
Labelling of Various Macromolecules Using Positron ...167031/FULLTEXT01.pdf · Labelling of Various...
58
Comprehensive Summaries of Uppsala Dissertations from the Faculty of Science and Technology 605 _____________________________ _____________________________ Labelling of Various Macromolecules Using Positron Emitting 76 Br and 68 Ga Synthesis and Characterisation BY ULRIKA YNGVE ACTA UNIVERSITATIS UPSALIENSIS UPPSALA 2001
Transcript of Labelling of Various Macromolecules Using Positron ...167031/FULLTEXT01.pdf · Labelling of Various...

Comprehensive Summaries of Uppsala Dissertationsfrom the Faculty of Science and Technology 605
_____________________________ _____________________________
Labelling of Various Macromolecules Using Positron Emitting 76Br and 68Ga
Synthesis and Characterisation
BY
ULRIKA YNGVE
ACTA UNIVERSITATIS UPSALIENSISUPPSALA 2001

Dissertation for the Degree of Doctor of Philosophy in Organic Chemistry presentedat Uppsala University in 2001
ABSTRACT
Yngve, U. 2001. Labelling of Various Macromolecules Using Positron Emitting 76Brand 68Ga. Synthesis and Characterisation. Acta Universitatis UpsaliensisComprehensive Summaries of Uppsala Dissertations from the Faculty of Science andTechnology 605. 58pp. Uppsala. ISBN 91-554-4939-5
Different prosthetic groups containing a trialkylstannyl- and an electrophilic grouphave been synthesised and labelled with the accelerator produced 76Br (T1/2=16 h)through oxidative bromination. The labelled prosthetic groups were conjugated toamino-containing macromolecules such as proteins and 5´-modified oligonucleotides.
N-Succinimidyl 4-[76Br]bromobenzoate 14 was synthesised in 65 % radio-chemical yield and was conjugated to 5´-hexylamino-modified phosphodiester andphosphorothioate oligonucleotides in 12-19 % isolated radiochemical yield. Thestability of the 76Br-oligonucleotide-conjugates in vivo in rats was investigated. Nodegradation from the 5´-end, resulting in labelled, low molecular weight compoundswas detected. Compound 14 has also been used for labelling of different proteins in23-61% radiochemical yield.
N-Succinimidyl-5-[76Br]bromo-3-pyridinecarboxylate 17 and methyl-4-[76Br]bromobenzimidate 15 were synthesised from the corresponding trimethyl-stannyl-compound in 25% and 40 % yield respectively. Compounds 14 and 17 wereconjugated to ε-Boc-octreotide in 55 and 50% isolated radiochemical yieldrespectively after microwave heating. Compound 15 did not react with octreotideunder the conditions investigated. The two 76Br-labelled octreotide derivativesshowed different lipophilicity and different binding-properties to tissue frommeningiomas.
Hyaluronic acid, a polysaccharide, was modified with tyramine and labelledby oxidative bromination using 76Br in 10% radiochemical yield.
The generator produced 68Ga (T1/2=68 min) was used to label octreotide andoligonucleotides modified with the metal chelating group 1,4,7,10-tetraazacyclo-dodecane-1,4,7,10-tetraacetic acid (DOTA). 68Ga-DOTA-octreotide was isolated in65% radiochemical yield and a phosphorothioate 68Ga-DOTA-oligonucleotide wasisolated in 35% radiochemical yield after 30 min synthesis time.
Compound 14 was reacted with 3-aminomethylbenzylamine to give compound18. The specific radioactivity of 18 was determined to be 36 GBq/µmol by measuringthe ratio between the mass-peaks for the 76Br and 79Br-compounds using packed-capillary LC-MS.
Ulrika Yngve, Department of Organic Chemistry, Institute of Chemistry, UppsalaUniversity, Box 531, SE-751 21 Uppsala, Sweden
Ulrika Yngve 2001
ISSN 1104-232XISBN 91-554-4939-5
Printed in Sweden by Eklundshofs Grafiska AB, Uppsala 2001

Till Jonas

Papers included in this thesis:
This thesis is based on the following papers, which are referred to in the text by theirroman numerals.
I Synthesis of N-Succinimidyl 4-[76Br]Bromobenzoate and its Use inConjugation Labelling of MacromoleculesYngve, U.; Hedberg, E.; Lövqvist, V.; Tolmachev, V.; Långström, B. ActaChem. Scand. 1999, 53, 508-512.
II Synthesis and Characterization of 76Br-Labeled Phosphodiester andPhosphorothioate OligonucleotidesYngve, U.; Hedberg, E.; Wu, F.; Lendvai, G.; Bergström, M.; Långström, B.Submitted to Bioconj. Chem., Unpublished work copyright (2001) AmericanChemical Society
III Determination of Specific Radioactivity for 76Br Labelled CompoundsMeasuring the Ratio between 76Br and 79Br Using Packed CapillaryLiquid Chromatography Mass SpectrometryForngren, B. H.; Yngve, U.; Forngren, T.; Långström, B. Nucl. Med. Biol.2000, 27, 851-853.
IV Distribution of 76Br-Labeled Antisense Oligonucleotides of DifferentLength Determined Ex Vivo in RatsWu, F.; Yngve, U.; Hedberg, E.; Honda, M.; Lu, L.; Eriksson, B.; Watanabe,Y.; Bergström, M.; Långström, B. Eur. J. Pharm. Sci. 2000, 10, 179-186.
V 68Ga-Labelling of DOTA-OligonucleotidesYngve, U.; Långström, B. Preliminary manuscript
VI Labelling of Octreotide using 76Br-Prosthetic GroupsYngve,U.; Khan, T. S.; Bergström, M.; Långström, B. Submitted to J.Labelled Compd. Radiopharm. 2001
VII 68Ga-labelling of DOTA-OctreotideYngve, U.; Mäcke, H. R.; Långström, B. Submitted
VIII Appendix: Supplementary MaterialYngve, U.
Reprints were made with permission from the publishers.
I have been responsible for planning and carrying out all synthetic and labellingchemistry, including the chemical characterization of the labelled products, in paperI-VII. In papers I and II this work was done in collaboration with E. Hedberg. TheLC-MS analyses in paper III were carried out by B. Forngren. I wrote papers I-IIIand V-VII, paper II together with E. Hedberg and paper III together with B.Forngren. My contribution to paper IV was, except for the labelling synthesis, toperform the metabolic studies and I took part in writing the paper.

Contents:
Abstract 2Papers Included In the Thesis 4List of Abbreviations 6
1. Introduction 7Imaging technology in vivo and in vitro 7Macromolecules as tracers 8
2. Oligonucleotides 9Antisense oligonucleotides 10
Backbone modified oligonucleotides 11
3. Radiolabelling Synthesis for Use with Macromolecules 12Radionuclides used in PET 12Specific radioactivity 13Biological aspects 14Experimental considerations 14Labelling of macromolecules using halogens 15Labelling using metal chelates 18Oligonucleotides 19Octreotide 21Hyaluronic acid 22
4. Radiolabelling Using 76Br 23Production of [76Br]bromide 23Synthesis of stannyl-compounds as prosthetic groups I, VI, VIIIA 23Oxidative bromination of stannylated precursors I, VI, VIIIA 26Determination of specific radioactivity I, III 28Conjugation labelling 30Oligonucleotides I, II, VIIIA 31Proteins I, VI, VIIIC 33Peptides VII 35Labelling of Hyaluronic Acid VIIIB 37
5. Labelling Using 68Ga 39Production of 68Ga 39Chelators used with Ga 39DOTA-OctreotideVII 40DOTA-OligonucleotidesV 41
6. Characterisation of Labelled Oligonucleotides 43Analysis using LC-MS II 43Metabolite analysis IV 44Distribution of 76Br-oligonucleotides in vivo and in vitro IV 46Characterisation of hybridisation properties of labelledoligonucleotidesIV
47
7. Conclusions 49
Acknowledgements 50References 52

Abbreviations
β+ PositronBoc tert-ButyloxycarbonylBSA Bovine serum albuminChloramine-T N-Chloro-p-toluenesulphonamide sodium salt
CPB Cetylpyridinium bromide, 1-hexadecylpyridinium bromideDMF N,N-DimethylformamideDMSO Dimethyl sulphoxideDNA Deoxyribonucleic acid
DOTA 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid
DOTATOC 4,7,10-Tricarboxymethyl-1,4,7,10-tetraazacyclododecan-1-yl-acetyl-D-Phe-Cys-Tyr-D-Trp-Lys-Thr-Cys-L-threoninol (DOTA-D-Phe1-Tyr3-Octreotide)
EC Electron captureEDC 1-Ethyl-3(3-dimethylaminopropyl)carbodiimideESI Electrospray ionisationHPLC High-performance liquid chromatographyHSA Human serum albuminLC Liquid chromatographyMRI Magnetic resonance imagingmRNA Messenger-RNAMS Mass spectrometryODN OligodeoxynucleotidePCR Polymerase chain reactionPd(PPh3)4 Tetrakis(triphenylphosphine)palladium(0)PET Positron emission tomographyRNA Ribonucleic acidSODN Phosphorothioate oligodeoxynucleotideSPE Solid phase extractionSPECT Single photon emission tomographySulfo-NHS 1-Hydroxy-2,5-dioxo-3-pyrrolidinesulfonic acidTEAA Triethylammonium acetateTFA Trifluoroacetic acidTyr TyrosineUV Ultraviolet

7
1. Introduction
Imaging technology in vivo and in vitro
The first tracer-experiment was performed by Hevesy in 1923 using a radioactive lead
salt to detect the uptake in plants.1 Since then the technology has developed in for
example the areas of detection devices and synthesis of tracer compounds.
One detection technique is autoradiographic imaging where the samples, for example
slices of tissues or slices of small animals injected with a radiolabelled tracer, are
placed on photographic films or reusable phosphor imager screens to image the
distribution of radioactivity.2 This technique is suitable for the detection of β-particles
with high precision and sensitivity. An alternative way to investigate the distribution
of radioactivity is to take out samples, for example different organs from a rat, and
measure the γ- radiation in a well-counter.
Imaging of living subjects requires non-invasive approaches where the distribution of
a substance can be measured by external detectors. Magnetic Resonance Imaging3
(MRI) is based on the detection of stable nuclides while techniques such as Positron
Emission Tomography4 (PET) and Single Photon Emission Computed Tomography5
(SPECT) detect the decay of radioactive nuclides. A main differences between these
techniques are related to resolution and sensitivity.3
The PET technique is based on the use of positron emitting radionuclides that are
neutron deficient and decays by the emission of a positron, the anti-matter of an
electron. After a few millimetres in tissue, the positron encounters an electron, the
particles are annihilated and two 511 keV photons (γ-radiation) are emitted in
opposite directions. These anti-parallel photons are detected in coincidence by the
PET-camera and the distribution of radioactivity in a living subject can be measured
with high precision and quantified as a function of time and region.

8
Macromolecules as tracers
Proteins and peptides play an important role in many diseases and can therefore be
used as probes or diagnostic tools if radiolabelled. This technique has been exploited
in the radiolabelling of antibodies and analogues of the peptide somatostatin for the
diagnosis and treatment of tumours. Oligonucleotides are currently investigated for
treatment of cancers and viral infections.6
There are a limited number of biogenic radionuclides available (such as 3H, 11C, 13N,14C, 15O, 32P, 33P, 35S) and their usefulness in synthesis of tracer compounds are set by
chemical properties, half-life, decay-properties and availability. Common
radionuclides for imaging processes involving macromolecules, such as antibodies
and proteins, are isotopes of iodine7 or various radiometals (e.g. 99mTc) attached via
chelating groups8 and mostly using SPECT as imaging modality. Usually, analogues
of the molecule of interest are prepared to introduce the label since these elements are
not frequently found in biomolecules. Halogens can be introduced in molecules
containing activated aromatic rings, for example the amino acid tyrosine, by oxidative
halogenation. Another option is to conjugate a labelled molecule, a so-called
prosthetic group, to the macromolecule. As a part of the characterisation, biological
assays or other tests need to be performed to confirm that the properties of the
analogue do not differ substantially from the original molecule.
The aim of the work presented in this thesis is the development of synthetic methods
for radioactive labelling of macromolecules. Oligonucleotides, proteins and peptides
have been labelled with the accelerator produced positron emitting nuclide 76Br
(T1/2=16 h) by the use of prosthetic groups. The study also includes labelling of
oligonucleotides and a peptide using a chelating agent for the generator produced 68Ga
(T1/2=68 min).
The synthesis of the prosthetic groups, radiolabelling and conjugation conditions are
described. With the emphasis on the oligonucleotides, the labelled molecules have
been subjected to chemical characterisation as well as characterisation of the
behaviour in vivo or in vitro.
9
2. Oligonucleotides
Recently the human genetic code has been resolved and a revolution in drug design
based on the genetic code can be expected within the pharmaceutical industry. A
following step would be the identification and understanding of the proteins expressed
in organisms, a more problematic work due to the complexity of the proteins as
compared to nucleic acids.9
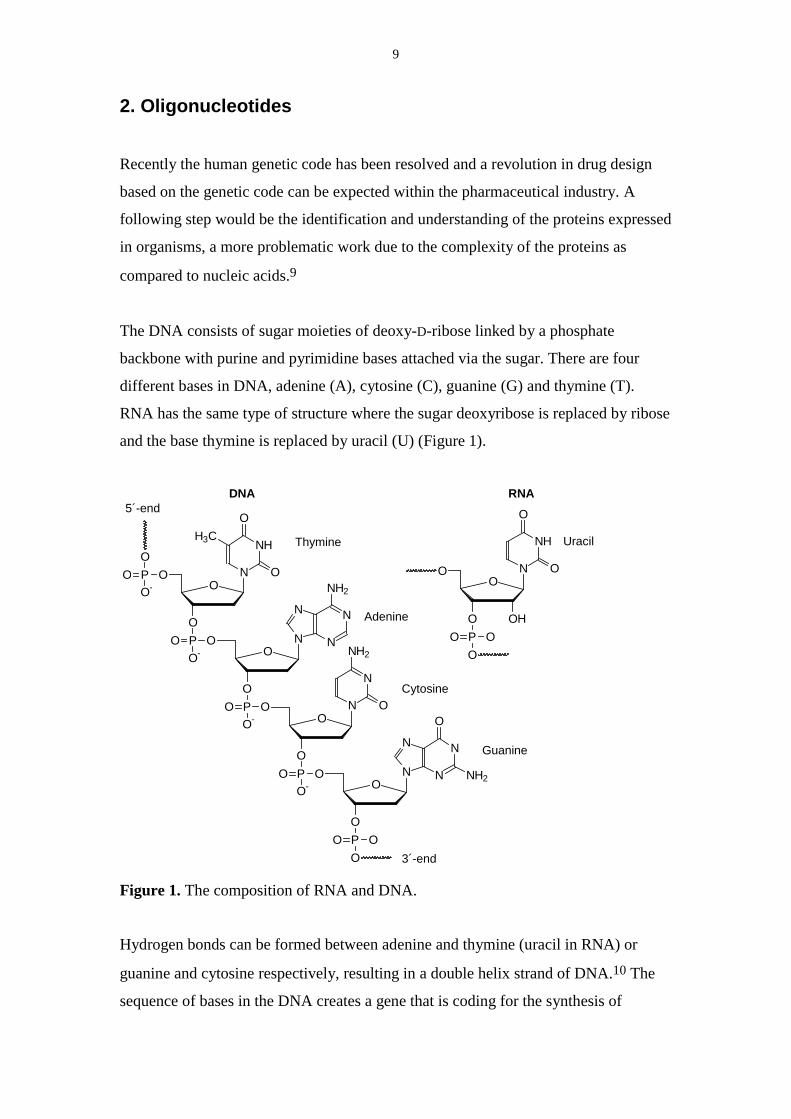
The DNA consists of sugar moieties of deoxy-D-ribose linked by a phosphate
backbone with purine and pyrimidine bases attached via the sugar. There are four
different bases in DNA, adenine (A), cytosine (C), guanine (G) and thymine (T).
RNA has the same type of structure where the sugar deoxyribose is replaced by ribose
and the base thymine is replaced by uracil (U) (Figure 1).
O
OPOO
O
N
N
N
N
O
NH2
ON
N
O
OPOO-
O
NH2
ON
NH
O
O
H3C
OPOO-
O
OPOO-
O
O
OPOO-
O
N
N
N
N
NH2O
N
NH
O
OPOO
O
O
O
OH
Thymine
Adenine
Cytosine
Guanine
Uracil
5´-end
3´-end
DNA RNA
Figure 1. The composition of RNA and DNA.
Hydrogen bonds can be formed between adenine and thymine (uracil in RNA) or
guanine and cytosine respectively, resulting in a double helix strand of DNA.10 The
sequence of bases in the DNA creates a gene that is coding for the synthesis of

10
proteins. The double helix partly uncoils and a complementary RNA-string,
pre-messenger-RNA, is built and transformed into messenger-RNA (mRNA). The
mRNA migrates into the cell where the ribosomes can read the encoded information
and assemble proteins from amino acids, in the so-called translation.
Antisense oligonucleotides
An antisense oligonucleotide is a synthetic single-stranded DNA that has a base
composition that is complementary to a part of DNA or RNA (the so called sense
strand).11 An oligonucleotide that is antisense to the mRNA can hybridise selectively
through Watson-Crick base pairing with the mRNA to form a double helix and inhibit
the protein synthesis that is encoded by that mRNA. There are two possible suggested
mechanisms, one is where this double-strand is prevented from transferring
information to the ribosome, thus protein synthesis is inhibited (Figure 2). The other
mode of action of the antisense oligonucleotide is that the mRNA-oligonucleotide
duplex can be recognised by degrading enzymes, RNase H, which destroy the mRNA.
The oligonucleotide is then released and may hybridise with the next mRNA molecule
(Figure 2).
Figure 2. Proposed mechanisms for inhibition of protein-synthesis by antisenseoligonucleotides.
Pro-messenger RNA
mRNA
Antisense oligonucleotide
RibosomeTranslation Protein-
synthesis
No translation
DNA
Enzymes
Breakdown of mRNA, release ofantisense oligonucleotide No translation
Ribosome

11
Antisense drugs are expected to inhibit the synthesis of the disease-causing protein
encoded by the target mRNA, and thereby intervene in an early stage of the disease-
causing process. There is a potential for antisense drugs to be more selective than
traditional drugs due to the multiple points of interaction between the mRNA and the
antisense oligonucleotide. When the sequence of the mRNA is known and
understood, the design of the drug should be straightforward. Statistically a 15-17
base sequence is enough to avoid unintentional complete match of the mRNA.12
Backbone modified oligonucleotides
In reality, there are many factors that complicate the use of antisense drugs.
Phosphodiester oligonucleotides are readily degraded by nuclease enzymes and have a
half-life of less than 30 minutes in the cell.11 One strategy of avoiding degradation
and to achieve more stable oligonucleotides would be to modify the phosphate
backbone (Figure 3). Replacing one of the non-bridging oxygens with sulphur gives
phosphorothioate oligonucleotides which are more stable towards attack by nucleases
and increases the half-life in serum to up to 24 hours. However, phosphorothioate
oligonucleotides show higher unspecific binding than phosphodiester oligonucleotides
to proteins.13 Methylphosphonate oligonucleotides are compounds where a methyl
group has replaced one of the non-bridging oxygens (Figure 3). The success of
phosphorothioate oligonucleotides as potential therapeutic agents is due to the fact
that they can be recognised by RNase H, while this mechanism does not work for
methylphosphonate oligonucleotides (Figure 2).14 Phosphorothioate and
methylphosphonate oligonucleotides are reported to hybridise with lower affinity than
phosphodiester oligonucleotides.15 One explanation is that phosphorothioate and
methylphosphonate oligonucleotides are synthesised as a mixture of disasteromers.11
O
O
HO Base
PX OO
X= O-, Phosphodiester DNAX= S-, Phosphorothioate DNAX= CH3, Methylphosphonate DNA
Figure 3. Backbone modified oligonucleotides.
12
3. Radiolabelling Synthesis for Use with Macromolecules
Radionuclides used in PET
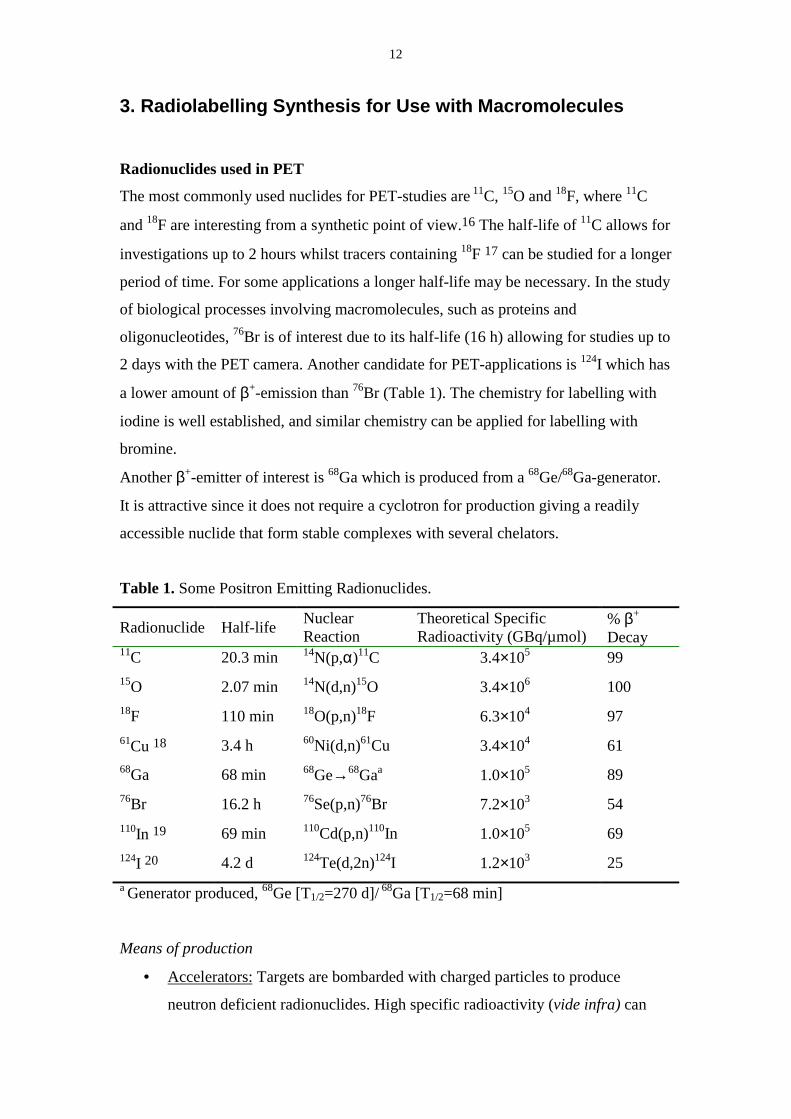
The most commonly used nuclides for PET-studies are 11C, 15O and 18F, where 11C
and 18F are interesting from a synthetic point of view.16 The half-life of 11C allows for
investigations up to 2 hours whilst tracers containing 18F 17 can be studied for a longer
period of time. For some applications a longer half-life may be necessary. In the study
of biological processes involving macromolecules, such as proteins and
oligonucleotides, 76Br is of interest due to its half-life (16 h) allowing for studies up to
2 days with the PET camera. Another candidate for PET-applications is 124I which has
a lower amount of β+-emission than 76Br (Table 1). The chemistry for labelling with
iodine is well established, and similar chemistry can be applied for labelling with
bromine.
Another β+-emitter of interest is 68Ga which is produced from a 68Ge/68Ga-generator.
It is attractive since it does not require a cyclotron for production giving a readily
accessible nuclide that form stable complexes with several chelators.
Table 1. Some Positron Emitting Radionuclides.
Radionuclide Half-life NuclearReaction
Theoretical SpecificRadioactivity (GBq/µmol)
% β+
Decay11C 20.3 min 14N(p,α)11C 3.4×105 9915O 2.07 min 14N(d,n)15O 3.4×106 10018F 110 min 18O(p,n)18F 6.3×104 9761Cu 18 3.4 h 60Ni(d,n)61Cu 3.4×104 6168Ga 68 min 68Ge→68Gaa 1.0×105 8976Br 16.2 h 76Se(p,n)76Br 7.2×103 54110In 19 69 min 110Cd(p,n)110In 1.0×105 69124I 20 4.2 d 124Te(d,2n)124I 1.2×103 25a Generator produced, 68Ge [T1/2=270 d]/ 68Ga [T1/2=68 min]
Means of production
• Accelerators: Targets are bombarded with charged particles to produce
neutron deficient radionuclides. High specific radioactivity (vide infra) can

13
often be achieved since the production of many radionuclides is no carrier
added.21
• Reactors: In a reactor the target is bombarded by neutrons to form
radionuclides with an excess of neutrons.22
• Generators: Generators consist of a parent nuclide (produced by reactors or
accelerators) that decays to form the daughter radioisotope. One way to
separate the nuclides is to adsorb the parent nuclide on to a solid support in
such a fashion that the daughter nuclide can be eluted from the support.23
Specific radioactivity
Specific radioactivity is defined as the amount of radioactivity per unit mass and can
also be expressed as radioactivity per mole. The theoretical specific radioactivity can
be calculated from the half-life of the nuclide, N=A0× T1/2/ln2. Due to the isotopic
dilution from for example equipment, target materials and chemicals used, the
specific radioactivity achieved is usually not equal to the theoretical value. In order to
avoid isotopic dilution, the reaction conditions should be considered to minimise the
used of contaminating chemicals and materials. Examples of contaminating sources
are glass ware when working with radiometals or the atmosphere when working with
[11C]CO2. Also, in order to maximise the specific radioactivity, the synthesis time
should be minimised.
Effective specific radioactivity can be discussed when there are chemical impurities in
the final product-preparation that have biological activity towards the same system as
the desired product. Receptor binding assays have been designed for the
determination of the effective specific radioactivity. This has previously been
described for 16α-[77Br]-bromoestradiol, a steroid, where the effective specific
radioactivity was found to be about five times lower than the specific radioactivity. 24
Unlabelled impurities with similar biological properties as the labelled substance can
also be referred to as pseudo carriers, for example when Cl is incorporated instead of76Br and not removed in the purification process. Another example, discussed later in
this thesis, is the specific radioactivity of labelled proteins. In this case, the specific
radioactivity is determined by the amount of protein used in the labelling reaction,
since it is difficult to separate the unlabelled protein from the labelled by
chromatographic means.

14
Biological aspects
Some biological aspects are important to take into consideration when designing the
tracer molecules.
• To minimise the radiation dose to the subject undergoing a PET-investigation
and maximise the signal measured, the half-life of the radionuclide used
should match the biological half-life of the process studied.
• To obtain sufficient radioactivity for measurements at low concentrations of
the tracer, the specific radioactivity has to be high. For oligonucleotides, the
specific radioactivity required has been estimated to be 100 µCi/µg12, which
for a 30 mer oligonucleotide correspond to approximately 40 MBq/nmol.
• The measurement of radioactive decay does not discriminate between different
chemical species. Therefore the position of the label should be considered
regarding the metabolism of the molecule. Alternatively the position of the
label can be selected so that the cleavage of the label visualises a metabolic
process.25, 26 Analysis of plasma using chromatographic techniques can be of
value for interpretation of the PET-data if the tracer is metabolised.27, 28
• When prosthetic groups are used to introduce the label, the prosthetic group
should be chosen so that the biological activity is altered as little as possible.
The design of the prosthetic group can also be considered to increase the
stability of the molecule against degrading processes29 or to facilitate
excretion30.14, 31
Experimental considerations
The short half-life and the damaging radiation from the radionuclides used, put some
demands on the experimental methods used.
• From a radiation safety point of view, the time the chemist is exposed to the
radiation should be minimised. Considering the radiation dose to the chemist
and the possible application for the labelled compound in clinical
investigations, automatisation of the synthesis and purification procedures is
desirable.
• Large excess of reagents are used compared to the labelled precursor, giving
pseudo-first-order kinetics and fast reactions.32 The label should be introduced

15
as late as possible in the synthetic scheme to maximise the specific
radioactivity in the final product.
• Liquid chromatography (LC) and solid phase extraction (SPE) are routinely
used for purification of the labelled products. LC is also used for identification
and concentration determinations using UV-detection and radiometric
detection to match the retention time of the labelled compound with the
retention time of characterised standards. LC-MS is an important method for
characterisation of the products because of the high sensitivity and selectivity
and the possibility to identify a compound according to the molecular
weight.33
Labelling of macromolecules using halogens
Different radioactive isotopes of iodine are commonly used for the labelling of
proteins and antibodies because of its availability and range of decay properties. So
far these tracers have mostly been applied to SPECT and in vitro detection techniques.
Radiohalogens form chemically stable covalent bonds with the aromatic groups in the
proteins and high specific radioactivity can be achieved.31
OH
CH2
NHNHR'
OR
O
OH
CH2
NHNHR'
OR
O
X*
NaX*oxidant
X* CO
LG
NHNHR'''
O
NH2
R''
O
NHNHR'''
O
NH
R''
O
O*X
a)
b)
Figure 4. X*= Br, I, At, LG = leaving groupa) Direct halogenation of tyrosine-residue. b) Conjugation labelling at lysine residue.

16
A straightforward and well-established method to label proteins with iodine is through
oxidative halogenation on the tyrosine residues (Figure 4). 34-36 Bromine has a higher
oxidation potential than iodine but oxidative bromination of proteins have been
performed using enzymes as oxidants37, 38 or by the use of chloramine-T 39. The
methods usually gives high incorporation yields of the halogen but the oxidative
conditions can affect the activity of some proteins and it might be difficult to separate
the enzymes from the labelled product.40 Proteins labelled by oxidative iodination on
tyrosine can sometimes suffer from dehalogenation in vivo.
In the labelling of oligonucleotides and carbohydrates, for example hyaluronic acid,
there is no established method for direct halogenation so the molecules therefore have
to be derivatised prior to labelling.41 Conjugation labelling by the use of prosthetic
groups can be beneficial when direct labelling is not possible due to the lack of
tyrosine-residues or when the compound is sensitive to oxidative conditions. The use
of prosthetic groups is an established method for radiolabelling of proteins31,
derivatisation of oligonucleotides14 and it has also been used for the labelling of
peptides42, 43.
One drawback with the use of conjugation labelling is that the reaction is performed
in two steps, often leading to lower radiochemical yields compared to the direct
labelling. The advantage with conjugate labelling, however, is the mild reaction
conditions and also the increased stability towards in vivo dehalogenation caused by
enzymes. The labelling method is also applicable to a wide range of substrates.
Prosthetic groups for conjugation labelling
In the design of prosthetic groups, metal-halogen exchange reactions are reliable and
can be used for introducing a radiohalogen in aromatic or vinylic substrates under
oxidative conditions.44-48 Trialkylstannyl compounds are commonly used for this
metal-halogen exchange reaction due to high yields and good regiospecificity in the
halogenations.47 However, other metallic compounds such as organosilanes,
organogermanes, organomercurials and organothallates have also been used for the
introduction of radiohalogens.48

17
Activated esters of N-hydroxysuccinimide are good electrophiles for conjugation to
nucleophiles such as amines, forming stable amide-bonds with for example lysine-
residues or the N-terminal end in proteins and peptides or with amino-modified
oligonucleotides (Figure 5c).49 The first prosthetic group used for conjugation
labelling, iodinated 3-(4-hydroxyphenyl) propionic acid N-hydroxysuccinimide ester,
also referred to as the Bolton-Hunter reagent, is now commercially available.50
NH CS
NH RNH CNH
R
R CO
O N
O
OR NCS
R CNH
OCH3
NH2
NH CO
R
a b c
Figure 5. Reaction of an amino-containing molecule with a) methyl-iminoesters, b)isothiocyanates and c) N-hydroxysuccinimidyl esters.
Iminoesters (imidates) are reported to form amidine bonds when reacted with amines
(Figure 5a) and may therefore be useful as prosthetic groups for labelling of
proteins.31, 51, 52, 53-55 Iminoesters are reported to be less reactive towards amines
than succinimidyl-esters but they are also less sensitive to hydrolysis. One advantage
with amidines is that they are protonated at physiological pH and that the original
charge on the protein is thereby retained.
Isothiocyanates are commonly used electrophiles for reaction with amines (Figure
5b). The thiourea bond formed may be more stable than that of the amide bond
formed from reaction of amines with activated esters (Figure 5c).56
Apart from the electrophile, the “R-group” (Figure 5) can also be varied in the
prosthetic group. This feature has been used in the design of N-succinimidyl 5-iodo-3-
pyridine-carboxylate as a prosthetic group57, which has been proven to be
advantageous in that the metabolite iodonicotinic acid is cleared from the tissue faster
than iodobenzoic acid (from the use of N-succinimidyl iodobenzoate as prosthetic
group) due to its reduced lipophilicity.58

18
Labelling using metal chelates
Many different radiometals with a large spectrum of decay-properties are available
from either generator systems or by cyclotron production.59, 60 These metals can be
used in the synthesis of radiopharmaceuticals containing a chelating functionality. A
few positron-emitting radiometals are presented in Table 1.
A chelating agent should be covalently attached to the molecule of interest prior to
reaction with the metal. Alternatively the chelating agent is labelled before
conjugation.61 Some commonly used chelating agents are presented in Figure 6. The
chelator can be attached to the molecule of interest via the carboxylate-group or the
chelator can be functionalised at a carbon for further reaction (SCN-TETA, see
Figure 6).8 Functionalisation of the carbon-skeleton requires a multiple step synthetic
scheme, while the carboxyl groups can be coupled to amines in one step using
carbodiimide chemistry.62
DFO
(CH2)2 CO
NH (CH2)5 NOH
C (CH2)2 C NHO O
(CH2)5 NOH
CO
CH3CO
N(CH2)5H2NOH
N N
CO2H
HO2C
HO2CN
CO2H
CO2H
CPTA
N
N N
N
OH
O
DOTA
NN
N N
CO2H
CO2H
HO2C
HO2C
N NCO2H
CO2H
HO2C
HO2C EDTA
DTPA
N
N N
N
CO2H
CO2HHO2C
HO2C
SCN
SCN-TETA
Figure 6. Some metal-chelators used for radiopharmaceuticals.

19
For many applications it would be advantageous with a labelling protocol that can be
applied for kit-type preparations.41 Due to the high efficacy of many metal chelators,
a small amount of the precursor is mixed with the radiometal giving high yields. It is
often sufficient to use solid phase extraction or gel-filtration to separate the unreacted
metal, if any, from the product. Since only small amounts of the chelator can be used
in order to achieve high specific radioactivity, the reactions are very sensitive to
contamination from other metals often found in trace amounts in buffers, glassware
etc.
For use in vivo and in vitro the metal chelates have to be stable towards translocation
of the metal to serum proteins, e.g. transferrin, or to degradation at physiological pH.
The complexes also have to be stable to competition form the cations found in serum
(Ca2+, Zn2+, Mg2+).63 The stability of the metal complex is to a larger extent
determined by the kinetic inertness (rate of dissociation) rather than of its
thermodynamic stability.64 The stability of the complex can be measured by
incubation in plasma, and samples can be analysed for degradation products. Cyclic
chelators often form more stable metal complexes than open chain ligands, which can
be explained by the high degree of preorganisation.64 Macrocyclic chelators (DOTA,
SCN-TETA) form complexes with high stability with a variety of metal ions.
Sometimes the incorporation of the metal is slow and trace metals can compete in the
reaction.65
Oligonucleotides
Oligonucleotides can be modified in the purine or pyrimidine bases, sugar-moiety or
in the 5´- or 3´-end to give a handle for introduction of a radioactive or stable labelled
group (Figure 7). End-modifications might also increase the stability of the
oligonucleotide from degradation by exonucleases.66
The label can be introduced by a conjugate technique where the oligonucleotide has
been modified with a linker that can be reacted with a radioactive labelled prosthetic
group.41 Other groups can be used to trace the behaviour of the oligonucleotide, for
example fluorescent groups67 or biotin68. Many prosthetic groups used in the
labelling of proteins contain an electrophilic group. The same approach as used for

20
labelling of peptides and proteins can then be used for oligonucleotides containing a
nucleophilic group (Figure 7), regardless of chain length, base composition or
backbone modifications.
O
O
O Base
P-X OO
PO
X-
X = O, S
H2N
Figure 7. 5´-Hexylamino-modified oligonucleotide.
Radiohalogens
Oligonucleotides with different backbone modifications containing a
phosphorothioate monoester in the 3´-position have been conjugated to N-(4-
[18F]fluorobenzyl)-2-bromoacetamide.69, 70 The method has since been modified to
include 76Br and 125I-labelling.71 Labelling of 5´-aminohexyl-oligonucleotides has
been achieved using 18F using N-succinimidyl [18F]fluorobenzoate72 or 4-
([18F]fluoromethyl)phenyl isothiocyanate73 and it also has been radioiodinated using
4-methoxyphenyl isothiocyanate56. N-Succinimidyl 4-tributylstannylbenzoate has
been preconjugated to an amino-modified oligonucleotide followed by labelling of the
conjugate with 125I.74 Oxidative iodination has also been performed on
oligonucleotides modified with a tyramine-group in the 5´-position.75 A stannyl-
derivative of uracil has been utilised in the automated synthesis of oligonucleotides to
produce a stannylated oligonucleotide derivative, which could be radioiodinated.76
Enzymatic methods have been also been used to incorporate 125I-labelled cytosine.77
Radiometals
Oligonucleotides (ODN) have been labelled with 99mTc by conjugating a chelating
agent to the 5´-position.78, 79. The chelator CPTA has been attached to the 5´-end of
oligonucleotides using phosphoramidite chemistry on an automated DNA-synthesiser
and the CPTA-oligonucleotide was labelled with 64Cu or 99mTc.80 Using DTPA as a
chelator, oligonucleotides with amino-modifications in different positions have been
labelled with 111In.81, 82 The DTPA-ODN has also been used for labelling with 67Ga

21
83, 99mTc and 153Sm.84 Other approaches for labelling of oligonucleotides with 111In is
the linkage of isothiocyanobenzyl-EDTA to oligonucleotides where deoxyuracil
containing an amino-linker was used as a substitute for deoxythymine.85 This allowed
for oligonucleotides with more than one linker per molecule, which could be used to
achieve higher specific radioactivity.
Octreotide
Due to the interest in the role of
somatostatin receptors for
tumour diagnosis and treatment,
methods have been developed to
radiolabel analogues of
octreotide, a synthetic
octapeptide that has shown high
selectivity for the somatostatin
receptor. 111In-DTPA-(D)Phe1-
octreotide (Octreoscan ) and 123I-Tyr3-octreotide are routinely used for clinical
applications.86
Radiohalogens
Both N-succinimidyl 3-iodobenzoate 42 and N-succinimidyl 4-[18F]fluorobenzoate 43
have been used to label peptides. The [18F]fluoro-, [131I]iodo- and [211At]astato-
analogues of octreotide have previously been prepared by conjugation to the
corresponding N-succinimidyl 4-halobenzoate 87, N-succinimidyl 5-halopyridine-3-
carboxylate 87, 4-nitrophenyl 2-[18F]fluoropropionate 88 or 4-[18F]fluorobenzoic
acid 89. Direct iodination on Tyr-3-octreotide has also been performed.90
Radiometals
Different metal chelating ligands have been coupled to different derivatives of
octreotide and subsequently radiolabelled using 64Cu,91 67Ga,92 68Ga,93 111In,94
99mTc,95 86Y,96 and 90Y 97, 98.
NN
NHO
HOO
O
O
S
H2NN
NN
O
O
ON
OS
NH2
HO
NH
Octreotide

22
Hyaluronic acid
Hyaluronic acid is a linear polysaccharide composed of disaccharide units of N-
acetyl-D-glucosamine and D-glucuronic acid. Hyaluronic acid has been
radioiodinated by reacting tyramine with the aldehyde terminal followed by oxidative
iodination of the tyramine-group.99 The specific radioactivity obtained is limited by
the 1:1 molar ratio of tyramine to hyaluronic acid, yielding a maximum of one
radioiodine per polysaccharide. The high molecular weight hyaluronic acid has been
labelled with iodine after cyanogen bromide activation of the polysaccharide followed
by conjugation to tyrosine and radioiodination.100 Due to the possibility of multiple
sites of labelling in one molecule, higher specific radioactivity can be obtained.
Hydrazinolytic N- deacetylation of the amide-functionality gave access to an amine
that could be derivatised with tyrosine-cellobiose and subsequently iodinated.101
Hyaluronic acid has also been labelled with the short-lived isotope 11C (T1/2= 20 min)
using [11C]CNBr.102
Preconjugation of a prosthetic group was the only option available for the high-
molecular weight hyaluronic acid. Coupling of diamines to the carboxylic acid moiety
of the hyaluronic acid to by carbodiimide activation to introduce a nucleophile in the
molecule has proven to be unsuccessful.103 Hydrazides could be coupled to fragments
of hyaluronic acid due to their better nucleophilicity at a pH around 4-5.104 For
reasonable yields in conjugation reactions with a labelled electrophile about 70 nmol
amine/100 µl is required. This corresponds to 230 amino-modifications/ hyaluronic
acid molecules (Mw=1,5×106) at a concentration of 5 mg/ml.

23
4. Radiolabelling using 76Br
Due to its partial decay by positrons and its half-life of 16 h, 76Br is an attractive
nuclide for labelling of oligonucleotides and proteins for possible applications in PET.
Electrophilic bromine has been used for bromination of activated aromatic rings such
as tyrosine. The use of halogen-demetalation reactions are advantageous due to high
regioselectivity and its efficacy even on deactivated systems. Bromine-labelling by
nucleophilic substitution can be used, but there can, however, be a problem with
decreasing effective specific activity if the leaving group is a halogen. For aromatic
substrates, other reactions can used, for example the Sandmeyer reaction.105
Production of [76Br]bromide
The radionuclide 76Br is produced by proton irradiation of a [76Se]selenium enriched
Cu2Se-pellet using a low-energy cyclotron.106 The 76Br is recovered from the target-
pellet using a thermo-chromatographic method and it is obtained in water or ethanol.
In order to obtain reproducible yields in the labelling reaction, the water solution of
[76Br]bromide is purified using a C-18 SPE-disc to remove lipophilic components.76Br has previously been produced indirectly by the EC-decay of cyclotron produced76Kr (T1/2= 14.6 h)107, 108 or by a more direct route using the 75As(3He, 2n)76Br
nuclear reaction109.
Synthesis of stannyl-compounds as prosthetic groups I, VI, VIIIA
There are two major routes for the preparation of aromatic stannylated compounds,
either by nucleophilic attack of a lithiated compound on trialkylstannyl chloride or by
using a palladium(0)-catalysed coupling reaction between an aromatic or vinylic
halide and hexaalkyldistannane (Figure 8).
R' = CH3, (CH2)3CH3
ClSnR'3
BuLi
Sn2R'6, Pd(0)
R SnR'3
R LiX R
Figure 8. Two routes to aromatic stannyl-compounds.

24
Due to the milder reaction conditions, the route using palladium(0) catalysis was
preferred for introducing the stannyl functionality when synthesising the prosthetic
groups used in paper I, VI and VIIIA. However in the synthesis of N-succinimidyl-5-
trimethylstannyl-3-pyridinecarboxylate 13, the published method using n-butyl
lithium was applied.57
N-Succinimidyl 4-trialkylstannylbenzoate 4 I
N-Succinimidyl trialkylstannylbenzoates have previously been labelled with 211At 110,77Br 111 and different isotopes of iodine 112, 113. Several procedures have been
published for the preparation of N-succinimidyl trialkylstannylbenzoates.29-30, 112-114
These methods were modified in the synthesis of succinimidyl 4-trimethylstannyl-
benzoate 4a. Recently, a two-step route to 4a has been described, where iodobenzoic
acid was stannylated followed by esterfication.115
R3Sn CO
OCH2CH3I C
O
OCH2CH3
Sn2R6Pd(PPh3)4toluene
NaOH (aq)Ethanol
R3Sn CO
OHDisuccinimidylcarbonatePyridineAcetonitrile
R3Sn CO
O N
O
O
1 2
3 4a) R= Me4b) R= Bu
Scheme 1.
As described in paper I, ethyl 4-iodobenzoate was stannylated using hexamethylditin
and tetrakis(triphenyl-phosphine)palladium(0) [Pd(PPh3)4] as a catalyst to give
compound 2 in 93% yield.113, 114 The stannylbenzoate 2 was hydrolysed followed by
esterfication with N,N’-disuccinimidylcarbonate to give the desired 4a (16%)
(Scheme 1).30 The tributylstannyl analogue 4b was synthesised using the same
method.
Methyl-4-trimethylstannylbenzimidate 7VI
For halogenation of proteins, methyl-4-trimethylstannylbenzimidate has been
suggested as prosthetic group.116 Methyl-4-bromobenzimidate 6 was prepared from

25
4-bromobenzonitrile using the Pinner reaction where methanol was used both as a
solvent and a reactant (Scheme 2) and isolated in 74 % yield.117 When diethyl ether
was used as solvent in the Pinner reaction a lower yield was obtained.118 Base
catalysed conversion of bromobenzonitrile to the iminoester119 also resulted in low
yields. The methyl-4-bromobenzimidate was stannylated using the same conditions as
for compound 2 giving methyl-4-trimethylstannylbenzimidate 7 in 40% yield.
Sn2(CH3)6Pd(PPh3)4tolueneBr C N (CH3)3Sn C
NH
OCH3
5 7
Br CNH
OCH3
6
1) HCl(g)MeOH2) K2CO3
Scheme 2.
4-Tributylstannylphenylisothiocyanate 10VIIIA
Synthesis of 4-tributylstannylaniline 9 was achieved using triethylamine as solvent
and Pd(0) as catalyst in a low yield (16%) (Scheme 3).120 The 4-tributylstannylanilin
was transformed to the corresponding isothiocyanate 10 using thiocarbonyl-
diimidazole, a thiophosgene equivalent, in 83% yield (Scheme 3).121 Attempts were
made to synthesise the 4-trimethylstannylaniline using the same method as for the 4-
tributylstannylaniline but without success.
CHCl3
N NNN
S
Sn2Bu6Pd(PPh3)4Et3N
Bu3Sn NCSBu3Sn NH2Br NH2
9 108
Scheme 3.
N-Succinimidyl-5-trimethylstannyl-3-pyridinecarboxylate 13VI
Preparation of N-succinimidyl-5-trimethylstannyl-3-pyridinecarboxylate 13 was
carried out using n-butyl lithium and trimethylstannyl chloride (Scheme 4).57

26
CH3 SO2N-Na+
Cl
Chloramine-T
N-HydroxysuccinimideDCCCH2Cl2
1) n-BuLi (2 eq)2) (CH3)3SnCl (2.2 eq)
N
(CH3)3Sn CO
OSn(CH3)3
N
CO
OHBr
N
(CH3)3Sn CO
O N
O
O
11 12
13
Scheme 4.
Oxidative bromination of stannylated precursors I, VI, VIIIA
[76Br]Bromide can be oxidised to electrophilic bromine using
for example chloramine-T or peracetic acid as the
oxidant.105 The stannyl-compounds 4, 7, 10 and 13 were
brominated with 76Br, using chloramine-T as the oxidant.
N-Succinimidyl 4-[76Br]bromobenzoate 14 I
Bromination of 4a using chloramine-T as oxidant and ethanol as solvent gave isolated
yields of 76BrNHS 14 around 65% (Scheme 5). Other oxidants, such as N-
chlorosuccinimide, tert-butyl hydroperoxide and IODO-BEADS (polymer-bound
chloramine-T) were tested using 4a as the substrate for bromination, but these
oxidants gave lower yields than chloramine-T.
CO
O N
O
O
76BrMe3Sn CO
O N
O
O
4a 1476BrNHS
[76Br]Br-
Chloramine-T HOAC, EtOH70°C, 10 min
Scheme 5.
The purity of the stannyl-compound is of importance for the labelling yield since a
large excess of stannyl-compound is used compared to the halogen. Even small
amounts of impurities can give large amounts of labelled side-products. For example,
iodination of tributylstannyl 4-benzoic acid is reported to be much faster than the
iodination of the succinimidylester.116

27
The N-succinimidyl 4-trimethylstannylbenzoate 4a was chosen over N-succinimidyl
4-tributylstannylbenzoate 4b as substrate for radiobromination since recrystallisation
was possible, giving higher purity and higher labelling yields. It has also been
reported that the trimethylstannyl compound is more reactive than the tributylstannyl
compound in halogenation reactions.112
The [76Br]bromide-solution was purified in order to remove lipophilic compounds that
interfered in the labelling reaction. The labelling yields in the synthesis of 14 could be
increased from 10-35% to 65% by purification of the [76Br]bromide-solution through
a C-18 SPE disc.
N
76Br CO
O N
O
O1776BrNNHS
76Br NCS
1676BrNCS
76Br C
OCH3
NH
1576BrIm
Methyl-4-[76Br]bromobenzimidate 15 VI
Methyl-4-trimethylstannylbenzimidate 7 was brominated using chloramine-T in
methanol/ acetic acid at 40°C for 20 min to give 76BrIm 15 in 40 % yield after LC-
purification. Due to the low reactivity of 76BrIm 15 in conjugation to amines, the
labelling conditions were not further optimised. The identity of 76BrIm was confirmed
using LC-MS after a synthesis with carrier added bromide.
4-[76Br]Bromophenylisothiocyanate 16 VIIIA
The compound 76BrNCS 16 was synthesised using the same conditions as in the
synthesis of 76BrNHS 14 (Scheme 5) and could be isolated in 32% yield using
reversed phase chromatography. The compound was poorly retained on a silica
column using hexane as the eluent and the peaks from the stannyl-compound and 16
overlapped to some extent. The amount of radioactivity in the vessel decreased when
the aqueous solvent was removed from purified 16 by evaporation. It has been
reported that radioiodinated 4-methoxyphenylisothiocyanate is volatile when heating
above 45°C.56

28
N-Succinimidyl-5-[76Br]bromo-3-pyridinecarboxylate 17 VI
Radiobromination of N-succinimidyl-5-trimethylstannyl-3-pyridinecarboxylate 13
gave a 25% yield using chloramine-T as the oxidant. The reaction proceeded at room
temperature but with a lower yield compared to compound 4a. The [76Br]bromide was
consumed and a lipophilic labelled by-product was formed. This by-product was not
formed in the synthesis of 14 using the same batch of [76Br]bromide, indicating
impurities in compound 13. Compound 17 was isolated using straight phase HPLC
with either a mobile phase containing ethyl acetate: hexane: acetic acid 30:70:2 or 2%
methanol in dichloromethane. A drawback with the buffer containing acetic acid was
the difficulty in removing all acid when evaporating the solvent, giving a lower yield
in the following conjugation reaction. An increased amount of bromonicotinic acid
due to hydrolysis of 17, was observed.
Determination of specific radioactivity I, III
The bromide produced in the Cu276Se-target is expected to be of high specific
radioactivity due to the assumed carrier free production. The carrier bromide in the
labelling reaction most likely has its origin from the solvents and chemicals used.
Ethanol and chloramine-T are for example reported to contain varying amounts of
bromide.45, 122
The specific radioactivity of [76Br]bromide was determined using ion-exchange
chromatography with conductivity detection and was compared to the specific
radioactivity of the labelled 76BrNHS 14 determined by UV-detection.I It was found
that the specific radioactivity of the 76Br-ethanol solution was higher than the specific
radioactivity for 76BrNHS 14 as expected (180 vs. 86 GBq/µmol in this case). In the
determination of the specific radioactivity of 76BrNHS, the amount of pseudo-carrier
N-succinimidyl 4-chlorobenzoate in the purified product was included because of its
similar reactivity towards amines as 14. Chlorination can be a side-reaction when
using chlorine-containing oxidants.46, 123
The effective specific radioactivity of the 76Br-labeled oligonucleotides was assumed
to be of the same magnitude as the specific radioactivity of 76BrNHS. This is based on
the assumption that the chemical purity of isolated 76Br-labelled oligonucleotides is of
the same magnitude, with respect to unlabelled analogues, as the chemical purity of

29
the BrNHS since unconjugated oligonucleotides are efficiently separated from the
labelled oligonucleotides (see further chapter 6.)
LC-MS in determination of specific radioactivity III
For substances synthesised with high specific radioactivity and for compounds that
show low UV-absorption, the sensitivity obtained by UV-detection is not always
enough for determination of the specific radioactivity. Electrospray ionisation mass
spectrometry (ESI-MS) can be used as a complementary technique to UV-detection to
improve the sensitivity and selectivity in determination of specific radioactivity, for
substances that are readily ionised.33 Overlapping peaks in the UV-trace can be
resolved using the mass spectrometer as a detector since it is possible to detect one
mass at a time. When there is a limited amount of sample available, sensitivity can be
gained if columns with small inner diameters are used in combination with large
injection volumes and on-column focusing of the analyte since the mass spectrometer
behaves as a concentration sensitive detector.
To investigate whether the sensitivity obtained by LC-ESI-MS was sufficient to
measure the the mass for a 76Br-compound, 76BrNHS was conjugated to
3-aminomethyl-benzylamine to get a compound 18 that was readily ionised by ESI.
N-(3-Aminomethylbenzyl)-4-[76Br]bromobenzamide 18 was synthesised in 90%
radiochemical yield (Scheme 7). Analysis of 2 MBq of the crude product of 18 made
it possible to detect the mass for the 76Br and the 79Br-compound respectively. On-
column focusing was obtained by injecting the sample in a non-eluting solvent
containing reduced amounts of acid and organic modifier.
CH2Cl2, RT 40 min18
14
NC
O
O O
O
76BrCH2NH2
H2NCH2
76Br C NHCH2
CH2NH2O
Scheme 7.
The specific radioactivity was determined by calculating the peak area ratios of the
isotopes of compound 18 at m/z 316 and m/z 319, corresponding to the protonated
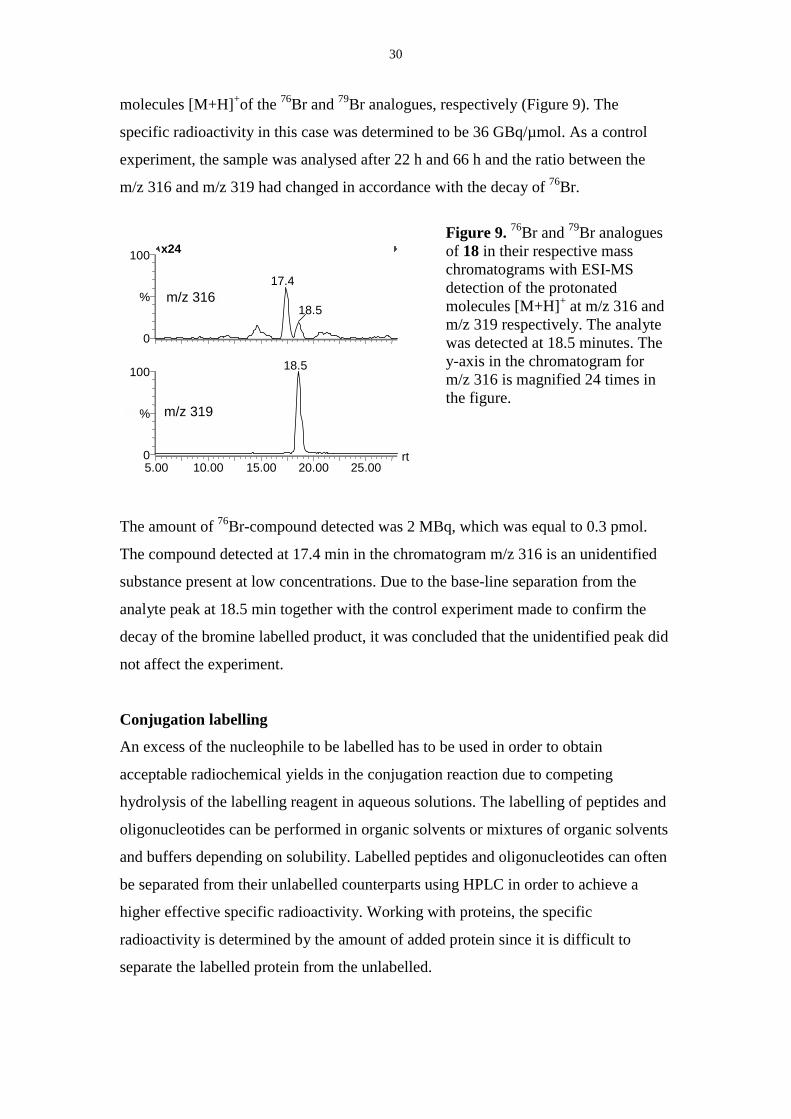
30
molecules [M+H]+of the 76Br and 79Br analogues, respectively (Figure 9). The
specific radioactivity in this case was determined to be 36 GBq/µmol. As a control
experiment, the sample was analysed after 22 h and 66 h and the ratio between the
m/z 316 and m/z 319 had changed in accordance with the decay of 76Br.
Figure 9. 76Br and 79Br analoguesof 18 in their respective masschromatograms with ESI-MSdetection of the protonatedmolecules [M+H]+ at m/z 316 andm/z 319 respectively. The analytewas detected at 18.5 minutes. They-axis in the chromatogram form/z 316 is magnified 24 times inthe figure.
The amount of 76Br-compound detected was 2 MBq, which was equal to 0.3 pmol.
The compound detected at 17.4 min in the chromatogram m/z 316 is an unidentified
substance present at low concentrations. Due to the base-line separation from the
analyte peak at 18.5 min together with the control experiment made to confirm the
decay of the bromine labelled product, it was concluded that the unidentified peak did
not affect the experiment.
Conjugation labelling
An excess of the nucleophile to be labelled has to be used in order to obtain
acceptable radiochemical yields in the conjugation reaction due to competing
hydrolysis of the labelling reagent in aqueous solutions. The labelling of peptides and
oligonucleotides can be performed in organic solvents or mixtures of organic solvents
and buffers depending on solubility. Labelled peptides and oligonucleotides can often
be separated from their unlabelled counterparts using HPLC in order to achieve a
higher effective specific radioactivity. Working with proteins, the specific
radioactivity is determined by the amount of added protein since it is difficult to
separate the labelled protein from the unlabelled.
5.00 10.00 15.00 20.00 25.00rt0
100
%
0
100
%
18.5
x24
17.4
18.5
m/z 319
m/z 316
31
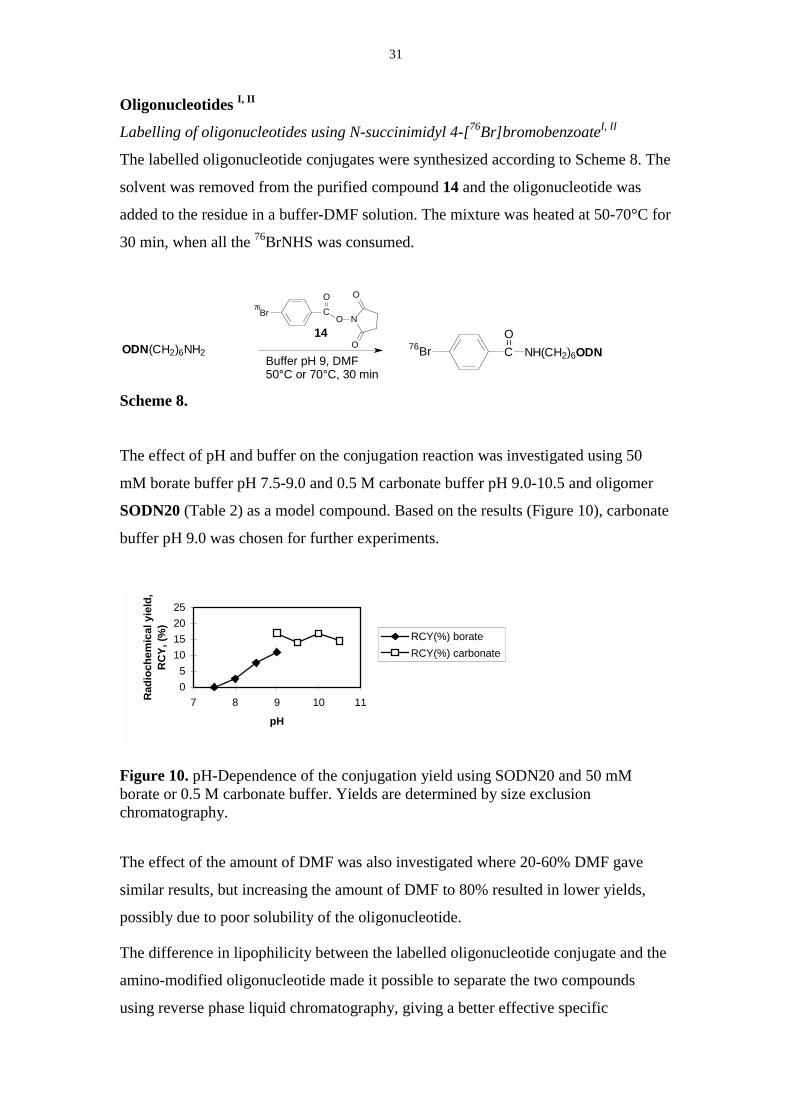
Oligonucleotides I, II
Labelling of oligonucleotides using N-succinimidyl 4-[76Br]bromobenzoateI, II
The labelled oligonucleotide conjugates were synthesized according to Scheme 8. The
solvent was removed from the purified compound 14 and the oligonucleotide was
added to the residue in a buffer-DMF solution. The mixture was heated at 50-70°C for
30 min, when all the 76BrNHS was consumed.
76Br CO
O N
O
OODN(CH2)6NH2Buffer pH 9, DMF50°C or 70°C, 30 min
76Br C NH(CH2)6ODNO14
Scheme 8.
The effect of pH and buffer on the conjugation reaction was investigated using 50
mM borate buffer pH 7.5-9.0 and 0.5 M carbonate buffer pH 9.0-10.5 and oligomer
SODN20 (Table 2) as a model compound. Based on the results (Figure 10), carbonate
buffer pH 9.0 was chosen for further experiments.
05
10152025
7 8 9 10 11
pH
Rad
ioch
emic
al y
ield
, R
CY,
(%)
RCY(%) borateRCY(%) carbonate
Figure 10. pH-Dependence of the conjugation yield using SODN20 and 50 mMborate or 0.5 M carbonate buffer. Yields are determined by size exclusionchromatography.
The effect of the amount of DMF was also investigated where 20-60% DMF gave
similar results, but increasing the amount of DMF to 80% resulted in lower yields,
possibly due to poor solubility of the oligonucleotide.
The difference in lipophilicity between the labelled oligonucleotide conjugate and the
amino-modified oligonucleotide made it possible to separate the two compounds
using reverse phase liquid chromatography, giving a better effective specific

32
radioactivity. Characterisation of the labelled oligonucleotides is further discussed in
chapter 6. The use of a large excess of oligonucleotide was necessary in order to get
an acceptable yield in the conjugation reaction. Higher concentration of
oligonucleotide gave higher conjugation yields using the 18F-analogue of 14.72
Table 2. Sequences of oligodeoxynucleotides used.Length Sequence, X = hexylamino-linker Abbreviation
Phosphorothioate oligonucleotides
6-mer 5´-XCACCTT-3´ SODN6
12-mer 5´-XCACCTTAGTGTC-3´ SODN12
20-mer 5´-XCACCTTAGTGTCCCCTTTTG-3´ SODN20
30-mer 5´- XCACCTTAGTGTCCCCTTTTGTCATAGGGCT-3´ SODN30
Phosphodiester oligonucleotides
20-mer 5´-XCACCTTAGTGTCCCCTTTTG-3´ ODN20
30-mer 5´- XCACCTTAGTGTCCCCTTTTGTCATAGGGCT-3´ ODN30
30-mer 5´-AGCCCTATGACAAAAGGGGACACTAAGGTG-3´ ODNsense
The conjugation yields were found to vary with the length of the oligomers (Table 3).
In the case of SODN30 conjugation yields increased somewhat (from 6-8% to 12-
15% radiochemical yield) when the oligomer was denatured at 90°C for 10 min prior
to conjugation or if the reaction mixture was heated at 70°C in the conjugation
reaction (Table 3).II This increase in conjugation yield might be due to the possibility
for the 30 mer to form a cyclic dimer at lower temperatures and thereby limiting the
access to the amino-linker.
In Table 3, there are large discrepancies between the isolated yields and the analytical
yields in for example entry 1 and 2. The concentration of DMF was higher in the
samples used for preparative LC compared to the analytical samples. When the
concentration of organic modifier in the injecting solvent is higher than in the mobile
phase, retention can be poor for parts of the injecting plug. The use of larger injection
volumes eliminated this problem as more buffer could be added to the sample.

33
Table 3. Radiochemical yields of labelled ODNs and SODNs using 14.a
Entry Sequence Radiochemical
yield (%)b
Radiochemical
purity (%)
n
1 76Br-SODN6 37-40 (18-19) >99 2
2 76Br-SODN12 33-40 (5-18) >99 3
3 76Br-SODN20 23-27 (15-19) >99 4
5 76Br-SODN30 (10-12) >99 10
6 76Br-ODN20 18(12) >99 1
7 76Br-ODN30 (6-12) >99 2aReaction conditions: 100 µl 30:35:35 DMF:0.5 M carbonate buffer pH 9.0:H2O,50°C (70° for ODN30 and SODN 30), 30 min. bParentheses refer to isolated radiochemical yieldswhereas other yields are based on analyses of the crude reaction mixture by analytical LC.
Labelling of oligonucleotides using 4-[76Br]bromophenylisothiocyanate VIIIA
An 18 mer oligonucleotide (5´-X ACG GAG GCA TCG CTT AGC (X= hexylamino-
linker)) (10 OD, 70 nmol) was dissolved in DMF and borate buffer pH 8 and added to
the dried compound 16. The mixture was heated at 40°C for 30 min and isolated in 14
% radiochemical yield using gel filtration liquid chromatography. Most of the
remaining radioactivity was stuck to walls of the reaction vial, probably due to poor
water-solubility of compound 16.
Proteins
Labelling of proteins has to be performed in aqueous solution in order not to denature
the protein and at a pH where the lysine-residues are partly deprotonated
Since a protein can contain many lysine-residues, there can be many differently
labelled species in the product. For imaging purposes it is important that the labelled
molecules behave in the same manner as the unlabelled. Therefore the function of the
labelled protein-conjugate has to be investigated as a part of the characterisation.
Conjugation to a lysine-residue in the active site of the molecule might affect the
activity of the protein or peptide.
34
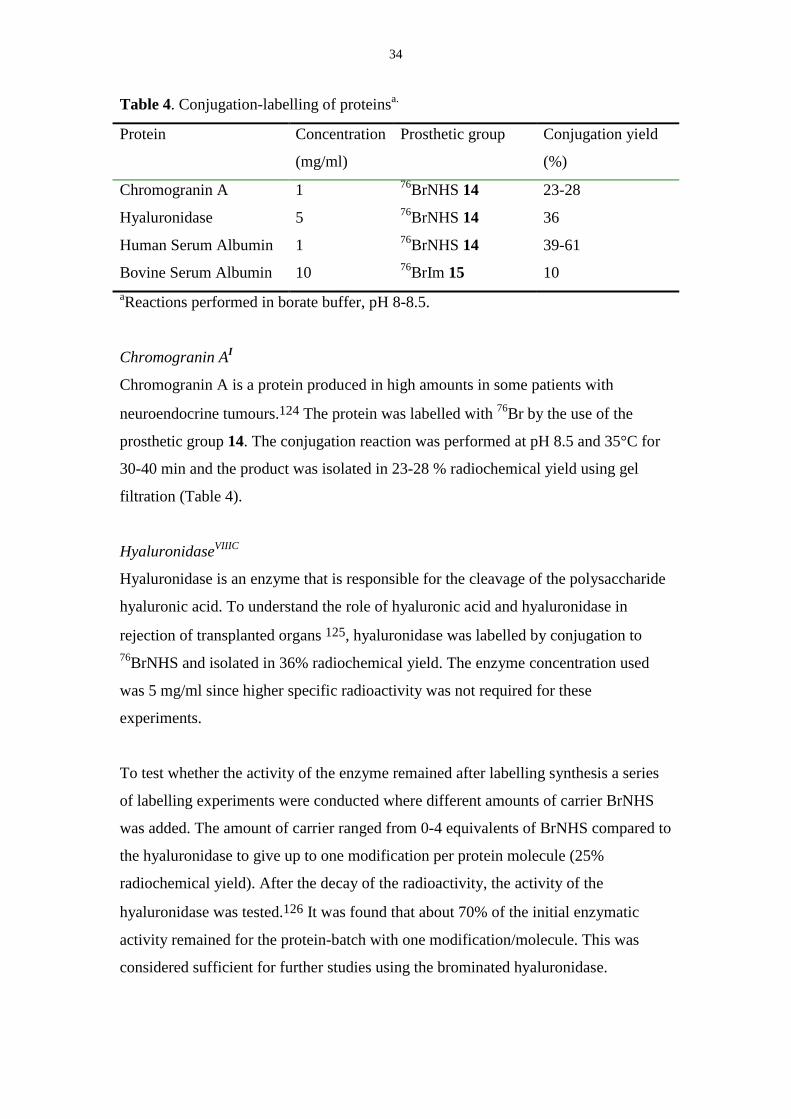
Table 4. Conjugation-labelling of proteinsa.
Protein Concentration
(mg/ml)
Prosthetic group Conjugation yield
(%)
Chromogranin A 1 76BrNHS 14 23-28
Hyaluronidase 5 76BrNHS 14 36
Human Serum Albumin 1 76BrNHS 14 39-61
Bovine Serum Albumin 10 76BrIm 15 10aReactions performed in borate buffer, pH 8-8.5.
Chromogranin AI
Chromogranin A is a protein produced in high amounts in some patients with
neuroendocrine tumours.124 The protein was labelled with 76Br by the use of the
prosthetic group 14. The conjugation reaction was performed at pH 8.5 and 35°C for
30-40 min and the product was isolated in 23-28 % radiochemical yield using gel
filtration (Table 4).
HyaluronidaseVIIIC
Hyaluronidase is an enzyme that is responsible for the cleavage of the polysaccharide
hyaluronic acid. To understand the role of hyaluronic acid and hyaluronidase in
rejection of transplanted organs 125, hyaluronidase was labelled by conjugation to76BrNHS and isolated in 36% radiochemical yield. The enzyme concentration used
was 5 mg/ml since higher specific radioactivity was not required for these
experiments.
To test whether the activity of the enzyme remained after labelling synthesis a series
of labelling experiments were conducted where different amounts of carrier BrNHS
was added. The amount of carrier ranged from 0-4 equivalents of BrNHS compared to
the hyaluronidase to give up to one modification per protein molecule (25%
radiochemical yield). After the decay of the radioactivity, the activity of the
hyaluronidase was tested.126 It was found that about 70% of the initial enzymatic
activity remained for the protein-batch with one modification/molecule. This was
considered sufficient for further studies using the brominated hyaluronidase.
35
AlbuminI, VI
76BrIm 15 was used to label bovine serum albumin (BSA). Using a high concentration
of BSA (10 mg/ml in borate buffer pH 8.7) the yield, as determined by size exclusion
chromatography, was 10%. This showed that the reactivity of 76BrIm 15 is lower
towards amines than that of 76BrNHS 14 which gave conjugation yields of 39-61%
using a tenfold lower protein concentration (Table 4).
Peptides
The resemblance of proteins and peptides makes it possible to use the same
methodology for labelling. Due to the smaller size of the peptide the effect of the
conjugate group might be larger than that for proteins. Protective groups can be
introduced in the peptide to direct the positioning of the label, avoiding conjugation to
amino acids in the active part of the peptide. Conjugation labelling of peptides can be
performed in organic solvents or mixtures of organic solvents and buffer depending
on solubility to decrease the competing hydrolysis of the prosthetic group. As a
consequence of the change in properties of the peptides by conjugate labelling, such
as lipophilicity, it is often possible to separate the labelled peptide-conjugate and the
peptide by liquid chromatography, thus giving a higher effective specific radioactivity
(Figure 11).
Figure 11. LC-chromatogram showing the analysis of the crude product of 76Br-labelled octreotide 19 using a C-18 column and a gradient of 10 mM TFA in waterand acetonitrile.
OctreotideVI
Protection of the amine-group in the ε-lysine-residue of octreotide to avoid
conjugation to this position is necessary since it is in the active site of the molecule.
Reaction conditions can be optimised to ensure selective conjugation to the N-
terminal amine in presence of lysine-residues due to the difference in pKa.49, 127
0 5 10 15 20 25 30 35
76Br-octreotide 19octreotide
UV-trace
radioactivity
retention time (min)

36
Since ε-Boc-protected octreotide was available and the deprotection could be
performed quantitatively without additional purification steps, the ε-Boc-octreotide
was used.
Labelling of octreotide using N-succinimidyl 4-[76Br]bromobenzoate 14
Conjugation of Boc-Octreotide and 76BrNHS 14 was performed in CH2Cl2, to
facilitate removal of the Boc-group by the use of TFA. In combination with
microwave heating (10 W, 30 min) a radiochemical yield of 70-90 % was achieved as
determined by HPLC-analysis (from 76BrNHS). After deprotection in TFA/CH2Cl2,
[76Br]bromobenzoyl-octreotide 19 was isolated in 55 % radiochemical yield by HPLC
calculated from 14 (Scheme 10).
Conjugation of 76BrNHS to ε-Boc-Octreotide using the same conditions as for
oligonucleotides (carbonate or borate buffer pH 9, DMF, 50°C 30 min) gave low
conjugation yields even at relatively high concentrations of Octreotide. A
concentration of 10 mg/ml (8 µmol/ml) was used, which is a tenfold increase of the
nucleophile-concentration as compared to the oligonucleotide-concentration in
corresponding reactions. Conjugation in pure organic solvents such as DMF and
DMSO also gave yields below 10 %. Even though organic bases were added to ensure
deprotonation of the free amine, the conjugation yields did not exceed 25 %.
Conventional heating as well as microwave heating was applied using DMF as
solvent.
Labelling of octreotide using N-succinimidyl-5-[76Br]bromo-3-pyridinecarboxylate 17
Since the same leaving group is present in compounds 17 and 14 (Scheme 9), similar
conditions for conjugation to octreotide were used and the octreotide-conjugate 20
was isolated in a 50 % radiochemical yield. The pyridine-moiety in compound 17
makes the conjugation-product 20 less lipophilic than 19, which is expected to
improve the binding-properties.

37
N
76Br CO
NH Octreotide
CO
76Br NH Octreotide
1) Boc-octreotide in CH2Cl2MW 10W, 30 min
2) TFA, RT, 30 min
N
76Br CO
O N
O
O
CO
O N
O
O
76Br
17
14 19
20
Scheme 9.
In preliminary experiments, the binding properties of octreotide conjugates 19 and 20
to meningioma tissue were investigated using autoradiography. High specific binding
was observed for 20, while 19 mostly showed unspecific binding. This might be
explained by the different lipophilicity of the peptide-conjugates.
Octreotide labelling using methyl-4-[76Br]bromobenzimidate 15
The compound 15 was investigated for conjugation to octreotide as an alternative to
17. The aim was to obtain a less lipophilic octreotide-conjugate than 19. The
conditions used for conjugating 76BrNHS 14 to octreotide (dichloromethane as solvent
and microwave heating) were investigated but no product was formed and most of the76BrIm 15 was left unreacted. Other solvents such as DMF, DMSO and combinations
of organic solvents and buffers were investigated without positive results. Using an
excess of stable BrIm 6 in methanol/dichloromethane and heating at 70° over night
gave conjugation to octreotide as identified by LC-ESI-MS. The use of these solvents
in the labelling reaction,however, proved unsuccessful.
Hyaluronic acid VIIIB
Labelled high-molecular weight hyaluronic acid (mean Mw=1.5×106g/mol) was
needed for distribution studies over 2-3 days using PET. For this application, the
specific radioactivity was of minor importance. Labelling of a hyaluronic acid-
tyramine conjugate was explored due to the simplicity of the method.99 The method
38
has the drawback of low specific radioactivity but the advantage that the hyaluronic
acid is subjected to minimum derivatisation.
n
OHO
HOOCO
OH
OHOCH2
HONH
O
O
OO
HOOC
OH
OHO
HOCH2
HOOH
NH
O
H
O
n
OHO
HOOCO
OH
OHOCH2
HONH
O
O
OO
HOOC
OH
OHO
HOCH2
HOOH
NH
O
OHNHCH2CH2
1)
2) NaCNBH3
OHH2NCH2CH2
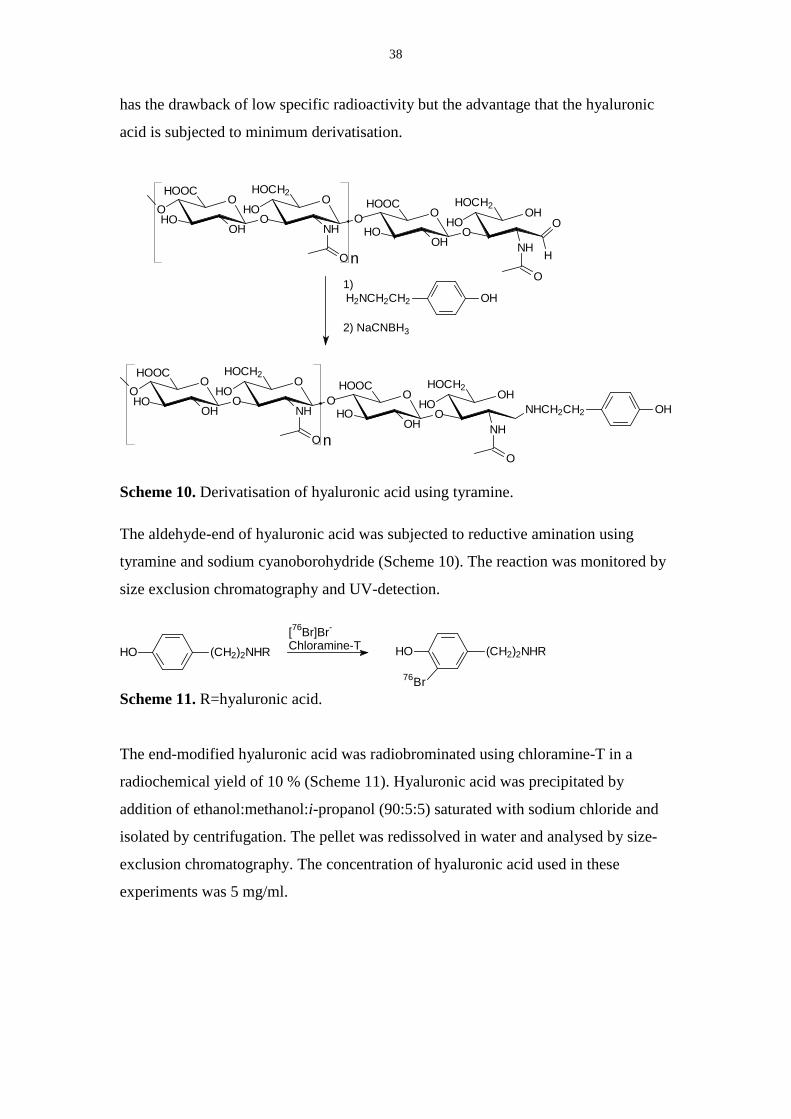
Scheme 10. Derivatisation of hyaluronic acid using tyramine.
The aldehyde-end of hyaluronic acid was subjected to reductive amination using
tyramine and sodium cyanoborohydride (Scheme 10). The reaction was monitored by
size exclusion chromatography and UV-detection.
(CH2)2NHRHO
76Br
[76Br]Br-
Chloramine-T(CH2)2NHRHO
Scheme 11. R=hyaluronic acid.
The end-modified hyaluronic acid was radiobrominated using chloramine-T in a
radiochemical yield of 10 % (Scheme 11). Hyaluronic acid was precipitated by
addition of ethanol:methanol:i-propanol (90:5:5) saturated with sodium chloride and
isolated by centrifugation. The pellet was redissolved in water and analysed by size-
exclusion chromatography. The concentration of hyaluronic acid used in these
experiments was 5 mg/ml.

39
5. Labelling using 68Ga
There are currently two radioisotopes of gallium that are commonly used in nuclear
medicine, 67Ga (T1/2=78 h, decay by EC) and 68Ga (T1/2=68 min, decay by β+ 89%).
Another isotope, 66Ga (T1/2=9.4 h) decays to 57 % by β+ and has potential for use in
PET.
The oxidation state of Ga is +III, and the complexing ligands used are dominated by
those where oxygen, nitrogen or sulphur act as donor atoms. Insoluble Ga(OH)3 is the
predominant species in the pH-interval 3.0-9.5. The labelling reactions involving Ga3+
are therefore generally performed in a buffer containing weakly coordinating ligands
such as acetate and citrate to keep the Ga3+ in solution. Ga3+ can then be transferred to
complexes of higher stability.59 For many applications, the Ga-citrate-complex is too
stable for rapid transchelation. The Ga-complexes used as radiopharmaceuticals
should be stable towards hydrolysis and should also be more stable than the Ga(III)-
transferrin complex to avoid transchelation in vivo.59, 128
Production of 68Ga68Ga is available from 68Ge (T1/2=270.8 d) in a generator-system.128 The 68Ge is
attached to an ion-exchange column and the 68Ga can be eluted in 0.1 M hydrochloric
acid.129 67Ga is commercially available and is routinely used in nuclear medicine for
imaging. A method for producing 66Ga and 68Ga by bombardment of Zn-targets has
been proposed.130
Chelators used with Ga
The chelator 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid, DOTA, has
been shown to form stable complexes with Ga as shown in the labelling of DOTA0-D-
Phe1-Tyr3-octreotide (DOTATOC).92 The chelating agent, in this case DOTA, can be
attached to the molecule of interest by modification of the carbon skeleton or by using
the carboxyl groups. 131 Other chelators used are DTPA132 and DFO
(desferrioxamine) 93 (Figure 6). The chelator has to be tribasic in order to satisfy the
40
nuclear charge of gallium and it needs to be hexadentate in order to form
coordinatively saturated complexes.133
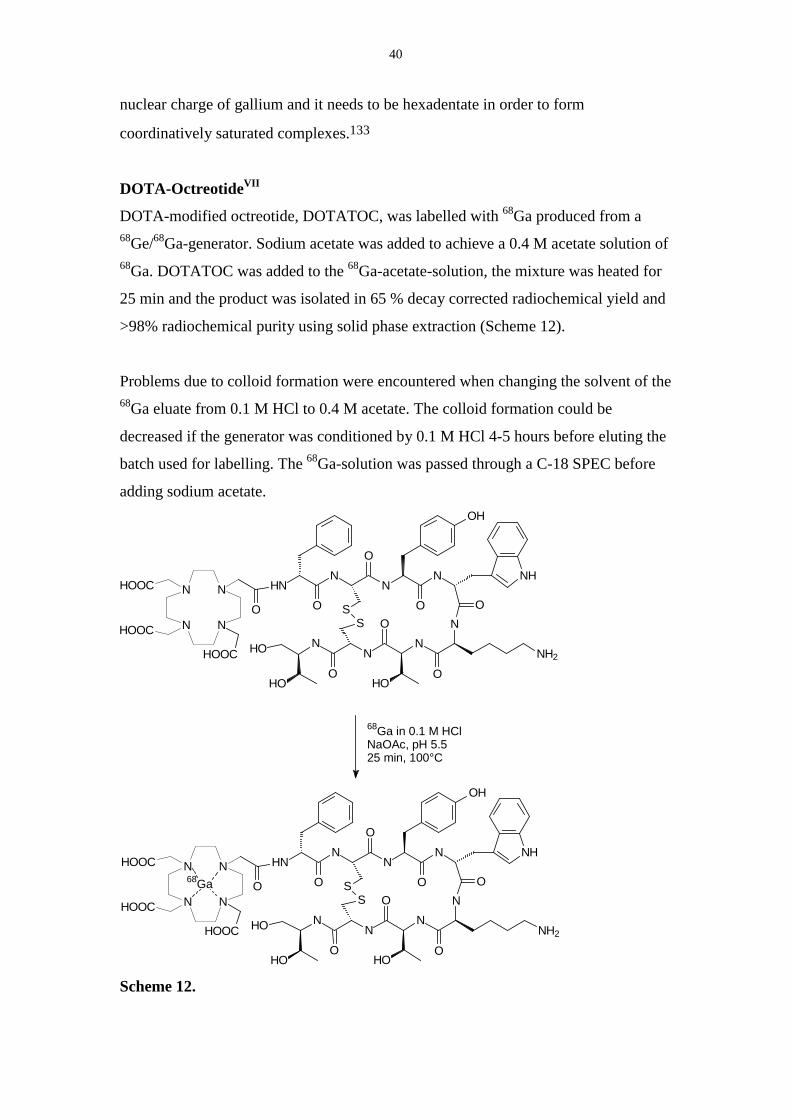
DOTA-OctreotideVII
DOTA-modified octreotide, DOTATOC, was labelled with 68Ga produced from a68Ge/68Ga-generator. Sodium acetate was added to achieve a 0.4 M acetate solution of68Ga. DOTATOC was added to the 68Ga-acetate-solution, the mixture was heated for
25 min and the product was isolated in 65 % decay corrected radiochemical yield and
>98% radiochemical purity using solid phase extraction (Scheme 12).
Problems due to colloid formation were encountered when changing the solvent of the68Ga eluate from 0.1 M HCl to 0.4 M acetate. The colloid formation could be
decreased if the generator was conditioned by 0.1 M HCl 4-5 hours before eluting the
batch used for labelling. The 68Ga-solution was passed through a C-18 SPEC before
adding sodium acetate.
NN
N NHOOC
HOOC
NN
NHO
HOO
O
O
S
HNN
NN
O
O
ON
OS
NH2
HO
NH
O
HOOC
OH
68Ga in 0.1 M HClNaOAc, pH 5.525 min, 100°C
68GaNN
N NHOOC
HOOC
NN
NHO
HOO
O
O
S
HNN
NN
O
O
ON
OS
NH2
HO
NH
O
HOOC
OH
Scheme 12.

41
The identity of the Ga-DOTATOC was confirmed by LC-MS after a synthesis with
carrier-added GaNO3. Peaks corresponding to Fe-DOTATOC and Zn-DOTATOC
could be detected from a no carrier added synthesis with 68Ga, due to contamination
from buffers and reaction vessels.
The 68Ga-DOTATOC showed high specific binding to somatostain-receptor positive
tissues such as meningiomas and brain cortex. This was demonstrated by blocking
experiments in frozen section autoradiography (Figure 13). The uptake in tissue-
samples from meningiomas and rat and pig brain could be blocked by 1 µM
octreotide in the incubation solution.
a)
b)
Figure 13. Frozen section autoradiography of a) uptake of 68Ga-DOTATOC in
selected tissues, b) uptake of 68Ga-DOTATOC with added octreotide (1 µM).
DOTA-OligonucleotidesV
The chelating agent DOTA was coupled to the phosphorothioate oligonucleotide
(SODN20 and SODN30, Table 2), using the method described by Lewis for coupling
of DOTA to proteins.62 The oligonucleotide was reacted with the N-hydroxy-
sulfosuccinimide ester of DOTA, prepared in situ using the water-soluble EDC
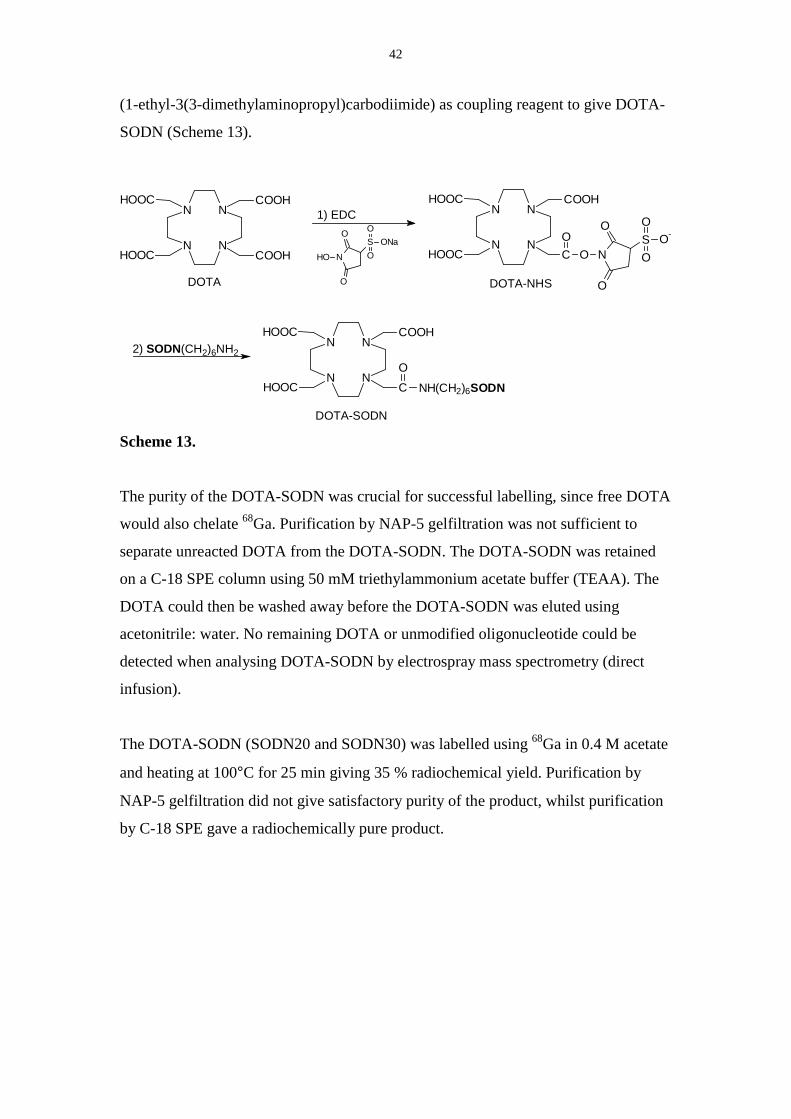
42
(1-ethyl-3(3-dimethylaminopropyl)carbodiimide) as coupling reagent to give DOTA-
SODN (Scheme 13).
NN
N N
COOH
HOOC
HOOC
COOH
NN
N N
COOH
HOOC
HOOC
C NH(CH2)6SODNO
DOTA
DOTA-SODN
NN
N N
COOH
HOOC
HOOC
C O NS O-O
O
O
O
O
2) SODN(CH2)6NH2
DOTA-NHS
1) EDC
NHO
SO
OONa
O
O
Scheme 13.
The purity of the DOTA-SODN was crucial for successful labelling, since free DOTA
would also chelate 68Ga. Purification by NAP-5 gelfiltration was not sufficient to
separate unreacted DOTA from the DOTA-SODN. The DOTA-SODN was retained
on a C-18 SPE column using 50 mM triethylammonium acetate buffer (TEAA). The
DOTA could then be washed away before the DOTA-SODN was eluted using
acetonitrile: water. No remaining DOTA or unmodified oligonucleotide could be
detected when analysing DOTA-SODN by electrospray mass spectrometry (direct
infusion).
The DOTA-SODN (SODN20 and SODN30) was labelled using 68Ga in 0.4 M acetate
and heating at 100°C for 25 min giving 35 % radiochemical yield. Purification by
NAP-5 gelfiltration did not give satisfactory purity of the product, whilst purification
by C-18 SPE gave a radiochemically pure product.

43
6. Characterisation of Labelled Oligonucleotides
Analysis using LC and ESI-MS II
In analysis of oligonucleotides with reverse phase chromatography, TEAA buffer is
used due to its ion-pairing properties, which increase the retention. In this study
polystyren divinylbenzene columns were used for separation and identification of the
oligonucleotides. Using this method, the 76Br-labelled oligomers could be separated
from the unlabelled oligomers.
Oligonucleotides can be analysed by mass spectrometry using electrospray ionisation.
Due to the polyanionic nature of the oligonucleotides, there is a distribution of
different charged species (M-nH+)n-. The polyanionic backbone can form adducts with
sodium and potassium and thereby decrease the sensitivity due to many different
charged species. The presences of a strong organic base such as triethylamine or
piperidine suppresses the formation of sodium and potassium adducts and increases
the sensitivity.134, 135 The TEAA buffer system, however, is not compatible with the
use of ESI-MS.134 Thus, to enable ESI-MS analysis, the ODN-samples had to be
desalted using gel filtration or dialysis. Buffer-systems compatible with ESI-MS have
been suggested where the LC-separation is good.134
In order to characterise the 76Br-labelled oligonucleotides, syntheses with carrier
added BrNHS were performed and the products were isolated according to the
radiotrace in the HPLC-chromatogram. The products were desalted, concentrated and
dissolved in piperidine/imidazole buffer and i-propanol prior to analysis by ESI-MS
using direct infusion. The distribution of ions with different charges could then be
used to calculate the molecular mass of the compound. An example is showed in
Figure 13. Oligonucleotides conjugated to the chelator DOTA were also identified by
the use of ESI-MS.
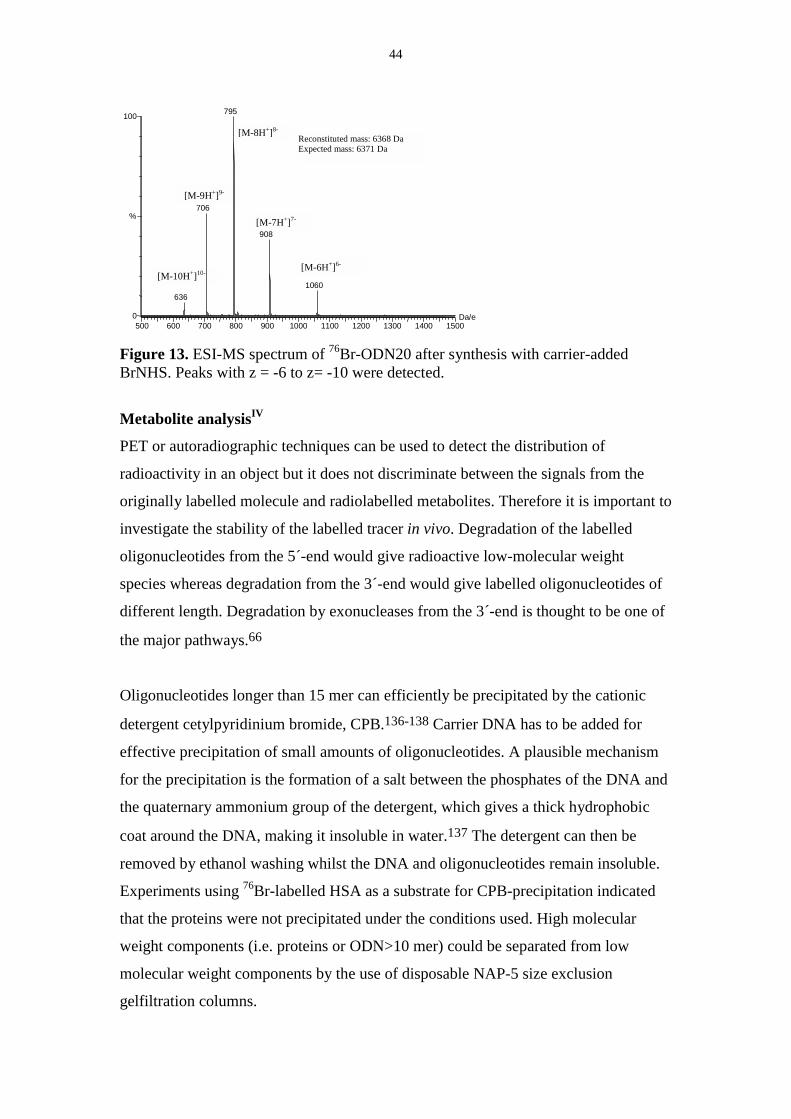
44
[M-9H+]9-
[M-10H+]10-
[M-8H+]8-
500 600 700 800 900 1000 1100 1200 1300 1400 1500Da/e 0
100
%
795
706
636
908
1060
Reconstituted mass: 6368 Da Expected mass: 6371 Da
[M-6H+]6-
[M-7H+]7-
Figure 13. ESI-MS spectrum of 76Br-ODN20 after synthesis with carrier-addedBrNHS. Peaks with z = -6 to z= -10 were detected.
Metabolite analysisIV
PET or autoradiographic techniques can be used to detect the distribution of
radioactivity in an object but it does not discriminate between the signals from the
originally labelled molecule and radiolabelled metabolites. Therefore it is important to
investigate the stability of the labelled tracer in vivo. Degradation of the labelled
oligonucleotides from the 5´-end would give radioactive low-molecular weight
species whereas degradation from the 3´-end would give labelled oligonucleotides of
different length. Degradation by exonucleases from the 3´-end is thought to be one of
the major pathways.66
Oligonucleotides longer than 15 mer can efficiently be precipitated by the cationic
detergent cetylpyridinium bromide, CPB.136-138 Carrier DNA has to be added for
effective precipitation of small amounts of oligonucleotides. A plausible mechanism
for the precipitation is the formation of a salt between the phosphates of the DNA and
the quaternary ammonium group of the detergent, which gives a thick hydrophobic
coat around the DNA, making it insoluble in water.137 The detergent can then be
removed by ethanol washing whilst the DNA and oligonucleotides remain insoluble.
Experiments using 76Br-labelled HSA as a substrate for CPB-precipitation indicated
that the proteins were not precipitated under the conditions used. High molecular
weight components (i.e. proteins or ODN>10 mer) could be separated from low
molecular weight components by the use of disposable NAP-5 size exclusion
gelfiltration columns.

45
Control experiments where the labelled oligonucleotides were homogenated with
kidney tissue were performed (referred to as “Control” in Figure 14) to test the
recovery of oligonucleotides from the CPB-precipitation and NAP gel filtration,
which was found to be about 80 %. Blank experiments without addition of kidney
gave the same results, indicating that the oligonucleotides were not metabolised in the
homogenation process.
Homogenated samples of kidney and liver from rats injected with 76Br-SODN20 and76Br-SODN30 were analysed using CPB precipitation and gel filtration. About 80 %
of the radioactivity from the homogenates was recovered in the precipitate (Figure
14). Gel filtration showed similar results as approximately 80 % of the radioactivity
was found in the high-molecular fraction (Figure 14).
The precipitates were redissolved and analysed by reversed phase HPLC. Fractions
were collected and measured for radioactivity. The samples from the liver and kidney
showed the same elution profile as the control sample. In the separation of
oligonucleotides that differed by only one mer in length, for example metabolites
degraded from the 3´-end, ion exchange chromatography has previously been reported
to be superior to reverse phase chromatography.139
Figure 14. Results fromanalysis of homogenatedtissue from rats injectedwith 76Br-SODN20 or76Br-SODN30.
CPB-precipitation
0255075
100
Kidney
20
Liver
20
Contro
l 20
Kidney
30
Liver
30
Contro
l 30%
of t
otat
raio
activ
ity
average stdev.
NAP-5 gelfiltration
0255075
100
Kidney
20
Liver
20
Contro
l 20
Kidney
30
Liver
30
Contro
l 30
% o
f tot
atl r
adio
activ
ity
average stdev
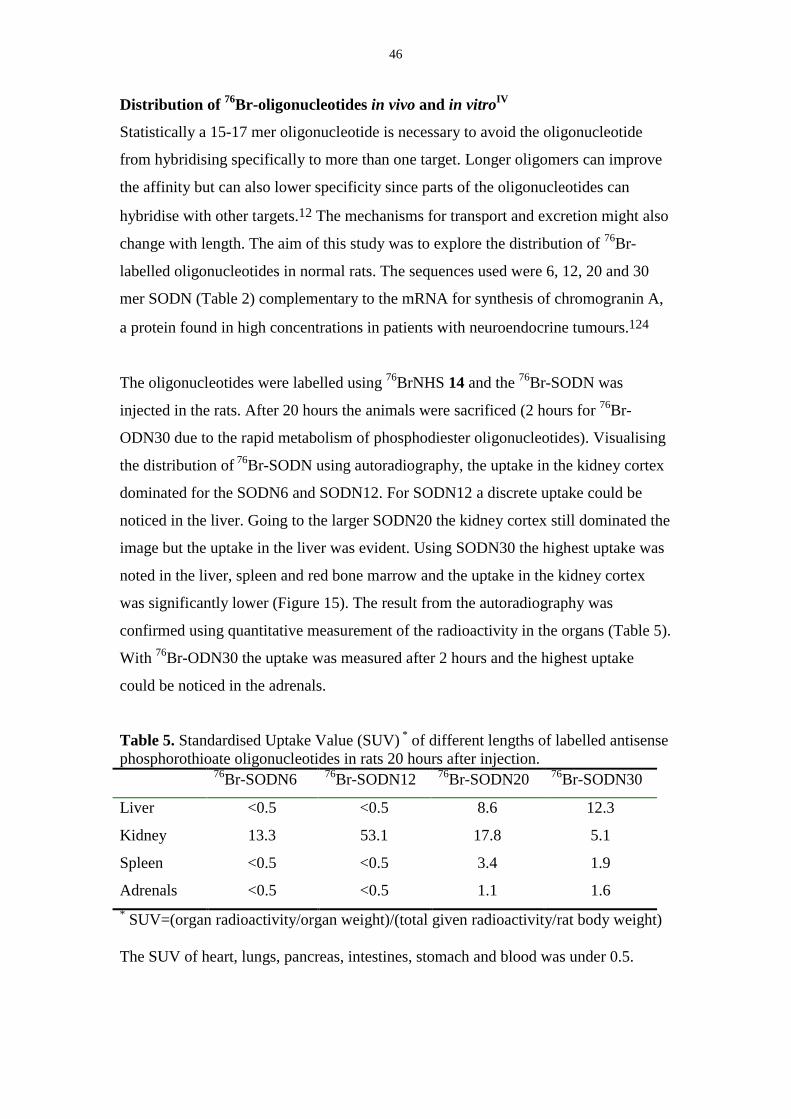
46
Distribution of 76Br-oligonucleotides in vivo and in vitroIV
Statistically a 15-17 mer oligonucleotide is necessary to avoid the oligonucleotide
from hybridising specifically to more than one target. Longer oligomers can improve
the affinity but can also lower specificity since parts of the oligonucleotides can
hybridise with other targets.12 The mechanisms for transport and excretion might also
change with length. The aim of this study was to explore the distribution of 76Br-
labelled oligonucleotides in normal rats. The sequences used were 6, 12, 20 and 30
mer SODN (Table 2) complementary to the mRNA for synthesis of chromogranin A,
a protein found in high concentrations in patients with neuroendocrine tumours.124
The oligonucleotides were labelled using 76BrNHS 14 and the 76Br-SODN was
injected in the rats. After 20 hours the animals were sacrificed (2 hours for 76Br-
ODN30 due to the rapid metabolism of phosphodiester oligonucleotides). Visualising
the distribution of 76Br-SODN using autoradiography, the uptake in the kidney cortex
dominated for the SODN6 and SODN12. For SODN12 a discrete uptake could be
noticed in the liver. Going to the larger SODN20 the kidney cortex still dominated the
image but the uptake in the liver was evident. Using SODN30 the highest uptake was
noted in the liver, spleen and red bone marrow and the uptake in the kidney cortex
was significantly lower (Figure 15). The result from the autoradiography was
confirmed using quantitative measurement of the radioactivity in the organs (Table 5).
With 76Br-ODN30 the uptake was measured after 2 hours and the highest uptake
could be noticed in the adrenals.
Table 5. Standardised Uptake Value (SUV) * of different lengths of labelled antisensephosphorothioate oligonucleotides in rats 20 hours after injection.
76Br-SODN6 76Br-SODN12 76Br-SODN20 76Br-SODN30
Liver <0.5 <0.5 8.6 12.3
Kidney 13.3 53.1 17.8 5.1
Spleen <0.5 <0.5 3.4 1.9
Adrenals <0.5 <0.5 1.1 1.6* SUV=(organ radioactivity/organ weight)/(total given radioactivity/rat body weight)
The SUV of heart, lungs, pancreas, intestines, stomach and blood was under 0.5.

47
Figure 15. Ex-vivo autoradiography of rat injected with 76Br-SODN30, 76Br-SODN20
and 76Br-SODN12.
Characterisation of the Hybridisation-Properties of Labelled OligonucleotidesIV
The impact of the labelling procedure on the oligonucleotide probes was investigated
using hybridisation to a complementary 30 mer oligonucleotide in solution. The
results were analysed using gel electrophoresis and the results were visualised by
ethidium bromide staining and autoradiography. It was shown that the labelled
phosphodiester 76Br-ODN30 was able to hybridise to its complementary ODNsense
(Table 2) whilst the phosphorothioate 76Br-SODN30 could not hybridise under the
conditions used (Figure 16).
To further confirm the ability of the probes to hybridise specifically to a longer
sequence single-stranded DNA, reverse-transcription-polymerase chain reaction (RT-
PCR) was used. RNA was isolated from tissue from adrenals, kidney, liver, heart and
lungs in rat. The RNA was transcribed using reverse transcriptase and parts of the
mRNA coding for chromogranin was amplified by PCR to obtain a single stranded
230 mer DNA-fragment with the same sequence as part of mRNA (except that uridine
is replaced by deoxythymine). Two oligonucleotide sequences were used as primers
in the PCR-reaction, one with the same sequence as the labelled oligonucleotides. The
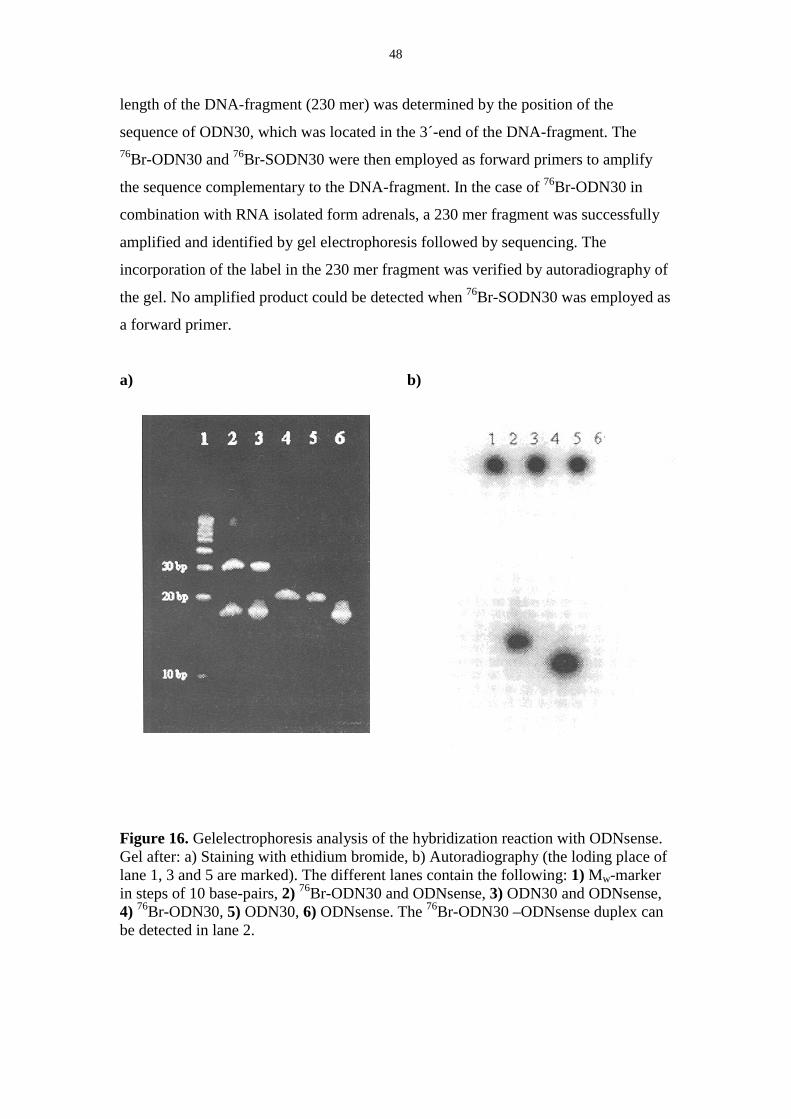
48
length of the DNA-fragment (230 mer) was determined by the position of the
sequence of ODN30, which was located in the 3´-end of the DNA-fragment. The76Br-ODN30 and 76Br-SODN30 were then employed as forward primers to amplify
the sequence complementary to the DNA-fragment. In the case of 76Br-ODN30 in
combination with RNA isolated form adrenals, a 230 mer fragment was successfully
amplified and identified by gel electrophoresis followed by sequencing. The
incorporation of the label in the 230 mer fragment was verified by autoradiography of
the gel. No amplified product could be detected when 76Br-SODN30 was employed as
a forward primer.
a) b)
Figure 16. Gelelectrophoresis analysis of the hybridization reaction with ODNsense.Gel after: a) Staining with ethidium bromide, b) Autoradiography (the loding place oflane 1, 3 and 5 are marked). The different lanes contain the following: 1) Mw-markerin steps of 10 base-pairs, 2) 76Br-ODN30 and ODNsense, 3) ODN30 and ODNsense,4) 76Br-ODN30, 5) ODN30, 6) ODNsense. The 76Br-ODN30 –ODNsense duplex canbe detected in lane 2.

49
7. Conclusions
Macromolecules such as oligonucleotides, proteins and peptides could be labelled
with 76Br by conjugation to a labelled prosthetic group containing an electrophilic
functionality. The N-Succinimidylesters, isothiocyanates and iminoesters were
investigated as electrophiles in the conjugation reaction, where the best results were
obtained with the succinimidylesters. 76Br could be introduced by halogenation-
demetallation reactions using trimethylstannyl- or tributylstannyl-compounds as
substrates. For some of the labelled macromolecule-conjugates, biological properties
such as binding to selected tissue, enzymatic activity or ability to hybridise, have been
investigated to see whether the prosthetic group affected these properties.
The advantage of using 76Br instead of 18F is the possibility to, in a simple fasion,
oxidise bromine and obtain the labelled prosthetic group in a one step reaction, while
the use of nucleophilic fluorine often requires a multi-step synthesis.
Another option for labelling of macromolecules is to use a radiometal in combination
with a chelating agent. Using this method, oligonucleotides and a peptide containing
the chelating agent DOTA could be labelled with 68Ga. The advantage with this
labelling strategy is that the reaction is performed in one step and that the only
purification necessary is solid phase extraction. Since the 68Ga is obtained from a
generator-system and the protocol to label these DOTA-macromolecules is suitable
for kit type preparations.

50
Acknowledgement:
Efter de här åren så är det några jag skulle vilja tacka extra mycket för hjälp, stöd och
uppmuntran. Tack också till er som bidragit till den kunskap och möjlighet till utveckling som
jag fått ta del av. Ett sort TACK till:
Professor Bengt Långström för förmågan att alltid hitta nya lösningar och för en aldrig
sinande entusiasm.
Dr Elisabeth Hedberg, Lisa, för introduktion till oligonukleotider och brommärkningar samt
för det samarbete som följde. Din orädda attityd till det mesta och ditt goda humör var en
tillgång på labbet. Tack för visat intresse (både för våra oligosar och skvaller) även efter du
”blev doktor”.
Dr Benita Forngren för samarbete med att bestämma specifik radioaktivitet. Tack också för all
hjälp med mass spektrometern och kromatografi.
Dr Vladimir Tolmachev för alla sena kvällar och tidiga mornar du överfört dina kunskaper om
bromproduktion samt för de brombatcher du producerat. Tack även för alla konstruktiva
förslag och diskussioner rörande gallium-inmärkningar.
Dr Feng Wu for collaboration with the oligonucleotides. Thank you for your patience when
synthesis failed, was delayed or when the amount of radioactivity was not what you expected.
Doc. Mats Bergström, Michinari Honda, Shaheena Tanweera Khan och Gabor Lendvai för
gott samarbete.
Doc. Gunnar Antoni, dr Margareta Björkman, dr Elisabeth Hedberg, dr Koichi Kato, dr Jonas
Malmström och fil. lic. Linda Samuelsson för synpunkter på utkast till avhandlingen. Dr.
Nicholas Power for liguistic corrections.

51
Personal på Kemikum, framförallt Gunnar, Leffe, Vicke, Tomas, Tatti och Eva för assistans
med allehanda saker. Personal på PET-centrum: Jocke och Kalle för hjälp med cyklotronen
och bromproduktioner samt Lotta och Pernilla för diverse inköp. Och tack till er som tryckt
”beam off” så jag fått gå hem lite tidigare.
Tobias, Benita, Lisa, Sven Åke, Koichi, Pelle, Johanna och Per för mycket träskkaffe,
diskussionerom stort och smått, lunch och lite jobb också.
Föredetta och nuvarande medlemmar av grupp BLå för trevligt arbetsklimat. Samt alla ni
andra på Kemikum, Glunten och PET-centrum som bidragit till att arbetsdagarna blev lite
roligare.
C. F. Liljevalchs, Anna Whitlocks och Acta Chemica Scandinavica stipendiefonder samt
Sigurd och Elsa Goljes Minne och Knut och Alice Wallenbergs stiftelser för generösa bidrag
till konferensresor.
Till er som sett till att det funnits ett liv utanför Kemikum:
”Flickorna från V-Dala”: Ulrika, Emma, Eva och Sandra. Tack för allt kul på nationen (och
annat också) och för andra perspektiv på tillvaron. Ett speciellt tack till Ulrika för vänskap
och stöd under hela studietiden och för att sedan bli min referens till ”verkligheten”.
”Flickorna från Uppsala”: Eva-Lotta, Åsa och Stina, för en vänskap som förhoppningsvis
alltid kommer att finnas, även om det ibland blir långt mellan träffarna.
Mamma och Pappa för det sjävförtroende och den självständighet ni gett mig, det har alltid
varit bra att ha.
Liselott, min allra bästa lillasyster.
Farmor för att du ständigt bevisar att ingenting är omöjligt.
Jonas, för ditt orubbliga lugn i alla situationer. Tack för all kärlek, stöd och uppmuntran. De
här åren hade inte varit lika roliga om jag inte haft dig att dela dem med.

52
References:1. Hevesy, G. (1923) The Absorbtion and Translocation of Lead by Plants. Biochem. J. 17, 439-
445.2. Sihver, S., Sihver, W., Bergström, M., Höglund, A. U., Sjöberg, P., Långström, B., and
Watanabe, Y. (1999) Quantitative Autoradiography with Short-Lived Positron EmissionTomography Tracers: A Study on Muscarinic Acetylcholine Receptors with N-[11C]Methyl-4-Piperidylbenzilate. J. Pharm. Exp. Ther. 290, 917-922.
3. Rudin, M., Beckmann, N., Porszasz, R., Reese, T., Bochelen, D., and Sauter, A. (1999) InVivo Magnetic Resonance Methods in Pharmaceutical Research: Current Status andPerspectives. NMR in Biomedicine 12, 69-97.
4. Ter-Pogossian, M. M., Positron Emission Tomography (PET), in Principles in NuclearMedicine, H.N.J. Wagner, Z. Szabo, and J.W. Buchanan, Editors. 1995, W. B. Saunders:Philadelphia. p. 342-346.
5. Jaszczak, R. J., and Tsui, B. M. W., Single Photon Emission Computed Tomography (SPECT),in Principles in Nuclear Medicine, H.N. Wagner, Z. Szabo, and J.W. Buchanan, Editors.1995, W. B. Saunders: Philadelphia. p. 317-328.
6. Akhtar, S., and Agrawal, S. (1997) In Vivo Studies with Antisense Oligonucleotides. TrendsPharmacol. Sci. 18, 12-17.
7. Lever, J. R., Radioiodinated Compounds, in Principles in Nuclear Medicine, H.N. Wagner, Z.Szabo, and J.W. Buchanan, Editors. 1995, W.B. Saunders: Philadelphia. p. 199-213.
8. Brunner, U. K., Renn, O., Li, M., and Meares, C. F., Radiometals and Their Chelates, inPrinciples of Nuclear Medicine, H.N. Wagner, Z. Szabo, and J.W. Buchanan, Editors. 1995,Saunders: Philadelphia. p. 220-229.
9. Service, R. F. (2000) Can Celera Do It Again? Science 287, 2136-2138.10. Watson, J. D., and Crick, F. H. C. (1953) Molecular Structure of Nucleic Acids. Nature 171,
737-738.11. Milligan, J. F., Matteucci, M. D., and Martin, J. C. (1993) Current Concepts in Antisense
Drug Design. J. Med. Chem. 36, 1923-1937.12. Hnatowich, D. J. (1999) Antisense and Nuclear Medicine. J. Nucl. Med. 40, 693-703.13. Agrawal, S. (1996) Antisense Oligonucleotides: Toward Clinical Trials. Trends Biotechnol.
14, 376-387.14. Goodchild, J. (1990) Conjugates of Oligonucleotides and Modified Oligonucleotides: A
Review of Their Synthesis and Properties. Bioconj. Chem. 1, 165-186.15. Kibler-Herzog, L., Zon, G., Uznanski, B., Whittier, G., and Wilson, W. D. (1991) Duplex
Stabilities of Phosphorothioate, Methylphosphonate and RNA Analogs of Two DNA 14-Mers.Nucl. Acid Res. 19, 2979-2986.
16. Fowler, J. S., and Wolf, A. P. (1997) Working Against Time: Rapid Radiotracer Synthesis andImaging the Human Brain. Acc. Chem. Res. 30, 181-188.
17. Kilbourn, M. R., Fluorine-18 Labeling of Radiopharmaceuticals, . 1990, National ResearchCouncil: Washington D.C.
18. Tolmachev, V., Lundqvist, H., and Einarsson, L. (1998) Production of 61Cu from a NaturalNickel Target. Appl. Radiat. Isot. 49, 79-81.
19. Lundqvist, H., Tolmachev, V., Bruskin, A., Einarsson, L., and Malmborg, P. (1995) RapidSeparation of 110In from Enriched Cd Targets by Thermal Diffusion. Appl. Radiat. Isot. 46,859-863.
20. Knust, E. J., Dutschka, K., and Weinreich, R. (2000) Preparation of 124I solutions afterThermodestillation of Irradiated 124TeO2 Targets. Appl. Radiat. Isot. 52, 181-184.
21. Nozaki, T., and Hazue, M., Accelerators, in Principles in Nuclear Medicine, H.N. Wagner, Z.Szabo, and J.W. Buchanan, Editors. 1995, W. B. Saunders: Philadelphia. p. 144-150.
22. Knapp, F. F., and Mirzadeh, S., Reactors, in Principles of Nuclear Medicine, H.N. Wagner, Z.Szabo, and J.W. Buchanan, Editors. 1995, W. B. Saunders: Philadelphia. p. 135-144.
23. Knapp, F. F. J., Brihaye, C., and Callahan, A. P., Generators, in Principles of NuclearMedicine, H.N. Wagner, Z. Szabo, and J.W. Buchanan, Editors. 1995, W. B. Saunders:Philadelphia. p. 150-165.
24. Senderoff, S. G., McElvany, K. D., Carlson, K. E., Heiman, D. F., Katzenellenbogen, J. A.,and Welch, M. J. (1982) Methodology for the Synthesis and Specific Activity Determinationof 16α-[77Br]-Bromoestradiol-17β and 16α-[77Br]-11β-methoxyestradiol-17β, Two EstrogenReceptor Binding Radiopharmaceuticals. Int. J. Appl. Radat. Isot. 33, 545-557.

53
25. Kihlberg, T., Valind, S., and Långström, B. (1994) Synthesis of [1-11C], [2-11C], [1-11C](2H3), [2-11C](2H3)Acetate for In Vivo Studies of Myocardium Using PET. Nucl. Med.Biol. 21, 1067-1072.
26. Bjurling, P., Watanabe, Y., Tokushige, M., Oda, T., and Långström, B. (1989) Syntheses of β-11C-Labelled L-Tryptophane and 5-Hydroxy-L-Tryptophane using a Multi-EnzymaticReaction Route. J. Chem. Soc. Perkin Tran. 1331-1334.
27. Torstenson, R., Tedroff, J., Hartvig, P., Fasth, K., and Långström, B. (1999) A Comparison ofC-11-Labeled L-DOPA and L-Fluorodopa as Positron Emission Tomography Tracers for thePresynaptic Dopaminergic System. J. Cereb. Blood Flow Metab. 19, 1142-1149.
28. Forngren, B. H., Tyrefors, N., Markides, K. E., and Långström, B. (2000) Determination ofRaclopride in Human Plasma by On-Column Focusing Packed Capillary LiquidChromatography-Electrospray Ionisation-Mass Spectrometry. J. Chromatogr. B. 748, 189-195.
29. Zalutsky, M. R., and Narula, A. S. (1987) A Method for the Radiohalogenation of ProteinsResulting in a Decreased Thyroid Uptake of Radioiodine. Appl. Radiat. Isot. 38, 1051-1055.
30. Arano, Y., Wakisaka, K., Ohmomo, Y., Uezono, T., Mukai, T., Motonari, H., Shiono, H.,Sakahara, H., Konishi, J., Tanaka, C., and Yokoyama, A. (1994) Maleimidoethyl 3-(Tri-n-butylstannyl)hippurate: A Useful Radioiodination Reagent for Protein RadiopharmaceuticalsTo Enhance Target Selective Radioactivity Localization. J. Med. Chem. 37, 2609-2618.
31. Wilbur, D. S. (1992) Radiohalogenation of Proteins: An Overview Of Radionuclides,Labeling Methods, and Reagents for Conjugate Labeling. Bioconj. Chem. 3, 433-470.
32. Långström, B., Kihlberg, T., Bergström, M., Antoni, G., Björkman, M., Forngren, B. H.,Forngren, T., Hartvig, P., Markides, K., Yngve, U., and Ögren, M. (1999) CompoundsLabelled with Short-Lived β+−Emitting Radionuclides and Some Applications in LifeSciences. The Importance of Time as a Parameter. Acta Chem. Scand. 53, 651-669.
33. Hyllbrant, B., Tyrefors, N., Markides, K. E., and Långström, B. (1999) On the Use of LiquidChromatography with Radio- and Ultraviolet Absorbance Detection Coupled to MassSpectrometry for Improved Sensitivity and Selectivity in Determination of SpecificRadioactivity of Radiopharmaceuticals. J. Pharm. Biomed. Anal. 20, 493-501.
34. Hunter, W. M., and Greenwood, F. C. (1962) Preparation of Iodine-131 Labelled HumanGrowth Hormone of High Specific Activity. Nature 194, 495-496.
35. Fraker, P. J., and Speck Jr, J. C. (1978) Protein and Cell Membrane Iodinations with aSparingly Soluble Chloramide, 1, 3, 4, 6-Tetrachloro-3a, 6a-Diphenylglycouril. Biochem.Biophys. Res. Com. 80, 849-857.
36. Mather, S. J. (1986) Radioiodinated Monoclonal Antibodies: a Critical Review. Appl. Radiat.Isot. 37, 727-733.
37. McElvany, K., Barnes, J. W., and Welch, M. J. (1980) Characterization of Bromine-77-Labeled Proteins Prepared Using Bromoperoxidase. J. Nucl. Med. 21, 953-960.
38. McElvany, K., Barnes, J. W., and Welch, M. J. (1980) Characterization of Bromine-77-Labeled Proteins Prepared Using Myeloperoxidase. Int. J. Appl. Radiat. Isot. 31, 679-688.
39. Sundin, J., Tolmachev, V., Koziorowski, J., Carlsson, J., Lundqvist, H., Welt, S., Larson, S.,and Sundin, A. (1999) High Yield Direct 76Br-Bromination of Monoclonal Antibodies UsingChloramine-T. Nucl. Med. Biol. 26, 923-929.
40. Cole, W. C., and Jhingran, S. G. (1993) Chloramine-T Induced Binding of MonoclonalAntibody B72.3 to Concanavalin-A. Nucl. Med. Biol. 20, 649-655.
41. Piwnica-Worms, D. (1994) Making Sense Out of Antisense: Challenges of Imaging GeneTranslation with Radiolabeled Oligonucleotides. J. Nucl. Med. 35, 1064-1066.
42. Garg, P. K., Alston, K. L., Welsh, P. C., and Zalutsky, M. R. (1996) Enhanced Binding andInertness to Dehalogenation of α-Melanotropic Peptides Labeled Using N-Succinimidyl 3-Iodobenzoate. Bioconj. Chem. 7, 233-239.
43. Vaidyanathan, G., and Zalutsky, M. R. (1995) Fluorine-18 Labeled Chemotactic Peptides: APotential Approach for the PET Imaging of Bacterial Infection. Nucl. Med. Biol. 22, 759-764.
44. Adam, M. J. (1986) The Demetallation Reaction in Radiohalogen Labelling: Synthesis ofBromine and Fluorine Labelled Compounds. Appl. Radiat. Isot. 37, 811-815.
45. Adam, M. J., Ruth, T. J., Homma, Y., and Pate, B. D. (1985) Radiobromination of AromaticCompounds by Cleavage of Aryl-Tin Bonds. Int J. Appl. Radiat. Isot. 36, 935-937.
46. Moerlein, S. M., and Coenen, H. H. (1985) Regiospecific No-Carrier-AddedRadiobromination and Radioiodination of Aryltrimethyl Group IVb Organometallics. J.Chem. Soc. Perkin. Trans. I 1941-1947.

54
47. Ali, H., and van Lier, J. E. (1996) Synthesis of Radiopharmaceuticals via OrganotinIntermediates. Synthesis 4, 423-445.
48. Kabalka, G. W., and Varma, R. S. (1989) The Synthesis of Radiolabeled Compounds viaOrganometallic Intermediates. Tetrahedron 45, 6601-6621.
49. Hermanson, G. T. (1996) Bioconjugate Techniques. 1996, San Diego: Academic Press.50. Bolton, A. E., and Hunter, W. M. (1973) The Labelling of Proteins to High Specifc
Radioactivities by Conjugation to a 125I-Containing Acylating Agent. Biochem. J. 133, 529-539.
51. Hunter, M. J., and Ludwig, M. L. (1962) The Reaction of Imidoesters with Proteins andRelated Small Molecules. J. Am. Chem. Soc. 84, 3491-3503.
52. Wood, F. T., Wu, M. M., and Gerhart, J. C. (1975) The Radioactive Labeling of Proteins withan Iodinated Amidination Reagent. Anal. Biochem. 69, 339-349.
53. Praissman, M., Praissman, L., Kent, S. B. H., and Berkowitz, J. M. (1981) Preparation andCharacterization of a Biologically Active Gastrin Derivative Modified with an 125I-LabeledImidoester. Anal. Biochem. 115, 287-296.
54. Tolan, D. R., Lambert, J. M., boileau, G., Fanning, T. G., Kenny, J. W., Vassos, A., and Traut,R. R. (1980) Radioiodination of Microgram Quantities of Ribosomal Proteins fromPolyacrylamide Gels. Analytical Biochemistry 103, 101-109.
55. Kilbourn, M. R., Dence, C. S., Welch, M. J., and Mathias, C. J. (1987) Fluorine-18 Labelingof Proteins. J. Nucl. Med. 28, 462-470.
56. Dewanjee, M. K., Ghafouripour, A. K., Werner, R. K., Serafini, A. N., and Sfakianakis, J. N.(1991) Development of Sensitive Radioiodinated Anti-Sense Oligonucleotide Probes byConjugation Technique. Bioconj. Chem. 2, 195-200.
57. Garg, S., Garg, P. K., and Zalutsky, M. R. (1991) N-Succinimidyl 5-(Trialkylstannyl)-3-Pyridinecarboxylates: A New Class of Reagents for Protein Radioiodination. Bioconj. Chem.2, 50-56.
58. Vaidyanathan, G., Zalutsky, M. R., and DeGrado, T. R. (1998) Iodopyridine-for-IodobenzeneSubstitution for Use with Low Molceular Weight Radiopharmaceuticals: Application to m-Iodobenzylguanidine. Bioconj. Chem. 9, 758-764.
59. Anderson, C. J., and Welch, M. J. (1999) Radiometal-Labeled Agents (Non-Technetium) forDiagnostic Imaging. Chem. Rev. 99, 2219-2234.
60. Heeg, M. J., and Jurisson, S. S. (1999) The Role of Inorganic Chemistry in the Developmentof Radiometal Agents for Cancer Therapy. Acc. Chem. Res. 32, 1053-1060.
61. Guhlke, S., Schaffland, A., Zamora, P. O., Sartor, J., Diekmann, D., Bender, H., Knapp, F. F.,and Biersack, H.-J. (1998) 188Re and 99mTc-MAG3 as Prosthetic groups for Labeling Aminesand Peptides: Approach with Pre- and Postconjugate Labeling. Nucl. Med. Biol. 25, 621-631.
62. Lewis, M. R., Raubitschek, A., and Shively, J. E. (1994) A Facile, Water-Soluble Method forModification of Proteins with DOTA. Use of Elevated Temperature and Optimized pH toAchieve High Specific Activity and High Chelate Stability in RadiolabeledImmunoconjugates. Bioconj. Chem. 5, 565-576.
63. Broan, C. J., Cox, J. P. L., Craig, A. S., Kataky, R., Parker, D., Harrison, A., Randall, A. M.,and Ferguson, G. (1991) Structure and Solution Stability of Indium and Gallium Complexesof 1,4,7-Triazacyclononanetriacetate and of Yttrium Complexes of 1,4,7,10-Tetraazacyclododecanetetraacetate and Related Ligands: Kinetically Stable Complexes forUse in Imaging and Radioimmunotherapy. X-Ray Molecular Structure of the Indium andGallium Complexes of 1,4,7-Triazacyclononane-1,4,7-triacetic Acid. J. Chem. Soc. PerkinTrans. 2 87-99.
64. Liu, S., and Edwards, D. S. (2001) Bifunctional Chelators for Therapeutic LanthanideRadiopharmaceuticals. Bioconj. Chem. 12, 7-34.
65. Lewis, M. R., and Shively, J. E. (1998) Maleimidocysteineamido-DOTA Derivatives: NewReagents for Radiometal Chelate Conjugation to Antibody Sulfhydryl Groups Undergo pH-Dependent Cleavage Reactions. Bioconj. Chem. 9, 72-86.
66. Crooke, S. T. (1992) Therapeutic Applications of Oligonucleotides. Annu. Rev. Pharmacol.Toxicol. 32, 329-376.
67. Hagmar, P., Tong, G., Haralambidis, J., Sawyer, W. H., and Davidson, B. E. (1995) Synthesisand Characterisation of Fluorescent Oligonucleotides. Effect of Internal Labelling on ProteinRecognition. Biochim. Biophys. Acta 1244, 259-268.
68. Eckstein, F., ed. Oligonucleotides and Analogues, A Practical Approach. The PracticalApproach Series, ed. D. Rickwood and B.D. Hames. 1991, Oxford University Press: Oxford.

55
69. Dollé, F., Hinnen, F., Vaufrey, F., Tavitian, B., and Christian, C. (1997) A General Methodfor Labeling Oligodeoxynucleotides with 18F for In Vivo PET Imaging. J. Labelled Compd.Radiopharm. 39, 319-330.
70. Kühnast, B., Dollé, F., Hinnen, F., Crouzel, C., and Tavitian, B. (2000) Fluorine-18 Labelingof Oligonucleotides Bearing Chemically-Modified Ribose-Phosphate Backbones. J. LabelledCompd. Radiopharm. 43, 837-848.
71. Kühnast, B., Dollé, F., Terrazzino, S., Rousseau, B., Loc'h, C., Vaufrey, F., Hinnen, F.,Doignon, I., Pillon, F., David, C., Crouzel, C., and Tavitian, B. (2000) General Method toLabel Antisense Oligonucleotides with Radioactive Halogens for Pharmacological andImaging Studies. Bioconj. Chem. 11, 627-636.
72. Hedberg, E., and Långström, B. (1998) 18F-Labelling of Oligonucleotides Using Succinimido4-[18F]Fluorobenzoate. Acta Chem. Scand. 52, 1034-1039.
73. Hedberg, E., and Långström, B. (1997) Synthesis of 4-([18F]Fluoromethyl)phenylIsothiocyanate and its Use in Labelling Oligonucleotides. Acta Chem. Scand. 51, 1236-1240.
74. Reed, M. W., Panyutin, I. G., Hamlin, D., Lucas, D. D., and Wilbur, D. S. (1997) Synthesis of125I-Labeled Oligonucleotides from Tributylstannylbenzamide Conjugates. Bioconj. Chem. 8,238-243.
75. Cammilleri, S., Sangrajrang, S., Persereau, B., Brixy, F., Calvo, F., Bazin, H., and Magdelnat,H. (1996) Biodistribution of Iodine-125 Tyramine Trasforming Growth Factor α AntisenseOligonucleotide in Arythmic Mice with a Human Mammary Tumour Xenograft FollowingIntratumoral Injection. Eur. J. Nucl. Med. 23, 448-452.
76. Dougan, H., Hobbs, J. B., Weitz, J. I., and Lyster, D. M. (1997) Synthesis and Radioiodinationof a Stannyl Oligodeoxyribonucleotide. Nucleic Acids Res. 25, 2897-2901.
77. Lewis, M. E., Arentzen, R., and F. Baldino, J. (1986) Rapid, High-Resolution In SituHybridization Histochemistry With Radioiodinated Synthetic Oligonucleotides. J. Neurosci.Res. 16, 117-124.
78. Hnatowich, D. J., Winnard Jr, P., Virzi, M., Fogarasi, T., Sano, T., Smith, C. L., Cantor, C. R.,and Rusckowski, M. (1995) Technetium-99m Labeling of DNA Oligonucleotides. J. Nucl.Med. 36, 2306-2314.
79. Winnard, P., Chang, F., Rusckowski, M., Mardirossian, G., and Hnatowich, D. J. (1997)Preparation and Use of NHS-MAG3 for Technetium-99m Labeling of DNA. Nucl. Med. andBiol. 24, 425-432.
80. Wagner, S., Eisenhut, M., Eritja, R., and Oberdorfer, F. (1997) Synthesis of Copper-64 andTechnetium-99m Labeled Oligonucleotides with Macrocyclic Ligands. Nucleosides andNucleotides 16, 1789-1792.
81. Dewanjee, M. K., Ghafouripour, A. K., Kapadvanjwala, M., Dewanjee, S., Serafini, A. N.,Lopez, D. M., and Sfakianakis, G. N. (1994) Noninvasive Imaging of c-myc OncogeneMessenger RNA with Indium-111-Antisense Probes in a Mammary Tumor-Bearing MouseModel. J. Nucl. Med. 35, 1054-1063.
82. Karamychev, V. N., Panyutin, I. G., Kim, M.-K., Le, N., Paik, C. H., Carrasquillo, J. A.,Reed, M. W., and Neumann, R. D. (2000) DNA Cleavage by 111In-LabeledOligdeoxynucleotides. J. Nucl. Med. 41, 1093-1101.
83. Dewanjee, M. K., Ghafouripour, A. K., Boothe, T., Mallin, W., Willem, L., Ganju-Krishan,A., Subramanian, M., Hanna, M., and Kapadvanjwala, M. Gallium-67 Labelling of AntisenseDeoxynucleotide (GAASDON) Probes and Uptake in Leukemic Cells (P388). in TenthInternational Symposium on Radiopharmaceutical Chemistry. 1993. Kyoto, Japan: JohanWiley.
84. Dewanjee, M. K., Ghafouripour, A. K., Willem, L., Ganju-Krishan, A., Kapadvanjwala, M.,Serafini, A. N., and Sfakianakis, G. N. Effect of Radionuclide on Membrane-Transport andIntracellular Hybridization of c-myc Probe in Cultured Murine Leukemic Cells. in TenthInternational Symposium of Radiopharmaceutical Chemistry. 1993. Kyoto, Japan: JohnWiley.
85. Fujibayashi, Y., Yoshimi, E., Waki, A., Sakahara, H., Saga, T., Konishi, J., Yonekura, Y., andYokoyama, A. (1999) A Novel 111In-Labeled Antisense DNA Probe with Multi-ChelatingSites (MSC-Probe) Showing High Specific Radioactivity and Labeling Efficiency. Nucl. Med.Biol. 26, 17-21.
86. Krenning, E. P., Kwekkebom, D. J., Bakker, W. H., Breeman, W. A. P., Kooji, P. P. M., Oei,H. Y., Hagen, M. v., Postema, P. T. E., Jong, M. d., Reubi, J. C., Visser, T. J., Reijs, A. E. M.,Hofland, L. J., Koper, J. W., and Lamberts, S. W. J. (1993) Somatostatin Receptor

56
Scintigraphy with [111In-DTPA-D-Phe1] and [123I-Tyr3]-Octreotide: the RotterdamExperience with more than 1000 Patients. E. J. Nucl. Med. 20, 716-731.
87. Vaidyanathan, G., Affleck, D., Welsh, P., Srinivasan, A., Schmidt, M., and Zalutsky, M.(2000) Radioiodination and Astatination of Octreotide by Conjugation Labeling. Nucl. Med.Biol. 27, 329-337.
88. Guhlke, S., Wester, H.-J., Bruns, C., and Stöcklin, G. (1994) (2-[18F]Fluoropropionyl-(D)phe1)-Octreotide, a Potential Radiopharmaceutical for Quantitative Somatostatin ReceptorImaging with PET: Synthesis, Radiolabeling, In Vitro Validation and Biodistribution in Mice.Nucl. Med. Biol. 21, 819-825.
89. Hostetler, E. D., Edwards, W. B., Anderson, C. J., and Welch, M. J. (1999) Synthesis of 4-[18F]Fluorobenzoyl Octreotide and Biodistribution in Tumor-Bearing Lewis Rats. J. LabelledCompd. Radiopharm. 42, Suppl. 1, 720-722.
90. Bakker, W. H., Krenning, E. P., Breeman, W. A., Koper, J. W., Kooij, P. P., Reubi, J.-C.,Klijn, J. G., Visser, T. J., Docter, R., and Lamberts, S. W. (1990) Receptor Scintigraphy witha Radioiodinated Somatostatin Analogue: Radiolabeling, Purification, Biological Activity andIn Vivo Application in Animals. J. Nucl. Med. 31, 1501-1509.
91. Anderson, C. J., Pajeau, T. S., Edwards, B. W., Sherman, E. L. C., Rogers, B. E., and Welch ,M. J. (1995) In Vitro and In Vivo Evaluation of Copper-64-Octreotide Conjugates. J. Nucl.Med. 36, 2315-2325.
92. Heppeler, A., Froidevaux, S., Mäcke, H. R., Jermann, E., Béhé, M., Powell, P., and Henning,M. (1999) Radiometal-Labelled Macrocyclic Chelator-Derivatised Somatostatin Analoguewith Superb Tumour-Targeting Properties and Potential for Receptor-Mediated InternalRadiotherapy. Chem. Eur. J. 5, 1974-1981.
93. Smith-Jones, P. M., Stolz, B., Burns, C., Albert, R., Reist, H. W., Fridrich, R., and Mäcke, H.M. (1994) Gallium-67/Gallium-68-[DFO]-Octreotide- A Potential Radiopharmaceutical forPET Imaging of Somatostatin Receptor-Positive Tumors: Synthesis and Radiolabeling InVitro and Preliminary In Vivo Studies. J. Nucl. Med. 35, 317-325.
94. Kwekkebom, D. J., Kooij, P. P., Bakker, W. H., Mäcke, H. R., and Krenning, E. P. (1999)Comparison of 111In-DOTA-Tyr3-Octreotide and 111In-DTPA-Octreotide in the SamePatients: Biodistribution, Kinetics, Organ and Tumor Uptake. J. Nucl. Med. 40, 762-767.
95. Decristoforo, C., and Mather, S. J. (1999) Preparation, 99mTc-Labeling, and in VitroCharacterization of HYNIC and N3S Modified RC-160 and [Tyr3]Octreotide. Bioconj. Chem.10, 431-438.
96. Rösch, F., Herzog, H., Stolz, B., Brockmann, J., Kohle, M., Muhlensiepen, H., Marbach, P.,and Muller Gartner, H. W. (1999) Uptake Kinetics of the Somatostatin Receptor Ligand[86Y]DOTA-DPhe1- Tyr3-Octreotide ([86Y]SMT487) Using Positron Emission Tomographyin Non-Human Primates and Calculation of Radiation Doses of the 90Y-Labelled Analogue.Eur. J. Nucl. Med. 26, 358-366.
97. Otte, A., Jermann, E., Behe, M., Goetze, M., Bucher, H. C., Roser, H. W., Heppeler, A.,Mueller Brand, J., and Maecke, H. R. (1997) DOTATOC: A Powerful New Tool forReceptor-Mediated Radionuclide Therapy. Eur. J. Nucl. Med. 24, 792-5.
98. de Jong, M., Bakker, W. H., Krenning, E. P., Breeman, W. A., van der Pluijm, M. E., Bernard,B. F., Visser, T. J., Jermann, E., Behe, M., Powell, P., and Macke, H. R. (1997) Yttrium-90and Indium-111 Labelling, Receptor Binding and Biodistribution of [DOTA0,D-Phe1,Tyr3]Octreotide, a Promising Somatostatin Analogue for Radionuclide Therapy. Eur. J.Nucl. Med. 24, 368-71.
99. Orlando, P., Binaglia, L., De Feo, A., Trenta, R., and Trevesi, R. (1995) An Improved Methodfor Hyaluronic Acid Radioiodination. J. Labelled Compd. Radiopharm. 36, 855-859.
100. Gustafson, S., Björkman, T., and Westlin, J.-E. (1994) Labelling of High Molecular WeightHyaluronan With 125I-Tyrosine: Studies in Vitro and in Vivo in the Rat. GlycoconjugateJournal 11, 608-613.
101. Dahl, L. B., Laurent, T. C., and Smedsröd, B. (1988) Preparation of Biologically IntactRadioiodinated Hyaluronan of High Specific Radioactivity: Coupling of 125I-Tyramine-Cellobiose to Aminogroups after Partial N-Deacetylation. Anal. Biochem. 175, 397-407.
102. Westerberg, G., Bergström, M., Gustafson, S., Lindqvist, U., Sundin, A., and Långström, B.(1995) Labelling of Polysaccharides Using [11C]Cyanogen Bromide. In Vivo and In VitroEvaluation of 11C-Hyaluronan Uptake Kinetics. Nucl. Med. Biol. 22, 251-256.
103. Kuo, J.-w., Swann, D. A., and Prestwich, G. D. (1991) Chemical Modification of HyaluronicAcid by Carbodiimides. Bioconj. Chem. 2, 232-241.

57
104. Pouyani, T., and Prestwich, G. D. (1994) Functionalized Derivatives of Hyaluronic AcidOligosaccharides: Drug Carriers and Novel Biomaterials. Bioconj. Chem. 5, 339-347.
105. Mazière, B., and Loc´h, C. (1986) Radiopharmaceuticals Labelled with Bromine Isotopes.Appl. Radiat. Isot. 37, 703-713.
106. Tolmachev, V., Lövqvist, A., Einarsson, L., Schultz, J., and Lundqvist, H. (1998) Productionof 76Br by a Low-Energy Cyclotron. Appl. Rad. Isot. 49, 1537-1540.
107. Qaim, S. M., Stöcklin, G., and Weinreich, R. (1977) Excitation Functions for the Formation ofNeutron Deficient Isotopes of Bromine and Krypton via High-Energy Deuteron InducedReactions on Bromine: Production of 77Br, 76Br and 79Kr. Appl. Radiat. Isot. 28, 947-953.
108. de Jong, D., Kooiman, H., and Veenboer, J. T. (1979) 76Br and 77Br from Decay of CyclotronProduced 76Kr and 77Kr. Int. J. Appl. Radiat. Isot. 30, 786-788.
109. Mazière, B., Loc´h, C., Hantraye, P., Guillon, R., Duquesnoy, N., Soussaline, F., Naquet, R.,Comar, D., and Mazière, M. (1984) 76Br-Bromospiroperidol: A New Tool for Quantitative In-Vivo Imaging of Neuroleptic Receptors. Life Science 35, 1349-1356.
110. Hadley, S. W., Wilbur, D. S., Gray, M.-A., and Atcher, R. W. (1991) Astatine-211 Labelingof an Antimelanoma Antibody and Its Fab Fragment Using N-Succinimidyl p-Astatobenzoate:Comparisons in Vivo with the p-[125I]Iodobenzoyl Conjugate. Bioconj. Chem. 2, 171-179.
111. Wilbur, D. S., and Hylarides, M. D. (1991) Radiolabeling of a Monoclonal Antibody with N-Succinimidyl para-[77Br]Bromobenzoate. Nucl. Med. Biol. 18, 363-365.
112. Garg, P. K., Archer Jr, G. E., Bigner, D. D., and Zalutsky, M. R. (1989) Synthesis ofRadioiodinated N-Succinimidyl Iodobenzoate: Optimization for Use in Antibody Labelling.Appl. Radiat. Isot. 40, 485-490.
113. Khawli, L. A., and Kassis, A. I. (1989) Synthesis of 125I Labeled N-Succinimidyl p-Iodobenzoate for Use in Radiolabeling Antibodies. Nucl. Med. Biol. 16, 727-733.
114. Wilbur, D. S., Hadley, S. W., Hylarides, M. D., Abrams, P. G., Beaumier, P. A., Morgan, C.A., Reno, J. M., and Fritzberg, A. R. (1989) Development of a Stable Radioiodinating Reagentto Label Monoclonal Antibodies for Radiotherapy of Cancer. J. Nucl. Med. 30, 216-226.
115. Koziorowski, J., Henssen, C., and Weinreich, R. (1998) A New Convenient Route toRadioiodinated N-Succinimidyl 3- and 4-Iodobenzoate, Two Reagents for Radioiodination ofProteins. Appl. Radiat. Isot. 49, 955-959.
116. Wilbur, S. D., Jones, D. S., and Fritzberg, A. R. (1986) Synthesis and Radioiodination ofsome Aryltin Compounds for Radiolabeling of Monoclonal Antibodies. J. Labelled Compd.Radiopharm. 23, 1304 (abstract).
117. Derkach, G. I., Lepesa, A. M., and Kirsanov, A. V. (1962) Alkyl Esters of N-Dialkoxyphophoryliminocarboxylic acids. Zh. Obshch. Khim. 32, 171-4.
118. Leschke, C., Elz, S., Garbarg, M., and Schunack, W. (1995) Synthesis and Histamine H1Receptor Agonist Activity of a Series of 2-Phenylhistamines, 2-Heteroarylhistamines, andAnalogues. J. Med. Chem. 38, 1287-1294.
119. Schaefer, F. C., and Peters, G. A. (1961) Base-Catalyzed Reaction of Nitriles with Alcohols.A Convenient Route to Imidates and Amidine Salts. J. Org. Chem. 26, 412-417.
120. Ram, S., and Buchsbaum, D. J. (1992) Development of 3-Iodophenylisothiocyanate forRadioiodination of Monoclonal Antibodies. Appl. Radiat. Isot. 43, 1387-1391.
121. Sato, M., and Stammer, C. H. (1976) Synthesis of N-Hydroxythiourea. J. Med. Chem. 19, 336-337.
122. Adam, M. J., Wilbur, D. S., Ruth, T. J., and Garcia, S. R. (1984) Sources of Carrier Brominein High Specific Activity Radiobromination Reactions as Determined by Neutron ActivationAnalysis. J. Labelled Compd. Radiopharm. 21, 1138-1139.
123. Suehiro, M., Iwamoto, M., Arai, I., and Nozaki, T. (1990) Bromination, No-Carrier-AddedRadiobromination and Simultaniously Occuring Chlorination by Chloramine T. Appl. Radiat.Isot. 41, 439-447.
124. Stridsberg, M., Hellman, U., Wilander, E., Lundqvist, G., Hellsing, K., and Öberg, K. (1993)Fragments of Chromogranin A are Present in the Urine of Patients with Carcinoid Tumours:Development of a Specific Radioimmunoassay for Chromogranin A and its Fragments. J.Endocrinology 139, 329-337.
125. Johnsson, C., Hällgren, R., Lindblom, L.-O., and Tufveson, G. (1999) Edema TreatmentDuring Cardiac Allograft Rejection. J. Heart and Lung Transplant 18, 1238-1242.
126. Richman, P. G., and Baer, H. (1980) A Convenient Plate Assay for the Quantitation ofHyaluronidase in Hymenoptera Venoms. Anal. Biochem. 109, 376-381.

58
127. Wester, H. J., Guhlke, S., and Stöklin, G. (1994) Regioselective [18F]Fluoropropionylation ofPeptides and Proteins in Aqueous Solution. J. Labelled Compd. Radiopharm. 35, 297-299.
128. Green, M. A., and Welch, M. J. (1989) Gallium Radiopharmaceutical Chemistry. Nucl. Med.Biol. 16, 435-448.
129. Schuhmacher, J., and Maier-Borst, W. (1981) A New 68Ge/68Ga Radioisotope GeneratorSystem for Production of 68Ga in Dilute HCl. Int. J. Appl. Radiat. Isot. 32, 31-36.
130. Tolmachev, V., and Lundqvist, H. (1996) Rapid Separation of Gallium from Zink Targets bythermal Diffusion. Appl. Radiat. Isot. 47, 297-299.
131. Li, M., and Meares, C. F. (1993) Synthesis, Metal Chelate Stability Studies, and EnzymeDigestion of a Peptide-Linked DOTA Derivative and Its Corresponding RadiolabeledImmunoconjugates. Bioconj. Chem. 4, 275-283.
132. Otsuka, F. L., Welch, M. J., Kilbourne, M. R., Dence, C. S., Dilley, W. G., and Jr, S. L. W.(1991) Antibody Fragments Labeled with Fluorine-18 and Gallium-68: In Vivo Comparisonwith Indium-111 and Iodine-125-labeled Fragments. Nucl. Med. Biol. 18, 813-816.
133. Parker, D. (1990) Tumour Targeting with Radiolabelled Macrocycle-Antibody Conjugates.Chem. Soc. Rev. 19, 271-291.
134. Apffel, A., Chakel, J. A., Fischer, S., Lichtenwalter, K., and Hancock, W. S. (1997) Analysisof Oligonucleotides by HPLC-Electrospray Ionization Mass Spectrometry. Anal. Chem. 69,1320-1325.
135. Greig, M., and Griffey, R. H. (1995) Utility of Organic Bases for Improved Electrospray MassSpectrometry of Oligonucleotides. Rapid Commun. in Mass Spectrom. 9, 97-102.
136. Tavitian, B., Terrazino, S., Kühnast, B., Marzabal, S., Stettler, O., Dollé, F., Deverre, J.-R.,Jobert, A., Hinnen, F., Bendriem, B., Crouzel, C., and Di Giamberardion, L. (1998) In VivoImaging of Oligonucleotides with Positron Emission Tomography. Nature Medicine 4, 467-471.
137. Geck, P., and Nász, I. (1983) Concentrated, Digestable DNA after HydroxylapatiteChromatography with Cetylpyridinium Bromide Precipitation. Anal. Biochem. 135, 264-268.
138. Sambrook, J., Fritsch, E. F., and Maniatis, T. (1989)Molecular Cloning: A LaboratoryManual. 1989, Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press.
139. Metelev, V., and Agrawal, S. (1992) Ion-Exchange High-Performance LiquidChromatography Analysis of Oligodeoxyribonucleotide Phosphorothioates. Anal. Biochem.200, 342-346.


















