
Linux, Remote Computing, and NMR Calculations on a Linux Cluster ‘n1’ at UNCW.
24
Linux, Remote Computing, and NMR Calculations on a Linux Cluster ‘n1’ at UNCW
-
Upload
haley-kings -
Category
Documents
-
view
226 -
download
0
Transcript of Linux, Remote Computing, and NMR Calculations on a Linux Cluster ‘n1’ at UNCW.

Linux, Remote Computing, and NMR Calculations on
a Linux Cluster ‘n1’ at UNCW

Objectives
Certain carbocations may exist as structures in which the positive charge is localized mainly on one carbon (classical) or spread over several carbons (non-classical) due to bridging or other delocalization.
Examples of such carbocations are shown on the following slides. Many others exist.
NMR has been useful in determining the structures of these carbocations.

Rates of solvolysis
OTs
&
OAc
HOAc
k1
OTs OAc
HOAc
k2 k2 / k1 = 10 11
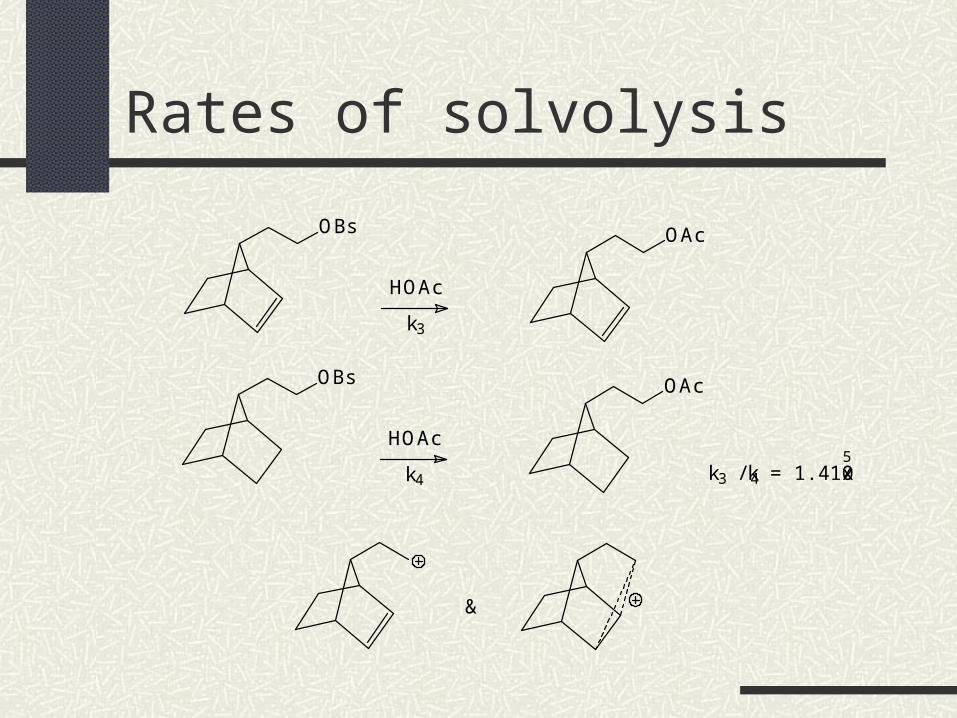
Rates of solvolysis
OBs
&
HOAc
k3
OAc
OBs
HOAc
k4
OAc
k3 / k4 = 1.4 x 10 5

In this lab we will do calculations to predict the lowest energy structure for a given carbocation. We will also calculate the NMR shielding values of the carbon atoms in the cations. NMR shielding values are related to chemical shifts.

The difference in the sum of the carbon atom isotropic shielding values between the parent hydrocarbon and the cation (which is equal to the difference in the sum of their chemical shifts) has been correlated to the nature of the carbocation (classical or non-classical).If the absolute difference (hydrocarbon parent - cation) is >350 ppm, the cation is considered to be classical; if the difference is <250 ppm, it is considered to be non-clasical.

NMR Isotropic Shielding Values
TMSH+
shielding values chemical shifts
0 ppm
(Gaussian mistakenly reports these as Isotropic Shielding Tensors)
Differences in shielding values = differences in chemical shifts

Ethane/ethyl/bridged ethenium ion
Isotropic Shielding Values (in ppm )
Difference from 388: 358 262 (classical) (~non-classical)
CH3 CH3 CH3 CH2 CH2 CH2
H
ethane ethyl cation bridged ethenium ion
194 + 194 = 388
148 + (-118) = 30
61+ 61 = 122

Objectives…
Because NMR calculations require large basis sets for accuracy, you will use a computer faster than a pc; these calculations will be performed remotely on a 4 dual-node processor Linux cluster in the CIS building at UNCW.This machine is a Linux-based platform, so you will need to learn how to do remote computing and a few Linux commands.You will also learn how to use Gaussian 03, a widely used quantum chemistry computational software package.

Logging on to a remote computer
We’ll use puTTy (icon in Applications folder) for secure connections to logon to:
n1.dobo.uncw.edu (this is our Linux cluster, called ‘n1’ for short)
Login (on n1):martin
password: (I’ll enter this)

Unix Commands
Useful Unix commands:pwd (tells you what directory you are in)
mkdir NMR (makes a new directory named NMR)
ls –la (gives a complete listing of files in directory)
cd .. (changes directories to the next higher directory)
cd ~ (changes directories to your home, or login dir.)
cp filename1.dat filename2.dat (makes a copy of a file, giving it a different filename)

Other useful Unix commands
cp filename1.dat dirname1/filename2.dat (makes a copy of a file, saves it in a different directory and gives it a different filename)
mv filename1.dat dirname2/filename1.dat (moves a file to a different directory, keeping the same name)
mv filename1.dat dirname2/filename2.dat (moves a file to a different directory, and changes the filename)
rm filename1.dat (removes (deletes) a file) rmdir dirname1 (removes (deletes) a directory; it must be
empty first; could use rm dirname1 –r, but this is dangerous
exit (or logout) (logs you off the computer)

vi text editor commands
vi filename.dat (opens filename.dat in text editor; must be in same directory as file, or else must use pathname/filename)
i (enters insert mode; Esc gets you out of any mode) a (enters append mode; Esc gets you out of any mode) x (enters delete mode; Esc gets you out of any mode) r (single character overstrike mode; upper case R is for
multiple character overstrike mode) dd (deletes entire line)ZZ (exits vi editor, saving changes):q! (exits vi editor without saving changes)

Editing an input file for Gaussian
Gaussian 03 requires input files with a .dat (or .com) extensionThe file must be in a certain format, shown on the following slideSpacing is critical (blank spaces or no spaces and blank lines must be exactly as called for)The molecule description (Cartesian coordinates or Z-matrix) is free-form; any number of spaces between adjacent columns is OK

Format of Gaussian input file
%chk=/tmp/yourdirname/filename.chk# HF/6-31++G** nosymm opt freq(blank line)(title or any comments you wish to add go here)(blank line)1 1 (charge & multiplicity )[molecular specification…XYZ coordinates or Z-matrix goes here](blank line)--Link1--%chk=/tmp/yourdirname/filename.chk# HF/6-31++G** geom=allcheckpoint nosymm pop=npa nmr(blank line)
(the red section is a link to an nmr job)

Submitting a Gaussian job
An executable script called xg03 has been written to automate the submission process for Gaussian 03 calculations.
xg03 filename.dat

Checking on a Gaussian job
After a job has been submitted, note the .job and .out files in your directory; an .error file indicates a submitted job; it will remain empty unless an error occurs.A copy of the .out file may be viewed at any time without interrupting the calculation; type cat filename.out to scroll through the file very quickly to the end; use the elevator bar to go back into the file.Alternatively, type more filename.out and the spacebar to view the file one screen at a time

Objectives for Tuesday’s lab
Logon to the accountCreate a directory for your scratch (temporary) files:
mkdir /tmp/425_nhmMake a copy of the H.dat file to a file having your initials in the filename:
cp H.dat Hnhm.datUse the vi editor to edit the new file Hnhm.dat

Objectives for Tuesday’s lab
vi Hnhm.dat (use vi editing commands to change the directory and filename in the first line)
submit Hnhm.dat for Gaussian 03 calculation.
xg03 Hnhm.dat
When the job is finished, examine the output file:
more Hnhm.out
(This simple calculation will allow you to see if your calculations are running properly.)

Objectives for Tuesday’s lab
Build a model of each of your isomeric carbocations in TitanOptimize each of them using semi-empirical MO theory (AM1 or PM3). If this changes the structure from what you want, re-build the structure using constraints if needed and skip the semi-empirical MO optimization step.Save the structures as .pdb files on your flashdrive. The rest of the lab will be done in Dr. Martin’s office.Open the files in GaussView; save them as Cartesian Coordinate files (with a .dat extension). Note the atom numbering. A sketch or a copy of the structure with atom numbers will be helpful.

Objectives for Tuesday’s lab…
Edit the .dat files using Notepad on a pc (easiest) or vi on ‘n1’ to conform to the Gaussian 03 format used by n1. (see handout)Use WINSCP3 to copy the files to an account on n1 (our Linux cluster).More simply, use puTTY to access n1, then create a new file in vi (vi newfilename.dat), then copy and paste your .dat file from Notepad.Submit them by typing the command:
xg03 filename.dat

Visualizing Gaussian results
Transmit a copy of the .out file from n1 to the local pc; this is done using WINSCP3.
Use GaussView to open Gaussian .out files and render models of structures in various renderings.
Observe each structure and make measurements on each structure as needed to determine if it is a classical or non-classical carbocation.

Objectives for the Next lab
Repeat the entire process, this time modeling the parent hydrocarbon (alkane).Perform the data analysis, including calculating the difference in the sums of the chemical shielding values between the parent hydrocarbon and each carbocation structure.Visualize the carbocations, copy the structure to put in your report, and make measurements as needed to determine whether each is classical or non-classical.

Objectives for the Next lab…
Also obtain the NMR Isotropic Shielding values and the npa charges on the carbon atoms in each structure. These can be done using the vi editor and some commands (e.g., grep) as indicated in the lab handout.Analyze the data (NMR shielding values, npa charges, energies, bond lengths and bond angles) to determine whether the carbocation you modeled exists as a classical or non-classical structure, according to the HF/6-31++G** calculations.


















