
Metabolism of Hemoglobin and Bile Pigments · METABOLISM OF HEMOGLOBIN AND BILE PIGMENTS 115 'Amino...
12
A nnals of C linical L aboratory S cience , Vol. X, No. 2 Copyright © 1971, Institute for Clinical Science Metabolism of Hemoglobin and Bile Pigments JAMES M. ORTEN, Ph.D. Department of Biochemistry, Wayne State University School of Medicine Detroit, MI 48201 This presentation is designed to review the catabolic aspects of the metabolism of hemoglobin. Emphasis will be placed pri- marily on observations during the past few years which have expanded knowledge in this field considerably and have elucidated further the classical concepts of the degra- dation of hemoglobin. Some details, how- ever, still remain uncertain. Review arti- cles1, 6’ 8i n ’ 20, 21,23, 27 cover the earlier liter- ature. The catabolism of hemoglobin will be discussed in this paper under the following four major headings: I —The release and transport of “free” hemoglobin and heme II — The conversion of hemoglobin into biliverdin and bilirubin III —The transport, conjugation, and bil- iary excretion of bilirubin IV —The fate of bilirubin conjugates in the intestine I. The Release and Transport of “Free” Hemoglobin and Heme The breakdown of hemoglobin into bile pigments and their derivatives begins pri- marily with the phagocytizing of its form- erly protective but now worn-out erythro - cyte in the reticuloendothelial cells, mainly in the liver and spleen, and the release of “free” hemoglobin. In man this occurs on the average of 120 days after the erythro- cyte is formed, as shown by classical isotopic labeling studies14’16 to be described later. Any hemoglobin released from erythrocytes directly into the circulation because of trauma or other reasons is bound immedi- ately by haptoglobin, a plasma a2-globulin, and presumably is picked up by the reticuloendothelial system and then con- verted into bile pigments. This device prevents the renal loss of hemoglobin and particularly its iron which must be retained and conserved by the body. As shown by the early classical studies of Whipple and Mann and by recent inves- tigations9 using erythrocytes labeled with 51chromium or 59iron, the reticuloendothel- ial cells primarily of the liver, spleen and bone marrow are the principal sites of the removal and beginning steps in the degra- dation of hemoglobin. However, the process may occur more slowly in most other tis- sues in which the extravasation of blood may occur. This is manifest by the fine play of colors which develop in bruises or in a “black eye.” In the reticuloendothelial cells, hemoglobin is converted to heme and then into bile pigments. About 85 percent of the bile pigment formed in man is derived from senescent erythrocytes. In addition, some may be derived from the destruction of immature Presented at the Applied Seminar on Chemical Hematology, November, 1970. 113
Transcript of Metabolism of Hemoglobin and Bile Pigments · METABOLISM OF HEMOGLOBIN AND BILE PIGMENTS 115 'Amino...

A n n a l s o f C l i n i c a l L a b o r a t o r y S c i e n c e , Vol. X, No. 2C o p y r i g h t © 1971, I n s t i t u t e f o r C l i n i c a l S c i e n c e
Metabolism of Hemoglobin and Bile Pigments
JAMES M. ORTEN, Ph.D.
D epartm ent of Biochemistry, W ayne State University School of M edicine
Detroit, M I 48201
This presentation is designed to review the catabolic aspects of the metabolism of hemoglobin. Emphasis will be placed primarily on observations during the past few years which have expanded knowledge in this field considerably and have elucidated further the classical concepts of the degradation of hemoglobin. Some details, however, still remain uncertain. Review articles1, 6’ 8i n ’ 20, 21,23, 27 cover the earlier literature.
The catabolism of hemoglobin will be discussed in this paper under the following four major headings:
I — The release and transport of “free” hemoglobin and heme
II — The conversion of hemoglobin into biliverdin and bilirubin
III — The transport, conjugation, and biliary excretion of bilirubin
IV — The fate of bilirubin conjugates inthe intestine
I. The Release and Transport of “Free” Hemoglobin and Heme
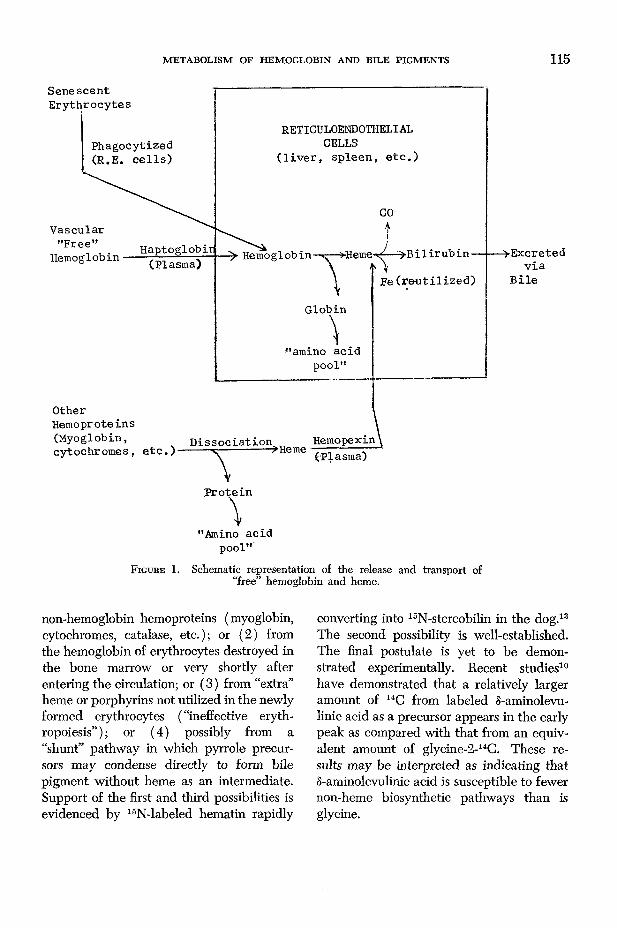
The breakdown of hemoglobin into bile pigments and their derivatives begins primarily with the phagocytizing of its formerly protective but now worn-out erythrocyte in the reticuloendothelial cells, mainly in the liver and spleen, and the release of
“free” hemoglobin. In man this occurs on the average of 120 days after the erythrocyte is formed, as shown by classical isotopic labeling studies14’ 16 to be described later. Any hemoglobin released from erythrocytes directly into the circulation because of traum a or other reasons is bound immediately by haptoglobin, a plasma a2-globulin, and presumably is picked up by the reticuloendothelial system and then converted into bile pigments. This device prevents the renal loss of hemoglobin and particularly its iron which must be retained and conserved by the body. As shown by the early classical studies of Whipple and Mann and by recent investigations9 using erythrocytes labeled with 51chromium or 59iron, the reticuloendothelial cells primarily of the liver, spleen and bone marrow are the principal sites of the removal and beginning steps in the degradation of hemoglobin. However, the process may occur more slowly in most other tissues in which the extravasation of blood may occur. This is manifest by the fine play of colors which develop in bruises or in a “black eye.” In the reticuloendothelial cells, hemoglobin is converted to heme and then into bile pigments.
About 85 percent of the bile pigment formed in man is derived from senescent erythrocytes. In addition, some may be derived from the destruction of immature
Presented at the A pp lied Seminar on Chem ical Hematology, Novem ber, 1970.
113

114 ORTEN
erythrocytes in the bone marrow, especially in certain pathological conditions such as pernicious anemia.16 In the average adult male approximately 8 g of hemoglobin is thus catabolized daily, resulting in the formation of some 300 mg of bile pigments. Of this amount, about 100 to 250 mg is excreted in the feces as stercobilin and 1 to 2 mg in the urine as urobilin. The fate of the remainder is uncertain; part may be metabolized by other pathways and eliminated.
In addition to hemoglobin, some bile pigment formation from non-hemoglobin hemoproteins occurs. These sources include such hemoproteins as myoglobin ( “muscle hemoglobin”), the cytochromes, catalase, and tryptophan pyrrolase. I t is estimated that these substances are responsible for about 15 percent of the total bilirubin formed in the body. The site of the degradation of these non-hemoglobin hemoproteins to heme is uncertain, bu t any “free” heme from them apparently is firmly bound immediately by a scavenger plasma /}-glycoprotein called hemopexin.19 This binding mechanism prevents the renal loss of heme-iron, as is true of the hemoglobin- haptoglobin complex. It also may prevent the interference by heme of certain essential enzyme functions.18 The heme-hemo- pexin complex apparently is picked up selectively by the reticuloendothelial cells and the porphyrin moiety is converted into bile pigments.
The release of “free” hemoglobin from senescent erythrocytes and other sources and of free heme and their transport to reticuloendothelial cells is shown schematically in figure 1.
In their classical studies in the late 1940’s, London, Shemin, Rittenberg and their coworkers conclusively demonstrated that bile pigments are derived from other sources as well as from senescent erythrocytes.
Glycine labeled with 15N was administered to normal hum an subjects14-16 and to patients w ith pernicious anemia15 or congenital porphyria.17 Isotope concentrations were determined in hemin prepared periodically from blood samples and in stercobilin prepared from feces. In normal subjects (figure 2 ), senescent erythrocytes were destroyed at an average of 120 days, the 1BN content of hemin began to decrease. The 15N level of stercobilin, on the other hand, increased proportionally with the decrease in the labeling of hemin. This work was confirmed and extended by other investigators using glycine-2-14C in various experimental animals and establishes beyond question the fact that the major origin of the bile pigments is senescent erythrocytes. The work also verifies the indications of earlier investigations that the average life span of the erythrocyte in normal adult m an is 120 days and for the normal adult woman is about 109 days. 16 Subsequent work by these investigators demonstrated that the average life of the erythrocyte is considerably shorter than normal in certain pathological conditions such as pernicious anemia15 and in hemolytic diseases like sickle cell anemia.16
I t is evident further from figure 2, that a rapid rise in 15N-labeling of stercobilin occurred during the first few days after the administration of 15N-glycine followed by a precipitous decrease. This is the so-called “early peak” in bile pigment formation after an isotopically labeled precursor of heme is administered. This peak can not be attributed to bile pigment formation from the degradation of hemoglobin from senescent erythrocytes. The earlier16 as well as the current explanation is that the early peak, amounting to some 10 to 20 percent of the total labeled bile pigment, is from several sources, mostly non-erythrocytic. These include (1) bilirubin formation from
METABOLISM OF HEMOGLOBIN AND BILE PIGMENTS 115
'Amino acid pool"
F ig u r e 1. Schematic representation of the release and transport of “free” hemoglobin and heme.
non-hemoglobin hemoproteins (myoglobin, cytochromes, catalase, etc.); or (2) from the hemoglobin of erythrocytes destroyed in the bone marrow or very shortly after entering the circulation; or (3) from “extra” heme or porphyrins not utilized in the newly formed erythrocytes ( “ineffective eryth- ropoiesis” ); or (4) possibly from a “shunt” pathway in which pyrrole precursors may condense directly to form bile pigment without heme as an intermediate. Support of the first and third possibilities is evidenced by 15N-labeled hematin rapidly
converting into 15N-stercobilin in the dog.13 The second possibility is well-established. The final postulate is yet to be demonstrated experimentally. Recent studies10 have demonstrated that a relatively larger amount of 14C from labeled 8-aminolevu- linic acid as a precursor appears in the early peak as compared with that from an equivalent amount of glycine-2-14C. These results may be interpreted as indicating that S-aminolevulinic acid is susceptible to fewer non-heme biosynthetic pathways than is glycine.

116 ORTEN
II. The Conversion of Hemoglobin into Biliverdin and Bilirubin
The steps involved in the conversion of hemoglobin and heme into bile pigments in reticuloendothelial cells have been the subject of extensive investigation during the past several decades. Conflicting views are held and complete elucidation of the pathways involved has not been attained as yet. Also, different nomenclatures and terminology have served to somewhat confuse the problem. Predominating current concepts follow.
In contrast to the earlier view, based primarily on in vitro studies, it is no longer believed that the cleavage of the porphyrin
tetrapyrrole ring occurs while it is attached to the globin moiety of hemoglobin, forming the so-called choleglobin. Rather, current evidence indicates that heme first dissociates from the a-and /?-chains of “globin” and then converts to biliverdin and bilirubin. Recrystallized hemin has been found to be readily converted to bile pigment.13 Heme readily dissociates from the a-and ^-chains of globin with relatively mild changes in environment such as PH or ionic concentration.
The first major step in the conversion of heme into bile pigment is the opening of the tetrapyrrole porphyrin ring system by the oxidation of the a-methene bridge car-
(/>COLÜ<JXUJ
in
\ -ZLl )Oc rLJQ_
OI -<
TIME IN DAYSF ig u r e 2. The atom percent 15N excess in hemin and stercobilin o f a normal man after
the start of feeding 18N-labeled glycine for two days. (From the studies of London, W est, Shemin and Rittenberg.16)

METABOLISM OF HEMOGLOBIN AND BILE PIGMENTS 117
bon atom to form carbon monoxide (figure 3). This unique reaction in a biological system is apparently the sole source of the “endogenous” carbon monoxide found in man and animals. Recent studies25,20 have demonstrated that this reaction occurs primarily in the microsomal fraction of the liver and spleen (reticuloendothelial cells apparently). The reaction requires an enzyme system composed of a mixed-function oxygenase, called heme oxygenase, and has an absolute requirement for NADPH ( “TPNH”) and molecular oxygen. Cytochrome P-450 may serve as the terminal oxidase, as is the case in the oxidation of certain endogenous steroids and drugs by a similar mixed function oxidase system. The reaction is inhibited by carbon monoxide.
Evidence that the oxidation of the a- methene bridge carbon atom to carbon monoxide is involved in the opening of the tetrapyrrole porphyrin ring to form bili- verdin is obtained not only from considerations of the structural formulae of these compounds (figures 3 and 4) and from isotopic studies.4 If 14C-labeled hemoglobin, in erythrocytes or in solution, is injected into dogs, 14C-labeled carbon monoxide subsequently appears in the blood as 14C- carbon monoxide hemoglobin (carboxy
hemoglobin). Similar results were obtained after the administration of a labeled precursor of heme ( glycine-2-14C ). The label appeared in carbon monoxide as the 14C-heme was released from destroyed erythrocytes, formed at the time the 14C-glycine was administered. Interesting in this connection is the fact that blood carbon monoxide levels (carboxy hemoglobin) vary with the degree of hemolysis of erythrocytes, being significantly elevated in patients with hemolytic anemia.24 Indeed, Cobum4 has suggested that since carbon monoxide production parallels bile pigment formation, the determination of blood carbon monoxide levels may be more valuable in investigations of hemoglobin catabolism than measurements of bile pigments. The procedures for determining carbon monoxide production are apparently relatively simple.
Recent studies6 have demonstrated that certain drugs, such as phénobarbital, which induce the formation of the hepatic microsomal enzymes, cytochrome P-450, and possibly cytochrome B-5, cause as much as a 100 percent increase in carbon monoxide production in man. Apparently the increased catabolism of the newly-formed cytochromes is responsible for the increased carbon monoxide production. This indicates that non-hemoglobin hemoprotein, like he
H h h H
PROTOPORPHYRIN BILIRUBINF ig u r e 3 . O x id a t iv e c o n v e r s io n o f h e m e ( p r o to p o r p h y r in ) to b i l iv e r d in - b i l i ru b in .

118 ORTEN
moglobin, are catabolized to bile pigments involving the oxidative opening of the tetrapyrrole porphyrin ring with carbon monoxide production.
Endogenous carbon monoxide is apparently metabolized slowly in the body partly by oxidation to carbon dioxide by a poorly understood mechanism which may involve cytochrome oxidase. Some carbon monoxide may be exhaled from the lungs in gaseous form. There is no evidence that hydration
occurs to produce formate or that there are exchanges with formate stores.
Further strong evidence that the oxidation of the a-methene bridge carbon atom is dependent on the presence of molecular oxygen was reported recently by Marver and his associates. An hepatic microsomal system containing heme and incubated in the presence of labeled 180 2 was found to produce lsO-labeled carbon monoxide.
The opening of the porphyrin ring of
Bile Pigments Chemistry
M
H O
M
H O
M
H O
N
V M H
= C -----
V M H
= C -----
\
P P H
---- C =N
biliverdin
P P H2C
\
MH2C -
bilirubin
P PH2
— c ----
M M H
---- C =
M M H
----- C =
M M H2
----- C -----
vO H
N
NH
NH
V
O H
E
O H
mesobilirubinog en
M
O HH N
H2C -
M
\
P P H2
— c ----N /
H
M M H2
----- C -----\ t h
O H
M
O H
stercobilinogen (urobilinogen)
M
c 2
P P H
-— C =
u i i
N N HOH
stercobilin (urobilin)
The side groups in the bile pigments are as follows:
M = — CH.j (methyl)P = — CH 2 CH 2 C O O H (propionic acid)V = — C H — CH 2 (vinyl)E = — CH 2CH:t (ethyl)
F ig u r e 4 . Chemical structures of some major bile pigments and their derivatives.

M ETABOLISM OF HEMOGLOBIN AND BILE PIGMENTS 119
heme by the oxidation of the a-methene bridge apparently results in the transient formation of an iron-containing intermediate termed “verdohemin” by some investigators. However, the iron binding appears to be labilized by the cleavage of the tetrapyrrole ring and it dissociates spontaneously, being picked up by transferrin, a plasma protein with a relatively higher iron-binding affinity. Thus the iron is transported by the plasma either to the bone marrow for reutilization for the synthesis of new hemoglobin or to the liver or spleen where it is stored as ferritin for future use. Little or no iron is lost by excretion.
The oxidative opening of the porphyrin ring of heme and the removal of iron results in the formation of the first major bile pigment, biliverdin (figure 4 ). However, it is apparently reduced immediately to bilirubin by means of the enzyme, biliverdin reductase, NADPH serving as a cofactor. Two hydrogen atoms are added, one to the original y-methene bridge and one to the nitrogen atom of the adjacent pyrrole ring (ring c) as shown in figure 4. The conversion of biliverdin to bilirubin seems to be rapid and practically complete since bilirubin can be readily demonstrated in hepatic microsome systems whereas biliverdin cannot. The conversion of hemoglobin and heme to bilirubin (and carbon monoxide) is nearly quantitative.
The principal steps in the conversion of hemoglobin to bilirubin in hepatic micro- somes are summarized in the schematic representation in figure 5.
III. The Transport, Conjugation and Biliary Excretion of Bilirubin
Bilirubin formed as described apparently diffuses from the reticuloendothelial cells and is immediately bound mainly by plasma albumin and possibly to a lesser extent by a- and /3-globulins. One mole of plasma albumin can bind two moles of bili
rubin.6 It is transported in this form at a concentration normally of 0.5 to 1 mg per 100 ml blood plasma or serum. The “indirect” van den Bergh reaction is given. The plasma biliburin is selectively taken up from the albumin complex by the liver parenchymal cells, possibly involving active transport and a specific transport protein in the membrane of the cell.6 The process of the removal of bilirubin from the plasma by the liver is rapid, some 65 percent of a tracer dose of 14C-bilirubin injected into rats being removed within 5 minutes. Approximately 85 percent of this amount is found in the supernatant of liver homog- enates.6
The next step in the metabolism of bilirubin is its conjugation with glucuronic acid, as the uridine diphosphate derivative ( UDP-glucuronic acid), in the endoplasmic reticulum of the liver parenchymal cell. This reaction is catalyzed by the enzyme, bili- rubin-UDP glucuronyl transferase. Bilirubin glucuronide is formed. I t is the ester formed between the propionic acid side chains of bilirubin (figure 4) and the carbon-1- hydroxyl groups of 2 moles of glucuronic acid. Most of the conjugate formed is the diglucuronide, although smaller amounts of the monoglucuronide are also found. This reaction is shown schematically in figure 6.
There is evidence that a small amount of bilirubin is conjugated with sulfate, as “active sulfate” ( 3'-phosphoadenosine-5'- phosphosulfate or PAPS), involving the enzyme sulfokinase. Bilirubin sulfate is formed.
The conjugated forms of bilirubin are water-soluble. Thus the “direct” van den Bergh reaction is given. Being water- soluble, they are readily excreted from the liver in the bile possibly bound loosely to protein and are discharged into the intestine for elimination from the body. If the blood level of bilirubin glucuronide becomes high,

120 OBTEN
as may occur in obstruction of the bile ducts and a “damming back” of bile pigments, excretion of conjugated (direct) bilirubin may occur in the urine. Determination of the amount of direct and indirect (unconjugated) bilirubin appearing in the blood and urine are extremely valuable in the differential diagnosis of different types of jaundice. In general, high direct-reacting bilirubin levels in the plasma and urine are indicative of obstructive jaundice whereas high plasma (not urine) levels of indirect-reacting bilirubin indicate jaundice due to excessive hemolysis of erythrocytes and hemolytic anemia or a reduced capacity of the liver to conjugate and excrete bilirubin as in congenital hyperbilirubinemia, cirrhosis and infectious hepatitis. Since the excretion of bilirubin glucuronide is maximal in hemolytic jaundice, the excretion of fecal (stercobilin) and urinary (urobilin- ogen-urobilin) bile pigments will also be increased.
Since the biosynthesis of the enzyme, bilirubin-UDP-glucuronyl transferase, is genetically controlled like all other enzymes, “inborn error” in bilirubin metabolism may result from its deficiency due to hereditary defects. This occurs occasionally in man and is termed congenital hyperbilirubinemia. I t may occur in infancy resulting in the Crigler-Najjar syndrome w ith severe jaundice, brain damage (kernicterus, with staining of the basal ganglia), and even death. High levels of indirect-reacting bili
rubin are found. A second type of congenital hyperbilirubinemia is seen clinically. It is more chronic and less severe with lower serum bilirubin levels and without kernicterus. I t is transmitted as an autosomal dominant character. Black and Billing3 have recently shown by direct assay that there is a decrease in the activity of glucuronyl transferase in liver specimens from 11 patients with Gilbert’s syndrome, congenital hemolytic jaundice. Lowered transferase activity was also found in Wilson s disease but an increased level was observed in cases of biliary obstruction.
Hyperbilirubinemias (indirect) may also be found transiently in new-born infants, particularly in prematures. It has been termed a “physiologic jaundice.” Apparently a delayed or inadequate hepatic synthesis of bilirubin-UDP-glucuronyl transferase is at fault. Normally the synthesis of this enzyme and the capacity of the liver to conjugate bilirubin increases rapidly during the first few days after birth. Interesting recent studies indicate that exposing these infants to a special fluorescent light — a so-called “bilirubin reduction lamp,” results in a reduction of the hyperbilirubinemia. Apparently the excess bilirubin is degraded to less toxic products, possibly di- and mono-pyrrole derivatives12 that can be metabolized or excreted in the urine. If the hyperbilirubinemia can be controlled for a period of time, the liver may attain a normal rate of formation of glucuronyl
Hemoglobin •D isso c ia tio n
T<=C - and ^ -
chains
->• Heme
HemeOxygenase r i
».-----> LVerdoheminJ—«r—^ B iliv e rd in -NADPH \ \
1 FeV ( r e u t i l i z e d )CO
B iliv e rd in re d u c ta se
NAD PH B iliru b in
Amino a c id s ( r e u t i l i z e d )
F i g u r e 5. Schematic representation of the conversion of hemoglobin to bilirubin in an hepatic microsomal system (reticuloendothelial cells).

M ETABOLISM OF HEMOGLOBIN AND BILE PIGM ENTS 121
transferase and adequate conjugation and excretion of bilirubin will be maintained without therapy.
Impaired bilirubin conjugation and an associated moderate hyperbilirubinemia may also occur in normal individuals during fasting2 or in patients with Gilbert’s disease during periods of low caloric intake.5 A lowered availability of glucuronic acid, which is formed in the body from glucose, may be responsible for the decreased conjugation of bilirubin and hence its decreased excretion in the bile.
While a number of drugs and hormones allegedly decrease the capacity of the liver to transport or conjugate bilirubin, a few are capable of increasing the ability of the liver microsomes to form glucuronides. Phénobarbital is a notable example.6 D ramatic effects of this drug in reducing serum bilirubin levels have been observed in infants w ith chronic, non-hemolytic, unconjugated hyperbilirubinemia. However, phénobarbital appears to have little or no effect in genetically determined deficiency of glucuronyl transferase, as in the classical Crigler-Najjar syndrome. The mechanism of action of phénobarbital is uncertain at this time. Its administration may stimulate an increased formation of glucuronyl transferase, since this enzyme is involved in the conjugation (detoxication) of phénobarbital with glucuronic acid.
IV. The Fate of Bilirubin Conjugates in the Intestine
The excretion of conjugated bilirubin in the bile from the hepatic parenchymal cell is believed to involve an active transport mechanism.6 Once in the intestine, bilirubin glucuronide (or sulfate) is hydrolyzed by intestinal or bacterial enzymes. The residual “free” bilirubin is then converted to a variety of derivatives by reduction by bacterial enzymes and the autoxidation of some of these products. The
B i l i r u b in
IJDP-Glucuronic Acid
UDP-
B i l i r u b in Glucuronide
B i l i r u b inUDP-GlucuronylT ransferase
V
Excreted v ia b i l e
Small I n t e s t in e
F ig u r e 6. Schematic representation of the conjugation of bilirubin in endoplasmic reticulum of the hepatic parenchymal cell.
identity of the reduction products of bilirubin is still one of the most uncertain areas of bile pigment metabolism. Few of the products have been isolated and characterized beyond question and the field is further confused by varying nomenclatures and terminologies. The schematic diagram in figure 7 summarizes the principal products of bilirubin degradation by bacterial enzymes in the intestine and their ultimate conversion to stercobilinogen and sterco- bilin, the final excretory products in feces and urobilinogen and urobilin, the terminal degradation products excreted in urine. The structural formulae of the major bile pigments found in the intestine are given in figure 4.
Part of the urobilinogen formed as shown in the diagram is reabsorbed from the intestine and is either reoxidized to bilirubin and reexcreted in the bile or excreted in the urine and converted by autoxidation to urobilin. A normal adult excretes approximately 1 to 2 mg of urobilin in the urine daily and as much as 150 to 250 m g of stercobilin in the feces. This accounts for about 80 percent of the heme liberated

122 ORTEN
VExcreted in Urine. Excreted in Feces
F ig u h e 7. Schematic representation of some of the principal products formed by bacterial enzyme action on bilirubin conjugates in the intestine. The excretory fates of the end-products are shown.
from the daily destruction of erythrocytes in man. The fate of the remaining 20 percent is as yet uncertain.
Some investigators believe that fecal stercobilinogen and stercobilin are identical with urinary urobilinogen and urobilin whereas others believe they are isomers.
Further studies of their chemical structures will be required to resolve these divergent opinions.
The semi-quantitative determination of urobilinogen in urine by Ehrlich’s aldehyde reagent is a valuable clinical laboratory procedure in the differential diagnosis of

M ETABOLISM OF HEMOGLOBIN AND BILE PIGM ENTS 123
diseases of bile pigment metabolism. In general, it tends to be high in hemolytic types of jaundice and low in obstructive types.
Thus the clinical laboratory determination of several of the products of the metabolism of hemoglobin and the bile pigments are invaluable guides in the diagnosis of a number of diseases that afflict mankind.
General Summary
The catabolism of hemoglobin entails its release primarily from senescent erythrocytes which are phagocytized by reticuloendothelial cells, normally after approximately 120 days of service. Any hemoglobin released by other processes is bound immediately by haptoglobin and transferred to the R-E-cells. Heme from non-hemo- globin hemoproteins is similarly bound by hemopexin and transferred to R-E-cells for degradation.
In the R-E cells, primarily of the liver and spleen, hemoglobin readily dissociates the a- and /3-chains being hydrolyzed subsequently and their constituent amino acids are re-utilized. The heme moiety, together with that from other hemoproteins, is cleaved by a mixed function oxygenase system comprised of heme oxygenase, NADPH, and 0 2, at the a-methene bridge to form carbon monoxide. W ith the opening of the porphyrin ring, the iron of heme is labilized and removed for reuse or storage. The bile pigment, biliverdin, is then transiently formed bu t is immediately converted to bilirubin in the presence of biliverdin reductase and NADPH.
The bilirubin formed is removed from the R-E-cells, bound by plasma albumin, and transported to the hepatic parenchymal for final disposition. It is first conjugated with glucuronic acid in the presence of bilirubin- UDP-glucuronyl transferase to form water- soluble bilirubin glucuronide which is then
excreted into the intestine in bile, possibly in a protein-bound form.
In the intestine, the bilirubin conjugate is hydrolyzed and bilirubin is reduced by bacterial enzymes into a variety of bile pigment derivatives, including mesobilirubino- gen, stercobilinogen, and urobilinogen. Stercobilinogen and urobilinogen differ little structurally, probably being isomeric forms. Stercobilinogen is autoxidized to stercobilin and excreted in the feces. Some urobilinogen is reabsorbed and part is reoxidized to bilirubin in the liver and excreted in the bile. A small part is excreted by the kidney into the urine after autoxi- dation to urobilin.
The catabolism of hemoglobin into the various bile pigments and their derivatives yields intermediates which are of tremendous importance today in the clinico-chemi- cal recognition and diagnosis of a number of diseases in man.
References
1. A r i a s , I. M.: Hepatic aspects of bilirubin metabolism. Ann. Rev. Med. 1 7 :257-274, 1 9 6 6 .
2 . B a r r e t t , P.: Hyperbilirubinemia of fasting: Studies in man and in the congenitally jaundiced (G unn) rat. Gastroenterology 58: 9 2 6 , 1 9 7 0 .
3. B l a c k , M. a n d B e l l in g , B . H.: Hepatic bilirubin UDP-glucuronyl transferase activity in liver disease and Gilbert’s syndrome. N ew Eng. J. Med. 280: 1266-1271,1969.
4. C o b u r n , R. F.: Endogenous carbon monoxide production. N ew Eng. J. Med. 282: 207-209, 1970.
5 . F e l s h e r , B . F . , R i c h a r d , D., a n d R e d e k e r , A. G.: Caloric intake and degree of hyperbilirubinemia in Gilbert’s syndrome. N ew Eng. J. Med. 2 8 3 : 170-172,1970.
6. G a r t n e r , L. M. a n d A r ia s , I. M.: Formation, transport, metabolism and excretion of bilirubin. N ew Eng. J. Med. 280: 1339-1345,1969.
7. G a r t n e r , L. M. a n d A r ia s , I. M.: Pharmacologic and genetic determinants of disordered bilirubin transport and metabolism in the liver. Ann. NY Acad. Sci. 151: 833-841,1968.

124 ORTEN
8. G r a y , C. H.: Bile Pigments in Health and Disease, Charles C Thomas, Springfield, IL.,1961.
9. H u g h e s -J o n e s , N . C . a n d C h e n e y , B.: The Use of BiCr and 59F e as red cell labels to determine the fate of normal erythrocytes in the rat. Clin. Sci. 20: 323-332, 1961.
10. I s r a e l s , L. G., Ya m a m o t o , T., Sk a n d e r b e g , J . a n d Z iptj’r s k y , A.: Shunt bilirubin: Evidence for two components. Science 1 3 9 :1054- 1055, 1963.
11. L e s t e r , R. a n d S c h m id , R.: Bilirubin metabolism. N ew Eng. J. Med. 270: 779-786, 1964.
12. L e s t e r , R. a n d T r o x l e r , R. F.: N ew light on neonatal jaundice. N ew Eng. J. Med. 280: 779-780, 1969.
13. L o n d o n , I. M.: The conversion of hematin to bile pigment. J. Biol. Chem. 1 8 4 :373-375, 1950.
14. L o n d o n , I. M., S h e m i n , D ., W e s t , R ., a n d R it t e n b e r g , D .: Hem e synthesis and red blood cell dynamics in normal humans and in subjects w ith polycythemia vera, sickle-cell anemia, and pernicious anemia. J. Biol. Chem. 179: 463-484, 1949.
15. L o n d o n , I. M., a n d W e s t , R.: The formation of bile pigments in pernicious anemia. J. Biol. Chem. 184: 359-364, 1950.
16. L o n d o n , I . M., W e s t , R ., S h e m i n , D., a n d R it t e n b e r g , D.: On the origin of bile pigment in normal man. J. Biol. Chem. 184: 351- 358, 1950.
17. L o n d o n , I. M., W e s t , R ., S h e m i n , D . a n d
R it t e n b e r g , D.: Porphyrin formation andhemoglobin metabolism in congenital porphyria. J. Biol. Chem. 184: 365-371, 1950.
18. M u i x e r - E b e r iia r d , U. a n d B a s h o r e , R .: Bilirubin, albumin and hemopexin in amniotic fluid. N ew Eng. J. Med. 282; 1163-1167,1970.
19. M u l l e r - E b e r h a r d , U., L i e m , H . H . , H a m -
s t e i n , A., et al.: Studies on the disposal of intravascular heme in the rabbit. J. Lab. Clin. M e d . 73:210-218, 1969.
20. O r t e n , J. M. a n d N e u h a u s , O. W .: Biochemistry, 8th ed., pp. 681-686, C. V. Mosby Co., St. Louis, MO., 1970.
21. R o b in s o n , S. H.: The origins of bilirubin. N ew Eng. J. Med. 279: 143-149, 1969.
22. S c h m i d , R . : Hyperbilirubinemia. Metabolic Basis o f Inherited Disease, pp. 871-902, 2nd ed., Stanbury, J. B ., Wyngaarden, J. B., and Fredrickson, D . S ., eds., McGraw-Hill, N ew York, 1966.
23. Ibid ., Jaundice and bilirubin metabolism. Bull. NY Acad. Med. 35: 755-764, 1959.
24. S jö s t r a n d , T.: Endogenous formation of carbon monoxide in man under normal and pathological conditions. Scand. J. Clin. Lab. Invest. 1: 201-214, 1949.
25. S n y d e r , A. L. a n d Sc h m id , R .: The conversion of hemoglobin to bile pigment in the rat. J. Lab. Clin. Med. 65: 817-824, 1965.
26. T e n h u n e n , R ., M a r v e r , H. S ., a n d S c h m id , R .: The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc. Nat. Acad. Sci. 61; 748-755, 1968.
27. W i t h , T.: Bile Pigments: Chemical, Biological and Clinical Aspects, Academic Press, N ew York, 1968.
28. Y a m a m o t o , T., Sk a n d e r b e g , J., Z ip u r s k y , A., a n d I s r a e l s , L. G.: The early-appearing bilirubin: Evidence for two components. J. Clin. Invest. 44: 31-41, 1965.


















