
Microfluidic reactor for the radiosynthesis of PET radiotracers
8
Applied Radiation and Isotopes 64 (2006) 325–332 Microfluidic reactor for the radiosynthesis of PET radiotracers J.M. Gillies a, , C. Prenant a,b , G.N. Chimon a,b , G.J. Smethurst a , W. Perrie c , I. Hamblett a , B. Dekker a , J. Zweit a,b a Cancer Research-UK/UMIST Radiochemical Targeting and Imaging Group, Paterson Institute for Cancer Research, Manchester M20 4BX, UK b School of Chemical Engineering and Analytical Sciences, University of Manchester, PO Box 88, Manchester M60 1QD, UK c Department of Engineering, University of Liverpool, Liverpool L69 3GH, UK Received 13 January 2005; accepted 30 August 2005 Abstract Here we show the first application of a microfabricated reaction system to PET radiochemistry, we term ‘‘microfluidic PET’’. The short half-life of the positron emitting isotopes and the trace chemical quantities used in radiolabelling make PET radiochemistry amenable to miniaturisation. Microfluidic technologies are capable of controlling and transferring tiny quantities of liquids which allow chemical and biochemical assays to be integrated and carried out on a small scale. Such technologies provide distinct advantages over current methods of PET radiochemical synthesis. To demonstrate ‘‘proof of principle’’ we have investigated the radiohalogenation of small and large molecular weight molecules using the microfluidic device. These reactions involved the direct radioiodination of the apoptosis marker Annexin V using iodine-124, the indirect radioiodination of the anti-cancer drug doxorubicin from a tin-butyl precursor and the radiosynthesis of 2-[ 18 F]FDG from a mannose triflate precursor and fluorine-18 and hence provide a test bed for microfluidic reactions. We demonstrate the rapid radioiodination of the protein Annexin V (40% radiochemical yield within 1 min) and the rapid radiofluorination of 2-[ 18 F]FDG (60% radiochemical yield within 4 s) using a polymer microreactor chip. Chromatographic analysis showed that the labelling efficiency of the unoptimised microfluidic chip is comparable to conventional PET radiolabelling reactions. r 2005 Elsevier Ltd. All rights reserved. Keywords: Molecular imaging; Pet radiochemistry; Microfluidics; Nanotechnology 1. Introduction Molecular positron emission tomography (PET) imaging allows the study of molecular and cellular processes associated with diseases including cancer (Gambhir, 2002; Massoud and Gambhir, 2003; Reader and Zweit, 2001). PET is an in vivo molecular imaging technique based on the external detection of biomolecules labelled with positron emitting isotopes. The development of micro- fluidic PET radiochemistry has the potential to further advance molecular PET imaging, which is emerging as an important technology in the post genomic era of molecular medicine. The short half-life of the positron emitting isotopes and the trace chemical quantities used in radiolabelling make PET radiochemistry amenable to miniaturisation. Microfluidic, technologies, also known as ‘‘lab on a chip’’ technologies, are capable of controlling and transfer- ring quantities of liquids which allow chemical and biochemical assays to be integrated and carried out on a small scale (Regenfuss et al., 1985; Mitchell, 2001; Ramsey, 1999). Such technologies provide distinct advantages over current methods of PET radiochemical synthesis. Signifi- cantly, radiochemical reactions on a microfabricated device (chip) can be easily shielded and will not need the space and resources required for conventional hot cell synthesis. Secondly, it provides scope for an integrated total system (synthesis, purification and analysis). Thirdly, due to the ARTICLE IN PRESS www.elsevier.com/locate/apradiso 0969-8043/$ - see front matter r 2005 Elsevier Ltd. All rights reserved. doi:10.1016/j.apradiso.2005.08.007 Corresponding author. Radiochemical Targeting and Imaging, Pater- son Institute for Cancer Research, Christie Hospital NHS Trust, Wilmslow Road, Manchester M20 4BX, UK. Tel.: +44 161 446 3150; fax: +44 161 446 3109. E-mail address: [email protected] (J.M. Gillies).
-
Upload
jm-gillies -
Category
Documents
-
view
216 -
download
2
Transcript of Microfluidic reactor for the radiosynthesis of PET radiotracers

ARTICLE IN PRESS
0969-8043/$ - se
doi:10.1016/j.ap
�Correspondson Institute
Wilmslow Roa
fax: +44161 44
E-mail addr
Applied Radiation and Isotopes 64 (2006) 325–332
www.elsevier.com/locate/apradiso
Microfluidic reactor for the radiosynthesis of PET radiotracers
J.M. Gilliesa,�, C. Prenanta,b, G.N. Chimona,b, G.J. Smethursta, W. Perriec,I. Hambletta, B. Dekkera, J. Zweita,b
aCancer Research-UK/UMIST Radiochemical Targeting and Imaging Group, Paterson Institute for Cancer Research, Manchester M20 4BX, UKbSchool of Chemical Engineering and Analytical Sciences, University of Manchester, PO Box 88, Manchester M60 1QD, UK
cDepartment of Engineering, University of Liverpool, Liverpool L69 3GH, UK
Received 13 January 2005; accepted 30 August 2005
Abstract
Here we show the first application of a microfabricated reaction system to PET radiochemistry, we term ‘‘microfluidic PET’’. The
short half-life of the positron emitting isotopes and the trace chemical quantities used in radiolabelling make PET radiochemistry
amenable to miniaturisation. Microfluidic technologies are capable of controlling and transferring tiny quantities of liquids which allow
chemical and biochemical assays to be integrated and carried out on a small scale. Such technologies provide distinct advantages over
current methods of PET radiochemical synthesis. To demonstrate ‘‘proof of principle’’ we have investigated the radiohalogenation of
small and large molecular weight molecules using the microfluidic device. These reactions involved the direct radioiodination of the
apoptosis marker Annexin V using iodine-124, the indirect radioiodination of the anti-cancer drug doxorubicin from a tin-butyl
precursor and the radiosynthesis of 2-[18F]FDG from a mannose triflate precursor and fluorine-18 and hence provide a test bed for
microfluidic reactions. We demonstrate the rapid radioiodination of the protein Annexin V (40% radiochemical yield within 1min)
and the rapid radiofluorination of 2-[18F]FDG (60% radiochemical yield within 4 s) using a polymer microreactor chip.
Chromatographic analysis showed that the labelling efficiency of the unoptimised microfluidic chip is comparable to conventional
PET radiolabelling reactions.
r 2005 Elsevier Ltd. All rights reserved.
Keywords: Molecular imaging; Pet radiochemistry; Microfluidics; Nanotechnology
1. Introduction
Molecular positron emission tomography (PET) imagingallows the study of molecular and cellular processesassociated with diseases including cancer (Gambhir, 2002;Massoud and Gambhir, 2003; Reader and Zweit, 2001).PET is an in vivo molecular imaging technique based onthe external detection of biomolecules labelled withpositron emitting isotopes. The development of micro-fluidic PET radiochemistry has the potential to furtheradvance molecular PET imaging, which is emerging as an
e front matter r 2005 Elsevier Ltd. All rights reserved.
radiso.2005.08.007
ing author. Radiochemical Targeting and Imaging, Pater-
for Cancer Research, Christie Hospital NHS Trust,
d, Manchester M20 4BX, UK. Tel.: +44 161 446 3150;
6 3109.
ess: [email protected] (J.M. Gillies).
important technology in the post genomic era of molecularmedicine. The short half-life of the positron emittingisotopes and the trace chemical quantities used inradiolabelling make PET radiochemistry amenable tominiaturisation.Microfluidic, technologies, also known as ‘‘lab on a
chip’’ technologies, are capable of controlling and transfer-ring quantities of liquids which allow chemical andbiochemical assays to be integrated and carried out on asmall scale (Regenfuss et al., 1985; Mitchell, 2001; Ramsey,1999). Such technologies provide distinct advantages overcurrent methods of PET radiochemical synthesis. Signifi-cantly, radiochemical reactions on a microfabricated device(chip) can be easily shielded and will not need the space andresources required for conventional hot cell synthesis.Secondly, it provides scope for an integrated total system(synthesis, purification and analysis). Thirdly, due to the

ARTICLE IN PRESSJ.M. Gillies et al. / Applied Radiation and Isotopes 64 (2006) 325–332326
rapid and thorough mixing achieved in miniaturisedreactors (Fletcher et al., 2002), the speed and radiochemicalyield of radiochemical syntheses could be enhanced.Finally, the photolithographic fabrication of the micro-fabricated device allows the manufacture of complex, yetrelatively inexpensive and disposable devices (Stuernstromand Roeraade, 1998; Lin et al., 2001; Tsai and Lin, 2001).
Currently, radiosynthesis of compounds labelled withpositron emitting radioisotopes are carried out in lead-shielded ‘‘hot cells’’ using automated systems in order toprevent radiation exposure to the operators. Such auto-mated systems carry out a range of operations such asheating, cooling, transfer of liquids and gaseous reagents,mixing, evaporation and distillation. Essentially, theyperform all the operations used by radiochemists. Thereare essentially two types of systems currently in operation(Luthra et al., 1994; Crouzel et al., 1987; Crouzel et al.,1993). These are either static systems, e.g., the GEMSTracerLab MX FDG Synthesizer, in which switchingvalves and transfer lines made from inert materials areused to transfer reagents around the system, or roboticsystems, e.g. SYNTHIA (Bjurling et al., 1996), in which arobot performs some or most of the operations. Thesesystems are generally designed to handle total liquidvolumes in the range of 0.2–0.5mL.
Regardless of the automation approach, all of theexisting automated radiosynthesis systems, static and/orrobotic, share some common features. To ensure practicaltransfer of reagents etc. around the system, small volumesof liquids, usually of the order of 500 mL, are used assolvents and, usually, only mg amounts of organicprecursors for radiolabelling are used. Carbon-11 andfluorine-18 labelled compounds produced by these systemsare usually associated with only micrograms of stable‘‘carrier’’. The other hardware used in systems e.g. HPLCpumps, columns, heaters, rotary evaporators etc. are quitespace consuming. In contrast to this, however, in allradiosyntheses the number of radioactive atoms ormolecules involved is vanishingly small. We are, therefore,faced with the dilemma of having to use a fairly largeautomated system to handle minute amounts of radioactivematerials. The automated systems associated parameters(e.g. volumes, timing, temperatures, etc.), are already verywell defined, making the transformation to microfabricateddevices relatively straightforward, in principle. Given thesmall amounts of reagents, especially the starting concen-tration of radioisotope, involved in the radiosynthesis ofcompounds of clinical interest, it was hypothesised thatminiaturisation of automated radiosynthesis systems isfeasible.
Miniaturisation of chemical systems started in the lastdecade with the development of a number of microanaly-tical systems. These devices combine sensors, actuators andmicrofluidic elements to create a micro total analysissystem (mTAS) (Manz and Widmer, 1990; Kopp et al.,1998; Hadd et al., 1997). The devices consisted ofphotolithographically etched microstructures on silicon
substrates. Each structure performed an individual task inorder to complete the chemical synthesis and analysis, e.g.reagent mixing or separation systems. Each structure waslinked with a channel to the next structure in order to allowthe flow of reactants and products from one part of themicrofabricated device to the next.At present, the simplest microfabricated systems are
microreactors which correspond to a single operation.Most of these operations perform mixing or separation.The majority of the present microfluidic structures actuallybelong to this group (Ramsey, 1999). There are a numberof examples of microreactors used for chemical reactions.For example, Chambers, et.al. has developed a micro-reactor in which elemental fluorine has been used to allowselective fluorination of organic compounds (Chambersand Spink, 1999).Microfabrication technology would appear extremely
attractive for application in PET radiochemistry, since anumber of important advantages could be gained. Thepossibility of performing multiple radiosyntheses from onebatch of labelling agent would be simplified. Potentially,more rapid radiochemistry could be performed usingminiaturised devices not only for radiosyntheses, but alsofor purification and analysis. This would lead to improvedspecific radioactivity of radioligands and would allowexploration of the chemistry with positron emitters whichhave extremely short half-lives, e.g. nitrogen-13 (t1/2 ¼9.96min) and even oxygen-15 (t1/2 ¼ 2.04min). Suchdevelopments could allow the possible radiolabelling of agreatly expanded range of compounds. An additionaladvantage would be that, since the devices could be mass-produced they could be discarded after use, thus over-coming a major bottleneck by removing the need to cleanthe system after each radiosynthesis.However, before the microfabricated synthesis and/or
analysis device could be developed, it was necessary todecide what type of radiosynthesis was to be attempted onthe microfabricated system. The purpose being to usemicrofabricated devices to carry out a range of aqueous ororganic-based radiolabelling reactions on model com-pounds, in order to establish a simple radiolabelling anddetection of the radiolabelled products as ‘‘proof ofprinciple’’. The proof of principle being that radiochem-istry on a microfabricated device was feasible. Therefore,the aims of this work were to develop a microfabricateddevice for the radiolabelling of commonly used PETradioligands.
2. Experimental
2.1. Microreactor fabrication
On-chip experiments were performed using a polymer-based vortex microreactor designed in-house and manu-factured by Epigem Ltd. UK (Fig. 1). The microreactorconsisted of three layers of polycarbonate bonded togetherusing an SU-8, UV-sensitive polymer, which formed the

ARTICLE IN PRESS
Fig. 1. Polycarbonate/SU-8 microfluidic reactor with baseplate and connectors. This is the experimental apparatus used for radiosynthesis of
[99mTc]MAA and [99mTc]HDP, [124I]Annexin V and [124I]doxorubicin. Fig. 1 shows the microfabricated reactor mounted onto its base plate and connected
to the three reagent inlets and one reagent outlet.
J.M. Gillies et al. / Applied Radiation and Isotopes 64 (2006) 325–332 327
reactor chamber and the 100 mm microfluidic channels. Themicrofluidic device was then connected via ferrules to abase plate (Fig. 1). The baseplate allowed the directconnection of three reagent reservoirs via 1/16th PEEKtubing to the microfluidic device.
2.2. On-chip radiosynthesis of [99mTc]macroagregated
albumen (MAA)
Two 1mL hypodermic syringes were primed with thefollowing solutions, respectively, [99mTc]sodium pertechne-tate (75MBqmL�1) and a [99mTc]human albumen micro-agregates kit (CIS Bio International, USA) containinghuman albumen as microaggregates (Kondo et al., 1998)(2.0mg), stannous chloride (0.2mg), human albumen(7.0mg) and sodium chloride (8.7mg). Each syringe wasthen attached to the chip and the solutions were pumped ata total flow rate of approximately 250 mLmin�1 at ambienttemperature. Fractions were collected at 1 s intervals intoEppendorf tubes. The crude reaction mixture was analysedby ITLC (Macherey–Nagel Polygram Sil G/UV254 silicagel plates:100% methylethylketone) and analysed usingan Instant Imager electronic autoradiography system(Packard, USA).
2.3. On-chip radiosynthesis of [99mTc]HDP oxidronate
Two 1mL hypodermic syringes were primed with thefollowing solutions, respectively, [99mTc]sodium pertechne-tate (75MBqmL�1) and a [99mTc]Technescan HDP kit(Lary et al., 1998) (Mallinckrodt, Holland) containingsodium oxidronate (3.0mg), stannous chloride (0.24mg),gentisic acid (0.84mg) and sodium chloride (30.0mg). Each
syringe was then attached to the chip and the solutionswere pumped at a total flow rate of, approximately,250 mLmin�1 at ambient temperature. Fractions werecollected at 1 s intervals into Eppendorf tubes. The crudereaction mixture was analysed by ITLC (Silica Gel60F254:90% acetone) and analysed using an Instant Imagerelectronic autoradiography system (Packard, USA).
2.4. On-chip radioiodination of doxorubicin using iodine-124
Three 50 mL Hamilton syringes (Hamilton, USA) wereprimed with the following solutions, respectively, DOXSn(Hadfield et al., 2001) (50 mL, 25 nmol) in MeOH/AcOH99:1, N-chlorosuccinamide in MeOH (50 mL), aqueous PBS(1 mL, 0.1M) and [124I]NaI (2 mL, 3.96MBq) in 50 mLMeOH /H2O 2.3:1. Each syringe was then attached to thechip, and the solutions were pumped at a flow rate of15 mLmin�1, giving a total flow rate of approximately40 mLmin�1 at ambient temperature. Fractions werecollected at 1min intervals into Eppendorf tubes contain-ing 10 mL sodium metabisulphite (0.5 mg mL�1, 2.6 mmol).Each fraction was analysed by radio-HPLC (Mobilephase—100% Acetonitrile; Column—Bondaclone-C18,1mLmin�1) and analysed using both UV and a Bioscanflow-count detector.
2.5. On-chip radioiodination of Annexin V using iodine-124
Three 50 mL Hamilton syringes were primed with thefollowing solutions, respectively, Iodogen in acetonitrilesolution (50 mL, 1mgmL�1), Annexin V (Blankenberget al., 1998; Keen et al., 2003) (10 mL, 1mgmL�1) and[124I]NaI (50 mL, 2.0MBq). Each syringe was then attached
ARTICLE IN PRESSJ.M. Gillies et al. / Applied Radiation and Isotopes 64 (2006) 325–332328
to the chip, and the solutions were pumped at a total flowrate of approximately 40 mLmin�1 at ambient temperature.Fractions were collected at 1min intervals into Eppendorftubes containing 10mL sodium metabisulphite (0.5 mg mL�1,2.6mmol). The crude reaction mixture was analysed byradio-TLC (Silica Gel 60F254—mobile phase—5% trichloro-acetic acid) and analysed using an Instant Imager electro-nic autoradiography system (Packard, USA).
2.6. Multi-chip radiosynthesis of 2-[18F]FDG
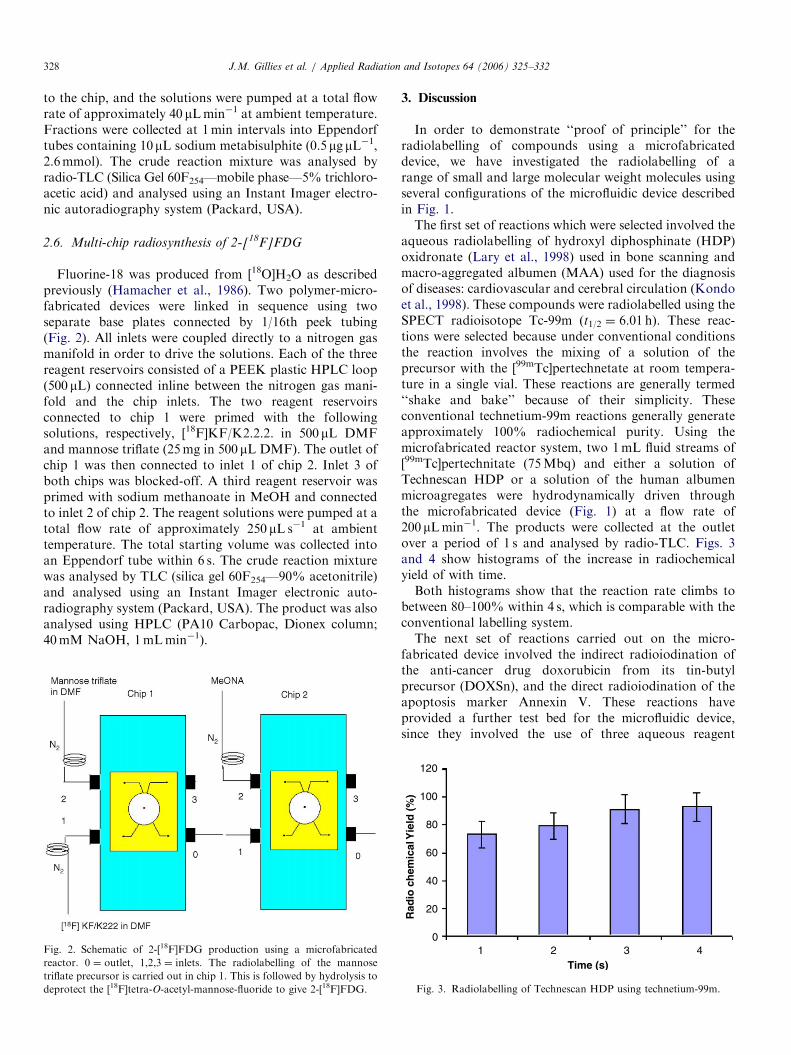
Fluorine-18 was produced from [18O]H2O as describedpreviously (Hamacher et al., 1986). Two polymer-micro-fabricated devices were linked in sequence using twoseparate base plates connected by 1/16th peek tubing(Fig. 2). All inlets were coupled directly to a nitrogen gasmanifold in order to drive the solutions. Each of the threereagent reservoirs consisted of a PEEK plastic HPLC loop(500 mL) connected inline between the nitrogen gas mani-fold and the chip inlets. The two reagent reservoirsconnected to chip 1 were primed with the followingsolutions, respectively, [18F]KF/K2.2.2. in 500 mL DMFand mannose triflate (25mg in 500 mL DMF). The outlet ofchip 1 was then connected to inlet 1 of chip 2. Inlet 3 ofboth chips was blocked-off. A third reagent reservoir wasprimed with sodium methanoate in MeOH and connectedto inlet 2 of chip 2. The reagent solutions were pumped at atotal flow rate of approximately 250 mL s�1 at ambienttemperature. The total starting volume was collected intoan Eppendorf tube within 6 s. The crude reaction mixturewas analysed by TLC (silica gel 60F254—90% acetonitrile)and analysed using an Instant Imager electronic auto-radiography system (Packard, USA). The product was alsoanalysed using HPLC (PA10 Carbopac, Dionex column;40mM NaOH, 1mLmin�1).
Fig. 2. Schematic of 2-[18F]FDG production using a microfabricated
reactor. 0 ¼ outlet, 1,2,3 ¼ inlets. The radiolabelling of the mannose
triflate precursor is carried out in chip 1. This is followed by hydrolysis to
deprotect the [18F]tetra-O-acetyl-mannose-fluoride to give 2-[18F]FDG.
3. Discussion
In order to demonstrate ‘‘proof of principle’’ for theradiolabelling of compounds using a microfabricateddevice, we have investigated the radiolabelling of arange of small and large molecular weight molecules usingseveral configurations of the microfluidic device describedin Fig. 1.The first set of reactions which were selected involved the
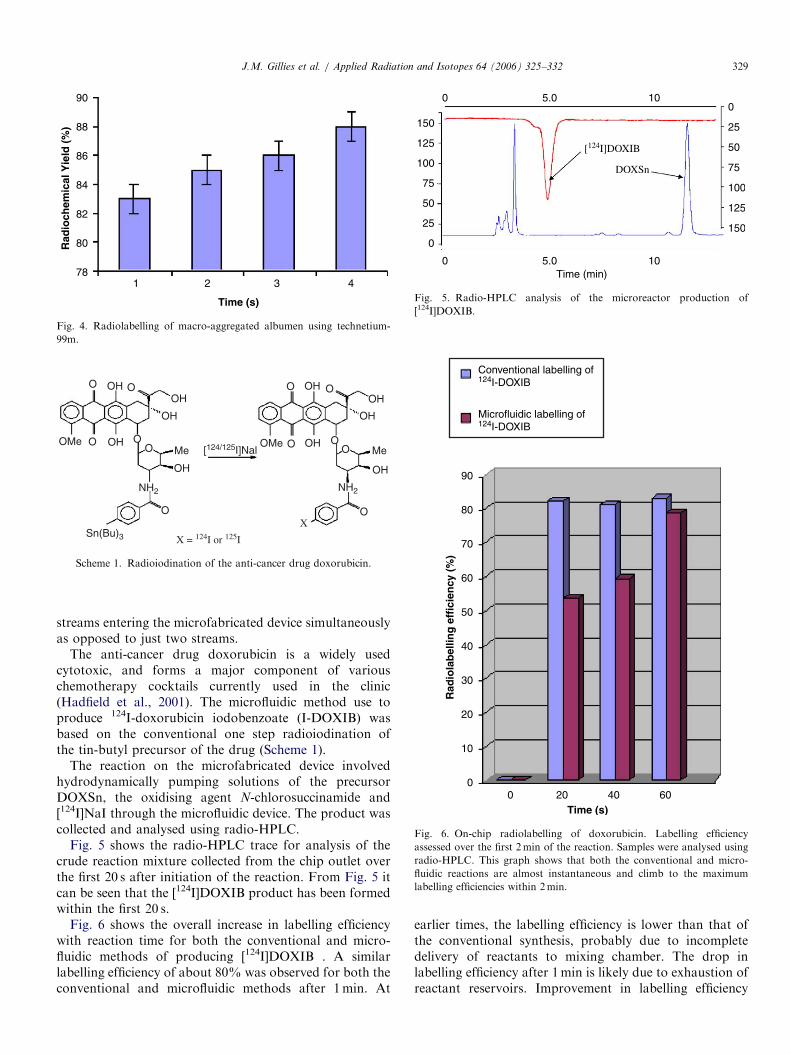
aqueous radiolabelling of hydroxyl diphosphinate (HDP)oxidronate (Lary et al., 1998) used in bone scanning andmacro-aggregated albumen (MAA) used for the diagnosisof diseases: cardiovascular and cerebral circulation (Kondoet al., 1998). These compounds were radiolabelled using theSPECT radioisotope Tc-99m (t1/2 ¼ 6.01 h). These reac-tions were selected because under conventional conditionsthe reaction involves the mixing of a solution of theprecursor with the [99mTc]pertechnetate at room tempera-ture in a single vial. These reactions are generally termed‘‘shake and bake’’ because of their simplicity. Theseconventional technetium-99m reactions generally generateapproximately 100% radiochemical purity. Using themicrofabricated reactor system, two 1mL fluid streams of[99mTc]pertechnitate (75Mbq) and either a solution ofTechnescan HDP or a solution of the human albumenmicroagregates were hydrodynamically driven throughthe microfabricated device (Fig. 1) at a flow rate of200 mLmin�1. The products were collected at the outletover a period of 1 s and analysed by radio-TLC. Figs. 3and 4 show histograms of the increase in radiochemicalyield of with time.Both histograms show that the reaction rate climbs to
between 80–100% within 4 s, which is comparable with theconventional labelling system.The next set of reactions carried out on the micro-
fabricated device involved the indirect radioiodination ofthe anti-cancer drug doxorubicin from its tin-butylprecursor (DOXSn), and the direct radioiodination of theapoptosis marker Annexin V. These reactions haveprovided a further test bed for the microfluidic device,since they involved the use of three aqueous reagent
120
100
80
60
40
20
01 2 3 4
Time (s)
Rad
io c
hem
ical
Yie
ld (
%)
Fig. 3. Radiolabelling of Technescan HDP using technetium-99m.
ARTICLE IN PRESS
90
88
86
84
82
80
781 2 3 4
Time (s)
Rad
ioch
emic
al Y
ield
(%
)
Fig. 4. Radiolabelling of macro-aggregated albumen using technetium-
99m.
O OH O
O
O
O
OH
OH
OH
OHOMeMe
NH2
Sn(Bu)3 X = 124I or 125I
XO
O
NH2
OH
OH
OHOH
OH
MeO
O O
OMe[124/125I]NalO O
Scheme 1. Radioiodination of the anti-cancer drug doxorubicin.
150
125
100
75
50
25
0
0 5.0 10Time (min)
0 5.0 100
25
50
75
100
125
150
[124I]DOXIB
DOXSn
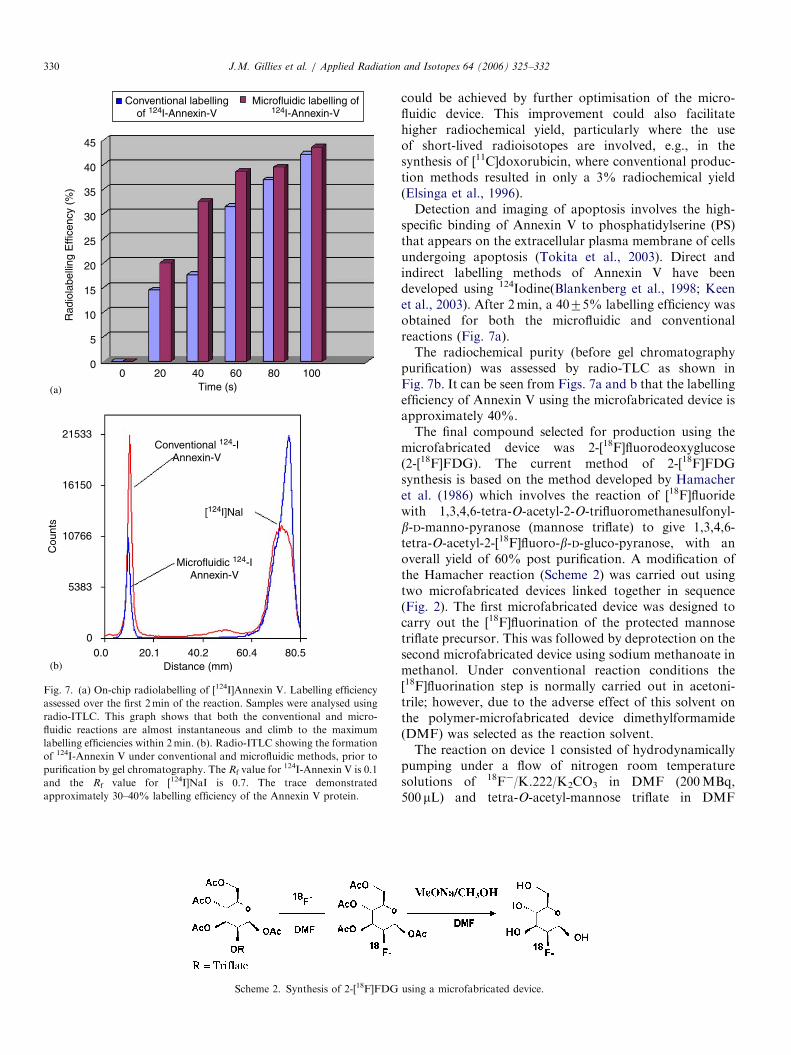
Fig. 5. Radio-HPLC analysis of the microreactor production of
[124I]DOXIB.
90
80
70
60
50
40
30
20
10
00 20 40 60
Time (s)
Conventional labelling of124I-DOXIB
Microfluidic labelling of124I-DOXIB
Rad
iola
bel
ling
eff
icie
ncy
(%
)
Fig. 6. On-chip radiolabelling of doxorubicin. Labelling efficiency
assessed over the first 2min of the reaction. Samples were analysed using
radio-HPLC. This graph shows that both the conventional and micro-
fluidic reactions are almost instantaneous and climb to the maximum
labelling efficiencies within 2min.
J.M. Gillies et al. / Applied Radiation and Isotopes 64 (2006) 325–332 329
streams entering the microfabricated device simultaneouslyas opposed to just two streams.
The anti-cancer drug doxorubicin is a widely usedcytotoxic, and forms a major component of variouschemotherapy cocktails currently used in the clinic(Hadfield et al., 2001). The microfluidic method use toproduce 124I-doxorubicin iodobenzoate (I-DOXIB) wasbased on the conventional one step radioiodination ofthe tin-butyl precursor of the drug (Scheme 1).
The reaction on the microfabricated device involvedhydrodynamically pumping solutions of the precursorDOXSn, the oxidising agent N-chlorosuccinamide and[124I]NaI through the microfluidic device. The product wascollected and analysed using radio-HPLC.
Fig. 5 shows the radio-HPLC trace for analysis of thecrude reaction mixture collected from the chip outlet overthe first 20 s after initiation of the reaction. From Fig. 5 itcan be seen that the [124I]DOXIB product has been formedwithin the first 20 s.
Fig. 6 shows the overall increase in labelling efficiencywith reaction time for both the conventional and micro-fluidic methods of producing [124I]DOXIB . A similarlabelling efficiency of about 80% was observed for both theconventional and microfluidic methods after 1min. At
earlier times, the labelling efficiency is lower than that ofthe conventional synthesis, probably due to incompletedelivery of reactants to mixing chamber. The drop inlabelling efficiency after 1min is likely due to exhaustion ofreactant reservoirs. Improvement in labelling efficiency
ARTICLE IN PRESS
45
40
35
30
25
20
15
10
5
00 20 40 60 80 100
Rad
iola
belli
ng E
ffice
ncy
(%)
Time (s)
21533
16150
10766
5383
00.0 20.1 40.2 60.4 80.5
Distance (mm)
Conventional 124-IAnnexin-V
[124I]Nal
Microfluidic 124-IAnnexin-V
Cou
nts
Conventional labellingof 124I-Annexin-V
Microfluidic labelling of124I-Annexin-V
(a)
(b)
Fig. 7. (a) On-chip radiolabelling of [124I]Annexin V. Labelling efficiency
assessed over the first 2min of the reaction. Samples were analysed using
radio-ITLC. This graph shows that both the conventional and micro-
fluidic reactions are almost instantaneous and climb to the maximum
labelling efficiencies within 2min. (b). Radio-ITLC showing the formation
of 124I-Annexin V under conventional and microfluidic methods, prior to
purification by gel chromatography. The Rf value for124I-Annexin V is 0.1
and the Rf value for [124I]NaI is 0.7. The trace demonstrated
approximately 30–40% labelling efficiency of the Annexin V protein.
Scheme 2. Synthesis of 2-[18F]FDG
J.M. Gillies et al. / Applied Radiation and Isotopes 64 (2006) 325–332330
could be achieved by further optimisation of the micro-fluidic device. This improvement could also facilitatehigher radiochemical yield, particularly where the useof short-lived radioisotopes are involved, e.g., in thesynthesis of [11C]doxorubicin, where conventional produc-tion methods resulted in only a 3% radiochemical yield(Elsinga et al., 1996).Detection and imaging of apoptosis involves the high-
specific binding of Annexin V to phosphatidylserine (PS)that appears on the extracellular plasma membrane of cellsundergoing apoptosis (Tokita et al., 2003). Direct andindirect labelling methods of Annexin V have beendeveloped using 124Iodine(Blankenberg et al., 1998; Keenet al., 2003). After 2min, a 4075% labelling efficiency wasobtained for both the microfluidic and conventionalreactions (Fig. 7a).The radiochemical purity (before gel chromatography
purification) was assessed by radio-TLC as shown inFig. 7b. It can be seen from Figs. 7a and b that the labellingefficiency of Annexin V using the microfabricated device isapproximately 40%.The final compound selected for production using the
microfabricated device was 2-[18F]fluorodeoxyglucose(2-[18F]FDG). The current method of 2-[18F]FDGsynthesis is based on the method developed by Hamacheret al. (1986) which involves the reaction of [18F]fluoridewith 1,3,4,6-tetra-O-acetyl-2-O-trifluoromethanesulfonyl-b-D-manno-pyranose (mannose triflate) to give 1,3,4,6-tetra-O-acetyl-2-[18F]fluoro-b-D-gluco-pyranose, with anoverall yield of 60% post purification. A modification ofthe Hamacher reaction (Scheme 2) was carried out usingtwo microfabricated devices linked together in sequence(Fig. 2). The first microfabricated device was designed tocarry out the [18F]fluorination of the protected mannosetriflate precursor. This was followed by deprotection on thesecond microfabricated device using sodium methanoate inmethanol. Under conventional reaction conditions the[18F]fluorination step is normally carried out in acetoni-trile; however, due to the adverse effect of this solvent onthe polymer-microfabricated device dimethylformamide(DMF) was selected as the reaction solvent.The reaction on device 1 consisted of hydrodynamically
pumping under a flow of nitrogen room temperaturesolutions of 18F�/K.222/K2CO3 in DMF (200MBq,500 mL) and tetra-O-acetyl-mannose triflate in DMF
using a microfabricated device.
ARTICLE IN PRESS
579
436
292
149
6
0.0O
20.1 40.2 60.4 80.5FDistance (mm)
18F-
2-[18F]FDG [18F]tetra-O-acetyl-mannose
Cou
nts
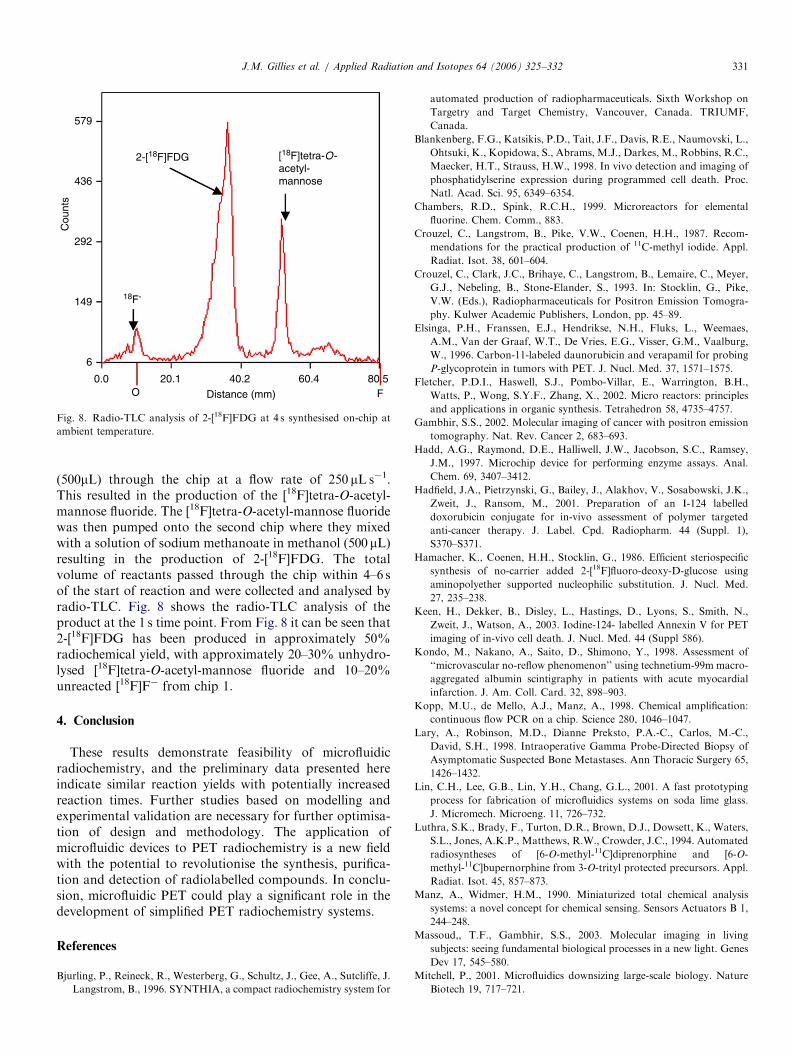
Fig. 8. Radio-TLC analysis of 2-[18F]FDG at 4 s synthesised on-chip at
ambient temperature.
J.M. Gillies et al. / Applied Radiation and Isotopes 64 (2006) 325–332 331
(500mL) through the chip at a flow rate of 250 mL s�1.This resulted in the production of the [18F]tetra-O-acetyl-mannose fluoride. The [18F]tetra-O-acetyl-mannose fluoridewas then pumped onto the second chip where they mixedwith a solution of sodium methanoate in methanol (500 mL)resulting in the production of 2-[18F]FDG. The totalvolume of reactants passed through the chip within 4–6 sof the start of reaction and were collected and analysed byradio-TLC. Fig. 8 shows the radio-TLC analysis of theproduct at the 1 s time point. From Fig. 8 it can be seen that2-[18F]FDG has been produced in approximately 50%radiochemical yield, with approximately 20–30% unhydro-lysed [18F]tetra-O-acetyl-mannose fluoride and 10–20%unreacted [18F]F� from chip 1.
4. Conclusion
These results demonstrate feasibility of microfluidicradiochemistry, and the preliminary data presented hereindicate similar reaction yields with potentially increasedreaction times. Further studies based on modelling andexperimental validation are necessary for further optimisa-tion of design and methodology. The application ofmicrofluidic devices to PET radiochemistry is a new fieldwith the potential to revolutionise the synthesis, purifica-tion and detection of radiolabelled compounds. In conclu-sion, microfluidic PET could play a significant role in thedevelopment of simplified PET radiochemistry systems.
References
Bjurling, P., Reineck, R., Westerberg, G., Schultz, J., Gee, A., Sutcliffe, J.
Langstrom, B., 1996. SYNTHIA, a compact radiochemistry system for
automated production of radiopharmaceuticals. Sixth Workshop on
Targetry and Target Chemistry, Vancouver, Canada. TRIUMF,
Canada.
Blankenberg, F.G., Katsikis, P.D., Tait, J.F., Davis, R.E., Naumovski, L.,
Ohtsuki, K., Kopidowa, S., Abrams, M.J., Darkes, M., Robbins, R.C.,
Maecker, H.T., Strauss, H.W., 1998. In vivo detection and imaging of
phosphatidylserine expression during programmed cell death. Proc.
Natl. Acad. Sci. 95, 6349–6354.
Chambers, R.D., Spink, R.C.H., 1999. Microreactors for elemental
fluorine. Chem. Comm., 883.
Crouzel, C., Langstrom, B., Pike, V.W., Coenen, H.H., 1987. Recom-
mendations for the practical production of 11C-methyl iodide. Appl.
Radiat. Isot. 38, 601–604.
Crouzel, C., Clark, J.C., Brihaye, C., Langstrom, B., Lemaire, C., Meyer,
G.J., Nebeling, B., Stone-Elander, S., 1993. In: Stocklin, G., Pike,
V.W. (Eds.), Radiopharmaceuticals for Positron Emission Tomogra-
phy. Kulwer Academic Publishers, London, pp. 45–89.
Elsinga, P.H., Franssen, E.J., Hendrikse, N.H., Fluks, L., Weemaes,
A.M., Van der Graaf, W.T., De Vries, E.G., Visser, G.M., Vaalburg,
W., 1996. Carbon-11-labeled daunorubicin and verapamil for probing
P-glycoprotein in tumors with PET. J. Nucl. Med. 37, 1571–1575.
Fletcher, P.D.I., Haswell, S.J., Pombo-Villar, E., Warrington, B.H.,
Watts, P., Wong, S.Y.F., Zhang, X., 2002. Micro reactors: principles
and applications in organic synthesis. Tetrahedron 58, 4735–4757.
Gambhir, S.S., 2002. Molecular imaging of cancer with positron emission
tomography. Nat. Rev. Cancer 2, 683–693.
Hadd, A.G., Raymond, D.E., Halliwell, J.W., Jacobson, S.C., Ramsey,
J.M., 1997. Microchip device for performing enzyme assays. Anal.
Chem. 69, 3407–3412.
Hadfield, J.A., Pietrzynski, G., Bailey, J., Alakhov, V., Sosabowski, J.K.,
Zweit, J., Ransom, M., 2001. Preparation of an I-124 labelled
doxorubicin conjugate for in-vivo assessment of polymer targeted
anti-cancer therapy. J. Label. Cpd. Radiopharm. 44 (Suppl. 1),
S370–S371.
Hamacher, K., Coenen, H.H., Stocklin, G., 1986. Efficient steriospecific
synthesis of no-carrier added 2-[18F]fluoro-deoxy-D-glucose using
aminopolyether supported nucleophilic substitution. J. Nucl. Med.
27, 235–238.
Keen, H., Dekker, B., Disley, L., Hastings, D., Lyons, S., Smith, N.,
Zweit, J., Watson, A., 2003. Iodine-124- labelled Annexin V for PET
imaging of in-vivo cell death. J. Nucl. Med. 44 (Suppl 586).
Kondo, M., Nakano, A., Saito, D., Shimono, Y., 1998. Assessment of
‘‘microvascular no-reflow phenomenon’’ using technetium-99m macro-
aggregated albumin scintigraphy in patients with acute myocardial
infarction. J. Am. Coll. Card. 32, 898–903.
Kopp, M.U., de Mello, A.J., Manz, A., 1998. Chemical amplification:
continuous flow PCR on a chip. Science 280, 1046–1047.
Lary, A., Robinson, M.D., Dianne Preksto, P.A.-C., Carlos, M.-C.,
David, S.H., 1998. Intraoperative Gamma Probe-Directed Biopsy of
Asymptomatic Suspected Bone Metastases. Ann Thoracic Surgery 65,
1426–1432.
Lin, C.H., Lee, G.B., Lin, Y.H., Chang, G.L., 2001. A fast prototyping
process for fabrication of microfluidics systems on soda lime glass.
J. Micromech. Microeng. 11, 726–732.
Luthra, S.K., Brady, F., Turton, D.R., Brown, D.J., Dowsett, K., Waters,
S.L., Jones, A.K.P., Matthews, R.W., Crowder, J.C., 1994. Automated
radiosyntheses of [6-O-methyl-11C]diprenorphine and [6-O-
methyl-11C]bupernorphine from 3-O-trityl protected precursors. Appl.
Radiat. Isot. 45, 857–873.
Manz, A., Widmer, H.M., 1990. Miniaturized total chemical analysis
systems: a novel concept for chemical sensing. Sensors Actuators B 1,
244–248.
Massoud,, T.F., Gambhir, S.S., 2003. Molecular imaging in living
subjects: seeing fundamental biological processes in a new light. Genes
Dev 17, 545–580.
Mitchell, P., 2001. Microfluidics downsizing large-scale biology. Nature
Biotech 19, 717–721.

ARTICLE IN PRESSJ.M. Gillies et al. / Applied Radiation and Isotopes 64 (2006) 325–332332
Ramsey, J.M., 1999. The burgeoning power of the shrinking laboratory.
Nature Biotech 17, 1061–1062.
Reader, A.J., Zweit, J., 2001. Developments in whole-body molecular
imaging in live subjects. Trends Pharmacol. Sci. 22, 604–607.
Regenfuss, P., Clegg, R.M., Fulwyler, M.J., Barrantes, F.J., Jovin, T.M.,
1985. Mixing liquids in microseconds. Rev. Sci. Instrum. 56, 283–290.
Stuernstrom, M., Roeraade, J., 1998. Methods for fabrication of
microfluidic systems in glass. J. Micromech. Microeng. 8, 33–38.
Tokita, N., Hasegawa, S., Maruyama, K., Izumi, T., Blankenberg, F.G.,
Tait, J.F., Strauss, H.W., Nishimura, T., 2003. 99mTc–Hynic–Annexin
V imaging to evaluate inflammation and apoptosis in rats with
autoimmune myocarditis. Eur. J. Nucl. Med. Mol. Imaging. 30,
232–238.
Tsai, J.H., Lin, L., 2001. Micro to macro fluidic interconnects with
an integrated polymer sealant. J. Micromech. Microeng. 11,
577–581.


















