Levulinic acid hydrogenolysis on Al2O3-based Ni-Cu bimetallic catalysts
7
Chinese Journal of Catalysis 35 (2014) 656–662 available at www.sciencedirect.com journal homepage: www.elsevier.com/locate/chnjc Article (Special Issue on the 2nd International Congress on Catalysis for Biorefineries (CatBior 2013)) Levulinic acid hydrogenolysis on Al 2 O 3 ‐based Ni‐Cu bimetallic catalysts Iker Obregón *, Eriz Corro, Urko Izquierdo, Jesus Requies, Pedro L. Arias Department of Chemical and Environmental Engineering, School of Engineering, University of the Basque Country, UPV/EHU, Alameda Urquijo s/n, 48013 Bilbao, Spain ARTICLE INFO ABSTRACT Article history: Received 8 December 2013 Accepted 24 January 2014 Published 20 May 2014 Inexpensive γ‐alumina‐based nickel‐copper bimetallic catalysts were studied for the hydrogenolysis of levulinic acid, a key platform molecule for biomass conversion to biofuels and other valued chemicals, into γ‐valerolactone as a first step towards the production of 2‐methyltetrahydrofurane. The activities of both monometallic and bimetallic catalysts were tested. Their textural and chemical characteristics were determined by nitrogen physisorption, elemental analysis, temperature‐pro‐ grammed ammonia desorption, and temperature‐programmed reduction. The monometallic nickel catalyst showed high activity but the highest by‐product production and significant amounts of carbon deposited on the catalyst surface. The copper monometallic catalyst showed the lowest activity but the lowest carbon deposition. The incorporation of the two metals generated a bimetal‐ lic catalyst that displayed a similar activity to that of the Ni monometallic catalyst and significantly low by‐product and carbon contents, indicating the occurrence of important synergetic effects. The influence of the preparation method was also examined by studying impregnated‐ and sol‐gel‐derived bimetallic catalysts. A strong dependency on the preparation procedure and calcina‐ tion temperature was observed. The highest activity per metal atom was achieved using the sol‐gel‐derived catalyst that was calcined at 450 °C. High reaction rates were achieved; the total levulinic acid conversion was obtained in less than 2 h of reaction time, yielding up to 96% γ‐valerolactone, at operating temperature and pressure of 250 °C and 6.5 MPa hydrogen, respec‐ tively. © 2014, Dalian Institute of Chemical Physics, Chinese Academy of Sciences. Published by Elsevier B.V. All rights reserved. Keywords: Levulinic acid γ‐Valerolactone Hydrogenolysis Biomass Catalyst Copper Nickel 1. Introduction Owing to diminishing oil reserves, increasing energy de‐ mands from emerging economies, and increasing global warming concerns, researchers have focused their attention on biomass‐derived chemicals to replace non‐renewable energy resources [1–5]. The suitability of biomass‐derived products, such as liquid fuels, is being established by currently used first‐generation biofuels (bioethanol and biodiesel) that are easily produced from edible biomass [5,6]. However, this leads to expensive feedstocks and competition with the food market. In contrast, second‐generation biofuels are derived from non‐edible, inexpensive, and abundant lignocellulosic biomass. However, their production is more challenging than that of first‐generation biofuels [6]. Levulinic acid (LA) is a key chemical towards the develop‐ ment of these new biofuels because it can be produced in good yields at both laboratory scale [7,8] and semi‐industrial scale * Corresponding author. Tel: +34‐946‐017297; Fax: +34‐946‐014179; E‐mail: [email protected] This work was supported by the UPV/EHU, Spanish Ministry of Economy and Competitiveness CARBIOCAT (Project Ref. CTQ2012‐38204‐C03‐03) and the Basque Government Predoc Training Programme and Department of Education and University (Project Ref. GIC 10/31 University). DOI: 10.1016/S1872‐2067(14)60051‐6 | http://www.sciencedirect.com/science/journal/18722067 | Chin. J. Catal., Vol. 35, No. 5, May 2014
Transcript of Levulinic acid hydrogenolysis on Al2O3-based Ni-Cu bimetallic catalysts

ChineseJournalofCatalysis35(2014)656–662
a v a i l a b l e a t www . s c i e n c e d i r e c t . c om
j o u r n a l h omep a g e : www . e l s e v i e r . c om / l o c a t e / c h n j c
Article (Special Issue on the 2nd International Congress on Catalysis for Biorefineries (CatBior 2013))
LevulinicacidhydrogenolysisonAl2O3‐basedNi‐Cubimetalliccatalysts
IkerObregón*,ErizCorro,UrkoIzquierdo,JesusRequies,PedroL.AriasDepartmentofChemicalandEnvironmentalEngineering,SchoolofEngineering,UniversityoftheBasqueCountry,UPV/EHU,AlamedaUrquijos/n,48013Bilbao,Spain
A R T I C L E I N F O
A B S T R A C T
Articlehistory:Received8December2013Accepted24January2014Published20May2014
Inexpensiveγ‐alumina‐basednickel‐copperbimetalliccatalystswerestudiedforthehydrogenolysisof levulinic acid, a key platform molecule for biomass conversion to biofuels and other valuedchemicals,intoγ‐valerolactoneasafirststeptowardstheproductionof2‐methyltetrahydrofurane.Theactivitiesofbothmonometallicandbimetalliccatalystsweretested.Theirtexturalandchemicalcharacteristicswere determined by nitrogen physisorption, elemental analysis, temperature‐pro‐grammedammoniadesorption,andtemperature‐programmedreduction.Themonometallicnickelcatalyst showed high activity but the highest by‐product production and significant amounts ofcarbon deposited on the catalyst surface. The copper monometallic catalyst showed the lowestactivitybutthelowestcarbondeposition.Theincorporationofthetwometalsgeneratedabimetal‐liccatalystthatdisplayedasimilaractivitytothatoftheNimonometalliccatalystandsignificantlylowby‐productandcarboncontents,indicatingtheoccurrenceofimportantsynergeticeffects.Theinfluence of the preparation method was also examined by studying impregnated‐ andsol‐gel‐derivedbimetalliccatalysts.Astrongdependencyonthepreparationprocedureandcalcina‐tion temperature was observed. The highest activity per metal atom was achieved using thesol‐gel‐derived catalyst thatwas calcined at 450 °C.High reaction rateswere achieved; the totallevulinic acid conversion was obtained in less than 2 h of reaction time, yielding up to 96%γ‐valerolactone,atoperating temperatureandpressureof250°Cand6.5MPahydrogen,respec‐tively.
©2014,DalianInstituteofChemicalPhysics,ChineseAcademyofSciences.PublishedbyElsevierB.V.Allrightsreserved.
Keywords:Levulinicacidγ‐ValerolactoneHydrogenolysisBiomassCatalystCopperNickel
1. Introduction
Owing to diminishing oil reserves, increasing energy de‐mands from emerging economies, and increasing globalwarmingconcerns,researchershavefocusedtheirattentiononbiomass‐derived chemicals to replace non‐renewable energyresources [1–5]. The suitability of biomass‐derived products,such as liquid fuels, is being established by currently usedfirst‐generation biofuels (bioethanol and biodiesel) that are
easilyproducedfromediblebiomass[5,6].However,thisleadstoexpensivefeedstocksandcompetitionwiththefoodmarket.In contrast, second‐generation biofuels are derived fromnon‐edible,inexpensive,andabundantlignocellulosicbiomass.However, their production is more challenging than that offirst‐generationbiofuels[6].
Levulinicacid (LA) isakeychemical towards thedevelop‐mentofthesenewbiofuelsbecauseitcanbeproducedingoodyields at both laboratory scale [7,8] and semi‐industrial scale
*Correspondingauthor.Tel:+34‐946‐017297;Fax:+34‐946‐014179;E‐mail:[email protected]/EHU,SpanishMinistryofEconomyandCompetitivenessCARBIOCAT(ProjectRef.CTQ2012‐38204‐C03‐03)andtheBasqueGovernmentPredocTrainingProgrammeandDepartmentofEducationandUniversity(ProjectRef.GIC10/31University).DOI:10.1016/S1872‐2067(14)60051‐6|http://www.sciencedirect.com/science/journal/18722067|Chin.J.Catal.,Vol.35,No.5,May2014

IkerObregónetal./ChineseJournalofCatalysis35(2014)656–662
byacidhydrolysisof lignocellulosicmaterials andurban resi‐dues[6,9,10].In2004,theUSDepartmentofEnergyclassifiedLAasa“topten”bio‐basedproductobtainedfrombiorefinerycarbohydrates in accordancewith characteristics such as po‐tentialasaplatformchemicalandbuildingblock,petrochemi‐cal replacement capability, and potential production scale‐up[10].
LAcanbeconvertedintosuitableandsustainablefuelssuchas γ‐valerolactone (GVL) and 2‐methyltetrahydrofuran(MTHF), as shown in Scheme 1. GVL possesses many of theproperties of an ideal liquid both for energy and chemicalproductmanufacture,asreported[11],andshowssimilarbe‐havior to ethanol when added to gasoline. MTHF is a well‐
known fuel additive that canbe added to gasoline in propor‐tionsofupto70vol%[12]intheabsenceofenginemodifica‐tionorusedaspart ofnew fuel formulations for flexible fuelengines, as reported by Paul [13] and the US Department ofEnergy [14].However, direct productionofMTHF fromLA israrelyreported.Toourknowledgeonlytwopublishedreportsaddressed this issue: Elliot and Frye [15] achieved an 89.8%MTHFyieldusingoperatingconditionsof242°Cand10MPaH2, and a Pd(5%)–Re(5%)/C catalyst. Upare and co‐workers[16]reportedamaximumMTHFyieldof89%usingoperatingconditionsof265°Cand2.5MPaH2andaCu(72%)‐Ni(8%)/
SiO2catalyst.Inbothcases,1,4‐dioxanewasusedasasolvent.Incontrast,GVLproductioniswidelyreportedandcarried
outundermilderconditions.Carbon‐basedrutheniumcatalystsareusedbymanyauthorsunderreactionconditionsthatvaryfrom25to215°Candfrom1.2to5.5MPahydrogen[17–20]insolvents such as 1,4‐dioxane or methanol. Temperatures of70–150°Candpressuresof0.5–3.5MPaH2havebeensuccess‐fullyusedwhenwaterisemployedasasolvent[21–23].OthernoblemetalsondifferentsupportshavealsobeenreportedtoeffectivelyactivatethereactionprocesssuchasPtovertitania,silica,andzirconia[24–26],Auoverzirconia[27],andIrsup‐ported on carbon nanotubes [28]. However, reports usingnon‐noblemetals forLAhydrogenationarerare.GVLproduc‐tion in liquidphase [29,30]usingcopper supportedonCr2O3,Al2O3, or ZrO2 has been reportedbut requires harsher condi‐tions, i.e., temperatures of 200–265 °C, pressures of 3.0–7.0MPaH2,andlongreactiontimesof5–10h.
Non‐noblemetalcatalystsaregenerallypreferredowingtotheirhigheravailabilityand lowerprice thataremoreappro‐priate for large‐scale operations such as biofuel production[29,30]. Hence, this work investigates the use of sustainablenon‐noblemetal‐based catalysts for the production of LA de‐rivatives.
2. Experimental
2.1. Catalystpreparation
A series of catalystswere prepared by either thewet im‐pregnation method (WI) or the sol‐gel (SG) method. A com‐mercialγ‐Al2O3support(AlfaAesar)wasusedfortheWI‐basedmethod.Nickel(II)nitratehexahydrate(98.5%,Sigma‐Aldrich)and copper(II) nitrate hemipentahydrate (98%, Alfa Aesar)wereusedasmetalprecursorsalts,andaluminumisopropox‐ide(AIP,98%,Aldrich)wasusedasanaluminaprecursor fortheSG‐basedpreparation.
TheWImethodconsistedofmixing1gofthesupportwith9mL water and calculated amounts of the precursor salt in arotaryvacuumevaporator.Themixturewasstirredovernightat90r/minandheatedto65°C,andthemoisturewasremovedundervacuum.
To prepare the SG‐based catalyst, the method previouslydescribed by Gandarias et al. [31]was employed. Briefly, thedesiredamountofAIPwasdissolvedinwateratanAIP/watermassratioof1:9at40°CwhilemaintainingthepHofthesolu‐tionbetween3.8and4.2,adjustedwith1mol/LHNO3solution.Themetal precursor salt solutionswere dissolved in an ade‐quateamountofethanol,andtheresultingsolutionwasaddeddropwise to theAIP solution. Themixturewas stirred for 30minat40°Candthensonicated for30min.Themixturewasallowedtositat50°Cfor48h,followedbyovernightdryingat105 °C. The obtained products were ground (particle size:0.42–0.50mm)andcalcinedunderairflowatthedesiredtem‐perature(300or450°C)for1h(heatingrate:2°C/min).Themethods are referred to as SG 300 and SG 450 based on thecalcinationtemperatureused.
Prior to conducting the activity tests, the catalysts under‐wentreductioninatubularfurnaceat650°Cfor1h(heatingrate:10°C/min)under75mL/minH2flow.Followingcoolingto room temperature, the catalysts were stored under isooc‐tanetoavoidcontactwithair.
2.2. Catalystcharacterization
Elementalanalysisofthecatalystswascarriedoutbyinduc‐tivelycoupledplasmaatomicemissionspectroscopy(ICP‐AES)on a Perkin Elmer Optima 2000 OV. Samples were digestedwith amixture of nitric, hydrochloric, and hydrofluoric acidsusinganETHOS1AdvancedMicrowaveDigestionSystem.
Surfacearea,porevolume,andporesizedistributionweredetermined by N2 physisorption at –196 °C on an Autosorb
OH
O
OOH
OH
O
O O
O O
OH
OH
O
Levulinic acid
4-Hydroxyvaleric acid
-Angelicolactone
-Valerolactone1,4-Pentanediol 2-Methyl
tetrahydrofuran Scheme1.Levulinicacidconversionpathintoγ‐valerolactoneand2‐methyltetrahydrofuran.

IkerObregónetal./ChineseJournalofCatalysis35(2014)656–662
1C‐TCD (Quantachrome). Prior to analysis, all samples weredegassedat250°Cand10Pafor3h.Thesurfaceareawascal‐culatedusingtheBrunauer‐Emmett‐Teller(BET)method,andporesizedistributionwascalculatedusingtheBarrett‐Joyner‐
Halenda(BJH)methodontheN2desorptioncurve. Temperature‐programmedreduction (TPR)of thesamples
was performed on an Autosorb 1C‐TCD (Quantachrome). Re‐ductionofthecalcinedcatalystsampleswasconductedunderagaseousmixtureof5vol%hydrogeninargon(gasflowrate:50mL/min)at1100°C(heatingrate:10°C/min).Hydrogencon‐sumption was monitored on a thermal conductivity detector(TCD).
Thermogravimetric analyses (TGA) of the catalysts wereperformedonaMettlerToledoTGA/SDTA851usingpureoxy‐genasanoxidizingagent.Thecatalystswereheatedat800°Cataheatingrateof5°C/min.
Ammoniatemperature‐programmeddesorption(NH3‐TPD)analyseswereperformedonthesameapparatususedforcon‐ducting the TPR analyses. The samples were placed in aU‐shapedquartz reactor and reducedunder a 50mL/minH2flowat650°Cfor1h(heatingrate:10°C/min).Then,thesam‐pleswere cooled to 250 °CunderHe flow andmaintained atthistemperaturefor30minfordegassing.Thedegassedsam‐pleswerethencooledto100°CunderflowingHepriortosat‐urationwithNH3in10vol%NH3/He,followedbypurgingwithHe (50mL/min) at 100 °C for 30min. The temperaturewasthen raised to900 °C at aheating rateof10 °C/min.Thede‐sorbedNH3wasmonitoredonaTCDdetector.
2.3. Activitytests
Theexperimentswerecarriedout instainlesssteelstirredautoclaves(AutoclaveEngineers,300mL)at250°Cfor6hat6.5MPaH2. The reactorwas loadedwith 1 g of the catalyst,heatedtothereactiontemperatureundernitrogen,andpurgedwith hydrogen. Then the feed mixture (200 mL, 5 wt% LAaqueoussolution(98%,Aldrich))wasintroducedfromthefeedcylinder. The reactionmixturewas stirred at 600 r/min andsampleswereperiodicallyextractedfromthereactortoassessthereactionkinetics.
The samples were analyzed on an Agilent Technologies6890NGCgaschromatographequippedwithaflameionizationdetector and a thermal conductivity detector. The GC wasequippedwithaDB‐1column(60m×530µm×5µm,AgilentTechnologies), and heliumwas used as the carrier gas at 9.2psi.The temperatureprogramwas setas follows:40 °C for5min,10°C/minheatingrampfor16min,and250°Cfor5minusinga30°C/minramp.Productidentificationandquantifica‐
tion were performed by calibration against commerciallyavailable pure chemicals. The resultswere further confirmedbyGC‐MSanalysis.
3. Resultsanddiscussion
3.1. Catalystcharacterization
Table 1 presents the textural properties and the experi‐mentally determinedmetal contents of the calcined catalysts.TheexperimentallydeterminedmetalcontentsoftheWI‐pre‐pared catalysts were comparable with the theoretical metalcontents.Nicontentswereeitherhigherorlowerthanthethe‐oretical values, whereas Cu contentswere consistently lowerthan the expected contents. The SG‐based bimetallic catalystsfeaturedacomparableNi‐to‐CuratiowiththatoftheWI‐basedbimetallic catalyst despite the lower metal contents of theSG‐basedbimetalliccatalysts.
The monometallic and bimetallic catalysts supported oncommercial alumina prepared by the metallic impregnationprocessfeaturedlowersurfaceareas(~150m2/g)thanthatofthebarecommercialaluminasupport(~230m2/g)atagivenmetal loading. Pore volumewas also lower, but the decreasewasmoresignificant fortheNi‐containingcatalystbya factoroftwo.TheadditionofCuonlyresultedinaporevolumelossof25%, suggesting that i) Ni preferentially deposited onto thepores of the alumina support, whereas Cu deposited on theouter surface of the alumina support or ii) Cu obstructedsmaller pores,whereasNi deposited on larger pores. Consid‐ering the differences in the average pore radius between theWI‐preparedcatalystsandthealuminasupport,thefirstoptionismore likelybecause incorporationofNidecreasedtheporeradius, whereas Cu addition enlarged the average pore size.Regardless, the effects associatedwithNi deposition seem toprevailoverthechangesinducedbyCudeposition.
UsingtheSGmethod,ahighersurfaceareawasachievedata calcination temperatureof300 °C.However,a reducedsur‐faceareawasobtainedatahighercalcination temperatureof450 °C despite the lowermetal loading of the correspondingcatalyst.Calcinationhadaminimaleffectontheporevolumeofthe final catalysts. However, at a higher calcination tempera‐ture,porevolumeof the finalbimetalliccatalystwasapproxi‐matelythreetimeslowerthanthatofthecatalystcounterpartpreparedby the impregnationmethod.Also, ahigher calcina‐tiontemperatureenlargedtheaverageporesize.
Figure1showstheresultsoftheTPRanalysisofthethreebimetallic catalysts. Important differences are highlighted re‐gardingthecompositionandreducibilityofthesurfacemetallic
Table1Elementalandtexturalcharacterizationofthecalcinedcatalysts.
Catalyst Ni(wt%) Cu(wt%) Ni+Cu(wt%) Ni/Curatio Surfacearea(m2/g) Porevolume(cm3/g) Poreradius(nm)γ‐Al2O3 0 0 0 0 227.2 0.81 6.81Ni/Al2O3(WI) 36.11 0 36.11 ∞ 146.9 0.35 4.64Cu/Al2O3(WI) 0 30.01 30.01 0 152.9 0.58 7.55Ni‐Cu/Al2O3(WI) 19.62 12.86 32.48 1.53 151.0 0.36 4.76Ni‐Cu/Al2O3(SG300) 13.26 8.88 22.14 1.49 186.1 0.20 2.06Ni‐Cu/Al2O3(SG450) 8.56 5.5 14.06 1.56 132.4 0.16 4.87

IkerObregónetal./ChineseJournalofCatalysis35(2014)656–662
species.Thestrongnickel‐aluminainteractions,correspondingtopeaksobservedabove600°CintheSG450catalyst,formingnickelaluminates,wereabsentintheSG300catalyst.Thisin‐dicates that these interactions are strongly dependent on thetemperature[32].However,theWIbimetalliccatalystfeaturesreduction peaks above 600 °C that are associatedwith thesenickelspecies[31]evenatacalcinationtemperatureof300°C.ThisdiscrepancymaybeattributedtothehighernickelcontentintheWIcatalyst(asopposedtothatintheSGcatalyst)andthedifferenttypesofaluminasupport;thesol‐gel‐derivedaluminacalcined at 300 °C is not fully converted to γ‐alumina [33].Thus, the differences between the supports can instigate theformationofdifferentspecies[32].
Copper species are reduced at temperatures ranging from245to340°C.Theincreaseinthereductionpeak’smaximumtemperature is related to the copper loading of the catalyst.Highermetalloadingspromoteagglomerationandformationoflargercrystallites thatrequirehigher temperatures for reduc‐tion[34].Additionally,copperadditioninducesashifttolowertemperaturesinthereductionpeaksascribedtoweaklyinter‐acting nickel by hydrogen spillover effects [34] that facilitatethereductionreactionofNi2+toNi0andnucleatetheformationof metallic Ni particles. This reduction took place at the lowtemperaturesintheSGcatalystcalcinedat300°C,asindicatedby the overlapping peaks because fewer nickel aluminateswereformed.
The catalysts prepared by impregnation and the bare alu‐mina support were further characterized by NH3‐TPD to de‐terminetheacidityofthecatalysts.TheprofilesarepresentedinFig.2.
AsobservedinFig.2,thebareγ‐Al2O3supportexhibitsthehighestacidity,displayingasmallpeakcenteredat200°Candabroadpeakbetween300and700°Cwithitsmaximumat410°C.The incorporationofNi resulted inaminoracidity loss inthe low‐temperatureregion.Considerablereduction inaciditywas observed in the high‐temperature region. However, im‐portant desorption peaks centered at 400 °C were also ob‐served.Ingeneral,Cumonometalliccatalystshowedhighacid‐
ity loss across the temperature range studied. The bimetalliccatalyst featured an intermediate acidity profile. However, itdisplayed a higher acidity loss in the low‐temperature regionwhen compared with that of other catalysts. In thehigh‐temperature region, the bimetallic catalyst showed twosmall peakswith an appreciable shift to higher temperatureswhencomparedwiththepeakofthemonometallicNicatalyst.Theseobservationswereattributed to the co‐existenceof theCuandNispecies.
3.2. Catalystactivity
The hydrogenolysis activities of the five catalysts weremeasuredtowardstheconversionofLAintoα‐angelicolactone(AL), GVL, andMTHF in an aqueousmedium under 6.5MPahydrogen.First, a seriesof experimentswereperformed, em‐ployingthemonometallicandbimetalliccatalystspreparedbythe WI method to determine the most hydrogenating activephase forthisreactionandevaluatethesynergeticeffectaris‐ingfromtheco‐existenceoftheCuandNimetalsinthebime‐talliccatalyst.TheseresultsaresummarizedinFig.3.
Figure3showsthattheNiandNi‐Cucatalystsarethemostactive systems and produced a maximum GVL yield of
0 100 200 300 400 500 600 700 800
WI
339423
467
649 721
H2 c
onsu
mpt
ion
SG 300
297 401525
T/oC
SG 450
243 284629 732
Fig. 1. TPR profiles of the calcined bimetallic catalysts Ni‐Cu/Al2O3
preparedbydifferentmethodsandassociatedpeakdecomposition.
100 200 300 400 500 600 700 800 900
Al2O3
Ni/Al2O3(WI) Cu/Al2O3(WI) Ni-Cu/Al2O3(WI)
TC
D s
igna
l
T/oC
Fig.2.NH3‐TPDprofilesofγ‐Al2O3,Ni/Al2O3,Cu/Al2O3,andNi‐Cu/Al2O3catalystspreparedbyWI.
0 50 100 150 200 250 300 3500
20
40
60
80
100
YG
VL/%
Time (min)
Ni/Al2O3(WI) Cu/Al2O3(WI) Ni-Cu/Al2O3(WI)
Fig.3.GVL yield as a function of reaction time plots of theNi/Al2O3,Cu/Al2O3,andNi‐Cu/Al2O3catalystspreparedbyWI.

IkerObregónetal./ChineseJournalofCatalysis35(2014)656–662
91%–92%after 2 h of reaction, corresponding to an LA con‐versionof~100%. In contrast,Cu catalyst exhibiteda signifi‐cantly lower activity, achieving a non‐asymptotic maximumGVLyieldof66%after6hofreactiontime,correspondingtoanLAconversionof75%.Despitetheloweractivityofthecoppercatalyst,thepresenceofcopperseemstoinhibitcatalystdeac‐tivation based on the low AL yields obtained with theCu‐containingcatalysts.ALyieldsof~0,1.6%,and4.1%wereobtainedwith the Cumonometallic, bimetallic, andNimono‐metalliccatalysts,respectively.BecauseAL isproducedbyac‐id‐catalyzeddehydrationofLA,ALdetectionindicatestheonsetof activity lossbecause active catalysts easily convertLA intoGVL.ALisawell‐knowncokeprecursor[35];hence,ALdetec‐tioncanberelatedtotheproductionofcokeontheusedcata‐lysts. AL concentration became significant at reaction timesgreaterthan3h.Atthesereactiontimes,LAconcentrationwasnegligiblebecausecompleteconversionofLAwasachievedatreactiontimesshorter than2h.Hence, thedetectedAL isbe‐lievedtobeduetothepreviouslyformedGVLorALdesorptedfromthecatalystsurface.Todate,thereversibleAL↔GVLreac‐tionhasnotbeenreportedintheliterature.
Theinfluenceofthepreparationmethodonthecatalyticac‐tivityofthebimetalliccatalystswastheninvestigated.Theac‐tivityofthetwobimetalliccatalystspreparedbytheSGmethodand calcined at twodifferent temperatures (300 and 450 °C)wasexamined.Figure4showstheresults.
AsobservedinFig.4,allstudiedcatalystsachievedcompleteLA conversion in less than 2 h with comparable GVL yields(~90%). The catalyst prepared by WI displayed the highestactivity at the early stages of the reaction. However, bothSG‐prepared catalysts calcined at different temperaturesachieved superior GVL yields (SG 450 catalyst: 92%; SG 300catalyst:96%;WIcatalyst:91%)atthelaterstagesofthereac‐tion.Moreover, the SG catalysts achieved a slightly higherALproduction(2.4%)whencomparedwiththatoftheWIcatalyst(1.6%)—these yields were considerably lower than that ob‐
tainedfortheNi/Al2O3(WI)catalyst.Consideringthelargedif‐ferences in the totalmetal loading between these three cata‐lysts,thedifferentyieldsobtainedareattributedtothehigherdispersion obtained by the SGmethod when compared withthat achieved by theWI method [36]. The higher dispersionenables a more intimate contact between the acidic and hy‐drogenating active sites,which results in a faster hydrogena‐tion of the intermediate product AL, consequently reducingcarbondepositiononthecatalystsurface.Thelatterprocesseswerefurtherinvestigatedbythermogravimetricanalysisofthecalcined and the used catalysts. The results are presented inFig.5.
The fresh catalysts (Fig. 5(a–c)) show a water desorptionpeak between 50 and 100 °C and additional peaks at highertemperatures depending on the type of catalyst studied. Theadditionalpeaks canbe associatedwith thedecompositionofthemetal precursor salts. It isworth noting that these peaksare shifted to lower temperatures for the SG catalyst, which
0 50 100 150 200 250 300 3500
20
40
60
80
100
YG
VL/%
Time (min)
Ni-Cu/Al2O3(WI) Ni-Cu/Al2O3(SG 450) Ni-Cu/Al2O3(SG 300)
Fig.4.GVLyieldasafunctionofreactiontimeplotsoftheNi‐Cu/Al2O3
bimetalliccatalystspreparedbySGandWI.
88
90
92
94
96
98
100
828486889092949698
100
828486889092949698
100
0 200 400 600 80094
96
98
100
0 200 400 600 80086
88
90
92
94
96
98
100
0 200 400 600 800828486889092949698
100
m /%
m /%
m /%
m /%
m /%
m /%
(a)
-0.10
-0.08
-0.06
-0.04
-0.02
0.00
0.02
0.04
0.06
Der
ivat
ive
Der
ivat
ive
Der
ivat
ive
Der
ivat
ive
Der
ivat
ive
Der
ivat
ive
(b)
-0.10
-0.08
-0.06
-0.04
-0.02
0.00
0.02
0.04
0.06
(c)
-0.10
-0.08
-0.06
-0.04
-0.02
0.00
0.02
0.04
0.06
T /oC
(d)
-0.12-0.10-0.08-0.06-0.04-0.020.000.020.04
T /oC
(e)
-0.24-0.21-0.18-0.15-0.12-0.09-0.06-0.030.000.03
T /oC
(f)
-0.10
-0.08
-0.06
-0.04
-0.02
0.00
0.02
0.04
Fig.5.TGAcurvesoffreshcatalysts(a)Cu/Al2O3(WI),(b)Ni/Al2O3(WI)and(c)Ni‐Cu/Al2O3(SG300)and(d–f)correspondingtousedcatalysts.

IkerObregónetal./ChineseJournalofCatalysis35(2014)656–662
mayindicateagreaterextentofdispersionand/orexposureofthemetalparticles.
Theprofilesoftheusedcatalystsdiffersignificantlyamongthemselvesandfromthoseofthefreshcatalysts.Thethermo‐gram(Fig.5(d))ofthespentCumonometalliccatalystfeatureda water desorption peak at 50 °C, a reaction product (LAand/orGVL)desorptionpeakat220°C,andalargeweightgainpeakat~450°CthatisduetooxidationofmetallicCutoCuO.At higher temperatures (500 °C), a sharp and considerableweightlosspeakisobservedduetothecombustionofcarbondepositedonthesurfaceofthecatalyst[36,37].Theprofile(Fig.5(e)) of the Nimonometallic catalyst is comparablewith thethermogram of the Cu monometallic catalyst, except for thesharpweightlossat300°C.Also,theNimonometalliccatalystshowed a lowerweight increase at 450 °C because Ni formsspeciesthataremoredifficulttoreduce,suchasnickelalumi‐nates, which are not reduced upon activation of the catalystpriortotheactivitytestandhencethesespeciescannotbeoxi‐dizedfurther.
The profile (Fig. 5(f)) of the SG‐prepared catalyst exhibitsthesamedesorptionpeaks;however,theydifferfromthoseofthe WI‐prepared catalysts in regards to the weight increasefeaturesassociatedwithmetaloxidation.MetaloxidationisnotevidencedintheprofileoftheSG‐preparedcatalyst,suggestingthehighdispersionoftheparticlesandconsequentlytheexist‐ence of smaller particles and greater particle exposure. Theused SG catalysts were stored in the presence of air. Hence,oxidationof theSGcatalysts is expected tooccurprior to theTGAtreatment.Thecarboncontentsofthecatalystswereesti‐mated based on two assumptions owing to the interferingpresence of themetal oxidation peak. The first estimation isbasedontheareaofthepeakassociatedwithcarboncombus‐tion, whereas the second estimation is based on the weightdifferencebetweenthepre‐andpost‐combustionstateswherethemetaloxidationpeak isunaccounted for.Theresults,pre‐sentedinTable2,areconsistentwiththepreviouslydiscussedhypothesis that correlates the deposited carbon amountwiththeincreasedALyieldsobservedintheWI‐preparedcatalysts.Becausethecarboncontentsestimatedforthethreebimetalliccatalystsaresimilar,theestimationmethodcanbeextendedtotheSGcatalysts.
The measured deposited carbon contents are in goodagreementwith thepreviously discussed catalyst aciditypro‐files. The deposited carbon amount is clearly related to thepreviouslydiscussedhigh‐temperatureacidityshowninFig.2;the Ni monometallic catalyst showed the highest amount ofstrong acid sites, whereas Cu monometallic catalyst showed
negligibleamountsofthesetypeofacidsitesandthebimetalliccatalystfeaturedanintermediatelevelofacidity.
Tofurthercomparetheactivitiesofthefivepreparedcata‐lysts,theinitialreactionrateofLAconversionpermoleofmet‐alwascalculated.ThisparameterwasestimatedusingtheLAconversionobtainedatthefirstsamplingtime(30min)andthetotalmetal content of each catalyst, asmeasured by ICP‐AES(Table1).TheresultsaredisplayedinFig.6.
Themetal‐normalized initial reaction ratesof thedifferentcatalystsdiffersignificantly.However,thetrendiscomparable.First,theWI‐basedCumonometalliccatalystwastheleastac‐tive, displaying < 0.05 molLA molmetal–1 min–1, whereas theWI‐basedNimonometalliccatalystwasfivetimesmoreactive.In terms of activity per total metal content, only a small in‐creasewasnoticedbetweentheWINimonometallicandNi‐Cubimetalliccatalysts.However, thedifferencesintheindividualmetalcontentsweresignificantlylargerinthepresenceofthetwometals.Forinstance,theactivityrelatedtoNiincreasedby50% and the activity related to Cu was more than 14 timeshigherwhencomparedwiththatoftheWImono‐andbimetal‐liccatalysts.InregardstotheSGcatalysts,thecalcinationtem‐perature plays an important role. SG 300 catalyst achieved alower activity than that of theWI bimetallic catalyst. In con‐trast,SG450catalystshowedthehighestactivity,achievingupto0.67molLAmolNi–1min–1and1.14molLAmolCu–1min–1.Thisactivity increase isclearevidenceof thesynergeticeffectsex‐ertedbyCuandNi,affordingahighlyefficientcatalystintermsof metal loading—similar conversion and selectivity areachievedwithsignificantlylowmetalcontents.
4. Conclusions
Inexpensive alumina‐based Ni‐Cu catalysts were success‐fullypreparedforthehydrogenolysisof levulinicacidto formγ‐valerolactone,achievinghighyieldsofupto96%.Theactivi‐ties of themonometallic and bimetallic catalystswere tested.Theco‐existenceofthemetalssupportedonaluminaaffordedenhanced positive effects. Additionally, a higher activity permetal atom was observed for the sol‐gel‐prepared catalyst
Table2CarboncontentoftheusedcatalystsestimatedfromTGA.
CatalystCarboncontent(%)
Basedonpeakarea BasedonweightdifferenceNi/Al2O3(WI) 5.7 4.5Cu/Al2O3(WI) 4.1 2.0Ni‐Cu/Al2O3(WI) 5.1 3.7Ni‐Cu/Al2O3(SG300) 4.8 4.7Ni‐Cu/Al2O3(SG450) 3.6 4.0
0.00.10.20.30.40.50.60.70.80.91.01.11.2
Init
ial r
eact
ion
rate
(m
olL
A m
olm
etal1
min
1)
molLA molNi + Cu1 min1
molLA molNi1 min1
molLA molCu1 min1
(1) (2) (3) (4) (5)Catalyst
Fig. 6. Metal‐normalized reaction rates of the catalysts. (1) Cu/Al2O3(WI); (2)Ni/Al2O3(WI); (3)Ni‐Cu/Al2O3(WI); (4)Ni‐Cu/Al2O3(SG300);(5)Ni‐Cu/Al2O3(SG450).

IkerObregónetal./ChineseJournalofCatalysis35(2014)656–662
when comparedwith that of the catalysts prepared by awetimpregnation procedure. This difference can be attributed tothe balanced amounts of acid andmetallic sites. TGA assess‐ment of the carbon deposition on the catalytic surfaces re‐vealedthatcopperplaysakeyroleininhibitingcarbondeposi‐tiononthecatalystdespitethe loweractivityofCucomparedwiththatofNi.Cu‐Ni/Al2O2catalystsarepromisingcandidatesforhydrogenolysisreactionsinthebiorefineryindustry.
References
[1] Serrano‐RuizJC,DumesicJA.EnergyEnvironSci,2011,4:83[2] British Petroleum. BP Statistical Review of World Energy June
2013.London:BP,2013[3] SorrellS,SpeirsJ,BentleyR,BrandtA,MillerR.GlobalOilDeple‐
tion, An Assessment of the Evidence for a Near‐Term Peak inGlobalOilProduction.London:UKENERGYRESEARCHCENTRE,2009
[4] IPCC,2001.ClimateChange2001:TheScientificBasis.Contribu‐tionofWorkingGroupItotheThirdAssessmentReportoftheIn‐tergovernmentalPanelonClimateChange.Cambridge:CambridgeUniversityPress,2001
[5] AlonsoDM,BondJQ,DumesicJA.GreenChem,2010,12:1493[6] HayesDJ.CatalToday,2009,145:138[7] MascalM,NikitinEB.GreenChem,2010,12:370[8] MascalM,NikitinEB.ChemSusChem,2009,2:859[9] HayesDJ,FitzpatrickSW,HayesMHB,RossJRH.In:KammB,
GruberPReds.Biorefineries‐IndustrialProcessesandProducts.Weinheim:Wiley‐VCHVerlagGmbH,2006.139
[10] BozellJJ,MoensL,ElliottDC,WangY,NeuenscwanderGG,Fitz‐patrick S W, Bilski R J, Jarnefeld J L. Resour Conserv Recycling,2000,28:227
[11] Horváth I T, Mehdi H, Fabos V, Boda L, Mika L T. Green Chem,2008,10:238
[12] HuberGW,IborraS,CormaA.ChemRev,2006,106:4044[13] PaulSF.USPatent6309430B1.2001[14] USDepartmentofEnergy.In:FederalRegistered.AlternativeFuel
TransportationPrograme;P‐SeriesFuels;FinalRule.Washington,NW:U.S.GovernmentPrintingOffice,1999.26822
[15] ElliottDC,FryeJG.USPatent5883266A.1999[16] UparePP,LeeJM,HwangYK,HwangDW,LeeJH,HalligudiSB,
HwangJS,ChangJS.ChemSusChem,2011,4:1749[17] ManzerLE.USPatent6617464B2.2003[18] YanZP,LinL,LiuSJ.EnergyFuels,2009,23:3853[19] GongY,LinL,YanZP.BioResources,2011,6:686[20] Al‐ShaalMG,WrightWRH,PalkovitsR.GreenChem, 2012,14:
1260[21] GallettiAMR,AntonettiC,DeLuiseV,MartinelliM.GreenChem,
2012,14:688[22] Serrano‐RuizJC,BradenDJ,WestRM,DumesicJA.ApplCatalB,
2010,100:184[23] Serrano‐RuizJC,WangD,DumesicJA.GreenChem,2010,12:574[24] AyoubPM,LangeJP.WOPatent2008142127A1.2008[25] LangeJP,PriceR,AyoubPM,LouisJ,PetrusL,ClarkeL,Gosselink
H.AngewChemIntEd,2010,49:4479[26] HaanRJ,LangeJP.WOPatent2011015645A2.2011[27] DuXL,HeL,ZhaoS,LiuYM,CaoY,HeHY,FanKN.AngewChem
IntEd,2011,50:7815[28] DuXL,LiuYM,WangJQ,CaoY,FanKN.ChinJCatal,2013,34:
993[29] HengneAM,RodeCV.GreenChem,2012,14:1064[30] YanK,LiaoJY,WuX,XieXM.RSCAdv,2013,3:3853[31] GandariasI,RequiesJ,AriasPL,ArmbrusterU,MartinA.JCatal,
2012,290:79[32] SalagreP,FierroJLG,MedinaF,SueirasJE.JMolCatalA,1996,
106:125[33] PriyaGK,PadmajaP,WarrierKGK,DamodaranAD,AruldhasG.J
MaterSciLett,1997,16:1584[34] Ashok J, SubrahmanyamM,VenugopalA. Int JHydrogenEnergy,
2008,33:2704[35] LuoWH, Deka U, Beale AM, van Eck E R H, Bruijnincx P C A,
WeckhuysenBM.JCatal,2013,301:175[36] ElDoukkaliM,IriondoA,AriasPL,CambraJF,GandariasI,Barrio
VL.IntJHydrogenEnergy,2012,37:8298[37] ShamsiA,BaltrusJP,SpiveyJJ.ApplCatalA,2005,293:145
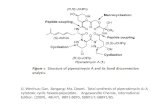
GraphicalAbstract
Chin.J.Catal.,2014,35:656–662 doi:10.1016/S1872‐2067(14)60051‐6
LevulinicacidhydrogenolysisonAl2O3‐basedNi‐Cubimetalliccatalysts
IkerObregón*,ErizCorro,UrkoIzquierdo,JesusRequies,PedroL.AriasUniversityoftheBasqueCountry,Spain
The influence of the metallic phase and preparation methods of alumi‐na‐based copper‐nickel catalysts on the hydrogenolysis of levulinic acid toproduce γ‐valerolactone was studied. High yields of up to 96% wereachieved. Al2O3
Ni Cu NiNi CuCoke Ni
H2
H H
OH
O
O O O
O O