
Metabolic Therapy for Age-Dependent Impaired Wound Healing
244
University of South Florida Scholar Commons Graduate eses and Dissertations Graduate School 3-16-2016 Metabolic erapy for Age-Dependent Impaired Wound Healing Shannon Lynn Kesl Follow this and additional works at: hp://scholarcommons.usf.edu/etd Part of the Physiology Commons is Dissertation is brought to you for free and open access by the Graduate School at Scholar Commons. It has been accepted for inclusion in Graduate eses and Dissertations by an authorized administrator of Scholar Commons. For more information, please contact [email protected]. Scholar Commons Citation Kesl, Shannon Lynn, "Metabolic erapy for Age-Dependent Impaired Wound Healing" (2016). Graduate eses and Dissertations. hp://scholarcommons.usf.edu/etd/6104
Transcript of Metabolic Therapy for Age-Dependent Impaired Wound Healing

University of South FloridaScholar Commons
Graduate Theses and Dissertations Graduate School
3-16-2016
Metabolic Therapy for Age-Dependent ImpairedWound HealingShannon Lynn Kesl
Follow this and additional works at: http://scholarcommons.usf.edu/etd
Part of the Physiology Commons
This Dissertation is brought to you for free and open access by the Graduate School at Scholar Commons. It has been accepted for inclusion inGraduate Theses and Dissertations by an authorized administrator of Scholar Commons. For more information, please [email protected].
Scholar Commons CitationKesl, Shannon Lynn, "Metabolic Therapy for Age-Dependent Impaired Wound Healing" (2016). Graduate Theses and Dissertations.http://scholarcommons.usf.edu/etd/6104

Metabolic Therapy for Age-Dependent Impaired Wound Healing
by
Shannon Lynn Kesl
A dissertation submitted in partial fulfillment of the requirements for the degree of
Doctor of Philosophy Department of Molecular Physiology and Pharmacology
Morsani College of Medicine University of South Florida
Co-Major Professor: Dominic D’Agostino, Ph.D. Co-Major Professor: Mack Wu, M.D.
Lisa Gould, M.D., Ph.D. Thomas Taylor-Clark, Ph.D.
Paula Bickford, Ph.D. Kenneth Ugen, Ph.D.
Date of Approval: December 09, 2015
Keywords: Aging, Chronic Wounds, Ketosis, Exogenous Ketone Supplementation
Copyright © 2016, Shannon Lynn Kesl

ACKNOWLEDGEMENTS
The research included in this dissertation was made possible by support from our funding
sources, including the James A. Haley Veterans Hospital’s Merit Review, the Department of
Molecular Pharmacology and Physiology, Scivation Inc., and the Office of Naval Research
(ONR).
I would like to express my heartfelt gratitude to everyone who has supported me in my
pursuit of my doctoral degree. Firstly, I want to acknowledge the contribution from all of the
ratticans that have sacrificed their lives to further scientific research.
To my mentor, Dr. Dominic D’Agostino thank you for your relentless encouragement,
guidance, and always going above and beyond for me no matter what was going on around you.
You are one of the hardest working people I know. Thank you for always being just an email or
phone call away, and always working diligently through the night for the betterment of our lab,
my career, and my life. Thank you for instilling in me your passion and drive for helping others
through our research and for helping me to realize why our job as scientists is so important. To
my Co-mentor, Dr. Mack Wu, thank you for investing in me when I needed someone and for
your continuous direction and support. To Dr. Lisa Gould, thank you for believing in me as your
first Ph.D. student, for always being there even when we are far apart, for giving me an
opportunity to see the clinical aspect of our research, and especially for challenging me and
making me a better scientist.

To the members of my dissertation committee, Dr. Thomas Taylor-Clark, Dr. Paula
Bickford, Dr. Kenneth Ugen, Dr. Patrick Bradshaw for their scientific guidance and support over
the past five plus years. To my outside chair, Dr. Stanley Stevens Jr., thank you for stepping in
last minute and handling everything efficiently. I wish I had gotten to know you earlier in my
career, but am hopeful for a continued relationship in the future.
To my lab family, Carol Landon, Dr. Jay Dean, Geoffrey Ciarlone, Jacob Sherwood, Dr.
Christopher Rogers, Dr. Csilla Ari, Andrew Koutnik, and Nathan Ward, thank you for making
work such an entertaining but reassuring environment even when I needed to order something
last minute, we have had a midnight time point, or when I have forced you to listen to musicals. I
will be forever grateful for the time we have shared. To Dr. Andrea Trujillo for your continued
guidance and support, thank you for teaching me so much. I want to give a special recognition
and thank you to my wonderful friend and colleague, Dr. Angela Poff, from my whole heart I
wish to thank you for your unwavering love, support, and encouragement, none of this would
have been possible without you.
To my school family, Dr. Franklin Poff, Randi McCallian, Dr. Chase Lambert, and Dr.
Jillian Whelan, who have always been there with wine, excellent dinners, wine, relaxing
company, wine, ridiculous conversation, and wine to ease the stress of this journey. To Shelby
Evans and Marilyn Rodriguez, who always listened when I gushed about science even when they
didn’t understand and thought it was boring, thank you for being there whether near or far. To
Myshell (Michelle) Jung, thank you for being a source of constant inspiration and love even
when we didn’t get to see each other every day or get to braid hair during incubation times. To
my friends and family who have always loved and encouraged me throughout my life, Beth
Webster, Joshua Smith, Katie Rader, Chad Rader, Harper Rader, June Guy, Merle Guy, John

Fry, Michelle Adkins, Kyle Adkins, Travis Adkins, Amanda Fox, James Fox, Keith Waldron,
Jeanette Kesl, and Robert Kesl. To my Presley Rader for always being enthusiastic to learn about
science, watching your face light up when you looked into a microscope for the first time
restored my wonderment for science. To my hippo and chicken, thank you for your
unconditional love and for always being there to melt my stress away with snuggles on the
couch.
To my best friend, my heart, my husband, Jason, I know it has been hard being apart for
the majority of my dissertation work and prep, but know that I felt your unconditional love and
support even from Germany. Thank you for the countless times of reassurance, encouragement,
laughter, and inspiration. I look forward to many more years of our life together. Last but not
least to my mother, Jennifer Fry, thank you for always believing in me and pushing me to
achieve my dreams. None of this would have been possible without your continual words of
encouragement, your unconditional love, and your unwavering support no matter where life has
taken me or how long it has taken me to get there. I am forever grateful.

i
TABLE OF CONTENTS
List of Tables ................................................................................................................................. iv List of Figures ..................................................................................................................................v List of Abbreviations .................................................................................................................... vii Abstract ........................................................................................................................................ xiii Chapter 1: Wound Healing Physiology ...........................................................................................1
1.1. Chapter Synopsis .....................................................................................................1 1.2. Skin Morphology .....................................................................................................2 1.3. Wound Classification and Closure Types ................................................................2 1.4. Acute Wound Healing Cascade ...............................................................................4
1.4.1. Hemostasis ...................................................................................................5 1.4.2. Inflammation ................................................................................................5 1.4.3. Proliferation .................................................................................................7 1.4.4. Maturation ..................................................................................................10
1.5. Cellular Energy Metabolism ..................................................................................10 1.5.1. Catabolism .................................................................................................11 1.5.2. Anabolism ..................................................................................................13
1.6. Wound Healing Metabolism ..................................................................................14 1.7. Nutrition and Wound Healing ................................................................................15 1.8. Chronic Wounds ....................................................................................................17
1.8.1. Classification of Chronic Wounds .............................................................18 1.8.2. Current Standard of Care, Therapies and Their Disadvantages .................20
1.8.2.1. TIME and Amputation .................................................................21 1.8.2.2. Adjunctive Therapies ...................................................................22
1.9. Age Influence on Chronic Wounds ........................................................................26 1.9.1. Aged Related Changes in Skin Morphology .............................................26 1.9.2. Age Related Changes in the Wound Healing Cascade ..............................27
1.10. Closing Remarks ....................................................................................................30 1.11. References for Chapter 1 .......................................................................................31
Chapter 2: Oral Ketone Supplementation as a Novel Therapy for Age-Dependent Delay Wound
Healing .........................................................................................................................46 2.1. Chapter Synopsis ...................................................................................................46

ii
2.2. Ketone Body Synthesis and Metabolism ...............................................................47 2.3. Review of Ketogenic Diet and Therapeutic Uses ..................................................48
2.3.1. Potential Concerns and Side Effects ..........................................................49 2.4. Exogenous Ketone Supplementation .....................................................................50
2.4.1. 1,3-Butanediol ............................................................................................51 2.4.2. MCT Oil .....................................................................................................52 2.4.3. βHB Salt .....................................................................................................54 2.4.4. Combination BMS+MCT ..........................................................................55 2.4.5. Ketone Esters .............................................................................................56 2.4.6. Potential Concerns and Side Effects: Ketoacidosis ...................................57
2.5. Potential Physiology Affected to Enhance Wound Healing ..................................58 2.5.1. Ketones and Inflammation .........................................................................59 2.5.2. Ketones and ROS .......................................................................................60 2.5.3. Ketones and Blood Flow and Angiogenesis ..............................................62 2.5.4. Ketones and Metabolism ............................................................................63 2.5.5. Ketones Suppress Blood Glucose via Increasing Insulin Sensitivity ........64
2.6. Ischemic Wound Healing Model ...........................................................................66 2.7. Use of Primary Human Dermal Fibroblasts in vitro ..............................................69 2.8. Central Hypothesis and Project Goals ...................................................................70 2.9. Closing Remarks ....................................................................................................71 2.10. References for Chapter 2 .......................................................................................72
Chapter 3: Establishing Therapeutic Ketosis (Metabolic Therapy) with Exogenous Ketone
Supplementation ..........................................................................................................86 3.1. Chapter Synopsis ...................................................................................................86 3.2. Effects of Exogenous Ketone Supplementation on Blood Ketones, Glucose,
Triglycerides, and Lipoprotein Levels in Sprague-Dawley Rats ...........................87 3.2.1. Ketone supplementation causes little to no change in triglycerides and
lipoproteins ................................................................................................88 3.2.2. Therapeutic levels of hyperketonemia suppress blood glucose levels .......90 3.2.3. Effects of ketone supplementation on organ weight and body weight
percentage ..................................................................................................92 3.3. Effect of Sustaining Dietary Ketosis Through Exogenous Ketone
Supplementation on the Hippocampal and Serum Metabolome of Sprague-Dawley Rats .........................................................................................................103 3.3.1. Oral administration of ketone supplements elevates blood ketone levels,
increases Krebs cycle intermediates, MCFAs, antioxidants, and adenosine in serum and hippocampal tissue and implications for wound healing ...103
3.4. References for Chapter 3 .....................................................................................113
Chapter 4: Enhancing Wound Healing with Metabolic Therapy ................................................123 4.1. Chapter Synopsis .................................................................................................123 4.2. Dietary Ketone Supplementation Increases Blood Flow and Wound Closure in an
Ischemic Wound Model in Young and Aged Fischer Rats ..................................124

iii
4.2.1. Aged rats metabolize exogenous ketone supplements differently than young rats .................................................................................................125
4.2.2. Hypoglycemic effect of hyperketonemia attenuated with age .................127 4.2.3. Ketone supplementation increases blood flow in both young and aged
rats ............................................................................................................128 4.2.4. Ketone supplementation accelerates wound closure in young and aged
rats ............................................................................................................129 4.2.5. Food- integrated ketone supplementation did not elicit weight loss in
young and aged rats .................................................................................131 4.3. Closing Remarks ..................................................................................................133 4.4. References for Chapter 4 .....................................................................................139
Chapter 5: Potential Mechanisms for Exogenous Ketone Supplementation to Enhance Wound
Healing .......................................................................................................................144 5.1. Chapter Synopsis .................................................................................................144 5.2. Potential Mechanisms of Action for Exogenous Ketone Enhancement of Ischemic
Wound Healing in Young and Aged Fisher Rats .................................................145 5.2.1. Effects of ketone supplementation on inflammation ...............................146 5.2.2. Effects of ketone supplementation on ROS production ...........................150 5.2.3. Effects of ketone supplementation on angiogenesis ................................152 5.2.4. Effects of ketone supplementation on metabolism ..................................153
5.3. Closing Remarks ..................................................................................................154 5.4. References for Chapter 5 .....................................................................................165
Chapter 6: Discussion: Implications for Wound Healing ............................................................172 6.1. Chapter Synopsis .................................................................................................172 6.2. Implications for Wound Healing and Future Directions ......................................172 6.3. References for Chapter 6 .....................................................................................176
Appendix A: Methods and Materials ...........................................................................................179 Appendix B: Copyright Permissions ...........................................................................................198 Appendix C: Published Manuscripts ...........................................................................................202 About the Author ............................................................................................................... End Page

iv
LIST OF TABLES
Table 3.1 Global metabolism profile of serum and hippocampal tissue following chronic
administration of KE and BMS+MCT .................................................................109 Table A.1 Caloric density of standard rodent chow and dose of ketone supplements .........181

v
LIST OF FIGURES
Figure 3.1 Effects of ketone supplementation on triglycerides and lipoproteins ....................96 Figure 3.2 Effects of ketone supplementation on blood βHB .................................................... 97 Figure 3.3 Effects of ketone supplementation on blood glucose ................................................... 98 Figure 3.4 Relationship between blood ketone and glucose levels ............................................... 99 Figure 3.5 Effects of ketone supplementation on organ weight ..............................................100 Figure 3.6 Effects of ketone supplementation on body weight .................................................... 101 Figure 3.7 Effects of ketone supplementation on basal blood ketone and basal blood glucose
levels ....................................................................................................................102 Figure 3.8 Ketone supplementation elevates blood ketone levels in serum and hippocampal
tissues ...................................................................................................................110 Figure 3.9 Ketone supplementation increases medium chain fatty acids in serum and
hippocampal tissue ...............................................................................................110 Figure 3.10 Ketone supplementation increases Kreb’s cycle intermediates ...........................111 Figure 3.11 Ketone supplementation increases antioxidants ..................................................112 Figure 3.12 Ketone supplementation increase Adenosine ......................................................112 Figure 4.1 Effects of ketone supplementation on blood ketone and blood glucose levels ....134 Figure 4.2 Relationship between blood ketone and blood glucose levels attenuated with age ........................................................................................................................134 Figure 4.3 Ketone supplementation increases blood flow in both young and aged rats .......135 Figure 4.4 Ketone supplementation enhances wound closure time in young and aged rats .......................................................................................................................136

vi
Figure 4.5 Ketone supplementation elevates blood ketones levels but does not affect blood glucose levels .......................................................................................................137
Figure 4.6 Body and Spleen Weights ....................................................................................138 Figure 5.1 Heterogeneity of Aged Ischemic Wound Healing ...............................................155 Figure 5.2 Effects of ketone supplementation on pro-inflammatory and anti-inflammatory
cytokines ..............................................................................................................156 Figure 5.3 Effects of ketone supplementation on growth factors and leptin .........................157 Figure 5.4 Ketone supplementation does not affect M1 to M2 ratio of day 7 ischemic wounds
in aged rats ...........................................................................................................158 Figure 5.5 Ketone supplementation decreases ROS production ...........................................159 Figure 5.6 Ketone supplementation does not affect Antioxidants SOD2 and NQO1 in vitro
and ex vivo ............................................................................................................160 Figure 5.7 Ketone supplementation does not significantly increase blood vessel density in
day 7 aged ischemic wounds ................................................................................161 Figure 5.8 Ketone supplementation increases migration in young and aged HDFs .............162 Figure 5.9 Ketone supplementation increases proliferation in young and aged HDFs .........163 Figure 5.10 Ketone supplementation decreases lactate levels in young and aged HDFs .......164

vii
LIST OF ABBREVIATIONS
α-SMA ................................................................................................... Alpha-smooth muscle actin βHB ................................................................................................................ Beta-hydroxybutyrate AcAc ............................................................................................................................. Acetoacetate AD ..................................................................................................................... Alzheimer’s disease ADH .......................................................................................................... Aldehyde dehydrogenase ADP ............................................................................................................. Adenosine diphosphate ALS .................................................................................................... Amyotrophic lateral sclerosis AMP ....................................................................................................... Adenosine monophosphate ARE.......................................................................... Antioxidant or electrophilic response element ATP .............................................................................................................. Adenosine triphosphate BD ................................................................................................................... R, S-1, 3- Butanediol BMI ....................................................................................................................... Body Mass Index BMS ....................................................................................................................... Na+/K+ βHB Salt BMS+MCT ....................................................... Na+/K+ βHB Salt: Medium Chain Triglyceride Oil CAT...................................................................................................................................... Catalase CD31 ..................................................................................................... Cluster of differentiation 31 CD68 ..................................................................................................... Cluster of differentiation 68 CD206 ................................................................................................. Cluster of differentiation 206

viii
CMS .............................................................................. Centers for Medicare & Medicaid Services CNS ............................................................................................................ Central Nervous System CRP .................................................................................................................. Cysteine-rich protein CVD .............................................................................................................. Cardiovascular disease DHE ....................................................................................................................... Dihydroethidium DMEM .................................................................................... Dulbecco’s Modified Eagle Medium ECM .................................................................................................................. Extracellular matrix EGF .......................................................................................................... Endothelial growth factor EMT ........................................................................................................... Electromagnetic therapy eNOS ............................................................................................. Endothelial nitric oxide synthase ET-1 .............................................................................................................................. Endothelin-1 ETC ............................................................................................................. Electron transport chain FADH2 .................................................................................................. Flavin adenine dinucleotide FDA ........................................................................................... US Food and Drug Administration FGF ............................................................................................................. Fibroblast growth factor FOXO3A .............................................................................................................. Forkhead box O3a G6P .................................................................................................................. Glucose-6-phosphate G6PDH .................................................................................... Glucose-6-phosphate dehydrogenase GC/MS ............................................................................ Gas chromatography - mass spectrometry GI ............................................................................................................................. Gastrointestinal GLUT ................................................................................................................. Glucose transporter GNG ....................................................................................................................... Gluconeogenesis GSH .................................................................................................................. Reduced glutathione

ix
GSSG ............................................................................................................... Oxidized glutathione H2O2 .................................................................................................................... Hydrogen peroxide HBOT ..................................................................................................... Hyperbaric oxygen therapy HDAC ................................................................................................................ Histone deacetylase HDACI ................................................................................................ Histone deacetylase inhibitor HDF ............................................................................................. Primary human dermal fibroblast HDL ........................................................................................................... High density lipoprotein HKE ............................................................................................................ High Dose Ketone Ester I-CAM ............................................................................................. Intercellular adhesion molecule IFN-γ ..................................................................................................................... Interferon gamma IGF-1 ..................................................................................................... Insulin like growth factor-1 IL-1 ............................................................................................................................... Interleukin-1 IL-1β ..................................................................................................................... Interleukin-1 beta IL-4 ............................................................................................................................... Interleukin-4 IL-6 ............................................................................................................................... Interleukin-6 IL-8 ............................................................................................................................... Interleukin-8 IL-10 ........................................................................................................................... Interleukin-10 IL-13 ........................................................................................................................... Interleukin-13 IL-18 ........................................................................................................................... Interleukin-18 IRF-5 .................................................................................................. Interferon regulatory factor-5 KD .............................................................................................................................. Ketogenic diet KE ......................................................................... R, S-1, 3-butanediol diacetoacetate ketone ester KEAP-1 ................................................................................. Kelch-like ECH-associated protein -1

x
LBM ......................................................................................................................... Lean body mass LC-MS/MS .............................................. Liquid chromatography with tandem mass spectrometry LCFA .............................................................................................................. Long chain fatty acid LDH .............................................................................................................. Lactate dehydrogenase LDL ............................................................................................................. Low density lipoprotein LKE ................................................................................................................ Low dose ketone ester LPS ..................................................................................................................... Lipopolysaccharide M1 .................................................................................................... Pro-inflammatory macrophage M2 ................................................................................................... Anti-inflammatory macrophage MCFA ........................................................................................................ Medium chain fatty acid MCT ........................................................................................................ Medium chain triglyceride MCP-1 ..................................................................................... Monocyte chemoattractant protein-1 MDA ...................................................................................................................... Malondialdehyde Mg-ATP .................................................................................... Magnesium adenosine triphosphate MMP ........................................................................................................... Matrix metallopeptidase MnSOD ........................................................................................ Manganese superoxide dismutase MRC ................................................................................................ Mitochondrial respiratory chain MT2....................................................................................................................... Metallothionein-2 mtDNA .............................................................................................................. Mitochondrial DNA NAD+ .......................................................................... Oxidized nicotinamide adenine dinucleotide NADH ......................................................................... Reduced nicotinamide adenine dinucleotide NADPH .................................................................... Nicotinamide adenine dinucleotide phosphate NLRP3 ............................................................................... NLR family, pyrin domain containing 3

xi
NO .................................................................................................................................. Nitric oxide NOS ................................................................................................................. Nitric oxide synthase NPWT ....................................................................................... Negative Pressure Wound Therapy NQO1 .................................................................................. NAD(P)H dehydrogenase [Quinone]-1 Nrf2 ................................................................................ Nuclear factor (erythroid-derived 2)-like 2 O2
- ......................................................................................................................... Superoxide anion PAI-1 ............................................................................................ Plasminogen activator inhibitor-1 PCOS .................................................................................................... Poly cystic ovary syndrome PDGF ................................................................................................. Platelet-derived growth factor PDH ........................................................................................................... Pyruvate dehydrogenase PEM ...................................................................................................... Protein energy malnutrition PHMB ............................................................................................... Polyhexamethylene Biguanide PO2 .......................................................................................................... Partial pressure of oxygen PMN ................................................................................................. Polymorphonuclear leukocytes PPP ........................................................................................................ Pentose phosphate pathway QUICKI ........................................................................ Quantitative insulin-sensitivity check index RANTES ........................................ Regulated on activation, normal T cell expressed and secreted ROS ............................................................................................................ Reactive oxygen species S-BHB .............................................................................. Na+/Ca+2-β-hydroxybutyrate mineral salt S/MCT ........................ Na+/Ca+2-β-hydroxybutyrate mineral salt: Medium Chain Triglyceride Oil SAD ............................................................................................................. Standard American diet SD ................................................................................................................................ Standard diet SOD ............................................................................................................... Superoxide dismutase

xii
SS ............................................................................................................................. Skin substitutes T2D ............................................................................................................ Type 2 diabetes mellitus TBOOH .............................................................................................. Tert-butyl hydrogen peroxide TBI ................................................................................................................ Traumatic brain injury TGF-β ............................................................................................ Transforming growth factor-beta TIME ........................................................................... Tissue, infection, moisture, and wound edge TNF-α ................................................................................................... Tumor necrosis factor-alpha VCO ..................................................................................................................... Virgin coconut oil VEGF ......................................................................................... Vascular endothelial growth factor

xiii
ABSTRACT
Chronic wounds represent an under-acknowledged socioeconomic epidemic, affecting 1.8
million new patients per year and costing the US health care system upwards of $25 billion
annually. This substantial cost is rapidly growing due to a disproportionate occurrence in the
ever-aging population. Key features associated with age-related impairment of wound healing
include limited energy and nutrient exchange, unremitting inflammations, increased reactive
oxygen species (ROS), and diminished blood flow. Most chronic wound therapies target specific
molecular mechanisms; however, there are often multiple mitigating factors that prevent normal
wound closure. This is likely one reason most wound therapies are minimally effective. In the
standard American diet, carbohydrates are broken down for fuel (glucose). While fasting,
starvation, and calorie or carbohydrate restriction, beta-oxidation of stored fats in the liver
produces ketone bodies (primarily acetoacetate (AcAc) and β-hydroxybutyrate (βHB) to serve as
energy metabolites for extra-hepatic tissues. In addition to enhancing metabolic physiology,
ketone bodies have recently been discovered to have signaling properties that are independent of
their function as energy metabolites. Here we present the evidence for a novel method of
inducing therapeutic ketosis via exogenous ketone supplementation to promote enhanced
ischemic wound healing in young and aged Fischer 344 rats. Preliminary mechanistic studies
demonstrated that exogenous ketone supplementation enhanced wound healing via increasing
proliferation and migration, decreasing lactate production, and decreasing ROS production as

xiv
well as affecting inflammatory cytokines and growth factors. We conclude that exogenous
ketone supplementation will be an effective, cost efficient, low toxicity therapy to promote
enhancement of wound healing in an aged population.

1
CHAPTER 1: WOUND HEALING PHYSIOLOGY
1.1. Chapter Synopsis
The following is a brief review of skin morphology, wound classification and types of
wound healing, and the classic wound healing cascade. It is important to understand the
complexity of the orchestration in conjunction with the magnitude of participants and the vast
number of interactions it takes to heal a wound effectively. Even though most of the time wound
healing proceeds to completion without complication, the majority of this chapter addresses what
happens when things do not follow the standard progression. The current topical approach to
wound healing treatments has failed not only in reversing chronic wound stagnancies but has
cost the US alone billions of dollars in health care costs. There is a dire need to discover novel
and effective treatments for chronic wounds. The aged population has a high prevalence of
chronic wounds due to comorbidities in combination with physiological changes of aging.
Increasing evidence shows that unresolved inflammation, increased reactive oxygen species
(ROS), diminished blood flow, and decreased metabolism and ATP production are key
physiological features associated with age-related impairment of chronic wound healing. In this
chapter, we concentrate on the importance of these factors as well as how age affects them in a
wound-healing environment. Additionally, these are the four physiological features that we will
seek to target in our proposed treatment, which will be discussed in later chapters. A good
understanding of the information presented in this chapter will be critical to interpreting the

2
rationale behind our proposed therapies and the data represented in the subsequent chapters of
this dissertation.
1.2. Skin Morphology
The skin is the largest organ in the human body, comprising up to 12-15% of total body
weight and covering 1.5-2m2 of surface area [1]. There are three distinct layers of the skin: the
epidermis, the dermis, and the hypodermis. The epidermis is the avascular outermost layer,
primarily populated by keratinocytes, which regulates body temperature, absorbs nutrients, and
defends against pathogens. The dermis, the thicker layer deep to the epidermis, contains an
extracellular matrix (ECM) that provides tensile strength and flexibility. It also includes
fibroblasts, nerve endings, hair follicles, sweat glands, sebaceous glands, apocrine glands,
lymphatic vessels, and blood vessels. Though they are only present in the dermis, the blood
vessels supply nutrients to both the dermis and the epidermis [2-4]. The deepest layer, the
hypodermis, is mainly made up of adipose tissue and serves as an anchor, permitting most areas
of the skin to move freely over the deeper tissues [1]. These distinct layers each play a unique
role in wound healing and add to the heterogeneity of wound classification.
1.3. Wound Classification and Closure Types
Wound classification takes into account the nature and the severity of the wound to
determine an appropriate treatment plan. First, the wound is classified as acute or chronic. Acute
wounds achieve sustained restoration of structure and function in an orderly and timely

3
reparative manner. In contrast, a chronic wound does not proceed through the healing process in
a timely fashion and does not advance to closure [5]. Discussion of the classification of chronic
wounds and treatment protocols will occur later in this chapter. The rest of this section will focus
mostly on acute wound closure.
Initially, acute wounds are classified as being open or closed. Open or penetrating
wounds are those with exposed underlying tissue and/or organs. These include stab, gunshot, and
surgical wounds. Closed or non-penetrating wounds do not break through the skin. These include
abrasions, lacerations, contusions, and concussions. There are also some wounds that do not fit
into either of these categories; these include thermal (burns or frostbite) and chemical wounds
[6]. Each type of wound has an additional classification for severity. For example, open surgical
wounds are further classified into four different classes based on surgical location and level of
contamination [6].
Wound healing, or cicatrization, is the process by which tissues restore anatomical
structure and function after an injury [7, 8]. Factors that affect the wound closure process include
the type of wound, size, depth, location, the age of wound, presence of infection, condition of the
patient, and urgency of closure [9]. Wound depth is classified as partial thickness or full
thickness. Partial thickness wounds are shallow and involve epidermal loss and partial loss of
the dermal layer. Full thickness wounds involve a total loss of the epidermal and dermal layers
and extend to at least the subcutaneous tissue layer and possibly as deep as the fascia, muscle
layer, and the bone. Partial thickness wounds heal primarily by re-epithelialization from the
remaining dermis with minimal scar formation. There are three methods of open wound closure:
primary, secondary, and tertiary repair [5, 7, 9]. In primary closure or first intention, the wounds
are sealed immediately with simple suturing, skin graft placement, or flap closure. Wound edges

4
are brought close together, and the wound heals spontaneously with scar formation oriented
along the Langer’s lines (collagen fiber alignment within the dermis) [5]. Closure by secondary
or spontaneous intention involves no active intent to seal the wound. This type of healing is
associated with large, highly contaminated wounds, coupled with extensive tissue loss. They
close by the natural wound-healing cascade, which results in contraction and increased scar
formation. Failure to heal within a month can lead to a chronic state [5]. Wound closure by
tertiary intention or delayed primary closure occurs when wounds are infected or contain foreign
debris and cannot be closed until the complications are resolved. A contaminated wound is
initially treated by various methods for several days to control infection. Once the wound is
ready for closure, surgical intervention allows the wound to heal by first intention. Again, failure
to heal within a month can lead to a chronic state. Primary or tertiary wound healing is preferred
to secondary since secondary involves a more severe wound contraction and scar formation.
1.4. Acute Wound Healing Cascade
All wounds undergo the same basic steps of repair regardless of tissue type or nature of
the injury. Full thickness wounds damage many structures within in the skin. These include
epidermal keratinocytes and appendages (sweat glands, sebaceous glands, and hair follicles), the
basement membrane, and dermal fibroblasts and appendages (ECM, nerves, and blood vessels)
[10]. To heal the gamut of damaged structures, the classic wound-healing model consists of four
distinct but overlapping phases: hemostasis, inflammation, proliferation, and maturation [11-14].

5
1.4.1. Hemostasis
Hemostasis or the coagulation phase is the initiator of healing and occurs immediately
following an injury [15, 16]. Ruptured cells and damaged blood vessels activate the blood-
clotting cascade, which promotes platelet activation, adhesion, and aggregation at the site of
injury to form a fibrin clot. This fibrin clot prevents exsanguination, creates a protective layer to
minimize infection and further injury, and provides a provisional matrix of support as the wound
heals [3, 7, 16-18]. Activated platelets are a major source of growth factors that play a significant
role in the subsequent phases of wound healing [10].
1.4.2. Inflammation
Inflammation, the next phase of wound healing, begins within the first 24 hours of injury
and can last up to two weeks in the normal wound healing cascade [16]. Physically,
inflammation is associated with signs of redness, swelling, heat, and pain caused by the release
of histamine and other active amines by mast cells [16]. The priority of the inflammatory phase
is to neutralize infection and prepare the wound bed for repair; therefore, it is not the active stage
of wound closure. Nevertheless, it is the most critical phase of the wound-healing cascade.
Successful wound repair requires effective and timely resolution of inflammatory responses to
activate subsequent steps of the wound healing process. Incomplete resolution of the
inflammatory phase leads to a chronic state [8, 19].
The initial vasoconstriction needed to prevent excess blood loss is closely followed by
vasodilation and increased capillary permeability. The increased capillary permeability and

6
chemotactic factors facilitate diapedesis of neutrophils into the wound bed. Likewise, there is an
influx of pro-inflammatory cytokines (IL-1, IL-6, IL-8, and TNF-α) and growth factors (PDGF,
TGF-β, IGF-1, and FGF) into the wound bed [16]. Polymorphonuclear neutrophils (PMNs) are
the most abundant cells in the wound site during the first two days post initial wounding [8].
They generate a respiratory burst to destroy phagocytized bacteria by the release of reactive
oxygen species (ROS) [2, 10, 20, 21]. Electrons donated by the reduced form of nicotinamide
adenine dinucleotide phosphate (NADPH) are transported across the membrane into lysosomes,
and superoxide anion (O2-) is produced [22]. O2- is bactericidal but is also toxic to neutrophils
and surrounding viable tissue if not stringently regulated. In healthy young cells, downstream
antioxidant enzymes such as superoxide dismutase (SOD), glutathione peroxidase, and catalase
mitigate ROS production by catalyzing the formation of H2O2 [23-25]. Neutrophils utilize
myeloperoxidase to combine H2O2 with Cl- to form hypochlorite, which plays a supplementary
role in eliminating bacteria [3]. Additionally, neutrophils release high levels of proteases
(elastase, neutrophil collagenase, and neutrophil collagenase MMP-8) and remove necrotic
debris and foreign material [16]. Once the neutrophils complete their function, usually after 2-3
days, they are extruded in the eschar (the crust containing dead cells and degraded products of
the wound) or phagocytized by macrophages.
Monocytes from the bloodstream infiltrate into avascular hypoxic areas of the wound site
and mature into macrophages by stimulation from pro-inflammatory cytokines produced by the
neutrophils [3, 26]. Macrophages in the wound bed can exhibit two distinct functional
phenotypes: M1 (classically activated, pro-inflammatory) and M2 (alternatively activated, anti-
inflammatory). Early during the inflammation phase, macrophages that are activated by
lipopolysaccharide (LPS) or inflammatory cytokines like interferon gamma (IFN-γ) continue the

7
job of neutrophils by phagocytizing bacteria and damaged tissue. Additionally, the M1
macrophages release pro-inflammatory cytokines such as tumor necrosis factor alpha (TNF-α)
and interleukin-6 (IL-6) [3]. Later during inflammation, initiated by the phagocytosis of
apoptotic cells, M1 macrophages phenotypically convert to M2 macrophages [27]. M2
macrophages, activated by interleukin-4 (IL-4) and interleukin-13 (IL-13), play a critical role in
the resolution of inflammation. Additionally, they are a predominant component of the transition
to the proliferative phase by promoting angiogenesis, tissue remodeling, and repair [3, 28-35].
Lucas et al. demonstrated that a depletion of macrophages during the inflammatory phase
significantly delayed wound repair in a mouse model; thus, they demonstrated the critical need
for macrophages for wound healing completion [36].
1.4.3. Proliferation
Until now, the wound bed has been prepping for the active repair phase by preventing
exsanguination, accomplishing wound debridement, and controlling the bacterial load.
Proliferation or the reparative phase is subdivided into different stages including granulation
tissue formation, collagen deposition, re-epithelialization, and angiogenesis [7, 15, 16]. The
initiation of the proliferative phase is evident by the infiltration of dermal fibroblasts into the
wound bed. Fibroblasts in the non-wounded dermis are typically dormant and sparsely
populated; therefore, fibroblasts are recruited from neighboring uninjured cutaneous tissues and
migrate to the wound bed [16]. A concentration gradient provided by chemotactic growth
factors, cytokines, and chemokines in conjunction with the alignment of collagen fibrils in the
provisional ECM govern the direction of fibroblast migration [16, 37]. Once the fibroblasts have

8
reached the wound bed, they anchor in the scaffold of the interim ECM and begin to proliferate
and synthesize granulation tissue components such as collagen, elastin, and proteoglycans. The
creation of granulation tissue, named for its irregular grainy appearance, slowly replaces the
temporary fibrin clot. Initially collagen, predominantly Type III collagen, is laid down in
irregular bundles providing moderate stability and tensile strength. In the later part of
proliferation, fibroblasts assume the myofibroblast phenotype, expressing α-smooth muscle actin
(α-SMA) [2, 38]. The myofibroblast phenotype enables the wound to contract but is dependent
upon growth factors PDGF and TGF-β [39].
Simultaneously, the epidermis must replace the initial wound seal, the fibrin clot, with a
new epithelial barrier to protect the newly forming ECM. Growth factors released during the
healing process stimulate the migration and proliferation of keratinocytes. Actively migrating
cells are incapable of proliferating; therefore, the neo-epithelium is created using new cells from
the proliferating basal layer of the epithelium near the wound margin. The normal cuboidal basal
epithelial cells flatten in shape and begin to migrate as a monolayer over the newly deposited
granulation tissue. The epithelial cells move in a tumbling fashion (also known as the epithelial
tongue) until the edges establish contact and form a confluent sheet. Once the newly formed
monolayer of keratinocytes covers the wound’s surface, migration stops, and the sheet enters a
proliferative state to re-establish the stratified layers of the epidermis and restore barrier function.
This process has to happen succinctly with ECM foundation or else the wound can easily re-
open.
As dermal and epidermal cells migrate and proliferate within the wound bed, there is a
substantial need for adequate blood supply for nutrient delivery and gas and metabolite
exchange. Due to vascular disruption, ischemia, and high oxygen consumption by metabolically

9
active cells, the microenvironment of the early wound is hypoxic, oxygen depleted. Compared to
normal tissue PO2 levels of 45–50 mm Hg, wound oxygen tension is 6 –7 mm Hg after five days
[40]. An inadequately replenished oxygen supply suppresses wound-healing processes; as an
example, fibroblasts can survive in a low-oxygen state but cannot replicate or synthesize [16,
41]. Angiogenesis restores tissue nutrient exchange, reestablishes microcirculation, and increases
oxygen tension to 30 – 40 mm Hg. Ischemia and hypoxia are critical inducers of angiogenic
factors to the wound site [40]. Binding of angiogenic factors such as VEGF, FGF, and TGF-β
cause microvascular endothelial cells of the blood vessels next to the wound site to migrate in the
same fashion as the fibroblasts. Once in the wound bed, the microvascular endothelial cells
proliferate to form buds or sprouts. As sprouts grow and encounter other sprouts, they develop a
cleft that subsequently becomes the lumen of the new vessel and complete a new vascular loop.
This process continues until the capillary system is sufficiently repaired for nutrient delivery and
oxygenation [16]. VEGF is noted to be the most potent angiogenic factor for its effects on
multiple components of the angiogenesis cascade: VEGF is an endothelial cell mitogen,
chemotactic agent, and inducer of vascular permeability [3, 42]. However, the angiogenic effect
of VEGF appears to be dependent on nitric oxide (NO). It has been shown that VEGF increases
NO production by up-regulating endothelial NOS (eNOS). Conversely, it has been shown that
when blocking eNOS, VEGF angiogenic properties such as endothelial migration and mitogen
activities are prevented [43, 44].

10
1.4.4. Maturation
The maturation or remodeling phase is different from the previous phases in that it does
not have a time sensitive pressure and can take from months to years to complete. During this
phase cell proliferation slows, protein synthesis decreases, and most endothelial cells,
macrophages, and fibroblasts undergo apoptosis or exit the wound. The goal of the remodeling
phase is to convert the new ECM, with a tensile strength of only 25-30% compared to normal
skin, to the a final scar with maximum tensile strength. Though tensile strength increases, the
maximum a wound can achieve is 80% of unwounded skin’s tensile strength. Collagen is
remodeled, and Type III collagen that is predominant in the proliferation stage is replaced by
stronger Type I collagen. The collagen is further strengthened by cross-links between collagen
fibrils. The final result is a scar that is more brittle and less elastic than normal skin. It should be
noted that scar tissue lacks the appendages (sweat glands) seen in the undamaged skin. Thus,
scar represents wound repair rather than regeneration.
1.5. Cellular Energy Metabolism
Cellular energy metabolism extracts a utilizable form of energy, adenosine triphosphate
(ATP) from the nutrition we eat via an elaborate set of biochemical reactions. There are two
categories of metabolic pathways: catabolism and anabolism. Catabolism includes the digestion
of ingested macromolecules (carbohydrates and fats) into smaller components (sugars and fatty
acids), which are either fully metabolized for energy or stored for later use. As a result of these
pathways, the energy released (7.3 kCal/mole) from the hydrolysis of the tertiary (ATP to ADP)
and secondary (ADP to AMP) phosphate bonds is used to execute all biologically necessary

11
reactions in the body. In contrast, anabolism uses the newly created ATP to synthesize essential
biomolecules such as proteins, lipids, and nucleic acids (DNA and RNA). The ratio between
anabolism and catabolism is an important factor in determining wound-healing progression,
which will be discussed later in this section.
1.5.1. Catabolism
In a standard American diet (SAD), ingested carbohydrates are metabolized into
monosaccharides, predominantly glucose, as the main fuel source for ATP. Glucose is
transferred into the cellular cytosol by low affinity, high capacity membrane transporters
(GLUTs). While in the cytosol, the process of glycolysis, ten enzymatic steps involving a
number of intermediates and specific enzymes, converts the 6-carbon glucose into two molecules
of 3-carbon pyruvate. Glycolysis results in the net gain of 2 ATP (2 ATP are invested in
reactions 1-5 and 4 ATP are produced in reactions 6-10) and the formation of 2 molecules of
NADH, an electron carrier that will eventually play an important role in the production of
additional ATP. Glycolysis is the only pathway to produce ATP without the presence of oxygen.
The flux through glycolysis is enough to sustain both ATP generation and fueling of the
biosynthetic pathways. Additionally, glycolytic intermediates can funnel into multiple
biosynthetic pathways. As an example glucose-6-phospahte (G6P) can enter the pentose
phosphate pathway, which generates NADPH to maintain antioxidant capacity and nucleic acid
synthesis.
The fate of pyruvate is dependent on the cellular oxygen concentration. In the absence of
oxygen, lactate dehydrogenase (LDH) converts pyruvate to lactate in a process known as

12
anaerobic respiration or fermentation. Coupled to this reaction are the formation of another ATP
and the oxidation of NADH to NAD+, which is cycled back to glycolysis. In the presence of
oxygen, the cells will use aerobic respiration. The two molecules of pyruvate are shuttled into the
mitochondrial matrix where they are oxidized into acetyl-CoA by the pyruvate dehydrogenase
complex (PDH). Two additional NADH molecules are created during this step. Acetyl-CoA is
joined to oxaloacetate, beginning the Krebs cycle. The Krebs cycle or citric acid cycle
completely oxidizes the remaining carbons from glucose, generates additional electron carriers (6
NADH and 2 FADH2) to feed the electron transport chain (ETC), and generates 2 ATP. As of
now, these metabolic pathways have synthesized a net gain of only 4 ATP, 10 NADH, and 2
FADH2. The electron carriers are shuttled to ETC, in the inner mitochondrial membrane for
oxidative phosphorylation. NADH and FADH2 are oxidized by the components of the ETC and
their electrons are passed between the ETC complexes in a series of oxidation/reduction
reactions. The final electron acceptor of the ETC is molecular oxygen, which reduces to H2O.
During the transfer of electrons through the ETC, H+ protons are pumped into the inner
mitochondrial membrane space creating an electrochemical gradient. This electrochemical
gradient drives the proton motive force used to synthesize ATP. Thus, the ETC generates 32-34
ATP per glucose molecule (3 ATP per NADH and 2 ATP per FADH2).
Dietary fats and stored fats are both metabolized via lipolysis to release their glycerol
backbone and their fatty acid side chains. The free fatty acids undergo β-oxidation to produce
acetyl-CoA. The metabolic fate of acetyl-CoA is the same as the previously described steps of
aerobic respiration from glucose. Each round of the β-oxidation cycle reduces the length of the
free fatty acid’s aliphatic tail by two carbon atoms and produces one NADH and one FADH2.
This process continues until the entire chain is cleaved into acetyl-CoA units. The ATP yield for

13
every β-oxidation cycle is 17 ATP (3 per NADH, 2 per FADH2, and 12 for the acetyl-CoA/Krebs
cycle). Thus, the total ATP yield will depend on chain length. Two ATP are required to activate
the fatty acid; hence a net ATP yield for palmitic acid (C16:0) is 113 ATP.
1.5.2. Anabolism
Anabolic or biosynthetic pathways utilize the newly created energy to fuel the creation of
necessary biomolecules. These include glycogenesis (glycogen synthesis), gluconeogenesis
(glucose synthesis), protein synthesis, and the pentose phosphate pathway. The main
biosynthetic pathways to focus on for this dissertation are the pentose phosphate pathway (PPP)
and gluconeogenesis. As mentioned previously, the first step of glycolysis produces glucose 6-
phosphate (G6P), which can be shuttled to the PPP, an anabolic pathway parallel to glycolysis
that generates the reducing agent NADPH and synthesizes 5-carbon sugars. The PPP accounts
for approximately 60% of NADPH production in the cells. It is important to note that NADPH is
the reducing agent consumed during biosynthetic reactions, whereas NADH is a reducing agent
generated in energy-yielding reactions. NADPH is used for fatty acid synthesis and helps fight
oxidative stress by reducing the antioxidant glutathione via glutathione reductase. The ribose
sugars generated from the PPP are utilized in the synthesis of nucleotides and nucleic acids.
Gluconeogenesis (GNG) is a series of eleven enzyme-catalyzed reactions to synthesize
glucose from non-carbohydrate sources including pyruvate, amino acids (alanine and glutamine),
lactate, and glycerol. The system is a reciprocal of glycolysis, which together creates a feedback
mechanism to normalize and maintain blood glucose levels. While most steps in gluconeogenesis

14
are the exact reverse of glycolysis, there are three reactions that require different catalytic
enzymes to make them more kinetically favorable.
In general, normal metabolism is hormone-mediated to alter energy production to meet
energy need and to restore daily macronutrient balance, especially proteins, through the catabolic
and anabolic pathways [45-50]. This balanced metabolic profile significantly changes in the
presence of a wound.
1.6. Wound Healing Metabolism
The presence of any substantial wound increases metabolic energy demands from 30-
50% above normal requirements to produce the necessary biomolecules for increased cellular
proliferation and migration of the various cell types during wound healing [17, 51]. Cuthbertson
famously described the metabolic response to injury as having an “ebb” phase, the period of
traumatic shock or hypometabolism during the first few hours or days after injury and a “flow”
or hypermetabolic phase (proliferation) that may last weeks to months depending on the
complexity of the wound [17, 52, 53].
It has been well established that non-wounded epidermal cells, as well as wounded skin,
are mainly dependent upon carbohydrate metabolism via glycolysis and the PPP rather than the
Krebs cycle for their energy requirement [54-59]. In a hallmark study, Im et al. showed a 4-fold
increase of glycolytic enzymes of wounded skin compared to normal skin [56]. The contribution
of the Krebs cycle to glucose metabolism was reduced in wounded skin compared to normal skin
(8% to 4%) demonstrating a metabolic shift to the PPP and other biosynthetic pathways, needed

15
for repairing the wound. This metabolic shift towards glycolysis was confirmed by the
observations that the rate of nucleotide formation was higher in wounded skin compared to
normal skin and a 7-fold increase in the PPP enzymatic activities. Furthermore, glucose-6-
phosphate dehydrogenase (G6PDH) was increased 7-fold, corresponding to the increased
contribution to the PPP [56]. Additionally, there was evidence that lactate production was tripled
in day three post-wounding, further confirming the higher flux through glycolysis and
subsequently anaerobic fermentation. Even though glycolysis is less efficient at creating ATP
compared to flux through the TCA cycle, the rate of ATP formation was doubled after three days
of wound healing. The ATP production was seen as a gradient with higher concentrations of
ATP in the migrating epithelium compared to the marginal proliferating epithelium. The study
showed a decrease in energy production per unit of glucose in wounded skin compared to normal
skin, as would be expected in the switch to glycolytic metabolism compared to aerobic
respirations. The inefficiency of glycolysis forming ATP compared to the ETC is overcome by
the increased metabolic rate of wounded skin. It has been shown that elevated activities of
glycolytic enzymes may contribute to increased cellular demand for energy during healing of
acute wounds [51, 59, 60].
1.7. Nutrition and Wound Healing
Nutritional status is extremely critical for effective wound healing [61-81]. As mentioned
earlier in this chapter, maintenance of a balanced anabolism: catabolism ratio is fundamental to
optimizing the wound healing process. Since healing a wound is crucial to surviving, a wound
requires high priority of the circulating nutrients in the blood stream. Increased macronutrient

16
intake is required to keep up with the catabolic losses and to allow wound-healing processes. The
American Society for Parenteral and Enteral Nutrition and Wound Healing recommends
approximately 30 to 35 kcal/kg/d for optimal wound healing, which is roughly 1.2-1.5 times
more than a non-wounded patient would ingest [82]. These macronutrients are predominantly
used as fuel; however, they also have direct effects essential for healing [64]. It should be noted
that individual energy needs depend on age, gender, nutritional status, basal metabolic rate, body
mass index (BMI), comorbid conditions, activity level, the stress of illness, severity and number
of wounds, and wound size of that patient, which supports the heterogeneous classification of
wounds [17, 83].
Macronutrients (fats, proteins, and carbohydrates) provide the energy for all body
functions and are major building blocks for the reparative process. In a healing wound, the influx
of inflammatory cells, as well as fibroblasts, use carbohydrates as an energy source. Additionally
carbohydrates have been shown to have a variety of non-energy related roles (structural,
lubricant, immunologic, hormonal, and enzymatic) [17]. Fats and their derived lipid components
provide the building blocks for the cell membrane and certain inflammatory mediators, behave as
signaling molecules, control tissue growth, wound remodeling, and collagen and extracellular
matrix production [84-91]. Proteins are predominantly metabolized into amino acids and
peptides for the purpose of protein synthesis; only 5% of protein metabolism is used for energy
[83]. Protein derived amino acids provide the major structural building blocks of all proteins in
the body, notably collagen. Amino acids are necessary for the cell membrane, enzymes, and
cytokine production in the healing wound [64].
When dietary nutrients are not adequate, or if the catabolic state outweighs the anabolic
state, the wound can proficiently use substrates available from the rest of the body for energy

17
production, resulting in what is called protein-energy malnutrition (PEM). The body catabolizes
the muscle, skin, and bone to support the synthesis of proteins, inflammatory cells, and collagen
needed to fight infection and repair the wound, causing a loss of lean body mass (LBM). As an
individual loses more LBM, wound healing is more likely to be delayed. A loss of more than
15% LBM impairs wound healing, a 30% LBM loss stops wound healing, and a loss of 40%
LBM typically results in death [64, 92-95]. As will be important later, a body in starvation
(ketosis) has been shown to be muscle sparing, attenuating the catabolic effects [83]. LBM loss
becomes vital in older adults as many suffer from pre-existing malnutrition and PEM.
1.8. Chronic Wounds
Chronic wounds represent an under-acknowledged socioeconomic epidemic, affecting
1.8 million new patients per year in the US. [96]. The care of these chronic wounds costs the US
health care system at least $25 billion annually. This substantial cost is rapidly growing due to
increased healthcare costs, an aging population, an increase in diabetes and obesity, and the
expanding need for wound care for veterans [96-98]. As more of our population ages (>65
years), projected to be 20% of the population by 2030, there is a critical need to develop
effective wound healing treatments to alleviate the considerable medical, social, and economic
burden facing the country [99].

18
1.8.1. Classification of Chronic Wounds
Chronic wounds are defined as wounds that have failed to proceed through and orderly
and timely reparative process to produce anatomic and functional integrity. All wound types
have the potential to become chronic, and as such, chronic wounds are traditionally divided by
etiology. A prolonged inflammatory stage, persistent infections, drug-resistant microbial
biofilms, and the inability of cells to respond to reparative signals are pathophysiological features
of all chronic wounds [8, 100, 101]. The Wound Healing Society’s Chronic Wound Care
Guidelines divide chronic wound into four different phenotypes: vascular ulcers (venous and
arterial), pressure ulcers, and diabetic ulcers.
The venous ulcer accounts for more than half of ulcer cases and 70-90% of ulcers found
on the lower limbs [102]. Venous ulcers affect approximately 1.69% of older adults or 600,000
patients total in the US annually [96, 103]. They are associated with deep vein thrombosis,
varicose veins, and venous hypertension. Physically, venous ulcers have skin that is shiny,
smooth, with minimal to no hair, are superficial, shallow, irregularly shaped, and are
accompanied by pain and edema. Less common than venous ulcers, arterial ulcers affect
approximately 100,000 Americans annually [104]. Arterial ulcers occur from hypertension,
atherosclerosis, and thrombosis, where the reduced blood supply leads to an ischemic state.
Commonly, they are full thickness wounds with a punched out appearance and smooth edges. As
compared to the venous ulcers that have prevalence on the lower limbs (between the knee and
ankle), arterial ulcers usually occur more distal such as the tip of the toe and over distal bony
prominences.

19
Pressure ulcers, also known as bedsores, are a result of prolonged pressure, friction and
shear from body weight and usually occur at a bony prominence such as the sacrum or heel.
Clinically, they present with redness, itching, blistering, warmth, swelling, and discoloration of
the overlying skin. They are prevalent in any patient that is wheelchair bound, bedridden, or
suffering from immobility or sensory neuropathies [105]. 2.5 million pressure ulcers are treated
in the US per year [106]. The cost of treating one pressure ulcer is estimated to be as much as
$70,000, totaling to an estimated $11 billion a year for pressure ulcer care [107, 108]. About
15% of patients in an acute care facility and up to 29% of patients in long-term care will
experience a pressure ulcer [96]. According to the Centers for Medicare & Medicaid Services
(CMS), there were a staggering 257, 412 preventable pressure ulcers in 2007 [109]. Additionally,
pressure ulcers can be a major source of infection, leading to complications and even death.
Development of a pressure ulcer increases mortality rate by 7.23% [110]. Over the last ten years
nearly 60,000 deaths occurred annually from hospital-acquired pressure ulcers [96]. Even though
many resources have been devoted to rectify these overwhelming statistics, it is still extremely
challenging and costly to prevent pressure ulcers [111-114].
As of October 2014, 29.1 million people in the United States have diabetes mellitus of
which 8.1 million people are undiagnosed [115]. Diabetic ulcers are a common complication in
uncontrolled diabetes, resulting from impaired immune function, ischemia (due to poor blood
circulation from high glucose content as well as vascular insufficiencies), and neuropathy. It is
estimated that up to 25% of all diabetics will develop an ulcer in their lifetime [116]. In 2007, a
single foot ulcer cost between $7,439 and $20,622, totaling to a $9 billion expense per year
[117].

20
1.8.2. Current Standard of Care, Alternative Therapies, and Their Disadvantages
Identifying and treating the underlying etiology of chronic wounds such as venous
insufficiency, arterial perfusion, diabetes, or continuous pressure as well as systemic factors such
as age, nutritional status, immunosuppression, and infection is key to successful wound healing
therapies. However, the establishment of effective wound healing therapies has been
unsuccessful due to the magnitude of these underlying pathogenic factors and the heterogeneity
across and within wound types [118]. Furthermore, it is important to emphasize that no two
wounds are exactly the same; thus, a treatment that could work for one patient may not be
effective for another.
Most current wound therapies being studied target a specific molecular or cellular
mechanism where there are often multiple mitigating factors that prevent normal wound closure.
This is likely one reason most wound therapies are minimally effective. Many growth factor
supplements tested in animal models appeared to be promising therapeutic agents promoting
wound healing [119]; however, human clinical trials have not been very successful [120, 121].
Additionally, there is a multitude of topical therapies (antimicrobials, dressings, silver-containing
products) that are commercially available, with minimal data to support their effectiveness in
promoting wound healing [122].
In vitro models have aided in the understanding of basic biology of wound healing; however,
more complex models to simulate the wound environment (multiple cell types) in conjunction
with comorbid conditions have not been developed. Many studies in wound repair have relied on
rodent models; however, skin morphology and the mechanisms of repair are markedly different
between rodents and humans [118, 123]. Additionally, some pre-clinical studies have used acute

21
wounds in young animals and not appropriately aged animals, demonstrating that there are no
animal models that comprehensively mimic human chronic wounds [124].
1.8.2.1. TIME and Amputation
Standard clinical therapies for non-healing wounds use the wound bed preparation
concept, which focuses on optimizing the condition of the wound bed to encourage normal
healing. In 2002, the acronym TIME was developed as a practical guideline for wound
management. TIME encompasses four main components of wound bed preparation: Tissue
management (debridement), control of Infection and inflammation, manage Moisture, and the
Edge of wound advancement [125, 126].
Debridement is the surgical removal of necrotic, damaged, or infected tissue, biofilms,
exudate, and debris to improve the healing of the remaining healthy tissue [127-132]. The goal of
debridement is to transition an excessively inflamed chronic wound to more of an acute profile,
promoting resolution of inflammation [122, 133, 134]. Chronic wounds are well characterized as
being stuck in the inflammatory phase with a marked increase in degrading proteases such as
matrix metalloproteinases (MMPs) and elastases and persistence of inflammatory cells [126,
129, 135-137]. Resolution of prolonged inflammation renews tissue healing, lessens exudate, and
reduces bioburden. It is important to distinguish between normal inflammation associated with
physiological healing compared to excessive inflammation caused by underlying adverse
etiologies and infection. Inflammation and infection are controlled using antimicrobials
(disinfectants, antiseptics, and antibiotics) in a variety of applications including silver dressings,
iodine dressings, PHMB dressings, honey, and surfactants [126]. Balanced moisture is essential

22
for the action of growth factors, cytokines, and cell migration. Excessive moisture damages
surrounding skin and insufficient moisture inhibits cellular activities and leads to eschar (scab)
formation [137]. Wound edge advancement assessment determines if wound contraction or
epithelialization is progressing and evaluates the effectiveness of the wound treatment. An
increasing range of treatment modalities has been proposed to improve wound edge advancement
including electromagnetic therapy (EMT), laser therapy, ultrasound therapy, systemic oxygen
therapy, and negative pressure wound therapy (NPWT) [126].
Amputation is the final and last course of action against chronic wounds (when they
occur on an extremity). This is done to prevent the spread of infection to healthy tissue.
Amputations are classified as either major (an amputation of the leg above or below the knee) or
minor (amputation of toes or forefoot) [138]. Many diabetic patients eventually must undergo
lower extremity amputations as a result of infection brought on by untreated foot ulcers. Diabetic
foot ulcers are responsible for 20% of diabetes-related hospitalization and underlie up to 80% of
non-traumatic amputations per year. It is estimated that 12% of individuals with a foot ulcer will
require amputation. Mortality following amputation is 13-40% at one year, 35-65% at three
years, and 39-80% at five years, which is worse than most malignancies. Once amputation
occurs, 50% of patients will develop an ulcer in the contralateral limb within five years [117].
1.8.2.2. Adjunctive Therapies
Even with the standard care practices of wound healing, there is a multitude of treatment
options and modalities. As previously mentioned, most of these treatments have limited research
data to prove effectiveness and many have controversial clinical results. Adjunctive treatments

23
for wound healing include negative pressure therapy, hyperbaric oxygen therapy (HBOT),
dermal substitutes, growth factor supplementation therapy, and stem cell therapy [139, 140].
Negative Pressure Wound Therapy (NPWT), vacuum-assisted wound therapy or
commonly known as a wound VAC®, has become a popular adjunctive therapy[141-143].
Negative pressure is applied to the wound via foam or gauze dressing moistened with saline,
which is then covered with a transparent adhesive sheet attached to a vacuum pump. NPWT has
been shown to manage wound drainage, reduce edema, reduce the bio-burden of
microorganisms, increase wound perfusion, and loosen the exudate [126]. This therapy in
combination with debridement has been shown to promote granulation tissue formation,
contraction and epithelialization [135]. However, a systematic review and meta-analysis of 21
studies found no clear evidence that wounds heal either better or worse with NPWT compared to
standard of care [144]. Additionally, the mechanism of action for NPWT is poorly understood,
and few studies have focused on the efficacy of this therapy in older adults [118].
Hyperbaric oxygen therapy (HBOT), originally utilized for decompression illness, has
been an adjunctive therapy for wound healing for more than two decades. It involves placing a
patient in a sealed chamber where 100% oxygen is pressurized to 1.5-3 atmospheres absolute
(ATA) for 60-120 minutes over a course of multiple treatments [139]. There isn’t an optimized
protocol for treatment and the mechanism of action for HBOT to improve wound healing is
poorly understood. However, it is speculated that HBOT helps reduce edema via
vasoconstriction and fluid absorption, increases oxygen perfusion aiding collagen elongation and
deposition, and stimulates epithelial progenitor cells and stem cell release for vasculogenesis
[139]. Published systematic reviews have reported mix results regarding the efficacy of HBOT,
as observational studies have shown a significant advantage of HBOT for wound healing;

24
however, six randomized trials did not show a benefit [145]. There are some promising animal
data suggesting that HBOT is effective for all ages; however, there are no studies specifically
focused on the efficacy of HBOT in older adults [146, 147].
Both biosynthetic skin substitutes (synthetic) and cultured autologous-engineered skin
(natural) are available to provide the protective barrier of skin when placed over a wound [148-
154]. The synthetic substitutes are made of an acellular material, and the natural substitutes
consist of cultures allogeneic or autologous cell suspensions or sheets used alone or with a
dermal matrix. The purpose of engineered skin products is to expedite wound healing by serving
as a scaffold for cellular ingrowth, stimulating the release of growth factors from the wound bed,
and restoring the functional properties of the skin [154, 155]. The skin substitutes (SS) can be
either permanent or temporary, depending on design and composition. It’s important to note that
although each of skin substitutes has their advantages and have applications, none of them can
fully simulate native skin. Both types of SS have been shown to be quite effective in increasing
the speed of wound healing, decreasing recurrence, reducing the number of required surgical
procedures, and reducing patient hospitalization time [148, 153, 155, 156]. Nevertheless, the
commercially available skin substitutes have several limitations such as reduced vascularization,
scarring, failure to integrate, poor mechanical integrity, and immune rejection. Furthermore, the
cost associated with the use of the current skin substitutes is very high. For example, it is
estimated that the cost for each 1% body surface areas covered by the commercially available
Epicel ™ is more than $13,000 [157]. As it relates to this project, the effect of the patient age on
skin substitutes is poorly characterized [118].
It is well characterized that growth factors play a substantial role in both healing and non-
healing wounds. Wound healing products have been designed to accelerate repair by augmenting

25
or modulating these growth factors in hopes of mediating inflammation and promoting
angiogenesis. Among all the possible growth factors, platelet-derived growth factor (PDGF) has
been approved by the Food and Drug Administration (FDA) for the treatment of diabetic foot
ulcers [4]. PDGF has been shown to stimulate granulation, increased odds of wound healing and
decrease rates of amputations in diabetic patients [158-165]. Growth factors EGF and FGF have
also been studied as topical wound healing therapies; however, the heterogeneity of wounds,
applications, patient management protocols, and clinical trial outcomes has limited their use to
outside of the USA [122].
Stem cells have the unique capability to differentiate into the variety of tissues in the
body. Stem cells derived from various sources including bone marrow, adipose, peripheral and
umbilical blood, in addition to vascular progenitor cells have been shown to migrate towards the
wound site and contribute to the wound healing process [166-168]. These cells have been to
shown to increase angiogenesis, increase granular tissue formation, enhance wound healing, and
reduce scarring [166, 168-172]. Even though stem cell research seems promising, the use of stem
cells is hampered by immunogenicity, potential for malignancy, pluripotency maintenance,
ethical consideration, and limited supply [118]. Additionally, harvesting bone marrow derived
stem cells is painful and leads to donor site morbidity [173].
The field of wound care is ever expanding with advances in technology. However, there
is still no superior substitute to allowing the body to heal systemically on its own accord in
conjunction with reconstruction using patients’ own tissues and carefully thought-out surgical
procedures. The therapy proposed in the next chapter of this dissertation focuses on exploiting
the body’s systemic physiology to enhance wound healing.

26
1.9. Age Influence on Chronic Wounds
Chronic wounds disproportionally affect older adults. 72% of hospitalized patients with
pressure ulcers were 65 and older. Most chronic wounds are associated with co-morbid
conditions prevalent in older adults, including cardiovascular disease, unrelieved pressure,
obesity, or diabetes mellitus [96]. However, Wicke and associates in 2009 demonstrated that age
was an independent risk factor for delayed wound closure. By the age of 70, the frequency of
wound closure was significantly lower than the younger counterparts [174]. It should be noted
that the effect of aging on wound repair is primarily a temporal delay; a change in the quality of
wound closure has not been reported [175]. The exact mechanisms responsible for this increased
risk remain unknown. In this dissertation we focus on four physiological attributes that we feel
lead to the prevalence of chronic wounds in older adults: unresolved inflammation, unmitigated
ROS production, decreased angiogenesis/blood flow, and decreased metabolism.
1.9.1. Age-Related Changes in Skin Morphology
Though the skin is incredibly durable, like all other systems, it eventually succumbs to
the inevitable effects of aging. The skin is also the most visible indicator of age. Clinically,
drying, roughness, sagging, wrinkling, and alterations in pigmentation characterize aged skin.
Physiological changes in the skin include both structural and biochemical changes.
Morphologically, the skin is characterized by atrophy due to the flattening of the
epidermis/dermis junction and a decrease of epidermal and dermal cellular content, collagen

27
disorganization, a decrease in microcirculation, and an increase in time for keratinocyte
maturation [4, 176-179]. The thickness of the skin decreases about 6.4% per decade on average.
Keratinocytes change shape as the skin ages, becoming shorter and fatter while corneocytes
become bigger as a result of decreased epidermal turnover. The cellular content of the dermis
(fibroblasts, mast cells, and macrophages) and protein content (collagen) are reduced with age.
The collagen and elastin that remain in the dermis are less organized, resulting in a decreased
elasticity of the skin [180]. Enzymatically active melanocytes decrease at a rate of 8-20% per
decade, resulting in the irregular pigmentation of elderly skin. Alterations in the aging skin not
only have an impact on wound healing but also make the skin more susceptible to injury. The
flattening of the epidermis/dermis junction results in less resistance to shearing forces and
increased vulnerability to insult as well as increased separation.
1.9.2. Age-Related Changes in the Wound Healing Cascade
Aging affects every phase of wound healing [176]. It has been demonstrated that aged
wounds have decreased levels and response to growth factors, reduced microcirculation,
diminished cell proliferation and migration, and diminished ECM secretion [181-183]. Aging is
also associated with delays in macrophage and T cell infiltration, angiogenesis, and re-
epithelialization [118]. It has been hypothesized that age-associated disadvantages in healing
may arise from overexpression of matrix metalloproteinases, which normally aid in
phagocytosis, angiogenesis, cell migration during epidermal restoration, and tissue remodeling.
Protease expression and activity are elevated in older human adults, which leads to the
perpetuation of the pro-inflammatory state and inhibits progression to the reparative phase [184].

28
Inflammation is a major contributing factor in wound chronicity and the aging
phenotype. As previously mentioned, chronic wounds are characterized as being stuck in the
inflammatory phase. Without resolution, the wound cannot advance to the reparative phase. This
inability to resolve inflammation is believed to be the most significant delaying factor in the
healing of chronic wounds [185]. It has been established that aged individuals have a consistent
low-grade inflammation marked by elevated cytokines like TNF-α, IL-6, and CRP, even when
the individual is healthy, or infection is absent [186]. This is in direct contrast to young
individuals, who only produce elevated levels of cytokines in response to injury or infection.
This low-grade inflammation has been implicated in the higher prevalence of co-morbidities
such as cardiovascular disease and diabetes in older adults. It has been suggested that
inflammation may be a common underlying cause of several age-related diseases or could be a
common pathway by which multiple diseases lead to disability and adverse outcomes in older
adults [186]. This already heightened inflammatory response is worsened in the presence of a
wound. It has been shown that there are increased numbers of neutrophils with prolongation of
the pro-inflammatory state as well as a delay in macrophage recruitment [185]. Additionally, a
study by Mahbub and colleagues demonstrated that M1 macrophages fail to transition to the M2
phenotype, perpetuating the inflamed state in an aged population [187].
ROS are produced in the healing wound as a bactericidal tool; however, when the levels
are not quenched by downstream antioxidants elevated ROS levels transcend the beneficial effect
and cause mitochondrial dysfunction, DNA damage, lipid peroxidation, and even cell death [188,
189]. Additionally, ROS are produced as a normal by-product of oxidative phosphorylation;
thus in a wound environment cells are hit with ROS from neutrophilic respiratory bursts as well
as from enhanced energy metabolism necessary to heal the wound [23, 190, 191]. The energy

29
metabolism shift to anaerobic respiration during wound healing likely protects the mitochondria
from over usage and damage related to ROS production [55]. Skin mitochondria in the human
aged population demonstrated an increased incidence of mitochondrial DNA (mtDNA)
mutations leading to mitochondrial dysfunction [192, 193]. Electron microscope images of aged
mitochondria show disorganization and degeneration via absent cristae, vacuolation, and
swelling [194]. Additionally, it has been determined that both superoxide anion and H2O2 are
increased in aged dermal fibroblasts [195]. Several studies have shown that the aged phenotype
has decreased levels of antioxidants to quench the increased ROS [196-199]. Treiber et al.
demonstrated that the skin of manganese superoxide dismutase (MnSOD, SOD2) deficient mice
was phenotypically similar to aged animals with characteristics such as atrophy of dermal
connective tissue, muscle fibers, and subcutaneous fat, loosely packaged collagen fibrils,
damaged mitochondria, and decreased cellular proliferation [200].
It has been demonstrated that angiogenesis is decreased with age because of reduced
levels of the angiogenic factors: FGF, VEGF, and TGF-β, impaired vasodilation, proliferation
and migration of angiogenic, microvascular endothelial cells. It is important to note that the
coexistence of hypertension, diabetes, smoking, and hypercholesterolemia exacerbates the
inherent effects of aging that are detrimental to angiogenesis [176, 201].
The metabolic challenge of a wound is exacerbated in elderly patients, who have a higher
prevalence of malnutrition combined with reduced total energy expenditure, basal metabolic rate,
and protein synthesis [17]. Without appropriate energy (ATP) levels, wound healing is
significantly impaired. The ATP deficiency creates an imbalance of energy production versus
utilization. To compensate, anaerobic respiration increases in the wound bed, which only
produces 2 ATP per glucose molecule as compared to the potential 38 ATP made from normal

30
aerobic respiration [41]. Gupta et al. showed that key metabolic enzymes were decreased in aged
rats [51]. It has been illustrated that mitochondria become larger and less numerous with age,
mitochondrial respiratory chain (MRC) enzyme activities decrease as well as mitochondrial
membrane potential [202]. Furthermore, there is a significant reduction in mitochondrial
bioenergetic capacity with advancing age, which has been shown in numerous animal models
and recently in a study with human volunteers [202, 203].
1.10. Closing Remarks
In summary, there is come controversy regarding whether chronological age alone affects
wound healing. However, it is well documented that there is increased ROS production with age,
leading to cellular damage, reduced blood flow leading to the lessening of nutrient exchange,
decreased metabolic activity and ATP production at the wound bed, and unresolved chronic
inflammation. It is easily deduced how these factors may lead to increased prevalence of chronic
wounds in older adults. Most approaches to accelerate wound healing focus on delivery of
growth factors by topical application. The discouraging results of focusing on single molecular
targets are not surprising since wound healing is the product of a complex set of interactions
between numerous factors. It is here that we aim to switch the approach to optimizing the
body’s physiology to reach the wound bed systemically as well as targeting multiple facets of
impaired wound healing with one therapy- exogenous ketone supplementation, which will be
discussed further in the next chapter.

31
1.11. References for Chapter 1
1. Martini FH: Chapter 5: The Integumentary System. In: Fundalmentals of Anatomy and Physiology. Edited by Berriman L. San Francisco, CA: Pearson Education Inc, Publishing as Benjamin Cummings; 2006: 153-178.
2. Gurtner G, Werner S, Barrandon Y, Longaker M: Wound repair and regeneration. Nature 2008, 453(7193):314-321.
3. Mahdavian Delavary B, van der Veer W, van Egmond M, Niessen F, Beelen R: Macrophages in skin injury and repair. Immunobiology 2011, 216(7):753-762.
4. Thomas D, Burkemper N: Aging skin and wound healing. Clinics in geriatric medicine 2013, 29(2).
5. Atiyeh BS, Ioannovich J, Al-Amm CA, El-Musa KA: Management of acute and chronic open wounds: the importance of moist environment in optimal wound healing. Current pharmaceutical biotechnology 2002, 3(3):179-195.
6. Different Types of Wounds [http://www.woundcarecenters.org/article/wound-basics/different-types-of-wounds]
7. Ethridge RT, MD, PhD; Mimi Leong, MD; Linda G. Phillips, MD: Chapter 8: Wound Healing. In: Textbook of Surgery: The Biological Basis of Modern Surgical Practice. 18th edn. Philadelphia, PA: Saunders Elsevier; 2007: 191-216.
8. Eming SA, Krieg T, Davidson JM: Inflammation in wound repair: molecular and cellular mechanisms. The Journal of investigative dermatology 2007, 127(3):514-525.
9. Wound Closure [http://www.medstudentlc.com/page.php?id=66]
10. Tanya JS, Paul M: Wound repair at a glance. Journal of Cell Science 2009.
11. Bennett NT, Schultz GS: Growth factors and wound healing: biochemical properties of growth factors and their receptors. American journal of surgery 1993, 165(6):728-737.
12. Bennett NT, Schultz GS: Growth factors and wound healing: Part II. Role in normal and chronic wound healing. American journal of surgery 1993, 166(1):74-81.
13. Lawrence WT: Physiology of the acute wound. Clinics in plastic surgery 1998, 25(3):321-340.
14. Cordeiro J, Jacinto A: The role of transcription-independent damage signals in the initiation of epithelial wound healing. Nature reviews Molecular cell biology 2013, 14(4):249-262.

32
15. Demidova-Rice TN, Hamblin MR, Herman IM: Acute and impaired wound healing: pathophysiology and current methods for drug delivery, part 1: normal and chronic wounds: biology, causes, and approaches to care. Advances in skin & wound care 2012, 25(7):304-314.
16. Gloria A C, Robert F. Diegelmann, Gregory S. Schultz: Cellular and Molecular Regulation of Wound Healing. In: Wound Healing. Edited by Anna F. Falabella RSK. Boca Raton, FL: Taylor and Francis Group; 2005: 17-37.
17. Molnar JA: Nutrition and wound healing. CRC 2006.
18. Gailit J, Clark RA: Wound repair in the context of extracellular matrix. Current opinion in cell biology 1994, 6(5):717-725.
19. Mast BA, Schultz GS: Interactions of cytokines, growth factors, and proteases in acute and chronic wounds. Wound repair and regeneration : official publication of the Wound Healing Society [and] the European Tissue Repair Society 1996, 4(4):411-420.
20. Clark RF: Overview and General Considerations of Wound Repair. In: The Molecular and Cellular Biology of Wound Repair. Edited by Clark RAF, Henson PM: Springer US; 1988: 3-33.
21. Martin P: Wound healing--aiming for perfect skin regeneration. Science (New York, NY) 1997, 276(5309):75-81.
22. Ushio-Fukai M: Redox signaling in angiogenesis: role of NADPH oxidase. Cardiovascular research 2006, 71(2):226-235.
23. Ray PD, Huang B-WW, Tsuji Y: Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cellular signalling 2012, 24(5):981-990.
24. Schäfer M, Werner S: Oxidative stress in normal and impaired wound repair. Pharmacological research : the official journal of the Italian Pharmacological Society 2008, 58(2):165-171.
25. Sies H: Oxidative stress: from basic research to clinical application. The American journal of medicine 1991, 91(3C).
26. Willenborg S, Eming SA: Macrophages - sensors and effectors coordinating skin damage and repair. Journal der Deutschen Dermatologischen Gesellschaft = Journal of the German Society of Dermatology : JDDG 2014, 12(3):214.
27. Khallou-Laschet J, Varthaman A, Fornasa G, Compain C, Gaston A-TT, Clement M, Dussiot M, Levillain O, Graff-Dubois S, Nicoletti A et al: Macrophage plasticity in experimental atherosclerosis. PloS one 2009, 5(1).
28. Gordon S: Alternative activation of macrophages. Nature reviews Immunology 2003, 3(1):23-35.

33
29. Xue L, Liu X-s: [Advancement in the research of the role of macrophages in wound healing]. Zhonghua shao shang za zhi = Zhonghua shaoshang zazhi = Chinese journal of burns 2013, 29(1):62-64.
30. Ploeger D, Hosper N, Schipper M, Koerts J, de Rond S, Bank R: Cell plasticity in wound healing: paracrine factors of M1/ M2 polarized macrophages influence the phenotypical state of dermal fibroblasts. Cell communication and signaling : CCS 2013, 11(1):29.
31. Zhang Y, Choksi S, Chen K, Pobezinskaya Y, Linnoila I, Liu Z-GG: ROS play a critical role in the differentiation of alternatively activated macrophages and the occurrence of tumor-associated macrophages. Cell research 2013.
32. Frank S, Hübner G, Breier G, Longaker MT, Greenhalgh DG, Werner S: Regulation of vascular endothelial growth factor expression in cultured keratinocytes. Implications for normal and impaired wound healing. The Journal of biological chemistry 1995, 270(21):12607-12613.
33. Goerdt S, Orfanos CE: Other functions, other genes: alternative activation of antigen-presenting cells. Immunity 1999, 10(2):137-142.
34. Knighton D, Hunt T, Scheuenstuhl H, Halliday B, Werb Z, Banda M: Oxygen tension regulates the expression of angiogenesis factor by macrophages. Science 1983.
35. Porcheray F, Viaud S, Rimaniol ACC, Léone C, Samah B, Dereuddre-Bosquet N, Dormont D, Gras G: Macrophage activation switching: an asset for the resolution of inflammation. Clinical and experimental immunology 2005, 142(3):481-489.
36. Lucas T, Waisman A, Ranjan R, Roes J, Krieg T, Müller W, Roers A, Eming SA: Differential roles of macrophages in diverse phases of skin repair. Journal of immunology (Baltimore, Md : 1950) 2010, 184(7):3964-3977.
37. Greiling D, Clark RA: Fibronectin provides a conduit for fibroblast transmigration from collagenous stroma into fibrin clot provisional matrix. Journal of cell science 1997, 110 ( Pt 7):861-870.
38. Opalenik SR, Davidson JM: Fibroblast differentiation of bone marrow-derived cells during wound repair. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 2005, 19(11):1561-1563.
39. Macri L, Clark RA: Tissue engineering for cutaneous wounds: selecting the proper time and space for growth factors, cells and the extracellular matrix. Skin pharmacology and physiology 2008, 22(2):83-93.
40. Tonnesen M, Feng X, Clark R: Angiogenesis in wound healing. The journal of investigative dermatology Symposium proceedings / the Society for Investigative Dermatology, Inc [and] European Society for Dermatological Research 2000, 5(1):40-46.

34
41. Chiang B, Essick E, Ehringer W, Murphree S, Hauck M, Li M, Chien S: Enhancing skin wound healing by direct delivery of intracellular adenosine triphosphate. American journal of surgery 2007, 193(2):213-218.
42. Bao P, Kodra A, Tomic-Canic M, Golinko M, Ehrlich H, Brem H: The role of vascular endothelial growth factor in wound healing. The Journal of surgical research 2009, 153(2):347-358.
43. Luo J-d, Chen A: Nitric oxide: a newly discovered function on wound healing. Acta pharmacologica Sinica 2005, 26(3):259-264.
44. Schwentker A, Billiar T: Nitric oxide and wound repair. The Surgical clinics of North America 2003, 83(3):521-530.
45. Demling R, De Santi L: Closure of the "non-healing wound" corresponds with correction of weight loss using the anabolic agent oxandrolone. Ostomy/wound management 1998, 44(10):58.
46. Ferrando AA, Wolfe RR: Restoration of hormonal action and muscle protein. Critical care medicine 2007, 35(9 Suppl):4.
47. Korth O, Bosy-Westphal A, Zschoche P, Glüer CC, Heller M, Müller MJ: Influence of methods used in body composition analysis on the prediction of resting energy expenditure. European journal of clinical nutrition 2007, 61(5):582-589.
48. Paddon-Jones D: Interplay of stress and physical inactivity on muscle loss: Nutritional countermeasures. The Journal of nutrition 2006, 136(8):2123-2126.
49. Schutz Y: The adjustment of energy expenditure and oxidation to energy intake: the role of carbohydrate and fat balance. International journal of obesity and related metabolic disorders : journal of the International Association for the Study of Obesity 1993, 17 Suppl 3.
50. Swinburn BA, Ravussin E: Energy and macronutrient metabolism. Baillière's clinical endocrinology and metabolism 1994, 8(3):527-548.
51. Gupta A, Manhas N, Raghubir R: Energy metabolism during cutaneous wound healing in immunocompromised and aged rats. Molecular and cellular biochemistry 2004, 259(1-2):9-14.
52. Cuthbertson D: Metabolic changes. Journal of Clinical Pathology 1970, s3-4(1).
53. Cuthbertson DP: Second annual Jonathan E. Rhoads Lecture. The metabolic response to injury and its nutritional implications: retrospect and prospect. JPEN Journal of parenteral and enteral nutrition 1979, 3(3):108-129.
54. Im MJ, Freshwater MF, Hoopes JE: Enzyme activities in granulation tissue: Energy for collagen synthesis. The Journal of surgical research 1976, 20(2):121-125.

35
55. Im MJ, Hoopes JE: Energy metabolism in healing skin wounds. The Journal of surgical research 1970, 10(10):459-464.
56. Im MJ, Hoopes JE: Enzyme activities in the repairing epithelium during wound healing. The Journal of surgical research 1970, 10(4):173-179.
57. Nguyen DT, Keast D: Maximal activities of glutaminase, citrate synthase, hexokinase, 6-phosphofructokinase and lactate dehydrogenase in skin of immune-competent Balb/c and immune-deficient Balb/c (nu/nu) mice during wound healing. The International journal of biochemistry 1991, 23(5-6):589-593.
58. Decker RH: Nature and regulation of energy metabolism in the epidermis. The Journal of investigative dermatology 1971, 57(6):351-363.
59. Keast D, Nguyen T, Newsholme EA: Maximal activities of glutaminase, citrate synthase, hexokinase, phosphofructokinase and lactate dehydrogenase in skin of rats and mice at different ages. FEBS Letters 1989, 247(1).
60. Gupta A, Raghubir R: Energy metabolism in the granulation tissue of diabetic rats during cutaneous wound healing. Molecular and cellular biochemistry 2005, 270(1-2):71-77.
61. Arnold M, Barbul A: Nutrition and wound healing. Plastic and reconstructive surgery 2006, 117(7 Suppl).
62. Benati G, Bertone MS: Nutrition and Wound Healing. Measurements in Wound Healing 2013.
63. Brown KL, Phillips TJ: Nutrition and wound healing. Clinics in dermatology 2010.
64. Demling RH: Nutrition, anabolism, and the wound healing process: an overview. Eplasty 2009, 9.
65. Dryden SV, Shoemaker WG, Kim JH: Wound management and nutrition for optimal wound healing. Atlas of the oral and maxillofacial surgery clinics of North America 2013, 21(1):37-47.
66. Guo S, Dipietro LA: Factors affecting wound healing. Journal of dental research 2010, 89(3):219-229.
67. Jacobs DO, Lara TM: Nutrition, Metabolism, and Wound Healing in the Elderly. Principles and Practice of Geriatric Surgery 2001:65.
68. Kavalukas SL, Barbul A: Nutrition and wound healing: an update. Plastic and reconstructive surgery 2011.
69. Kiyama T, Witte MB, Thornton FJ: The route of nutrition support affects the early phase of wound healing. … and Enteral Nutrition 1998.

36
70. Mathus-Vliegen EM: Old age, malnutrition, and pressure sores: an ill-fated alliance. The journals of gerontology Series A, Biological sciences and medical sciences 2004, 59(4):355-360.
71. Mazzotta MY: Nutrition and wound healing. Journal of the American Podiatric Medical Association 1994, 84(9):456-462.
72. Medlin S: Nutrition for wound healing. British journal of nursing (Mark Allen Publishing) 2012, 21(12).
73. Molnar JA: Nutrition and wound healing. Nutrition and wound healing 2006.
74. Posthauer ME: The role of nutrition in wound care. Advances in skin & wound care 2006, 19(1):43.
75. Ruberg RL: Role of nutrition in wound healing. The Surgical clinics of North America 1984, 64(4):705-714.
76. Shepherd AA: Nutrition for optimum wound healing. Nursing standard (Royal College of Nursing (Great Britain) : 1987) 2003, 18(6):55-58.
77. Sherman AR, Barkley M: Nutrition and wound healing. Journal of wound care 2011.
78. Stechmiller JK: Understanding the role of nutrition and wound healing. Nutrition in clinical practice : official publication of the American Society for Parenteral and Enteral Nutrition 2010, 25(1):61-68.
79. Stotts NA, Washington DF: Nutrition: a critical component of wound healing. AACN clinical issues in critical care nursing 1990, 1(3):585-594.
80. Wild T, Rahbarnia A, Kellner M, Sobotka L, Eberlein T: Basics in nutrition and wound healing. Nutrition 2010.
81. Williams JZ, Barbul A: Nutrition and wound healing. The Surgical clinics of North America 2003, 83(3):571-596.
82. Stechmiller J: Understanding the role of nutrition and wound healing. Nutrition in clinical practice : official publication of the American Society for Parenteral and Enteral Nutrition, 25(1):61-68.
83. Demling R: Nutrition, anabolism, and the wound healing process: an overview. Eplasty 2009, 9.
84. Breslow RA, Hallfrisch J, Guy DG, Crawley B, Goldberg AP: The importance of dietary protein in healing pressure ulcers. Journal of the American Geriatrics Society 1993, 41(4):357-362.
85. Calder PC: Dietary fatty acids and the immune system. Lipids 1999, 34 Suppl:40.

37
86. Fliers E, Kreier F, Voshol PJ, Havekes LM, Sauerwein HP, Kalsbeek A, Buijs RM, Romijn JA: White adipose tissue: getting nervous. Journal of neuroendocrinology 2003, 15(11):1005-1010.
87. MacKay D, Miller AL: Nutritional support for wound healing. Alternative medicine review : a journal of clinical therapeutic 2003, 8(4):359-377.
88. Mora S, Pessin JE: An adipocentric view of signaling and intracellular trafficking. Diabetes/metabolism research and reviews 2002.
89. Randle PJ: Regulatory interactions between lipids and carbohydrates: the glucose fatty acid cycle after 35 years. Diabetes/metabolism reviews 1998, 14(4):263-283.
90. Wanders RJ, Tager JM: Lipid metabolism in peroxisomes in relation to human disease. Molecular aspects of medicine 1998, 19(2):69-154.
91. Zhang X, Young HA: PPAR and immune system--what do we know? International immunopharmacology 2002, 2(8):1029-1044.
92. Moore FD: Getting well: the biology of surgical convalescence. Annals of the New York Academy of Sciences 1958, 73(2):387-400.
93. Tatti P, Barber A: Nutritional Treatment of Diabetic Foot Ulcers-A Key to Success. Nutritional Treatment of Diabetic Foot Ulcers-A Key to Success 2011.
94. Wernerman J, Brandt R, Strandell T, Allgén LG, Vinnars E: The effect of stress hormones on the interorgan flux of amino acids and on the concentration of free amino acids in skeletal muscle. Clinical nutrition (Edinburgh, Scotland) 1985, 4(4):207-216.
95. Piers LS, Soares MJ, Frandsen SL, O'Dea K: Indirect estimates of body composition are useful for groups but unreliable in individuals. International journal of obesity and related metabolic disorders : journal of the International Association for the Study of Obesity 2000, 24(9):1145-1152.
96. Sen C, Gordillo G, Roy S, Kirsner R, Lambert L, Hunt T, Gottrup F, Gurtner G, Longaker M: Human skin wounds: a major and snowballing threat to public health and the economy. Wound repair and regeneration : official publication of the Wound Healing Society [and] the European Tissue Repair Society 2009, 17(6):763-771.
97. Mokdad AH, Ford ES, Bowman BA, Nelson DE, Engelgau MM, Vinicor F, Marks JS: The continuing increase of diabetes in the US. Diabetes care 2001, 24(2):412.
98. Wild S, Roglic G, Green A, Sicree R, King H: Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes care 2004, 27(5):1047-1053.
99. Administration on Aging [http://www.aoa.gov/AoARoot/Aging_Statistics/index.aspx]

38
100. Edwards R, Harding KG: Bacteria and wound healing. Current opinion in infectious diseases 2004, 17(2):91-96.
101. Wolcott RD, Rhoads DD, Dowd SE: Biofilms and chronic wound inflammation. Journal of wound care 2008, 17(8):333-341.
102. Fife C, Walker D, Thomson B, Carter M: Limitations of Daily Living Activities in Patients With Venous Stasis Ulcers Undergoing Compression Bandaging: Problems With the Concept of Self-bandaging. Wounds: a compendium of clinical research and practice 2007, 19(10):255-257.
103. Abbade LP, Lastória S: Venous ulcer: epidemiology, physiopathology, diagnosis and treatment. International journal of dermatology 2005, 44(6):449-456.
104. Sieggreen MY, Kline RA: Arterial insufficiency and ulceration: diagnosis and treatment options. Advances in skin & wound care 2004, 17(5 Pt 1):242.
105. Defloor T: The risk of pressure sores: a conceptual scheme. Journal of clinical nursing 1999, 8(2):206-216.
106. Reddy M, Gill SS, Rochon PA: Preventing pressure ulcers: a systematic review. JAMA 2006, 296(8):974-984.
107. Gordon MD, Gottschlich MM, Helvig EI, Marvin JA, Richard RL: Review of evidenced-based practice for the prevention of pressure sores in burn patients. The Journal of burn care & rehabilitation 2003, 25(5):388-410.
108. Kuhn BA, Coulter SJ: Balancing the pressure ulcer cost and quality equation. Nursing economic$ 1991, 10(5):353-359.
109. for a: Medicare program; changes to the hospital inpatient prospective payment systems and fiscal year 2008 rates. Federal … 2007.
110. Niezgoda JA, Mendez-Eastman S: The effective management of pressure ulcers. Advances in skin & wound care 2005, 19 Suppl 1:3-15.
111. Conine TA, Hershler C, Daechsel D, Peel C, Pearson A: Pressure ulcer prophylaxis in elderly patients using polyurethane foam or Jay wheelchair cushions. International journal of rehabilitation research Internationale Zeitschrift für Rehabilitationsforschung Revue internationale de recherches de réadaptation 1994, 17(2):123-137.
112. Höök O, Gabrielsson L, Lagerman U: Prophylaxis and treatment of decubitus ulcers with a rocking bed. Scandinavian journal of rehabilitation medicine 1981, 14(1):33-37.
113. Lippert-Grüner M: Gluteal neuromuscular stimulation in therapy and prophylaxis of recurrent sacral pressure ulcers. Spinal cord 2003, 41(6):365-366.

39
114. Torrance C: Pressure sores: pathogenesis, prophylaxis and treatments. 2. Predisposing factors: the'at-risk'patient. Nursing times 1981.
115. National Diabetes Statistics Report: Estimates of Diabetes and Its Burden in the United States [http://www.cdc.gov/diabetes/data/statistics/2014statisticsreport.html]
116. Singh N, Armstrong DG, Lipsky BA: Preventing foot ulcers in patients with diabetes. JAMA 2005, 293(2):217-228.
117. Jeffcoate WJ, Harding KG: Diabetic foot ulcers. The Lancet 2003.
118. Gould L, Abadir P, Brem H, Carter M, Conner-Kerr T, Davidson J, DiPietro L, Falanga V, Fife C, Gardner S et al: Chronic wound repair and healing in older adults: current status and future research. Journal of the American Geriatrics Society 2015, 63(3):427-438.
119. Pierce G, Mustoe T: Pharmacologic enhancement of wound healing. Annual review of medicine 1995, 46:467-481.
120. Mustoe T, Cutler N, Allman R, Goode P, Deuel T, Prause J, Bear M, Serdar C, Pierce G: A phase II study to evaluate recombinant platelet-derived growth factor-BB in the treatment of stage 3 and 4 pressure ulcers. Archives of surgery (Chicago, Ill : 1960) 1994, 129(2):213-219.
121. Richard J, Parer-Richard C, Daures J, Clouet S, Vannereau D, Bringer J, Rodier M, Jacob C, Comte-Bardonnet M: Effect of topical basic fibroblast growth factor on the healing of chronic diabetic neuropathic ulcer of the foot. A pilot, randomized, double-blind, placebo-controlled study. Diabetes care 1995, 18(1):64-69.
122. Frykberg RG, Banks J: Challenges in the Treatment of Chronic Wounds. Advances in wound care 2015, 4(9):560-582.
123. Trujillo AN, Kesl SL, Sherwood J, Wu M, Gould LJ: Demonstration of the rat ischemic skin wound model. Journal of visualized experiments : JoVE 2015(98).
124. Wu L, Xia Y, Roth S, Gruskin E, Mustoe T: Transforming growth factor-beta1 fails to stimulate wound healing and impairs its signal transduction in an aged ischemic ulcer model: importance of oxygen and age. The American journal of pathology 1999, 154(1):301-309.
125. Schultz GS, Sibbald GR, Falanga V, Ayello EA, Dowsett C, Harding K, Romanelli M, Stacey MC, Teot L, Vanscheidt W: Wound bed preparation: a systematic approach to wound management. Wound Repair and Regeneration 2003, 11(s1).
126. Leaper DJ, Schultz G, Carville K, Fletcher J, Swanson T, Drake R: Extending the TIME concept: what have we learned in the past 10 years?(*). International wound journal 2012, 9 Suppl 2:1-19.

40
127. Wolcott RD, Kennedy JP, Dowd SE: Regular debridement is the main tool for maintaining a healthy wound bed in most chronic wounds. Journal of wound care 2009, 18(2):54-56.
128. Steed DL, Donohoe D, Webster MW, Lindsley L: Effect of extensive debridement and treatment on the healing of diabetic foot ulcers. Diabetic Ulcer Study Group. Journal of the American College of Surgeons 1996, 183(1):61-64.
129. Ousey K, McIntosh C: Understanding wound bed preparation and wound debridement. British journal of community nursing 2010, 15(3).
130. Cardinal M, Eisenbud DE, Armstrong DG, Zelen C, Driver V, Attinger C, Phillips T, Harding K: Serial surgical debridement: a retrospective study on clinical outcomes in chronic lower extremity wounds. Wound repair and regeneration 2009, 17(3):306-311.
131. Donohoe D, Webster MW, Lindsley L: Effect of extensive debridement and treatment on the healing of diabetic foot ulcers. Diabetic Ulcer Study Group. Journal of the American … 1996.
132. Panuncialman J, Falanga V: The science of wound bed preparation. Surgical Clinics of North America 2009.
133. Schultz GS, Sibbald RG, Falanga V: Wound bed preparation: a systematic approach to wound management. Wound repair and … 2003.
134. Gamelli R, Schultz G, Tenenhaus M: Toward a common language: surgical wound bed preparation and debridement. Wound repair and … 2006.
135. Dowsett C: Exudate management: a patient-centred approach. Journal of wound care 2008, 17(6):249-252.
136. Trengove NJ, Stacey MC, Macauley S, Bennett N, Gibson J, Burslem F, Murphy G, Schultz G: Analysis of the acute and chronic wound environments: the role of proteases and their inhibitors. Wound Repair and Regeneration 1999, 7(6).
137. Sibbald RG, Goodman L, Woo KY, Krasner DL, Smart H, Tariq G, Ayello EA, Burrell RE, Keast DH, Mayer D et al: Special considerations in wound bed preparation 2011: an update©. Advances in skin & wound care 2011, 24(9):415.
138. O'Reilly D, Pasricha A, Campbell K, Burke N, Assasi N, Bowen JM, Tarride J-EE, Goeree R: Hyperbaric oxygen therapy for diabetic ulcers: systematic review and meta-analysis. International journal of technology assessment in health care 2013, 29(3):269-281.
139. Murphy P, Evans G: Advances in wound healing: a review of current wound healing products. Plastic surgery international, 2012:190436.

41
140. Ennis W, Lee C, Meneses P: A biochemical approach to wound healing through the use of modalities. Clinics in dermatology 2007, 25(1):63-72.
141. Desai KK, Hahn E, Pulikkotill B, Lee E: Negative Pressure Wound Therapy. Clinics in Plastic Surgery 2012, 39(3).
142. Suess JJ, Kim PJ, Steinberg JS: Negative pressure wound therapy: evidence-based treatment for complex diabetic foot wounds. Current diabetes reports 2006, 6(6):446-450.
143. Vig S, Dowsett C, Berg L, Caravaggi C, Rome P, Birke-Sorensen H, Bruhin A, Chariker M, Depoorter M, Dunn R et al: Evidence-based recommendations for the use of negative pressure wound therapy in chronic wounds: Steps towards an international consensus. Journal of Tissue Viability 2011, 20.
144. Peinemann F, Sauerland S: Negative-pressure wound therapy: systematic review of randomized controlled trials. Deutsches Ärzteblatt international 2011, 108(22):381-389.
145. Eskes AM, Ubbink DT, Lubbers MJ, Lucas C, Vermeulen H: Hyperbaric oxygen therapy: solution for difficult to heal acute wounds? Systematic review. World journal of surgery 2011, 35(3):535-542.
146. Bonomo SR, Davidson JD, Tyrone JW, Lin X, Mustoe TA: Enhancement of wound healing by hyperbaric oxygen and transforming growth factor beta3 in a new chronic wound model in aged rabbits. Archives of surgery (Chicago, Ill : 1960) 2000, 135(10):1148-1153.
147. Gomez CR, Knutson GJ, Clifton KB, Schreiber CA, Vuk-Pavlović S: Age-dependent response of murine female bone marrow cells to hyperbaric oxygen. Biogerontology 2012, 13(3):287-297.
148. Boyce ST, Warden GD: Principles and practices for treatment of cutaneous wounds with cultured skin substitutes. The American Journal of Surgery 2002, 183(4).
149. Halim A, Khoo T, Shah J: Biologic and synthetic skin substitutes: An overview. Indian Journal of Plastic Surgery 2010, 43(3).
150. Jones I, Currie L, Martin R: A guide to biological skin substitutes. British journal of plastic surgery 2002, 55(3):185-193.
151. MacNeil S: Progress and opportunities for tissue-engineered skin. Nature 2007, 445(7130):874-880.
152. Schulz JT, Tompkins RG, Burke JF: Artificial skin. Annual review of medicine 2000, 51:231-244.

42
153. Supp DM, Boyce ST: Engineered skin substitutes: practices and potentials. Clinics in dermatology 2005, 23(4):403-412.
154. Varkey M, Ding J, Tredget EE: Advances in Skin Substitutes—Potential of Tissue Engineered Skin for Facilitating Anti-Fibrotic Healing. Journal of Functional Biomaterials 2015, 6(3).
155. Shankaran V, Brooks M, Mostow E: Advanced therapies for chronic wounds: NPWT, engineered skin, growth factors, extracellular matrices. Dermatologic therapy 2013, 26(3):215-221.
156. Boyce ST, Kagan RJ, Yakuboff KP, Meyer NA, Rieman MT, Greenhalgh DG, Warden GD: Cultured skin substitutes reduce donor skin harvesting for closure of excised, full-thickness burns. Annals of surgery 2002, 235(2):269-279.
157. Rue LW, Cioffi WG, McManus WF, Pruitt BA: Wound closure and outcome in extensively burned patients treated with cultured autologous keratinocytes. The Journal of trauma 1993, 34(5):662.
158. Hom DB, Manivel JC: Promoting healing with recombinant human platelet-derived growth factor--BB in a previously irradiated problem wound. The Laryngoscope 2003, 113(9):1566-1571.
159. Margolis DJ, Bartus C, Hoffstad O, Malay S, Berlin JA: Effectiveness of recombinant human platelet-derived growth factor for the treatment of diabetic neuropathic foot ulcers. Wound repair and regeneration : official publication of the Wound Healing Society [and] the European Tissue Repair Society 2005, 13(6):531-536.
160. Shackelford DP, Fackler E, Hoffman MK, Atkinson S: Use of topical recombinant human platelet-derived growth factor BB in abdominal wound separation. American journal of obstetrics and gynecology 2002, 186(4):701-704.
161. Steed DL: Clinical evaluation of recombinant human platelet-derived growth factor for the treatment of lower extremity diabetic ulcers. Diabetic Ulcer Study Group. Journal of vascular surgery 1995, 21(1):71.
162. Perry BH, Sampson AR, Schwab BH: … safety of becaplermin (recombinant human platelet‐derived growth factor‐BB) in patients with nonhealing, lower extremity diabetic ulcers: a combined analysis of four … . Wound Repair and … 1999.
163. Steed DL: Clinical evaluation of recombinant human platelet–derived growth factor for the treatment of lower extremity diabetic ulcers. Journal of Vascular Surgery 1995.
164. Steed DL: Clinical evaluation of recombinant human platelet-derived growth factor for the treatment of lower extremity ulcers. Plastic and reconstructive surgery 2006.

43
165. Wieman TJ: Clinical efficacy of becaplermin (rhPDGF-BB) gel. The American journal of surgery 1998.
166. Fu X, Fang L, Li X, Cheng B, Sheng Z: Enhanced wound-healing quality with bone marrow mesenchymal stem cells autografting after skin injury. Wound repair and regeneration : official publication of the Wound Healing Society [and] the European Tissue Repair Society 2006, 14(3):325-335.
167. Cottler-Fox MH, Lapidot T, Petit I, Kollet O, DiPersio JF, Link D, Devine S: Stem cell mobilization. Hematology / the Education Program of the American Society of Hematology American Society of Hematology Education Program 2003:419-437.
168. Fu S, Liesveld J: Mobilization of hematopoietic stem cells. Blood reviews 2000, 14(4):205-218.
169. Carmeliet P, Luttun A: The emerging role of the bone marrow-derived stem cells in (therapeutic) angiogenesis. Thromb Haemost 2001.
170. Kwon DS, Gao X, Liu YB: Treatment with bone marrow‐derived stromal cells accelerates wound healing in diabetic rats. International wound … 2008.
171. Laverdet B, Micallef L, Lebreton C, Mollard J: Use of mesenchymal stem cells for cutaneous repair and skin substitute elaboration. Pathologie … 2014.
172. Wu Y, Huang S, Enhe J, Ma K, Yang S, Sun T, Fu X: Bone marrow‐derived mesenchymal stem cell attenuates skin fibrosis development in mice. International Wound Journal 2014, 11(6).
173. Dreifke MB, Jayasuriya AA, Jayasuriya AC: Current wound healing procedures and potential care. Materials science & engineering C, Materials for biological applications 2015, 48:651-662.
174. Wicke C, Bachinger A, Coerper S, Beckert S, Witte M, Königsrainer A: Aging influences wound healing in patients with chronic lower extremity wounds treated in a specialized Wound Care Center. Wound repair and regeneration : official publication of the Wound Healing Society [and] the European Tissue Repair Society 2009, 17(1):25-33.
175. Ashcroft GS, Horan MA, Ferguson MW: Aging is associated with reduced deposition of specific extracellular matrix components, an upregulation of angiogenesis, and an altered inflammatory response in a murine incisional wound healing model. The Journal of investigative dermatology 1997, 108(4):430-437.
176. Gosain A, DiPietro L: Aging and wound healing. World journal of surgery 2004, 28(3):321-326.
177. Makrantonaki E, Zouboulis C: Molecular mechanisms of skin aging: state of the art. Annals of the New York Academy of Sciences 2007, 1119:40-50.

44
178. Kurban RS, Bhawan J: Histologic changes in skin associated with aging. The Journal of dermatologic surgery and oncology 1990, 16(10):908-914.
179. Gilchrest BA, Murphy GF, Soter NA: Effect of chronologic aging and ultraviolet irradiation on Langerhans cells in human epidermis. The Journal of investigative dermatology 1982, 79(2):85-88.
180. Lavker RM, Zheng PS, Dong G: Aged skin: a study by light, transmission electron, and scanning electron microscopy. The Journal of investigative dermatology 1987, 88(3 Suppl).
181. Bryant R: Acute and Chronic Wounds: Current Management Concepts. Philadelphia, PA: Elsevier; 2011.
182. Pittman J: Effect of aging on wound healing: current concepts. Journal of wound, ostomy, and continence nursing : official publication of The Wound, Ostomy and Continence Nurses Society / WOCN 2007, 34(4):412.
183. Sgonc R, Gruber J: Age-related aspects of cutaneous wound healing: a mini-review. Gerontology 2013, 59(2):159-164.
184. Ashcroft GS, Horan MA, Herrick SE, Tarnuzzer RW, Schultz GS, Ferguson MW: Age-related differences in the temporal and spatial regulation of matrix metalloproteinases (MMPs) in normal skin and acute cutaneous wounds of healthy humans. Cell and tissue research 1997, 290(3):581-591.
185. Valacchi G, Zanardi I, Sticozzi C, Bocci V, Travagli V: Emerging topics in cutaneous wound repair. Annals of the New York Academy of Sciences 2012, 1259:136-144.
186. Ferrucci L, Corsi A, Lauretani F, Bandinelli S, Bartali B, Taub D, Guralnik J, Longo D: The origins of age-related proinflammatory state. Blood 2005, 105(6):2294-2299.
187. Mahbub S, Deburghgraeve C, Kovacs E: Advanced age impairs macrophage polarization. Journal of interferon & cytokine research : the official journal of the International Society for Interferon and Cytokine Research 2012, 32(1):18-26.
188. Murphy M: How mitochondria produce reactive oxygen species. The Biochemical journal 2009, 417(1):1-13.
189. Schreml S, Szeimies R, Prantl L, Karrer S, Landthaler M, Babilas P: Oxygen in acute and chronic wound healing. The British journal of dermatology, 163(2):257-268.
190. Aw TY: Tissue oxidative stress revisited: significance of ROS and redox signaling. Seminars in cell & developmental biology 2012, 23(7):721.
191. Murphy MP: How mitochondria produce reactive oxygen species. The Biochemical journal 2009, 417(1):1-13.

45
192. Miquel J: Can Antioxidant Diet Supplementation Protect against Age‐related Mitochondrial Damage? Annals of the New York Academy of Sciences 2002.
193. Yang JH, Lee HC, Lin KJ, Wei YH: A specific 4977-bp deletion of mitochondrial DNA in human ageing skin. Archives of dermatological research 1994, 286(7):386-390.
194. Savitha S, Panneerselvam C: Mitochondrial membrane damage during aging process in rat heart: potential efficacy of L-carnitine and DL alpha lipoic acid. Mechanisms of ageing and development 2006, 127(4):349-355.
195. Roy S, Khanna S, Nallu K, Hunt T, Sen C: Dermal wound healing is subject to redox control. Molecular therapy : the journal of the American Society of Gene Therapy 2006, 13(1):211-220.
196. Kozina L, Borzova I, Arutiunov V, Ryzhak G: [The role of oxidative stress in skin aging]. Advances in gerontology = Uspekhi gerontologii / Rossiĭskai͡ a akademii͡ a nauk, Gerontologicheskoe obshchestvo 2012, 25(2):217-222.
197. Masaki H: Role of antioxidants in the skin: anti-aging effects. Journal of dermatological science 2010, 58(2):85-90.
198. Holbrook N, Ikeyama S: Age-related decline in cellular response to oxidative stress: links to growth factor signaling pathways with common defects. Biochemical pharmacology 2002, 64(5-6):999-1005.
199. Moor AN, Tummel E, Prather JL, Jung M, Lopez JJ, Connors S, Gould LJ: Consequences of age on ischemic wound healing in rats: altered antioxidant activity and delayed wound closure. Age (Dordrecht, Netherlands) 2014, 36(2):733-748.
200. Treiber N, Maity P, Singh K, Ferchiu F, Wlaschek M, Scharffetter-Kochanek K: The role of manganese superoxide dismutase in skin aging. Dermato-endocrinology 2012, 4(3):232-235.
201. Reed M, Edelberg J: Impaired angiogenesis in the aged. Science of aging knowledge environment : SAGE KE 2004, 2004(7).
202. Bratic I, Trifunovic A: Mitochondrial energy metabolism and ageing. Biochim Biophys Acta, 1797(6-7):961-967.
203. 4-105-1-PB.pdf.

46
CHAPTER 2: ORAL KETONE SUPPLEMENTATION AS A NOVEL THERAPEUTIC
FOR DELAYED WOUND HEALING
2.1. Chapter Synopsis
In the previous chapter, I provided a thorough overview of chronic wounds and the non-
efficacious therapies that are currently available, especially for the aged population. Here I
provide a rationalization for the use of an oral exogenous ketone supplement as a novel therapy
for wound healing. The ketogenic diet has been used since the 1920s for refractory epilepsy in
children. Though it has been proven to be effective, it is hard to maintain and is limited in a
clinical setting. Recently, it has been shown that ketone bodies themselves may confer
therapeutic benefits, paving the way for an exogenous ketone supplement that does not require
dietary restriction. Ketones have been shown to decrease inflammation, decrease ROS levels,
increase blood flow, and increase ATP hydrolysis and increase metabolism. As previously
described, these are the four underlying age-dependent features we will utilize to determine the
efficacy of our treatment. For these reasons, we hypothesize that exogenous ketone
supplementation will augment wound healing and we propose to test their effects in vivo using
an ischemic wound model in young and aged Fischer 344 rats and in vitro using patient-derived
primary human dermal fibroblasts isolated from discarded skin.
Portions of this chapter have previously been published in “Kesl SL, Poff AM, Ward NP,

47
Fiorelli TN, Ari C, Van Putten AJ, Sherwood JW, Arnold P, D’Agostino DP. Effects of
Exogenous Ketone Supplementation on Blood Ketone, Glucose, Triglyceride, and Lipoprotein
Levels in Sprague-Dawley Rats. Nutrition and Metabolism (2016)”, Poff AM, Kesl SL,
D’Agostino DP “Ketone Supplementation for Health and Disease” Book Chapter, Oxford Press
(Submitted), and “ Trujilio AN*, Kesl SL*, Sherwood J, Wu M, Gould LJ. (2014)
Demonstration of the Rat Ischemic Wound Model. Journal of Visualized Experiments: JoVE ”. *
Indicates that both authors contributed equally to the work. Copies of these articles may be found
in Appendix C. See Appendix B for copyright permissions. Materials and methods presented in
this chapter can be found in Appendix A.
2.2. KetoneBodySynthesisandMetabolism
Ketone bodies are naturally elevated to serve as alternative metabolic substrates for extra-
hepatic tissues during the prolonged reduction of glucose availability, suppression of insulin,
and depletion of liver glycogen, such as occurs during starvation, fasting, vigorous exercise,
calorie restriction, or the ketogenic diet (KD). As blood glucose levels drop, the body goes
through a metabolic shift from glucose-based metabolism towards fatty acid oxidation and
hepatic ketogenesis to generate ATP. As a result, hepatic cells synthesize necessary glucose
through gluconeogenesis (GNG) and β-oxidation metabolizes fats (dietary and stored) to form
Acetyl CoA. During GNG, the oxaloacetate stores are depleted, causing the Acetyl CoA from
fatty acid oxidation to accumulate and not enter the Krebs cycle. The excess Acetyl CoA
produces the ketone bodies beta-hydroxybutyrate (βHB) and acetoacetate (AcAc) via a 3-step
enzymatic process known as ketogenesis. From the liver, these substrates are transported

48
systemically to extra-hepatic tissues (i.e. muscle, brain, and skin) where Acetyl CoA is
reconstructed and used to generate ATP via the Krebs cycle and the ETC [1, 2]. Additionally,
AcAc can be decarboxylated to produce the third ketone body, acetone. Although acetone has
been considered mainly to be a metabolic by-product that is eliminated via the lungs and urine, it
has recently been shown to play an important role in the anticonvulsant properties of the
ketogenic diet [3-7].
2.3. Review of Ketogenic Diet and Therapeutic Uses
The classical ketogenic diet (KD) consists of a 4:1 ratio of fat to protein and carbohydrate
combined, with 80-90% of total calories derived from fat [8]. For most patients this equates to
eating less than 50 grams of carbohydrates and around 100 grams of protein a day [9]. Actual
carbohydrate and protein intake must be individually optimized to maintain therapeutic ketone
production. The macronutrient ratio of the KD induces the metabolic shift towards hepatic
ketogenesis, elevating AcAc and βHB in the blood. Compared to other low carb diets like the
Atkins diet, the ketogenic diet requires only adequate protein. Excess protein intake can fuel
GNG, resulting in glucose synthesis that disables ketogenesis. It is important to emphasize that
not all low carbohydrate diets are ketogenic. The ketogenic diet is really a high-fat diet, and not a
high-protein diet.
Emerging evidence supports the therapeutic potential of the ketogenic diet (KD) for a
variety of disease states, leading investigators to research methods of harnessing the benefits of
nutritional ketosis without the dietary restrictions. The KD has been used as an effective non-
pharmacological therapy for pediatric intractable seizures since the 1920s [10-12]. In a study of

49
150 epileptic children who averaged 400 seizures per month and were on multiple anti-epileptic
medications, 60% reported a significant decrease in seizure frequency [12]. In addition to
epilepsy, the ketogenic diet has elicited significant therapeutic effects for weight loss and type-2
diabetes (T2D) [13]. Several studies have shown significant weight loss on a high fat, low
carbohydrate diet without significant elevations of serum cholesterol [14-21]. Another study
demonstrated the safety and benefits of long-term application of the KD in T2D patients. Patients
exhibited significant weight loss, reduction of blood glucose, and improvement of lipid markers
after eating a well-formulated KD for 56 weeks [22]. Recently, researchers have begun to
investigate the use of the KD as a treatment for acne, polycystic ovary syndrome (PCOS),
cancer, amyotrophic lateral sclerosis (ALS), traumatic brain injury (TBI) and Alzheimer’s
disease (AD) with promising preliminary results [23-35].
2.3.1. Potential Concerns and Side Effects
Although the KD has clear therapeutic potential, several factors limit the efficacy and
clinical utility of the KD for metabolic therapy. Patient compliance to the KD can be low due to
the severe dietary restriction, the diet being perceived as unpalatable, and intolerance to high-fat
ingestion. Maintaining ketosis can be difficult as consumption of even a small quantity of
carbohydrates or excess protein can rapidly inhibit ketogenesis [36, 37]. Furthermore, enhanced
ketone production and tissue utilization can take several weeks (keto-adaptation), and patients
may experience mild hypoglycemic symptoms during this transitional period [38]. One common
concern regarding the KD is its purported potential to increase the risk of atherosclerosis by
elevating blood cholesterol and triglyceride levels [39, 40]. This topic remains controversial as

50
some, but not all studies have demonstrated that the KD elevates blood levels of cholesterol and
triglycerides [41-46]. Kwitervich and colleagues demonstrated an increase in low-density
lipoprotein (LDL) and a decrease in high-density lipoprotein (HDL) in epileptic children fed the
classical KD for two years [8]. In this study, total cholesterol increased by ~130%, but then
stabilized at the elevated level over the 2-year period. A similar study demonstrated that the lipid
profile returned to baseline in children who remained on the KD for six years [47]. Children
usually stay on the diet for approximately two years then return to a diet of normal fat and
carbohydrate ingestion [48]. In contrast, the majority of recent studies have suggested that the
KD can lead to significant benefits in biomarkers of metabolic health, including blood lipid
profiles [49-56]. In these studies, the KD positively altered blood lipids, decreasing total
triglycerides and cholesterol while increasing the ratio of HDL to LDL [52-61]. Although, the
KD is well established in children, the diet has only recently been accepted as a strategy to
control seizures in adults. In 2014, Schoeler et al reported on the feasibility of the KD for adults,
concluding that 39% of individuals achieved > 50% reduction in seizure frequency, similar to
results seen in pediatric studies. Patients experienced similar gastrointestinal adverse events that
have been previously described in pediatric patients, but they did not lead to discontinuation of
the diet in any patient [62].
2.4. Exogenous Ketone Supplementation
Considering both the broad therapeutic potential and limitations of the KD, an oral ketone
supplement that could rapidly induce and sustain therapeutic ketosis without the need for dietary
restriction would be an attractive and practical alternative. Recent studies suggest that many of

51
the benefits of the KD are due to the effects of ketone body metabolism. Veech et al. have
summarized the potential therapeutic uses for ketone bodies, which will be discussed later in this
chapter [3, 63]. Furthermore, there is potential for an additive therapeutic effect between a
standard KD and an oral ketone supplement for patients consuming a low carbohydrate or
ketogenic diet. Additionally, in some scenarios, a ketone supplemented ketogenic diet may be
more effective than KD or ketone supplementation alone. Several natural and synthetic ketone
supplements capable of inducing nutritional ketosis have been identified. There are numerous
sources of ketones and ketogenic precursors being developed and tested, including medium chain
triglycerides, diols, salts, and esters that have been shown to elevate blood ketone levels without
dietary restriction. The investigation of natural and synthetic ketogenic precursors to establish
nutritional ketosis without the need for dietary restriction has revealed that each formulation has
distinct properties in terms of extent and duration of ketosis as well as metabolic effects. Most of
the developed ketone supplements are currently under investigation for safety and efficacy in a
number of disease states. For this dissertation, we focus on five ketogenic agents including 1, 3-
butanediol (BD), medium chain triglyceride oil (MCT), sodium/potassium-βHB mineral salt
(BMS), combination of BMS+MCT, and 1,3-butanediol acetoacetate diester/ ketone ester (KE).
2.4.1. 1,3-Butanediol
1,3-Butanediol (BD) is a FDA approved organic alcohol used as a food flavoring solvent, an
intermediate in the manufacture of certain polyester plasticizers, and a humectant for cosmetics
[64, 65]. When ingested orally, BD is metabolized by the liver via alcohol dehydrogenase (ADH)
to β-hydroxybutyraldehyde, which is rapidly oxidized to βHB by aldehyde dehydrogenase [66].

52
BD contributes approximately 6 kcal/g of energy and can produce dose-dependent millimolar
concentrations of ketones in the blood [67-69]. Extensive toxicology studies have concluded that
BD is safe with very little adverse health effects in humans or animals [70-73]. BD has been
investigated as a therapy to reduce brain damage following cerebral ischemia. BD attenuated
hypoxic damage via a cerebral protective effect mediated by brain ketone metabolism [74].
Additionally, ketosis induced via exogenous BD supplementation has been shown to increase
survival time of mice exposed to hypoxia in a stroke model [75]. BD supplementation has been
shown to significantly prolong the survival time in mice with metastatic cancer by 51% [26].
Recently, BD has been investigated therapeutically as a backbone of ketone mono- and di-esters,
which are discussed later in this chapter.
2.4.2. MCT Oil
Medium chain triglycerides (MCTs) contain a glycerol backbone esterified to medium-
chain fatty acids (MCFAs), which are fatty acids with hydrocarbon side chains 6 to 12 carbons in
length. MCFAs include caproic acid (C6:0, hexanoic acid), caprylic acid (C8:0, octanoic acid),
capric acid (C10:0, decanoic acid), and lauric acid (C12:0, dodecanoic acid). MCTs are naturally
found in coconut oil (15%), palm kernel oil (7.9%), cheese (7.3%), milk (6.9%), butter (6.8%),
and yogurt (6.6%) [76, 77]. Compared to long-chain fatty acids (LCFAs), MCFAs have a lower
melting point, smaller molecule size, and are less calorically dense (8.3 calories per gram versus
9.2). These distinct physiochemical properties allow MCTs to be absorbed directly into the
bloodstream through the portal vein without the need for bile or pancreatic enzymes for
degradation. Additionally, MCTs do not require carnitine to enter the mitochondria, but rather

53
quickly cross the double mitochondrial matrix where they are metabolized to acetyl co-A, and
subsequently, to ketone bodies. Thus, they are easily and rapidly digested, transferred to the
liver, and used for energy rather than stored as fat. In comparison, LCFA metabolism is much
slower requiring re-esterification in the small intestine, transport by chylomicrons via the
lymphatic and vascular systems, and oxidation in the liver for energy or storage. MCTs are
metabolized as rapidly as glucose but have roughly twice the energy density [78]. In the early
1980s, Dr. Vigen K. Babayan of the Nutrition Laboratory at Harvard University developed a
process to produce MCTs in large quantities [79]. These commercialized MCTs are acquired
through lipid fractionation from natural fats such as coconut oil and milk and are predominantly
comprised of C:8 and C:10 MCTs [80, 81].
Since the 1970s, MCTs have been used in a modification of the classical KD as an
alternative fat source [82]. The ketogenic properties of MCTs allow patients to eat less total fat
in their diet and include more carbohydrate and protein without sacrificing their nutritional
ketosis. Although MCTs have the potential to be an efficient and beneficial ketogenic fat, they
are currently limited in clinical use due to gastrointestinal (GI) side effects stimulated by the
large dose needed to induce ketonemia (greater than 40 g/day) [78]. The original modified KD
with MCT allowed 60% of its energy to be derived from MCTs; however, this amount caused GI
distress in some children [83-86]. For this reason, an additional modified KD with MCT was
developed using only 30% of its energy from MCTs; however, it induced much lower levels of
ketosis [87, 88].

54
2.4.3. βHB Salt
Originally, researchers attempted to administer βHB or AcAc in their free acid forms;
however, this was shown to be too expensive and ineffective at producing sustained ketosis.
Subsequently, it was suggested to buffer the free acid from of βHB with sodium salts, but this
causes potentially harmful sodium overload and mineral imbalance at therapeutic levels of
ketosis, and is largely ineffective at preventing seizures in animal models [89]. A study showed
that oral administration of Na+/βHB in doses from 80-900mg/kg/day elevated blood ketone
levels to 0.19-0.36 mM in children with acyl CoA dehydrogenase deficiency [90]. However, to
achieve the same level of ketosis a 70 kg man would need to ingest between 5.6 to 63 g/day,
costing up to $200 a day. Considering the potential effects of such a large sodium load, the costs
of the administration of Na+ /βHB salts to achieve ketosis made this approach unrealistic [63].
Recently, a βHB mineral salt was developed in collaboration with Savind Inc. These
ketone salts are balanced with minerals to prevent sodium overload and can include arginine,
potassium, calcium, magnesium, lithium, lysine, histidine, ornithine, creatine, agmatine
(guanidine), or citrulline. Maintaining an optimal sodium-mineral ratio should help offset any
potential adverse effects of sodium on blood pressure. It is speculated that this formulation will
be especially beneficial for elderly patients most susceptible to sodium-induced hypertension
[91]. The mineral salt has to be delivered as a concentrated liquid because potassium βHB is too
hygroscopic to isolate as a powder (it picks up water from the atmosphere and turns into a paste
at an astonishing rate, whether by itself or mixed with sodium βHB). Many people experience
the “low carb flu” during keto-adaptation (2-3 week phase characterized by enhancement of
ketone production and tissue utilization from being on a ketogenic diet) caused by glucose
withdrawal in the brain and a depletion of minerals, especially sodium and potassium, in the

55
plasma [38]. These symptoms can be attenuated or reversed with sufficient supplementation of
sodium, potassium, calcium, and magnesium.
Several mineral combinations are currently being tested for safety and efficacy. In a recent
case study, a 100 kg male subject was administered a 4% Na+/K+ βHB salt solution (containing
11 grams of sodium and 7.1 grams βHB) and his plasma levels of βHB were significantly
elevated 30-60 minutes after administration. After receiving the same dose for three days, the
patient had sustained elevated blood ketone levels from 15-120 minutes after administration.
Similar results were seen in a 70kg male who fasted three days prior to administration of the
ketone salt supplement [92]. In a 15-week study, Sprague-Dawley rats were administered
Na+/Ca+2-β-hydroxybutyrate mineral salt (S-BHB), 20% by weight (~25 g/kg/day) in their food
fed ad libitum. The S-BHB supplemented rats exhibited sustained elevated blood ketone levels
(1mM βHB) at 1, 4, 8, 10, and 13 weeks of chronic feeding which did not affect blood glucose
levels compared to controls (unpublished data).
2.4.4. Combination BMS+MCT
Considering the variety of ketogenic precursors available, researchers are investigating
unique combinations of the individual supplements in hopes of optimizing their benefits. A
combination of βHB salts and MCT oil has been administered in 1:1 to 1:2 mixtures. This
mixture allows for a lower dosing of the components compared to administering the individual
compounds, thus reducing potential for side effects (sodium-induced hypertension, GI side
effects, etc) and resulting in distinct synergistic blood ketone profile [92].

56
In a case study, a 100 kg male was administered a combination of a 4% Na+/K+ βHB salt
solution (containing 11 grams of sodium and 7.1 grams βHB) + 20 mL MCT oil. This
combination demonstrated higher elevation of blood ketone levels than either βHB salts or MCT
oil alone, starting at 15 minutes post consumption and lasting for 4 hours. Similar results were
observed in a 70-kg male who fasted 3 days prior to administration; however elevated blood
ketone levels were observed sooner after supplementation and were sustained for a considerable
longer time after administration (8 hours). This effect was accompanied by a reduction in blood
glucose, and a lower starting blood glucose concentration on each subsequent day of
supplementation [92]. In a 15-week study, Sprague-Dawley rats were administered a 1:1 mixture
of Na+/Ca+2-β-hydroxybutyrate mineral salt + Medium Chain Triglyceride Oil (S/MCT) (20%
by weight (~25 g/kg/day) in their food fed ad libitum. The combination-supplemented rats had
significantly sustained and elevated blood ketone levels as weeks 3, 4, 8, 10, and 13 without
significantly affecting blood glucose levels during the study (unpublished data).
2.4.5. Ketone Esters
Researchers have developed and investigated several synthetic ketone mono- and di-
esters to produce nutritional ketosis without dietary restriction. In these compounds, gastric
esterases rapidly liberate ketones as a free acid from a backbone molecule, which varies
depending upon the specific formulation, but is often R,S-1,3-butanediol. As previously
discussed, 1,3-BD is subsequently metabolized by the liver to produce βHB [4]. Thus, the ketone
esters currently available are unique amongst the aforementioned ketone supplements in that
many contain, and thus directly elevate, AcAc rather than only βHB.

57
In the late 1970s, Birkhahn et al. were the first to synthesize a monoester of glycerol and
AcAc (monoacetoacetin) for parenteral nutrition. These studies demonstrated that
monoacetoacetin induced hyperketonemia comparable to fasted rats at a dose of 50 g/kg per day
[93-95]. In attempts to increase the caloric density of monoacetoacetin, they synthesized both a
monoester and triester of glycerol and βHB. These esters are hydrolyzed to release free βHB,
thus elevating blood ketones. Later, Desrochers and colleagues synthesized mono- and di-esters
of 1,3-BD with either AcAc or βHB [96]. These and other esters developed by or in collaboration
with Henri Brunengraber and Richard Veech have been shown to induce various degrees of
hyperketonemia (2-7mM) in rats, mice, dogs, pigs, and humans [97-102]. Clarke and colleagues
demonstrated the safety of these ketone esters in rats and humans [103, 104]. Recently, a R,S-
1,3-butanediol acetoacetate diester (KE) was developed in collaboration with Savind Inc. A
single dose of the KE administered via intragastric gavage elevated both AcAc and βHB blood
levels to >3mM in rats [4]. In a 15-week study, the KE was administered to Sprague-Dawley rats
in a low dose (5%, 10 g/kg/day) (LKE) and a high dose (20%, 25 g/kg/day) (HKE) in food fed
ad libitum. Both doses significantly elevated blood ketone levels without affecting blood glucose
levels. Serum clinical chemistry of both LKE and HKE did not reveal any changes in any major
markers for kidney and liver function compared to standard diet fed rats (unpublished data).
2.4.6. Potential Concerns and Side Effects: Ketoacidosis
Ketoacidosis is one major concern for the use of oral ketone supplements without dietary
restriction. Ketoacidosis is normally only seen in patients with diabetes mellitus. In this
circumstance, the lack of insulin prevents glucose delivery to the tissues to be used as energy.

58
High glucose levels spill over into the urine, taking water and solutes (sodium and potassium)
with it in a process known as osmotic diuresis. The body senses that it is in starvation mode and
enhances b-oxidation of fatty acids for ketogenesis. The resulting ketone bodies are used as fuel.
Additionally, insulin acts as a negative feedback loop to ketogenesis. Any rise in insulin and/or
glucose normally inhibits ketogenesis immediately or excess ketones are expelled through
ketonuria. Without insulin, the body is overwhelmed by ketone bodies. Since the ketone bodies
are acidic, the uncontrolled elevation of ketone bodies (20-25 mM) drops the blood pH and can
lead to organ failure, cerebral edema, and even death. If not properly administered, oral ketone
supplementation does have the potential to induce ketoacidosis; however, optimal dosing
circumvents this danger. Oral ketone supplementation will be optimized to elevate blood ketone
levels to the 2-7 mM range, which is far below the elevation seen in ketoacidosis.
2.5. Potential Physiology Factors Affected to Enhance Wound Healing
It has been well established that optimized nutritional support aids wound healing. [105,
106]. Demling et al. stated that in order to optimize healing, a substrate that is more dependent
on intake rather than on the bodily breakdown of protein needs to be available [107]. In the
traditional classification, ketone bodies are known as a carrier of energy from the liver to extra-
hepatic tissue. However they can also be considered as a nutritional supplement and an anabolic
agent [108]. Indeed, Nair et al. concluded that βHB promotes protein synthesis in human beings
[109]. Recently, it has been shown that βHB possess a variety of signaling functions that may
regulate a broad range of cellular functions. [110-113]. It has been shown that βHB can regulate
cellular processes via histone deacetylase (HDAC) inhibition, binding to cell surface receptors,

59
and altering the levels of other regulatory metabolites including acetyl-CoA, succinyl-CoA, and
NAD+ [110]. These unique cellular and physiological properties support the assertion that
ketone bodies themselves confer many of the benefits associated with the KD. We hypothesize
that ketone bodies may augment wound healing via the suppression of inflammation, inhibition
of oxidative stress, enhancement of blood flow, and enhancement of metabolic efficiency.
Additionally, ketones may aid diabetic foot ulcer healing via modulation of blood glucose levels
and enhancement of insulin sensitivity.
2.5.1. Ketones and Inflammation
As mentioned in the previous chapter, chronic wounds are stuck in the inflammatory
phase. A therapy that promotes resolution of inflammation may push the chronic wound to a
more acute wound phenotype, therefore progressing to closure. Several studies have determined
that the KD reduces circulating inflammatory markers in animals and in humans [114-117].
Additionally, Ketone bodies themselves have been shown to decrease inflammatory biomarkers
including TNF-a, IL-6, IL-8, MCP-1, E-selectin, I-CAM, and PAI-1 [17, 118]. In an unpublished
study from the D’Agostino laboratory, rats were fed one of three exogenous ketone supplements
LKE, HKE, S-BHB, or S/MCT mixed into standard rodent chow at 5- 20% by weight for 15
weeks. Inflammatory profiling was performed on serum collected from the animals at the end of
the chronic feeding study and revealed decreases in several pro-inflammatory cytokines
including IL-1β, IL-6, IFN-γ, MCP-1, and RANTES.
Recently, Dixit and colleagues demonstrated in both in vitro and in vivo that βHB inhibits
the NLRP3 inflammasome, which controls activation of caspase-1 and release of the pro-

60
inflammatory cytokines IL-1β and IL-18 from M1 macrophages. The authors concluded that
elevating blood ketones could be a therapeutic option for patients with NLRP3-mediated chronic
inflammatory diseases [119]. It has been established that elevated levels of IL-1β are present in
wounds of diabetic humans and mice, consistent with increased inflammasome activity. Since
M1 macrophages are activated by IL-1β, the NLRP3 is thought to perpetuate this pro-
inflammatory phenotype, leading to impaired wound healing [120]. Two studies have shown that
blocking the activation of the inflammasome decreased pro-inflammatory cytokine levels and
accelerated wound healing in diabetic mice [120, 121]. This evidence supports the hypothesis
that oral ketone supplementation could produce similar effects on the inflammasome.
2.5.2. Ketones and ROS
Veech and others have determined some of the molecular mechanisms by which ketone
bodies suppress oxidative stress [122-124]. In non-wounded skin, the major source of free
radicals is the half-reduced semiquinone of co-enzyme Q in the electron transport chain of the
mitochondria. At physiological levels, ketone bodies increase the oxidation of co-enzyme Q,
decreasing mitochondrial oxygen radical generation, thereby reducing the amount of ROS
produced [3, 63, 124]. Additionally, ketone metabolism induces reduction of the mitochondrial
NAD+ and cytoplasmic NADP electron carriers. As previously mentioned in Chapter 1, NADH
is important for energy producing pathways and NADPH is necessary for the regeneration of the
endogenous antioxidant reduced glutathione (GSH) [125].
Eric Verdin and colleagues have recently demonstrated βHB is an endogenous and
specific inhibitor of class 1 histone deacetylases (HDACs) [126]. Elevated βHB levels by fasting,

61
calorie restriction, and exogenous administration increased global histone acetylation in mouse
tissues and induced the transcription of genes encoding oxidative resistance factors FOXO3A
and MT2 via selective depletion of HDAC 1 and HDAC2. Furthermore, the authors
demonstrated in vivo that exogenous ketone supplementation could prevent oxidative stress.
Mice were pretreated with βHB via a subcutaneous pump for 24 hours prior to receiving an
injection of paraquat, which induces the production and accumulation of ROS. Protein
carbonylation was suppressed by 54%, lipid peroxidation was completely suppressed, and
endogenous antioxidants mitochondrial superoxide dismutase (MnSOD, SOD2) and catalase
(CAT) were elevated in the renal tissue of βHB pre-treated mice.
Regulated ROS levels are critical for optimal wound healing. Normal oxidative
phosphorylation, transient hypoxia, and increased neutrophil and macrophage respiratory bursts
release ROS during wound healing. The increased level of ROS transcends the beneficial effect
and causes additional tissue damage and healing delay. This unquenched ROS production is
prevalent in the aged population. Recently, Moor et al demonstrated age-dependent deficiencies
in the glutathione and SOD2 antioxidant pathways. In an ischemic wound model, wounds from
aged rats had lower SOD2 protein and activity, decreased ratio of reduced/oxidized glutathione,
and decreased glutathione peroxidase activity [127]. These age-exaggerated insufficiencies lead
to excessive inflammation and impaired wound healing.
One of the hallmarks of aging is mitochondrial dysfunction [128]. Electron microscope
images of aged mitochondria show disorganization and degeneration via absent cristae,
vacuolation, and swelling. Treatment with lipoic acid and carnitine stimulates repair of
mitochondrial membranes, returns functionality to the mitochondria, and demonstrates a young
phenotype [129]. Bough et al showed increased mitochondrial biogenesis in rats maintained on a

62
KD for 4-6 weeks [130, 131]. Interestingly, exogenous ketone supplementation with a ketone
ester has been shown to induce mitochondrial biogenesis [132]. Mice in this study were fed a
diet from which approximately 30% of calories were derived from the D-β-hydroxybutyrate-R-
1,3-butanediol monoester for one month. The mitochondrial content and expression of electron
transport chain proteins were significantly increased in the intrascapular brown adipose tissue as
compared to control mice, although calorie intake was matched between the two groups. These
experiments support the hypothesis that ketone supplementation could have therapeutic benefits
for an aged population.
2.5.3. Ketones and Blood Flow and Angiogenesis
Restoration of blood flow via angiogenesis is critical for healing a wound, as well as the
integration of skin substitutes. Venous, arterial and diabetic blood flow insufficiencies are major
underlying contributors to chronic wound development. Additionally, most older patients have
hypertension or other comorbidities that affect blood flow. In a study by Hasselbalch and
colleagues, 8 volunteers (4 male, 4 female, 24±4 years of age, normal weight) were given an
infusion of Na+-βHB via antecubital vein at a rate of 4-5 mg/kg/min corresponding to an infusion
of ~350mL/hr for 3-3.5 hours. Global cerebral blood flow was measured via the Kety-Schmidt
technique; ketone supplementation showed a 39% increase in cerebral blood flow [133].
As noted in Chapter 1 of this dissertation, VEGF is the most potent angiogenic factor for
its effects on multiple components of the angiogenesis cascade: endothelial cell mitogenesis,
chemotaxis, and induction of vascular permeability [134, 135]. In addition to VEGF, endothelin-
1 (ET-1) is another cytokine produced in the response to hypoxia and is a crucial promoter of

63
cell proliferation, migration and chemotaxis, and angiogenesis in wound healing [136-138]. A
study by Isales and colleagues demonstrated that supplementation of mouse brain microvascular
endothelial cells with βHB and AcAc both independently increased ET-1 and had an added affect
when applied together. βHB but not AcAc increased VEGF levels [139]. Though this area needs
further exploration, this may be one mechanism that oral ketone supplementation may augment
wound healing.
2.5.4. Ketones and Metabolism
In the 1940s Henry Lardy demonstrated that βHB and AcAc had distinct energetic
efficiencies compared to 16 major carbohydrate, lipid, and intermediary metabolites in their
ability to increase bull sperm mobility while simultaneously decreasing oxygen consumption.
[140, 141]. Nearly 50 years later, Richard Veech and colleagues confirmed that ketone bodies
increased metabolic efficiency and elucidated the molecular mechanisms in the working perfused
rat heart [124, 142]. They demonstrated that supplementation of glucose-containing perfusate (10
mM glucose) with 5 mM ketones (4 mM βHB, 1 mM AcAc) increased cardiac hydraulic work
by approximately 25% while simultaneously reducing oxygen consumption [124]. Their study
demonstrated the ketone-mediated increase in metabolic efficiency was facilitated by a reduction
of the mitochondrial NAD couple and an oxidation of the coenzyme Q couple increasing the
energy span between the sites (mentioned previously to decrease ROS production). This results
in an increase in energy released by electrons in the ETC, causing more protons to be pumped
into the inner mitochondrial space. The electrochemical gradient is enhanced, hyperpolarizing
the cell, and increasing the ΔG0 (free enthalpy) of ATP hydrolysis; thus, increasing metabolic

64
efficiency. Additionally, ketone bodies were shown to cause a 16-fold elevation in acetyl-CoA
content and increased Krebs cycle intermediates. Furthermore, thermodynamic tables for heat of
combustion, calculated with bomb calorimeter experiments, show that βHB produces more
energy than glucose per carbon molecule [143].
Decreased availability of ATP negatively impacts nearly every aspect of the healing
process [144]. Decreased nutrient and oxygen exchange during wound healing limits ATP
production to fuel wound healing processes. Additionally, the skin predominantly uses anaerobic
respiration, which decreases the potential ATP production by 90% compared to aerobic
respiration [145-147]. Chiang and colleagues developed a new intracellular ATP delivery
technique in which highly fusogenic lipid vesicles (ATP-vesicles) are used to encapsulate
magnesium-ATP (Mg-ATP). When the vesicles come into contact with the cell membrane, they
fuse together and deliver the contents into the cytosol. Using eleven controlled-pairs of adult
nude mice, they demonstrated accelerated wound healing via enhanced development of
granulation tissue, more rapid re-epithelialization, and elevated VEGF levels [31, 63].
2.5.5. Ketones Suppress Blood Glucose via Increasing Insulin Sensitivity
Failure to heal in diabetic lower limbs is associated with hyperglycemia, insulin
resistance, tissue hypoxia, chronic inflammation, oxidative stress, impairment of the immune
system, and metabolic dysfunction [148, 149]. A study by Rubinstein et al. showed that once
diabetes was a controlled, diabetic foot ulcers in 11 of 15 patients healed in 4 to 13 weeks [150].
In addition, diabetic patients with glucose under 200 mg/dl showed a decrease in surgical site
infections after foot and ankle surgery [151]. Recent studies suggest that many of the benefits of

65
the KD are due to the effects of ketone body metabolism. Interestingly, in studies on T2D
patients, improved glycemic control, improved lipid markers, and retraction of insulin and other
medications occurred before weight loss became significant, suggesting physiological effects of
ketone metabolism [3, 63]. Exogenous ketone supplements may provide therapeutic benefits for
the underlying hyperglycemia and insulin resistance as reports have demonstrated that oral
ketone administration lowers blood glucose by increasing insulin sensitivity. This could be
critical for older adults as all have demonstrable insulin resistance, even if not diabetic. Richard
Veech has suggested this hypoglycemic effect is the result of ketones activating pyruvate
dehydrogenase (PDH), which enhances insulin-mediated glucose uptake and the production of
acetyl-CoA. In addition, the body regulates ketone production via ketonuria and ketone-induced
insulin release, which shuts off hepatic ketogenesis. The insulin from this process may increase
glucose disposal which, when coupled with PDH activation, could drive down blood glucose
levels. The administration of 5 mM ketone has been shown to increase acetyl-CoA production
16-fold in the glucose-perfused isolated rat heart. Additionally, in this model, ketones and insulin
increased cardiac hydraulic efficiency to a similar degree, approximately 25-35% [124].
Male rats were fed a standard diet with 30% of calories replaced with the R-3-
hydroxybutyrate-R-1,3-butanediol monoester for 14 days. The ketone ester-supplemented diet
induced nutritional ketosis (3.5mM βHB), and both plasma glucose and insulin were decreased
by approximately 50% [132]. Glucose was decreased from 5 mM to 2.8 mM, and insulin was
decreased from 0.54 ng/mL to 0.26 ng/mL. In a similar study by the same group, mice receiving
a KE diet exhibited a 73% increase in the Quantitative Insulin-Sensitivity Check Index
(QUICKI), a surrogate marker of insulin sensitivity, compared to control, calorie-matched mice
[132]. Fasting plasma glucose levels were not altered in these mice, but fasting plasma insulin

66
levels were reduced by approximately 85% in the KE-fed mice compared to controls,
demonstrating that exogenous ketones enhance insulin sensitivity [152].
The histone deacetylase inhibitor (HDACI) activity of βHB could also be beneficial in
T2D by altering the direct regulation of HDAC-dependent glucose metabolism and by inducing
resistance to oxidative stress. HDACs regulate the expression of genes encoding many metabolic
enzymes, and HDAC3 knockout animals exhibit reduced glucose and insulin. SAHA, a class I
HDAC inhibitor, has been shown to improve insulin sensitivity and increase oxidative
metabolism and metabolic rate in a mouse model of diabetes [153]. Butyrate, a short-chain fatty
acid that is structurally similar to β-hydroxybutyrate and also acts as a HDACI lowers blood
glucose and insulin levels and improves glucose tolerance and respiratory efficiency [154]. The
vascular dysfunction in T2D is thought to be caused by oxidative stress [155]. HDAC inhibition
prevents renal damage in mouse models of diabetic nephropathy through modulation of redox
mechanisms [156]. Therefore, βHB suppression of oxidative stress through HDAC inhibition
may help restore insulin sensitivity and manage complications of diabetes.
2.6. Ischemic Wound Healing Model
As mentioned previously, a chronic wound is by definition one that entirely fails to heal.
In this respect, there is no standardized animal model that reflects the human chronic wound
environment. However, animal models have been developed that attempt to mimic these
conditions for the purpose of furthering our understanding of the complexity of chronic wounds.
The rat species, often employed due to its wide availability, size and docile nature is used for
wound healing studies as it is large enough to provide a suitable skin area for incisional and

67
excisional wounding, imaging and tissue collection [157]. Yet, it should be noted that the skin of
a rat and a human are different morphologically, as rats are loose-skinned animals. This distinct
anatomical characteristic enhances wound contraction over re-epithelialization in rat integument
[157]. Additionally, the presence of a subcutaneous panniculus carnosus muscle in rats,
contributes to healing by both contraction and collagen formation [158, 159]. These very
important morphological distinctions were considered in the optimization of the following rat
ischemic skin wound model used for this dissertation. Additionally, specific modifications were
implemented to decrease wound contraction and reduce the influence of the panniculus carnosus
muscle [160].
In the rat ischemic wound model, a dorsal bi-pedicle 10.5 x 3 cm flap is surgically
created on day 0. A silicone sheet is placed under the panniculus carnosus fascia and above the
paraspinous muscles, limiting revascularization from the underlying tissue, which increases the
duration of flap ischemia. Two 6mm punches are created within the flap, extending just above
the fascia, creating ischemic wounds. Additionally, two 6 mm punches are created lateral to the
flap, where there isn’t silicone sheet, providing control non-ischemic wounds within the same
animal.
One of the main factors in the development of a chronic wound is localized tissue
ischemia (reduced blood flow) contributing to the inability to clear inflammation [160]. In 2005,
Dr. Lisa Gould developed and validated this model as a modification of the ischemic wound
model originally described by Schwartz et al. and subsequently used in modified form by Chen
et al. [161, 162]. The wound model was modified to test the induction of angiogenesis in the
wound bed.

68
Wound healing in rats has often been the subject of debate due to their ability to heal
infected wounds and high rate of inter-animal variability [160]. One of the original goals of the
model during its development was to decrease this variation. Modifications to the width of the
flap, reducing the number of wounds with specific placement (centered on the flap with
consistent cranio-caudal location) and introduction of a silicone sheet has accomplished this
goal. Wound healing by contraction has also been reduced and healing by epithelialization, as in
humans, is the measured outcome. Adaptation of the model to a different strain of rat, ie the
F344, has also proven successful and reproduces the degree of ischemia observed using Sprague
Dawley rats.
To achieve consistency with this model while performing multiple surgeries, it was found
that it is important to create the ischemic wounds prior to elevation of the flap for silicone sheet
placement [160]. Additionally, not punching through the panniculus carnosus fascia is critical to
provide a viable wound bed to remain over the silicone. The silicone acts not only to prevent
vascular regrowth but also as a “splint” that reduces wound contraction. The application of the
adhesive and dressings to prevent infection and maintain a moist environment for wound healing
is also important. Product choice can be what is preferred or used in the researcher’s animal
facility. However, it is not uncommon for some of the animals to be able to remove their
dressings, no matter what type of adhesive/dressing combination is used.
The bi-pedicled flap should remain viable throughout a time course of healing which is
approximately 28 days, depending on rat strain and other co-morbidities present. Rarely,
abscesses can form in the flap (particularly near sutures) and seromas may form under the flap.
Fluid can be drained and antibiotics administered if necessary. However, if the flap loses
viability and becomes necrotic it is recommended that that animal no longer be used. Wound

69
excision for biochemical analysis does introduce variability due to (1) some normal tissue must
be retained for support (2) the choice of tissue homogenization and preparation for isolation of
RNA, DNA or protein and (3) inherent inter-animal variability [127, 160, 163]. One could
consider this last point a limitation to the model. It was found that reducing the size of the flap
(<2.0 cm) or flap trauma can cause necrosis, indicating that minor variations in technical or
environmental factors such as temperature or stress levels, may also lead to biochemically
detectable variation between wound samples from one rat to another [160].
In summary, this model, with a longitudinal, bi-pedicle flap ranging from 2.0-3.0 cm in
width and a strategically placed silicone sheet, is a reliable model of prolonged tissue ischemia.
Once the user is adept at using the techniques to create a consistent ischemic wound, they should
be able to adapt it to additional ages and species of rodents (mice included). The excisional
wounds can be treated topically, or systemic treatments utilized to further explore the
mechanism(s) involved in chronic wound formation, exacerbated inflammatory responses,
aberrant angiogenesis and delayed wound closure.
2.7. Use of Primary Human Dermal Fibroblasts in vitro
Though immortal cells lines are more abundant and more cost efficient to maintain, in this
dissertation, we utilized primary human dermal fibroblasts isolated from discarded skin.
Compared to immortal cell lines, primary cell lines maintain a physiology that is more closely
identified to in vivo physiology. The caveat is that they must be used at early passage, generally
passage 2-8. Additionally, due to the heterogeneity of patients, the in vitro work was repeated in
at least two different patient-derived cell lines thereby reducing individual confounding. Also,

70
fibroblasts behave differently depending on the location from which they are isolated; therefore,
skin for these studies was collected from similar anatomic locations. The complex extracellular
environment, paracrine, and autocrine interactions and range of cell types involved in wound
repair limits in vitro models to confirm what we see in in vivo models. Additionally, since there
is such a gamut of players and interactions, by focusing on just one cell type we may miss some
of the potential mechanism for our therapy.
2.8. Central Hypothesis and Project Goals
There is a dire need to discover novel and effective treatments for chronic wounds. There
are often multiple mitigating factors that prevent normal wound closure leading to minimally
effective wound therapies as few multifactorial treatments are available.
Exogenous ketone supplements are being developed as an alternative or adjuvant method
of inducing therapeutic ketosis aside from the classic ketogenic diet. It seems clear that these
novel compounds have the potential to offer benefits for both healthy and diseased individuals
alike. It is likely that most, if not all, of the conditions which are known to benefit from the KD
would receive some benefit from exogenous ketone supplementation. Importantly, ketone
supplementation provides a tool for achieving therapeutic ketosis in patients who are unable,
unwilling, or uninterested in consuming a low carbohydrate or ketogenic diet. It may also help
circumvent some of the difficulties associated with KD therapy, as it allows for rapid induction
of ketosis in a dose-dependent fashion, which can be sustained with prolonged consumption.
Simultaneously, it could provide patients with the opportunity to reap the benefits of ketosis
without the practical and social difficulties of a highly restrictive diet.

71
Therefore, we hypothesize that exogenous ketone supplementation will improve metabolic
and physiological attributes to promote accelerated healing in the impaired wound environment
associated with aging.. To date, the majority of ketone-based metabolic enhancement strategies
have focused on enhancing brain and heart metabolism; thus, this dissertation proposes a novel
therapy, which will be evaluated in a cutaneous wound-healing model [164, 165]. Although this
dissertation focuses on the underlying features of the aging phenotype, similar molecular deficits
have been described in diabetes and other disorders [166], suggesting that administration of
ketones as an alternative fuel may have broader applications. The studies completed in this
dissertation provide an important step in the potential discovery of effective and novel treatments
for chronic wounds.
2.9. Closing Remarks
In this chapter I have provided scientific rationale for the potential utility of oral ketone
supplementation against age impaired wound healing. Approaching the healing wound from a
metabolic perspective provides an innovative opportunity to stimulate beneficial physiological
and cellular mechanisms that may be lacking or attenuated in the aged population. Chapters 3-5
will consist of the results and a detailed discussion of this dissertation work. A summary of the
findings, closing remarks, and major implications of this data will be discussed in Chapter 6.

72
2.10. References to Chapter 2
1. Cotter D, Schugar R, Crawford P: Ketone body metabolism and cardiovascular disease. American journal of physiology Heart and circulatory physiology 2013, 304(8):76.
2. Peter Attia MD: Ketosis – advantaged or misunderstood state? (Part I and II). In: The Eating Academy. 2012.
3. Veech RL, Chance B, Kashiwaya Y, Lardy HA, Cahill GF, Jr.: Ketone bodies, potential therapeutic uses. IUBMB Life 2001, 51(4):241-247.
4. D'Agostino D, Pilla R, Held H, Landon C, Puchowicz M, Brunengraber H, Ari C, Arnold P, Dean J: Therapeutic ketosis with ketone ester delays central nervous system oxygen toxicity seizures in rats. American journal of physiology Regulatory, integrative and comparative physiology 2013, 304(10):R829-836.
5. Gasior M, French A, Joy M, Tang R, Hartman A, Rogawski M: The anticonvulsant activity of acetone, the major ketone body in the ketogenic diet, is not dependent on its metabolites acetol, 1,2-propanediol, methylglyoxal, or pyruvic acid. Epilepsia 2007, 48(4):793-800.
6. Likhodii S, Nylen K, Burnham W: Acetone as an anticonvulsant. Epilepsia 2008, 49 Suppl 8:83-86.
7. Seymour K, Bluml S, Sutherling J, Sutherling W, Ross B: Identification of cerebral acetone by 1H-MRS in patients with epilepsy controlled by ketogenic diet. Magma (New York, NY) 1999, 8(1):33-42.
8. Kwiterovich P, Vining E, Pyzik P, Skolasky R, Freeman J: Effect of a high-fat ketogenic diet on plasma levels of lipids, lipoproteins, and apolipoproteins in children. JAMA : the journal of the American Medical Association 2003, 290(7):912-920.
9. Mancinelli K: The Ketogenic Diet: A Scientifically Proven Approach to Fast, Healthy Weight Loss: Ulysses Press; 2015.
10. Sirven J, Whedon B, Caplan D, Liporace J, Glosser D, O'Dwyer J, Sperling M: The ketogenic diet for intractable epilepsy in adults: preliminary results. Epilepsia 1999, 40(12):1721-1726.
11. Wilder R: The effect of ketonemia on the course of epilepsy. Mayo Clin Bulletin 1921, 2:307-308.
12. Thiele E: Assessing the efficacy of antiepileptic treatments: the ketogenic diet. Epilepsia 2003, 44 Suppl 7:26-29.

73
13. Paoli A, Rubini A, Volek JS, Grimaldi KA: Beyond weight loss: a review of the therapeutic uses of very-low-carbohydrate (ketogenic) diets. European journal of clinical nutrition 2013, 67(8):789-796.
14. Foster GD, Wyatt HR, Hill JO, McGuckin BG, C B, Mohemmed BS, Szapary PO, Rader DJ, Edman JS, Klein S: A randomized trial of a low-carbohydrate diet for obesity. The New England journal of medicine 2003, 348:2082-2090.
15. Westman EC, Feinman RD, Mavropoulos JC, Vernon MC, Volek JS, Wortman JA, Yancy WS, Phinney SD: Low-carbohydrate nutrition and metabolism. The American journal of clinical nutrition 2007(86):276-284.
16. Westman EC, Yancy WS, Edman JS, Tomlin KF, Perkins CE: Effect of 6-month adherence to a very low carbohydrate diet program. The American journal of medicine 2002, 113(1):30-36.
17. Forsythe C, Phinney S, Fernandez M, Quann E, Wood R, Bibus D, Kraemer W, Feinman R, Volek J: Comparison of low fat and low carbohydrate diets on circulating fatty acid composition and markers of inflammation. Lipids 2008, 43(1):65-77.
18. Boden G, Sargrad K, Homko C, Mozzoli M, Stein T: Effect of a low-carbohydrate diet on appetite, blood glucose levels, and insulin resistance in obese patients with type 2 diabetes. Annals of internal medicine 2005, 142(6):403-411.
19. Gumbiner B, Wendel J, McDermott M: Effects of diet composition and ketosis on glycemia during very-low-energy-diet therapy in obese patients with non-insulin-dependent diabetes mellitus. The American journal of clinical nutrition 1996, 63(1):110-115.
20. Nielsen J, Joensson E: Low-carbohydrate diet in type 2 diabetes: stable improvement of bodyweight and glycemic control during 44 months follow-up. Nutrition & metabolism 2008, 5:14.
21. Yancy W, Foy M, Chalecki A, Vernon M, Westman E: A low-carbohydrate, ketogenic diet to treat type 2 diabetes. Nutrition & metabolism 2005, 2:34.
22. Dashti HM, Al-Zaid NS, Mathew TC, Al-Mousawi M, Talib H, Asfar SK, Behbahani AI: Long term effects of ketogenic diet in obese subjects with high cholesterol level. Molecular and cellular biochemistry 2006, 286(1-2):1-9.
23. Maalouf M, Rho JM, Mattson MP: The neuroprotective properties of calorie restriction, the ketogenic diet, and ketone bodies. Brain Res Rev 2009, 59(2):293-315.
24. Mavropoulos JC, Yancy WS, Hepburn J, Westman EC: The effects of a low-carbohydrate, ketogenic diet on the polycystic ovary syndrome: a pilot study. Nutrition & metabolism 2004, 2:35.

74
25. Seyfried T, Flores R, Poff A, D'Agostino D: Cancer as a metabolic disease: implications for novel therapeutics. Carcinogenesis 2014, 35:515-527.
26. Poff AM, Ari C, Arnold P, Seyfried TN, D'Agostino DP: Ketone supplementation decreases tumor cell viability and prolongs survival of mice with metastatic cancer. International journal of cancer Journal international du cancer 2014, 135:1711-1720.
27. Poff A, Ari C, Seyfried T, D'Agostino D: The ketogenic diet and hyperbaric oxygen therapy prolong survival in mice with systemic metastatic cancer. PloS one 2013, 8(6):e65522.
28. Seyfried T, Shelton L: Cancer as a metabolic disease. Nutrition & metabolism, 7:7.
29. Fine E, Segal-Isaacson C, Feinman R, Herszkopf S, Romano M, Tomuta N, Bontempo A, Negassa A, Sparano J: Targeting insulin inhibition as a metabolic therapy in advanced cancer: a pilot safety and feasibility dietary trial in 10 patients. Nutrition (Burbank, Los Angeles County, Calif) 2012, 28(10):1028-1035.
30. Zhao Z, Lange D, Voustianiouk A, MacGrogan D, Ho L, Suh J, Humala N, Thiyagarajan M, Wang J, Pasinetti G: A ketogenic diet as a potential novel therapeutic intervention in amyotrophic lateral sclerosis. BMC neuroscience 2006, 7:29.
31. White H, Venkatesh B: Clinical review: ketones and brain injury. Critical care (London, England) 2011, 15(2):219.
32. Prins M: Cerebral metabolic adaptation and ketone metabolism after brain injury. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 2008, 28(1):1-16.
33. Henderson S, Vogel J, Barr L, Garvin F, Jones J, Costantini L: Study of the ketogenic agent AC-1202 in mild to moderate Alzheimer's disease: a randomized, double-blind, placebo-controlled, multicenter trial. Nutrition & metabolism 2009, 6:31.
34. Brownlow M, Benner L, D'Agostino D, Gordon M, Morgan D: Ketogenic diet improves motor performance but not cognition in two mouse models of Alzheimer's pathology. PloS one 2013, 8(9):e75713.
35. Kossoff EH, Hartman AL: Ketogenic diets: new advances for metabolism-based therapies. Current Opinion in Neurology 2012, 25:173-178.
36. Halevy A, Peleg-Weiss L, Cohen R, Shuper A: An update on the ketogenic diet, 2012. Rambam Maimonides medical journal 2012, 3(1):e0005.
37. Amari A, Grace N, Fisher W: Achieving and maintaining compliance with the ketogenic diet. Journal of applied behavior analysis 1995, 28(3):341-342.

75
38. Zhang Y, Kuang Y, LaManna J, Puchowicz M: Contribution of brain glucose and ketone bodies to oxidative metabolism. Advances in experimental medicine and biology 2013, 765:365-370.
39. McPherson P, McEneny J: The biochemistry of ketogenesis and its role in weight management, neurological disease and oxidative stress. Journal of physiology and biochemistry 2012, 68(1):141-151.
40. Jimmy Moore and Eric C. Westman M: Cholesterol Clarity: What the HDL is wrong with my numbers. Las Vegas,NV: Victory Belt Publishing Inc.; 2013.
41. Dekaban A: Plasma lipids in epileptic children treated with the high fat diet. Archives of neurology 1966, 15(2):177-184.
42. Chesney D, Brouhard B, Wyllie E, Powaski K: Biochemical abnormalities of the ketogenic diet in children. Clinical pediatrics 1999, 38(2):107-109.
43. Schwartz R, Boyes S, Aynsley-Green A: Metabolic effects of three ketogenic diets in the treatment of severe epilepsy. Developmental medicine and child neurology 1989, 31(2):152-160.
44. Katyal N, Koehler A, McGhee B, Foley C, Crumrine P: The ketogenic diet in refractory epilepsy: the experience of Children's Hospital of Pittsburgh. Clinical pediatrics 2000, 39(3):153-159.
45. Ellenbroek J, van Dijck L, Töns H, Rabelink T, Carlotti F, Ballieux B, de Koning E: Long-term ketogenic diet causes glucose intolerance and reduced beta and alpha cell mass but no weight loss in mice. American journal of physiology Endocrinology and metabolism 2014, 306(5):E552-E558.
46. Bergqvist A: Long-term monitoring of the ketogenic diet: Do's and Don'ts. Epilepsy Research 2012, 100(3):261-266.
47. Groesbeck D, Bluml R, Kossoff E: Long-term use of the ketogenic diet in the treatment of epilepsy. Developmental medicine and child neurology 2006, 48(12):978-981.
48. Patel A, Pyzik P, Turner Z, Rubenstein J, Kossoff E: Long-term outcomes of children treated with the ketogenic diet in the past. Epilepsia 2010, 51(7):1277-1282.
49. Brehm BJ, Seeley RJ, Daniels SR, D'Alessio DA: A randomized trial comparing a very low carbohydrate diet and a calorie-restricted low fat diet on body weight and cardiovascular risk factors in healthy women. The Journal of clinical endocrinology and metabolism 2003, 88(4):1617-1623.
50. Shai I, Schwarzfuchs D, Henkin Y, Shahar DR, Witkow S, Greenberg I, Golan R, Fraser D, Bolotin A, Vardi H et al: Weight loss with a low-carbohydrate, Mediterranean, or low-fat diet. The New England journal of medicine 2008, 359(3):229-241.

76
51. Volek JS, Phinney SD, Forsythe CE, Quann EE, Wood RJ, Puglisi MJ, Kraemer WJ, Bibus DM, Fernandez ML, Feinman RD: Carbohydrate restriction has a more favorable impact on the metabolic syndrome than a low fat diet. Lipids 2009, 44(4):297-309.
52. Feinman RD, Volek JS: Low carbohydrate diets improve atherogenic dyslipidemia even in the absence of weight loss. Nutrition & metabolism 2006, 3:24.
53. Sharman MJ, Gomez AL, Kraemer WJ, Volek JS: Very low-carbohydrate and low-fat diets affect fasting lipids and postprandial lipemia differently in overweight men. J Nutr 2004, 134(4):880-885.
54. Sharman MJ, Kraemer WJ, Love DM, Avery NG, Gomez AL, Scheett TP, Volek JS: A ketogenic diet favorably affects serum biomarkers for cardiovascular disease in normal-weight men. J Nutr 2002, 132(7):1879-1885.
55. Westman EC, Mavropoulos J, Yancy WS, Volek JS: A review of low-carbohydrate ketogenic diets. Current atherosclerosis reports 2003, 5(6):476-483.
56. Wood RJ, Volek JS, Davis SR, Dell'Ova C, Fernandez ML: Effects of a carbohydrate-restricted diet on emerging plasma markers for cardiovascular disease. Nutrition & metabolism 2006, 3:19.
57. Volek JS, Sharman MJ, Forsythe CE: Modification of lipoproteins by very low-carbohydrate diets. J Nutr 2005, 135(6):1339-1342.
58. Volek JS, Sharman MJ, Gomez AL, DiPasquale C, Roti M, Pumerantz A, Kraemer WJ: Comparison of a very low-carbohydrate and low-fat diet on fasting lipids, LDL subclasses, insulin resistance, and postprandial lipemic responses in overweight women. J Am Coll Nutr 2004, 23(2):177-184.
59. Volek JS, Sharman MJ: Cardiovascular and hormonal aspects of very-low-carbohydrate ketogenic diets. Obesity research 2004, 12 Suppl 2:115s-123s.
60. Volek JS, Sharman MJ, Gomez AL, Scheett TP, Kraemer WJ: An isoenergetic very low carbohydrate diet improves serum HDL cholesterol and triacylglycerol concentrations, the total cholesterol to HDL cholesterol ratio and postprandial pipemic responses compared with a low fat diet in normal weight, normolipidemic women. J Nutr 2003, 133(9):2756-2761.
61. Volek JS, Westman EC: Very-low-carbohydrate weight-loss diets revisited. Cleveland Clinic journal of medicine 2002, 69(11):849, 853, 856-848 passim.
62. Schoeler NE, Wood S, Aldridge V, Sander JW, Cross JH, Sisodiya SM: Ketogenic dietary therapies for adults with epilepsy: feasibility and classification of response. Epilepsy & behavior : E&B 2014, 37:77-81.

77
63. Veech R: The therapeutic implications of ketone bodies: the effects of ketone bodies in pathological conditions: ketosis, ketogenic diet, redox states, insulin resistance, and mitochondrial metabolism. Prostaglandins, leukotrienes, and essential fatty acids 2004, 70(3):309-319.
64. Falbe J, Bahrmann H, Lipps W: Alcohols, aliphatic. Ullmann's encyclopedia … 1985.
65. Budavari SE: The merck index: an encyclopedia of chemical, drugs, and biologicals. The merck index: an encyclopedia of chemical, drugs, and biologicals 1989.
66. Tate RL, Mehlman MA, Tobin RB: Metabolic fate of 1,3-butanediol in the rat: conversion to -hydroxybutyrate. The Journal of nutrition 1971, 101(12):1719-1726.
67. Drackley JK, Richard MJ, Young JW: In vitro production of beta-hydroxybutyrate from 1,3-butanediol by bovine liver, rumen mucosa, and kidney. Journal of dairy science 1990, 73(3):679-682.
68. Tobin RB, Mehlman MA, Parker M: Effect of 1,3-butanediol and propionic acid on blood ketones, lipids and metal ions in rats. The Journal of nutrition 1972, 102(8):1001-1008.
69. Desrochers S, David F, Garneau M, Jetté M, Brunengraber H: Metabolism of R- and S-1,3-butanediol in perfused livers from meal-fed and starved rats. The Biochemical journal 1992, 285 ( Pt 2):647-653.
70. Scala RA, Paynter OE: Chronic oral toxicity of 1, 3-butanediol. Toxicology and applied pharmacology 1967.
71. Dymsza HA: Nutritional application and implication of 1,3-butanediol. Federation proceedings 1975, 34(12):2167-2170.
72. Hess FG, Cox GE, Bailey DE, Parent RA, Becci PJ: Reproduction and teratology study of 1,3-butanediol in rats. Journal of applied toxicology : JAT 1981, 1(4):202-209.
73. Opitz K: Über die glykogenbildende Wirkung von Glykolen. Naunyn-Schmiedeberg's Archives of Pharmacology 1958.
74. Marie C, Bralet AM, Bralet J: Protective action of 1,3-butanediol in cerebral ischemia. A neurologic, histologic, and metabolic study. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 1987, 7(6):794-800.
75. Kirsch JR, d'Alecy LG, Mongroo PB: Butanediol induced ketosis increases tolerance to hypoxia in the mouse. Stroke 1980.
76. Karen M, Welma S: Effects of Medium-Chain Triglycerides on Weight Loss and Body Composition: A Meta-Analysis of Randomized Controlled Trials. Journal of the Academy of Nutrition and Dietetics 2015, 115(2).

78
77. Koji N, Teruyoshi Y: Medium-chain fatty acids: Functional lipids for the prevention and treatment of the metabolic syndrome. Pharmacological Research 2010, 61(3).
78. Stagey J B, Dondeena B, R.Armour F, Bruce R B: The New Dietary Fats in Health and Disease. Journal of the American Dietetic Association 1997, 97(3):280-286.
79. Bach AC, Babayan VK: Medium-chain triglycerides: an update. The American journal of clinical nutrition 1982, 36(5):950-962.
80. Babayan VK: Medium chain triglycerides and structured lipids. Lipids 1987.
81. Hashim SA, Tantibhedyangkul P: Medium chain triglyceride in early life: effects on growth of adipose tissue. Lipids 1987.
82. Huttenlocher PR, Wilbourn AJ, Signore JM: Medium-chain triglycerides as a therapy for intractable childhood epilepsy. Neurology 1971, 21(11):1097-1103.
83. Huttenlocher PR: Ketonemia and seizures: metabolic and anticonvulsant effects of two ketogenic diets in childhood epilepsy. Pediatric research 1976, 10(5):536-540.
84. Mak SC, Chi CS, Wan CJ: Clinical experience of ketogenic diet on children with refractory epilepsy. Acta paediatrica Taiwanica = Taiwan er ke yi xue hui za zhi 1999, 40(2):97-100.
85. Sills MA, Forsythe WI, Haidukewych D, MacDonald A, Robinson M: The medium chain triglyceride diet and intractable epilepsy. Archives of disease in childhood 1986, 61(12):1168-1172.
86. Trauner DA: Medium-chain triglyceride (MCT) diet in intractable seizure disorders. Neurology 1985, 35(2):237-238.
87. Ruby HS, Jane E, Bower BD, Aynsley-Green A: KETOGENIC DIETS IN THE TREATMENT OF EPILEPSY: SHORT-TERM CLINICAL EFFECTS. Developmental Medicine & Child Neurology 1989, 31(2):145-151.
88. Elizabeth GN, Hannah C, Ruby HS, Margaret SL, Nicole E, Georgiana F, Andrea W, Cross JH: A randomized trial of classical and medium-chain triglyceride ketogenic diets in the treatment of childhood epilepsy. Epilepsia 2009, 50(5):1109-1117.
89. Bough KJ, Rho JM: Anticonvulsant mechanisms of the ketogenic diet. Epilepsia 2007, 48(1):43-58.
90. Hove JLK, Grünewald S, Jaeken J, Demaerel P: D, L-3-hydroxybutyrate treatment of multiple acyl-CoA dehydrogenase deficiency (MADD). The Lancet 2003.
91. Veech RL: Ketone ester effects on metabolism and transcription. Journal of lipid research 2014, 55(10):2004-2006.

79
92. D'Agostino D, Arnold P, Kesl S: Compositions and methods for producing elevated and sustained ketosis. In: Compositions and methods for producing elevated and sustained ketosis. Google Patents; 2014.
93. Birkhahn RH, Border JR: Intravenous feeding of the rat with short chain fatty acid esters. II. Monoacetoacetin. The American journal of clinical nutrition 1978, 31(3):436-441.
94. Birkhahn RH, McMenamy RH, Border JR: Intravenous feeding of the rat with short chain fatty acid esters. I. Glycerol monobutyrate. The American journal of clinical nutrition 1977, 30(12):2078-2082.
95. Birkhahn RH, McMenamy RH, Border JR: Monoglyceryl acetoacetate: a ketone body-carbohydrate substrate for parenteral feeding of the rat. The Journal of nutrition 1979, 109(7):1168-1174.
96. Desrochers S, Quinze K, Dugas H, Dubreuil P, Bomont C, David F, Agarwal KC, Kumar A, Soloviev MV, Powers L et al: R,S-1,3-Butanediol Acetoacetate Esters, Potential Alternates to Lipid Emulsions for Total Parenteral-Nutrition. Journal of Nutritional Biochemistry 1995, 6(2):111-118.
97. Brunengraber H: Potential of ketone body esters for parenteral and oral nutrition. Nutrition (Burbank, Los Angeles County, Calif) 1997, 13(3):233-235.
98. Sylvain D, Khadijah Q, Hermann D, Pascal D, Catherine B, France D, Kamlesh CA, Alok K, Maxim VS, Lisa P et al: R,S-1,3-butanediol acetoacetate esters, potential alternates to lipid emulsions for total parenteral nutrition. The Journal of Nutritional Biochemistry 1995, 6(2).
99. Ciraolo ST, Previs SF, Fernandez CA, Agarwal KC, David F, Koshy J, Lucas D, Tammaro A, Stevens MP, Tserng KY: Model of extreme hypoglycemia in dogs made ketotic with (R,S)-1,3-butanediol acetoacetate esters. The American journal of physiology 1995, 269(1 Pt 1):75.
100. Puchowicz MA, Smith CL, Bomont C, Koshy J, David F, Brunengraber H: Dog model of therapeutic ketosis induced by oral administration of R,S-1,3-butanediol diacetoacetate. The Journal of nutritional biochemistry 2000, 11(5):281-287.
101. Desrochers S, Dubreuil P, Brunet J, Jetté M, David F, Landau BR, Brunengraber H: Metabolism of (R,S)-1,3-butanediol acetoacetate esters, potential parenteral and enteral nutrients in conscious pigs. The American journal of physiology 1995, 268(4 Pt 1):7.
102. Srivastava S, Kashiwaya Y, King MT, Baxa U, Tam J, Niu G, Chen X, Clarke K, Veech RL: Mitochondrial biogenesis and increased uncoupling protein 1 in brown adipose tissue of mice fed a ketone ester diet. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 2012, 26(6):2351-2362.

80
103. Clarke K, Tchabanenko K, Pawlosky R, Carter E, Knight N, Murray A, Cochlin L, King M, Wong A, Roberts A et al: Oral 28-day and developmental toxicity studies of (R)-3-hydroxybutyl (R)-3-hydroxybutyrate. Regulatory toxicology and pharmacology : RTP 2012, 63(2):196-208.
104. Clarke K, Tchabanenko K, Pawlosky R, Carter E, Todd King M, Musa-Veloso K, Ho M, Roberts A, Robertson J, Vanitallie T et al: Kinetics, safety and tolerability of (R)-3-hydroxybutyl (R)-3-hydroxybutyrate in healthy adult subjects. Regulatory toxicology and pharmacology : RTP 2012, 63(3):401-408.
105. Williams J, Barbul A: Nutrition and wound healing. The Surgical clinics of North America 2003, 83(3):571-596.
106. Wallace E: Feeding the wound: nutrition and wound care. British journal of nursing (Mark Allen Publishing) 1994, 3(13):662-667.
107. Demling R: Nutrition, anabolism, and the wound healing process: an overview. Eplasty 2009, 9.
108. Allison SP, Go VW: Metabolic issues of clinical nutrition, vol. 9: Karger Medical and Scientific Publishers; 2004.
109. Nair KS, Welle SL, Halliday D, Campbell RG: Effect of beta-hydroxybutyrate on whole-body leucine kinetics and fractional mixed skeletal muscle protein synthesis in humans. The Journal of clinical investigation 1988, 82(1):198-205.
110. Newman JC, Verdin E: Ketone bodies as signaling metabolites. Trends in endocrinology and metabolism: TEM 2014, 25(1):42-52.
111. Newman JC, Verdin E: β-hydroxybutyrate: Much more than a metabolite. Diabetes Research and Clinical Practice 2014, 106(2):173-181.
112. Shimazu T, Hirschey MD, Newman J, He W, Shirakawa K, Le Moan N, Grueter CA, Lim H, Saunders LR, Stevens RD et al: Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science (New York, NY) 2013, 339(6116):211-214.
113. Youm Y-H, Nguyen KY, Grant RW, Goldberg EL, Bodogai M, Kim D, D'Agostino D, Planavsky N, Lupfer C, Kanneganti TD et al: The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome–mediated inflammatory disease. Nature Medicine 2015, 21(3):263-269.
114. Cassandra EF, Stephen DP, Maria Luz F, Erin EQ, Richard JW, Doug MB, William JK, Richard DF, Jeff SV: Comparison of Low Fat and Low Carbohydrate Diets on Circulating Fatty Acid Composition and Markers of Inflammation. Lipids 2007, 43.

81
115. Sharman M, Volek J: Weight loss leads to reductions in inflammatory biomarkers after a very-low-carbohydrate diet and a low-fat diet in overweight men. Clinical science 2004, 107(4):365-369.
116. Torres-Gonzalez M, Volek JS, Leite JO, Fraser H, Luz Fernandez M: Carbohydrate restriction reduces lipids and inflammation and prevents atherosclerosis in Guinea pigs. J Atheroscler Thromb 2008, 15(5):235-243.
117. Paoli A, Moro T, Bosco G, Bianco A, Grimaldi KA, Camporesi E, Mangar D: Effects of n-3 polyunsaturated fatty acids (ω-3) supplementation on some cardiovascular risk factors with a ketogenic Mediterranean diet. Marine drugs 2015, 13(2):996-1009.
118. Ruskin D, Kawamura M, Masino S: Reduced pain and inflammation in juvenile and adult rats fed a ketogenic diet. PloS one 2009, 4(12):e8349.
119. Youm YH, Nguyen KY, Grant RW, Goldberg EL, Bodogai M, Kim D, D'Agostino D, Planavsky N, Lupfer C, Kanneganti TD et al: The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat Med 2015.
120. Mirza RE, Fang MM, Weinheimer-Haus EM, Ennis WJ, Koh TJ: Sustained inflammasome activity in macrophages impairs wound healing in type 2 diabetic humans and mice. Diabetes 2014, 63(3):1103-1114.
121. Bitto A, Altavilla D, Pizzino G, Irrera N, Pallio G, Colonna MR, Squadrito F: Inhibition of inflammasome activation improves the impaired pattern of healing in genetically diabetic mice. British journal of pharmacology 2014, 171(9):2300-2307.
122. Maalouf M, Sullivan PG, Davis L, Kim DY, Rho JM: Ketones inhibit mitochondrial production of reactive oxygen species production following glutamate excitotoxicity by increasing NADH oxidation. Neuroscience 2007, 145(1):256-264.
123. Kim do Y, Davis LM, Sullivan PG, Maalouf M, Simeone TA, van Brederode J, Rho JM: Ketone bodies are protective against oxidative stress in neocortical neurons. J Neurochem 2007, 101(5):1316-1326.
124. Kashiwaya Y, Sato K, Tsuchiya N, Thomas S, Fell DA, Veech RL, Passonneau JV: Control of glucose utilization in working perfused rat heart. The Journal of biological chemistry 1994, 269(41):25502-25514.
125. Maalouf M, Sullivan P, Davis L, Kim D, Rho J: Ketones inhibit mitochondrial production of reactive oxygen species production following glutamate excitotoxicity by increasing NADH oxidation. Neuroscience 2007, 145(1):256-264.
126. Shimazu T, Hirschey MD, Newman J, He W, Shirakawa K, Le Moan N, Grueter CA, Lim H, Saunders LR, Stevens RD et al: Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 2013, 339(6116):211-214.

82
127. Moor AN, Tummel E, Prather JL, Jung M, Lopez JJ, Connors S, Gould LJ: Consequences of age on ischemic wound healing in rats: altered antioxidant activity and delayed wound closure. Age (Dordrecht, Netherlands) 2014, 36(2):733-748.
128. Miquel J: Can Antioxidant Diet Supplementation Protect against Age‐related Mitochondrial Damage? Annals of the New York Academy of Sciences 2002.
129. Savitha S, Panneerselvam C: Mitochondrial membrane damage during aging process in rat heart: potential efficacy of L-carnitine and DL alpha lipoic acid. Mechanisms of ageing and development 2006, 127(4):349-355.
130. Bough K, Rho J: Anticonvulsant mechanisms of the ketogenic diet. Epilepsia 2007, 48(1):43-58.
131. Bough K, Wetherington J, Hassel B, Pare J, Gawryluk J, Greene J, Shaw R, Smith Y, Geiger J, Dingledine R: Mitochondrial biogenesis in the anticonvulsant mechanism of the ketogenic diet. Annals of neurology 2006, 60(2):223-235.
132. Srivastava S, Kashiwaya Y, King M, Baxa U, Tam J, Niu G, Chen X, Clarke K, Veech R: Mitochondrial biogenesis and increased uncoupling protein 1 in brown adipose tissue of mice fed a ketone ester diet. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 2012, 26(6):2351-2362.
133. Hasselbalch SG, Madsen PL, Hageman LP, Olsen KS, Justesen N, Holm S, Paulson OB: Changes in cerebral blood flow and carbohydrate metabolism during acute hyperketonemia. The American journal of physiology 1996, 270(5 Pt 1):51.
134. Bao P, Kodra A, Tomic-Canic M, Golinko M, Ehrlich H, Brem H: The role of vascular endothelial growth factor in wound healing. The Journal of surgical research 2009, 153(2):347-358.
135. Mahdavian Delavary B, van der Veer W, van Egmond M, Niessen F, Beelen R: Macrophages in skin injury and repair. Immunobiology 2011, 216(7):753-762.
136. Freinkel RK, Woodley DT: The biology of the skin. The biology of the skin 2001.
137. Rodriguez PG, Felix FN, Woodley DT, Shim EK: The role of oxygen in wound healing: a review of the literature. Dermatologic surgery : official publication for American Society for Dermatologic Surgery [et al] 2008, 34(9):1159-1169.
138. Guo S, Dipietro LA: Factors affecting wound healing. Journal of dental research 2010, 89(3):219-229.
139. Isales CM, Min L, Hoffman WH: Acetoacetate and beta-hydroxybutyrate differentially regulate endothelin-1 and vascular endothelial growth factor in mouse brain microvascular endothelial cells. Journal of diabetes and its complications 1999, 13(2):91-97.

83
140. Lardy HA, Phillips, P. H.: Studies of fat and carbohydrate oxidation in mammalian spermatozoa. Archives of Biochemistry 1945, 6:53-61.
141. Lardy H, Hansen RG, and Phillips PH: The metabolism of bovine epididymal spermatozoa. Archive of Biochemistry 1945(6):41-51.
142. Sato K, Kashiwaya Y, Keon C, Tsuchiya N, King M, Radda G, Chance B, Clarke K, Veech R: Insulin, ketone bodies, and mitochondrial energy transduction. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 1995, 9(8):651-658.
143. Cahill GF, Jr., Veech RL: Ketoacids? Good medicine? Trans Am Clin Climatol Assoc 2003, 114:149-161; discussion 162-143.
144. Wang DJ, Huang NN, Heppel LA: Extracellular ATP shows synergistic enhancement of DNA synthesis when combined with agents that are active in wound healing or as neurotransmitters. Biochemical and biophysical research communications 1990, 166(1):251-258.
145. Im MJC, Hoopes JE: Energy metabolism in healing skin wounds. Journal of Surgical Research 1970.
146. Almskog BA, Haljamäe H, Hasselgren PO: Local metabolic changes in skeletal muscle following high-energy missile injury. The Journal of … 1982.
147. Kristensen SR: A critical appraisal of the association between energy charge and cell damage. Biochimica et biophysica acta 1989, 1012(3):272-278.
148. Cychosz CC, Phisitkul P, Belatti DA, Wukich DK: Current Concepts Review: Preventive and Therapeutic Strategies for Diabetic Foot Ulcers. Foot & ankle international 2015.
149. Gau B-RR, Chen H-YY, Hung S-YY, Yang H-MM, Yeh J-TT, Huang C-HH, Sun J-HH, Huang Y-YY: The impact of nutritional status on treatment outcomes of patients with limb-threatening diabetic foot ulcers. Journal of diabetes and its complications 2015.
150. Rubinstein A, Pierce CE: Rapid healing of diabetic foot ulcers with meticulous blood glucose control. Acta diabetologica latina 1988, 25(1):25-32.
151. Sadoskas D, Suder NC, Wukich DK: Perioperative Glycemic Control and the Effect on Surgical Site Infections in Diabetic Patients Undergoing Foot and Ankle Surgery. Foot & ankle specialist 2015.
152. Kashiwaya Y, King MT, Veech RL: Substrate signaling by insulin: a ketone bodies ratio mimics insulin action in heart. The American journal of cardiology 1997, 80(3A):50A-64A.

84
153. Galmozzi A, Mitro N, Ferrari A, Gers E, Gilardi F, Godio C, Cermenati G, Gualerzi A, Donetti E, Rotili D et al: Inhibition of class I histone deacetylases unveils a mitochondrial signature and enhances oxidative metabolism in skeletal muscle and adipose tissue. Diabetes 2013, 62(3):732-742.
154. Gao Z, Yin J, Zhang J, Ward RE, Martin RJ, Lefevre M, Cefalu WT, Ye J: Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes 2009, 58(7):1509-1517.
155. Giacco F, Brownlee M: Oxidative stress and diabetic complications. Circ Res 2010, 107(9):1058-1070.
156. Advani A, Huang Q, Thai K, Advani SL, White KE, Kelly DJ, Yuen DA, Connelly KA, Marsden PA, Gilbert RE: Long-term administration of the histone deacetylase inhibitor vorinostat attenuates renal injury in experimental diabetes through an endothelial nitric oxide synthase-dependent mechanism. Am J Pathol 2011, 178(5):2205-2214.
157. Dorsett-Martin WA: Rat models of skin wound healing: a review. Wound repair and regeneration : official publication of the Wound Healing Society [and] the European Tissue Repair Society 2003, 12(6):591-599.
158. Davidson JM: Animal models for wound repair. Arch Dermatol Res 1998, 290 Suppl:S1-11.
159. Finn G, Magnus SA, Tonny K: Models for use in wound healing research: A survey focusing on in vitro and in vivo adult soft tissue. Wound Repair and Regeneration 2000, 8.
160. Gould LJ, Leong M, Sonstein J, Wilson S: Optimization and validation of an ischemic wound model. Wound repair and regeneration : official publication of the Wound Healing Society [and] the European Tissue Repair Society 2004, 13(6):576-582.
161. Schwarz DA, Lindblad WJ, Rees RR: Altered collagen metabolism and delayed healing in a novel model of ischemic wounds. Wound Repair Regen 1995, 3(2):204-212.
162. Chin C, Gregory SS, Melissa B, Paul DE, Steve T, Bruce AM: Molecular and mechanistic validation of delayed healing rat wounds as a model for human chronic wounds. Wound Repair and Regeneration 1999, 7.
163. Zhang Q, Gould LJ: Hyperbaric oxygen reduces matrix metalloproteinases in ischemic wounds through a redox-dependent mechanism. The Journal of investigative dermatology 2013, 134(1):237-246.

85
164. D'Agostino D, Pilla R, Held H, Landon C, Puchowicz M, Brunengraber H, Ari C, Arnold P, Dean J: Therapeutic ketosis with ketone ester delays central nervous system oxygen toxicity seizures in rats. American journal of physiology Regulatory, integrative and comparative physiology, 304(10):36.
165. Kashiwaya Y, Sato K, Tsuchiya N, Thomas S, Fell D, Veech R, Passonneau J: Control of glucose utilization in working perfused rat heart. The Journal of biological chemistry 1994, 269(41):25502-25514.
166. Wilson P, Anderson K, Kannel W: Epidemiology of diabetes mellitus in the elderly. The Framingham Study. The American journal of medicine 1986, 80(5A):3-9.

86
CHAPTER 3: ESTABLISHING THERAPEUTIC KETOSIS (METABOLIC THERAPY)
WITH EXOGENOUS KETONE SUPPLEMENTATION
3.1. Chapter Synopsis
In this chapter, we present data demonstrating the effects of five oral ketone supplements
on blood ketones, glucose, triglyceride, and lipoprotein levels in Sprague-Dawley rats. Though
each ketogenic precursor had a distinctive effect on each of the parameters, overall oral ketone
supplementation rapidly elevated blood ketone levels and suppressed blood glucose levels
without significantly affecting heart health biomarkers. Additionally we sent serum and
hippocampal samples from KE (5 g/kg) and BMS+MCT (10 g/kg) supplemented rats to
Metabolon Inc. to determine the effects that oral ketone supplementation would have on the
metabolome. Ketone supplements increased Kreb’s cycle intermediates, antioxidants, and
adenosine, which supports our hypothesis that oral ketone supplementation, will enhance wound
healing.
Portions of this chapter have previously been published in “Kesl SL, Poff AM, Ward NP,
Fiorelli TN, Ari C, Van Putten AJ, Sherwood JW, Arnold P, D’Agostino DP. Effects of
Exogenous Ketone Supplementation on Blood Ketone, Glucose, Triglyceride, and Lipoprotein
Levels in Sprague-Dawley Rats. Nutrition and Metabolism (2016)”. A copy of this article may
be found in Appendix C. See Appendix B for copyright permissions. Materials and methods

87
presented in this chapter can be found in Appendix A.
3.2. Effects of Exogenous Ketone Supplementation on Blood Ketones, Glucose,
Triglyceride, and Lipoprotein Levels in Sprague-Dawley Rats
Nutritional ketosis induced with the KD has proven effective for the metabolic
management of seizures and potentially other disorders [1-26]. We hypothesized that oral
administration of ketone supplements could safely produce sustained nutritional ketosis (>0.5
mM) without carbohydrate restriction. Thus, we tested the effects of 28-day administration of
five ketone supplements on blood glucose, ketones, and lipids in male Sprague-Dawley rats. The
supplements included: 1,3-butanediol (BD), a sodium/potassium β-hydroxybutyrate (βHB)
mineral salt (BMS), medium chain triglyceride oil (MCT), BMS+MCT 1:1 mixture, and 1,3
butanediol acetoacetate diester (KE). Rats received a daily 5-10g/kg dose of their respective
ketone supplement via intragastric gavage. Weekly whole blood samples were taken for analysis
of glucose and βHB at baseline and 0.5, 1, 4, 8, and 12 hrs post-gavage, or until βHB returned to
baseline. In this section, we present evidence that chronic administration of ketone supplements
can induce a state of nutritional ketosis without the need for dietary carbohydrate restriction with
little or no effect on lipid biomarkers. The notion that we can produce the therapeutic effects of
the KD with exogenous ketone supplementation is supported by our previous study which
demonstrated that acutely administered KE supplementation delays central nervous system
(CNS) oxygen toxicity seizures without the need for dietary restriction [27]. We propose that
exogenous ketone supplementation could provide an alternative method of attaining the
therapeutic benefits of nutritional ketosis, and as a means to further augment the therapeutic
potential of the KD.

88
3.2.1 Ketone supplementation causes little to no change in triglycerides and lipoproteins
One common concern regarding the KD is its purported potential to increase the risk of
atherosclerosis by elevating blood cholesterol and triglyceride levels [28, 29]. This topic
remains controversial as some, but not all studies have demonstrated that the KD elevates blood
levels of cholesterol and triglycerides [30-35]. Kwitervich and colleagues demonstrated an
increase in low-density lipoprotein (LDL) and a decrease in high-density lipoprotein (HDL) in
epileptic children fed the classical KD for two years [36]. In that study, total cholesterol
increased by ~130%, and stabilized at the elevated level over the 2-year period. A similar study
demonstrated that the lipid profile returned to baseline in children who remained on the KD for
six years [37]. Children typically remain on the diet for approximately two years then return to a
diet of common fat and carbohydrate ingestion [38]. The implications of these findings are
unclear, since the influence of cholesterol on cardiovascular health is controversial and
macronutrient sources of the diet vary per study. In contrast, more recent studies suggest that the
KD can actually lead to significant benefits in biomarkers of metabolic health, including blood
lipid profiles [39-46]. In these studies, the KD positively altered blood lipids, decreasing total
triglycerides and cholesterol while increasing the ratio of HDL to LDL [42-51]. Although, the
KD is well established in children, it has only recently been utilized as a strategy to control
seizures in adults. In 2014, Schoeler and colleagues reported on the feasibility of the KD for
adults, concluding that 39% of individuals achieved > 50% reduction in seizure frequency,
similar to the results reported in pediatric studies. Patients experienced similar gastrointestinal
adverse events to those previously described in pediatric patients, but they did not lead to
discontinuation of the diet in any patient [52].

89
With oral ketone supplementation, we observed a significant elevation in blood βHB
without dietary restriction and with little change in lipid biomarkers (Figure 3.1). Over the 4
week study, MCT-supplemented rats demonstrated decreased HDL compared to controls. No
significant changes were observed in any of the triglycerides or lipoproteins (HDL, LDL) with
any of the remaining exogenously applied ketone supplements. It should be noted that the rats
used for this study had not yet reached full adult body size [53]. Their normal growth rate and
maturation was likely responsible for the changes in triglyceride and lipoprotein levels observed
in the control animals over the 4 week study (baseline data not shown, no significant differences)
[54, 55]. Future studies are needed to investigate the effect of ketone supplementation on fully
mature and aged animals. Overall, our study suggests that oral ketone supplementation has little
effect on the triglyceride or lipoprotein profile after 4 weeks. However, it is currently unknown if
ketone supplementation would affect lipid biomarkers after a longer duration of consumption.
Further studies are needed to determine the effects of ketone supplements on blood triglyceride
and lipoproteins after chronic administration and as a means to further enhance the
hyperketonemia and improve the lipid profile of the clinically implemented (4:1) KD.
LDL is the lipoprotein particle that is most often associated with atherosclerosis. LDL
particles exist in different sizes: large molecules (Pattern A) or small molecules (Pattern
B). Recent studies have investigated the importance of LDL-particle type and size rather than
total concentration as being the source for cardiovascular risk [29]. Patients whose LDL particles
are predominantly small and dense (Pattern B) have a greater risk of cardiovascular disease
(CVD). It is thought that small, dense LDL particles are more able to penetrate the endothelium
and cause in damage and inflammation [56-59]. Volek et al. reported that the KD increased the
pattern and volume of LDL particles, which is considered to reduce cardiovascular risk [47].

90
Though we did not show a significant effect on LDL levels for ketone supplements, future
chronic feeding studies will investigate the effects of ketone supplementation on lipidomic
profile and LDL particle type and size.
3.2.2. Therapeutic levels of hyperketonemia suppress blood glucose levels
We demonstrated that therapeutic ketosis could be induced without dietary (calorie or
carbohydrate) restriction and that this acute elevation in blood ketones was significantly
correlated with a reduction in blood glucose (Figure 3.2-3.4). The BMS ketone supplement did
not significantly induce blood hyperketonemia or reduce glucose in the rats. The KE
supplemented rats trended towards reduced glucose levels; however, the lower dose of this agent
did not lower glucose significantly, as reported previously in acute response of mice (60). MCTs
have previously been shown to elicit a slight hypoglycemic effect by enhancing glucose
utilization in both diabetic and non-diabetic patients [60-62]. Kashiwaya et al. demonstrated that
both blood glucose and blood insulin decreased by approximately 50% in rats fed a diet where
30% of calories from starch were replaced with ketone esters for 14 days, suggesting that ketone
supplementation increases insulin sensitivity or reduces hepatic glucose output [63]. This
ketone-induced hypoglycemic effect has been previously reported in humans with IV infusions
of ketone bodies [64, 65]. Recently, Mikkelsen et al. showed that a small increase in βHB
concentration decreases glucose production by 14% in post-absorptive healthy males [66].
However, this has not been previously reported with any of the oral exogenous ketone
supplements we studied. Ketones are an efficient and sufficient energy substrate for the brain,
and will therefore prevent side effects of hypoglycemia when blood levels are elevated and the

91
patient is keto-adapted. Owen et al. most famously demonstrated this in 1967 wherein keto-
adapted patients (starvation induced therapeutic ketosis) were given 20 IU of insulin. The blood
glucose of fasted patients dropped to 1-2mM, but they exhibited no hypoglycemic symptoms due
to brain utilization of ketones for energy [67]. Therefore, ketones maintain brain metabolism and
are neuroprotective during severe hypoglycemia. The rats in the MCT group had a correlation of
blood ketone and glucose levels at week 4, whereas the combination of BMS+MCT produced a
significant hypoglycemic correlation both at baseline and at week 4. No hypoglycemic
symptoms were observed in the rats during this study. Insulin levels were not measured in this
study; however, future ketone supplementation studies should measure the effects of exogenous
ketones on insulin sensitivity with a glucose tolerance test. An increase in insulin sensitivity in
combination with our observed hypoglycemic effect has potential therapy implications for
glycemic control in T2D [68]. Furthermore, it should be noted that the KE metabolizes to both
AcAc and βHB in 1:1 ratio [27]. The ketone monitor used in this study only measures βHB as
levels of AcAc are more difficult to measure due to spontaneous decarboxylation to acetone;
therefore, the total ketone levels (βHB +AcAc) measured were likely higher, specifically for the
KE (14). Interestingly, the 10 g/kg dose produced a delayed blood βHB peak for ketone
supplements MCT and BMS+MCT. The higher dose of the ketogenic supplements elevated
blood levels more substantially, and thus reached their maximum blood concentration later due
to prolonged metabolic clearance. It must be noted that the dosage used in this study does not
translate to human patients, since the metabolic rate of rats is considerably higher. Future studies
will be needed to determine optimal dosing for human patients.

92
3.2.3. Effects of ketone supplementation on organ weight and body weight percentage
Ketone supplementation did not affect the size of the brain, lungs, kidneys or heart of
rats. As previously mentioned, the rats were still growing during the experimental time frame;
therefore, organ weights were normalized to body weight to determine if organ weight changed
independently to growth. There could be several reasons why ketones influenced liver and spleen
weight. The ratio of liver to body weight was significantly higher in the MCT supplemented
animals (Figure 3.5). MCTs are readily absorbed in the intestinal lumen and transported directly
to the liver via hepatic portal circulation. When given a large bolus, such as in this study, the
amount of MCTs in the liver will likely exceed the β-oxidation rate, causing the MCTs to be
deposited in the liver as fat droplets [69]. The accumulated MCT droplets in the liver could
explain the higher liver weight to body weight percentage observed with MCT supplemented
rats. Future toxicology and histological studies will be needed to determine the cause of the
observed hepatomegaly. It should be emphasized that the dose in this study is not optimized in
humans. We speculate that an optimized human dose would be lower due to the higher metabolic
rate of the rats and may not cause hepatomegaly or potential fat accumulation. Nutritional ketosis
achieved with the KD has been shown to decrease inflammatory markers such as TNF-α, IL-6,
IL-8, MCP-1, E-selectin, I-CAM, and PAI-1 [8, 70], which may account for the observed
decrease in spleen weight. As previously mentioned, Veech and colleagues demonstrated that
exogenous supplementation of 5mM βHB resulted in a 28% increase in hydraulic work in the
working perfused rat heart and a significant decrease in oxygen consumption [71-73]. Ketone
bodies have been shown to increase cerebral blood flow and perfusion [74]. Also, ketone bodies
have been shown to increase ATP synthesis and enhance the efficiency of ATP production [14,
68, 73]. It is possible that sustained ketosis results in enhanced cardiac efficiency and O2

93
consumption. Even though the size of the heart did not change for any of the ketone
supplements, further analysis of tissues harvested from the ketone-supplemented rats will be
needed to determine any morphological changes and to understand changes in organ size. It
should be noted that the Harlan standard rodent chow 2018 is nutritionally complete and
formulated with high-quality ingredients to optimize gestation, lactation, growth, and overall
health of the animals. The same cannot be said for the standard American diet (SAD). Therefore,
we plan to investigate the effects of ketone supplements administered with the SAD to determine
if similar effects will be seen when the micronutrient deficiencies and macronutrient profile
mimics what most Americans consume.
MCT oil has recently been used to induce nutritional ketosis although it produces dose-
dependent gastrointestinal (GI) side effects in humans that limit the potential for its use to
significantly elevate ketones (>0.5 mM). Despite these limitations, Azzam and colleagues
published a case report in which a 43-year-old-man had a significant decrease in seizure
frequency after supplementing his diet with 4 tablespoons of MCT oil twice daily [75]. An
attempt to increase his dosage to 5 tablespoons twice daily was halted by severe GI intolerance.
Henderson et al. observed that 20% of patients reported GI side effects with a 20g dose of
ketogenic agent AC-1202 in a double blind trial in mild to moderate Alzheimer’s patients [76].
We visually observed similar gastrointestinal side effects (loose stools) in the rats treated with
MCT oil in our study. Rats were closely monitored to avoid dehydration, and gastric motility
returned to normal between 12-24 hrs. Interestingly, the BMS+MCT supplement elevated βHB
similarly to MCT oil alone, without causing the adverse gastrointestinal effects seen in MCT-
supplemented rats. However, this could be due to the fact in a 10 g/kg dose of BMS+MCT, only
5 g/kg is MCT alone, which is less than the 10 g/kg dose that elicits the GI side effects. This

94
suggests that this novel combination may provide a more useful therapeutic option than MCT oil
alone, which is limited in its ability to elevate ketones in humans.
Exogenously delivered ketone supplements significantly altered rat weight gain for the
duration of the study (Figure 3.6). However, rats did not lose weight and maintained a healthy
range for their age. Rats have been shown to effectively balance their caloric intake to prevent weight
loss/gain [77-79]. Due to the caloric density of the exogenous ketone supplements (Table 3.1) it is
possible for the rats to eat less of the standard rodent chow and therefore fewer carbohydrates while
maintaining their caloric intake. Food intake was not measured for this study. However, if there
was a significant carbohydrate restriction there would be a significant change in basal blood
ketone and blood glucose levels. As the hallmark to the KD, carbohydrate restriction increases
blood ketone levels and reduces blood glucose levels. Neither an increase in basal blood ketone
levels nor a decrease in basal blood glucose levels was observed in this study (Figure 3.7).
Additionally, if there were an overall blood glucose decrease due to a change in food intake, this
would not explain the rapid reduction (within 30 minutes) in blood glucose correlated with an
elevation of blood ketone levels after an intragastric bolus of ketone supplement (Figures 3.2-
3.4).
Several studies have investigated the safety and efficacy of ketone supplements for disease
states such as AD and Parkinson’s disease, and well as for parenteral nutrition [68, 80-86]. Our
research demonstrates that several forms of dietary ketone supplementation can effectively
elevate blood ketone levels and achieve therapeutic nutritional ketosis without the need for
dietary carbohydrate restriction. We also demonstrated that ketosis achieved with exogenous
ketone supplementation can reduce blood glucose, and this is inversely associated with the blood
ketone levels. Although preliminary results are encouraging, further studies are needed to

95
determine if oral ketone supplementation can produce the same therapeutic benefits as the classic
KD in the broad-spectrum of disease states that are currently under investigation. Ketone
supplementation could be used as an alternative method for inducing ketosis in patients who
have previously had difficulty implementing the KD because of palatability issues, gall bladder
removal, liver abnormalities, or intolerance to fat. Additional experiments should be conducted
to see if ketone supplementation could be used in conjunction with the KD to assist and ease the
transition to nutritional ketosis and enhance the speed of keto-adaptation. In this study we have
demonstrated the ability of several ketone supplements to elevate blood ketone levels, providing
multiple options to induce therapeutic ketosis based on patient need. Though additional studies
are needed to determine the therapeutic potential of ketone supplementation, many patients that
previously were unable to benefit from the KD may now have an alternate method of achieving
therapeutic ketosis. Ketone supplementation may also represent a means to further augment
ketonemia in those responsive to therapeutic ketosis, especially in those individuals where
maintaining low glucose is important.
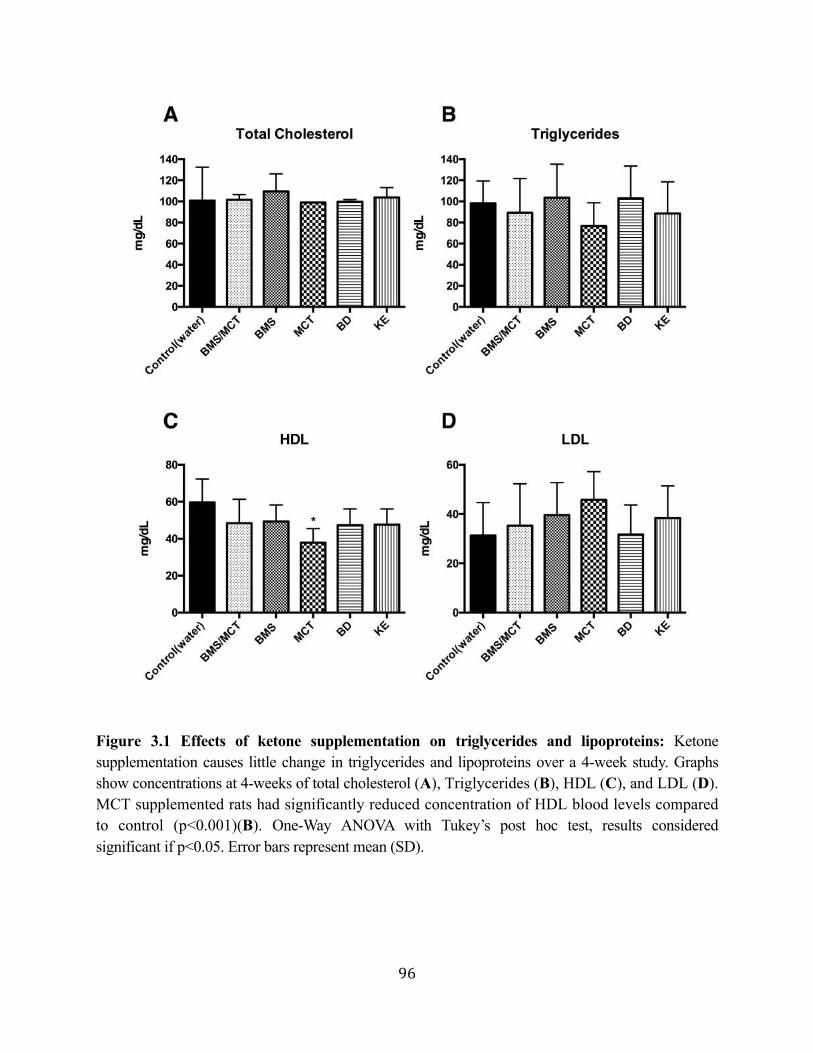
96
Figure 3.1 Effects of ketone supplementation on triglycerides and lipoproteins: Ketone supplementation causes little change in triglycerides and lipoproteins over a 4-week study. Graphs show concentrations at 4-weeks of total cholesterol (A), Triglycerides (B), HDL (C), and LDL (D). MCT supplemented rats had significantly reduced concentration of HDL blood levels compared to control (p<0.001)(B). One-Way ANOVA with Tukey’s post hoc test, results considered significant if p<0.05. Error bars represent mean (SD).
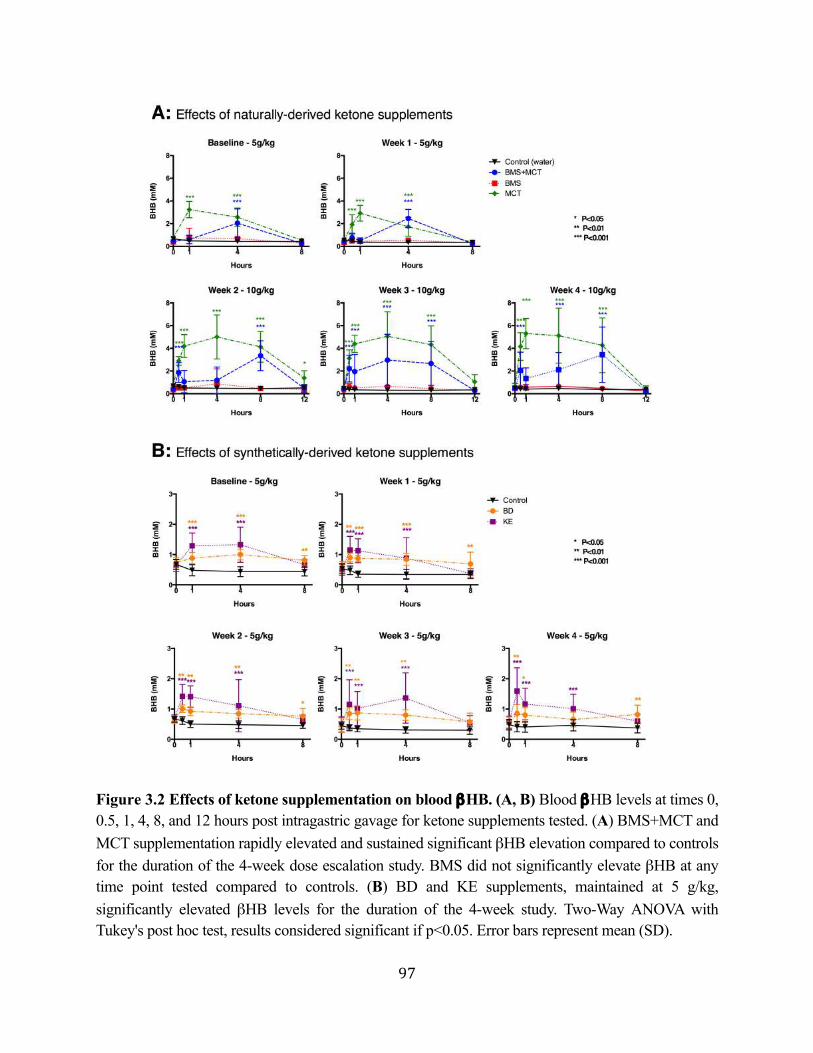
97
Figure 3.2 Effects of ketone supplementation on blood βHB. (A, B) Blood βHB levels at times 0, 0.5, 1, 4, 8, and 12 hours post intragastric gavage for ketone supplements tested. (A) BMS+MCT and MCT supplementation rapidly elevated and sustained significant βHB elevation compared to controls for the duration of the 4-week dose escalation study. BMS did not significantly elevate βHB at any time point tested compared to controls. (B) BD and KE supplements, maintained at 5 g/kg, significantly elevated βHB levels for the duration of the 4-week study. Two-Way ANOVA with Tukey's post hoc test, results considered significant if p<0.05. Error bars represent mean (SD).
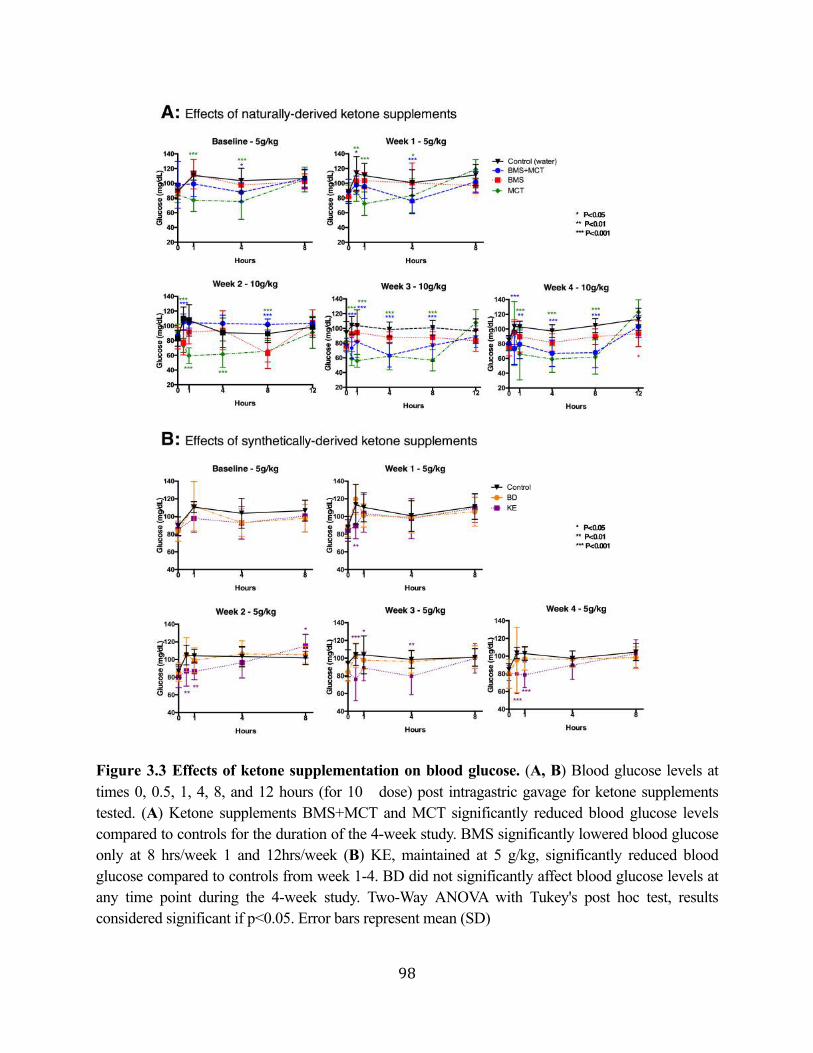
98
Figure 3.3 Effects of ketone supplementation on blood glucose. (A, B) Blood glucose levels at times 0, 0.5, 1, 4, 8, and 12 hours (for 10 dose) post intragastric gavage for ketone supplements tested. (A) Ketone supplements BMS+MCT and MCT significantly reduced blood glucose levels compared to controls for the duration of the 4-week study. BMS significantly lowered blood glucose only at 8 hrs/week 1 and 12hrs/week (B) KE, maintained at 5 g/kg, significantly reduced blood glucose compared to controls from week 1-4. BD did not significantly affect blood glucose levels at any time point during the 4-week study. Two-Way ANOVA with Tukey's post hoc test, results considered significant if p<0.05. Error bars represent mean (SD)
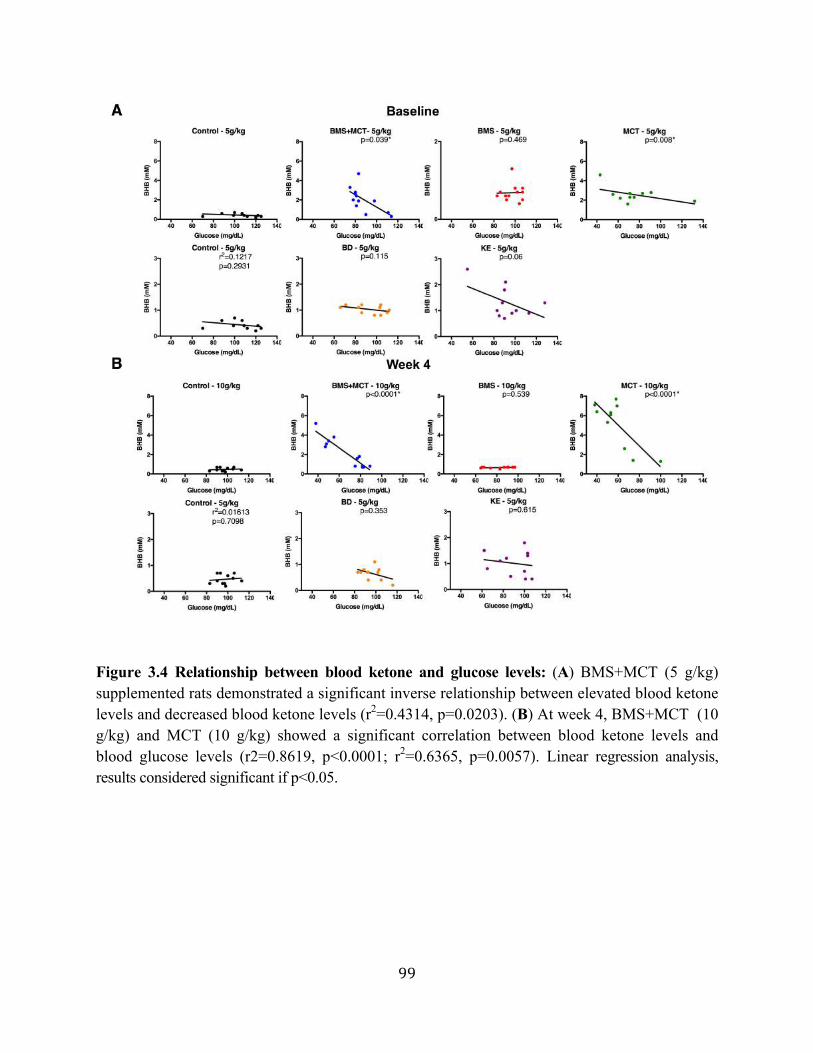
99
Figure 3.4 Relationship between blood ketone and glucose levels: (A) BMS+MCT (5 g/kg) supplemented rats demonstrated a significant inverse relationship between elevated blood ketone levels and decreased blood ketone levels (r2=0.4314, p=0.0203). (B) At week 4, BMS+MCT (10 g/kg) and MCT (10 g/kg) showed a significant correlation between blood ketone levels and blood glucose levels (r2=0.8619, p<0.0001; r2=0.6365, p=0.0057). Linear regression analysis, results considered significant if p<0.05.
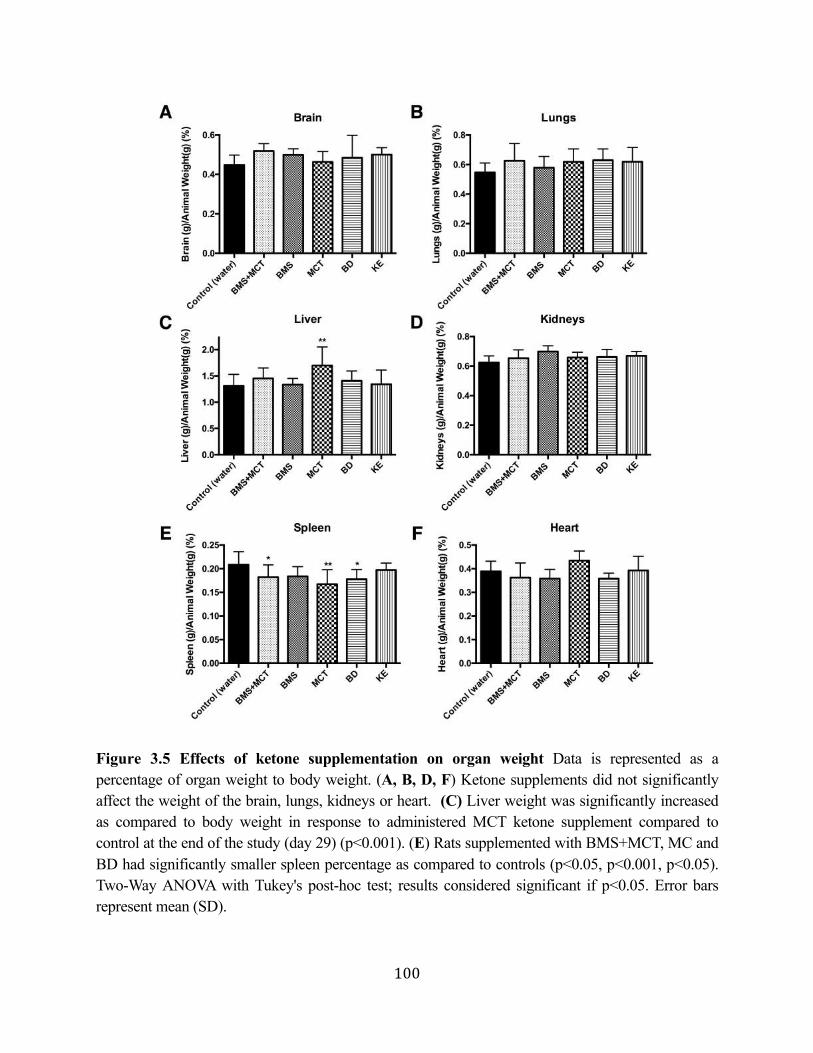
100
Figure 3.5 Effects of ketone supplementation on organ weight Data is represented as a percentage of organ weight to body weight. (A, B, D, F) Ketone supplements did not significantly affect the weight of the brain, lungs, kidneys or heart. (C) Liver weight was significantly increased as compared to body weight in response to administered MCT ketone supplement compared to control at the end of the study (day 29) (p<0.001). (E) Rats supplemented with BMS+MCT, MC and BD had significantly smaller spleen percentage as compared to controls (p<0.05, p<0.001, p<0.05). Two-Way ANOVA with Tukey's post-hoc test; results considered significant if p<0.05. Error bars represent mean (SD).
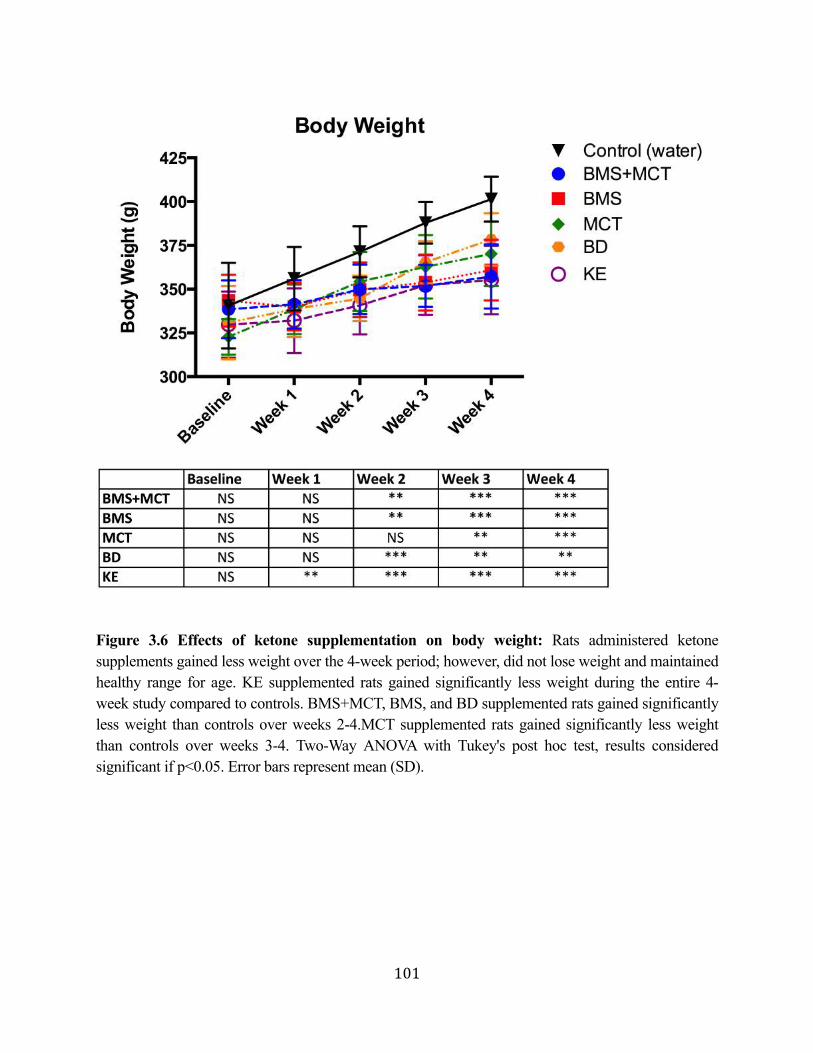
101
Figure 3.6 Effects of ketone supplementation on body weight: Rats administered ketone supplements gained less weight over the 4-week period; however, did not lose weight and maintained healthy range for age. KE supplemented rats gained significantly less weight during the entire 4-week study compared to controls. BMS+MCT, BMS, and BD supplemented rats gained significantly less weight than controls over weeks 2-4.MCT supplemented rats gained significantly less weight than controls over weeks 3-4. Two-Way ANOVA with Tukey's post hoc test, results considered significant if p<0.05. Error bars represent mean (SD).
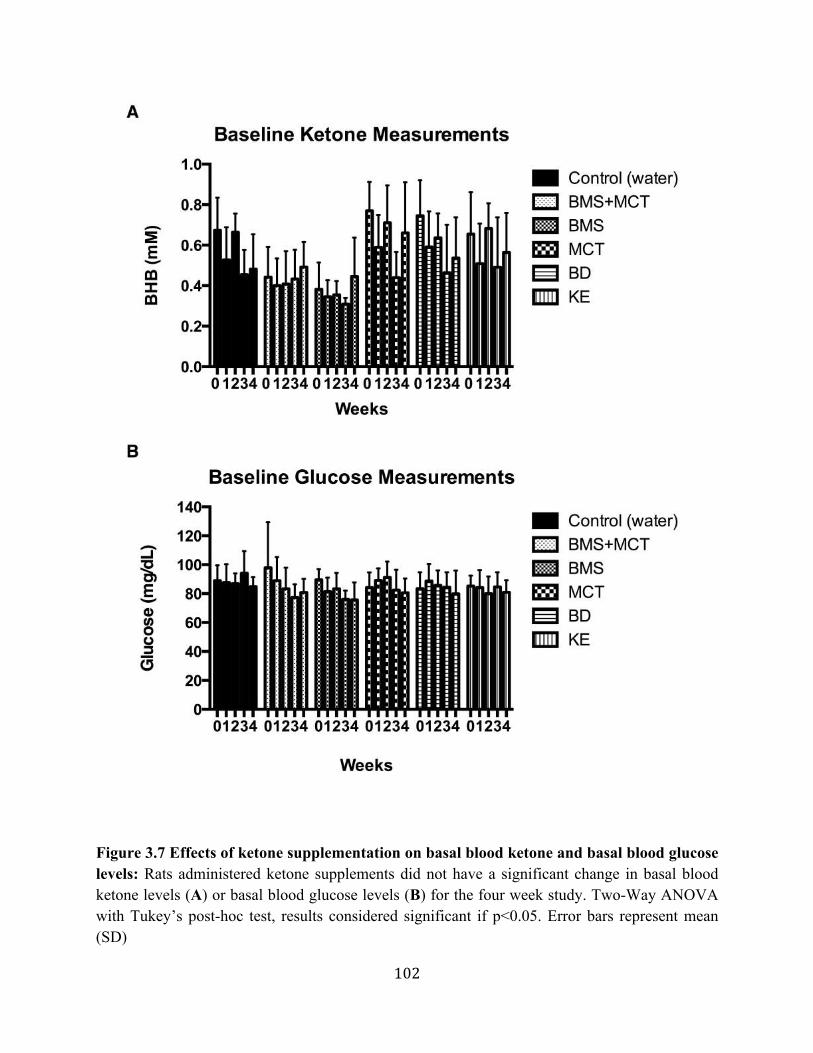
102
Figure 3.7 Effects of ketone supplementation on basal blood ketone and basal blood glucose levels: Rats administered ketone supplements did not have a significant change in basal blood ketone levels (A) or basal blood glucose levels (B) for the four week study. Two-Way ANOVA with Tukey’s post-hoc test, results considered significant if p<0.05. Error bars represent mean (SD)

103
3.3. Effect of Sustaining Dietary Ketosis Through Exogenous Ketone Supplementation on
the Serum and Hippocampal Metabolome of Sprague-Dawley rats
In the previous section, we reported that oral administration of ketone supplements
produced nutritional ketosis (>0.5 mM) without carbohydrate restriction and the effects of a 28-
day administration of five ketone supplements on blood glucose, ketones, and lipids in male
Sprague-Dawley rats. We hypothesized that the 28-day administration would affect metabolomic
markers. Serum (~300 µL) and hippocampal tissues for ketone supplements KE and BMS+MCT
were collected on day 28 at 4 hours post-intragastric gavage (peak ketone elevation). Metabolon
analyzed samples by liquid chromatography-tandem mass spectrometry (LC-MS/MS) and gas
chromatography-mass spectrometry (GC-MS). Both ketone supplements significantly increased
serum Krebs cycle intermediates: citrate, fumarate and malate. KE supplement significantly
increased alpha-ketoglutarate and BMS+MCT supplement significantly increased succinate in
the rat serum. Additionally, both supplements affected medium chain fatty acids, antioxidants,
and adenosine in both serum and hippocampal tissue. Although both forms of ketone
supplementation increased brain and blood ketone levels, the global metabolic profiles were
different.
3.3.1 Oral administration of ketone supplements elevates blood ketone levels, increases
Krebs cycle intermediates, MCFAs, antioxidants, and adenosine in serum and hippocampal
tissue and implications for wound healing
Global metabolomic profiling of serum from KE and BMS+MCT supplemented rats
exhibited a total of 142 out of 388 metabolites that were significantly changed from control for

104
KE-treated rats and 119 out of 388 metabolites were significantly altered compared to control for
BMS+MCT treated rats. Additionally, 12 of 290 metabolites were significantly changed in the
hippocampal tissue for KE-treated rats and 36 of 290 metabolites were significantly altered for
BMS+MCT treated rats (Table 3.1). These metabolic changes provide preliminary insight to
possible mechanisms by which ketones could enhance wound healing as well as potential
mechanisms to target other disease states.
Metabolomics analysis confirmed elevated ketone bodies βHB as well as AcAc in the
serum and elevated βHB in the hippocampus (Figure 3.8). βHB is a more stable molecule since
it cannot be decarboxylated to acetone; therefore, it is the predominant ketone body within the
blood stream and likely a reason only βHB was elevated in the hippocampus. It should be noted
that KE metabolizes to both βHB and AcAc as compared to BMS+MCT that only metabolizes to
βHB. However, both supplements elevated AcAc, KE showed a 9-fold increase and
interestingly, BMS+MCT demonstrated a 15-fold increase. It should be noted that KE was at a 5
g/kg dose and BMS+MCT was given at a 10 g/kg dose. Both ketone supplements demonstrated a
2:1 ratio of βHB: AcAc (KE 15.78:8.84, BMS+MCT 35.33:15.39) in the serum. Thus, total
blood ketones seen in our previous study were likely higher in the KE-treated and BMS+MCT-
treated animals. The commercially available blood ketone meters used in previous study offer the
most cost feasible method of measuring blood ketones, but are only able to measure βHB, not
AcAc. Additionally, BMS+MCT treated animals had significantly lower serum glucose (0.87,
p<0.05). This observation supports the hypoglycemic effect demonstrated in the previous study
as well as previous studies by Veech and colleagues [87, 88]. As discussed, this hypoglycemic
effect has been attributed to the enhancement insulin sensitivity, which may help diabetic foot
ulcers specifically. As discussed, MCFAs are one of the main components of MCT oil namely

105
caprylic acid (C8:0) and Capric acid (10:0); these medium chain fatty acids were significantly
elevated in the serum and hippocampal tissue of BMS+MCT supplemented rats (Figure 3.9).
This demonstrates that the MCT oil is being metabolized into its MCFAs, which are available in
the blood stream and tissues to be used for energy. Interestingly, KE supplemented rats also had
significantly elevated capric acid in the serum and hippocampal tissue.
Krebs cycle intermediates citrate, fumarate and malate were significantly elevated in the
serum of both KE-treated and BMS+MCT-treated animals (Figure 3.10). Additionally, KE
significantly elevated the intermediate α-ketoglutarate and BMS+MCT significantly elevated
succinate. These elevations were not seen in the hippocampal tissue. As discussed, non-wounded
skin predominantly uses anaerobic respiration with only about 30% going to the Krebs cycle; this
is exaggerated during wound healing when hypoxia due to tissue damage shifts the metabolic
profile to only using 4% Krebs cycle [89-92]. However, ketones have been shown to increase
blood flow; thus, increased blood flow would lead to potential increased oxygenation and the
restoration of the non-wounded skin metabolic profile. Even with a 30% Krebs cycle flux, an
increase in Acetyl CoA via ketolysis would produce a net increase in ATP production. Indeed,
Veech and colleagues demonstrated a 16-fold increase in acetyl CoA with a ketone ester
supplementation [88]. Acetyl co-A was significantly elevated via BMS+MCT supplementations
but not KE in the hippocampal tissue, supporting this finding. Levels of pyruvate and lactate
were unchanged in both ketone supplements in both the serum and hippocampal tissue
demonstrating that there wasn’t an increased flux of glycolysis and anaerobic respiration. Even
though there initially wouldn’t be sufficient oxygen for the wound bed to use the intermediates in
the ETC, Krebs cycle intermediates have other anabolic fates that would be important to wound
healing. Citrate is a precursor to fatty acid synthesis; α-ketoglutarate has been shown to scavenge

106
H2O2 in cell cultures and is an amino acid synthesis precursor [93]. Krebs cycle intermediates
were shown to be decreased in aged rats; this supplementation may restore these levels to those
of a young person or at least elevate them [94]. Significant amino acid synthesis is supported by
significant serum elevations in the amino acids glycine and serine. Additionally, KE-treated rats
had a significant increase in PPP intermediates ribose-5-phosphate and ribulose/xylulose-5-
phosphate demonstrating an increase flux through the pathway and potentially nucleic acid
synthesis needed for wound healing. From this data, we can speculate that if the ketones were
being used as energy a higher portion of the glucose can be shunted to the PPP instead of being
metabolized for energy.
Surprisingly, oxidative stress (oxidized glutathione, GSSG (serum)) and antioxidant
capacity (reduced glutathione (hippo), carnosine (serum), and anserine (serum)) were elevated,
suggesting a heightened state of oxidative stress and antioxidant defense with ketone
supplementation. As discussed by Moor and colleagues, age alone affects the levels of SOD2 and
glutathione antioxidant profile [95]. Without appropriate oxidative stress, neutrophil respiratory
bursts cannot effectively clear bacteria from the wound bed; however, without appropriate
antioxidant capacity, the reactive oxygen species become damaging to neighboring tissue. This
ketogenic elevation of both oxidative stress and antioxidant defense may help clear bacteria as
well as quench ROS to promote wound healing in an aged phenotype. Additionally, carnosine (β-
alanyl-L-histidine) and anserine (β-alanyl-N-methylhistidine) were elevated by KE and
BMS+MCT supplementation (Figure 3.11). Carnosine is a natural dipeptide widely and
abundantly distributed in excitable tissue. Although its physiologic role is not completely
understood, many beneficial actions have been attributed to carnosine, such as being an
antioxidant, antiglycating and ion-chelating agent, and a free-radical scavenger. Several studies

107
have shown that supplementation with carnosine accelerates wound healing [96-100]. Anserine
is a dipeptide containing beta alanine and histidine and has been shown to exhibit equal
antioxidant activity to carnosine by reducing the primary molecular products of lipid
peroxidation [101-103]. A study by Altavilla et al. demonstrated that reduction of lipid
peroxidation restores impaired VEGF expression leading to accelerated wound healing and
angiogenesis in a diabetic wound model [104]. This supports a possible mechanism of action for
ketone supplementation to augment wound healing. However, further experiments need to be
performed to optimize dose as too high of an oxidative state, which can be seen in the KE
supplementation can potentially be damaging, especially in an aged patient.
Commonly known for its role in energy transfer via phosphorylation, adenosine also
plays a role in the regulation of blood flow as a potent vasodilator. Adenosine was significantly
elevated in the serum via KE supplementation and trended towards significance for BMS+MCT;
however, BMS+MCT supplemented rats showed a significant elevation of adenosine in their
hippocampal tissue (KE not significant). Even though BMS+MCT did not show a significant
elevation in the blood stream, this suggests that the BMS+MCT supplemented rat tissues were
more efficient at using it; therefore a larger portion had already been sequestered in the
hippocampal tissue. High levels of AMP could reflect a metabolic insufficiency for the KE
supplement, which may be due to the high dose; further studies optimizing the dose of ketone
supplements are needed. Masino and colleagues have demonstrated that increased adenosine of
the A1 subtype plays a key role in the anticonvulsant success of ketogenic strategies [105-108].
Additionally, several studies have shown that topical adenosine application accelerates wound
healing via adenosine A2A receptors and promoting angiogenesis [109-117].

108
A recent study by Sood et al, was the first to document a global metabolomic profile of
diabetic and non-diabetic wounds, 7 days post injury. In non-diabetic mice, 88 of the 129
detected metabolites had a significant response to injury, 85 up regulated and 3 down-regulated.
In diabetic wounds, 81 metabolites had a significant response to injury with 76 up regulated and
5 down regulated. Interestingly, they found 62 unique metabolites that differed between the non-
diabetic and diabetic wound phenotype [118]. From their study, glycine was dysregulated in
diabetic wounds and both ketone supplements significantly increased glycine in the serum (1.3,
1.27 fold change). A recent metabolic profiling of cancer cells has correlated glycine with
increased cell proliferation; however, it remains to be determined whether glycine is essential for
cell proliferation during wound healing [119]. Two recent studies suggest that Kynurenine, a
kynurenate precursor, may play a role in both anti-inflammatory activity and fibroblast
proliferation during wound repair; additionally, it has been shown to be dysregulated in diabetic
wounds [120, 121]. KE supplemented rats showed an increase in kynurenate but didn’t reach
significance (2.61 fold) (0.05<p<0.10). Additionally, metabolite OH-phenylpyruvate was shown
to be dysregulated and KE supplementation significantly enhanced the levels 3.59 fold in the
serum. Little is known about the role during wound healing. Further experiments need to be
conducted to determine the meaning of this observation.
The metabolomics profiling data presented here help us to understand the metabolic
consequences of KE and BMS+MCT administration and offers insights into potential
mechanisms of action by which ketone supplementation may augment wound healing as well as
affect other disease states for future studies.
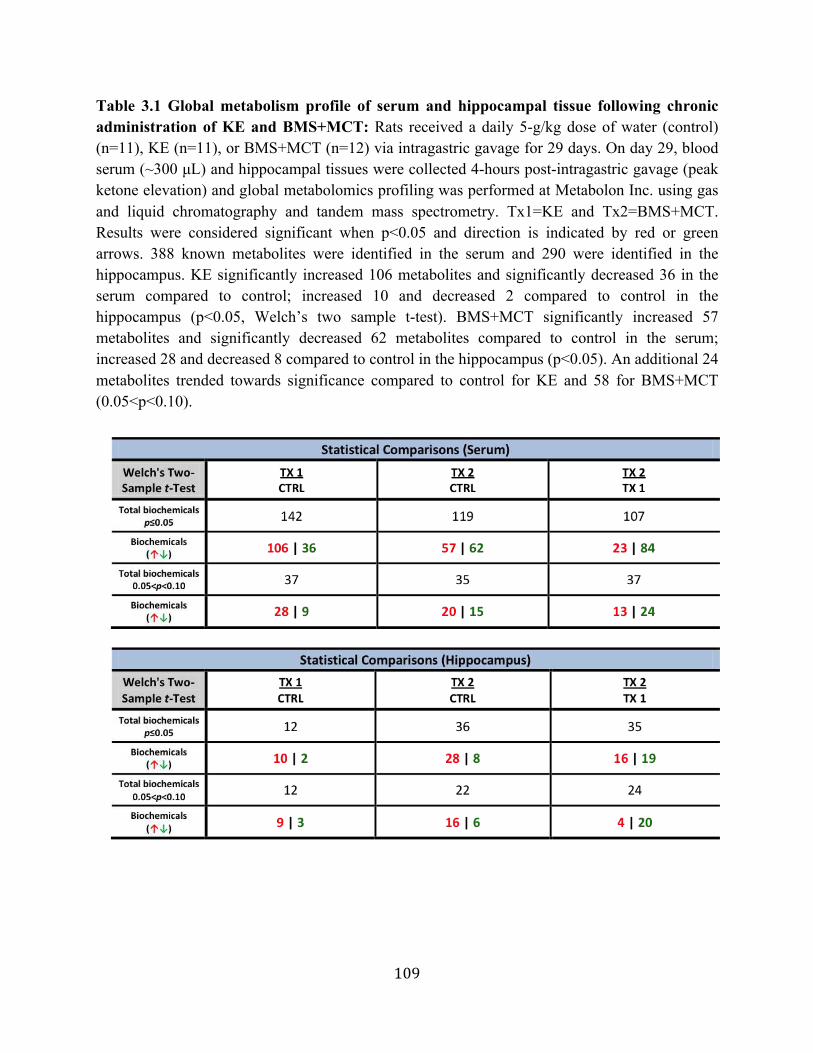
109
Table 3.1 Global metabolism profile of serum and hippocampal tissue following chronic administration of KE and BMS+MCT: Rats received a daily 5-g/kg dose of water (control) (n=11), KE (n=11), or BMS+MCT (n=12) via intragastric gavage for 29 days. On day 29, blood serum (~300 µL) and hippocampal tissues were collected 4-hours post-intragastric gavage (peak ketone elevation) and global metabolomics profiling was performed at Metabolon Inc. using gas and liquid chromatography and tandem mass spectrometry. Tx1=KE and Tx2=BMS+MCT. Results were considered significant when p<0.05 and direction is indicated by red or green arrows. 388 known metabolites were identified in the serum and 290 were identified in the hippocampus. KE significantly increased 106 metabolites and significantly decreased 36 in the serum compared to control; increased 10 and decreased 2 compared to control in the hippocampus (p<0.05, Welch’s two sample t-test). BMS+MCT significantly increased 57 metabolites and significantly decreased 62 metabolites compared to control in the serum; increased 28 and decreased 8 compared to control in the hippocampus (p<0.05). An additional 24 metabolites trended towards significance compared to control for KE and 58 for BMS+MCT (0.05<p<0.10).
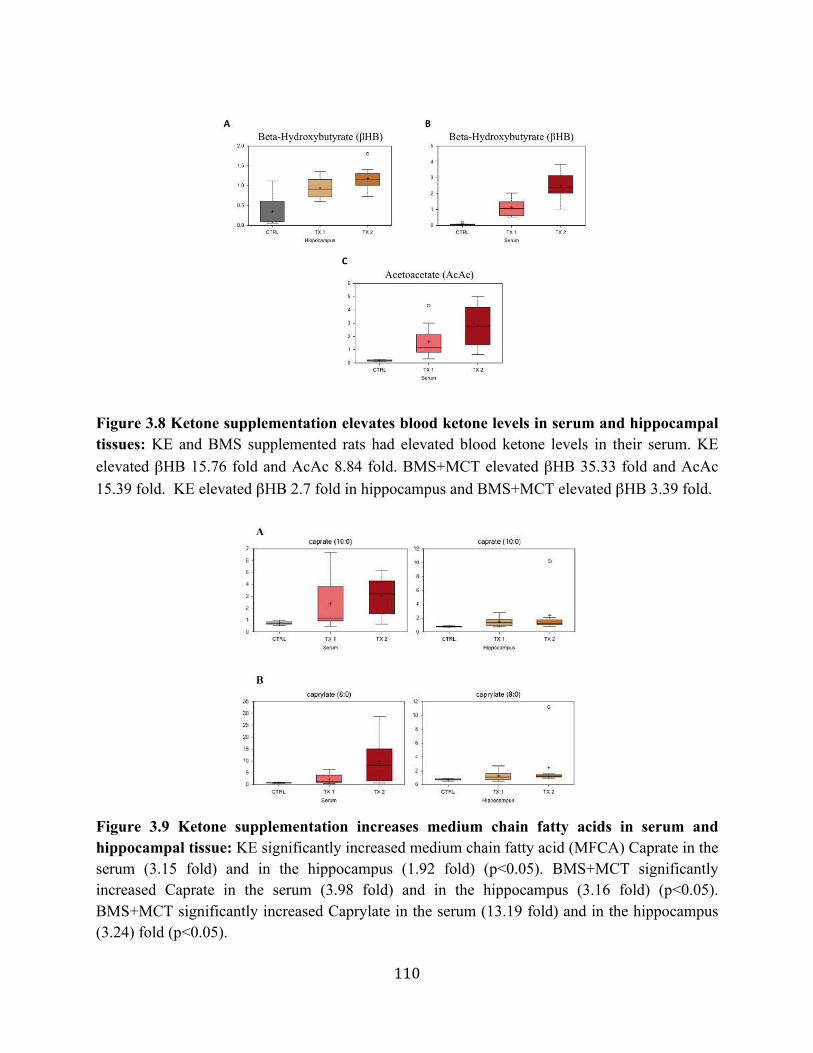
110
Figure 3.8 Ketone supplementation elevates blood ketone levels in serum and hippocampal tissues: KE and BMS supplemented rats had elevated blood ketone levels in their serum. KE elevated βHB 15.76 fold and AcAc 8.84 fold. BMS+MCT elevated βHB 35.33 fold and AcAc 15.39 fold. KE elevated βHB 2.7 fold in hippocampus and BMS+MCT elevated βHB 3.39 fold.
Figure 3.9 Ketone supplementation increases medium chain fatty acids in serum and hippocampal tissue: KE significantly increased medium chain fatty acid (MFCA) Caprate in the serum (3.15 fold) and in the hippocampus (1.92 fold) (p<0.05). BMS+MCT significantly increased Caprate in the serum (3.98 fold) and in the hippocampus (3.16 fold) (p<0.05). BMS+MCT significantly increased Caprylate in the serum (13.19 fold) and in the hippocampus (3.24) fold (p<0.05).
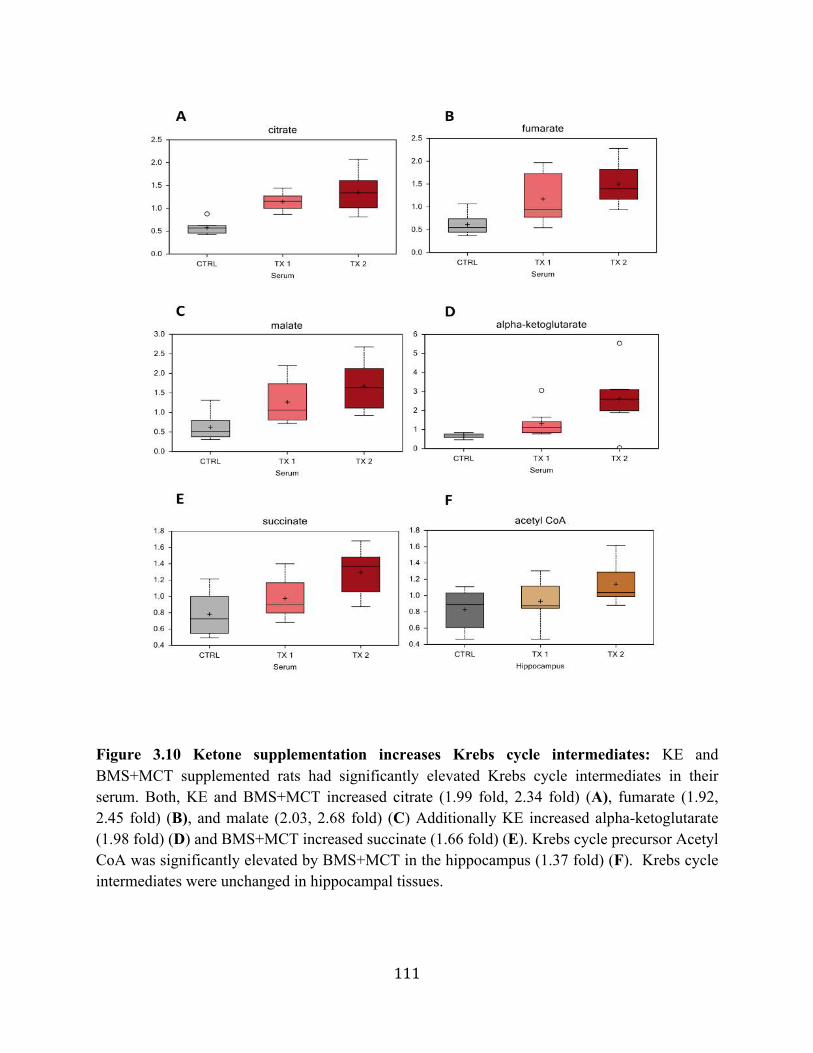
111
Figure 3.10 Ketone supplementation increases Krebs cycle intermediates: KE and BMS+MCT supplemented rats had significantly elevated Krebs cycle intermediates in their serum. Both, KE and BMS+MCT increased citrate (1.99 fold, 2.34 fold) (A), fumarate (1.92, 2.45 fold) (B), and malate (2.03, 2.68 fold) (C) Additionally KE increased alpha-ketoglutarate (1.98 fold) (D) and BMS+MCT increased succinate (1.66 fold) (E). Krebs cycle precursor Acetyl CoA was significantly elevated by BMS+MCT in the hippocampus (1.37 fold) (F). Krebs cycle intermediates were unchanged in hippocampal tissues.
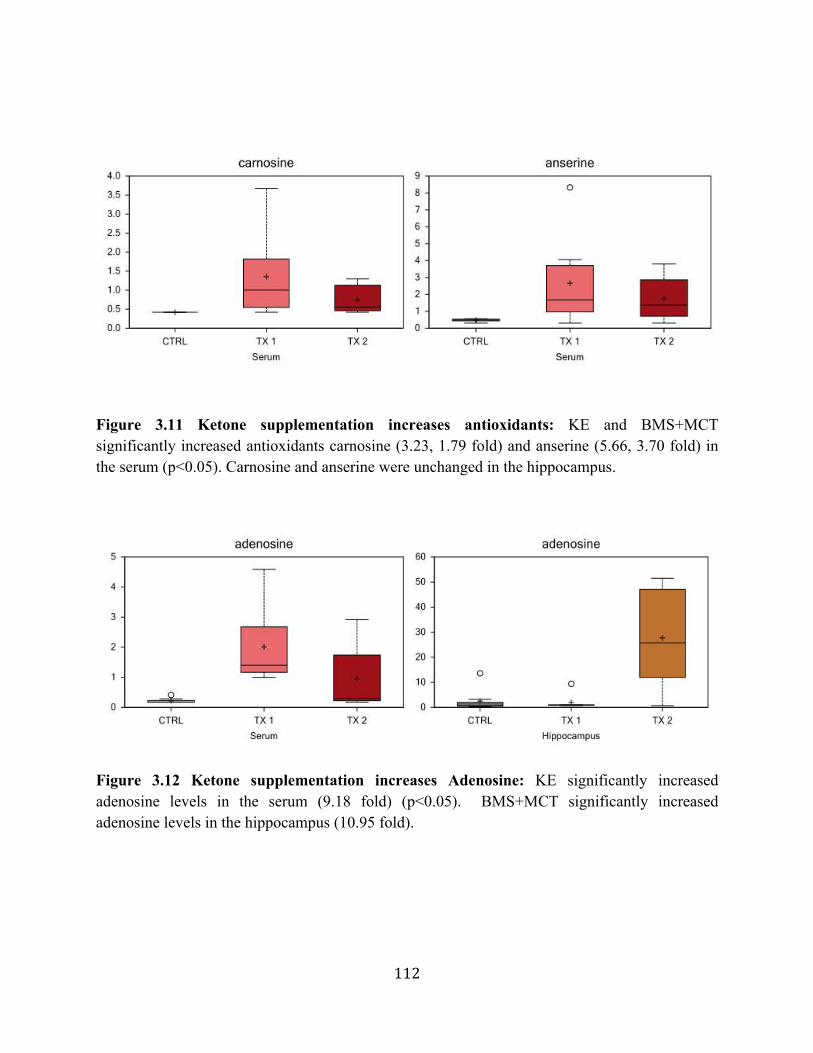
112
Figure 3.11 Ketone supplementation increases antioxidants: KE and BMS+MCT significantly increased antioxidants carnosine (3.23, 1.79 fold) and anserine (5.66, 3.70 fold) in the serum (p<0.05). Carnosine and anserine were unchanged in the hippocampus.
Figure 3.12 Ketone supplementation increases Adenosine: KE significantly increased adenosine levels in the serum (9.18 fold) (p<0.05). BMS+MCT significantly increased adenosine levels in the hippocampus (10.95 fold).

113
3.4. References for Chapter 3
1. Sirven J, Whedon B, Caplan D, Liporace J, Glosser D, O'Dwyer J, Sperling M: The ketogenic diet for intractable epilepsy in adults: preliminary results. Epilepsia 1999, 40(12):1721-1726.
2. Wilder R: The effect of ketonemia on the course of epilepsy. Mayo Clin Bulletin 1921, 2:307-308.
3. Thiele E: Assessing the efficacy of antiepileptic treatments: the ketogenic diet. Epilepsia 2003, 44 Suppl 7:26-29.
4. Paoli A, Rubini A, Volek JS, Grimaldi KA: Beyond weight loss: a review of the therapeutic uses of very-low-carbohydrate (ketogenic) diets. European journal of clinical nutrition 2013, 67(8):789-796.
5. Foster GD, Wyatt HR, Hill JO, McGuckin BG, C B, Mohemmed BS, Szapary PO, Rader DJ, Edman JS, Klein S: A randomized trial of a low-carbohydrate diet for obesity. The New England journal of medicine 2003, 348:2082-2090.
6. Westman EC, Feinman RD, Mavropoulos JC, Vernon MC, Volek JS, Wortman JA, Yancy WS, Phinney SD: Low-carbohydrate nutrition and metabolism. The American journal of clinical nutrition 2007(86):276-284.
7. Westman EC, Yancy WS, Edman JS, Tomlin KF, Perkins CE: Effect of 6-month adherence to a very low carbohydrate diet program. The American journal of medicine 2002, 113(1):30-36.
8. Forsythe C, Phinney S, Fernandez M, Quann E, Wood R, Bibus D, Kraemer W, Feinman R, Volek J: Comparison of low fat and low carbohydrate diets on circulating fatty acid composition and markers of inflammation. Lipids 2008, 43(1):65-77.
9. Boden G, Sargrad K, Homko C, Mozzoli M, Stein T: Effect of a low-carbohydrate diet on appetite, blood glucose levels, and insulin resistance in obese patients with type 2 diabetes. Annals of internal medicine 2005, 142(6):403-411.
10. Gumbiner B, Wendel J, McDermott M: Effects of diet composition and ketosis on glycemia during very-low-energy-diet therapy in obese patients with non-insulin-dependent diabetes mellitus. The American journal of clinical nutrition 1996, 63(1):110-115.
11. Nielsen J, Joensson E: Low-carbohydrate diet in type 2 diabetes: stable improvement of bodyweight and glycemic control during 44 months follow-up. Nutrition & metabolism 2008, 5:14.
12. Yancy W, Foy M, Chalecki A, Vernon M, Westman E: A low-carbohydrate, ketogenic diet to treat type 2 diabetes. Nutrition & metabolism 2005, 2:34.

114
13. Dashti HM, Al-Zaid NS, Mathew TC, Al-Mousawi M, Talib H, Asfar SK, Behbahani AI: Long term effects of ketogenic diet in obese subjects with high cholesterol level. Molecular and cellular biochemistry 2006, 286(1-2):1-9.
14. Maalouf M, Rho JM, Mattson MP: The neuroprotective properties of calorie restriction, the ketogenic diet, and ketone bodies. Brain Res Rev 2009, 59(2):293-315.
15. Mavropoulos JC, Yancy WS, Hepburn J, Westman EC: The effects of a low-carbohydrate, ketogenic diet on the polycystic ovary syndrome: a pilot study. Nutrition & metabolism 2004, 2:35.
16. Seyfried T, Flores R, Poff A, D'Agostino D: Cancer as a metabolic disease: implications for novel therapeutics. Carcinogenesis 2014, 35:515-527.
17. Poff AM, Ari C, Arnold P, Seyfried TN, D'Agostino DP: Ketone supplementation decreases tumor cell viability and prolongs survival of mice with metastatic cancer. International journal of cancer Journal international du cancer 2014, 135:1711-1720.
18. Poff A, Ari C, Seyfried T, D'Agostino D: The ketogenic diet and hyperbaric oxygen therapy prolong survival in mice with systemic metastatic cancer. PloS one 2013, 8(6):e65522.
19. Seyfried T, Shelton L: Cancer as a metabolic disease. Nutrition & metabolism, 7:7.
20. Fine E, Segal-Isaacson C, Feinman R, Herszkopf S, Romano M, Tomuta N, Bontempo A, Negassa A, Sparano J: Targeting insulin inhibition as a metabolic therapy in advanced cancer: a pilot safety and feasibility dietary trial in 10 patients. Nutrition (Burbank, Los Angeles County, Calif) 2012, 28(10):1028-1035.
21. Zhao Z, Lange D, Voustianiouk A, MacGrogan D, Ho L, Suh J, Humala N, Thiyagarajan M, Wang J, Pasinetti G: A ketogenic diet as a potential novel therapeutic intervention in amyotrophic lateral sclerosis. BMC neuroscience 2006, 7:29.
22. White H, Venkatesh B: Clinical review: ketones and brain injury. Critical care (London, England) 2011, 15(2):219.
23. Prins M: Cerebral metabolic adaptation and ketone metabolism after brain injury. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 2008, 28(1):1-16.
24. Henderson S, Vogel J, Barr L, Garvin F, Jones J, Costantini L: Study of the ketogenic agent AC-1202 in mild to moderate Alzheimer's disease: a randomized, double-blind, placebo-controlled, multicenter trial. Nutrition & metabolism 2009, 6:31.
25. Brownlow M, Benner L, D'Agostino D, Gordon M, Morgan D: Ketogenic diet improves motor performance but not cognition in two mouse models of Alzheimer's pathology. PloS one 2013, 8(9):e75713.

115
26. Kossoff EH, Hartman AL: Ketogenic diets: new advances for metabolism-based therapies. Current Opinion in Neurology 2012, 25:173-178.
27. D'Agostino D, Pilla R, Held H, Landon C, Puchowicz M, Brunengraber H, Ari C, Arnold P, Dean J: Therapeutic ketosis with ketone ester delays central nervous system oxygen toxicity seizures in rats. American journal of physiology Regulatory, integrative and comparative physiology 2013, 304(10):R829-836.
28. McPherson P, McEneny J: The biochemistry of ketogenesis and its role in weight management, neurological disease and oxidative stress. Journal of physiology and biochemistry 2012, 68(1):141-151.
29. Jimmy Moore and Eric C. Westman M: Cholesterol Clarity: What the HDL is wrong with my numbers. Las Vegas,NV: Victory Belt Publishing Inc.; 2013.
30. Dekaban A: Plasma lipids in epileptic children treated with the high fat diet. Archives of neurology 1966, 15(2):177-184.
31. Chesney D, Brouhard B, Wyllie E, Powaski K: Biochemical abnormalities of the ketogenic diet in children. Clinical pediatrics 1999, 38(2):107-109.
32. Schwartz R, Boyes S, Aynsley-Green A: Metabolic effects of three ketogenic diets in the treatment of severe epilepsy. Developmental medicine and child neurology 1989, 31(2):152-160.
33. Katyal N, Koehler A, McGhee B, Foley C, Crumrine P: The ketogenic diet in refractory epilepsy: the experience of Children's Hospital of Pittsburgh. Clinical pediatrics 2000, 39(3):153-159.
34. Ellenbroek J, van Dijck L, Töns H, Rabelink T, Carlotti F, Ballieux B, de Koning E: Long-term ketogenic diet causes glucose intolerance and reduced beta and alpha cell mass but no weight loss in mice. American journal of physiology Endocrinology and metabolism 2014, 306(5):E552-E558.
35. Bergqvist A: Long-term monitoring of the ketogenic diet: Do's and Don'ts. Epilepsy Research 2012, 100(3):261-266.
36. Kwiterovich P, Vining E, Pyzik P, Skolasky R, Freeman J: Effect of a high-fat ketogenic diet on plasma levels of lipids, lipoproteins, and apolipoproteins in children. JAMA : the journal of the American Medical Association 2003, 290(7):912-920.
37. Groesbeck D, Bluml R, Kossoff E: Long-term use of the ketogenic diet in the treatment of epilepsy. Developmental medicine and child neurology 2006, 48(12):978-981.
38. Patel A, Pyzik P, Turner Z, Rubenstein J, Kossoff E: Long-term outcomes of children treated with the ketogenic diet in the past. Epilepsia 2010, 51(7):1277-1282.

116
39. Brehm BJ, Seeley RJ, Daniels SR, D'Alessio DA: A randomized trial comparing a very low carbohydrate diet and a calorie-restricted low fat diet on body weight and cardiovascular risk factors in healthy women. The Journal of clinical endocrinology and metabolism 2003, 88(4):1617-1623.
40. Shai I, Schwarzfuchs D, Henkin Y, Shahar DR, Witkow S, Greenberg I, Golan R, Fraser D, Bolotin A, Vardi H et al: Weight loss with a low-carbohydrate, Mediterranean, or low-fat diet. The New England journal of medicine 2008, 359(3):229-241.
41. Volek JS, Phinney SD, Forsythe CE, Quann EE, Wood RJ, Puglisi MJ, Kraemer WJ, Bibus DM, Fernandez ML, Feinman RD: Carbohydrate restriction has a more favorable impact on the metabolic syndrome than a low fat diet. Lipids 2009, 44(4):297-309.
42. Feinman RD, Volek JS: Low carbohydrate diets improve atherogenic dyslipidemia even in the absence of weight loss. Nutrition & metabolism 2006, 3:24.
43. Sharman MJ, Gomez AL, Kraemer WJ, Volek JS: Very low-carbohydrate and low-fat diets affect fasting lipids and postprandial lipemia differently in overweight men. J Nutr 2004, 134(4):880-885.
44. Sharman MJ, Kraemer WJ, Love DM, Avery NG, Gomez AL, Scheett TP, Volek JS: A ketogenic diet favorably affects serum biomarkers for cardiovascular disease in normal-weight men. J Nutr 2002, 132(7):1879-1885.
45. Westman EC, Mavropoulos J, Yancy WS, Volek JS: A review of low-carbohydrate ketogenic diets. Current atherosclerosis reports 2003, 5(6):476-483.
46. Wood RJ, Volek JS, Davis SR, Dell'Ova C, Fernandez ML: Effects of a carbohydrate-restricted diet on emerging plasma markers for cardiovascular disease. Nutrition & metabolism 2006, 3:19.
47. Volek JS, Sharman MJ, Forsythe CE: Modification of lipoproteins by very low-carbohydrate diets. J Nutr 2005, 135(6):1339-1342.
48. Volek JS, Westman EC: Very-low-carbohydrate weight-loss diets revisited. Cleveland Clinic journal of medicine 2002, 69(11):849, 853, 856-848 passim.
49. Volek JS, Sharman MJ, Gomez AL, DiPasquale C, Roti M, Pumerantz A, Kraemer WJ: Comparison of a very low-carbohydrate and low-fat diet on fasting lipids, LDL subclasses, insulin resistance, and postprandial lipemic responses in overweight women. J Am Coll Nutr 2004, 23(2):177-184.
50. Volek JS, Sharman MJ: Cardiovascular and hormonal aspects of very-low-carbohydrate ketogenic diets. Obesity research 2004, 12 Suppl 2:115s-123s.

117
51. Volek JS, Sharman MJ, Gomez AL, Scheett TP, Kraemer WJ: An isoenergetic very low carbohydrate diet improves serum HDL cholesterol and triacylglycerol concentrations, the total cholesterol to HDL cholesterol ratio and postprandial pipemic responses compared with a low fat diet in normal weight, normolipidemic women. J Nutr 2003, 133(9):2756-2761.
52. Schoeler NE, Wood S, Aldridge V, Sander JW, Cross JH, Sisodiya SM: Ketogenic dietary therapies for adults with epilepsy: feasibility and classification of response. Epilepsy & behavior : E&B 2014, 37:77-81.
53. Sengupta P: The Laboratory Rat: Relating Its Age With Human's. International journal of preventive medicine 2013, 4(6):624-630.
54. Tsuchiya N, Harada Y, Taki M, Minematsu S, Maemura S, Amagaya S: [Age-related changes and sex differences on the serum chemistry values in Sprague-Dawley rats--I. 6-30 weeks of age]. Experimental animals / Japanese Association for Laboratory Animal Science 1995, 43(5):671-678.
55. Saito K, Ishikawa M, Murayama M, Urata M, Senoo Y, Toyoshima K, Kumagai Y, Maekawa K, Saito Y: Effects of sex, age, and fasting conditions on plasma lipidomic profiles of fasted Sprague-Dawley rats. PloS one 2013, 9(11).
56. Ellington A, Kullo I: Atherogenic lipoprotein subprofiling. Advances in clinical chemistry 2008, 46:295-317.
57. Mudd J, Borlaug B, Johnston P, Kral B, Rouf R, Blumenthal R, Kwiterovich P: Beyond low-density lipoprotein cholesterol: defining the role of low-density lipoprotein heterogeneity in coronary artery disease. Journal of the American College of Cardiology 2007, 50(18):1735-1741.
58. Sacks F, Campos H: Clinical review 163: Cardiovascular endocrinology: Low-density lipoprotein size and cardiovascular disease: a reappraisal. The Journal of clinical endocrinology and metabolism 2003, 88(10):4525-4532.
59. Wierzbicki A: Quality as well as quantity? Beyond low-density lipoprotein-cholesterol - the role of particle size. International journal of clinical practice 2007, 61(11):1780-1782.
60. Tantibhedhyangkul P, Hashim S, Van Itallie T: Effects of ingestion of long-chain triglycerides on glucose tolerance in man. Diabetes 1967, 16(11):796-799.
61. Eckel R, Hanson A, Chen A, Berman J, Yost T, Brass E: Dietary substitution of medium-chain triglycerides improves insulin-mediated glucose metabolism in NIDDM subjects. Diabetes 1992, 41(5):641-647.

118
62. Yost T, Erskine J, Gregg T, Podlecki D, Brass E, Eckel R: Dietary substitution of medium chain triglycerides in subjects with non-insulin-dependent diabetes mellitus in an ambulatory setting: impact on glycemic control and insulin-mediated glucose metabolism. Journal of the American College of Nutrition 1994, 13(6):615-622.
63. Kashiwaya Y, Pawlosky R, Markis W, King MT, Bergman C, Srivastava S, Murray A, Clarke K, Veech RL: A ketone ester diet increases brain malonyl-CoA and Uncoupling proteins 4 and 5 while decreasing food intake in the normal Wistar Rat. The Journal of biological chemistry 2010, 285(34):25950-25956.
64. Senior B, Loridan L: Direct regulatory effect of ketones on lipolysis and on glucose concentrations in man. Nature 1968, 219(5149):83-84.
65. Miles JM, Haymond MW, Gerich JE: Suppression of glucose production and stimulation of insulin secretion by physiological concentrations of ketone bodies in man. The Journal of clinical endocrinology and metabolism 1980, 52(1):34-37.
66. Kristian HM, Thomas S, Niels HS, Thomas G, Gerrit van H: Systemic, cerebral and skeletal muscle ketone body and energy metabolism during acute hyper-D-β-hydroxybutyrataemia in post-absorptive healthy males. The Journal of Clinical Endocrinology & Metabolism 2014.
67. Owen OE, Morgan AP, Kemp HG, Sullivan JM, Herrera MG, Cahill GF: Brain metabolism during fasting. The Journal of clinical investigation 1967, 46(10):1589-1595.
68. Veech R: The therapeutic implications of ketone bodies: the effects of ketone bodies in pathological conditions: ketosis, ketogenic diet, redox states, insulin resistance, and mitochondrial metabolism. Prostaglandins, leukotrienes, and essential fatty acids 2004, 70(3):309-319.
69. Papamandjaris AA, MacDougall DE, Jones PJ: Medium chain fatty acid metabolism and energy expenditure: obesity treatment implications. Life sciences 1997, 62(14):1203-1215.
70. Ruskin D, Kawamura M, Masino S: Reduced pain and inflammation in juvenile and adult rats fed a ketogenic diet. PloS one 2009, 4(12):e8349.
71. Kashiwaya Y, Sato K, Tsuchiya N, Thomas S, Fell D, Veech R, Passonneau J: Control of glucose utilization in working perfused rat heart. The Journal of biological chemistry 1994, 269(41):25502-25514.
72. Sato K, Kashiwaya Y, Keon C, Tsuchiya N, King M, Radda G, Chance B, Clarke K, Veech R: Insulin, ketone bodies, and mitochondrial energy transduction. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 1995, 9(8):651-658.

119
73. Veech RL, Chance B, Kashiwaya Y, Lardy HA, Cahill GF, Jr.: Ketone bodies, potential therapeutic uses. IUBMB Life 2001, 51(4):241-247.
74. Linde R, Hasselbalch S, Topp S, Paulson O, Madsen P: Global cerebral blood flow and metabolism during acute hyperketonemia in the awake and anesthetized rat. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 2006, 26(2):170-180.
75. Azzam R, Azar N: Marked Seizure Reduction after MCT Supplementation. Case reports in neurological medicine 2013, 2013:809151.
76. Henderson ST, Vogel JL, Barr LJ, Garvin F, Jones JJ, Costantini LC: Study of the ketogenic agent AC-1202 in mild to moderate Alzheimer's disease: a randomized, double-blind, placebo-controlled, multicenter trial. Nutrition & metabolism 2008, 6:31.
77. Corwin RL: Binge-type eating induced by limited access in rats does not require energy restriction on the previous day. Appetite 2004, 42(2):139-142.
78. Keenan KP, Ballam GC, Dixit R, Soper KA, Laroque P, Mattson BA, Adams SP, Coleman JB: The effects of diet, overfeeding and moderate dietary restriction on Sprague-Dawley rat survival, disease and toxicology. The Journal of nutrition 1997, 127(5 Suppl).
79. Keenan KP, Smith PF, Hertzog P, Soper K, Ballam GC, Clark RL: The Effects of Overfeeding and Dietary Restriction on Sprague-Dawley Rat Survival and Early Pathology Biomarkers of Aging. Toxicologic Pathology 1994.
80. Clarke K, Tchabanenko K, Pawlosky R, Carter E, Knight N, Murray A, Cochlin L, King M, Wong A, Roberts A et al: Oral 28-day and developmental toxicity studies of (R)-3-hydroxybutyl (R)-3-hydroxybutyrate. Regulatory toxicology and pharmacology : RTP 2012, 63(2):196-208.
81. Clarke K, Tchabanenko K, Pawlosky R, Carter E, Todd King M, Musa-Veloso K, Ho M, Roberts A, Robertson J, Vanitallie T et al: Kinetics, safety and tolerability of (R)-3-hydroxybutyl (R)-3-hydroxybutyrate in healthy adult subjects. Regulatory toxicology and pharmacology : RTP 2012, 63(3):401-408.
82. Kashiwaya Y, Bergman C, Lee J-H, Wan R, King M, Mughal M, Okun E, Clarke K, Mattson M, Veech R: A ketone ester diet exhibits anxiolytic and cognition-sparing properties, and lessens amyloid and tau pathologies in a mouse model of Alzheimer's disease. Neurobiology of Aging 2013, 34(6):1530-1539.
83. Birkhahn R, McCombs C, Clemens R, Hubbs J: Potential of the monoglyceride and triglyceride of DL-3-hydroxybutyrate for parenteral nutrition: synthesis and preliminary biological testing in the rat. Nutrition (Burbank, Los Angeles County, Calif) 1997, 13(3):213-219.

120
84. Puchowicz M, Smith C, Bomont C, Koshy J, David F, Brunengraber H: Dog model of therapeutic ketosis induced by oral administration of R,S-1,3-butanediol diacetoacetate. The Journal of nutritional biochemistry 2000, 11(5):281-287.
85. Brunengraber H: Potential of ketone body esters for parenteral and oral nutrition. Nutrition (Burbank, Los Angeles County, Calif) 1997, 13(3):233-235.
86. Desrochers S, Dubreuil P, Brunet J, Jetté M, David F, Landau BR, Brunengraber H: Metabolism of (R,S)-1,3-butanediol acetoacetate esters, potential parenteral and enteral nutrients in conscious pigs. The American journal of physiology 1995, 268(4 Pt 1):E660-E667.
87. Kashiwaya Y, King MT, Veech RL: Substrate signaling by insulin: a ketone bodies ratio mimics insulin action in heart. The American journal of cardiology 1997, 80(3A).
88. Kashiwaya Y, Sato K, Tsuchiya N, Thomas S, Fell DA, Veech RL, Passonneau JV: Control of glucose utilization in working perfused rat heart. The Journal of biological chemistry 1994, 269(41):25502-25514.
89. Im MJ, Freshwater MF, Hoopes JE: Enzyme activities in granulation tissue: Energy for collagen synthesis. The Journal of surgical research 1976, 20(2):121-125.
90. Im MJ, Hoopes JE: Enzyme activities in the repairing epithelium during wound healing. The Journal of surgical research 1970, 10(4):173-179.
91. Im MJ, Hoopes JE: Energy metabolism in healing skin wounds. The Journal of surgical research 1970, 10(10):459-464.
92. Im MJC, Hoopes JE: Energy metabolism in healing skin wounds. Journal of Surgical Research 1970.
93. Long L, Halliwell B: Artefacts in cell culture: α-Ketoglutarate can scavenge hydrogen peroxide generated by ascorbate and epigallocatechin gallate in cell culture media. Biochemical and Biophysical Research Communications 2011, 406(1).
94. Gupta A, Manhas N, Raghubir R: Energy metabolism during cutaneous wound healing in immunocompromised and aged rats. Molecular and cellular biochemistry 2004, 259(1-2):9-14.
95. Moor AN, Tummel E, Prather JL, Jung M, Lopez JJ, Connors S, Gould LJ: Consequences of age on ischemic wound healing in rats: altered antioxidant activity and delayed wound closure. Age (Dordrecht, Netherlands) 2014, 36(2):733-748.
96. Fitzpatrick DW, Fisher H: Carnosine, histidine, and wound healing. Surgery 1982, 91(1):56-60.
97. Nagai K, Suda T, Kawasaki K, Mathuura S: Action of carnosine and beta-alanine on wound healing. Surgery 1986, 100(5):815-821.

121
98. Roberts PR, Black K, Santamauro JT, Zaloga GP: Dietary Peptides Improve Wound Healing Following Surgery. Nutrition 1998, 14(3).
99. Sakae K, Yanagisawa H: Oral treatment of pressure ulcers with polaprezinc (zinc L-carnosine complex): 8-week open-label trial. Biological trace element research 2014, 158(3):280-288.
100. Ansurudeen I, Sunkari VG, Grünler J, Peters V, Schmitt CP, Catrina S-BB, Brismar K, Forsberg EA: Carnosine enhances diabetic wound healing in the db/db mouse model of type 2 diabetes. Amino acids 2012, 43(1):127-134.
101. Boldyrev AA, Dupin AM, Pindel EV, Severin SE: Antioxidative properties of histidine-containing dipeptides from skeletal muscles of vertebrates. Comparative biochemistry and physiology B, Comparative biochemistry 1988, 89(2):245-250.
102. Boldyrev AA, Severin SE: The histidine-containing dipeptides, carnosine and anserine: Distribution, properties and biological significance. Advances in Enzyme Regulation 1990, 30.
103. Kohen R, Yamamoto Y, Cundy KC, Ames BN: Antioxidant activity of carnosine, homocarnosine, and anserine present in muscle and brain. Proceedings of the National Academy of Sciences of the United States of America 1988, 85(9):3175-3179.
104. Altavilla D, Saitta A, Cucinotta D, Galeano M, Deodato B, Colonna M, Torre V, Russo G, Sardella A, Urna G et al: Inhibition of lipid peroxidation restores impaired vascular endothelial growth factor expression and stimulates wound healing and angiogenesis in the genetically diabetic mouse. Diabetes 2001, 50(3):667-674.
105. Masino SA, Geiger JD: The ketogenic diet and epilepsy: is adenosine the missing link? Epilepsia 2009, 50(2):332-333.
106. Masino SA, Kawamura M, Ruskin DN: Adenosine receptors and epilepsy: current evidence and future potential. International review of neurobiology 2014, 119:233-255.
107. Masino SA, Kawamura M, Wasser CD, Wasser CA, Pomeroy LT, Ruskin DN: Adenosine, ketogenic diet and epilepsy: the emerging therapeutic relationship between metabolism and brain activity. Current neuropharmacology 2009, 7(3):257-268.
108. Masino SA, Li T, Theofilas P, Sandau US, Ruskin DN, Fredholm BB, Geiger JD, Aronica E, Boison D: A ketogenic diet suppresses seizures in mice through adenosine A₁ receptors. The Journal of clinical investigation 2011, 121(7):2679-2683.
109. Chen W, Wu Y, Li L, Yang M, Shen L, Liu G, Tan J, Zeng W, Zhu C: Adenosine accelerates the healing of diabetic ischemic ulcers by improving autophagy of endothelial progenitor cells grown on a biomaterial. Scientific reports 2015, 5:11594.

122
110. Chiang B, Essick E, Ehringer W, Murphree S, Hauck MA, Li M, Chien S: Enhancing skin wound healing by direct delivery of intracellular adenosine triphosphate. American journal of surgery 2007, 193(2):213-218.
111. Cronstein BN: Adenosine receptors and wound healing. TheScientificWorldJournal 2004, 4:1-8.
112. Feoktistov I, Biaggioni I, Cronstein BN: Adenosine receptors in wound healing, fibrosis and angiogenesis: Springer; 2009.
113. Montesinos MC, Desai A, Chen J-FF, Yee H, Schwarzschild MA, Fink JS, Cronstein BN: Adenosine promotes wound healing and mediates angiogenesis in response to tissue injury via occupancy of A(2A) receptors. The American journal of pathology 2002, 160(6):2009-2018.
114. Montesinos MC, Desai-Merchant A, Cronstein BN: Promotion of Wound Healing by an Agonist of Adenosine A2A Receptor Is Dependent on Tissue Plasminogen Activator. Inflammation 2015.
115. Valls MD, Cronstein BN, Montesinos MC: Adenosine receptor agonists for promotion of dermal wound healing. Biochemical pharmacology 2009.
116. Valls MDD, Cronstein BN, Montesinos MC: Adenosine receptor agonists for promotion of dermal wound healing. Biochemical pharmacology 2009, 77(7):1117-1124.
117. Victor-Vega C, Desai A, Montesinos MC, Cronstein BN: Adenosine A2A receptor agonists promote more rapid wound healing than recombinant human platelet-derived growth factor (Becaplermin gel). Inflammation 2002, 26(1):19-24.
118. Sood RF, Gu H, Djukovic D, Deng L, Ga M, Muffley LA, Raftery D, Hocking AM: Targeted metabolic profiling of wounds in diabetic and non-diabetic mice. Wound repair and regeneration : official publication of the Wound Healing Society [and] the European Tissue Repair Society 2015.
119. Jain M, Nilsson R, Sharma S, Madhusudhan N, Kitami T, Souza AL, Kafri R, Kirschner MW, Clish CB, Mootha VK: Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science (New York, NY) 2012, 336(6084):1040-1044.
120. Li Y, Kilani RT, Rahmani-Neishaboor E, Jalili RB, Ghahary A: Kynurenine increases matrix metalloproteinase-1 and -3 expression in cultured dermal fibroblasts and improves scarring in vivo. The Journal of investigative dermatology 2014, 134(3):643-650.
121. Salimi Elizei S, Poormasjedi-Meibod M-SS, Li Y, Baradar Jalili R, Ghahary A: Effects of kynurenine on CD3+ and macrophages in wound healing. Wound repair and regeneration : official publication of the Wound Healing Society [and] the European Tissue Repair Society 2015, 23(1):90-97.

123
CHAPTER 4: ENHANCING WOUND HEALING WITH EXOGENOUS KETONE
SUPPLEMENTATION
4.1. Chapter Synopsis
In the previous chapter, we determined that exogenous ketone supplementation
significantly elevated blood ketone levels and reduced blood glucose levels in juvenile Sprague-
Dawley rats. In this chapter, we present data demonstrating that both BD and BMS+MCT
ketone supplements elicit age-dependent rapid elevation of blood ketone levels and reduction of
blood glucose levels in young (8 month) and aged (20 month) Fischer 344 rats similar to those
determined in Chapter 3. Additionally, we present data demonstrating the effects of food-
integrated oral ketone supplementation on an ischemic wound-healing model in young and aged
Fischer 344 rats. In the ischemic wound model, the flap placement on the dorsum prevents the
use of oral gavage; therefore, the ketone supplements were mixed into the food and fed ad
libitum. Nonetheless, this allowed continuous delivery of the ketone supplements in smaller
doses as the rats fed throughout the day. Compared to a bolus administration, multiple daily
doses or inclusion in the food may provide more sustained benefits. Additionally, we chose one
synthetic (BD) and one natural (BMS+MCT) ketone supplement for the following studies.
Though KE would seem to be a logical choice to continue with, the Fischer 344 rats refused to
eat the food mixed with this supplement; therefore, BD was used.

124
Portions of this chapter have been taken from a manuscript Poff AM, Kesl SL,
D’Agostino DP “Ketone Supplementation for Health and Disease” Book Chapter, Oxford Press
(Submitted). A copy of this article may be found in Appendix C. Materials and Methods
presented in this chapter may be found in Appendix A.
4.2. Dietary Ketone Supplementation Increases Blood Flow and Wound Closure in an
Ischemic Wound Model in Young and Aged Fischer Rats
It has been well established that the ketogenic diet (KD) induces “keto-adaptation”, a
physiologic state characterized by a shift away from glucose metabolism and towards fat and
ketone body metabolism. [1-3]. As discussed in previous chapters, there are many
physiological changes associated with sustained ketosis that may contribute to its multifaceted
therapeutic potential for many disease states including chronic wounds. Thus, we previously
measured blood glucose, ketones, lipids, and other biochemical metabolites in response to
chronic oral ketone administration to show physiological equivalence to KD [4]. Increasing
evidence shows that limited energy and nutrient exchange is associated with age-related
impairment of wound healing. We hypothesized that oral ketone supplementation without
dietary restriction would enhance wound closure in young and aged Fischer rats by improving
blood flow and supplying an alternative energy substrate. In our preliminary studies, we
measured the magnitude and duration of ketosis following administration of a single 6.5g/kg
dose of ketone precursors: 1,3-Butanediol (BD), Na+/K+ βHB salt and medium chain
triglyceride (MCT) oil 1:1 mixture (BMS+MCT), or water in young and aged Fischer 344 rats
(n=6). Substances were administered through an intragastric gavage, and whole blood samples

125
(10 µl) were acquired for analysis of glucose and βHB at 0, 0.5, 1, 1.5, 2, 4, 8, 12, and 24 hours
following administration. Following the creation of ischemic wounds, the ketogenic
supplements were added to a standard diet fed ad libitum for 28 days. Laser Doppler imaging of
the ischemic peri-wound tissue every seven days demonstrated significantly increased blood
flow in young rats (n=10) fed BD at day 14 and 28 (p<0.001) and BMS+MCT at day 7, 14, and
28 (p<0.01). In aged rats, blood flow was significantly increased in BD-fed at day 14 and
BMS+MCT-fed at days 7 and 14 (p<0.05). Wound size was significantly smaller in young rats
fed BD and BMS+MCT compared to control at 11 and 14 days following wound creation
(p<0.05). In aged rats, BD-fed wounds were significantly smaller at days 11 and 14 (p<0.05)
and in BMS+MCT-fed at days 11, 14, and 28 (p<0.05). Wound healing improved by three days
in aged BD-fed, seven days in young BMS+MCT-fed, and ten days in aged BMS+MCT-fed
compared to the healing time line of the control animals.
4.2.1 Aged rats metabolize exogenous ketone supplements differently than young rats
In Chapter 3, we studied if oral ketone administration could elicit similar physiological
effects as the KD by determining how blood glucose, ketones, and lipids and other biochemical
metabolites are affected by chronic ketone administration [4]. Here we present evidence that
chronic administration of ketone supplements can induce a state of nutritional ketosis without
the need for dietary carbohydrate restriction, enhance blood flow, and augment wound closure
in young and aged Fischer 344 rats.
Both young and aged rats exhibited elevated ketones within 30 minutes of the bolus
administration for both BD and BMS+MCT supplements and were sustained for 12 hours
(p<0.05). However, BD supplemented aged rats demonstrated a peak elevation of blood βHB

126
levels at 8 hours, which was delayed compared to their younger counterparts who had peaked
blood βHB levels at 4 hours post intragastric gavage. Additionally, aged animals supplemented
with either of the ketone supplements demonstrated a ~1.5x higher elevation of blood ΒHB
levels for the same dose compared to the young animals (Figures 4.1A, B). An additional set of
young and aged animals were tested with a 10 g/kg dose of their respective ketone supplements
and a similar pattern emerged, young and aged animals’ blood βHB levels both peaked at 12
hours post gavage (data not shown). Aged animals administered BD appeared sedated, had
increased blood BHB up to 8 mM, and hypoglycemia (<50 mg/dL). We speculated that these
sedative effects might be because BD is metabolized as an alcohol; thus, we decided that an
intragastric gavage at 10 g/kg was not optimal for aged animals. Young animals were able to
tolerate the dose.
The metabolic challenge of a wound is exacerbated in elderly patients [5]. Without
sufficient energy (ATP) levels, wound healing is significantly impaired. As discussed, the ATP
deficiency creates an imbalance of energy production versus utilization (the catabolism to
anabolism ratio) leading to loss of lean body mass (LBM) [6, 7]. Additionally, Gupta and
colleagues demonstrated that metabolic enzymes hexokinase, citrate synthase,
phosphofructokinase, and lactate dehydrogenase were decreased in aged rats. Additionally, the
metabolic enzyme glucose-6-phosphate dehydrogenase exhibited elevated activity and the
beginnings stages followed by a significant decreased activity in a later phase compared to
controls [8]. These age-dependent metabolic changes could explain the difference between the
young and aged rats’ response to acute ketone supplementation. However, ketones have been
shown to increase Kreb’s cycle intermediates, mitochondrial biogenesis, and enhance overall

127
metabolic efficiency; thus, this is one potential mechanism of how the prolonged ketone
administration could have enhanced wound healing.
4.2.2. Hypoglycemic effect of hyperketonemia attenuated with age
In Chapter 3, we demonstrated that at baseline and 4 weeks, 4 hours after intragastric
gavage with BMS+MCT (5 g/kg), the elevation of blood ketones was inversely correlated with
the reduction in blood glucose (r2=0.4314, p=0.0203, r2=0.8619, p<0.0001). This correlation
was not observed at any time point for BD supplemented rats [4]. Veech and colleagues have
demonstrated that administration of similar ketone supplement simultaneously decreased blood
glucose and blood insulin by approximately 50%. Our data show the same correlation between
elevated blood ketone levels and reduced blood glucose levels with exogenous BMS+MCT
supplementation in 8-month-old Fischer rats, supporting the work of Veech and confirming our
previous work (Figure 4.2). However, this hypoglycemic effect is not apparent in the 20-month
animals (Figure 4.2). As discussed, metabolic changes that occur during aging may play a role
in diminishing this relationship. Furthermore, insulin resistance has been shown to increase with
age [9]. Veech and colleagues have determined that ketone-induced hypoglycemia occurs via
increasing insulin sensitivity [10-12]. In the older adult, with pre-existing has insulin resistance,
the same ketone-induced effect on insulin sensitivity may not occur or it may not be enough to
cause hypoglycemia. It should be noted that even though there wasn’t a significant correlation
between blood ketone levels and blood glucose levels in aged ketone supplemented rats, there
was suppression of glucose based on the linear regression analysis (data not shown). This

128
observation supports the speculation that an age-dependent metabolic change diminishes the
significant correlation but doesn’t completely neutralize the relationship.
Additionally, hyperketonemia was observed throughout the 28-day wound healing study
in the BD supplemented young and aged rats, but was not noted in the BMS+MCT
supplemented rats (Figure 4.5A). In Chapter 3, we demonstrated that ketone supplementation
caused a sustained reduction in glucose over the time course of the intragastric gavage study;
however, this sustained reduction of glucose was not observed over the course of the wound
healing study (Figure 4.5B). In a follow-up chronic feeding study (15 weeks), food-integrated
ketone supplementation resulted in elevated blood ketone levels without affecting the blood
glucose levels throughout the study, which supports our findings in this study (data not shown,
unpublished data).
4.2.3. Ketone supplementation increases blood flow in both young and aged rats
Restoration of blood flow via angiogenesis is critical for healing a wound and to support
revascularization of grafts including tissue engineered skin substitutes. Venous, arterial, and
diabetic blood flow insufficiencies are major underlying contributors to chronic wound
development. Additionally, older patients have a greater prevalence of hypertension, diabetes,
and smoking, which exacerbate the age-dependent angiogenic insufficiencies [13, 14]. In this
study, oral ketone supplementation significantly increased blood flow in young and aged rats
supplemented with BD or BMS+MCT (Figure 5.3B). In the raw Laser Doppler data, darker
colors are indicative of low blood flow/ischemia and lighter colors demonstrate increased blood
flow. The increased blood flow in the ketone-supplemented rats is clearly visible in the raw data

129
(Figure 5.3A). This wound-healing model was created to induce ischemia and thus delay
wound healing [15, 16]. Further studies need to be conducted to measure oxygen concentrations
during wound healing to establish that increased blood flow reverses flap ischemia leading to
accelerated wound healing. In a study by Hasselbalch and colleagues, exogenous ketone
supplementation exhibited a 39% increase in cerebral blood flow [17]. Additionally, VEGF, a
potent angiogenic factor, has shown to be elevated with exogenous ketone supplementation [18-
20]. As discussed in Chapter 3, ketone supplementation has been determined to increase the
potent vasodilator, adenosine. These two mechanisms may explain how ketone supplementation
increased blood flow in the ischemic wound model.
4.2.4 Ketone supplementation accelerates wound closure in young and aged rats
As the population continues to age, there is a critical need to develop effective wound
healing therapies. In this study, we have demonstrated that exogenous ketone supplementation
enhanced wound closure by ten days (36% faster) in aged rats supplemented with BMS+MCT
and three days (10% faster) in aged rats supplemented with BD compared to control aged rats
(Figure 4.4). Additionally, young rats supplemented with BMS+MCT healed three days earlier
(14% faster) compared to standard diet fed young rats (Figure 4.4). Future studies are designed
to determine the wound healing effect of topical ketone administration alone and in combination
with the oral exogenous ketone therapy. As is, this therapy could allow patients to benefit from
nutritional ketosis without dietary restriction.
Studies by Nevin and colleagues investigated the influence of a topical application of
virgin coconut oil (VCO) on the healing of dermal wounds in young rats [21-23]. Their results

130
demonstrated a significant beneficial effect of VCO administration on intracellular and
extracellular matrix components compared to controls including increased cross-linking
collagen molecules indicating greater wound tensile strength and increased total DNA of the
granulation tissue. Additionally, VCO therapy induced greater antioxidant capacity (superoxide
dismutase 2, glutathione reductase, glutathione peroxidase) during wound healing, which led to
a decrease in lipid peroxides (MDA). They concluded that the wound healing property of VCO
might be due to its minor biologically active components and antimicrobial fatty acids. Previous
studies have demonstrated that food integrated coconut oil was able to eliminate bacterial
infection and stimulate the immune response [24]. Coconut oil is a natural source of medium
chain triglycerides (MCTs), which have been shown to modulate cellular proliferation, cell
signaling, and growth factor activities as well as elicit ketogenesis as seen in this study and our
previous study [4, 25-27].
The BMS contains potassium and other minerals to prevent sodium overload.
Maintaining an optimal sodium-mineral ratio should help offset any potential adverse effects of
sodium on blood pressure. For example, multiple studies have shown that potassium provides
an antihypertensive effect and protects cardiovascular damage in salt-sensitive hypertension
[28, 29]. It is speculated that this formulation will be especially beneficial for elderly patients
most susceptible to sodium-induced hypertension [11]. The dose of the salt solution is easily
adjusted to benefit the patients’ needs. In this study a 10% ketone salt solution is mixed in a 1:1
ratio with MCT oil, which allows for reduced dosing of each component compared to
administering the compounds individually. This reduces the potential for side effects (sodium-
induced hypertension, gastric side effects, etc.) and results in distinct synergistic blood ketone
profile [30]. A noteworthy observation from our previous study revealed that in rats, MCT alone

131
was more effective than BMS+MCT or BMS alone at inducing ketosis. However, preliminary
human data suggests that the BMS+MCT mixture is most effective at inducing ketosis and
MCT the least. This suggests that there is inter-species variability in the metabolic response to
ketone supplements, which will need to be further characterized to fully understand effects in
humans. In the rats, the BMS+MCT supplement elevated blood ketones similar to that of MCT
alone; however, the gastric side effects were not observed suggesting a potential method for
avoiding this unwanted adverse effect [31].
4.2.5. Food- integrated ketone supplementation did not elicit weight loss in young and
aged rats
The KD is hypothesized to induce weight loss by reducing appetite through the satiety
effect of ketone bodies, reducing lipogenesis and increasing lipolysis, and enhancing metabolic
efficiency with fat and ketone metabolism [32]. Recently, preliminary studies have shown that
exogenous ketone supplementation can also induce weight loss. The administrations of both
βHB and BD have been shown to decrease food intake in rats and pigmy goats [33-37].
Similarly, it is suggested that MCTs increase satiety, resulting in reduced food intake and
weight loss as a consequence of their rapid oxidation into ketone bodies [38-40]. MCTs may
further counteract fat deposition in adipocytes by increasing thermogenesis [41]. Several studies
in animals and humans have revealed increased energy expenditure and lipid oxidation with
MCTs compared to LCTs [42-51]. Ketone esters have also been shown to affect weight in
mice, rats, and humans [20, 52-55]. In Chapter 3, all five ketogenic supplements tested via
intragastric oral gavage (BD, KE, MCT, BMS, BMS+MCT) inhibited weight gain compared to

132
control animals [4]. Similarly, a 15-week chronic feeding study in which KE, BMS, or
BMS+MCT replaced approximately 20% of the diet by weight, fed ad libitum, led to reduced
weight gain compared to control animals (data not shown, unpublished data).
Even though weight loss has been noted in a variety of studies, significant weight loss
was not observed in either young or aged rats of this study (Figure 4.6A). To improve post-
anesthetic appetite, thereby limiting the initial weight loss that is standard with an invasive
surgery, the rats were fasted for 18 hours before surgery. . Weight loss was noted during the
first week after surgery, but did not reach significance in any group, and was similar across all
groups demonstrating that it wasn’t a ketone-mediated effect. Weight was maintained in all
groups for the remainder of the study. This initial fasting may be a reason that the initial weight
loss during the first week post-surgery did not reach significance. In the previously discussed
studies, rats were Sprague Dawley and were either juvenile or still in a growth phase, whereas
in this study the rats were mature or elderly Fischer 344s. Age and strain variability may
account for the lack of weight loss in this study compared to previously discussed experiments.
Particularly in elderly patients where malnutrition may already be present, it is critical to
prevent the wound from parasitizing substrates from the rest of the body for energy production,
resulting in what is called protein-energy malnutrition (PEM). The body catabolizes the
muscle, skin, and bone to support the synthesis of proteins, inflammatory cells, and collagen
needed to fight infection and repair the wound, causing a loss of lean body mass (LBM). As an
individual loses more LBM, wound healing is more likely to be delayed. A loss of more than
15% LBM impairs wound healing, a 30% LBM loss stops wound healing, and a loss of 40%
LBM typically results in death [56-60]. A body in ketosis has been shown to be muscle sparing
as it is adapted to using the readily available fat stores, attenuating the catabolic effects [7]. The

133
weight maintenance seen in this wound healing study may reflect this protein-sparing effect.
Moreover, if ketosis enhances insulin sensitivity, glucose uptake by the insulin-sensitive
skeletal muscle should be increased. Together, these effects would help support muscle tissue
health and function, suggesting another possible mechanism of ketone supplementation in
diminishing LBM and PEM.
4.3 Closing Remarks
It has been debated if chronological age alone affects wound healing. However, with the
aged population showing decreased blood flow leading to the decline of nutrient exchange,
decreased metabolic activity and ATP production at the wound bed, it is clear how these factors
may result in an exacerbation of chronic wounds in older adults. The discouraging results from
studies focusing on single molecular targets are not surprising since wound healing is the
outcome of a complex set of interactions between numerous factors. This is likely one reason
most wound therapies are minimally effective. Our approach utilizes endogenous physiology to
reach the wound bed systemically with one therapy, exogenous ketone supplementation, that
has multiple downstream effects.. Even though there is much work still to be done, this
approach shows promising results as a potential wound therapy.
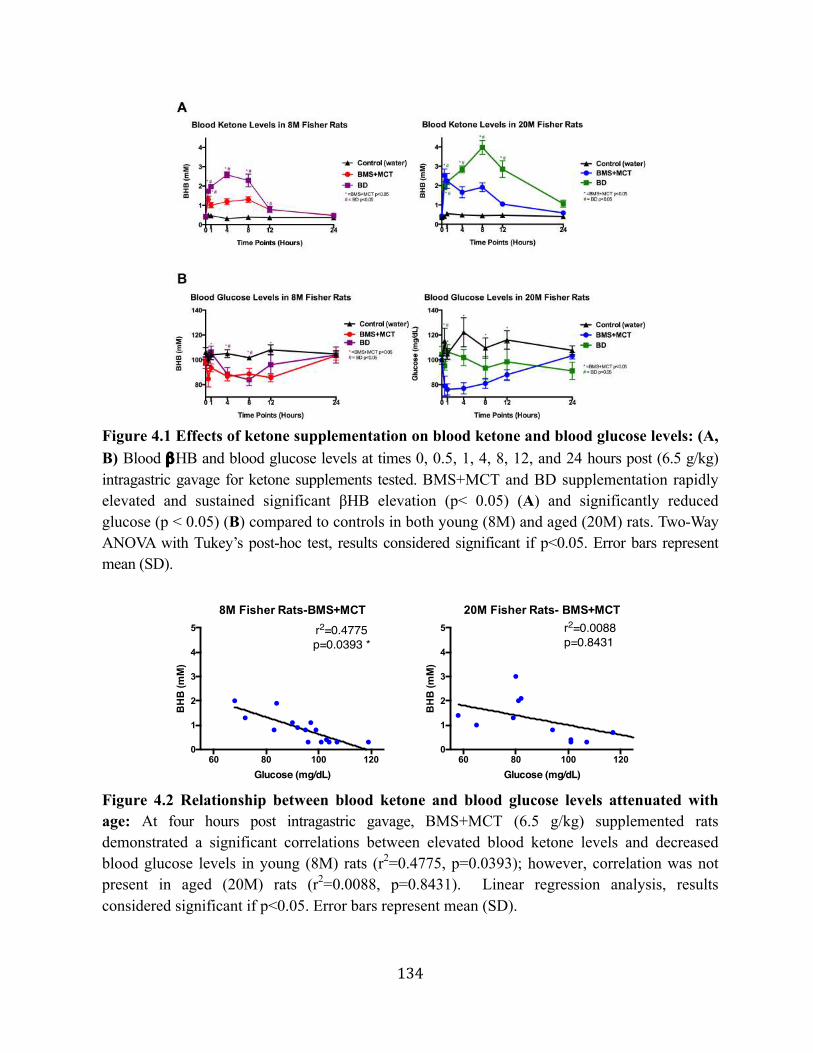
134
Figure 4.1 Effects of ketone supplementation on blood ketone and blood glucose levels: (A, B) Blood βHB and blood glucose levels at times 0, 0.5, 1, 4, 8, 12, and 24 hours post (6.5 g/kg) intragastric gavage for ketone supplements tested. BMS+MCT and BD supplementation rapidly elevated and sustained significant βHB elevation (p< 0.05) (A) and significantly reduced glucose (p < 0.05) (B) compared to controls in both young (8M) and aged (20M) rats. Two-Way ANOVA with Tukey’s post-hoc test, results considered significant if p<0.05. Error bars represent mean (SD).
Figure 4.2 Relationship between blood ketone and blood glucose levels attenuated with age: At four hours post intragastric gavage, BMS+MCT (6.5 g/kg) supplemented rats demonstrated a significant correlations between elevated blood ketone levels and decreased blood glucose levels in young (8M) rats (r2=0.4775, p=0.0393); however, correlation was not present in aged (20M) rats (r2=0.0088, p=0.8431). Linear regression analysis, results considered significant if p<0.05. Error bars represent mean (SD).
8M Fisher Rats-BMS+MCT
60 80 100 1200
1
2
3
4
5
Glucose (mg/dL)
BH
B (m
M)
r2=0.4775p=0.0393 *
20M Fisher Rats- BMS+MCT
60 80 100 1200
1
2
3
4
5
Glucose (mg/dL)
BH
B (m
M)
r2=0.0088p=0.8431
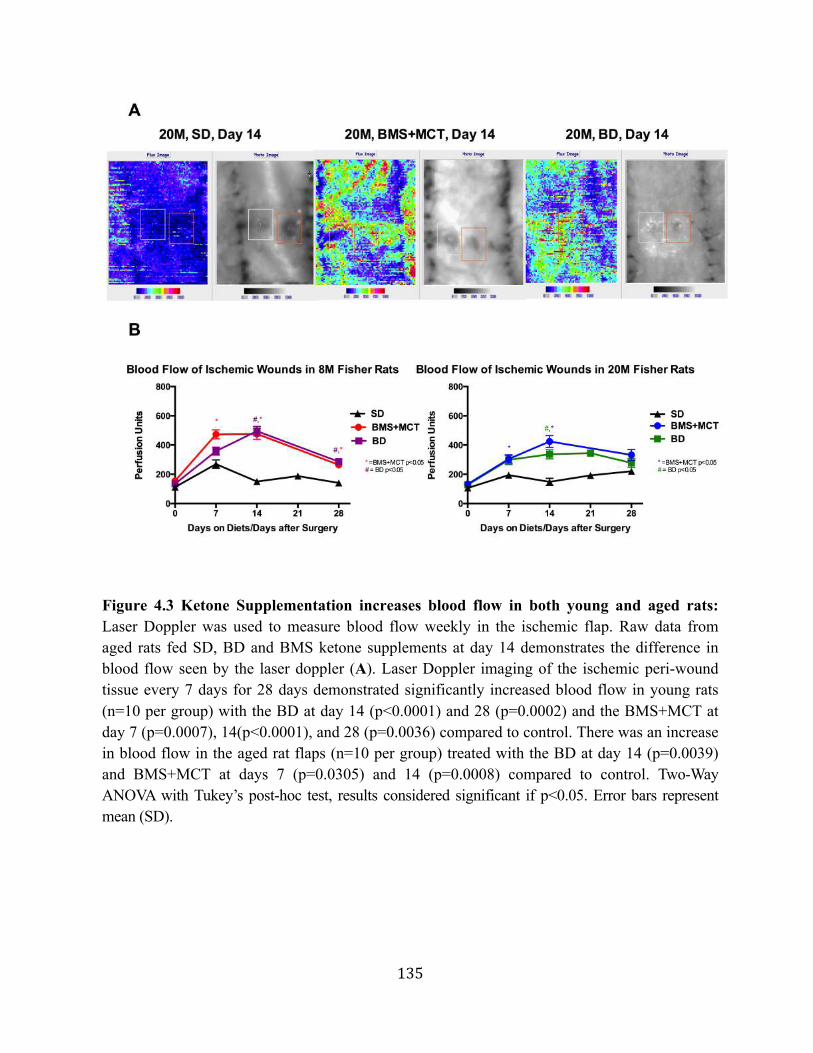
135
Figure 4.3 Ketone Supplementation increases blood flow in both young and aged rats: Laser Doppler was used to measure blood flow weekly in the ischemic flap. Raw data from aged rats fed SD, BD and BMS ketone supplements at day 14 demonstrates the difference in blood flow seen by the laser doppler (A). Laser Doppler imaging of the ischemic peri-wound tissue every 7 days for 28 days demonstrated significantly increased blood flow in young rats (n=10 per group) with the BD at day 14 (p<0.0001) and 28 (p=0.0002) and the BMS+MCT at day 7 (p=0.0007), 14(p<0.0001), and 28 (p=0.0036) compared to control. There was an increase in blood flow in the aged rat flaps (n=10 per group) treated with the BD at day 14 (p=0.0039) and BMS+MCT at days 7 (p=0.0305) and 14 (p=0.0008) compared to control. Two-Way ANOVA with Tukey’s post-hoc test, results considered significant if p<0.05. Error bars represent mean (SD).
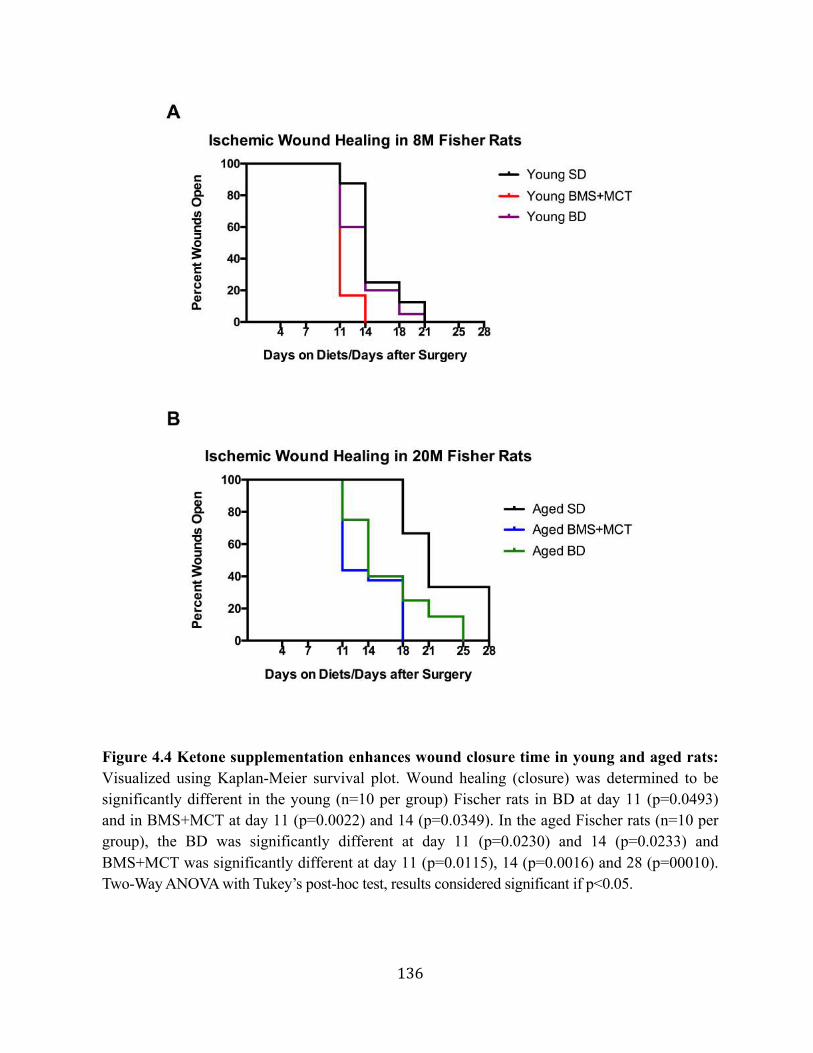
136
Figure 4.4 Ketone supplementation enhances wound closure time in young and aged rats: Visualized using Kaplan-Meier survival plot. Wound healing (closure) was determined to be significantly different in the young (n=10 per group) Fischer rats in BD at day 11 (p=0.0493) and in BMS+MCT at day 11 (p=0.0022) and 14 (p=0.0349). In the aged Fischer rats (n=10 per group), the BD was significantly different at day 11 (p=0.0230) and 14 (p=0.0233) and BMS+MCT was significantly different at day 11 (p=0.0115), 14 (p=0.0016) and 28 (p=00010). Two-Way ANOVA with Tukey’s post-hoc test, results considered significant if p<0.05.
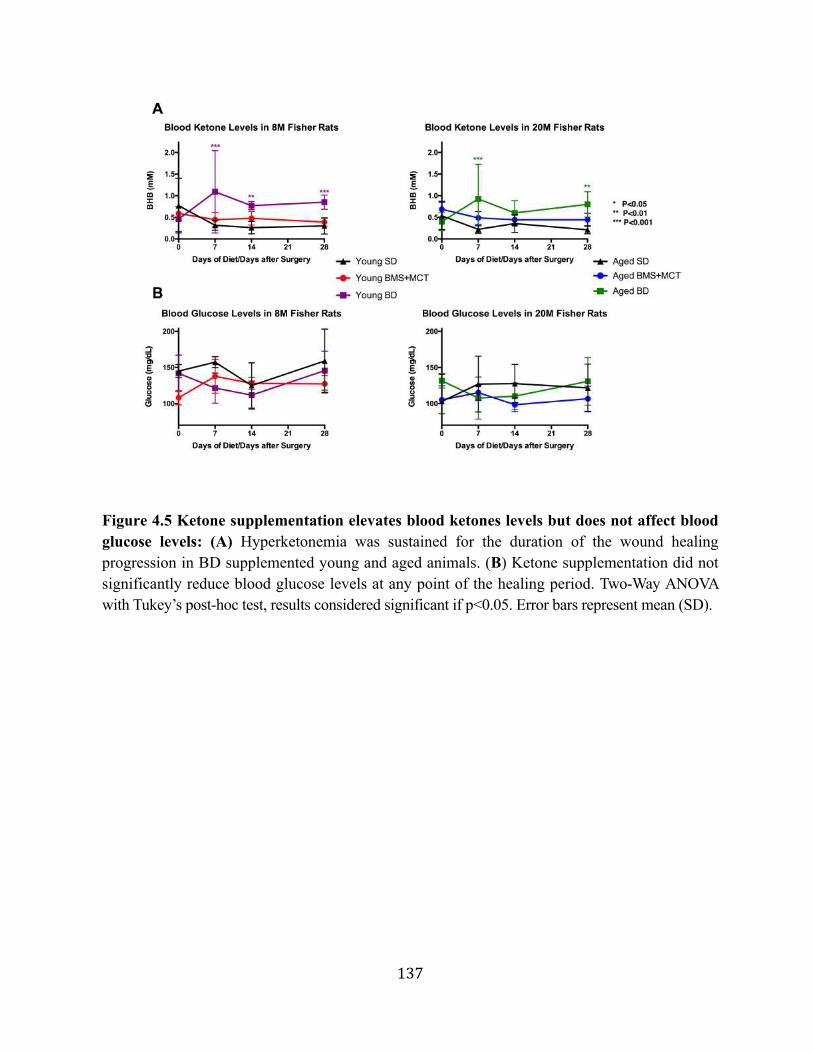
137
Figure 4.5 Ketone supplementation elevates blood ketones levels but does not affect blood glucose levels: (A) Hyperketonemia was sustained for the duration of the wound healing progression in BD supplemented young and aged animals. (B) Ketone supplementation did not significantly reduce blood glucose levels at any point of the healing period. Two-Way ANOVA with Tukey’s post-hoc test, results considered significant if p<0.05. Error bars represent mean (SD).
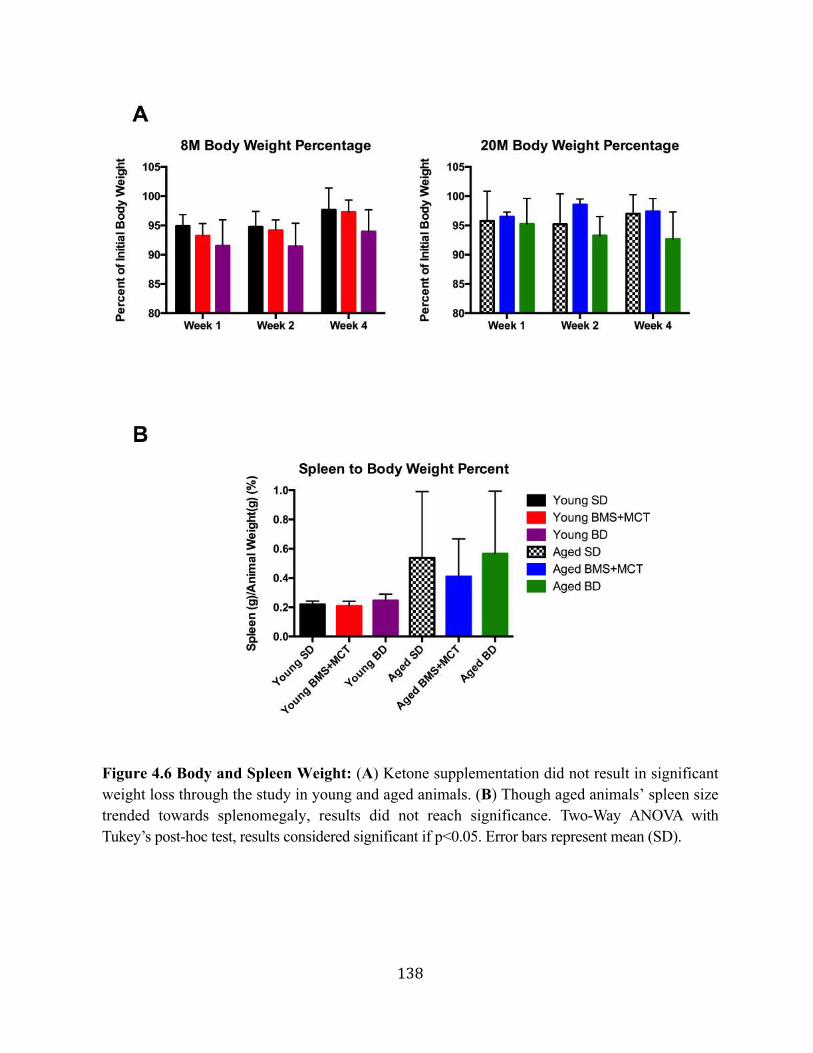
138
Figure 4.6 Body and Spleen Weight: (A) Ketone supplementation did not result in significant weight loss through the study in young and aged animals. (B) Though aged animals’ spleen size trended towards splenomegaly, results did not reach significance. Two-Way ANOVA with Tukey’s post-hoc test, results considered significant if p<0.05. Error bars represent mean (SD).

139
4.3. ReferencesforChapter4
1. Halevy A, Peleg-Weiss L, Cohen R, Shuper A: An update on the ketogenic diet, 2012. Rambam Maimonides medical journal 2012, 3(1):e0005.
2. Amari A, Grace N, Fisher W: Achieving and maintaining compliance with the ketogenic diet. Journal of applied behavior analysis 1995, 28(3):341-342.
3. Zhang Y, Kuang Y, LaManna J, Puchowicz M: Contribution of brain glucose and ketone bodies to oxidative metabolism. Advances in experimental medicine and biology 2013, 765:365-370.
4. Kesl S, Poff, A., Ward, N., Fiorelli, T., Ari, C., Van Putten, A., Sherwood, J., Arnold, P., D'Agostino, D.P: Effects of Exogenous Ketone Supplementation on Blood Ketone, Glucose, Triglyceride, and Lipoprotein Levels in Sprague-Drawley Rats. Nutrition & Metabolism (Accepted with Revisions) 2015.
5. Molnar JA: Nutrition and wound healing. CRC 2006.
6. Chiang B, Essick E, Ehringer W, Murphree S, Hauck M, Li M, Chien S: Enhancing skin wound healing by direct delivery of intracellular adenosine triphosphate. American journal of surgery 2007, 193(2):213-218.
7. Demling R: Nutrition, anabolism, and the wound healing process: an overview. Eplasty 2009, 9.
8. Gupta A, Manhas N, Raghubir R: Energy metabolism during cutaneous wound healing in immunocompromised and aged rats. Molecular and cellular biochemistry 2004, 259(1-2):9-14.
9. Park MH, Kim DH, Lee EK, Kim ND, Im DS, Lee J, Yu BP, Chung HY: Age-related inflammation and insulin resistance: a review of their intricate interdependency. Archives of pharmacal research 2014, 37(12):1507-1514.
10. Veech R: The therapeutic implications of ketone bodies: the effects of ketone bodies in pathological conditions: ketosis, ketogenic diet, redox states, insulin resistance, and mitochondrial metabolism. Prostaglandins, leukotrienes, and essential fatty acids 2004, 70(3):309-319.
11. Veech RL: Ketone ester effects on metabolism and transcription. Journal of lipid research 2014, 55(10):2004-2006.
12. Veech RL, Chance B, Kashiwaya Y, Lardy HA, Cahill GF, Jr.: Ketone bodies, potential therapeutic uses. IUBMB Life 2001, 51(4):241-247.
13. Reed M, Edelberg J: Impaired angiogenesis in the aged. Science of aging knowledge environment : SAGE KE 2004, 2004(7).

140
14. Gosain A, DiPietro L: Aging and wound healing. World journal of surgery 2004, 28(3):321-326.
15. Gould L, Leong M, Sonstein J, Wilson S: Optimization and validation of an ischemic wound model. Wound repair and regeneration : official publication of the Wound Healing Society [and] the European Tissue Repair Society 2005, 13(6):576-582.
16. Trujillo AN, Kesl SL, Sherwood J, Wu M, Gould LJ: Demonstration of the rat ischemic skin wound model. Journal of visualized experiments : JoVE 2015(98).
17. Hasselbalch SG, Madsen PL, Hageman LP, Olsen KS, Justesen N, Holm S, Paulson OB: Changes in cerebral blood flow and carbohydrate metabolism during acute hyperketonemia. The American journal of physiology 1996, 270(5 Pt 1):51.
18. Bao P, Kodra A, Tomic-Canic M, Golinko M, Ehrlich H, Brem H: The role of vascular endothelial growth factor in wound healing. The Journal of surgical research 2009, 153(2):347-358.
19. Mahdavian Delavary B, van der Veer W, van Egmond M, Niessen F, Beelen R: Macrophages in skin injury and repair. Immunobiology 2011, 216(7):753-762.
20. Isales CM, Min L, Hoffman WH: Acetoacetate and beta-hydroxybutyrate differentially regulate endothelin-1 and vascular endothelial growth factor in mouse brain microvascular endothelial cells. Journal of diabetes and its complications 1999, 13(2):91-97.
21. Nevin KG, Rajamohan T: Effect of topical application of virgin coconut oil on skin components and antioxidant status during dermal wound healing in young rats. Skin pharmacology and physiology 2010, 23(6):290-297.
22. Nevin KG, Rajamohan T: Beneficial effects of virgin coconut oil on lipid parameters and in vitro LDL oxidation. Clinical biochemistry 2004, 37(9):830-835.
23. Nevin KG, Rajamohan T: Virgin coconut oil supplemented diet increases the antioxidant status in rats. Food Chemistry 2006, 99(2).
24. de Pablo MA, Puertollano MA, Gálvez A, Ortega E, Gaforio JJ, Alvarez de Cienfuegos G: Determination of natural resistance of mice fed dietary lipids to experimental infection induced by Listeria monocytogenes. FEMS immunology and medical microbiology 2000, 27(2):127-133.
25. Bandyopadhyay U, Das D, Banerjee RK: Reactive oxygen species: oxidative damage and pathogenesis. Current science 1999, 77(5):658-666.
26. Jiang WG, Hiscox S, Hallett MB, Scott C, Horrobin DF, Puntis MC: Inhibition of hepatocyte growth factor-induced motility and in vitro invasion of human colon cancer cells by gamma-linolenic acid. British journal of cancer 1995, 71(4):744-752.

141
27. Rose DP, Connolly JM, Liu XH: Effects of linoleic acid on the growth and metastasis of two human breast cancer cell lines in nude mice and the invasive capacity of these cell lines in vitro. Cancer research 1994, 54(24):6557-6562.
28. Ando K, Matsui H, Fujita M, Fujita T: Protective effect of dietary potassium against cardiovascular damage in salt-sensitive hypertension: possible role of its antioxidant action. Current vascular pharmacology 2010, 8(1):59-63.
29. Skrabal F, Auböck J, Hörtnagl H: Low sodium/high potassium diet for prevention of hypertension: probable mechanisms of action. Lancet (London, England) 1981, 2(8252):895-900.
30. D'Agostino D, Arnold P, Kesl S: Compositions and methods for producing elevated and sustained ketosis. In: Compositions and methods for producing elevated and sustained ketosis. Google Patents; 2014.
31. Kesl S, Poff, A., Ward, N., Fiorelli, T., Ari, C., Van Putten, A., Sherwood, J., Arnold, P., D'Agostino, D.: Effects of Oral Ketone Supplementation on Blood Ketone, Glucose, Triglyceride, and Lipoprotein Levels in Sprague-Drawley Rats. Nutrition & Metabolism (Accepted with Revisions) 2015.
32. Paoli A: Ketogenic diet for obesity: friend or foe? International journal of environmental research and public health 2014, 11(2):2092-2107.
33. Arase K, Fisler JS, Shargill NS, York DA, Bray GA: Intracerebroventricular infusions of 3-OHB and insulin in a rat model of dietary obesity. The American journal of physiology 1988, 255(6 Pt 2):81.
34. Carpenter RG, Grossman SP: Plasma fat metabolites and hunger. Physiology & behavior 1983, 30(1):57-63.
35. Davis RJ, Brand MD, Martin BR: The effect of insulin on plasma-membrane and mitochondrial-membrane potentials in isolated fat-cells. The Biochemical journal 1981, 196(1):133-147.
36. Langhans W, Wiesenreiter F, Scharrer E: Different effects of subcutaneous D,L-3-hydroxybutyrate and acetoacetate injections on food intake in rats. Physiology & behavior 1983, 31(4):483-486.
37. Rossi R, Dörig S, Del Prete E, Scharrer E: Suppression of feed intake after parenteral administration of D-beta-hydroxybutyrate in pygmy goats. Journal of veterinary medicine A, Physiology, pathology, clinical medicine 2000, 47(1):9-16.
38. Poppitt SD, Strik CM, MacGibbon AKH, McArdle BH, Budgett SC, McGill AT: Fatty acid chain length, postprandial satiety and food intake in lean men. Physiology & Behavior 2010, 101(1):161-167.

142
39. Krotkiewski M: Value of VLCD supplementation with medium chain triglycerides. International journal of obesity and related metabolic … 2001.
40. Wymelbeke VV, Himaya A: Influence of medium-chain and long-chain triacylglycerols on the control of food intake in men. The American journal … 1998.
41. Dulloo AG: The search for compounds that stimulate thermogenesis in obesity management: from pharmaceuticals to functional food ingredients. Obesity reviews 2011, 12(10):866-883.
42. Bach AC, Babayan VK: Medium-chain triglycerides: an update. The American journal of clinical nutrition 1982, 36(5):950-962.
43. Karen M, Welma S: Effects of Medium-Chain Triglycerides on Weight Loss and Body Composition: A Meta-Analysis of Randomized Controlled Trials. Journal of the Academy of Nutrition and Dietetics 2015, 115(2).
44. Baba N, Bracco EF, Hashim SA: Enhanced thermogenesis and diminished deposition of fat in response to overfeeding with diet containing medium chain triglyceride. The American journal of clinical … 1982.
45. Crozier G, Bois-Joyeux B, Chanez M, Girard J, Peret J: Metabolic effects induced by long-term feeding of medium-chain triglycerides in the rat. Metabolism 1987.
46. Dulloo AG, Fathi M, Mensi N: Twenty-four-hour energy expenditure and urinary catecholamines of humans consuming low-to-moderate amounts of medium-chain triglycerides: a dose-response …. European journal of clinical … 1996.
47. Ferreira L, Lisenko K, Barros B: Influence of medium‐chain triglycerides on consumption and weight gain in rats: a systematic review. Journal of animal … 2014.
48. Scalfi L, Coltorti A, Contaldo F: Postprandial thermogenesis in lean and obese subjects after meals supplemented with medium-chain and long-chain triglycerides. The American journal of clinical … 1991.
49. Seaton TB, Welle SL, Warenko MK: Thermic effect of medium-chain and long-chain triglycerides in man. The American journal … 1986.
50. St-Onge MP, Bourque C, Jones PJH, Ross R: Medium-versus long-chain triglycerides for 27 days increases fat oxidation and energy expenditure without resulting in changes in body composition in overweight …. International journal of … 2003.
51. St‐Onge MP, Ross R, Parsons WD: Medium‐chain triglycerides increase energy expenditure and decrease adiposity in overweight men. Obesity … 2003.

143
52. Srivastava S, Kashiwaya Y, King MT, Baxa U, Tam J, Niu G, Chen X, Clarke K, Veech RL: Mitochondrial biogenesis and increased uncoupling protein 1 in brown adipose tissue of mice fed a ketone ester diet. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 2012, 26(6):2351-2362.
53. Kashiwaya Y, Bergman C, Lee J-HH, Wan R, King MT, Mughal MR, Okun E, Clarke K, Mattson MP, Veech RL: A ketone ester diet exhibits anxiolytic and cognition-sparing properties, and lessens amyloid and tau pathologies in a mouse model of Alzheimer's disease. Neurobiology of aging 2013, 34(6):1530-1539.
54. Kashiwaya Y, Pawlosky R, Markis W, King MT, Bergman C, Srivastava S, Murray A, Clarke K, Veech RL: A ketone ester diet increases brain malonyl-CoA and Uncoupling proteins 4 and 5 while decreasing food intake in the normal Wistar Rat. The Journal of biological chemistry 2010, 285(34):25950-25956.
55. Hashim SA, VanItallie TB: Ketone body therapy: from the ketogenic diet to the oral administration of ketone ester. Journal of lipid research 2014, 55(9):1818-1826.
56. Demling RH: Nutrition, anabolism, and the wound healing process: an overview. Eplasty 2009, 9.
57. Moore FD: Getting well: the biology of surgical convalescence. Annals of the New York Academy of Sciences 1958, 73(2):387-400.
58. Tatti P, Barber A: Nutritional Treatment of Diabetic Foot Ulcers-A Key to Success. Nutritional Treatment of Diabetic Foot Ulcers-A Key to Success 2011.
59. Wernerman J, Brandt R, Strandell T, Allgén LG, Vinnars E: The effect of stress hormones on the interorgan flux of amino acids and on the concentration of free amino acids in skeletal muscle. Clinical nutrition (Edinburgh, Scotland) 1985, 4(4):207-216.
60. Piers LS, Soares MJ, Frandsen SL, O'Dea K: Indirect estimates of body composition are useful for groups but unreliable in individuals. International journal of obesity and related metabolic disorders : journal of the International Association for the Study of Obesity 2000, 24(9):1145-1152.

144
CHAPTER 5: POTENTIAL MECHANISMS FOR EXOGENOUS KETONE
SUPPLEMENTATION TO ENHANCE WOUND HEALING
5.1. Chapter Synopsis
In this chapter, we present data defining some potential mechanisms by which exogenous
ketone supplementation augments ischemic wound healing. We focus on four main age
dependent factors: inflammation, ROS production, angiogenesis, and metabolism. Since ketone
supplementation enhanced wound healing, we hypothesized that there would be measurable
physiological changes in wound healing as early three days post-wounding. Periwound tissue
was harvested at day three and day seven post-wounding for mechanistic analysis. Additionally
mechanistic studies were conducted using primary human dermal fibroblast (HDFs) to confirm
ex vivo observations. From these studies we conclude that exogenous ketone supplementation
decreases ROS production, increases migration and proliferation, and decreases lactate
production. Ketone supplementation did not affect inflammatory markers or antioxidants SOD2
and NQO1. Though there were similarities in effects of ketone supplements, BD and BMS+MCT
elicited distinctive mechanistic profiles. We propose further studies at later time points of wound
healing to determine if exogenous ketone supplementation will affect wound healing at a later
phase than we originally hypothesized.

145
5.2. Potential Mechanisms of Action for Exogenous Ketone Enhancement of Ischemic
Wound Healing in Young and Aged Fisher Rats
In the previous chapter, we reported that oral ketone supplementation without dietary
restriction enhanced wound closure and increased blood flow in young and aged Fisher 344 rats.
We hypothesized that exogenous ketone supplementation promoted wound healing via
enhancement of physiological factors such as increasing proliferation, advancing migration,
reducing ROS production, and resolving inflammation. Experiments in vitro with young and
aged primary human dermal fibroblasts supplemented with 5mM βHB for 72 hours ahead of an
oxidative stimulus (100μM tert-butyl-hydrogen peroxide) resulted in significantly decreased
cellular ROS production, enhanced cell migration, and augmented cellular proliferation (p<0.05).
Comparing peri-wound tissue lysates isolated from rats fed 1,3 Butanediol (BD), βHB NA+/K+
salt mixed with MCT oil in a 1:1 ratio (BMS+MCT), or standard diet (SD) we show that ketone
administration during wound healing induced changes in cytokines, including a significant
elevation in epidermal growth factor (EGF) at day 7 of wound healing compared to control
(p<0.05). BMS+MCT supplementation in aged rats significantly decreased tumor necrosis
factor- alpha (TNF-α) levels on day 7 compared to control (p<0.05). Furthermore, ketone
supplementation significantly reduced leptin levels in ischemic wound tissue lysates in both
young and aged rats. Changes in pro-inflammatory (IRF-5 labeled, M1) to anti-inflammatory
(CD206 labeled, M2) macrophage ratio and blood vessel density were demonstrated by
immunohistochemistry. We conclude that ketone supplementation in vivo and in vitro modifies
systemic physiology to enhance wound closure.

146
5.2.1 Effects of ketone supplementation on inflammation
Inflammation is a key regulatory step in the progression of wound healing. Chronic
wounds are stuck in the inflammatory phase characterized by a proteolytic environment in which
pro-inflammatory cells, cytokines, and chemokines inhibit the normal progression of wound
healing [1]. Pro-inflammatory cytokines including IL-6, IL-1β, and TNF-α are upregulated
during the inflammatory phase of wound healing [2]. IL-6 is produced by neutrophils and
monocytes and has been shown to be important in initiating the healing response, has mitogenic
and proliferative effects on keratinocytes, and is chemoattractive to neutrophils [3-8]. TNF-α
alone has been shown to enhance wound healing in a concentration-dependent manner. Low
levels of TNF-α can promote wound healing indirectly by stimulating inflammation and
macrophage-produced growth factors. However, at higher levels, TNF-α has a detrimental effect
on wound healing by suppressing ECM synthesis and TIMPs while increasing MMPs, inhibiting
re-epithelialization. Additionally, levels of TNF-α and IL-1β are elevated in chronic wounds and
have been shown to work synergistically and perpetuate each other’s expression to amplify the
pro-inflammatory environment [9-11].
Exogenous ketone supplementation did not affect pro-inflammatory markers IL-6 or IL-
1β in young or aged periwound tissue lysates on day three or day seven of wound healing
(Figure 5.2). BMS+MCT supplementation significantly reduced TNF-α levels in periwound
tissue lysates of aged rats after seven days of wound healing compared to aged controls.
Significant changes were not observed in selected anti-inflammatory cytokines including IL-4,
IL-10, or IL-13 (Figure 5.2). In an unpublished study from our laboratory, rats were fed one of
three exogenous ketone supplements (1, 3-butanediol diacetoacetate ester (KE), Na+/Ca+2-β-
hydroxybutyrate mineral salt (ΒHB-S), or a 1:1 mixture of Na+/Ca+2-β-hydroxybutyrate mineral

147
salt: Medium Chain Triglyceride Oil (S/MCT) mixed into standard rodent chow at 5-20% by
weight for 15 weeks. Inflammatory profiling was performed on serum collected from the rats at
the end of the chronic feeding study and revealed decreases in several pro-inflammatory and
anti-inflammatory cytokines including IL-1β, IL-6, IFN-γ, MCP-1, RANTES, IL-4, IL-10, IL-13.
These findings suggest that ketone supplementation may suppress total inflammation and not
affect pro-inflammatory or anti-inflammatory markers independently; however, further
experiments need to be conducted to confirm this observation. Because ketone supplementation
hastened wound healing, we hypothesized that we would see early resolution of inflammation.
Further studies need to be conducted on subsequent days of wound healing to see if ketone
supplementation affects inflammatory markers after day 7. Epidermal growth factor (EGF) is
secreted by platelets, macrophages and fibroblasts to accelerate epithelialization and increase the
tensile strength of wounds [12-15]. Topical EGF administration has shown promising results for
venous ulcers and diabetic ulcers; however, the heterogeneity of wounds, modes of
administration, patient management protocols, and varied clinical trial outcomes has limited
clinical application to outside of the USA [16]. Exogenous ketone supplementation with BD
elevated EGF levels in day seven periwound tissue lysates of young rats (Figure 5.3B).
Leptin is a hormone that works in opposition of ghrelin to regulate energy homeostasis by
inhibiting hunger and establishing satiety. The KD has been demonstrated to slow weight gain by
altering leptin levels. Juvenile rats fed a KD for two weeks had slower weight gain, had higher
leptin levels, and lower insulin levels compared to those fed an SD [17]. We demonstrated a
similar slowing of weight gain in juvenile Sprague-Dawley rats in Chapter 3, consistent with this
observation. In the same study, they demonstrated that calorie restriction did not elicit the same
leptin elevation as the KD. However, Kolacynski and colleagues investigated the responses of

148
leptin following a short term fasting, demonstrating that leptin levels declined following a 12
hours fast. Re-feeding prompted leptin levels to rise and return to normal values within 24 hours
[18]. They demonstrated a reverse relationship between levels of βHB and leptin. Small amounts
of glucose added to inhibit hepatic ketogenesis was enough to prevent the fall of leptin.
However, exogenous ketone administration did not reduce leptin, causing the team to conclude
that the relationship was due to a physiological response to ketogenesis and not ketone bodies
themselves.
An increasing number of studies have determined that leptin has a broad spectrum of
physiological effects in the wound-healing environment. Leptin has been shown to directly cause
T cells and monocytes to release pro-inflammatory cytokines, to produce angiogenesis in
endothelial cells, and to induce neo-vascularization in corneal cells. Leptin production has been
documented in a variety of non-adipose cells including placental trophoblasts, mammary
epithelial cells, gastric fundic mucosa, and ovarian follicle cells [19-29]. In a study by Murad and
colleagues, it was demonstrated that cells within an incisional dermal wound produced a burst of
leptin shortly after surgery [30]. The study concluded that leptin might have a multifunctional
role during wound healing and serves a critical functional role as an autocrine/paracrine regulator
of normal wound healing.
As discussed previously, leptin is produced locally in the wound bed. In our experiment
leptin was only measured in the wound bed tissue lysates, not in circulating blood serum. BD and
BMS+MCT significantly reduced leptin levels in ischemic wound tissue lysates in young
animals compared to controls on day three post-wounding but this observation was no longer
apparent after seven days of wound healing (Figure 5.3C). In aged animals, leptin levels were
decreased in both ketone-supplemented groups on both day three and day seven post-wounding

149
(Figure 5.3C). This decrease in leptin levels in the ischemic wound bed may play a role to the
enhanced wound healing via exogenous ketone supplementation. The circulating blood serum
levels may give more insight to the data. If these measurements were in systemic blood serum, it
may indicate that the rats were not eating the food-integrated ketone supplements, as leptin levels
were elevated in the SD. Additionally, this would suggest that the elevations in blood ketone
levels would be due to caloric restriction and not ingestion of the supplement. Further
experiments monitoring food intake would be required to decipher this observation. Further
experiments need to be conducted to determine the full impact of decreasing leptin levels with
ketone supplementation.
As discussed in Chapter 1, macrophages in the wound bed can exhibit two distinct
functional phenotypes: M1 (classically activated, pro-inflammatory) and M2 (alternatively
activated, anti-inflammatory). During the early inflammatory phase, macrophages that are
activated by lipopolysaccharide (LPS) or inflammatory cytokines like interferon gamma (IFN-γ)
continue the job of neutrophils by phagocytizing bacteria and damaged tissue. Additionally, the
M1 macrophages release pro-inflammatory cytokines such as TNF-α and IL-6 [31]. During the
late inflammatory phase, initiated by the phagocytosis of apoptotic cells, M1 macrophages
phenotypically convert to M2 macrophages [32]. M2 macrophages, activated by interleukin-4
(IL-4) and interleukin-13 (IL-13), play a critical role in the resolution of inflammation.
Additionally, they facilitate transition to the proliferative phase by promoting angiogenesis,
tissue remodeling, and repair [31, 33-40]. In this study, we did not see an effect of exogenous
ketone supplementation on IFN-γ, IL-4, or IL-13 as previously hypothesized (Figure 5.2).
Additionally, exogenous ketone supplementation did not elicit a significant change in M1 to M2
phenotype in day seven aged ischemic wounds (Figure 5.4). This observation supports the

150
inflammatory array data and further supports that the aged ischemic wounds were still in a pro-
inflammatory environment seven days post-wounding. Heterogeneity seen in the wound healing
in this model is consistent with patient heterogeneity seen in the clinical setting and accounts for
the lack of statistical significance in the immunohistochemical analysis (Figure 5.1). In general,
ketone supplemented wounds demonstrated more epithelial advancement, granulation tissue
formation, and fewer labeled macrophages (Figure 5.1, Figure 5.4). Further studies need to be
conducted to confirm these histological observations.
5.2.2 Effects of ketone supplementation on ROS production
As discussed in previous chapters ROS production is an important bactericidal
component in acute wound healing. However, in a chronic wound environment, ROS production
isn’t properly mitigated, leading to mitochondrial dysfunction, DNA damage, lipid peroxidation,
and even cell death, which delays wound healing [41, 42]. Ketones have been shown to decrease
ROS production through a variety of physiological effects [43-46]. Additionally, ketones have
been shown to decrease oxidative stress by increasing the downstream antioxidant glutathione
via the Nrf2-ARE pathway [47]. Nrf2 is the primary transcription factor that responds to
oxidative stress. Under normal conditions, Nrf2 is targeted for proteasomal degradation by the
E3 ubiquitin ligase, Keap1. Oxidants and electrophiles disrupt the Nrf2/Keap1 binding allowing
Nrf2 to translocate to the nucleus, bind DNA at the antioxidant or electrophilic response element
(ARE), and transcribe downstream antioxidants including SOD2, NAD (P) H: quinone
oxidoreductase (NQO1) and enzymes for glutathione synthesis [48-50]. Additionally, Nrf2 has
been shown to play a role in wound healing as disruption of Nrf2 has shown to impair the

151
angiogenic capacity of endothelial cells [51]. It has been shown that Nrf2 and antioxidant
capacity are decreased with age, leading to increased oxidative stress [52, 53]. Recently, Moor
and colleagues demonstrated age-dependent deficiencies in the glutathione and SOD2
antioxidant pathways. In an ischemic wound model, wounds from aged rats had lower SOD2
protein expression and activity, decreased ratio of reduced/oxidized glutathione, and decreased
glutathione peroxidase activity [54]. These age-exaggerated insufficiencies lead to excessive
inflammation and impaired wound healing. New data indicate that nutrient intake, energy
metabolism, and ROS are linked at the nuclear level via the Nrf2/ARE pathway [55]. The KD
has been shown to increase glutathione synthesis via the Nrf2/ARE pathway [56-59].
Additionally, the age-dependent deficit in Nrf2 has been reversed with lipoic acid
supplementation [60]. Lipoic acid is derived from the MCFA caprylic acid (a main component of
MCT oil); thus we hypothesized that exogenous ketone supplementation would result in similar
effects.
In this study, we were unable to measure Nrf2 levels via Western blot due to inefficient
antibodies; the downstream antioxidants SOD2 and NQO1, measured by Western blot analysis,
were not significantly altered by exogenous ketone supplementation in either primary human
dermal fibroblasts (HDFs) or ischemic wounds (Figure 5.6). Western blot sensitivity may not be
able to detect the subtle changes in these antioxidants during the first few days of wound healing;
thus, further experiments need to be conducted to determine antioxidant levels. However, DHE
and Mitosox Red labeled ROS production in primary human dermal fibroblasts was significantly
reduced with 5mM βHB supplementation (Figure 5.5). Metabolomic studies demonstrated that
ketone supplementation increased antioxidants carnosine and anserine. Further studies are

152
needed to investigate other antioxidant pathways and determine how ketone supplementation is
suppressing ROS production in a wound-healing environment.
5.2.3. Effects of ketone supplementation on angiogenesis
As a continuation of Chapter 4, we sought to delineate if the observed increase in blood
flow occurred via enhanced angiogenesis or vasodilatation. Immunohistochemical analysis of
CD31 blood vessel density demonstrated a trend of increased blood vessels in the ischemic
wounds of aged rats supplemented with BD and BMS+MCT seven days post-wounding;
however, this observation did not reach significance (Figure 5.7). As discussed, VEGF is a
potent angiogenic factor. Periwound tissue lysates were analyzed for VEGF levels, revealing a
trend of elevation of VEGF on day seven of wound compared to day three in both young and
aged animals though this did not reach significance. Ketone supplementation did not affect levels
of VEGF in this study, (Figure 5.2) but in the unpublished chronic feeding study, in which rats
were ketone supplemented via food-integration for 15-weeks, low dose KE significantly
increased serum (systemic) VEGF levels. As discussed in the metabolomics data in Chapter 3,
exogenous ketone supplementation leads to an increase in the potent vasodilator adenosine.
These observations suggest that the increased blood flow may be due to increased vasodilation
and not angiogenesis. Further studies need to be conducted to determine the impact of
vasodilation on ketone supplemented wound healing and to determine the exact mechanisms of
the observed increase in blood flow.

153
5.2.4. Effects of ketone supplementation on metabolism
Proliferation and migration represent crucial processes by which epidermal and dermal
layers restore anatomical integrity to the wound site. During the formation of granulation tissue
in the dermis, platelets, monocytes, and other cellular blood constituents release various peptide
growth factors, stimulating fibroblasts to migrate into the wound site where they proliferate to
reconstitute the various connective tissue components [61]. We investigated if ketone
supplementation enhanced proliferation and migration of primary human dermal fibroblasts;
indeed, 5mM HB supplementation elicited a significant increase in migration and proliferation
in both young and aged HDFs (Figure 5.8 and Figure 5.9). Further experiments need to be
conducted using keratinocytes of the epidermal layer to examine if ketone supplementation also
increases proliferation and migration in those cells. These further studies will help determine if
ketone supplementation affects all layers of the wound healing process or the dermal layer
independently. Additionally, the enhanced proliferation and migration may explain the enhanced
granulation tissue detected in the histological analysis (Figure 5.1, Figure 5.4).
Proliferating and migrating cells require more nutrients and energy production to perform
their functions; thus, an increase in these physiological factors may be a reflection of a higher
metabolic state. As discussed in Chapter 1, one of the fates of pyruvate is lactate production and
is indicative of anaerobic respiration. The hypoxic wound environment shifts to higher anaerobic
respiration and thus higher lactate production. Throughout the last decade, wound-healing
studies have revealed that high lactate levels (10-15 mM) are characteristic in healing, stimulate
collagen deposition, and enhance angiogenesis [62-66]. Subsequent studies demonstrated that
lactate has several sources in wounds, remains elevated even in the presence of oxygen, and
intense neutrophil respiratory bursts drive lactate and ROS accumulation through aerobic

154
glycolysis (Warburg effect) [64, 67-71]. These studies have concluded that lactate is produced in
both aerobic and anaerobic environments during tissue repair and the presence of both lactate
and oxygen is required for optimal wound healing. However, it is the quantity of lactate that is
critically significant [72]. Systemic lactate concentration rarely exceeds 10 mM due to tight
metabolic control; accumulation exceeding this level is associated with a poor wound-healing
prognosis [73]. When lactate levels reach ~20 mM it becomes deleterious to the wound bed as
high lactate levels reduce cell viability, diminish cell motility, and decrease polymerized
cytoskeletal actin, which impedes angiogenesis, fibroplasia, and wound contraction [74]. Aging
further complicates this environment as cells of older patients have shown decreased sensitivity
to growth factor and cytokine stimulation, diminished antioxidant capacity, and reduced cell
motility and proliferation, causing their tissues to be very susceptible to hypoxic and oxidant
stress and worsening patient survival outcome [75-78]. In this study, we demonstrated the 5 mM
βHB supplementation in young and aged HDFs significantly reduced lactate production (Figure
5.10). Further research needs to be conducted to investigate lactate levels in the wound bed with
exogenous ketone supplementation and explore lactate production in keratinocytes of the
epidermal layer to determine the full impact of ketone supplementation on lactate production
during wound healing.
5.3. Closing Remarks
In summary, we have just begun to elicit some of the mechanisms by which exogenous
ketone supplementation impacts ischemic wound healing. As ketone supplementation represents
a multifaceted tool, it is likely that multiple mechanisms are acting simultaneously to provide a
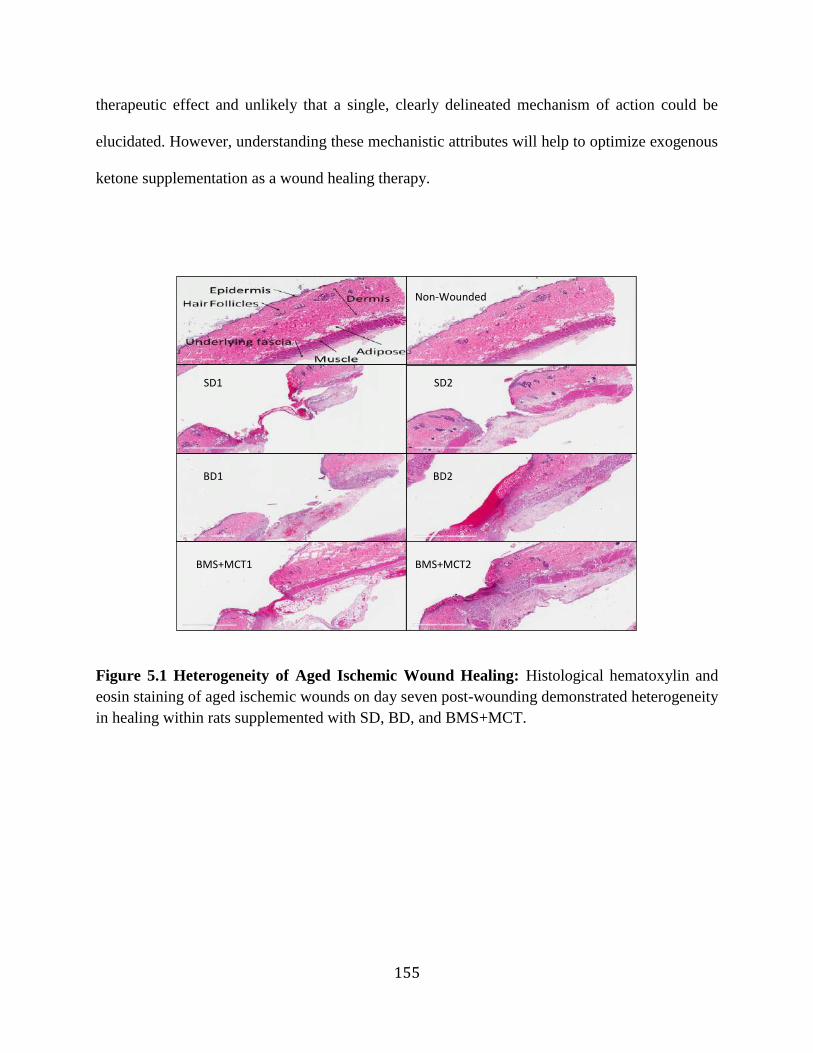
155
therapeutic effect and unlikely that a single, clearly delineated mechanism of action could be
elucidated. However, understanding these mechanistic attributes will help to optimize exogenous
ketone supplementation as a wound healing therapy.
Figure 5.1 Heterogeneity of Aged Ischemic Wound Healing: Histological hematoxylin and
eosin staining of aged ischemic wounds on day seven post-wounding demonstrated heterogeneity
in healing within rats supplemented with SD, BD, and BMS+MCT.
SD1 SD2
BD1 BD2
BMS+MCT1 BMS+MCT2
Non-Wounded
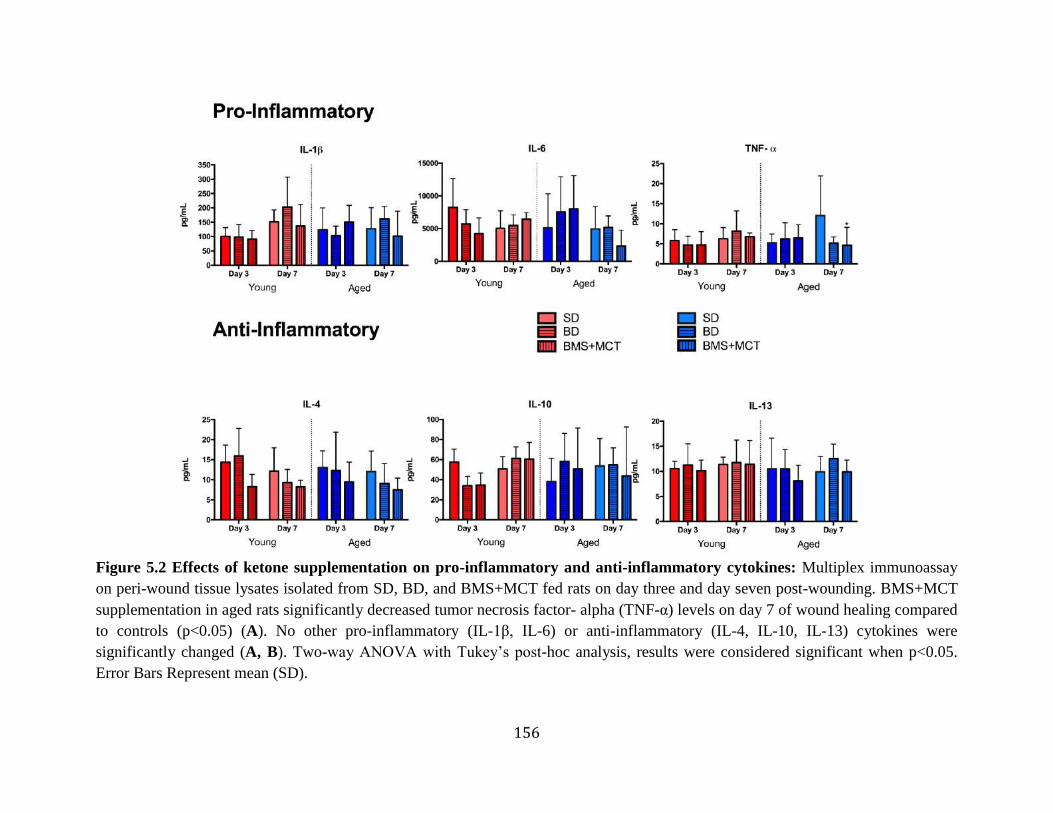
156
Figure 5.2 Effects of ketone supplementation on pro-inflammatory and anti-inflammatory cytokines: Multiplex immunoassay
on peri-wound tissue lysates isolated from SD, BD, and BMS+MCT fed rats on day three and day seven post-wounding. BMS+MCT
supplementation in aged rats significantly decreased tumor necrosis factor- alpha (TNF-α) levels on day 7 of wound healing compared
to controls (p<0.05) (A). No other pro-inflammatory (IL-1β, IL-6) or anti-inflammatory (IL-4, IL-10, IL-13) cytokines were
significantly changed (A, B). Two-way ANOVA with Tukey’s post-hoc analysis, results were considered significant when p<0.05.
Error Bars Represent mean (SD).
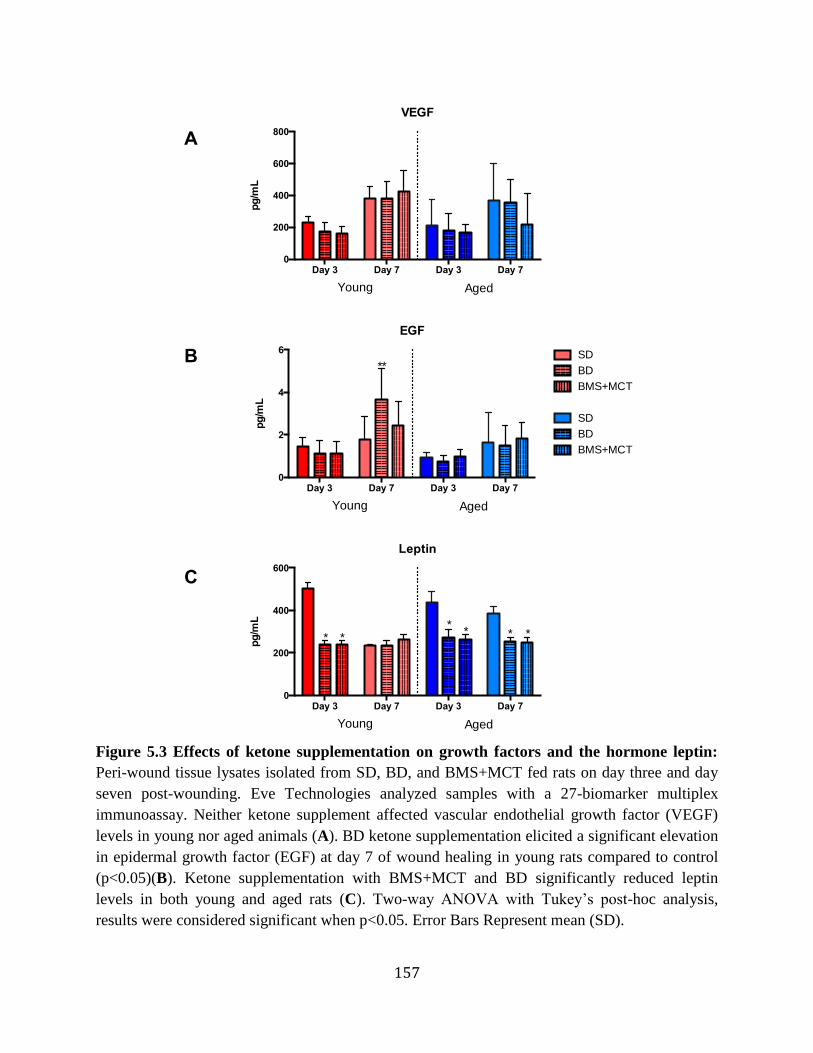
157
Figure 5.3 Effects of ketone supplementation on growth factors and the hormone leptin:
Peri-wound tissue lysates isolated from SD, BD, and BMS+MCT fed rats on day three and day
seven post-wounding. Eve Technologies analyzed samples with a 27-biomarker multiplex
immunoassay. Neither ketone supplement affected vascular endothelial growth factor (VEGF)
levels in young nor aged animals (A). BD ketone supplementation elicited a significant elevation
in epidermal growth factor (EGF) at day 7 of wound healing in young rats compared to control
(p<0.05)(B). Ketone supplementation with BMS+MCT and BD significantly reduced leptin
levels in both young and aged rats (C). Two-way ANOVA with Tukey’s post-hoc analysis,
results were considered significant when p<0.05. Error Bars Represent mean (SD).
VEGF
Day 3 Day 7 Day 3 Day 70
200
400
600
800
pg
/mL
Young Aged
EGF
Day 3 Day 7 Day 3 Day 70
2
4
6
pg
/mL
Young Aged
**
Leptin
Day 3 Day 7 Day 3 Day 70
200
400
600
pg
/mL
Young Aged
* **
* * *
A
B
C
SD
BD
BMS+MCT
SD
BD
BMS+MCT
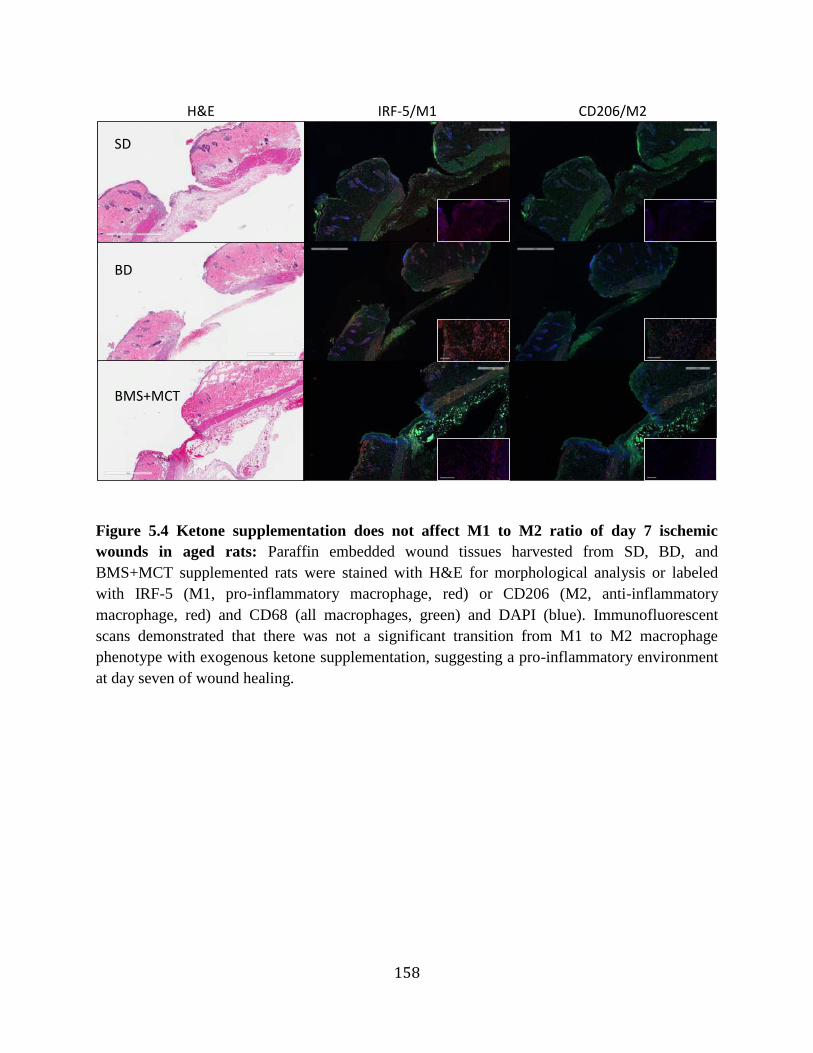
158
Figure 5.4 Ketone supplementation does not affect M1 to M2 ratio of day 7 ischemic
wounds in aged rats: Paraffin embedded wound tissues harvested from SD, BD, and
BMS+MCT supplemented rats were stained with H&E for morphological analysis or labeled
with IRF-5 (M1, pro-inflammatory macrophage, red) or CD206 (M2, anti-inflammatory
macrophage, red) and CD68 (all macrophages, green) and DAPI (blue). Immunofluorescent
scans demonstrated that there was not a significant transition from M1 to M2 macrophage
phenotype with exogenous ketone supplementation, suggesting a pro-inflammatory environment
at day seven of wound healing.
H&E IRF-5/M1 CD206/M2
SD
BD
BMS+MCT
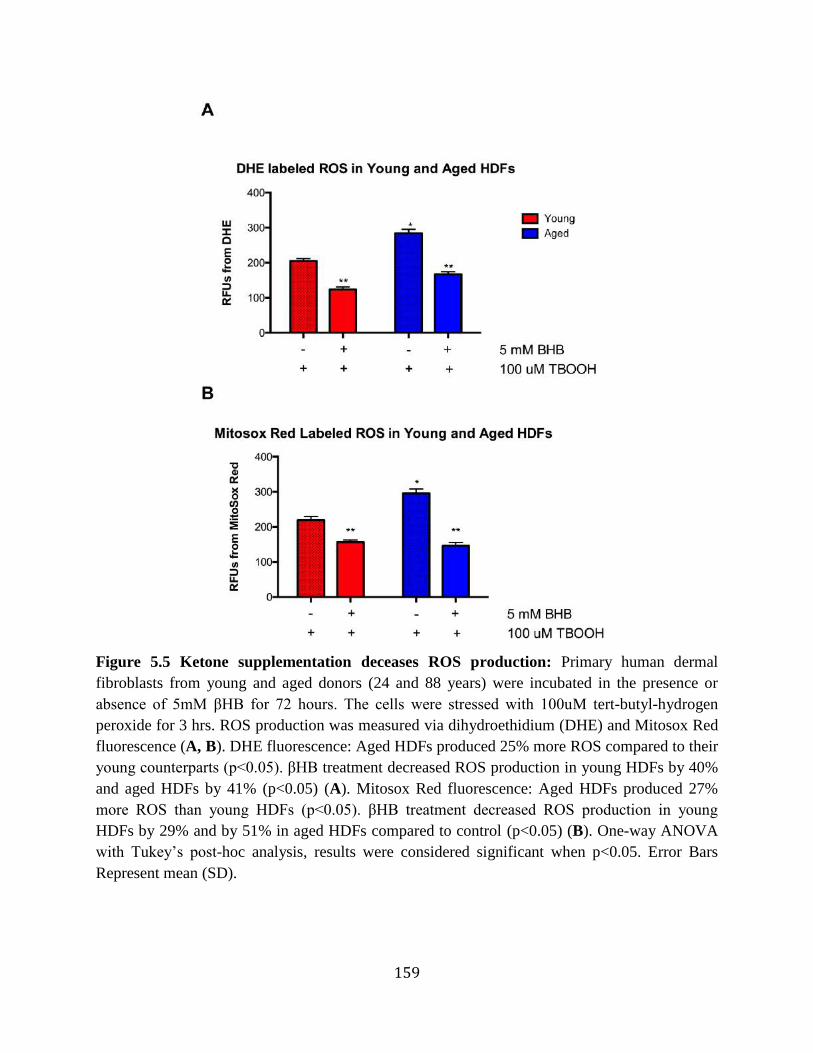
159
Figure 5.5 Ketone supplementation deceases ROS production: Primary human dermal
fibroblasts from young and aged donors (24 and 88 years) were incubated in the presence or
absence of 5mM βHB for 72 hours. The cells were stressed with 100uM tert-butyl-hydrogen
peroxide for 3 hrs. ROS production was measured via dihydroethidium (DHE) and Mitosox Red
fluorescence (A, B). DHE fluorescence: Aged HDFs produced 25% more ROS compared to their
young counterparts (p<0.05). βHB treatment decreased ROS production in young HDFs by 40%
and aged HDFs by 41% (p<0.05) (A). Mitosox Red fluorescence: Aged HDFs produced 27%
more ROS than young HDFs (p<0.05). βHB treatment decreased ROS production in young
HDFs by 29% and by 51% in aged HDFs compared to control (p<0.05) (B). One-way ANOVA
with Tukey’s post-hoc analysis, results were considered significant when p<0.05. Error Bars
Represent mean (SD).
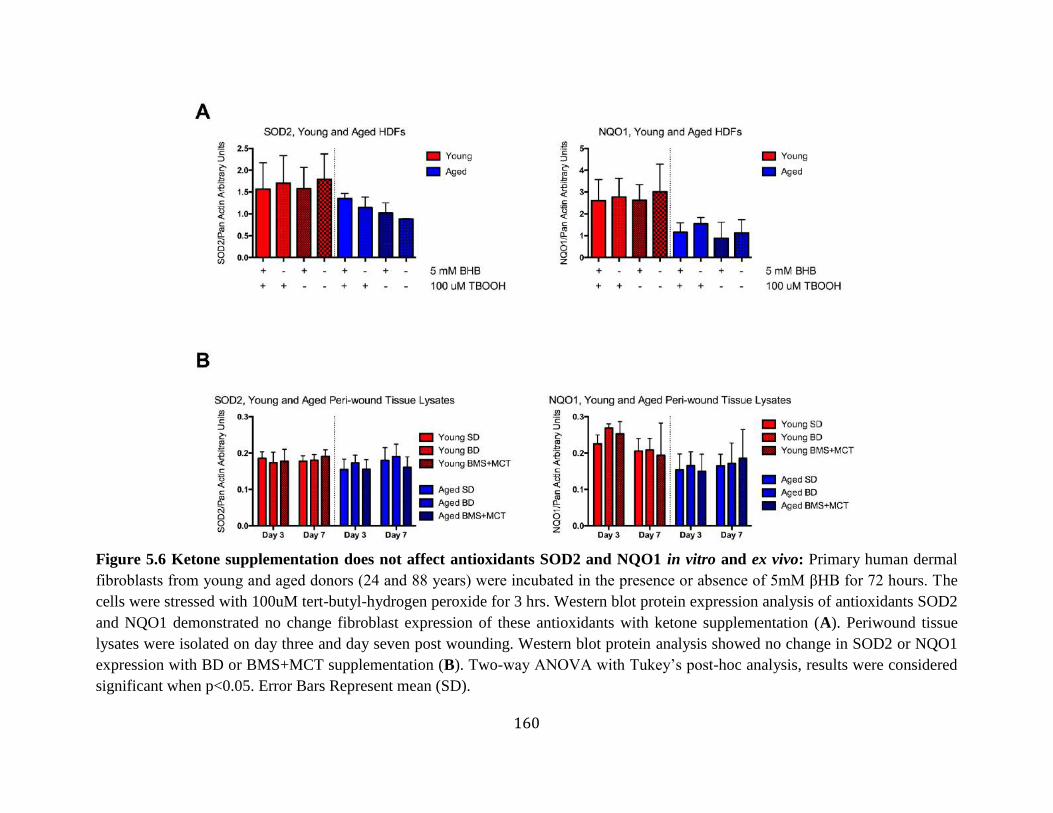
160
Figure 5.6 Ketone supplementation does not affect antioxidants SOD2 and NQO1 in vitro and ex vivo: Primary human dermal
fibroblasts from young and aged donors (24 and 88 years) were incubated in the presence or absence of 5mM βHB for 72 hours. The
cells were stressed with 100uM tert-butyl-hydrogen peroxide for 3 hrs. Western blot protein expression analysis of antioxidants SOD2
and NQO1 demonstrated no change fibroblast expression of these antioxidants with ketone supplementation (A). Periwound tissue
lysates were isolated on day three and day seven post wounding. Western blot protein analysis showed no change in SOD2 or NQO1
expression with BD or BMS+MCT supplementation (B). Two-way ANOVA with Tukey’s post-hoc analysis, results were considered
significant when p<0.05. Error Bars Represent mean (SD).
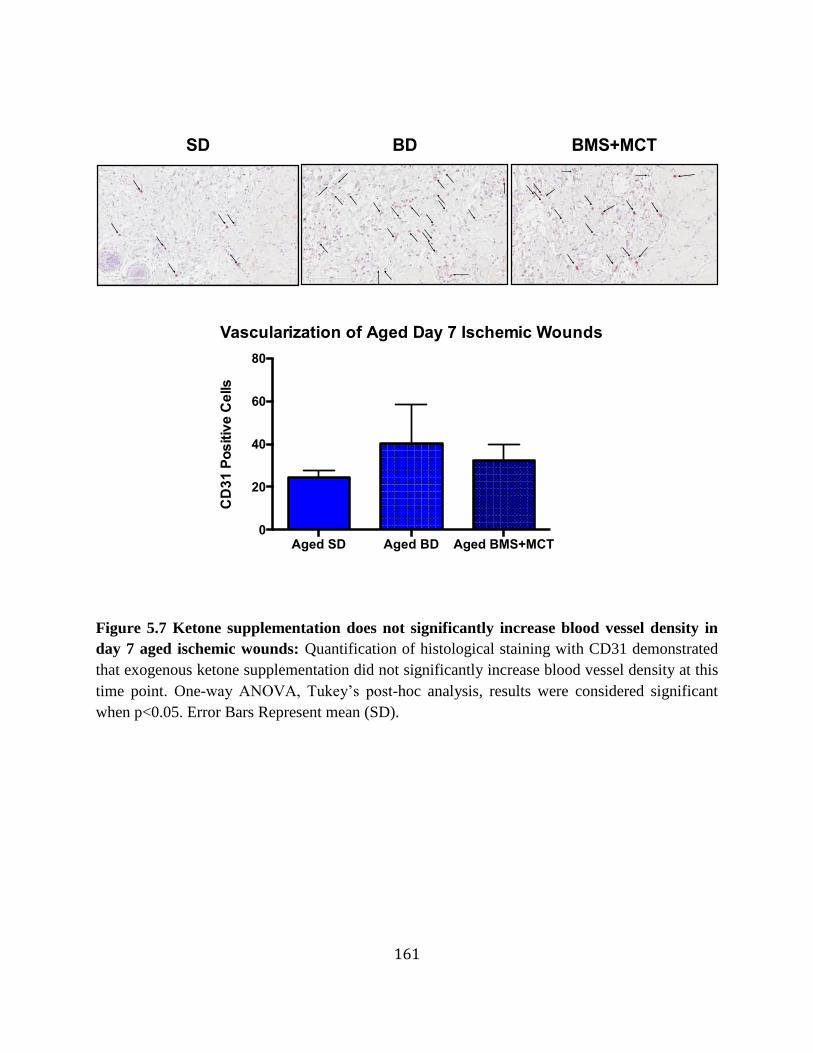
161
Figure 5.7 Ketone supplementation does not significantly increase blood vessel density in
day 7 aged ischemic wounds: Quantification of histological staining with CD31 demonstrated
that exogenous ketone supplementation did not significantly increase blood vessel density at this
time point. One-way ANOVA, Tukey’s post-hoc analysis, results were considered significant
when p<0.05. Error Bars Represent mean (SD).
Vascularization of Aged Day 7 Ischemic Wounds
Aged SD Aged BD Aged BMS+MCT0
20
40
60
80
CD
31
Po
sitiv
e C
ells
SD BD BMS+MCT

162
Figure 5.8 Ketone supplementation increases migration in young and aged HDFs: Young
and aged HDFs were grown to confluence in 0.2% FBS 5mM glucose DMEM media, cells were
treated with or without the presence of 5mM βHB for 72 Hrs, cells were scratched with 200uL
yellow pipette tip. After 18 hours, migrated cells were counted using Dapi staining. Migration
was increased in both young and aged HDFs compared to the controls (p=0.0260 and p=0.0004)
(A, B). One-way ANOVA, Tukey’s post-hoc analysis, results were considered significant when
p<0.05. Error Bars Represent mean (SD).
Ketones Increase Migration in Young and Aged HDFs
0
10
20
30
40
50
Ce
ll N
um
be
r
Young
Aged*
*
+
+-
+ +
+-
+
5 mM BHB
100 uM TBOOH
A
B
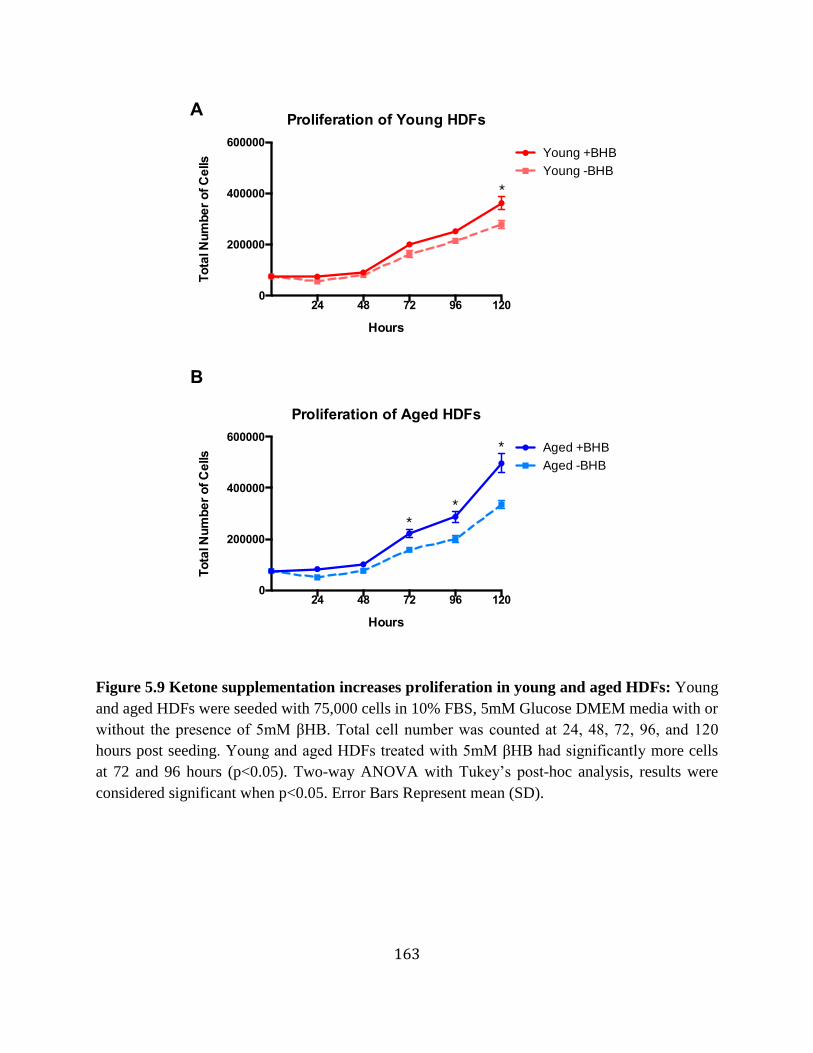
163
Figure 5.9 Ketone supplementation increases proliferation in young and aged HDFs: Young
and aged HDFs were seeded with 75,000 cells in 10% FBS, 5mM Glucose DMEM media with or
without the presence of 5mM βHB. Total cell number was counted at 24, 48, 72, 96, and 120
hours post seeding. Young and aged HDFs treated with 5mM βHB had significantly more cells
at 72 and 96 hours (p<0.05). Two-way ANOVA with Tukey’s post-hoc analysis, results were
considered significant when p<0.05. Error Bars Represent mean (SD).
Proliferation of Young HDFs
24 48 72 96 1200
200000
400000
600000
Hours
To
tal N
um
be
r o
f C
ells
Young +BHB
Young -BHB
*
Proliferation of Aged HDFs
24 48 72 96 1200
200000
400000
600000
Hours
To
tal N
um
be
r o
f C
ells
Aged +BHB
Aged -BHB
**
*
A
B
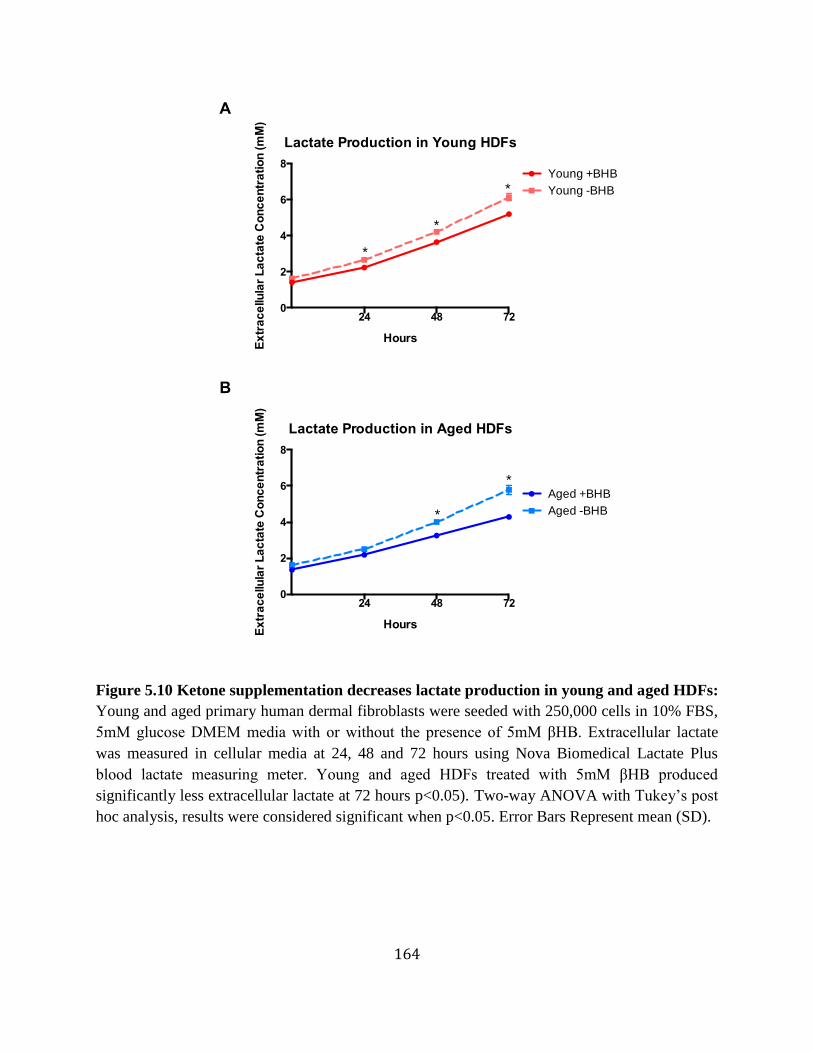
164
Figure 5.10 Ketone supplementation decreases lactate production in young and aged HDFs:
Young and aged primary human dermal fibroblasts were seeded with 250,000 cells in 10% FBS,
5mM glucose DMEM media with or without the presence of 5mM βHB. Extracellular lactate
was measured in cellular media at 24, 48 and 72 hours using Nova Biomedical Lactate Plus
blood lactate measuring meter. Young and aged HDFs treated with 5mM βHB produced
significantly less extracellular lactate at 72 hours p<0.05). Two-way ANOVA with Tukey’s post
hoc analysis, results were considered significant when p<0.05. Error Bars Represent mean (SD).
Lactate Production in Young HDFs
24 48 720
2
4
6
8
Ex
tra
ce
llula
r L
ac
tate
Co
nc
en
tra
tio
n (m
M)
Hours
Young +BHB
Young -BHB
*
*
*
Lactate Production in Aged HDFs
24 48 720
2
4
6
8
Hours
Ex
tra
ce
llula
r L
ac
tate
Co
nc
en
tra
tio
n (m
M)
Aged +BHB
Aged -BHB *
*
A
B

165
5.4. References for Chapter 5:
1. Barrientos S, Stojadinovic O, Golinko MS, Brem H, Tomic‐Canic M: PERSPECTIVE
ARTICLE: Growth factors and cytokines in wound healing. Wound Repair and
Regeneration 2008, 16(5).
2. Singer AJ, Clark RA: Cutaneous wound healing. The New England journal of medicine
1999, 341(10):738-746.
3. Di Vita G, Patti R, D'Agostino P, Caruso G, Arcara M, Buscemi S, Bonventre S, Ferlazzo
V, Arcoleo F, Cillari E: Cytokines and growth factors in wound drainage fluid from
patients undergoing incisional hernia repair. Wound repair and regeneration : official
publication of the Wound Healing Society [and] the European Tissue Repair Society
2006, 14(3):259-264.
4. Finnerty CC, Herndon DN, Przkora R, Pereira CT, Oliveira HM, Queiroz DMM, Rocha
AM, Jeschke MG: Cytokine expression profile over time in severely burned pediatric
patients. Shock (Augusta, Ga) 2006, 26(1):13-19.
5. Gallucci RM, Sloan DK, Heck JM, Murray AR, O'Dell SJ: Interleukin 6 indirectly
induces keratinocyte migration. The Journal of investigative dermatology 2004,
122(3):764-772.
6. Grellner W, Georg T, Wilske J: Quantitative analysis of proinflammatory cytokines
(IL-1beta, IL-6, TNF-alpha) in human skin wounds. Forensic science international
2000, 113(1-3):251-264.
7. Peschen M, Grenz H, Brand-Saberi B, Bunaes M, Simon JC, Schöpf E, Vanscheidt W:
Increased expression of platelet-derived growth factor receptor alpha and beta and
vascular endothelial growth factor in the skin of patients with chronic venous
insufficiency. Archives of dermatological research 1998, 290(6):291-297.
8. Sato M, Sawamura D, Ina S, Yaguchi T, Hanada K, Hashimoto I: In vivo introduction
of the interleukin 6 gene into human keratinocytes: induction of epidermal
proliferation by the fully spliced form of interleukin 6, but not by the alternatively
spliced form. Archives of dermatological research 1999, 291(7-8):400-404.
9. Tarnuzzer RW, Schultz GS: Biochemical analysis of acute and chronic wound
environments. Wound repair and regeneration : official publication of the Wound
Healing Society [and] the European Tissue Repair Society 1996, 4(3):321-325.
10. Wallace HJ, Stacey MC: Levels of tumor necrosis factor-alpha (TNF-alpha) and
soluble TNF receptors in chronic venous leg ulcers--correlations to healing status.
The Journal of investigative dermatology 1998, 110(3):292-296.

166
11. Mast BA, Schultz GS: Interactions of cytokines, growth factors, and proteases in
acute and chronic wounds. Wound repair and regeneration : official publication of the
Wound Healing Society [and] the European Tissue Repair Society 1996, 4(4):411-420.
12. Schultz G, Rotatori DS, Clark W: EGF and TGF-alpha in wound healing and repair.
Journal of cellular biochemistry 1991, 45(4):346-352.
13. Shiraha H, Glading A, Gupta K, Wells A: IP-10 inhibits epidermal growth factor-
induced motility by decreasing epidermal growth factor receptor-mediated calpain
activity. The Journal of cell biology 1999, 146(1):243-254.
14. Brown GL, Curtsinger L, Brightwell JR, Ackerman DM, Tobin GR, Polk HC, George-
Nascimento C, Valenzuela P, Schultz GS: Enhancement of epidermal regeneration by
biosynthetic epidermal growth factor. The Journal of experimental medicine 1986,
163(5):1319-1324.
15. Brown GL, Curtsinger LJ, White M, Mitchell RO, Pietsch J, Nordquist R, von
Fraunhofer A, Schultz GS: Acceleration of tensile strength of incisions treated with
EGF and TGF-beta. Annals of surgery 1988, 208(6):788-794.
16. Frykberg RG, Banks J: Challenges in the Treatment of Chronic Wounds. Advances in
wound care 2015, 4(9):560-582.
17. Thio LL, Erbayat-Altay E, Rensing N, Yamada KA: Leptin contributes to slower
weight gain in juvenile rodents on a ketogenic diet. Pediatric research 2006,
60(4):413-417.
18. Kolaczynski JW, Ohannesian JP, Considine RV, Marco CC, Caro JF: Response of leptin
to short-term and prolonged overfeeding in humans. The Journal of clinical
endocrinology and metabolism 1996, 81(11):4162-4165.
19. Bado A, Levasseur S, Attoub S, Kermorgant S, Laigneau JP, Bortoluzzi MN, Moizo L,
Lehy T, Guerre-Millo M, Le Marchand-Brustel Y et al: The stomach is a source of
leptin. Nature 1998, 394(6695):790-793.
20. Cao R, Brakenhielm E, Wahlestedt C, Thyberg J, Cao Y: Leptin induces vascular
permeability and synergistically stimulates angiogenesis with FGF-2 and VEGF.
Proceedings of the National Academy of Sciences of the United States of America 2001,
98(11):6390-6395.
21. Gainsford T, Willson TA, Metcalf D, Handman E, McFarlane C, Ng A, Nicola NA,
Alexander WS, Hilton DJ: Leptin can induce proliferation, differentiation, and
functional activation of hemopoietic cells. Proceedings of the National Academy of
Sciences of the United States of America 1996, 93(25):14564-14568.

167
22. Henson MC, Castracane VD, O'Neil JS, Gimpel T, Swan KF, Green AE, Shi W: Serum
leptin concentrations and expression of leptin transcripts in placental trophoblast
with advancing baboon pregnancy. The Journal of clinical endocrinology and
metabolism 1999, 84(7):2543-2549.
23. Hoggard N, Hunter L, Duncan JS, Williams LM, Trayhurn P, Mercer JG: Leptin and
leptin receptor mRNA and protein expression in the murine fetus and placenta.
Proceedings of the National Academy of Sciences of the United States of America 1997,
94(20):11073-11078.
24. Lord GM, Matarese G, Howard JK, Baker RJ, Bloom SR, Lechler RI: Leptin modulates
the T-cell immune response and reverses starvation-induced immunosuppression.
Nature 1998, 394(6696):897-901.
25. Masuzaki H, Ogawa Y, Sagawa N, Hosoda K, Matsumoto T, Mise H, Nishimura H,
Yoshimasa Y, Tanaka I, Mori T et al: Nonadipose tissue production of leptin: leptin as
a novel placenta-derived hormone in humans. Nature medicine 1997, 3(9):1029-1033.
26. Santos-Alvarez J, Goberna R, Sánchez-Margalet V: Human leptin stimulates
proliferation and activation of human circulating monocytes. Cellular immunology
1999, 194(1):6-11.
27. Señarís R, Garcia-Caballero T, Casabiell X, Gallego R, Castro R, Considine RV, Dieguez
C, Casanueva FF: Synthesis of leptin in human placenta. Endocrinology 1997,
138(10):4501-4504.
28. Sierra-Honigmann MR, Nath AK, Murakami C, García-Cardeña G, Papapetropoulos A,
Sessa WC, Madge LA, Schechner JS, Schwabb MB, Polverini PJ et al: Biological action
of leptin as an angiogenic factor. Science (New York, NY) 1998, 281(5383):1683-1686.
29. Smith-Kirwin SM, O'Connor DM, De Johnston J, Lancey ED, Hassink SG, Funanage
VL: Leptin expression in human mammary epithelial cells and breast milk. The
Journal of clinical endocrinology and metabolism 1998, 83(5):1810-1813.
30. Murad A, Nath AK, Cha S-T, Demir E, Flores-Riveros J, Sierra-Honigmann RM: Leptin
is an autocrine/paracrine regulator of wound healing. The FASEB Journal 2003.
31. Mahdavian Delavary B, van der Veer W, van Egmond M, Niessen F, Beelen R:
Macrophages in skin injury and repair. Immunobiology 2011, 216(7):753-762.
32. Khallou-Laschet J, Varthaman A, Fornasa G, Compain C, Gaston A-TT, Clement M,
Dussiot M, Levillain O, Graff-Dubois S, Nicoletti A et al: Macrophage plasticity in
experimental atherosclerosis. PloS one 2009, 5(1).

168
33. Gordon S: Alternative activation of macrophages. Nature reviews Immunology 2003,
3(1):23-35.
34. Xue L, Liu X-s: [Advancement in the research of the role of macrophages in wound
healing]. Zhonghua shao shang za zhi = Zhonghua shaoshang zazhi = Chinese journal
of burns 2013, 29(1):62-64.
35. Ploeger D, Hosper N, Schipper M, Koerts J, de Rond S, Bank R: Cell plasticity in
wound healing: paracrine factors of M1/ M2 polarized macrophages influence the
phenotypical state of dermal fibroblasts. Cell communication and signaling : CCS
2013, 11(1):29.
36. Zhang Y, Choksi S, Chen K, Pobezinskaya Y, Linnoila I, Liu Z-GG: ROS play a critical
role in the differentiation of alternatively activated macrophages and the occurrence
of tumor-associated macrophages. Cell research 2013.
37. Frank S, Hübner G, Breier G, Longaker MT, Greenhalgh DG, Werner S: Regulation of
vascular endothelial growth factor expression in cultured keratinocytes.
Implications for normal and impaired wound healing. The Journal of biological
chemistry 1995, 270(21):12607-12613.
38. Goerdt S, Orfanos CE: Other functions, other genes: alternative activation of antigen-
presenting cells. Immunity 1999, 10(2):137-142.
39. Knighton D, Hunt T, Scheuenstuhl H, Halliday B, Werb Z, Banda M: Oxygen tension
regulates the expression of angiogenesis factor by macrophages. Science 1983.
40. Porcheray F, Viaud S, Rimaniol ACC, Léone C, Samah B, Dereuddre-Bosquet N,
Dormont D, Gras G: Macrophage activation switching: an asset for the resolution of
inflammation. Clinical and experimental immunology 2005, 142(3):481-489.
41. Murphy M: How mitochondria produce reactive oxygen species. The Biochemical
journal 2009, 417(1):1-13.
42. Schreml S, Szeimies R, Prantl L, Karrer S, Landthaler M, Babilas P: Oxygen in acute
and chronic wound healing. The British journal of dermatology, 163(2):257-268.
43. Veech R: The therapeutic implications of ketone bodies: the effects of ketone bodies
in pathological conditions: ketosis, ketogenic diet, redox states, insulin resistance,
and mitochondrial metabolism. Prostaglandins, leukotrienes, and essential fatty acids
2004, 70(3):309-319.
44. Veech RL, Chance B, Kashiwaya Y, Lardy HA, Cahill GF, Jr.: Ketone bodies, potential
therapeutic uses. IUBMB Life 2001, 51(4):241-247.

169
45. Shimazu T, Hirschey M, Newman J, He W, Shirakawa K, Le Moan N, Grueter C, Lim H,
Saunders L, Stevens R et al: Suppression of Oxidative Stress by β-Hydroxybutyrate,
an Endogenous Histone Deacetylase Inhibitor. Science (New York, NY) 2013, 339:211-
214.
46. Youm Y-H, Nguyen KY, Grant RW, Goldberg EL, Bodogai M, Kim D, D'Agostino D,
Planavsky N, Lupfer C, Kanneganti TD et al: The ketone metabolite β-
hydroxybutyrate blocks NLRP3 inflammasome–mediated inflammatory disease.
Nature Medicine 2015, 21(3):263-269.
47. Milder J, Patel M: Modulation of oxidative stress and mitochondrial function by the
ketogenic diet. Epilepsy Research, 100(3):295-303.
48. Maher J, Yamamoto M: The rise of antioxidant signaling--the evolution and hormetic
actions of Nrf2. Toxicology and applied pharmacology 2010, 244(1):4-15.
49. Kim J, Cha Y-N, Surh Y-J: A protective role of nuclear factor-erythroid 2-related
factor-2 (Nrf2) in inflammatory disorders. Mutation research 2010, 690(1-2):12-23.
50. Nguyen T, Nioi P, Pickett C: The Nrf2-antioxidant response element signaling
pathway and its activation by oxidative stress. The Journal of biological chemistry
2009, 284(20):13291-13295.
51. Valcarcel-Ares M, Gautam T, Warrington J, Bailey-Downs L, Sosnowska D, de Cabo R,
Losonczy G, Sonntag W, Ungvari Z, Csiszar A: Disruption of Nrf2 signaling impairs
angiogenic capacity of endothelial cells: implications for microvascular aging. The
journals of gerontology Series A, Biological sciences and medical sciences 2012,
67(8):821-829.
52. Sykiotis GP, Bohmann D: Stress-activated cap'n'collar transcription factors in aging
and human disease. Sci Signal, 3(112):re3.
53. Shih P-H, Yen G-C: Differential expressions of antioxidant status in aging rats: the
role of transcriptional factor Nrf2 and MAPK signaling pathway. Biogerontology
2007, 8(2):71-80.
54. Moor AN, Tummel E, Prather JL, Jung M, Lopez JJ, Connors S, Gould LJ:
Consequences of age on ischemic wound healing in rats: altered antioxidant activity
and delayed wound closure. Age (Dordrecht, Netherlands) 2014, 36(2):733-748.
55. Vomhof-Dekrey E, Picklo M: The Nrf2-antioxidant response element pathway: a
target for regulating energy metabolism. The Journal of nutritional biochemistry 2012,
23(10):1201-1206.

170
56. Jarrett SG, Milder JB, Liang L-PP, Patel M: The ketogenic diet increases
mitochondrial glutathione levels. Journal of neurochemistry 2008, 106(3):1044-1051.
57. Milder JB, Liang L-PP, Patel M: Acute oxidative stress and systemic Nrf2 activation
by the ketogenic diet. Neurobiology of disease 2010, 40(1):238-244.
58. Schäfer M, Farwanah H, Willrodt A-HH, Huebner AJ, Sandhoff K, Roop D, Hohl D,
Bloch W, Werner S: Nrf2 links epidermal barrier function with antioxidant defense.
EMBO molecular medicine 2012, 4(5):364-379.
59. Yu Z-WW, Li D, Ling W-HH, Jin T-RR: Role of nuclear factor (erythroid-derived 2)-
like 2 in metabolic homeostasis and insulin action: A novel opportunity for diabetes
treatment? World journal of diabetes 2012, 3(1):19-28.
60. Suh J, Shenvi S, Dixon B, Liu H, Jaiswal A, Liu R-M, Hagen T: Decline in
transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis,
which is reversible with lipoic acid. Proceedings of the National Academy of Sciences
of the United States of America 2004, 101(10):3381-3386.
61. Schreier T, Degen E, Baschong W: Fibroblast migration and proliferation during in
vitro wound healing. A quantitative comparison between various growth factors and
a low molecular weight blood dialysate used in the clinic to normalize impaired
wound healing. Research in experimental medicine Zeitschrift für die gesamte
experimentelle Medizin einschliesslich experimenteller Chirurgie 1993, 193(4):195-205.
62. Hunt TK: The effect of varying ambient oxygen tensions on wound metabolism and
collagen synthesis. Surg Gynecol Obstet 1972.
63. Hunt TK, Linsey M, Sonne M, Jawetz E: Oxygen tension and wound infection. In:
Oxygen tension and wound infection: 1972; 1972: 47.
64. Hunt TK, Conolly WB, Aronson SB, Goldstein P: Anaerobic metabolism and wound
healing: an hypothesis for the initiation and cessation of collagen synthesis in
wounds. American journal of surgery 1978, 135(3):328-332.
65. Jensen JA, Hunt TK, Scheuenstuhl H: Effect of lactate, pyruvate, and pH on secretion
of angiogenesis and mitogenesis factors by macrophages. … investigation; a journal
… 1986.
66. Knighton DR, Silver IA, Hunt TK: Regulation of wound-healing angiogenesis-effect of
oxygen gradients and inspired oxygen concentration. Surgery 1981, 90(2):262-270.
67. Mastrofrancesco B, Henry W, Albina JE: Evidence for aerobic glycolysis in λ-
carrageenan-wounded skeletal muscle. … of Surgical Research 1984.

171
68. Warburg O: On the origin of cancer cells. Science 1956.
69. Weinhouse S, Warburg O, Burk D, Schade AL: On respiratory impairment in cancer
cells. Science 1956.
70. Fazli M, Bjarnsholt T, Kirketerp‐Møller K, Jørgensen A, Andersen C, Givskov M,
Tolker‐Nielsen T: Quantitative analysis of the cellular inflammatory response
against biofilm bacteria in chronic wounds. Wound Repair and Regeneration 2011,
19(3).
71. Hopf HW, Rollins MD: Wounds: an overview of the role of oxygen. Antioxidants &
redox signaling 2007, 9(8):1183-1192.
72. Trabold O, Wagner S, Wicke C, Scheuenstuhl H, Hussain ZM, Rosen N, Seremetiev A,
Becker HD, Hunt TK: Lactate and oxygen constitute a fundamental regulatory
mechanism in wound healing. Wound Repair and Regeneration 2003, 11(6).
73. Bakker J, Gris P, Coffernils M, Kahn RJ, Vincent JL: Serial blood lactate levels can
predict the development of multiple organ failure following septic shock. American
journal of surgery 1996, 171(2):221-226.
74. Britland S, Ross-Smith O, Jamil H, Smith AG, Vowden K, Vowden P: The lactate
conundrum in wound healing: clinical and experimental findings indicate the
requirement for a rapid point-of-care diagnostic. Biotechnology progress 2012,
28(4):917-924.
75. Wu L, Xia YP, Roth SI, Gruskin E, Mustoe TA: Transforming growth factor-beta1
fails to stimulate wound healing and impairs its signal transduction in an aged
ischemic ulcer model: importance of oxygen and age. The American journal of
pathology 1999, 154(1):301-309.
76. Xia YP, Zhao Y, Tyrone JW, Chen A, Mustoe TA: Differential activation of migration
by hypoxia in keratinocytes isolated from donors of increasing age: implication for
chronic wounds in the elderly. The Journal of investigative dermatology 2001,
116(1):50-56.
77. Mogford JE, Sisco M, Bonomo SR, Robinson AM, Mustoe TA: Impact of aging on
gene expression in a rat model of ischemic cutaneous wound healing. The Journal of
surgical research 2004, 118(2):190-196.
78. Tandara AA, Kloeters O, Kim I, Mogford JE, Mustoe TA: Age effect on HSP70:
decreased resistance to ischemic and oxidative stress in HDF. The Journal of surgical
research 2006, 132(1):32-39.

172
CHAPTER 6: IMPLICATIONS FOR WOUND HEALING
6.1. Chapter Synopsis
In this chapter, we describe the major findings of this work based on the detailed
discussion of the data in previous chapters. This chapter will discuss the major implications for
wound healing as well as future directions for the project.
6.2. Implications for Wound Healing
The major goals of this dissertation work were to establish a method of sustaining
nutritional ketosis via exogenous ketone administration, determine the healing capabilities of
exogenous ketone supplementation in an aged rat population, and to acquire data of potential
mechanisms of action. As mentioned earlier, there is a dire need for a therapy that can accelerate
wound healing and efficiently restore normal tissue functionality. Chronic wounds, as discussed
in Chapter 1, impact several million people annually and is a billion dollar growing market. The
therapy presented within this dissertation exhibits great potential to accelerate cutaneous wound
healing closure and to be used as a multifaceted tool to stimulate wound repair and regeneration
in poor healing tissues especially chronic wounds in the aged population.
The KD has proven effective for the metabolic management of seizures and potentially
numerous other disorders, but is limited clinically [1-26]. Exogenous ketone supplements are

173
primarily being developed as an alternative or adjuvant method of inducing therapeutic ketosis to
free patients from the restrictiveness of the KD. In Chapter 3, we were able to clearly
demonstrate that nutritional ketosis can be established with exogenous ketone supplementation
without negatively affecting blood triglyceride and lipoprotein profiles. Importantly, exogenous
ketone supplementation provides a tool for achieving ketosis in patients that are unable,
unwilling, or uninterested in consuming a low carbohydrate or ketogenic diet. It may also help
circumvent some of the difficulties associated with KD therapy initiation and maintenance, as it
allows for rapid induction of ketosis in a dose-dependent fashion, eases the transition to
nutritional ketosis, enhances the speed of keto-adaptation, and easily sustains ketosis with
prolonged consumption. Simultaneously, it could provide patients with the opportunity to reap
the benefits of ketosis without the practical and social difficulties of a highly restrictive diet.
Additionally, we demonstrated the ability of several ketone supplements to elevate blood ketone
levels, providing multiple options to induce therapeutic ketosis based on patient need. Many
patients that previously were unable to benefit from the KD may now have an alternative method
of achieving therapeutic ketosis. Ketone supplementation may also represent a means to further
augment ketonemia in those that respond to the ketogenic diet, especially in those individuals
where maintaining low glucose and insulin through carbohydrate restriction is important.
In Chapter 3, we demonstrated the exogenous ketone supplementation produced a
hypoglycemic effect, which in Chapter 4, was no longer apparent in the aged rats. Further
experiments need to be performed to determine what role age has explicitly on the observed
hypoglycemic effect. Additionally, further experiments need to be conducted to determine the
role that insulin sensitivity has in the age-dependent diminished hypoglycemic effect. As Chapter
3’s hypoglycemic effect was the result of an intragastric bolus and Chapter 4 transitioned to

174
food-integrated administration, future studies need to be conducted to determine the effects of
administration on the observed hypoglycemic effect.
It should be noted that in these studies, rats were administered high doses due to their
higher basal metabolic rate (5.6x faster than humans). Future studies are needed to optimize
doses for humans. Preliminary data from the D’Agostino laboratory demonstrates that a 100 kg
male can obtain elevated ketone levels at a 0.4 g/kg dose compared to the 5 g/kg and 10 g/kg
doses that were administered to the rats. Follow up studies are currently being conducted to
determine the safety and toxicity profile of the ketone supplements KE and BMS as BD is
already FDA approved and MCT is a naturally occurring food source. Clarke and colleagues
have already demonstrated the safety of a similar ketone ester administration in humans; thus, we
don’t anticipate difficulty with the feasibility of using ketone supplements in humans [27].
However, one obstacle to overcome in the synthetic ketone supplements tested in this project is
their palatability. Both BD and KE are not pleasing to taste and will need to be optimized for
flavor without sacrificing their functionality. Additionally, future studies are needed to determine
the best route of administration for ketone supplements in patients, it may be more suitable to
have the supplements available in a variety of applications including parenteral nutrition,
integrated into food that the patients could ingest, meal replacement drinks, and oral pills.
As described in Chapter 2, exogenous ketone supplementation has the potential to
simultaneously target several underlying etiologies associated with age-impaired wound healing,
including unrelieved inflammation, unquenched ROS production, insufficient blood flow, and
dysfunctional metabolism. This is very different from most current wound therapies being
studied, which target a specific molecular or cellular mechanism. Because of the complexity of
wound healing, and the fact that there are often multiple mitigating factors that prevent normal

175
wound closure, most wound therapies are minimally effective. We hypothesize that using
exogenous ketone supplementation, as a component of standard care would target the
multifaceted etiologies underling most chronic wounds in the aged population. In Chapter 4, we
demonstrated that both BD and BMS+MCT ketone supplement therapies were effective in
enhancing wound healing in aged animals and the BMS+MCT also enhanced wound healing in
young animals. Further studies are needed to confirm the optimal dosing protocol to maximize
wound-healing efficacy.
Additionally, we elucidated some potential mechanisms of actions by which this novel
proposed therapy was able to significantly enhance wound healing. We demonstrated that
exogenous ketone supplementation altered the serum and hippocampal metabolome including
increasing Kreb’s cycle intermediates, antioxidants carnosine and anserine, and the potent
vasodilator adenosine. Further metabolomic studies need to be conducted to determine the
effects of age on ketone supplementation changes to the metabolome. Furthermore, young and
aged metabolomic studies of the wound healing environment with and without ketone
supplementation would offer great insights to the wound healing process. Mechanistically, we
demonstrated that ketone supplementation increases fibroblast proliferation and migration,
reduces ROS production, reduces lactate production, and increases blood flow. However, we did
not demonstrate a signifcant effect of ketone supplementation on antioxidants SOD2 or NQO1,
further studies need to be conducted to determine if the administration of ketone supplements
elicit the same physiological changes in other antioxidants. Further investigation into exploring
potential mechanisms of action are needed to fully understand the impact exogenous ketone
supplementation has on wound healing, specifically in an aged population. However, the
complexity of wound healing and the multifaceted targets of ketone bodies may constrain

176
elucidating a single, clearly delineated mechanism of action.
Moreover, futures studies will explore a topical application of ketone bodies. Even
though there was a significant increase in blood flow, this may not efficient for some patients;
thus, a direct application of the ketones or a combination of both therapies may be more
beneficial. Base on the results included in this dissertation we propose that exogenous ketone
supplementation could offer a novel therapeutic option for chronic wounds, especially in the
aging population.
6.3. References for Chapter 6
1. SirvenJ,WhedonB,CaplanD,LiporaceJ,GlosserD,O'DwyerJ,SperlingM:Theketogenicdietforintractableepilepsyinadults:preliminaryresults.Epilepsia1999,40(12):1721-1726.
2. WilderR:Theeffectofketonemiaonthecourseofepilepsy.MayoClinBulletin1921,2:307-308.
3. ThieleE:Assessingtheefficacyofantiepileptictreatments:theketogenicdiet.Epilepsia2003,44Suppl7:26-29.
4. PaoliA,RubiniA,VolekJS,GrimaldiKA:Beyondweightloss:areviewofthetherapeuticusesofvery-low-carbohydrate(ketogenic)diets.Europeanjournalofclinicalnutrition2013,67(8):789-796.
5. FosterGD,WyattHR,HillJO,McGuckinBG,CB,MohemmedBS,SzaparyPO,RaderDJ,EdmanJS,KleinS:Arandomizedtrialofalow-carbohydratedietforobesity.TheNewEnglandjournalofmedicine2003,348:2082-2090.
6. WestmanEC,FeinmanRD,MavropoulosJC,VernonMC,VolekJS,WortmanJA,YancyWS,PhinneySD:Low-carbohydratenutritionandmetabolism.TheAmericanjournalofclinicalnutrition2007(86):276-284.
7. WestmanEC,YancyWS,EdmanJS,TomlinKF,PerkinsCE:Effectof6-monthadherencetoaverylowcarbohydratedietprogram.TheAmericanjournalofmedicine2002,113(1):30-36.

177
8. ForsytheC,PhinneyS,FernandezM,QuannE,WoodR,BibusD,KraemerW,FeinmanR,VolekJ:Comparisonoflowfatandlowcarbohydratedietsoncirculatingfattyacidcompositionandmarkersofinflammation.Lipids2008,43(1):65-77.
9. BodenG,SargradK,HomkoC,MozzoliM,SteinT:Effectofalow-carbohydratedietonappetite,bloodglucoselevels,andinsulinresistanceinobesepatientswithtype2diabetes.Annalsofinternalmedicine2005,142(6):403-411.
10. GumbinerB,WendelJ,McDermottM:Effectsofdietcompositionandketosisonglycemiaduringvery-low-energy-diettherapyinobesepatientswithnon-insulin-dependentdiabetesmellitus.TheAmericanjournalofclinicalnutrition1996,63(1):110-115.
11. NielsenJ,JoenssonE:Low-carbohydratedietintype2diabetes:stableimprovementofbodyweightandglycemiccontrolduring44monthsfollow-up.Nutrition&metabolism2008,5:14.
12. YancyW,FoyM,ChaleckiA,VernonM,WestmanE:Alow-carbohydrate,ketogenicdiettotreattype2diabetes.Nutrition&metabolism2005,2:34.
13. DashtiHM,Al-ZaidNS,MathewTC,Al-MousawiM,TalibH,AsfarSK,BehbahaniAI:Longtermeffectsofketogenicdietinobesesubjectswithhighcholesterollevel.Molecularandcellularbiochemistry2006,286(1-2):1-9.
14. MaaloufM,RhoJM,MattsonMP:Theneuroprotectivepropertiesofcalorierestriction,theketogenicdiet,andketonebodies.BrainResRev2009,59(2):293-315.
15. MavropoulosJC,YancyWS,HepburnJ,WestmanEC:Theeffectsofalow-carbohydrate,ketogenicdietonthepolycysticovarysyndrome:apilotstudy.Nutrition&metabolism2004,2:35.
16. SeyfriedT,FloresR,PoffA,D'AgostinoD:Cancerasametabolicdisease:implicationsfornoveltherapeutics.Carcinogenesis2014,35:515-527.
17. PoffAM,AriC,ArnoldP,SeyfriedTN,D'AgostinoDP:Ketonesupplementationdecreasestumorcellviabilityandprolongssurvivalofmicewithmetastaticcancer.InternationaljournalofcancerJournalinternationalducancer2014,135:1711-1720.
18. PoffA,AriC,SeyfriedT,D'AgostinoD:Theketogenicdietandhyperbaricoxygentherapyprolongsurvivalinmicewithsystemicmetastaticcancer.PloSone2013,8(6):e65522.
19. SeyfriedT,SheltonL:Cancerasametabolicdisease.Nutrition&metabolism,7:7.

178
20. FineE,Segal-IsaacsonC,FeinmanR,HerszkopfS,RomanoM,TomutaN,BontempoA,NegassaA,SparanoJ:Targetinginsulininhibitionasametabolictherapyinadvancedcancer:apilotsafetyandfeasibilitydietarytrialin10patients.Nutrition(Burbank,LosAngelesCounty,Calif)2012,28(10):1028-1035.
21. ZhaoZ,LangeD,VoustianioukA,MacGroganD,HoL,SuhJ,HumalaN,ThiyagarajanM,WangJ,PasinettiG:Aketogenicdietasapotentialnoveltherapeuticinterventioninamyotrophiclateralsclerosis.BMCneuroscience2006,7:29.
22. WhiteH,VenkateshB:Clinicalreview:ketonesandbraininjury.Criticalcare(London,England)2011,15(2):219.
23. PrinsM:Cerebralmetabolicadaptationandketonemetabolismafterbraininjury.Journalofcerebralbloodflowandmetabolism:officialjournaloftheInternationalSocietyofCerebralBloodFlowandMetabolism2008,28(1):1-16.
24. HendersonS,VogelJ,BarrL,GarvinF,JonesJ,CostantiniL:StudyoftheketogenicagentAC-1202inmildtomoderateAlzheimer'sdisease:arandomized,double-blind,placebo-controlled,multicentertrial.Nutrition&metabolism2009,6:31.
25. BrownlowM,BennerL,D'AgostinoD,GordonM,MorganD:KetogenicdietimprovesmotorperformancebutnotcognitionintwomousemodelsofAlzheimer'spathology.PloSone2013,8(9):e75713.
26. KossoffEH,HartmanAL:Ketogenicdiets:newadvancesformetabolism-basedtherapies.CurrentOpinioninNeurology2012,25:173-178.
27. ClarkeK,TchabanenkoK,PawloskyR,CarterE,ToddKingM,Musa-VelosoK,HoM,RobertsA,RobertsonJ,VanitallieTetal:Kinetics,safetyandtolerabilityof(R)-3-hydroxybutyl(R)-3-hydroxybutyrateinhealthyadultsubjects.Regulatorytoxicologyandpharmacology:RTP2012,63(3):401-408.

179
APPENDIX A: MATERIALS AND METHODS
A.1 Ethics Statement
All animal studies were approved by and performed within strict adherence to the
University of South Florida and James A. Haley Veterans Hospital’s Institutional Animal Care
and Use Committee (IACUC) protocols R00006 and V4251.
A.2 Materials and Methods for Experiments included in Chapter 3: Establishing
Therapeutic Ketosis (Metabolic Therapy with Exogenous Ketone Supplementation)
A.2.1 Synthesis and Formulation of Ketone Supplements
KE was synthesized as previously described [1]. BMS is a novel agent
(sodium/potassium- βHB mineral salt) supplied as a 50% solution containing approximately
375mg/g of pure βHB and 125 mg/g of sodium/potassium. Both KE and BMS were developed
and synthesized in collaboration with Savind Inc. Pharmaceutical grade MCT oil (~65% caprylic
triglyceride; 45% capric triglyceride) was purchased from Now Foods (Bloomingdale, IL). BMS
was formulated in a 1:1 ratio with MCT at the University of South Florida (USF), yielding a final

180
mixture of 25% water, 25% pure βHB mineral salt and 50% MCT. BD was purchased from
Sigma-Aldrich (Prod # B84785, Milwaukee, WI).
A.2.2. Daily Gavage to Induce Dietary Ketosis
Animal procedures were performed in accordance with the University of South Florida
Institutional Animal Care and Use Committee (IACUC) guidelines (Protocol #0006R). Juvenile
male Sprague-Dawley rats (275-325 g, Harlan Laboratories) were randomly assigned to one of
six study groups: control (water, n=11), BD (n=11), KE (n=11), MCT (n=10), BMS (n=11), or
BMS+MCT (n=12). Caloric density of standard rodent chow and dose of ketone supplements are
listed in Table 1. On days 1-14, rats received a 5 g/kg body weight dose of their respective
treatments via intragastric gavage. Dosage was increased to 10 g/kg body weight for the second
half of the study (days 15-28) for all groups except BD and KE to prevent excessive
hyperketonemia (ketoacidosis). Each daily dose of BMS would equal ~1000-1500 mg of βHB,
depending on the weight of the animal. Intragastric gavage was performed at the same time
daily, and animals had ad libitum access to standard rodent chow 2018 (Harlan Teklad) for the
duration of the study. The macronutrient ratio the standard rodent chow was 62.2%, 23.8% and
14% of carbohydrates, protein and fat respectively.
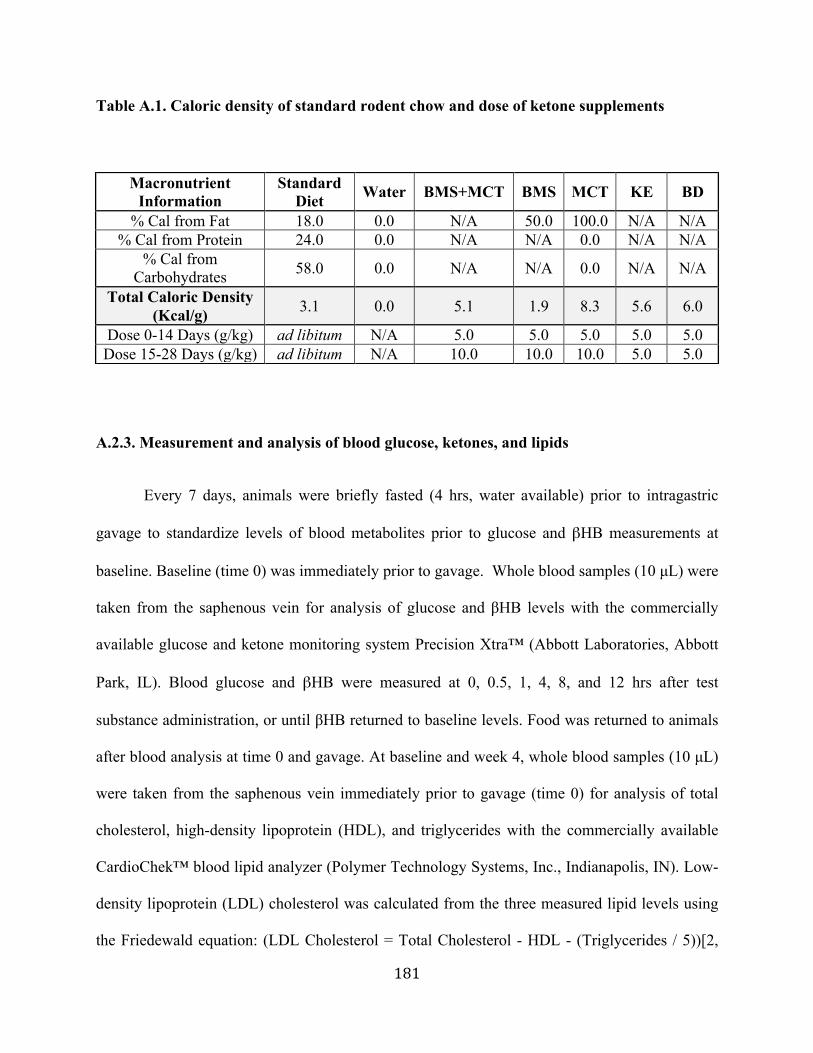
181
Table A.1. Caloric density of standard rodent chow and dose of ketone supplements
A.2.3. Measurement and analysis of blood glucose, ketones, and lipids
Every 7 days, animals were briefly fasted (4 hrs, water available) prior to intragastric
gavage to standardize levels of blood metabolites prior to glucose and βHB measurements at
baseline. Baseline (time 0) was immediately prior to gavage. Whole blood samples (10 µL) were
taken from the saphenous vein for analysis of glucose and βHB levels with the commercially
available glucose and ketone monitoring system Precision Xtra™ (Abbott Laboratories, Abbott
Park, IL). Blood glucose and βHB were measured at 0, 0.5, 1, 4, 8, and 12 hrs after test
substance administration, or until βHB returned to baseline levels. Food was returned to animals
after blood analysis at time 0 and gavage. At baseline and week 4, whole blood samples (10 µL)
were taken from the saphenous vein immediately prior to gavage (time 0) for analysis of total
cholesterol, high-density lipoprotein (HDL), and triglycerides with the commercially available
CardioChek™ blood lipid analyzer (Polymer Technology Systems, Inc., Indianapolis, IN). Low-
density lipoprotein (LDL) cholesterol was calculated from the three measured lipid levels using
the Friedewald equation: (LDL Cholesterol = Total Cholesterol - HDL - (Triglycerides / 5))[2,
Macronutrient Information
Standard Diet Water BMS+MCT BMS MCT KE BD
% Cal from Fat 18.0 0.0 N/A 50.0 100.0 N/A N/A % Cal from Protein 24.0 0.0 N/A N/A 0.0 N/A N/A
% Cal from Carbohydrates 58.0 0.0 N/A N/A 0.0 N/A N/A
Total Caloric Density (Kcal/g) 3.1 0.0 5.1 1.9 8.3 5.6 6.0
Dose 0-14 Days (g/kg) ad libitum N/A 5.0 5.0 5.0 5.0 5.0 Dose 15-28 Days (g/kg) ad libitum N/A 10.0 10.0 10.0 5.0 5.0

182
3]. Animals were weighed once per week to track changes in body weight associated with
hyperketonemia.
A.2.4. Organ Weight and Collection
On day 29, rats were sacrificed 4-8 hrs after intragastric gavage which correlated to the
time range where the most significantly elevated blood βHB levels were observed. Brain, lungs,
liver, kidneys, spleen and heart were harvested, weighed (AWS-1000 1kg portable digital scale
(AWS, Charleston, SC)), and flash-frozen in liquid nitrogen or preserved in 4%
paraformaldehyde for future analysis.
A.2.5. Statistics
All data are presented as the mean ± standard deviation (SD). Data analysis was
performed using GraphPad PRISM™ version 6.0a and IBM SPSS Statistics 22.0. Results were
considered significant when p<0.05. Triglyceride and lipoprotein profile data were analyzed
using One-Way ANOVA. Blood ketone and blood glucose were compared to control at the
applicable time points using a Two-Way ANOVA. Correlation between blood βHB and glucose
levels in ketone supplemented rats was compared to controls using ANCOVA analysis. Organ
and body weights were analyzed using One-Way ANOVA. Basal blood ketone and blood
glucose levels were analyzed using Two-Way ANOVA. All mean comparisons were carried out
using Tukey’s multiple comparisons post-hoc test.

183
A.2.6. Metabolomics Profiling of KE and BMS+MCT Treated Rats
On day 29, approximately 500 uL of whole blood was collected from the saphenous vein
of control, KE, and BMS+MCT-treated animals 4 hours post-intragastric gavage. Serum was
separated by centrifugation using Microtainer© Tubes with Serum Separator (Becton
Dickinson). Serum was transferred to screw-to cryovials and flash frozen in liquid nitrogen.
Frozen serum samples were shipped to Metabolon, Inc. for global metabolomics profiling using
liquid chromatography-tandem mass spectrometry (LC-MS/MS) and gas chromatography-mass
spectrometry (GC-MS). The concentration of 388 named biochemicals of known identity was
analyzed in the serum samples. Welch’s two-sample t-test was used to determine which
metabolites differed significantly in concentration between ketone supplemented animals and
controls. Results were considered significant when p<0.05.
A.3. Materials and Methods for Experiments included in Chapter 4: Enhancing Wound
Healing With Metabolic Therapy
A.3.1. Synthesis and Formulation of Ketone Precursors
BMS is a novel agent (sodium/potassium- βHB mineral salt) supplied as a 50% solution
containing approximately 375mg/g of pure βHB and 125 mg/g of sodium/potassium. Both KE
and BMS were developed and synthesized in collaboration with Savind Inc. Pharmaceutical

184
grade MCT oil (~65% caprylic triglyceride) was purchased from Now Foods (Bloomingdale,
IL). BMS was mixed in a 1:1 ratio with MCT at the University of South Florida (USF), yielding
25% water, 25% pure βHB mineral salt and 50% MCT. BD was purchased from Sigma-Aldrich
(Prod # B84785, Milwaukee, WI).
A.3.2 Animal Gavage to Induce Dietary Ketosis
Animal procedures were performed in accordance with the University of South Florida
Institutional Animal Care and Use Committee (IACUC) guidelines (Protocol #V4251) and
abided by all requirements of the Animal Welfare Act and the Guide for Care and Use of
Laboratory Animals. Healthy 8 month (young) and 20 month (aged) male Fisher 344 rats (Harlan
Laboratories and NIA) were randomly assigned to one of three groups: Control (water, n≥6), BD
(n≥6), or BMS+MCT (n≥6). Rats received one 6.5 g/kg dose of their respective treatments via
intragastric gavage. Animals had ad libitum access to standard rodent chow 2018 (Harlan
Teklad) for duration of the study. Whole blood samples (10 µL) were taken from the saphenous
vein for analysis of glucose and βHB levels with the commercially available glucose and ketone
monitoring system Precision Xtra™ (Abbott Laboratories, Abbott Park, IL). The Precision
Xtra™ ketone monitoring system only measures βHB blood levels; therefore, total blood ketone
levels would be higher than measured. Blood glucose and βHB were measured at 0, 0.5, 1, 4, 8,
12, and 24 hrs after test substance administration.

185
A.3.3. Surgical Procedure
Healthy 8 month (young) and 20 month (aged) Fisher 344 rats (National Institutes of
Aging, Bethesda, MD) were utilized to create and ischemic wound model adapted from our
previous work [4-6]. In all Fisher 344 rats, a dorsal bi-pedicle 10.5x3 cm flap was surgically
created on day 0 (time of wounding), in which a silicone sheet was placed under the panniculus
carnosus fascia and above the paraspinous muscles, limiting revascularization and causing the
flap to become ischemic. Two 6mm punch biopsies created full thickness wounds extending just
above the fascia within the flap (ischemic wounds) and two 6mm punches were created laterally
to ischemic flap giving the control (non-ischemic) wounds in the same animal.
A.3.4. Diet Administration
The day before flap creation, animals are fasted overnight (~18hrs) to enable initiation of
the diet immediately upon waking from surgery. On the day of ischemic flap creation (day 0),
young and aged rats were randomly assigned to one of three study groups: Control standard diet
(SD), BD or BMS+MCT. Control rats were fed a combination of standard rodent chow (2018
Teklad Global 18% Protein Rodent Diet, Harlan Laboratories), water, and peanut butter mixed
into a pudding and fed ad libitum. Rats in BD group received a pudding diet of standard rodent
chow, peanut butter, water, 1% saccharine, and 20% 1,3-Butanediol by weight ad libitum. Rats
in the BMS+MCT group received a pudding diet of standard rodent chow, peanut butter, water,
1% saccharine, and 10% MCT oil by weight and 10% βHB mineral salt solution (10% βHB) by
weight ad libitum. Diets were continuously replaced to maintain freshness and to allow rat to
feed ad libitum.

186
A.3.5. βHB, Glucose, and Weight Measurements
Every 7 days, whole blood samples (10 µL) were taken from the saphenous vein for
analysis of glucose and βHB levels with the commercially available glucose and ketone
monitoring system Precision Xtra™ (Abbott Laboratories, Abbott Park, IL) to confirm that the
dietary ingestion of BD and BMS+MCT induced continuous ketosis in the blood. Rats were
weighed 2x weekly with scales (Mittler Toledo SB16001) available in the Morsani College of
Medicine vivarium. Blood and weight measurements were taken at the same time of day each
week to limit variation based on normal metabolism fluctuations.
A.3.6. Analysis of Blood Flow and Wound Size
Every 7 days, while under anesthesia, Laser Doppler scanning was used to measure blood
flow over the course of the study. On days 3, 7, 10, 14, 21, 25, and 28 animals were anesthetized,
both ischemic and non-ischemic wounds were digitally photographed, and wound sizes were
determined (Bamboo Create v5, Wacom Co, Ltd, and Image J 64 Version 10.2).
A.3.7. Extraction of Tissue
On day 28 of the study, the rats were euthanized by carbon dioxide (CO2) asphyxiation
where periwound tissue (tissue surrounding the wound) including the wound, spleen and liver
were surgically harvested. Portions of the spleen, liver and wounds were flash frozen by liquid

187
nitrogen, formalin-fixed, or preserved in optimal cutting temperature (OCT) freezing media for
future analysis.
A.3.8. Statistics
All data are presented as the mean ± standard deviation (SD). Data analysis was performed using
GraphPad PRISM™ version 6.0a. Results were considered significant when p<0.05. Blood
ketone and blood glucose were compared to controls at the applicable time points using a Two-
Way ANOVA. Correlation between blood βHB and glucose levels was determined by linear
regression analysis. Blood flow and wound healing progression were analyzed compared to
controls at the applicable time points using Two-Way ANOVA. Organ and body weights were
analyzed using One-Way ANOVA. All mean comparisons were carried out using Tukey’s
multiple comparisons post-hoc test.
A.4. Materials and Methods for Experiments included in Chapter 5: Potential Mechanisms
for Exogenous Ketone Supplementation to Enhance Wound Healing
A.4.1. Cytokine Array
After 3 days and 7 days post-wounding, young and aged Fischer rats were humanely
euthanized and per-wound tissue was harvested and preserved in liquid nitrogen. 500 mg of
periwound tissue was measured and razor blade minced on a petri dish in 400 µL of RIPA Lysis

188
Buffer (Sigma Chemical Co, St. Louis, MO) with 10 µL / mL of Halt protease and phosphatase
inhibitor cocktail (Pierce, Rockford, IL). Minced tissue with liquid was placed into lysing
matrix-A tubes (MP Biomedical, Santa Ana, CA). An additional 400 µL of RIPA Lysis Buffer
was used to wash petri dish and added to the tube. Tissues were homogenized for 20 seconds
using FastPrep®-24 Instrument (MP Biomedical, Santa Ana, CA). Tubes were centrifuged at
16,000xg for 10 minutes, supernatants were transferred to a clean 1.5 mL conical tube, and
lysates were sonicated at low power setting and centrifuged at 16,000xg for 10 minutes. The
supernatant of the lysates was frozen at -80° for future use. Protein concentration of the lysate
supernatant was determined using Pierce BCA protein Assay Kit (Rockford, IL) reading
absorbance at 540 nanometers. 50µg of samples were sent to Eve Technologies Inc. (Calgary,
Alberta, Canada), where they were analyzed on Bio-Plex 200 Systems (Bio-Rad Laboratories,
Inc., Hercules, CA) via Rat Cytokine Array/ Chemokine Array 27-Plex/ Discovery Assay, a
multiplex assay that measures twenty-seven inflammatory biomarkers in a single microwell
(Millipore MILLIPLEX®, Billerica, MA).
A.4.2. Macrophage Profile Analysis with Immunofluorescence of CD68, IRF-5, CD206
After 3 days and 7 seven days post-wounding, young and aged Fischer 344 rats were
humanely euthanized and peri-wound tissue, livers, and spleen were harvested and preserved in
10% neutral, paraformaldehyde or phosphate buffered formalin. The peri-wound tissue was
paraffin-embedded, cut into 5-micron sections, and mounted onto microscope slides at the USF
Morsani College of Medicine Histology Core. One set of slides was stained with hematoxylin
and eosin and mounted with cover slips for morphological analysis. Another set of slides was co-

189
probed with either interferon regularity factor-5 (IRF-5) to label M1 pro-inflammatory
macrophages or CD206 to label M2 anti-inflammatory macrophages and CD68 to label all
macrophages. These slides were rehydrated through a series of xylene and ethanol washes, and
antigen was retrieved via standard heat-mediated citrate buffer method. Slides were incubated in
0.3% H2O2 in dH2O to inhibit endogenous peroxide activity. Immunofluorescent analysis of the
slides was performed using the VECTASTAIN Elite ABC Kit (Vector Laboratories Inc.,
Burlingame, CA). Slides were blocked for 60 minutes in normal anti-rabbit blocking serum, and
then incubated in anti-IRF-5 antibody (1:100, ab175317, Abcam) or anit-CD206 antibody
(1:100, ab64693, Abcam) over night at 4 degrees in a humidified container. Slides were then
incubated in ALEXA Fluor 568 donkey anti-rabbit secondary antibody (Life Technologies,
Eugene, OR) in normal blocking serum for 30 mints at room temperature in the dark, followed
by VECTASTAIN ABC reagent containing avidin and biotinylated horseradish peroxidase
solution for 30 minutes at room temperature in the dark. Slides were blocked for 60 minutes in
normal anti-mouse blocking serum in the dark, and then incubated in anti-CD68 antibody (1:25,
ab955, Abcam) over night at 4 degrees in a dark humidified container. Slides were then
incubated in ALEXA Fluor 488 donkey anti-mouse secondary antibody (Life Technologies,
Eugene, OR) in normal anti-mouse blocking serum for 30 mints at room temperature in the dark,
followed by VECTASTAIN ABC reagent containing avidin and biotinylated horseradish
peroxidase solution for 30 minutes at room temperature. Slides were wet mounted with
VECTASHIELD mounting media with DAPI (Vector Laboratories Inc., Burlingame, CA) and
sealed with clear nail polish.

190
A.4.3. Cellular ROS production
DHE
Cells were grown on 6-well poly-d lysine coated plates in DMEM +Glutamax 4.5g/L
glucose media with 10% FBS and 1% penicillin and streptomycin until 50-60% confluent,
appropriate wells were grown with or without the addition of 5 mM βHB to the media for 72
hours. After 72 hours, they were then serum starved for 18 hrs with DMEM phenol red free 4.5
g/L glucose + L-Glutamine media with 0.2% FBS and 1% penicillin and streptomycin. At the
end of the 18 hours, cells were subjected to either 100uM tBOOH (Sigma Chemical Co, St.
Louis, MO) in serum starve media for three hours or media only. At the end of three hours, the
media was taken off and the cells were treated with 5 µM Dihydroethidium (DHE) in DMSO
(Life Technologies, Eugene, OR) dissolved in serum starve media for 30min. The cells were put
into Hank’s BSS and fluorescence was read on the plate reader at [528, 617] excitation and
emission. 3 wells in each grouping did not receive the DHE and the average RFUs of these wells
were used as a blank to normalize the readings. The control values have an N=3 (total N=9) and
the 100 µM has an N=6 (total N=18). Graph and statistics were created using GraphPad
PRISM™ for Mac OSX version 6.0a, statistical significance was determined using a one-way
ANOVA with Tukey’s post-hoc analysis.
Mitosox Red
Cells were grown on 6-well poly-d lysine coated plates in DMEM +Glutamax 4.5g/L
glucose media with 10% FBS and 1% penicillin and streptomycin until 50-60% confluent,

191
appropriate wells were grown with or without the addition of 5 mM βHB to the media for 72
hours. After 72 hours, they were then serum starved for 18 hrs with DMEM phenol red free 4.5
g/L glucose + L-Glutamine media with 0.2% FBS and 1% penicillin and streptomycin. Cells
were subjected to 100uM tBOOH (Sigma Chemical Co, St. Louis, MO) in serum starve media
for three hours or media only. At the end of three hours, the media was taken off and the cells
were treated with 1 µM Mitosox Red (Life Technologies, Eugene, OR) in Hank’s Balanced Salt
Solution (with magnesium and calcium) for 10 min. The cells were put into fresh Hank’s BSS
and fluorescence was read on the plate reader at [510, 580] excitation and emission. 3 wells in
each grouping did not receive the Mitosox Red and the average RFUs of these wells were used
as a blank to normalize the readings. The control values have an N=3 (total N=12) and the 100
uM has an N=6 (total N=24). Graph and statistics were created using GraphPad PRISM™ for
Mac OSX version 6.0a, statistical significance was determined using a one-way ANOVA with
Tukey’s post-hoc analysis.
A.4.4. Western Blot Analysis for in vitro and ex vivo SOD2 and NQO1
In Vitro
Cells were grown on 100 mm poly-d lysine coated petri dishes in DMEM +Glutamax 4.5
g/L glucose media with 10% FBS and 1% penicillin and streptomycin until cells were around 50-
60% confluent. Cells were changed to DMEM phenol red free 5mM glucose + L-Glutamine
media with 10% FBS and 1% penicillin and streptomycin and appropriate cells were treated with
5mM βHB for 72 hours. Media was replaced every 24 hours. After 72 hours, appropriate cells
were subjected to 100uM tBOOH (Sigma Chemical Co, St. Louis, MO) or media only. At the

192
end of three hours, the media was taken off, cells were washed with 1mL 1x cold PBS, then
incubated with 300 uL per well of Triton X-100 Lysis Buffer [containing 50mM Tris-HCl pH
7.4,150 mM NaCl, 5mM EDTA and 1% Triton-X 100], with 10 uL/ mL of Halt protease and
phosphatase inhibitor cocktail (Pierce, Rockford, IL) for 30 minutes at 4° Celsius. The cells were
scrape-harvested, and three wells were pooled for each sample. The lysates were sonicated at low
power setting and centrifuged at 16,000xg for 10 minutes. The supernatant of the lysates was
frozen at -80° for future use. Protein concentration of the lysate supernatant was determined
using Pierce BCA protein Assay Kit (Rockford, IL) reading absorbance at 540 nanometers. 20
µg of samples were separated on a 4-20% SDS gel and transferred to nitrocellulose membrane.
Membrane was blocked with 5% Milk in TBS-T (50 mM Tris HCl, 150 mM NaCl and 0.1%
Tween 20, pH 7.5) and then probed with anti-SOD2 antibody (1:2500, ab13533, Abcam), anti-
NQO1 antibody (1:500, ab34173, Abcam) followed by anti Pan-Actin antibody (1:3000, Cell
Signaling, Boston, MA) as a loading control and Anti-Rabbit IgG, HRP-linked (1:3000, Cell
Signaling, Boston, MA) as secondary antibody. Membrane was developed using Pierce ECL
Western Blotting Substrate (Rockford, IL). Quantitative densitometry was determined using
Image J software version 1.45s and graph and statistics were determined using GraphPad
PRISM™ for Mac OSX version 6.0a. Data in graph is represented as an N=3 for each group.
Ex vivo
After 3 days and 7 days post-wounding, young and aged Fischer rats were humanely
euthanized and per-wound tissue was harvested and preserved in liquid nitrogen. 500 mg of
periwound tissue was measured and razor blade minced on a petri dish in 400 µL of RIPA Lysis

193
Buffer (Sigma Chemical Co, St. Louis, MO) with 10 µL / mL of Halt protease and phosphatase
inhibitor cocktail (Pierce, Rockford, IL). Minced tissue with liquid was placed into lysing
matrix-A tubes (MP Biomedical, Santa Ana, CA). An additional 400 µL of RIPA Lysis Buffer
was used to wash petri dish and added to the tube. Tissues were homogenized for 20 seconds
using FastPrep®-24 Instrument (MP Biomedical, Santa Ana, CA). Tubes were centrifuged at
16,000xg for 10 minutes, supernatants were transferred to a clean 1.5 mL conical tube, and
lysates were sonicated at low power setting and centrifuged at 16,000xg for 10 minutes. The
supernatant of the lysates was frozen at -80° for future use. Protein concentration of the lysate
supernatant was determined using Pierce BCA protein Assay Kit (Rockford, IL) reading
absorbance at 540 nanometers. 20 µg of samples were separated on a 4-20% SDS gel and
transferred to nitrocellulose membrane. Membrane was blocked with 5% Milk in TBS-T (50 mM
Tris HCl, 150 mM NaCl and 0.1% Tween 20, pH 7.5) and then probed with anti-SOD2 antibody
(1:2500, ab13533, Abcam), anti-NQO1 antibody (1:500, ab34173, Abcam) followed by anti Pan-
Actin antibody (1:3000, Cell Signaling, Boston, MA) as a loading control and Anti-Rabbit IgG,
HRP-linked (1:3000, Cell Signaling, Boston, MA) as secondary antibody. Membrane was
developed using Pierce ECL Western Blotting Substrate (Rockford, IL). Quantitative
densitometry was determined using Image J software version 1.45s and graph and statistics were
determined using GraphPad PRISM™ for Mac OSX version 6.0a. Data in graph is represented
as an N=6 for each group.

194
A.4.5 Immunohistochemistry Analysis of Periwound Tissue Vascularization
After 3 days and 7 seven days post-wounding, young and aged Fischer 344 rats were
humanely euthanized and peri-wound tissue, livers, and spleen were harvested and preserved in
10% neutral, phosphate buffered formalin. The peri-wound tissue was paraffin-embedded, cut
into 5-micron sections, and mounted onto microscope slides at the USF Morsani College of
Medicine Histology Core. One set of slides was stained with hematoxylin and eosin and mounted
with cover slips for morphological analysis. Another set of slides was probed for the endothelial
cell marker CD31 to visualize blood vessels. These slides were rehydrated through a series of
xylene and ethanol washes, and antigen was retrieved via standard heat-mediated citrate buffer
method. Slides were incubated in 0.3% H2O2 in dH2O to inhibit endogenous peroxide activity.
Immunohistochemical analysis of the slides was performed using the VECTASTAIN Elite ABC
Kit (Vector Laboratories Inc., Burlingame, CA). Slides were blocked for 60 minutes in normal
blocking serum, and then incubated in anti-CD31 antibody (1:25, ab28364, Abcam) over night at
4 degrees Celsius in a humidified container. Slides were then incubated in biotinylated anti-
rabbit secondary antibody in normal blocking serum for 30 mints at room temperature, followed
by VECTASTAIN ABC reagent containing avidin and biotinylated horseradish peroxidase
solution for 30 minutes at room temperature. Slides were developed using the VECTOR
NovaRED Peroxidase (HRP) Substrate Kit (Vector Laboratories Inc., Burlingame, CA). Slides
were counterstained with hematoxylin, washed, mounted with coverslips, sealed with clear nail
polish, and visualized and scanned with light microscope via Moffitt Core Facilities. The entire
periwound tissue was analyzed for blood vessels. The number of CD31+ blood vessels in the
peri wound tissue were counted between control and treated groups as a measure of wound

195
vascularization. Statistical analysis was performed via one-way ANOVA with Tukey’s post-hoc
analysis. Results were considered significant when p<0.05.
A.4.6 Migration Analysis of young and aged HDFs
Cells were grown on 6-well poly-d lysine coated plates in DMEM +Glutamax 4.5g/L
glucose media with 10% FBS and 1% penicillin and streptomycin until 75-80% confluent. Cells
were changed to DMEM phenol red free 5mM glucose + L-Glutamine media with 10% FBS and
1% penicillin and streptomycin and appropriate cells were treated with 5mM βHB for 72 hours.
Media was replaced every 24 hours. After 72 hours, cells were then serum starved for 18 hrs
with DMEM phenol red free 4.5 g/L glucose + L-Glutamine media with 0.2% FBS and 1%
penicillin and streptomycin. There was not a need to use mytomycin to stop proliferation since
used the serum free media. After 18 hours, two scratches were created per well using a 200 µL
pipette tip. Wells were rinsed with 1mL RT Hank’s BBS to remove floating and loose cells.
Wells were visualized using phase contrast microscopy and a picture was taken for baseline
comparison. After 18 hours, cells were fixed with 4% paraformaldehyde for 15 minutes, rinsed
with RT 1x PBS, and a cover slip was placed in the well using VECTASHIELD mounting media
with DAPI (Vector Laboratories Inc., Burlingame, CA) and sealed with clear nail polish. Wells
were visualized using phase contrast and fluorescent microscopy, and number of cells that
migrated into scratch area was counted. Statistical analysis was performed via one-way ANOVA
with Tukey’s post-hoc analysis. Results were considered significant when p<0.05.

196
A.4.7. Proliferation Analysis of young and aged HDFs
75,000 young and aged HDF cells were seeded on 6-well poly-d lysine coated plates in
DMEM phenol red free 5mM glucose + L-Glutamine media with 10% FBS and 1% penicillin
and streptomycin with or without the presence of 5 mM βHB. Media was replaced every 24
hours. Total cell number was counted at 24, 48, 72, 96, and 120 hours post seeding. At given
time point, media was removed and cells were washed with 1 mL sterile RT 1x PBS, cells were
suspended using 250 µL (0.25% Trypsin in Hank’s Phenol Red BBS) and 750 µL of appropriate
media (with or with out βHB). Total cell density was measured using hemocytometry. Two-way
ANOVA with Tukey’s post-hoc analysis, results were considered significant when p<0.05. Error
Bars Represent mean (SD).
A.4.8. Lactate Quantification of young and aged HDFs
Young and aged primary HDFs were seeded with 250,000 cells on 6-well poly-d lysine
coated plates in DMEM phenol red free 5mM glucose + L-Glutamine media with 10% FBS and
1% penicillin and streptomycin with or without the presence of 5 mM βHB. Extracellular lactate
was measured in 10 µL of cellular media at 24, 48 and 72 hours post-seeding using Nova
Biomedical© Lactate Plus (Nova Biomedical, Waltham, MA) blood lactate measuring meter.
Two-way ANOVA with Tukey’s post-hoc analysis, results were considered significant when
p<0.05. Error Bars Represent mean (SD).

197
A.5. References for Appendix A
1. D'AgostinoD,PillaR,HeldH,LandonC,PuchowiczM,BrunengraberH,AriC,ArnoldP,DeanJ:Therapeuticketosiswithketoneesterdelayscentralnervoussystemoxygen toxicity seizures in rats. American journal of physiology Regulatory,integrativeandcomparativephysiology2013,304(10):R829-836.
2. WarnickG,KnoppR,FitzpatrickV,BransonL:Estimatinglow-densitylipoproteincholesterolbytheFriedewaldequationisadequateforclassifyingpatientsonthe basis of nationally recommended cutpoints. Clinical chemistry 1990,36(1):15-19.
3. FriedewaldW, Levy R, FredricksonD:Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparativeultracentrifuge.Clinicalchemistry1972,18(6):499-502.
4. Moor AN, Tummel E, Prather JL, Jung M, Lopez JJ, Connors S, Gould LJ:Consequencesof ageon ischemicwoundhealing in rats: alteredantioxidantactivity and delayed wound closure. Age (Dordrecht, Netherlands) 2014,36(2):733-748.
5. Gould L, Leong M, Sonstein J, Wilson S: Optimization and validation of anischemicwoundmodel.Woundrepairandregeneration:officialpublicationoftheWoundHealing Society [and] the EuropeanTissue Repair Society2005,13(6):576-582.
6. Trujillo AN, Kesl SL, Sherwood J, Wu M, Gould LJ: Demonstration of the ratischemicskinwoundmodel.Journalofvisualizedexperiments:JoVE2014(98).

198
APPENDIX B: COPYRIGHT PERMISSIONS
B.1 Copyright Permissions for: Trujilio AN*, Kesl SL*, Sherwood J, Wu M, Gould LJ. (2014)
Demonstration of the Rat Ischemic Wound Model. Journal of Visualized Experiments: JoVE ”.
*Indicates that both authors contributed equally in the work.
Permission for inclusion of the aforementioned publication was granted via email (see below).
B.2 Copy Right Permissions for: Kesl SL, Poff AM, Ward NP, Fiorelli TN, Ari C, Van Putten
AJ, Sherwood JW, Arnold P, D’Agostino DP. Effects of Exogenous Ketone Supplementation on
Blood Ketone, Glucose, Triglyceride, and Lipoprotein Levels in Sprague-Dawley Rats. Nutrition
and Metabolism (2016).

199
Skip to main content
MenuSearch Search
Publisher main menuJournalsSubmissionsPublishing servicesAbout
My Account
Follow BioMed Central
TwitterFacebook
Copyright and LicenseCopyright on any open access article in a journal published by BioMed Central is retained by theauthor(s).Authors grant BioMed Central a license to publish the article and identify itself as the original publisher.Authors also grant any third party the right to use the article freely as long as its integrity is maintainedand its original authors, citation details and publisher are identified.The Creative Commons Attribution License 4.0 formalizes these and other terms and conditions ofpublishing articles.In accordance with our Open Data policy, the Creative Commons CC0 1.0 Public Domain Dedicationwaiver applies to all published data in BioMed Central open access articles.
Where an author is prevented from being the copyright holder (for instance in the case of US governmentemployees or those of Commonwealth governments), minor variations may be required. In such cases thecopyright line and license statement in individual articles will be adjusted, for example to state ‘© 2016 Crowncopyright’. Authors requiring a variation of this type should inform BioMed Central during or immediately aftersubmission of their article.
Exceptions to copyright policyOur policy pages provide details concerning copyright and licensing for articles which were previouslypublished under policies that are different from the above. For instance, occasionally BioMed Central may co-publish articles jointly with other publishers, and different licensing conditions may then apply. In all suchcases, however, access to these articles is free from fees or any other access restrictions.
Skip to main content
MenuSearch Search
Publisher main menuJournalsSubmissionsPublishing servicesAbout
My Account
Follow BioMed Central
TwitterFacebook
License agreementIn submitting an article to any of the journals published by BioMed Central I certify that;
1. I am authorized by my co-authors to enter into these arrangements2. I warrant, on behalf of myself and my co-authors, that:
the article is original, has not been formally published in any other peer-reviewed journal, is notunder consideration by any other journal and does not infringe any existing copyright or any otherthird party rights; I am/we are the sole author(s) of the article and have full authority to enter into this agreement andin granting rights to BioMed Central are not in breach of any other obligation;the article contains nothing that is unlawful, libellous, or which would, if published, constitute abreach of contract or of confidence or of commitment given to secrecy;I/we have taken due care to ensure the integrity of the article. To my/our - and currently acceptedscientific - knowledge all statements contained in it purporting to be facts are true and any formulaor instruction contained in the article will not, if followed accurately, cause any injury, illness ordamage to the user.
3. I, and all co-authors, agree that the article, if editorially accepted for publication, shall be licensed underthe Creative Commons Attribution License 4.0. In line with BioMed Central's Open Data Policy, dataincluded in the article shall be made available under the Creative Commons 1.0 Public DomainDedication waiver, unless otherwise stated. If the law requires that the article be published in the publicdomain, I/we will notify BioMed Central at the time of submission, and in such cases not only the databut also the article shall be released under the Creative Commons 1.0 Public Domain Dedication waiver.For the avoidance of doubt it is stated that sections 1 and 2 of this license agreement shall apply andprevail regardless of whether the article is published under Creative Commons Attribution License 4.0 orthe Creative Commons 1.0 Public Domain Dedication waiver.

200
[End of BioMed Central’s license agreement]_________________________________________________________
Explanatory notes regarding BioMed Central’s license agreementAs an aid to our authors, the following paragraphs provide some brief explanations concerning the CreativeCommons licenses that apply to the articles published in BioMed Central-published journals and the rationalefor why we have chosen these licenses.
The Creative Commons Attribution License (CC BY), of which CC BY 4.0 is the most recent version, wasdeveloped to facilitate open access as defined in the founding documents of the movement, such as the 2003Berlin Declaration. Open access content has to be freely available online, and through licensing their workunder CC BY authors grant users the right to unrestricted dissemination and re-use of the work, with only theone proviso that proper attribution is given to authors. This liberal licensing is best suited to facilitate thetransfer and growth of scientific knowledge. The Open Access Scholarly Publishers Association (OASPA)therefore strongly recommends the use of CC BY for the open access publication of research literature, andmany research funders worldwide either recommend or mandate that research they have supported be publishedunder CC BY. Examples for such policies include funders as diverse as the Welcome Trust, the AustralianGovernments, the European Commission’s Horizon 2020 framework programme, or the Bill & Melinda GatesFoundation.
The default use of the Creative Commons 1.0 Public Domain Dedication waiver (CC0 or CC zero) for datapublished within articles follows the same logic, facilitating maximum benefit and the widest possible re-use ofknowledge. It is also the case that in some jurisdictions copyright does not apply to data. CC0 waives allpotential copyrights, to the extent legally possible, as well as the attribution requirement. The waiver applies todata, not to the presentation of data. If, for instance, a table or figure displaying research data is reproduced, CCBY and the requirement to attribute applies. Increasingly, however, new insights are possible through the use ofbig data techniques, such as data mining, that harness the entire corpus of digital data. In such cases attributionis often technically infeasible due to the sheer mass of the data mined, making CC0 the most suitable licensingtool for research outputs generated from such innovative techniques.
It is important to differentiate between legal requirements and community norms. It is first and foremost acommunity norm, not a law, that within the scientific community attribution mostly takes the form of citation. Itis also a community norm that researchers are expected to refer to their sources, which usually takes the form ofcitation. Across all cases of research reuse (including data, code, etc), community norms will apply as isappropriate for the situation: researchers will cite their sources where it is feasible, regardless of the applicablelicense. CC0 therefore covers those instances that lie beyond long-established community norms. The overalleffect, then, of CC0 for data is to enable further use, without any loss of citations. For further explanation, werecommend you refer to our Open Data page.
In the following, we provide the licenses’ summaries as they can be found on the Creative Commonswebsite. The Creative Commons Attribution License 4.0 provides the following summary (where ‘you’ equals‘the user’):
You are free to:
Share — copy and redistribute the material in any medium or formatAdapt — remix, transform, and build upon the material for any purpose, even commercially. The licensorcannot revoke these freedoms as long as you follow the license terms

201
Under the following terms:
Attribution— you must give appropriate credit, provide a link to the license, and indicate if changes weremade. You may do so in any reasonable manner, but not in any way that suggests the licensor endorsesyou or your useNo additional restrictions—you may not apply legal terms or technological measures that legally restrictothers from doing anything the license permits
NoticesYou do not have to comply with the license for elements of the material in the public domain or where your useis permitted by an applicable exception or limitation.
No warranties are given. The license may not give you all of the permissions necessary for your intended use.For example, other rights such as publicity, privacy, or moral rights may limit how you use the material.Please note: For the terms set in italics in the summary above further details are provided on the CreativeCommons web page from which the summary is taken (http://creativecommons.org/licenses/by/4.0/).The Creative Commons 1.0 Public Domain Dedication waiver provides the following summary:
No copyrightThe person who associated a work with this deed has dedicated the work to the public domain by waiving all ofhis or her rights to the work worldwide under copyright law, including all related and neighbouring rights, to theextent allowed by law.
You can copy, modify, distribute and perform the work, even for commercial purposes, all without askingpermission. See Other information below.
Other information
In no way are the patent or trademark rights of any person affected by CC0, nor are the rights that otherpersons may have in the work or in how the work is used, such as publicity or privacy rightsUnless expressly stated otherwise, the person who associated a work with this deed makes no warrantiesabout the work, and disclaims liability for all uses of the work, to the fullest extent permitted byapplicable law
When using or citing the work, you should not imply endorsement by the author or the affirmer.
Please note: for the terms set in italics in the summary above further details are provided on the CreativeCommons web page from which the summary is taken (http://creativecommons.org/publicdomain/zero/1).
© 2016 BioMed Central Ltd unless otherwise stated. Part of Springer Science+Business Media.
By continuing to use this website, you agree to our Terms and Conditions, Privacy statement and Cookiespolicy.

202
APPENDIX C: PUBLISHED MANUSCRIPTS
This appendix contains the original publications for the following references:
Trujilio AN*, Kesl SL*, Sherwood J, Wu M, Gould LJ. (2014) Demonstration of the Rat
Ischemic Wound Model. Journal of Visualized Experiments: JoVE. * Indicates that both authors
contributed equally to the work.
Kesl SL, Poff AM, Ward NP, Fiorelli TN, Ari C, Van Putten AJ, Sherwood JW, Arnold P,
D’Agostino DP. Effects of Exogenous Ketone Supplementation on Blood Ketone, Glucose,
Triglyceride, and Lipoprotein Levels in Sprague-Dawley Rats. Nutrition and Metabolism. 2016.

203

204

205
206

207
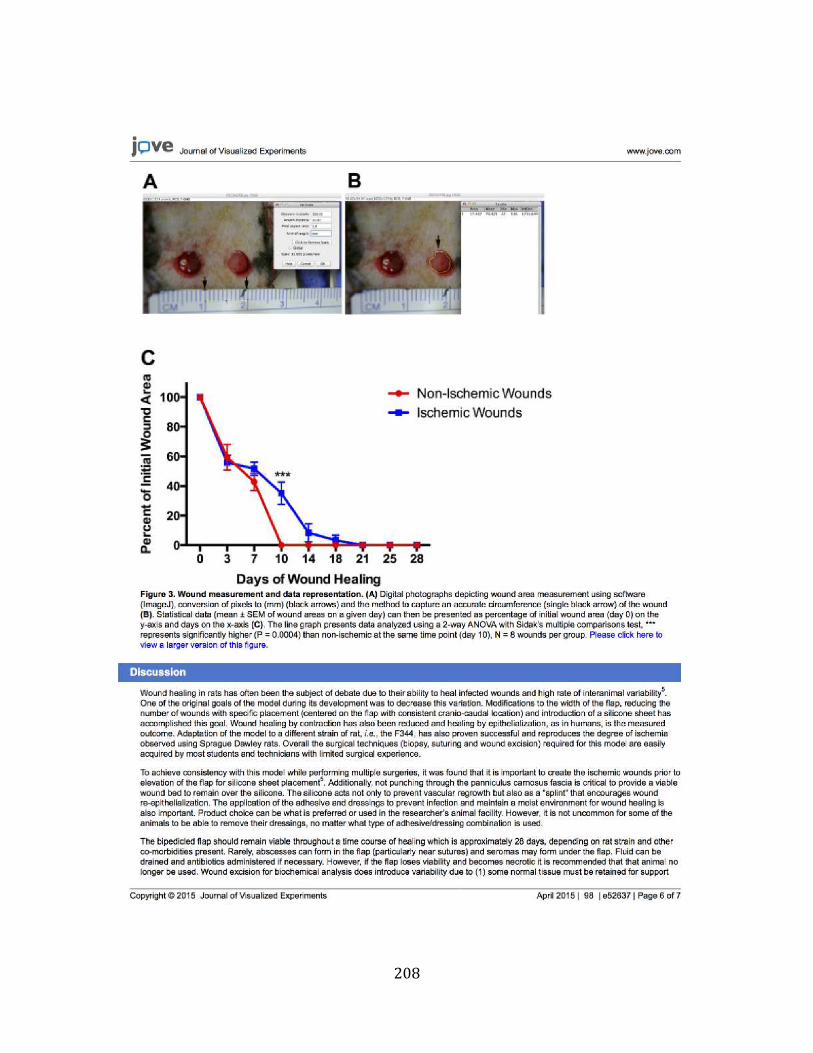
208

209

210
RESEARCH Open Access
Effects of exogenous ketonesupplementation on blood ketone, glucose,triglyceride, and lipoprotein levels inSprague–Dawley ratsShannon L. Kesl1*, Angela M. Poff1, Nathan P. Ward1, Tina N. Fiorelli1, Csilla Ari1, Ashley J. Van Putten1,Jacob W. Sherwood1, Patrick Arnold2 and Dominic P. D’Agostino1
Abstract
Background: Nutritional ketosis induced by the ketogenic diet (KD) has therapeutic applications for many diseasestates. We hypothesized that oral administration of exogenous ketone supplements could produce sustainednutritional ketosis (>0.5 mM) without carbohydrate restriction.
Methods: We tested the effects of 28-day administration of five ketone supplements on blood glucose, ketones,and lipids in male Sprague–Dawley rats. The supplements included: 1,3-butanediol (BD), a sodium/potassium β-hydroxybutyrate (βHB) mineral salt (BMS), medium chain triglyceride oil (MCT), BMS + MCT 1:1 mixture, and 1,3butanediol acetoacetate diester (KE). Rats received a daily 5–10 g/kg dose of their respective ketone supplement viaintragastric gavage during treatment. Weekly whole blood samples were taken for analysis of glucose and βHB atbaseline and, 0.5, 1, 4, 8, and 12 h post-gavage, or until βHB returned to baseline. At 28 days, triglycerides, totalcholesterol and high-density lipoprotein (HDL) were measured.
Results: Exogenous ketone supplementation caused a rapid and sustained elevation of βHB, reduction of glucose,and little change to lipid biomarkers compared to control animals.
Conclusions: This study demonstrates the efficacy and tolerability of oral exogenous ketone supplementation ininducing nutritional ketosis independent of dietary restriction.
Keywords: Ketogenic diet, Ketone ester, Ketone supplement, Appetite, β-hydroxybutyrate, Hyperketonemia,Triglycerides
BackgroundEmerging evidence supports the therapeutic potential ofthe ketogenic diet (KD) for a variety of disease states,leading investigators to research methods of harnessingthe benefits of nutritional ketosis without the dietaryrestrictions. The KD has been used as an effective non-pharmacological therapy for pediatric intractable sei-zures since the 1920s [1–3]. In addition to epilepsy, theketogenic diet has elicited significant therapeutic effects
for weight loss and type-2 diabetes (T2D) [4]. Severalstudies have shown significant weight loss on a high fat,low carbohydrate diet without significant elevations ofserum cholesterol [5–12]. Another study demonstratedthe safety and benefits of long-term application of theKD in T2D patients. Patients exhibited significant weightloss, reduction of blood glucose, and improvement oflipid markers after eating a well-formulated KD for56 weeks [13]. Recently, researchers have begun toinvestigate the use of the KD as a treatment for acne,polycystic ovary syndrome (PCOS), cancer, amyotrophiclateral sclerosis (ALS), traumatic brain injury (TBI) andAlzheimer’s disease (AD) with promising preliminaryresults [14–26].
* Correspondence: [email protected] of Molecular Pharmacology and Physiology, Morsani College ofMedicine, University of South Florida, 12901 Bruce B. Downs Blvd. MDC8,Tampa, FL 33612, USAFull list of author information is available at the end of the article
© 2016 Kesl et al. Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, andreproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link tothe Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver(http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
Kesl et al. Nutrition & Metabolism (2016) 13:9 DOI 10.1186/s12986-016-0069-y

211
The classical KD consists of a 4:1 ratio of fat to pro-tein and carbohydrate, with 80–90 % of total calories de-rived from fat [27]. The macronutrient ratio of the KDinduces a metabolic shift towards fatty acid oxidationand hepatic ketogenesis, elevating the ketone bodiesacetoacetate (AcAc) and β-hydroxybutyrate (βHB) in theblood. Acetone, generated by decarboxylation of AcAc, hasbeen shown to have anticonvulsant properties [28–32].Ketone bodies are naturally elevated to serve as alternativemetabolic substrates for extra-hepatic tissues during theprolonged reduction of glucose availability, suppression ofinsulin, and depletion of liver glycogen, such as occursduring starvation, fasting, vigorous exercise, calorierestriction, or the KD. Although the KD has cleartherapeutic potential, several factors limit the efficacyand utility of this metabolic therapy for widespreadclinical use. Patient compliance to the KD can be lowdue to the severe dietary restriction - the diet beinggenerally perceived as unpalatable - and intolerance tohigh-fat ingestion. Maintaining ketosis can be difficultas consumption of even a small quantity of carbohydratesor excess protein can rapidly inhibit ketogenesis [33, 34].Furthermore, enhanced ketone body production and tissueutilization by the tissues can take several weeks (keto-adap-tation), and patients may experience mild hypoglycemicsymptoms during this transitional period [35].Recent studies suggest that many of the benefits of the
KD are due to the effects of ketone body metabolism.Interestingly, in studies on T2D patients, improved gly-cemic control, improved lipid markers, and retraction ofinsulin and other medications occurred before weightloss became significant. Both βHB and AcAc have beenshown to decrease mitochondrial reactive oxygen species(ROS) production [36–39]. Veech et al. have summarizedthe potential therapeutic uses for ketone bodies [28, 40].They have demonstrated that exogenous ketones favorablyalter mitochondrial bioenergetics to reduce the mitochon-drial NAD couple, oxidize the co-enzyme Q, and increasethe ΔG’ (free enthalpy) of ATP hydrolysis [41]. Ketonebodies have been shown to increase the hydraulic effi-ciency of the heart by 28 %, simultaneously decreasingoxygen consumption while increasing ATP production[42]. Thus, elevated ketone bodies increase metabolic effi-ciency and as a consequence, reduce superoxide productionand increase reduced glutathione [28]. Sullivan et al. demon-strated that mice fed a KD for 10–12 days showed increasedhippocampal uncoupling proteins, indicative of decreasedmitochondrial-produced ROS [43]. Bough et al. showed anincrease of mitochondrial biogenesis in rats maintained on aKD for 4–6 weeks [44, 45]. Recently, Shimazu et al. reportedthat βHB is an exogenous and specific inhibitor of class Ihistone deacetylases (HDACs), which confers protectionagainst oxidative stress [38]. Ketone bodies have also beenshown to suppress inflammation by decreasing the
inflammatory markers TNF-a, IL-6, IL-8, MCP-1, E-selectin,I-CAM, and PAI-1 [8, 46, 47]. Therefore, it is thought thatketone bodies themselves confer many of the benefits asso-ciated with the KD.Considering both the broad therapeutic potential and
limitations of the KD, an oral exogenous ketone supple-ment capable of inducing sustained therapeutic ketosiswithout the need for dietary restriction would serve as apractical alternative. Several natural and synthetic ketonesupplements capable of inducing nutritional ketosis havebeen identified. Desrochers et al. elevated ketone bodiesin the blood of pigs (>0.5 mM) using exogenous ketonesupplements: (R, S)-1,3 butanediol and (R, S)-1,3butanediol-acetoacetate monoesters and diester [48]. In2012, Clarke et al. demonstrated the safety and efficacyof chronic oral administration of a ketone monoester ofR-βHB in rats and humans [49, 50]. Subjects maintainedelevated blood ketones without dietary restriction andexperienced little to no adverse side effects, demonstratingthe potential to circumvent the restrictive diet typicallyneeded to achieve therapeutic ketosis. We hypothesized thatexogenous ketone supplements could produce sustainedhyperketonemia (>0.5 mM) without dietary restriction andwithout negatively influencing metabolic biomarkers, suchas blood glucose, total cholesterol, HDL, LDL, and triglycer-ides. Thus, we measured these biomarkers during a 28-dayadministration of the following ketone supplements in rats:naturally-derived ketogenic supplements included mediumchain triglyceride oil (MCT), sodium/potassium -βHBmineral salt (BMS), and sodium/potassium -βHB mineralsalt +medium chain triglyceride oil 1:1 mixture (BMS +MCT) and synthetically produced ketogenic supplementsincluded 1, 3-butanediol (BD), 1, 3-butanediol acetoace-tate diester/ ketone ester (KE).
MethodsSynthesis and formulation of ketone supplementsKE was synthesized as previously described [29]. BMS isa novel agent (sodium/potassium- βHB mineral salt)supplied as a 50 % solution containing approximately375 mg/g of pure βHB and 125 mg/g of sodium/potas-sium. Both KE and BMS were developed and synthesizedin collaboration with Savind Inc. Pharmaceutical gradeMCT oil (~65 % caprylic triglyceride; 45 % capric trigly-ceride) was purchased from Now Foods (Bloomingdale,IL). BMS was formulated in a 1:1 ratio with MCT at theUniversity of South Florida (USF), yielding a final mix-ture of 25 % water, 25 % pure βHB mineral salt and50 % MCT. BD was purchased from Sigma-Aldrich(Prod # B84785, Milwaukee, WI).
Daily gavage to induce dietary ketosisAnimal procedures were performed in accordance withthe University of South Florida Institutional Animal
Kesl et al. Nutrition & Metabolism (2016) 13:9 Page 2 of 15

212
Care and Use Committee (IACUC) guidelines (Protocol#0006R). Juvenile male Sprague–Dawley rats (275–325 g,Harlan Laboratories) were randomly assigned to one ofsix study groups: control (water, n = 11), BD (n = 11), KE(n = 11), MCT (n = 10), BMS (n = 11), or BMS +MCT(n = 12). Caloric density of standard rodent chow anddose of ketone supplements are listed in Table 1. Ondays 1–14, rats received a 5 g/kg body weight dose oftheir respective treatments via intragastric gavage.Dosage was increased to 10 g/kg body weight for thesecond half of the study (days 15–28) for all groupsexcept BD and KE to prevent excessive hyperketone-mia (ketoacidosis). Each daily dose of BMS would equal~1000–1500 mg of βHB, depending on the weight of theanimal. Intragastric gavage was performed at the same timedaily, and animals had ad libitum access to standardrodent chow 2018 (Harlan Teklad) for the duration ofthe study. The macronutrient ratio the standard rodentchow was 62.2, 23.8 and 14 % of carbohydrates, proteinand fat respectively.
Measurement and analysis of blood glucose, ketones,and lipidsEvery 7 days, animals were briefly fasted (4 h, wateravailable) prior to intragastric gavage to standardizelevels of blood metabolites prior to glucose and βHBmeasurements at baseline. Baseline (time 0) was imme-diately prior to gavage. Whole blood samples (10 μL)were taken from the saphenous vein for analysis ofglucose and βHB levels with the commercially avail-able glucose and ketone monitoring system PrecisionXtra™ (Abbott Laboratories, Abbott Park, IL). Bloodglucose and βHB were measured at 0, 0.5, 1, 4, 8,and 12 h after test substance administration, or untilβHB returned to baseline levels. Food was returned toanimals after blood analysis at time 0 and gavage. Atbaseline and week 4, whole blood samples (10 μL)were taken from the saphenous vein immediatelyprior to gavage (time 0) for analysis of total choles-terol, high-density lipoprotein (HDL), and triglycerideswith the commercially available CardioChek™ bloodlipid analyzer (Polymer Technology Systems, Inc., In-dianapolis, IN). Low-density lipoprotein (LDL)
cholesterol was calculated from the three measuredlipid levels using the Friedewald equation: (LDL Choles-terol = Total Cholesterol - HDL - (Triglycerides/5)) [51,52]. Animals were weighed once per week to trackchanges in body weight associated with hyperketonemia.
Organ weight and collectionOn day 29, rats were sacrificed via deep isofluraneanesthesia, exsanguination by cardiac puncture, and de-capitation 4–8 h after intragastric gavage, which correlatedto the time range where the most significantly elevatedblood βHB levels were observed. Brain, lungs, liver,kidneys, spleen and heart were harvested, weighed (AWS-1000 1 kg portable digital scale (AWS, Charleston, SC)),and flash-frozen in liquid nitrogen or preserved in 4 %paraformaldehyde for future analysis.
StatisticsAll data are presented as the mean ± standard deviation(SD). Data analysis was performed using GraphPadPRISM™ version 6.0a and IBM SPSS Statistics 22.0. Re-sults were considered significant when p < 0.05. Trigly-ceride and lipoprotein profile data were analyzed usingOne-Way ANOVA. Blood ketone and blood glucosewere compared to control at the applicable time pointsusing a Two-Way ANOVA. Correlation between bloodβHB and glucose levels in ketone supplemented rats wascompared to controls using ANCOVA analysis. Organand body weights were analyzed using One-WayANOVA. Basal blood ketone and blood glucose levelswere analyzed using Two-Way ANOVA. All mean com-parisons were carried out using Tukey’s multiple com-parisons post-hoc test.
ResultsEffect of ketone supplementation on triglycerides andlipoproteinsBaseline measurements showed no significant changesin triglycerides or the lipoproteins (data not shown).Data represent triglyceride and lipoprotein concentra-tions measured after 4 weeks of daily exogenous ketonesupplementation. No significant change in total choles-terol was observed at 4 weeks for any of the ketonetreatment groups compared to control. (Fig. 1a). No
Table 1 Caloric density and dose of ketone supplementsMacronutrient Information Standard Diet Water BMS + MCT BMS MCT KE BD
% Cal from Fat 18.0 0.0 50.0 N/A 100.0 N/A N/A
% Cal from Protein 24.0 0.0 N/A N/A 0.0 N/A N/A
% Cal from Carbohydrates 58.0 0.0 N/A N/A 0.0 N/A N/A
Total Caloric Density (Kcal/g) 3.1 0.0 5.1 1.9 8.3 5.6 6.0
Dose 0–14 Days (g/kg) ad libitum N/A 5.0 5.0 5.0 5.0 5.0
Dose 15–28 Days (g/kg) ad libitum N/A 10.0 10.0 10.0 5.0 5.0
Kesl et al. Nutrition & Metabolism (2016) 13:9 Page 3 of 15
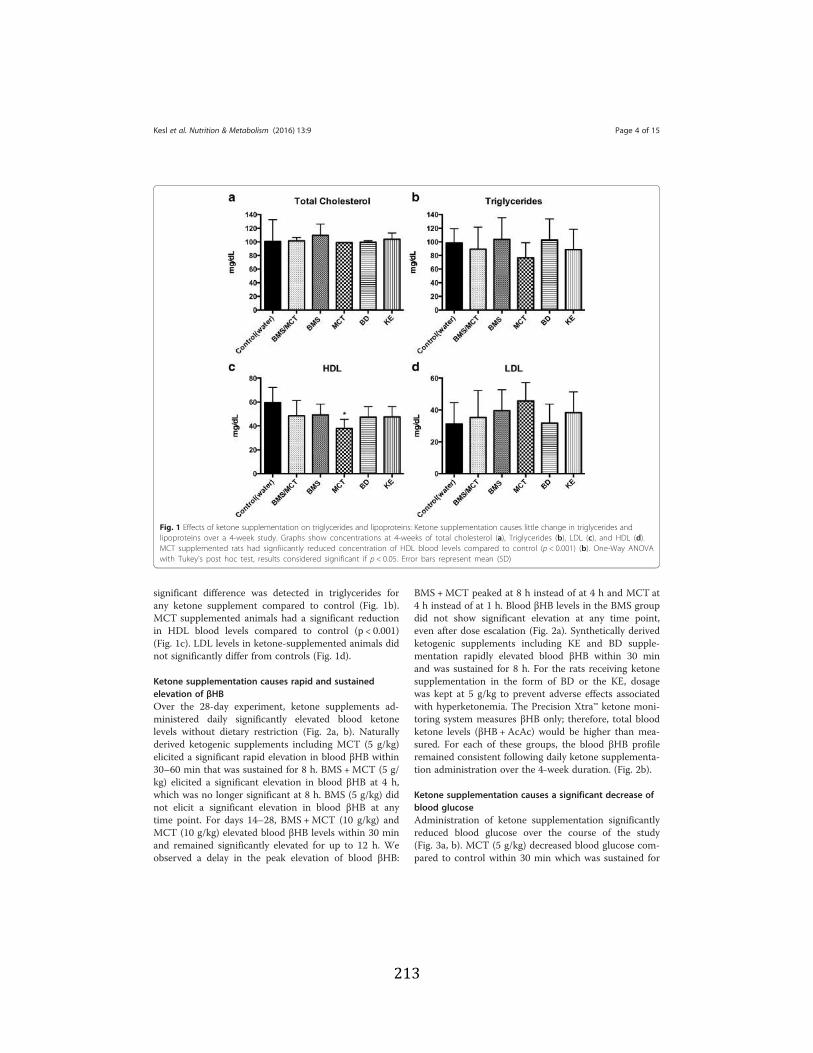
213
significant difference was detected in triglycerides forany ketone supplement compared to control (Fig. 1b).MCT supplemented animals had a significant reductionin HDL blood levels compared to control (p < 0.001)(Fig. 1c). LDL levels in ketone-supplemented animals didnot significantly differ from controls (Fig. 1d).
Ketone supplementation causes rapid and sustainedelevation of βHBOver the 28-day experiment, ketone supplements ad-ministered daily significantly elevated blood ketonelevels without dietary restriction (Fig. 2a, b). Naturallyderived ketogenic supplements including MCT (5 g/kg)elicited a significant rapid elevation in blood βHB within30–60 min that was sustained for 8 h. BMS +MCT (5 g/kg) elicited a significant elevation in blood βHB at 4 h,which was no longer significant at 8 h. BMS (5 g/kg) didnot elicit a significant elevation in blood βHB at anytime point. For days 14–28, BMS +MCT (10 g/kg) andMCT (10 g/kg) elevated blood βHB levels within 30 minand remained significantly elevated for up to 12 h. Weobserved a delay in the peak elevation of blood βHB:
BMS +MCT peaked at 8 h instead of at 4 h and MCT at4 h instead of at 1 h. Blood βHB levels in the BMS groupdid not show significant elevation at any time point,even after dose escalation (Fig. 2a). Synthetically derivedketogenic supplements including KE and BD supple-mentation rapidly elevated blood βHB within 30 minand was sustained for 8 h. For the rats receiving ketonesupplementation in the form of BD or the KE, dosagewas kept at 5 g/kg to prevent adverse effects associatedwith hyperketonemia. The Precision Xtra™ ketone moni-toring system measures βHB only; therefore, total bloodketone levels (βHB +AcAc) would be higher than mea-sured. For each of these groups, the blood βHB profileremained consistent following daily ketone supplementa-tion administration over the 4-week duration. (Fig. 2b).
Ketone supplementation causes a significant decrease ofblood glucoseAdministration of ketone supplementation significantlyreduced blood glucose over the course of the study(Fig. 3a, b). MCT (5 g/kg) decreased blood glucose com-pared to control within 30 min which was sustained for
Fig. 1 Effects of ketone supplementation on triglycerides and lipoproteins: Ketone supplementation causes little change in triglycerides andlipoproteins over a 4-week study. Graphs show concentrations at 4-weeks of total cholesterol (a), Triglycerides (b), LDL (c), and HDL (d).MCT supplemented rats had signfiicantly reduced concentration of HDL blood levels compared to control (p < 0.001) (b). One-Way ANOVAwith Tukey’s post hoc test, results considered significant if p < 0.05. Error bars represent mean (SD)
Kesl et al. Nutrition & Metabolism (2016) 13:9 Page 4 of 15
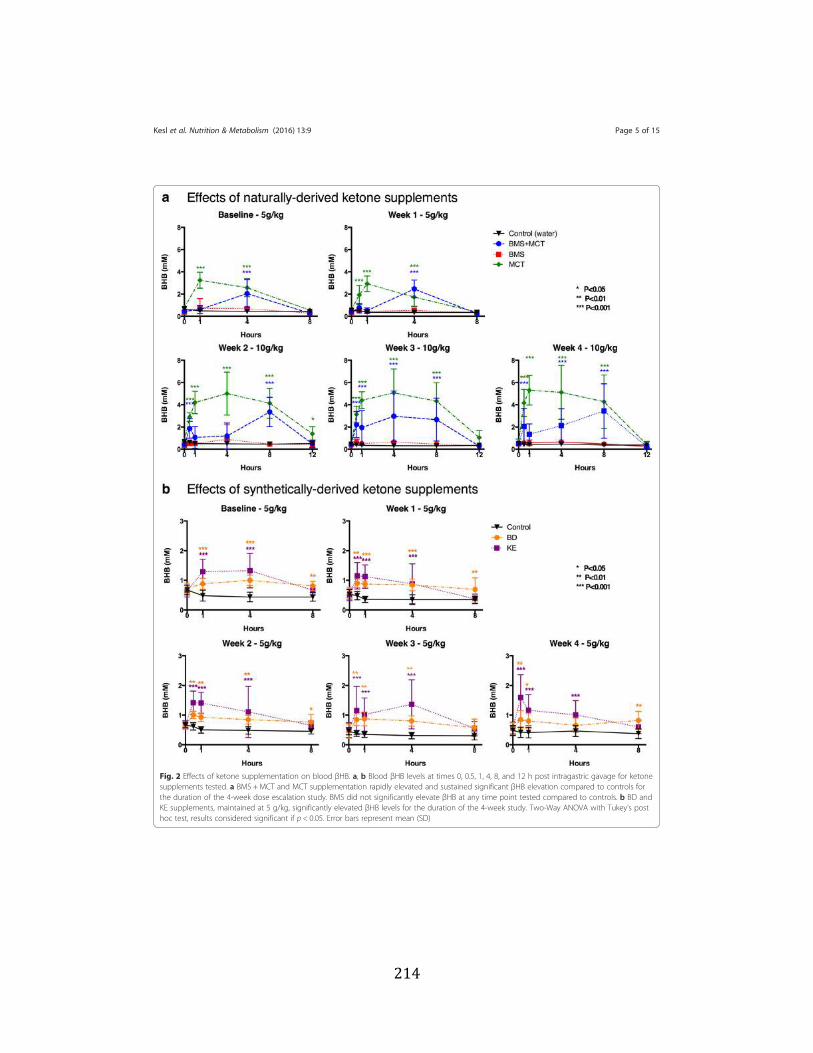
214
Fig. 2 Effects of ketone supplementation on blood βHB. a, b Blood βHB levels at times 0, 0.5, 1, 4, 8, and 12 h post intragastric gavage for ketonesupplements tested. a BMS +MCT and MCT supplementation rapidly elevated and sustained significant βHB elevation compared to controls forthe duration of the 4-week dose escalation study. BMS did not significantly elevate βHB at any time point tested compared to controls. b BD andKE supplements, maintained at 5 g/kg, significantly elevated βHB levels for the duration of the 4-week study. Two-Way ANOVA with Tukey’s posthoc test, results considered significant if p < 0.05. Error bars represent mean (SD)
Kesl et al. Nutrition & Metabolism (2016) 13:9 Page 5 of 15
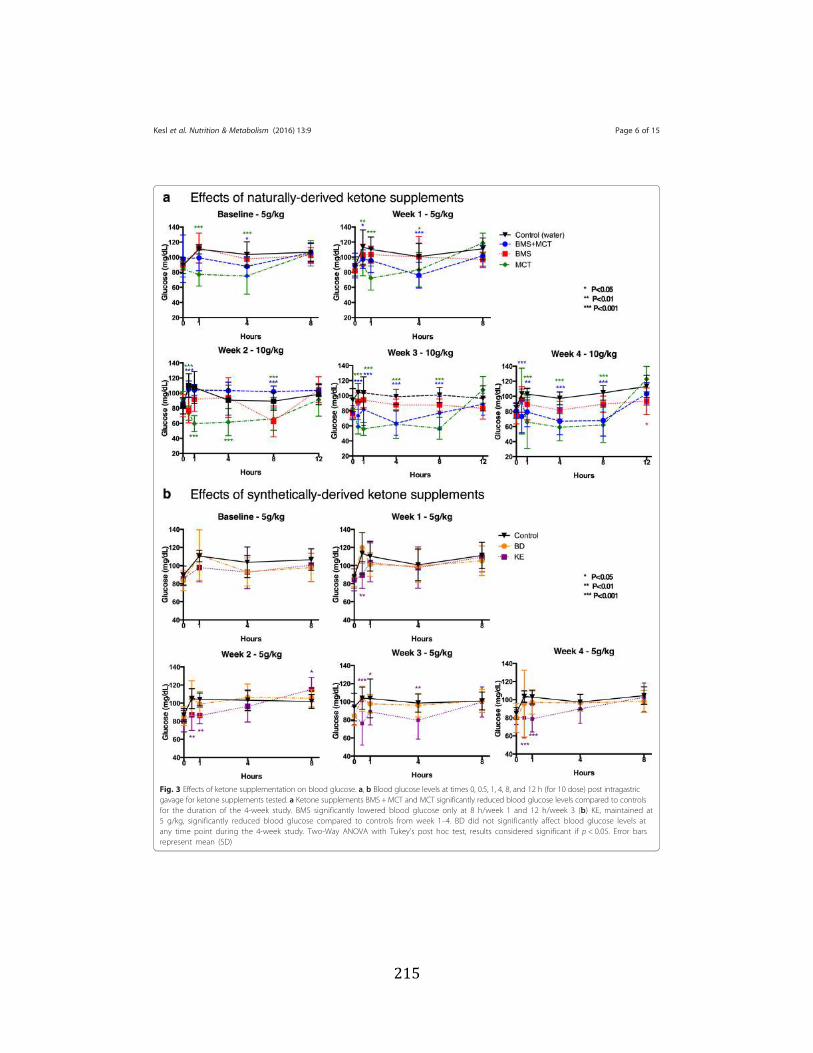
215
Fig. 3 Effects of ketone supplementation on blood glucose. a, b Blood glucose levels at times 0, 0.5, 1, 4, 8, and 12 h (for 10 dose) post intragastricgavage for ketone supplements tested. a Ketone supplements BMS +MCT and MCT significantly reduced blood glucose levels compared to controlsfor the duration of the 4-week study. BMS significantly lowered blood glucose only at 8 h/week 1 and 12 h/week 3 (b) KE, maintained at5 g/kg, significantly reduced blood glucose compared to controls from week 1–4. BD did not significantly affect blood glucose levels atany time point during the 4-week study. Two-Way ANOVA with Tukey’s post hoc test, results considered significant if p < 0.05. Error barsrepresent mean (SD)
Kesl et al. Nutrition & Metabolism (2016) 13:9 Page 6 of 15
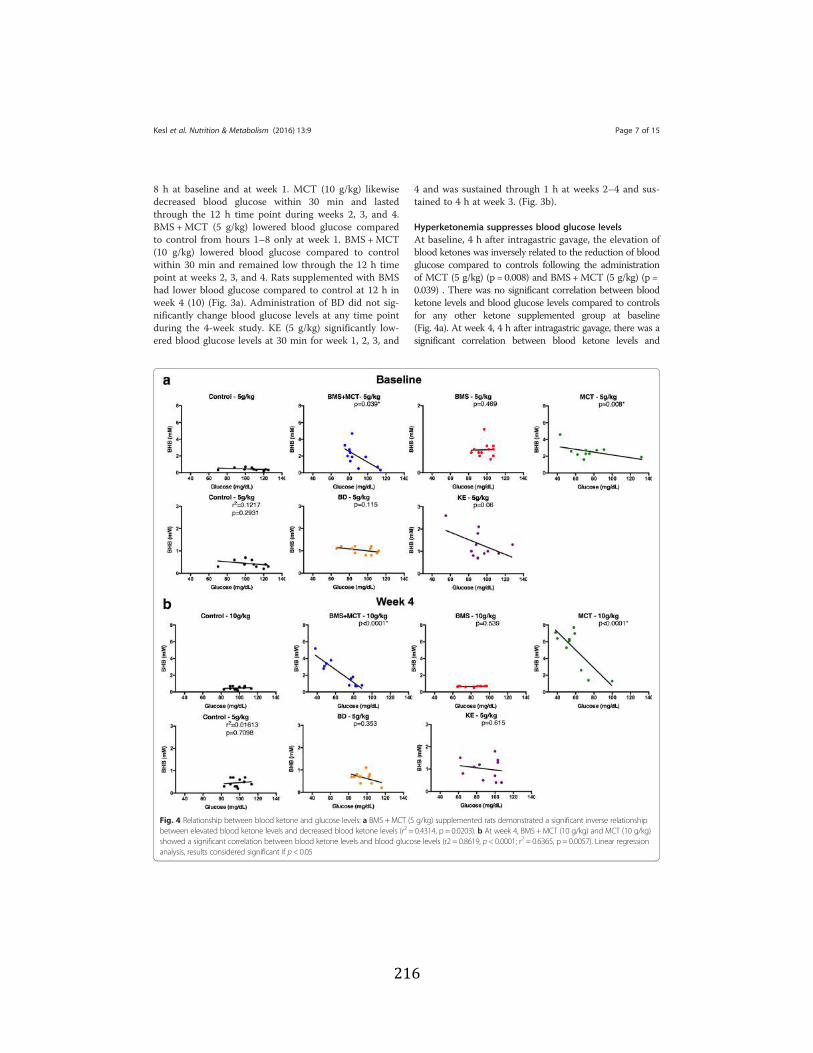
216
8 h at baseline and at week 1. MCT (10 g/kg) likewisedecreased blood glucose within 30 min and lastedthrough the 12 h time point during weeks 2, 3, and 4.BMS +MCT (5 g/kg) lowered blood glucose comparedto control from hours 1–8 only at week 1. BMS +MCT(10 g/kg) lowered blood glucose compared to controlwithin 30 min and remained low through the 12 h timepoint at weeks 2, 3, and 4. Rats supplemented with BMShad lower blood glucose compared to control at 12 h inweek 4 (10) (Fig. 3a). Administration of BD did not sig-nificantly change blood glucose levels at any time pointduring the 4-week study. KE (5 g/kg) significantly low-ered blood glucose levels at 30 min for week 1, 2, 3, and
4 and was sustained through 1 h at weeks 2–4 and sus-tained to 4 h at week 3. (Fig. 3b).
Hyperketonemia suppresses blood glucose levelsAt baseline, 4 h after intragastric gavage, the elevation ofblood ketones was inversely related to the reduction of bloodglucose compared to controls following the administrationof MCT (5 g/kg) (p = 0.008) and BMS+MCT (5 g/kg) (p =0.039) . There was no significant correlation between bloodketone levels and blood glucose levels compared to controlsfor any other ketone supplemented group at baseline(Fig. 4a). At week 4, 4 h after intragastric gavage, there was asignificant correlation between blood ketone levels and
Fig. 4 Relationship between blood ketone and glucose levels: a BMS +MCT (5 g/kg) supplemented rats demonstrated a significant inverse relationshipbetween elevated blood ketone levels and decreased blood ketone levels (r2 = 0.4314, p = 0.0203). b At week 4, BMS +MCT (10 g/kg) and MCT (10 g/kg)showed a significant correlation between blood ketone levels and blood glucose levels (r2 = 0.8619, p < 0.0001; r2 = 0.6365, p = 0.0057). Linear regressionanalysis, results considered significant if p < 0.05
Kesl et al. Nutrition & Metabolism (2016) 13:9 Page 7 of 15
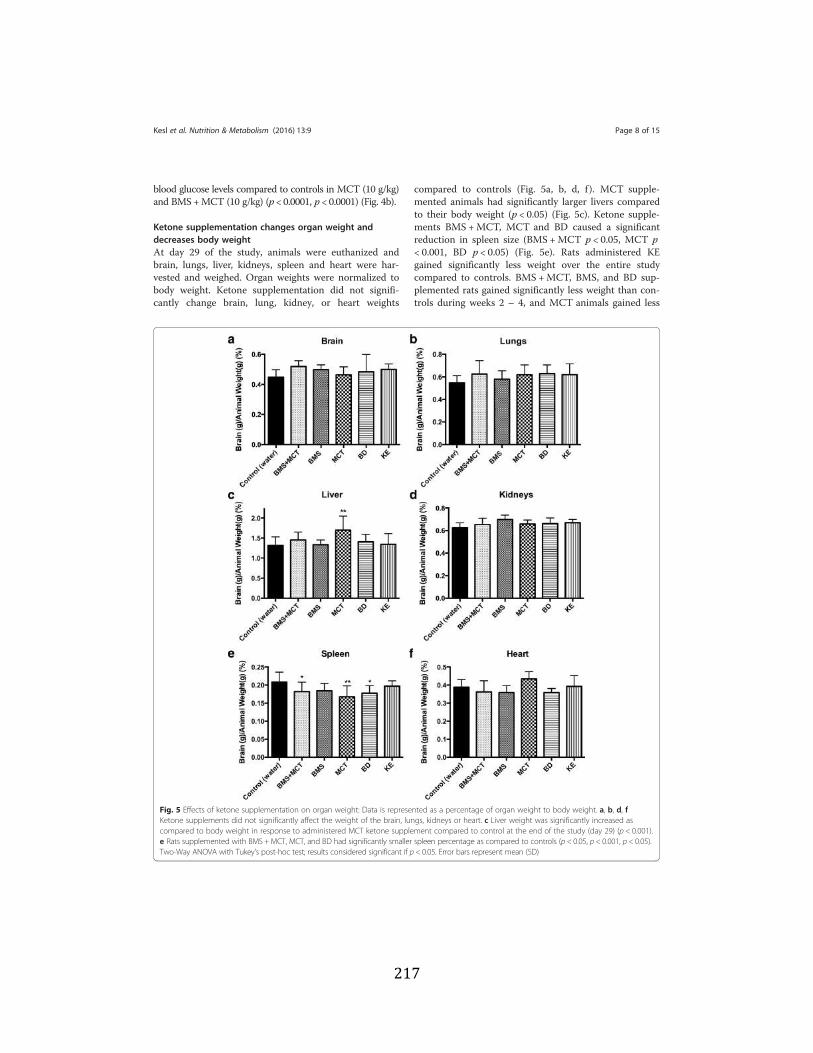
217
blood glucose levels compared to controls in MCT (10 g/kg)and BMS+MCT (10 g/kg) (p < 0.0001, p < 0.0001) (Fig. 4b).
Ketone supplementation changes organ weight anddecreases body weightAt day 29 of the study, animals were euthanized andbrain, lungs, liver, kidneys, spleen and heart were har-vested and weighed. Organ weights were normalized tobody weight. Ketone supplementation did not signifi-cantly change brain, lung, kidney, or heart weights
compared to controls (Fig. 5a, b, d, f ). MCT supple-mented animals had significantly larger livers comparedto their body weight (p < 0.05) (Fig. 5c). Ketone supple-ments BMS +MCT, MCT and BD caused a significantreduction in spleen size (BMS +MCT p < 0.05, MCT p< 0.001, BD p < 0.05) (Fig. 5e). Rats administered KEgained significantly less weight over the entire studycompared to controls. BMS +MCT, BMS, and BD sup-plemented rats gained significantly less weight than con-trols during weeks 2 – 4, and MCT animals gained less
Fig. 5 Effects of ketone supplementation on organ weight: Data is represented as a percentage of organ weight to body weight. a, b, d, fKetone supplements did not significantly affect the weight of the brain, lungs, kidneys or heart. c Liver weight was significantly increased ascompared to body weight in response to administered MCT ketone supplement compared to control at the end of the study (day 29) (p < 0.001).e Rats supplemented with BMS +MCT, MCT, and BD had significantly smaller spleen percentage as compared to controls (p < 0.05, p < 0.001, p < 0.05).Two-Way ANOVA with Tukey’s post-hoc test; results considered significant if p < 0.05. Error bars represent mean (SD)
Kesl et al. Nutrition & Metabolism (2016) 13:9 Page 8 of 15
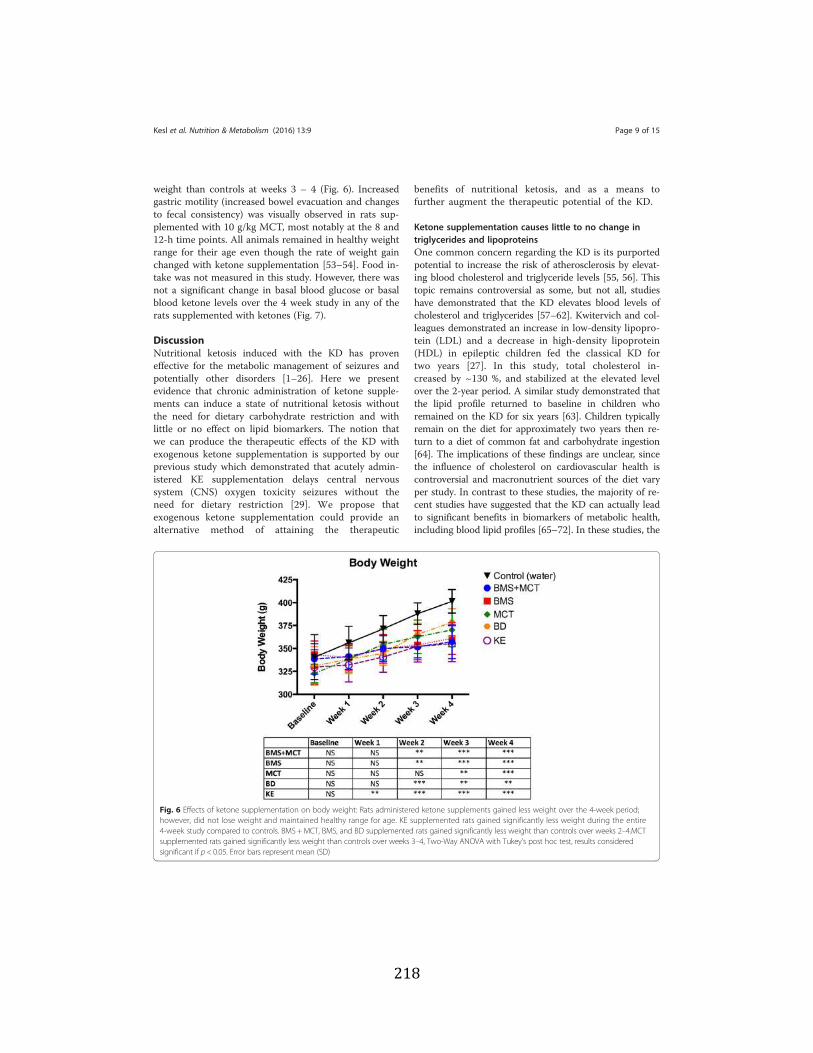
218
weight than controls at weeks 3 – 4 (Fig. 6). Increasedgastric motility (increased bowel evacuation and changesto fecal consistency) was visually observed in rats sup-plemented with 10 g/kg MCT, most notably at the 8 and12-h time points. All animals remained in healthy weightrange for their age even though the rate of weight gainchanged with ketone supplementation [53–54]. Food in-take was not measured in this study. However, there wasnot a significant change in basal blood glucose or basalblood ketone levels over the 4 week study in any of therats supplemented with ketones (Fig. 7).
DiscussionNutritional ketosis induced with the KD has proveneffective for the metabolic management of seizures andpotentially other disorders [1–26]. Here we presentevidence that chronic administration of ketone supple-ments can induce a state of nutritional ketosis withoutthe need for dietary carbohydrate restriction and withlittle or no effect on lipid biomarkers. The notion thatwe can produce the therapeutic effects of the KD withexogenous ketone supplementation is supported by ourprevious study which demonstrated that acutely admin-istered KE supplementation delays central nervoussystem (CNS) oxygen toxicity seizures without theneed for dietary restriction [29]. We propose thatexogenous ketone supplementation could provide analternative method of attaining the therapeutic
benefits of nutritional ketosis, and as a means tofurther augment the therapeutic potential of the KD.
Ketone supplementation causes little to no change intriglycerides and lipoproteinsOne common concern regarding the KD is its purportedpotential to increase the risk of atherosclerosis by elevat-ing blood cholesterol and triglyceride levels [55, 56]. Thistopic remains controversial as some, but not all, studieshave demonstrated that the KD elevates blood levels ofcholesterol and triglycerides [57–62]. Kwitervich and col-leagues demonstrated an increase in low-density lipopro-tein (LDL) and a decrease in high-density lipoprotein(HDL) in epileptic children fed the classical KD fortwo years [27]. In this study, total cholesterol in-creased by ~130 %, and stabilized at the elevated levelover the 2-year period. A similar study demonstrated thatthe lipid profile returned to baseline in children whoremained on the KD for six years [63]. Children typicallyremain on the diet for approximately two years then re-turn to a diet of common fat and carbohydrate ingestion[64]. The implications of these findings are unclear, sincethe influence of cholesterol on cardiovascular health iscontroversial and macronutrient sources of the diet varyper study. In contrast to these studies, the majority of re-cent studies have suggested that the KD can actually leadto significant benefits in biomarkers of metabolic health,including blood lipid profiles [65–72]. In these studies, the
Fig. 6 Effects of ketone supplementation on body weight: Rats administered ketone supplements gained less weight over the 4-week period;however, did not lose weight and maintained healthy range for age. KE supplemented rats gained significantly less weight during the entire4-week study compared to controls. BMS +MCT, BMS, and BD supplemented rats gained significantly less weight than controls over weeks 2–4.MCTsupplemented rats gained significantly less weight than controls over weeks 3–4, Two-Way ANOVA with Tukey’s post hoc test, results consideredsignificant if p < 0.05. Error bars represent mean (SD)
Kesl et al. Nutrition & Metabolism (2016) 13:9 Page 9 of 15
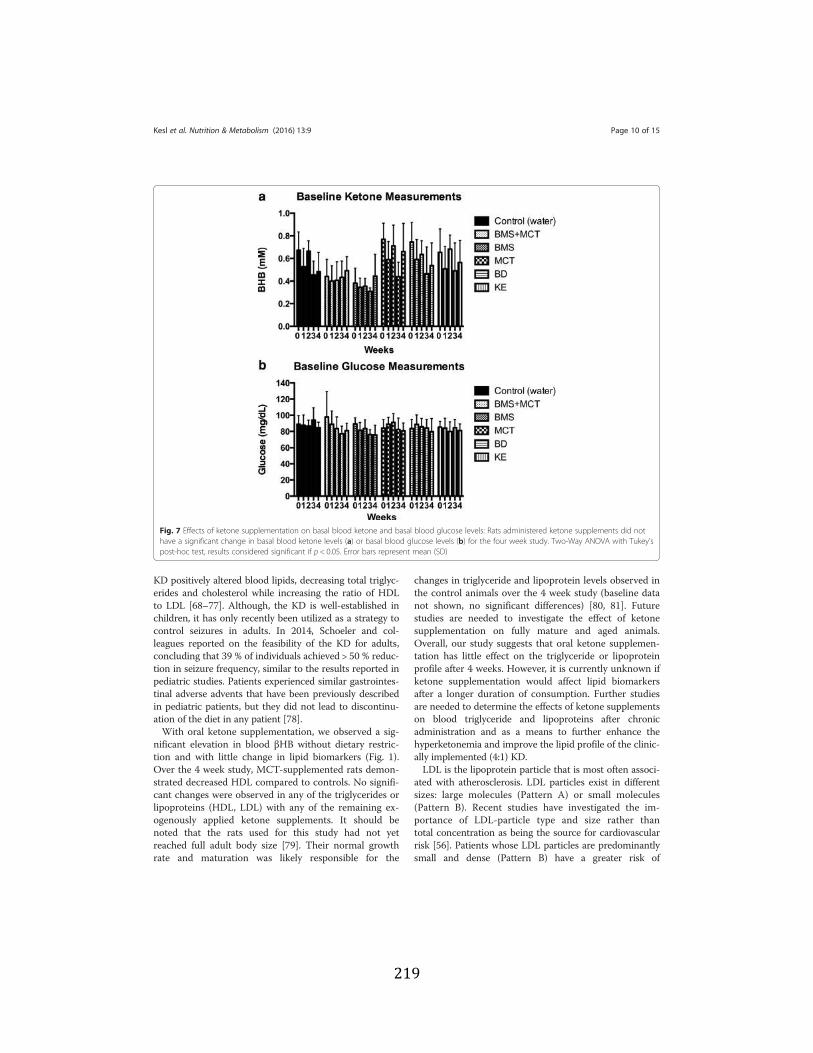
219
KD positively altered blood lipids, decreasing total triglyc-erides and cholesterol while increasing the ratio of HDLto LDL [68–77]. Although, the KD is well-established inchildren, it has only recently been utilized as a strategy tocontrol seizures in adults. In 2014, Schoeler and col-leagues reported on the feasibility of the KD for adults,concluding that 39 % of individuals achieved > 50 % reduc-tion in seizure frequency, similar to the results reported inpediatric studies. Patients experienced similar gastrointes-tinal adverse advents that have been previously describedin pediatric patients, but they did not lead to discontinu-ation of the diet in any patient [78].With oral ketone supplementation, we observed a sig-
nificant elevation in blood βHB without dietary restric-tion and with little change in lipid biomarkers (Fig. 1).Over the 4 week study, MCT-supplemented rats demon-strated decreased HDL compared to controls. No signifi-cant changes were observed in any of the triglycerides orlipoproteins (HDL, LDL) with any of the remaining ex-ogenously applied ketone supplements. It should benoted that the rats used for this study had not yetreached full adult body size [79]. Their normal growthrate and maturation was likely responsible for the
changes in triglyceride and lipoprotein levels observed inthe control animals over the 4 week study (baseline datanot shown, no significant differences) [80, 81]. Futurestudies are needed to investigate the effect of ketonesupplementation on fully mature and aged animals.Overall, our study suggests that oral ketone supplemen-tation has little effect on the triglyceride or lipoproteinprofile after 4 weeks. However, it is currently unknown ifketone supplementation would affect lipid biomarkersafter a longer duration of consumption. Further studiesare needed to determine the effects of ketone supplementson blood triglyceride and lipoproteins after chronicadministration and as a means to further enhance thehyperketonemia and improve the lipid profile of the clinic-ally implemented (4:1) KD.LDL is the lipoprotein particle that is most often associ-
ated with atherosclerosis. LDL particles exist in differentsizes: large molecules (Pattern A) or small molecules(Pattern B). Recent studies have investigated the im-portance of LDL-particle type and size rather thantotal concentration as being the source for cardiovascularrisk [56]. Patients whose LDL particles are predominantlysmall and dense (Pattern B) have a greater risk of
Fig. 7 Effects of ketone supplementation on basal blood ketone and basal blood glucose levels: Rats administered ketone supplements did nothave a significant change in basal blood ketone levels (a) or basal blood glucose levels (b) for the four week study. Two-Way ANOVA with Tukey’spost-hoc test, results considered significant if p < 0.05. Error bars represent mean (SD)
Kesl et al. Nutrition & Metabolism (2016) 13:9 Page 10 of 15

220
cardiovascular disease (CVD). It is thought that small,dense LDL particles are more able to penetrate the endo-thelium and cause in damage and inflammation [82–85].Volek et al. reported that the KD increased the patternand volume of LDL particles, which is considered to re-duce cardiovascular risk [73]. Though we did not show asignificant effect on LDL levels for ketone supplements,future chronic feeding studies will investigate the effectsof ketone supplementation on lipidomic profile and LDLparticle type and size.
Therapeutic levels of hyperketonemia suppress bloodglucose levelsWe demonstrated that therapeutic ketosis could beinduced without dietary (calorie or carbohydrate) restric-tion and that this acute elevation in blood ketones wassignificantly correlated with a reduction in blood glucose(Figs. 2, 3 and 4). The BMS ketone supplement did notsignificantly induce blood hyperketonemia or reducedglucose in the rats. The KE supplemented rats trendedtowards reduced glucose levels; however, the lower doseof this agent did not lower glucose significantly, as re-ported previously in acute response of mice [59]. MCTshave previously been shown to elicit a slight hypoglycemiceffect by enhancing glucose utilization in both diabeticand non-diabetic patients [86–88]. Kashiwaya et al. dem-onstrated that both blood glucose and blood insulindecreased by approximately 50 % in rats fed a diet where30 % of calories from starch were replaced with ketone es-ters for 14 days, suggesting that ketone supplementationincreases insulin sensitivity or reduced hepatic glucoseoutput [89]. This ketone-induced hypoglycemic effect hasbeen previously reported in humans with IV infusions ofketone bodies [90, 91]. Recently, Mikkelsen et al. showedthat a small increase in βHB concentration decreases glu-cose production by 14 % in post-absorptive health males[92]. However, this has not been previously reported withany of the oral exogenous ketone supplements we studied.Ketones are an efficient and sufficient energy substrate forthe brain, and will therefore prevent side effects ofhypoglycemia when blood levels are elevated and the pa-tient is keto-adapted. This was most famously demon-strated by Owen et al. in 1967 wherein keto-adaptedpatients (starvation induced therapeutic ketosis) weregiven 20 IU of insulin. The blood glucose of fasted pa-tients dropped to 1–2 mM, but they exhibited nohypoglycemic symptoms due to brain utilization of ke-tones for energy [93]. Therefore, ketones maintain brainmetabolism and are neuroprotective during severehypoglycemia. The rats in the MCT group had a correl-ation of blood ketone and glucose levels at week 4,whereas the combination of BMS +MCT produced a sig-nificant hypoglycemic correlation both at baseline and atweek 4. No hypoglycemic symptoms were observed in the
rats during this study. Insulin levels were not measured inthis study; however, future ketone supplementation stud-ies should measure the effects of exogenous ketones oninsulin sensitivity with a glucose tolerance test. Anincrease in insulin sensitivity in combination with ourobserved hypoglycemic effect has potential therapy impli-cations for glycemic control in T2D [40]. Furthermore, itshould be noted that the KE metabolizes to both AcAcand βHB in 1:1 ratio [29]. The ketone monitor used in thisstudy only measures βHB as levels of AcAc are more diffi-cult to measure due to spontaneous decarboxylation toacetone; therefore, the total ketone levels (βHB+AcAc)measured were likely higher, specifically for the KE [14].Interestingly, the 10 g/kg dose produced a delayed bloodβHB peak for ketone supplements MCT and BMS +MCT.The higher dose of the ketogenic supplements elevatedblood levels more substantially, and thus reached theirmaximum blood concentration later due to prolongedmetabolic clearance. It must be noted that the dosage usedin this study does not translate to human patients, sincethe metabolic physiology of rats is considerably higher.Future studies will be needed to determine optimal dosingfor human patients.
Effects of ketone supplementation on organ weight andbody weight percentageKetone supplementation did not affect the size of thebrain, lungs, kidneys or heart of rats. As previously men-tioned, the rats were still growing during the experimen-tal time frame; therefore, organ weights were normalizedto body weight to determine if organ weight changed in-dependently to growth. There could be several reasonswhy ketones influenced liver and spleen weight. The ra-tio of liver to body weight was significantly higher in theMCT supplemented animals (Fig. 5). MCTs are readilyabsorbed in the intestinal lumen and transported directlyto the liver via hepatic portal circulation. When given alarge bolus, such as in this study, the amount of MCTsin the liver will likely exceed the β-oxidation rate, caus-ing the MCTs to be deposited in the liver as fat droplets[94]. The accumulated MCT droplets in the liver couldexplain the higher liver weight to body weight percentageobserved with MCT supplemented rats. Future toxicologyand histological studies will be needed to determine thecause of the observed hepatomegaly. It should be empha-sized that the dose in this study is not optimized inhumans. We speculate that an optimized human dosewould be lower and may not cause hepatomegaly or po-tential fat accumulation. Nutritional ketosis achieved withthe KD has been shown to decrease inflammatory markerssuch as TNF-α, IL-6, IL-8, MCP-1, E-selectin, I-CAM, andPAI-1 [8, 46], which may account for the observeddecrease in spleen weight. As previously mentioned,Veech and colleagues demonstrated that exogenous
Kesl et al. Nutrition & Metabolism (2016) 13:9 Page 11 of 15

221
supplementation of 5 mM βHB resulted in a 28 % increasein hydraulic work in the working perfused rat heart and asignificant decrease in oxygen consumption [28, 41, 42].Ketone bodies have been shown to increase cerebral bloodflow and perfusion [95]. Also, ketone bodies have beenshown to increase ATP synthesis and enhance the effi-ciency of ATP production [14, 28, 40]. It is possible thatsustained ketosis results in enhanced cardiac efficiencyand O2 consumption. Even though the size of theheart did not change for any of the ketone supple-ments, further analysis of tissues harvested from theketone-supplemented rats will be needed to determineany morphological changes and to understand changes inorgan size. It should be noted that the Harlan standardrodent chow 2018 is nutritionally complete and formu-lated with high-quality ingredients to optimize gestation,lactation, growth, and overall health of the animals. Thesame cannot be said for the standard American diet(SAD). Therefore, we plan to investigate the effects ofketone supplements administered with the SAD to deter-mine if similar effects will be seen when the micronutrientdeficiencies and macronutrient profile mimics what mostAmericans consume.MCT oil has recently been used to induce nutritional
ketosis although it produces dose-dependent gastrointes-tinal (GI) side effects in humans that limit the potentialfor its use to significantly elevate ketones (>0.5 mM).Despite these limitations, Azzam and colleagues pub-lished a case report in which a 43-year-old-man had asignificant decrease in seizure frequency after supple-menting his diet with 4 tablespoons of MCT oil twicedaily [96]. An attempt to increase his dosage to 5 table-spoons twice daily was halted by severe GI intolerance.Henderson et al. observed that 20 % of patients re-ported GI side effects with a 20 g dose of ketogenicagent AC-1202 in a double blind trial in mild to moderateAlzheimer’s patients [24]. We visually observed similargastrointestinal side effects (loose stools) in the ratstreated with MCT oil in our study. Rats were closely mon-itored to avoid dehydration, and gastric motility returnedto normal between 12–24 h. Interestingly, the BMS +MCT supplement elevated βHB similarly to MCT oilalone, without causing the adverse gastrointestinal effectsseen in MCT-supplemented rats. However, this could bedue to the fact in a 10 g/kg dose of BMS +MCT, only 5 g/kg is MCT alone, which is less than the 10 g/kg dose thatelicits the GI side effects. This suggests that this novelcombination may provide a more useful therapeuticoption than MCT oil alone, which is limited in its abilityto elevate ketones in humans.Exogenously delivered ketone supplements signifi-
cantly altered rat weight gain for the duration of thestudy (Fig. 6). However, rats did not lose weight andmaintained a healthy range for their age. Rats have been
shown to effectively balance their caloric intake to preventweight loss/gain [97–99]. Due to the caloric density of theexogenous ketone supplements (Table 1) it is possible forthe rats to eat less of the standard rodent chow and there-fore less carbohydrates while maintaining their caloricintake. Food intake was not measured for this study.However, if there was a significant carbohydrate restric-tion there would be a signifcant change in basal blood ke-tone and blood glucose levels. As the hallmark to the KD,carbohydrate restriction increases blood ketone levels andreduces blood glucose levels. Neither an increase in basalblood ketone levels nor a decrease in basal blood glucoselevels was observed in this study (Fig. 7). Additionally, ifthere were an overall blood glucose decrease due to achange in food intake, this would not explain therapid reduction (within 30 min) in blood glucose cor-related with an elevation of blood ketone levels afteran intragastric bolus of ketone supplement (Figs. 2, 3and 4).
ConclusionsSeveral studies have investigated the safety and efficacyof ketone supplements for disease states such as AD andParkinson’s disease, and well as for parenteral nutrition[40, 48–50, 100–103]. Our research demonstrates thatseveral forms of dietary ketone supplementation can ef-fectively elevate blood ketone levels and achieve deleted:therapeutic nutritional ketosis without the need for diet-ary carbohydrate restriction. We also demonstrated thatketosis achieved with exogenous ketone supplementationcan reduce blood glucose, and this is inversely associatedwith the blood ketone levels. Although preliminary resultsare encouraging, further studies are needed to determineif oral ketone supplementation can produce the sametherapeutic benefits as the classic KD in the broad-spectrum of KD-responsive disease states . Additionally,further experiments need to be conducted to see if the ex-ogenous ketone supplementation affects the same physio-logical features as the KD (i.e. ROS, inflammation, ATPproduction). Ketone supplementation could be used as analternative method for inducing ketosis in patientsuninterested in attempting the KD or those who have pre-viously had difficulty implementing the KD because ofpalatability issues, gall bladder removal, liver abnormal-ities, or intolerance to fat. Additional experiments shouldbe conducted to see if ketone supplementation could beused in conjunction with the KD to assist and ease thetransition to nutrition ketosis and enhance the speed ofketo-adaptation. In this study we have demonstrated theability of several ketone supplements to elevate bloodketone levels, providing multiple options to induce thera-peutic ketosis based on patient need. Though additionalstudies are needed to determine the therapeutic potentialof ketone supplementation, many patients that previously
Kesl et al. Nutrition & Metabolism (2016) 13:9 Page 12 of 15

222
were unable to benefit from the KD may now have analternate method of achieving therapeutic ketosis. Ketonesupplementation may also represent a means to furtheraugment ketonemia in those responsive to therapeuticketosis, especially in those individuals where maintaininglow glucose is important.
AbbreviationsAcAc: acetoacetate; ALS: amyotrophic lateral sclerosis; AD: Alzheimer’s;βHB: beta-hydroxybutyrate; BMS: sodium/potassium βHB mineral salt; BMS +MCT: BMS + MCT 1:1 mixture; BD: 1,3-butanediol; CNS: central nervoussystem; HDL: high density lipoprotein; HDACs: histone deacetylases; LDL: lowdensity lipoprotein; KD: ketogenic diet; KE: 1, 3-butanediol acetoacetate dies-ter/ketone ester; MCT: medium chain triglyceride oil; PCOS: polycystic ovarysyndrome; ROS: reactive oxygen species; SAD: standard American diet;TBI: traumatic brain injury; T2D: type-2 diabetes.
Competing interestsInternational Patent # PCT/US2014/031237, University of South Florida, D.P.D’Agostino, S. Kesl, P. Arnold, “Compositions and Methods for ProducingElevated and Sustained Ketosis”. P. Arnold (Savind) has received financialsupport (ONR N000140610105 and N000140910244) from D.P. D’Agostino(USF) to synthesize ketone esters. The remaining authors have no conflicts ofinterest.
Authors’ contributionsConceived and designed the experiments: SK, AP, NW, TF, DP. Performed theexperiments: SK, AP, NW, TF, CA, JS, AVP. Analyzed the data: SK, AP, DP.Contributed reagents/materials/analysis tools: PA. Helped draft themanuscript: SK, AP, NW, CA, DP. All authors read and approved the finalmanuscript.
AcknowledgementsThe authors would like to thank Savind Inc for manufacturing some of theketone supplements, Jay Dean for the use of his Hyperbaric ResearchLaboratory, and the ONR and Scivation Inc. for funding the project.
GrantsThis study was supported by the Office of Naval Research (ONR) GrantN000140610105 (DPD); and a Morsani College of Medicine Department ofMolecular Pharmacology and Physiology departmental grant.
Author details1Department of Molecular Pharmacology and Physiology, Morsani College ofMedicine, University of South Florida, 12901 Bruce B. Downs Blvd. MDC8,Tampa, FL 33612, USA. 2Savind Inc, 205 South Main Street, Seymore, IL61875, USA.
Received: 10 September 2015 Accepted: 28 January 2016
References1. Sirven J, Whedon B, Caplan D, Liporace J, Glosser D, O’Dwyer J, et al. The
ketogenic diet for intractable epilepsy in adults: preliminary results.Epilepsia. 1999;40(12):1721–6. doi:10.1111/j.1528-1157.1999.tb01589.x.
2. Wilder R. The effect of ketonemia on the course of epilepsy. Mayo ClinBulletin. 1921;2:307–8.
3. Thiele E. Assessing the efficacy of antiepileptic treatments: the ketogenicdiet. Epilepsia. 2003;44 Suppl 7:26–9. doi:10.1046/j.1528-1157.44.s7.4.x.
4. Paoli A, Rubini A, Volek JS, Grimaldi KA. Beyond weight loss: a review of thetherapeutic uses of very-low-carbohydrate (ketogenic) diets. Eur J Clin Nutr.2013;67(8):789–96. doi:10.1038/ejcn.2013.116.
5. Foster GD, Wyatt HR, Hill JO, McGuckin BG, Brill C, Mohemmed BS, et al. Arandomized trial of a low-carbohydrate diet for obesity. N Engl J Med. 2003;348:2082–90.
6. Westman EC, Feinman RD, Mavropoulos JC, Vernon MC, Volek JS, WortmanJA, et al. Low-carbohydrate nutrition and metabolism. Am J Clin Nutr. 2007;86:276–84.
7. Westman EC, Yancy WS, Edman JS, Tomlin KF, Perkins CE. Effect of 6-monthadherence to a very low carbohydrate diet program. Am J Med. 2002;113(1):30–6.
8. Forsythe C, Phinney S, Fernandez M, Quann E, Wood R, Bibus D, et al.Comparison of low fat and low carbohydrate diets on circulating fatty acidcomposition and markers of inflammation. Lipids. 2008;43(1):65–77. doi:10.1007/s11745-007-3132-7.
9. Boden G, Sargrad K, Homko C, Mozzoli M, Stein T. Effect of a low-carbohydrate diet on appetite, blood glucose levels, and insulin resistancein obese patients with type 2 diabetes. Ann Intern Med. 2005;142(6):403–11.
10. Gumbiner B, Wendel J, McDermott M. Effects of diet composition andketosis on glycemia during very-low-energy-diet therapy in obesepatients with non-insulin-dependent diabetes mellitus. Am J Clin Nutr.1996;63(1):110–5.
11. Nielsen J, Joensson E. Low-carbohydrate diet in type 2 diabetes: stableimprovement of bodyweight and glycemic control during 44 monthsfollow-up. Nutr Metab. 2008;5:14. doi:10.1186/1743-7075-5-14.
12. Yancy W, Foy M, Chalecki A, Vernon M, Westman E. A low-carbohydrate,ketogenic diet to treat type 2 diabetes. Nutr Metab. 2005;2:34. doi:10.1186/1743-7075-2-34.
13. Dashti HM, Al-Zaid NS, Mathew TC, Al-Mousawi M, Talib H, Asfar SK, et al.Long term effects of ketogenic diet in obese subjects with high cholesterollevel. Mol Cell Biochem. 2006;286(1–2):1–9. doi:10.1007/s11010-005-9001-x.
14. Maalouf M, Rho JM, Mattson MP. The neuroprotective properties of calorierestriction, the ketogenic diet, and ketone bodies. Brain Res Rev. 2009;59(2):293–315. doi:10.1016/j.brainresrev.2008.09.002.
15. Mavropoulos JC, Yancy WS, Hepburn J, Westman EC. The effects of alow-carbohydrate, ketogenic diet on the polycystic ovary syndrome: a pilotstudy. Nutr Metab. 2004;2:35. doi:10.1186/1743-7075-2-35.
16. Seyfried T, Flores R, Poff A, D’Agostino D. Cancer as a metabolic disease:implications for novel therapeutics. Carcinogenesis. 2014;35:515–27. doi:10.1093/carcin/bgt480.
17. Poff AM, Ari C, Arnold P, Seyfried TN, D’Agostino DP. Ketone supplementationdecreases tumor cell viability and prolongs survival of mice with metastaticcancer. Int J Cancer. 2014;135:1711–20. doi:10.1002/ijc.28809.
18. Poff A, Ari C, Seyfried T, D’Agostino D. The ketogenic diet and hyperbaricoxygen therapy prolong survival in mice with systemic metastatic cancer.PLoS ONE. 2013;8(6), e65522. doi:10.1371/journal.pone.0065522.
19. Seyfried T, Shelton L. Cancer as a metabolic disease. Nutr Metab. 2010;7:7.doi:10.1186/1743-7075-7-7.
20. Fine E, Segal-Isaacson C, Feinman R, Herszkopf S, Romano M, Tomuta N,et al. Targeting insulin inhibition as a metabolic therapy in advancedcancer: a pilot safety and feasibility dietary trial in 10 patients. Nutrition.2012;28(10):1028–35. doi:10.1016/j.nut.2012.05.001.
21. Zhao Z, Lange D, Voustianiouk A, MacGrogan D, Ho L, Suh J, et al. Aketogenic diet as a potential novel therapeutic intervention in amyotrophiclateral sclerosis. BMC Neurosci. 2006;7:29. doi:10.1186/1471-2202-7-29.
22. White H, Venkatesh B. Clinical review: ketones and brain injury. Crit Care.2011;15(2):219. doi:10.1186/cc10020.
23. Prins M. Cerebral metabolic adaptation and ketone metabolism after brain injury.J Cereb Blood Flow Metab. 2008;28(1):1–16. doi:10.1038/sj.jcbfm.9600543.
24. Henderson S, Vogel J, Barr L, Garvin F, Jones J, Costantini L. Study of theketogenic agent AC-1202 in mild to moderate Alzheimer’s disease: arandomized, double-blind, placebo-controlled, multicenter trial. Nutr Metab.2009;6:31. doi:10.1186/1743-7075-6-31.
25. Brownlow M, Benner L, D’Agostino D, Gordon M, Morgan D. Ketogenic dietimproves motor performance but not cognition in two mouse models ofAlzheimer’s pathology. PLoS ONE. 2013;8(9), e75713. doi:10.1371/journal.pone.0075713.g008.
26. Kossoff EH, Hartman AL. Ketogenic diets: new advances for metabolism-basedtherapies. Curr Opin Neurol. 2012;25:173–8. doi:10.1097/WCO.0b013e3283515e4a.
27. Kwiterovich P, Vining E, Pyzik P, Skolasky R, Freeman J. Effect of a high-fatketogenic diet on plasma levels of lipids, lipoproteins, and apolipoproteinsin children. JAMA. 2003;290(7):912–20. doi:10.1001/jama.290.7.912.
28. Veech RL, Chance B, Kashiwaya Y, Lardy HA, Cahill Jr GF. Ketone bodies,potential therapeutic uses. IUBMB Life. 2001;51(4):241–7. doi:10.1080/152165401753311780.
29. D’Agostino D, Pilla R, Held H, Landon C, Puchowicz M, Brunengraber H,et al. Therapeutic ketosis with ketone ester delays central nervous systemoxygen toxicity seizures in rats. Am J Physiol Regul Integr Comp Physiol.2013;304(10):R829–36. doi:10.1152/ajpregu.00506.2012.
Kesl et al. Nutrition & Metabolism (2016) 13:9 Page 13 of 15

223
30. Gasior M, French A, Joy M, Tang R, Hartman A, Rogawski M. The anticonvulsantactivity of acetone, the major ketone body in the ketogenic diet, is notdependent on its metabolites acetol, 1,2-propanediol, methylglyoxal, or pyruvicacid. Epilepsia. 2007;48(4):793–800. doi:10.1111/j.1528-1167.2007.01026.x.
31. Likhodii S, Nylen K, Burnham W. Acetone as an anticonvulsant. Epilepsia.2008;49 Suppl 8:83–6. doi:10.1111/j.1528-1167.2008.01844.x.
32. Seymour K, Bluml S, Sutherling J, Sutherling W, Ross B. Identification ofcerebral acetone by 1H-MRS in patients with epilepsy controlled byketogenic diet. Magma. 1999;8(1):33–42.
33. Halevy A, Peleg-Weiss L, Cohen R, Shuper A. An update on the ketogenicdiet, 2012. Rambam Maimonides Med J. 2012;3(1), e0005. doi:10.5041/RMMJ.10072.
34. Amari A, Grace N, Fisher W. Achieving and maintaining compliance withthe ketogenic diet. J Appl Behav Anal. 1995;28(3):341–2. doi:10.1901/jaba.1995.28-341.
35. Zhang Y, Kuang Y, LaManna J, Puchowicz M. Contribution of brain glucoseand ketone bodies to oxidative metabolism. Adv Exp Med Biol. 2013;765:365–70. doi:10.1007/978-1-4614-4989-8_51.
36. Maalouf M, Sullivan P, Davis L, Kim D, Rho J. Ketones inhibit mitochondrialproduction of reactive oxygen species production following glutamateexcitotoxicity by increasing NADH oxidation. Neuroscience. 2007;145(1):256–64. doi:10.1016/j.neuroscience.2006.11.065.
37. Milder J, Patel M. Modulation of oxidative stress and mitochondrial functionby the ketogenic diet. Epilepsy Res. 2012;100(3):295–303. doi:10.1016/j.eplepsyres.2011.09.021.
38. Shimazu T, Hirschey M, Newman J, He W, Shirakawa K, Le Moan N, et al.Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histonedeacetylase inhibitor. Science. 2013;339:211–4. doi:10.1126/science.1227166.
39. Kim DY, Davis L, Sullivan P, Maalouf M, Simeone T, van Brederode J, et al.Ketone bodies are protective against oxidative stress in neocortical neurons.J Neurochem. 2007;101(5):1316–26. doi:10.1111/j.1471-4159.2007.04483.x.
40. Veech R. The therapeutic implications of ketone bodies: the effects ofketone bodies in pathological conditions: ketosis, ketogenic diet, redoxstates, insulin resistance, and mitochondrial metabolism. ProstaglandinsLeukot Essent Fat Acids. 2004;70(3):309–19. doi:10.1016/j.plefa.2003.09.007.
41. Sato K, Kashiwaya Y, Keon C, Tsuchiya N, King M, Radda G, et al. Insulin, ketonebodies, and mitochondrial energy transduction. FASEB J. 1995;9(8):651–8.
42. Kashiwaya Y, Sato K, Tsuchiya N, Thomas S, Fell D, Veech R, et al. Control ofglucose utilization in working perfused rat heart. J Biol Chem. 1994;269(41):25502–14.
43. Sullivan P, Rippy N, Dorenbos K, Concepcion R, Agarwal A, Rho J. Theketogenic diet increases mitochondrial uncoupling protein levels andactivity. Ann Neurol. 2004;55(4):576–80. doi:10.1002/ana.20062.
44. Bough K, Rho J. Anticonvulsant mechanisms of the ketogenic diet. Epilepsia.2007;48(1):43–58. doi:10.1111/j.1528-1167.2007.00915.x.
45. Bough K, Wetherington J, Hassel B, Pare J, Gawryluk J, Greene J, et al.Mitochondrial biogenesis in the anticonvulsant mechanism of the ketogenicdiet. Ann Neurol. 2006;60(2):223–35. doi:10.1002/ana.20899.
46. Ruskin D, Kawamura M, Masino S. Reduced pain and inflammation injuvenile and adult rats fed a ketogenic diet. PLoS ONE. 2009;4(12), e8349.doi:10.1371/journal.pone.0008349.
47. Paoli A, Moro T, Bosco G, Bianco A, Grimaldi KA, Camporesi E, et al. Effectsof n-3 polyunsaturated fatty acids (ω-3) supplementation on somecardiovascular risk factors with a ketogenic Mediterranean diet. Marinedrugs. 2015;13(2):996–1009. doi:10.3390/md13020996.
48. Desrochers S, Dubreuil P, Brunet J, Jetté M, David F, Landau BR, et al.Metabolism of (R, S)-1,3-butanediol acetoacetate esters, potentialparenteral and enteral nutrients in conscious pigs. Am J Physiol. 1995;268(4 Pt 1):E660–7.
49. Clarke K, Tchabanenko K, Pawlosky R, Carter E, Knight N, Murray A, et al.Oral 28-day and developmental toxicity studies of (R)-3-hydroxybutyl (R)-3-hydroxybutyrate. Regul Toxicol Pharmacol. 2012;63(2):196–208. doi:10.1016/j.yrtph.2012.04.001.
50. Clarke K, Tchabanenko K, Pawlosky R, Carter E, Todd King M, Musa-Veloso K,et al. Kinetics, safety and tolerability of (R)-3-hydroxybutyl (R)-3-hydroxybutyrate in healthy adult subjects. Regul Toxicol Pharmacol. 2012;63(3):401–8. doi:10.1016/j.yrtph.2012.04.008.
51. Warnick G, Knopp R, Fitzpatrick V, Branson L. Estimating low-densitylipoprotein cholesterol by the Friedewald equation is adequate forclassifying patients on the basis of nationally recommended cutpoints. ClinChem. 1990;36(1):15–9.
52. Friedewald W, Levy R, Fredrickson D. Estimation of the concentration oflow-density lipoprotein cholesterol in plasma, without use of thepreparative ultracentrifuge. Clin Chem. 1972;18(6):499–502.
53. Harlan Laboratories Inc U. Sprague Dawley Outbred Rat. In: http://www.harlan.com/products_and_services/research_models_and_services/research_models/sprague_dawley_outbred_rat.hl. 2008. http://www.harlan.com/products_and_services/research_models_and_services/research_models/sprague_dawley_outbred_rat.hl. Accessed date January 30, 2014.
54. Inc TB. Sprague Dawley Rat. In: http://www.taconic.com/sd. 2014. http://www.taconic.com/user-assets/documents/spraguedawley_booklet.pdf.Accessed date January 30, 2014.
55. McPherson P, McEneny J. The biochemistry of ketogenesis and its role inweight management, neurological disease and oxidative stress. J PhysiolBiochem. 2012;68(1):141–51. doi:10.1007/s13105-011-0112-4.
56 Moore J, Eric C, Westman M. Cholesterol Clarity: What the HDL is WrongWith my Numbers. Las Vegas: Victory Belt Publishing Inc; 2013.
57 Dekaban A. Plasma lipids in epileptic children treated with the high fat diet.Arch Neurol. 1966;15(2):177–84.
58 Chesney D, Brouhard B, Wyllie E, Powaski K. Biochemical abnormalities ofthe ketogenic diet in children. Clin Pediatr. 1999;38(2):107–9. doi:10.1177/000992289903800207.
59. Schwartz R, Boyes S, Aynsley-Green A. Metabolic effects of three ketogenic dietsin the treatment of severe epilepsy. Dev Med Child Neurol. 1989;31(2):152–60.
60. Katyal N, Koehler A, McGhee B, Foley C, Crumrine P. The ketogenic diet inrefractory epilepsy: the experience of Children’s Hospital of Pittsburgh. ClinPediatr. 2000;39(3):153–9. doi:10.1177/000992280003900303.
61. Ellenbroek J, van Dijck L, Töns H, Rabelink T, Carlotti F, Ballieux B, et al.Long-term ketogenic diet causes glucose intolerance and reduced beta andalpha cell mass but no weight loss in mice. Am J Physiol Endocrinol Metab.2014;306(5):E552–8. doi:10.1152/ajpendo.00453.2013.
62. Bergqvist A. Long-term monitoring of the ketogenic diet: Do’s and Don’ts.Epilepsy Res. 2012;100(3):261–6. doi:10.1016/j.eplepsyres.2011.05.020.
63. Groesbeck D, Bluml R, Kossoff E. Long-term use of the ketogenic diet in thetreatment of epilepsy. Dev Med Child Neurol. 2006;48(12):978–81. doi:10.1017/s0012162206002143.
64. Patel A, Pyzik P, Turner Z, Rubenstein J, Kossoff E. Long-term outcomes ofchildren treated with the ketogenic diet in the past. Epilepsia. 2010;51(7):1277–82. doi:10.1111/j.1528-1167.2009.02488.x.
65. Brehm BJ, Seeley RJ, Daniels SR, D’Alessio DA. A randomized trial comparinga very low carbohydrate diet and a calorie-restricted low fat diet on bodyweight and cardiovascular risk factors in healthy women. J Clin EndocrinolMetab. 2003;88(4):1617–23. doi:10.1210/jc.2002-021480.
66. Shai I, Schwarzfuchs D, Henkin Y, Shahar DR, Witkow S, Greenberg I, et al.Weight loss with a low-carbohydrate, Mediterranean, or low-fat diet. N EnglJ Med. 2008;359(3):229–41. doi:10.1056/NEJMoa0708681.
67. Volek JS, Phinney SD, Forsythe CE, Quann EE, Wood RJ, Puglisi MJ, et al.Carbohydrate restriction has a more favorable impact on the metabolicsyndrome than a low fat diet. Lipids. 2009;44(4):297–309. doi:10.1007/s11745-008-3274-2.
68. Feinman RD, Volek JS. Low carbohydrate diets improve atherogenicdyslipidemia even in the absence of weight loss. Nutr Metab. 2006;3:24. doi:10.1186/1743-7075-3-24.
69. Sharman MJ, Gomez AL, Kraemer WJ, Volek JS. Very low-carbohydrate andlow-fat diets affect fasting lipids and postprandial lipemia differently inoverweight men. J Nutr. 2004;134(4):880–5.
70. Sharman MJ, Kraemer WJ, Love DM, Avery NG, Gomez AL, Scheett TP, et al.A ketogenic diet favorably affects serum biomarkers for cardiovasculardisease in normal-weight men. J Nutr. 2002;132(7):1879–85.
71. Westman EC, Mavropoulos J, Yancy WS, Volek JS. A review of low-carbohydrate ketogenic diets. Curr Atheroscler Rep. 2003;5(6):476–83.
72. Wood RJ, Volek JS, Davis SR, Dell’Ova C, Fernandez ML. Effects of acarbohydrate-restricted diet on emerging plasma markers for cardiovasculardisease. Nutr Metab. 2006;3:19. doi:10.1186/1743-7075-3-19.
73. Volek JS, Sharman MJ, Forsythe CE. Modification of lipoproteins by verylow-carbohydrate diets. J Nutr. 2005;135(6):1339–42.
74. Volek JS, Westman EC. Very-low-carbohydrate weight-loss diets revisited.Cleve Clin J Med. 2002;69(11):849. 53, 56–8 passim.
75. Volek JS, Sharman MJ, Gomez AL, DiPasquale C, Roti M, Pumerantz A, et al.Comparison of a very low-carbohydrate and low-fat diet on fasting lipids,LDL subclasses, insulin resistance, and postprandial lipemic responses inoverweight women. J Am Coll Nutr. 2004;23(2):177–84.
Kesl et al. Nutrition & Metabolism (2016) 13:9 Page 14 of 15

224
76. Volek JS, Sharman MJ. Cardiovascular and hormonal aspects of very-low-carbohydrate ketogenic diets. Obes Res. 2004;12 Suppl 2:115s–23. doi:10.1038/oby.2004.276.
77. Volek JS, Sharman MJ, Gomez AL, Scheett TP, Kraemer WJ. An isoenergeticvery low carbohydrate diet improves serum HDL cholesterol andtriacylglycerol concentrations, the total cholesterol to HDL cholesterol ratioand postprandial pipemic responses compared with a low fat diet innormal weight, normolipidemic women. J Nutr. 2003;133(9):2756–61.
78. Schoeler NE, Wood S, Aldridge V, Sander JW, Cross JH, Sisodiya SM.Ketogenic dietary therapies for adults with epilepsy: feasibility andclassification of response. Epilepsy & behavior : E&B. 2014;37:77–81. doi:10.1016/j.yebeh.2014.06.007.
79. Sengupta P. The laboratory Rat: relating its Age with Human’s. Int J PrevMed. 2013;4(6):624–30.
80. Tsuchiya N, Harada Y, Taki M, Minematsu S, Maemura S, Amagaya S. Age-relatedchanges and sex differences on the serum chemistry values in Sprague–Dawleyrats–I. 6–30 weeks of age. Exp Anim. 1995;43(5):671–8.
81. Saito K, Ishikawa M, Murayama M, Urata M, Senoo Y, Toyoshima K, et al.Effects of sex, age, and fasting conditions on plasma lipidomic profiles offasted Sprague–Dawley rats. PLoS ONE. 2013;9(11):e112266. doi:10.1371/journal.pone.0112266.
82. Ellington A, Kullo I. Atherogenic lipoprotein subprofiling. Adv Clin Chem.2008;46:295–317.
83. Mudd J, Borlaug B, Johnston P, Kral B, Rouf R, Blumenthal R, et al. Beyondlow-density lipoprotein cholesterol: defining the role of low-densitylipoprotein heterogeneity in coronary artery disease. J Am Coll Cardiol.2007;50(18):1735–41. doi:10.1016/j.jacc.2007.07.045.
84. Sacks F, Campos H. Clinical review 163: cardiovascular endocrinology:Low-density lipoprotein size and cardiovascular disease: a reappraisal. JClin Endocrinol Metab. 2003;88(10):4525–32. doi:10.1210/jc.2003-030636.
85. Wierzbicki A. Quality as well as quantity? Beyond low-density lipoprotein-cholesterol - the role of particle size. Int J Clin Pract. 2007;61(11):1780–2. doi:10.1111/j.1742-1241.2007.01571.x.
86. Tantibhedhyangkul P, Hashim S, Van Itallie T. Effects of ingestion of long-chaintriglycerides on glucose tolerance in man. Diabetes. 1967;16(11):796–9. doi:10.2337/diab.16.11.796.
87. Eckel R, Hanson A, Chen A, Berman J, Yost T, Brass E. Dietary substitution ofmedium-chain triglycerides improves insulin-mediated glucose metabolismin NIDDM subjects. Diabetes. 1992;41(5):641–7. doi:10.2337/diab.41.5.641.
88. Yost T, Erskine J, Gregg T, Podlecki D, Brass E, Eckel R. Dietary substitution ofmedium chain triglycerides in subjects with non-insulin-dependentdiabetes mellitus in an ambulatory setting: impact on glycemic controland insulin-mediated glucose metabolism. J Am Coll Nutr. 1994;13(6):615–22. doi:10.1080/07315724.1994.10718457.
89. Kashiwaya Y, Pawlosky R, Markis W, King MT, Bergman C, Srivastava S, et al.A ketone ester diet increases brain malonyl-CoA and Uncoupling proteins 4and 5 while decreasing food intake in the normal Wistar Rat. J Biol Chem.2010;285(34):25950–6. doi:10.1074/jbc.M110.138198.
90. Senior B, Loridan L. Direct regulatory effect of ketones on lipolysis and onglucose concentrations in man. Nature. 1968;219(5149):83–4.
91. Miles JM, Haymond MW, Gerich JE. Suppression of glucose production andstimulation of insulin secretion by physiological concentrations of ketone bodiesin man. J Clin Endocrinol Metab. 1980;52(1):34–7. doi:10.1210/jcem-52-1-34.
92. Kristian HM, Thomas S, Niels HS, Thomas G, Gerrit van H. Systemic, cerebraland skeletal muscle ketone body and energy metabolism during acutehyper-D-β-hydroxybutyrataemia in post-absorptive healthy males. J ClinEndocrin Metabol. 2014. doi:10.1210/jc.2014-2608
93. Owen OE, Morgan AP, Kemp HG, Sullivan JM, Herrera MG, Cahill GF. Brainmetabolism during fasting. J Clin Invest. 1967;46(10):1589–95. doi:10.1172/JCI105650.
94. Papamandjaris AA, MacDougall DE, Jones PJ. Medium chain fatty acidmetabolism and energy expenditure: obesity treatment implications. LifeSci. 1997;62(14):1203–15.
95. Linde R, Hasselbalch S, Topp S, Paulson O, Madsen P. Global cerebral bloodflow and metabolism during acute hyperketonemia in the awake andanesthetized rat. J Cereb Blood Flow Metab. 2006;26(2):170–80. doi:10.1038/sj.jcbfm.9600177.
96. Azzam R, Azar N. Marked seizure reduction after MCT supplementation.Case reports in neurological medicine. 2013;2013:809151. doi:10.1155/2013/809151.
97. Corwin RL. Binge-type eating induced by limited access in rats does notrequire energy restriction on the previous day. Appetite. 2004;42(2):139–42.doi:10.1016/j.appet.2003.08.010.
98. Keenan KP, Ballam GC, Dixit R, Soper KA, Laroque P, Mattson BA, et al. Theeffects of diet, overfeeding and moderate dietary restriction on Sprague–Dawley rat survival, disease and toxicology. J Nutr. 1997;127(5 Suppl):851S–6.
99. Keenan KP, Smith PF, Hertzog P, Soper K, Ballam GC, Clark RL. The effects ofoverfeeding and dietary restriction on Sprague–Dawley Rat survival andearly pathology biomarkers of aging. Toxicol Pathol. 1994. doi:10.1177/019262339402200308.
100. Kashiwaya Y, Bergman C, Lee J-H, Wan R, King M, Mughal M, et al. A ketoneester diet exhibits anxiolytic and cognition-sparing properties, and lessensamyloid and tau pathologies in a mouse model of Alzheimer’s disease.Neurobiol Aging. 2013;34(6):1530–9. doi:10.1016/j.neurobiolaging.2012.11.023.
101. Birkhahn R, McCombs C, Clemens R, Hubbs J. Potential of themonoglyceride and triglyceride of DL-3-hydroxybutyrate for parenteralnutrition: synthesis and preliminary biological testing in the rat. Nutrition.1997;13(3):213–9. doi:10.1016/s0899-9007(96)00404-2.
102. Puchowicz M, Smith C, Bomont C, Koshy J, David F, Brunengraber H.Dog model of therapeutic ketosis induced by oral administration of R,S-1,3-butanediol diacetoacetate. J Nutr Biochem. 2000;11(5):281–7.
103. Brunengraber H. Potential of ketone body esters for parenteral and oralnutrition. Nutrition. 1997;13(3):233–5.
• We accept pre-submission inquiries • Our selector tool helps you to find the most relevant journal• We provide round the clock customer support • Convenient online submission• Thorough peer review• Inclusion in PubMed and all major indexing services • Maximum visibility for your research
Submit your manuscript atwww.biomedcentral.com/submit
Submit your next manuscript to BioMed Central and we will help you at every step:
Kesl et al. Nutrition & Metabolism (2016) 13:9 Page 15 of 15

ABOUT THE AUTHOR
Shannon Kesl was born and raised in Springfield, Ohio. She attended Wittenberg
University in Springfield, Ohio until Spring 2007. In 2007, she moved to Tampa, FL to continue
her studies in Biomedical Sciences at the University of South Florida; she received her BS in fall
of 2008. In 2010, Shannon began her graduate training as a student in the Ph.D. Program in
Integrated Biomedical Sciences at the Morsani College of Medicine, University of South Florida.
In 2014, she married the love of her life and currently shares her heart with her two dogs Bailey
and Isa. Shannon has performed her dissertation research in the Laboratory of Nutritional and
Metabolic Medicine under the co-mentorship of Dr. Dominic D’Agostino and Dr. Mack Wu.
During her time as a graduate student, she presented her research at many national conferences,
won numerous travel awards, and was a member of ASN, AAA, WHS, and APS societies. She
received her Masters of Science in Medical Science in 2013, and successfully defended her
dissertation on December 09, 2015.


















