
MicroRNA-141-3p plays a role in human mesenchymal stem cell aging...
11
CORRECTION MicroRNA-141-3p plays a role in human mesenchymal stem cell aging by directly targeting ZMPSTE24 Kyung-Rok Yu, SeungHee Lee, Ji-Won Jung, In-Sun Hong, Hyung-Sik Kim, Yoojin Seo, Tae-hoon Shin and Kyung-Sun Kang There was an error published in J. Cell Sci. 126, 5422-5431. The affiliations for Ji-Won Jung and In-Sun Hong were given incorrectly. The correct affiliations are as shown below. Kyung-Rok Yu 1,2 , SeungHee Lee 1,3,4 , Ji-Won Jung 5 , In-Sun Hong 1,3 , Hyung-Sik Kim 1,3 , Yoojin Seo 1,3 , Tae-hoon Shin 1,3 and Kyung-Sun Kang 1,3 1 Adult Stem Cell Research Center, College of Veterinary Medicine, Seoul National University, Seoul, Korea 2 The Research Institute for Agriculture and Life Sciences, College of Agriculture and Life Sciences, Seoul National University, Seoul, Korea 3 The Research Institute for Veterinary Science, College of Veterinary Medicine, Seoul National University, Seoul, Korea 4 Institute for Stem Cell and Regenerative Medicine in Kang Stem Holdings, Biotechnology Incubating Center, Seoul National University, Seoul 151-742, Korea 5 Division of Intractable Diseases, Center for Biomedical Sciences, Korea National Institute of Health, Chungbuk, Republic of Korea We apologise to the authors and readers for any confusion that this error might have caused. ß 2014. Published by The Company of Biologists Ltd | Journal of Cell Science (2014) 127, 475 doi:10.1242/jcs.147645
Transcript of MicroRNA-141-3p plays a role in human mesenchymal stem cell aging...

CORRECTION
MicroRNA-141-3p plays a role in human mesenchymal stem cellaging by directly targeting ZMPSTE24
Kyung-Rok Yu, SeungHee Lee, Ji-Won Jung, In-Sun Hong, Hyung-Sik Kim, Yoojin Seo, Tae-hoon Shin andKyung-Sun Kang
There was an error published in J. Cell Sci. 126, 5422-5431.
The affiliations for Ji-Won Jung and In-Sun Hong were given incorrectly.
The correct affiliations are as shown below.
Kyung-Rok Yu1,2, SeungHee Lee1,3,4, Ji-Won Jung5, In-Sun Hong1,3, Hyung-Sik Kim1,3, Yoojin Seo1,3, Tae-hoon Shin1,3
and Kyung-Sun Kang1,3
1Adult Stem Cell Research Center, College of Veterinary Medicine, Seoul National University, Seoul, Korea2The Research Institute for Agriculture and Life Sciences, College of Agriculture and Life Sciences, Seoul National University, Seoul,Korea3The Research Institute for Veterinary Science, College of Veterinary Medicine, Seoul National University, Seoul, Korea4Institute for Stem Cell and Regenerative Medicine in Kang Stem Holdings, Biotechnology Incubating Center, Seoul NationalUniversity, Seoul 151-742, Korea5Division of Intractable Diseases, Center for Biomedical Sciences, Korea National Institute of Health, Chungbuk, Republic of Korea
We apologise to the authors and readers for any confusion that this error might have caused.
� 2014. Published by The Company of Biologists Ltd | Journal of Cell Science (2014) 127, 475 doi:10.1242/jcs.147645
475

Journ
alof
Cell
Scie
nce
MicroRNA-141-3p plays a role in human mesenchymalstem cell aging by directly targeting ZMPSTE24
Kyung-Rok Yu1,2,*, SeungHee Lee1,3,4,*, Ji-Won Jung3, In-Sun Hong1,5, Hyung-Sik Kim1,3, Yoojin Seo1,3,Tae-hoon Shin1,3 and Kyung-Sun Kang1,3,`
1Adult Stem Cell Research Center, College of Veterinary Medicine, Seoul National University, Seoul, Korea2The Research Institute for Agriculture and Life Sciences, College of Agriculture and Life Sciences, Seoul National University, Seoul, Korea3The Research Institute for Veterinary Science, College of Veterinary Medicine, Seoul National University, Seoul, Korea4Institute for Stem Cell and Regenerative Medicine in Kang Stem Holdings, Biotechnology Incubating Center, Seoul National University, Seoul 151-742, Korea5Division of Intractable Diseases, Center for Biomedical Sciences, Korea National Institute of Health, Chungbuk, Republic of Korea
*These authors contributed equally to this work`Author for correspondence ([email protected])
Accepted 12 September 2013Journal of Cell Science 126, 5422–5431� 2013. Published by The Company of Biologists Ltddoi: 10.1242/jcs.133314
SummaryHuman mesenchymal stem cell (hMSC) aging may lead to a reduced tissue regeneration capacity and a decline in physiologicalfunctions. However, the molecular mechanisms controlling hMSC aging in the context of prelamin A accumulation are not completely
understood. In this study, we demonstrate that the accumulation of prelamin A in the nuclear envelope results in cellular senescence andpotential downstream regulatory mechanisms responsible for prelamin A accumulation in hMSCs. We show for the first time thatZMPSTE24, which is involved in the post-translational maturation of lamin A, is largely responsible for the prelamin A accumulation
related to cellular senescence in hMSCs. Direct binding of miR-141-3p to the 39UTR of ZMPSTE24 transcripts was confirmed using a39UTR-luciferase reporter assay. We also found that miR-141-3p, which is overexpressed during senescence as a result of epigeneticregulation, is able to decrease ZMPSTE24 expression levels, and leads to an upregulation of prelamin A in hMSCs. This study providesnew insights into mechanisms regulating MSC aging and may have implications for therapeutic application to reduce age-associated
MSC pool exhaustion.
Key words: Mesenchymal stem cells, ZMPSTE24, Prelamin A accumulation, miRNA-141-3p, Histone modification, Aging
IntroductionAdvancing age is a major risk factor for developing many chronicdiseases, but the mechanisms that regulate the aging processremain largely unknown. Recent studies have revealed that aging
has been negatively correlated with the number and functionalactivities of tissue stem cells. Adult stem cells present inmammalian organs are essential for tissue generation,maintenance and injury repair throughout adult life. Similar to
other somatic cells, adult stem cells experience lifelong exposureto intrinsic and extrinsic factors, throughout the lifetime of theorganism, which lead to an age-associated decline in their
number and function (Janzen et al., 2006; Kasper et al., 2009;Rao and Mattson, 2001; Trosko, 2008).
It has been previously reported that the premature aging diseaseHutchinson–Gilford Progeria syndrome (HGPS) is caused by amutation in the LMNA gene (the corresponding protein is known as
lamin A). HGPS seems to mainly affect mesenchymal celllineages, and HGPS induces progerin-mediated mesenchymalstem cell pool exhaustion (Halaschek-Wiener and Brooks-Wilson,
2007) and mesenchymal lineage differentiation defects (Scaffidiand Misteli, 2006). These studies have shown that HGPSfibroblasts have a short replicative lifespan compared with their
wild-type counterparts. In HGPS fibroblasts, prematuresenescence is due to the toxic accumulation of progerin, amutant form of prelamin A (Bridger and Kill, 2004). In particular,
introduction of progerin, a mutant form of lamin A, into hMSCsaccelerated cellular senescence and caused adult stem celldysfunction (Scaffidi and Misteli, 2008). This study showed that
hMSCs expressing progerin display marked defects in growth,nuclear abnormalities and accelerated senescence, which closelyresemble the defects observed in cells from HGPS patients.
Lamin A, synthesized as a precursor named prelamin A,undergoes a multi-step maturation process, which includes
ZMPSTE24-mediated cleavage of the last 15 amino acids(Corrigan et al., 2005). Failure to cleave the last 15 aminoacids of prelamin A because of disruptions in either prelamin A
or ZMPSTE24 causes nuclear structural abnormalities, ashortened lifespan and multiple age-related phenotypes (Bergoet al., 2002; Pendas et al., 2002). Some reports have demonstrated
that ZMPSTE24 expression levels are downregulated in aged orsenescent human vascular smooth muscle cells and fibroblastcells (Ragnauth et al., 2010; Ukekawa et al., 2007), but the
molecular mechanisms that regulate ZMPSTE24 expressionduring hMSC aging have not been identified (Maraldi et al.,2011).
MiRNAs, small RNAs of ,22 nucleotides in length, areknown to perform important regulatory roles through the
repression of target mRNA translation by complementarybinding to the 39 untranslated region (UTR) (Bartel, 2004).More recent evidence has shown that several miRNAs (miR-371,
5422 Research Article

Journ
alof
Cell
Scie
nce
miR-369-5p, miR-29c, miR-499 and let-7f) are involved in theregulation of cellular senescence (Wagner et al., 2008). However,the effects of these miRNAs on ZMPSTE24 expression during in
vitro cellular senescence of hMSCs are unknown.
The dynamic balance between epigenetic elements, such asacetylation, methylation or phosphorylation, is important for the
maintenance of stemness and the regulation of cellularsenescence because these elements control chromatinmodification and transcriptional regulation (Bibikova et al.,
2008; Narita, 2007). Histone deacetylase (HDAC) and DNAmethyltransferase (DNMT) inhibitors induce senescence ofhMSCs by affecting p16INK4A and p21CIP1/WAF1 expression
(Jung et al., 2010; So et al., 2011). Moreover, non-codingRNAs as well as protein-coding RNAs can be regulated byepigenetic modifications (Delcuve et al., 2012).
In the study reported here, we assessed the impact ofdownregulation of ZMPSTE24 on the proliferation defects andDNA damage responses observed in hMSCs as they progress
toward replicative senescence. Furthermore, our results indicatethat ZMPSTE24 expression can be directly regulated by miR-141-3p during hMSC senescence. We anticipate that these results
might provide clues to understanding the biological relevance ofZMPSTE24 in hMSCs senescence and the aging process of adultstem cells.
ResultsSenescent hMSCs show increased prelamin Aaccumulation and progeria-like abnormalities in thenucleus
First, we evaluated the direct effects of replicative- and progerin-induced senescence on senescence-associated-b-galactosidase
(SA-b-gal) activity and hMSC proliferation. Both senescentconditions significantly increased SA-b-gal activity anddecreased hMSC proliferation (Fig. 1A,B). Replicative and
progerin-induced senescence clearly increased expression ofp16INK4A, a senescence marker, in hMSCs (Fig. 1C;supplementary material Fig. S1B). To determine whetherprelamin A accumulation is related to the physiological aging
of hMSCs, we investigated the accumulation levels of prelamin Ain senescent cells. Replicative senescence significantly increasedprelamin A accumulation compared with negative controls
(Fig. 1C). To support this observation, we subsequentlyinvestigated whether replicative and progerin-inducedsenescence were correlated with abnormal nuclear morphology
of hMSCs. We found that replicative and progerin-inducedsenescence increased the incidence of severely wrinkled nuclei inhMSCs compared with negative controls (Fig. 1D). To assess the
age-related accumulation of DNA damage under a prelamin Aaccumulation condition, we examined whether replicative andprogerin-induced senescence had any effect on the expression ofcH2AX as a general biomarker of DNA damage. As illustrated in
Fig. 1E, cH2AX expression was increased significantly underboth senescent conditions.
ZMPSTE24 inhibition induces cellular senescence withDNA damage accumulation
We found that expression of the LMNA gene was notsignificantly altered by serial in vitro passages (supplementary
material Fig. S1A). As illustrated in Fig. 1F and supplementarymaterial Fig. S1C, the expression of ZMPSTE24, which convertsprelamin A into mature lamin A, was clearly decreased by
replicative senescence in hMSCs. To further elucidate the role of
ZMPSTE24 as a regulator of prelamin A accumulation duringcellular senescence, hMSCs were transfected with ZMPSTE24siRNA. We found that transfection with ZMPSTE24 siRNA
efficiently increased prelamin A accumulation, which wasfollowed by an increase in p16INK4A and cH2AX expression(Fig. 1G). Furthermore, ZMPSTE24 knockdown increased SA-b-gal activity (Fig. 1H) and decreased proliferation (Fig. 1I;
supplementary material Fig. S1D) compared with negativecontrols, and these effects coincided with increased prelaminA-accumulation and cH2AX expression (Fig. 1J).
HDAC regulates ZMPSTE24 expression during replicativecellular senescence
The activities of DNMT and HDAC, regulatory factors of
epigenetic states and transcriptional activities, decreased duringcellular senescence in our present (Fig. 2A) and previous studies(Jung et al., 2010; So et al., 2011). To evaluate whether these
epigenetic factors are involved in the regulation of ZMPSTE24expression, we determined the effects of the DNMT inhibitor [5-azacytidine (5-AzaC); 2 mM] and two HDAC inhibitors [valproicacid (VPA; 4 mM) and sodium butyrate (NB; 2 mM)] on the
induction of senescence in hMSCs. As shown in Fig. 2B–E,inhibition of DNMT and HDAC induced cellular senescence andincreased p16INK4A expression. However, only HDAC inhibition
decreased ZMPSTE24 expression, which resulted in an increase inprelamin A expression (Fig. 2E) and nuclear abnormalities(Fig. 2F,G). Knockdown of HDAC1 and HDAC2 decreased
ZMPSTE24 expression, resulting in prelamin A accumulation(Fig. 2H), and treatment with specific inhibitors for HDAC1 andHDAC2 activities increased p16INK4A expression and SA-b-gal
activity (Fig. 2H,I). We also found that suppression of HDAC1and HDAC2 activity by specific inhibitors decreased proliferation(Fig. 2J; supplementary material Fig. S1E) and that these effectscoincided with increased nuclear abnormalities (Fig. 2K). Taken
together, these results indicate that the inhibition of HDACsuppresses ZMPSTE24 expression, which in turn induces prelaminA-mediated cellular senescence in hMSCs.
miR-141-3p regulates cellular senescence through theregulation of ZMPSTE24 expression
Epigenetic regulations, such as DNA methylation and histone
modifications, can affect the expressions of miRNAs as well asprotein-coding RNAs (Delcuve et al., 2012). Moreover,according to our previous study, the decrease of DNMT
activity affected the expression of polycomb-gene-targetingmiRNAs in replicative senescent hMSCs (So et al., 2011).Therefore, we hypothesized that the decrease of HDAC activitywould affect the expressions of ZMPSTE24-targeting miRNAs in
replicative senescent hMSCs. Using three periodicity andcorrelation algorithms (miRanda, TargetScan5.0 and PicTar),we identified three miRNAs (miR-124, miR-141-3p and miR-
182) as the most likely regulatory miRNAs affecting ZMPSTE24expression (Fig. 3A). Among the predicted miRNAs, miR-141-3p significantly reduced firefly luciferase activity in the reporter
containing the ZMPSTE24-39UTR compared with the controlRenilla luciferase activity, suggesting that miR-141-3p coulddirectly bind to the 39UTR of the ZMPSTE24 RNA (Fig. 3B,C).
To support this observation, we further investigated whethermiR-141-3p regulates ZMPSTE24 expression, prelamin Aaccumulation and nuclear abnormalities. The transfection of
miR-141-3p regulates ZMPSTE24 5423

Journ
alof
Cell
Scie
nce
hMSCs with miR-141-3p decreased ZMPSTE24 expression,
resulting in increased expression of prelamin A, p16INK4A and
cH2AX. Conversely, these effects were reversed by miR-141-3p
knockdown (Fig. 3D). In the miR-141-3p-transfected cells, MTT
assays and cell cycle analyses revealed that the growth
rate was reduced 48 hours post-transfection (supplementary
Fig. 1. Senescent hMSCs show a downregulation of ZMPSTE24 expression and an accumulation of prelamin A. (A) SA b-gal (b-gal) staining was performed
on the indicated group [early (p6)- and late (p20)-passaged hMSCs, WT and progerin-virus-infected hMSCs] to confirm the state of senescence. Phase-contrast
images are also shown. (B) The proliferation rate of the hMSCs treated as in A was measured using the MTT assay (**P,0.01). (C) Western blot analysis of prelamin A
and p16INK4A was performed on early- and late-passaged cells. After transfection of progerin-GFP, the protein expression levels of lamin A and p16INK4A were confirmed
as shown by western blot. (D) Prelamin A accumulation and the nuclear phenotype in early- and late-passaged cells are shown by immunocytochemistry. After
transfection of the lamin A-GFP and progerin-GFP vectors, GFP expression was detected by confocal microscopy. The graph represents the ratio of abnormal nuclei over
total nuclei (**P,0.01). (E) cH2AX expression as detected by immunocytochemistry. The graph represents the ratio of cells that contain more than three cH2AX foci
per nucleus (**P,0.01). (F) ZMPSTE24 and p16INK4A expression were confirmed in serially passaged cells by western blot analysis. (G) After inhibition of ZMPSTE24
by siRNA, the protein expression levels of prelamin A, ZMPSTE24, p16INK4A and cH2AX were confirmed by western blot analysis. (H) SA b-gal staining was
performed on ZMPSTE24-inhibited cells that showed b-galactosidase activity. (I) To confirm the effect of ZMPSTE24 inhibition on the cellular growth rate, the
cumulative population doubling rate was examined. (J) After inhibition of ZMPSTE24 by siRNA, cH2AX expression was determined by immunocytochemistry. The
graph represents the ratio of cells that contained more than three cH2AX foci per nucleus (**P,0.01). Scale bars: 10 mm.
Journal of Cell Science 126 (23)5424

Journ
alof
Cell
Scie
nce
material Fig. S3A,B). In contrast, the transfection of hMSCs with
anti-miR-141-3p yielded an increase in proliferation. The
correlation between the expression of ZMPSTE24 and miR-
141-3p was confirmed in five other hMSC cell lines
(supplementary material Fig. S2A), and the cells treated with
miR-141-3p showed changes in their nuclear morphology, with
many of the cells displaying folds in the nuclear envelope
(Fig. 3E). To determine whether these observations could be
extended to hMSCs derived from different sources, we analyzed
the effects of miR-141-3p on the nuclear morphology of hMSCs
derived from bone marrow and adipose tissue (supplementary
material Fig. S2B). The overexpression of miR-141-3p also
increased SA-b-gal activity and cH2Ax expression (Fig. 3F,G) in
these cells.
To demonstrate the long-term effect of miR-141-3p, we
infected hMSCs with a GFP-tagged miR-141-3p virus. The
virally infected hMSCs showed strong GFP expression, with
significantly elevated levels of miR-141-3p, as confirmed by
Fig. 2. HDAC inhibition induces cellular senescence in hMSCs through a decrease in ZMPSTE24 expression and an accumulation of prelamin A.
(A) Changes in DNMT1, DNMT3b, HDAC1 and HDAC2 expression levels during replicative cellular senescence were confirmed by western blot analysis.
(B,C) After the inhibition of DNMT by 5-azacytidine (5-AzaC), SA-b-gal staining (B) and western blot analysis (C) were performed. (D) After treatment of cells
with the HDAC inhibitors valproic acid (VPA) and sodium butyrate (NB) for 6 days, SA b-gal staining was performed. (E,F) After treatment of cells with VPA
and NB for the indicated times, the expression levels of prelamin A, ZMPSTE24 and p16INK4A were confirmed by western blot analysis (E) and
immunocytochemistry (F). (G) The graph represents the ratio of abnormal nuclei over total nuclei (**P,0.01). (H) After inhibition of HDAC1 and HDAC2 by
siRNA, the protein expression levels of HDAC1, HDAC2, prelamin A, ZMPSTE24 and p16INK4A were confirmed by western blot analysis. (I) After inhibition of
HDAC1 and HDAC2, cellular senescence was demonstrated by SA b-gal staining. (J) To confirm the effect of HDAC1 and HDAC2 inhibition on the cellular
growth rate, the cumulative population doubling rate was examined. (K) After inhibition of HDAC1 and HDAC2, the nuclear phenotype was evaluated by
immunocytochemistry. The graph represents the ratio of abnormal nuclei over total nuclei (**P,0.01). Scale bars: 10 mm.
miR-141-3p regulates ZMPSTE24 5425
Journ
alof
Cell
Scie
nce
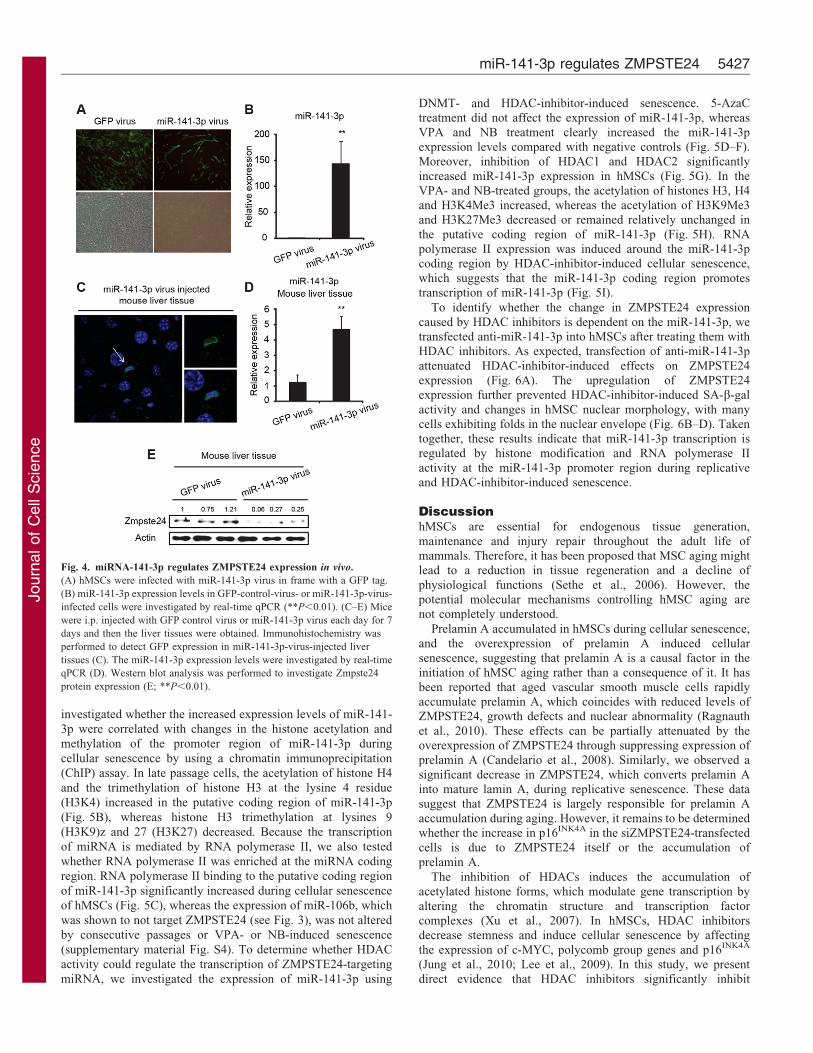
real-time qPCR (Fig. 4A,B). Using the MTT assay and a cell
cycle analysis, we also confirmed that proliferation was inhibited
after 5 days of miR-141-3p overexpression (supplementary
material Fig. S3D,E). To examine the role of miR-141-3p in
vivo, we delivered the miR-141-3p virus into mice by
intraperitoneal (i.p.) injection. Previously, it was reported that
using this method of delivery the organ with the highest level of
viral particles was the liver, presumably because this organ has a
relatively high percentage of blood-derived cells (Delgado et al.,
2008; Dismuke et al., 2009). Therefore, we obtained liver tissue 7
days after i.p. injection and performed immunohistochemistry
and western blot analyses. Because the viral vector was designed
to express GFP together with miR-141-3p, we used GFP
fluorescence to detect the overexpression of miR-141-3p in
liver tissue (Fig. 4C). Accordingly, we found that the expression
of miR-141-3p was increased and ZMPSTE24 expression was
reduced in the miR-141-3p-virus-injected liver (Fig. 4D,E).
These results indicate that ZMPSTE24 is the direct target of
miR-141-3p both in vitro and in vivo. Collectively, these results
support the hypothesis that miR-141-3p modulates cellular
senescence through the regulation of ZMPSTE24, which
correlates with the change in phenotype observed in the
siZMPSTE24-treated cells.
Activation of a histone marker at the miR-141-3p promoter
increases miR-141-3p expression
We hypothesized that miR141-3p may play a role in cellular
senescence of hMSCs by controlling ZMPSTE24 expression. The
expression levels of miR-141-3p were significantly increased
with consecutive passages (Fig. 5A). We subsequently
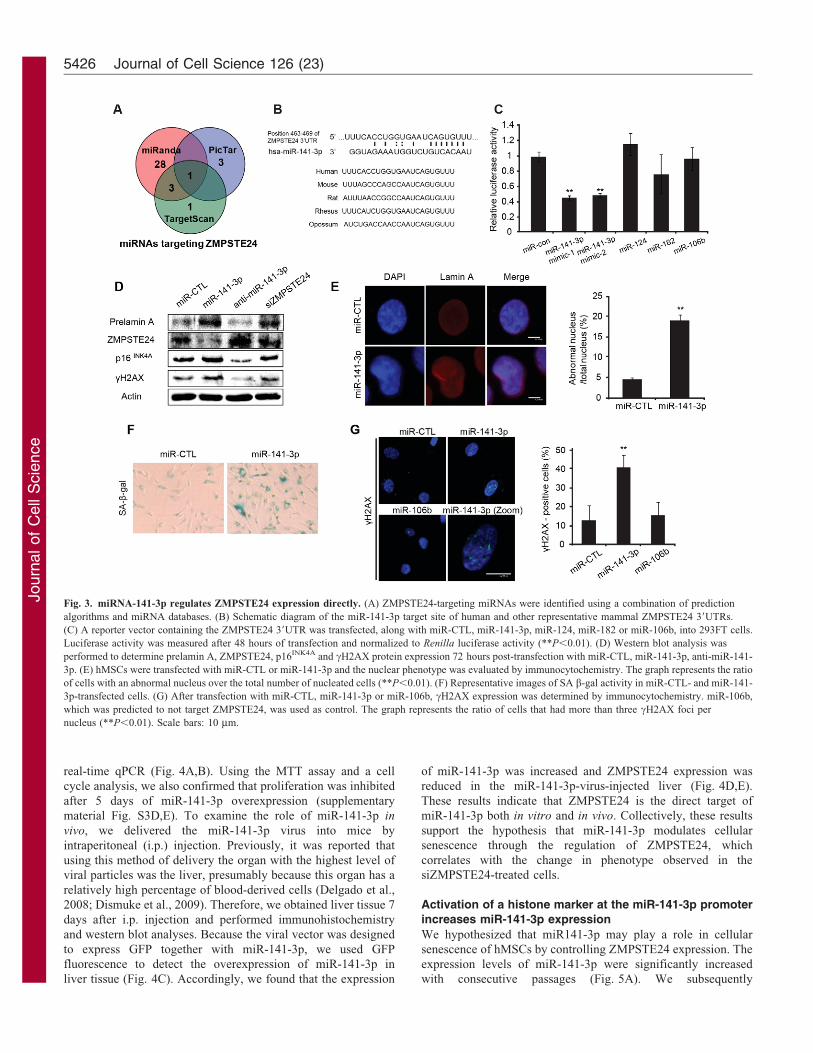
Fig. 3. miRNA-141-3p regulates ZMPSTE24 expression directly. (A) ZMPSTE24-targeting miRNAs were identified using a combination of prediction
algorithms and miRNA databases. (B) Schematic diagram of the miR-141-3p target site of human and other representative mammal ZMPSTE24 39UTRs.
(C) A reporter vector containing the ZMPSTE24 39UTR was transfected, along with miR-CTL, miR-141-3p, miR-124, miR-182 or miR-106b, into 293FT cells.
Luciferase activity was measured after 48 hours of transfection and normalized to Renilla luciferase activity (**P,0.01). (D) Western blot analysis was
performed to determine prelamin A, ZMPSTE24, p16INK4A and cH2AX protein expression 72 hours post-transfection with miR-CTL, miR-141-3p, anti-miR-141-
3p. (E) hMSCs were transfected with miR-CTL or miR-141-3p and the nuclear phenotype was evaluated by immunocytochemistry. The graph represents the ratio
of cells with an abnormal nucleus over the total number of nucleated cells (**P,0.01). (F) Representative images of SA b-gal activity in miR-CTL- and miR-141-
3p-transfected cells. (G) After transfection with miR-CTL, miR-141-3p or miR-106b, cH2AX expression was determined by immunocytochemistry. miR-106b,
which was predicted to not target ZMPSTE24, was used as control. The graph represents the ratio of cells that had more than three cH2AX foci per
nucleus (**P,0.01). Scale bars: 10 mm.
Journal of Cell Science 126 (23)5426
Journ
alof
Cell
Scie
nce
investigated whether the increased expression levels of miR-141-
3p were correlated with changes in the histone acetylation and
methylation of the promoter region of miR-141-3p during
cellular senescence by using a chromatin immunoprecipitation
(ChIP) assay. In late passage cells, the acetylation of histone H4
and the trimethylation of histone H3 at the lysine 4 residue
(H3K4) increased in the putative coding region of miR-141-3p
(Fig. 5B), whereas histone H3 trimethylation at lysines 9
(H3K9)z and 27 (H3K27) decreased. Because the transcription
of miRNA is mediated by RNA polymerase II, we also tested
whether RNA polymerase II was enriched at the miRNA coding
region. RNA polymerase II binding to the putative coding region
of miR-141-3p significantly increased during cellular senescence
of hMSCs (Fig. 5C), whereas the expression of miR-106b, which
was shown to not target ZMPSTE24 (see Fig. 3), was not altered
by consecutive passages or VPA- or NB-induced senescence
(supplementary material Fig. S4). To determine whether HDAC
activity could regulate the transcription of ZMPSTE24-targeting
miRNA, we investigated the expression of miR-141-3p using
DNMT- and HDAC-inhibitor-induced senescence. 5-AzaCtreatment did not affect the expression of miR-141-3p, whereas
VPA and NB treatment clearly increased the miR-141-3pexpression levels compared with negative controls (Fig. 5D–F).Moreover, inhibition of HDAC1 and HDAC2 significantly
increased miR-141-3p expression in hMSCs (Fig. 5G). In theVPA- and NB-treated groups, the acetylation of histones H3, H4and H3K4Me3 increased, whereas the acetylation of H3K9Me3and H3K27Me3 decreased or remained relatively unchanged in
the putative coding region of miR-141-3p (Fig. 5H). RNApolymerase II expression was induced around the miR-141-3pcoding region by HDAC-inhibitor-induced cellular senescence,
which suggests that the miR-141-3p coding region promotestranscription of miR-141-3p (Fig. 5I).
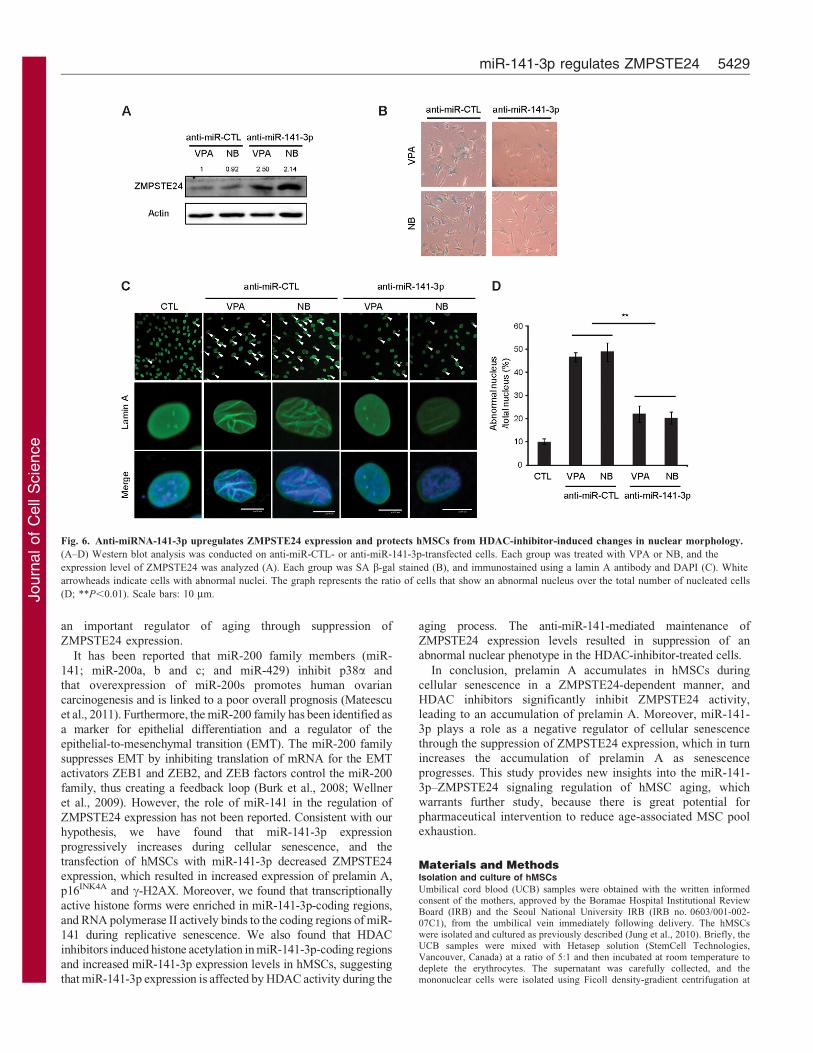
To identify whether the change in ZMPSTE24 expression
caused by HDAC inhibitors is dependent on the miR-141-3p, wetransfected anti-miR-141-3p into hMSCs after treating them withHDAC inhibitors. As expected, transfection of anti-miR-141-3p
attenuated HDAC-inhibitor-induced effects on ZMPSTE24expression (Fig. 6A). The upregulation of ZMPSTE24expression further prevented HDAC-inhibitor-induced SA-b-gal
activity and changes in hMSC nuclear morphology, with manycells exhibiting folds in the nuclear envelope (Fig. 6B–D). Takentogether, these results indicate that miR-141-3p transcription isregulated by histone modification and RNA polymerase II
activity at the miR-141-3p promoter region during replicativeand HDAC-inhibitor-induced senescence.
DiscussionhMSCs are essential for endogenous tissue generation,
maintenance and injury repair throughout the adult life ofmammals. Therefore, it has been proposed that MSC aging mightlead to a reduction in tissue regeneration and a decline of
physiological functions (Sethe et al., 2006). However, thepotential molecular mechanisms controlling hMSC aging arenot completely understood.
Prelamin A accumulated in hMSCs during cellular senescence,and the overexpression of prelamin A induced cellularsenescence, suggesting that prelamin A is a causal factor in the
initiation of hMSC aging rather than a consequence of it. It hasbeen reported that aged vascular smooth muscle cells rapidlyaccumulate prelamin A, which coincides with reduced levels ofZMPSTE24, growth defects and nuclear abnormality (Ragnauth
et al., 2010). These effects can be partially attenuated by theoverexpression of ZMPSTE24 through suppressing expression ofprelamin A (Candelario et al., 2008). Similarly, we observed a
significant decrease in ZMPSTE24, which converts prelamin Ainto mature lamin A, during replicative senescence. These datasuggest that ZMPSTE24 is largely responsible for prelamin A
accumulation during aging. However, it remains to be determinedwhether the increase in p16INK4A in the siZMPSTE24-transfectedcells is due to ZMPSTE24 itself or the accumulation of
prelamin A.
The inhibition of HDACs induces the accumulation ofacetylated histone forms, which modulate gene transcription by
altering the chromatin structure and transcription factorcomplexes (Xu et al., 2007). In hMSCs, HDAC inhibitorsdecrease stemness and induce cellular senescence by affecting
the expression of c-MYC, polycomb group genes and p16INK4A
(Jung et al., 2010; Lee et al., 2009). In this study, we presentdirect evidence that HDAC inhibitors significantly inhibit
Fig. 4. miRNA-141-3p regulates ZMPSTE24 expression in vivo.
(A) hMSCs were infected with miR-141-3p virus in frame with a GFP tag.
(B) miR-141-3p expression levels in GFP-control-virus- or miR-141-3p-virus-
infected cells were investigated by real-time qPCR (**P,0.01). (C–E) Mice
were i.p. injected with GFP control virus or miR-141-3p virus each day for 7
days and then the liver tissues were obtained. Immunohistochemistry was
performed to detect GFP expression in miR-141-3p-virus-injected liver
tissues (C). The miR-141-3p expression levels were investigated by real-time
qPCR (D). Western blot analysis was performed to investigate Zmpste24
protein expression (E; **P,0.01).
miR-141-3p regulates ZMPSTE24 5427

Journ
alof
Cell
Scie
nce
ZMPSTE24 activity, leading to an accumulation of prelamin A
after 6 days of treatment. We also demonstrate that, among the
multiple targets of HDAC inhibitors, HDAC1 and HDAC2 can
specifically affect the expression of ZMPSTE24 and prelamin A.
Importantly, our observations in hMSCs are consistent with those
of Miller et al., who recently reported that HDAC1 and HDAC2
are involved in the DNA damage response and are required for
double-stranded break repair (Miller et al., 2010). However, in
mouse Zmpste242/2 cells, treatment with HDAC inhibitors,
including NB, promoted repair protein recruitment to DNA
damage sites and substantially ameliorated aging-associated
phenotypes both in vitro and in vivo (Krishnan et al., 2011).
Further studies are required to identify which factor causes the
different results in HDAC inhibitor treatment of Zmpste242/2
cells and MSCs. Epigenetic alterations of Zmpste242/2 cells,
including a notable decrease in the acetylation status of histone
H2B and H4, may be responsible for the different results.
Although some studies have reported that ZMPSTE24
expression decreases in senescent cells or tissues (Ragnauth
et al., 2010; Ukekawa et al., 2007), the molecular mechanisms
that modulate ZMPSTE24 expression during aging have not yet
been elucidated. miRNA is a potent regulator of various target
genes and the expression of nearly 60% of protein-coding RNAs
are affected by miRNAs (Friedman et al., 2009). However, a
significant number of miRNAs are regulated by epigenetic
modulations. A short exposure of the breast cancer cell line
SKBr3 to a HDAC inhibitor changed 40% of the miRNA
population significantly (Scott et al., 2006). Thus, we focused on
miRNA as a candidate regulator of ZMPSTE24, which is
controlled by HDAC activity during senescence processes. To
address this issue, we searched for miRNAs that target
ZMPSTE24 during the aging process. Combining the results
obtained using different predictive software prompted us to
consider that transcriptional activation of miR-141-3p could be
Fig. 5. Replicative- and HDAC-inhibitor-induced senescent cells show increased miR-141-3p expression with active histone markers at the miR-141-3p
promoter. (A) miR-141-3p expression levels were investigated following consecutive passages (**P,0.01). (B,C) ChIP analysis was performed on replicative
senescent cells, using antibodies targeted to the indicated proteins (AcetylH3, AcetylH4, H3K4Me3, H3K9Me3, H3K27Me3 and RNA polymerase II). The fold
enrichment of AcetylH3, AcetylH4, H3K4Me3, H3K9Me3, H3K27Me3 (B) and RNA polymerase II (C) proteins near the miR-141-3p genomic region were
investigated by real-time qPCR (*P,0.05; **P,0.01). (D) miR-141-3p expression levels were investigated following 5-AzaC treatment. (E,F) miR-141-3p
expression levels were investigated in HDAC inhibitor (VPA/NB)-treated cells. (G) After transfection with siHDAC1 and siHDAC2, the miR-141-3p expression
levels were investigated (**P,0.01). (H–I) ChIP analysis was performed on HDAC-inhibitor-treated cells, using antibodies targeted to the indicated proteins
(AcetylH3, AcetylH4, H3K4Me3, H3K9Me3, H3K27Me3 and RNA polymerase II). The fold enrichment of the AcetylH3, AcetylH4, H3K4Me3, H3K9Me3,
H3K27Me3 (H) and RNA polymerase II (I) proteins near the miR-141-3p genomic region was investigated by real-time qPCR. The graphs represent the relative
expression in cells treated with VPA or NB compared with control cells (**P,0.01).
Journal of Cell Science 126 (23)5428
Journ
alof
Cell
Scie
nce
an important regulator of aging through suppression of
ZMPSTE24 expression.
It has been reported that miR-200 family members (miR-
141; miR-200a, b and c; and miR-429) inhibit p38a and
that overexpression of miR-200s promotes human ovarian
carcinogenesis and is linked to a poor overall prognosis (Mateescu
et al., 2011). Furthermore, the miR-200 family has been identified as
a marker for epithelial differentiation and a regulator of the
epithelial-to-mesenchymal transition (EMT). The miR-200 family
suppresses EMT by inhibiting translation of mRNA for the EMT
activators ZEB1 and ZEB2, and ZEB factors control the miR-200
family, thus creating a feedback loop (Burk et al., 2008; Wellner
et al., 2009). However, the role of miR-141 in the regulation of
ZMPSTE24 expression has not been reported. Consistent with our
hypothesis, we have found that miR-141-3p expression
progressively increases during cellular senescence, and the
transfection of hMSCs with miR-141-3p decreased ZMPSTE24
expression, which resulted in increased expression of prelamin A,
p16INK4A and c-H2AX. Moreover, we found that transcriptionally
active histone forms were enriched in miR-141-3p-coding regions,
and RNA polymerase II actively binds to the coding regions of miR-
141 during replicative senescence. We also found that HDAC
inhibitors induced histone acetylation in miR-141-3p-coding regions
and increased miR-141-3p expression levels in hMSCs, suggesting
that miR-141-3p expression is affected by HDAC activity during the
aging process. The anti-miR-141-mediated maintenance of
ZMPSTE24 expression levels resulted in suppression of an
abnormal nuclear phenotype in the HDAC-inhibitor-treated cells.
In conclusion, prelamin A accumulates in hMSCs during
cellular senescence in a ZMPSTE24-dependent manner, and
HDAC inhibitors significantly inhibit ZMPSTE24 activity,
leading to an accumulation of prelamin A. Moreover, miR-141-
3p plays a role as a negative regulator of cellular senescence
through the suppression of ZMPSTE24 expression, which in turn
increases the accumulation of prelamin A as senescence
progresses. This study provides new insights into the miR-141-
3p–ZMPSTE24 signaling regulation of hMSC aging, which
warrants further study, because there is great potential for
pharmaceutical intervention to reduce age-associated MSC pool
exhaustion.
Materials and MethodsIsolation and culture of hMSCs
Umbilical cord blood (UCB) samples were obtained with the written informedconsent of the mothers, approved by the Boramae Hospital Institutional ReviewBoard (IRB) and the Seoul National University IRB (IRB no. 0603/001-002-07C1), from the umbilical vein immediately following delivery. The hMSCswere isolated and cultured as previously described (Jung et al., 2010). Briefly, theUCB samples were mixed with Hetasep solution (StemCell Technologies,Vancouver, Canada) at a ratio of 5:1 and then incubated at room temperature todeplete the erythrocytes. The supernatant was carefully collected, and themononuclear cells were isolated using Ficoll density-gradient centrifugation at
Fig. 6. Anti-miRNA-141-3p upregulates ZMPSTE24 expression and protects hMSCs from HDAC-inhibitor-induced changes in nuclear morphology.
(A–D) Western blot analysis was conducted on anti-miR-CTL- or anti-miR-141-3p-transfected cells. Each group was treated with VPA or NB, and the
expression level of ZMPSTE24 was analyzed (A). Each group was SA b-gal stained (B), and immunostained using a lamin A antibody and DAPI (C). White
arrowheads indicate cells with abnormal nuclei. The graph represents the ratio of cells that show an abnormal nucleus over the total number of nucleated cells
(D; **P,0.01). Scale bars: 10 mm.
miR-141-3p regulates ZMPSTE24 5429

Journ
alof
Cell
Scie
nce
1200 g for 20 minutes. The cells were washed twice in PBS and were seeded at adensity of 26105 to 26106 cells/cm2 on plates in growth medium consisting of D-medium (formula no. 78-5470EF, Gibco BRL, USA) containing EGM-2SingleQuot and 10% fetal bovine serum (Gibco BRL). After 3 days, the non-adherent cells were removed. For long-term culture, the cells were seeded at adensity of 46105 cells/10 cm plate, and the cells were subcultured upon reaching80,90% confluency.
Senescence-associated beta-galactosidase (SA b-gal) stainingThe SA b-gal staining was performed as previously described (Yu et al., 2013).The hMSCs were seeded on 6-well plates at a density of 16105 cells/well for late-passage cells and 56104 cells/well for early-passage cells. The cells wereincubated for 3 days until reaching the appropriate confluency. The cells were thenwashed twice with PBS and fixed with 0.5% glutaraldehyde in PBS (pH 7.2) for5 minutes at room temperature. The cells were then washed with PBS containingMgCl2 (pH 7.2, 1 mM MgCl2) and stained with X-gal solution [1 mg/ml X-gal,0.12 mM K3Fe(CN)6, 1 mM MgCl2 in PBS, pH 6.0] overnight at 37 C. The cellswere washed twice with PBS, and the images were captured using a microscope(IX70, Olympus, Japan).
MTT assayThe proliferative potential of the cells was measured using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, Sigma-Aldrich,USA) assay, which is based on the ability of live cells to convert a tetrazoliumsalt into purple formazan. hMSCs (20,000 per well) were seeded in 24-well plates.After 48 hours incubation, 50 ml MTT stock solution (5 mg/ml; Sigma) was addedto each well, and the plates were further incubated for 4 hours at 37 C. Thesupernatant was removed, and 200 ml of DMSO was added to each well tosolubilize the water-insoluble purple formazan crystals; the solution was thentransferred to a 96-well microplate for reading. The absorbance at a wavelength of540 nm was measured using an EL800 microplate reader (BIO-TEK Instruments,Winooski, VT, USA). All of the measurements were performed in triplicate.
Western blot analysisWestern blot analyses of lamin A, prelamin A, ZMPSTE24, HDAC1, HDAC2,DNMT1, DNMT3B, p16Ink4A, c-H2AX and b-actin were performed as previouslydescribed (Jung et al., 2005). The hMSCs were lysed with 50 mM Tris-HCl buffercontaining 0.1% Triton X-100 and freshly supplemented with a protease/phosphatase inhibitor cocktail. The proteins were then separated using 7.5–15%SDS-PAGE and transferred to nitrocellulose membranes at 350 mA for 5 hours.The primary antibodies used to detect each protein were as follows: lamin A(monoclonal, Abcam, 1:2500); prelamin A (polyclonal, Santa Cruz, 1:500);ZMPSTE24 (polyclonal, Abcam, 1:500); HDAC1 (monoclonal, Upstate, 1:2000);HDAC2 (monoclonal, Upstate, 1:2000); DNMT1 (polyclonal, BD, 1:1000);DNMT3A (polyclonal, Millipore, 1:1000); DNMT3B (polyclonal, Abcam,
1:1000); p16Ink4A (polyclonal, Abcam, 1:1000); c-H2AX (polyclonal, Abcam,1:1000) and b-actin (monoclonal, Cell-signaling, 1:5000). All of the antibodieswere used according to the manufacturer’s instructions, and the protein bands weredetected using an enhanced chemiluminescence detection kit (AmershamPharmacia Biotech, UK).
Viral packaging and cell infectionCells expressing GFP-wt-lamin A and GFP-progerin were obtained from Addgene(Cambridge, MA; Plasmid 17662, 17663), and the miR-141 lentiviral vector waspurchased from Genecopoeia (Rockville, MD; HmiR0181). Viral production andinfection were performed as previously described (Yu et al., 2012b). Using theVSVG-based package system or the Mission lentiviral packaging mix (Sigma,Ronkonkoma, NY, USA) 6 mg of vectors were added to tubes containing theFugene 6 transfection reagent (Roche, Basel, Switzerland). The plasmids weretransfected into 293FT cells; after 48 and 72 hours, the viral supernatants werefiltered through a 0.45 mm cellulose acetate filter and concentrated bycentrifugation at 50,000 g for 90 minutes. The viral supernatants were then usedto infect the human MSCs in the presence of polybrene at 5 mg/ml (Sigma).
RT-PCRThe total cellular RNA was extracted from cells using the TRIzol reagentTM
(Invitrogen, USA) according to the manufacturer’s instructions. The cDNA wassynthesized by adding the purified RNA and oligo(dT) primers to Accupower RTpremix (Bioneer, Korea), according to the manufacturer’s instructions. For themiRNA cDNA synthesis, the NCode VILO miRNA cDNA Synthesis Kit(Invitrogen, USA) was used.
Real-time quantitative PCRReal-time qPCR analyses were performed using SYBRH Green (AppliedBiosystems, USA) according to the manufacturer’s protocol. For the miRNAreal-time qPCR, the universal primers supplied with the NCode VILO miRNAcDNA Synthesis Kit (Invitrogen, USA) and miRNA-specific primers were used.
RPL13A was used as an internal control. All of the amplicons were analyzed usingthe Prism 7000 sequence detection system 2.1 software (Applied Biosystems,USA). The primer set sequences used for this study are listed in supplementarymaterial Table S1.
siRNA, mature miRNA and anti-miRNA transfection studies
Transient transfection assays were performed using specific, commerciallyavailable siRNAs for the inhibition of ZMPSTE24 (L-006104-00-0005), alongwith a non-targeting siRNA (D-001810-10; ON Target plus SMART pool,Dharmacon, USA). The inhibition or overexpression of the miRNAs was achievedby commercial antisense miRNAs or mature miRNAs for hsa-miR-141-3p with anappropriate miRNA precursor-negative control [mature miR-141-3p mimic-1 no.PM 10860 and anti-miR-141-3p inhibitor no. AM 10860, and miRNA precursor-negative control no. 1; Ambion, USA]. The siRNA, mature miRNA and anti-miRNA transfections were performed using the Dharmafect transfection reagent(Dharmacon) according to the manufacturer’s instructions. Briefly, the cells wereseeded at a concentration of 26104 cells/well, and the siRNA, miRNA and anti-miRNA-containing media (without the addition of antibiotics) were added whenthe cells reached 50–60% confluence. The cells were incubated with 50 nMsiRNA, 50 nM anti-miRNA or 50 nM mature miRNA for 48 hours. To investigatethe long-term effects of inhibition, the cells were subcultured 48–72 hoursfollowing the siRNA, anti-miRNA or mature miRNA transfection. Subculturedcells were stabilized for 24 hours and incubated with siRNA, anti-miRNA ormature miRNA, at the same concentrations, for 48–72 hours. After inhibition,RNA extraction and the subsequent real-time qPCR or SA b-gal staining wereperformed for genetic or characteristic analyses, respectively.
Immunocytochemistry
Cultured cells were fixed in 4% paraformaldehyde and permeabilized with 0.2% TritonX-100 (Sigma Aldrich, USA). The cells were then incubated with 10% normal goatserum (Zymed Laboratories Inc., USA) and stained with antibodies against lamin A(monoclonal, Abcam, 1:300), prelaimin A (polyclonal, Santa Cruz, 1:200), ZMPSTE24 (polyclonal, Abcam, 1:200), p16Ink4A (polyclonal, Abcam, 1:200), c-H2AX(polyclonal, Abcam, 1:200), followed by incubation for 1 hour with an Alexa-Fluor-488- or Alexa-Fluor-594-labeled secondary antibody (1:1000; Molecular Probes,USA). The nuclei were stained with Hoechst 33258 (1 mg/ml; 10 minutes), and theimages were captured using a confocal microscope (Eclipse TE200, Nikon, Japan).
Chromatin immunoprecipitation assays
The chromatin immunoprecipitation (ChIP) assays were performed according tothe manufacturer’s protocol (ChIP assay kit, Upstate Biotechnology, USA). Toprepare for the ChIP testing, the hMSCs (1–26107 cells per IP) were fixed with 1%formaldehyde for 10 minutes; the solution was then neutralized by the addition of1/20 volume of 2.5 M glycine for 5 minutes. The cells were washed with ice-coldPBS and scraped with SDS lysis buffer (1% SDS, 10 mM EDTA and 50 mM Tris,pH 8.1) containing protease inhibitors. The lysates were sonicated to shear theDNA to lengths between 200 and 1000 base pairs, and the samples werecentrifuged for 10 minutes at 15,000 g at 4 C to remove the insoluble material.The supernatant was diluted 10-fold in the ChIP dilution buffer [0.01% SDS, 1.1%Triton X-100, 1.2 mM EDTA, 16.7 mM Tris-HCl (pH 8.1) and 167 mM NaCl],and the chromatin was immunoprecipitated using antibodies, according to themanufacturer’s instructions. Real-time qPCR was performed at a final templatedilution of 1:50. The primer sequences used in the ChIP assays in this study aregiven in supplementary material Table S1.
Measurement of the proliferative potential and cell cycle distribution
The effects of replicative senescence, progerin, ZMPSTE24 inhibition and miR-141-3p inhibition on hMSC proliferation were measured using the MTT assay, asdescribed above (Jung et al., 2005).
Flow cytometry cell cycle analyses using propidium iodide staining was alsoperformed as previously described. Briefly, hMSCs in the exponential growthphase were transfected with siRNA or miRNA and then harvested bytrypsinization. The cells were washed with ice-cold PBS and then fixed with70% ethanol at 220 C and stained with 50 mg/ml of propidium iodide in thepresence of 100 mg/ml RNase A for 30 minutes. The cell cycle distribution wasanalyzed using the FACS Calibur system (Becton Dickinson, Franklin Lakes, NJ,USA).
Luciferase assays
For miRNA target validation, the entire 39UTR sequence of human ZMPSTE24was amplified by PCR and cloned into a T-vector (Promega, Madison, WI, USA,no. A1360). The 39UTR was subcloned into pmirGLO Dual-Luciferase vector(Promega, no. E1330) using the restriction enzymes, XhoI and SalI. The 293FTcells were seeded 24 hours prior to transfection in 24-well plates at 50%confluence. The control constructs and ZMPSTE24 39UTR reporter constructswere co-transfected along with 50 nM of the miRNAs (Ambion) usingDharmafect, following the manufacturer’s instruction. After 24 hours of
Journal of Cell Science 126 (23)5430

Journ
alof
Cell
Scie
nce
transfection, the firefly and Renilla luciferase activities were measured using aluminometer with Dual-Glo Luciferase Assay System (Promega, E2920). Thefirefly luminescence was normalized to the Renilla luminescence.
Statistical analysisAll of the experiments were conducted at least in triplicate, and the results areexpressed as the means 6 s.d. Statistical analyses were conducted using ananalysis of variance (ANOVA), followed by Duncan’s multiple range tests orStudent’s t-test. A value of P,0.05 was considered significant (*P,0.05;**P,0.01).
Author contributionsK.-R.Y.: conception and design, collection and/or assembly of data,data analysis and interpretation, manuscript writing. S.H.L.:conception and design, collection and/or assembly of data,manuscript writing. J.-W.J.: conception and design, manuscriptwriting. I.-S.H.: data analysis and interpretation, manuscript writing.H.-S.K.: collection and/or assembly of data, data analysis andinterpretation. Y.S., T.S.: data analysis and interpretation. K.-S.K.:conception and design, administrative support, final approval ofmanuscript, manuscript writing.
FundingThis work was supported by the Bio and Medical TechnologyDevelopment Program [grant number MEST 2010-0020265]; aNational Junior Research Fellowship from the National ResearchFoundation funded by the Ministry of Science, Information andCommunications Technology and Future Planning, Korea [grantnumber 2012H1A8002383]; the Research Institute for VeterinaryScience, Seoul National University; the Research Institute forAgriculture and Life Sciences, Seoul National University.
Supplementary material available online at
http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.133314/-/DC1
ReferencesBartel, D. P. (2004). MicroRNAs: genomics, biogenesis, mechanism, and function. Cell
116, 281-297.Bergo, M. O., Gavino, B., Ross, J., Schmidt, W. K., Hong, C., Kendall, L. V., Mohr,
A., Meta, M., Genant, H., Jiang, Y. et al. (2002). Zmpste24 deficiency in micecauses spontaneous bone fractures, muscle weakness, and a prelamin A processingdefect. Proc. Natl. Acad. Sci. USA 99, 13049-13054.
Bibikova, M., Laurent, L. C., Ren, B., Loring, J. F. and Fan, J. B. (2008). Unravelingepigenetic regulation in embryonic stem cells. Cell Stem Cell 2, 123-134.
Bridger, J. M. and Kill, I. R. (2004). Aging of Hutchinson-Gilford progeria syndromefibroblasts is characterised by hyperproliferation and increased apoptosis. Exp.
Gerontol. 39, 717-724.Burk, U., Schubert, J., Wellner, U., Schmalhofer, O., Vincan, E., Spaderna, S. and
Brabletz, T. (2008). A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 9, 582-589.
Candelario, J., Sudhakar, S., Navarro, S., Reddy, S. and Comai, L. (2008).Perturbation of wild-type lamin A metabolism results in a progeroid phenotype. Aging
Cell 7, 355-367.Corrigan, D. P., Kuszczak, D., Rusinol, A. E., Thewke, D. P., Hrycyna, C. A.,
Michaelis, S. and Sinensky, M. S. (2005). Prelamin A endoproteolytic processing invitro by recombinant Zmpste24. Biochem. J. 387, 129-138.
Delcuve, G. P., Khan, D. H. and Davie, J. R. (2012). Roles of histone deacetylases inepigenetic regulation: emerging paradigms from studies with inhibitors. Clin
Epigenetics 4, 5.Delgado, M., Toscano, M. G., Benabdellah, K., Cobo, M., O’Valle, F., Gonzalez-
Rey, E. and Martın, F. (2008). In vivo delivery of lentiviral vectors expressingvasoactive intestinal peptide complementary DNA as gene therapy for collagen-induced arthritis. Arthritis Rheum. 58, 1026-1037.
Dismuke, A. D., Kohn, A. D., Moon, R. T. and Wong, M. H. (2009). Lentiviral-mediated transgene expression can potentiate intestinal mesenchymal-epithelialsignaling. Biol. Proced. Online 11, 130-144.
Friedman, R. C., Farh, K. K., Burge, C. B. and Bartel, D. P. (2009). Mostmammalian mRNAs are conserved targets of microRNAs. Genome Res. 19, 92-105.
Halaschek-Wiener, J. and Brooks-Wilson, A. (2007). Progeria of stem cells: stem cellexhaustion in Hutchinson-Gilford progeria syndrome. J. Gerontol. A Biol. Sci. Med.
Sci. 62, 3-8.Janzen, V., Forkert, R., Fleming, H. E., Saito, Y., Waring, M. T., Dombkowski,
D. M., Cheng, T., DePinho, R. A., Sharpless, N. E. and Scadden, D. T. (2006).
Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a.Nature 443, 421-426.
Jung, J. W., Cho, S. D., Ahn, N. S., Yang, S. R., Park, J. S., Jo, E. H., Hwang, J. W.,
Jung, J. Y., Kim, S. H., Kang, K. S. et al. (2005). Ras/MAP kinase pathways areinvolved in Ras specific apoptosis induced by sodium butyrate. Cancer Lett. 225, 199-206.
Jung, J. W., Lee, S., Seo, M. S., Park, S. B., Kurtz, A., Kang, S. K. and Kang, K. S.
(2010). Histone deacetylase controls adult stem cell aging by balancing the expressionof polycomb genes and jumonji domain containing 3. Cell. Mol. Life Sci. 67, 1165-1176.
Kasper, G., Mao, L., Geissler, S., Draycheva, A., Trippens, J., Kuhnisch, J.,Tschirschmann, M., Kaspar, K., Perka, C., Duda, G. N. et al. (2009). Insights intomesenchymal stem cell aging: involvement of antioxidant defense and actincytoskeleton. Stem Cells 27, 1288-1297.
Krishnan, V., Chow, M. Z., Wang, Z., Zhang, L., Liu, B., Liu, X. and Zhou,
Z. (2011). Histone H4 lysine 16 hypoacetylation is associated with defective DNArepair and premature senescence in Zmpste24-deficient mice. Proc. Natl. Acad. Sci.
USA 108, 12325-12330.
Lee, S., Park, J. R., Seo, M. S., Roh, K. H., Park, S. B., Hwang, J. W., Sun, B., Seo, K.,Lee, Y. S., Kang, S. K. et al. (2009). Histone deacetylase inhibitors decreaseproliferation potential and multilineage differentiation capability of human mesenchymalstem cells. Cell Prolif. 42, 711-720.
Maraldi, N. M., Capanni, C., Cenni, V., Fini, M. and Lattanzi, G. (2011).Laminopathies and lamin-associated signaling pathways. J. Cell. Biochem. 112, 979-992.
Mateescu, B., Batista, L., Cardon, M., Gruosso, T., de Feraudy, Y., Mariani, O.,
Nicolas, A., Meyniel, J. P., Cottu, P., Sastre-Garau, X. et al. (2011). miR-141 andmiR-200a act on ovarian tumorigenesis by controlling oxidative stress response. Nat.
Med. 17, 1627-1635.
Miller, K. M., Tjeertes, J. V., Coates, J., Legube, G., Polo, S. E., Britton, S. and
Jackson, S. P. (2010). Human HDAC1 and HDAC2 function in the DNA-damageresponse to promote DNA nonhomologous end-joining. Nat. Struct. Mol. Biol. 17,1144-1151.
Narita, M. (2007). Cellular senescence and chromatin organisation. Br. J. Cancer 96,686-691.
Pendas, A. M., Zhou, Z., Cadinanos, J., Freije, J. M., Wang, J., Hultenby, K.,
Astudillo, A., Wernerson, A., Rodrıguez, F., Tryggvason, K. et al. (2002).Defective prelamin A processing and muscular and adipocyte alterations in Zmpste24metalloproteinase-deficient mice. Nat. Genet. 31, 94-99.
Ragnauth, C. D., Warren, D. T., Liu, Y., McNair, R., Tajsic, T., Figg, N., Shroff, R.,Skepper, J. and Shanahan, C. M. (2010). Prelamin A acts to accelerate smoothmuscle cell senescence and is a novel biomarker of human vascular aging. Circulation
121, 2200-2210.
Rao, M. S. and Mattson, M. P. (2001). Stem cells and aging: expanding thepossibilities. Mech. Ageing Dev. 122, 713-734.
Scaffidi, P. and Misteli, T. (2006). Lamin A-dependent nuclear defects in human aging.Science 312, 1059-1063.
Scaffidi, P. and Misteli, T. (2008). Lamin A-dependent misregulation of adult stemcells associated with accelerated ageing. Nat. Cell Biol. 10, 452-459.
Scott, G. K., Mattie, M. D., Berger, C. E., Benz, S. C. and Benz, C. C. (2006). Rapidalteration of microRNA levels by histone deacetylase inhibition. Cancer Res. 66,1277-1281.
Sethe, S., Scutt, A. and Stolzing, A. (2006). Aging of mesenchymal stem cells. Ageing
Res. Rev. 5, 91-116.
So, A. Y., Jung, J. W., Lee, S., Kim, H. S. and Kang, K. S. (2011). DNAmethyltransferase controls stem cell aging by regulating BMI1 and EZH2 throughmicroRNAs. PLoS ONE 6, e19503.
Trosko, J. E. (2008). Role of diet and nutrition on the alteration of the quality andquantity of stem cells in human aging and the diseases of aging. Curr. Pharm. Des.
14, 2707-2718.
Ukekawa, R., Miki, K., Fujii, M., Hirano, H. and Ayusawa, D. (2007). Accumulationof multiple forms of lamin A with down-regulation of FACE-1 suppresses growth insenescent human cells. Genes Cells 12, 397-406.
Wagner, W., Horn, P., Castoldi, M., Diehlmann, A., Bork, S., Saffrich, R., Benes,
V., Blake, J., Pfister, S., Eckstein, V. et al. (2008). Replicative senescence ofmesenchymal stem cells: a continuous and organized process. PLoS ONE 3, e2213.
Wellner, U., Schubert, J., Burk, U. C., Schmalhofer, O., Zhu, F., Sonntag, A.,
Waldvogel, B., Vannier, C., Darling, D., zur Hausen, A. et al. (2009). The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibitingmicroRNAs. Nat. Cell Biol. 11, 1487-1495.
Xu, W. S., Parmigiani, R. B. and Marks, P. A. (2007). Histone deacetylase inhibitors:molecular mechanisms of action. Oncogene 26, 5541-5552.
Yu, K. R., Yang, S. R., Jung, J. W., Kim, H., Ko, K., Han, D. W., Park, S. B., Choi,
S. W., Kang, S. K., Scholer, H. et al. (2012). CD49f enhances multipotency andmaintains stemness through the direct regulation of OCT4 and SOX2. Stem Cells 30,876-887.
Yu, K. R., Park, S. B., Jung, J. W., Seo, M. S., Hong, I. S., Kim, H. S., Seo, Y., Kang,
T. W., Lee, J. Y., Kurtz, A. et al. (2013). HMGA2 regulates the in vitro aging andproliferation of human umbilical cord blood-derived stromal cells through the mTOR/p70S6K signaling pathway. Stem Cell Res. 10, 156-165.
miR-141-3p regulates ZMPSTE24 5431
![Original Article MicroRNA-28-3p promotes fracture … promotes fracture healing through inhibition of Sox6 and ... Base () [9]. ... utilizing X- tremeGENE siRNA Transfection ...](https://static.fdocuments.us/doc/165x107/5b2827607f8b9a026e8b4b55/original-article-microrna-28-3p-promotes-fracture-promotes-fracture-healing-through.jpg)
![· Web view[Abstract] Objective To detect the expression of microRNA-338-3p (miR-338-3p) and MET transcriptional regulator MACC1 (MACC1) gene in different ovarian tissues, to analyze](https://static.fdocuments.us/doc/165x107/5e7a68666f8914127e1fd339/web-view-abstract-objective-to-detect-the-expression-of-microrna-338-3p-mir-338-3p.jpg)
















