited by pretreatment withMPC. Although the precise mechanism of this reversal isunclear, the protective effect of MPC against Ab1–42-induced neurotoxicity is likely to result from the reduc-tion of glutamate excitotoxicity and ROS.
In addition, we show that MPC could reduce gluta-mate-induced toxicity in SH-SY5Y neuroblastoma cells.Glutamate-mediated excitotoxicity plays a vital role inthe pathogenesis of neurodegenerative diseases such asAD and brain ischemia (Meldrum et al., 1990). Gluta-mate is an excitatory neurotransmitter and is related tothe mechanisms of learning and memory processing(Greenamyre, 1986; Cotman et al., 1987). However,overstimulation of N-methyl-D-aspartate (NMDA) recep-tors induced by glutamate can lead to increase in intracel-
lular Ca21 level and excitotoxicity, and subsequentlyresults in neuronal death (MacDermott et al., 1986; Nic-otera and Orrenius, 1992). Several studies have suggestedthat a strategy to modulate the excitotoxicity throughNMDA receptor antagonists might be beneficial for thetreatments of several brain diseases (Simon et al., 1984;Keyser et al., 1999). Here, we report that MPC protectedthe cultured cells against glutamate-induced neurotoxic-ity. The treatment of glutamate in the cell culturedecreased cell viability by about 50%. Glutamate-inducedcell death was reversed by pretreatment with MPC.
We also showed that MPC could reduce H2O2-induced toxicity, ROS production, cytochrome c releaseinto cytosol, and caspase 3 activation in SH-SY5Y neuro-blastoma cells. Because H2O2 may perturb the antioxidantdefense system in the cell and result in apoptotic cell death(Chandra et al., 2000; Bilici et al., 2001), an H2O2-induced toxicity model has been used for studying oxida-tive stress-induced neurodegeneration (Richter-Landsbergand Vollgraf, 1998). The accumulation of macromoleculardamage induced by ROS is the central causal factor thatpromotes the process of aging (Sohal, 2002). The oxida-tive modification of proteins by ROS is involved in thepathogenesis of both normal aging and neurodegenerativediseases (Beal, 2002; Suh and Checler, 2002). Moreover,brain tissue is highly vulnerable to oxidative stress becauseof its oxidative damage potential (Leutner et al., 2001;Floyd and Hensley, 2002). ROS can react with polyunsa-turated fatty acids to form lipid peroxides, and the accu-mulation of end-products of lipid peroxidation with agemay contribute to the aging process (Inal et al., 2001;Kasapoglu and Ozben, 2001; Leutner et al., 2001; Wick-ens, 2001; Montine et al., 2002). More powerful antioxi-dants such as Egb761, clotrimazole, dehydroepiandroster-one, melatonin, indole propionic acid, and DHED haveall been shown to inhibit oxidative stress-induced toxicityin various cell lines (Barlow-Walden et al., 1995; Caze-vieille et al., 1997; Chyan et al., 1999; Martin et al., 2000;Okatani et al., 2000; Isaev et al., 2002; Luo et al., 2002;Zhang et al., 2002; Suh et al., 2005). We described thatMPC protected the cultured cells against H2O2 bydecreasing ROS production, cytochrome c release intocytosol, and caspase 3 activation induced by H2O2.
Similar to the effects of vitamin E, pretreatment ofMPC reduced ROS level, cytochrome c release intocytosol, and caspase 3 activation and rescued SH-SY5Ycells from the subsequent H2O2-induced apoptotic celldeath. Recently, we determined that MPC was not adirect scavenger of hydroxyl radicals (data not shown).Further study on its detailed antioxidant mechanismsremains to be clarified. MPC might exert an neuropro-tective effects blocking ROS production, cytochrome crelease, or caspase 3 activation.
Summarizing our results, newly synthesized com-pound, MPC, attenuated learning and memory impairmentsin vivo dementia animal models, scopolamine-injectedor Ab1–42-infused rats. In addition, MPC showed neuro-protective effects against Ab1–42, glutamate, or H2O2 invitro neuronal cells, reducing ROS, cytochrome c re-
Neuroprotective Effects of MPC 2509
Journal of Neuroscience Research DOI 10.1002/jnr

lease into cytosol, and caspase 3 activation induced byH2O2. Taking these in vitro and in vivo results together,our study suggests that MPC is a potentially effectivecandidate against AD that is characterized by widespreadneuronal death and progressive decline of cognitivefunction.
ACKNOWLEDGMENTS
This study was supported by a National CreativeResearch Initiative Grant (2006–2009) from Ministry ofScience and Technology and in part by BK21 HumanLife Sciences. We thank Dr. Sung Ki Lim, chairman ofHanmi Pharmaceutical Company Limited for very help-ful discussion.
REFERENCES
Abdel-Wahab MH, El-Mahdy MA, Abd-Ellah MF, Helal GK, Khalifa F,
Hamada FM. 2003. Influence of p-coumaric acid on doxorubicin-
induced oxidative stress in rat’s heart. Pharmacol Res 48:461–465.
Barlow-Walden LR, Reiter RJ, Abe M, Pablos M, Menendez-Pelaez A,
Chen LD. 1995. Melatonin stimulates brain glutathione peroxidase ac-
tivity. Neurochem Int 26:497–502.
Bartus RT, Dean RL, Beer B, Lippa AS. 1982. The cholinergic hypoth-
esis of geriatric memory dysfunction. Science 217:408–414.
Bartus RT. 2000. On neurodegenerative diseases, models, and treatment
strategies: lessons learned and lessons forgotten a generation following
the cholinergic hypothesis. Exp Neurol 163:495–529.
Beal MF. 2002. Oxidatively modified proteins in aging and disease. Free
Radic Biol Med 32:797–803.
Behl C, Davis JB, Cole GM, Schubert D. 1992. Vitamin E protects
nerve cells from amyloid b protein toxicity. Biochem Biophys Res
Commun 186:944–950.
Behl C, Davis JB, Lesley R, Schubert D. 1994. Hydrogen peroxide
mediates amyloid beta protein toxicity. Cell 77:817–827.
Bilici M, Efe H, Koroglu MA, Uydu HA, Bekaroglu M, Deger O. 2001.
Antioxidative enzyme activities and lipid peroxidation in major depres-
sion: alteration by antidepressant treatments. J Affect Disord 64:43–51.
Cazevieille C, Safa R, Osborne NN. 1997. Melatonin protects primary
cultures of rat cortical neurons from NMDA excitotoxicity and hy-
poxia/reoxygenation. Brain Res 768:120–124.
Chandra J, Samali A, Orrenius S. 2000. Triggering and modulation of
apoptosis by oxidative stress. Free Radic Biol Med 29:323–333.
Choi JH, Kim IS, Kim JI, Kim DW, Yoon TH. 1993. Effect of reed
root extract (Phragmites communis) on physiological activity of SD
rats. Kor J Gerontol 3:109–115.
Choi JH, Kim DW, Kim KS, Kim CM, Baek YH. 1997a. Effect of reed
root extract (RRE) on learning and memory impairment animal model
SAMP8. 2. Feeding effect of RRE on oxygen radicals and their scav-
enger enzymes in SAMP8 brain. Kor J Gerontol 7:23–28.
Choi JH, Kim DW, Choi JS, Han YS, Baek YH. 1997b. Effect of reed
root extract (RRE) on learning and memory impairment animal model
SAMP8. 3. Feeding effect of RRE on neurotransmitters and their
metabolites in SAMP8 brain. Kor J Gerontol 7:29–36.
Choi JH, Kim IS, Kim DW, Kim JH, Han YS Kim HS. 1998. Effect of
p-coumaric acid on lipid metabolism in SAMP8 serum. Kor J Gerontol
8:99–104.
Chyan YJ, Poeggeler B, Omar RA, Chain DG, Frangione B, Ghiso J,
Pappolla MA. 1999. Potent neuroprotective properties against the Alz-
heimer beta-amyloid by an endogenous melatonin-related indole struc-
ture, indole-3-propionic acid. J Biol Chem 274:21937–21942.
Craft JM, Watterson DM, Frautschy SA, Van Eldik LJ. 2004. Aminopyri-
dazines inhibit b-amyloid induced glial activation and neuronal damage
in vivo. Neurobiol Aging 25:1283–1292.
Cotman CW, Monaghan DT, Ottersen OP, Storm-Mathisen J. 1987.
Anatomical organization of excitatory amino acid receptors and their
pathways. Trends Neurosci 10:273–279.
D’Hooge R, De Deyn PP. 2001. Applications of the Morris water maze
in the study of learning and memory. Brain Res Rev 36:60–90.
Dutar P, Bassant MH, Senut MC, Lamour Y. 1995. The septohippocam-
pal pathway: structure and function of a central cholinergic system.
Physiol Rev 75:393–427.
Etoh H, Murakami K, Yogoh T, Ishikawa H, Fukuyama Y, Tanaka H.
2004. Anti-oxidative compounds in barley tea. Biosci Biotechnol Bio-
chem 68:2616–2618.
Floyd RA, Hensley K. 2002. Oxidative stress in brain aging. Implications for
therapeutics of neurodegenerative diseases. Neurobiol Aging 23:795–807.
Gavrieli Y, Sherman Y, Ben-Sasson SA. 1992. Identification of pro-
grammed cell death via specific labeling of nuclear DNA fragmentation.
J Cell Biol 119:493–501.
Goodman Y, Mattson MP. 1994. Secreted forms of b-amyloid precursor
protein protect hippocampal neurons against amyloid b-peptide-induced oxidative injury. Exp Neurol 128:1–12.
Greenamyre JT. 1986. The role of glutamate in neurotransmission and in
neurologic disease. Arch Neurol 43:1058–1063.
Hardy JA. 1997. Amyloid, the presenilins and Alzheimer’s disease. Trends
Neurosci. 20:154–159.
Hyman BT, Damasio AR, Van Hoesen GW, Barnes CL. 1984. Alzhei-
mer’s disease: cell specific pathology isolates the hippocampal formation.
Science 225:1168–1170.
Inal ME, Kanbak G, Sunal E. 2001. Antioxidant enzyme activities and
malondialdehyde levels related to aging. Clin Chim Acta 305:75–80.
Isaev NK, Stelmashook EV, Dirnagl U, Andreeva NA, Manuhova L,
Vorobjev VS, Sharonova IN, Skrebitsky VG, Victorov IV, Katchanov
J, Weih M, Zorov DB. 2002. Neuroprotective effects of the antifungal
drug clotrimazole. Neuroscience 113:47–53.
Itoh A, Nitta A, Nadai M, Nishimura K, Hirose M, Hasegawa T, Nabe-
shima T. 1996. Dysfunction of cholingeric and dopaminergic neuronal
systems in b-amyloid protein-infused rats. J Neurochem 66:1113–1117.
Jhoo JH, Kim HC, Nabeshima T, Yamada K, Shin EJ, Jhoo WK, Kim
W, Kang KS, Ahn JS, Woo JI. 2004. b-Amyloid (1–42)-induced learn-
ing and memory deficits in mice: involvement of oxidative burdens in
the hippocampus and cerebral cortex. Behav Brain Res 155:185–196.
Kang D, Nishida J, Iyama A, Nakabeppu Y, Furuichi M, Fujiwara T,
Sekiguchi M, Takeshige K. 1995. Intracellular localization of 8-oxo-
dGTPase in human cells, with special reference to the role of the
enzyme in mitochondria. J Biol Chem 270:14659–14665.
Karczmar AG. 1993. Brief presentation of the story and present status of
studies of the vertebrate cholinergic system. Neuropsychopharmacology
9:181–199.
Kasapoglu M, Ozben T. 2001. Alterations of antioxidant enzymes and
oxidative stress markers in aging. Exp Gerontol 36:209–220.
Kato Y, Ogino Y, Aoki T, Uchida K, Kawasaki S, Osawa T. 1997. Phe-
nolic antioxidants prevent peroxynitrite-derived collagen modification
in vitro. J Agric Food Chem 45:3004–3009.
Keyser JD, Sulter G, Luiten PG. 1999. Clinical trials with neuroprotec-
tive drugs in acute ischaemic stroke: are we doing the right thing?
Trends Neurosci 22:535–560.
Kim YB, Oh SH, Sok DE, Kim MR. 2004. Neuroprotective effect of
maltol against oxidative stress in brain of mice challenged with kainic
acid. Nutr Neurosci 7:33–39.
Lee KG, Shibamoto T. 2000. Antioxidant properties of aroma compounds
isolated from soybeans andmung beans. J Agric Food Chem. 48:4290–4293.
Lee SJ, Lee KG. 2005. Inhibitory effects of volatile antioxidants found in
various beans on malonaldehyde formation in horse blood plasma. Food
Chem Toxicol 43:515–520.
Leutner S, Eckert A, Muller WE. 2001. ROS generation, lipid peroxida-
tion and antioxidant enzyme activities in the aging brain. J Neural
Transm 108:955–967.
2510 Shin et al.
Journal of Neuroscience Research DOI 10.1002/jnr

Lodovici M, Guglielmi F, Casalini C, Meoni M, Cheynier V, Dolara P.
2001. Antioxidant and radical scavenging properties in vitro of poly-
phenolic extracts from red wine. Eur J Nutr 40:74–77.
Luo Y, Smith JV, Paramasivam V, Burdick A, Curry K, Buford JP, Khan
I, Netzer WJ, Xu H, Butko P. 2002. Inhibition of amyloid-b aggrega-
tion and caspase-3 activation by the Ginkgo biloba extract Egb761.
PNAS 12197–12202.
MacDermott AB, Mayer ML, Westbrook GL, Smith SJ, Barker JL. 1986.
NMDA-receptor activation increases cytoplasmic calcium concentration
in cultured spinal cord neurons. Nature 32:519–522.
Mann DMA. 1985. The neuropathology of Alzheimer’s disease: a review
with pathogenetic, etiological and therapeutic considerations. Mech
Ageing Dev 31:213–255.
Mark RJ, Pang Z, Geddes JW, Uchida K, Mattson MP. 1997. Amyloid
b-peptide impairs glucose uptake in hippocampal and cortical neurons:
involvement of membrane lipid peroxidation. J Neurosci 17:1046–1054
Martin M, Macias M, Escames G, Reiter RJ, Agapito MT, Ortiz GG,
Acuna-Castroviejo D. 2000. Melatonin-induced increased activity of
the respiratory chain complexes I and IV can prevent mitochondrial
damage induced by ruthenium red in vivo. J Pineal Res 28:242–248.
Mattson MP. 1997. Cellular actions of b-amyloid precursor protein and
its soluble and fibrillogenic derivatives. Physiol Rev 77:1081–1132.
Meldrum B, Garthwaite J. 1990. Excitatory amino acid neurotoxicity and
neurodegenerative disease. Trends Pharmacol Sci 11:379–387.
Montine TJ, Neely MD, Quinn JF, Beal MF, Markesbery WR, Roberts
LJ, Morrow JD. 2002. Lipid peroxidation in aging brain and Alzhei-
mer’s disease. Free Radical Bio Med 33:620–626.
Nakamura S, Murayama N, Noshita T, Annoura H, Ohno T. 2001. Pro-
gressive brain dysfunction following intracerebroventricular infusion of
beta1–42-amyloid peptide. Brain Res 912:128–136.
Nicotera P, Orrenius S. 1992. Ca21 and cell death. Ann NY Acad Sci
648:17–27.
Nitta A, Fukuta T, Hasegawa T, Nabeshima T. 1997. Continuous infu-
sion of b-amyloid protein into cerebral ventricle induces learning impair-
ment and neuronal and morphological degeneration. Jpn J Pharmacol
73:51–57.
Niwa T, Doi U, Kato Y, Osawa T. 2001. Antioxidative Properties of
Phenolic Antioxidants Isolated from Corn Steep Liquor. J Agric Food
Chem 49:177–182.
Oka J-I, Suzuki E, Goto N, Kameyama T. 1999. Endogenous GLP-1
modulates hippocampal activity in b-amyloid protein-treated rats. Neu-
roreport 10:2961–2964.
Oka J-I, Suzuki E, Kondo Y. 2000. Endogenous GLP-1 is involved in
b-amyloid protein induced memory impairment and hippocampal neu-
ronal death in rats. Brain Res 878:194–198.
Okatani Y, Wakatsuki A, Kaneda C. 2000. Melatonin increases activities
of glutathione peroxidase and superoxide dismutase in fetal rat brain. J
Pineal Res 28:89–96.
Park CH, Lee YJ, Lee SH, Choi SH, Kim HS, Jeong SJ, Kim SS, and
Suh YH. 2000. Dehydroevodiamine _mHCl prevents impairment of
learning and memory and neuronal loss in rat models of cognitive dis-
turbance. J Neurochem 74:244–253.
Park CH, Choi SH, Koo JW, Seo JH, Kim HS, Jeong SJ, Suh YH.
2002. Novel cognitive improving and neuroprotective activities of Pol-
ygala tenuifolia Willdenow extract, BT-11. J Neurosci Res 70:484–92.
Paxinos G, Watson C. 1986. The rat brain in the stereotaxic coordinates.
New York: Academic Press.
Petkov VD, Kehayov R, Belcheva S, Konstantinova E, Petkov VV,
Getova D, Markovska V. 1993. Memory effects of standardized extracts
of Panax ginseng (G115), Ginkgo biloba (GK 501) and their combina-
tion Gincosan (PHL-00701). Planta Med 59:106–114.
Richter-Landsberg C, Vollgraf U. 1998. Mode of cell injury and death
after hydrogen peroxide exposure in cultured oligodendroglia cells. Exp
Cell Res 244:218–229.
Roth M, Tomlinson BE, Blessed G. 1966. Correlation between scores
for dementia and counts of senile plaques in cerebral gray matter of el-
derly subjects. Nature 206:109–110.
Satry PS, Rao KS. 2000. Apoptosis and the nervous system. J Neuro-
chem 74:1–20.
Scalbert A, Williamson G. 2000. Dietary intake and bioavailability of
polyphenols. J Nutr 130:2073S–2085S.
Shen Z, Wang G, Lin SZ. 1990. Two way shuttle box avoidance condi-
tioning and brain NADH in rats. Physiol Behav 48:515–517.
Simon RP, Swan JH, Griffiths T, Meldrum BS. 1984. Blockade of N-
methyl-D-aspartate receptors may protect against ischemic damage in
the brain. Science 226:850–852.
Sohal RS. 2002. Oxidative stress hypothesis of aging. Free Radic Biol
Med 33:573–574.
Stone WS, Croul CE, Gold PE. 1988. Attenuation of scopolamine-
induced amnesia in mice. Psychopharmacology 96:417–420.
Suh WH, Suslick KS, Suh YH. 2005. Therapeutic agents for Alzheimer’s
disease. Curr Med Chem CNS Agents 5:259–269.
Suh YH, Checler F. 2002. Amyloid precursor protein, presenilins, and
alpha-synuclein; molecular pathogenesis and pharmacological applica-
tions in Alzheimer’s disease. Pharmacol Rev 54:469–525.
Thompson CE. 1995. Apoptosis in the pathogenesis and treatment of dis-
ease. Science 267:1456–1462.
van der Staay FJ, Bouger PC. 2005. Effects of the cholinesterase inhibi-
tors donepezil and metrifonate on scopolamine-induced impairments in
the spatial cone field orientation task in rats. Behav Brain Res 56:1–10.
Wang RG, Zhu XZ. 2003. Subtoxic concentration of manganese synerg-
istically potentiates 1–14 methyl-4-phenylpyridinium-induced neuro-
toxicity in PC12 cells. Brain Res 961:131–138.
Wei A, Mura K, Shibamoto T. 2001. Antioxidative activity of volatile
chemicals extracted from beer. J Agric Food Chem 49:4097–4101.
Whitehouse PJ, Price DL, Struble RG, Clark AW, Coyle JT, DeLong
MR. 1982. Alzheimer’s disease in senile dementia: loss of neurones in
the basal forebrain. Science 215:1237–1239.
Wickens AP. 2001. Ageing and the free radical theory. Respir Physiol
128:379–391.
Winkler J, Suhr ST, Gage FH, Thal LJ, Fisher LJ. 1995. Essential role of
neocortical acetylcholine in spatial memory. Nature 375:484–487.
Wortwein G, Stackman RW, Walsh TJ. 1994. Vitamin E prevents the
place learning deficit and the cholinergic hypofunction induced by
AF64A. Exp Neurol 125:15–21.
Yamada K, Tanaka T, Han D, Senzaki K, Kameyama T, Nabeshima T.
1999. Protective effects of idebenone and a-tocopherol on b-amyloid-
(1–42)-induced learning and memory deficits in rats: implication of oxi-
dative stress in b-amyloid-induced neurotoxicity in vivo. Eur J Neuro-
sci 11:83–90.
Yang Y, Wang J, Xu C, Pan H, Zhang Z. 2006. Maltol inhibits apopto-
sis of human neuroblastoma cells induced by hydrogen peroxide. J Bio-
chem Mol Biol 39:145–149.
Yankner BA. 1996. Mechanisms of neuronal degeneration in Alzheimer’s
disease. Neuron 16:921–932.
Yankner BA, Dawes LR, Fisher S, Villa-Komaroff L, Oster-Granite ML,
Neve RL. 1989. Neurotoxicity of a fragment of the amyloid precursor
associated with Alzheimer’s disease. Science 245:417–420.
Zhang L, Li B, Ma W, Barker JL, Chang YH, Zhao W, Rubinow DR.
2002. Dehydroepiandrosterone (DHEA) and its sulfated derivative
(DHEAS) regulate apoptosis during neurogenesis by triggering the Akt
signaling pathway in opposing ways. Brain Res Mol Brain Res 98:58–66.
Neuroprotective Effects of MPC 2511
Journal of Neuroscience Research DOI 10.1002/jnr


















