

Michael Schroeder BioTechnological Center TU Dresden [email protected] Biotec Structure...
63
Michael Schroeder BioTechnological Center TU Dresden [email protected] www.biotec.tu-dresden.de Biotec Structure Alignment
-
Upload
clifton-walton -
Category
Documents
-
view
221 -
download
2
Transcript of Michael Schroeder BioTechnological Center TU Dresden [email protected] Biotec Structure...
By Michael Schroeder, Biotec 2
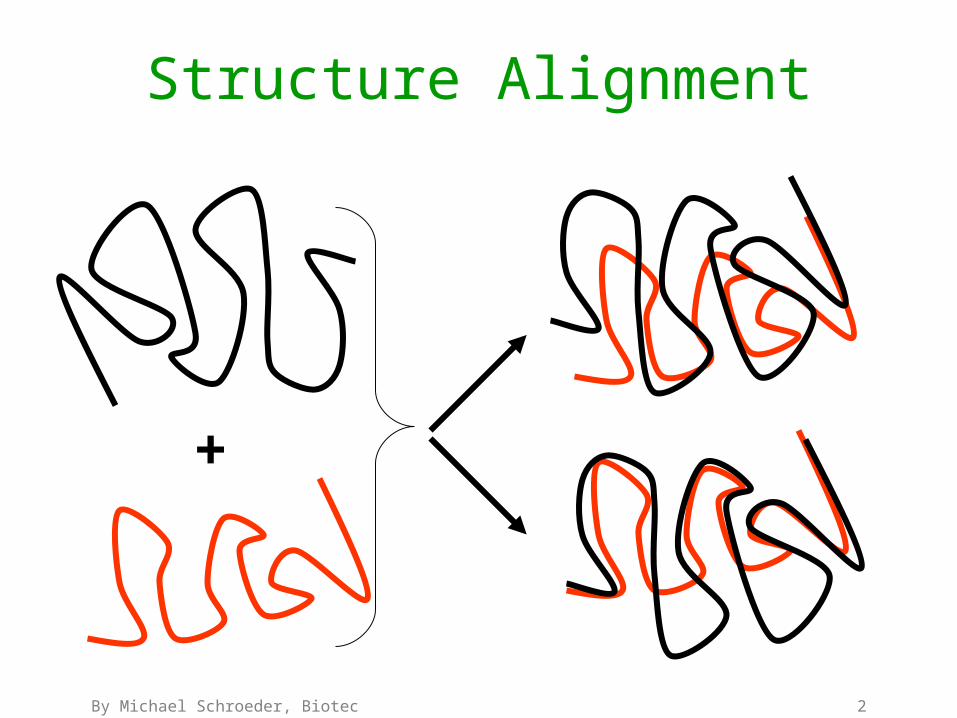
Structure Alignment
+

By Michael Schroeder, Biotec 3
Content
Motivation Some basics Double Dynamic Programming

By Michael Schroeder, Biotec 4
PART I: Motivation

By Michael Schroeder, Biotec 5
Motivation: Conformational changes
Upon ligand binding structures may change Structural alignment can highlight the changes

By Michael Schroeder, Biotec 6
GEFs
GAPs
Conformational changes: Small GTPases
Small GTPases act as molecular switches to control and regulate important functions and pathways within in cell
Activated by guanine nucleotide exchange factors (GEF)
Inactivated by GTPase activating proteins (GAP)

By Michael Schroeder, Biotec 7
G proteins: Conformational change in GTP and GDP bound state

By Michael Schroeder, Biotec 8
Open and closed conformation of cytrate synthase (1cts,5cts)
Open: oxalacetate, Closed: oxalacetate and co-enzyme A Loop between two helices moves by 6A and rotates by 28º, some atoms
move by 10A

By Michael Schroeder, Biotec 9

By Michael Schroeder, Biotec 10
Hinge motion in Lactoferrin (1lfh, 1lfg) Lactoferrin is an iron-binding protein found in
secretions such as milk or tears Rotation of 54º upon iron-binding

By Michael Schroeder, Biotec 11
Hinge motion in Lactoferrin (1lfh, 1lfg) Lactoferrin is an iron-binding protein found in
secretions such as milk or tears Rotation of 54º upon iron-binding

By Michael Schroeder, Biotec 12

By Michael Schroeder, Biotec 13
Motivation: (Distant) Relatives Sequence similarity may be low, but structural
similarity can still be high
Picture from www.jenner.ac.uk/YBF/DanielleTalbot.ppt

By Michael Schroeder, Biotec 14
Distant relatives
Globins occur widely Primary function: binding oxygen Assembly of helices surrounding haem group

By Michael Schroeder, Biotec 15
Relatives
Sperm whale myoglobin (2lh7) and Lupin leghaemoglobin (1mbd)

By Michael Schroeder, Biotec 16
Distant Relatives

By Michael Schroeder, Biotec 17
Relatives Actinidin (2act) and Papain (9pap) Sequence identity 49%, rmsd 0.77A Same family: Papain-like

By Michael Schroeder, Biotec 18
Relatives
Plastocyanin (5pcy) and azurin (2aza) Core of structure is conserved

By Michael Schroeder, Biotec 19
Relatives
Structure classifications like CATH and FSSP use structural alignments to identify superfamilies.

By Michael Schroeder, Biotec 20
Motivation: Convergent Evolution

By Michael Schroeder, Biotec 21
Sequence similarity: low
>1cse SubtilisinAQTVPYGIPLIKADKVQAQGFKGANVKVAVLDTGIQASHPDLNVVGGASFVAGEAYNTDGNGHGTHVAGTVAALDNTTGVLGVAPSVSLYAVKVLNSSGSGSYSGIVSGIEWATTNGMDVINMSLGGASGSTAMKQAVDNAYARGVVVVAAAGNSGNSGSTNTIGYPAKYDSVIAVGAVDSNSNRASFSSVGAELEVMAPGAGVYSTYPTNTYATLNGTSMASPHVAGAAALILSKHPNLSASQVRNRLSSTATYLGSSFYYGKGLINVEAAAQ>1acb ChymotrypsinCGVPAIQPVLSGLSRIVNGEEAVPGSWPWQVSLQDKTGFHFCGGSLINENWVVTAAHCGVTTSDVVVAGEFDQGSSSEKIQKLKIAKVFKNSKYNSLTINNDITLLKLSTAASFSQTVSAVCLPSASDDFAAGTTCVTTGWGLTRYTNANTPDRLQQASLPLLSNTNCKKYWGTKIKDAMICAGASGVSSCMGDSGGPLVCKKNGAWTLVGIVSWGSSTCSTSTPGVYARVTALVNWVQQTLAAN

By Michael Schroeder, Biotec 22
Structural similarity: low
1CSE:E, 1ACB:E
By Michael Schroeder, Biotec 23
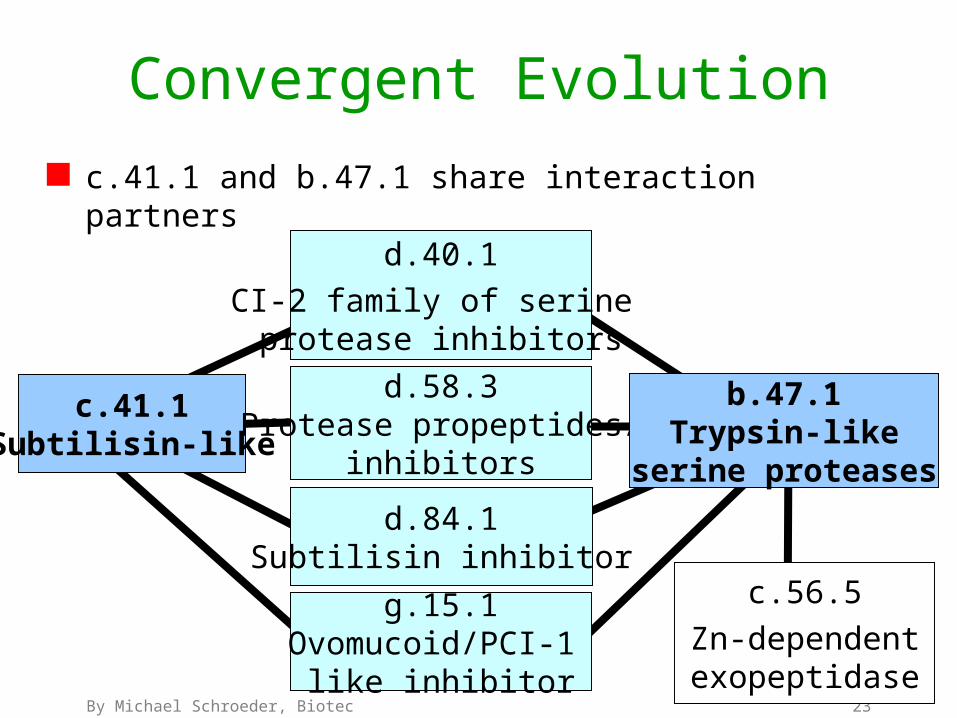
Convergent Evolution
c.41.1 and b.47.1 share interaction partners
c.41.1Subtilisin-like
d.58.3Protease propeptides/
inhibitors
d.84.1Subtilisin inhibitor
d.40.1
CI-2 family of serine protease inhibitors
b.47.1Trypsin-like
serine proteases
c.56.5
Zn-dependentexopeptidase
g.15.1Ovomucoid/PCI-1
like inhibitor

By Michael Schroeder, Biotec 24
Convergent Evolution
1OYV
4sgbOvomucoid/PCI-1 like inhibitor, g.15.1, topTrypsin-like serine proteases, b.47.1.2, bottom
1oyvOvomucoid/PCI-1 like inhibitor, g.15.1topSubtilisin like c.41.1bottom

By Michael Schroeder, Biotec 25
Aligned structures
1cseCI-2 family of serine proteases inhitors, d.40.1 topSubtilisin like c.41.1bottom
1acbCI-2 family of serine proteases inhitors, d.40.1 topTrypsin-like serine proteases, b.47.1.2, bottom
Convergent Evolution

By Michael Schroeder, Biotec 26
Catalytic Triad
>1cse SubtilisinAQTVPYGIPLIKADKVQAQGFKGANVKVAVLDTGIQASHPDLNVVGGASFVAGEAYNTDGNGHGTHVAGTVAALDNTTGVLGVAPSVSLYAVKVLNSSGSGSYSGIVSGIEWATTNGMDVINMSLGGASGSTAMKQAVDNAYARGVVVVAAAGNSGNSGSTNTIGYPAKYDSVIAVGAVDSNSNRASFSSVGAELEVMAPGAGVYSTYPTNTYATLNGTSMASPHVAGAAALILSKHPNLSASQVRNRLSSTATYLGSSFYYGKGLINVEAAAQ>1acb ChymotrypsinCGVPAIQPVLSGLSRIVNGEEAVPGSWPWQVSLQDKTGFHFCGGSLINENWVVTAAHCGVTTSDVVVAGEFDQGSSSEKIQKLKIAKVFKNSKYNSLTINNDITLLKLSTAASFSQTVSAVCLPSASDDFAAGTTCVTTGWGLTRYTNANTPDRLQQASLPLLSNTNCKKYWGTKIKDAMICAGASGVSSCMGDSGGPLVCKKNGAWTLVGIVSWGSSTCSTSTPGVYARVTALVNWVQQTLAAN

By Michael Schroeder, Biotec 27
Convergent evolution
A and B are native, C is viral
C
BA
A’
A CB C
Henschel et al., Bioinformatics 2006

By Michael Schroeder, Biotec 28
Comparison of Nef-SH3 and intra-chain interaction of catalytic domain and SH3 of Hck, PDBs: 1efn and 2hck
No evidence of homology between Nef and Kinase
HIV1-Nef
Kinase (Src Haematopoeitic cell kinase, Catalytic domain)
Fyn-SH3/Hck-SH3
HIV Nef mimics kinase in binding SH3
Henschel et al., Bioinformatics 2006

By Michael Schroeder, Biotec 29
Automatic calculation of equivalent residues
Apart from PxxP motif matches: Arg71/Lys249, Phe90/His289
Residues with equivalents are strictly conserved in HIV-Nef
Nef Kinase
Henschel et al., Bioinformatics 2006

By Michael Schroeder, Biotec 30
Caspase (red) P35 (yellow) IAP (green)
Upon infection cell starts apoptosis programme, p35 tries to stop it
Mimickry of baculovirus p35 and human inhibitor of apoptosis
Henschel et al., Bioinformatics 2006

By Michael Schroeder, Biotec 31
HIV capsid protein (yellow)
Cyclophilin (red, green)
Cyclophilin A restricts HIV infectivity
Upon mutation of cyclophilin or inhibition with cyclophorin, infectivity goes up >100 (Towers, Nature Medicine, 2003)
Mimickry of Capsids and Cyclophilin
Henschel et al., Bioinformatics 2006

By Michael Schroeder, Biotec 32
PART II: Some basics

By Michael Schroeder, Biotec 33
What do we need?
To main operations to align structures: Translation Rotation
How to evaluate a structural alignment? Root mean square deviation, rmsd

By Michael Schroeder, Biotec 34
Basic Operations: Translation

By Michael Schroeder, Biotec 35
Basic Operations: Translation

By Michael Schroeder, Biotec 36
Basic Operations: Translation

By Michael Schroeder, Biotec 37
Basic Operations: Rotation

By Michael Schroeder, Biotec 38
Root Mean Square Deviation What is the distance between two points a with
coordinates xa and ya and b with coordinates xb and yb? Euclidean distance:
d(a,b) = √ (xa--xb )2 + (ya -yb )2
And in 3D?
a
b

By Michael Schroeder, Biotec 39
Root Mean Square Deviation
In a structure alignment the score measures how far the aligned atoms are from each other on average
Given the distances di between n aligned atoms, the root mean square deviation is defined as
rmsd = √ 1/n ∑ di2

By Michael Schroeder, Biotec 40
Quality of Alignment and Example Unit of RMSD => e.g. Ångstroms
Identical structures => RMSD = “0” Similar structures => RMSD is small (1 – 3 Å) Distant structures => RMSD > 3 Å

By Michael Schroeder, Biotec 41
PART III: Dynamic Programming

By Michael Schroeder, Biotec 42
A very simple algorithm…
…to align identical structures with conformational changes
Generate a sequence alignment (not necessary if both sequences are really 100% identical)
Compute center of mass for both structures Move both structures so that the centers of mass are
the origin Compute the angle between all aligned residues Rotate structure by median of all angles

By Michael Schroeder, Biotec 43
A very simple algorithm…
…to align identical structures with conformational changes
Generate a sequence alignment (not necessary if both sequences are really 100% identical)
Compute center of mass for both structures Move both structures so that the centers of mass are
the origin Compute the angle between all aligned residues Rotate structure by median of all angles
Question: How?Assume n atoms
(x1,y1,z1) to (xn,yn,zn)(for one structure)

By Michael Schroeder, Biotec 44
A very simple algorithm…
…to align identical structures with conformational changes
Generate a sequence alignment (not necessary if both sequences are really 100% identical)
Compute center of mass for both structures Move both structures so that the centers of mass are
the origin Compute the angle between all aligned residues Rotate structure by median of all angles
Question: How?
Question: How?Assume n atoms(x1,y1,z1) to (xn,yn,zn:)Center of mass (xCoM,yCoM,zCoM) = (1/n n
i=1 xi , 1/n ni=1 yi 1/n n
i=1 zi )

By Michael Schroeder, Biotec 45
A very simple algorithm…
…to align identical structures with conformational changes
Generate a sequence alignment (not necessary if both sequences are really 100% identical)
Compute center of mass for both structures Move both structures so that the centers of mass are
the origin Compute the angle between all aligned residues Rotate structure by median of all angles
For all i: do xi:= xi-xCoM, yi:= yi-yCoM, yi:= yi-yCoM,
Question: How?Assume n atoms (x1,y1,z1) to (xn,yn,zn:)Center of mass (xCoM,yCoM,zCoM) = (1/n n
i=1 xi , 1/n ni=1 yi 1/n n
i=1 zi

By Michael Schroeder, Biotec 46
A very simple algorithm…
…to align identical structures with conformational changes
Generate a sequence alignment (not necessary if both sequences are really 100% identical)
Compute center of mass for both structures Move both structures so that the centers of mass are
the origin Compute the angle between all aligned residues Rotate structure by median of all angles
Why median andnot mean?

By Michael Schroeder, Biotec 47
A refinement: Alternating alignment and superposition
1. P = initial alignment (e.g. based on sequence alignment)
2. Superpose structures A and B based on P 3. Generate distance-based scoring matrix R from
superposition 4. Use dynamic programming to align A and B using
scoring matrix R 5. P‘ = new alignment derived from dynamic
programming step 6. If P‘ is different from P then go to step 2 again

By Michael Schroeder, Biotec 48
Distance-based scoring matrix Let d(Ai, Bj) be the Euclidean distance between Ai and Bj
Let t be the upper distance limit for residues to be rewarded
The scoring matrix R is defined as follows:
R(Ai, Bj) = 1 / d(Ai, Bj) - 1 / t
if R(Ai, Bj) > max. score then R(Ai, Bj) = max. score
The gap/mismatch penalty is set to 0

By Michael Schroeder, Biotec 49
Let d(Ai, Bj) be the Euclidean distance between Ai and Bj
Let t be the upper distance limit for residues to be rewarded
The scoring matrix R is defined as follows:
R(Ai, Bj) = 1 / d(Ai, Bj) - 1 / t
if R(Ai, Bj) > max. score then R(Ai, Bj) = max. score
The gap/mismatch penalty is set to 0
Distance-based scoring matrix
What size doesPAM have?
What size doesR have?

By Michael Schroeder, Biotec 50
Example
R(Ai, Bj) = 1/d(Ai, Bj) - 1/t for t=1/10 and max. score =2

By Michael Schroeder, Biotec 51
Part IV: Double dynamic programming (chapter 9)

By Michael Schroeder, Biotec 52
Doube dynamic programming
Goal: Simultaniously align and superpose structures Double dynamic programming is a heuristic which
tries to achieve goal Implemented as part of SSAP (used e.g. by CATH)

By Michael Schroeder, Biotec 53
Idea of double dynamic programming
Use two levels of dynamic programming: High level, which
summarises low level DP
Low level, which generates alignment based on assumption that ai and bj are part of an optimal alignment

By Michael Schroeder, Biotec 54
Low level matrix
ijR is the low level scoring matrix assuming the pair ai and bj are aligned
ijRkl is the score showing how well ak fits onto bl under the constraint that ai and bj are aligned
Perform dynamic programming for all pairs i,j using ijR with constraint that optimal alignment includes (i,j)

By Michael Schroeder, Biotec 55

By Michael Schroeder, Biotec 56

By Michael Schroeder, Biotec 57
Questions: How was max. score set in this example?

By Michael Schroeder, Biotec 58

By Michael Schroeder, Biotec 59

By Michael Schroeder, Biotec 60

By Michael Schroeder, Biotec 61

By Michael Schroeder, Biotec 62

By Michael Schroeder, Biotec 63
Summary
Structural alignments are useful to study conformational changes, to classify domains into families (DDP is used in CATH), to study proteins with distant relationships and hence low sequence similarity
Algorithms Basic operations: translate and rotate Simple algorithm based on dynamic programming Double dynamic programming:
low-level programming using substitution matrix based residue distance
Aggregation of best paths for high-level programming


















