
Membrane protein structure prediction Hydrophobicity analysis and the positive-inside rule
9
.I . Mol. Bid. (1992) 225, 487494 Membrane Protein Structure Prediction Gunnar von Heijne Department of Molecular Biology Karolinska Institute Center for 8tructural Biochemistry NOV T’A f. S-141 57 Huddinge. Suleden (Received 14 October 1991; accepted 20 Jan,uary 1992) A ne w strategy for predicting the topology of bacterial inner membran e proteins is propo sed on the basis of hydrophobicity analysis , automatic gener ation of a set of possible topolog ies and ranking of these accordi ng to the positive-inside rule. A straightforward implementati on with no attempts at optimiza tion predict’s the correct topology for 23 out of 24 inner membrane proteins with experim entally determined topologies , and cborr ectl y identifies 135 transme mbrane segments with only one overprediction. Keywords: membrane protein: protein struc t’urr; prediction 1. Introduction The predicti on of protein structure from sequenc e is a central problem in molecular biology. For globular proteins, even secondary structure predic- tion is fraught with di fficul ties and not ve ry reliab le (Kabsch & Sander. 1983); however, recent progress in the field of membra ne protei n assembl y and folding has fuelled hopes that a solution to the folding problem may be within reach for t,his impor- tant class of proteins (von Heij ne & Manoi l, 1990). The basic structura l building-block in plasma membrane proteins of both prokaryotic and eukary- otic cells is the apolar, often sli ghtly amphipathic, transmembrane a-helix (Jennings, 1989; Pattu s, 1990), and the arguably most important event during the biogenesis of an integral membrane pro- tein is the inse rtion of its t ransmembr ane segment(s) into the lipid bilayer. Once this has been accomplished, the basic topology of the molecule is defined. What t’hen remains is for the trans- membrane helices to assemble into a membrane- embedded helix bundle and for the polar segmen ts exposed outside t#he bilayer to fold into their prop er tertiary structure. It should be noted that the situa- tion m ay be ve ry different for outer membran e proteins m Gram negative bacteria , where the cano- nical structural princip le is that of a large, antipar- allel P-barrel rather than a helical bundle (Jeanteur et al., 1991; Weiss et al., 1991); this latter type of protein will not’ be considered furth er here. Thus. at least for multi-spanning (polytopic) membrane prot.eins with most of their m ass buried within the bilayer in the form of transme mbrane helices, the insertion event is decisive . For structure prediction, this me ans that a good part of the problem is solv ed if the transmembrane organiza - tion of the chain can be eff ect ive ly calculated from the amino acid sequence. At the present state o f the art. this is best attempted using some method of hydrophobicity analysis , where t,he amino acid sequence is scanned to locate segments rich in apolar residues. T ypical ly, such an analysis will iden trfy some segments of such high average hydro- phoblalty that they can be considered “certain” transmembrane segments, but will also produce one or more candidat e segments of intermedi ate average hydrophobicity that cannot be confidently predicted as transmembran e. Clearly, the ultimate hydrophobicity analysis method wou ld be able to discriminate perfe ctly between transmembrane and non-membrane segrnents in any protei n chain; this might be accom- plished b y bett’er algorithms and better hydrophobi- cit y scales. Here, however, I suggest a complementary approac h: to “bootstr ap” the output from a standard hydrophobicity analysis by a subsequen t step of charge-bias analys ts t’hat ranks all possible structures on the basis of their confor- mit y with the positive-inside rule; i.e. on the observation that positi vely charg ed amino acids are manifold more abund ant in cytoplas mic, as compar ed to periplasmic, segments of integr al membrane proteins (von Heijne, 1986; von Heijne & Gavel, 1988; Gavel et aE., 1991) and that) po sitive ly charged residues can be used to manipulate the 0022-“836/92/100487-08 $03.00/O 187 c 1992 Academic Press Limited
-
Upload
biosynthesis -
Category
Documents
-
view
219 -
download
0
Transcript of Membrane protein structure prediction Hydrophobicity analysis and the positive-inside rule

8/6/2019 Membrane protein structure prediction Hydrophobicity analysis and the positive-inside rule
http://slidepdf.com/reader/full/membrane-protein-structure-prediction-hydrophobicity-analysis-and-the-positive-inside 1/8
.I . Mol. Bid. (1992) 225, 487494
Membrane Protein Structure Prediction
Hydrophobicity Analysis and the Positive-inside Rule
Gunnar von Heijne
Department of Molecular Biology
Karolinska Institute Center for 8tructural BiochemistryNOVT’Af. S-141 57 Huddinge. Suleden
(Received 14 October 1991; accepted 20 Jan,uary 1992)
A new strategy for predicting the topology of bacterial inner membrane proteins is proposed
on the basis of hydrophobicity analysis , automatic generation of a set of possible topologies
and ranking of these according to the positive-inside rule. A straightforward
implementation with no attempts at optimization predict’s the correct topology for 23 out of
24 inner membrane proteins with experimentally determined topologies, and cborrectly
identifies 135 transmembrane segments with only one overprediction.
Keywords: membrane protein: protein struct’urr; prediction
1. IntroductionThe prediction of protein structure from sequence
is a central problem in molecular biology. For
globular proteins, even secondary structure predic-
tion is fraught with di fficul ties and not very reliable
(Kabsch & Sander. 1983); however, recent progress
in the field of membrane protein assembly and
folding has fuelled hopes that a solution to the
folding problem may be within reach for t,his impor-
tant class of proteins (von Heijne & Manoil, 1990).
The basic structura l building-block in plasma
membrane proteins of both prokaryotic and eukary-
otic cells is the apolar, often slightly amphipathic,
transmembrane a-helix (Jennings, 1989; Pattus,
1990), and the arguably most important event
during the biogenesis of an integral membrane pro-
tein is the insertion of its transmembrane
segment(s) into the lipid bilayer. Once this has been
accomplished, the basic topology of the molecule is
defined. What t’hen remains is for the trans-
membrane helices to assemble into a membrane-
embedded helix bundle and for the polar segments
exposed outside t#he bilayer to fold into their proper
tertiary structure. It should be noted that the situa-
tion may be very different for outer membrane
proteins m Gram negative bacteria, where the cano-
nical structural principle is that o f a large, antipar-
allel P-barrel rather than a helical bundle (Jeanteur
et al., 1991; Weiss et al., 1991); this latter type of
protein will not’ be considered further here.
Thus. at least for multi-spanning (polytopic)
membrane prot.eins with most of their mass buried
within the bilayer in the form of transmembrane
helices, the insertion event is decisive. For structure
prediction, this means that a good part of the
problem is solved if the transmembrane organiza-
tion of the chain can be eff ect ive ly calculated from
the amino acid sequence. At the present state of the
art. this is best attempted using some method of
hydrophobicity analysis , where t,he amino acid
sequence is scanned to locate segments rich in
apolar residues. Typical ly, such an analysis will
identrfy some segments of such high average hydro-
phoblalty that they can be considered “certain”
transmembrane segments, but will also produce one
or more candidate segments of intermediate average
hydrophobicity that cannot be confidently
predicted as transmembrane.
Clearly, the ultimate hydrophobicity analysis
method would be able to discriminate perfectly
between transmembrane and non-membrane
segrnents in any protein chain; this might be accom-
plished by bett’er algorithms and better hydrophobi-
cit y scales. Here, however, I suggest a
complementary approach: to “bootstrap” the
output from a standard hydrophobicity analysis by
a subsequent step of charge-bias analysts t’hat ranks
all possible structures on the basis of their confor-
mit y with the positive-inside rule; i.e. on the
observation that positi vely charged amino acids are
manifold more abundant in cytoplasmic, as
compared to periplasmic, segments of integral
membrane proteins (von Heijne, 1986; von Heijne &
Gavel, 1988; Gavel et aE., 1991) and that) positive ly
charged residues can be used to manipulate the
0022-“836/92/100487-08 $03.00/O187
c 1992 Academic Press Limited

8/6/2019 Membrane protein structure prediction Hydrophobicity analysis and the positive-inside rule
http://slidepdf.com/reader/full/membrane-protein-structure-prediction-hydrophobicity-analysis-and-the-positive-inside 2/8
Figure 1. Sliding window used in the hydrophobicity
analysis. For a given window-position, the hydrophobi-ci ty value hi for each residue in the window is multiplied
by the corresponding window-weight, wi, and the sumover the window is taken. wi is given by u+ = {i/S for
I<i<n-q+l; (n-q+l)/S for (n-q+l)<i<(n+q+l):(%+2--Q/S for (n+q+l)<i<2n+l}. where S =
(1 + n)’ -q2 is a normalization factor to make.
2ntl-C~wi=li=l
In the calculations reported here, n = 10 and q = 5
topology of such proteins (Boyd & Beckwith, 1990:
Dalbey, 1990; von Heijne & Manoil, 1990). As
shown below, this strategy leads to a considerable
improvement in prediction accuracy. at least for
bacterial inner membrane proteins.
2. Methods
(a) Collection of sequence data
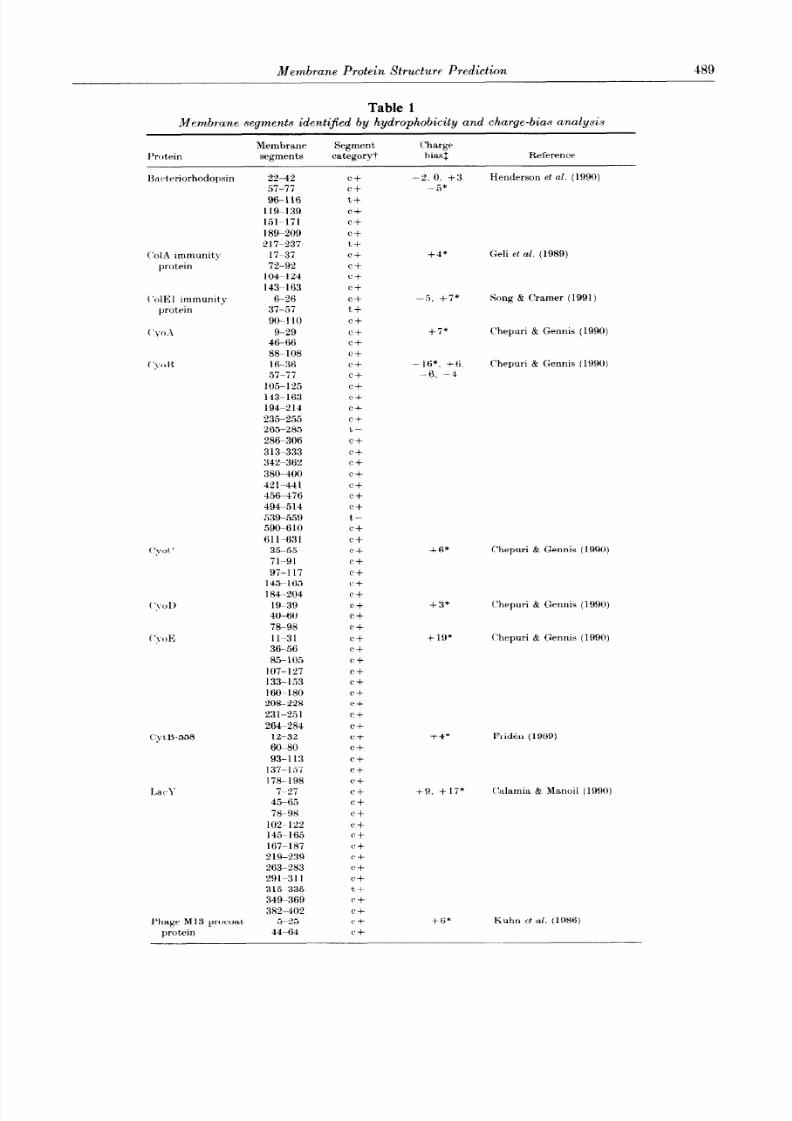
Twenty-four bacterial inner membrane proteins of
known sequence and with experimentally well-character-ized topologies were collected f rom the literature (Table
1). In most cases, the topology has been determined byfusion protein analysis (Manoil & Beckwith, 1986).
Although this method seems to be a very reliable way ofobtaining topological information, it, should be kept in
mind that the 3-dimensional structure has been deter-mined for only 3 of the proteins listed in Table 1, namely
bacteriorhodopsin, and the recation center L and Msubunits.
(b) Hydrophobic ity analysis
Hydrophobic ity analysis was carried out using theGES-scale (Engelman et al., 1986) and a trapezoid slidingwindow composed o f a central, ll-residue rectangular
section and 2 flanking wedge-like sections, each 5 residueslong (Fig.1). This window-shape was chosen to combine
the favorable noise-reduction of a triangular window
(Claverie & Daulmiere, 1991) with a physically morerealistic rectangular window representing the centralapolar part of a lipid bilayer. In this way, one obtains a
physically reasonable, sof t transition from the apolarinterior to the polar surface of the membrane.
From the hydrophobicity profile, candidate trans-membrane segments were extracted automatically by a
procedure that 1st identifies the highest peak, records its
average hydrophobicity (H), and removes the part of theprofile that corresponds to the 21 residues in this segment.This is repeated until either no stretch longer than 20residues remains, or until no peak higher than the lowercutof f ((H) = 0.5) remains. All peaks with (H) 2 1.0 areconsidered “certain” transmembrane segments, and allwith 95 I(H) < 1.0 are considered putative candidates.
-4 -3 -2 -I 0 I 2
<l-f4
Figure 2. Peak-height distributions derived fromhydrophobicity analysis of 92 bacterial inner membrane
proteins ( q ), 25 periplasmic Escherichia coli proteins (+).and the 135 transmembrane segments identified in the 24bacterial inner membrane proteins discussed in t,he t,ext
(ml.
These cutoff values were chosen on t,he basis of an initialanalysis of the bimodal peak-height distribution for the
whole sequence sample obtained with no cutoff s (Fig. 2:
see von Heijne, 1986).Finally, all possible topologies that include the certain
transmembrane segments and either include or exclude
each of the candidate segments were automatically gener-
ated. a,nd the difference in the number of posnivelycharged amino acid residues (Arg and Lys ) between the 2sides of each structure was calculated. If the penultimate
N-terminal amino acid was neither Arg, Lys, Leu. Phe norHe (i.e. residues that block f-Met removal in prokaryotes
(Flinta et al., 1986)) an additional posi tive charge repre-
senting the free N-terminal amino group was added to the1st polar segment. Polar segments longer than 70 residueswere not included in the charge-bias calculation, since
t,heir content of charged residues does not seem to be
dependent on their cytoplasmic or periplasmic location(von Heijne & Gavel, 1988). The possible topologies were
then ranked in decreasing order o f charge bias, and theirorientation wa s predicted as the one with the more highlycharged side facing the cytoplasm. A program called
TOP-PRED (TOPology PREDiction program. writt!en in
THINK Pascal for Macintosh computers) implementingthese steps is available upon request.
3. Results
(a) The positive-inside rule holds for all exposed
loops in bacterial inner membrane proteins
The positive-inside rule was originally suggested
by the observation that positi vely charged amino
acid residues (Arg and Lys ) are much more ahun-
dant in cytoplasmic as compared to periplasmic
regions of bacterial inner membrane proteins (von
Heijne, 1986), and has since been confirmed by a
number of experimental as well as further stat’istlcal
studies (von Heijne, 1989; Boyd & Beckwith, 1990;
Dalbey, 1990; Nilsson & von Heijne, 1990; von
Heijne & Manoil, 1990). However, to be able t’o
make use of this rule for prediction purposes. it was
8/6/2019 Membrane protein structure prediction Hydrophobicity analysis and the positive-inside rule
http://slidepdf.com/reader/full/membrane-protein-structure-prediction-hydrophobicity-analysis-and-the-positive-inside 3/8
Membrane Protein Structure Prediction
Table 1
489
Membrane segments identi$ed by hydrophobicity and charge-bias analysis
I’rotrin
Membrane Segment Charge
segments categoryt biasf Reference
lkc~trriorhodopsin 22-42
57-77
96116
119-139
151-171
189-209
217-237
C’olA immun it\ 17-37
protein 72-92
104-124
143-163
(‘olE1 immunity G-26
protein 37-57
90-110
(‘yoA 9-29
4C-8688-108
(‘,td< 16-36
57-77
105-125
143-163
194-214
235-255
265-285
286-306
313-333
342-362
380-400
G-441
456-476
494-514
539-559
590-6 10
611-631
( ‘yd ’ 35-55
71-91
97-l 17
1455165
184-204
19-39
40-60
78-98
11-31
36-56
85-105
107-127
133-153
160-180
208-228
231-2.51
264-284
CyW558 12-32
6G80
93-l 13
137-1,57
178-198
‘i-27
45-65
78-98
102~12%
145p I65
167YlRi
219-239263- 283
291L311
315.-335
34%369
382-402
Phage Ml3 prowat 5-25
protein 44-64
C+
C+
t, +
C+
C+
C+
t+
C+
c+
c+
c+
c+
t+
c+
c+
c+C+
c+
c+
c+
c+
c+
c+
t,-
c+
c+
Cf
c+
c+
C+
c+
t-
C+
C+
C+
C+
c+
C+
C+
C+
C+
C+
c+
c+
C+
C+
C+
(‘+
C+
C+
C-t
C+
c+
Cf
C+
c +
C+
c+
C+
C’ +
C+
(‘+
c+C+
C+
tt
C+
Cf
Cf
Cf
-5, +7* Song & (Iramer ( 1991)
-IS* +6.
-6,‘-4
Chepuri & Gennis (1990)
-2. 0. +3
-5*
+4*
+7*
+6*
+ :3*
+ lo*
+4*
+6*
Henderson et al. (1990)
Geli ct al. (1989)
Chepuri & Gennis (1990)
Chepuri & Gennis (1990)
(‘hepuri & Gennis (1990)
C’hepuri & Gennis (1990)
Fridkn (1989)
Kuhn et al. (1986)
+9, +17* (klamia & Manoil (1990)
8/6/2019 Membrane protein structure prediction Hydrophobicity analysis and the positive-inside rule
http://slidepdf.com/reader/full/membrane-protein-structure-prediction-hydrophobicity-analysis-and-the-positive-inside 4/8
Light-harvesting
complex LH 1
Light-harvesting
complex LH:!
Keac%ion crntrr
complex
L-subunit
I’hpT
PI* Rohrw & Kuhn (1990)
OX. -2 Moore $2 Miura (1987)
-I. +9* Munoa rf (11. ( I ! , ! , ] )
+4* Ihws (19%)
+1* Ihws ( I9X:i)
+x*. t-" Michel rt /I/. (1986)
+9*. +:I
+fi* Schatz rt al. ( 19X9)
+?A*. +x .Ikiyarna & Ito (1987)
+ 1:3*
+27* Lloyd & Kadnrr (1990)

8/6/2019 Membrane protein structure prediction Hydrophobicity analysis and the positive-inside rule
http://slidepdf.com/reader/full/membrane-protein-structure-prediction-hydrophobicity-analysis-and-the-positive-inside 5/8
Membrane Protein Structure Prediction 491
Table 1
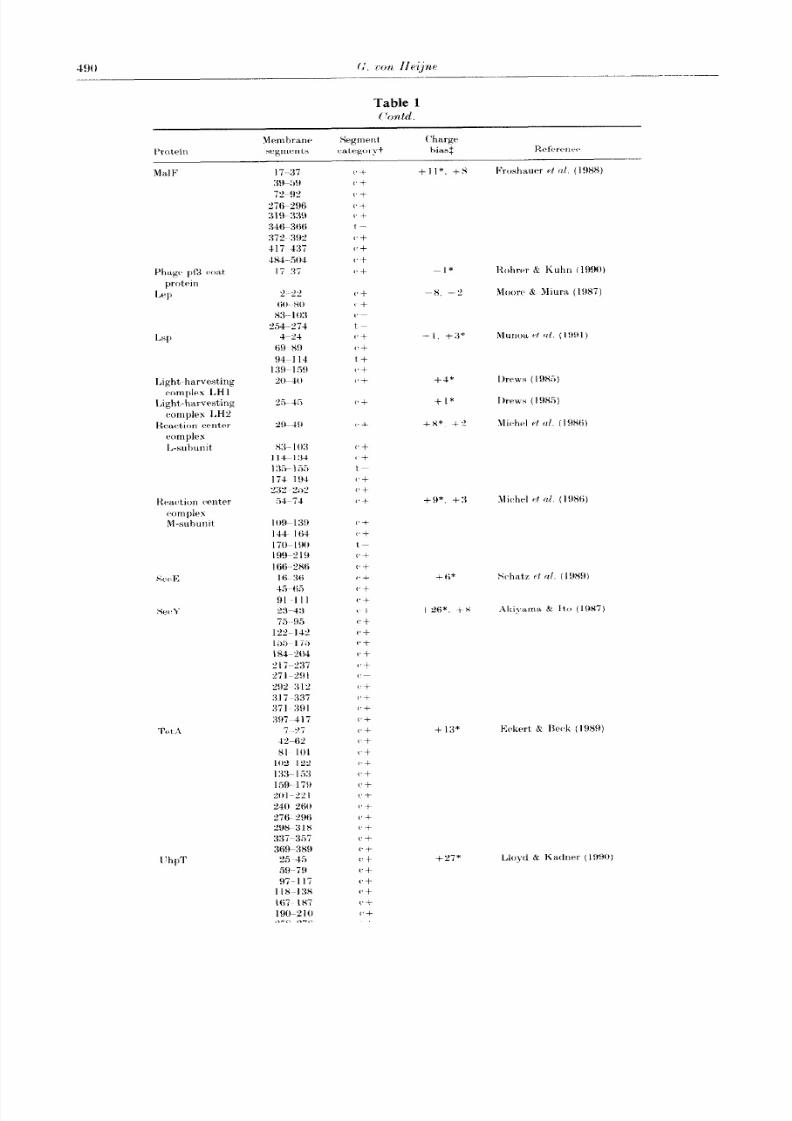
C’ontd.
Membrane Segment Charge
Protein segme nts categoryt bias: Reference
299-319 (‘+
38tk34ti c-+
351-371 C+
405-425 C’+
427447 (‘+
t’trr~F: 11-31 Cf -2* Drckers-Hebestreit, C
59-79 c + Altendorf (1986)
tr. certain; t, tentative; +, a membrane segmen t present in the known structure; -, a membrane
segmen t not present in the known structure.
$The t,otai difference in the number of Arg+I,ys residues between thr 2 sides of the structure for all
po&hlr topologies generated as described in Methods are given, with the value corresponding to the
kuown topology marked with an asterisk. Positive values correspond to topologies where the S
terminus is predicted to be cytoplasm ic. negative values to topologies predicted to have the X t~rrminu~
import,ant to establish that it, holds for all segmentsof a transmembrane protein, and not just for theN-terminal or (‘-t,erminal ends, say.
To this end. 24 bacterial inner membrane proteinsof known sequence and with experimentallymapped topologies were analyzed (seeTable 1). Foreach protein, the number of Arg and Lys residues neach cytoplasmic and periplasmic segment wasrecorded, and t,hr counts were pooled according tothe position of the segment (counting from the r\;terminus) and its cytoplasmic or periplasmic loca-tion. As shown in Figure 3: the bias in the distribu-tion of’ Arg+ Lys is equally strong throughout, thesequences; .e. all cytoplasmic loops have a similarlyhigh content (- IS”/,) whereas all periplasmic loopshave a low content (-SOL). It is thus clear that allparts of a multi-spanning inner membrane protein[*onform to the positive-inside rule.
(b) Thr positiwinxide rule can be used to improrr
thw prediction of transmembrane sepents
Since the positive-inside rule apparently appliesto any part of a bacterial inner membrane protein.it should provide a strong crit,erion for testingwhether a putative transmembrane topology islikely to be correct or not. One is thus led t’oconsider the following procedure. First, make a listof all possible transmembrane segments in the pro-t)ein based on some standard method of hydrophobi-city analysis and a liberal cutoff criterion; second.decide which of these candidat’es are certain and
which must be regarded as tentative based on a
more stringent cutoff criterion; third, construct’ allpossible transmembrane st’ructures always includingthr cert,ain candidates but either including or
excluding each of the tentative segment#s: andfourth, rank these structures according to theirdegree of bias in the dist’ribution of positivelycshargrd residues. The rationale behind the last st,ep.is that any structure with an incorrect number oftransmembrane segments would have one or morepolar domains placed on the wrong side of the
membrane, and hence would be likely to have asmaller charge bias than the corre& structure.
As an example. consider the 1,acY protein. whichis known to have a cytoplasmic N terminus followedby 12 transmembrane segments (Calamia B Manoil,1990). The hgdrophobicity plot shown in Figure4(a). predicts 11 certain segments and one putativecandidate. Thus, two topologies can be generated,one excluding and one including the putative candi-date (Fig. 4(b)). The former has a bias in the distri-bution of positively charged residues of nine, thelatt’er of 17, and it is clear from the Figure that theC-terminal part of the second. correct model has amuch better correspondence with the positive-insiderule.
To test this strategy more thoroughly, hydropho-bicity analysis was carried out on the 24 proteinslisted in Table 1 as described in -Methods, with alower cutoff set to (H) = 0.5 and a higher cutoff set
Segment positlon
Figure 3. Mean number of Lys+ hrg in v)-toplasmic
(stippled bars) and periplasm ic (open bars) polar segme nts
as a function of the position of the segment (1st cyto-
plasm ic segment. 2nd cytoplasm ic segmen t. rt,c., counting
from the N terminus) in a samp le of 24 bac*twial inner
membra,ne prot,eins with known topology.

8/6/2019 Membrane protein structure prediction Hydrophobicity analysis and the positive-inside rule
http://slidepdf.com/reader/full/membrane-protein-structure-prediction-hydrophobicity-analysis-and-the-positive-inside 6/8
2
I
A 0
$v
-I u v
1 li
U I
-2
-3
-41 I I 1
0 100 200
Posltion
(a)
300 400
A+ = 9
A+=17
2 3 5 4 2 2 2
(b)
Figure 4. (a) Hydrophobicity plot for the SecY protein.The upper and lower cutoffs are marked. A tentative
transmembrane segment with a mean hydrophobicitfalling between the 2 cutof fs is marked by an arrow.
(b) Two possible topologies for the SecY protein based on
the hydrophobicity plot. The putative transmembranesegment is shown in black. The number of Arg+Lysresidues is shown next to each polar segment. IVote that
t)hr correct, alternative (bottom, including the putativetransmembrane segment) has a much higher charge-bias
than the incorrect. one.
to (H) = 1.0 (i.e. t’he criterion for tentative trans-membrane segments was 0.5 _< H) < 1.0). Fourteenprot’eins were predicted to contain only certaintransmembrane segments with no additional tenta-tive candidat,es; all of these turned out to be correctand their orientation in the membrane was correctlypredicted by the positive-inside rule in all cases.Forthe remaining ten proteins, the correct structurehad the highest charge bias (highest differencehetween the number of periplasmic and cytoplasmicArg+ Lys) in eight cases Table 1). An extra. incor-rect certain transmembrane segment ((H) = 1.06)
was predicted for the SecY protein; closer inspectionrevealed that this resulted from a “shoulder” on ahigher, somewhat broad peak. Inclusion of thissegment gave a topology with a charge bias of eight.and its exclusion gave a charge bias of 26. Thus.
Thus. using the positive-inside rule as :I J(‘r(‘en.
t)he transmcmhranr topologies of 22 (or. wit)h somegootl-will. 23) out of 21 proteins were (*orrec+lJ
predicted. atld all of the 135 transrnrm t)ranc>segments wTt‘re dentif ied with onl?- oru’ ovtq)rerl icn-
tion (I,tlf)).
(c) Possibilities for improvements in thv
hydrophobicity scnlrCs
In the a,bove analysis. the standard (JES hydra-phobicity scale (Engelman rt nl.. 1986) based rriti-matrly on physico-chemical caonsiderationswas usedand no at,tempt at, optimization was made. Withthis scale and through application of t,he positivr-
inside rule. 135 t’ransmembrnne segments werecorrectI\- identified in t.hr 24 proteins analvzrd.From t,hese data, we derived a new. statistically
based scale by comparing the amino acid frequen-cies fi M in the cent,ral I 1 -residue stretch of t,ht>transmembrane segments with the amino acidfrequencies f;” m the non-meml)raneous srgrnent#s(taking the membrane-embedded segments to hf. 17residues long): Ai = In(fiM/fiA), Table 2. \Vith a11upper cutoff of O$.i and a lower of - 0. I and t hts
Table 2Hydrophobicity scak bused on 735 transmemhrane
segments from 24 bacterial inner ,membrane proteins

8/6/2019 Membrane protein structure prediction Hydrophobicity analysis and the positive-inside rule
http://slidepdf.com/reader/full/membrane-protein-structure-prediction-hydrophobicity-analysis-and-the-positive-inside 7/8
Memhranr Protein Strwtuw Prediction 493
same sliding window as above, this scale seems ’operform slightfly better than the original GES-scale,with 18 out of the 24 proteins (including SecY) nowbeing predicted to contain only certain trans-membrane segments (a.11correct). Again. ranking
based on the positive-inside rule produced thecorrecst topolop~ for iLlI but one (I,ep) of theremaining prot,elns.
4. Discussion
Two important conclusions can be drawn fromt.he data present,ed here: first., the posit’ive-insiderule applies with equal strength to all parts ofpolytopic bacxterial transmembrane proteins, and.second. the positive-inside rule can very efficientlybe used to rank possible predicted transmembranc
topologies and thus in a sense t’o bootstrap thest.andard hydrophobicit? analysis.
The first, c*onclusionhas certain mechanistic impli-rations, and suggests a mode of assembly ofbacterial membrane proteins where locally formed“helical hairpins” composed of two neighboringhydrophobic- helices and a connecting loopcontaining few positively charged residues insertinto the membrane more or less independently oft*he remaining parts of the polypeptide chain(Engelman & Steitz, 1981; von Heijne, 1986).A similar model is also suggested by recent experi-mental data on the t,opological consequences of
deleting one or more transmembrane segment.(Yatnane et al., 1990; Kibi et al.. 1991: Ehrmann 8:Krckwit’h. 1991: McGovern et al.. 1991). This is incontrast t.0 it “linear” insertion model, where themost S-terminal transmembrane segment(s) insertfirst. and the rest of the chain simply follows thetopological dictate of this first insertion (Vessels &
Spiess. 1988: Hartmann et al., 1989). It may be.though. t’hat the latter model more accuratelydescribes membrane protein assembly into the endo-plasmic reticulum membrane, which is t’hought tobe obligatlorily co-translational and hence likely toproceed in an K to C-terminal direction (High et al..
1991). It should be noted in this regard that the biasin i he dist,ribnt.ion of positively charged residues.although clearly t,here, is nevertheless less marked in
eukary-otic plasma membrane proteins (von Heijne& Gavel. 198X).
For predi&m purposes, a combination of hydro-phobicity analysis and charge-bias analysis clearl>improves the reault,s. almost to the point where veqlittle experiment.al work would have t.o be done tocoonfirm a predicted t,opologg. In this study. thetopology of 9.5(& of the prot.eins was correctlypredi&ed I)!- a fully automatic method. even whenthe initial hydrophobicity analysis method had notbeen consc~iou+,- optimized. Thus, the topology ofbacterial inner membrane proteins seems to berather straightforward to predict direc+ from theirsequence. setting the stage for attempts t,o developmethods for modeling their fIlli three-dimensionalstructure.
This work was supported hv grants from the Swedish
Natural Sciences Research &unc:il and thr BwrdishBoard fo r Technical Development.
References
Akiyama. Y. & Ito. K. (1987). Topology analysis of’ theSrcY protein. an integral membrane protrin involved
in protein rlxport in Eschrrichitr roli. h’.MBO ,I. 6.
3465 -3470.Bibi. IX.. Verner. (i.. (‘hang. (‘. 1.. & Kabwk. H. R.
(1991). Organization and stabil ity of a polytopicmembrane protein-deletion analysis of’ the Iac+osepermease of EarhfwZchiu coli. Pror Sat. ;Icrrd. S ri..
l’.S.A 88. i2’il-727s.
Rilgin. X.. Lee. .I. 1.. Zhu. H.. J)alt)r!-. Ii. & van Heijnr,(;. (1990). Mapping of c.atal~ticalI~ importantdomains in Eschrrichin roli leadel. prl~t~tiase, EURO
,J. 9. %il’i m”iP?.Hoyt i. I). d l+ckwith. ,I. (1990). The role of c,harped
amino acids in the localieatiorl of ~rc*rt~trd andmemhraneproteins. Cell. 62. 1031 IOXX.
(‘alamia. .J. k Manoil. (‘. (lR!)O). ],a(. lwrmcas(z of
Esrhvrichicz co/i&topology and srquen~e elementspromoting membrane insertion I’rvc .V///. .-Iwcl. Sci..
l‘.S.d. 87. -4937-4941.(‘hrpuri. \‘. & (irnnis, R. 13. (1990). Tbr use of gene
fusions to d&ermine the topology of all of the sub-
units of t,hr cytochrome o terminal oxidase wmplexof Ewherichin coli. J. Biol. C’h~m. 265. 12978P1ZM6.
(‘la.verir. .1.-M. & Daulmiere, (‘. (1991). Smoot.hing profiles
with sliding windows: better to wear A hat! /‘.-1 f?I f)S
7. 113-115.I)albe>-. R,. E. ( 1990). Positively c*hargtad wsidues are
important determinants of tncmbranr proWin topo-logy. Twnda Biochem. Sci. 15. %54-?.5i.
T)rc~kers-H~bestreit. (:. & r\Itendorf. K. (1986).r\ccrssibilit> of F, suhunit)s from KsrhPrirhirr coli
ATI- synthase. Ew. ./. HiochPv. 161. ~~T,-~Sl.Drrna. ( :. (l!P&). Structure and func+ionnl organization
of light-harvesting complexes and phot.oc~hrnricaIreaction c,enters in membranes of l~hototrophic~
bacteria. N icrobiol. Hev. 49. Z-70.
Eckert. R. & Reck, C. F. (1989). Topology of the trans-poson TnlO-encoded tetracycline resistance protein
within the inner membrane of Escherichia col i .
J Bid Chum. 264. 11633-11670.
Ehrmann. 1cl. & Reckwith. *J. (1991). I’ropw insertion of acomplex mt\mhranr prot.rin in the absrnw 01‘ itsamino-terminal export. signal. .I. /1;0/ (‘him. 266.
165‘3W 1651’i.. I.<Engelman. I). 11 8: St,ritz. T. A. (1981 i. The spontaneous
insertion of proteins int,o and across membranes: The
helical hairpin hypothesis. (‘~11. 23. -411-4ZE:ngcelman. I) . 11.. Steitz. T. A. Xr C:oldtnan. ,1. (19X6).
Identify ing nonpolar transbilayrr hrlicbrh in amino
wid srquenc~es of mrmhrane proteins. .~lnrclc. Rw.Biophys. Bicyhys. C’h.Pm. 15. 4”l -8X1.
Flinta. t‘.. Pwsson, R.. ,Jiirnvall . H. & \-on Hrijne. (G.(1986). Sequence determinants of cytosolicX-terminal protein processing!. L:crr. .J. Hiockrm. 154.
193 196.Fridin. H. (1989). Studies on the c~ytoc~hrornr B-558 sub-
unit of HacilZ?Ls su6ti l is succ~inate:yuino~~r oxidorr-duct,ase. Phi) t’hesis. und IrniversitJ-. C;wedrn.
Froshauer. S.. Green. G. X.. Boyd. I).. Mc(:overn, K. &
Rrckw-ith. .I. (1988). (>metic ana.lysis of t.hrmembrane insertion and t.opolog?- of MaIF. a cyto-

8/6/2019 Membrane protein structure prediction Hydrophobicity analysis and the positive-inside rule
http://slidepdf.com/reader/full/membrane-protein-structure-prediction-hydrophobicity-analysis-and-the-positive-inside 8/8
plasmic membrane protein of Exherichicl co/i. J, .Ilo/
Riol. 200. WI 511.(:avc~l. E’.. Steppuhn. J . Herrmann, R. & van Heijntx. ( :.
(1991). The posit,&-inside rule applies to thvlakoi t]membrane proeeins. FEBA’ L&w. 282, 11 46.
(ieli. \‘ .. Katy. I).. l’attus. F. & Lazdunski. (‘. (1989).Topology and fun&ion of the integral membrane
protein conferring immunity to colicin A. Tklol.
Microbid. 3. 679. 687.
Hartmann, E., Rapoport, T. A. 8r Lodish, H. F. (1989).
I’rrdic*ting t)hr orient’ation of eukaryotic membraneproteins. Pror. ,Vat. A cad. sc i., 1’JS.d. 86.r,786X790.
Henderson. R.. Baldwin, J. M.. (‘eska, T. A.. Zemlin. F..
Beckmann. E. & Downing, K. H. (1990). 4 model forthe structure of bacteriorhodopsin based on highrrsolut,ion electron cqo-mivroscsopy. -1. No/. Hiol.
213. 899-929.
High. S.. Flint. X. & Dobberstein, K. (1991).Rtyuirrments for the membrane insertion of signal-
anchor type proteins. J. Call. Riol. 113. 2.5 34.Jeant,rur. I).. Lakey. J. H. & Pattus, F. (1991). The
ba&erial porin superfamily-sequence alignment andstructure prediction. Mol. Microhiol. 5, %]53%2164.
.lennings, M. I,. (1989). Topography of membrane pro-teins. A~anu. Krv. Biochem. 58, 999.-1oP7.
Ka1~sc.h. M’. & Sander. C. (1983). How good are predic-tions of’ protein secondary structure? FERS Ltittux.
155. 179-182.Kuhn. A.. Kreil. (:. & Wickner. W. (1986). Both hydro-
phobic~ domains of Ml3 provoat are required toinitiate membrane insertion. ENRO. .I. 5.
3681~ 3685.
T~loytl. A. I>. & Kadnrr. R. ,J. (1990). Topology of the
h’sch~richia roli &pT sugar-phosphate transporteranalyzed by using TnphoA fusions. ./. Bactrriol. 172.
I6H8~1693.Manoil. (‘. & Beckwith, J. (1986). A genetic approach to
analyzing membrane protein topology. Science. 233.
1403-140x.Mv(:ovrrn. K.. Ehrmann. M. $ Heekwith. .J. (1991).
I)ec*oding signals for membrane protein assemhll
using alkaline phosphatase fusions. EA’RO ./. 10.
2773 %7X”.Michrl. H ., IVryer. K. A.. Gruenberg, H., Dunger, I..
Orsterhelt, D. B Lottspeich, F. (1986). The ‘light’
and .medium’ subunits of the photosynthetic reaca-
tion ventre from tlhodopseudomonacs kdis: isolation
of the genes. nucleotide and amino avid seyuenc.r.F;MKO .I. 5. 1149~1158.
l\loorv. K. E:. R Miura. S. (19X71. .\ small hytlrc~l~hohit~
domain anc*hors Ieadt?r pr])t,ittascl t,o the (.yto])lastllic .tnrmbranr of Escl~~rirhirr ~oli. J Iiiol C’he,~/ 262.
8XO6~~881:1.
Mrmoa. I’. .I Miller. K. 1V.. ISeers. It.. (:raharrl. Xl. & \$‘I].H ( ‘. ( 199 I ). Membrane topology of /fschrrichirr co/;
prolipoprot,ein signal peptidase (signal pt’])tidasr II ).
.I. Hid. C’hPm. 266. 17667 1767”.Nilssolt. 1. &I. 8r van Heijne. (;. (1990). Fine-tuning thr
topology of a polytopic, mrmbrane ])rotrin. l<ol~~ of
positix-taly and nrgativrly c.hargetl residuc~s. c r/l. 62.1135 1111.
I’attus. F. (1990). Mrmbranr prot,eitr struc%urv t ‘err.
Opi,/ion (‘41. Biol. 2, 68lm 68.Y
Rohrer. *I. 8: Kuhn. A. (1990). The furtvtion of’ a leaderprptidr in transloczat)ing caharged amino acsy l resitluvsacross a mrmbranr. Srirwr. 250, 141 8 1121
Schatz. 1’. .] liiggs. 1’. I).. .Jaq. L\.. Fath. hl. .I. &
Brvkwith. ,I. (1989). Thr sr~E gene t~nc~odvs anintegral membrane protein required for protein
export in Eschrrichicr r~ol;. (:etrP.s lk1dop 3.
IO3;i 1Ol-c.
Song, H. 1’. & (‘ranter. L\‘. A. (1991). ,IlrmbranrTopography of’ (‘oNI gent’ producnts- the immrmit~
protein .I. IZnctrriol. 173, 835-2948.van Hrijntx. (:. (1986). ‘I? IP distribution of positivt)l?
caharged residues in bacterial inner mc~rnbratx~ ]m,~teins (*orrelates with thtb tram-membranr topology.
ENI~O ./ . 5, 3021 -3027.van Heijne. (:. (1989). C’ontrol of topology ant1 moth. of
assrtnbl?. of a polytopiv membrane protein by posi-tiv rl! charged rrsidurs. Srrt~r~ ~lonrlor/ 1, 341.
15tibkr,x.van Hri,jncs. (:. & (:aveI, Y. (19%). ‘I’c,])ogrnic. signals in
integral mC~mt)rattt~ proteins. E/c/. . ./. fjioChr,rr/. 174.671 6iX.
van Heijnr. (:. & Manoil. C’. (1990). Membrane proteins
--from sequence to structure. Protein En,g . 4.
109- I I”.
MTeiss. M. S.. Kreuscsh, A.. Schiltz. E., Nested. I’.. \Yt~lt~r.iv. . \Vrckrssrr. J. & Schulz. (:. E. (1991). ‘l ’hv strucmturn of porin from Rl~odobfrct~~r rupxulatcf at I .X,i
resolution. PEES Lrtters. 280. 379 382.
Vessels, H. 1’. & Spiess. M. (1988). Insertion of a multi-spanning membrane protein occurs sequentially and
requires only one signal sequence. (‘ell, 55, 61- 70.Yamant,. K.. Akivama. \‘.. Ito. K. & Mizushim:i. S.
(1990). ;\ positi vely vhargrd regiotl is a dett~tmirtatrt
of the orientat,ion of c~ytoplastnic membrane lnvteinsin Esrhrrichin coli. ./. Niol. C’hun. 265. “1 I66 11 171.
Edifrd by K. IT. J!!atthru~.s


















