
Microsoft Word - Guion final MyN Oct 2010.doc - uam.es practic… · Web viewMicrosoft Word -...
29
METABOLISMO GUIÓN DE PRÁCTICAS 2º Curso Página 1
-
Upload
truongdieu -
Category
Documents
-
view
216 -
download
0
Transcript of Microsoft Word - Guion final MyN Oct 2010.doc - uam.es practic… · Web viewMicrosoft Word -...

METABOLISMOGUIÓN DE PRÁCTICAS
2º CursoCrado en Ciencias de la AlimentaciónCurso 2011-2012Profesores: Beatriz Pardo y Félix Hernández
Dpto de Biología MolecularFacultad de CienciasUAM
Página 1

NORMAS DE LABORATORIOOrganización:
1 Las prácticas se realizan por pareja, pero cada alumno entregará una ficha con sus datos a lo/a(s) profesore/a(s). 2 Habrá una explicación previa cada día por parte de los profesores 3 Es importante saber que se está haciendo en cada momento y para qué, si existen dudas hay que preguntar. Concentración. 4 Es importante el compañerismo dentro del laboratorio
Antes de realizar las prácticas:
1 Estudiar el guión que corresponde antes del comienzo de cada práctica. 2 Se pasará lista para comprobar la asistencia a las clases (siendo obligatoria la aistencia para aprobar la asignatura), por lo que se ruega puntualidad
Durante las prácticas:
1 Es obligatorio del uso de bata dentro del laboratorio. 2 Utilizar gafas de protección cuando sea necesario y se indique por los profesores. 3 Segregar correctamente los residuos 4 Está completamente prohibido comer y beber en los laboratorios así como la utilización de móviles durante el desarrollo de las prácticas 5 Está prohibido pipetear con la boca 6 El puesto de trabajo ha de mantenerse limpio durante el desarrollo de las prácticas y limpiarlo completamente al finalizar
Después de la realización de las prácticas:
1 Dejar el puesto de trabajo limpio y ordenado 2 Controlar el material del que sois responsables (pipetas, rotuladores, etc.) 3 Limpieza del material utilizado durante la práctica 4 Limpieza de las zonas comunes (centrifugas, baños, pilas, balanzas, etc) 5 Completar el cuaderno de prácticas para entregarlo al final de las mismas cuando los profesores lo indiquen.
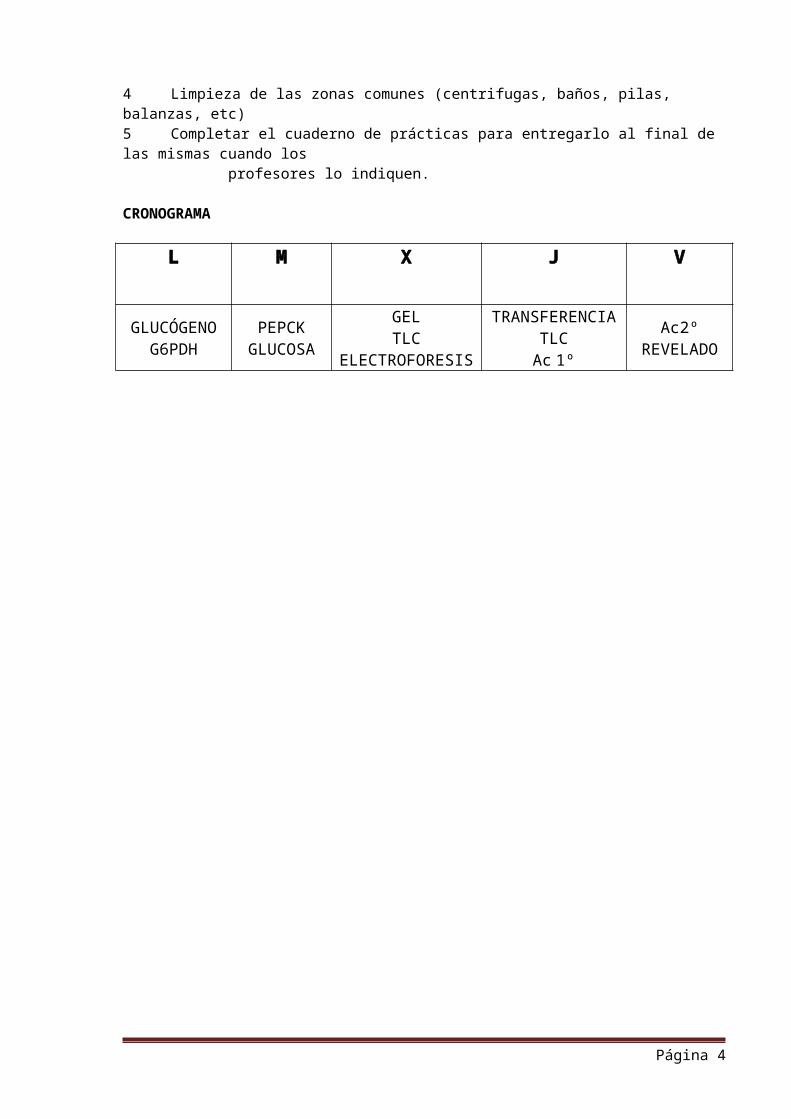
CRONOGRAMA
L M X J V
GLUCÓGENO PEPCK GEL TRANSFERENCIA Ac2º
Página 2

G6PDH GLUCOSA TLCELECTROFORESIS
TLCAc 1º REVELADO
Página 3
PRÁCTICA I: DETECCIÓN DE AMINOÁCIDOS POR CROMATOGRAFÍA EN CAPA FINA (TLC)
INTRODUCCIÓN
Las enfermedades metabólicas hereditarias son trastornos bioquímicos de origen genético debidos a un defecto específico en la estructura o función de las moléculas de proteínas. El origen es siempre una modificación en el gen que codifica la síntesis de una determinada proteína. La proteína deficiente puede afectar el funcionamiento de una vía metabólica cualquiera, produciéndose un aumento de los metabolitos anteriores a dicho bloqueo, que dependiendo de su toxicidad afectarán en mayor o menor grado a la severidad de la enfermedad. Estos metabolitos acumulados en fluidos biológicos (sangre y orina principalmente), sirven en la mayoría de los casos para el diagnóstico de la enfermedad.
La cromatografía en capa fina es uno de los métodos más utilizados para el diagnóstico de las enfermedades metabólicas dada su versatilidad en la identificación de metabolitos tales como aminoácidos, azúcares, nucleótidos, ácidos grasos etc.
La cromatografía en capa fina es un caso particular de cromatografía de reparto, donde la fase estacionaria (agua) se adsorbe en el soporte sólido que constituye la capa fina y que podrá ser cualquier material que pueda reducirse a polvo y formar una capa uniforme (por ejemplo celulosa). La fase móvil (disolvente orgánico saturado en agua) fluye a través de la fase estacionaria. Los componentes de la mezcla se separarán si los coeficientes de reparto (K) entre los disolventes son lo suficientemente distintos, denominándose a la movilidad de un compuesto respecto a la del disolvente “Rf”. Estos valores dependen de la naturaleza del compuesto, del soporte del disolvente y de la temperatura.
Una de las aplicaciones más extendida, es su utilización como método de selección masiva de fenilcetonuria, así como de cualquier aminoacidopatía. La detección de aminoácidos se puede realizar tanto en orina como en sangre o suero impregnado en papel. Entre los errores hereditarios del metabolismo que se pueden diagnosticar se encuentran: la Hiperglicinemia, Metioninemia, Sindrome de Fanconi, Tirosinemia y Jarabe de Arce entre otros. También es posible la detección de algunas acidurias orgánicas que impliquen la elevación de forma secundaria de algún aminoácido. Este es el caso de la Acidemia Propiónica en la cual los pacientes presentan niveles elevados de Gly en orina.
Otra aplicación de la técnica es el control bioquímico del tratamiento de los fenilcetonúricos mediante medida de la fenilalanina en sangre impregnada en papel. En este caso se utilizan patrones con diferentes concentraciones de fenilalanina.
OBJETIVOS
Identificación de una mezcla de aminoácidos por cromatografía en capa fina (TLC) utilizando aminoácidos patrón.
MATERIALES Y REACTIVOS
-Aminoácidos patrón (Fenilalanina, Tirosina, Leucina, Isoleucina, Glutámico, Prolina Glicina y Valina) -Cubetas cromatográficas. -Estufa de 120ºC -Cromatografía: n-butanol: acetona: acido acético: agua (35:35:10:20) -Muestras problema (A y B) -Solución de tinción: 200 mg ninhidrina en 100 ml etanol 70%.
Página 4

MÉTODO
EMPLEAR GUANTES DURANTE TODA LA PRÁCTICA
Aplicación de las muestras:Sobre una cromatoplaca de celulosa aplicar 2 µl de las disoluciones patrón de aminoácidos, así como de las muestras problema. Todas las muestras deben ser aplicadas a 2 cm del borde inferior de la cromatoplaca y distantes entre sí aproximadamente 2 cm. Marcar los puntos de aplicación con lápiz.
Desarrollo cromatográfico:
- Se introduce la placa en el eluyente que constituye la fase móvil (n-butanol, acetona, acido acético, agua, 35:35:10:20) y se deja correr de forma ascendente hasta que el frente llegue al borde superior de la placa (dos veces consecutivas es recomendable para una mejor separación cromatográfica dejando secar la placa entre ambos desarrollos).
- Para obtener Rf reproducibles es imprescindible tener la atmósfera del tanque saturada con la fase móvil, ya que en caso contrario puede evaporarse disolvente de la capa fina, aumentando los Rf.
Revelado: - Terminado el desarrollo se seca la placa con aire frío hasta la eliminación total de
los disolventes. - Se tiñe por inmersión de la placa en la solución de ninhidrina durante 1 minuto
aproximadamente y se seca en una estufa a 120°C hasta la aparición de las zonas teñidas (aproximadamente 5 min).
RESULTADOS
Una vez visualizadas las manchas:
1. Fotografiar la cromatoplaca2. Calcular los Rf de aminoácidos patrón y los problema. Anotar también la forma de las manchas, el color y su intensidad.3. Identificar los aminoácidos de las muestras problema.
Página 5

PRACTICA II: ANÁLISIS DE LA ADAPTACIÓN DEL METABOLISMO GLUCÍDICO HEPÁTICO EN RESPUESTA AL AYUNO/REALIMENTACIÓN Y DIABETES
INTRODUCCIÓN
Las principales rutas del metabolismo glucídico empiezan o terminan en la glucosa.
La glucosa es un compuesto orgánico muy abundante en la naturaleza que es utilizado por el organismo como fuente de energía. Además de energía, el metabolismo de la glucosa proporciona productos intermedios para otras vías metabólicas. Por tanto, un aporte de glucosa constante es esencial para el mantenimiento de las funciones específicas de los distintos órganos y vendrá determinado por el volumen y la composición de la ingesta así como por la capacidad de almacenamiento y regulación de algunos tejidos. En este sentido el hígado tiene un papel regulador esencial ya que es el órgano que recibe y procesa las sustancias nutritivas procedentes de la ingesta y que, junto con el tejido adiposo, es capaz de almacenar sustancias de reserva en forma de triglicéridos y glucógeno. La coordinación de las funciones de estos órganos se lleva a cabo mediante finos sistemas de regulación hormonal. Todos estos mecanismos, en su conjunto, permiten una adecuación del organismo a las distintas condiciones metabólicas.
La glucosa puede seguir en el hígado tres caminos principales:
1.- Síntesis de glucógeno cuya función será proveer una reserva de carbohidratos para mantener los niveles plasmáticos de glucosa cuando sea necesario. 2.- Glucolisis, que tiene una doble función: provisión de piruvato para su oxidación en la mitocondria y obtención de energía y formación de precursores para procesos biosintéticos (lipogénesis). 3.- Ciclo de las pentosas fosfato para la generación de poder reductor en forma de NADPH,necesario para la síntesis de ácidos grasos y esteroides, así como pentosas fosfato para lasíntesis de ácidos nucleicos.
Por tanto, tras la ingesta:
se elevan los niveles de glucosa en sangre se sintetiza glucógeno se favorece la síntesis de ácidos grasos, acumulándose en forma de triglicéridos
Página 6

en el tejido adiposo
Todos estos procesos están dirigidos por una razón Insulina/Glucagón elevada que actúa directamente favoreciendo la glucogenosíntesis y la lipogénesis.
En situaciones de ayuno más o menos prolongado:
se movilizan las reservas de glucosa acumuladas en forma de glucógeno tanto en hígado como en músculo.
se degradan los triglicéridos almacenados en el tejido adiposo que darán lugar a ácidos grasos que a su vez actuarán como sustratos energéticos alternativos
se sintetiza glucosa por gluconeogénesis a partir de metabolitos intermediarios. La escasez de glucosa característica de este estado provoca una inversión de la razón Insulina/Glucagón que dispara la cascada de reajustes metabólicos.
Todos estos procesos están regulados hormonalmente por las siguientes hormonas: insulina y glucagón. La insulina actúa favoreciendo la glucogenogénesis y la lipogénesis. Por su parte, el glucagón estimula el efecto contrario es decir, la producción de glucosa a partir de glucógeno. Al producirse la realimentación en un organismo ayunado vuelve a elevarse la razón insulina/glucagón con lo que se reestablecen los parámetros metabólicos originales.
Finalmente, hay situaciones en las que la razón insulina/glucagón está alterada en situaciones patológicas como es el caso de la Diabetes Mellitus cuyo origen es la deficiencia de Insulina. En una situación de diabetes, los bajos niveles de insulina originan una disminución en la síntesis de enzimas claves para la glucólisis y por tanto disminuye la glucólisis. Entonces, en situaciones de diabetes:
aumentan los niveles plasmáticos de glucosa. se moviliza el glucógeno para la provisión de glucosa al interior de la célula. se activa la gluconeogénesis con el mismo fin. se movilizan las reservas lipídicas para dar lugar a ácidos grasos que actúen como
sustratos energéticos ante la imposibilidad de utilización de la glucosa por los tejidos periféricos (músculo esquelético y tejido adiposo).
se producirá proteolisis cuando se agoten las reservas lipídicas.
OBJETIVOS
Analizar el efecto de la dieta y de la carencia de insulina sobre varios parámetros metabólicos en rata.
En concreto se van a utilizar 4 grupos de ratas (control, sometidas a ayuno, sometidas a ayuno y posteriormente realimentadas y diabéticas). Los parámetros metabólicos que vamos a utilizar son:
1) actividad de la glucosa-6 P deshidrogenasa en hígado (G6PDH) 2) actividad de la fosfoenol piruvato carboxiquinasa en hígado (PEPCK) 3) glucógeno hepático 4) glucosa sanguínea
MATERIALES Y MÉTODOS
1. Tratamiento de los animales
Página 7

Se utilizan ratas macho (250-300 g) que tienen en todo momento libre acceso a la bebida. Estos animales de dividen en 4 grupos:
1) CONTROLES (C): disfrutan de libre acceso a una dieta comercial estándar rica en hidrato de carbono.2) AYUNADAS (A): 48 horas antes del sacrificio se les retira el alimento. 3) AYUNADAS y REALIMENTADAS (AR): se les retira el alimento durante 48 horas. Posteriormente se les alimenta durante 24 horas con su dieta normal y un suplemento en el agua de glucosa 20mM. 4) DIABÉTICAS (D): La diabetes se induce mediante la inyección por vía intraperitoneal de estreptozotocina a una dosis de 65 mg/kg peso. La estreptozotocina destruye selectivamente las células ß del páncreas, encargadas de la producción de insulina. Los animales tienen en todo momento acceso a la comida.
2. Preparación de las muestras2.1. Plasma: Se tomarán muestras de sangre de los distintos lotes de animales. Con objeto de obtener plasma se ajusta el volumen de sangre a 0.1% de heparina. Después de tener 30 minutos las muestras en hielo, se centrifugan 10 min x 10,000 rpm en un rotor SS34 a 4ºC. Los plasmas son aspirados con una pipeta pasteur, alicuoteados en fracciones de 1 ml, rotulados y congelados a -20ºC hasta el momento de emplearlos en la determinación de glucosa.2.2. Actividades enzimáticas: tanto la PEPCK como la G6PDH son enzimas citosólicas. Se hará un extracto hepático 1:10 (p:v) en un tampón 50 mM Tris/HCl, pH 7.4; 50 mM KCl; 1 mM DTT; 0.25 M sacarosa. Para ello se extraen los hígados, se lavan en solución salina y se trocean. A continuación se homogeneiza en el tampón descrito (2.5 g en 25 ml) y se centrifugan a 15.000 rpm durante 1 hora a 4ºC. Los sobrenadantes se guardan en fracciones de 2 ml debidamente rotulados y se congelan a -20ºC para la determinación de PEPCK y G6PDH en los días posteriores.2.3. Glucógeno: se harán extractos de hígado 1:10 (p/v) en HClO4 al 2%. Los extractos se mantienen en agitación durante 24 h a 4ºC con objeto de desnaturalizar todas las proteínas y solubilizar el glucógeno. Después de centrifugar 10 min a 10.000 rpm a 4ºC, el sobrenadante se guarda en fracciones de 2 ml congelado a -20ºC.
Contamos con muestras de PLASMA, FRACCIÓN CITOSÓLICA DE HÍGADO y GLUCÓGENO de todos lois grupos de animales. Todas estas muestras serán preparadas por los profesores.
1.-DETERMINACIÓN DE LA ACTIVIDAD G6PDH EN EXTRACTO HEPÁTICO
La glucosa-6-P deshidrogenasa (G6PDH) es la enzima que cataliza una de las reacciones de la vía de las pentosas fosfato. Como se ha indicado anteriormente en este guión, el ciclo de las pentosas fosfato genera poder reductor en forma de NADPH necesario para la síntesis de ácidos grasos y esteroides, así como pentosas fosfato para la síntesis de ácidos nucleicos.
Cuando hay síntesis de ácidos grasos hay niveles altos de G6PDH y por tanto de NADPH. La determinación de la actividad G6PDH se realizará mediante la medida espectrofotométrica de la aparición de NADPH a 340 nm (el NADP+ no absorbe a 340 nm, mientras que el NADPH sí lo hace). Por tanto la actividad G6PDH viene dada por los nmoles
Página 8

de NADPH/min formados.
Protocolo: MANTENER TODAS LAS MUESTRAS EN HIELO DURANTE LA PRÁCTICA
1.- preparar 2 cubetas de espectrofotómetro: una para el blanco y otra para la muestra. 2.- añadir 2,8 ml de TAMPÓN A (0,1 M Trietanolamina pH 7,4, 2,5 mM MgCl2, 0,26 mM NADP) a cada cubeta.3.- añadir 0,2 ml de extracto de la fracción citosólica de la muestra problema a cada cubeta. 4.- añadir a la cubeta blanco 0,1 ml de H2O, ajustar el espectrofotómetro (este va a ser el tiempo cero, t=0). Mantener los tubos en incubación en baño a 37º C y realizar una medida cada minuto durante 10 minutos.. 5.- añadir a la segunda cubeta 0,1 ml de glucosa-6P (66mM) y agitar (tapar la cubeta conparafilm). Medir la absorbancia a t=0. Mantener los tubos a 37º C y realizar una medida cadaminuto durante 10 minutos. (t=1, t=2,…,t=10). 5.- Repetir con las demás muestras
Cálculo de la actividad enzimática:
La actividad enzimática es universalmente representada por las Unidades Internacionales. 1.0 Unidad de enzima se define como la cantidad de enzima que transforma 1 µmol de sustrato por minuto a 25 ºC. Así el término actividad se refiere a las unidades totales de enzima en solución. Cuando la actividad se refiere a la cantidad de proteína o gramo de tejido se denomina actividad específica. La actividad específica es el número de unidades
Página 9

de enzima por miligramo de proteína total, siendo una medida de la pureza de la proteína. Las unidades en las que se expresa la actividad específica de una enzima son: µmol de sustrato transformado/min/mg de proteína a una temperatura y pH determinado. Pero también puede reflejarse como cambio de incremento de la DO por minuto para expresar una actividad relativa.
- Representar en una gráfica Abs (DO) frente a tiempo (min) las rectas de la muestra blanco y problema. - Determinar la pendiente de ambas rectas. - Sustraer a la pendiente de la recta problema, la pendiente de la recta blanco. ∆ DO/min específica = ∆ DO/min total(problema) -∆ DO/min inespecífica (blanco) Teniendo en cuenta la ecuación: DO=c x ε x l donde: c= concentración de NADPH; ε = coeficiente de extinción molar (el NADPH tiene un ε a 340 nm de 6.22 x 106 cm2 mol-1); l longitud del paso de luz (1 cm).
Resultados1. Representación de absorbancia en función del tiempo para cada una de las muestras determinadas.2. Calcular la actividad G6PDH de cada muestra en µmol/min/mg de proteína, sabiendo que las muestras utilizadas se encuentran a una concentración de 1 mg/ml de proteína utilizando albúmina de suero de vaca como patrón.3. Explica como prepararías 40 ml de la mezcla de reacción y 1 ml de 66 mM de βD-glucosa-6P.4. ¿A qué concentración de βD-glucosa-6P y NADP se realiza el ensayo?
2.-DETERMINACIÓN DE LA ACTIVIDAD PEPCK EN EXTRACTO HEPÁTICO
El fosfoenolpiruvato (PEP) está implicado en la gluconeogénesis, es decir, en la producción de glucosa a partir de fuentes no hidrocarbonadas. Como se ha explicado anteriormente, durante un periodo de ayuno, la glucosa tiene que formarse a partir de sustratos alternativos con lo que se consigue mantener su concentración en la sangre.
En esta práctica se va a utilizar un sistema enzimático en el que la PEPCK endógena se acopla a la enzima Malato deshidrogenasa exógena. La mezcla de reacción (tampón B) contiene IDP, CO3H, NADH y MDH en exceso de tal forma que la reacción total está únicamente limitada por la actividad PEPCK. La actividad de la PEPCK se determina en base a la velocidad de desaparición de NADH.
Protocolo: MANTENER TODAS LAS MUESTRAS EN HIELO DURANTE LA PRÁCTICA
1.- preparar 2 cubetas de espectrofotómetro: una para el blanco y otra para la muestra. 2.- ajustar a cero el espectrofotómetro con una muestra de agua y NO volver a ajustarlo 3.- añadir 2,8 ml de TAMPÓN B (36 mM Tris-ClH pH 7,4, 1,18 mM MnCl2, 17,85 mM Na CO3H, 1,1 mM glutation reducido, 1,18 mM IDP, 0,14 mM NADH) a cada cubeta.4.- añadir 0,1 ml de extracto de la fracción citosólica de la muestra problema a cada cubeta. 5.- añadir 0,1ml de enzima MDH (1600U/litro). 6.- añadir a la cubeta blanco 0,2 ml de H2O (este va a ser tiempo cero, t=0), ANOTAR LA D O (debe estar en torno a 0.5-0.7; NO AJUSTAR A CERO). Mantener los tubos a 37º C.y
Página 10

medir la absorbancia cada minuto durante un periodo de 10 minutos (t=1, t=2,…,t=10). 7.- añadir a la segunda cubeta 0,2 ml de PEP (16,5 mM). Medir la absorbancia a t=0 y cadaminuto durante un periodo de 10 minutos (t=1, t=2,…,t=10). 8.- repetir con las demás muestras.
Cálculo de la actividad enzimática: Representar en una gráfica Abs frente a tiempo (min) la recta de la muestra blanco y la muestra problema. Determinar la pendiente de ambas rectas. Sustraer a la pendiente de la recta problema la pendiente de la recta control
∆ DO/min específica = ∆ DO/min total(problema) -∆ DO/min inespecífica (blanco)
Teniendo en cuenta la ecuación: DO=c x ε x l
donde: c= concentración de NADPH; ε = coeficiente de extinción molar (el NADPH tiene un ε a 340 nm de 6.22 x 106 cm2 mol-1); l longitud del paso de luz (1 cm).
Resultados1. Representación de absorbancia en función del tiempo para cada una de las muestras determinadas.2. Calcular la actividad PEPCK de cada muestra en µmol/min/mg de proteína, sabiendo que las muestras utilizadas se encuentran a una concentración de 1 mg/ml de proteína utilizando albúmina de suero de vaca como patrón.3. Explica como prepararías 40 ml del tampón B.4. ¿A qué concentración de PEP se realiza el ensayo?
Página 11

3.-DETERMINACIÓN DEL GLUCÓGENO HEPÁTICO El glucógeno se determina midiendo la cantidad de glucosa total hepática obtenida después de una hidrólisis con amiloglucosidasa que degrada totalmente el glucógeno hasta convertirlo en glucosa y sustrayendo a esta medida el valor obtenido para la glucosa hepática libre.
Protocolo:
1.- tomar 1 ml del sobrenadante de las muestras hepáticas desnaturalizadas con ácido perclórico y ponerlo en tubos graduados debidamente rotulados. 2.- añadir una gota de indicador de pH y neutralizar todos los tubos con KOH al 5%. 3.- anotar los volúmenes de cada tubo y dejar los tubos en hielo 5 minutos para que precipite el perclorato potásico. 4.- Centrifugar los tubos 5 min en la microcentrífuga (1,5 ml) 5.- separar una alícuota de 0,2 ml de cada muestra para la hidrólisis del glucógeno 6.- guardar el resto para determinar la glucosa libre (congelado a –20 ºC). 7.- realizar la hidrólisis de glucógeno con las alícuotas de 0,2 ml del paso 5: para hacer la hidrólisis hay que añadir a cada alícuota 3ml de acetato sódico 0,1 M pH 4, 75 y 0,2 ml de amiloglucosidasa (25 mg/ml)
8.- Incubar la reacción a 40º C durante una hora en un baño con agitación. 9.- Parar las reacciones añadiendo 1,8 ml de ClO4H al 2%. Transcurridos 10 min neutralizar las muestras con KOH al 10% en presencia de indicador de pH. 10.- anotar el volumen final de cada tubo, dejar en hielo durante 10 min y centrifugar las muestras 5 min en la microcentrífuga (1,5 ml es suficiente). Tomar 1,5 ml de los sobrenadantes en tubos eppendorf debidamente rotulados y congelar a –20º C para determinar glucosa total hepática.
4.-DETERMINACIÓN DE GLUCOSA
La cantidad de glucosa se determinará utilizando el ensayo enzimático de la glucosa oxidasa
Página 12

El peróxido de hidrógeno formado se descompone mediante la acción de la peroxidasa y el oxígeno liberado oxida a la orto-dianisidina reducida (D).
La orto-dianisidina oxidada tiene un pico de absorción a 440 nm. El coeficiente de extinción molar de esta sustancia varía con las condiciones del ensayo por lo cual siempre hay que utilizar una curva de glucosa patrón de concentración conocida.
Para esta práctica se usará la siguiente mezcla de reacción: tampón fosfato 0,5 M Tris 0,1 M pH 7,3, 1 U/ml glucosa oxidasa (0.062 mg7ml), 3,5 U/ml peroxidasa (0,02 mg/ml), 0,5 ml de orto-dianisidina (1% p/v en etanol al 96%) en 150 ml de mezcla de reacción.
Protocolo: 1.- Preparar los siguientes tubos, incubar a 37º C durante 1 hora y medir la absorbancia a 440 nm. Añadir la mezcla de reacción al mismo tiempo a todas las muestras: Inicio de la reacción.
GluLibre*: Para calcular la glucosa libre se usará el extracto del paso 6 del apartado “DETERMINACIÓN DELGLUCÓGENO HEPÁTICO”
GluTotal**: Para calcular la glucosa total se usará el extracto del paso 10 del apartado “DETERMINACIÓN DELGLUCÓGENO HEPÁTICO”
2.- Con la ayuda de la curva de calibración, calcular los mg/ml de glucosa presente en plasma e hígado (como glucosa total y glucosa libre). Tomar en cuenta las diluciones efectuadas en cada caso.
Cálculo de la concentración de glucosa:
Página 13

- - Determinar la pendiente de la recta. Interpolar los valores de DO de las muestras
problema para obtener su concentración. - La cantidad de glucosa procedente de la degradación del glucógeno se obtiene
restando al dato de glucosa total la glucosa libre.
Resultados1. Construir una curva patrón, representando absobancia frente a µmoles de glucosa. (OJO! Se añaden diferentes ml de una solución de gluicosa m0.4 mM en un volumen final de 5 ml). 2. Ajustar a una recta. Interpolar los valores de DO de las muestras problema .3. Calcular la concentración plasmática de glucosa en cada caso expresada en mg/100 ml y en mM.4. La cantidad de glucosa procedente de la degradación del glucógeno se obtiene restando al dato de glucosa total la glucosa libre.5. Calcular la glucosa libre hepática en µmol/mg de proteína6. Calcular la glucosa procedente del glucógeno en µmol/mg de proteína para cada caso.
5. DETERMINACIÓN DE LOS NIVELES PROTEÍCOS DE G3PDH POR WESTERN-BLOT
La actividad enzimática de una enzima depende de varios factores como son: disponibilidad de sus sustratos, los niveles proteícos de la enzima y, en muchos casos, las modificaciones posttraducionales de las mismas, siendo el ejemplo más característico la fosforilación específica de ciertos aminoácidos como Ser, Thr y Tyr llevada a cabo por diferentes proteínas quinasas y proteínas fosfatasas. La medida de actividad enzimática que hempos llevado a cabo previamente no es capaz de discriminar si el incremento o disminución de la actividad se debe a una modificación posttraduccional o a una disminución o incremento en los niveles proteícos de esa enzima. En esta práctica se determinaran los niveles proteícos de la enzima Gliceraldehido-3-fosfato deshidrogenasa (G3PDH) por western-blot. Para ello primero se separaran las proteínas por su peso molecular haciendo una una electroforesis en condiciones desnaturalizante en geles de poliacrilamida y posteriormente se transferirán estas proteína a una membrana de nitrocelulosa que se incubará con un anticuerpo específico para esta enzima.
La detección inmunológica de una enzima permite además llevar a cabo ciertos estudios cuando la cantidad de material biológico disponible es muy pequeña y no se puede llevar a cabo la determinación enzimática.
5.1.- ELECTROFORESIS EN GELES DE POLIACRILAMIDA-SDS. Introducción.
Página 14

El principio básico de la electroforesis es el movimiento que experimenta una partícula cargada en un determinado medio cuando se ve sometida a la acción de un campo eléctrico. Dada una partícula de carga neta Q sometida a un campo eléctrico de intensidad E, se moverá por la acción de una fuerza igual a:
F = Q E = Q V/d
siendo V la diferencia de potencial y d la distancia entre los electrodos.
Esta técnica es utilizada frecuentemente en el laboratorio para la separación de proteínas y ácidos nucleicos. En la figura 1 se presenta un dibujo del sistema de electroforesis de proteínas a utilizar en estas prácticas.
Uno de los medios de soporte más empleado en la electroforesis, es el gel de poliacrilamida. Este gel o red tridimensional se consigue mediante la polimerización lineal de la acrilamida, y el entrecruzamiento de cadenas provocado por la bisacrilamida. El gel se organiza en una serie de fibras que al entrecruzarse, y según el grado de entrecruzamiento, deja poros a través de los cuales circulan las proteínas a separar (figura 2.).
El tamaño de los poros depende de la concentración absoluta y de la proporción de acrilamida y bisacrilamida del gel. La polimerización de la acrilamida y bisacrilamida es promovida por radicales libres generados por el persulfato amónico y que son estabilizados por el TEMED. La poliacrilamida presenta la ventaja de ser a la vez criba y soporte, no presenta fenómenos de adsorción de tipo iónico ni flujo electro-osmótico, y ofrece la posibilidad de utilizar un amplio rango de pH.
Figura 1. Componentes del sistema de electroforesis vertical
Los pesos moleculares de las proteínas pueden ser determinados midiendo la movilidad en geles de poliacrilamida en presencia del detergente dodecilsulfato sódico (SDS). Esto es debido a que la cantidad de SDS, detergente aniónico, que se une a una proteína es proporcional al peso molecular del polipéptido y es independiente de su secuencia; a saturación, se unen aproximadamente 1,4 g.
Página 15

de detergente por gramo de proteína. Así, si una serie de proteínas de peso molecular conocido son sometidas a electroforesis y se separan en una serie de bandas, la representación gráfica de la distancia recorrida en función del logaritmo del peso molecular, da una línea recta. Este sistema de electroforesis en poliacrilamida-SDS se conoce como método de Laemmli.
Figura 2. Polimerización del gel de acrilamida
Tareas a realizar:
-Preparación del gel de SDS-poliacrilamida-Separación electroforética de las fracciones seleccionadas en gel del 10% SDS-poliacrilamida.
Materiales
A) Medios preparados o Acrilamida/bisacrilamida al 30/0.8% (p/c) o Tris-HCl 1M, pH 6.8 o Tris-HCl 1 M, pH 8.8 o SDS 10% (p/v) o Tampón fosfato 0.1 M, pH 6.2 o Persulfato amónico (APS) 10% (p/v) o Isobutanol saturado con agua destilada
o Tampón de electroforesis: SDS 0.1%, Tris 0.025M/glicina 0.192M, pH 8.3
Página 16

o Tampón de carga 2x: SDS 4% (p/v), glicerol 20% (v/v), 2-mercaptoetanol
10% (v/v), EDTA 15 mM, azul de bromofenol 0.008% (p/v), Tris-HCl 0.125 M, pH 6.8
B) Instrumentos y otros materiales
o TEMED. Importante: no se puede almacenar en tubos o botellas de plástico. o Cubetas de electroforesis, cristales, peines y fuentes o Marcadores de peso molecular de proteínas o Sistema de documentación de geles
Método
Preparación del gel de poliacrilamida
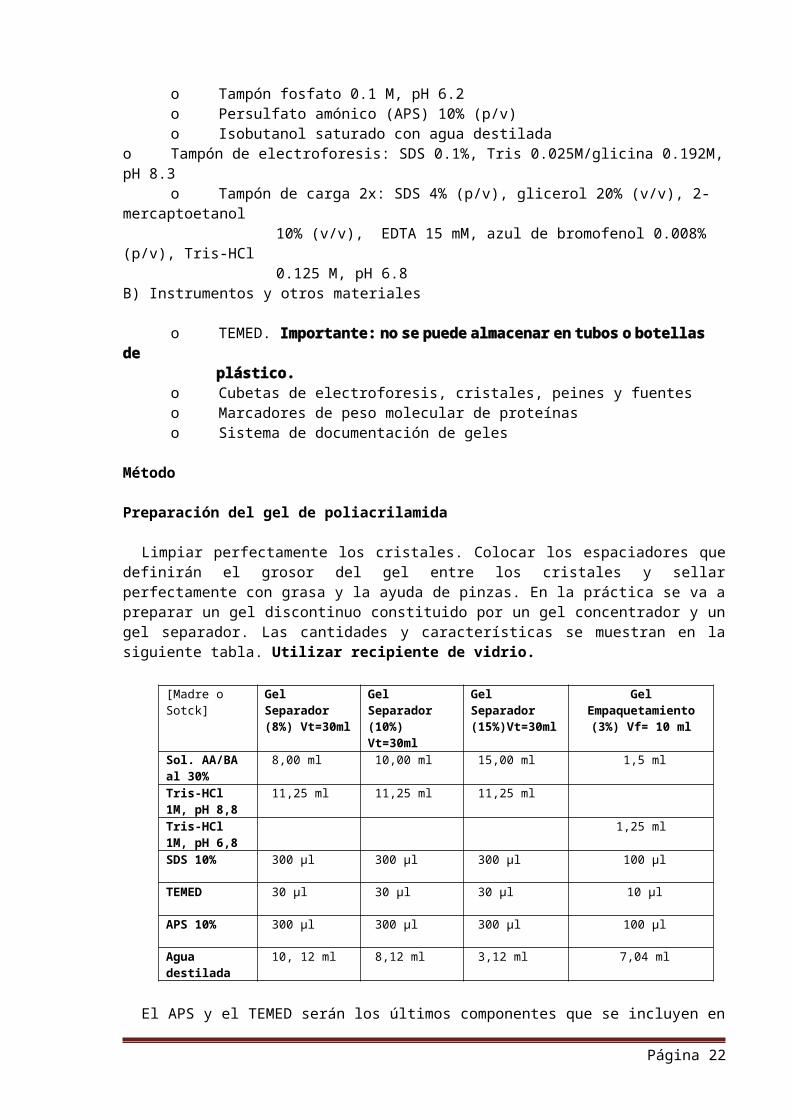
Limpiar perfectamente los cristales. Colocar los espaciadores que definirán el grosor del gel entre los cristales y sellar perfectamente con grasa y la ayuda de pinzas. En la práctica se va a preparar un gel discontinuo constituido por un gel concentrador y un gel separador. Las cantidades y características se muestran en la siguiente tabla. Utilizar recipiente de vidrio.
[Madre o Sotck]
Gel Separador (8%) Vt=30ml
Gel Separador (10%) Vt=30ml
Gel Separador (15%)Vt=30ml
Gel Empaquetamiento
(3%) Vf= 10 ml
Sol. AA/BA al 30%
8,00 ml 10,00 ml 15,00 ml 1,5 ml
Tris-HCl 1M, pH 8,8
11,25 ml 11,25 ml 11,25 ml
Tris-HCl 1M, pH 6,8
1,25 ml
SDS 10% 300 µl 300 µl 300 µl 100 µl
TEMED 30 µl 30 µl 30 µl 10 µl
APS 10% 300 µl 300 µl 300 µl 100 µl
Agua destilada
10, 12 ml 8,12 ml 3,12 ml 7,04 ml
El APS y el TEMED serán los últimos componentes que se incluyen en la mezcla.
1 Preparar primero la solución del gel separador y verter cuidadosamente entre los cristales hasta 1 cm por debajo del nivel que será ocupado por el peine formador de los pocillos. Colocar cuidadosamente un poco de isobutanol saturado con agua sobre la solución del gel y dejar polimerizar a temperatura ambiente. Una vez polimerizado, retirar el isobutanol lavando con agua destilada y preparar la solución del gel concentrador. Verter esta solución y colocar el peine. Dejar polimerizar a temperatura ambiente. 2 Cuando el gel concentrador haya polimerizado, retirar el peine e instalar el gel en el aparato de electroforesis siguiendo las instrucciones del Profesor. Rellenar las cámaras de los electrodos con el tampón de electroforesis.
Página 17

Separación electroforética de las proteínas
1. Mezclar 1:1 las muestras a desarrollar en la electroforesis con el tampón de carga. Calentar las muestras en baño de agua a ebullición durante 3 min, pasar entonces a hielo.
Las muestras hepáticas a desarrollar serán: a) Muestra Control, b) Muestra Ayunada, c) Muestra Ayunada y Realimentada, y d) Muestra Diabética. Se cargarán 5 y 10 µl de las muestras. También se cargaran patrones de peso molecular (mezcla de diferentes proteínas que se indican a continuación).
Marcadores coloreados:
1 Colocar las muestras cuidadosamente en los pocillos. Desarrollar la electroforesis a una intensidad constante de 15 mA durante toda la noche o a lo que se indique durante el día. 2 Parar la electroforesis cuando el azul de bromofenol esté a 1 cm del final del gel. Desmontar el gel y transferirlo a membransa de nitrocelulosa.
5.2.- TRANSFERENCIA DE PROTEÍNAS A MEMBRANAS (“WESTERN BLOT”).
Introducción.
Se trata de una técnica frecuentemente utilizada en el laboratorio, que consiste en la transferencia de proteínas desde un gel de poliacrilamida hasta una membrana o soporte sólido donde las proteínas quedan unidas. Esta transferencia se realiza mediante la aplicación de un campo eléctrico (figura 3.).
Página 18

Figura 3. Western-blot: transferencia de proteínas a una membrane y revelado con anticuerpos específicos.
Existen múltiples tipos de membranas o soportes con afinidad elevada por las proteínas. Uno de los tipos de membranas más utilizado es la nitrocelulosa,
que es el elegido para la realización de estas prácticas. El tipo de interacciones entre la nitrocelulosa y las proteínas no está totalmente definido, aunque es de muy alta afinidad. Sin embargo, la unión de las proteínas a la nitrocelulosa es bloqueada por aceites o grasas y, obviamente, por otras proteínas, por lo que se recomienda utilizar guantes para la manipulación de las hojas de nitrocelulosa.
Tareas a realizar:
- Transferencia de las proteínas a una membrana de nitrocelulosa - Tinción reversible de las proteínas con Rojo-Ponceau
Material
A) Medios preparados
o Tampón de transferencia: metanol al 20% (v/v); bicarbonato sódico 10mM,
carbonato sódico 2mM, pH 10. Tris-HCl 1M, pH 6.8 o Ponceau S: Ponceau S al 0.2% (p/v); ácido sulfosalicílico al 3% (p/v);
ácido tricloroacético al 3% (p/v).
B) Instrumentos y otros materiales
Página 19

o Cubetas de transferen cia y fuentes o Membranas de nitrocelulosa (0.45 micras de poro) o de PVDF o Metanol
Método:
Transferencia de proteínas a filtros de nitrocelulosa
Una vez desarrollada la electroforesis:
1 Incubar el gel para transferir en una bandeja durante 5-10 min con tampón de transferencia. 2 A continuación cortar una membrana de nitrocelulosa y dos filtros de papel 3MM con las dimensiones del gel. Sumergir estos filtros en el tampón de transferencia o, si se utiliza PVDF para la transferencia hay que incubar previamente el filtro de PVDF con metanol absoluto unos 30 segundos, y pasar a continuación a tampón de transferencia. Los filtros de PVDF, aunque más sensibles y resistentes a agentes orgánicos, presentan más problemas de inespecificidad (más fondo) y cuando se hace impermeable hay que tratarlos con metanol. 3 A continuación se colocarán en el aparato de transferencia de acuerdo con la disposición de "bocadillo" que se ilustra en la siguiente figura 4.
Figura 4.: Diagrama del montaje del gel de poliacrimalida para la transferencia
Es importante hacer una marca en el filtro de nitrocelulosa o PVDF que nos permita, inequívocamente, reconocer la orientación de las muestras con respecto al gel de poliacrilamida.
Una vez que el sistema de transferencia se encuentra colocado, conectar a la fuente de electroforesis con la polaridad adecuada, recordar que las proteínas a este pH estarán cargadas negativamente. La transferencia se realiza durante 1.5 horas a 0.8 amperios.
Tinción de proteínas fijadas sobre filtros de nitrocelulosa o PVDF.
Página 20

La tinción de las proteínas transferidas a nitrocelulosa o PVDF es utilizada para asegurarse de que la transferencia ha tenido lugar; sin embargo, es importante utilizar una tinción que no interfiera con los métodos de detección para los que posteriormente sea utilizado el filtro de nitrocelulosa o PVDF. Así, de los múltiples procedimientos existentes para la tinción de proteínas unidas a nitrocelulosa, sólo la tinción con Ponceau S es compatible con todos los métodos inmunológicos debido a que la tinción es reversible y desaparece fácilmente con lavados.
En nuestro caso concreto, una vez realizada la transferencia, poner el filtro de nitrocelulosa o PVDF en una bandeja. Cubrir el filtro con la solución de Ponceau S e incubar durante 5-10 min a temperatura ambiente. Lavar con agua destilada hasta que las bandas del marcador de peso molecular resulten patentes. Si se trabaja con filtros de PVDF y no se observa ninguna banda volver a incubar unos segundos con metanol y volver a teñir con el Rojo Ponceau. Macar entonces, con ayuda de un lápiz, la posición de las bandas correspondientes a los estándares de proteínas. En este momento el filtro está listo para realizarse la detección inmunológica.
PARA EL CUADERNO: 1 Fotografiar el filtro de nitrocelulosa teñido con el Rojo Ponceau e identificar e indicar qué hay en cada uno de los carriles y el tamaño de las proteínas estandar. 2 Comentar los resultados.
5.3.-DETECCIÓN INMUNOLÓGICA DE PROTEÍNAS
Introducción.
En ocasiones, cuando la cantidad de material biológico disponible es pequeña, muchos de los métodos habituales para la identificación de moléculas fracasan. Los procedimientos inmunológicos dan solución a estas dificultades, pues posibilitan la determinación de cantidades pequeñas de proteínas en mezclas complejas.
En esta práctica se trata de ilustrar la utilización de anticuerpos específicos para la identificación de proteínas de interés, previamente transferidas a una membrana de nitrocelulosa o PVDF.
Tareas a realizar:
-Incubación de las membranas con anticuerpo primario anti-G6PDH producido en conejo y con anticuerpo secundario unido a peroxidasa.
- Revelado con 4-cloro-naftol de la proteína problema.
Material
Página 21
A) Medios preparados
o PBS o 4-cloro-naftol a 3 mg/ml en methanol (se guarda a -20ºC) o Solución de bloqueo: leche en polvo al 5% (p/v),Tween-20 al 0.5%
(v/v) en PBS. o Solución de lavado: Tween-20 al 0.05% (v/v) en PBS
B) Instrumentos y otros materiales
o Cubetas o bandejas o Anticuerpo de conejo anti-G3PDH o Anticuerpo anti-IgGs de conejo (producido en cabra) acoplado a
peroxidasa o Sistema de documentación de geles
Método:
Incubación con anticuerpos
1 Colocar el filtro dentro de una bandeja. Añadir 10 ml de la solución de bloqueo e incubar de 30 min (a temperatura ambiente) a 12 horas (a 4ºC). 2 Transcurrido este tiempo, añadir 10 µl del primer anticuerpo (anti-G3PDH; 1/1000) e incubar durante 1 hora a temperatura ambiente. 3 A continuación, lavar 3 veces, 5 min cada una, con solución de lavado.Es importante que durante el proceso el filtro no se llegue a secar en ningún momento. 4 Una vez realizados los lavados, colocar 10 ml de la solución de bloqueo y añadir 10 µl de anticuerpo anti-IgGs de conejo unido a peroxidasa. Incubar de nuevo 1 h a temperatura ambiente. 5 Después de la incubación, volver a lavar 3 veces con la solución de lavado. Dejar el filtro en el último lavado y proceder a preparar la solución de revelado.
Revelado de anticuerpos unidos a peroxidasa El segundo anticuerpo está acoplado a la enzima peroxidasa, siendo esta
actividad enzimática la que se va a utilizar para detectar la unión antígeno-anticuerpo. La solución de revelado contiene el compuesto 4-cloro-naftol que al actuar la peroxidasa sobre él va a dar lugar a un producto insoluble de color azul-violeta.
Preparar la solución de revelado que contiene: - 4ml de 4-cloronaftol. - 20 ml de PBS. - 50 µl de H2O2 al 30%.
Añadir la solución de revelado, tras eliminar el líquido del último lavado de la bandeja del filtro de nitrocelulosa o PVDF, y esperar hasta que aparezca la coloración. Este proceso puede ser instantáneo o requerir de 15-30 min, dependiendo de la cantidad de antígeno unido al filtro. Cuando se considere que ha aparecido coloración suficiente, parar la reacción pasando el filtro a agua destilada, fotografiar y secar.
Página 22

PARA EL CUADERNO: 1 Fotografiar el resultado de la detección inmunológica e incluirlo en el cuaderno indicando qué hay en cada uno de los carriles y el tamaño de las proteínas estandar. 2 Comentar los resultados obtenidos y comparar con los resultados obtenidos con la tinción con Rojo Ponceau del filtro. 3 Comentar los resultados obtenidos y los esperados en el conjunto de estas cinco prácticas
RESULTADOS GLOBALES:
Realizar una tabla resumen con todos los parámetros metabólicos de las ratas control, ayunadas, ayunadas y realimentadas y diabéticas. Analizar los resultados.
Página 23


















