
Lost in folding space? Comparing four variants of the thermodynamic model for RNA secondary...
19
RESEARCH ARTICLE Open Access Lost in folding space? Comparing four variants of the thermodynamic model for RNA secondary structure prediction Stefan Janssen 1 , Christian Schudoma 2 , Gerhard Steger 3* and Robert Giegerich 1* Abstract Background: Many bioinformatics tools for RNA secondary structure analysis are based on a thermodynamic model of RNA folding. They predict a single, “optimal” structure by free energy minimization, they enumerate near- optimal structures, they compute base pair probabilities and dot plots, representative structures of different abstract shapes, or Boltzmann probabilities of structures and shapes. Although all programs refer to the same physical model, they implement it with considerable variation for different tasks, and little is known about the effects of heuristic assumptions and model simplifications used by the programs on the outcome of the analysis. Results: We extract four different models of the thermodynamic folding space which underlie the programs RNAFOLD, RNASHAPES, and RNASUBOPT. Their differences lie within the details of the energy model and the granularity of the folding space. We implement probabilistic shape analysis for all models, and introduce the shape probability shift as a robust measure of model similarity. Using four data sets derived from experimentally solved structures, we provide a quantitative evaluation of the model differences. Conclusions: We find that search space granularity affects the computed shape probabilities less than the over- or underapproximation of free energy by a simplified energy model. Still, the approximations perform similar enough to implementations of the full model to justify their continued use in settings where computational constraints call for simpler algorithms. On the side, we observe that the rarely used level 2 shapes, which predict the complete arrangement of helices, multiloops, internal loops and bulges, include the “true” shape in a rather small number of predicted high probability shapes. This calls for an investigation of new strategies to extract high probability members from the (very large) level 2 shape space of an RNA sequence. We provide implementations of all four models, written in a declarative style that makes them easy to be modified. Based on our study, future work on thermodynamic RNA folding may make a choice of model based on our empirical data. It can take our implementations as a starting point for further program development. Background Motivation A wide variety of bioinformatics tools exist, which help to analyze RNA secondary structure based on an experi- mentally supported, thermodynamic model of RNA fold- ing [1]. Typical tasks performed by such tools are • prediction of a single, “optimal” structure of mini- mal free energy, • computation of near-optimal structures, either by complete enumeration up to a certain energy thresh- old, or by sampling from the folding space, • computation of base pair probabilities and dot plots, • computation of representative structures of differ- ent abstract shapes, or • computation of Boltzmann probabilities, either of individual structures, or accumulated over all struc- tures of the same abstract shape. * Correspondence: [email protected]; [email protected] bielefeld.de 1 Faculty of Technology, Bielefeld University, 33615 Bielefeld, Germany 3 Institut für Physikalische Biologie, Heinrich-Heine-Universität Düsseldorf, 40204 Düsseldorf, Germany Full list of author information is available at the end of the article Janssen et al. BMC Bioinformatics 2011, 12:429 http://www.biomedcentral.com/1471-2105/12/429 © 2011 Janssen et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
-
Upload
stefan-janssen -
Category
Documents
-
view
212 -
download
0
Transcript of Lost in folding space? Comparing four variants of the thermodynamic model for RNA secondary...

RESEARCH ARTICLE Open Access
Lost in folding space? Comparing four variants ofthe thermodynamic model for RNA secondarystructure predictionStefan Janssen1, Christian Schudoma2, Gerhard Steger3* and Robert Giegerich1*
Abstract
Background: Many bioinformatics tools for RNA secondary structure analysis are based on a thermodynamicmodel of RNA folding. They predict a single, “optimal” structure by free energy minimization, they enumerate near-optimal structures, they compute base pair probabilities and dot plots, representative structures of differentabstract shapes, or Boltzmann probabilities of structures and shapes. Although all programs refer to the samephysical model, they implement it with considerable variation for different tasks, and little is known about theeffects of heuristic assumptions and model simplifications used by the programs on the outcome of the analysis.
Results: We extract four different models of the thermodynamic folding space which underlie the programsRNAFOLD, RNASHAPES, and RNASUBOPT. Their differences lie within the details of the energy model and thegranularity of the folding space. We implement probabilistic shape analysis for all models, and introduce the shapeprobability shift as a robust measure of model similarity. Using four data sets derived from experimentally solvedstructures, we provide a quantitative evaluation of the model differences.
Conclusions: We find that search space granularity affects the computed shape probabilities less than the over- orunderapproximation of free energy by a simplified energy model. Still, the approximations perform similar enoughto implementations of the full model to justify their continued use in settings where computational constraints callfor simpler algorithms. On the side, we observe that the rarely used level 2 shapes, which predict the completearrangement of helices, multiloops, internal loops and bulges, include the “true” shape in a rather small number ofpredicted high probability shapes. This calls for an investigation of new strategies to extract high probabilitymembers from the (very large) level 2 shape space of an RNA sequence. We provide implementations of all fourmodels, written in a declarative style that makes them easy to be modified. Based on our study, future work onthermodynamic RNA folding may make a choice of model based on our empirical data. It can take ourimplementations as a starting point for further program development.
BackgroundMotivationA wide variety of bioinformatics tools exist, which helpto analyze RNA secondary structure based on an experi-mentally supported, thermodynamic model of RNA fold-ing [1]. Typical tasks performed by such tools are
• prediction of a single, “optimal” structure of mini-mal free energy,• computation of near-optimal structures, either bycomplete enumeration up to a certain energy thresh-old, or by sampling from the folding space,• computation of base pair probabilities and dotplots,• computation of representative structures of differ-ent abstract shapes, or• computation of Boltzmann probabilities, either ofindividual structures, or accumulated over all struc-tures of the same abstract shape.
* Correspondence: [email protected]; [email protected] of Technology, Bielefeld University, 33615 Bielefeld, Germany3Institut für Physikalische Biologie, Heinrich-Heine-Universität Düsseldorf,40204 Düsseldorf, GermanyFull list of author information is available at the end of the article
Janssen et al. BMC Bioinformatics 2011, 12:429http://www.biomedcentral.com/1471-2105/12/429
© 2011 Janssen et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative CommonsAttribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction inany medium, provided the original work is properly cited.

From a macroscopic point of view, all theseapproaches are based on the same thermodynamicmodel, but when checking in detail, this does not hold.Algorithms for different tasks make certain assumptionsabout the folding space, where little is known to whichextent these assumptions influence the outcome of theanalysis.The present study is designed to fill this gap. We
explicate the details of four different models of the RNAfolding space, named NoDangle, OverDangle, Micro-State and MacroState. They capture four different mod-els of the folding space, as they are implemented in theprograms RNAFOLD[2], RNASHAPES[3], and RNASU-BOPT[4].1 We compare the outcome of predictionsfrom the different models, and evaluate them againstthree data sets derived from experimentally provedstructures.
Goals of the evaluationThe goal of this study is not to define a “correct” or“best” way of modeling the RNA folding space. Differentdefinitions may retain their merits in the light of differ-ent computational constraints. We want to explicate thedifferences in the results which are due to the choice ofa particular model. Aside being interesting in its ownright, this allows future algorithms designers to make awell-founded choice of the model they base their workon.How to compare the performance of different models?
A first idea would be to evaluate them with respect toprediction of the structure of minimum free energy(MFE; for details see below), using a reference set oftrusted structures. This has been done occasionally[1,5], and we will include such an evaluation here forthe sake of completeness. However, MFE structure pre-diction is notorious in the sense that a slight offset inenergy can lead to a radically different structure. This isa consequence of the underlying thermodynamic model,and not due to its inadequate implementation. For amore robust evaluation, we need a measure which con-stitutes a more comprehensive characteristic of the over-all folding space of an RNA molecule, includingevidence for competing near-optimal structures of sig-nificant structural variation.Abstract shapes of RNA [3,6] provide such a measure.
This approach provides two essential types of analysis:(1) to compute a handsome set of representative, near-optimal structures, which are different enough to be ofinterest, and (2) to compute shape probabilities, whichaccumulate individual Boltzmann probabilities over allstructures of the same shape. The shape probability is arobust measure of structural well-definedness, and incontrast to folding energy, it is independent of base
composition and meaningful for comparing foldings ofdifferent sequences with similar length.Types (1) and (2) of abstract shape analysis are
achieved by different algorithms, using different modelsof the folding space, in the program RNASHAPES. Asimilar situation prevails within the Vienna RNA pack-age, where different models of the folding space areused with various functions of RNAFOLD and RNASU-BOPT under different parameter settings.For our evaluation, we implement probabilistic shape
analysis in four different ways, three of which closelycorrespond to the folding space models implementedfor MFE prediction in RNAFOLD2, and two of whichcorrespond to the algorithms used in RNASHAPES.This set of programs will allow us to derive observationsabout the underlying folding space models.
MethodsIn this section, we recall the definitions underlying thethermodynamic model of RNA folding, and then pro-ceed to specify four different implementations of thismodel.
The thermodynamic modelFree energy and partition functionStructure formation of a single-stranded nucleic acidsequence x–from an unfolded, random coil structure cinto the folded structure s–is a standard equilibriumreaction with temperature-dependent free energy �G0
T
and equilibrium constant KT:
c � s
KT =[s][c]
�G0T = −RT ln KT .
The number of possible secondary structures of a sin-gle sequence, i. e. the folding space F(x) of x, growsexponentially with the sequence length n [7,8]. Thesepossible structures si of a single sequence coexist insolution with concentrations dependent on their freeenergies ΔG0(si); that is, each structure is present as afraction psi according to its Boltzmann probability
psi = exp(−�G0
T(si)RT
)/Q
given by its molar Boltzmann weight
exp(−�G0T(si)/(RT)) and the partition function Q for
the ensemble of all possible structures
Q =∑
all structures si∈F(x)
exp(−�G0
T(si)RT
).
Janssen et al. BMC Bioinformatics 2011, 12:429http://www.biomedcentral.com/1471-2105/12/429
Page 2 of 19

The structure of lowest free energy is called the (ther-modynamically) optimal structure or structure of mini-mum free energy (MFE).RNA secondary structures are conveniently repre-
sented as dot-bracket strings, such as
“((.((((..(((...))).....((.((.....))...)).))))))” (1)
where matched parentheses indicate a base pair anddots indicate unpaired bases.Abstract shapesMany of the possible structures differ from each otherby only tiny structural rearrangements like addition orremoval of a base pair, or a slight shift in position of asmall bulge loop. Structures can be pooled according totheir abstract shape. Generally, an abstract shape givesinformation about the arrangement of structural ele-ments such as helices, but no concrete base pairs [3,6].The MFE structure within each shape class is called“shrep”, which is short for shape representative struc-ture. The partition function Qp for the ensemble of allstructures of shape p is
Qp =∑
all structures si∈p
exp(−�G0
T(si)RT
).
Of course, the structures from all shape classes sumup to the ensemble of all structures:
Q =∑
all shape classes p
Qp
and the probability of shape p is
Prob(p) = Qp/Q .
Shape abstraction can be defined in various ways.RNASHAPES provides shape abstraction functions π1,..., π5 which implement different levels of abstraction,with π5 being the most abstract. Shapes can be repre-sented as strings, similar to structure representations,where a single pair of square brackets marks a helix (ofany length), and an underscore marks a stretch ofunpaired bases, also of any length. Levels of abstractiondiffer in the amount of information they retain aboutunpaired regions. The above RNA structure (1) ismapped to shape strings on abstraction levels 2 and 5 asfollows:
π2 : “[ [[][ [] ]]]”
π5 : “[[][]]”
Both shapes indicate that the structure is a so-calledY-shape, a multiloop with a two-way branch. This mostabstract view is conveyed by abstraction level 5. The lessabstract level 2 shape indicates, in addition, that the
outer stem is interrupted by a bulge on the 5’ side, andthat the 3’ branch inside the multiloop is interrupted byan internal loop. For a detailed definition of shapeabstraction levels, see [9].Implementing the basic energy model - no dangling basesIn the usual approximation, the free energy of an indivi-dual structure s is the sum of the energetic contribu-tions of all structural elements of s:
�G0T,s =
∑helices j
�G0T,j +
∑loops k
�G0T,k
with energy of an individual helix:
�G0T,helix =
∑base pairstacks m
�G0T,m.
That is, the energy of a helix depends only on its typeof base pairs (G:C, C:G, A:U, U:A, G:U, U:G) stackingon its neighboring base pair [10]. The minimum lengthof a helix is two base pairs (one base pair stack). Single(lonely) pairs should not exist. The energy of a loopdepends on its type (hairpin loop closed by a helix,internal and bulge loop closed by two helices, and mul-tiloop or junction closed by more than two helices), thesequence(s) of loop nucleotides, and type of closing basepair(s). That is, the free energy of a given secondarystructure s is obtained by decomposition of s into itsstructural elements and summation of values obtainedby respective calls of the elementary energy functions ofthese elements as listed in Table 1. With the exampleshown in Figure 1, this would be three calls to sr_energy
for the three base pair stacks (5′AC3′
3′UG5′,
5′CC3′
3′GG5′, and
5′XY3′
3′YX5′),
a call to termau_energy for the terminal5′A
3′Upair, and a
call to bl_energy for a bulge loop with sequence 5’N–N3’
and closing pairs5′C
3′Gand
5′Y
3′X.
Implementing the full energy model - with dangling basesIn addition to the basic energy model described above,unpaired bases at the end of a helix can stabilize thehelix by stacking on the terminal base pair [11-13]3.Introducing dangling bases effectively refines our
notion of structure. Any secondary structure, as definedsolely by its set of base pairs, can now have several var-iants according to different choices of dangling bases.Such refinement can be reflected in our structure repre-sentation by replacing certain dot symbols by “d”, indi-cating a base dangling onto a helix to its left, and “b”for a base dangling onto a helix to its right. For exam-ple, a structure like
“((..((...)).((...)).))”
Janssen et al. BMC Bioinformatics 2011, 12:429http://www.biomedcentral.com/1471-2105/12/429
Page 3 of 19

now has dangle variants such as
“((d.((...))b((...))b))”
“((.b((...))b((...))b))”
“((db((...))b((...))b))”
“((..((...))d((...))b))”
“((..((...))b((...)).))”
and 31 more. Each end of a helix can have danglingbases, except an end which leads to the hairpin loop. Inthis case, energy contributions from dangling bases arealready incorporated in the energy parameters for theloops.Given a concrete secondary structure, it is no problem
to consider all possible dangles and compute the opti-mal energy for this structure. The program RNAEVALfrom the Vienna Package can be used for this purpose.
However, for structure prediction from a primary RNAsequence, dangle means trouble, as we shall see shortly.
Modeling folding spaces with tree grammarsTree representation of structuresAll approaches using the thermodynamic model areimplemented via dynamic programming. Recursively,structures are composed from smaller substructures. Sucha dynamic programming algorithm always has an underly-ing grammar, which describes all the candidates in thefolding space of a given RNA sequence. Hence, by extract-ing the grammars behind different algorithms, we can ana-lyze the differences in their respective folding space in aprecise way, and without obscuring implementation detail.The grammars we use are tree grammars. Non-term-
inal symbols designate different components of second-ary structure, such as a stacking region or a bulge loop.Function symbols in the tree grammar are used to indi-cate how structures are built up from smaller compo-nents. For example, a snippet of a tree structure such asshown in Figure 1 designates at its bottom an unpairedstretch of one or more bases (r), 5’ of a closed substruc-ture of any type. This situation is indicated by the func-tion symbol bl, which stands for “bulge left”. Theunpaired stretch and the substructure is surrounded bytwo stacking (C:G) base pairs, and enclosed in yetanother base pair, added by function sr, which extends a“stacking region”. These functions can be seen as actualconstructors of a tree-like data structure, representingsecondary structures. They can (and will) also be seenas functions, which all call upon the energy functions ofthe thermodynamic model, to compute either free ener-gies or their corresponding Boltzmann weights. We canalso interpret them as functions which count base pairsin the structure they build, or compose the dot-bracketstring for that structure, compute their abstract shape,and so on. Modeling structures as trees built from func-tions that can be interpreted in different ways providesa uniform and flexible formalism for many purposes.
Table 1 Elementary functions in the basic thermodynamic energy model
Function Description
sr_energy The most important source for stabilizing an RNA secondary structure is stacking of two (or more) base pairs.
termau_energy A base pair A:U at the terminal end of a stacking region adds less stabilizing energy than within a stacking region.
hl_energy Stabilizing contribution for the loop-closing base pair stack plus destabilizing contribution for the hairpin loop region plus bonusenergy for special loop sequence (e. g. extrastable tetra loops).
bl_energy Analog to hl_energy, but for a destabilizing loop region bulged out to the left.
br_energy Symmetric case to bl_energy.
il_energy Analog to hl_energy, but with two destabilizing loop regions.
ml_energy Since a multiloop of x stems is less stable than x adjacent stems, it gets a penalty.
ul_energy Each stem in a multiloop gets an initial penalty.
ss_energy Regions of unpaired bases could get penalized, but we set this value to zero.
sbase_energy Same as ss_energy, but for a single unpaired base.
Figure 1 Example on structure representations. A sequence,shown in A), folds into a structure that is represented by the threeequivalent illustrations in B-D). The structure consists of a helix withthree base pairs (ACC paired with GGU), a bulge loop (N–N; Nmeaning aNy nucleotide), and a helix with two base pairs formedby any complementary nucleotides. The dashes designate omittedsequence stretches. The structure in B) is in dot-bracket notation;that is, dots mark unpaired nucleotides and pairs of opening andclosing brackets mark a base pair. The structure in C) is the usualsquiggly representation. D) is the tree representation of the samestructure: a stacked region (sr) is formed by an A:U pair stacked ontop a bulge loop (bl) including two stacking pairs (C:G/C:G) and aloop region with one or more residues (r) on the left (5’) side. Thehelix continues with a “closed” structural element (which is definedas any substructure starting with a base stack).
Janssen et al. BMC Bioinformatics 2011, 12:429http://www.biomedcentral.com/1471-2105/12/429
Page 4 of 19

From tree grammars to folding algorithmsTree grammars modeling the folding space of RNAessentially constitute executable code. They can be lit-erally transcribed into a language supporting the alge-braic dynamic programming technique [14]. We use thelanguage GAP-L as provided in the recent Bellman’sGAP programming system [15,16]. This approach isessential for the study at hand. It takes from us not onlythe burden to implement and debug dynamic program-ming recurrences for each of the four algorithms. It alsoguarantees that the different algorithms correctly imple-ment their respective models, share the energy model,are implemented with the same degree of optimization,and are independent of the programming skills of abunch of graduate students.Grammars and their relation to established structureprediction programsWe will present four grammars, NoDangle, OverDangle,MicroState and MacroState. The first three implementthe folding space of RNAFOLD used with options -d0,-d2, and -d1, respectively. The grammars MicroStateand MacroState implement the folding space of RNA-SHAPES in its two functions. All four grammars willthen be empowered with shape abstraction, and areused in our evaluation for computing shape probabilitiesunder the different models.All grammars use the same energy parameters, but in
a different way. The 16 functions of the energy model,as specified in Tables 1 and 2, are used in differentcombinations by the evaluation functions in the gram-mars. For example, in all grammars the function mlcalls the model function termau_energy, sr_energy, andml_energy. Table 3 provides the cross-referencesbetween the energy functions in our programs to bedescribed below, and the energy functions of the ther-modynamic model.
Model NoDangleNoDangle is our grammar incorporating the elementaryenergy model, without considering dangling bases at all.It corresponds to the model underlying RNAFOLDwhen used with option -noLP -d04. It is also used in
RNASUBOPT. We give a narrative explanation of howthis grammar works.Each complete structure is a struct, i. e. it is derived
from the axiom of the grammar (see Figure 2). It mighthave leading unpaired bases (sadd), hold one or moreclosed substructures (non-terminal dangle, functioncadd), or just end with the empty word (nil). A dangleis a closed substructure whose directly neighbored basesmight dangle onto the stack of base pairs. We keep thename dangle for consistency with the other grammars,but no dangle energies are considered in NoDangle; thefunction drem simply passes on the energy of its closedsubstructure, which may include a penalty for a terminalA:U pair if appropriate.A closed substructure is a stack of base pairs which
eventually leads to one of five structural motifs: hairpinloop (hairpin), bulge to the left (leftB), bulge to theright (rightB), internal loop (iloop) or multiloop. Themultiloop is a concatenation (ml_comps and ml_comps1)of two or more substructures, embraced by one closingstack. Note that all motifs have at least two closing basepairs which form a stack. This implements the conven-tion of disallowing lonely pairs. The helix initiated bytwo closing pairs can be elongated by sr. A region (r) isa non-empty stretch of unpaired bases (b), whose lengthcan be further constrained, e. g. to be at most 30 bases(r30) for internal loops or at least 3 bases (r3) for a hair-pin loop.The algebra functions drem and ml control the dan-
gling behavior, which is the only difference betweenNoDangle and OverDangle. In NoDangle, they do notmake any dangling energy contributions at all.
Model OverDangleOverDangle is the grammar which considers danglingbase energies in a simplified form. It corresponds toRNAFOLD called with options -noLP -d25. The gram-mar itself is identical to NoDangle (cf. Figure 2). It com-putes the same folding space, but evaluates energiesdifferently. It assumes an energy contribution from dan-gling bases on every side of a helix, even if a base is notavailable for dangling, for example because it is itself
Table 2 Energy functions for dangling bases
Function Description
dl_energy A single base left of a closed substructure can dangle onto this stack and thus might further stabilize it.
dr_energy Symmetric case to dl_energy.
ext_mismatch_energy Two bases left and right of a stack, which do not form a basepair (they mismatch), can dangle from both sides to the stack.
dli_energy A multiloop is closed by one stack. A single base at the inside of the multiloop and directly next to the closing stack mightdangle from left onto this stack. The energy values are the same as dr_energy, but for a reversed subsequence.
dlr_energy Symmetric case to dli_energy.
ml_mismatch_energy Two bases on both inner sides of a multiloop closing stack may dangle from inside onto this stack, but do not form abasepair (mismatch).
Janssen et al. BMC Bioinformatics 2011, 12:429http://www.biomedcentral.com/1471-2105/12/429
Page 5 of 19

Table 3 Cross-reference between the energy functions in our programs, and which energy contributions (modelfunctions) they call upon.
Function Used in evaluation function
NoDangle OverDangle MicroState MacroState
termauenergy ml ml ml ml
mldl mldl
mldr mldr
mldlr mldlr
mladl
mladr
mladlr
mldladr
mladldr
drem drem drem drem
edl edl
edr edr
edlr edlr
dl_energy edl edl
ambd
ambd’
acomb
mladl
mladlr
mladldr
dr_energy edr edr
ambd
ambd’
acomb
mladr
mladlr
mldladr
ext_mismatch_energy drem edlr edlr
dli_energy mldl mldl
mldladr
mladl
mladlr
mladldr
dri_energy mldr mldr
mladldr
mladr
mladlr
mldladr
ml_mismatch_energy ml mldlr mldlr
sr_energy sr sr sr sr
hl hl hl hl
bl bl bl bl
br br br br
il il il il
ml ml ml ml
mldl mldl
mldr mldr
mldlr mldlr
mladldr
Janssen et al. BMC Bioinformatics 2011, 12:429http://www.biomedcentral.com/1471-2105/12/429
Page 6 of 19
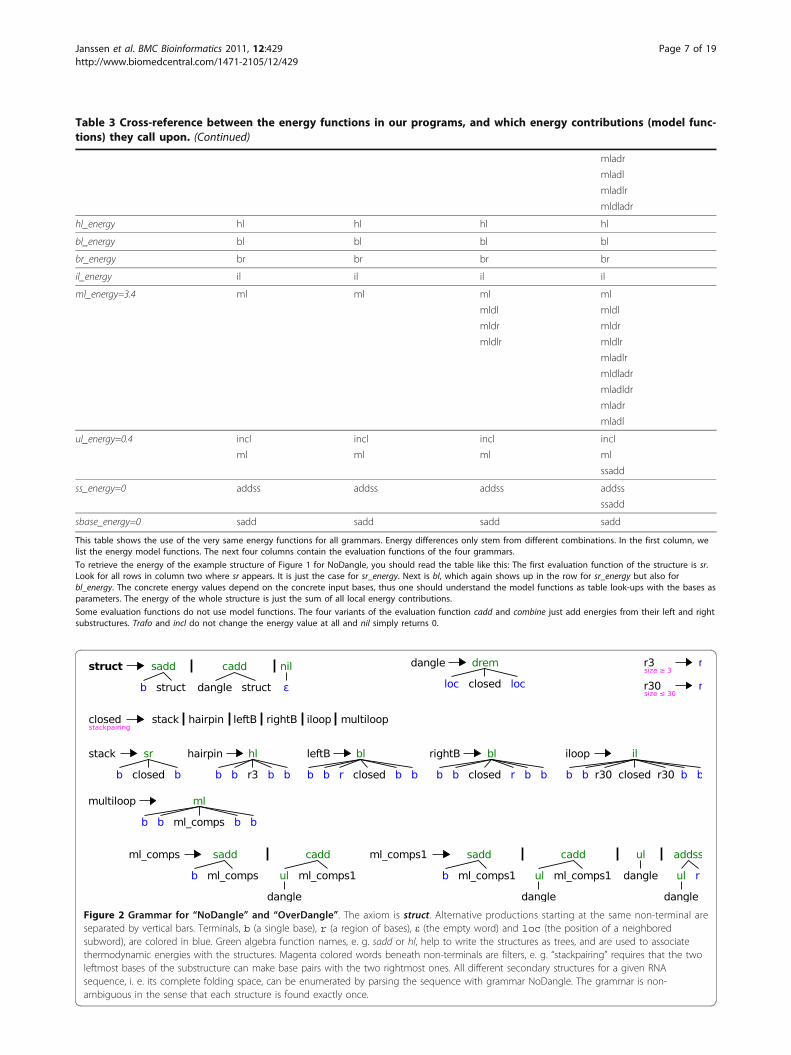
Figure 2 Grammar for “NoDangle” and “OverDangle”. The axiom is struct. Alternative productions starting at the same non-terminal areseparated by vertical bars. Terminals, b (a single base), r (a region of bases), ε (the empty word) and loc (the position of a neighboredsubword), are colored in blue. Green algebra function names, e. g. sadd or hl, help to write the structures as trees, and are used to associatethermodynamic energies with the structures. Magenta colored words beneath non-terminals are filters, e. g. “stackpairing” requires that the twoleftmost bases of the substructure can make base pairs with the two rightmost ones. All different secondary structures for a given RNAsequence, i. e. its complete folding space, can be enumerated by parsing the sequence with grammar NoDangle. The grammar is non-ambiguous in the sense that each structure is found exactly once.
Table 3 Cross-reference between the energy functions in our programs, and which energy contributions (model func-tions) they call upon. (Continued)
mladr
mladl
mladlr
mldladr
hl_energy hl hl hl hl
bl_energy bl bl bl bl
br_energy br br br br
il_energy il il il il
ml_energy=3.4 ml ml ml ml
mldl mldl
mldr mldr
mldlr mldlr
mladlr
mldladr
mladldr
mladr
mladl
ul_energy=0.4 incl incl incl incl
ml ml ml ml
ssadd
ss_energy=0 addss addss addss addss
ssadd
sbase_energy=0 sadd sadd sadd sadd
This table shows the use of the very same energy functions for all grammars. Energy differences only stem from different combinations. In the first column, welist the energy model functions. The next four columns contain the evaluation functions of the four grammars.
To retrieve the energy of the example structure of Figure 1 for NoDangle, you should read the table like this: The first evaluation function of the structure is sr.Look for all rows in column two where sr appears. It is just the case for sr_energy. Next is bl, which again shows up in the row for sr_energy but also forbl_energy. The concrete energy values depend on the concrete input bases, thus one should understand the model functions as table look-ups with the bases asparameters. The energy of the whole structure is just the sum of all local energy contributions.
Some evaluation functions do not use model functions. The four variants of the evaluation function cadd and combine just add energies from their left and rightsubstructures. Trafo and incl do not change the energy value at all and nil simply returns 0.
Janssen et al. BMC Bioinformatics 2011, 12:429http://www.biomedcentral.com/1471-2105/12/429
Page 7 of 19

engaged in another helix, or already dangling there. Thealgebra functions drem and ml control the danglingbehavior, which is the only difference between NoDan-gle and OverDangle. In OverDangle drem and mlalways adds dangling energies for left and right dangles.This is why the production using drem uses two locsymbols: loc recognizes the empty word, and returns itsposition in the sequence. These positions are used bydrem to look at the two bases to the left and right ofthe closed substructure.This “overdangling” model is used because a correct
treatment of dangles is much more complicated, as weshall see below. As a plausibility argument in favor ofthis heuristic, one may say that when a base is over-dangled, for example between two adjacent helices, aswith the midpoint in “((...)).((...))“, this can beseen as a bonus for co-axial stacking of the two helices.Including full co-axial stacking could be considered as afurther refinement of the folding space beyond theMicroState model, which will be described below. Still,due to overdangling, the MFE energy value computedmay be smaller than actually assigned by the thermody-namic model to the underlying structure. Partition func-tion computations in RNAFOLD use the OverDangleapproach, and so does RNASUBOPT with option -d2(and even -d1, but see below).Would we use both NoDangle and OverDangle to
produce a list of all structures in the folding space,sorted by free energy, these lists would hold the samestructures, but in a different order. The true MFE struc-ture (under the full model with correct dangles) will benear the front of each list, but it is not guaranteed tocome out on first place. Our next two grammars aredesigned to achieve this goal.
Model MicroStateGrammar MicroState is a grammar which refines ourmodel of a secondary structure. It corresponds to RNA-FOLD -noLP -d16 and is used in the 2004 release ofRNASHAPES[3] for the computation of representativestructures of different shape.
MicroState has separate rules for a helix end with twobases, one base or no base dangling onto it (see Figure3). These four cases compete with each other for mini-mum free energy. If surrounding bases are already basepaired, only the drem case applies (no dangles). If it isdecided (say) that the left neighboring base dangles ontothe helix, then this base is not available for also danglingon another helix. In this way, grammar MicroState cor-rectly finds the structure of minimal free energy, andcould, in principle, also explicitly report the optimaldangles, as in “..b((...))d((...))...“.All variants of the same secondary structure, augmen-
ted with different dangles, are now separate members ofthe folding space. In contrast to the classical model,accounting only for base pairs, we call them “micro-states”. Let us derive a rough estimate of this foldingspace enlargement. The size of the folding space for asequence of length n grows asymptotically with a · bn ·n -3/2, with b = 1.44358 and a = 3.45373 [8]. A structurehas, on average, k(n) helices, where k grows with n.Each helix end has up to four ways to play with thedangles, but helix ends in hairpin loops do not count.Directly adjacent helices further reduce the number ofdangling alternatives.Let us, for simplicity, assume that an helix has 4 dan-
gle variants on average. Then, the above formulachanges for the number of microstates to a · 4k(n) · bn ·n -3/2. An empirical measurement is shown in Figure 4.From the measurements, and for their particular datasequences and lengths, we can estimate k(n) ≈ n
15 . Fora sequence of length 100, for example, we see anincrease by a factor of 104. Clearly, this is a substantialenlargement of the folding space, and different struc-tures are affected to a different extent. (For example, theopen structure (no base pairs) gives rise to only onemicrostate.)This enlargement of the search space is not a problem
for MFE structure prediction. The dynamic program-ming algorithm derived from the grammar MicroStateonly does a constant amount of extra work compared toNoDangle and OverDangle. But a severe problem arises
Figure 3 Grammar MicroState extends the rules of grammars NoDangle or OverDangle for the non-terminal symbols “dangle” and“multiloop”. Instead of just one way, we now have four alternatives to dangle bases onto a closed substructure: Both neighboring bases donot dangle (drem and ml), only the left neighbored base dangles onto the stack (edl and mldl), only the right one (edr and mldr), or both ones(edlr and mldlr).
Janssen et al. BMC Bioinformatics 2011, 12:429http://www.biomedcentral.com/1471-2105/12/429
Page 8 of 19

with the desire to investigate near-optimal structures.The roughly 4k microstates of an optimal structure withk helices crowd the near-optimal folding space, whilerepresenting the same structure in the non-danglingsense. Enumerating suboptimals returns a tremendousamount of useless information. RNASUBOPT thereforeuses OverDangle for enumeration, even when option-d1 is specified. Afterwards, it re-evaluates the energy ofpredicted structures using correct dangling. Hence, theranking of structures may change. Occasionally, weobserve that the energy of the true MFE structure is somuch above the energy of other, overdangled structuresthat it falls above the energy threshold for enumerationand is not returned at all.7
The second problem arises with computations that arebased on Boltzmann statistics. The partition function Qsums up the Boltzmann-weighted energies of all mem-bers in the folding space. Each secondary structure con-tributes to the partition function as many times as it hasmicrostates, hence the result would be skewed towardsstructures with many microstates. The significance ofthis bias is hard to judge8, and up to this study, it couldnot be evaluated empirically. For this reason, RNAFOLDdoes not support partition function computation withthe MicroState model (option -d1).Fortunately, the partition function with correct dan-
gles, avoiding overdangling as well as explosion of thefolding space, can also be computed. To keep the fold-ing space simple, we need a more sophisticated gram-mar: MacroState.
Model MacroStateGrammar MacroState (see Figure 5) follows the overallpattern of the other grammars, but is much morerefined. This grammar was designed originally with the2006 release of RNASHAPES[6] to compute completeprobabilistic shape analysis. Its rules are written torecord and distinguish the situation where a helix (1)ends with a base pair, (2) already has a single unpairedbase to its right or left, or (3) has several unpaired baseson either side. No dangle energies are added in cases (1)and (3), and in case (2), all possible dangle variants (upto four microstates) are evaluated and minimized overwhile considering the corresponding macrostate. Thisleads to a much larger number of non-terminal symbolsand functions in the grammar. MacroState has 25 non-terminal symbols and 32 functions, compared toNoDangle with 11 non-terminals and 12 functions.The important feature of MacroState is that for any
sequence, it defines the identical folding space asNoDangle. This is hard to believe when just looking atthe grammar, but has been shown in [6], and is furtherdemonstrated by the measurements shown in Figure 4.The size of the folding space, as defined by MacroState,agrees with that of NoDangle and OverDangle not onlyon average, but also on each individual sequence.What is the effect of using either MicroState or
MacroState? Does it really matter? Table 4 shows anextreme example of how the choice of the state spaceaffects the computed probabilities:In this example, 40% of the probability mass is shifted
by switching models, causing the order of the two top-ranking shapes to be reversed. To find out whether thissituation is the exception or the rule is a main motiva-tion of this study.
Results & DiscussionData setsThe four data sets used in this study, DARTS, FR3D:3A,FR3D:4A, and RNAstrand:91 are based on RNA 3Dstructure data sets prepared in the context of previouslypublished studies.Structures drawn from PDBWe examined three datasets - DARTS, FR3D:3A, andFR3D:4A- based on RNA 3D structural data sets pre-pared in the context of previously published studies. Allthree original data sets were created in order to reflectthe currently available structural repertoire of RNAmolecules as given by structures solved experimentallyby X-ray and NMR analysis.The DARTS set was used for the analysis and classifi-
cation of RNA tertiary structures in [17]. It was builtfrom all structures available in the March 2007 versionof the Protein Data Bank (PDB) [18,19]. The DARTSdata set is available at http://bioinfo3d.cs.tau.ac.il/
Figure 4 Growth of folding spaces for all four grammars. Weused uniformly distributed random sequences, with step-size 5 bp.The number of secondary structures heavily depends on sequencecomposition, thus we took the average over 100 sequences perdata point. Curves for “MacroState” and “OverDangle” are not visible,because they are perfectly overlayed by “NoDangle”, i. e. all threefolding spaces have exactly the same size.
Janssen et al. BMC Bioinformatics 2011, 12:429http://www.biomedcentral.com/1471-2105/12/429
Page 9 of 19

DARTS and contains 244 structures. The creation ofthis data set involved dedicated structural comparisonsto ensure pairwise structural and sequence variability.Unfortunately, the DARTS database is not updated any-more and therefore is limited to data deposited in thePDB before March 2007.
Figure 5 “MacroState” grammar. The color code is identical to Figure 2. The basic structure of the “MacroState” grammar is inherited from theprevious three grammars, but it has a more complex distinction of cases for dangling bases. “MacroState” has to consider all the differentdangling situations as in “MicroState”, but its search space is restricted to the k(n)-times smaller folding space of the input sequence. To achievethese contradicting goals, dangling alternatives do not exist as search space candidates but are implicitly examined within the evaluationalgebra. The grammar has to ensure that a substructure is of a defined dangling type whenever its energy or partition function value is used inan algebra evaluation function. We know that any helix derivated from nodg has no unpaired bases to its left or right, while helices from edgl,edgr or edglr have exactly one unpaired base dangling from left, right or exactly two unpaired bases dangling from both sides, respectively. In allfour cases, there is no unpaired base left for a further dangling. Care must be taken, where we can not be sure if e. g. the leftmost unpairedbase of a block_dl derivation is free to dangle to some helix to its left. The unpaired base would be available for a dangling if we use ssadd, butis occupied in incl situations. This uncertainty is passed to every calling function, but with a clever grammar design we can at least ensure thatits type does not change. For example every mc1 or mcadd2 derivation contains one or more helices with one or more unpaired bases at its 5’end and definitely no unpaired base at its 3’ end. Furthermore mc2 and mcadd1 always have no unpaired bases to both sides, mc3 or mcadd4have one or more unpaired bases only at its 3’ end and finally mc4 or mcadd3 are known to have one or more unpaired bases to both ends.The benefit of these distinctions can be demonstrated with the multiloop functions mldl and mladl. The important base is the one that isdirectly left to the mc1 or mc2 substructure. In principle, it can either dangle to the left, that is the closing stem of the multiloop, or the right,that is the leftmost helix within the multiloop. Actually, for mldl our base of interest can only dangle to the left, because every mc1 derivationalready has at least one further base in front of the first inner helix. For mladl we truly have an ambiguous situation, where the base of interestcould dangle to one of both sides. Please note that mldl and mladl correspond to two different dot-bracket structures. mldl handles macrostatesof the type “((...“ including microstates “((...“ and “((d..“, whereas mladl handles macrostates of type “((.((...“ and includes themicrostates “((.((...“, “((d((...“, and “((b((...“. The mfe algebra function locally chooses the variant with the better free energy,even if a global analysis would reveal that the locally worse structure would become MFE in the end. This constitutes a rare case where the MFEstructure may be missed. Our partition function algebra correctly keeps track of these situations.
Table 4 Extreme probability shift example
GACCAAAGCCUUUGUCCCACAAAUUGCGAUCGCGUCGCGGAGC
MacroState prob. MicroState prob. shape class
58.44% 32.58% [][]
29.32% 63.43% [[][]]
12.24% 03.99% []
Janssen et al. BMC Bioinformatics 2011, 12:429http://www.biomedcentral.com/1471-2105/12/429
Page 10 of 19

The two FR3D data sets [20,21] are representative setsbased on all RNA X-ray structures with a resolution of upto 3 Å (246 structures containing 653 chains) and up to 4Å (293 structures containing 764 chains), respectively, thatwere contained in the PDB in 2010. Both sets contain onerepresentative structure for each group of RNA structuresfound similar (or identical) according to the employedsequence (> 95% identity) and structural (cf. [21]) similar-ity cutoffs. Both data sets FR3D:3A and FR3D:4A are avail-able as weekly updated lists at http://rna.bgsu.edu/FR3D.The FR3D data sets were created taking recently solvedstructures into consideration and therefore represent thecurrently known RNA 3D structural space. Here, theFR3D:3A set is restricted to structures that have beensolved at a better resolution and may therefore be morereliable than structures contained in the FR3D:4A set. Inturn, the FR3D:4A set has a less strict resolution cutoffand therefore contains more structures.From PDB structures to “gold” structuresIn order to generate the data sets for this study, wedownloaded all 3D structures contained in the originaldata sets from the PDB and extracted the secondarystructures of each RNA chain using the stereo-geometri-cal information encoded within the atomic coordinates.Each chain was processed with the base pair annotationsoftware tool MC-Annotate [22] resulting in a list of allintramolecular contacts in the chain. For this study, weonly used base pair interactions that are formallyinvolved in secondary structure formation, namely thecis Watson-Crick (cWC) base pairs (G:C, C:G, A:U, U:A,G:U, U:G). All other interactions, such as non-canonicalbase pairs, base stackings, and base-backbone interac-tions were ignored since they are not part of the sec-ondary structure. The secondary structure of an RNAchain could then be reconstructed directly from theordered list of canonical base pairs. In a next step, this“preliminary” structure was scanned for lonely basepairs and pseudoknot interactions. Since lonely basepairs are thermodynamically unstable in a secondarystructure, they were removed from the list. Due to thefact that there is no unique solution to remove the knot(s) from a pseudoknotted structure, these structures areunusable for the purpose of our study. Therefore, struc-tures containing pseudoknots larger than one base pair,were also discarded. We consider the set of structuresreduced in this way as the set of “gold” structures. Theyconstitute our standard of truth, but we are reluctant tocall them “true” structures, not only because of ourremoval of information, but also since structures in cris-tallo may be different from structures in vivo9.Our gold data sets resulting from DARTS, FR3D:3A,
and FR3D:4A consist of 147, 111, and 136 structures,respectively.
As a final detail: in a few cases, FR3D:3A andFR3D:4A contain the same sequence, with differentresolution in 3D and with different secondary structurederived from it. No secondary structure prediction pro-gram can be expected to be correct in both cases.A data set derived from RNAstrand Aside from thesedata sets, we also created a data set RNAstrand:91with 91 structures from the RNAstrand database [23].Since RNAstrand was designed as a source of validatedstructures, with an eye on the evaluation of RNA-related bioinformatics tools, it will be interesting toobserve if the findings on this data set agree with theothers.Overall, we shall find that our four data sets deliver
consistent sets of results. Therefore, the text of this arti-cle will discuss only selected measurements in detail,with the other ones given in the additional file 1, as wellas all four raw data sets in additional file 2.
Evaluation of models for MFE structure predictionWhile our main interest is in the effect of the chosenmodel on the partition function based computations, wehere evaluate the four grammars with respect to predic-tion of a single MFE structure.Evaluation setupIn evaluating models with respect to MFE structure pre-diction, we include not only our programs NoDangleand OverDangle, MicroState and MacroState, but alsothe folding programs UNAFOLD and RNAFOLD, whichour readers are rightfully curious about because of theirpractical importance. Turner’99 parameters [1] wereused throughout10. These parameters are derived frommelting experiments, with a few exceptions. Multiloopparameters such as ml_energy in Turner’99 are notderived from experiment, but are optimized from struc-ture data to be used in conjunction with the MicroStatemodel. Out of competition, we also include CEN-TROIDFOLD, which goes beyond strict energy minimi-zation by producing a near-optimal ensemble ofstructures and choosing the eventual, single-structureprediction based on this sample.Relative performance of programs of different origin
is, however, not our main interest here. Mainly, the eva-luation should support that our four grammars faithfullyreproduce the behavior of the models underlying RNA-FOLD with options -d0, -d1, and -d2, as postulated atthe outset of this study.The data set in this evaluation is DARTS. Evaluation
results are summarized in Figure 6. We use an asym-metric base pair distance for comparison, as explainedwith Figure 6, where one structure (row entry) is treatedas the prediction, the other as the reference (columnentry).
Janssen et al. BMC Bioinformatics 2011, 12:429http://www.biomedcentral.com/1471-2105/12/429
Page 11 of 19

Observations from MFE prediction experimentConsistency of implementations Naturally, comparingthe results from the same tool leads to entries of zerobase pair distance in the diagonal of Figure 6. The off-diagonal zero entries, however, are quite remarkable.When two different algorithms perfectly agree in theirMFE predictions on the complete data set, this providesstrong evidence that they both faithfully implement thesame thermodynamic model of the folding space in each
of its variants. In particular, this shows that our gram-mars NoDangle/OverDangle and MicroState indeed cap-ture the analysis computed by RNAFOLD with options-d0/d2 and -d1. The perfect zeroes might even makeour reader suspicious! Occasionally, there must be two(or more) co-optimal structures of minimal free energy,and it is not formally defined which one a programshould return in this situation. Hence, it is accidentalwhether or not two different programs, implemented by
reference1:pd 2:go 3:RN 4:No 5:UN 6:RN 7:Ma 8:Mi 9:UN 10:RN 11:Ov 12:Ce 13:Ce
prediction
1: pdb structure 0 44
2: gold structure 30 0
3: RNAfold -d0 657 633 0 0 224 324 317 317 483 418 417 441 530
4: NoDangle 631 605 0 0 188 286 284 284 463 362 362 415 496
5: UNAfold –nodangle 701 676 222 186 0 429 418 418 367 416 411 491 554
6: RNAfold -d1 562 531 312 278 423 0 0 0 271 173 171 330 447
7: MacroState 552 521 305 272 412 0 0 0 262 171 169 322 433
8: MicroState 552 521 305 272 412 0 0 0 262 171 169 322 433
9: UNAfold 593 564 471 451 356 265 256 256 0 294 287 412 485
10: RNAfold -d2 608 572 427 375 428 190 188 188 320 0 0 364 479
11: OverDangle 606 570 426 375 423 188 186 186 313 0 0 360 469
12: CentroidFold McCaskill 278 254 244 216 296 126 124 124 243 142 140 0 196
13: CentroidFold CONTRAfold 375 353 372 337 393 293 284 284 346 307 299 234 0
Figure 6 Comparison of different MFE prediction programs. Dataset: we use the 147 sequences from the DARTS set, except pdb1ajt1B,pdb1kod1A, pdb1koc1A, pdb1lpw1B and pdb1t4x1B, which crashed under UNAFOLD. Together, all according “PDB” structures contain 1,614base pairs. All “gold” structures have 1,593 base pairs. Distance: One base pair set, i.e. secondary structure, is the reference (R: table columns),the other one is the prediction (P: table rows). Traditional base pair distance is defined as|R \P| + |P \R|. Following [34], we decide to allowadditional base pairs in the prediction, as long as they are compatible with the reference, i.e. both bases are unpaired and the additional basepair does not introduce a pseudoknot in the reference. The set of compatible base pairs is P-c = P\{(a, b)|(a, b) ∉ R Λ (a, b) compatible to R}.Then, our asymmetric base pair distance is: |R \P| + |P-c \R|. Table values are the sums of base pair distances for all 142 sequences. In the case ofco-optimal results, the one with the smallest distance to the reference is chosen. Our distance function is rather strict and does not allow basepair slippage. If a gold base pair (i, j) is mispredicted as (i + 1, j), this contributes a distance of 2. Programs: for each RNA sequence we calledthe programs with the following command line options: RNAFOLD (version 1.8.5): echo sequence | RNAfold -noPS -noLP -dX, where Xis 0, 1 or 2. UNAFOLD (version 3.8): hybrid-ss-min –suffix = DAT –mfold –NA=RNA –tmin = 37 –tinc = 1 –tmax = 37–sodium = 1 –magnesium = 0 –noisolate –nodangle tmpseqfile >/dev/null && ct2b.pl tmpseqfile.ct, with andwithout the –nodangle switch, where “tmpseqfile” is a fasta file containing the sequence and “ct2b.pl” is a small Perl script from the ViennaPackage, which converts RNA structures from “connect” to “dot-bracket” format. CENTROIDFOLD (version v0.0.9): centroid_fold–engine=X tmpseqfile, where X is the source of base pair probabilities and is either computed by RNAFOLD (McCaskill) or byCONTRAFOLD. Our ADP implementation of the four grammars “NoDangle”, “OverDangle”, “MicroState” and “MacroState” get the sequence astheir sole input. The binaries can be built with the source code from the additional file 3 and the Bellman’s GAP compiler.
Janssen et al. BMC Bioinformatics 2011, 12:429http://www.biomedcentral.com/1471-2105/12/429
Page 12 of 19

different programmers, make the same choice. Wetherefore have designed our new programs to report allco-optimal solutions in such a situation, and thenchoose the structure closest to the RNAFOLD predic-tion. This always delivered a perfect match.We apply the same technique of safe-guarding against
co-optimals when comparing to a database structure.Note that in practice, when predicting structure for anovel RNA, the users of a structure prediction programhave no reference structure to resort to. In this case,reporting all co-optimal structures makes them aware ofthe ambiguity of the situation, and leaves them with thechoice to make. This is somewhat preferable to quietlyreporting a single MFE structure, selected from severalby implementation peculiarities.The perfect agreement of MacroState with the MFE
prediction of RNAFOLD -d1 as well as with MicroStatedemonstrates that MacroState in fact computes theenergy model of the other two programs, while avoiding(as explained above) their explosion of the state space.Taken together, these consistency results shows that wehave correct programs set up for our second experi-ment, where we will evaluate the effect of the chosenenergy and state space model on partition functioncalculations.Quality of MFE predictions Overall, the quality of MFEpredictions compared to “real” structures is moderatewhen measured on the individual base pair level, witherrors11 ranging from 16% to 21% for the gold structures.This is expected and well-known. It is the reason whyresearchers have developed more advanced techniques,such as structure sampling, complete enumeration, orshape abstraction. The PDB structures contain base pairswhich by definition are not predicted - non-standardpairs, 3D interactions, pseudoknots, and lonely pairs. Asexplained above, the data set of gold structures has beencleaned up in these respects, and as expected, the predic-tions come closer, but deviations are still considerable.The gold structures are best predicted by MacroState
and MicroState (distance 521) and RNAFOLD -d1 (dis-tance 531). The small difference is accidental and arisesfrom the rare case where RNAFOLD picks an unluckychoice from several co-optimal structures.Performance of different dangling models Comparingthe full dangling model (MicroState, MacroState) to itsupper and lower approximations NoDangle and Over-Dangle, we find that its proper implementation pays off.It reduces the accumulated distance by about 14% overNoDangle, and by 9% over OverDangle. Similar percen-tages apply for RNAFOLD option -d1 versus -d0 and-d2. This also shows that OverDangle approximates thecorrect model better than NoDangle and justifies its useas a substitute for the full model in partition function
calculations with RNAFOLD and RNASUBOPT, wherethe grammar MacroState is not available.unafold performance The two versions of UNAFOLDconsistently score a bit worse against the gold structuresthan all other programs. Compared to each other, wealso observe that the distance is improved by consider-ing dangling energies, here by 17%. Otherwise, the twoUNAFOLD versions cluster with the NoDangle/Micro-State groups, as they should12.Looking deeper into the near-optimal folding spaceWe included CENTROIDFOLD[24] as a representativeof methods which, in contrast to the above programs,look deeper into the Boltzmann ensemble of near-opti-mal structures. Our evaluation shows that the extraeffort is well spent. CENTROIDFOLD comes closest tothe good structures, and with respect to the single struc-ture predictors, it corresponds best with the group ofRNAFOLD -d1, MacroState and MicroState.
Evaluating models for partition function and relatedcomputationsWe will explain our evaluations in detail based on ourlargest data set, DARTS. Results on the other data setsare obtained in an analogous way and are summarizedin the end of this section.Evaluation CriteriaIn this section, we apply probabilistic shape analysis toour data set. We are interested in the difference of per-formance of the four models NoDangle, OverDangle,MicroState and MacroState. For simplicity, we call theabstract shape of the reference structure the “referenceshape”, and refer to the most likely predicted shape asthe “dominant shape”, although its actual dominancewithin the Boltzmann ensemble will not be strong ifthere is another shape with similar probability. Theshape string of the reference shape of sequence s isobtained by a call to RNAshapes -t l -D “s“, where 1is one of the five shape abstraction levels.We ask the following questions:
• What are the differences in the shape probabilitiescomputed with each of the four models?• How is the difference affected by the shapeabstraction level considered?
Since we do observe significant differences in modelbehavior, we also ask which model comes closer to thetruth:
• To what extend does the dominant shape agreewith the reference shape?• What is the median (or the 75% and 90% quantile)of the reference shape among the predicted shapes?
Janssen et al. BMC Bioinformatics 2011, 12:429http://www.biomedcentral.com/1471-2105/12/429
Page 13 of 19

Finally, we consider
• What are the runtime or memory trade-offs forcomputing with different models?
Evaluation method Shape probabilities do not make astructure prediction per se. They provide holistic infor-mation by assigning probabilities to all shapes in thefolding space of a sequence x. It is our responsibilityhow we interpret theses data. The hope is, of course, tofind the biologically functional structure among thehigh-probability shapes, to find two high probabilityshapes for a riboswitch, to use lack of any shape withhigh probability as an indicator of absence of a well-defined structure, and so on. Such analysis goes beyondshape probabilities, and takes into account the concreteshreps returned for each shape.Independent of what the shape probabilities will be
used for, we want to focus on the agreement betweenthe four grammars. To measure this, we use the shapeprobability shift (SPS). For a given sequence x, all gram-mars will report the same shape classes, but with differ-ent probabilities. Let P (x) be the shape space, i. e. theset of all shape classes for x, and ProbG(p) the shapeprobability of p under grammar G. The shape probabil-ity shift for x and grammars A and B is defined as:
SPSA,B(x) =12·
∑p∈P(x)
| ProbA(p)− ProbB(p) | (2)
Note that 0 ≤ SPS(x) ≤ 1, where the extreme case of 1would only be achieved when all shapes with positiveprobability by grammar A have zero probability bygrammar B and vice versa. The SPS can be interpretedas the overall probability mass that moves betweenshapes.We chose the SPS measure because of this nice inter-
pretation. We also evaluated two alternative measures.The squared distance of base pair probability matrices iscorrelated with the SPS by a factor around 0.83 at shapelevel 5 and not much lower on less abstract shape levels.The Kullback-Leibler divergence turned out to be unsui-table for the purpose, as it is not symmetric and bothversions (KL(x, y) versus KL(y, x)) show the poorest cor-relation among all methods tested. Details of this inves-tigation of alternatives are given in additional file 1.Observations The values in Figure 7 are average SPS13
over all x Î DARTS, which is the largest of our datasets.First, consider shape abstraction level 5. We find that
models MacroState and MicroState show the mostagreement, where the SPS is around 3.7%. MacroStateshows a significant SPS against the others, strongestagainst NoDangle (9.6%) but also against OverDangle(5.7%). A SPS in this range means that while in many
cases, the predicted dominant shape will be the samefor all models, this need not hold in general.This justifies the question which of the model finds
the gold shape as the dominant shape more often (seebelow). By the way: the dominant shape and the shapeof the MFE structure agree for MacroState in 143 out of147 cases.Let us next turn from level 5 to decreasinging levels of
abstraction. Moving to abstraction levels 4, 3, 2, and 1,the number of shapes increases with each step, whileeach shape class holds a smaller number of structures.The overall relationship between the models on levels 4through 1 is consistent with what we observe for level 5.Overall, the SPS values increase. A closer inspection ofthe raw data shows that SPS values actually decrease foreach individual shape, but due to the larger number of(smaller) shifts, their sum increases. Evidence is pro-vided in Figure 8.Dominant shape is gold shape? The values in Table 5show the ratios of correct shape predictions vs. the sizeof the testset, which is 147 in the case of DARTS. Weobserve the following:The best ratio of agreement of dominant shape and
gold shape is 82.3%. The fact that this value is not higheris the reason which makes investigators look into severalhigh-probability shapes and their shreps in practice.Comparing the models, we find that there is no clearwinner, with a margin of only 2.7% between the best andthe worst performer. (Moreover, the first position variesover our data sets.) Here, MacroState finds agreementmost often, with a 0.7% margin over MicroState and 1.3%margin over NoDangle. OverDangle performs worst(79.6%), but not hopeless when we consider that one willlook at a number of top-ranking shapes anyway.Thus, the more interesting question is how the gold
shape is placed among the predicted shapes - cf. Table6. We investigate this aspect by compiling a list of rank(pgold) for all 147 testsequences, sorting this list ascend-ingly and report the median (50%), the 75%, and the90% quantile of the list, as well as the complete list(100%). For example, the value 2 for MacroState inshape abstraction level 5 in the 90% column means that,if we decide to take only the top two shapes for closerstudy, the gold shape is among them in 90% of thecases. Three top shapes are suffice to reach this cover-age with MicroState and OverDangle. Overall, theadvantage of MacroState appears marginal over theother grammars on level 5, and appears somewhat ran-domized for weaker abstraction levels.An unexpected observation is the strong performance
of shape level 2. Considering the 75% quartile, 3 shapessuffice to find the gold shape, independent of the modelchosen. We will return to this observation in theConclusion.
Janssen et al. BMC Bioinformatics 2011, 12:429http://www.biomedcentral.com/1471-2105/12/429
Page 14 of 19

Ma Mi Ov No
Ma 0.000 0.037 0.057 0.096
Mi 0.037 0.000 0.054 0.128
Ov 0.057 0.054 0.000 0.141
No 0.096 0.128 0.140 0.000
shape level 5
Ma Mi Ov No
Ma 0.000 0.043 0.066 0.122
Mi 0.043 0.000 0.070 0.150
Ov 0.066 0.070 0.000 0.161
No 0.122 0.150 0.160 0.000
shape level 4
Ma Mi Ov No
Ma 0.000 0.047 0.071 0.132
Mi 0.047 0.000 0.078 0.162
Ov 0.071 0.078 0.000 0.169
No 0.132 0.162 0.168 0.000
shape level 3
Ma Mi Ov No
Ma 0.000 0.048 0.072 0.137
Mi 0.048 0.000 0.081 0.167
Ov 0.072 0.081 0.000 0.172
No 0.137 0.168 0.171 0.000
shape level 2
Ma Mi Ov No
Ma 0.000 0.082 0.113 0.196
Mi 0.084 0.000 0.152 0.252
Ov 0.113 0.152 0.000 0.206
No 0.196 0.250 0.204 0.000
shape level 1
Ma = MacroStatesMi = MicroStatesOv = OverDangleNo = NoDangle
Figure 7 Model similarity: shape probability shift.
Ma Mi Ov No
Ma 0.000 0.014 0.019 0.037
Mi 0.013 0.000 0.017 0.046
Ov 0.014 0.015 0.000 0.043
No 0.040 0.055 0.057 0.000
shape level 5
Ma Mi Ov No
Ma 0.000 0.009 0.006 0.027
Mi 0.009 0.000 0.011 0.032
Ov 0.005 0.010 0.000 0.028
No 0.026 0.034 0.032 0.000
shape level 4
Ma Mi Ov No
Ma 0.000 0.009 0.005 0.026
Mi 0.008 0.000 0.010 0.031
Ov 0.004 0.010 0.000 0.026
No 0.025 0.033 0.030 0.000
shape level 3
Ma Mi Ov No
Ma 0.000 0.006 0.002 0.017
Mi 0.006 0.000 0.007 0.022
Ov 0.002 0.006 0.000 0.018
No 0.017 0.021 0.018 0.000
shape level 2
Ma Mi Ov No
Ma 0.000 0.005 0.001 0.012
Mi 0.005 0.000 0.006 0.017
Ov 0.001 0.005 0.000 0.012
No 0.012 0.016 0.012 0.000
shape level 1
Ma = MacroStatesMi = MicroStatesOv = OverDangleNo = NoDangle
Figure 8 Model similarity: average shape probability shift per shape.
Janssen et al. BMC Bioinformatics 2011, 12:429http://www.biomedcentral.com/1471-2105/12/429
Page 15 of 19
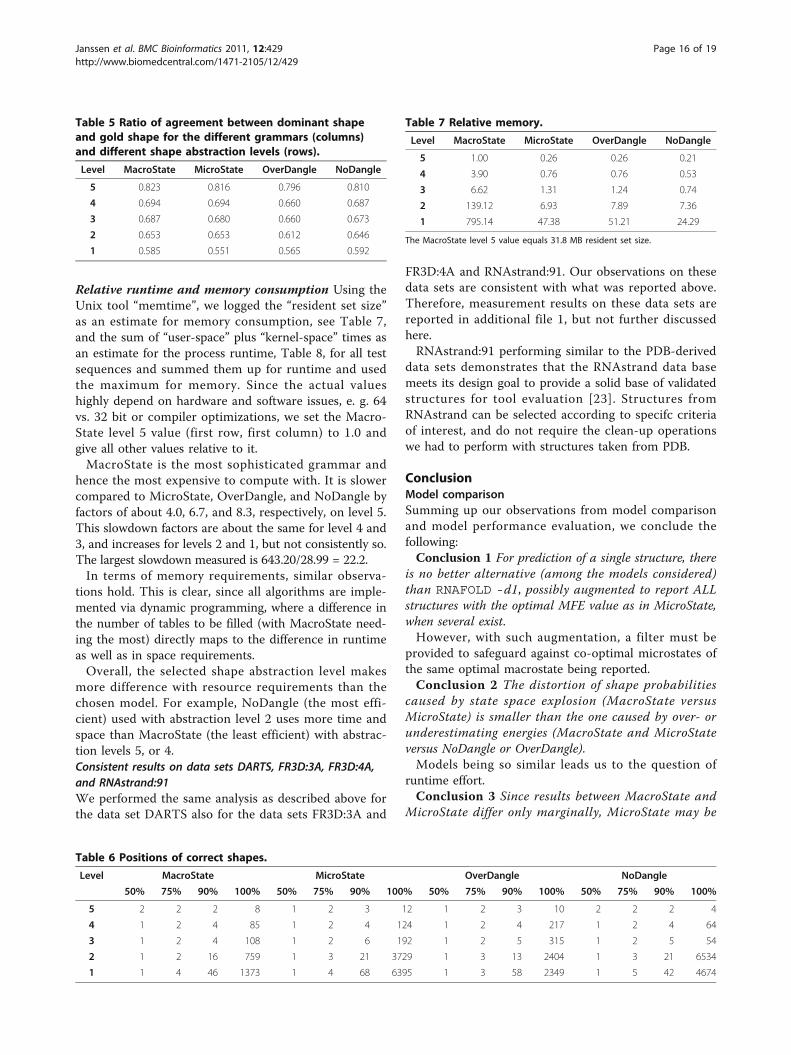
Relative runtime and memory consumption Using theUnix tool “memtime”, we logged the “resident set size”as an estimate for memory consumption, see Table 7,and the sum of “user-space” plus “kernel-space” times asan estimate for the process runtime, Table 8, for all testsequences and summed them up for runtime and usedthe maximum for memory. Since the actual valueshighly depend on hardware and software issues, e. g. 64vs. 32 bit or compiler optimizations, we set the Macro-State level 5 value (first row, first column) to 1.0 andgive all other values relative to it.MacroState is the most sophisticated grammar and
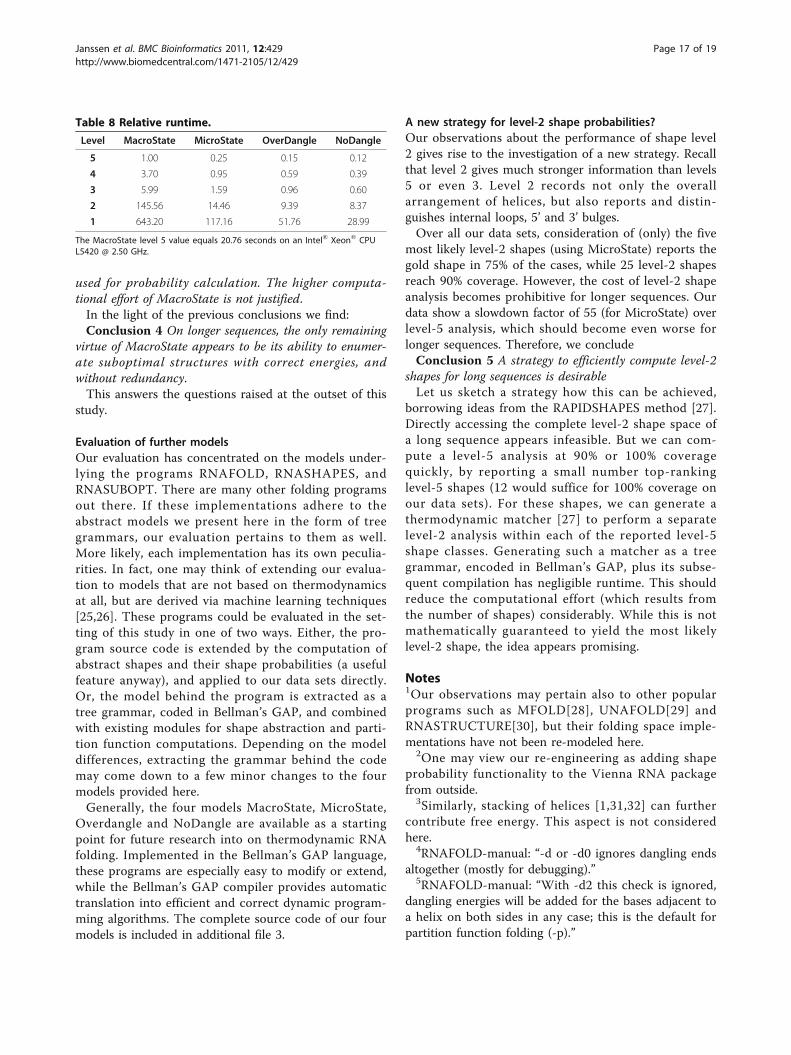
hence the most expensive to compute with. It is slowercompared to MicroState, OverDangle, and NoDangle byfactors of about 4.0, 6.7, and 8.3, respectively, on level 5.This slowdown factors are about the same for level 4 and3, and increases for levels 2 and 1, but not consistently so.The largest slowdown measured is 643.20/28.99 = 22.2.In terms of memory requirements, similar observa-
tions hold. This is clear, since all algorithms are imple-mented via dynamic programming, where a difference inthe number of tables to be filled (with MacroState need-ing the most) directly maps to the difference in runtimeas well as in space requirements.Overall, the selected shape abstraction level makes
more difference with resource requirements than thechosen model. For example, NoDangle (the most effi-cient) used with abstraction level 2 uses more time andspace than MacroState (the least efficient) with abstrac-tion levels 5, or 4.Consistent results on data sets DARTS, FR3D:3A, FR3D:4A,and RNAstrand:91We performed the same analysis as described above forthe data set DARTS also for the data sets FR3D:3A and
FR3D:4A and RNAstrand:91. Our observations on thesedata sets are consistent with what was reported above.Therefore, measurement results on these data sets arereported in additional file 1, but not further discussedhere.RNAstrand:91 performing similar to the PDB-derived
data sets demonstrates that the RNAstrand data basemeets its design goal to provide a solid base of validatedstructures for tool evaluation [23]. Structures fromRNAstrand can be selected according to specifc criteriaof interest, and do not require the clean-up operationswe had to perform with structures taken from PDB.
ConclusionModel comparisonSumming up our observations from model comparisonand model performance evaluation, we conclude thefollowing:Conclusion 1 For prediction of a single structure, there
is no better alternative (among the models considered)than RNAFOLD -d1, possibly augmented to report ALLstructures with the optimal MFE value as in MicroState,when several exist.However, with such augmentation, a filter must be
provided to safeguard against co-optimal microstates ofthe same optimal macrostate being reported.Conclusion 2 The distortion of shape probabilities
caused by state space explosion (MacroState versusMicroState) is smaller than the one caused by over- orunderestimating energies (MacroState and MicroStateversus NoDangle or OverDangle).Models being so similar leads us to the question of
runtime effort.Conclusion 3 Since results between MacroState and
MicroState differ only marginally, MicroState may be
Table 5 Ratio of agreement between dominant shapeand gold shape for the different grammars (columns)and different shape abstraction levels (rows).
Level MacroState MicroState OverDangle NoDangle
5 0.823 0.816 0.796 0.810
4 0.694 0.694 0.660 0.687
3 0.687 0.680 0.660 0.673
2 0.653 0.653 0.612 0.646
1 0.585 0.551 0.565 0.592
Table 6 Positions of correct shapes.
Level MacroState MicroState OverDangle NoDangle
50% 75% 90% 100% 50% 75% 90% 100% 50% 75% 90% 100% 50% 75% 90% 100%
5 2 2 2 8 1 2 3 12 1 2 3 10 2 2 2 4
4 1 2 4 85 1 2 4 124 1 2 4 217 1 2 4 64
3 1 2 4 108 1 2 6 192 1 2 5 315 1 2 5 54
2 1 2 16 759 1 3 21 3729 1 3 13 2404 1 3 21 6534
1 1 4 46 1373 1 4 68 6395 1 3 58 2349 1 5 42 4674
Table 7 Relative memory.
Level MacroState MicroState OverDangle NoDangle
5 1.00 0.26 0.26 0.21
4 3.90 0.76 0.76 0.53
3 6.62 1.31 1.24 0.74
2 139.12 6.93 7.89 7.36
1 795.14 47.38 51.21 24.29
The MacroState level 5 value equals 31.8 MB resident set size.
Janssen et al. BMC Bioinformatics 2011, 12:429http://www.biomedcentral.com/1471-2105/12/429
Page 16 of 19
used for probability calculation. The higher computa-tional effort of MacroState is not justified.In the light of the previous conclusions we find:Conclusion 4 On longer sequences, the only remaining
virtue of MacroState appears to be its ability to enumer-ate suboptimal structures with correct energies, andwithout redundancy.This answers the questions raised at the outset of this
study.
Evaluation of further modelsOur evaluation has concentrated on the models under-lying the programs RNAFOLD, RNASHAPES, andRNASUBOPT. There are many other folding programsout there. If these implementations adhere to theabstract models we present here in the form of treegrammars, our evaluation pertains to them as well.More likely, each implementation has its own peculia-rities. In fact, one may think of extending our evalua-tion to models that are not based on thermodynamicsat all, but are derived via machine learning techniques[25,26]. These programs could be evaluated in the set-ting of this study in one of two ways. Either, the pro-gram source code is extended by the computation ofabstract shapes and their shape probabilities (a usefulfeature anyway), and applied to our data sets directly.Or, the model behind the program is extracted as atree grammar, coded in Bellman’s GAP, and combinedwith existing modules for shape abstraction and parti-tion function computations. Depending on the modeldifferences, extracting the grammar behind the codemay come down to a few minor changes to the fourmodels provided here.Generally, the four models MacroState, MicroState,
Overdangle and NoDangle are available as a startingpoint for future research into on thermodynamic RNAfolding. Implemented in the Bellman’s GAP language,these programs are especially easy to modify or extend,while the Bellman’s GAP compiler provides automatictranslation into efficient and correct dynamic program-ming algorithms. The complete source code of our fourmodels is included in additional file 3.
A new strategy for level-2 shape probabilities?Our observations about the performance of shape level2 gives rise to the investigation of a new strategy. Recallthat level 2 gives much stronger information than levels5 or even 3. Level 2 records not only the overallarrangement of helices, but also reports and distin-guishes internal loops, 5’ and 3’ bulges.Over all our data sets, consideration of (only) the five
most likely level-2 shapes (using MicroState) reports thegold shape in 75% of the cases, while 25 level-2 shapesreach 90% coverage. However, the cost of level-2 shapeanalysis becomes prohibitive for longer sequences. Ourdata show a slowdown factor of 55 (for MicroState) overlevel-5 analysis, which should become even worse forlonger sequences. Therefore, we concludeConclusion 5 A strategy to efficiently compute level-2
shapes for long sequences is desirableLet us sketch a strategy how this can be achieved,
borrowing ideas from the RAPIDSHAPES method [27].Directly accessing the complete level-2 shape space ofa long sequence appears infeasible. But we can com-pute a level-5 analysis at 90% or 100% coveragequickly, by reporting a small number top-rankinglevel-5 shapes (12 would suffice for 100% coverage onour data sets). For these shapes, we can generate athermodynamic matcher [27] to perform a separatelevel-2 analysis within each of the reported level-5shape classes. Generating such a matcher as a treegrammar, encoded in Bellman’s GAP, plus its subse-quent compilation has negligible runtime. This shouldreduce the computational effort (which results fromthe number of shapes) considerably. While this is notmathematically guaranteed to yield the most likelylevel-2 shape, the idea appears promising.
Notes1Our observations may pertain also to other popularprograms such as MFOLD[28], UNAFOLD[29] andRNASTRUCTURE[30], but their folding space imple-mentations have not been re-modeled here.
2One may view our re-engineering as adding shapeprobability functionality to the Vienna RNA packagefrom outside.
3Similarly, stacking of helices [1,31,32] can furthercontribute free energy. This aspect is not consideredhere.
4RNAFOLD-manual: “-d or -d0 ignores dangling endsaltogether (mostly for debugging).”
5RNAFOLD-manual: “With -d2 this check is ignored,dangling energies will be added for the bases adjacent toa helix on both sides in any case; this is the default forpartition function folding (-p).”
Table 8 Relative runtime.
Level MacroState MicroState OverDangle NoDangle
5 1.00 0.25 0.15 0.12
4 3.70 0.95 0.59 0.39
3 5.99 1.59 0.96 0.60
2 145.56 14.46 9.39 8.37
1 643.20 117.16 51.76 28.99
The MacroState level 5 value equals 20.76 seconds on an Intel® Xeon® CPUL5420 @ 2.50 GHz.
Janssen et al. BMC Bioinformatics 2011, 12:429http://www.biomedcentral.com/1471-2105/12/429
Page 17 of 19

6RNAFOLD-manual: “With -d1 only unpaired basescan participate in at most one dangling end, this is thedefault for mfe folding but unsupported for the partitionfunction folding.”
7A larger threshold will always help. However, onecannot tell whether this situation has occurred.
8Whether or not it is adequate in partition functioncomputations to split a secondary structure into severalmicrostates is an unresolved dispute among experts (M.Zuker, personal communication).
9This can be evaluated by experimental techniques[33], but sufficient data are not yet available.
10While in press, Turner’2004 energy parametersbecame available. Results for all evaluations are listed inadditional file 4.
11It is not obvious how to convert our absolute dis-tances into error rates. Remember that a mispredictedbase pair can contribute a distance of 2 (cf. Figure 6).Assuming that predictions hold about the same numberof base pairs as the gold structures (1593), the intervalof possible distance scores is [0, 3186], from which theabove percentages are derived.
12We also looked at four further UNAFOLD variantsin dangle and no-dangle mode. Their behavior deviatesconsiderably, which is explained by differences in theimplemented energy model (M. Zuker, personalcommunication).
13In theory, these tables should be symmetric. We seea small asymmetry on the last decimal position in eightcases. This results from the fact that our programs - forbetter speed - ignore shapes with an initial probabilityless that 10-6. This means our resulting shape lists arenor perfectly identical in the low probability tail, andtogether with rounding errors, this leads to discrepan-cies ≤ 0.002.
Additional material
Additional file 1: Measurements on Data Sets FR3D:3A, FR3D:4Aand RNAstrand:91. File “supplement.pdf” contains detailed results forthe three mentioned data sets FR3D:3A, FR3D:4A and RNAstrand:91,which have not been shown in the main paper. We also provide fourVenn diagrams to demonstrate overlaps between the data sets.
Additional file 2: Data Sets. Archive “datasets.tgz” contains all four datasets DARTS, FR3D:3A, FR3D:4A and RNAstrand:91 as FASTA like files.Format description is given in additional file 1: “supplement.pdf”.
Additional file 3: Source Code of all models. The archive “fold-grammars.tgz” hold source code for all four models (NoDangle,OverDangle, MicroState and MacroState) in the Advanced DynamicProgramming language Bellman’s GAP. Please see the enclosed readmefile for further instructions on how to compile binaries.
Additional file 4: Evaluation results for Turner 2004 energyparameters. File “turner2004.pdf” contains results for all our evaluations,but computed with the more recent Turner 2004 energy parameter set,which became available while our manuscript was in press.
AcknowledgementsThanks go to Michael Zuker for comments on the energy model and theUNAFOLD program, and to Georg Sauthoff for support with the Bellman’sGAP system. Additional thanks go to Craig Zirbel for providing the FR3D:3Adata set.We acknowledge support of the publication fee by DeutscheForschungsgemeinschaft and the Open Access Publication Funds ofBielefeld University.We also thank the anonymous reviewers for helpful comments on themanuscript.
Author details1Faculty of Technology, Bielefeld University, 33615 Bielefeld, Germany.2Bioinformatics Group, Max Planck Institute of Molecular Plant Physiology,14476 Potsdam, Germany. 3Institut für Physikalische Biologie, Heinrich-Heine-Universität Düsseldorf, 40204 Düsseldorf, Germany.
Authors’ contributionsSJ suggested to tackle the problem of model discrepancies empirically. SJ,RG and GS designed the study. CS prepared the data sets derived from 3Dstructural data, and GS provided background on the thermodynamic model.SJ implemented the four models and ran the evaluations. All authors closelycooperated in interpreting the results and writing the manuscript. Allauthors read and approved the final manuscript.
Received: 10 June 2011 Accepted: 3 November 2011Published: 3 November 2011
References1. Mathews D, Sabina J, Zuker M, Turner D: Expanded sequence dependence
of thermodynamic parameters improves prediction of RNA secondarystructure. J Mol Biol 1999, 288:911-940.
2. Hofacker IL, Fontana W, Stadler PF, Bonhoeffer SL, Tacker M, Schuster P:Fast Folding and Comparison of RNA Secondary Structures. MonatshChem 1994, 125:167-188.
3. Giegerich R, Voß B, Rehmsmeier M: Abstract shapes of RNA. Nucleic AcidsResearch 2004, 32(16):4843.
4. Wuchty S, Fontana W, Hofacker IL, Schuster P: Complete suboptimalfolding of RNA and the stability of secondary structures. Biopolymers1999, 49(2):145-165.
5. Dowell R, Eddy S: Evaluation of several lightweight stochastic context-free grammars for RNA secondary structure prediction. BMCBioinformatics 2004, 5:71[http://www.biomedcentral.com/1471-2105/5/71].
6. Voß B, Giegerich R, Rehmsmeier M: Complete probabilistic analysis of RNAshapes. BMC Biology 2006, 4:5.
7. Waterman M: Introduction to computational biology. Maps, sequences andgenomes London: Chapman & Hall; 1995.
8. Nebel M, Scheid A: On quantitative effects of RNA shape abstraction.Theory in Biosciences 2009, 128:211-225, [10.1007/s12064-009-0074-z].
9. Janssen S, Reeder J, Giegerich R: Shape based indexing for faster searchof RNA family databases. BMC Bioinformatics 2008, 9:131+.
10. Borer P, Dengler B, Tinoco I Jr, Uhlenbeck O: Stability of ribonucleic aciddouble-stranded helices. J Mol Biol 1974, 86:843-853.
11. Burkard M, Kierzek R, Turner D: Thermodynamics of unpaired terminalnucleotides on short RNA helixes correlates with stacking at helixtermini in larger RNAs. J Mol Biol 1999, 290:967-982.
12. Ohmichi T, Nakano S, Miyoshi D, Sugimoto N: Long RNA dangling end haslarge energetic contribution to duplex stability. J Am Chem Soc 2002,124:10367-10372.
13. Liu J, Zhao L, Xia T: The dynamic structural basis of differentialenhancement of conformational stability by 5’- and 3’-dangling ends inRNA. Biochemistry 2008, 47:5962-5975.
14. Giegerich R, Meyer C, Steffen P: A discipline of dynamic programmingover sequence data. Science of Computer Programming 2004, 51(3):215-263.
15. Giegerich R, Sautho G: Yield grammar analysis in the Bellman’s GAPcompiler. Proceedings of the Eleventh Workshop on Language Descriptions,Tools and Applications LDTA 2011, ACM; 2011.
16. Sauthoff G, Janssen S, Giegerich R: Bellman’s GAP - A DeclarativeLanguage for Dynamic Programming. 13th International ACM SIGPLANSymposium on Principles and Practice of Declarative Programming, PPDP2011, ACM 2011.
Janssen et al. BMC Bioinformatics 2011, 12:429http://www.biomedcentral.com/1471-2105/12/429
Page 18 of 19

17. Abraham M, Dror O, Nussinov R, Wolfson H: Analysis and classification ofRNA tertiary structures. RNA 2008, 14(11):2274.
18. Berman H, Westbrook J, Feng Z, Gilliland G, Bhat T, Weissig H, Shindyalov I,Bourne P: The protein data bank. Nucleic Acids Res 2000, 28:235-242.
19. Rose P, Beran B, Bi C, Bluhm W, Dimitropoulos D, Goodsell D, Prlic A,Quesada M, Quinn G, Westbrook J, Young J, Yukich B, Zardecki C,Berman H, Bourne P: The RCSB Protein Data Bank: redesigned web siteand web services. Nucleic Acids Res 2010, 39:D392-401.
20. Sarver M, Zirbel CL, Stombaugh J, Mokdad A, Leontis NB: FR3D: findinglocal and composite recurrent structural motifs in RNA 3D structures. JMath Biol 2008, 56(1-2):215-252.
21. Stombaugh J, Zirbel CL, Westhof E, Leontis NB: Frequency and isostericityof RNA base pairs. Nucleic Acids Res 2009, 37(7):2294-2312.
22. Gendron P, Lemieux S, Major F: Quantitative analysis of nucleic acidthree-dimensional structures. J Mol Biol 2001, 308(5):919-936.
23. Andronescu M, Bereg V, Hoos H, Condon A: RNA STRAND: The RNASecondary Structure and Statistical Analysis Database. BMC Bioinformatics2008, 9:340.
24. Hamada M, Kiryu H, Sato K, Mituyama T, Asai K: Prediction of RNAsecondary structure using generalized centroid estimators. Bioinformatics2009, 25(4):465-473.
25. Do CB, Woods DA, Batzoglou S: CONTRAfold: RNA secondary structureprediction without physics-based models. Bioinformatics 2006, 22(14):e90-98.
26. Andronescu M, Condon A, Hoos H, Mathews DH, Murphy KP:Computational approaches for RNA energy parameter estimation. RNA2010, 16(12):2304-2318.
27. Janssen S, Giegerich R: Faster computation of exact RNA shapeprobabilities. Bioinformatics 2010, 26(5):632-639.
28. Zuker M: Mfold web server for nucleic acid folding and hybridizationprediction. Nucleic Acids Res 2003, 31:3406-3415.
29. Markham NR, Zuker M: UNAFold: software for nucleic acid folding andhybridization. Methods in molecular biology (Clifton, N.J.) 2008, 453:3-31.
30. Reuter J, Mathews D: RNAstructure: software for RNA secondary structureprediction and analysis. BMC Bioinformatics 2010, 11:129.
31. Walter A, Turner D, Kim J, Lyttle M, Müller P, Mathews D, Zuker M: Coaxialstacking of helixes enhances binding of oligoribonucleotides andimproves predictions of RNA folding. Proc Nat Acad Sci USA 1994,91:9218-9222.
32. Xia T, SantaLucia J, Burkard M, Kierzek R, Schroeder S, Jiao X, Cox C,Turner D: Thermodynamic parameters for an expanded nearest-neighbormodel for formation of RNA duplexes with Watson-Crick base pairs.Biochemistry 1998, 37:14719-14735.
33. Gherghe C, Shajani Z, Wilkinson K, Varani G, Weeks K: Strong correlationbetween SHAPE chemistry and the generalized NMR order parameter(S2) in RNA. J Am Chem Soc 2008, 130(37):12244-5.
34. Gardner P, Giegerich R: A comprehensive comparison of comparativeRNA structure prediction approaches. BMC Bioinformatics 2004, 5:140.
doi:10.1186/1471-2105-12-429Cite this article as: Janssen et al.: Lost in folding space? Comparing fourvariants of the thermodynamic model for RNA secondary structureprediction. BMC Bioinformatics 2011 12:429.
Submit your next manuscript to BioMed Centraland take full advantage of:
• Convenient online submission
• Thorough peer review
• No space constraints or color figure charges
• Immediate publication on acceptance
• Inclusion in PubMed, CAS, Scopus and Google Scholar
• Research which is freely available for redistribution
Submit your manuscript at www.biomedcentral.com/submit
Janssen et al. BMC Bioinformatics 2011, 12:429http://www.biomedcentral.com/1471-2105/12/429
Page 19 of 19

















