
Mitochondrial Oxidative Stress in Aortic Stiffening With...
30
Mitochondrial Oxidative Stress in Aortic Stiffening With Age The Role of Smooth Muscle Cell Function Rui-Hai Zhou, Aleksandr E. Vendrov, Igor Tchivilev, Xi-Lin Niu, Kimberly C. Molnar, Mauricio Rojas, Jacqueline D. Carter, Haiyan Tong, George A. Stouffer, Nageswara R. Madamanchi, Marschall S. Runge Objective—Age-related aortic stiffness is an independent risk factor for cardiovascular diseases. Although oxidative stress is implicated in aortic stiffness, the underlying molecular mechanisms remain unelucidated. Here, we examined the source of oxidative stress in aging and its effect on smooth muscle cell (SMC) function and aortic compliance using mutant mouse models. Methods and Results—Pulse wave velocity, determined using Doppler, increased with age in superoxide dismutase 2 (SOD2) / but not in wild-type, p47phox / and SOD1 / mice. Echocardiography showed impaired cardiac function in these mice. Increased collagen I expression, impaired elastic lamellae integrity, and increased medial SMC apoptosis were observed in the aortic wall of aged SOD2 / versus wild-type (16-month-old) mice. Aortic SMCs from aged SOD2 / mice showed increased collagen I and decreased elastin expression, increased matrix metalloproteinase-2 expression and activity, and increased sensitivity to staurosporine-induced apoptosis versus aged wild-type and young (4-month-old) SOD2 / mice. Smooth muscle -actin levels were increased with age in SOD2 / versus wild-type SMCs. Aged SOD2 / SMCs had attenuated insulin-like growth factor-1-induced Akt and Forkhead box O3a phosphorylation and prolonged tumor necrosis factor-–induced Jun N-terminal kinase 1 activation. Aged SOD2 / SMCs had increased mitochondrial superoxide but decreased hydrogen peroxide levels. Finally, dominant-negative Forkhead box O3a overexpression attenuated staurosporine-induced apoptosis in aged SOD2 / SMCs. Conclusion—Mitochondrial oxidative stress over a lifetime causes aortic stiffening, in part by inducing vascular wall remodeling, intrinsic changes in SMC stiffness, and aortic SMC apoptosis. ( Arterioscler Thromb Vasc Biol . 2012;32:745-755.) Key Words: aging blood pressure reactive oxygen species signal transduction vasodilation A dvancing age is the major risk factor for cardiovascular disease (CVD) morbidity and mortality. With aging, central arteries stiffen (and dilate) as a result of physiological remodeling arising from the fracture of elastin lamellae from repetitive pulsations and also from endothelial dysfunction, chronic low-grade inflammation, and altered vascular smooth muscle tone. 1,2 Aortic stiffening is the principal cause of CVD with age in people without atherosclerosis, 1 including in- creased systolic and pulse pressures, increased left ventricular hypertrophy and diastolic dysfunction, and congestive heart failure. 3 Carotid-femoral pulse wave velocity (PWV), a direct noninvasive measure of the thoracic and abdominal aortic stiffness, is correlated with higher CVD events and is an independent predictor of coronary heart disease and stroke. 4 Despite the strong epidemiological and biological connection of age to CVD risk, the molecular mechanisms responsible for age-related vascular dysfunction have yet to be elucidated. Although advancing age is an unmodifiable risk factor for CVD, it might be possible to target specific molecular signals as an approach to limiting age-related CVD risk. Oxidative stress has been implicated in vascular dysfunc- tion, whether as a result of CVD or aging or both. 5–7 The free radical theory of aging, first proposed by Harman more than 50 years ago, 8 suggested that increased reactive oxygen species (ROS) generation underlies many features of aging. Prior studies have indicated that increased vascular ROS generation results in decreased compliance, as measured by PWV. 9,10 Recent studies suggest that mitochondrial dysfunc- tion plays an important role in aging and impairing vascular function. 11,12 Received on: April 1, 2011; final version accepted on: December 12, 2011. From the McAllister Heart Institute (R.-H.Z., A.E.V., I.T., X.-L.N., K.C.M., M.R., G.A.S., N.R.M., M.S.R.), Division of Cardiology (R.-H.Z., G.A.S., M.S.R.), Department of Medicine, University of North Carolina at Chapel Hill, Chapel Hill, NC; Environmental Public Health Division, National Health and Environmental Effects Research Laboratory, US Environmental Protection Agency, Research Triangle Park, NC (J.D.C., H.T.). Dr Zhou and Dr Vendrov contributed equally to this work. The online-only Data Supplement is available with this article at http://atvb.ahajournals.org/lookup/suppl/doi:10.1161/ATVBAHA.111.243121/-/DC1. Correspondence to Marschall S. Runge, MD, PhD, Department of Medicine, 125 MacNaider Hall, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599-7005. E-mail [email protected] © 2011 American Heart Association, Inc. Arterioscler Thromb Vasc Biol is available at http://atvb.ahajournals.org DOI: 10.1161/ATVBAHA.111.243121 745 by guest on June 24, 2018 http://atvb.ahajournals.org/ Downloaded from by guest on June 24, 2018 http://atvb.ahajournals.org/ Downloaded from by guest on June 24, 2018 http://atvb.ahajournals.org/ Downloaded from by guest on June 24, 2018 http://atvb.ahajournals.org/ Downloaded from by guest on June 24, 2018 http://atvb.ahajournals.org/ Downloaded from by guest on June 24, 2018 http://atvb.ahajournals.org/ Downloaded from by guest on June 24, 2018 http://atvb.ahajournals.org/ Downloaded from by guest on June 24, 2018 http://atvb.ahajournals.org/ Downloaded from by guest on June 24, 2018 http://atvb.ahajournals.org/ Downloaded from by guest on June 24, 2018 http://atvb.ahajournals.org/ Downloaded from by guest on June 24, 2018 http://atvb.ahajournals.org/ Downloaded from by guest on June 24, 2018 http://atvb.ahajournals.org/ Downloaded from by guest on June 24, 2018 http://atvb.ahajournals.org/ Downloaded from by guest on June 24, 2018 http://atvb.ahajournals.org/ Downloaded from by guest on June 24, 2018 http://atvb.ahajournals.org/ Downloaded from by guest on June 24, 2018 http://atvb.ahajournals.org/ Downloaded from by guest on June 24, 2018 http://atvb.ahajournals.org/ Downloaded from by guest on June 24, 2018 http://atvb.ahajournals.org/ Downloaded from by guest on June 24, 2018 http://atvb.ahajournals.org/ Downloaded from by guest on June 24, 2018 http://atvb.ahajournals.org/ Downloaded from
Transcript of Mitochondrial Oxidative Stress in Aortic Stiffening With...

Mitochondrial Oxidative Stress in Aortic StiffeningWith Age
The Role of Smooth Muscle Cell Function
Rui-Hai Zhou, Aleksandr E. Vendrov, Igor Tchivilev, Xi-Lin Niu, Kimberly C. Molnar,Mauricio Rojas, Jacqueline D. Carter, Haiyan Tong, George A. Stouffer,
Nageswara R. Madamanchi, Marschall S. Runge
Objective—Age-related aortic stiffness is an independent risk factor for cardiovascular diseases. Although oxidative stressis implicated in aortic stiffness, the underlying molecular mechanisms remain unelucidated. Here, we examined thesource of oxidative stress in aging and its effect on smooth muscle cell (SMC) function and aortic compliance usingmutant mouse models.
Methods and Results—Pulse wave velocity, determined using Doppler, increased with age in superoxide dismutase 2(SOD2)�/� but not in wild-type, p47phox�/� and SOD1�/� mice. Echocardiography showed impaired cardiac functionin these mice. Increased collagen I expression, impaired elastic lamellae integrity, and increased medial SMC apoptosiswere observed in the aortic wall of aged SOD2�/� versus wild-type (16-month-old) mice. Aortic SMCs from agedSOD2�/� mice showed increased collagen I and decreased elastin expression, increased matrix metalloproteinase-2expression and activity, and increased sensitivity to staurosporine-induced apoptosis versus aged wild-type and young(4-month-old) SOD2�/� mice. Smooth muscle �-actin levels were increased with age in SOD2�/� versus wild-typeSMCs. Aged SOD2�/� SMCs had attenuated insulin-like growth factor-1-induced Akt and Forkhead box O3aphosphorylation and prolonged tumor necrosis factor-�–induced Jun N-terminal kinase 1 activation. Aged SOD2�/�
SMCs had increased mitochondrial superoxide but decreased hydrogen peroxide levels. Finally, dominant-negativeForkhead box O3a overexpression attenuated staurosporine-induced apoptosis in aged SOD2�/� SMCs.
Conclusion—Mitochondrial oxidative stress over a lifetime causes aortic stiffening, in part by inducing vascular wall remodeling,intrinsic changes in SMC stiffness, and aortic SMC apoptosis. (Arterioscler Thromb Vasc Biol. 2012;32:745-755.)
Key Words: aging � blood pressure � reactive oxygen species � signal transduction � vasodilation
Advancing age is the major risk factor for cardiovasculardisease (CVD) morbidity and mortality. With aging,
central arteries stiffen (and dilate) as a result of physiologicalremodeling arising from the fracture of elastin lamellae fromrepetitive pulsations and also from endothelial dysfunction,chronic low-grade inflammation, and altered vascular smoothmuscle tone.1,2 Aortic stiffening is the principal cause of CVDwith age in people without atherosclerosis,1 including in-creased systolic and pulse pressures, increased left ventricularhypertrophy and diastolic dysfunction, and congestive heartfailure.3 Carotid-femoral pulse wave velocity (PWV), a directnoninvasive measure of the thoracic and abdominal aorticstiffness, is correlated with higher CVD events and is anindependent predictor of coronary heart disease and stroke.4
Despite the strong epidemiological and biological connection
of age to CVD risk, the molecular mechanisms responsiblefor age-related vascular dysfunction have yet to be elucidated.Although advancing age is an unmodifiable risk factor forCVD, it might be possible to target specific molecular signalsas an approach to limiting age-related CVD risk.
Oxidative stress has been implicated in vascular dysfunc-tion, whether as a result of CVD or aging or both.5–7 The freeradical theory of aging, first proposed by Harman more than50 years ago,8 suggested that increased reactive oxygenspecies (ROS) generation underlies many features of aging.Prior studies have indicated that increased vascular ROSgeneration results in decreased compliance, as measured byPWV.9,10 Recent studies suggest that mitochondrial dysfunc-tion plays an important role in aging and impairing vascularfunction.11,12
Received on: April 1, 2011; final version accepted on: December 12, 2011.From the McAllister Heart Institute (R.-H.Z., A.E.V., I.T., X.-L.N., K.C.M., M.R., G.A.S., N.R.M., M.S.R.), Division of Cardiology (R.-H.Z., G.A.S.,
M.S.R.), Department of Medicine, University of North Carolina at Chapel Hill, Chapel Hill, NC; Environmental Public Health Division, National Healthand Environmental Effects Research Laboratory, US Environmental Protection Agency, Research Triangle Park, NC (J.D.C., H.T.).
Dr Zhou and Dr Vendrov contributed equally to this work.The online-only Data Supplement is available with this article at http://atvb.ahajournals.org/lookup/suppl/doi:10.1161/ATVBAHA.111.243121/-/DC1.Correspondence to Marschall S. Runge, MD, PhD, Department of Medicine, 125 MacNaider Hall, University of North Carolina at Chapel Hill, Chapel
Hill, NC 27599-7005. E-mail [email protected]© 2011 American Heart Association, Inc.
Arterioscler Thromb Vasc Biol is available at http://atvb.ahajournals.org DOI: 10.1161/ATVBAHA.111.243121
745
by guest on June 24, 2018http://atvb.ahajournals.org/
Dow
nloaded from
by guest on June 24, 2018http://atvb.ahajournals.org/
Dow
nloaded from
by guest on June 24, 2018http://atvb.ahajournals.org/
Dow
nloaded from
by guest on June 24, 2018http://atvb.ahajournals.org/
Dow
nloaded from
by guest on June 24, 2018http://atvb.ahajournals.org/
Dow
nloaded from
by guest on June 24, 2018http://atvb.ahajournals.org/
Dow
nloaded from
by guest on June 24, 2018http://atvb.ahajournals.org/
Dow
nloaded from
by guest on June 24, 2018http://atvb.ahajournals.org/
Dow
nloaded from
by guest on June 24, 2018http://atvb.ahajournals.org/
Dow
nloaded from
by guest on June 24, 2018http://atvb.ahajournals.org/
Dow
nloaded from
by guest on June 24, 2018http://atvb.ahajournals.org/
Dow
nloaded from
by guest on June 24, 2018http://atvb.ahajournals.org/
Dow
nloaded from
by guest on June 24, 2018http://atvb.ahajournals.org/
Dow
nloaded from
by guest on June 24, 2018http://atvb.ahajournals.org/
Dow
nloaded from
by guest on June 24, 2018http://atvb.ahajournals.org/
Dow
nloaded from
by guest on June 24, 2018http://atvb.ahajournals.org/
Dow
nloaded from
by guest on June 24, 2018http://atvb.ahajournals.org/
Dow
nloaded from
by guest on June 24, 2018http://atvb.ahajournals.org/
Dow
nloaded from
by guest on June 24, 2018http://atvb.ahajournals.org/
Dow
nloaded from
by guest on June 24, 2018http://atvb.ahajournals.org/
Dow
nloaded from

Many pro- and antioxidant enzymes regulate ROS levels incells. Of these, the superoxide dismutase (SOD) family is themost studied antioxidant system and has been previouslyimplicated in CVD.13–15 SODs convert superoxide to producehydrogen peroxide (H2O2), which is further degraded byeither catalase or glutathione peroxidase. One member of theSOD family, manganese SOD (SOD2), is present in mito-chondria. Deletion of the SOD2 gene results in early postnatallethality in mice.16,17 SOD2-deficient (SOD2�/�) mice areviable but demonstrate increased susceptibility to oxidativestress, diminished mitochondrial function, and enhanced sen-sitivity to apoptosis.18,19 In an atherosclerotic background(apolipoprotein E knockout), SOD2 deficiency results inaccelerated atherosclerosis20 and endothelial dysfunction inmice.21 In addition, decreased expression/activity of SOD2with age was implicated in vascular aging.22
In the present study, we investigated the effect of oxidativestress in aging-associated increase in aortic stiffness usingmutant mouse models. Our data indicate that prolongedexposure to increased mitochondrial oxidative stress de-creases aortic compliance and induces cardiac dysfunction.Specifically, we elucidate the significance of lifelong SOD2deficiency on the phenotype, function, and molecular signal-ing pathways in aortic smooth muscle cells (SMCs) and howthese events regulate aortic wall homeostasis and aorticstiffening.
Materials and MethodsAortic PWVArterial compliance was determined as described by Hartley et al.23
In brief, mice were anesthetized with inhaled isoflurane (1% in O2)and fixed in a supine position on the temperature-controlled ECGboard (THM100, Indus Instruments). Body temperature was main-tained at 37°C and monitored with a rectal probe. Blood flowvelocity was recorded using a 20-MHz pulsed Doppler probe at thelevels of aortic arch and at the abdominal aorta. Data were analyzedusing an Indus Instruments Doppler Signal Processing Workstation.Aortic PWV was calculated by dividing separation distance (40 mm)by difference in pulse wave arrival time in respect to ECG R-peaks.
EchocardiographyMice were anesthetized with inhaled isoflurane (1% in O2) and fixedin a supine position on the ECG temperature-controlled board.Ultrasound biomicroscopy was performed using VisualSonics Vevo660 equipped with a 30-MHz probe. Ultrasonic images of leftventricle were acquired at the long axis using M-mode. Measure-ments of interventricular septum, posterior wall thickness, andventricle internal diameter at systole and diastole were taken. Valuesof ejection fraction, end diastolic volume, and myocardial mass werederived using VisualSonics Vevo 660 software.
Blood PressureSystolic and diastolic blood pressure was measured as described inthe online-only Data Supplement.
Vascular Relaxation in Isolated MouseAortic RingsRelaxation of isolated mouse aortic rings was measured as describedin the online-only Data Supplement.
Cell Culture and MaterialsMouse aortic SMCs were isolated from young (4 months) and aged(16 months) wild-type and SOD2�/� mice (C57BL/6J) as describedpreviously24 (see online-only Data Supplement).
Histology, Immunohistochemistry,and ImmunofluorescenceImmunohistochemistry and immunofluorescence studies were per-formed as previously described.25
Western Blot AnalysisPreparation of cell extracts and Western blot analysis was performedas described previously.26
Quantitative Real-Time PolymeraseChain ReactionQuantitative analysis of mRNA expression of target genes wasperformed using total RNA extracted from cells and tissues. Reversetranscription was performed using the TaqMan Reverse Transcrip-tion Reagents Kit (Applied Biosystems). Real-time polymerase chainreaction was performed in quadruplicate with TaqMan Gene Expres-sion Assays for mouse collagen I (Mm01302043_gl), elastin(Mm00514670_ml), matrix metalloproteinase-2 (MMP-2)(Mm00439508_ml), and 18S rRNA (Hs99999901_sl) using an ABIPrism 7900 HT Sequence Detection System according to manufac-turer’s recommended protocol. Target gene mRNA expression wasnormalized to 18S rRNA expression. Individual gene expression inSOD2�/� aortic SMCs was calculated relative to that in wild-typeusing REST2008 (Relative Expression Software Tool).27
Gelatin ZymographyMMP-2 activity was assayed by gelatin zymography (see Methods inthe online-only Data Supplement).
Adenovirus Infection of Aortic SMCsA replication-defective adenoviral vector expressing dominant-negative Forkhead box O3a (DN-FOXO3a) was obtained fromVector BioLabs. DN-FoxO3a, constructed by deletion of the trans-activation domain from the C-terminus,28 had a heme agglutinin tagat the N-terminus and expressed green fluorescent protein. Adeno-virus expressing only green fluorescent protein was used as anegative control. Mouse aortic SMCs were cultured to 80% to 90%confluence before adenoviral infection. Infections were performedusing a multiplicity of infection of 100, and the infection efficiencywas typically greater than 90%. Measurement of proteins of interestwas made in cells harvested 36 hours after viral infection.
Detection of Mitochondrial SuperoxideMitochondrial superoxide levels in aortic SMCs were detected asdescribed in the Methods in the online-only Data Supplement.
H2O2 MeasurementAortic SMCs extracellular H2O2 levels were determined using theAmplex Red assay (Invitrogen) (see Methods in the online-only DataSupplement).
Statistical AnalysisData presented graphically are shown as mean�SE from at least 3independent experiments. All data were tested for normality usingthe Kolmogorov-Smirnov test and were analyzed by 1-wayANOVA, and post hoc analysis was performed using the Newman-Keuls test. To account for multiple comparisons, arterial complianceand cardiac function data were analyzed by 1-way ANOVA followedby the Ryan-Einot-Gabriel-Welsch multiple-range test with an over-all ��0.05 (SSPS software, version 19.0).
ResultsAortic Compliance, Cardiac Function, andVasorelaxation Are Decreased With Age inSOD2�/� MiceTo examine the interactive effect of oxidative stress, diet, andaging on vascular health, we measured aortic compliance in
746 Arterioscler Thromb Vasc Biol March 2012
by guest on June 24, 2018http://atvb.ahajournals.org/
Dow
nloaded from
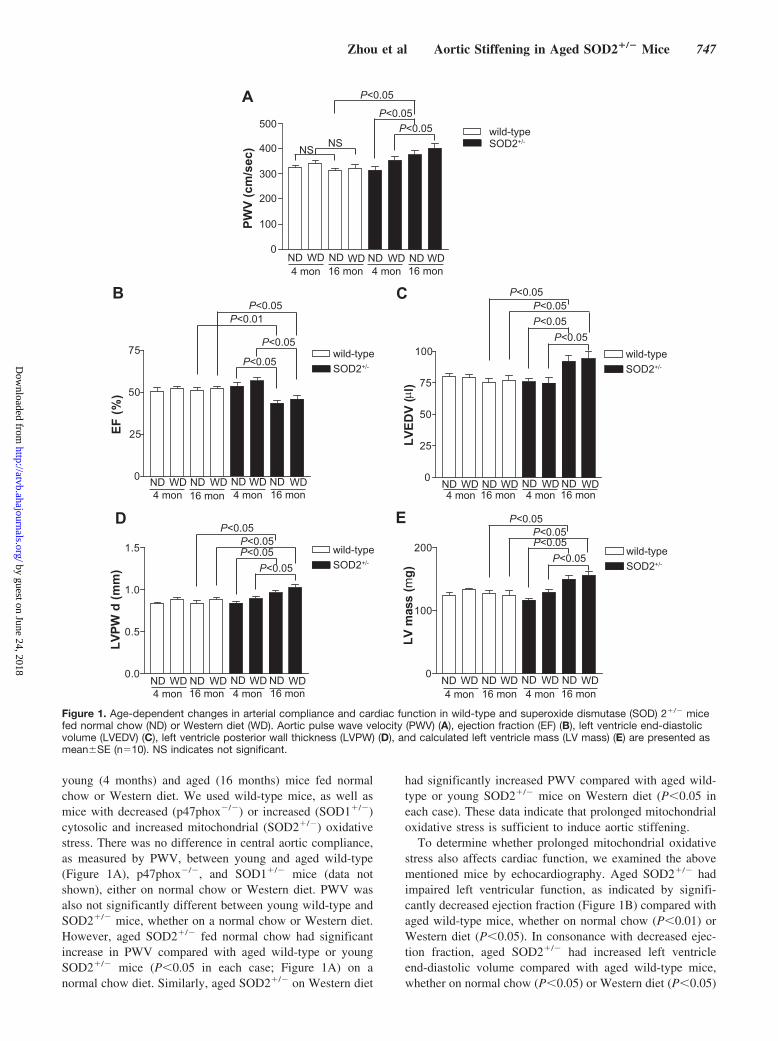
young (4 months) and aged (16 months) mice fed normalchow or Western diet. We used wild-type mice, as well asmice with decreased (p47phox�/�) or increased (SOD1�/�)cytosolic and increased mitochondrial (SOD2�/�) oxidativestress. There was no difference in central aortic compliance,as measured by PWV, between young and aged wild-type(Figure 1A), p47phox�/�, and SOD1�/� mice (data notshown), either on normal chow or Western diet. PWV wasalso not significantly different between young wild-type andSOD2�/� mice, whether on a normal chow or Western diet.However, aged SOD2�/� fed normal chow had significantincrease in PWV compared with aged wild-type or youngSOD2�/� mice (P�0.05 in each case; Figure 1A) on anormal chow diet. Similarly, aged SOD2�/� on Western diet
had significantly increased PWV compared with aged wild-type or young SOD2�/� mice on Western diet (P�0.05 ineach case). These data indicate that prolonged mitochondrialoxidative stress is sufficient to induce aortic stiffening.
To determine whether prolonged mitochondrial oxidativestress also affects cardiac function, we examined the abovementioned mice by echocardiography. Aged SOD2�/� hadimpaired left ventricular function, as indicated by signifi-cantly decreased ejection fraction (Figure 1B) compared withaged wild-type mice, whether on normal chow (P�0.01) orWestern diet (P�0.05). In consonance with decreased ejec-tion fraction, aged SOD2�/� had increased left ventricleend-diastolic volume compared with aged wild-type mice,whether on normal chow (P�0.05) or Western diet (P�0.05)
wild-typeSOD2+/-
0
100
200
300
400
500
ND NDNDNDWD WDWDWD4 mon 4 mon16 mon 16 mon
PWV
(cm
/sec
)
A
B C
D E
P<0.05P<0.05
NSNS
wild-typeSOD2+/-
EF (%
)
ND NDNDNDWD WDWDWD4 mon 4 mon16 mon 16 mon
P<0.05
P<0.05
0
25
50
75
P<0.01P<0.05
ND NDNDNDWD WDWDWD4 mon 4 mon16 mon 16 mon
P<0.05P<0.05 wild-type
SOD2+/-
0.0
0.5
1.0
1.5
LVPW
d (m
m)
P<0.05P<0.05
wild-typeSOD2+/-
ND NDNDNDWD WDWDWD4 mon 4 mon16 mon 16 mon
P<0.05P<0.05
0
25
50
75
100
LVED
V (µ
l)
P<0.05P<0.05
ND NDNDNDWD WDWDWD4 mon 4 mon16 mon 16 mon
P<0.05P<0.05
wild-typeSOD2+/-
0
100
200
LV m
ass
(mg)
P<0.05P<0.05
P<0.05
Figure 1. Age-dependent changes in arterial compliance and cardiac function in wild-type and superoxide dismutase (SOD) 2�/� micefed normal chow (ND) or Western diet (WD). Aortic pulse wave velocity (PWV) (A), ejection fraction (EF) (B), left ventricle end-diastolicvolume (LVEDV) (C), left ventricle posterior wall thickness (LVPW) (D), and calculated left ventricle mass (LV mass) (E) are presented asmean�SE (n�10). NS indicates not significant.
Zhou et al Aortic Stiffening in Aged SOD2�/� Mice 747
by guest on June 24, 2018http://atvb.ahajournals.org/
Dow
nloaded from

(Figure 1C). Ejection fraction and left ventricle end-diastolicvolume in aged SOD2�/� were significantly different fromyoung SOD2�/� mice, irrespective of the diet. Left ventricleposterior wall thickness (Figure 1D) and left ventricle mass(Figure 1E) also increased in aged SOD2�/� compared withaged wild-type and young SOD2�/� mice, independent ofdiet. Together, these data suggest that long-term exposure toincreased mitochondrial oxidative stress causes adverse ef-fects on vascular health, as evidenced by increased arterialstiffening and impaired cardiac function.
Because blood pressure is an important determinant ofPWV,29 we measured changes in blood pressure with aging.No significant difference was observed in systolic bloodpressure between wild-type and SOD2�/� mice (Table I inthe online-only Data Supplement). Diet and age had no effect;however, SOD2 deficiency significantly increased diastolicblood pressure (Table I in the online-only Data Supplement),indicating that enhanced diastolic blood pressure associatedwith prolonged mitochondrial oxidative stress may contributeto aortic stiffening. To determine the interaction of age andSOD2 deficiency on SMC function, we measured nitroglyc-erine-induced relaxation of phenylephrine-preconstricted tho-racic aortic rings. Wild-type mice had decreased vascularrelaxation with age at 10�7 mol/L nitroglycerine (P�0.01versus young) (Figure I in the online-only Data Supplement).At this concentration, young and aged SOD2�/� had im-
paired vascular relaxation compared with young wild-typemice (P�0.001). No significant difference was observed innitroglycerine-induced relaxation between SOD2�/� andaged wild-type mice. However, at 10�6 mol/L nitroglycerine,SOD2�/� had impaired aortic relaxation compared with agedwild-type mice (P�0.01, young SOD2�/� versus aged wild-type; P�0.05, aged SOD2�/� versus aged wild-type). SOD2deficiency had impaired vascular relaxation independent ofage. These data indicate that aging in general and increasedmitochondrial oxidative stress in particular impair vascularSMC function and hence vascular relaxation.
Collagen Levels Are Increased and Elastin Levelsand Integrity Are Decreased With Age in theAortic Wall and SMCs of SOD2�/� MiceBecause a decrease in the elastin/collagen ratio is associatedwith increase in aortic stiffness30 and increased aortic oxida-tive stress is correlated with extensive collagen depositionand elastin degradation and decline in aortic compliance,31 weexamined aortic collagen and elastin expression in the aorticwall of wild-type and SOD2�/� mice by immunohistochem-istry. Collagen I expression was increased in the media ofaged SOD2�/� compared with aged wild-type mice (Figure2A). The elastic laminae in the media were normal in agedwild-type mice, but their integrity was compromised withruptures in aged SOD2�/� mice (Figure 2A). No perceptible
Collagen I
Elastin
GAPDH
16-mon Wild-type 16-mon SOD2+/-
Collagen I
Elastin
wild-type wild-typeSOD2+/- SOD2+/-
4-mon 16-mon
wild-typeSOD2+/-
4-mon 16-mon
Col
lage
n I
(fold
cha
nge)
0
2.0
4.0
6.0
8.0 wild-typeSOD2+/-
4-mon 16-mon
Ela
stin
(fold
cha
nge)
0
0.5
1.0
A B
C MMP-2
wild-typeSOD2+/-
4-mon 16-mon0
2.5
5.0
7.5
MM
P-2
act
ivity
(fold
cha
nge)
4-mon 4-mon16-mon 16-monwild-type SOD2+/-
P<0.01 P<0.01
P<0.05
P<0.001 P<0.001
100 µm
Figure 2. Superoxide dismutase 2 (SOD2) deficiency increased collagen I synthesis and disrupted elastic laminae in aortas of agedmice, increased collagen levels and decreased elastin levels in aged aortic smooth muscle cells (SMCs), and enhanced matrixmetalloproteinase-2 (MMP-2) activity in the SMCs of young and aged mice. A, Representative sections from fresh frozen aortas werestained for collagen I and elastin. B, Aortic SMC lysates were analyzed by Western blotting with anti-collagen I, anti-elastin, and anti-GAPDH antibodies. Densitometric analysis of collagen I and elastin levels is shown in the lower panels (mean�SE, n�3). C, Represen-tative gelatin zymogram showing MMP-2 activity in aortic SMC lysates. Densitometric analysis of MMP-2 activity is shown in the lowerpanel (mean�SE, n�3).
748 Arterioscler Thromb Vasc Biol March 2012
by guest on June 24, 2018http://atvb.ahajournals.org/
Dow
nloaded from

increase in collagen I or ruptures in elastic lamina wereobserved in the aortas of young SOD2�/� mice (data notshown). Because increased calcification is implicated inaortic stiffening,32 we examined calcium deposition in theaortic sections. We did not detect any calcium deposition orfocal calcification in aged wild-type or SOD2�/� mice.
To determine whether the changes in aortic collagenexpression and elastin integrity represent the intrinsic effectof SOD2 deficiency, we examined collagen I and elastinexpression in SMCs. Real-time reverse transcription–poly-merase chain reaction analysis showed a significant increasein collagen I mRNA levels in aged SOD2�/� compared withaged wild-type aortic SMCs (1.7�0.1-fold increase;P�0.01). In contrast, elastin mRNA levels were significantlylower in aged SOD2�/� SMCs (2.5�0.1-fold decrease versusaged wild-type; P�0.001). Increase in collagen I mRNAlevels was followed by a significant increase in collagen Iprotein levels in aged SOD2�/� SMCs (2.2-fold increaseversus aged wild-type; P�0.01; Figure 2B). Similarly,elastin protein levels were decreased nearly 7-fold in agedSOD2�/� SMCs compared with aged wild-type SMCs(P�0.05; Figure 2B). Taken together, these data suggestthat prolonged exposure to mitochondrial oxidative stressduring aging induces structural changes in the arterial wallby regulating collagen levels, as well as elastin synthesisand degradation.
MMP-2 Expression and Activity Are Increased inSOD2�/� Aortic SMCsMMP-2 is a critical regulator of extracellular matrix degra-dation and age-associated vascular remodeling33 and has beenimplicated in arterial stiffening.34 A 3.2�0.8-fold increase inMMP-2 mRNA expression was observed in aged SOD2�/�
compared with aged wild-type SMCs (P�0.001) as deter-mined by real-time reverse transcription–polymerase chainreaction. MMP-2 activity was significantly increased(P�0.01) in both young and aged SOD2�/� compared withwild-type SMCs (Figure 2C). These data suggest that mito-chondrial oxidative stress activates signaling pathways in-volved in MMP-2 expression and activity.
Prolonged SOD2 Deficiency Renders Aortic SMCsSusceptible to Apoptosis and ImpairsAntiapoptotic Akt PathwayA decrease in arterial medial SMC number and vascularremodeling with aging has been attributed to increasedapoptosis,35 and we and others have shown that increasedmitochondrial oxidative stress is an important regulator ofSMC apoptosis.36,37 As shown in Figure 3A, immunofluores-cence staining for the cleaved form of caspase-3, a member ofthe caspase superfamily that initiates apoptotic events, isincreased in medial SMCs of aged SOD2�/� mice. Cleavedcaspase-3 was barely detectable in young SOD2�/� mice(data not shown) and not observed in the aortic walls of either
16-mon wild-type 16-mon SOD2+/-
CleavedCaspase-3
α-SMA
Merged
Caspase-3
Cleaved caspase-3
PARPCleaved PARP
β-actin
A
B
Staurosporine - + - + - + - +wild-typeSOD2+/-wild-type SOD2+/-
4-mon 16-mon
DAPI
CleavedCaspase-3
100 100 µm
α-SMA
DAPI Merged
wild
-type
SO
D2+/
-
DAPI TUNEL Merge
0
25
50
75 wild-typeSOD2+/-
% T
UN
EL-p
ositi
ve c
ells P<0.001
C
Figure 3. Superoxide dismutase 2 (SOD2) deficiency enhanced medial smooth muscle cell (SMC) apoptosis in the aorta of aged miceand sensitized aortic SMCs from aged mice to staurosporine-induced apoptosis. A, Dual immunofluorescent staining of cleavedcaspase-3 (red) and �-smooth muscle actin (green) demonstrated colocalization (yellow) in SMCs. Nuclei were counterstained with4�,6-diamidino-2-phenylindole (DAPI) (blue). B, Lysates from aortic SMCs treated or without 1.0 �mol/L staurosporine for 6 hours wereanalyzed by Western blotting with anti-caspase-3, anti-poly(ADP-ribose) polymerase (anti-PARP), and anti-�-actin antibodies. Datashown represent an experiment that was repeated at least twice with similar results. C, Aged SMCs treated with staurosporine(0.1 �mol/L) for 12 hours were analyzed by fluorescent (green) terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL)staining. Nuclei were stained with DAPI (blue). Quantitation of apoptotic cells is presented as the percentage of TUNEL-positive cells ineach field of view (mean�SE, n�3).
Zhou et al Aortic Stiffening in Aged SOD2�/� Mice 749
by guest on June 24, 2018http://atvb.ahajournals.org/
Dow
nloaded from

young or aged wild-type mice. Similarly, we did not find anyapoptosis in the hearts of either aged wild-type or SOD2�/�
mice (data not shown).To determine whether the increased apoptosis of medial
SMCs in aged SOD2�/� reflects the intrinsic effect of SOD2deficiency, we examined cleaved caspase-3 levels in aorticSMCs of young and aged wild-type and SOD2�/� miceexposed to staurosporine, a well-known inducer of apoptosisin a wide spectrum of cells, by Western analysis. Althoughnot observed in untreated cells, cleaved caspase-3 levels weresignificantly increased in aged SOD2�/� compared with agedwild-type SMCs following staurosporine treatment (Figure3B). Cleaved caspase-3 was not detected in young wild-typeand barely detectable in young SOD2�/� SMCs treated withstaurosporine. Activated caspase-3 proteolytically cleavesand inactivates many proteins, including the nuclear enzymepoly(ADP-ribose) polymerase (PARP), involved in cell via-bility, and cleaved PARP is a more specific marker ofapoptosis. A significant increase in cleaved PARP levels inresponse to staurosporine treatment was observed inSOD2�/� compared with wild-type SMCs (Figure 3B). Con-sistent with this, aged SOD2�/� SMCs treated with stauro-sporine had significantly higher number of terminal deoxy-nucleotidyl transferase dUTP nick-end labeling–positive cellscompared with aged wild-type (Figure 3C).
To determine whether prolonged exposure to mitochon-drial oxidative stress also impairs other cell survival path-ways, we investigated the activation of protein kinase B/Akt,which preserves mitochondrial integrity and protects againstapoptosis,38 in aged wild-type and SOD2�/� SMCs treatedwith and without insulin-like growth factor-1 (IGF-1). Aktphosphorylation increased significantly at 3 hours (3.7-fold,P�0.001) and remained elevated at 6 hours after IGF-1treatment in aged wild-type SMCs (Figure 4A). The increasein Akt phosphorylation in aged SOD2�/� compared withuntreated cells was much less robust at both 3 and 6 hours(2-fold increase) and significantly less than in aged wild-type(P�0.01 versus aged wild-type at 3 hours). Forkhead box O(FoxO) transcription factors are important downstream tar-gets of Akt, and FoxO3a has been implicated in SMCapoptosis.39 IGF-1 significantly increased FoxO3a phosphory-lation at both 3 hours (3.7-fold increase) and 6 hours (4.1-foldincrease) after treatment in aged wild-type SMCs (Figure4B). In contrast, the increase in FoxO3a phosphorylationfollowing IGF-1 treatment was significantly less in agedSOD2�/� SMCs (P�0.001 versus aged wild-type at both 3and 6 hours). Attenuation of FoxO3a phosphorylation in agedSOD2�/� SMCs was also observed in cells treated withangiotensin II, thrombin, and platelet-derived growth factor,indicating the intrinsic effect of SOD2 deficiency on SMCapoptosis (data not shown). Less robust stimulation of Akt inaged SOD2�/� SMCs was not associated with a decreasedproliferative response to IGF-1 (data not shown), indicatingthat SMCs of various phenotypes can coexist in the arterialwall during remodeling. Together, these data suggest thatprolonged exposure to increased mitochondrial oxidativestress during aging affects cell viability by impairing survivaland activating apoptotic signaling pathways.
Stress-Activated Protein Kinase 1/c-JunN-terminal Kinase 1 Activity and �-SmoothMuscle Actin Levels Are Increased in AgedSOD2�/� Aortic SMCsAn inhibition of Akt and increase in caspase activity result inc-Jun N-terminal kinase 1 (JNK1) activity,40 which inducesthe mitochondrial death pathway leading to apoptosis.41 It isknown that circulating tumor necrosis factor-� (TNF-�)levels are increased with aging in both animals and humans.42
Therefore, we investigated TNF-�-induced activation ofJNK1 in young and aged wild-type and SOD2�/� SMCs. Asshown in Figure 4C, JNK1 phosphorylation was significantlyincreased in young SOD2�/� compared with young wild-typeSMCs after 3 hours of TNF-� treatment (P�0.01). In agedwild-type SMCs, JNK1 phosphorylation was increased sig-nificantly at 3 hours after TNF-� treatment (P�0.05). Incontrast to young SOD2�/� and aged wild-type SMCs, agedSOD2�/� SMCs had sustained JNK1 activation throughoutthe TNF-� treatment (P�0.001). TNF-�-induced JNK1phosphorylation was also significantly higher in agedSOD2�/� compared with young SOD2�/� (P�0.05) andaged wild-type (P�0.01) SMCs 3 hours after the treatment.
Increased smooth muscle �-actin levels and intrinsic SMCstiffness are a mechanism for increased aortic stiffening withaging.43 Consistent with the data shown in Figure 3A, �-actinlevels were increased 2.8-fold with aging in wild-type and3.5-fold in young SOD2�/� compared with young wild-typeSMCs (Figure 4D). However, aged SOD2�/� had signifi-cantly higher �-actin levels compared with young SOD2�/�
(5.4-fold versus 3.5-fold) and aged wild-type SMCs (5.4-foldversus 2.8-fold). Collectively, these data indicate concurrentactivation of apoptotic signaling pathways and alterations inarterial wall structure and SMC cytoskeleton.
Basal Mitochondrial Superoxide Levels AreIncreased and Basal and IGF-1-Induced H2O2Levels Are Decreased With Aging in SOD2�/�
Aortic SMCsRecent evidence indicates that H2O2 activates the phosphati-dylinositol-3-kinase/Akt pathway and promotes cell sur-vival.44 To determine whether impaired cell survival path-ways in aged SOD2�/� SMCs are mediated by changes inROS levels, we measured superoxide and H2O2 levels inyoung and aged wild-type and SOD2�/� cells. First, weinvestigated colocalization of MitoTracker Green FM, amitochondria-selective dye, with MitoSOX Red, asuperoxide-sensitive fluorescent dye using confocal micros-copy (Figure 5A). Compared with young wild-type, youngSOD2�/� SMCs showed bright yellow fluorescence in mito-chondria because of colocalization of MitoTracker Green andMitoSOX Red, indicating increased mitochondrial superox-ide production. Similarly, aged SOD2�/� had more mito-chondrial superoxide levels, as shown by yellow/orangefluorescence in mitochondria compared with that in agedwild-type and young SOD2�/� SMCs.
To determine the effect of prolonged SOD2 deficiency onH2O2 levels, we measured basal and IGF-1-induced H2O2
levels in aged wild-type and SOD2�/� SMCs by Amplex Redassay (Figure 5B). H2O2 levels were significantly lower (31%
750 Arterioscler Thromb Vasc Biol March 2012
by guest on June 24, 2018http://atvb.ahajournals.org/
Dow
nloaded from

wild-typeSOD2+/-
0
1.0
2.0
3.0
4.0
5.0
IGF-1 (h) 0 3 6 0 3 6
Akt
pho
spho
ryla
tion
(fold
cha
nge)
Akt
pAkt (Ser473)
IGF-1 (h) 0 3 6 0 3 6wild-type SOD2+/-
pFOXO3a (Thr32)FoxO3a
IGF-1(h) 0 3 6 0 3wild-type SOD2+/-
6
wild-typeSOD2+/-
0IGF-1 (h) 0 3 6 0 3 6
1.0
2.0
3.0
4.0
5.0
FoxO
3a p
hosp
hory
latio
n(fo
ld c
hang
e)
pJNK1(Thr183/Tyr185)
JNK1
0 3 6 0 3 6 0 3 6 0 3 6TNF-α (h)wild-type wild-typeSOD2+/- SOD2+/-
4-mon 16-monwild-typeSOD2+/-
0 3 6 0 3 6 0 3 6 0 3 64-mon 16-mon
pJN
K1
leve
ls(fo
ld c
hang
e)
0
1.0
2.0
3.0
α-actin
GAPDH
wild-typeSOD2+/-
α-a
ctin
leve
ls(fo
ld c
hang
e)
01.02.0
3.0
4.0
5.0
6.0
A B
C
D
TNF-α (h)
wild-type SOD2+/- SOD2+/-wild-type4-mon 16-mon
4-mon 16-mon
P<0.001P<0.001P<0.001
P<0.05
P<0.01P<0.05
P<0.001P<0.05
P<0.001
P<0.001P<0.001
Figure 4. Superoxide dismutase 2 (SOD2) deficiency decreased insulin-like growth factor-1 (IGF-1)–induced Akt and FoxO3a phosphor-ylation and increased tumor necrosis factor-� (TNF-�)–induced c-Jun N-terminal kinase 1 (JNK1) phosphorylation in aortic smoothmuscle cells (SMCs) from aged mice and �-actin levels in SMCs from young and aged mice. A and B, Lysates from growth-arrestedand IGF-1 (100 ng/mL)–treated aged SMCs were analyzed by Western blotting with anti-phosphospecific Akt or Akt (A) or anti-phosphospecific FoxO3a or FoxO3a antibodies (B). C, Lysates from growth-arrested and TNF-� (100 ng/mL)–treated SMCs were ana-lyzed by Western blotting with anti-phosphospecific JNK1 or JNK1 antibodies. A to C, Densitometric analysis of phosphorylated pro-teins is shown in the lower panels (mean�SE, n�3). D, Representative Western blot of SMC lysates probed with anti-�-actin orGAPDH antibodies. Densitometric analysis of �-actin levels is shown in the lower panel (mean�SE, n�3).
Zhou et al Aortic Stiffening in Aged SOD2�/� Mice 751
by guest on June 24, 2018http://atvb.ahajournals.org/
Dow
nloaded from

decrease, P�0.001) in aged SOD2�/� SMCs compared withaged wild-type SMCs. IGF-1 treatment significantly in-creased H2O2 levels in wild-type cells (30% increase,P�0.001) but had no such effect in SOD2�/� SMCs. Theseresults indicate that decreased H2O2 levels, caused by SOD2deficiency, impair Akt activity and aortic SMC survival inaged mice via enhanced FoxO3a activation.
Downregulation of FoxO3a Activity DecreasesStaurosporine-Induced Apoptosis in AgedAortic SMCsBecause staurosporine, which inhibits Akt45 and activatesFoxO3a,46 increased cleaved caspase-3 and PARP levels, weinvestigated whether alteration in Akt/FoxO3a signalingpathway contributes to increased apoptosis in aged SOD2�/�
SMCs. Adenoviral overexpression of DN-FoxO3a signifi-cantly decreased (58%, P�0.001) cleaved PARP levels inaged SOD2�/� compared with cells transfected with controlvirus (Figure 6). Collectively, our data suggest that prolongedexposure to increased mitochondrial oxidative stress duringaging in SOD2�/� SMCs increases apoptosis by modulatingAkt/FoxO3a signaling pathway.
DiscussionIn this study, we provide evidence that (1) SOD2 deficiencyover a lifetime is sufficient to induce aortic stiffening,decrease aortic compliance, and cause cardiac dysfunction;(2) aortic stiffening with aging in SOD2�/� mice is associ-
ated with structural changes in the aortic wall, with increasedcollagen content and ruptures in elastin laminae; (3) SOD2deficiency increases collagen I expression, decreases elastinexpression, and increases MMP-2 expression and activity inaged SMCs; (4) SOD2 deficiency over a lifetime increasesmedial SMC apoptosis in aged mice and sensitizes SMCs tostaurosporine-induced increase in cleaved caspase-3 andcleaved PARP levels; (5) prolonged SOD2 deficiency inSMCs activates JNK1 in response to TNF-� treatment; (6)prolonged SOD2 deficiency impairs cell survival as observedby decreased Akt and increased FoxO3a activation in re-sponse to IGF-1 treatment; and (7) increased �-actin levels inSOD2�/� SMCs are integral to increased aortic stiffness withaging. It was previously established that SOD2�/� mice havean �50% reduction in SOD2 activity in all tissues comparedwith the wild-type mice, and the decrease in enzyme activitydoes not cause any compensatory upregulation of other majorcomponents of mitochondrial antioxidant defense system.18
Although impairment of cardiac function was reported in6-month-old TRE/SOD2�/� mice,47 our finding is the first toimplicate increased mitochondrial oxidative stress over alifetime as the source of aortic stiffening and cardiac dys-function in SOD2�/� mice. Specifically, we provide evidenceof how molecular signaling pathways initiated by increasedmitochondrial oxidative stress in aortic SMCs contribute toaortic stiffening.
Aortic stiffness is a complex phenomenon that arises fromstructural alterations in the aortic wall, impaired endothelialfunction, increased smooth muscle tone, phenotypic modula-tion of adventitial fibroblasts to myofibroblasts, and chroniclow-grade inflammation.2 The scaffolding proteins collagen
wild-type SOD2+/-
4 mon
16 mon
16-mon wild-type16-mon SOD2+/-
H2O
2 lev
els
0
0.5
1.0
1.5
(fold
cha
nge)
A
B
IGF-1 - -+ +
P<0.001P<0.001P<0.001
10 10 µm
Figure 5. Mitochondrial superoxide generation was increasedand extracellular H2O2 levels were decreased with age in aorticsmooth muscle cells (SMCs) from superoxide dismutase 2(SOD2)�/� mice. A, Confocal laser-scanning microscopy show-ing colocalization of mitochondria-targeting fluorescent probeMitoSOX Red with the mitochondria-selective dye MitoTrackerGreen. Yellow fluorescence indicates localization of superoxidein mitochondria. B, H2O2 production was measured using theAmplex Red fluorescence assay (mean�SE, n�9).
PARPCleaved PARP
HA
β-actinStaurosporine - + +-
AdGFP AdDN-FoxO3a
16-mon SOD2+/-
- + - +
Cle
aved
PA
RP
leve
ls(fo
ld c
hang
e)
0
1.0
2.0
3.0
4.0
Staurosporine - + +-AdGFP ++
AdDN-FoxO3a ++--
P<0.001 P<0.001
- -
Figure 6. Dominant-negative Forkhead box O3a (DN-FoxO3a)overexpression attenuated the increase in cleaved poly(ADP-ribose) polymerase (PARP) levels induced by staurosporine inaortic smooth muscle cells (SMCs) from aged superoxide dis-mutase 2 (SOD2)�/� mice. Aged aortic SMCs infected with ade-novirus expressing-green fluorescent protein (AdGFP) or dominantnegative Forkhead box O3a (AdDN-FoxO3a) were either untreatedor treated with 1.0 �mol/L staurosporine for 6 hours, and celllysates were analyzed by Western blotting with anti-PARP, anti-HA,or anti-�-actin antibodies. Densitometric analysis of �-actin levelsis shown in the lower panel (mean�SE, n�3).
752 Arterioscler Thromb Vasc Biol March 2012
by guest on June 24, 2018http://atvb.ahajournals.org/
Dow
nloaded from

and elastin provide the structural integrity of the aortic wall,and our results show that increased mitochondrial oxidativestress over a lifetime increases the collagen content andruptures and decreases the elastin in the aorta. The changes inaortic collagen and elastin levels were accompanied byincreased expression and activity of MMP-2 in agedSOD2�/� aortic SMCs. Similar to this, Dasgupta et al48 andothers33 reported both age- and redox-regulated increase inMMP expression and activity. Although the increase inMMP-2 expression and activity should decrease collagenlevels at first glance, increased collagen I levels are observedin SMCs under oxidative stress conditions.49 In fact, anincrease in interstitial and perivascular collagen is ob-served in cardiac MMP-2 transgenic mice.50 Nevertheless,activation of MMP-2 is strongly correlated with elastic fiberfragmentation, disorganization, and increased stiffness of thearterial vasculature.34 Endothelial dysfunction and inflamma-tion may contribute to increased aortic stiffening in agedSOD2�/�, as endothelial dysfunction is increased in apolipo-protein E�/� mice that are deficient in SOD2,21 and proin-flammatory cytokine production is upregulated with in-creased mitochondrial ROS levels.51
Our results showing increased aortic stiffness in agedSOD2�/� mice accompanied by ventricular dysfunction aresupported by several cross-sectional studies that reported apositive association between age-related aortic stiffness andventricular dysfunction.52 Aortic stiffening increases leftventricular afterload by inducing earlier return of reflectedwaves in the late systole and causes left ventricle hypertrophyand ventricular dysfunction. Interestingly, the impairment ofaortic relaxation and increased diastolic blood pressure inSOD2�/� mice precede increased PWV and Doppler abnor-malities in heart function. Furthermore, mitochondrial oxida-tive stress–induced coupling of vascular-ventricular dysfunc-tion is supported by the observation of impaired heartfunction with lifelong reduction of SOD2.47
Increased apoptosis of SMCs in the aortic media andincreased sensitivity to staurosporine-induced apoptosis inaged SOD2�/� mouse SMCs observed in the present inves-tigation are consistent with the concept that medial SMCapoptosis is an important contributor to age-associated vas-cular remodeling and loss of aortic elasticity.35 The propen-sity of aged SOD2�/� aortic SMCs to apoptosis is underlinedby impaired activation of Akt and increased activation ofFoxO3a in response to IGF-1 treatment. Akt is a negativeregulator of FoxO3a transcription factor, which in the ab-sence of Akt-mediated phosphorylation induces the expres-sion of genes involved in apoptosis.53 Interestingly, an in-crease in MMP-2 and MMP-9 activities was observed invascular cells following FoxO3a activation.54 Because theseMMPs do not contain a consensus binding site for forkheadfactors, activated FoxO3a may regulate MMP-2 activityindirectly, including via activation of MMP-3.
Activated MMP-2 induces apoptosis by stimulating JNKactivity, as well as cytochrome c release.41 Inhibition of Aktsignaling has been shown to induce JNK activity and promotethe cleavage of caspase-3 in SMCs.40 JNK activation, in turn,initiates mitochondrial apoptotic pathway via Bax-dependentrelease of cytochrome c.55 Alternatively, aged SOD2�/�
aortic SMCs could undergo apoptosis in the absence ofAkt-mediated phosphorylation of apoptosis regulatory pro-teins Bad and Bax, which suggests that Akt-JNK cross-talk isan important determinant of aged SMC apoptosis.40 Ourobservation that DN-FoxO3a overexpression attenuatescleaved PARP levels is consistent with the regulatory role ofAkt/FoxO3a signaling in aged SOD2�/� aortic SMC apoptosis.
Calcium channel blockers and angiotensin II receptorantagonists are used to treat large artery stiffening.43 Thesedrugs affect vascular SMC tone, which suggests that age-associated vascular stiffening is partly regulated by intrinsicmechanical properties of these cells. Our data showingsignificantly increased �-actin levels in aged SOD2�/� com-pared with aged wild-type SMCs are in agreement with thereport of Qiu et al43 that smooth muscle �-actin is a keydeterminant of vascular SMC stiffness during aging. In-creases in �-actin levels and MMP-2 activity were observedin young SOD2�/� compared with young wild-type SMCs,and yet the aortic stiffening and cardiac dysfunction areevident only in aged SOD2�/� mice, which suggests athreshold for mitochondrial oxidative stress to affect struc-tural and biochemical changes in the SMC and aorta and tocause a phenotypic effect. Our observation that H2O2 levelsare decreased in SOD2�/� SMCs is consistent with similarfindings in SOD2-deficient and knockout mice.56,57 Exoge-nous H2O2 stimulates Akt phosphorylation in many celltypes, including vascular SMCs.44,58 Therefore, it is conceiv-able that low H2O2 levels in aged SOD2�/� SMCs impair cellsurvival and promote apoptosis by downregulating Akt sig-naling and activating FoxO3a.
In summary, our data provide insight into the molecularmechanisms by which increased mitochondrial oxidativestress promotes aortic stiffening associated with aging. Al-tered ROS metabolism in the mitochondria over a lifetime notonly enhances collagen secretion and intrinsic stiffness ofaortic medial SMCs but also affects redox signaling to induceSMC apoptosis, all of which contribute to aortic stiffening. Itwould be worth determining whether strategies aimed atregulating mitochondrial oxidative stress have a therapeuticeffect against aortic stiffening and its pathophysiologicalsequelae.
Sources of FundingThis work was supported by National Institutes of Health GrantsHL-57352 and AG 024282. Dr Zhou is a cardiology fellow sup-ported by National Institutes of Health T32 Training GrantHL083828-04.
DisclosuresNone.
References1. O’Rourke MF, Hashimoto J. Mechanical factors in arterial aging: a
clinical perspective. J Am Coll Cardiol. 2007;50:1–13.2. Laurent S, Boutouyrie P. Recent advances in arterial stiffness and wave
reflection in human hypertension. Hypertension. 2007;49:1202–1206.3. Chue CD, Townend JN, Steeds RP, Ferro CJ. Arterial stiffness in chronic
kidney disease: causes and consequences. Heart. 2010;96:817–823.4. Mitchell GF, Hwang SJ, Vasan RS, Larson MG, Pencina MJ, Hamburg
NM, Vita JA, Levy D, Benjamin EJ. Arterial stiffness and cardiovascularevents: the Framingham Heart Study. Circulation. 2010;121:505–511.
Zhou et al Aortic Stiffening in Aged SOD2�/� Mice 753
by guest on June 24, 2018http://atvb.ahajournals.org/
Dow
nloaded from

5. Griendling KK, FitzGerald GA. Oxidative stress and cardiovascular inju-ry: part II: animal and human studies. Circulation. 2003;108:2034–2040.
6. Sindler AL, Fleenor BS, Calvert JW, Marshall KD, Zigler ML, Lefer DJ,Seals DR. Nitrite supplementation reverses vascular endothelial dys-function and large elastic artery stiffness with aging. Aging Cell. 2011;10:429–437.
7. Heitzer T, Schlinzig T, Krohn K, Meinertz T, Munzel T. Endothelialdysfunction, oxidative stress, and risk of cardiovascular events in patientswith coronary artery disease. Circulation. 2001;104:2673–2678.
8. Harman D. Aging: a theory based on free radical and radiation chemistry.J Gerontol. 1956;11:298–300.
9. Delles C, Zimmerli LU, McGrane DJ, Koh-Tan CH, Pathi VL, McKayAJ, Steedman T, Dargie HJ, Hamilton CA, Dominiczak AF. Vascularstiffness is related to superoxide generation in the vessel wall.J Hypertens. 2008;26:946–955.
10. Noma K, Goto C, Nishioka K, Jitsuiki D, Umemura T, Ueda K, KimuraM, Nakagawa K, Oshima T, Chayama K, Yoshizumi M, Liao JK, HigashiY. Roles of rho-associated kinase and oxidative stress in the pathogenesisof aortic stiffness. J Am Coll Cardiol. 2007;49:698–705.
11. Edgar D, Shabalina I, Camara Y, Wredenberg A, Calvaruso MA,Nijtmans L, Nedergaard J, Cannon B, Larsson NG, Trifunovic A.Random point mutations with major effects on protein-coding genes arethe driving force behind premature aging in mtDNA mutator mice. CellMetab. 2009;10:131–138.
12. Doughan AK, Harrison DG, Dikalov SI. Molecular mechanisms of an-giotensin II-mediated mitochondrial dysfunction: linking mitochondrialoxidative damage and vascular endothelial dysfunction. Circ Res. 2008;102:488–496.
13. Kinouchi H, Epstein CJ, Mizui T, Carlson E, Chen SF, Chan PH. Atten-uation of focal cerebral ischemic injury in transgenic mice overexpressingCuZn superoxide dismutase. Proc Natl Acad Sci U S A. 1991;88:11158–11162.
14. Harrison CM, Pompilius M, Pinkerton KE, Ballinger SW. Mitochondrialoxidative stress significantly influences atherogenic risk and cytokine-in-duced oxidant production. Environ Health Perspect. 2011;119:676–681.
15. Van Deel ED, Lu Z, Xu X, Zhu G, Hu X, Oury TD, Bache RJ, DunckerDJ, Chen Y. Extracellular superoxide dismutase protects the heart againstoxidative stress and hypertrophy after myocardial infarction. Free RadicBiol Med. 2008;44:1305–1313.
16. Li Y, Huang TT, Carlson EJ, Melov S, Ursell PC, Olson JL, Noble LJ,Yoshimura MP, Berger C, Chan PH, Wallace DC, Epstein CJ. Dilatedcardiomyopathy and neonatal lethality in mutant mice lacking manganesesuperoxide dismutase. Nat Genet. 1995;11:376–381.
17. Lebovitz RM, Zhang H, Vogel H, Cartwright J Jr, Dionne L, Lu N, HuangS, Matzuk MM. Neurodegeneration, myocardial injury, and perinataldeath in mitochondrial superoxide dismutase-deficient mice. Proc NatlAcad Sci U S A. 1996;93:9782–9787.
18. Van Remmen H, Williams MD, Guo Z, Estlack L, Yang H, Carlson EJ,Epstein CJ, Huang TT, Richardson A. Knockout mice heterozygous forSod2 show alterations in cardiac mitochondrial function and apoptosis.Am J Physiol Heart Circ Physiol. 2001;281:H1422–H1432.
19. Williams MD, Van Remmen H, Conrad CC, Huang TT, Epstein CJ,Richardson A. Increased oxidative damage is correlated to altered mito-chondrial function in heterozygous manganese superoxide dismutaseknockout mice. J Biol Chem. 1998;273:28510–28515.
20. Ballinger SW, Patterson C, Knight-Lozano CA, Burow DL, Conklin CA,Hu Z, Reuf J, Horaist C, Lebovitz R, Hunter GC, McIntyre K, Runge MS.Mitochondrial integrity and function in atherogenesis. Circulation. 2002;106:544–549.
21. Ohashi M, Runge MS, Faraci FM, Heistad DD. MnSOD deficiencyincreases endothelial dysfunction in ApoE-deficient mice. ArteriosclerThromb Vasc Biol. 2006;26:2331–2336.
22. Li M, Chiu JF, Mossman BT, Fukagawa NK. Down-regulation ofmanganese-superoxide dismutase through phosphorylation of FOXO3aby Akt in explanted vascular actin muscle cells from old rats. J BiolChem. 2006;281:40429–40439.
23. Hartley CJ, Taffet GE, Michael LH, Pham TT, Entman ML. Noninvasivedetermination of pulse-wave velocity in mice. Am J Physiol. 1997;273:H494–H500.
24. Moon SK, Thompson LJ, Madamanchi N, Ballinger S, PapaconstantinouJ, Horaist C, Runge M. S., Patterson C. Aging, oxidative responses, andproliferative capacity in cultured mouse aortic smooth muscle cells. Am JPhysiol Heart Circ Physiol. 2001;280:H2779–H2788.
25. Zhou RH, Pesant S, Cohn HI, Eckhart AD. Enhanced sterol responseelement-binding protein in postintervention restenotic blood vessels plays
an important role in vascular smooth muscle proliferation. Life Sci.2008;82:174–181.
26. Madamanchi NR, Li S, Patterson C, Runge MS. Thrombin regulatesvascular smooth muscle cell growth and heat shock proteins via theJAK-STAT pathway. J Biol Chem. 2001;276:18915–18924.
27. Pfaffl MW, Horgan GW, Dempfle L. Relative expression software tool(REST) for groupwise comparison and statistical analysis of relativeexpression results in real-time PCR. Nucleic Acids Res. 2002;30:e36.
28. Skurk C, Maatz H, Kim HS, Yang J, Abid MR, Aird WC, Walsh K. TheAkt-regulated forkhead transcription factor FOXO3a controls endothelialcell viability through modulation of the caspase-8 inhibitor FLIP. J BiolChem. 2004;279:1513–1525.
29. Nurnberger J, Dammer S, Opazo Saez A, Philipp T, Schafers RF. Dia-stolic blood pressure is an important determinant of augmentation indexand pulse wave velocity in young, healthy males. J Hum Hypertens.2003;17:153–158.
30. Osborne-Pellegrin M, Labat C, Mercier N, Challande P, Lacolley P.Changes in aortic stiffness related to elastic fiber network anomalies inthe Brown Norway rat during maturation and aging. Am J Physiol HeartCirc Physiol. 2010;299:H144–H152.
31. Steed MM, Tyagi N, Sen U, Schuschke DA, Joshua IG, Tyagi SC.Functional consequences of the collagen/elastin switch in vascularremodeling in hyperhomocysteinemic wild-type, eNOS�/�, andiNOS�/� mice. Am J Physiol Lung Cell Mol Physiol. 2010;299:L301–L311.
32. Ng K, Hildreth CM, Phillips JK, Avolio AP. Aortic stiffness is associatedwith vascular calcification and remodeling in a chronic kidney disease ratmodel. Am J Physiol Renal Physiol. 2011;300:F1431–F1436.
33. Wang M, Lakatta EG. Altered regulation of matrix metalloproteinase-2 inaortic remodeling during aging. Hypertension. 2002;39:865–873.
34. Chung AW, Yang HH, Kim JM, Sigrist MK, Chum E, Gourlay WA,Levin A. Upregulation of matrix metalloproteinase-2 in the arterial vas-culature contributes to stiffening and vasomotor dysfunction in patientswith chronic kidney disease. Circulation. 2009;120:792–801.
35. Sawabe M. Vascular aging: from molecular mechanism to clinical sig-nificance. Geriatr Gerontol Int. 2010;10(suppl. 1):S213–S220.
36. Tchivilev I, Madamanchi NR, Vendrov AE, Niu XL, Runge MS. Iden-tification of a protective role for protein phosphatase 1c�1 against oxi-dative stress-induced vascular smooth muscle cell apoptosis. J Biol Chem.2008;283:22193–22205.
37. Lee JY, Jung GY, Heo HJ, Yun MR, Park JY, Bae SS, Hong KW, LeeWS, Kim CD. 4-Hydroxynonenal induces vascular smooth muscle cellapoptosis through mitochondrial generation of reactive oxygen species.Toxicol Lett. 2006;166:212–221.
38. Miyamoto S, Murphy AN, Brown JH. Akt mediated mitochondrial pro-tection in the heart: metabolic and survival pathways to the rescue.J Bioenerg Biomembr. 2009;41:169–180.
39. Park KW, Kim DH, You HJ, Sir JJ, Jeon SI, Youn SW, Yang HM, SkurkC, Park YB, Walsh K, Kim HS. Activated forkhead transcription factorinhibits neointimal hyperplasia after angioplasty through induction ofp27. Arterioscler Thromb Vasc Biol. 2005;25:742–747.
40. Suhara T, Kim HS, Kirshenbaum LA, Walsh K. Suppression of Aktsignaling induces Fas ligand expression: involvement of caspase and Junkinase activation in Akt-mediated Fas ligand regulation. Mol Cell Biol.2002;22:680–691.
41. Menon B, Singh M, Ross RS, Johnson JN, Singh K. �-Adrenergicreceptor-stimulated apoptosis in adult cardiac myocytes involves MMP-2-mediated disruption of �1 integrin signaling and mitochondrialpathway. Am J Physiol Cell Physiol. 2006;290:C254–C261.
42. Csiszar A, Wang M, Lakatta EG, Ungvari Z. Inflammation and endothe-lial dysfunction during aging: role of NF-�B. J Appl Physiol. 2008;105:1333–1341.
43. Qiu H, Zhu Y, Sun Z, Trzeciakowski JP, Gansner M, Depre C, ResuelloRR, Natividad FF, Hunter WC, Genin GM, Elson EL, Vatner DE,Meininger GA, Vatner SF. Short communication: vascular smoothmuscle cell stiffness as a mechanism for increased aortic stiffness withaging. Circ Res. 2010;107:615–619.
44. Sadidi M, Lentz SI, Feldman EL. Hydrogen peroxide-induced Akt phos-phorylation regulates Bax activation. Biochimie. 2009;91:577–585.
45. Hill MM, Andjelkovic M, Brazil DP, Ferrari S, Fabbro D, Hemmings BA.Insulin-stimulated protein kinase B phosphorylation on Ser-473 is inde-pendent of its activity and occurs through a staurosporine-insensitivekinase. J Biol Chem. 2001;276:25643–25646.
754 Arterioscler Thromb Vasc Biol March 2012
by guest on June 24, 2018http://atvb.ahajournals.org/
Dow
nloaded from

46. Zhang Y, Xing D. PUMA promotes Bax activation in a FoxO3a-dependent manner in STS-induced apoptosis. J Innov Opt Health Sci.2010;3:31–38.
47. Loch T, Vakhrusheva O, Piotrowska I, Ziolkowski W, Ebelt H, Braun T,Bober E. Different extent of cardiac malfunction and resistance to oxi-dative stress in heterozygous and homozygous manganese-dependentsuperoxide dismutase-mutant mice. Cardiovasc Res. 2009;82:448–457.
48. Dasgupta J, Kar S, Van Remmen H, Melendez JA. Age-dependentincreases in interstitial collagenase and MAP kinase levels are exac-erbated by superoxide dismutase deficiencies. Exp Gerontol. 2009;44:503–510.
49. Bachem MG, Wendelin D, Schneiderhan W, Haug C, Zorn U, Gross HJ,Schmid-Kotsas A, Grunert A. Depending on their concentration oxidizedlow density lipoproteins stimulate extracellular matrix synthesis or induceapoptosis in human coronary artery smooth muscle cells. Clin Chem LabMed. 1999;37:319–326.
50. Bergman MR, Teerlink JR, Mahimkar R, Li L, Zhu BQ, Nguyen A, DahiS, Karliner JS, Lovett DH. Cardiac matrix metalloproteinase-2 expressionindependently induces marked ventricular remodeling and systolic dys-function. Am J Physiol Heart Circ Physiol. 2007;292:H1847–H1860.
51. Bulua AC, Simon A, Maddipati R, Pelletier M, Park H, Kim KY, SackMN, Kastner DL, Siegel RM. Mitochondrial reactive oxygen speciespromote production of proinflammatory cytokines and are elevated inTNFR1-associated periodic syndrome (TRAPS). J Exp Med. 2011;208:519–533.
52. Abhayaratna WP, Srikusalanukul W, Budge MM. Aortic stiffness for thedetection of preclinical left ventricular diastolic dysfunction: pulse wavevelocity versus pulse pressure. J Hypertens. 2008;26:758–764.
53. Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ,Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival byphosphorylating and inhibiting a Forkhead transcription factor. Cell.1999;96:857–868.
54. Lee HY, You HJ, Won JY, Youn SW, Cho HJ, Park KW, Park WY, SeoJS, Park YB, Walsh K, Oh BH, Kim HS. Forkhead factor, FOXO3a,induces apoptosis of endothelial cells through activation of matrix met-alloproteinases. Arterioscler Thromb Vasc Biol. 2008;28:302–308.
55. Papadakis ES, Finegan KG, Wang X, Robinson AC, Guo C, Kayahara M,Tournier C. The regulation of Bax by c-Jun N-terminal protein kinase(JNK) is a prerequisite to the mitochondrial-induced apoptotic pathway.FEBS Lett. 2006;580:1320–1326.
56. Lee S, Van Remmen H, Csete M. Sod2 overexpression preservesmyoblast mitochondrial mass and function, but not muscle mass withaging. Aging Cell. 2009;8:296–310.
57. Morten KJ, Ackrell BA, Melov S. Mitochondrial reactive oxygen speciesin mice lacking superoxide dismutase 2: attenuation via antioxidanttreatment. J Biol Chem. 2006;281:3354–3359.
58. Ushio-Fukai M, Alexander RW, Akers M, Yin Q, Fujio Y, Walsh K,Griendling KK. Reactive oxygen species mediate the activation of Akt/protein kinase B by angiotensin II in vascular smooth muscle cells. J BiolChem. 1999;274:22699–22704.
Zhou et al Aortic Stiffening in Aged SOD2�/� Mice 755
by guest on June 24, 2018http://atvb.ahajournals.org/
Dow
nloaded from

Madamanchi and Marschall S. RungeMauricio Rojas, Jacqueline D. Carter, Haiyan Tong, George A. Stouffer, Nageswara R. Rui-Hai Zhou, Aleksandr E. Vendrov, Igor Tchivilev, Xi-Lin Niu, Kimberly C. Molnar,
Cell FunctionMitochondrial Oxidative Stress in Aortic Stiffening With Age: The Role of Smooth Muscle
Print ISSN: 1079-5642. Online ISSN: 1524-4636 Copyright © 2011 American Heart Association, Inc. All rights reserved.
Greenville Avenue, Dallas, TX 75231is published by the American Heart Association, 7272Arteriosclerosis, Thrombosis, and Vascular Biology
doi: 10.1161/ATVBAHA.111.2431212011;
2012;32:745-755; originally published online December 22,Arterioscler Thromb Vasc Biol.
http://atvb.ahajournals.org/content/32/3/745World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://atvb.ahajournals.org//subscriptions/
at: is onlineArteriosclerosis, Thrombosis, and Vascular Biology Information about subscribing to Subscriptions:
http://www.lww.com/reprints
Information about reprints can be found online at: Reprints:
document. Question and AnswerPermissions and Rightspage under Services. Further information about this process is available in the
which permission is being requested is located, click Request Permissions in the middle column of the WebCopyright Clearance Center, not the Editorial Office. Once the online version of the published article for
can be obtained via RightsLink, a service of theArteriosclerosis, Thrombosis, and Vascular Biologyin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on June 24, 2018http://atvb.ahajournals.org/
Dow
nloaded from

http://atvb.ahajournals.org/content/suppl/2013/10/17/ATVBAHA.111.243121.DC2 http://atvb.ahajournals.org/content/suppl/2011/12/22/ATVBAHA.111.243121.DC1
Data Supplement (unedited) at:
http://atvb.ahajournals.org//subscriptions/
at: is onlineArteriosclerosis, Thrombosis, and Vascular Biology Information about subscribing to Subscriptions:
http://www.lww.com/reprints
Information about reprints can be found online at: Reprints:
document. Question and AnswerPermissions and Rightspage under Services. Further information about this process is available in the
which permission is being requested is located, click Request Permissions in the middle column of the WebCopyright Clearance Center, not the Editorial Office. Once the online version of the published article for
can be obtained via RightsLink, a service of theArteriosclerosis, Thrombosis, and Vascular Biologyin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on June 24, 2018http://atvb.ahajournals.org/
Dow
nloaded from

SUPPLEMENTAL METHODS
Mice and Diet All animal experimental procedures were performed in compliance with protocols approved by University of North Carolina IACUC according to NIH guidelines. Mice were maintained at 22°C
with a 12-hour light/dark cycle and given free access to food and water. Mice on high-fat diet were maintained on standard rodent chow until 1 (young) or 13 months (old) of age and then fed a Western-type diet (TD88137, Harlan Teklad, Indianapolis) for 12 weeks. Blood pressure Systolic and diastolic blood pressure was measured in conscious mice daily for 5 days using volume-pressure recording system (CODA 6, Kent Scientific). Mice were acclimated to the warmed (32°C) restrainer for 10-15 min and blood pressure was recorded during 10 acclamation followed by 20 measurement cycles.
Vascular relaxation in isolated mouse aortic rings Mouse aortas were dissected and cleaned from adventitia. Segments of mouse aortas approximately 2-3 mm in length were placed in Krebs-Hensleit buffer (in mmol/L; 120 NaCl, 25 NaHCO3, 11 glucose, 1.8 CaCl2, 4.7 KCl, 1.2 MgSO4, 1.2 KH2PO4). The aorta segments were then mounted in a Radnoti 4-unit organ bath system (Radnoti Glass Technology Inc.). Each reservoir held 20 mL of buffer and was aerated constantly with 95% O2 + 5% CO2 gas mixture. After 15-20 min stabilization, the baseline tension of the rings was adjusted to 700 mg. All rings were pre-constricted with 1 µmol/L phenylephrine and then response to nitroglycerine (NTG); 10-8 to 10-5 mol/L) was recorded. The degree of relaxation was expressed as a percentage of maximum constriction induced by phenylephrine. Cell Culture and Materials Aortic smooth muscle cells (SMC) were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 10% (v/v) fetal bovine serum (FBS). Primary aortic SMC cultures from 4-6 mice were pooled and used at passages 5-10 for all experiments. Antibodies were obtained from the following commercial sources: Akt, phosphorylated Akt, FoxO3a, caspase-3 and cleaved caspase-3 (Asp175) (5A1E) antibodies were purchased from Cell Signaling; phosphorylated FoxO3a, collagen type I and elastin from Abcam; β-actin, α-tubulin, and hemagglutinin (HA) antibodies were obtained from Sigma. HRP-conjugated secondary antibodies against mouse or rabbit IgG were also purchased from Sigma. Rhodamine-conjugated AffiniPure donkey anti-rabbit IgG, normal mouse sera and donkey sera were purchased from Jackson ImmunoResearch. Phenylephrine was purchased from Sigma and nitroglycerin was supplied by Dupont. Histology, Immunohistochemistry, and Immunofluorescence Abdominal aortas from WT and SOD2+/- mice were embedded in OCT compound (Tissue-Tek), and snap-frozen in liquid nitrogen. Aortic sections were stained using rabbit anti-collagen type I antibody while rabbit IgG was used as a negative control. Immunostaining was performed using the Vector VIP peroxidase substrate kit (Vector Laboratories) following manufacturer recommendations. Sections were counterstained with methylgreen. Aortic cross-sections were stained with Verhoeff’s stain for delineating elastic laminae. For immunofluorescence studies, aortic sections were stained with rabbit anti-cleaved caspase-3 antibody followed by rhodamine-conjugated donkey anti-rabbit secondary antibody. Nuclei were counter-stained with DAPI and permanently mounted with VectaMount Mounting Medium (Vector Laboratories). Photographs were taken using Nikon Eclipse E800 microscope. Fresh frozen aortic sections were stained by von Kossa method for visualization of calcium deposits.

Zhou, Aortic Stiffening In Aged SOD2+/- Mice
2
Gelatin Zymography Cell lysates were diluted in Tris-glycine SDS sample buffer containing 62.5 mmol/L Tris-HCl (pH 6.8), 10% glycerol, 2% SDS (w/v), and 0.025 % bromophenol blue for gel electrophoresis. Samples were resolved on 10% denaturing polyacrylamide gels containing 0.1% gelatin (Bio-Rad) with Tris-glycine SDS running buffer (0.29% Tris base, 1.44% glycine and 0.1% SDS). Gels were then incubated in zymogram renaturing buffer containing 2.5% (v/v) Triton X-100 with gentle agitation for 30 min at room temperature. Renaturing buffer was then removed and replaced with zymogram developing buffer containing 0.121% Tris base, 0.63%, Tris-HCl, 1.17% NaCl, 0.074% CaCl2 and 0.02% Brij 35. After incubation for 30 min at room temperature with gentle agitation, gels were then placed in fresh zymogram developing buffer and incubated overnight at 37°C. Gels were stained with 0.5% (w/v) Coomassie Blue R-250 for 30 min and then destained with solution containing methanol:acetic acid:water (5:1:4). Areas of protease activity appeared as clear bands – signifying protease digestion of substrate – set against a dark blue background. The gels were scanned and the bands densities were measured using ImageJ. Detection of Mitochondrial Superoxide Aortic SMC grown in glass bottom dishes were washed with Hanks' balanced salt solution and incubated with 5 μmol/L MitoSOX Red and 1 μmol/L MitoTracker Green FM (Molecular Probes) at 37 °C for 10 min. Excess stains were removed, and cells were imaged using Olympus FV500 confocal laser-scanning microscopy. MitoTracker Green FM was visualized at an excitation of 490 nm and an emission of 516 nm, whereas MitoSOX Red was visualized at an excitation of 560 nm and an emission of 600 nm. MitoTracker Green FM preferentially translocates to the mitochondria. MitoSOX Red accumulates in mitochondria and exhibits bright red fluorescence upon oxidation and subsequent binding to mitochondrial DNA. H2O2 Measurement Extracellular H2O2 levels were determined using Amplex Red assay (Invitrogen). In the presence of horseradish peroxidase, N-acetyl-3,7-dihydroxyphenoxazine (Amplex Red) reacts with H2O2 to produce the fluorescent molecule resorufin. Mouse aortic SMC were grown on 24-well plates to subconfluent condition and were starved in 0.5 ml OPTI-MEM for 24 h. To measure H2O2 production, IGF-1 was added to a final concentration of 100 ng/ml. After 30 min incubation, 50 µL of media were transferred to a 96-well plate containing Amplex Red reaction mixture (50 μmol/L Amplex Red and 0.1 U/mL horseradish peroxidase). After 30 min incubation, fluorescence was measured at an excitation wavelength of 560 nm and emission wavelength of 590 nm.

Zhou, Aortic Stiffening In Aged SOD2+/- Mice
3
Suppl Table I. Systolic and diastolic blood pressure was measured in 4- and 16-month-old wild-type and SOD2+/- mice fed normal chow (ND) or Western diet (WD) (mean±SE, n=15).
Suppl Figure I: Vascular relaxation is impaired with age and SOD2 deficiency in mice. Relaxation (% maximum response) of aortic rings, precontracted with 1 µmol/L phenylephrine (PE), to NTG treatment (6-8 mice and 12-15 rings were used per each group). *P<0.01 aged vs young wild-type; **P<0.001 young and aged SOD2+/- vs young wild-type; ***P<0.01 young SOD2+/- vs aged wild-type; ****P<0.05 aged SOD2+/- vs aged wild-type mice.
Systolic BP Diastolic BP
ND-4 mon HF-4 mon ND-16mon
HF-16mon
ND-4 mon HF-4 mon ND-16 mon
HF-16mon
Wild-type
136.7±2.7 130.0±2.8 129.0±3.1 132.0±4.9 102.8± 2.1
99.6± 2.4 101.5± 1.3
100.1± 2.7
SOD2+/- 138.4± 3.6
133.4± 3.0
131.7± 3.4 132.6± 3.5 115.7± 3.2*
109.5± 3.2*
109.4± 2.7*
111.1± 3.2*
*P<0.01 *P<0.05 *P<0.05 *P<0.05

23
산화 스트레스가 혈관노화의 특징적 소견인 대동맥 경직도 증가의 주범이다.
김 광 일 교수분당서울대학교병원 노인병내과
Summary
배경
연령증가에 따른 대동맥 경직도 증가는 심혈관질
환의 독립적인 위험인자이다. 산화 스트레스가 대
동맥 경직도 증가와 관련되어 있지만 이를 뒷받침
하는 분자생물학적 기전에 대해서는 밝혀져 있지
않다. 우리는 노화에 의한 산화 스트레스의 유발
원인을 찾고, 산화 스트레스가 혈관 평활근세포의
기능과 대동맥 순응도에 미치는 영향을 유전자변
형 생쥐모델로 연구하였다.
방법 및 결과
Superoxide dismutase 2 (SOD2)+/- 생쥐에서
도플러를 이용하여 측정한 대동맥 맥파속도가
연령에 비례하여 증가하였으나 야생형 생쥐,
p47phox-/-, SOD1+/- 생쥐에서는 증가하지 않았다.
SOD2+/- 생쥐에서 심초음파 검사를 시행하였을
때 심장기능의 저하 소견이 관찰되었다. 고령의
SOD2+/- 생쥐 대동맥벽에서 콜라젠 I 발현이
증가하였고 elastic lamellae integrity 손상, 중막의
혈관 평활근세포의 세포고사가 관찰되었다.
고령의 SOD2+/- 생쥐 대동맥 혈관 평활근세포에서
4개월령 SOD2+/- 생쥐나 고령의 야생형 생쥐와
비교하였을 때 콜라젠 I 증가, elastin 감소,
matrix metalloproteinase-2 발현과 활성도 증가,
세포고사의 증가가 관찰되었다. 고령의 SOD2+/-
생쥐에서는 야생형에 비해 smooth muscle
α-actin이 증가하였다. 고령의 SOD2+/- 혈관
평활근세포는 인슐린 유사 성장인자(insulin-like
growth factor-1)에 의한 Akt와 Forkhead box
O3a의 인산화를 감소시켰고 tumor necrosis
factor-α에 의한 Jun N-terminal kinase 1
활성화를 지속시켰다. 고령의 SOD2+/- 생쥐 혈관
평활근세포는 미토콘드리아의 superoxide가
증가되어 있으나 hydrogen peroxide는 감소되어
있었다. 마지막으로 dominant-negative Forkhead
box O3a의 과발현은 고령의 SOD2+/- 생쥐의
혈관 평활근세포에서 staurosporine에 의한
세포고사를 감소시켰다.
결론
미토콘드리아의 산화 스트레스가 혈관벽의 재형성과
혈관 평활근세포의 경직도 증가, 그리고 대동맥
혈관 평활근세포의 세포고사를 초래하여 대동맥
경직도를 증가시킨다.

24
Commentary
연령이 증가함에 따라 혈관벽의 특징적인 소견이
관찰되며 이러한 혈관노화는 고혈압, 당뇨병, 고지
혈증 등의 다른 위험인자와 독립적으로 심혈관질
환의 위험을 증가시키는 위험인자임이 잘 알려져
있다.
노화에 의해 혈관에서 나타나는 가장 현저한 변화
는 혈관 내경의 증가와 혈관벽의 비후이다. 혈관의
내막과 중막(intima-medial thickness, IMT) 두께
는 연령에 비례하여 증가하며 이러한 변화는 심혈
관, 뇌혈관질환 발생과 유의한 상관관계가 있다. 또
한 혈관벽에서 탄성섬유(elastin fiber)가 감소되고
콜라젠(collagen)의 침착이 증가되며 석회화가 동
반되어 혈관의 탄성이 감소된다. 한편, 연령이 증가
함에 따라 혈관 평활근세포의 증식이 증가하고 세
포외기질 생성이 증가하는 것도 혈관벽의 경직도
증가의 원인이 된다. 이러한 혈관노화에 의해 특히
대동맥과 같은 큰 혈관의 경직도가 증가하게 되어
좌심실의 후부하가 증가된다.
한편, 노화에 의해 혈관의 구조적인 변화뿐 아니
라 혈관내피세포의 기능이 저하되며 혈관내피세
포에서 생성되는 산화질소(nitric oxide, NO)에 의
한 혈관내피세포 의존성 혈관 확장이 저하되어 혈
관 긴장도가 증가하게 되나 혈관내피세포 비의존
성 혈관 확장에는 큰 변화가 없다. 혈관내피세포의
기능 저하 기전으로는 내피세포 노화에 의한 산화
질소의 생성 감소와 산화 스트레스 증가에 의해
내피세포에서 생성된 산화질소가 peroxynitrite로
변환되며 eNOS의 uncoupling 및 항산화 효소의
감소 등이 제시되고 있다.1
혈관벽의 비후와 혈관내피세포의 기능저하로 인
한 대동맥의 경직도 증가는 동맥경화와는 무관하
게 노화에 의해 진행하며 이러한 변화는 좌심실의
후부하를 증가시켜 심비대를 유발하고 고혈압과
죽상동맥경화의 진행을 초래한다. 즉, 노화에 따른
혈관의 변화는 그 자체로 심혈관질환의 원인이 되
며 진행을 촉진하는 위험인자로서 심혈관질환의
발생을 줄이기 위해서는 혈관노화의 진행을 막는
새로운 치료법에 대한 관심을 기울여야 할 것으로
생각된다.
이와 같이 혈관노화의 특징적인 소견에 대해서는
잘 알려져 있으나 혈관노화의 기전에 대한 연구는
아직까지 많이 진행되지 못해 혈관노화의 근본적
인 원인과 기전에 대해 잘 밝혀져 있지 못하였다.
본 논문은 유전자변형 생쥐모델을 이용하여 미토
콘드리아의 산화 스트레스 증가가 혈관노화의 특
징적인 소견을 발현하는 것을 확인하였다. 또한 미
토콘드리아의 산화 스트레스 증가가 콜라젠 생성
증가, 혈관 평활근세포의 세포고사 촉진 등의 기전
에 의해 혈관노화를 발현하는 것을 확인하여 향후
혈관노화에 대한 근본적인 치료적 접근을 위해서
는 미토콘드리아의 산화 스트레스를 감소시키는
전략적 방법을 개발해야 한다는 점을 제시하였다.
산화 스트레스에 의한 세포 손상은 지난 50년간
노화 이론에서 중요하게 생각되고 있는 기전으로
산화 스트레스의 생성이 증가하거나 방어 기제에
장애가 있는 경우 지속적으로 산화 스트레스에 의
해 세포가 손상되어 세포의 수명이 단축되고 궁극
적으로 개체의 수명이 줄어들게 된다는 이론이다.
따라서 산화 스트레스에 대한 방어 기제에 관여하
는 유전적 영향에 대해 많은 관심이 있어왔다.2 즉,
산소를 매개로 한 에너지 대사를 이용하는 개체에
서는 필연적으로 생성되는 산소 라디칼의 효과적
인 제거가 중요하며 따라서 산화 스트레스에 대한
효율적인 방어 시스템이 에너지 대사 과정 중 지속
적으로 생성되는 산소 라디칼에 의한 세포 손상과
이에 기인한 노화에 중요한 역할을 할 것으로 생각

25
되어진다.
산소 라디칼의 효과적인 제거를 위해서는 과산화
이온을 제거하는 SOD (superoxide dismutase),
catalase, glutathione peroxidase 등의 항산화 효
소계와 항산화 물질인 vitamin C, E, uric acid 등
이 중요한 역할을 한다. 이중 SOD는 대표적인 항
산화 효소로서 3개의 isoform이 존재하며 주로
세포질과 핵 내에 존재하는 Cu/ZnSOD (SOD1),
미토콘드리아에 존재하는 MnSOD (SOD2), 세
포 밖으로 분비되어 세포 표면에 heparan sulfate
proteoglycan과 결합하여 존재하는 EcSOD
(SOD3)로 구분된다.
그런데 산화 스트레스에 대한 방어 기제를 항진시
킴으로써 노화를 억제할 수 있는가에 대해서는 논
란이 있어 왔다. 초파리에서 Cu/ZnSOD를 과발현
시키면 수명이 연장된다고 하는 초기의 보고가 있
었으나 이후 Cu/ZnSOD 발현을 조절한 생쥐 모델
에서 수명 연장의 효과가 없다고 보고되어 차이를
보인다.3,4 그러나 미토콘드리아에서 생성되는 산
화 스트레스 조절에 관여하는 MnSOD의 유전자
발현 억제 생쥐 모델에서 미토콘드리아의 기능 저
하와 세포고사가 증가하여 노화가 촉진된다는 결
과를 고려하면 산화 스트레스에 의한 노화 현상
은 결국 세포 내에서 산화 스트레스에 가장 취약
한 곳에서 증가된 산화 스트레스에 대해 적절하게
대처하지 못해 발생하는 것으로 생각되며 특히 세
포의 노화에 있어 미토콘드리아의 손상이 중요하
다는 점을 시사한다.5 특히 미토콘드리아는 그 자
체의 유전 정보를 가지고 있으며 산화 스트레스가
많이 생성되는 장소에 존재한다라는 특성과 함께
손상에 대한 복구 시스템이 핵 내 유전 정보에 비
해 불완전하여 유전 정보의 손상에 취약하다. 따
라서 미토콘드리아의 산화 스트레스를 감소시키
는 것이 향후 혈관노화의 진행을 억제할 수 있는
새로운 치료전략으로 고려될 수 있을 것으로 기대
된다.
REFERENCESBrandes RP, Fleming I, Busse R. Endothelial aging. 1. Cardiovasc Res. 2005;66:286-294.Johnson FB, Sinclair DA, Guarente L. Molecular biology of 2. aging. Cell. 1999;96:291-302.Huang TT, Carlson EJ, Gillespie AM, Shi Y, Epstein CJ. 3. Ubiquitous overexpression of CuZn superoxide dismutase does not extend life span in mice. J Gerontol A Biol Sci Med Sci. 2000;55:B5-9.Parkes TL, El ia AJ, Dick inson D, Hil l iker AJ, Phi l l ips 4. JP, Boulianne GL. Extension of Drosophila l i fespan by overexpression of human SOD1 in motorneurons. Nat Genet. 1998;19:171-174.Kokoszka JE, Coskun P, Esposito LA, Wallace DC. Increased 5. mitochondrial oxidative stress in the Sod2(+/-) mouse results in the age-related decline of mitochondrial function culminating in increased apoptosis. Proc Natl Acad Sci U S A. 2001;98:2278-2283.

Mitochondrial Oxidative Stress in Aortic StiffeningWith Age
The Role of Smooth Muscle Cell Function
Rui-Hai Zhou, Aleksandr E. Vendrov, Igor Tchivilev, Xi-Lin Niu, Kimberly C. Molnar,Mauricio Rojas, Jacqueline D. Carter, Haiyan Tong, George A. Stouffer,
Nageswara R. Madamanchi, Marschall S. Runge
Objective—Age-related aortic stiffness is an independent risk factor for cardiovascular diseases. Although oxidative stressis implicated in aortic stiffness, the underlying molecular mechanisms remain unelucidated. Here, we examined thesource of oxidative stress in aging and its effect on smooth muscle cell (SMC) function and aortic compliance usingmutant mouse models.
Methods and Results—Pulse wave velocity, determined using Doppler, increased with age in superoxide dismutase 2(SOD2)�/� but not in wild-type, p47phox�/� and SOD1�/� mice. Echocardiography showed impaired cardiac functionin these mice. Increased collagen I expression, impaired elastic lamellae integrity, and increased medial SMC apoptosiswere observed in the aortic wall of aged SOD2�/� versus wild-type (16-month-old) mice. Aortic SMCs from agedSOD2�/� mice showed increased collagen I and decreased elastin expression, increased matrix metalloproteinase-2expression and activity, and increased sensitivity to staurosporine-induced apoptosis versus aged wild-type and young(4-month-old) SOD2�/� mice. Smooth muscle �-actin levels were increased with age in SOD2�/� versus wild-typeSMCs. Aged SOD2�/� SMCs had attenuated insulin-like growth factor-1-induced Akt and Forkhead box O3aphosphorylation and prolonged tumor necrosis factor-�–induced Jun N-terminal kinase 1 activation. Aged SOD2�/�
SMCs had increased mitochondrial superoxide but decreased hydrogen peroxide levels. Finally, dominant-negativeForkhead box O3a overexpression attenuated staurosporine-induced apoptosis in aged SOD2�/� SMCs.
Conclusion—Mitochondrial oxidative stress over a lifetime causes aortic stiffening, in part by inducing vascular wall remodeling,intrinsic changes in SMC stiffness, and aortic SMC apoptosis. (Arterioscler Thromb Vasc Biol. 2012;32:745-755.)
Key Words: aging � blood pressure � reactive oxygen species � signal transduction � vasodilation
Advancing age is the major risk factor for cardiovasculardisease (CVD) morbidity and mortality. With aging,
central arteries stiffen (and dilate) as a result of physiologicalremodeling arising from the fracture of elastin lamellae fromrepetitive pulsations and also from endothelial dysfunction,chronic low-grade inflammation, and altered vascular smoothmuscle tone.1,2 Aortic stiffening is the principal cause of CVDwith age in people without atherosclerosis,1 including in-creased systolic and pulse pressures, increased left ventricularhypertrophy and diastolic dysfunction, and congestive heartfailure.3 Carotid-femoral pulse wave velocity (PWV), a directnoninvasive measure of the thoracic and abdominal aorticstiffness, is correlated with higher CVD events and is anindependent predictor of coronary heart disease and stroke.4
Despite the strong epidemiological and biological connection
of age to CVD risk, the molecular mechanisms responsiblefor age-related vascular dysfunction have yet to be elucidated.Although advancing age is an unmodifiable risk factor forCVD, it might be possible to target specific molecular signalsas an approach to limiting age-related CVD risk.
Oxidative stress has been implicated in vascular dysfunc-tion, whether as a result of CVD or aging or both.5–7 The freeradical theory of aging, first proposed by Harman more than50 years ago,8 suggested that increased reactive oxygenspecies (ROS) generation underlies many features of aging.Prior studies have indicated that increased vascular ROSgeneration results in decreased compliance, as measured byPWV.9,10 Recent studies suggest that mitochondrial dysfunc-tion plays an important role in aging and impairing vascularfunction.11,12
Received on: April 1, 2011; final version accepted on: December 12, 2011.From the McAllister Heart Institute (R.-H.Z., A.E.V., I.T., X.-L.N., K.C.M., M.R., G.A.S., N.R.M., M.S.R.), Division of Cardiology (R.-H.Z., G.A.S.,
M.S.R.), Department of Medicine, University of North Carolina at Chapel Hill, Chapel Hill, NC; Environmental Public Health Division, National Healthand Environmental Effects Research Laboratory, US Environmental Protection Agency, Research Triangle Park, NC (J.D.C., H.T.).
Dr Zhou and Dr Vendrov contributed equally to this work.The online-only Data Supplement is available with this article at http://atvb.ahajournals.org/lookup/suppl/doi:10.1161/ATVBAHA.111.243121/-/DC1.Correspondence to Marschall S. Runge, MD, PhD, Department of Medicine, 125 MacNaider Hall, University of North Carolina at Chapel Hill, Chapel
Hill, NC 27599-7005. E-mail [email protected]© 2011 American Heart Association, Inc.
745
26

27
Many pro- and antioxidant enzymes regulate ROS levels incells. Of these, the superoxide dismutase (SOD) family is themost studied antioxidant system and has been previouslyimplicated in CVD.13–15 SODs convert superoxide to producehydrogen peroxide (H2O2), which is further degraded byeither catalase or glutathione peroxidase. One member of theSOD family, manganese SOD (SOD2), is present in mito-chondria. Deletion of the SOD2 gene results in early postnatallethality in mice.16,17 SOD2-deficient (SOD2�/�) mice areviable but demonstrate increased susceptibility to oxidativestress, diminished mitochondrial function, and enhanced sen-sitivity to apoptosis.18,19 In an atherosclerotic background(apolipoprotein E knockout), SOD2 deficiency results inaccelerated atherosclerosis20 and endothelial dysfunction inmice.21 In addition, decreased expression/activity of SOD2with age was implicated in vascular aging.22
In the present study, we investigated the effect of oxidativestress in aging-associated increase in aortic stiffness usingmutant mouse models. Our data indicate that prolongedexposure to increased mitochondrial oxidative stress de-creases aortic compliance and induces cardiac dysfunction.Specifically, we elucidate the significance of lifelong SOD2deficiency on the phenotype, function, and molecular signal-ing pathways in aortic smooth muscle cells (SMCs) and howthese events regulate aortic wall homeostasis and aorticstiffening.
Materials and MethodsAortic PWVArterial compliance was determined as described by Hartley et al.23
In brief, mice were anesthetized with inhaled isoflurane (1% in O2)and fixed in a supine position on the temperature-controlled ECGboard (THM100, Indus Instruments). Body temperature was main-tained at 37°C and monitored with a rectal probe. Blood flowvelocity was recorded using a 20-MHz pulsed Doppler probe at thelevels of aortic arch and at the abdominal aorta. Data were analyzedusing an Indus Instruments Doppler Signal Processing Workstation.Aortic PWV was calculated by dividing separation distance (40 mm)by difference in pulse wave arrival time in respect to ECG R-peaks.
EchocardiographyMice were anesthetized with inhaled isoflurane (1% in O2) and fixedin a supine position on the ECG temperature-controlled board.Ultrasound biomicroscopy was performed using VisualSonics Vevo660 equipped with a 30-MHz probe. Ultrasonic images of leftventricle were acquired at the long axis using M-mode. Measure-ments of interventricular septum, posterior wall thickness, andventricle internal diameter at systole and diastole were taken. Valuesof ejection fraction, end diastolic volume, and myocardial mass werederived using VisualSonics Vevo 660 software.
Blood PressureSystolic and diastolic blood pressure was measured as described inthe online-only Data Supplement.
Vascular Relaxation in Isolated MouseAortic RingsRelaxation of isolated mouse aortic rings was measured as describedin the online-only Data Supplement.
Cell Culture and MaterialsMouse aortic SMCs were isolated from young (4 months) and aged(16 months) wild-type and SOD2�/� mice (C57BL/6J) as describedpreviously24 (see online-only Data Supplement).
Histology, Immunohistochemistry,and ImmunofluorescenceImmunohistochemistry and immunofluorescence studies were per-formed as previously described.25
Western Blot AnalysisPreparation of cell extracts and Western blot analysis was performedas described previously.26
Quantitative Real-Time PolymeraseChain ReactionQuantitative analysis of mRNA expression of target genes wasperformed using total RNA extracted from cells and tissues. Reversetranscription was performed using the TaqMan Reverse Transcrip-tion Reagents Kit (Applied Biosystems). Real-time polymerase chainreaction was performed in quadruplicate with TaqMan Gene Expres-sion Assays for mouse collagen I (Mm01302043_gl), elastin(Mm00514670_ml), matrix metalloproteinase-2 (MMP-2)(Mm00439508_ml), and 18S rRNA (Hs99999901_sl) using an ABIPrism 7900 HT Sequence Detection System according to manufac-turer’s recommended protocol. Target gene mRNA expression wasnormalized to 18S rRNA expression. Individual gene expression inSOD2�/� aortic SMCs was calculated relative to that in wild-typeusing REST2008 (Relative Expression Software Tool).27
Gelatin ZymographyMMP-2 activity was assayed by gelatin zymography (see Methods inthe online-only Data Supplement).
Adenovirus Infection of Aortic SMCsA replication-defective adenoviral vector expressing dominant-negative Forkhead box O3a (DN-FOXO3a) was obtained fromVector BioLabs. DN-FoxO3a, constructed by deletion of the trans-activation domain from the C-terminus,28 had a heme agglutinin tagat the N-terminus and expressed green fluorescent protein. Adeno-virus expressing only green fluorescent protein was used as anegative control. Mouse aortic SMCs were cultured to 80% to 90%confluence before adenoviral infection. Infections were performedusing a multiplicity of infection of 100, and the infection efficiencywas typically greater than 90%. Measurement of proteins of interestwas made in cells harvested 36 hours after viral infection.
Detection of Mitochondrial SuperoxideMitochondrial superoxide levels in aortic SMCs were detected asdescribed in the Methods in the online-only Data Supplement.
H2O2 MeasurementAortic SMCs extracellular H2O2 levels were determined using theAmplex Red assay (Invitrogen) (see Methods in the online-only DataSupplement).
Statistical AnalysisData presented graphically are shown as mean�SE from at least 3independent experiments. All data were tested for normality usingthe Kolmogorov-Smirnov test and were analyzed by 1-wayANOVA, and post hoc analysis was performed using the Newman-Keuls test. To account for multiple comparisons, arterial complianceand cardiac function data were analyzed by 1-way ANOVA followedby the Ryan-Einot-Gabriel-Welsch multiple-range test with an over-all ��0.05 (SSPS software, version 19.0).
ResultsAortic Compliance, Cardiac Function, andVasorelaxation Are Decreased With Age inSOD2�/� MiceTo examine the interactive effect of oxidative stress, diet, andaging on vascular health, we measured aortic compliance in
746

28
young (4 months) and aged (16 months) mice fed normalchow or Western diet. We used wild-type mice, as well asmice with decreased (p47phox�/�) or increased (SOD1�/�)cytosolic and increased mitochondrial (SOD2�/�) oxidativestress. There was no difference in central aortic compliance,as measured by PWV, between young and aged wild-type(Figure 1A), p47phox�/�, and SOD1�/� mice (data notshown), either on normal chow or Western diet. PWV wasalso not significantly different between young wild-type andSOD2�/� mice, whether on a normal chow or Western diet.However, aged SOD2�/� fed normal chow had significantincrease in PWV compared with aged wild-type or youngSOD2�/� mice (P�0.05 in each case; Figure 1A) on anormal chow diet. Similarly, aged SOD2�/� on Western diet
had significantly increased PWV compared with aged wild-type or young SOD2�/� mice on Western diet (P�0.05 ineach case). These data indicate that prolonged mitochondrialoxidative stress is sufficient to induce aortic stiffening.
To determine whether prolonged mitochondrial oxidativestress also affects cardiac function, we examined the abovementioned mice by echocardiography. Aged SOD2�/� hadimpaired left ventricular function, as indicated by signifi-cantly decreased ejection fraction (Figure 1B) compared withaged wild-type mice, whether on normal chow (P�0.01) orWestern diet (P�0.05). In consonance with decreased ejec-tion fraction, aged SOD2�/� had increased left ventricleend-diastolic volume compared with aged wild-type mice,whether on normal chow (P�0.05) or Western diet (P�0.05)
wild-typeSOD2+/-
0
100
200
300
400
500
ND NDNDNDWD WDWDWD4 mon 4 mon16 mon 16 mon
PWV
(cm
/sec
)
A
B C
D E
P<0.05P<0.05
NSNS
wild-typeSOD2+/-
EF (%
)
ND NDNDNDWD WDWDWD4 mon 4 mon16 mon 16 mon
P<0.05
P<0.05
0
25
50
75
P<0.01P<0.05
ND NDNDNDWD WDWDWD4 mon 4 mon16 mon 16 mon
P<0.05P<0.05 wild-type
SOD2+/-
0.0
0.5
1.0
1.5
LVPW
d (m
m)
P<0.05P<0.05
wild-typeSOD2+/-
ND NDNDNDWD WDWDWD4 mon 4 mon16 mon 16 mon
P<0.05P<0.05
0
25
50
75
100
LVED
V (µ
l)
P<0.05P<0.05
ND NDNDNDWD WDWDWD4 mon 4 mon16 mon 16 mon
P<0.05P<0.05
wild-typeSOD2+/-
0
100
200
LV m
ass
(mg)
P<0.05P<0.05
P<0.05
Figure 1. Age-dependent changes in arterial compliance and cardiac function in wild-type and superoxide dismutase (SOD) 2�/� micefed normal chow (ND) or Western diet (WD). Aortic pulse wave velocity (PWV) (A), ejection fraction (EF) (B), left ventricle end-diastolicvolume (LVEDV) (C), left ventricle posterior wall thickness (LVPW) (D), and calculated left ventricle mass (LV mass) (E) are presented asmean�SE (n�10). NS indicates not significant.
� /� 747

29
(Figure 1C). Ejection fraction and left ventricle end-diastolicvolume in aged SOD2�/� were significantly different fromyoung SOD2�/� mice, irrespective of the diet. Left ventricleposterior wall thickness (Figure 1D) and left ventricle mass(Figure 1E) also increased in aged SOD2�/� compared withaged wild-type and young SOD2�/� mice, independent ofdiet. Together, these data suggest that long-term exposure toincreased mitochondrial oxidative stress causes adverse ef-fects on vascular health, as evidenced by increased arterialstiffening and impaired cardiac function.
Because blood pressure is an important determinant ofPWV,29 we measured changes in blood pressure with aging.No significant difference was observed in systolic bloodpressure between wild-type and SOD2�/� mice (Table I inthe online-only Data Supplement). Diet and age had no effect;however, SOD2 deficiency significantly increased diastolicblood pressure (Table I in the online-only Data Supplement),indicating that enhanced diastolic blood pressure associatedwith prolonged mitochondrial oxidative stress may contributeto aortic stiffening. To determine the interaction of age andSOD2 deficiency on SMC function, we measured nitroglyc-erine-induced relaxation of phenylephrine-preconstricted tho-racic aortic rings. Wild-type mice had decreased vascularrelaxation with age at 10�7 mol/L nitroglycerine (P�0.01versus young) (Figure I in the online-only Data Supplement).At this concentration, young and aged SOD2�/� had im-
paired vascular relaxation compared with young wild-typemice (P�0.001). No significant difference was observed innitroglycerine-induced relaxation between SOD2�/� andaged wild-type mice. However, at 10�6 mol/L nitroglycerine,SOD2�/� had impaired aortic relaxation compared with agedwild-type mice (P�0.01, young SOD2�/� versus aged wild-type; P�0.05, aged SOD2�/� versus aged wild-type). SOD2deficiency had impaired vascular relaxation independent ofage. These data indicate that aging in general and increasedmitochondrial oxidative stress in particular impair vascularSMC function and hence vascular relaxation.
Collagen Levels Are Increased and Elastin Levelsand Integrity Are Decreased With Age in theAortic Wall and SMCs of SOD2�/� MiceBecause a decrease in the elastin/collagen ratio is associatedwith increase in aortic stiffness30 and increased aortic oxida-tive stress is correlated with extensive collagen depositionand elastin degradation and decline in aortic compliance,31 weexamined aortic collagen and elastin expression in the aorticwall of wild-type and SOD2�/� mice by immunohistochem-istry. Collagen I expression was increased in the media ofaged SOD2�/� compared with aged wild-type mice (Figure2A). The elastic laminae in the media were normal in agedwild-type mice, but their integrity was compromised withruptures in aged SOD2�/� mice (Figure 2A). No perceptible
Collagen I
Elastin
GAPDH
16-mon Wild-type 16-mon SOD2+/-
Collagen I
Elastin
wild-type wild-typeSOD2+/- SOD2+/-
4-mon 16-mon
wild-typeSOD2+/-
4-mon 16-mon
Col
lage
n I
(fold
cha
nge)
0
2.0
4.0
6.0
8.0 wild-typeSOD2+/-
4-mon 16-mon
Ela
stin
(fold
cha
nge)
0
0.5
1.0
A B
C MMP-2
wild-typeSOD2+/-
4-mon 16-mon0
2.5
5.0
7.5
MM
P-2
act
ivity
(fold
cha
nge)
4-mon 4-mon16-mon 16-monwild-type SOD2+/-
P<0.01 P<0.01
P<0.05
P<0.001 P<0.001
100 µm
Figure 2. Superoxide dismutase 2 (SOD2) deficiency increased collagen I synthesis and disrupted elastic laminae in aortas of agedmice, increased collagen levels and decreased elastin levels in aged aortic smooth muscle cells (SMCs), and enhanced matrixmetalloproteinase-2 (MMP-2) activity in the SMCs of young and aged mice. A, Representative sections from fresh frozen aortas werestained for collagen I and elastin. B, Aortic SMC lysates were analyzed by Western blotting with anti-collagen I, anti-elastin, and anti-GAPDH antibodies. Densitometric analysis of collagen I and elastin levels is shown in the lower panels (mean�SE, n�3). C, Represen-tative gelatin zymogram showing MMP-2 activity in aortic SMC lysates. Densitometric analysis of MMP-2 activity is shown in the lowerpanel (mean�SE, n�3).
748
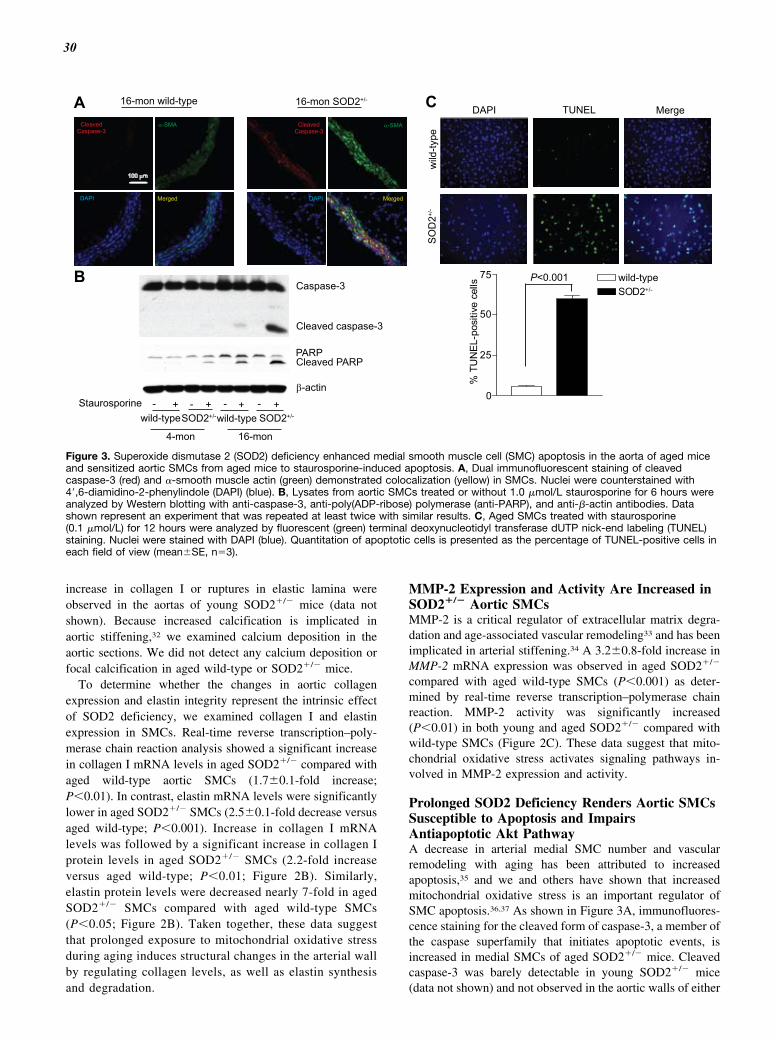
30
increase in collagen I or ruptures in elastic lamina wereobserved in the aortas of young SOD2�/� mice (data notshown). Because increased calcification is implicated inaortic stiffening,32 we examined calcium deposition in theaortic sections. We did not detect any calcium deposition orfocal calcification in aged wild-type or SOD2�/� mice.
To determine whether the changes in aortic collagenexpression and elastin integrity represent the intrinsic effectof SOD2 deficiency, we examined collagen I and elastinexpression in SMCs. Real-time reverse transcription–poly-merase chain reaction analysis showed a significant increasein collagen I mRNA levels in aged SOD2�/� compared withaged wild-type aortic SMCs (1.7�0.1-fold increase;P�0.01). In contrast, elastin mRNA levels were significantlylower in aged SOD2�/� SMCs (2.5�0.1-fold decrease versusaged wild-type; P�0.001). Increase in collagen I mRNAlevels was followed by a significant increase in collagen Iprotein levels in aged SOD2�/� SMCs (2.2-fold increaseversus aged wild-type; P�0.01; Figure 2B). Similarly,elastin protein levels were decreased nearly 7-fold in agedSOD2�/� SMCs compared with aged wild-type SMCs(P�0.05; Figure 2B). Taken together, these data suggestthat prolonged exposure to mitochondrial oxidative stressduring aging induces structural changes in the arterial wallby regulating collagen levels, as well as elastin synthesisand degradation.
MMP-2 Expression and Activity Are Increased inSOD2�/� Aortic SMCsMMP-2 is a critical regulator of extracellular matrix degra-dation and age-associated vascular remodeling33 and has beenimplicated in arterial stiffening.34 A 3.2�0.8-fold increase inMMP-2 mRNA expression was observed in aged SOD2�/�
compared with aged wild-type SMCs (P�0.001) as deter-mined by real-time reverse transcription–polymerase chainreaction. MMP-2 activity was significantly increased(P�0.01) in both young and aged SOD2�/� compared withwild-type SMCs (Figure 2C). These data suggest that mito-chondrial oxidative stress activates signaling pathways in-volved in MMP-2 expression and activity.
Prolonged SOD2 Deficiency Renders Aortic SMCsSusceptible to Apoptosis and ImpairsAntiapoptotic Akt PathwayA decrease in arterial medial SMC number and vascularremodeling with aging has been attributed to increasedapoptosis,35 and we and others have shown that increasedmitochondrial oxidative stress is an important regulator ofSMC apoptosis.36,37 As shown in Figure 3A, immunofluores-cence staining for the cleaved form of caspase-3, a member ofthe caspase superfamily that initiates apoptotic events, isincreased in medial SMCs of aged SOD2�/� mice. Cleavedcaspase-3 was barely detectable in young SOD2�/� mice(data not shown) and not observed in the aortic walls of either
16-mon wild-type 16-mon SOD2+/-
CleavedCaspase-3
α-SMA
Merged
Caspase-3
Cleaved caspase-3
PARPCleaved PARP
β-actin
A
B
Staurosporine - + - + - + - +wild-typeSOD2+/-wild-type SOD2+/-
4-mon 16-mon
DAPI
CleavedCaspase-3
100 µm
α-SMA
DAPI Merged
wild
-type
SO
D2+/
-
DAPI TUNEL Merge
0
25
50
75 wild-typeSOD2+/-
% T
UN
EL-p
ositi
ve c
ells P<0.001
C
Figure 3. Superoxide dismutase 2 (SOD2) deficiency enhanced medial smooth muscle cell (SMC) apoptosis in the aorta of aged miceand sensitized aortic SMCs from aged mice to staurosporine-induced apoptosis. A, Dual immunofluorescent staining of cleavedcaspase-3 (red) and �-smooth muscle actin (green) demonstrated colocalization (yellow) in SMCs. Nuclei were counterstained with4�,6-diamidino-2-phenylindole (DAPI) (blue). B, Lysates from aortic SMCs treated or without 1.0 �mol/L staurosporine for 6 hours wereanalyzed by Western blotting with anti-caspase-3, anti-poly(ADP-ribose) polymerase (anti-PARP), and anti-�-actin antibodies. Datashown represent an experiment that was repeated at least twice with similar results. C, Aged SMCs treated with staurosporine(0.1 �mol/L) for 12 hours were analyzed by fluorescent (green) terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL)staining. Nuclei were stained with DAPI (blue). Quantitation of apoptotic cells is presented as the percentage of TUNEL-positive cells ineach field of view (mean�SE, n�3).
Zhou et al Aortic Stiffening in Aged SOD2�/� Mice 749

31
increase in collagen I or ruptures in elastic lamina wereobserved in the aortas of young SOD2�/� mice (data notshown). Because increased calcification is implicated inaortic stiffening,32 we examined calcium deposition in theaortic sections. We did not detect any calcium deposition orfocal calcification in aged wild-type or SOD2�/� mice.
To determine whether the changes in aortic collagenexpression and elastin integrity represent the intrinsic effectof SOD2 deficiency, we examined collagen I and elastinexpression in SMCs. Real-time reverse transcription–poly-merase chain reaction analysis showed a significant increasein collagen I mRNA levels in aged SOD2�/� compared withaged wild-type aortic SMCs (1.7�0.1-fold increase;P�0.01). In contrast, elastin mRNA levels were significantlylower in aged SOD2�/� SMCs (2.5�0.1-fold decrease versusaged wild-type; P�0.001). Increase in collagen I mRNAlevels was followed by a significant increase in collagen Iprotein levels in aged SOD2�/� SMCs (2.2-fold increaseversus aged wild-type; P�0.01; Figure 2B). Similarly,elastin protein levels were decreased nearly 7-fold in agedSOD2�/� SMCs compared with aged wild-type SMCs(P�0.05; Figure 2B). Taken together, these data suggestthat prolonged exposure to mitochondrial oxidative stressduring aging induces structural changes in the arterial wallby regulating collagen levels, as well as elastin synthesisand degradation.
MMP-2 Expression and Activity Are Increased inSOD2�/� Aortic SMCsMMP-2 is a critical regulator of extracellular matrix degra-dation and age-associated vascular remodeling33 and has beenimplicated in arterial stiffening.34 A 3.2�0.8-fold increase inMMP-2 mRNA expression was observed in aged SOD2�/�
compared with aged wild-type SMCs (P�0.001) as deter-mined by real-time reverse transcription–polymerase chainreaction. MMP-2 activity was significantly increased(P�0.01) in both young and aged SOD2�/� compared withwild-type SMCs (Figure 2C). These data suggest that mito-chondrial oxidative stress activates signaling pathways in-volved in MMP-2 expression and activity.
Prolonged SOD2 Deficiency Renders Aortic SMCsSusceptible to Apoptosis and ImpairsAntiapoptotic Akt PathwayA decrease in arterial medial SMC number and vascularremodeling with aging has been attributed to increasedapoptosis,35 and we and others have shown that increasedmitochondrial oxidative stress is an important regulator ofSMC apoptosis.36,37 As shown in Figure 3A, immunofluores-cence staining for the cleaved form of caspase-3, a member ofthe caspase superfamily that initiates apoptotic events, isincreased in medial SMCs of aged SOD2�/� mice. Cleavedcaspase-3 was barely detectable in young SOD2�/� mice(data not shown) and not observed in the aortic walls of either
16-mon wild-type 16-mon SOD2+/-
CleavedCaspase-3
α-SMA
Merged
Caspase-3
Cleaved caspase-3
PARPCleaved PARP
β-actin
A
B
Staurosporine - + - + - + - +wild-typeSOD2+/-wild-type SOD2+/-
4-mon 16-mon
DAPI
CleavedCaspase-3
100 µm
α-SMA
DAPI Merged
wild
-type
SO
D2+/
-
DAPI TUNEL Merge
0
25
50
75 wild-typeSOD2+/-
% T
UN
EL-p
ositi
ve c
ells P<0.001
C
Figure 3. Superoxide dismutase 2 (SOD2) deficiency enhanced medial smooth muscle cell (SMC) apoptosis in the aorta of aged miceand sensitized aortic SMCs from aged mice to staurosporine-induced apoptosis. A, Dual immunofluorescent staining of cleavedcaspase-3 (red) and �-smooth muscle actin (green) demonstrated colocalization (yellow) in SMCs. Nuclei were counterstained with4�,6-diamidino-2-phenylindole (DAPI) (blue). B, Lysates from aortic SMCs treated or without 1.0 �mol/L staurosporine for 6 hours wereanalyzed by Western blotting with anti-caspase-3, anti-poly(ADP-ribose) polymerase (anti-PARP), and anti-�-actin antibodies. Datashown represent an experiment that was repeated at least twice with similar results. C, Aged SMCs treated with staurosporine(0.1 �mol/L) for 12 hours were analyzed by fluorescent (green) terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL)staining. Nuclei were stained with DAPI (blue). Quantitation of apoptotic cells is presented as the percentage of TUNEL-positive cells ineach field of view (mean�SE, n�3).
Zhou et al Aortic Stiffening in Aged SOD2�/� Mice 749
young or aged wild-type mice. Similarly, we did not find anyapoptosis in the hearts of either aged wild-type or SOD2�/�
mice (data not shown).To determine whether the increased apoptosis of medial
SMCs in aged SOD2�/� reflects the intrinsic effect of SOD2deficiency, we examined cleaved caspase-3 levels in aorticSMCs of young and aged wild-type and SOD2�/� miceexposed to staurosporine, a well-known inducer of apoptosisin a wide spectrum of cells, by Western analysis. Althoughnot observed in untreated cells, cleaved caspase-3 levels weresignificantly increased in aged SOD2�/� compared with agedwild-type SMCs following staurosporine treatment (Figure3B). Cleaved caspase-3 was not detected in young wild-typeand barely detectable in young SOD2�/� SMCs treated withstaurosporine. Activated caspase-3 proteolytically cleavesand inactivates many proteins, including the nuclear enzymepoly(ADP-ribose) polymerase (PARP), involved in cell via-bility, and cleaved PARP is a more specific marker ofapoptosis. A significant increase in cleaved PARP levels inresponse to staurosporine treatment was observed inSOD2�/� compared with wild-type SMCs (Figure 3B). Con-sistent with this, aged SOD2�/� SMCs treated with stauro-sporine had significantly higher number of terminal deoxy-nucleotidyl transferase dUTP nick-end labeling–positive cellscompared with aged wild-type (Figure 3C).
To determine whether prolonged exposure to mitochon-drial oxidative stress also impairs other cell survival path-ways, we investigated the activation of protein kinase B/Akt,which preserves mitochondrial integrity and protects againstapoptosis,38 in aged wild-type and SOD2�/� SMCs treatedwith and without insulin-like growth factor-1 (IGF-1). Aktphosphorylation increased significantly at 3 hours (3.7-fold,P�0.001) and remained elevated at 6 hours after IGF-1treatment in aged wild-type SMCs (Figure 4A). The increasein Akt phosphorylation in aged SOD2�/� compared withuntreated cells was much less robust at both 3 and 6 hours(2-fold increase) and significantly less than in aged wild-type(P�0.01 versus aged wild-type at 3 hours). Forkhead box O(FoxO) transcription factors are important downstream tar-gets of Akt, and FoxO3a has been implicated in SMCapoptosis.39 IGF-1 significantly increased FoxO3a phosphory-lation at both 3 hours (3.7-fold increase) and 6 hours (4.1-foldincrease) after treatment in aged wild-type SMCs (Figure4B). In contrast, the increase in FoxO3a phosphorylationfollowing IGF-1 treatment was significantly less in agedSOD2�/� SMCs (P�0.001 versus aged wild-type at both 3and 6 hours). Attenuation of FoxO3a phosphorylation in agedSOD2�/� SMCs was also observed in cells treated withangiotensin II, thrombin, and platelet-derived growth factor,indicating the intrinsic effect of SOD2 deficiency on SMCapoptosis (data not shown). Less robust stimulation of Akt inaged SOD2�/� SMCs was not associated with a decreasedproliferative response to IGF-1 (data not shown), indicatingthat SMCs of various phenotypes can coexist in the arterialwall during remodeling. Together, these data suggest thatprolonged exposure to increased mitochondrial oxidativestress during aging affects cell viability by impairing survivaland activating apoptotic signaling pathways.
Stress-Activated Protein Kinase 1/c-JunN-terminal Kinase 1 Activity and �-SmoothMuscle Actin Levels Are Increased in AgedSOD2�/� Aortic SMCsAn inhibition of Akt and increase in caspase activity result inc-Jun N-terminal kinase 1 (JNK1) activity,40 which inducesthe mitochondrial death pathway leading to apoptosis.41 It isknown that circulating tumor necrosis factor-� (TNF-�)levels are increased with aging in both animals and humans.42
Therefore, we investigated TNF-�-induced activation ofJNK1 in young and aged wild-type and SOD2�/� SMCs. Asshown in Figure 4C, JNK1 phosphorylation was significantlyincreased in young SOD2�/� compared with young wild-typeSMCs after 3 hours of TNF-� treatment (P�0.01). In agedwild-type SMCs, JNK1 phosphorylation was increased sig-nificantly at 3 hours after TNF-� treatment (P�0.05). Incontrast to young SOD2�/� and aged wild-type SMCs, agedSOD2�/� SMCs had sustained JNK1 activation throughoutthe TNF-� treatment (P�0.001). TNF-�-induced JNK1phosphorylation was also significantly higher in agedSOD2�/� compared with young SOD2�/� (P�0.05) andaged wild-type (P�0.01) SMCs 3 hours after the treatment.
Increased smooth muscle �-actin levels and intrinsic SMCstiffness are a mechanism for increased aortic stiffening withaging.43 Consistent with the data shown in Figure 3A, �-actinlevels were increased 2.8-fold with aging in wild-type and3.5-fold in young SOD2�/� compared with young wild-typeSMCs (Figure 4D). However, aged SOD2�/� had signifi-cantly higher �-actin levels compared with young SOD2�/�
(5.4-fold versus 3.5-fold) and aged wild-type SMCs (5.4-foldversus 2.8-fold). Collectively, these data indicate concurrentactivation of apoptotic signaling pathways and alterations inarterial wall structure and SMC cytoskeleton.
Basal Mitochondrial Superoxide Levels AreIncreased and Basal and IGF-1-Induced H2O2Levels Are Decreased With Aging in SOD2�/�
Aortic SMCsRecent evidence indicates that H2O2 activates the phosphati-dylinositol-3-kinase/Akt pathway and promotes cell sur-vival.44 To determine whether impaired cell survival path-ways in aged SOD2�/� SMCs are mediated by changes inROS levels, we measured superoxide and H2O2 levels inyoung and aged wild-type and SOD2�/� cells. First, weinvestigated colocalization of MitoTracker Green FM, amitochondria-selective dye, with MitoSOX Red, asuperoxide-sensitive fluorescent dye using confocal micros-copy (Figure 5A). Compared with young wild-type, youngSOD2�/� SMCs showed bright yellow fluorescence in mito-chondria because of colocalization of MitoTracker Green andMitoSOX Red, indicating increased mitochondrial superox-ide production. Similarly, aged SOD2�/� had more mito-chondrial superoxide levels, as shown by yellow/orangefluorescence in mitochondria compared with that in agedwild-type and young SOD2�/� SMCs.
To determine the effect of prolonged SOD2 deficiency onH2O2 levels, we measured basal and IGF-1-induced H2O2
levels in aged wild-type and SOD2�/� SMCs by Amplex Redassay (Figure 5B). H2O2 levels were significantly lower (31%
750

32
wild-typeSOD2+/-
0
1.0
2.0
3.0
4.0
5.0
IGF-1 (h) 0 3 6 0 3 6
Akt
pho
spho
ryla
tion
(fold
cha
nge)
Akt
pAkt (Ser473)
IGF-1 (h) 0 3 6 0 3 6wild-type SOD2+/-
pFOXO3a (Thr32)FoxO3a
IGF-1(h) 0 3 6 0 3wild-type SOD2+/-
6
wild-typeSOD2+/-
0IGF-1 (h) 0 3 6 0 3 6
1.0
2.0
3.0
4.0
5.0
FoxO
3a p
hosp
hory
latio
n(fo
ld c
hang
e)
pJNK1(Thr183/Tyr185)
JNK1
0 3 6 0 3 6 0 3 6 0 3 6TNF-α (h)wild-type wild-typeSOD2+/- SOD2+/-
4-mon 16-monwild-typeSOD2+/-
0 3 6 0 3 6 0 3 6 0 3 64-mon 16-mon
pJN
K1
leve
ls(fo
ld c
hang
e)
0
1.0
2.0
3.0
α-actin
GAPDH
wild-typeSOD2+/-
α-a
ctin
leve
ls(fo
ld c
hang
e)
01.02.0
3.0
4.0
5.0
6.0
A B
C
D
TNF-α (h)
wild-type SOD2+/- SOD2+/-wild-type4-mon 16-mon
4-mon 16-mon
P<0.001P<0.001P<0.001
P<0.05
P<0.01P<0.05
P<0.001P<0.05
P<0.001
P<0.001P<0.001
Figure 4. Superoxide dismutase 2 (SOD2) deficiency decreased insulin-like growth factor-1 (IGF-1)–induced Akt and FoxO3a phosphor-ylation and increased tumor necrosis factor-� (TNF-�)–induced c-Jun N-terminal kinase 1 (JNK1) phosphorylation in aortic smoothmuscle cells (SMCs) from aged mice and �-actin levels in SMCs from young and aged mice. A and B, Lysates from growth-arrestedand IGF-1 (100 ng/mL)–treated aged SMCs were analyzed by Western blotting with anti-phosphospecific Akt or Akt (A) or anti-phosphospecific FoxO3a or FoxO3a antibodies (B). C, Lysates from growth-arrested and TNF-� (100 ng/mL)–treated SMCs were ana-lyzed by Western blotting with anti-phosphospecific JNK1 or JNK1 antibodies. A to C, Densitometric analysis of phosphorylated pro-teins is shown in the lower panels (mean�SE, n�3). D, Representative Western blot of SMC lysates probed with anti-�-actin orGAPDH antibodies. Densitometric analysis of �-actin levels is shown in the lower panel (mean�SE, n�3).
Zhou et al Aortic Stiffening in Aged SOD2�/� Mice 751

33
wild-typeSOD2+/-
0
1.0
2.0
3.0
4.0
5.0
IGF-1 (h) 0 3 6 0 3 6
Akt
pho
spho
ryla
tion
(fold
cha
nge)
Akt
pAkt (Ser473)
IGF-1 (h) 0 3 6 0 3 6wild-type SOD2+/-
pFOXO3a (Thr32)FoxO3a
IGF-1(h) 0 3 6 0 3wild-type SOD2+/-
6
wild-typeSOD2+/-
0IGF-1 (h) 0 3 6 0 3 6
1.0
2.0
3.0
4.0
5.0
FoxO
3a p
hosp
hory
latio
n(fo
ld c
hang
e)
pJNK1(Thr183/Tyr185)
JNK1
0 3 6 0 3 6 0 3 6 0 3 6TNF-α (h)wild-type wild-typeSOD2+/- SOD2+/-
4-mon 16-monwild-typeSOD2+/-
0 3 6 0 3 6 0 3 6 0 3 64-mon 16-mon
pJN
K1
leve
ls(fo
ld c
hang
e)
0
1.0
2.0
3.0
α-actin
GAPDH
wild-typeSOD2+/-
α-a
ctin
leve
ls(fo
ld c
hang
e)
01.02.0
3.0
4.0
5.0
6.0
A B
C
D
TNF-α (h)
wild-type SOD2+/- SOD2+/-wild-type4-mon 16-mon
4-mon 16-mon
P<0.001P<0.001P<0.001
P<0.05
P<0.01P<0.05
P<0.001P<0.05
P<0.001
P<0.001P<0.001
Figure 4. Superoxide dismutase 2 (SOD2) deficiency decreased insulin-like growth factor-1 (IGF-1)–induced Akt and FoxO3a phosphor-ylation and increased tumor necrosis factor-� (TNF-�)–induced c-Jun N-terminal kinase 1 (JNK1) phosphorylation in aortic smoothmuscle cells (SMCs) from aged mice and �-actin levels in SMCs from young and aged mice. A and B, Lysates from growth-arrestedand IGF-1 (100 ng/mL)–treated aged SMCs were analyzed by Western blotting with anti-phosphospecific Akt or Akt (A) or anti-phosphospecific FoxO3a or FoxO3a antibodies (B). C, Lysates from growth-arrested and TNF-� (100 ng/mL)–treated SMCs were ana-lyzed by Western blotting with anti-phosphospecific JNK1 or JNK1 antibodies. A to C, Densitometric analysis of phosphorylated pro-teins is shown in the lower panels (mean�SE, n�3). D, Representative Western blot of SMC lysates probed with anti-�-actin orGAPDH antibodies. Densitometric analysis of �-actin levels is shown in the lower panel (mean�SE, n�3).
Zhou et al Aortic Stiffening in Aged SOD2�/� Mice 751
decrease, P�0.001) in aged SOD2�/� SMCs compared withaged wild-type SMCs. IGF-1 treatment significantly in-creased H2O2 levels in wild-type cells (30% increase,P�0.001) but had no such effect in SOD2�/� SMCs. Theseresults indicate that decreased H2O2 levels, caused by SOD2deficiency, impair Akt activity and aortic SMC survival inaged mice via enhanced FoxO3a activation.
Downregulation of FoxO3a Activity DecreasesStaurosporine-Induced Apoptosis in AgedAortic SMCsBecause staurosporine, which inhibits Akt45 and activatesFoxO3a,46 increased cleaved caspase-3 and PARP levels, weinvestigated whether alteration in Akt/FoxO3a signalingpathway contributes to increased apoptosis in aged SOD2�/�
SMCs. Adenoviral overexpression of DN-FoxO3a signifi-cantly decreased (58%, P�0.001) cleaved PARP levels inaged SOD2�/� compared with cells transfected with controlvirus (Figure 6). Collectively, our data suggest that prolongedexposure to increased mitochondrial oxidative stress duringaging in SOD2�/� SMCs increases apoptosis by modulatingAkt/FoxO3a signaling pathway.
DiscussionIn this study, we provide evidence that (1) SOD2 deficiencyover a lifetime is sufficient to induce aortic stiffening,decrease aortic compliance, and cause cardiac dysfunction;(2) aortic stiffening with aging in SOD2�/� mice is associ-
ated with structural changes in the aortic wall, with increasedcollagen content and ruptures in elastin laminae; (3) SOD2deficiency increases collagen I expression, decreases elastinexpression, and increases MMP-2 expression and activity inaged SMCs; (4) SOD2 deficiency over a lifetime increasesmedial SMC apoptosis in aged mice and sensitizes SMCs tostaurosporine-induced increase in cleaved caspase-3 andcleaved PARP levels; (5) prolonged SOD2 deficiency inSMCs activates JNK1 in response to TNF-� treatment; (6)prolonged SOD2 deficiency impairs cell survival as observedby decreased Akt and increased FoxO3a activation in re-sponse to IGF-1 treatment; and (7) increased �-actin levels inSOD2�/� SMCs are integral to increased aortic stiffness withaging. It was previously established that SOD2�/� mice havean �50% reduction in SOD2 activity in all tissues comparedwith the wild-type mice, and the decrease in enzyme activitydoes not cause any compensatory upregulation of other majorcomponents of mitochondrial antioxidant defense system.18
Although impairment of cardiac function was reported in6-month-old TRE/SOD2�/� mice,47 our finding is the first toimplicate increased mitochondrial oxidative stress over alifetime as the source of aortic stiffening and cardiac dys-function in SOD2�/� mice. Specifically, we provide evidenceof how molecular signaling pathways initiated by increasedmitochondrial oxidative stress in aortic SMCs contribute toaortic stiffening.
Aortic stiffness is a complex phenomenon that arises fromstructural alterations in the aortic wall, impaired endothelialfunction, increased smooth muscle tone, phenotypic modula-tion of adventitial fibroblasts to myofibroblasts, and chroniclow-grade inflammation.2 The scaffolding proteins collagen
wild-type SOD2+/-
4 mon
16 mon
16-mon wild-type16-mon SOD2+/-
H2O
2 lev
els
0
0.5
1.0
1.5
(fold
cha
nge)
A
B
IGF-1 - -+ +
P<0.001P<0.001P<0.001
10 µm
Figure 5. Mitochondrial superoxide generation was increasedand extracellular H2O2 levels were decreased with age in aorticsmooth muscle cells (SMCs) from superoxide dismutase 2(SOD2)�/� mice. A, Confocal laser-scanning microscopy show-ing colocalization of mitochondria-targeting fluorescent probeMitoSOX Red with the mitochondria-selective dye MitoTrackerGreen. Yellow fluorescence indicates localization of superoxidein mitochondria. B, H2O2 production was measured using theAmplex Red fluorescence assay (mean�SE, n�9).
PARPCleaved PARP
HA
β-actinStaurosporine - + +-
AdGFP AdDN-FoxO3a
16-mon SOD2+/-
- + - +
Cle
aved
PA
RP
leve
ls(fo
ld c
hang
e)
0
1.0
2.0
3.0
4.0
Staurosporine - + +-AdGFP ++
AdDN-FoxO3a ++--
P<0.001 P<0.001
- -
Figure 6. Dominant-negative Forkhead box O3a (DN-FoxO3a)overexpression attenuated the increase in cleaved poly(ADP-ribose) polymerase (PARP) levels induced by staurosporine inaortic smooth muscle cells (SMCs) from aged superoxide dis-mutase 2 (SOD2)�/� mice. Aged aortic SMCs infected with ade-novirus expressing-green fluorescent protein (AdGFP) or dominantnegative Forkhead box O3a (AdDN-FoxO3a) were either untreatedor treated with 1.0 �mol/L staurosporine for 6 hours, and celllysates were analyzed by Western blotting with anti-PARP, anti-HA,or anti-�-actin antibodies. Densitometric analysis of �-actin levelsis shown in the lower panel (mean�SE, n�3).
752

34
and elastin provide the structural integrity of the aortic wall,and our results show that increased mitochondrial oxidativestress over a lifetime increases the collagen content andruptures and decreases the elastin in the aorta. The changes inaortic collagen and elastin levels were accompanied byincreased expression and activity of MMP-2 in agedSOD2�/� aortic SMCs. Similar to this, Dasgupta et al48 andothers33 reported both age- and redox-regulated increase inMMP expression and activity. Although the increase inMMP-2 expression and activity should decrease collagenlevels at first glance, increased collagen I levels are observedin SMCs under oxidative stress conditions.49 In fact, anincrease in interstitial and perivascular collagen is ob-served in cardiac MMP-2 transgenic mice.50 Nevertheless,activation of MMP-2 is strongly correlated with elastic fiberfragmentation, disorganization, and increased stiffness of thearterial vasculature.34 Endothelial dysfunction and inflamma-tion may contribute to increased aortic stiffening in agedSOD2�/�, as endothelial dysfunction is increased in apolipo-protein E�/� mice that are deficient in SOD2,21 and proin-flammatory cytokine production is upregulated with in-creased mitochondrial ROS levels.51
Our results showing increased aortic stiffness in agedSOD2�/� mice accompanied by ventricular dysfunction aresupported by several cross-sectional studies that reported apositive association between age-related aortic stiffness andventricular dysfunction.52 Aortic stiffening increases leftventricular afterload by inducing earlier return of reflectedwaves in the late systole and causes left ventricle hypertrophyand ventricular dysfunction. Interestingly, the impairment ofaortic relaxation and increased diastolic blood pressure inSOD2�/� mice precede increased PWV and Doppler abnor-malities in heart function. Furthermore, mitochondrial oxida-tive stress–induced coupling of vascular-ventricular dysfunc-tion is supported by the observation of impaired heartfunction with lifelong reduction of SOD2.47
Increased apoptosis of SMCs in the aortic media andincreased sensitivity to staurosporine-induced apoptosis inaged SOD2�/� mouse SMCs observed in the present inves-tigation are consistent with the concept that medial SMCapoptosis is an important contributor to age-associated vas-cular remodeling and loss of aortic elasticity.35 The propen-sity of aged SOD2�/� aortic SMCs to apoptosis is underlinedby impaired activation of Akt and increased activation ofFoxO3a in response to IGF-1 treatment. Akt is a negativeregulator of FoxO3a transcription factor, which in the ab-sence of Akt-mediated phosphorylation induces the expres-sion of genes involved in apoptosis.53 Interestingly, an in-crease in MMP-2 and MMP-9 activities was observed invascular cells following FoxO3a activation.54 Because theseMMPs do not contain a consensus binding site for forkheadfactors, activated FoxO3a may regulate MMP-2 activityindirectly, including via activation of MMP-3.
Activated MMP-2 induces apoptosis by stimulating JNKactivity, as well as cytochrome c release.41 Inhibition of Aktsignaling has been shown to induce JNK activity and promotethe cleavage of caspase-3 in SMCs.40 JNK activation, in turn,initiates mitochondrial apoptotic pathway via Bax-dependentrelease of cytochrome c.55 Alternatively, aged SOD2�/�
aortic SMCs could undergo apoptosis in the absence ofAkt-mediated phosphorylation of apoptosis regulatory pro-teins Bad and Bax, which suggests that Akt-JNK cross-talk isan important determinant of aged SMC apoptosis.40 Ourobservation that DN-FoxO3a overexpression attenuatescleaved PARP levels is consistent with the regulatory role ofAkt/FoxO3a signaling in aged SOD2�/� aortic SMC apoptosis.
Calcium channel blockers and angiotensin II receptorantagonists are used to treat large artery stiffening.43 Thesedrugs affect vascular SMC tone, which suggests that age-associated vascular stiffening is partly regulated by intrinsicmechanical properties of these cells. Our data showingsignificantly increased �-actin levels in aged SOD2�/� com-pared with aged wild-type SMCs are in agreement with thereport of Qiu et al43 that smooth muscle �-actin is a keydeterminant of vascular SMC stiffness during aging. In-creases in �-actin levels and MMP-2 activity were observedin young SOD2�/� compared with young wild-type SMCs,and yet the aortic stiffening and cardiac dysfunction areevident only in aged SOD2�/� mice, which suggests athreshold for mitochondrial oxidative stress to affect struc-tural and biochemical changes in the SMC and aorta and tocause a phenotypic effect. Our observation that H2O2 levelsare decreased in SOD2�/� SMCs is consistent with similarfindings in SOD2-deficient and knockout mice.56,57 Exoge-nous H2O2 stimulates Akt phosphorylation in many celltypes, including vascular SMCs.44,58 Therefore, it is conceiv-able that low H2O2 levels in aged SOD2�/� SMCs impair cellsurvival and promote apoptosis by downregulating Akt sig-naling and activating FoxO3a.
In summary, our data provide insight into the molecularmechanisms by which increased mitochondrial oxidativestress promotes aortic stiffening associated with aging. Al-tered ROS metabolism in the mitochondria over a lifetime notonly enhances collagen secretion and intrinsic stiffness ofaortic medial SMCs but also affects redox signaling to induceSMC apoptosis, all of which contribute to aortic stiffening. Itwould be worth determining whether strategies aimed atregulating mitochondrial oxidative stress have a therapeuticeffect against aortic stiffening and its pathophysiologicalsequelae.
Sources of FundingThis work was supported by National Institutes of Health GrantsHL-57352 and AG 024282. Dr Zhou is a cardiology fellow sup-ported by National Institutes of Health T32 Training GrantHL083828-04.
DisclosuresNone.
References1. O’Rourke MF, Hashimoto J. Mechanical factors in arterial aging: a
clinical perspective. J Am Coll Cardiol. 2007;50:1–13.2. Laurent S, Boutouyrie P. Recent advances in arterial stiffness and wave
reflection in human hypertension. Hypertension. 2007;49:1202–1206.3. Chue CD, Townend JN, Steeds RP, Ferro CJ. Arterial stiffness in chronic
kidney disease: causes and consequences. Heart. 2010;96:817–823.4. Mitchell GF, Hwang SJ, Vasan RS, Larson MG, Pencina MJ, Hamburg
NM, Vita JA, Levy D, Benjamin EJ. Arterial stiffness and cardiovascularevents: the Framingham Heart Study. Circulation. 2010;121:505–511.
Zhou et al Aortic Stiffening in Aged SOD2�/� Mice 753

35
and elastin provide the structural integrity of the aortic wall,and our results show that increased mitochondrial oxidativestress over a lifetime increases the collagen content andruptures and decreases the elastin in the aorta. The changes inaortic collagen and elastin levels were accompanied byincreased expression and activity of MMP-2 in agedSOD2�/� aortic SMCs. Similar to this, Dasgupta et al48 andothers33 reported both age- and redox-regulated increase inMMP expression and activity. Although the increase inMMP-2 expression and activity should decrease collagenlevels at first glance, increased collagen I levels are observedin SMCs under oxidative stress conditions.49 In fact, anincrease in interstitial and perivascular collagen is ob-served in cardiac MMP-2 transgenic mice.50 Nevertheless,activation of MMP-2 is strongly correlated with elastic fiberfragmentation, disorganization, and increased stiffness of thearterial vasculature.34 Endothelial dysfunction and inflamma-tion may contribute to increased aortic stiffening in agedSOD2�/�, as endothelial dysfunction is increased in apolipo-protein E�/� mice that are deficient in SOD2,21 and proin-flammatory cytokine production is upregulated with in-creased mitochondrial ROS levels.51
Our results showing increased aortic stiffness in agedSOD2�/� mice accompanied by ventricular dysfunction aresupported by several cross-sectional studies that reported apositive association between age-related aortic stiffness andventricular dysfunction.52 Aortic stiffening increases leftventricular afterload by inducing earlier return of reflectedwaves in the late systole and causes left ventricle hypertrophyand ventricular dysfunction. Interestingly, the impairment ofaortic relaxation and increased diastolic blood pressure inSOD2�/� mice precede increased PWV and Doppler abnor-malities in heart function. Furthermore, mitochondrial oxida-tive stress–induced coupling of vascular-ventricular dysfunc-tion is supported by the observation of impaired heartfunction with lifelong reduction of SOD2.47
Increased apoptosis of SMCs in the aortic media andincreased sensitivity to staurosporine-induced apoptosis inaged SOD2�/� mouse SMCs observed in the present inves-tigation are consistent with the concept that medial SMCapoptosis is an important contributor to age-associated vas-cular remodeling and loss of aortic elasticity.35 The propen-sity of aged SOD2�/� aortic SMCs to apoptosis is underlinedby impaired activation of Akt and increased activation ofFoxO3a in response to IGF-1 treatment. Akt is a negativeregulator of FoxO3a transcription factor, which in the ab-sence of Akt-mediated phosphorylation induces the expres-sion of genes involved in apoptosis.53 Interestingly, an in-crease in MMP-2 and MMP-9 activities was observed invascular cells following FoxO3a activation.54 Because theseMMPs do not contain a consensus binding site for forkheadfactors, activated FoxO3a may regulate MMP-2 activityindirectly, including via activation of MMP-3.
Activated MMP-2 induces apoptosis by stimulating JNKactivity, as well as cytochrome c release.41 Inhibition of Aktsignaling has been shown to induce JNK activity and promotethe cleavage of caspase-3 in SMCs.40 JNK activation, in turn,initiates mitochondrial apoptotic pathway via Bax-dependentrelease of cytochrome c.55 Alternatively, aged SOD2�/�
aortic SMCs could undergo apoptosis in the absence ofAkt-mediated phosphorylation of apoptosis regulatory pro-teins Bad and Bax, which suggests that Akt-JNK cross-talk isan important determinant of aged SMC apoptosis.40 Ourobservation that DN-FoxO3a overexpression attenuatescleaved PARP levels is consistent with the regulatory role ofAkt/FoxO3a signaling in aged SOD2�/� aortic SMC apoptosis.
Calcium channel blockers and angiotensin II receptorantagonists are used to treat large artery stiffening.43 Thesedrugs affect vascular SMC tone, which suggests that age-associated vascular stiffening is partly regulated by intrinsicmechanical properties of these cells. Our data showingsignificantly increased �-actin levels in aged SOD2�/� com-pared with aged wild-type SMCs are in agreement with thereport of Qiu et al43 that smooth muscle �-actin is a keydeterminant of vascular SMC stiffness during aging. In-creases in �-actin levels and MMP-2 activity were observedin young SOD2�/� compared with young wild-type SMCs,and yet the aortic stiffening and cardiac dysfunction areevident only in aged SOD2�/� mice, which suggests athreshold for mitochondrial oxidative stress to affect struc-tural and biochemical changes in the SMC and aorta and tocause a phenotypic effect. Our observation that H2O2 levelsare decreased in SOD2�/� SMCs is consistent with similarfindings in SOD2-deficient and knockout mice.56,57 Exoge-nous H2O2 stimulates Akt phosphorylation in many celltypes, including vascular SMCs.44,58 Therefore, it is conceiv-able that low H2O2 levels in aged SOD2�/� SMCs impair cellsurvival and promote apoptosis by downregulating Akt sig-naling and activating FoxO3a.
In summary, our data provide insight into the molecularmechanisms by which increased mitochondrial oxidativestress promotes aortic stiffening associated with aging. Al-tered ROS metabolism in the mitochondria over a lifetime notonly enhances collagen secretion and intrinsic stiffness ofaortic medial SMCs but also affects redox signaling to induceSMC apoptosis, all of which contribute to aortic stiffening. Itwould be worth determining whether strategies aimed atregulating mitochondrial oxidative stress have a therapeuticeffect against aortic stiffening and its pathophysiologicalsequelae.
Sources of FundingThis work was supported by National Institutes of Health GrantsHL-57352 and AG 024282. Dr Zhou is a cardiology fellow sup-ported by National Institutes of Health T32 Training GrantHL083828-04.
DisclosuresNone.
References1. O’Rourke MF, Hashimoto J. Mechanical factors in arterial aging: a
clinical perspective. J Am Coll Cardiol. 2007;50:1–13.2. Laurent S, Boutouyrie P. Recent advances in arterial stiffness and wave
reflection in human hypertension. Hypertension. 2007;49:1202–1206.3. Chue CD, Townend JN, Steeds RP, Ferro CJ. Arterial stiffness in chronic
kidney disease: causes and consequences. Heart. 2010;96:817–823.4. Mitchell GF, Hwang SJ, Vasan RS, Larson MG, Pencina MJ, Hamburg
NM, Vita JA, Levy D, Benjamin EJ. Arterial stiffness and cardiovascularevents: the Framingham Heart Study. Circulation. 2010;121:505–511.
Zhou et al Aortic Stiffening in Aged SOD2�/� Mice 753
5. Griendling KK, FitzGerald GA. Oxidative stress and cardiovascular inju-ry: part II: animal and human studies. Circulation. 2003;108:2034–2040.
6. Sindler AL, Fleenor BS, Calvert JW, Marshall KD, Zigler ML, Lefer DJ,Seals DR. Nitrite supplementation reverses vascular endothelial dys-function and large elastic artery stiffness with aging. Aging Cell. 2011;10:429–437.
7. Heitzer T, Schlinzig T, Krohn K, Meinertz T, Munzel T. Endothelialdysfunction, oxidative stress, and risk of cardiovascular events in patientswith coronary artery disease. Circulation. 2001;104:2673–2678.
8. Harman D. Aging: a theory based on free radical and radiation chemistry.J Gerontol. 1956;11:298–300.
9. Delles C, Zimmerli LU, McGrane DJ, Koh-Tan CH, Pathi VL, McKayAJ, Steedman T, Dargie HJ, Hamilton CA, Dominiczak AF. Vascularstiffness is related to superoxide generation in the vessel wall.J Hypertens. 2008;26:946–955.
10. Noma K, Goto C, Nishioka K, Jitsuiki D, Umemura T, Ueda K, KimuraM, Nakagawa K, Oshima T, Chayama K, Yoshizumi M, Liao JK, HigashiY. Roles of rho-associated kinase and oxidative stress in the pathogenesisof aortic stiffness. J Am Coll Cardiol. 2007;49:698–705.
11. Edgar D, Shabalina I, Camara Y, Wredenberg A, Calvaruso MA,Nijtmans L, Nedergaard J, Cannon B, Larsson NG, Trifunovic A.Random point mutations with major effects on protein-coding genes arethe driving force behind premature aging in mtDNA mutator mice. CellMetab. 2009;10:131–138.
12. Doughan AK, Harrison DG, Dikalov SI. Molecular mechanisms of an-giotensin II-mediated mitochondrial dysfunction: linking mitochondrialoxidative damage and vascular endothelial dysfunction. Circ Res. 2008;102:488–496.
13. Kinouchi H, Epstein CJ, Mizui T, Carlson E, Chen SF, Chan PH. Atten-uation of focal cerebral ischemic injury in transgenic mice overexpressingCuZn superoxide dismutase. Proc Natl Acad Sci U S A. 1991;88:11158–11162.
14. Harrison CM, Pompilius M, Pinkerton KE, Ballinger SW. Mitochondrialoxidative stress significantly influences atherogenic risk and cytokine-in-duced oxidant production. Environ Health Perspect. 2011;119:676–681.
15. Van Deel ED, Lu Z, Xu X, Zhu G, Hu X, Oury TD, Bache RJ, DunckerDJ, Chen Y. Extracellular superoxide dismutase protects the heart againstoxidative stress and hypertrophy after myocardial infarction. Free RadicBiol Med. 2008;44:1305–1313.
16. Li Y, Huang TT, Carlson EJ, Melov S, Ursell PC, Olson JL, Noble LJ,Yoshimura MP, Berger C, Chan PH, Wallace DC, Epstein CJ. Dilatedcardiomyopathy and neonatal lethality in mutant mice lacking manganesesuperoxide dismutase. Nat Genet. 1995;11:376–381.
17. Lebovitz RM, Zhang H, Vogel H, Cartwright J Jr, Dionne L, Lu N, HuangS, Matzuk MM. Neurodegeneration, myocardial injury, and perinataldeath in mitochondrial superoxide dismutase-deficient mice. Proc NatlAcad Sci U S A. 1996;93:9782–9787.
18. Van Remmen H, Williams MD, Guo Z, Estlack L, Yang H, Carlson EJ,Epstein CJ, Huang TT, Richardson A. Knockout mice heterozygous forSod2 show alterations in cardiac mitochondrial function and apoptosis.Am J Physiol Heart Circ Physiol. 2001;281:H1422–H1432.
19. Williams MD, Van Remmen H, Conrad CC, Huang TT, Epstein CJ,Richardson A. Increased oxidative damage is correlated to altered mito-chondrial function in heterozygous manganese superoxide dismutaseknockout mice. J Biol Chem. 1998;273:28510–28515.
20. Ballinger SW, Patterson C, Knight-Lozano CA, Burow DL, Conklin CA,Hu Z, Reuf J, Horaist C, Lebovitz R, Hunter GC, McIntyre K, Runge MS.Mitochondrial integrity and function in atherogenesis. Circulation. 2002;106:544–549.
21. Ohashi M, Runge MS, Faraci FM, Heistad DD. MnSOD deficiencyincreases endothelial dysfunction in ApoE-deficient mice. ArteriosclerThromb Vasc Biol. 2006;26:2331–2336.
22. Li M, Chiu JF, Mossman BT, Fukagawa NK. Down-regulation ofmanganese-superoxide dismutase through phosphorylation of FOXO3aby Akt in explanted vascular actin muscle cells from old rats. J BiolChem. 2006;281:40429–40439.
23. Hartley CJ, Taffet GE, Michael LH, Pham TT, Entman ML. Noninvasivedetermination of pulse-wave velocity in mice. Am J Physiol. 1997;273:H494–H500.
24. Moon SK, Thompson LJ, Madamanchi N, Ballinger S, PapaconstantinouJ, Horaist C, Runge M. S., Patterson C. Aging, oxidative responses, andproliferative capacity in cultured mouse aortic smooth muscle cells. Am JPhysiol Heart Circ Physiol. 2001;280:H2779–H2788.
25. Zhou RH, Pesant S, Cohn HI, Eckhart AD. Enhanced sterol responseelement-binding protein in postintervention restenotic blood vessels plays
an important role in vascular smooth muscle proliferation. Life Sci.2008;82:174–181.
26. Madamanchi NR, Li S, Patterson C, Runge MS. Thrombin regulatesvascular smooth muscle cell growth and heat shock proteins via theJAK-STAT pathway. J Biol Chem. 2001;276:18915–18924.
27. Pfaffl MW, Horgan GW, Dempfle L. Relative expression software tool(REST) for groupwise comparison and statistical analysis of relativeexpression results in real-time PCR. Nucleic Acids Res. 2002;30:e36.
28. Skurk C, Maatz H, Kim HS, Yang J, Abid MR, Aird WC, Walsh K. TheAkt-regulated forkhead transcription factor FOXO3a controls endothelialcell viability through modulation of the caspase-8 inhibitor FLIP. J BiolChem. 2004;279:1513–1525.
29. Nurnberger J, Dammer S, Opazo Saez A, Philipp T, Schafers RF. Dia-stolic blood pressure is an important determinant of augmentation indexand pulse wave velocity in young, healthy males. J Hum Hypertens.2003;17:153–158.
30. Osborne-Pellegrin M, Labat C, Mercier N, Challande P, Lacolley P.Changes in aortic stiffness related to elastic fiber network anomalies inthe Brown Norway rat during maturation and aging. Am J Physiol HeartCirc Physiol. 2010;299:H144–H152.
31. Steed MM, Tyagi N, Sen U, Schuschke DA, Joshua IG, Tyagi SC.Functional consequences of the collagen/elastin switch in vascularremodeling in hyperhomocysteinemic wild-type, eNOS�/�, andiNOS�/� mice. Am J Physiol Lung Cell Mol Physiol. 2010;299:L301–L311.
32. Ng K, Hildreth CM, Phillips JK, Avolio AP. Aortic stiffness is associatedwith vascular calcification and remodeling in a chronic kidney disease ratmodel. Am J Physiol Renal Physiol. 2011;300:F1431–F1436.
33. Wang M, Lakatta EG. Altered regulation of matrix metalloproteinase-2 inaortic remodeling during aging. Hypertension. 2002;39:865–873.
34. Chung AW, Yang HH, Kim JM, Sigrist MK, Chum E, Gourlay WA,Levin A. Upregulation of matrix metalloproteinase-2 in the arterial vas-culature contributes to stiffening and vasomotor dysfunction in patientswith chronic kidney disease. Circulation. 2009;120:792–801.
35. Sawabe M. Vascular aging: from molecular mechanism to clinical sig-nificance. Geriatr Gerontol Int. 2010;10(suppl. 1):S213–S220.
36. Tchivilev I, Madamanchi NR, Vendrov AE, Niu XL, Runge MS. Iden-tification of a protective role for protein phosphatase 1c�1 against oxi-dative stress-induced vascular smooth muscle cell apoptosis. J Biol Chem.2008;283:22193–22205.
37. Lee JY, Jung GY, Heo HJ, Yun MR, Park JY, Bae SS, Hong KW, LeeWS, Kim CD. 4-Hydroxynonenal induces vascular smooth muscle cellapoptosis through mitochondrial generation of reactive oxygen species.Toxicol Lett. 2006;166:212–221.
38. Miyamoto S, Murphy AN, Brown JH. Akt mediated mitochondrial pro-tection in the heart: metabolic and survival pathways to the rescue.J Bioenerg Biomembr. 2009;41:169–180.
39. Park KW, Kim DH, You HJ, Sir JJ, Jeon SI, Youn SW, Yang HM, SkurkC, Park YB, Walsh K, Kim HS. Activated forkhead transcription factorinhibits neointimal hyperplasia after angioplasty through induction ofp27. Arterioscler Thromb Vasc Biol. 2005;25:742–747.
40. Suhara T, Kim HS, Kirshenbaum LA, Walsh K. Suppression of Aktsignaling induces Fas ligand expression: involvement of caspase and Junkinase activation in Akt-mediated Fas ligand regulation. Mol Cell Biol.2002;22:680–691.
41. Menon B, Singh M, Ross RS, Johnson JN, Singh K. �-Adrenergicreceptor-stimulated apoptosis in adult cardiac myocytes involves MMP-2-mediated disruption of �1 integrin signaling and mitochondrialpathway. Am J Physiol Cell Physiol. 2006;290:C254–C261.
42. Csiszar A, Wang M, Lakatta EG, Ungvari Z. Inflammation and endothe-lial dysfunction during aging: role of NF-�B. J Appl Physiol. 2008;105:1333–1341.
43. Qiu H, Zhu Y, Sun Z, Trzeciakowski JP, Gansner M, Depre C, ResuelloRR, Natividad FF, Hunter WC, Genin GM, Elson EL, Vatner DE,Meininger GA, Vatner SF. Short communication: vascular smoothmuscle cell stiffness as a mechanism for increased aortic stiffness withaging. Circ Res. 2010;107:615–619.
44. Sadidi M, Lentz SI, Feldman EL. Hydrogen peroxide-induced Akt phos-phorylation regulates Bax activation. Biochimie. 2009;91:577–585.
45. Hill MM, Andjelkovic M, Brazil DP, Ferrari S, Fabbro D, Hemmings BA.Insulin-stimulated protein kinase B phosphorylation on Ser-473 is inde-pendent of its activity and occurs through a staurosporine-insensitivekinase. J Biol Chem. 2001;276:25643–25646.
754

36
46. Zhang Y, Xing D. PUMA promotes Bax activation in a FoxO3a-dependent manner in STS-induced apoptosis. J Innov Opt Health Sci.2010;3:31–38.
47. Loch T, Vakhrusheva O, Piotrowska I, Ziolkowski W, Ebelt H, Braun T,Bober E. Different extent of cardiac malfunction and resistance to oxi-dative stress in heterozygous and homozygous manganese-dependentsuperoxide dismutase-mutant mice. Cardiovasc Res. 2009;82:448–457.
48. Dasgupta J, Kar S, Van Remmen H, Melendez JA. Age-dependentincreases in interstitial collagenase and MAP kinase levels are exac-erbated by superoxide dismutase deficiencies. Exp Gerontol. 2009;44:503–510.
49. Bachem MG, Wendelin D, Schneiderhan W, Haug C, Zorn U, Gross HJ,Schmid-Kotsas A, Grunert A. Depending on their concentration oxidizedlow density lipoproteins stimulate extracellular matrix synthesis or induceapoptosis in human coronary artery smooth muscle cells. Clin Chem LabMed. 1999;37:319–326.
50. Bergman MR, Teerlink JR, Mahimkar R, Li L, Zhu BQ, Nguyen A, DahiS, Karliner JS, Lovett DH. Cardiac matrix metalloproteinase-2 expressionindependently induces marked ventricular remodeling and systolic dys-function. Am J Physiol Heart Circ Physiol. 2007;292:H1847–H1860.
51. Bulua AC, Simon A, Maddipati R, Pelletier M, Park H, Kim KY, SackMN, Kastner DL, Siegel RM. Mitochondrial reactive oxygen speciespromote production of proinflammatory cytokines and are elevated inTNFR1-associated periodic syndrome (TRAPS). J Exp Med. 2011;208:519–533.
52. Abhayaratna WP, Srikusalanukul W, Budge MM. Aortic stiffness for thedetection of preclinical left ventricular diastolic dysfunction: pulse wavevelocity versus pulse pressure. J Hypertens. 2008;26:758–764.
53. Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ,Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival byphosphorylating and inhibiting a Forkhead transcription factor. Cell.1999;96:857–868.
54. Lee HY, You HJ, Won JY, Youn SW, Cho HJ, Park KW, Park WY, SeoJS, Park YB, Walsh K, Oh BH, Kim HS. Forkhead factor, FOXO3a,induces apoptosis of endothelial cells through activation of matrix met-alloproteinases. Arterioscler Thromb Vasc Biol. 2008;28:302–308.
55. Papadakis ES, Finegan KG, Wang X, Robinson AC, Guo C, Kayahara M,Tournier C. The regulation of Bax by c-Jun N-terminal protein kinase(JNK) is a prerequisite to the mitochondrial-induced apoptotic pathway.FEBS Lett. 2006;580:1320–1326.
56. Lee S, Van Remmen H, Csete M. Sod2 overexpression preservesmyoblast mitochondrial mass and function, but not muscle mass withaging. Aging Cell. 2009;8:296–310.
57. Morten KJ, Ackrell BA, Melov S. Mitochondrial reactive oxygen speciesin mice lacking superoxide dismutase 2: attenuation via antioxidanttreatment. J Biol Chem. 2006;281:3354–3359.
58. Ushio-Fukai M, Alexander RW, Akers M, Yin Q, Fujio Y, Walsh K,Griendling KK. Reactive oxygen species mediate the activation of Akt/protein kinase B by angiotensin II in vascular smooth muscle cells. J BiolChem. 1999;274:22699–22704.
Zhou et al Aortic Stiffening in Aged SOD2�/� Mice 755


















