
Mechanism-based pain management in chronic pancreatitis ...
30
Full Terms & Conditions of access and use can be found at https://www.tandfonline.com/action/journalInformation?journalCode=ierj20 Expert Review of Clinical Pharmacology ISSN: 1751-2433 (Print) 1751-2441 (Online) Journal homepage: https://www.tandfonline.com/loi/ierj20 Mechanism-based pain management in chronic pancreatitis - is it time for a paradigm shift? Louise Kuhlmann, Søren S. Olesen, Anne E Olesen, Lars Arendt-Nielsen & Asbjørn M. Drewes To cite this article: Louise Kuhlmann, Søren S. Olesen, Anne E Olesen, Lars Arendt- Nielsen & Asbjørn M. Drewes (2019): Mechanism-based pain management in chronic pancreatitis - is it time for a paradigm shift?, Expert Review of Clinical Pharmacology, DOI: 10.1080/17512433.2019.1571409 To link to this article: https://doi.org/10.1080/17512433.2019.1571409 Accepted author version posted online: 21 Jan 2019. Submit your article to this journal View Crossmark data
Transcript of Mechanism-based pain management in chronic pancreatitis ...

Full Terms & Conditions of access and use can be found athttps://www.tandfonline.com/action/journalInformation?journalCode=ierj20
Expert Review of Clinical Pharmacology
ISSN: 1751-2433 (Print) 1751-2441 (Online) Journal homepage: https://www.tandfonline.com/loi/ierj20
Mechanism-based pain management in chronicpancreatitis - is it time for a paradigm shift?
Louise Kuhlmann, Søren S. Olesen, Anne E Olesen, Lars Arendt-Nielsen &Asbjørn M. Drewes
To cite this article: Louise Kuhlmann, Søren S. Olesen, Anne E Olesen, Lars Arendt-Nielsen & Asbjørn M. Drewes (2019): Mechanism-based pain management in chronicpancreatitis - is it time for a paradigm shift?, Expert Review of Clinical Pharmacology, DOI:10.1080/17512433.2019.1571409
To link to this article: https://doi.org/10.1080/17512433.2019.1571409
Accepted author version posted online: 21Jan 2019.
Submit your article to this journal
View Crossmark data

Accep
ted M
anus
cript
1
Publisher: Taylor & Francis
Journal: Expert Review of Clinical Pharmacology
DOI: 10.1080/17512433.2019.1571409
Review
Mechanism-based pain management in chronic pancreatitis - is it time for a paradigm shift?
Louise Kuhlmann1,2,3, Søren S. Olesen1,3, Anne E Olesen1,3 Lars Arendt-Nielsen4 & Asbjørn M. Drewes1,3
1. Centre for Pancreatic Diseases and Mech-Sense, Department of Gastroenterology and Hepatology, Aalborg University Hospital, Aalborg, Denmark
2. Department of Internal Medicine, North Denmark Regional Hospital, Hjørring, Denmark
3. Department of Clinical Medicine, Aalborg University, Aalborg, Denmark
4. Center for Sensory-Motor Interaction, School of Medicine, Aalborg University, Aalborg, Denmark
Corresponding author:
Asbjørn Mohr Drewes
Mech-Sense & Centre for Pancreatic Diseases, Department of Gastroenterology and Hepatology,
Clinical Institute, Aalborg University Hospital, Mølleparkvej 4, 9000 Aalborg, Denmark
Telephone: +45 97663562, Fax: +45 97666507
E-mail: [email protected]

Accep
ted M
anus
cript
2
Abstract
Introduction
Pain is the most common symptom in chronic pancreatitis and treatment remains a challenge.
Management of visceral pain in general, is only sparsely documented, and treatment in the clinic is
typically based on empirical knowledge from somatic pain conditions. This may be problematic, as
many aspects of the neurobiology differ significantly from somatic pain, and organs such as the gut
and liver play a major role in tolerability to analgesics. On the other hand, clinical awareness and new
methods for quantitative assessment of pain mechanisms, will likely increase our understanding of
the visceral pain system and guide more individualized pain management.
Areas covered
This review includes an overview of known pain mechanisms in chronic pancreatitis and how to
characterize them using quantitative sensory testing. The aim is to provide a mechanism-oriented
approach to analgesic treatment, including treatment of psychological factors affecting pain
perception and consideration of side effects in the management plan.
Expert opinion
A mechanism-based examination and profiling of pain in chronic pancreatitis will enable investigators
to provide a well-substantiated approach to effective management. This mechanisms-based,
individualized regime will pave the road to better pain relief and spare the patient from unnecessary
trial-and-error approaches and unwanted side effects.
Keywords: Chronic pancreatitis; Chronic Pain; Pain Management; Pain Measurements;
Neurophysiology; Analgesics; Symptom Assessment

Accep
ted M
anus
cript
3
1. Introduction
Chronic pancreatitis (CP) is a fibro-inflammatory disease where the pancreatic parenchyma is
progressively replaced by fibrous connective tissue, potentially leading to exocrine and endocrine
pancreatic insufficiency. The most common symptom in CP is abdominal pain which is present in
about 70% of the patients. The typical description of the pain is a constant dull ache in the
epigastrium with referral to the back (including referred muscle hyperalgesia) that often increases
with food intake, but it can manifest in a variety of ways, ranging from patients with limited,
intermittent pain to those with constant, intense, and severe pain [1]. Previously it was believed that
pancreatic pain would “burn-out” as the disease process evolved with destruction of the pancreatic
tissue [2]. Today the “burn-out” theory is regarded obsolete as evidence against it has been provided
in large retrospective studies [3,4]. Thus, it is not advisable with a “wait-and-see” approach to
patients with on-going pain. Unfortunately, management of pancreatic pain remains a considerable
therapeutic challenge [5]. It is known from other chronic pain conditions that the longer the pain
persists and the stronger it is, the more effect pain has on the central sensitization processes and the
more difficult is to treat [6].
Different management regimes for visceral pain are only sparsely documented as compared with its
somatic counterpart. As a consequence, the approach used in treatment of somatic pain is typically
used as framework for visceral pain management, with analgesic use based on the World Health
Organization (WHO) ladder [7,8]. Although visceral, somatic, neuropathic and inflammatory chronic
pain conditions share common mechanistic features, visceral and somatic pain has several important
differences that should be considered when initiating analgesic treatment [6]. Hence, visceral pain is
more diffuse and difficult to localize. This can lead to malpractice, as failing to localize the origin of the
pain can hinder for example surgical treatment. It is also accompanied by symptoms arising from the
autonomic and enteric nervous system that may need specific management, including nausea and
gastrointestinal disturbances [9]. Chronic visceral pain also induce peripheral and central sensitization
more frequent than in somatic pain conditions [10]. Finally, when drug absorption and metabolism is
considered, the gut and liver are of major importance, but they are often malfunctioning in CP due to
steatorrhea, bile duct stenosis, duodenal stenosis and comorbidities including alcoholic liver disease
[11]. These organs are also the main targets to side effects.
As a new dimension, characterization of the pain mechanisms underlying painful CP can theoretically
facilitate individualized treatment targeting the involved mechanism, and thereby enable
personalized pharmacological treatment, improve patient outcome, and reduce unwanted side
effects. Therefore, the aim of this paper is to review the involved pain mechanisms in chronic

Accep
ted M
anus
cript
4
pancreatic pain and discuss the future of mechanism-based analgesic treatment. This will be done by
evaluating studies that have used quantitative sensory testing (QST) to phenotype/profile patients
and evaluate treatment response.
2. Pain mechanisms and quantitative sensory testing
There are several reasons for chronic pain in patients with chronic pancreatitis, for review see [5]. The
pain can be nociceptive, inflammatory and/or neuropathic and thereby arise from an actual or
threatened damage to the tissues. This can occur due to a number of complications to CP, and many
of them are treatable. The initial step in treating pancreatic pain is to examine whether any of these
complications are present and treat appropriately. If there are no anatomical complications, or if
treatment of these complications does not relieve the patient’s symptoms, the pain could be of
predominantly neuropathic origin and a different approach must be taken. In case of no obvious
organic identifiable reasons but the patients show hyperalgesic reactions, the new descriptor
neuroplastic pain may be used [12]. Like all other kinds of chronic pain, pancreatic pain can affect the
peripheral and central nervous system leading to neuropathy and sensitization. For example,
exposure to several chemical agents released following cellular damage in the pancreas, including H+,
K+, inflammatory molecules and trypsin, lead to increased spontaneous activity and excitability
manifested as peripheral sensitization. These peripheral changes result in an ongoing and vigorous
nociceptive input to the spinal cord that may result in an altered function of the central pain
pathways due to neuroplasticity (central sensitization) [13,14].
***Figure 1 near here***
Sensitization is characterized by hyperalgesia, where pain detection threshold is decreased compared
to that in patients without chronic pain. The hyperalgesia can be detected either locally as seen in
peripheral sensitization or generalized as seen in central widespread sensitization. General
sensitization can also induce allodynia, where even stimuli that normally does not induce pain, can
feel painful. This is obviously difficult to assess from structures as the pancreas but often those
chronic visceral pain conditions manifest somatic hyperalgesia and allodynia in the referred somatic
areas [15]. Although hypothetical, symptoms such as postprandial pain may be a manifestation of
allodynia [16].

Accep
ted M
anus
cript
5
In central pain processing, there is a closely regulated balance between descending excitatory and
inhibitory drives. In chronic pain patients It has been suggested that an imbalance between inhibitory
and facilitatory descending pain modulatory systems can occur, favoring increased gain of incoming
nociceptive signals [17]. Multiple studies have shown, that the balance between descending pain
inhibition and facilitation is disturbed in patients with chronic pain and it has been proposed as a
significant factor in the chronification of pain [18–21]. Similar modulatory pathways can perhaps also
play a role in “temporal summation” of pain. Temporal summation is the perception of increased pain
as a response to repetitive nociceptive stimulations with the same intensity due to a wind-up effect in
the spinal dorsal horn [22]. A train of stimuli with a frequency of 0.33 Hz or higher induces cumulative
depolarizations of the post-synaptic membrane thus resulting in wind-up of action potentials [23,24].
Temporal summation is facilitated in case of central sensitization [25]. Central sensitization can in
some cases be detected without the presence of enhanced temporal summation, which shows that
the interaction between the two is a complex interplay [26].
The function of pain processing and some of the underlying mechanisms can be characterized using
QST. QST is comprised of a variety of stimulation modalities, at specific anatomical structures and
using various evaluation methods. Well defined stimulations of skin and muscle are widely used, as
the structures are easily accessible. Modalities may include mechanical stimulation (including touch,
pinprick, and pressure) as well as thermal and electrical stimulation. In contrast, visceral QST, with
stimulation of the gastrointestinal tract or other internal organs are unpleasant to the patient due to
the invasive nature of the stimulus and more comprehensive and time consuming to conduct [27].
Rectal distension with an electric “Barostat” is occasionally used clinically in patients with e.g.,
irritable bowel syndrome, but beyond this visceral QST has mainly been limited to research settings
[28,29].
On the other hand, central pain processing is the same whether the pain is visceral or somatic of
origin. Hence, convergence between somatic and visceral nerves makes it possible to use somatic QST
as a proxy of central referred pain mechanisms in the context of visceral pain [30]. The QST protocol
we use for patients with CP is shown in figure 1. There is a static part that determines pain detection
and pain tolerance thresholds to accurately calibrated phasic and tonic stimuli, and a dynamic part
that determines the function of e.g. descending pain modulation and temporal summation. It has
been shown to be sensitive for quantifying sensory abnormalities on an individual patient level and be
predictive for outcome of management [31–33]. The dynamic parts of the QST paradigm is designed
to evaluate changes in pain perception due to descending modulation and temporal summation. The
descending modulation can be studied by the conditioned pain modulation (CPM) paradigm, where a
painful conditioning stimulus at a remote area, attenuates pain responses to a test pain stimulus.

Accep
ted M
anus
cript
6
***Figure 1 near here***
Figure 1 shows the involved pain mechanisms that can be evaluated in a QST examination for CP
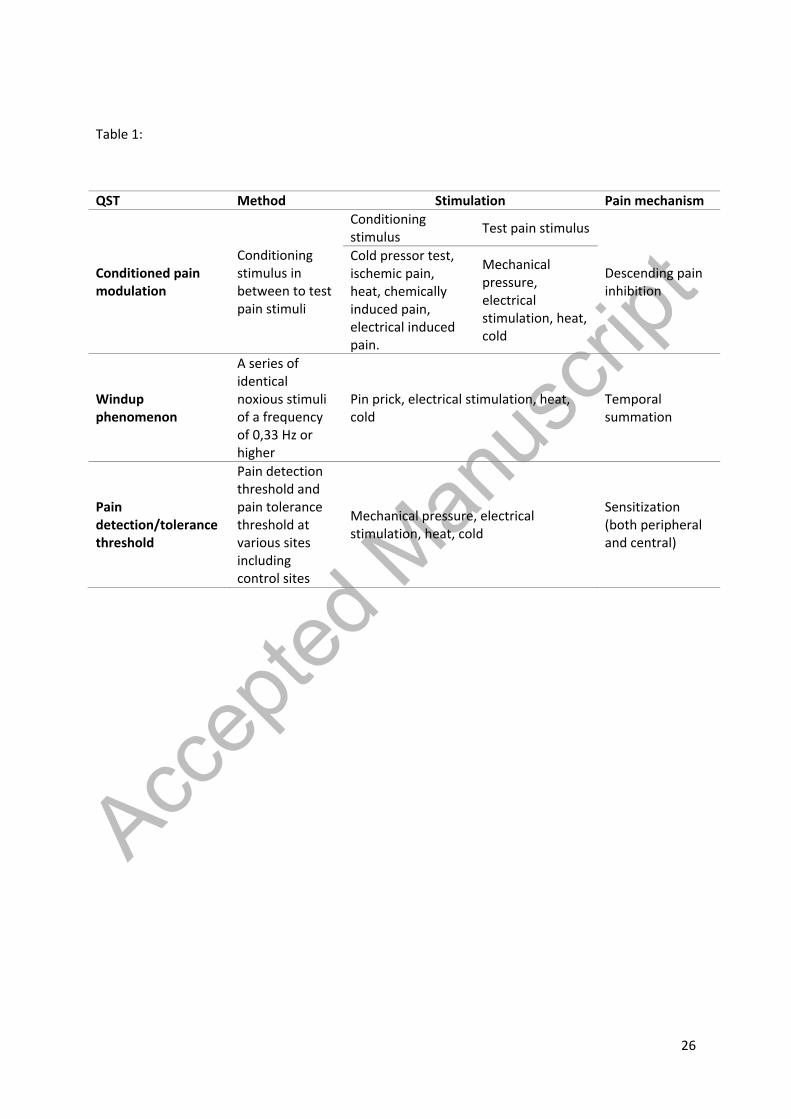
patients and Table 1 shows an overview of the different QST parameters:
- Segmental and generalized hyperalgesia. Sensitization of the nervous system can be explored
by examining pressure pain detection and tolerance thresholds and comparing data with data
from age and gender matched healthy volunteers. The pain detection threshold shall be
assessed from several anatomical locations, including the pancreatic dermatome (Th10) and
control dermatomes (e.g. C5, L4 etc.). Figure 2 shows an example of pressure pain detection
thresholds in a patient with generalized hyperalgesia compared to a healthy control
population [34].
- CPM. This can be assessed by applying a painful conditioning stimulus in between two
identical test pain stimuli and the difference in the result of the two stimuli would then be a
measure of the CPM efficacy [35]. The CPM effect can be expressed both as a relative and an
absolute value. The conditioning stimulus can for example be the cold pressor test, where the
extremity is exposed to cold water as used in our protocol. Ischemic pain, chemically induced
pain, heat, and electrical induced pain can also be used. The test pain stimuli can for example
be mechanical pressure, electrical stimulation, heat stimulation, and cold stimulation. There is
currently no consensus how CPM should be assessed and unfortunately CMP seems to vary
between repeated assessments and between patients/volunteers [36].
- Temporal summation is evoked by a series of e.g. 5 painful stimuli delivered by one per
second [37]. Temporal summation magnitude is the difference between the sensory rating of
the first and last stimuli. The stimulation can e.g. be heat, cold, mechanical pressure or
electrical stimulation. Again, there is no golden standard how to assess temporal summation.
***Table 1 and Figure 2 near here***
3. Mechanism-oriented pain treatment
3.1. Initial approach
When treating a patient with painful CP, the primary focus should be on treating the causality of pain
in a mechanistic way. Patients with CP can have several complications that cause pain, including

Accep
ted M
anus
cript
7
pancreatic pseudocysts, peptic ulcers, pancreatic cancer, duodenal stenosis, and duct obstruction due
to stones or stenosis [38,39]. Nutrition should be optimized, as high fat foods can provoke pancreatic
pain in lack of pancreatic enzymes, and patients should be advised against smoking and drinking [1].
Whether pancreatic enzymes works as an analgesic is debatable, please see the appendix in Drewes
2017 for clarification [5]. Alcohol is a risk factor for CP, and cessation of alcohol consumption has
been shown to be protective against developing pancreatic dysfunction as well as recurrent
pancreatitis [40]. Smoking is an independent risk factor for CP and more than 80% of patients with
chronic pancreatitis are smokers [41]. Tobacco can potentiate alcohol toxicity in a dose-dependent
way, and a recent study has shown that smoking cessation increases the chance of successful
outcome of pain relieving surgery [5,41,42].
Although treatment of the aforementioned complications along with successful smoking and alcohol
cessation may result in pain relief in a proportion of patients, many patients will require additional
pain management [43]. The patients could at this point possibly benefit from a mechanism-oriented
pharmacological treatment of malfunctions in the pain processing pathways. If this approach fails to
provide sufficient analgesia, a multidisciplinary approach adding alternative treatments including e.g.,
spinal cord stimulation and acupuncture may be useful.
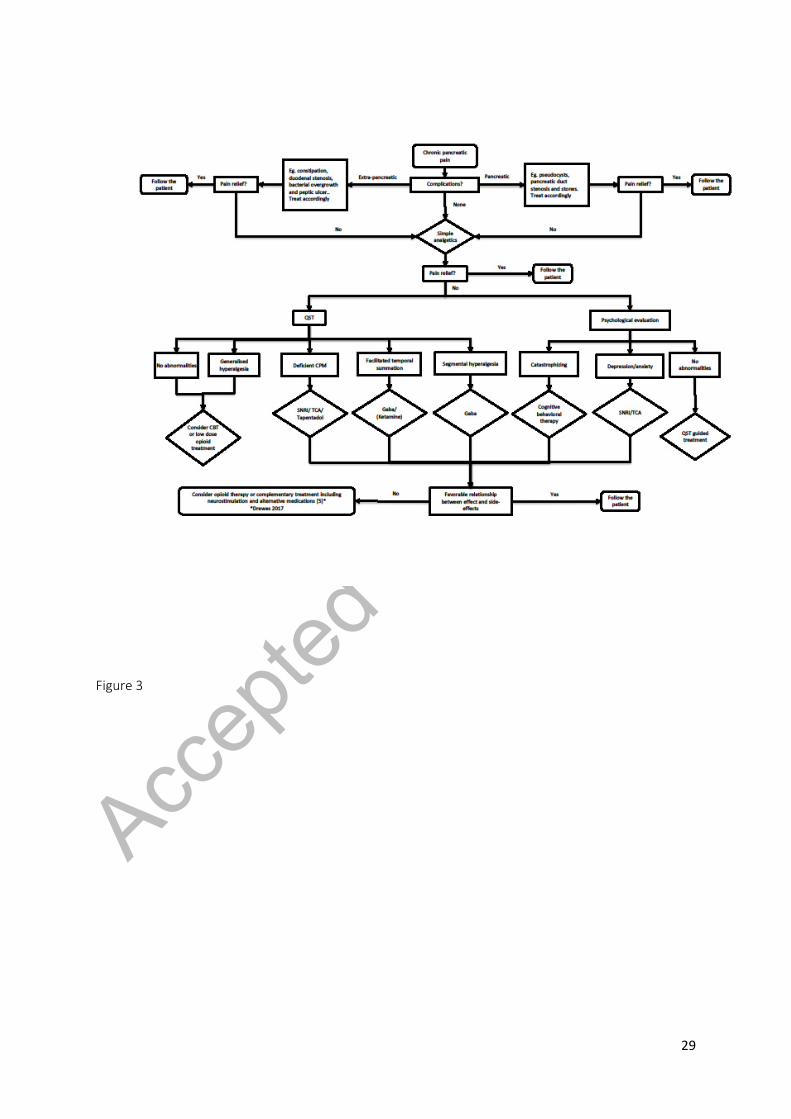
Although not validated, an example of a mechanism-based treatment algorithm is presented in figure
3, and the rationale explained below.
***Figure 3 near here***
3.2. Neurophysiological evaluation of the response to analgesics
3.2.1. Sensitization Sensitization is an important factor in the chronification of pain and a study has shown that treatment
can be guided from this feature. N-methyl-D-aspartate (NMDA)-receptor antagonists, tricyclic
antidepressant agents (TCA) and gabapentinoids have often been used to treat patients with
sensitization and their potential use in patients with chronic pancreatic pain with sensitization will be
reviewed here. Gabapentinoids is a group of anticonvulsant agents that acts by binding to the α2-δ
unit of voltage-gated calcium channels in the central neural pathways, whereby it reduces the release
of excitatory neurotransmitters [44]. The group includes gabapentin and pregabalin.
Olesen et al. examined the effect of pregabalin on painful CP and found that lower electrical pain
detection threshold at the abdominal pancreatic area had high accuracy to separate responders from

Accep
ted M
anus
cript
8
non-responders [45]. The classification accuracy was 80.6%, which is significantly above the effect of
random selection. From the same study, Bouwense et al. reported that treatment with pregabalin in
patients with CP leads to a greater increase in electrical pain thresholds at the C5 dermatome than
placebo treatment, but not in the pancreatic “viscerotome” (dorsal and ventral T10 dermatome)
suggesting a possible prevention on spreading hyperalgesia to higher segmental levels [46].
Pregabalin did also increase pain detection threshold and tolerance threshold in rectal stimulation
[47]. This supports findings in previous studies where pregabalin reduces visceral hyperalgesia
[48,49].
Many studies have examined the effect of ketamine on different types of pain. Ketamine is a NMDA
receptor antagonist and thereby a possible treatment modality for central sensitization. Arendt-
Nielsen et al. likewise found that ketamine increased the temporal summation pain threshold in
healthy volunteers compared to placebo [50]. Several studies have tried to unravel the long-term
effect of different administration forms of ketamine in patients suffering from various pain
syndromes, but with inconsistent results. Currently large studies are ongoing in Australia examining
the effects of low dose ketamine in managing and preventing chronic pain [51]. Generally the studies
showed that infusion of ketamine provides immediate analgesic effect, which attenuates over time
[52,53]. In CP, Bouwense et al. found that ketamine increases the sum of pressure pain detection
threshold immediately after infusion, but the effect was not significant one hour after infusion [54].
TCA probably relieves pain through multimodal mechanisms of action, involving inhibition of
serotonin and noradrenaline reuptake, blockage of sodium channels as well as blocking of the NMDA-
receptors. The effects on pain thresholds are therefore likely to attenuate central sensitization.
Enggaard et al. examined TCA’s effect on QST results in healthy volunteers and found that it increased
pressure pain thresholds and decreased temporal summation [55]. It has also been examined in
patients with painful polyneuropathy by Holbech et al., who found that pain intensity decreased
significantly and that it improved both temporal summation by mechanical repetitive stimuli as well
as cold pain [56]. TCA has not been tested in CP patients, but as the affected pain mechanisms of CP
resembles those affected by other chronic pain conditions, it makes sense to use findings in other
conditions when knowledge is limited in CP.
3.2.2. Impaired descending pain inhibition
Descending pain inhibition is an important pain processing pathway and it is decreased in many
chronic pain conditions [57]. Olesen et al. has shown that conditioned pain modulation function is
reduced in patients with painful CP compared to healthy volunteers [21]. Treatment targeted this pain

Accep
ted M
anus
cript
9
mechanisms could therefore be used to manage pancreatic pain. Bouwense et al. has looked at the
effect of pregabalin compared with placebo on CPM function in CP patients [58]. They found that
pregabalin responders (patients with a decrease of more than 30% on their average daily pain
intensity after 3 weeks of pregabalin treatment) had a significant increase in CPM function from
baseline measures. This indicates that pregabalin could have a direct anti-facilitatory effect on CPM or
an indirect reduction of the transmission in the ascending pain pathways [58]. Even though this was a
posthoc analysis, data are supported from preclinical studies, studies in healthy volunteers and from
different patient categories than CP. From these studies, we can be inspired to find other treatments
that could be helpful when treating pancreatic pain. As serotonin, noradrenaline, and opioid peptides
are important neurotransmitters in relation to descending pain inhibition, many studies have
examined the effect of serotonin and norepinephrine reuptake inhibitors (SNRI), TCA, and opioids in
relation to defective pain modulation.
In two rat studies, TCA and low dose of opioids, respectively, have been shown to enhance
endogenous pain modulation [59,60]. The noradrenergic system plays an important role in
endogenous pain modulation and is influenced by TCA [60]. The authors of the opioid study
speculated that opioids target supraspinal structures such as the periaqueductal grey and
ventromedial medulla and thereby enhance descending modulation [59]. In humans, studies of
opioids effects on descending modulation are more contradictory [61–64]. Olesen et al. examined
morphine’s effect on different experimental pain models in healthy volunteers and did not find that
morphine increased CPM effect [65]. Arendt-Nielsen et al. found that buprenorphine and fentanyl
both increased the CPM function significantly compared to placebo in healthy volunteers after 72
hours treatment [63]. Hermans et al. found that morphine had no effect on CPM in patients with
either rheumatoid arthritis or fibromyalgia combined with evidence of central sensitization [66].
However, the study only consisted of a single subcutaneous injection of morphine and does not tell us
anything about the long-term effects of morphine treatment. Tapentadol, which has a dual effect on
opioid receptors and noradrenalin reuptake, was examined in healthy volunteers in two studies, and it
was shown not to affect CPM or experimentally induced heat, cold or mechanical hyperalgesia
[61,64]. On the other hand, in patients with diabetic neuropathy where the pain system had
undergone pathological changes, Niesters et al. found that tapentadol increased CPM function after 4
weeks of treatment [62]. Tapentadol both activates the µ-opioid receptor as well as inhibits neuronal
norepinephrine reuptake. It therefore makes sense also to examine whether norepinephrine reuptake
inhibitors also increase CPM function in patients. This has been done by Yarnitsky et al. who has found
that CPM function predicts treatment effect of duloxetine [67].

Accep
ted M
anus
cript
10
3.2.3. Temporal summation
Chronic pain can induce enhanced temporal summation. The NMDA receptor is also believed to play a
key role in this mechanism through a release of both peptides and glutamate that may lead to hyper
excitability of spinothalamic tract neurons, and this characterizes central sensitization [23,68].
Different opioids have shown to inhibit temporal summation, including codeine and morphine
[55,69]. Anticonvulsants drugs including the gabapentinoids and carbamazepine have also been
shown to inhibit temporal summation in healthy subjects [50,70,71]. Amitriptyline (TCA) is thought to
block the NMDA receptor, and this would theoretically lead to a decrease in temporal summation.
However, a study surprisingly found that it significantly increased temporal summation, and
speculates that this probably is caused by amitriptyline’s augmentation of glutamate release, making
it less effective as a NMDA receptor antagonist [70].
As expected from a pathophysiological level, other NMDA antagonists are found to inhibit temporal
summation. Ketamine was found to decrease temporal summation in healthy volunteers [50]. In
another study, the effect of dextromethorphan on temporal summation was examined in patients
suffering from abdominal pain due to irritable bowel syndrome. Here, the drug decreased temporal
summation from a pathological baseline limit [72]. The authors did not report the effect of
dextromethorphan on the patients’ pain ratings, and one could question whether the effect was
clinically relevant.
3.2.4. Choosing analgesic treatment
In the clinic, many patients with severe pain will have several malfunctions when evaluating their QST
results. An example from our clinic is that there is a substantial overlap of between abnormal
mechanisms in 56% of the patients. This can be a challenge, because several treatments could
potentially be beneficial. In preliminary results from our pain clinic, 57% of the patients have deficient
CPM, 52% have segmental hyperalgesia and 43% of the patients have temporal summation. In this
case, we suggest to treat the most deviant mechanism first and if management is insufficient to retest
the algorithm on treatment and add supplementary therapies depending on the new findings.
However, the algorithm can also be a supportive tool in the management strategy, and treatment
shall always be based on the patients and doctors’ preferences, side-effect profile etc.
If the results of the QST examination is conflicting, it may make sense to use a more traditional
analgesic strategy instead of the QST-guided regimen.

Accep
ted M
anus
cript
11
We are not able to say whether this method for choosing analgesic treatment is more cost-effective
than using the WHO treatment ladder, as this will require a large number of treatments in different
combinations. However, analgesic treatment costs only represent less than 1% of the socio-economic
burden of chronic pain, and a small increase or decrease of the treatment cost will therefore seldomly
have a significant impact on the total chronic pain related cost [73].
3.3. Psychological evaluation
The patients’ mental condition has great influence on the experience of pain as well as treatment
outcomes [74]. Pain catastrophizing is when a patient describes the pain in exaggerated terms,
ruminates on it and feels increasingly helpless about the situation. Several studies have shown that
high pain catastrophizing predicts poorer treatment outcome in various chronic pain conditions
[75,76]. At this stage, it is not clear which of the individual factors composing the manifestation of
catastrophizing can be modulated by analgesics and e.g., cognitive therapy.
Depression is closely related to pain symptoms and studies have found that it has a significant
influence on quality of life in patients with chronic pain [77]. The response to pharmacological pain
treatment is also closely linked to the presence of depression [78,79]. Psychological interventions as
mindfulness and cognitive-behavioral therapy can have a positive influence on catastrophizing scores
and can raise the patients’ quality of life [80–82]. Pharmacological antidepressant treatments can also
decrease pain intensity, however it is not clear whether this effect is based on the improvement of
mental health rather than direct effects on malfunctioning pain system as discussed above [83].
3.4. Complementary treatment
Besides pharmacological management, many other pain treatment modalities exist. Acupuncture has
been used to alleviate pain for many years, although the scientific results have been inconsistent [84–
86]. It is difficult to examine the result of acupuncture in a randomized placebo-controlled study, as
sham treatments are difficult to control. Hence, sham acupuncture needles are only different from
actual acupuncture needles by the fact that it does not puncture the skin, but the tactile stimulation is
still present and can possibly induce some of the effects from acupuncture [87]. However, in a sham
controlled study examining the analgesic effect of acupuncture on painful CP, an effect on pain
intensity was seen, although a short lasting response limits its clinical use [84].

Accep
ted M
anus
cript
12
Different kinds of neuromodulation have also been evaluated. Spinal cord stimulation has shown
promising results in different types of chronic pain including CP [88,89], but as numbness during
stimulation can be felt, no real sham controlled studies have been done. The mechanisms involved
remains unclear, but several effects including blockage of nerve conduction, release of inhibitory
neuromodulators, and direct stimulation on the dorsal column pathways has been proposed [89]. A
study examining transcranial magnetic stimulation on the secondary somatosensory cortex in patients
with CP found a significant decline in pain ratings after treatment. The effect was correlated to an
increase in glutamate and N-acetyl-aspartate levels in the somatosensory cortices, and low baseline
glutamate level was predictive for a positive treatment outcome [90]. This field is current under
development as high frequency stimulators have been proposed to exert more specific effects [91].
Vagal nerve stimulation has been examined for a number of conditions with chronic visceral pain
often with good results [92,93]. No clearly defined mechanism has been discovered, but theories
point to anti-inflammatory effects in conjunction with pain modulatory effects [94]. Juel et al. has
examined acute accentuation of vagal tone on the auricular vagal branch in CP patients, but found no
effect on experimental pain thresholds. [95]. This may relate to the short stimulation period, as only
60 minutes stimulation was performed. It could also be because the analgesic potency of this type of
stimulation was not powerful enough to induce measurable effects. Further studies are needed
where longer stimulation periods with multiple stimulations are performed, and perhaps the
stimulation should be in closer relation to the vagal nerve than just the auricular branch.
At this level, complementary treatment cannot be used in a mechanism-based treatment, as the
effect on different pain processing mechanisms is unknown and the effects are not validated.
3.5. Gastrointestinal side effects of analgesics
No treatment with analgesics is without risks, and due to changes in exocrine pancreatic function,
motility and gut blood supply, patients with CP are especially prone to develop gastrointestinal
adverse effects, for details see [96]. Acetaminophen is generally well tolerated, but its treatment
carries a risk of acute liver failure if the instructed dosages are not complied. Furthermore long term
use of acetaminophen may cause medicine overuse headache [97]. NSAIDs is linked to non-variceal
upper gastrointestinal bleeding, lower gastrointestinal bleeding, gut perforation and strictures, and
protein-losing enteropathy. There are several risk factors for developing gastrointestinal side effects
to NSAID use including high age, female sex, smoking, former peptic ulcer, and concurrent use of
steroids, anticoagulants, or selective serotonin reuptake inhibitors, and in these cases NSAID
treatment should be avoided [98]. The risk of peptic ulcer seems to be especially high in CP patients

Accep
ted M
anus
cript
13
likely due to compromised blood supply of the stomach after attacks of acute pancreatitis as well as
increased infection rates with H Pylory [99].
Opioids, both weak and strong, have a potent pain-relieving effect, but are associated with addiction
and major side effects where the gastrointestinal includes anorexia, nausea, vomiting and
constipation [100,101]. In this context, new drugs such as peripherally-acting µ-opioid receptor
antagonists have proven valuable in the treatment of opioid-induced bowel dysfunction as they block
the µ-opioid receptors locally in the gut without affecting the central analgesic effect. [102].
Although long-term use of opioids may be necessary in CP and can provide potent pain control, it can
also lead to opioid-induced hyperalgesia, which is characterized by a paradox of increased perception
of pain, without a progression in the disease or opioid withdrawal [103].
Adjuvant analgesics such as antidepressants and anticonvulsants, are all known for having a long list
of potential gastrointestinal side effects, some more severe than others. These include abdominal
pain, diarrhea, constipation, dyspepsia, nausea, vomiting, hepatitis, and liver failure. They should
typically be started in a low dose and titrated up, to find an optimal balance between beneficial
effects and side effects [104]. Therefore, the risks of potential side effects shall be considered in the
algorithm proposed in figure 3, and often this limits dosing and optimized pain control.
4. Conclusion
Pain management in CP remains a significant challenge, and no single analgesic is the best match for
every patient. To achieve optimal pain management, quantitative assessment of affected pain
mechanisms provides a platform for individualized and multimodal pain management, which may lead
to a more rational and precise use of analgesics. This is exemplified in figure 3 where a suggestion for
a mechanism-based approach for pain management is illustrated. The flowchart is based on proposed
mechanisms mainly found in basic cross-sectional studies. Although different treatment studies in CP
and other painful conditions have confirmed the value of the different steps in the algorithm, it needs
to be tested further as an overall concept in clinical practice, preferably in a randomized fashion. This
will hopefully give data from mechanistic clinical studies, where analgesic treatments are supported
by the different steps in the algorithm, allowing us to adapt it to the results. Other features such as
genetic profiling, drug kinetics and assessment of the autonomic response may be important as well,
but the suggested tests can be used bedside with minimal costs."

Accep
ted M
anus
cript
14
5. Expert opinion
Pain treatment in CP has for years mainly been based on knowledge of somatic pain. Although the
mechanisms (and hence the effect of analgesics) are to some degree similar, there are also
differences. An increasing number of mechanism-based studies on pain treatment has elucidated a
number of relevant mechanisms that can be treated with specific analgesics. Our knowledge is
expanding, but there are still significant gaps that need to be explored. Quantitative sensory testing
can be used to characterize pain processing and delineate relevant pain mechanisms on an individual
patient basis, but many QST protocols used in clinical studies are quite extensive and not easily
transferred to the clinic. There is therefore a need for a simple and feasible QST examination protocol
with appropriate reference material. The QST protocol should include examinations providing
information on the presence of segmental and widespread hyperalgesia, temporal summation
(serving as proxies for central sensitization), and conditioned pain modulation as shown in figure 1.
From this protocol, the ultimate goal is to develop a treatment algorithm such as proposed in figure 3,
which will enable us to tailor the best-suited treatment plan for each patient. Before this is possible,
additional drugs should be examined to establish their effect on pain mechanisms. This could include
more studies on e.g. ketamine, cannabinoids, opioids, and antidepressant agents.
Treatment of psychological factors and the accompanying effect on pain ratings and quality-of-life
should be documented. This could include both pharmacological management as well as cognitive
behavioral therapy and help to evoke beneficial coping mechanisms. Quality of life is equally as
important as pain ratings when assessing effect of treatment, as complete pain relief often is a too
ambitious goal, and quality of life can be enhanced despite minor changes in pain intensity.
6. Article highlights
• Although pain is the most frequent symptom in chronic pancreatitis, pain management is an
ongoing challenge
• In chronic pain conditions, several pain mechanisms can be affected, and pain can therefore
affect each individual differently
• Although still in a developmental phase, somatic quantitative sensory testing can be used to
diagnose visceral pain mechanisms
• Many analgesics are targeting specific pain mechanisms
• By individualizing pain treatment, pain control is achieved faster, and the patient need not
test several analgesics, each accompanied by different side effects

Accep
ted M
anus
cript
15
• There are several psychological factors that influence pain perception, and these should be
treated alongside the pain to achieve sufficient pain control
Funding
This paper has been solely funded by the salary of the involved author from North Denmark Regional
Hospital and Aalborg University.
Declaration of Interest
The authors have no relevant affiliations or financial involvement with any organization or entity with
a financial interest in or financial conflict with the subject matter or materials discussed in the
manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert
testimony, grants or patents received or pending, or royalties.
Reviewer Disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.

Accep
ted M
anus
cript
16
References Papers of special note have been highlighted as:
* of interest
** of considerable interest
[1] Lieb JG, Forsmark CE. Review article: pain and chronic pancreatitis. Aliment. Pharmacol. Ther.
[Internet].29:706–19 (2009).
[2] Ammann RW, Buehler H, Muench R, Freiburghaus AW, Siegenthaler W. Differences in the
natural history of idiopathic (Nonalcoholic) and alcoholic chronic pancreatitis. a comparative
long-term study of 287 patients. Pancreas.2:368–77 (1987).
[3] Mullady DK, Yadav D, Amann ST, et al. Type of pain, pain-associated complications, quality of
life, disability and resource utilisation in chronic pancreatitis: A prospective cohort study. Gut.
60:77–84 (2011).
[4] Lankisch PG, Löhr-Happe A, Otto J CW. Natural course in chronic pancreatitis. Pain, exocrine
and endocrine pancreatic insufficiency and prognosis of the disease. Digestion. 54:148–55
(1993).
[5] Drewes AM, Bouwense SAW, Campbell CM, et al. Guidelines for the understanding and
management of pain in chronic pancreatitis. Pancreatology [Internet]. 17:720–31 (2017).*
* A comprehensive review of the newest research in pancreatic pain
[6] Arendt-Nielsen L, Morlion B, Perrot S, et al. Assessment and manifestation of central
sensitisation across different chronic pain conditions. Eur. J. Pain (United Kingdom). 22:216–41
(2018).
[7] Olesen AE, Farmer AD, Olesen SS, Aziz Q, Drewes AM. Management of chronic visceral pain.
Pain Manag. [Internet]. 6:469–86 (2016).
[8] Vargas-Schaffer G. Is the WHO analgesic ladder still valid? Twenty-four years of experience.
Can. Fam. Physician. 56:514–7 (2010)
[9] Cervero F. Visceral versus somatic pain: Similarities and differences. Dig. Dis. 27:3–10 (2010).
[10] Gebhart GF. Visceral pain — peripheral sensitisation. Gut. 47:54–5 (2000).
[11] Majumder S, Chari ST. Chronic pancreatitis. Lancet. 387:1957–66 (2016).
[12] Kosek E, Cohen M, Baron R, et al. Do we need a third mechanistic descriptor for chronic pain

Accep
ted M
anus
cript
17
states? Pain. 157:1382–6 (2016).
[13] Talukdar R, Reddy DN. Pain in chronic pancreatitis: managing beyond the pancreatic duct.
World J. Gastroenterol. 19:6319–28 (2013).
[14] Olesen S, Krauss T, Demir I, et al. Towards a Neurobiological Understanding of Pain in Chronic
Pancreatitis: Mechanisms and Implications for Treatment. Pain Reports. 0:1–9 (2017).*
* A review concerning the neurobiological mechanisms of pancreatic pain
[15] Jarrell J, Malekzadeh L, Yang H, Arendt-Nielsen L. The Significance of Cutaneous Allodynia in a
Woman With Chronic Pelvic Pain. J. Obstet. Gynaecol. Canada. 37:628–32 (2015).
[16] Drewes AM, Krarup AL, Detlefsen S, Malmstrøm M-L, Dimcevski G, Funch-Jensen P. Pain in
chronic pancreatitis: the role of neuropathic pain mechanisms. Gut. 57:1616–27 (2008).
[17] Ossipov MH, Morimura K, Porreca F. Descending pain modulation and chronification of pain.
Curr. Opin. Support. Palliat. Care. 8:143–151 (2014).*
* A comprehensive article concerning descending pain modulation
[18] Albu S, Gomez-Soriano J, Avila-Martin G, Taylor J. Deficient conditioned pain modulation after
spinal cord injury correlates with clinical spontaneous pain measures. Pain. 156:260–72
(2015).
[19] Wilder-Smith CH, Li X, Shen L, Cao Y, Ho KY, Wong RK. Dysfunctional endogenous pain
modulation in patients with functional dyspepsia. Neurogastroenterol. Motil. 26:489–98
(2014).
[20] King CD, Wong F, Currie T, Mauderli AP, Fillingim RB, Riley 3rd JL. Deficiency in endogenous
modulation of prolonged heat pain in patients with irritable bowel syndrome and
temporomandibular disorder. Pain. 143:172–8 (2009).
[21] Olesen SS, Brock C, Krarup AL, et al. Descending inhibitory pain modulation is impaired in
patients with chronic pancreatitis. Clin. Gastroenterol. Hepatol. 8:724–730 (2010).*
* An article showing how patients with CP differ from healthy controls by having deficient pain
modulation
[22] Cervero F, Wolstencroft the late JH. A positive feedback loop between spinal cord nociceptive
pathways and antinociceptive areas of the cat’s brain stem. Pain. 20:125–38 (1984).
[23] Eide PK. Wind-up and the NMDA receptor complex from a clinical perspective. Eur. J. Pain.
4:5–15 (2000).

Accep
ted M
anus
cript
18
[24] Price DD, Hu JW, Dubner R, Gracely RH. Peripheral suppression of first pain and central
summation of second pain evoked by noxious heat pulses. Pain. Feb, 3.:57–68 (1977).
[25] Herrero JF, Laird JMA, Lopez-Garcia JA. Wind-up of spinal cord neurones and pain sensation:
Much ado about something? Prog. Neurobiol. 61:169–203 (2000).
[26] Laird JM, de la Rubia PG, Cervero F. Excitability changes of somatic and viscero-somatic
nociceptive reflexes in the decerebrate-spinal rabbit: role of NMDA receptors. J. Physiol.
489:545–55 (1995).
[27] Brock C, Arendt-Nielsen L, Wilder-Smith O, Drewes AM. Sensory testing of the human
gastrointestinal tract. World J. Gastroenterol. 15:151–9 (2009).
[28] Staahl C, Drewes AM. Experimental human pain models: A review of standardised methods for
preclinical testing of analgesics. Basic Clin. Pharmacol. Toxicol. 95:97–111 (2004).
[29] Bouin M, Plourde V, Boivin M, et al. Rectal distention testing in patients with irritable bowel
syndrome: Sensitivity, specificity, and predictive values of pain sensory thresholds.
Gastroenterology. 122:1771–7 (2002).
[30] O’Neill S, O’Neill L. Improving QST reliability - More raters, tests, or occasions? A multivariate
generalizability study. J. Pain [Internet]. 16:454–62 (2015).
[31] Rolke R, Baron R, Maier C, et al. Quantitative sensory testing in the German Research Network
on Neuropathic Pain (DFNS): standardized protocol and reference values. Pain. 123:231–43
(2006).
[32] Petersen KK, Graven-Nielsen T, Simonsen O, Laursen MB, Arendt-Nielsen L. Preoperative pain
mechanisms assessed by cuff algometry are associated with chronic postoperative pain relief
after total knee replacement. Pain. 157:1400–6 (2016).
[33] Petersen KK, Arendt-Nielsen L, Simonsen O, Wilder-Smith O, Laursen MB. Presurgical
assessment of temporal summation of pain predicts the development of chronic postoperative
pain 12 months after total knee replacement. Pain. 156:55–61 (2015).
[34] Kuhlmann L, Olesen SS, Grønlund D, Olesen AE, Asbjørn Mohr Drewes. Sensory testing can
objectify pain in chronic pancreatitis. Pancreatology. 18:S110–1 (2018).
[35] Olesen AE, Andresen T, Staahl C, Drewes AM. Human Experimental Pain Models for Assessing
the Therapeutic Efficacy of Analgesic Drugs. Pharmacol. Rev. 64:722–79 (2012).
[36] Vaegter HB, Petersen KK, Mørch CD, Imai Y, Arendt-Nielsen L. Assessment of CPM reliability:

Accep
ted M
anus
cript
19
Quantification of the within-subject reliability of 10 different protocols. Scand. J. Pain. 18: 729-
37 (2018)
[37] Edwards RR, Ness TJ, Weigent DA, Fillingim RB. Individual differences in diffuse noxious
inhibitory controls (DNIC): Association with clinical variables. Pain. 106:427–37 (2003).
[38] Howell DA. Pancreatic stones: Treat or ignore? Can. J. Gastroenterol. 13:461–5 (1999).
[39] Puylaert M, Kapural L, Van Zundert J, et al. Pain in chronic pancreatitis. Pain Pract. 11:492–505
(2011).
[40] Samokhvalov A V., Rehm J, Roerecke M. Alcohol Consumption as a Risk Factor for Acute and
Chronic Pancreatitis: A Systematic Review and a Series of Meta-analyses. EBioMedicine.
2:1996–2002 (2015).
[41] Edderkaoui M, Thrower E. Smoking and Pancreatic Disease. J. Cancer Ther. 4:34–40 (2013).
[42] Bordaçahar B, Couvelard A, Vullierme MP, et al. Predicting the efficacy of surgery for pain
relief in patients with alcoholic chronic pancreatitis. Surg. (United States). 164:1064–70 (2018).
[43] Andrén-Sandberg A, Hoem D, Gislason H. Pain management in chronic pancreatitis. Eur. J.
Gastroenterol. Hepatol. 14:957–70 (2002).
[44] Giladi H, Choinière M, Fitzcharles M-A, Ware MA, Tan X, Shir Y. Pregabalin for chronic pain:
does one medication fit all? Curr. Med. Res. Opin. [Internet]. 31:1403–11 (2015).
[45] Olesen SS, Graversen C, Bouwense SAW, van Goor H, Wilder-Smith OHG, Drewes AM.
Quantitative sensory testing predicts pregabalin efficacy in painful chronic pancreatitis. PLoS
One [Internet]. 8:e57963 (2013). Available from:
http://www.ncbi.nlm.nih.gov/pubmed/23469256.**
** A study that proved that QST can predict the efficacy of pregabalin in CP patients
[46] Bouwense S a. W, Olesen SS, Drewes AM, Poley J-W, van Goor H, Wilder-Smith OHG. Effects of
pregabalin on central sensitization in patients with chronic pancreatitis in a randomized,
controlled trial. PLoS One [Internet]. 7:e42096 (2012). Available from:
http://www.ncbi.nlm.nih.gov/pubmed/22879908.
[47] Olesen SS, Graversen C, Olesen AE, et al. Randomised clinical trial: Pregabalin attenuates
experimental visceral pain through sub-cortical mechanisms in patients with painful chronic
pancreatitis. Aliment. Pharmacol. Ther. 34:878–87 (2011).
[48] Houghton LA, Fell C, Whorwell PJ, Jones I, Sudworth DP, Gale JD. Effect of a second-generation

Accep
ted M
anus
cript
20
α2δ ligand (pregabalin) on visceral sensation in hypersensitive patients with irritable bowel
syndrome. Gut. 56:1218–25 (2007).
[49] Million M, Wang L, Adelson DW, Roman F, Diop L, Taché Y. Pregabalin decreases visceral pain
and prevents spinal neuronal activation in rats [15]. Gut. 56:1482–4 (2007).
[50] Arendt-Nielsen L, Mansikka H, Staahl C, et al. A translational study of the effects of ketamine
and pregabalin on temporal summation of experimental Pain. Reg. Anesth. Pain Med. 36:585–
91 (2011).
[51] Schug SA, Peyton P. Does perioperative ketamine have a role in the prevention of chronic
postsurgical pain: the ROCKet trial. Br. J. Pain. 11:166–8 (2017).
[52] Dahan A, Olofsen E, Sigtermans M, et al. Population pharmacokinetic-pharmacodynamic
modeling of ketamine-induced pain relief of chronic pain. Eur. J. Pain. 15:258–67 (2011).
[53] Zekry O, Gibson SB, Aggarwal A. Subanesthetic, Subcutaneous Ketamine Infusion Therapy in
the Treatment of Chronic Nonmalignant Pain. J. Pain Palliat. Care Pharmacother. 30:91–8
(2016).
[54] Bouwense SAW, Buscher HCJL, van Goor H, Wilder-Smith OHG. S-ketamine modulates
hyperalgesia in patients with chronic pancreatitis pain. Reg. Anesth. Pain Med. [Internet].
36:303–7 (2011).
[55] Enggaard TP, Poulsen L, Arendt-Nielsen L, et al. The analgesic effect of codeine as compared to
imipramine in different human experimental pain models. Pain. 92:277–82 (2001).
[56] Holbech J V., Bach FW, Finnerup NB, Brøsen K, Jensen TS, Sindrup SH. Imipramine and
pregabalin combination for painful polyneuropathy: A randomized controlled trial. Pain.
156:958–66 (2015).
[57] Yarnitsky D. Role of endogenous pain modulation in chronic pain mechanisms and treatment.
Pain. 156:24–31 (2015).
[58] Bouwense SA, Olesen SS, Drewes AM, van Goor H, Wilder-Smith OH. Pregabalin and placebo
responders show different effects on central pain processing in chronic pancreatitis patients. J.
Pain Res. 8:375–86 (2015).
[59] Bars D Le, Chitour D, Kraus E, Clot AM, Dickenson AH, Besson JM. The effect of systemic
morphine upon diffuse noxious inhibitory controls (DNIC) in the rat: evidence for a lifting of
certain descending inhibitory controls of dorsal horn convergent neurones. Brain Res.

Accep
ted M
anus
cript
21
215:257–74 (1981).
[60] Matsuoka H, Suto T, Saito S, Obata H. Amitriptyline, but Not Pregabalin, Reverses the
Attenuation of Noxious Stimulus-Induced Analgesia after Nerve Injury in Rats. Anesth. Analg.
123:504–10 (2016).
[61] Martini C, Velzen M Van, Aarts L, Dahan A. A Randomized Controlled Trial on the Effect of
Tapentadol and Morphine on Conditioned Pain Modulation in Healthy Volunteers. 10:1–12
(2015).
[62] Niesters M, Proto PL, Aarts L, Sarton EY, Drewes AM, Dahan A. Tapentadol potentiates
descending pain inhibition in chronic pain patients with diabetic polyneuropathy. Br. J.
Anaesth. [Internet]. 113:148–56 (2014).
[63] Arendt-Nielsen L, Andresen T, Malver LP, Oksche A, Mansikka H, Drewes AM. A Double-blind,
Placebo-controlled Study on the Effect of Buprenorphine and Fentanyl on Descending Pain
Modulation. Clin. J. Pain. 28:623–7 (2012).
[64] Förster M, Helfert S, Dierschke R, et al. Evaluation of the antihyperalgesic effect of tapentadol
in two human evoked pain models – the TapCapMentho pilot trial. Expert Opin.
Pharmacother.. 17:1717–25 (2016).
[65] Olesen AE, Brock C, Sverrisdóttir E, Larsen IM, Drewes AM. Sensitivity of quantitative sensory
models to morphine analgesia in humans. 717–26 (2014).
[66] Hermans L, Nijs J, Calders P, et al. Influence of Morphine and Naloxone on Pain Modulation in
Rheumatoid Arthritis, Chronic Fatigue Syndrome/Fibromyalgia, and Controls: A Double-Blind,
Randomized, Placebo-Controlled, Cross-Over Study. Pain Pract. 18:418–30 (2018).
[67] Yarnitsky D, Granot M, Nahman-Averbuch H, Khamaisi M, Granovsky Y. Conditioned pain
modulation predicts duloxetine efficacy in painful diabetic neuropathy. Pain. 153:1193–8
(2012).*
* A study that proves that QST can predict the efficacy of duloxetine in diabetic neuropathy
[68] Willis W, Sluka K, Rees H, Westlund K. Cooperative mechanisms of neurotransmitter action in
central nervous sensitization. Prog. Brain Res. 110:151–66 (1996).
[69] Guan Y, Berzan J, Meyer RA, Raja SN. Windup in Dorsal Horn Neurons Is Modulated by
Endogenous Spinal -Opioid Mechanisms. J. Neurosci. 26:4298–307 (2006).
[70] Harding LM, Kristensen JD, Baranowski AP. Differential effects of neuropathic analgesics on

Accep
ted M
anus
cript
22
wind-up-like pain and somatosensory function in healthy volunteers. Clin. J. Pain. 21:127–32
(2005).
[71] Arendt-Nielsen L, Frøkjær JB, Staahl C, et al. Effects of Gabapentin on Experimental Somatic
Pain and Temporal Summation. Reg. Anesth. Pain Med. 32:382–8 (2007).
[72] Zhou Q, Price DD, Callam CS, Woodruff MA, Verne GN. Effects of the N-methyl-D-aspartate
Receptor on Temporal Summation of Second Pain (Wind-up) in Irritable Bowel Syndrome. J
Pain. 12:297–303 (2011).
[73] Gustavsson A, Bjorkman J, Ljungcrantz C, et al. Socio-economic burden of patients with a
diagnosis related to chronic pain - Register data of 840,000 Swedish patients. Eur. J. Pain.
16:289–99 (2012).
[74] Kristiansen FL, Olesen AE, Brock C, et al. The role of pain catastrophizing in experimental pain
perception. Pain Pract. 14:E136-45 (2014).
[75] Wertli MM, Burgstaller JM, Weiser S, Steurer J, Kofmehl R, Held U. Influence of catastrophizing
on treatment outcome in patients with nonspecific low back Pain: A systematic review. Spine.
39:263–73 (2014).
[76] Mankovsky T, Lynch M, Clark A, Sawynok J, Sullivan MJ. Pain catastrophizing predicts poor
response to topical analgesics in patients with neuropathic pain. Pain Res. Manag. 17:10–4
(2012).
[77] Orenius T, Koskela T, Koho P, et al. Anxiety and depression are independent predictors of
quality of life of patients with chronic musculoskeletal pain. J. Health Psychol. 18:167–75
(2013).
[78] Fishbain D, Cole B, Lewis J, Gao J. Does pain interfere with antidepressant depression
treatment response and remission in patients with depression and pain? An evidence-based
structured review. Pain Med. 15:1522–39 (2014).
[79] Bair MJ, Robinson RL, Katon W, Kroenke K. Depression and Pain Comorbidity. Arch. Intern.
Med. 163:2433 (2003).
[80] Buhrman M, Nilsson-Ihrfelt E, Jannert M, Ström L, Andersson G. Guided internet-based
cognitive behavioural treatment for chronic back pain reduces pain catastrophizing: A
randomized controlled trial. J. Rehabil. Med. 43:500–5 (2011).

Accep
ted M
anus
cript
23
[81] Davis M, Zautra A, Wolf L, Tennen H, Yeung E. Mindfulness and Cognitive-Behavioral
Interventions for Chronic Pain: Differential Effects on Daily Pain Reactivity and Stress
Reactivity. J Consult Clin Psychol. 83:24–35 (2015).
[82] Teh C, Zaslavsky A. Effect of depression treatment on chronic pain outcomes. Psychosom.
Med. 72:61–7 (2010).
[83] Dharmshaktu P, Tayal V, Kalra BS. Efficacy of antidepressants as analgesics: A review. J. Clin.
Pharmacol. 52:6–17 (2012).
[84] Juel J, Liguori S, Liguori A, et al. Acupuncture for pain in chronic pancreatitis: A single-blinded
randomized crossover trial. Pancreas. 46:170–6 (2017).
[85] Tobbackx Y, Meeus M, Wauters L, et al. Does acupuncture activate endogenous analgesia in
chronic whiplash-associated disorders? A randomized crossover trial. Eur. J. Pain. 17:279–89
(2013).
[86] Zheng Z, Feng SJQ, Costa C Da, Li CG, Lu D, Xue CC. Acupuncture analgesia for temporal
summation of experimental pain: A randomised controlled study. Eur. J. Pain. 14:725–31
(2010).
[87] Chae Y, Lee YS, Enck P. How placebo needles differ from placebo pills? Front. Psychiatry. 9:1–9
(2018).
[88] Kapural L, Cywinski JB, Sparks DA. Spinal cord stimulation for visceral pain from chronic
pancreatitis. Neuromodulation. 14:423-6 (2011).
[89] Kapural L. Spinal cord stimulation for intractable chronic pain. Curr. Pain Headache Rep.
18:18–23 (2014).
[90] Fregni F, Potvin K, Dasilva D, et al. Clinical effects and brain metabolic correlates in non-
invasive cortical neuromodulation for visceral pain. Eur J Pain. 15:53–60 (2011).
[91] Gill JS, Asgerally A, Simopoulos TT. High Frequency Spinal Cord Stimulation at 10 kHz for the
Treatment of Complex Regional Pain Syndrome: A Case Series of Patients with or without
Previous Spinal Cord Stimulator Implantation. Pain Pract. [Epub ahead of print]. (2018);
Available from: http://doi.wiley.com/10.1111/papr.12739.
[92] Maier A, Scheele D, Hurlemann R, Kinfe TM. Spotlight on cervical vagus nerve stimulation for
the treatment of primary headache disorders : a review. 11: 1613–25 (2018).
[93] Sedan O, Sprecher E, Yarnitsky D. Vagal stomach afferents inhibit somatic pain perception.

Accep
ted M
anus
cript
24
113:354–9 (2005).
[94] Chakravarthy K, Chaudhry H, Williams K, Christo PJ. Review of the Uses of Vagal Nerve
Stimulation in Chronic Pain Management. Curr. Pain Headache Rep. 19:54 (2015).
[95] Juel J, Brock C, Olesen SS, et al. Acute physiological and electrical accentuation of vagal tone
has no effect on pain or gastrointestinal motility in chronic pancreatitis. J. Pain Res. 10:1347–
55 (2017).
[96] Olesen AE, Brokjaer A, Fisher IW, Larsen IM. Pharmacological challenges in chronic
pancreatitis. World J. Gastroenterol. 19:7302–7 (2013).
[97] Diener H-C, Holle D, Dresler T, Gaul C. Chronic headache due to overuse of analgesics and anti-
migraine agents. Dtsch. Aerzteblatt Online. 115: 365-70 (2018)
[98] Dall M, Schaffalitzky de Muckadell OB, Lassen AT, Hansen JM, Hallas J. An Association Between
Selective Serotonin Reuptake Inhibitor Use and Serious Upper Gastrointestinal Bleeding. Clin.
Gastroenterol. Hepatol. 7:1314–21 (2009).
[99] Chebli JMF, de Souza AFM, Gaburri PD, et al. Prevalence and Pathogenesis of Duodenal Ulcer
in Chronic Alcoholic Pancreatitis. J. Clin. Gastroenterol. 35:71–4 (2002).
[100] Brock C, Olesen SS, Olesen AE, Frøkjaer JB, Andresen T, Drewes AM. Opioid-induced bowel
dysfunction: pathophysiology and management. Drugs [Internet]. 72:1847–65 (2012).
[101] De Schepper HU, Cremonini F, Park MI, Camilleri M. Opioids and the gut: Pharmacology and
current clinical experience. Neurogastroenterol. Motil. 16:383–94 (2004).
[102] Farmer AD, Drewes AM, Chiarioni G, et al. Pathophysiology and Management of Opioid-
Induced Constipation: European Expert Consensus Statement. United Eur. Gastroenterol. J.
0:1-14 (2019)
[103] Brush DE. Complications of Long-Term Opioid Therapy for Management of Chronic Pain: The
Paradox of Opioid-Induced Hyperalgesia. J. Med. Toxicol. 8:387–92 (2012).
[104] Mitra R, Jones S. Adjuvant analgesics in cancer pain: A review. Am. J. Hosp. Palliat. Med.
29:70–9 (2012).

Accep
ted M
anus
cript
25
Figure legends
Figure 1. Illustration of the bedside method used for objective assessment of the pain system in a
patient with chronic pancreatitis without the use of visceral stimulations. Due to convergence
between afferents from the pancreas and those of the skin in the Th10 dermatome (abdomen and
back), any increased afferent barrage from the pancreas may result in local neuronal sensitization at
this level. This will result in a lowering of the pain threshold (hyperalgesia) to experimental pain
stimuli of the skin and deep tissue (QST 1). In the left insert showing the dermatomes, this is
illustrated as white circles, whereas the remaining test sites are illustrated as black circles. If the
sensitization spreads along the neuraxis there will also be a lowering of pain thresholds in other areas
(QST 2; black circles in the dermatome-figure).
The efficacy of inhibitory bulbo-spinal descending pathways (black arrow) is tested as a change in the
pain threshold to pressure stimulation before and after the descending pathways are activated with a
tonic heterotopic stimulus. Finally, neuronal sensitization is also assessed as an increased response to
repeated pinprick stimuli (temporal summation). This is illustrated in the right insert.
Figure 2. A fictive example of pressure pain detection thresholds in a patient with generalized
sensitization compared with preliminary data from a healthy reference population.
Figure 3. If smoking cessation and alcohol abstinence do not provide pain relief, the flowchart
exemplifies a mechanism-based treatment algorithm that can be used to guide management of pain
in patients with chronic pancreatitis. Simple analgesics include non-steroidal anti-inflammatory drugs
(NSAID) and acetaminophen, complementary treatments include acupuncture, vagal nerve
stimulations etc. Abbreviations: Gaba = gabapentinoids, QST = Quantitative Sensory Testing, SNRI =
Serotonin and Norepinephrine Reuptake Inhibitor, TCA = Tricyclic Antidepressants
Table 1. An overview of QST modalities studied in this review including which pain mechanism the test
is designed to examine.
Accep
ted M
anus
cript
26
Table 1:
QST Method Stimulation Pain mechanism
Conditioned pain modulation
Conditioning stimulus in between to test pain stimuli
Conditioning stimulus Test pain stimulus
Descending pain inhibition
Cold pressor test, ischemic pain, heat, chemically induced pain, electrical induced pain.
Mechanical pressure, electrical stimulation, heat, cold
Windup phenomenon
A series of identical noxious stimuli of a frequency of 0,33 Hz or higher
Pin prick, electrical stimulation, heat, cold
Temporal summation
Pain detection/tolerance threshold
Pain detection threshold and pain tolerance threshold at various sites including control sites
Mechanical pressure, electrical stimulation, heat, cold
Sensitization (both peripheral and central)

Accep
ted M
anus
cript
27
Figure 1

Accep
ted M
anus
cript
28
Figure 2
Accep
ted M
anus
cript
29
Figure 3


















