M. Alducin, R. Díez Muiño, J. I. Juaristi. Contents Lecture 1 INTRODUCTION: SURFACE CHEMISTRY AND...
62
M. Alducin, R. Díez Muiño, J. I. Juaristi
-
Upload
audra-richard -
Category
Documents
-
view
221 -
download
3
Transcript of M. Alducin, R. Díez Muiño, J. I. Juaristi. Contents Lecture 1 INTRODUCTION: SURFACE CHEMISTRY AND...

M. Alducin, R. Díez Muiño, J. I. Juaristi

Contents• Lecture 1
INTRODUCTION: SURFACE CHEMISTRY AND HETEROGENEOUS CATALYSIS• Lecture 2
MOLECULAR STRUCTURE• Lecture 3
ELECTRONIC AND STRUCTURAL PROPERTIES OF SURFACES• Lecture 4
ELEMENTARY CHEMICAL PROCESSES AT SURFACES: POTENTIAL ENERGY SURFACES • Lecture 5
THEORY OF GAS/SURFACE DYNAMICS• Lecture 6
COMPUTATIONAL METHODS TO SIMULATE GAS/SURFACE DYNAMICS

Lectures on Molecular Dynamics at Surfaces:
Friday May 7th: 9.00 --> 11.00theoretical background
Tuesday May 11th : 9.00 --> 11.00Wednesday May 12th : 9.00 --> 11.00Friday May 14th : 9.00 --> 11.00
1st exercise: analysis of N2/W(110) PES2nd exercise: dissociation dynamics of N2 on W(110)

Lectures on Molecular Dynamics at Surfaces:
Friday May 7th: 9.00 --> 11.00theoretical background
Tuesday May 11th : 9.00 --> 12.001st exercise: analysis of N2/W(110) PES
Friday May 14th : 9.00 --> 12.002nd exercise: dissociation dynamics of N2 on W(110)
2nd option!!

I.- Theoretical background

Simulation (computer experiments):
Experiment: a system is subjected to measurements and a result, in numerical form, is obtained.
Theory: In the past, a model of the system was constructed and later validated by checking its ability to describe the behavior of the system in some limit cases (simplification understanding).
Simulation: The advent of powerful computational resources has brought the possibility to reduce the approximations, reproduce the complexity of experimental conditions, and accurately compare with experimental results (study of regions not accesible experimentally).

Physical and chemical processes at surfaces are dynamical in nature
-Theoretically, static properties are usually accurately described (ground state): Density Functional Theory, Quantum Chemistry, Quantum Monte Carlo, etc.
- Dynamic properties still require further theoretical development (may involve excited states)

gas/surface dynamics
dissociative adsorption
molecularadsorptiondesorption
some elementary reactive processes at surfaces
from the fundamental point of view, the goal is to understand how solid surfaces and nanostructures can be used to promote gas-phase chemical reactions

Langmuir-HinshelwoodRecombination
Eley-Rideal recombination
gas/surface dynamics

Physical and chemical processes at surfaces are dynamical in nature
-Theoretically, static properties are usually accurately described (ground state): Density Functional Theory, Quantum Chemistry, Quantum Monte Carlo, etc.
- Dynamic properties still require further theoretical development (may involve excited states)

Lawrence Livermore National Laboratory

Molecular Dynamics
Molecular dynamics is a computer simulation technique in which the time evolution of a set of interacting atoms is followed by integrating their equations of motion.

Some nomenclature
Classical Molecular Dynamics Simulation: A model of inteeractions between atoms is supplied as input before a simulation can be carried out.
Quantum Molecular Dynamics Simulation: They do not require any a priori knowledge of interatomic interactions. Only the laws of quantum mechanics are used.
Classical dynamics of nuclei
Quantum dynamics of nuclei

gas/surface dynamics
dissociative adsorption
molecularadsorptiondesorption
some elementary reactive processes at surfaces
from the fundamental point of view, the goal is to understand how solid surfaces and nanostructures can be used to promote gas-phase chemical reactions

Langmuir-HinshelwoodRecombination
Eley-Rideal recombination
gas/surface dynamics

The adiabatic approximationA physical system remains in its instantaneous eigenstate if a given
perturbation is acting on it slowly enough and if there is a gap between the eigenvalue and the rest of the Hamiltonian's spectrum
Sketch: Potential energy curves of a neutral diatomic molecule R and its negative ion R-

The adiabatic approximation

Validity of the adiabatic approximation
Notes from Ballentine’s book

Ballentine’s book

Ballentine’s book

Ballentine’s book

Molecular Dynamics
Molecular dynamics is a computer simulation technique in which the time evolution of a set of interacting atoms is followed by integrating their equations of motion.



Born – Oppenheimer approximation

Born – Oppenheimer approximation

The Born-Oppenheimer approximation



Quantum dynamics of nuclei are also possible (time-dependent wave-packet propagation, for instance)… but numerically VERY demanding!

Quantum dynamics of nuclei are also possible (time-dependent wave-packet propagation, for instance)… but numerically VERY demanding!
Díaz et al., PRB 72, 035401 (2005)
Dissociation probability of H2/Pd(111)



Classical dynamics: In practice…

http://www.mrflip.com/resources/MDPackages.html
A list of molecular dynamics packages, for those interested:
Molecular Dynamics

Classical Molecular Dynamics
Potential Energy Surface (PES)The PES provides the energy (and its derivatives!) for all positions of the nuclei Ri.
It is a 3N dimensional function!

Classical Molecular Dynamics
Potential Energy Surface (PES)The PES provides the energy (and its derivatives!) for all positions of the nuclei Ri.
It is a 3N dimensional function!
Frozen Surface: In our case, everything is reduced to a 6D problem!

How to calculate the forces (the PES)?
‘Classical’ calculation of the PES: - Force fields, parametrizations, etc.
Complete quantum calculation of the PES (previous to the dynamics): - Ab-initio methods (DFT, HF, etc.) with ensuing fitting
On-the-fly methods: - Ab-initio molecular dynamics - Car-Parrinello

How to calculate the forces (the PES)?
‘Classical’ calculation of the PES: - Force fields, parametrizations, etc.
Complete quantum calculation of the PES (previous to the dynamics): - Ab-initio methods (DFT, HF, etc.) with ensuing fitting
On-the-fly methods: - Ab-initio molecular dynamics - Car-Parrinello
All of them are adiabatic (ground-state) methods! Non-adiabatic methods (TDDFT, for instance) required for excited systems.

Excitation of electron-hole pairs in a metal surface (no gap for excitations)
Non-adiabatic processes
bulk metal
electrons at the surface can be excited

description of electronic excitations by a friction coefficient
classical equations of motion
mi(d2ri/dt2)=-dV(ri,rj)/d(ri) – h(ri)(dri/dt)
for each atom “i” in the molecule
friction coefficient
adiabaticforce:
6D DFT PES
- damping of adsorbate vibrations: Persson and Hellsing, PRL49, 662 (1982)
- dynamics of atomic adsorption Trail, Bird, et al., JCP119, 4539 (2003)
previously used for:friction coefficient:
effective medium approximation
n(z)
zn0
bulk metal
n0
h=n0kFstr(kF)
effective medium: FEG with electronic density n0
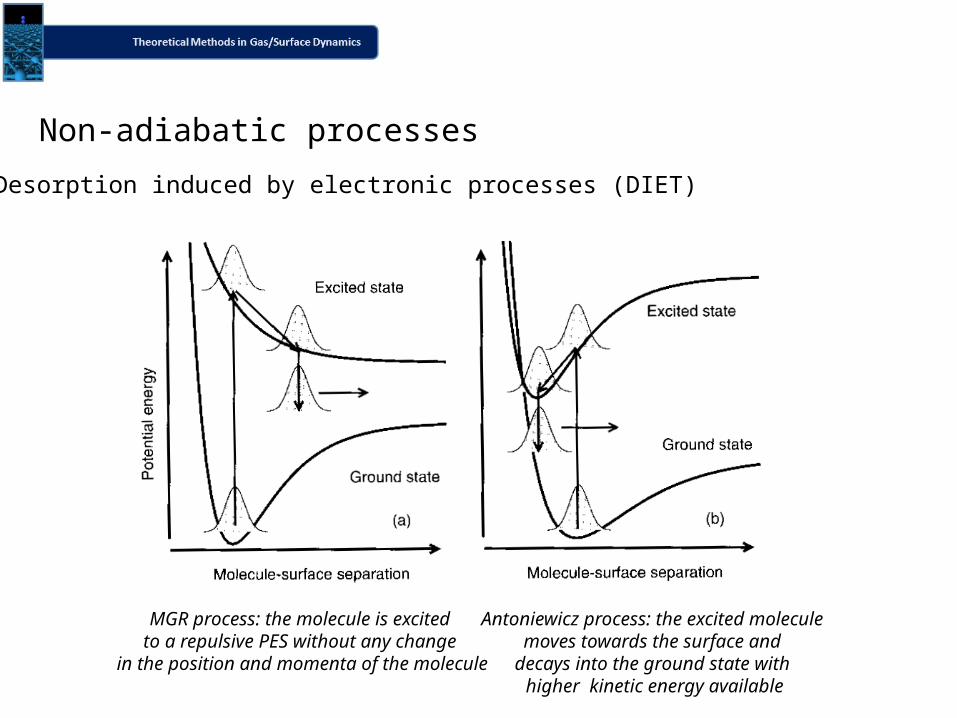
Desorption induced by electronic processes (DIET)
Non-adiabatic processes
MGR process: the molecule is excited to a repulsive PES without any change
in the position and momenta of the molecule
Antoniewicz process: the excited molecule moves towards the surface and
decays into the ground state with higher kinetic energy available

II.- Particular case: dissociation of diatomic molecules on metal surfaces

centro de física de materialesCFM
What is catalysis? The effect produced in facilitating a chemical reaction, by the presence of a substance, which itself undergoes no permanent change.
A+B P direct reactionA+B+C P+C catalyzed reaction
A and B are reactantsC is the catalyst
P is the reaction product
A+B
P
gas/solid interfaces and heterogeneous catalysis

centro de física de materialesCFM
What is catalysis? The effect produced in facilitating a chemical reaction, by the presence of a substance, which itself undergoes no permanent change.
A+B P direct reactionA+B+C P+C catalyzed reaction
A and B are reactantsC is the catalyst
P is the reaction product
A+B
P
gas/solid interfaces and heterogeneous catalysis
Heterogeneous catalysis: The catalyst is in a different phase solid surfaces.

centro de física de materialesCFM
What is catalysis? The effect produced in facilitating a chemical reaction, by the presence of a substance, which itself undergoes no permanent change.
A+B
P
gas/solid interfaces and heterogeneous catalysis
Heterogeneous catalysis: The catalyst is in a different phase solid surfaces.
The chinese symbol for catalyst
is the same as the one for marriage broker
(matchmaker)

centro de física de materialesCFM
ammonia synthesis: 3H2(g) + N2 (g) ↔ 2NH3(g) (catalyzed by Fe surface)
global context: chemical industry
world production: 130 million tons (year 2000),
~200US$/ton

centro de física de materialesCFM
Catalysis in car industry:
In car engines, CO, NO, and NO2 are formed.
Catalytic converters reduce such emissions by adsorbing CO and NO onto a catalytic surface, where the gases undergo a redox reaction.
CO2 and N2 are desorbed from the surface and emitted as relatively harmless gases:
2CO + 2NO → 2CO2 + N2
global context: car industry

Experimental techniques in Gas-Surface dynamics
Molecular beam techniques
Sticking CoefficientThe sticking coefficient, S , is a measure of the fraction of incident molecules which adsorb upon the surface i.e. it is a probability and lies in the range 0 - 1 , where the limits correspond to no adsorption and complete adsorption of all incident molecules respectively.
In general, S depends upon many variables i.e. S = f ( surface coverage , temperature, crystal face .... )
QEi
Sticking coefficient of O2 on Ag(111):dependence on energy and polar incidence angle

Experimental techniques in Gas-Surface dynamics
Molecular beam techniques
Molecular beam scattering studies of H2–D2 exchange on Pt332 surface, showing that atomic steps on metal surfacesbreak chemical bonds, in this case, hydrogen-hydrogen bonds, with unit reaction probability
(a) schematic defining the geometry of the incident angle polar and azimuthal of the molecular beam with respect to a stepped surface.
(b) HD production as a function of angle of incidence of the molecular beam normalized to the incident D2 intensity.

x
y
z
surface unit cell
q
j
XY
Z
QEi
Incidence conditions are fixed: (Ei, )Q
Monte-Carlo sampling on the internal degrees of freadom: (X, Y, q, j) and on (parallel velocity)
Born-Oppenheimer approximation
Frozen surface approximation 6D PES: V(X, Y, Z, r, q, j)
Calculation of the Potential Energy Surface (PES)
Classical trajectory calculations
Dissociative dynamics of molecules on surfaces: Theoretical calculations

The test system for our exercises will be N2 on W surfaces. Why?

W: Wolfram or Tungsten
W was first isolated in 1783 by Fausto and Juan José Elhuyar, two Spanish chemists working
in the Basque Country.
The Elhuyar brothers named the element 'volfram', from the mineral
from which it was extracted. But the name 'tungsten',
of Swedish origin, is the one that has prevailed.
centro de física de materialesCFM

stick
ing
coeffi
cien
t S0
0.25 0.50 0.75 1.000.00
0.25
0.50
0.75
1.00
W(100)
T=800K
T=300K
T=100K
impact energy Ei (eV)
0.25 0.50 0.75 1.000.00
0.25
0.50
0.75
1.00
W(110)
T=800K
no threshold threshold
normal incidence
Rettner et al. (1988)Beutl et al. (1997)
Pfnür et al. (1986)Rettner et al. (1990)
measurements of N2 dissociation on W surfaces
centro de física de materialesCFM

surface face and reactivity
- surface roughness (work function)- unique active sites at the surface
two possible reasons for the difference in reactivityover different faces:
the rate-limiting step in ammonia formation is the dissociative adsorption of N2 on the surface
centro de física de materialesCFM

surface face and reactivity
- surface roughness (work functions)- unique active sites at the surface
two possible reasons for the difference in reactivityover different faces:
the rate-limiting step in ammonia formation is the dissociative adsorption of N2 on the surface
most reactive surfaces have C7 sites
(seven nearest neighbors)
centro de física de materialesCFM

W(100) W(110)
adsorption energyDFT = 7.4 eV
Exp. = 6.6-7 eVadsorption distance
DFT = 0.63 Å
adsorption energyDFT = 6.8 eVExp. = 6.6 eV
adsorption distanceDFT = 1.15 Å
final state features: N adsorption on W
centro de física de materialesCFM

stick
ing
coeffi
cien
t S0
0.25 0.50 0.75 1.000.00
0.25
0.50
0.75
1.00
W(100)
T=800K
T=300K
T=100K
impact energy Ei (eV)
0.25 0.50 0.75 1.000.00
0.25
0.50
0.75
1.00
W(110)
T=800K
no threshold threshold
normal incidence
Rettner et al. (1988)Beutl et al. (1997)
Pfnür et al. (1986)Rettner et al. (1990)
measurements of N2 dissociation on W surfaces
activation barrier?
centro de física de materialesCFM

6D potential energy surface (PES) of N2/W(110)
- DFT - GGA (PW91) calculation with VASP
- Plane-wave basis set and US pseudopotentials
- periodic supercell: 5-layer slab and 2x2 surface cell
- 30 configurations = 5610 ab-initio values
- interpolation through the corrugation reducing procedure [Busnengo et al., JCP 112, 7641 (2000)]
centro de física de materialesCFM

q=45o
j=270o
q=90o
j 125oq=90o
j 54o
q=0o
Definition of System of coordinates for N2/W(110)
centro de física de materialesCFM

1 1 .2 1 .4 1 .6 1 .8 2 2 .2
1 .5
2
2 .5
3
3 .5
4
1 1 .2 1 .4 1 .6 1 .8 2 2 .2
1 .5
2
2 .5
3
3 .5
4
1 1 .2 1 .4 1 .6 1 .8 2 2 .2
1 .5
2
2 .5
3
3 .5
4
1 1 .2 1 .4 1 .6 1 .8 2 2 .2
1 .5
2
2 .5
3
3 .5
4
q=45o
j=270o
q=90o
j 125oq=90o
j 54o
q=0o
r(Å) r(Å)
r(Å)r(Å)
Z(Å)
Z(Å)
Z(Å)
Z(Å)
some elbow plots
for the N2/W(110)
system
distance between contour lines = 0.2eV
E<0
E>0E=0
centro de física de materialesCFM

classical dynamics
- classical trajectory method
- adiabatic description (no dissipation)
centro de física de materialesCFM





![AIN, E… - Ripollet Rock Festival - Noticias Olla/numeros/laolla54.pdf · Arctica, Rhapsody On Fire o Stratovarius. Disco auto-gestionado compuesto por once temas. . [Víctor Muiño]](https://static.fdocuments.us/doc/165x107/5a7e03097f8b9ae9398e1d0d/ain-e-ripollet-rock-festival-ollanumeroslaolla54pdfarctica-rhapsody.jpg)












