
Kursanleitung zum Botanischen Großpraktikum I, Teil B ... · 2-Synthese mit der Clark-Elektrode A....
58
Department Biologie I Botanik Kursanleitung zum Botanischen Großpraktikum I, Teil B Basisveranstaltung Crystal structure of the dimeric Mo-containing nitrate-reducing N-terminal fragment of eukaryotic nitrate reductase in: Fischer et al. (2005) Plant Cell 17
Transcript of Kursanleitung zum Botanischen Großpraktikum I, Teil B ... · 2-Synthese mit der Clark-Elektrode A....

Department Biologie I Botanik
Kursanleitung zum Botanischen Großpraktikum I, Teil B
Basisveranstaltung
Crystal structure of the dimeric Mo-containing nitrate-reducing N-terminal fragment of eukaryotic nitrate reductasein: Fischer et al. (2005) Plant Cell 17

Inhaltsverzeichnis
Seite
I Bestimmung der photosynthetischen O2-Synthese mit der Clark-Elektrode 3
II Funktionsänderungen der Microbodies bei der Umstellung des Stoffwechsels während der Keimung fetthaltiger Samen
15
III Anreicherung und Charakterisierung von Phosphofructokinase aus Karotten 19
IV Induktion der Nitratreduktase in Rettichkotyledonen 29
V Induktion der Synthese von -Amylase in den Aleuronzellen des Gersten-korns durch Gibberellin
32
VI Phytochrom als Signalgeber der Flavonolakkumulation in Senfkeimlingen 36
VII Verzögerung der Seneszenz abgeschnittener Haferblätter durch Kinetin 40
VIII Reversible Entfaltung eines Proteins am Beispiel des Phycocyanin 42
IX Der Pasteureffekt bei der Hefe 53
Abbildungsverzeichnis
I.1 Einige Strukturen und Reaktionen der im Versuch verwendeten Reagenzien 3
I.2 Redox-Reaktionen an den Elektroden des Clark-Oxygraphen 4
I.3 Lichtfluß-Effektkurve 4
I.4 Schematische Darstellung photosynthetischer Reaktionen an der Thylakoid-membran
6
I.5 Vorbereitung der Clark-Elektrode 8
II.1 Der Umbau von Fett in Kohlenhydrat im Fettspeichergewebe von Samen 18
III.1 Extinktionsspektren von NAD und NADH 28
V.1 Schematische Darstellung einer Getreidekaryopse während der Keimung 35
VI.1 Untergruppen der Flavonoide und ihre Ableitung vom Flavan-Grundgerüst 37
Tabellenverzeichnis
I.1 Eigenschaften einiger typischer Elektronen-Akzeptoren („Hill-Reagenzien“) 3
I.2 Sauerstoffkonzentration in luftgesättigtem Wasser 14
VIII.1 Ammoniumsulfatfällung 50
2

Versuch I: Bestimmung der photosynthetischen O2-Synthese mit der Clark-Elektrode
A. Theorie:
Die lichtabhängige Bildung von Sauerstoff (O2) durch isolierte Thylakoide wird als „Hill-
Reaktion“ bezeichnet. Sie wurde 1939 erstmals von Robert Hill beschrieben und stellte die
Grundlage für die Beweisführung dar, daß
O2 ohne gleichzeitige CO2-Fixierung gebildet werden kann,
O2 aus H2O und nicht aus CO2 gebildet wird,
die Enzyme der Photosynthese im Chloroplasten lokalisiert sind,
in der primären Lichtreaktion der Transfer eines Elektrons gegen ein chemisches
Energiegefälle erfolgt,
zwei Reaktionszentren an der Photosynthese beteiligt sind.
Die „Hill-Reaktion“ erfordert den Zusatz von Elektronen-Akzeptoren (A), sogenannten „Hill-
Reagenzien“ (Tabelle I.1). Die Gesamtreaktionsgleichung für den lichtabhängigen linearen
Elektronentransport von Wasser zu A ist:
h
H2O + A AH2 + ½ O2
Aus der Reaktionsgleichung folgt, daß die Oxidation des Wassers indirekt über die
Reduktion des Elektronen-Akzeptors zu AH2 oder direkt durch die Freisetzung von Sauerstoff
O2 gemessen werden kann. Im Pflanzenphysiologischen Grundkurs wurde die Reduktion
(AH2) des im oxidierten Zustand blauen Elektronen-Akzeptors 2,6-Dichlorophenolindophenol
(DCPIP, Abb. I.1) bei 600 nm im Photometer als Entfärbung bestimmt. Hier werden wir die
Sauerstoff-Bildung als µmol O2/h mg Chl mit der Clark-Elektrode bestimmen (Abb. I.2).
Tabelle I.1: Eigenschaften einiger typischer Elektronen-Akzeptoren („Hill-Reagenzien“).
Akzeptor Em 7 (mV) max (nm) (10-3M-1cm-1) MW Löslichkeit
DCPIP +217 590 ox. 16 290 Ethanol
Kaliumhexacyanoferrat +430 420 ox. 1.04 329 Wasser
NADP+ -320 340 red. 6.22 743 Wasser (pH 7.0)
Abb. I.1: Einige Strukturen und Reaktionen der im Versuch verwendeten Reagenzien.
3
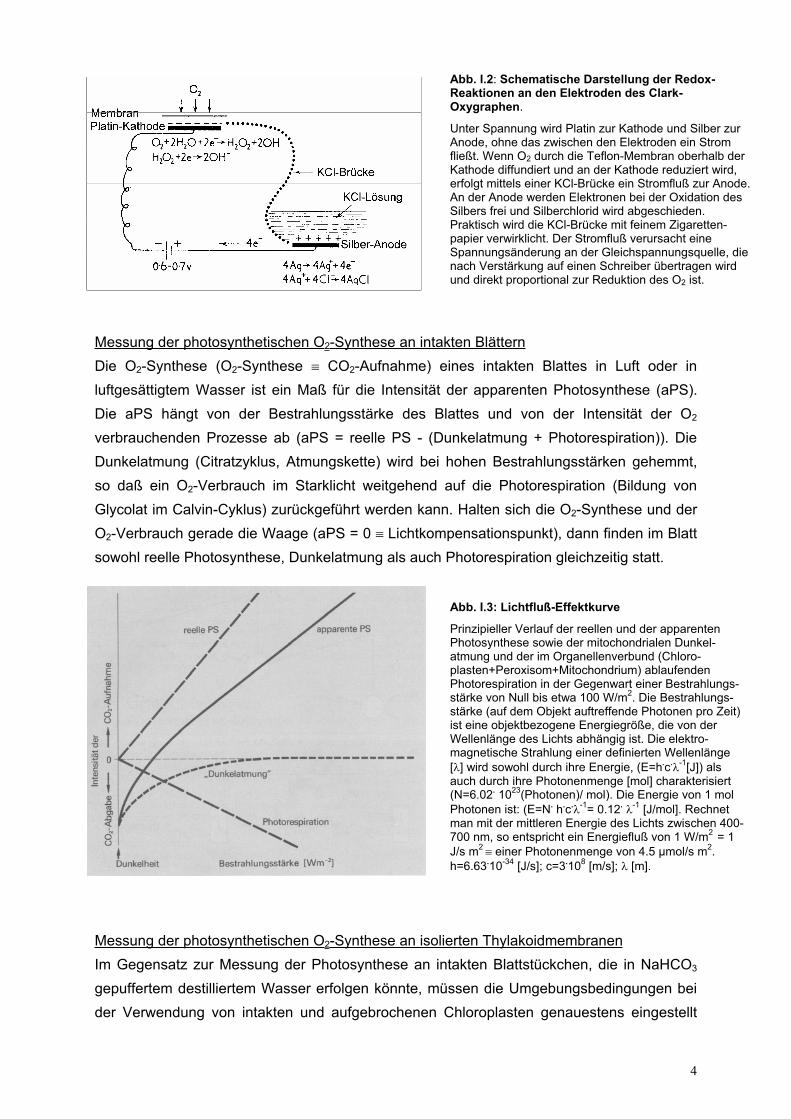
Abb. I.2: Schematische Darstellung der Redox-Reaktionen an den Elektroden des Clark-Oxygraphen.
Unter Spannung wird Platin zur Kathode und Silber zur Anode, ohne das zwischen den Elektroden ein Strom fließt. Wenn O2 durch die Teflon-Membran oberhalb der Kathode diffundiert und an der Kathode reduziert wird, erfolgt mittels einer KCl-Brücke ein Stromfluß zur Anode. An der Anode werden Elektronen bei der Oxidation des Silbers frei und Silberchlorid wird abgeschieden. Praktisch wird die KCl-Brücke mit feinem Zigaretten-papier verwirklicht. Der Stromfluß verursacht eine Spannungsänderung an der Gleichspannungsquelle, die nach Verstärkung auf einen Schreiber übertragen wird und direkt proportional zur Reduktion des O2 ist.
Messung der photosynthetischen O2-Synthese an intakten Blättern
Die O2-Synthese (O2-Synthese CO2-Aufnahme) eines intakten Blattes in Luft oder in
luftgesättigtem Wasser ist ein Maß für die Intensität der apparenten Photosynthese (aPS).
Die aPS hängt von der Bestrahlungsstärke des Blattes und von der Intensität der O2
verbrauchenden Prozesse ab (aPS = reelle PS - (Dunkelatmung + Photorespiration)). Die
Dunkelatmung (Citratzyklus, Atmungskette) wird bei hohen Bestrahlungsstärken gehemmt,
so daß ein O2-Verbrauch im Starklicht weitgehend auf die Photorespiration (Bildung von
Glycolat im Calvin-Cyklus) zurückgeführt werden kann. Halten sich die O2-Synthese und der
O2-Verbrauch gerade die Waage (aPS = 0 Lichtkompensationspunkt), dann finden im Blatt
sowohl reelle Photosynthese, Dunkelatmung als auch Photorespiration gleichzeitig statt.
Abb. I.3: Lichtfluß-Effektkurve
Prinzipieller Verlauf der reellen und der apparenten Photosynthese sowie der mitochondrialen Dunkel-atmung und der im Organellenverbund (Chloro-plasten+Peroxisom+Mitochondrium) ablaufenden Photorespiration in der Gegenwart einer Bestrahlungs-stärke von Null bis etwa 100 W/m2. Die Bestrahlungs-stärke (auf dem Objekt auftreffende Photonen pro Zeit) ist eine objektbezogene Energiegröße, die von der Wellenlänge des Lichts abhängig ist. Die elektro-magnetische Strahlung einer definierten Wellenlänge [ ] wird sowohl durch ihre Energie, (E=h.c. -1[J]) als auch durch ihre Photonenmenge [mol] charakterisiert (N=6.02. 1023(Photonen)/ mol). Die Energie von 1 mol Photonen ist: (E=N. h.c. -1= 0.12. -1 [J/mol]. Rechnet man mit der mittleren Energie des Lichts zwischen 400-700 nm, so entspricht ein Energiefluß von 1 W/m2 = 1 J/s m2 einer Photonenmenge von 4.5 µmol/s m2.h=6.63.10-34 [J/s]; c=3.108 [m/s]; [m].
Messung der photosynthetischen O2-Synthese an isolierten Thylakoidmembranen
Im Gegensatz zur Messung der Photosynthese an intakten Blattstückchen, die in NaHCO3
gepuffertem destilliertem Wasser erfolgen könnte, müssen die Umgebungsbedingungen bei
der Verwendung von intakten und aufgebrochenen Chloroplasten genauestens eingestellt
4

werden, um die physiologische Aktivität der Chloroplasten zu erhalten. Das bedeutet, daß
neben einer definierten Pufferkonzentration auch die Salzkonzentrationen und die Art des
Salzes genau eingestellt werden müssen.
A.T. Jagendorf hat den Photosyntheseprozess in der Thylakoidmembran treffend mit dem
‚Fressen von Salzsäure‘ beschrieben. Damit spielte er auf die hohen Aufnahmeraten von H+
und Cl—-Ionen aus dem Stroma ins Lumen des Thylakoids an, die während des
photosynthetischen Elektronentransports an der Thyakoidmembran des Chloroplasten
gemessen werden. Deshalb muß ein photosynthetisches Meßsystem stets für genügend
Nachschub an H+ und Cl—-Ionen auf der stromalen Seite der Thylakoidmembran sorgen.
Insgesamt führt der Ionen-Transport nur zu einer geringen elektrischen Potentialdifferenz
( ) (im Lumen positiv) aber zu einer großen Differenz in der Protonenkonzentration ( pH).
Der Rücktransport der H+ ins Stroma folgt dem Potentialunterschied zwischen Stroma (pH
8.0) und Lumen (pH 5.0), der bei sättigender Bestrahlungsintensität erreicht wird, und kann
analog dem Antrieb eines Wasserrades nebst Turbine durch einen Wasserfall, zum Antrieb
der enzymatischen ATP-Synthese aus ADP und Phosphat verwendet werden
(Chemiosmotische Theorie). Die Synthese von ATP ist somit an den Protonenfluß vom
Lumen zum Stroma gekoppelt. Diese Kopplung von Protonenfluß und ATP-Synthese wird
gemessen, wenn bei Sättigung des Enzyms mit den Substraten ADP und Phosphat die
Lichtintensität und damit der Protonenfluß verringert wird, wenn die Substrate subsaturierend
angeboten werden, oder wenn den Protonen ein Abfluß durch die Thylakoidmembran mit
geringerem Widerstand als durch den CF0-Kanal der ATPase angeboten wird. In jedem
dieser Fälle sinkt die Synthese von ATP. Der Chloroplast hält deshalb analog dem
Speichersee in einem Wasserkraftwerk den Gehalt an ADP und Phosphat hoch, um den
Stoffwechsel im Bedarfsfall rasch mit neusynthetisiertem ATP zu versorgen.
Für die zur ATP-Bildung nötige Konzentrierung von Protonen im Lumen sorgt der
lichtabhängige Elektronentransport zwischen H2O und NADP+ und das geringe Volumen des
Thylakoidlumen. Für den Transport von Elektronen muß gewährleistet sein, daß genügend
Donor-Moleküle mit einem negativen, und Akzeptor-Moleküle, mit einem positiveren
Redoxpotential vorhanden sind. Fehlt einer der Redoxpartner, so stoppt der Elektronen-
transport, folglich der H+-Transport und somit die ATP-Synthese. Der Protonentransport ist
somit an den lichtabhängigen Elektronentransport gekoppelt.
Als universelles Donor-Molekül steht der Photosynthese H2O zur Verfügung, das zu ½ O2
und 2 H+ oxidiert wird. Als Endakzeptor wird NADP+ verwendet, das nach Reduktion sehr
vielseitig im anabolen Stoffwechsel eingesetzt werden kann und somit schnellstens wieder
im reoxidierten Zustand als photosynthetischer Elektronen-Endakzeptor zur Verfügung steht.
Leider geht bei der Isolation von Thylakoiden der nicht an die Membran gebundene
Redoxfaktor Ferredoxin (Fd, Abb. I.4) verloren. Deshalb muß einer isolierten Thylakoid-
fraktion der natürliche oder ein künstlicher Elektronenakzeptor wieder zugesetzt werden, um
Elektronentransport und damit photosynthetische Oxidation des Wassers zu erreichen. Die
photosynthetische Syntheserate von O2 ist somit ein Maß für die Rate des Elektronen-
5

transports und ist an die Bildung des Protonengradienten und die Synthese von ATP
gekoppelt.
Abb. I.4: Schematische Darstellung photosynthetischer Reaktionen an der Thylakoid-membran.
Dargestellt ist die Lichtabsorption durch die Lichtsammelkomplexe des PSII und PSI, der lichtabhängige lineare (nichtzyklische) Transport von e— von H20 zu NADP+ in der Thylakoidmembran, der Ionen- und Protonen-Transport ins Thylakoidlumen, das ladungsabhängige ( ) und das protonenabhängige ( pH) Membran-potential, die Akkumulation von Protonen im Lumen und die Synthese von ATP durch die CFO/CFI-ATP Synthase. Der zyklische Elektronenfluß zwischen PSI und PSII und der pseudozyklische Elektronenfluß von Ferredoxin (Fd) auf Sauerstoff (O2) sind gestrichelt dargestellt.
Literatur
Hill, R. (1937) Oxygen production by isolated chloroplasts. Nature 139, 881-882
Delieu, T. and Walker, D.A. (1972) An improved cathode for the measurement of photosynthetic oxygen evolution by isolated chloroplasts. New Phytologist 71,201-225
Schopfer, P. und Brennicke, A. (1999) Pflanzenphysiologie, Springer
Schopfer, P. (1989) Experimentelle Pflanzenphysiologie. Einführung in die Anwendungen, Springer
Lawlor, D.W. (1990) Photosynthese: Stoffwechsel-Kontrolle-Physiologie, Thieme, Stuttgart, New York
Tevini, M., und Häder, D.-P. (1985) Allgemeine Photobiologie, Thieme, Stuttgart, New York
Eine dynamische Vorstellung zum Elektronen-Transfer Prozess während der Photosynthese liefert ein Quick-Time Video, das im Internet unter http://ampere.scale.uiuc.edu/ unter Photosynthesis & Time angesehen werden kann.
6

B. Aufgabe:
Anhand von intakten Blättern sollen die Eckwerte der Lichtfluß/Effekt-Kurve bestimmt werden
(Abb. I.3). Dazu werden die Dunkelatmung von Blattmaterial und die aPS unter hoher
Bestrahlungsstärke bestimmt, abschließend wird der Lichtkompensationspunkt ermittelt.
Die photosynthetische O2-Synthese wird mit isolierten Thylakoidmembranen bestimmt. Um
die Kopplung von Elektronentransport und Wasserspaltung kennenzulernen, wird anhand
der photosynthetischen Oxidation des Wassers die maximale Elektronentransportrate in der
Gegenwart von Entkopplern der Photophosphorylierung gemessen.
C. Reagenzien:
KCl-Lösung (gesättigt)
Na2S2O4 (Natriumdithionit, Pulver)
Folgende Puffer und Medien müssen rechtzeitig hergestellt und ggf. gekühlt werden:
500 mM Tricin-Puffer (N-Tris(hydroxymethyl)methylglycin) (50 ml, 4°C) Tricin-Lösung mit 2 N KOH auf pH 8.0 einstellen
100 mM K-Phosphat-Puffer (KPi) (10 ml, 4°C) 100 mM K2HPO4-Lösung und 100 mM KH2PO4-Lösung herstellen, K2HPO4-
Lösung vorlegen und mit KH2PO4-Lösung auf pH 8,0 einstellen
100 mM K3[Fe(CN)6]-Lösung (Kaliumhexacyanoferrat(III), 5 ml, 4°C)
50 mM ADP in 100 mM KPi-Puffer, pH 8.0 (1 ml, bei –20°C lagern)
100 mM CH3NH3Cl-Lösung (Methylammoniumchlorid, 10 ml, 4°C, frisch herstellen)
1% Rinderserumalbumin-Lösung (w/v) (5 ml, bei –20°C lagern)
80% Aceton (v/v) (100 ml)
1 M NaHCO3-Lösung (10 ml)
Isolationsmedium (150 ml, 4°C)
50 mM Tricin-Puffer, pH 8.0 300 mM Saccharose 1 mM MgCl2
Lysemedium (10 ml, 4°C)
50 mM Tricin-Puffer, pH 8.0 5 mM MgCl2
Inkubationsmedium (10 ml, 4°C)
50 mM Tricin-Puffer, pH 8.0 600 mM Saccharose 10 mM MgCl2100 mM KCl
7

D. Versuchsdurchführung:
1. Die Clark-Elektrode
Für die Bestimmung der O2-Konzentration müssen die Oberflächen der Elektroden fettfrei
sein und die Silber-Anode sollte keine schwarze Oxidhaut oder Silberchlorid-Ablagerungen
aufweisen, sondern silbrig glänzen. Anode eventuell unter Anleitung reinigen. Eine saubere,
luftblasenfrei montierte Elektrode (Abb. I.5) kann wochenlang funktionsfähig sein. Einfachster
Funktionstest ist das An- und Abschalten des Magnetrührers im luftgesättigten Wasser der
Reaktionskammer. Stoppt die Rotation des Rührfischchens in der Meßkammer, so führt die
Sauerstoffzehrung der Kathode sofort zur Abnahme der O2-Konzentration an der Membran
und somit zum Abfall der Meßwertanzeige. Die Reaktionszeit für 90% Anzeigeänderung
sollte kleiner 30 s bleiben.
Abb. I.5: Vorbereitung der Elektrode
Die anodische Elektrolyt-Mulde im Elektrodenblock (siehe a und b) mit gesättigter KCl-Lösung füllen, und die aus dem Elektrodenblock ragende Platin-Kathode mit gesättigter KCl-Lösung bedecken. Ein rundes Stückchen Zigarettenpapier ausschneiden ( 2 cm) und so über die Kathode legen, daß es die KCl-Lösung ansaugt. Über das feuchte Papier ein rundes Stück Teflonfolie faltenfrei auflegen ( Membran 2 cm). Folie und Papier mit O-Ring (a) fixieren, indem der O-Ring in die Mulde gedrückt wird, ohne daß sich Luft-blasen unter Folie und Papier befinden. Äußere Ring-dichtung auf den Elektrodenblock legen, Temperier-mantel (b) aufsetzen und von unten anschrauben, bis äußere Ringdichtung von unten dunkel sichtbar wird. Dann Meßapparatur von oben vorsichtig auf den Magnetrührer setzen. Überschüssiges KCl seitlich ab-saugen. In die Meßkammer Wasser einfüllen und auf Dichtigkeit prüfen. Wenn keine Luftblasen unter der Folie sind und die Kammer dicht ist, Gleich-Spannungs-quelle anschließen und Elektrode kalibrieren.
a
Kalibrierung
Die Kalibrierung hat zum Ziel, den Schreiberausschlag des Meßgeräts ( mV) durch
Bezugnahme auf bekannte Sauerstoff-Konzentrationen ( mol O2/ml) zu eichen, um den
unbekannten Sauerstoff-Konzentrationen der Meßlösungen einen Wert ( mol O2) zuordnen
zu können. Im Praktikum ist eine relative Kalibrierung ausreichend. Hierzu wird Wasser
verwendet, dem der Sauerstoff chemisch entzogen wird (Nullpunkt) und Luft-gesättigtes
Wasser, dessen Sauerstoffgehalt von der Temperatur abhängig ist (siehe auch Tabelle I.2 im
Anhang).
Vor dem Start der relativen Kalibrierung alle Geräte elektrisch anschalten, Reaktionskammer
mit H2Odest spülen und 1 ml H2Odest in die Meßkammer einfüllen.
Überprüfung der Voreinstellungen an den Geräten und Durchführung der Kalibrierung:
a. Sind alle Geräte elektrisch angeschlossen und eingeschaltet?
8

b. Ist der Thermostat mit Wasser gefüllt?
c. Wird temperiertes Wasser durch den Kühlmantel der Reaktionskammer gepumpt?
d. Ist 1 ml H2Odest in die Reaktionskammer eingefüllt? Wenn mehr als 1 ml, dann
Reaktionskammer absaugen. Mit Wasserstrahlvakuum und Absaugschlauch (!
weiches dünnes Gummiende) Reaktionskammer aussaugen. Danach 1 ml O2-ge-
sättigtes Wasser einfüllen. Dazu Wasser in einem Kolben oder Becherglas einige Zeit
auf dem Magnetrührer stark rühren.
e. Laptop-Computer zur Steuerung des Oxygraphen hochfahren, Einzelplatzbetrieb
wählen, weitere Anfragen mit „return“-Taste bestätigen, unter C:\BATCH>win
eingeben und mit „return“ bestätigen. Im Oxygraph-Fenster doppelklicken auf
„Oxygraph V1.10“.
f. Im Programm den Menüpunkt „Stirrers“ wählen. Am Oxygraphen überprüfen, ob sich
der Magnetrührer dreht. Wenn nein, Magnetrührerfunktion am Oxygraph-Kontroll-
kasten (weiße Box, auf der der Oxygraph steht) den Tastenschalter ganz rechts
drücken (Grüne Kontroll LED leuchtet).
g. Einen einfachen Funktionstest durchführen: Meßprogramm starten, einige Zeit auf-
zeichnen bis die Basislinie stabil ist, dann den Rührer ausschalten. Die Schreiberlinie
muß abfallen, warum?
h. „Calibrate“ anklicken, „New Calibration“ wählen und den Programmanweisungen
folgend die Kalibrierung durchführen. Zur O2-Sättigung gerührtes Wasser benutzen
(siehe d.), O2-freies Wasser chemisch herstellen (Zugabe einer bepuderten Spatel-
spitze Dithionitpulver) oder durch Spülen mit N2 aus Druckluftflasche (Assistent holen
und Verwendung der Druckflasche erläutern lassen).
i. Meßkammer gründlich (5x) mit H2Odest aus der Plastikspülflasche spülen, also 5 mal
mit H2Odest befüllen und wieder aussaugen. Darauf achten, daß Rührfischchen nicht
abgesaugt wird. Sollte es herausgesaugt werden, aus dem Schlauchende nehmen
und nach dem Wiederbefüllen der Reaktionskammer mit Wasser zurückgeben.
j. Messprogramm vorbereiten: Reaktionskammer mit 1 ml O2-gesättigtem H2Odest be-
füllen, Verschlußkolben in die Meßkammer einführen und Schreiberanstieg
wenigstens 2 min bis zum Erreichen eines stabilen Sättigungswerts aufzeichnen
lassen.
2. Messung der photosynthetischen O2-Synthese an intakten Blättern
Allgemeine Versuchsvorbereitungen vor jedem Versuch:
a. Ein Blattstück mit einer Fläche von etwa 2.5 cm2 aus einem im Dunkeln gehaltenen
Blattmaterial ausschneiden.
b. Blattstück auf der Feinwaage wiegen und Frischgewicht notieren.
9

c. Blattstück mit einer Rasierklinge auf dem Boden oder Deckel einer Plastik-Petrischale
in etwa 1 mm2 große Stückchen zerschneiden.
d. Überprüfen ob das Reaktionsgefäß der Elektrode mit 1 ml H2Odest beschickt ist.
e. Magnetrührer anschalten und überprüfen, ob sich Rührfisch dreht.
f. Das gesamte zerhäckselte Blattmaterial in das Reaktionsgefäß geben.
g. Überprüfen, ob der Rührfisch das gesamte Blattmaterial im Reaktionsgefäß bewegt.
h. Messprogramm mit „Start“-Button starten. „Overwrite current data?“ Mit „OK“
bestätigen. Daten werden in Channel 1 geschrieben.
i. Alle Manipulationen protokollieren.
j. Verschlußkolben einsetzen und das Gefäß mit schwarzem Tuch abdunkeln.
Dunkelatmung und apparente Photosynthese:
a. Dunkelatmung mindestens 2 min lang aufzeichnen.
b. Diaprojektor in 15 cm Entfernung vom Reaktionsgefäß aufstellen (Distanz Frontlinse-
Reaktionskammer) und Diaprojektorlicht anschalten.
c. Das Tuch entfernen und weitere 2 min die Lichtatmung messen.
d. Anschließend den Verschlußkolben herausziehen, 100 µl der NaHCO3-Lösung
zusetzen und den Verschlußkolben schnell wieder einsetzen.
e. Mindestens 8 min aufzeichnen, dann das Messprogramm stoppen, Daten speichern.
f. Reaktionskammer aussaugen und mit H2Odest 3x spülen.
g. 1 ml H2Odest einfüllen und Verschlußkolben einsetzen.
h. Auswertung (siehe E.): den Zeitpunkt der maximalen Steigung der Schreiberlinie
bestimmen und errechnen, welche Meßzeit nach Zugabe der NaHCO3-Lösung nötig
ist, um die maximale Steigung = maximale O2-Entwicklung zu erreichen.
Lichtkompensationspunkt:
a. Auf der Arbeitsbank ein mindestens 135 cm langes Klebeband anbringen, das an der
Reaktionskammer beginnt und mit 15 cm Einteilung versehen wird.
b. Wenn ein Lichtquantenfluxmeter vorhanden ist, kann die Abhängigkeit der Be-
strahlungsstärke vom Abstand der Lichtquelle zur Reaktionskammer (Distanz
Frontlinse-Reaktionskammer) bestimmt werden. Hierzu die Bestrahlungsstärke in 15
cm-Schritten (135 bis 15 cm) direkt vor und hinter der Meßapparatur ablesen, den
Mittelwert aus beiden Messungen bilden und auf einem Millimeterpapier darstellen.
Nimmt die Intensität der Strahlung (I) mit dem Quadrat des Abstandes (x) nach I(x) = I
1/x2 ab? Beachte: Die Intensität des Deckenlichts an der Meßkammer beeinflußt die
Messung! Abdunkeln!
c. Allgemeine Versuchsvorbereitungen durchführen.
d. Diaprojektor auf Position 135 cm stellen und anschalten.
10

e. Das Tuch entfernen und für 2 min die Lichtatmung messen.
f. Den Verschlußkolben herausziehen, 100 µl der NaHCO3-Lösung zusetzen und den
Verschlußkolben wieder einsetzen.
g. Gemäß vorherigem Versuch die errechnete Zeit bis zum Erreichen der maximalen
Syntheserate aufzeichnen lassen, dann für weitere 2 min aufzeichnen.
h. Den Diaprojektor 15 cm näher an die Reaktionskammer rücken, 2 min aufzeichnen
lassen.
i. Vorhergehenden Punkt wiederholen, bis 15 cm Entfernung erreicht sind.
j. Messung beenden, Daten speichern, Verschlußkolben herausziehen, Reaktions-
kammer aussaugen, mit H2Odest 3x spülen, 1 ml H2Odest einfüllen und Verschluß-
kolben einsetzen.
k. Die Abhängigkeit der O2-Entwicklung (Y-Achse) Steigung der Aufzeichnung ( y/ x-
Werte) von dem Abstand bzw. der Bestrahlungsstärke (X-Achse) grafisch darstellen.
Können sie aus der Aufzeichnung den Lichtkompensationspunkt direkt erkennen?
Änderung der apparenten Photosynthese unter O2-Mangel:
a. 100 ml Duran-Glasflasche mit H2Odest füllen und mindestens 5 min mit N2-Gas aus der
Druckflasche (bitte nur nach Anleitung) spülen. Deckel auf Glasflasche aufschrauben
und dicht verschließen.
b. Allgemeine Versuchsvorbereitungen durchführen, aber Punkt d. ändern: 1 ml H2Odest
aus Reaktionskammer entfernen und durch H2Odest ersetzen, das mit N2-Gas gespült
wurde.
c. Allgemeine Versuchsvorbereitungen Punkte e-j durchführen.
d. Mindestens 2 min aufzeichnen lassen und festhalten, daß die Dunkelatmung des
Blattmaterials gemessen wird.
e. Diaprojektor in der Entfernung vom Reaktionsgefäß aufstellen, die als Licht-
kompensationspunkt bestimmt wurde und Diaprojektor anschalten.
f. Das Tuch entfernen und 2 min die Lichtatmung messen.
g. Anschließend den Verschlußkolben herausziehen, 100 µl der NaHCO3-Lösung
zusetzen und den Verschlußkolben wieder einsetzen.
h. Mindestens 2 min die apparente Photosynthese im Lichtkompensationspunkt
messen.
i. Diaprojektor in 15 cm (bzw. in der letzten noch meßbaren) Entfernung vom
Reaktionsgefäß aufstellen und 2 min die apparente Photosynthese im
Lichtsättigungsspunkt messen.
j. Die Bestimmung der apparenten Photosynthese stoppen, Daten speichern,
Reaktionskammer aussaugen und mit H2Odest 3x spülen, 1 ml H2Odest einfüllen und
Verschlußkolben einsetzen.
11

3. Messung der photosynthetischen O2-Synthese an isolierten Thylakoidmembranen
Vor Versuchsbeginn Eis holen und Kühlzentrifuge einschalten.
Isolierung von Thylakoidmembranen (unbedingt immer kühl halten):
a. Spinatblätter (50 g) mit dem Messer klein schneiden, in vorgekühlten Glasmixbecher
einfüllen, mit 120 ml Isolationsmedium versetzen und 4 x 5 sec homogenisieren (nur
Teilhomogenisierung, sonst nur Chloroplastenbruchstücke!).
b. Homogenat durch mehrere Lagen Gaze in ein gekühltes Becherglas pressen
(Einweghandschuhe).
c. Durch 100 µm Nylontuch in Stahlbecher filtrieren und 1 min bei 2000 x g (4.000 Upm)
zentrifugieren.
d. Überstand (Chloroplastenbruchstücke) verwerfen.
e. Niederschlag in den Stahlbechern mit insgesamt 5 ml Isolationsmedium mit einem
Glasstab suspendieren.
f. Den Inhalt der Becher vereinigen; 0.1 ml für die mikroskopische Untersuchung der
intakten Chloroplasten abzweigen; 1 min bei 1 000 x g (3 000 Upm) zentrifugieren,
den Überstand wieder verwerfen.
g. Niederschlag mit 3 ml Lysemedium mit einem Glasstab suspendieren, 5 min auf Eis
stehen lassen (Lyse der Chloroplastenhülle durch osmotischen Schock).
h. Mit 3 ml Inkubationsmedium versetzen und vorsichtig mischen, bis keine Ver-
klumpung mehr sichtbar ist; anschließend durch 100 µm Nylontuch filtrieren.
i. Zweimal je 0.1 ml abzweigen (für die spätere Chlorophyllbestimmung und für die
mikroskopische Untersuchung). Der Rest wird für die Messung der Hill-Reaktion
verwendet.
Chlorophyllbestimmung:
Die Chlorophyllkonzentration (nmol Chl/ml; mg/ml) ist ein häufig verwendetes Bezugssystem
bei photosynthetischen Messungen, um die Ergebnisse vergleichen zu können. Die
zurückgestellten 0,1 ml der Thylakoidsuspension werden mit 19,9 ml 80% Aceton versetzt,
geschüttelt, abzentrifugiert und die Extinktion der Lösung bei 645 nm (Chl b) und 663 nm
(Chl a) im Spektralphotometer gegen 80% Aceton in Glasküvetten gemessen.
Die Gesamtchlorophyllmenge ergibt sich für die Thylakoidsuspension nach der empirischen
Formel: (E645 x 20,2 + E663 x 8,02) 1000 x A x B = Chl a+b [mg]
A = Verdünnungsfaktor: Vol. Lösemittel Aceton Vol. der vermessenen Thylakoidsuspension
B = Gesamtvolumen der Thylakoidsuspension in ml
Mikroskopie:
Die isolierten intakten und die aufgebrochenen Chloroplasten können mit Ölimmersion
mikroskopiert werden, möglichst im Phasenkontrast.
12

Messung:
Pipettierschema
Lösung Versuchsansatz 1 2 3 4
1 H2O 650 µl 550 µl 450 µl 450 µl 2 Inkubations-Lsg. 750 µl 750 µl 750 µl 750 µl 3 Rinderserum-Lsg. 50 µl 50 µl 50 µl 50 µl 4 K3[Fe(CN)6] -Lsg. 100 µl 100 µl 100 µl 5 ADP/Phosphat-Puffer 100 µl 6 CH3NH3Cl-Lsg. 100 µl
Thylakoidsuspension 50 µl 50 µl 50 µl 50 µl
a. Gemäß vorstehender Pipettieranleitung nur die Lösungen 1-6 in 4 Eppendorfgefäßen
mit 2 ml Volumen vorpipettieren und Gefäße auf Eis lagern.
b. Überprüfen, ob das Reaktionsgefäß der Elektrode mit H2Odest befüllt ist.
c. Verschlußkolben herausnehmen, H2Odest absaugen und 1.3 ml aus Eppendorfgefäß 1
einfüllen.
d. Magnetrührer anschalten und überprüfen, ob sich Rührfisch dreht.
e. 50 µl der Thylakoidsuspension in die Reaktionskammer einpipettieren, Verschluß-
kolben einsetzen und das Gefäß mit schwarzem Tuch abdunkeln.
f. Programm starten.
g. Die Dunkelatmung für 2 min aufzeichnen.
h. Diaprojektor in 15 cm Entfernung vom Reaktionsgefäß aufstellen (Distanz Frontlinse-
Reaktionskammer) und Diaprojektorlicht anschalten.
i. Das Tuch entfernen und 5 min die O2-Syntheserate messen.
j. Bestimmung der apparenten Photosynthese stoppen. Verschlußkolben heraus-
nehmen, Reaktionskammer aussaugen und mit H2Odest 3x spülen.
k. Daten auswerten und ausdrucken.
l. 1.3 ml aus Eppendorfgefäß 2 einfüllen und Versuchsansatz 2 wie unter Punkt d-k
beschrieben durchführen. Ebenso mit Versuchsansätzen 3 und 4 verfahren.
E. Auswertung
Die Änderung der O2-Entwicklung pro Zeit lässt sich unter dem Programmpunkt „Tools“ unter
Anwählen von „Show oxygen meters“ bestimmen. Werte gemäß Anzeige ablesen. Als
Bezugsgröße für die pflanzliche O2-Aufnahme/-Abgabe pro Zeit wird meistens der
Chlorophyllgehalt, die Fläche des Blattmaterials oder das Frischgewicht (mol Chl, cm2, g FG)
gewählt und der Relativwert auf eine Stunde hochgerechnet ( mol O2/h mol Chl, mol O2/h
cm2 oder mol O2/h g FG). Die Messergebnisse sollen grafisch aufbereitet werden. Welche
Achsenbezeichnungen müssen dabei jeweils gewählt werden?
Versuch 2: Der Lichtkompensationspunkt soll grafisch ermittelt werden.
13

Versuch 3: Die O2-Syntheseraten der Ansätze 1-4 berechnen. Muß die Dunkelatmung bei
der Berechung der O2-Syntheseraten berücksichtigt werden? Die Werte miteinander
vergleichen! Warum werden unterschiedliche Werte erhalten?
Fragen zu den Versuchen:
Warum müssen die Blattstücke vor der O2-Messung und der Chlorophyll-Bestimmung
zerhäckselt werden?
Wieviele intakte Zellen befinden sich etwa in einem 1 mm2 großen Blattstückchen?
Wieviele Chloroplasten sind etwa pro Zelle vorhanden?
Warum ist der Zusatz von NaHCO3 für das Gelingen des Versuches wichtig?
Welche Bezugssysteme für die Bestimmung der O2-Aufnahme/-Abgabe sind sinnvoll?
Welche Bedeutung hat der Lichtkompensationspunkt für die Pflanze?
Gibt es neben der Chlorophyllkonzentration andere Bezugssysteme, um die Ergebnisse von
unterschiedlichen photosynthetischen Experimenten vergleichen zu können?
Welche Bedeutung hat die Zentrifugation bei der Isolation von Thylakoidmembranen?
Welche Funktion hat Saccharose im Isolations-Medium?
Welche Bedeutung hat der pH im Inkubations-, Lyse-, und Isolationsmedium für das
Funktionieren der Hill-Reaktion?
F. Anhang
Tabelle I.2: Sauerstoffkonzentration in luftgesättigtem Wasser.
Die Gleichgewichts-Konzentration von Sauerstoff in Luft-gesättigtem Wasser hängt bei dem im Labor verfügbaren destillierten Wasser neben der Temperatur auch vom Luftdruck ab, der hier vernachlässigt wird. Die in ppm (parts per million=Anteil der Substanz in 106 Teilen der Gesamtsubstanz z.B. 1 ppm 1mg/kg oder 1 mm3/l) angegebenen Werte lassen sich durch Division mit der O2-Molmasse (32 µg O2/ml = 1 mM) in die hier verwendete Konzentrationsangabe µmol/ml (mM) umrechnen, wenn man berücksichtigt, daß 1 ml Wasser 1 g wiegt und somit die Angabe 1 ppm (1µg/g) auch als 1 µg O2/ml Wasser gelesen werden kann. Tabellenwerte nach Seidell and Linke (1965) Am. Chem. Soc. Div. Grad. Res. 2, p.1228
14

Versuch II: Funktionsänderungen der Microbodies bei der Umstellung des Stoff-
wechsels während der Keimung fetthaltiger Samen
A. Theorie:
Keimblätter von Sonnenblume (Helianthus annuus) enthalten Fett als Speichersubstanz.
Während der Keimung wird das Reservefett 'mobilisiert', d.h. es wird in ein Kohlenhydrat,
nämlich Saccharose, umgewandelt, welches in die wachsenden Teile des Embryos
transportiert wird. Im wachsenden Embryo wird die Saccharose zur Energiegewinnung und
für Synthesen verbraucht oder nach Umwandlung vorübergehend als Stärke gespeichert.
Beim Abbau des Reservefetts werden durch Lipasen die Fettsäuren freigesetzt. Die
Fettsäuren werden durch die ß-Oxidation zu Acetyl-CoA abgebaut, welches über die
Reaktionen des Glyoxylsäurezyklus in Succinat umgesetzt wird. Alle Enzyme für die ß-
Oxidation und den Glyoxylsäurezyclus sind in einem besonderen Organell enthalten und von
der übrigen Zelle abgegrenzt (kompartimentiert), in den Glyoxysomen. Diese stellen einen
spezialisierten Typ der Microbodies dar.
Der Glyoxylsäurezyklus besteht aus der Aufeinanderfolge der Enzymreaktionen der Citrat-
Synthase - Aconitase - Isocitratlyase - Malatsynthase - Malatdehydrogenase. Succinat kann
in den Glyoxysomen nicht weiterverarbeitet werden. Es wird in den Mitochondrien über
Fumarsäure in Äpfelsäure umgesetzt. Nach Umwandlung der Äpfelsäure in Oxalessigsäure
wird letztere unter Verbrauch von ATP und Freisetzung von CO2 durch das Enzym
Carboxykinase in Phosphoenolpyruvat umgesetzt. Phosphoenolpyruvat kann die Reaktionen
der Gluconeogenese durchlaufen und so über Phosphoglycerinsäure und Hexosephosphate
schließlich in Saccharose umgewandelt werden. Die Reaktionsfolge, die von der Oxalsäure
ausgeht, läuft zunächst im Cytoplasma, in den letzten Schritten (sehr wahrscheinlich) in den
Plastiden ab.
Wenn das Reservefett aufgebraucht ist, verschwinden die Enzyme des Glyoxylsäurezyklus
wieder und zumindest ein großer Teil der Glyoxysomen wird abgebaut oder umgebaut.
Wenn die Pflanze im Licht angezogen wird, ergrünen die Kotyledonen. Sie differenzieren
sich vom Speicherorgan zum Photosynthese treibenden Blatt um. Auch die ergrünten
Kotyledonen enthalten weiterhin Microbodies. Diese haben jetzt aber eine andere Funktion
und werden Peroxisomen genannt. Diese Organellen enthalten Enzyme, die für den weiteren
Umsatz der Glykolsäure sorgen, welche unter normalen Bedingungen in großer Menge
während der Photosynthese in den Chloroplasten gebildet wird (Glykolsäureoxidase und
Hydroxypyruvatreduktase). Der Verbrauch von O2 und die Abgabe von CO2 beim Umsatz der
Glykolsäure treten als sog. Lichtatmung in Erscheinung.
Literatur
Schopfer, P. und Brennicke, A. (1999) Pflanzenphysiologie, Springer
15

Lehninger, A., Nelson, D., Cox, M. (1993) Principles of Biochemistry, Worth Publishers
Buchanan, B., Gruissem, W., Jones, R. (2000) Biochemistry & Molecular Biology of Plants, American Society of Plant Physiologists
B. Aufgabe:
Bestimmen Sie durch die Messung von Leitenzymen die Funktionsänderung der Microbodies
in keimenden Sonnenblumensamen.
C. Reagenzien:
Je 25 Sonnenblumensamen (Helianthus annuus) auf feuchtem Vermiculite aussäen und
unter verschiedenen Bedingungen keimen lassen:
1 Schale 3 Tage Dunkel
1 Schale 5-7 Tage Licht (Dauerlicht)
1 Schale 5-7 Tage Dunkel
Folgende Lösungen werden gestellt:
0.5 M Tris/HCl, pH 7.5
0.1 M K-Phosphat-Puffer (KPi), pH 7.0
0.1 M K-Phosphat-Puffer (KPi), pH 7.5
0.5 M MgCl2 1% Triton X-100
Direkt vor den Enzymmessungen müssen jeweils folgende Lösungen hergestellt werden:
KPi-Puffer mit 10 mM H2O2 (kaltstellen) 160 µl 30% H2O2 zu 100 ml 0.1 M KPi-Puffer, pH 7.0
0.5 M Triethanolamin/HCl, pH 7.8
0.1 M Cysteinhydrochlorid (10 ml)
0.1 M Phenylhydrazinhydrochlorid, pH 6.8 (20 ml)
0.8 M Isocitrat (Na-Salz, 1 ml)
0.1 M Glutathion, oxidiert (5 ml)
12 mM Flavinmononukleotid (1 ml)
0.1 M Glykolsäure, neutralisiert mit NaOH (10 ml)
D. Versuchsdurchführung:
Präparation der löslichen Proteinfraktionen
Je 1 g junge etiolierte, 3 g alte etiolierte, und 3 g alte ergrünte Kotyledonen sowie 3 g Blätter
werden gezählt und dann jeweils im vorgekühlten Mörser mit etwas Quarzsand und 2 ml 50
mM Tris/HCl-Puffer, pH 7.5, zu einem feinen Brei zerrieben. Die Homogenate werden
quantitativ in Plastik-Zentrifugenröhrchen überführt. Dazu werden weitere 5 ml Puffer
verwendet. Dann wird gemischt und bei 8 000 rpm 10 min zentrifugiert. Mit einer Pipette wird
16

der Überstand (unter der Fettschicht) abgenommen, in ein graduiertes Reagenzglas
überführt, mit Tris/HCl-Puffer auf 10 ml aufgefüllt und auf Eis aufbewahrt. Es werden für
jedes Präparat die Aktivitäten von drei Leitenzymen bestimmt.
1. Katalase (Leitenzym für Peroxisomen/Glyoxysomen)
Der Umsatz von H2O2 ( 240 = 36 M-1 cm-1) wird durch die Abnahme der Extinktion bei 240 nm
während der ersten 2 min im Spektralphotometer bestimmt. Gemessen wird parallel in
Quarzküvetten, wobei der Testansatz mit einer Doppelbestimmung durchgeführt wird.
Gegebenenfalls muß der Proteinextrakt 1:10 verdünnt werden.
Testansatz (2x) Kontrollansatz0.1 M KPi + H2O2 990 µl0.1 M KPi, pH 7.0 990 µl Proteinextrakt 10 µl 10 µl
2. Isocitratlyase (Leitenzym für Glyoxysomen)
Die gebildete Glyoxylsäure wird als Phenylhydrazon an der Zunahme der Extinktion bei
324 nm ( = 1.7 x 104 M-1 cm-1) 10 min lang gemessen. Beim Zusammenpipettieren muß die
angegebene Reihenfolge unbedingt eingehalten werden. Mit jedem Proteinextrakt wird
mindestens eine Doppelbestimmung durchgeführt.
Testansatz (2x) KontrollansatzProteinextrakt 50 µl 50 µl0.1 M KPi-Puffer, pH 7.5 810 µl 810 µl 0.5 M MgCl2 20 µl 20 µl0.1 M Cysteinhydrochlorid (frisch) 50 µl 50 µl 0.1 M Phenylhydrazinhydrochlorid, pH 6.8 (frisch) 50 µl 50 µl mischen, temperieren (RT), Reaktion starten mit 0.8 M Isocitrat 20 µl H2O 20 µl
3. Glykolsäureoxidase (Leitenzym für Peroxisomen)
Die gebildete Glyoxylsäure wird wie oben, aber nur 5 min lang in Doppelbestimmungen
gemessen. Die Reagenzien vom FMN an werden erst unmittelbar vor dem Start der
Reaktion zugegeben. Der Start der Reaktion erfolgt durch Zugabe des Substrates
(Glykolsäure). Der Kontrollansatz wird mit H2O statt Glykolsäure durchgeführt.
Testansatz (2x) KontrollansatzH2O 195 µl 215 µl50 mM Triethanolaminpuffer-HCl, pH 7.8 600 µl 600 µl 0.05 % (v/v) Triton-X-100 in H2O 15 µl 15 µl0.1 M oxidiertes Glutathion 50 µl 50 µl für etwa 4 min inkubieren 12 mM Flavinmononukleotid (FMN) 20 µl 20 µl Proteinextrakt 50 µl 50 µl0.1 M Phenylhydrazinhydrochlorid, pH 6.8 (frisch) 50 µl 50 µl 0.1 M Glykolsäure (neutralisiert) 20 µl
17

E. Auswertung
In den untersuchten Entwicklungsstadien sind die Enzymaktivitäten entsprechend dem
Lambert-Beerschen-Gesetz (siehe auch Seite 38) in µMol umgesetztes Substrat/min pro
Kotyledon/Blatt bzw. pro Gramm Frischgewicht zu berechnen und darzustellen.
Abb. II.1:
18

Versuch III: Anreicherung und Charakterisierung von Phosphofructokinase aus
Karotten
A. Theorie:
Der glykolytische Abbau beginnt mit der Umwandlung von Hexose in 2 Moleküle Triose-
phosphat. Hierbei wird die Oxidationsstufe des Kohlenhydrats nicht verändert, jedoch ist ATP
zur Phosphorylierung erforderlich. Das Schlüsselenzym der Glykolyse ist die 6-Phospho-
fructokinase (PFK), welches die irreversible Umwandlung von Fructose-6-Phosphat (F-6-P)
in Fructose-1,6-bisphosphat (FbP) katalysiert. Das spezifisch auf F-6-P eingestellte
tetramere Enzym mit einem Molekulargewicht von ca. 360.000 wird allosterisch reguliert.
Wenn z.B. die Energieladung der Zelle hoch ist oder andere Brennstoffe zur
Energiegewinnung (z.B. Fettsäuren) zur Verfügung stehen, dann wird die PFK allosterisch
gehemmt, was die Abschaltung der Glykolyse zur Folge hat. Kriterien für die
Charakterisierung von Enzymen sowie für Eigenschaften von allosterischen Enzymen bzw.
Modelle für die allosterische Enzymregulation müssen der Literatur entnommen werden.
Literatur
Kosaku, U. (1979) Phosphofructokinase. In: Advances in Enzymology Vol. 48, 193-244, John Wiley & Sons, New York
Dennis, T.D. and Coultate, T.P. (1966) Phosphofructokinase, a regulatory enzyme in plants. Biochim. Biophys. Res. Commun. 25, 187-191
B. Aufgabe:
Aus Karotten soll die PFK isoliert und durch Ammoniumsulfatfällung angereichert werden.
Mit der angereicherten Enzymfraktion sind mit Hilfe eines gekoppelten enzymatischen Tests
die Michaelis-Menten-Werte (KM-Werte) für die Substrate F-6-P und ATP zu ermitteln und die
allosterische Hemmung durch hohe ATP-Konzentrationen aufzuzeigen.
C. Reagenzien:
Extraktionspuffer (50 mM Imidazol/EDTA-HCl, pH 7.8; 4°C)
0.52 g Imidazol 112 mg Titriplex III (EDTA) 3 ml 0.5 M MgCl2 in 140 ml H2Odest lösen, mit 6 N HCl auf pH 7.8 einstellen,
auf 150 ml auffüllen
10 g Polyvinylpoly- in 100 ml dieses Puffers suspendieren (zur Adsorption pyrrolidon von phenolischen Substanzen)
0.4 ml 2-Mercaptoethanol direkt vor der Extraktion zur Suspension zugeben (SH-Gruppenschutz)
50 mM Imidazol-HCl, pH 7.0 (4°C)
1.7 g Imidazol in 480 ml H2Odest lösen, mit 6 N HCl auf pH 7.0 einstellen, auf 500 ml auffüllen
19

Biuret-Reagenz (wird gestellt)
0.3 g CuSO4 x H2O 1.2 g Kalium-Natriumtartrat in etwas H2Odest lösen 6 g festes NaOH zugegeben und mit H2Odest auf 250 ml auffüllen
MgCl2-Lösung (50 mM, 50 ml, 4°C)
5 ml 0.5 M MgCl2 mit 50 mM Imidazol-Puffer, pH 7.0, verdünnen
Fructose-6-phosphat-Lösung (F-6-P, 5 mM, 10 ml, -20°C)
17 mg F-6-P (Dinatriumsalz x 2 H2O) in 50 mM Imidazol-Puffer, pH 7.0, lösen
Fructose-1,6-bisphosphat-Lösung (F-1,6-bP, 5 mM, 5 ml, -20°C)
13.8 mg F-1,6-bP (Trinatriumsalz x 8 H2O) in 50 mM Imidazol-Puffer, pH 7.0, lösen
ATP/MgCl2-Lösung (25 mM, 20 ml, -20°C)
275.6 mg ATP (Dinatriumsalz) in ca. 10 ml 50 mM MgCl2-Lösung lösen, mit 0.2 N NaOH auf pH 7.0 einstellen und mit MgCl2 auf 20 ml auffüllen
NADH-Lösung (1 mM, 10 ml, erst am Tag der Enzymmessung herstellen, im Eisbad lagern)
7.0 mg NADH (Dinatriumsalz) in 50 mM Imidazol-Puffer, pH 7.0, lösen
GDH/TIM-Suspension
Das Enzymgemisch GDH/TIM wird am Versuchstag im Verhältnis 1:20 verdünnt,dazu werden 0.1 ml Enzymgemisch (vorher vorsichtig schütteln!) mit 1.9 ml 50 mM Imidazol-Puffer, pH 7.0, verdünnt. Aufbewahrung im Eisbad
Aldolase-Suspension
Das Enzym wird am Versuchstag im Verhältnis 1:20 verdünnt, dazu werden 0.1 ml Aldolase-Suspension (vorher vorsichtig schütteln) mit 1.9 ml 50 mM Imidazol-Puffer, pH 7.0, verdünnt. Aufbewahrung im Eisbad
D. Durchführung:
Anreicherung und Reinigung des Enzyms
1. Hinweise für das Arbeiten mit Enzymen:
Viele biochemische Substanzen zeigen eine große Empfindlichkeit gegenüber pH-Ände-
rungen und höheren Temperaturen. Im besonderen Ausmaß trifft man diese Empfindlichkeit
bei katalytisch aktiven Proteinen, den Enzymen. Beim Arbeiten mit Enzymen sollte man
besonders beachten: Enzyme müssen immer mit großer Sorgfalt und Sauberkeit behandelt
werden und immer in der Kälte (Eisbad, Kühlraum, Kühlschrank, Kühltruhe) gehalten
werden. Viele Proteine verlieren ihre enzymatische Aktivität in der Wärme (Zimmer-
temperatur), bei Zusatz von organischen Lösungsmitteln, beim Schäumen der Lösung, bei
geringen pH-Verschiebungen oder sogar beim Verdünnen. Auch sehr kleine Mengen von
Verunreinigungen, z.B. Schwermetalle, oder Sauerstoff können zur vollständigen Inaktivitie-
20

rung des Enzyms führen. Die zum Spülen der Glasgeräte üblicherweise verwendeten Deter-
gentien haben oft schädliche Wirkung. Daher soll man stets mit H2Odest gespülte Geräte
verwenden. Da Proteinlösungen ein ideales Nährmedium für Mikroorganismen darstellen,
empfiehlt es sich, die Enzymlösungen immer in der Kälte oder aber eingefroren zu halten,
um das Wachstum von Bakterien zu verzögern.
Die große pH-Abhängigkeit der Enzymaktivität bedingt, daß man immer den pH-Wert der
Lösungen kontrollieren muß. Wegen denaturierend wirkender Oberflächenspannungseffekte
soll man die Enzymlösung nie schütteln oder unter Schaumbildung rühren. Enzymatisch
aktive Proteine werden durch anwesende Proteasen oft rasch inaktiviert. Andererseits
können andere im Rohextrakt vorhandene Enzyme durch ihr Zusammenspiel bestimmte
Reaktionsabläufe vortäuschen. Nur durch gut durchdachte Kontrollversuche läßt sich eine
bestimmte Enzymaktivität im Rohextrakt einwandfrei nachweisen. Zusatz von Fremd-
proteinen übt bisweilen auf die untersuchte Enzymaktivität eine stabilisierende Wirkung aus
(Schutzproteine z.B. Rinderserumalbumin). Nach der Abtrennung der überschüssigen
Fremdproteine wird das Enzym oft instabil.
2. Proteinfällung mit Ammoniumsulfat:
Beim Lösen von Ammoniumsulfat werden Wassermoleküle an die Salzionen gebunden. Je
mehr Ammoniumsulfat in der Lösung vorhanden ist, desto weniger „freies Wasser“ steht als
Lösungsmittel zur Verfügung, und die Löslichkeit von Proteinen wird herabgesetzt. Wegen
seiner großen Löslichkeit hat sich Ammoniumsulfat als Fällungsmittel gut bewährt. Seine
Löslichkeit hängt von der Temperatur ab, worauf man immer zu achten hat. Das
Ammoniumsulfat kann fest oder als gesättigte Lösung zugesetzt werden. Im ersten Fall wird
auch bei höheren Ammoniumsulfatkonzentrationen das Volumen der Lösung und damit die
Verdünnung klein gehalten. Man gibt das feste und fein pulverisierte Ammoniumsulfat
langsam unter Rühren zu. Man wartet mit der Zugabe von weiterem Salz so lange, bis das
vorher zugegebene sich gelöst hat. Setzt man Ammoniumsulfat in Form der gesättigten
Lösung zu, so muß man auch hier die Lösung nach Zugabe länger stehen lassen, damit sich
das ausgefallene Protein in ein echtes Gleichgewicht mit der überstehenden Lösung setzen
kann. Andernfalls sind die Ergebnisse schlecht reproduzierbar. Als Salz einer starken Säure
und einer schwachen Base reagiert Ammoniumsulfat schwach sauer. Durch Zugabe von
verdünntem Ammoniak zur Lösung während der Fällung oder zur gesättigten Ammonium-
sulfatlösung kann man einen neutralen pH-Wert einhalten.
Die Niederschläge der Ammoniumsulfatfällung werden durch Zentrifugation vom Überstand
getrennt. Die abzentrifugierten Niederschläge löst man wie folgt auf: Nach Zugabe von wenig
Puffer versucht man, durch Rühren mit einem Glasstab das Protein in Lösung zu bringen;
wird nur eine teilweise Lösung erreicht, so ist Puffer in kleinen Portionen zuzusetzen, bis sich
das Protein im gewünschten Volumen gelöst hat. Von denaturiertem und nicht mehr
löslichem Protein trennt man durch Zentrifugation ab.
21

3. Herstellung des Rohextraktes und fraktionierte Ammoniumsulfatfällung:
100 g gewaschene und geschälte Karotten (keine eingefroren gelagerten Karotten ver-
wenden!) werden mit einem Messer in Scheiben geschnitten und im gekühltem Mixer mit 100
ml Extraktionspuffer (wie unter C angegeben mit Mercaptoethanol) homogenisiert. Das
Homogenat wird durch Gaze gepreßt (Einweghandschuhe!) und der Extrakt bei 4°C für 10
min bei 12 000 Upm (ca. 15 000 g) zentrifugiert. Das Volumen des Überstandes (Rohextrakt)
wird auf 110 ml eingestellt; 10 ml davon werden im Kühlschrank aufbewahrt.
Der Rest des Rohextraktes wird mit Ammoniumsulfat fraktioniert gefällt. Hierzu wird der
Rohextrakt in einem Becherglas (250 ml, hohe Form) in ein Eisbad gestellt und mit einem
Magnetrührer langsam gerührt. Dazu wird langsam fein zerriebenes Ammoniumsulfat zuge-
geben (nach jeder Spatelspitze warten und rühren!) bis zur Sättigung von 35% (je 100 ml
Rohextrakt werden 20.9 g Ammoniumsulfat zugesetzt). Dann wird noch 15 min gerührt und
anschließend zentrifugiert (10 min, 12 000 Upm, 4°C). Nach dem Zentrifugieren wird der
Überstand vorsichtig in ein Becherglas dekantiert (Eisbad) und der Rückstand dann mit 7 ml
50 mM Imidazol-Puffer, pH 7.0, und 15 µl Mercaptoethanol gelöst. Mit dem Überstand wird
dann eine weitere Ammoniumsulfatfällung bis 55% Sättigung (weitere 12.9 g Salz je 100 ml
Ausgangsvolumen) vorgenommen. Nach dem Zentrifugieren (10 min, 12 000 Upm, 4°C) wird
erneut dekantiert, das Volumen des Überstandes bestimmt und davon 20 ml aufgehoben.
Der Rückstand wird wiederum in 7 ml Imidazol-Puffer, pH 7.0, und 15 µl Mercaptoethanol
gelöst.
Alle Fraktionen werden noch einmal zentrifugiert (10 min bei 12 000 Upm) und die
Überstände im Kühlschrank bei 4°C aufgehoben. Auf diese Weise erhält man 3 Protein-
fraktionen, von denen die Aktivität der 6-Phosphofructokinase bestimmt werden soll:
Fraktion 1: Proteine, die bis 35% Sättigung an Ammoniumsulfat ausfallen
Fraktion 2: Proteine, die zwischen 35% und 55% Sättigung an Ammoniumsulfat ausfallen
Fraktion 3: Proteine, die im Überstand verbleiben
Um die Aktivität der 6-Phosphofructokinase in diesen Fraktionen zu stabilisieren, müssen
jeden Tag pro ml Extrakt 10 µl Mercaptoethanol zugegeben werden. Extrakte im Kühlschrank
aufbewahren, nicht einfrieren.
4. Proteinbestimmung:
Zur quantitativen Proteinbestimmung wird die Biuret-Reaktion verwendet. Für die Aufnahme
einer Eichkurve werden 50 mg Serumalbumin in 5 ml H2Odest gelöst. Von dieser Lösung
werden 0.1; 0.2; 0.3; 0.4; 0.5; 0.6 und 0.7 ml auf 1.0 ml mit H2Odest aufgefüllt, mit 5 ml Biuret-
Reagenz versetzt, gemischt und 30 min stehen gelassen. Dann wird die Extinktion im Foto-
meter bei 540 nm gegen die Mischung von 1 ml demin. Wasser und 5 ml Biuret-Reagenz
gemessen. Von den Proteinextrakten werden je 0.5 und 1 ml (Rohextrakt), 0.2 und 0.5 ml
22

(Fraktion 1), 0.2 und 0.5 ml (Fraktion 2) bzw. 4 ml (Überstand = Fraktion 3) je mit 10%
Trichloressigsäure (0.2 ml pro ml Extrakt) versetzt und gemischt (Achtung mit Trichloressig-
säure in Berührung gekommene Glaswaren sofort und sorgfältig spülen!). Die ausgefallenen
Proteine werden in der Heraeus Megafuge (rote Becher, 5 min, RT, maximale Drehzahl)
abzentrifugiert und der Überstand vollständig dekantiert. Der Niederschlag wird in 1 ml
H2Odest suspendiert (Vortex) und damit dann wie oben der Biuret-Test durchgeführt. Vor der
fotometrischen Bestimmung muß zur Entfernung ungelöster Bestandteile erneut 5 min in der
Tischzentrifuge zentrifugiert werden.
Bestimmung der Enzymaktivität
1. Allgemeine Hinweise:
Die Aktivität eines Enzyms wird durch kinetische Messung der katalysierten Reaktion
bestimmt.
Die Anfangsgeschwindigkeit v der Reaktion:
Enzym A + B C + D k1
errechnet sich aus der Geschwindigkeitsgleichung, solange die Konzentration der Reaktions-
produkte C und D noch klein gegenüber A und B ist:
v = k1 x [A] x [B]
Die Geschwindigkeitskonstante k1 hängt von der Art des Enzyms und dessen Konzentration
ab. Bei hohen Konzentrationen von A und B ändern sich diese wenig, so daß die
Anfangsgeschwindigkeit v proportional der Konstanten k1 ist und damit der eingesetzten
Enzymmenge. Bei der Untersuchung einer Reaktion sind daher zunächst die Substrat-
konzentrationen festzulegen, bei denen die Reaktionsgeschwindigkeit proportional zur
eingesetzten Enzymmenge ist. Auch beim Vorliegen einer komplizierten Reaktionskinetik
kann man Bedingungen ermitteln, bei denen die Reaktionsgeschwindigkeit in einem
gewissen Konzentrations- und Zeitintervall nur von der Enzymkonzentration abhängt. Ist die
Geschwindigkeit der Reaktion bei einer bestimmten Enzymkonzentration sehr hoch, so
nimmt die Substratkonzentration rasch ab und es ist möglich, daß in diesem Bereich die Ge-
schwindigkeit nicht mehr allein von der Enzymkonzentration abhängt. Man muß daher den
Gültigkeitsbereich des Aktivitätstests prüfen und gegebenenfalls die Enzymlösung ent-
sprechend verdünnen. Hierzu wird der Lösung unmittelbar vor dem Aktivitätstest
Serumalbumin als Schutzprotein zugegeben, damit das verdünnte Enzym nicht während der
Reaktion inaktiviert wird. Will man mehrere kinetische Messungen - etwa zur Berechnung der
Michaelis-Konstanten - miteinander kombinieren, so muß zu allen Proben Enzym gleicher
23

Aktivität verwendet werden. Die Geschwindigkeitskonstante k1 ist nämlich zur Konzentration
an katalytisch aktivem Enzym proportional und nicht allgemein zur Proteinkonzentration.
Praktisch bedeutet dies, daß man alle Messungen mit dem Enzym gleicher Qualität in einem
Zug ohne Unterbrechung durchführen sollte.
2. Testprinzip:
Die Aktivität der PFK wird mit Hilfe des folgenden gekoppelten enzymatischen Tests
photometrisch bestimmt:
Bei einem Überschuß an NADH und genügender Konzentration der Hilfsenzyme (Aldolase,
TIM, GDH) verläuft die Reaktionskette:
F-1,6-bP + 2 NADH 2 Glycerin-3-phosphat + 2 NAD+
schnell ab. Die Gleichgewichtskonstante dieser Reaktion beträgt Kc = 7.75 x 104 M bei 20°C
und pH 7, das Gleichgewicht liegt also auf der rechten Seite. Die Geschwindigkeit der
Gesamtreaktion:
F-6-P + ATP + 2 NADH 2 Glycerin-3-P + 2 NAD+ + ADP
wird bei diesen Bedingungen also ganz von dem ersten Reaktionsschritt bestimmt. Dessen
Reaktionsgeschwindigkeit ist aber ein Maß für die Aktivität der PFK. Da NADH im Gegensatz
zu NAD+ ein Absorptionsmaximum bei 340 nm hat, kann der Reaktionsablauf am Fotometer
durch die Extinktionsabnahme bei dieser Wellenlänge beobachtet werden (siehe Anhang).
Aus der Extinktionsdifferenz errrechnet sich nach dem Lambert-Beerschen-Gesetz der
NADH-Verbrauch pro min nach der Formel:
µmol E/min x V (Test) ———— = ———————— ml x min x d x V (Einsatz)
V (Test) = Endvolumen der Flüssigkeit in der Küvette in ml V (Einsatz) = eingesetzte Menge des Enzyms in ml
Fructose-6-phosphat Fructose-1ATP ADP
PF,6-bisphosphat
Gl
K
A
ycerinaldehyd-3-phosphatDih
LD
TIMydroxyacetonphosphat
2 Glycerin-3-phosphat
GDH
2 NADH
2 NAD+ PFK Phosphofructokinase ALD Aldolase TIM Triosephosphat-Isomerase GDH Glycerin-3-phosphat-Dehydrogenase
24

E = Extinktionsdifferenz d = Schichtdicke in cm = Extinktionskoeffizient von NADH
340 = Extinktionskoeffizient von NADH bei 340 nm = 6.3 x 103 l x mol-1 x cm-1
= 6.3 ml x µmol-1 x cm-1
Aus dem NADH-Verbrauch läßt sich die durch die PFK gebildete Menge an F-1,6-bP
berechnen, da 2 Mole NADH einem Mol F-1,6-bP entsprechen. Die Enzymaktivität der PFK
wird in µKatal (1 µkat = 1 µMol F-1,6-bP/sec) angegeben. Die Angabe der Enzymaktivität in
Units (1 U = 1 µMol/min) ist nach den SI-Regeln nicht mehr zulässig.
3. Durchführung des Tests auf enzymatische Aktivität:
Zur Bestimmung müssen drei verschiedene Tests durchgeführt werden. Zunächst wird in
einem Kontrollversuch die Aktivität der zugesetzten Hilfsenzyme überprüft (Test 1). In einem
Test ohne Zusatz von Fructose-6-phosphat bestimmt man die Aktivität an NADH-Oxidase
(Test 2), während in Test 3 die Aktivität der PFK ermittelt wird. Test 2 und 3 müssen für den
Rohextrakt und die 3 Fraktionen durchgeführt werden. Zu jedem Test wird ein Leerwert
benötigt, die Zusammensetzung bitte mit den Assistenten besprechen.
Da im Rohextrakt NADH-Oxidase und PFK nebeneinander vorliegen, muß hier die Aktivität
aus der Differenz von Test 3 und Test 2 ermittelt werden. Bei sorgfältigem Arbeiten wird die
PFK nach der fraktionierten Ammoniumsulfatfällung vorwiegend in Fraktion 2 gefunden.
Die Hilfsenzyme werden unmittelbar vor Ansetzen der Versuchsreihe verdünnt (siehe
Seite 20) und ebenso wie ATP- und NADH-Lösungen im Eisbad aufbewahrt. Alle Lösungen
bis auf die Hilfsenzyme werden einpipettiert und für 5 min bei Raumtemperatur stehen
gelassen. Die Küvetten werden nach der Zugabe der letzten Hilfsenzymlösung (= Start der
Reaktion) sofort mit Parafilm verschlossen, zweimal gekippt und rasch in das Fotometer
(Amersham, Ultrospec 3100) gestellt.
Folgende Lösungen werden in eine Halbmikroküvette pipettiert (alle Angaben in µl):
Lösungen Test 1
(Hilfsenzyme) Lösungen Test 2
(NADH-Oxidase)
Test 3 (PFK + NADH-
Oxidase)
MgCl2 80 MgCl2 80 80 F-1,6-bP 200 F-1,6-bP - - F-6-P - F-6-P - 200 ATP 40 ATP 40 40 NADH 100 NADH 100 100 Imidazol-Puffer, pH 7.0
540 Imidazol-Puffer, pH 7.0
690 490
Karottenextrakt - Karottenextrakt 50 50 TIM/GDH 20 TIM/GDH 20 20 Aldolase 20 Aldolase 20 20
25

Vorher muß im Hauptmenü des Ultrospec die Karteikarte „Methods“ gewählt werden, dort ist
im Programm 1 „Kinetics“ ein Meßprotokoll gespeichert. Über die Taste „mode“ wählt man
durch die Pfeil-Tasten die Karteikarte „Timing“, hier muß die Anzahl der Meßproben (ohne
Leerwert, maximal 7) eingetragen werden. Durch die „stop“-Taste kommt man zurück ins
Meßprogram, gestartet wird durch die Tasten „run“ und „enter“. Abhängig von der Anzahl der
Proben wird alle 10 bis 15 sec ein Meßwert über den Zeitraum von 5 min aufgezeichnet. Die
jeweiligen Steigungen der Kurven jeder einzelnen Meßprobe wird mit der Funktion „post-run“
in „mode“ so bearbeitet, daß jeweils nur die linearen Bereiche zur automatischen Be-
rechnung verwendet werden, bevor die Grafiken über die „print“-Funktion ausgegeben
werden.
Enzymkinetik
Charakteristische Größen für ein Enzym sind die Michaelis-Konstante (KM) und die maximale
Umsatzgeschwindigkeit Vmax für ein bestimmtes Substrat. Vmax ist dann erreicht, wenn die
Substratkonzentration so hoch ist, daß alle Enzymmoleküle mit Substrat belegt sind. Eine
weitere Zugabe von Substrat ergibt dann keine Erhöhung der Umsatzgeschwindigkeit. Die
Michaelis-Konstante gibt an, bei welcher Substratkonzentration die Reaktion mit halb-
maximaler Geschwindigkeit abläuft. Sie ist ein Maß für die Affinität des Enzyms zum
Substrat.
Die Bestimmung von KM und Vmax erfolgt durch die Feststellung der Umsatzgeschwindigkeit
in Abhängigkeit von der Substratkonzentration. Da PFK zwei Substrate umsetzt, sollen die
Werte sowohl für ATP als auch für F-6-P bestimmt werden, wobei jeweils die Konzentration
eines Substrats konstant gehalten wird, während die Konzentration des anderen Substrats
variiert wird. Um außerdem die allosterische Hemmung durch ATP nachzuweisen, wird eine
weitere kinetische Messung bei einer höheren ATP-Konzentration durchgeführt.
1. Durchführung des Tests:
Alle Messungen werden mit derjenigen Fraktion durchgeführt, die die höchste Enzymaktivität
enthält. Zunächst werden in je eine Halbmikroküvette folgende Lösungen pipettiert:
80 µl MgCl2-Lösung
200 µl F-6-P-Lösung
100 µl NADH-Lösung
Messung der ATP-Abhängigkeit der PFK:
Die ATP-Konzentrationen in der Küvette sollen 10; 6; 4; 2; 1; 0.5; 0.4; 0.3; 0.2 und 0.1 mM
sein. Die Menge an einzusetzender ATP-Lösung (25 mM) wird wie folgt ausgerechnet: Um
aus einer 25 mM Lösung eine 10 mM Lösung zu erhalten, muß man die Ausgangslösung auf
das 2.5-fache verdünnen. Das Endvolumen in der Küvette soll 1 ml betragen, also muß 400
µl der Ausgangslösung eingesetzt werden. Das Gesamtvolumen sowohl der bereits
26

einpipettierten Lösungen als auch der noch zuzugebenden Enzymlösung beträgt 440 µl. Bei
Zugabe von 400 µl 25 mM ATP-Lösung ergibt sich ein Gesamtvolumen von 840 µl, so daß
noch 160 µl 50 mM Imidazol-Puffer, pH 7.0, zugegeben werden muß, um das verlangte
Endvolumen zu erhalten. Für eine 1 mM ATP-Konzentration gibt man 40 µl der 25 mM ATP-
Lösung und 520 µl Puffer in die Küvette. Vor Versuchsbeginn ist eine Tabelle vorzulegen, die
für die entsprechenden ATP-Konzentrationen die zu pipettierenden Mengen an ATP und
Puffer enthält.
Nachdem diese Lösungen vorbereitet sind, werden die Enzyme zugegeben:
20 µl Karottenextrakt (Fraktion 2, ggf. verdünnen)
20 µl TIM/GDH
20 µl Aldolase
Danach wird die Küvette sofort in das Fotometer gestellt und über einen Zeitraum von 5
Minuten gemessen (u.U. kann eine 5-minütige Vorinkubation bei RT ohne Hilfsenzyme auch
hier hilfreich sein).
Messung der F-6-P-Abhängigkeit der PFK:
In eine Küvette werden pipettiert:
80 µl MgCl2-Lösung
40 µll ATP-Lösung
100 µl NADH-Lösung
Die Konzentrationen an F-6-P in der Küvette sollen 2; 1; 0.6; 0,4; 0.2; 0.15; 0.1; 0.06; 0.04
und 0.02 mM sein. Diese Mengen an 5 mM F-6-P-Lösung und Puffer sind wie oben zu
berechnen (Tabelle!). Dann werden die gleichen Mengen Enzymlösung wie bei der ATP-
Abhängigkeit zugegeben und die Messungen analog durchgeführt.
Messung der F-6-P-Abhängigkeit der PFK bei Hemmung durch ATP:
Hierzu werden die gleichen Messungen durchgeführt wie bei der Messung der F-6-P-
Abhängigkeit der PFK. Jedoch werden 240 µl ATP-Lösung eingesetzt und die Puffermenge
entsprechend reduziert.
E. Auswertung
Anreicherung der PFK:
Schon während der Durchführung der Anreicherung stellt man sich eine Tabelle zusammen,
die den Arbeitsschritt, das Volumen der Lösung, den Proteingehalt (mg/ml), die Gesamt-
menge an Protein, die Enzymaktivität und den Anreicherungsfaktor enthält.
Die spezifische Enzymaktivität der PFK wird in µkat dividiert durch den Proteingehalt der
Enzymlösung berechnet. Die Einheit für die spezifische Enzymaktivität ist µkat/kg Protein. Da
das Enzym in einem Proteingemisch vorliegt, ist die spezifische Enzymaktivität ein Maß für
27

dessen Reinheit. Während der Anreicherung des Enzyms durch fraktionierte Ammonium-
sulfatfällung, d.h. bei der Entfernung von Fremdprotein, sollte daher die spezifische
Enzymaktivität steigen.
Enzymkinetik:
Trägt man die Wertepaare V (µmol/ml x min) = f [S] in ein Diagramm ein, so erhält man eine
Kurve, die bei idealem Verlauf für isosterische Enzyme der Michaelis-Menten-Gleichung
Vmax x [S] V = ————— entspricht. KM + [S]
Aus dieser Zeichnung werden Vmax und KM annähernd bestimmt. Eine genauere Bestimmung
ergibt die Auswertung nach Lineweaver und Burk. Dazu wird die Michaelis-Menten-
Gleichung umgeformt, zu:
1 1 KM 1 —— = —— + —— x —— V Vmax Vmax [S]
Trägt man also 1/V = f (1/[S]) auf, so erhält man eine Gerade. Der Schnittpunkt der Geraden
mit der Ordinate ergibt den Wert 1/Vmax und der Schnittpunkt mit der Abszisse ergibt den
Wert -1/KM. Hieraus erfolgt die genaue Berechnung von KM und Vmax. Die Ergebnisse aus
den Kinetik-Versuchen sind sofort im Michaelis-Menten- und Lineweaver-Burk-Diagramm
darzustellen, um gegebenenfalls fehlerhafte Messungen feststellen zu können, die dann
sofort zu wiederholen sind. KM und Vmax sind zu ermitteln und der Kurvenverlauf ist zu
diskutieren.
E. Anhang
Abb. III.1: Extinktionsspektrum von NAD+ und NADH (NADP+ und NADPH besitzen fast identische Spektren). Zur Konzentrationsmessung benutzt man entweder 340 = 6.3 x 103 l mol-1 cm-1 (Gipfelwert) oder 366 = 3.5 x 103 l mol-1 cm-1 (starke Emmissionslinie der Hg-Dampflampe).
28

Versuch IV: Induktion der Nitratreduktase in Rettichkotyledonen
A. Theorie:
Der Stickstoffgehalt der meisten höheren Pflanzen stammt zum überwiegenden Teil aus dem
Boden und wird von der Wurzel in Form von anorganischen Stickstoffverbindungen (NH4+
oder NO3-) aufgenommen. Als Akzeptor für Ammonium dient das Glutamat, das in einer
ATP-abhängigen Reaktion zum Glutamin fixiert wird. Vom Glutamin wird dann das aktivierte
Ammonium auf -Ketoglutarat übertragen (GOGAT-System), wobei reduziertes Ferredoxin
als Elektronendonator bei Pflanzen benötigt wird. Der auf diese Weise gebundene
Aminostickstoff kann mit Hilfe von Transaminasen von Glutamat aus auf andere -
Ketosäuren übertragen werden und so zur Bildung aller benötigten Aminosäuren
herangezogen werden. Aus dem Boden aufgenommenes Nitrat muß von der Pflanze erst zu
NH4+ reduziert werden, bevor es verwendet werden kann. Diese Reaktionsschritte werden
von den Enzymen Nitratreduktase und Nitritreduktase katalysiert. Diese Enzymaktivitäten
finden sich außer in Laubblättern auch in Wurzeln, so dass neben der photosynthetischen
Nitratreduktion auch das Nitrat in nicht grünen Geweben reduziert werden kann. Die Nitrat-
und Nitrit-Reduktase sind durch ihre Substrate induzierbar. Die Zunahme der Aktivität von
Nitratreduktase in nitratbehandelten Pflanzengeweben beruht auf einer echten Enzym-
induktion: Hemmstoffe der RNS- sowie der Proteinbiosynthese hemmen auch die Bildung
des Enzyms. Andererseits wird die Enzymbiosynthese aber auch durch überschüssiges NH4+
oder ausreichendes Angebot von Aminosäuren reprimiert. In Keimwurzeln von Mais
verschwindet vorhandene Nitratreduktaseaktivität unter solchen nicht-induktiven Be-
dingungen mit Halbwertszeiten von 2 bis 3 Stunden.
Literatur:
Hartmann, T. (1982) Die Ammoniumassimilation im N-Stoffwechsel der Pflanzen. Biologie in unserer Zeit 12, 9-12
Heldt, H.W. (1999) Pflanzenbiochemie, 2. Auflage
B. Aufgabe:
Kotyledonen von Rettichkeimlingen (Raphanus sativus) werden in Nitratlösung inkubiert. Aus
den Kotyledonen wird zu verschiedenen Zeiten nach Beginn der Inkubation ein Rohextrakt
hergestellt, in welchem die Aktivität der Nitratreduktase bestimmt wird.
C. Reagenzien:
150 mM KN03-Lösung (100 ml) 50 mM KN03-Lösung (300 ml)
Extraktionsmedium (100 ml)
0.1 M Imidazol
29

1 mM EDTA Imidazol und EDTA lösen, mit 6 N HCl auf pH 7.8 einstellen
Kaliumphosphatpuffer (KPi), pH 7.6
0.1 M KH2PO4-Lösung 20 ml herstellen 0.1 M K2HPO4 -Lösung 100 ml herstellen, K2HPO4-Lösung in 250 ml Becherglas
gießen, mit Rührstäbchen auf den Magnetrührer stellen. Am pH-Meter durch Zugabe der KH2PO4-Lösung den pH auf 7.6 eingestellen
6 mM NADH-Lösung (erst unmittelbar vor dem Verbrauch ansetzen, bei 4°C aufbewahren)
NADH in 0.5 ml KPi, pH 7.6, lösen und 4.5 ml H2Odest
zugeben
Sulfanilsäure-Reagenz (in brauner Flasche lichtgeschützt aufbewahren. Vorsicht: Haut-berührung vermeiden, möglicherweise cancerogen!)
1 g Sulfanilsäure in 100 ml verdünnter HCl (75 ml H2Odest + 25 ml konz. HCl) lösen
NED-Reagenz (in brauner Flasche lichtgeschützt aufbewahren)
20 mg N-(1-Naphthyl)-ethylendiamin-hydrochlorid in 100 ml H2Odest lösen
40 µM NaNO2-Lösung (Nitritstandardlösung für die Eichkurve, 1 l)
D. Versuchsdurchführung:
1. Inkubation des Pflanzenmaterials:
Rettichsamen in feuchtem Vermiculit einsäen; im Brutschrank 2 Tage bei 25°C keimen
lassen, dann im Gewächshaus 3 Tage mit Zusatzlicht kultivieren. Beim Induktionsversuch
werden für jeden Ansatz 1.5 g Kotyledonen (ca. 50 Keimlinge) abgeschnitten und in 25 ml
einer 50 mM KNO3-Lösung in einem 100 ml Erlenmeyerkolben nach zweimaliger Vakuum-
Infiltration (Saugflasche, je 2 min) auf der Schüttelmaschine bei Zimmertemperatur mit
Zusatzbeleuchtung inkubiert.
Insgesamt werden 4 Ansätze (= 4 Erlenmeyerkolben) vorbereitet und nach 0, 1, 2 und 3 ½
Stunden Inkubationszeit werden die Kotyledonen aus je einem Kolben gesammelt und
daraus der Rohextrakt hergestellt. Hinweis: Mit der längsten Inkubationszeit zuerst beginnen!
2. Herstellung des Rohextraktes:
Die Kotyledonen werden mit demin. Wasser gewaschen (Teesieb), mit Filterpapier gut ab-
getrocknet und dann in einem Becherglas im Eis vorgekühlt. Die kalten Kotyledonen werden
in einer gekühlten Reibschale mit 5 ml kaltem Extraktionspuffer mit dem Pistill zu einem
homogenen Brei zerrieben. Dieser wird durch Gaze (vierfache Lage) oder Nylontuch gepreßt
(Einweghandschuhe) und dann 10 min bei 7 000 Upm in der Kühlzentrifuge zentrifugiert. Aus
dem Überstand (= Rohextrakt) wird dann die Aktivität der Nitratreduktase bestimmt.
30

3. Messen der Enzymaktivität:
Die Nitratreduktase katalysiert die Übertragung zweier Elektronen von NADH auf das Nitrat;
das dabei entstandene Nitrit wird mit einer spezifischen Farbreaktion nachgewiesen. Die
Reduktion des Nitrits zum NH4+ läuft unter den hier vorgebenen Testbedingungen nicht ab,
obwohl das Enzym Nitritreduktase im Rohextrakt vorkommt: An dieser Reaktion ist nämlich
Ferredoxin als Elektronendonator beteiligt; dieses liegt jedoch im angegebenen Testgemisch
nicht in reduzierter Form und genügender Konzentration vor.
Test auf Nitratreduktaseaktivität:
Folgende Lösungen in 15 ml Falcongefäße (oder Zentrifugengläser) pipettieren:
1.0 ml 100 mM KPi, pH 7.6
0.3 ml H2O
0.1 ml 150 mM KN03-Lösung
0.1 ml 6 mM NADH-Lösung
0.5 ml Rohextrakt bzw. denaturierter Rohextrakt (= Start der Reaktion), mischen!
20 min bei 30°C im Wasserbad inkubieren.
Für jede Enzymmessung ist ein Leerwert zu bestimmen, der in seiner Zusammensetzung
dem vorherigen Test entspricht, bei dem aber das Enzym im Rohextrakt zuvor durch 2 min
Erhitzen auf ca. 90°C inaktiviert wurde.
Nitritnachweis:
1.0 ml Sulfanilsäure-Reagenz und 1.0 ml NED-Reagenz zupipettieren und 15 min bei
Zimmertemperatur reagieren lassen. Danach 10 min zentrifugieren (4 000 Upm, Hereaus
Megafuge, braune Becher für Falcongefäße, rote Becher für Zentrifugengläser, jeweils inkl.
Gummiadaptoren) und anschließend im Spektralphotometer bei 538 nm die Extinktion gegen
den Leerwert bestimmen. Danach Lösungen im Sonderabfall (unter dem Abzug) entsorgen.
Aufstellung einer Eichkurve:
Proben von 0; 0.1; 0.2; 0.4; 0.6; 0.8, 1.0 und 2.0 ml der Nitritstandardlösung auf 2 ml mit
H2Odest auffüllen, mit je 1.0 ml Sulfanilsäure und 1.0 ml NED-Reagenz versetzen, gut
mischen und nach 15 min (direktes Sonnenlicht vermeiden) die Extinktion im
Spektralphotometer bei 538 nm gegen den Nullwert bestimmen. Die Extinktion ist als
Funktion der eingesetzten Nitritmenge auf Millimeterpapier aufzutragen.
E. Auswertung
1. Mit Hilfe der Eichkurve ist die in den Proben vorhandene Nitritmenge zu errechnen.
2. Die Aktivität der Nitratreduktase (µmol Nitrit gebildet pro Gramm Frischgewicht) als
Funktion der Inkubationszeit ist in ein Diagramm einzutragen.
31

Versuch V: Induktion der Synthese von -Amylase in den Aleuronzellen des
Gerstenkorns durch Gibberellin
A. Theorie:
Keimende Samen produzieren hydrolytische Enzyme zur Mobilisierung ihrer gespeicherten
Reservestoffe. Die de novo Synthese hydrolytischer Enzyme beruht bei der Gerste auf der
Wirkung von Gibberellin. Das Gerstenkorn (Karyopse) besteht aus dem stärkereichen
Endosperm, das von einer peripheren Aleuronschicht umgeben wird und an einer Seite das
Scutellum des Keimlings abgrenzt. Ein Modell einer Karyopse ist im Kurssaal aufgestellt
(siehe auch Abb. V.1 im Anhang). Mit der Wasseraufnahme bei der Samenquellung gibt der
Keimling Gibberelline (GA1 und GA3) ab, die dann in der Aleuronschicht die Synthese von -
Amylase (und weiteren hydrolytischen Enzymen) induzieren. Diese Enzyme werden in das
Endospermgewebe sezerniert und bauen dort die Reservekohlenhydrate ab. Die Abbau-
produkte werden von Scutellum resorbiert und dem wachsenden Keimling zugeführt.
Das vom Embryo ausgehende "Signal" kann in embryofreien Kornhälften (oder auch
isolierten Aleuronzellen) durch exogen zugesetzte Gibberellinsäure ersetzt werden. Damit
steht gleichzeitig ein spezifischer Nachweis-Test für Gibberellinsäuren zur Verfügung. Die
Synthese der -Amylase kann durch anaerobe Bedingungen, Dinitrophenol, durch
Hemmstoffe der DNA-Transkription (Actinomycin D), und durch Hemmung der Translation
(Cycloheximid) sowie durch Aminosäureantagonisten wie p-Fluorphenylalanin gehemmt
werden. Diese Ergebnisse lassen den Schluß zu, daß durch die Wirkung des Phytohormons
Gibberellinsäure eine de novo Synthese von hydrolytischen Enzymen über Gendere-
primierung induziert wird.
Literatur
Schopfer, P. und Brennicke, A. (1999) Pflanzenphysiologie, Springer
B. Aufgabe:
Es soll gezeigt werden, daß Gibberellin die Synthese von -Amylase und Maltase im
embryofreien Gerstenkornhälften induzieren kann.
C. Reagenzien:
Natriumhypochloritlösung (0.5%, 50 ml)
CaCl2-Lösung (0.2 M, 1 ml)
Chloramphenicol-Lösung (2 mg/10 ml)
Cycloheximid-Lösung (2 mg/10 ml)
32

Gibberellinsäurelösung (GA3)
10 mg GA3 mit 50 µl Ethanol in Eppendorf-Reaktionsgefäß lösen, 0.25 ml 0,1 N KOH
und 1.75 ml H2Odest zugeben. Von dieser Stammlösung die Verdünnungen herstellen.
Acetatpuffer, pH 4.8 (50 ml)
10 mM Na-Acetat-Lösung mit einigen Tropfen konz. Essigsäure am pH-Meter auf pH 4.8 einstellen
Pflanzenmaterial:
Ca. 100 Gerstenkörner (Hordeum vulgare) werden morgens in einem 100 ml Becherglas mit
30 ml einer 0.5% Natriumhypochlorit-Lösung 3 min desinfiziert. Nach gründlichem Waschen
der Körner mit Leitungswasser werden diese mit dem Spalt nach unten in einer mit feuchtem
Filterpapier ausgelegten und abgedeckten Petrischale im Brutschrank bei 25-27°C für 18 bis
24 h inkubiert.
D. Versuchsdurchführung
1. Inkubation der Gerstenkörner in Gibberellinsäure-Lösung:
Folgende Proben sind anzusetzen:
1-5: Embryofreie Kornhälften mit GA3 in unterschiedlichen Konzentrationen
6: Embryofreie Kornhälften ohne GA3
7: Embryofreie Kornhälften mit GA3 und Cycloheximid
8: Embryohaltige Kornhälften ohne GA3
Dazu sind in 8 Erlenmeyerkolben (50 ml) jeweils folgende Lösungen zu pipettieren:
1.7 ml Acetatpuffer, pH 4.8
0.1 ml CaCl2-Lösung (Amylase wird durch Ca2+ aktiviert)
0.1 ml Chloramphenicol-Lösung (Hemmung von Bakterienwachstum)
Folgende Zusätze werden einpipettiert:
1-5: Je 0.1 ml GA3-Lösung folgender Konzentrationen: 5; 5 x 10-1; 5 x 10-2; 5 x 10-3 und 5
x 10-4 mg/ml
6: 0.1 ml H2Odest
7: 0.1 ml GA3 (0.5 mg/ml) + 0.1 ml Cycloheximidlösung
8: 0.1 ml H2Odest
Die gequollenen Gerstenkörner (nur Körner verwenden, die die Bildung einer Keimwurzel
zeigen) werden mit einer Rasierklinge quer halbiert und die embryohaltigen bzw. embryo-
freien Kornhälften getrennt in je einer feuchten Kammer gesammelt. Vor der Inkubation
werden pro Ansatz 5 Kornhälften gewogen und die embryofreien in die Erlenmeyer-Kölbchen
1-7 und die embryohaltigen in das Kölbchen 8 gegeben. Alle Kölbchen werden verschlossen
und 24 h geschüttelt.
33

2. Enzymatische Glucosebestimmung durch den „UV-Test“:
Testprinzip:
HK1) Glucose + ATP Glucose-6-phosphat + ADP
G6P-DH
2) Glucose-6-phosphat + NADP+ Gluconat-6-phosphat + NADPH + H+
HK = Hexokinase G6P-DH = Glucose-6-phosphat-Dehydrogenase
Die während der Reaktion gebildete NADPH-Menge ist der Glucosemenge äquivalent, die
Konzentration von NADPH wird aufgrund der Absorption bei 340 nm fotometrisch bestimmt
(siehe Abbildung III.1, Seite 28).
Fertig-Reagenzien des „UV-Testkits“:
Lösung 1 enthält Puffersubstanz, NADP, ATP, und MgSO4
Suspension 2 enthält HK und G6P-DH
hergestellt entsprechend der ausliegenden Anleitung (stabil jeweils mindestens 4 Wochen bei Lagerung im Kühlschrank)
Nach der Inkubation der Kornhälften wird die von Hydrolasen freigesetzte Glucose
enzymatisch bestimmt. Dazu werden die Kornhälften aus der Lösung, die verworfen wird,
herausgenommen und in einer kleinen Reibschale mit 2 ml H2Odest und wenig Seesand
homogenisiert. Das Homogenat wird in ein Zentrifugenglas abgegossen und die Reibschale
mit 2 x 1 ml H2Odest nachgewaschen. Nach 5 min Zentrifugation (Tischzentrifuge, volle
Umdrehungszahl) wird mit H2Odest genau auf 6 ml aufgefüllt und gemischt.
Testansatz:
Leerwert
(1x)
Probe
(8x)
Lösung 1 1 .0 ml 1 .0 ml
Probelösung - 0 .8 ml
H20dest 2 .0 ml 1 .2 ml
Suspension 2 20 µl 20 µl
Leerwert und Probe(n) mischen, 3 min inkubieren und die Extinktion bei 340 nm gegen Luft
messen (E1). Suspension 2 zugeben, mischen, 15 min inkubieren und wiederum die
Extinktionen bestimmen (E2). Für Leerwert und Probe(n) die Extinktionsdifferenzen be-
34

stimmen und die Exktinktionsdifferenz des Leerwertes von der Extinktionsdifferenz der Probe
abziehen.
E = (E2 – E1)Probe – (E2 – E1)Leerwert
Berechnung:
V x MG c = x E [g/l]
x d x v
V = Testvolumen [ml] v = Probevolumen [ml] MG = Molekulargewicht der Glucose 180 bzw. 198 [g/mol] d = Schichtdicke [cm]
= Extinktionskoeffizient von NADPH bei 340 nm: 6.3 x 103 [l x mol-1 x cm-1]
E. Auswertung:
Die Beziehungen zwischen dem Gehalt an Glucose, ausgedrückt in µg Glucose pro mg
Frischgewicht, und der Gibberellinkonzentration ist graphisch darzustellen.
F. Anhang:
Abb. V.1: Schematische Darstellung einer Getreidekaryopse während der Keimung. Aus: Schopfer & Brennicke: Pflanzenphysiologie, 5. Aufl., Springer Verlag, 1999
35

Versuch VI: Phytochrom als Signalgeber der Flavonolakkumulation in Senfkeimlingen
A. Theorie:
Im belichteten Senfkeimling (Weißer Senf, Sinapis alba) produziert der Flavonoid-
Biosyntheseweg neben einer Reihe von Anthocyanen (alle mit Cyanidin als Aglycon) auch
Flavonole (mit Kaempferol und Quercetin als Aglyca). Diese Flavonoide sind in den
Kotyledonen auf die Epidermis beschränkt. Damit gehorcht die Photomorphogenese einem
räumlichen Kompetenzmuster, das zu einem entsprechenden räumlichen Differenzierungs-
muster führt. Die Intensität der Flavonoidsynthese ist mit der Menge der physiologisch
aktiven Form von Phytochrom (Pfr) korreliert, welche durch eine bestimmte Belichtung in den
kompetenten Geweben des Keimlings aus der inaktiven Form Pr erzeugt wird. Im Dunkeln
werden praktisch keine Flavonoide gebildet, was für die Messung kleiner Lichteffekte
besonders günstig ist. Zur experimentellen Induktion der phytochromabhängigen
Flavonoidsynthese wählt man häufig nicht induktive Lichtpulse mit hellrotem Licht, sondern
eine Dauer-Dunkelrot (DR)-Bestrahlung, welche die Hochintensitätsreaktion des Phyto-
chroms anregt. Unter diesen Bedingungen ist eine zwar niedrige, aber weitgehend
konstante, hochwirksame Menge an Pfr aktiv, welche eine konstante Stimulation der
Photomorphogenese liefert. Man erzeugt also, im Gegensatz zur Pulsbestrahlung, stationäre
Induktionsbedingungen; der zeitliche Verlauf der photomorphogenetischen Prozesse wird
daher nicht durch Veränderungen der Pfr-Menge kompliziert.
Literatur
Schopfer, P. und Brennicke, A. (1999) Pflanzenphysiologie, Springer.
Schopfer, P. (1989) Experimentelle Pflanzenphysiologie. Einführung in die Anwendungen, Springer.
B. Aufgabe:
Es soll in den Kotyledonen von Senfkeimlingen die Induktion von Flavonolen durch
Phytochrom quantitativ untersucht werden.
C. Reagenzien:
Extraktions- und Hydrolysemedium: 2 N HCl
Ethylacetat (Essigsäureethylester)
NH3-Lösung (konz.)
Boratpuffer (0.1 M an Borat, pH 8.9), wird gestellt
Methanol (70 Vol.%)
Laufmittel: Eisessig (50 Vol.%, 100 ml)
Cellulose-Dünnschichtplatten
Quercetin (Fa. Roth), eine Standardlösung wird gestellt
36

2
3
O1
46
5
78
4´
5´
3´
6´
2´
1´
CA
B
Flavan
CA
B
R = H, Kaempferol
R = OH, Quercetin
R
OH
OH
OOH
OH O
CA
B
Cyanidin
C
O
O
H
H H
Flavanon
C
O
O
H
H H
Flavon
C
O
OH
H
H H
Chalkon
C
Isoflavon
H
O
O
H
H
C
Flavonol
OH
O
O
H
H H
C
Anthocyanidin
OH
O+
H
H H
OH
OH
OH
OH
OH O+
Abb. VI.1: Untergruppen der Flavonoide und ihre Ableitung vom Flavan-Grundgerüst. Kaempferol, Quercetin und Cyanidin sind Inhaltsstoffe von Senf.
D. Versuchsdurchführung:
Die Anzucht der Senfkeimlinge (je 6 g Samen) erfolgt im Dunkeln auf mehreren Lagen
feuchtem Filterpapier in einer Kunststoffdose. Die Keimlinge werden entweder 98 h im
Dunkeln bzw. 68 h im Dunkeln und 30 h im DR gehalten. (Aussaat mittags, DR-Beleuchtung
morgens am 3. Tag nach Aussaat für 30 h -> Ernte der Kotyledonen mittags am 4. Tag nach
Aussaat, Einfrieren in fl. N2, Lagern bei –80°C)
1. Extraktion und Hydrolyse
Die Flavonoide werden aus jeweils 1 g Kotyledonen von Dunkel- bzw. DR-Keimlingen in
einem Schraubdeckelröhrchen (10 ml, Pyrex) mit 4 ml HCl-Lösung (2 N) 60 min lang bei
100°C im Wasserbad extrahiert und gleichzeitig hydrolysiert. Nach dem Abkühlen wird der
Extrakt 10 min zentrifugiert (Heraeus-Megafuge, Rotor 2704, rote Becher), der Überstand mit
einer Pasteurpipette abgesaugt und in einem neuen Pyrexröhrchen mit 1 ml Ethylacetat
kräftig geschüttelt. Nach kurzer Zentrifugation trennt sich eine leicht gefärbte Oberphase von
einer roten Unterphase ab. Die Oberphase wird in ein 15 ml Falcongefäß überführt und die
verbleibende Unterphase nochmals mit 1 ml Ethylacetat extrahiert. Die Oberphasen werden
37

vereinigt, durch Abblasen mit N2 auf ca. 200 bis 300 µl eingeengt und können üN im
Kühlschrank aufbewahrt werden.
2. Chromatographie
Die eingeengten Oberphasen beider Proben werden mit einer Glaskapillare portionsweise
als 4 cm breites Band auf die Auftragslinie einer Cellulose-Dünnschichtplatte aufgetragen.
Daneben werden zweimal je 1 µg Quercetin als Vergleichssubstanz (s. E) aufgetragen. Die
Chromatographie erfolgt mit dem unter C. genannten Laufmittel. Wenn die Front etwa 12 cm
weit gewandert ist (nach 5 - 6 h), wird die Chromatographie abgebrochen und die Laufmittel-
front markiert. Die Platte wird im Abzug üN zum Trocknen stehen gelassen.
Die unter UV-Licht hellgelbe Bande bei Rf = 0.1 - 0.2 (die im NH3-Dampf deutlicher sichtbar
wird) enthält die unter diesen Bedingungen gemeinsam laufenden Flavonole Quercetin und
Kaempferol. Außerdem treten bei größeren Rf -Werten einige gefärbte oder unter UV-Licht
sichtbare Banden auf, die auf verschiedene Zimtsäurederivate, Anthocyanidine und andere
Verbindungen zurückgehen. Die Umrisse der Substanzflecken werden unter UV-Licht mit
einem weichen Bleistift nachgezeichnet und können anschließend durchgepaust werden.
3. Messung
Die Celluloseschichten mit den Flavonolbanden der Kotyledonenextrakte und der
Vergleichssubstanz werden getrennt mit einem Spatel herausgekratzt, das Pulver jeweils in
ein 2 ml-Mikroreaktionsgefäß überführt, mit 1 ml 70 Vol.% Methanol versetzt, durch Schütteln
extrahiert und der Extrakt durch Zentrifugieren geklärt. Der Überstand wird in eine
Quarzküvette gefüllt, das Absorptionsspektrum (220 - 450 nm) registriert und die Extinktion
bei = 370 nm abgelesen. Nach Zugabe von 5 µl alkalischem Boratpuffer tritt eine
charakteristische Verschiebung des langwelligen Absorptionsgipfels ( 370 nm) um ungefähr
15 nm zu längeren Wellenlängen ein (bathochrome Verschiebung). Die Extinktion im
Scheitelpunkt des Gipfels (vor oder nach Boratbehandlung) kann als relatives Maß für die
Flavonoidkonzentration im Extrakt verwendet werden.
E. Quantitative Auswertung:
Nach dem LAMBERT-BEERschen Gesetz ist die Extinktion (E) einer gelösten Substanz
direkt proportional zur Konzentration der Substanz (c) bei konstanter Schichtdicke (d) der
Probenlösung. Es gilt: E = c d.
Der Extinktionskoeffizient ( , Einheit: M-1 cm-1) hängt von der Lichtqualität ab. Er wird daher
stets für eine bestimmte Wellenlänge (monochromatisches Licht) angegeben und ist in
dieser Form ( ) eine wichtige Stoffkonstante des Pigments, welche dessen „Absorptions-
stärke“ bei der Wellenlänge charakterisiert.
38

Der molare Extinktionskoeffizient bei = 370 nm für Quercetin soll ermittelt werden. Mit Hilfe
dieses Wertes soll die Menge des unter D.2/3 isolierten Quercetins berechnet werden. Es
wird eine Eichkurve zur quantitativen Bestimmung von Quercetin bei 370 nm in 70%
Methanol erstellt. Dazu wird eine Standardlösung mit einer genau bekannten Menge von 2 -
3 mg Quercetin in 100 ml 70 Vol.% Methanol verwendet. Mit dieser Stammlösung werden
schrittweise Verdünnungen eingestellt und von allen Lösungen die Extinktionen in
Quarzküvetten bei 370 nm gemessen. Der Extinktionskoeffizient 370 wird aus der Eichkurve
graphisch ermittelt.
Unter Verwendung des 370 und der unter D.3 ermittelten Extinktion bei 370 nm wird die
Menge des Quercetins pro g Frischgewicht berechnet.
39

Versuch VII: Verzögerung der Seneszenz abgeschnittener Haferblätter durch Kinetin
A. Theorie:
Cytokinine sind Phytohormone, die in pflanzlichen Geweben in verschiedene physiologische
Prozesse eingreifen. Vor allem stimulieren sie die Zellteilungsaktivität und verzögern die
Seneszenz (Alterung) der Gewebe; daneben beeinflussen sie noch Differenzierungs-
vorgänge und die Samenkeimung.
Beim Altern pflanzlicher Gewebe sind eine Vielzahl verschiedenster katabolischer Stoff-
wechselprozesse beteiligt. Ein deutliches Symptom der Seneszenz grüner Gewebe (z.B der
Blätter) ist das Vergilben (Abbau von Chlorophyll). Dieser Abbau und andere Alterungs-
vorgänge können durch eine Applikation von Kinetin (6-Furfurylaminopurin) hinausgezögert
werden.
Literatur
Matile, P. und Kräutler, B. (1995) Wie und warum bauen Pflanzen das Chlorophyll ab? Chemie in unserer Zeit 29, 298-306
B. Aufgabe:
Die im Lebenslauf von Blättern natürlich eintretende Seneszenz kann künstlich durch
schlechte Ernährungsbedingungen ausgelöst werden. Schneidet man also die Blätter von
einer Pflanze ab und hält sie im Dunkeln, so tritt Chlorophyllabbau ein. Dieser Abbau und
seine Verzögerung durch Kinetin soll gezeigt werden.
C. Reagenzien:
Citrat-Phosphatpuffer, pH 4.7
0.25 g Citronensäure und 0.46 g Na2HPO4 x 2 H2O in 200 ml H2Odest lösen, diese Lösung sollte einen pH-
Wert von 4.7 aufweisen. Der pH-Wert kann durch Zugabe von etwas Citronensäure bzw. Phosphat korrigiert werden
0.5 ml Tween 80 (Detergenz) hinzupipettieren und im Meßzylinder auf 250 ml auffüllen
Kinetinlösung
10 mg Kinetin (= 6-Furfurylaminopurin) in einigen Tropfen 1 N NaOH in einem graduierten Reagenzglas in Lösung bringen, auf 10 ml mit demin. Wasser verdünnen und davon 1 ml im Meßkolben mit Citrat-Phosphatpuffer auf 100 ml auffüllen; dies ergibt eine Endkonzentration von 10 µg/ml Kinetin.
Pflanzenmaterial:
Hafer (Avena sativa) wird in Schalen in Erde ausgesät und im Gewächshaus bis zur
Entwicklung von Primärblättern von ca. 12 cm Länge herangezogen (etwa 1 Woche).
40

D. Versuchsdurchführung:
Möglichst gleich große Blättchen werden ca. 6 cm unter der Spitze mit einer scharfen Rasier-
klinge abgeschnitten, auf einer mit Millimeterpapier unterlegten Glasscheibe auf genau 5 cm
gekürzt und in einer feuchten Kammer auf einer Plastikleiste aufbewahrt. Die feuchte
Kammer stellt man sich durch Ausschlagen einer Plastikdose mit wassergetränktem
Filterpapier her. Die Wassersättigung ist täglich zu überprüfen!
Auf 25 Blättchen wird Kinetinlösung aufgesprüht. Weitere 25 Blättchen erhalten lediglich
Pufferlösung (Wasserkontrollen). Je 5 Blättchen werden sofort zur Extraktion von Chlorophyll
herangezogen.
Die feuchte Kammer wird verschlossen und im Dunklen bei 25°C gehalten. Nach 24, 48, 72
und 96 h werden von Kontrollen und Proben jeweils 5 Blättchen entnommen und deren
Chlorophyllgehalt bestimmt. Die Entnahme hat möglichst schnell zu geschehen, damit die
restlichen Blättchen nicht zu lange dem Licht ausgesetzt werden.
Zur Bestimmung des Chlorophyllgehalts 5 Blättchen auf der Analysenwaage wiegen. Mit
einer Schere in möglichst kleine Stückchen zerschneiden und in einer kleinen Reibschale mit
2 ml 80% Aceton und etwas Sand zu einem homogenen Brei verreiben; dann weitere 3 ml
80% Aceton zugeben. Den Brei quantitativ in ein Zentrifugenglas überführen und mit 1-2 ml
80% Aceton nachspülen (die Reibschale darf nicht mehr grün sein). In der Tischzentrifuge
bei voller Drehzahl für 5 min zentrifugieren; den Überstand in ein graduiertes Zentrifugenglas
dekantieren. (Achtung, direktes Sonnenlicht vermeiden, da Chlorophyll zerstört wird). Sofern
der Rückstand noch grün ist, diesen erneut in 1-2 ml 80% Aceton aufnehmen, am Whirlmix
gut durchmischen und erneut zentrifugieren. Die vereinigten Überstände im graduierten
Zentrifugenglas auf 10 ml mit 80% Aceton auffüllen, mischen und sofort im Spektralfotometer
die Extinktion bei 663 nm und 645 nm gegen 80% Aceton bestimmen (Glasküvetten!).
E. Auswertung:
Berechnen Sie den Gehalt von Chlorophyll a bzw. b und a+b pro g Frischgewicht und tragen
Sie die Werte in einem Diagramm als Funktion der Zeit auf.
Chlorophyll a (µg/ml) = 12,7 x E663 - 2,69 x E645
Chlorophyll b (µg/ml) = 22,9 x E645 - 4,8 x E663
41

Versuch VIII: Reversible Entfaltung eines Proteins am Beispiel des Phycocyanin
A. Einführung:
Proteine sind lineare, unverzweigte Makromoleküle. Im nativen Zustand nehmen sie eine
sehr komplexe, wohldefinierte dreidimensionale Struktur an, die durch vielfache schwache
Wechselwirkungen (u.a. H+-, Salz-, Disulfid-Brücken, hydrophobe Interaktionen) bestimmt
werden. Diese dreidimensionale Struktur ist beweglich aber doch so regelmäßig, dass man
Proteine kristallisieren und ihre Struktur durch Röntgendiffraktion bestimmen kann. Eine
Sammlung solcher Strukturen ist in der Protein-Datenbank (http://www.rcsb.org/pdb/) zu
finden. Ein einfaches Programm zum Betrachten kann man bei der Expasy Datenbank
herunterladen (http://us.expasy.org/spdbv/text/download.htm).
In der Regel steckt die gesamte Information für die dreidimensionale Struktur (Sekundär- und
Tertiärstruktur) eines Proteins in seiner Sequenz (Primärstruktur). Viele Proteine falten sich
spontan nach oder bereits während der Translation. Aber auch wenn Chaperone benötigt
werden, so haben diese vor allem eine Hilfsfunktion, indem sie falsch gefaltete Proteine
wieder partiell entfalten und ihnen damit eine weitere „Chance“ geben. Spontan faltende
Proteine können häufig auch in vitro reversibel durch Zugabe von geeigneten
Denaturierungsmitteln entfaltet werden, und finden dann nach Entfernen derselben innerhalb
von Sekunden wieder in den nativen Zustand zurück. Als Denaturierungsmittel eignen sich
Detergenzien und sog. chaotrope Substanzen, welche die Wasserstruktur stören und vor
allem Salz- und Wasserstoffbrücken aufbrechen, aber auch erhöhte Temperaturen.
Insbesondere wurden Harnstoff und Guanidinium-Hydrochlorid für viele reversible
Entfaltungsstudien verwendet, da sie Proteine nicht irreversibel modifizieren und leicht
wieder durch Dialyse oder Verdünnen entfernt werden können.
Die Regeln für die Faltung sind bisher nur ansatzweise verstanden. Es ist beispielsweise
bisher auch unter Einsatz von Höchstleistungsrechnern nicht möglich, aus der Sequenz
eines Proteins seine dreidimensionale Struktur vorherzusagen. Die reversible Faltung geeig-
neter Modellproteine wird deshalb statisch und zeitaufgelöst intensiv untersucht. Dabei kann
der Faltungszustand durch eine Vielzahl physiko-chemischer und biochemischer Parameter
verfolgt werden. Bei Enzymen kann man die Aktivität messen, man kann die Sekundär- und
Tertiärstruktur mittels Circulardichroismus oder Infrarotspektroskopie bestimmen, oder über
die Fluoreszenz von Tryptophan Aufschlüsse über die Hydrophobizität seiner Umgebung
erhalten. Man kann auch zusätzliche Sonden in das Protein einbringen, z.B. Farbstoffe
welche ihre Fluoreszenz bei der Faltung verändern, weil sie dabei protoniert werden oder in
eine spezielle Umgebung im Inneren des Proteins gelangen.
Es gibt auch Proteine, welche bereits durch natürliche post-translationale Modifikationen mit
solchen Sonden versehen sind. Ein Beispiel dafür sind die Phycobiliproteine, photosyntheti-
sche Antennenproteine aus Cyanobakterien, Rotalgen und Cryptophyceen, welche kovalent
42

mit linearen Tetrapyrrol-Chromophoren verknüpft sind, die im Bereich der „Grünlücke“ des
Chlorophylls absorbieren. Die Absorption, Fluoreszenz und optische Aktivität dieser Chromo-
phore ändern sich drastisch, wenn das Protein entfaltet wird: Die Absorption nimmt bei der
Renaturierung um einen Faktor fünf ab, die Fluoreszenz sogar auf ein Zehntausendstel.
Zudem ist diese Entfaltung reversibel, wobei die nativen Eigenschaften der Chromophore
vollständig wiederhergestellt werden können. Man kann daher über die spektroskopischen
Eigenschaften der Chromophore die De- und Renaturierung sehr gut verfolgen, z.T. bereits
mit bloßem Auge.
Literatur
Kyte, J. (1995) Structure in Protein Chemistry, Chapter 13, Garland
Laemmli, U.K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680 - 685
B. Aufgabe:
Im vorliegenden Versuch wird zunächst das Biliprotein Phycocyanin (PC) aus dem
sprühgetrockneten Cyanobakterium Spirulina geitleri isoliert. Die Reinigung soll durch eine
SDS-Gelelektrophorese dokumentiert werden. Die stufenweise reversible Entfaltung mit
Harnstoff wird mittels Absorptionsspektroskopie verfolgt, die vollständige Entfaltung und
Rückfaltung mittels Absorption und Fluoreszenz. Außerdem kann auch noch die Entfaltung
mit anderen Reagenzien und durch Hitze untersucht werden.
In einem weiteren Versuchsteil soll die dreidimensionale Struktur des PC visualisiert werden.
Die Strukturdaten mehrerer PC sind in der Protein-Datenbank zu finden. Sie soll mit dem
Swiss-Protein Viewer so aufbereitet werden, dass die Tertiärstruktur eines Monomers und
die Lage und Konformation der Chromophore gut zu sehen sind.
C. Reagenzien:
1 M Kaliumphosphatpuffer (KPi) pH 7.3: 1.97 g KH2PO4 und 10.27 g K2HPO4 x 3 H2O oder 7.84 g K2HPO4 in 50 ml H2O lösen.
daraus Puffer mit den folgenden Ionenstärken herstellen: KPi200 200 mM 50 ml KPi80 80 mM 50 ml KPi50 50 mM 100 ml KPi10 10 mM 200 ml KPi5 5 mM 3 l
D. Versuchsdurchführung:
1. Isolierung von Phycocyanin
Allgemeines: Biliproteine sind lichtempfindlich, insbesondere im denaturierten Zustand. Wie
alle Proteine müssen sie gekühlt aufbewahrt werden. Deshalb immer im Dämmer- oder
Grünlicht und weitgehend auf Eis arbeiten. Der pH-Wert der Puffer soll bei 7.3 liegen.
43

Isolierungsprinzip: Nach der Extraktion von S. geitleri befinden sich die Biliproteine im
Überstand. Mit Hilfe einer fraktionierten Ammoniumsulfatfällung und einer Ionenaustausch-
Chromatographie wird aus dem Rohextrakt C-Phycocyanin aufgereinigt.
Zellaufschluss
3 g des sprühgetrockneten Spirulina Pulvers in 40 ml KPi50 aufnehmen und 15 min auf Eis
rühren. Anschließend für 30 min mit ca. 30 000 x g (Kühlzentrifuge, SS34-Rotor, 18 000
Upm) abzentrifugieren. Den Überstand auf Eis lagern, das Pellet nochmals mit 10 ml KPi50
extrahieren, abzentrifugieren, die kräftig blaugrünen Überstände vereinigen und eine Probe
von 100 µl für die gelelektrophoretischen Analyse abzweigen.
Fraktionierte Ammoniumsulfatfällung (ASF) (auf Eis arbeiten)
a. Volumen des Überstandes abmessen, für eine 30%-Fällung benötigte Ammonium-
sulfat-Menge (176 g/l, siehe Tabelle VIII.1) abwiegen.
b. Überstand auf Eis auf dem Magnetrührer vorsichtig rühren, alle 2 min eine kräftige
Spatelspitze des abgewogenen Ammoniumsulfats zugeben, danach noch 30 min
weiterrühren.
c. 30 min zentrifugieren (18 000 Upm), mit dem Überstand (Ü30) weiterarbeiten.
d. Volumen des Überstandes Ü30 bestimmen. Für eine 30% 70% Fällung benötigte
Ammoniumsulfat-Menge (273 g/l, siehe Tabelle) wie bei der vorangehenden Fällung
unter Rühren auf Eis portionsweise zugeben, danach noch 30 min rühren.
e. 30 min abzentrifugieren (18 000 Upm), jetzt mit dem Pellet (P70) weiterarbeiten.
Dialyse
a. 15 cm Dialyseschlauch (Servapor, 16 mm Durchmesser) in H2Odest legen, bis er
weich ist (mindestens 15 min).
b. Pellet vorsichtig (mit dem Potter oder einem Glasstab) in möglichst wenig KPi5 (ca. 5
ml) aufnehmen.
c. Eine Seite des Dialyseschlauches verknoten und Probe einfüllen.
d. Den Dialyseschlauch am anderen Ende so zuknoten, dass etwa 1 bis 2 cm ungefüllt
bleiben.
e. Schlauch in ein Becherglas mit 1 l KPi5 geben, am oberen Rand fixieren und unter
Rühren dialysieren (vor Licht geschützt, im Kühlschrank). Nach 3 h Puffer wechseln,
dann über Nacht weiter dialysieren. Vor der Chromatographie eine Probe für die
Gelelektrophorese entnehmen (100 µl).
Ionenaustauschchromatographie
Das Phycocyanin wird mittels einer Anionenaustausch-Chromatographie aufgereinigt. Dazu
wird zunächst das Material mit Puffer niedriger Ionenstärke äquilibriert, entgast, und eine
44

Säule gegossen. Nach dem Auftragen werden die verschiedenen Proteine mit einem
Gradienten von 5-200 mM KPi von der Säule eluiert. Die verschiedenen Proteine lösen sich
je nach ihrem isoelektrischem Punkt (pI) und Ladung bei einer anderen Ionenkonzentration
von der Säule und werden so fraktioniert. Die gefärbten Proteine werden in folgender
Reihenfolge eluiert: Cytochrom, Allophycocyanin (APC), PC, Chlorophyll-bindende Proteine
und entsprechend ihren Absorptionen zusammengefasst.
Säulenvorbereitung
a. 50 ml einer Suspension regenerierte DEAE-Cellulose (DE-52, Whatmann) in eine
Waschflasche überführen, absetzen lassen und den Überstand abgießen.
b. Säulenmaterial in so viel KPi5 aufnehmen, daß eine gut flüssige Suspension entsteht.
c. DEAE-Cellulose-Suspension mit einer Membran- oder Wasserstrahlpumpe
evakuieren. Vorsicht! Das Material schäumt leicht, in diesem Fall den Stopfen lupfen,
nie die Pumpe abstellen! Zum Entgasen immer Schutzbrille tragen!
d. 0.25 l KPi5 entgasen.
e. Glassäule vorbereiten: Glaswolle zum Verschließen mit einem Glasstab reinstopfen.
f. Das entgaste Säulenmaterial durch leichtes Umschütteln gleichmäßig suspendieren,
vorsichtig ohne Luftblasen (am Glasstab einlaufen lassen) in die Säule geben, und 15
min gut absitzen lassen.
g. Den Hahn öffnen, so dass 1 bis 2 ml/min abfließen.
h. Wenn die Flüssigkeit bis auf wenig mm über dem DEAE-Cellulose-Bett durchgelaufen
ist, vorsichtig wieder mit dem entgasten Puffer auffüllen, ohne dabei das
Säulenmaterial aufzuwirbeln. Achtung: Die Säule darf nie trocken laufen, es sollen
immer ca. 2-4 mm Puffer über dem Säulenmaterial stehen, und es dürfen keine
Luftblasen eingebracht werden. Diesen Vorgang so lange wiederholen, bis das 10-
fache des Säulenvolumens durchgelaufen ist.
Chromatographie
a. Den überstehenden Puffer bis auf wenige mm Überstand ablaufen lassen. Die
Betthöhe mit einem Filzschreiber markieren.
b. Probe vorsichtig auf die Säule auftragen, bis auf wenige mm Überstand ablaufen
lassen. Ab jetzt soll die Säule abgedunkelt werden!
c. Mit 5 ml Portionen Puffer (KPi5) auffüllen und wieder bis auf wenige mm Überstand
ablaufen lassen; diesen Vorgang ggf. wiederholen bis der Überstand farblos ist.
d. Nacheinander mit je 50 ml der Puffer KPi5, KPi80 und KPi200 spülen. Den Durchlauf ab
der Zugabe von KPi80 mit Hilfe eines Fraktionskollektors auffangen. Dazu je 50
Tropfen (entspricht ca. 2.5 ml) pro Fraktion sammeln. Achtung: für die elektro-
phoretische Analyse auch schon vom Durchlauf mit KPi5 eine Probe nehmen.
45

Die Auftrennung der Proteine dauert mehrere Stunden. Alle Proben auf Eis oder im
Kühlschrank aufbewahren und möglichst schnell vereinigen (s.u.).
Farblose Fraktionen können verworfen werden. Bei den gefärbten kann bereits durch die
farblichen Unterschiede (PC: blau, APC: türkis) mit dem bloßen Auge eine grobe Zuordnung
der einzelnen Fraktionen zu bestimmten Pigmenten erfolgen. An den farblichen Übergängen
zwischen den Fraktionen müssen Spektren gemessen werden, in der Regel jede dritte,
außer wenn große Sprünge auftreten. An Hand der Spektren (PC: max 605 – 620 nm, APC:
max = 640 – 655 nm) die zusammengehörenden Fraktionen zusammen fassen, und davon
jeweils ein abschließendes Spektrum messen. Davor allerdings von den interessanten
Fraktionen Proben für die Elektrophorese entnehmen, es können sechs verschiedene
ausgewählt werden.
Die zusammengefasste PC-Fraktion mit 70% Ammoniumsulfat fällen (472 g Ammonium-
sulfat/l, siehe Tabelle, Vorgehensweise siehe oben), die gefällten Proben als Suspensionen
im Kühlschrank aufbewahren.
2. Entfaltung mit Harnstoff
a. Eine Stammlösung (max. 5 ml) von PC mit einer Absorption von 10 cm-1 im roten
Maximum ( max 614 - 616 nm) wird in KPi10 angesetzt und dunkel aufbewahrt, da
der Chromophor im entfalteten Zustand leicht durch Licht ausbleicht.
b. Im selben Puffer wird eine Harnstofflösung (8 M) angesetzt. Dazu geben Sie zu
19.2 g festen Harnstoff 25 ml KPi10 und rühren magnetisch bis zur vollständigen
Lösung. Dies ergibt 40 ml der 8 M Harnstofflösung.
c. Aliquote (0.15 ml) der PC-Stammlösung werden 1:8 so mit geeigneten Mengen der
Harnstofflösung und des Puffers verdünnt, daß die Endlösungen 0, 1, 2, 3, 4, 5, 6
und 7 M Harnstoff enthalten. Stellen Sie dazu vor Versuchsbeginn ein Pipettier-
schema auf und besprechen es vor der Ausführung mit dem Assistenten. Wie können
Sie eine Lösung der gleichen PC-Konzentration erzeugen, die 8 M an Harnstoff ist?
Bereiten Sie auch diese Probe vor, und bewahren Sie alle Proben bis zur Messung
im Dunklen auf.
d. Messen Sie genau 15 min nach Zugabe des Harnstoffs die Absorptionsspektren im
Bereich 250 - 800 nm. Referenz = KPi10 (Harnstoff hat keine Absorption im unter-
suchten Spektralbereich).
e. Messen Sie anschließend die Fluoreszenz-Intensität (Emissionsspektrum 580 - 680
nm) bei Anregung im Maximum im nahen UV (350 nm). Beginnen Sie dabei mit der
nativen Probe (0 M Harnstoff). Das Spektralfluorimeter wird so eingestellt, daß es
80% Vollausschlag zeigt. Diese Parameter werden bei den folgenden Messungen
nicht mehr verändert. Wenn kein Spektralfluorimeter zur Verfügung steht kann die
46

Fluoreszenz durch Bestrahlung mit der UV-Handlampe (366 nm) oder dem UV-
Transilluminator angeregt und dokumentiert werden.
3. Rückfaltung
a. Geben Sie zu 1.25 ml der PC-Stammlösung (2a) 0.96 g Harnstoff und lösen ihn
vollständig. Notieren Sie das Endvolumen der Lösung, und berechnen sie daraus die
Molarität des Harnstoffs.
b. Aliquote (0.15 ml) der so bereiteten Stammlösung von denaturiertem PC werden 1:8
so mit der 8 M Harnstofflösung und Puffer verdünnt, daß die Endlösungen 1, 2, 3, 4,
5, 6 und 7 M an Harnstoff sind. Stellen Sie dazu ein Pipettierschema auf und
besprechen es vor der Ausführung mit dem Assistenten.
c. Messen Sie wie bei der Denaturierung die Absorption und Fluoreszenz der Proben.
4. Geschwindigkeit der Rückfaltung
a. Geben Sie 50 ml destilliertes Wasser in ein Becherglas und rühren Sie es
magnetisch. Beleuchten Sie das Glas in einem abgedunkelten Raum von der Seite
mit der UV-Lampe.
b. Pipettieren Sie 0.5 ml der denaturierten Stammlösung aus dem vorangehenden
Versuch in das gerührte Wasser, spritzen Sie dabei die gesamte Menge so schnell
wie möglich ein. Was passiert?
5. Denaturierungsversuche mit anderen Agenzien
Bei den folgenden Versuchen wird PC mit verschiedenen anderen Denaturierungsagenzien
behandelt. Ausgangspunkt ist dabei jeweils 0.15 ml der Stammlösung (2a) ggf. verdünnt mit
KPi10.
a. Wärme: Inkubieren Sie jeweils eine Probe bei 40 und 70oC im Wasserbad und
schätzen Sie visuell die Absorption und die Fluoreszenz. Wie kann man den Versuch
im Photometer machen?
b. Titrieren Sie eine weitere Probe mit einer 2%igen Lösung von SDS in Wasser, und
verfolgen die Reaktion im Photometer.
c. Versuchen Sie andere Denaturierungsagenzien wie Zn-Acetat, Ethanol, Dimethyl-
sulfoxid, Pril, etc.
6. Gelelektrophoretische Analyse der Proteinfraktionen
Mit Elektrophorese wird die Wanderung geladener Teilchen im elektrischen Feld bezeichnet.
Für die Trennung komplexer Proteingemische wird meist das Verfahren der Polyacrylamid-
Gelelektrophorese (PAGE) unter entweder nicht denaturierenden oder denaturierenden,
kontinuierlichen oder diskontinuierlichen Bedingungen verwendet. Bei der denaturierenden,
47

diskontinuierlichen PAGE nach Laemmli (1970) werden die zu trennenden Proteine mit dem
Detergenz SDS (Natriumdodecylsulfat) in Gegenwart reduzierender Agenzien wie ß-
Mercaptoethanol oder DTT (Dithiotreitol) denaturiert. Die diskontinuierliche PAGE benutzt
zwei Phasen, das niedrig konzentrierte Sammelgel und das höher konzentrierte Trenngel,
und zwei Puffersysteme, die sich in der Ionenzusammensetzung und dem pH-Wert unter-
scheiden. In einem begrenzten Bereich des Trenngels ist die Wanderungsstrecke des
Proteinmoleküls umgekehrt proportional zum Logarithmus seines Molekulargewichtes. SDS-
PAGE kann daher in diesem Bereich zu Molekulargewichtsbestimmungen herangezogen
werden. Der Nachweis der aufgetrennten Proteine kann in den Gelen selbst durch
verschiedene Färbereaktionen erfolgen, oder nach dem Transfer auf eine Membran
(„Westernblotting“) anhand der spezifischen Wechselwirkung mit Antikörpern.
Zur Herstellung von Polyacrylamidgelen werden Lösungen der Monomere Acrylamid und N,
N‘-Methylenbisacrylamid (Quervernetzer) in Gegenwart der Katalysatoren TEMED (N, N, N‘,
N‘-Tetramethylethylendiamin) und Ammoniumperoxodisulfat (APS) als Starter polymerisiert.
Die Polymerisation verläuft radikalisch. Acrylamid und Bis-Acrylamid sind toxisch (Haut-und
Nervengift). Daher ist der Kontakt mit den Substanzen unbedingt zu vermeiden!
Pipettierschema (für 2 Minigele, 0.75 mm dick):
Trenngel (15%) Sammelgel (5%) Volumen 10 ml 5 ml
H2O 5. 00 ml 3. 125 ml 3 M Tris/HCl, pH 8.8 1. 25 ml - 0.5 M Tris/HCl, pH 6.8 - 1. 25 ml 40% Acrylamid 3. 75 ml 0. 625 ml 10% SDS 100 µl 50 µl 10% APS 50 µl 25 µl
TEMED 5 µl 5 µl
Von den Proteinproben werden je 15 µl mit 5 µl des 4-fach konzentrierten Proteinproben-
puffers versetzt, 5 min gekocht und nach dem Abkühlen auf das Gel aufgetragen. Ein
Protein-Größenmarker wird parallel behandelt und aufgetragen. Die Gele laufen für 30 min
bei 50 Volt und ca. 1.25 h bei 150 Volt.
Für die Analyse der PC-Proteinfraktionen können drei verschiedene Nachweismethoden
angewendet werden: der in situ-Nachweis der Chromoproteine anhand der Eigenfarbe, der
Nachweis der Fluoreszenz der Biliproteine durch Behandlung mit Zinkacetat, sowie der
unspezifische Coomassieblau-Nachweis aller Proteine. Dafür werden die Gele für 10 bis 30
min in der Zinkacetatlösung geschwenkt und ggf. mit H20dest gewaschen (10 min). Durch
Bestrahlung mit 365 nm wird die Fluoreszenz der Biliproteine angeregt. Nach der
Dokumentation werden die Gele zwei- bis dreimal mit H20dest gewaschen und über Nacht in
der Coomassie-Färbelösung geschüttelt. Diese Gele müssen nicht entfärbt werden und
werden vor dem Trocknen nur kurz in Wasser geschwenkt
48

Proteinprobenpuffer (4-fach, wird gestellt)
625 mM Tris/HCl, pH 6.8 20% Glycerin 5% ß-Mercaptoethanol 10% SDS 3 mg/ml Bromphenolblau
Laufpuffer (10-fach, wird gestellt)
30 g Tris 144 g Glycin 10 g SDS (pH-Wert wird nicht eingestellt, beträgt etwa 8.3)
Zinkacetatlösung
1.3 M Zinkacetat
Kolloidale Coomassie-Färbelösung
0.1 g Coomassie Brilliant Blue G250 in 5 ml H2Odest lösen
10 g Ammoniumsulfat und 2 g H3PO4 (85%) in 80 ml H2Odest lösen
beide Lösungen mischen und auf 100 ml auffüllen
Zum Anfärben der Gele vier Volumen dieser Färbelösung mit einem Volumen Methanol mischen.
7. Visualisierung der Kristallstruktur eines C-Phycocyanins
Laden Sie sich im CIP-Raum die Röntgenstrukturdaten eines C-Phycocyanins von der
Protein-Struktur-Datenbank (http://www.rcsb.org/pdb/) aus dem Internet herunter und
speichern Sie sie lokal ab. Starten Sie ‚Rasmol’ (s. Anhang 2). Öffnen Sie damit die
heruntergeladene Datei und stellen das Phycocyanin so dar, dass die Sekundär- und
Tertiärstruktur des Proteins und die Chromophore gut zu erkennen sind (Farbausdruck über
das Netz, Assistent fragen).
E. Auswertung:
Protokollieren Sie alle Versuche.
Beschreiben Sie die Aufreinigung und belegen Sie sie mittels der Absorptionsspektren.
Erstellen Sie eine Graphik, in der Sie die Absorption von PC in Abhängigkeit vom
Harnstoffgehalt der Probe (0 – 8 M) auftragen.
Belegen Sie die Änderungen der Fluoreszenz beim Entfalten und Rückfalten durch Spektren
und Foto.
Vergleichen Sie die Denaturierung mit den anderen Reagenzien und durch Hitze mit der
durch Harnstoff mit Bezug auf Vollständigkeit der Entfaltung und Reversibilität.
49

F. Anhang:
Tab. VIII.1: Ammoniumsulfat-Mengen (in g/l) um von x% (linke Spalte) auf y% (Kopfzeile) Sättigung zu kommen (bei 4°C). Gesättigt = 100 %.
50

Anhang 2: RasMol: Eine Übersicht mit Beispielen
RasMol ist ein Programm zur Visualisierung von Biopolymeren (Proteine, Nukleinsäuren) auf der Basis von Strukturen mit atomarer Auflösung (Röntgen, NMR). Es ist über die University of Massachusetts, Amherst, frei erhältlich und kann per Internet geholt werden (http://www.umass.edu/microbio/rasmol/). Dies ist eine Kurzdarstellung der wichtigsten Befehle. Es sind jeweils nur einige repräsentative Beispiele angegeben, der volle Befehlsumfang kann im Hilfe Menü nachgelesen werden.
Beim Aufruf von Rasmol werden zwei Fenster geöffnet. Man landet im Darstellungsfenster. Es dient zur Darstellung der Moleküle und enthält gleichzeitig mehrere Pulldown-Menüs für globale Operationen. Daneben gibt es das Command-Line Fenster, welches eine detaillierte Bearbeitung einer Abbildung ermöglicht. Die sog. Command-Line Befehle können auch dann eingegeben werden, wenn das Darstellungsfenster aktiv ist, man sieht sie aber nur, wenn man beide Fenster nebeneinander laufen läßt. Unter diesen Umständen muss man zwar das Darstellungsfenster verkleinern, aber hat eine einfache Kontrolle über die eingegebenen Befehle, Fehlermeldungen oder Ergebnisse von Abfragen.
Mausbelegung zum Bewegen und Zoomen des dargestellten MolekülsLage und Vergrößerung des Moleküls sind mit der Maus manipulierbar. Die Tastenbelegungen sind auf die der Programme ‘Insight’ und ‘Quanta’ umstellbar.
Linke Maustaste und ziehen Drehen um x- oder y-Achse Rechte Maustaste und ziehen Verschieben in x/y-Ebene Shift+Linke Maustaste und ziehen Zoomen: Nach oben = größer, nach unten = kleiner Shift+Rechte Maustaste und ziehen Drehen um z-Achse Ctrl+ Linke Maustaste und ziehen Verschieben in z-Richtung
Globale Operationen aus den Menüs im DarstellungsfensterDurch Anclicken des Display Menüs läßt sich die Darstellung eines Moleküls in einfacher Weise verändern. Ebenso lassen sich im Colours Menü bestimmte Einfärbungen erzeugen, und im OptionsMenü lassen sich bestimmte Darstellungsweisen verändern. Mit dem Export Menü läßt sich eine Abbildung in verschiedenen Formaten abspeichern
File Menü: Load/Close/Print Laden/Beende einer Struktur-Datei, Drucken der Darstellung Display Menü: Wireframe Strichmodell, Striche = Bindungen, Knicke = Atome Display Menü: Spacefill Raumerfüllendes Modell (CPK), Atome als Kugeln Display Menü: Cartoon Einsichtige Darstellung der Sekundärstruktur (Helix, Faltblatt) Colours Menü: Temperature Einfärbung entsprechend Beweglichkeit/Unordnung der Atome Colours Menü: Chain Einfärbung entsprechend Zugehörigkeit zu Protein-Untereinheit Colours Menü: Shapely Einfärbung entsprechend Zugehörigkeit zu AS-Typ Options Menü: Stereo Darstellung als Stereo-Doppelbild Options Menü: Slab mode Darstellung wird in Z-Richtung auf Scheibchen beschränkt
Detaillierte Operationen über das RasMol Command-Line FensterFür viele Anwendungen reichen die globalen Operationen nicht aus, in diesen Fällen gibt es eine Viel-zahl von Möglichkeiten, bestimmte Teile des Moleküls auszuwählen oder die Darstellung auf bestimmte Teile zu beschränken, und die Darstellung dieser Teile dann in vielfältiger Weise zu bearbeiten (Färben, Darstellungsart, Strichstärke, Beleuchtung, etc.). Bei allen Befehlen wird nicht zwischen GROSS- und klein-Schreibung unterschieden. Durch Select <Atom/Gruppe> werden Atomen oder Gruppen zur Bearbeitung ausgewählt, aber alle anderen Atome/Gruppen werden weiter angezeigt und durch die Bearbeitungsbefehle nicht verändert. Select und Bearbeitungs-Operationen können nacheinander auf verschiedene Atome/Gruppen ange-wendet werden. Durch Restrict <Atom/Gruppe> werden nur die ausgewählten Atome/Gruppen angezeigt, und alle anderen ausgeblendet.
Definition von Atomen und GruppenFür eine aussagekräftige Darstellung ist eine geschickte Auswahl der Atome und Gruppen häufig der wichtigste und schwierigste Teil. RasMol erlaubt für die Select oder Restrict Befehle eine vielfältige Definition von Gruppen. Dabei gibt es vordefinierte Gruppen (z.B. für das Peptidgerüst, basische AS, ß-Faltblatt, s. Hilfe: Predefined sets), oder man kann Gruppen oder Atome individuell mittels ihrer Codierung (Kette, AS Nr., Atom Nr.) auswählen und über logische Operatoren (and/or/not) kombinieren (Hilfe: Atom expressions). Falls man die Codierung nicht kennt, kann man sie mit setpicking identify ermittlen. Häufig ist es dabei günstig, wenn man sich parallel den pdb-file in ein Textbearbeitungsprogramm lädt, um z.B. die Sequenz oder die Bezeichung für Kofaktoren nachlesen zu können. Falls man in einem Molekül bestimmte Gruppen (z.B. Kofaktoren, aktives Zentrum) öfter
51

bearbeiten will, kann man mit dem Kommando define eine Gruppe neu definieren und später immer wieder unter diesem Namen aufrufen.
* Alle Atome cys Atome in Cysteinen hoh Atome in Wassermolekülen as? Atome in Asparagin oder Asparaginsäure *120 Atome des AS-Restes 120 in allen Ketten *p Alle Atome in Kette P *.n? Stickstoff Atome cys.sg Schwefel in Cysteinen (allg. als Schwefel ‘g’ (= ) gekennzeichnet ser70.c? Kohlenstoff Atome in Serin-70 hem*p.fe Eisenatom im Häm von Kette P backbone and not helix Proteingerüst für ß-Faltblatt und Loops within( 8.0, ser70 ) Alle Atome im Abstand 8 Å von AS Serin 70 8, 12, 16, 20-28 Atome der AS Nr. 8, 12, 16 sowie 20-28 arg, his, lys Alle Atome der AS Arg, His und Lys SER70A oder SER70:1 Alle Atome der AS Ser in Position 70 in Kette A bzw. 1 Ser70A.C2 C-Atom 2 von Ser 70 in Kette A oxygen oder elemno=8 Sauerstoffatome elemno=12 Mg Atome (=Element der Ordnungszahl 12) define az 20-36 Definition AS 20-36 als Gruppe ‘az’, danach mit ‘select az’ aufrufbar.
Bearbeiten von Atomen/Gruppen:Nach der Auswahl kann die Darstellungsweise und Einfärbung der Atome/Gruppen in vielfältiger Weise verändert werden. Hilfe: Color schemes und Hilfe: Command reference.
color red Rot einfärben (Standardfarbe) color [255,165,40] Helles Orange (selbst definierte RGB-Farbe) spacefill 100 Atome werden als Kugeln mit Radius 0,4 Å (= 100/250) dargestellt. backbone 150 Das Peptidgerüst wird mit 0,6 Å (=150/250) dicken Stichen dargestellt wireframe 50 Strichzeichnung mit 0,4 Å (= 50/250) dicken Bindungen. ribbons 500 Darstellung des Peptid-Gerüsts als 2 Å (= 500/250) breites Band hbonds on/off Anzeige/Unterdrückung von H-Brücken ssbonds on/off Anzeige/Unterdrückung von Disilfid-Brücken
RasMol Parameterkontrollieren die Darstellungsweise von RasMol, z.b. Hintergrundfarbe, Beleuchtungsstärke, Verhalten der Maus, Schattenwurf, glänzende Oberflächen usw. Hilfe: Internal Parameters
set background white Weisser Hintergrund set ambient 100/50 Hellste (100) bzw. mäßig helle (50) Beleuchtung, starker/mittlerer Kontrast
Informationen über Atome/Gruppenlassen sich mit einer Reihe von set picking <Abfrage> abfragen, die Anzeige erfolgt im RasMolCommand Line Fenster.
set picking identify Codierung des angeclickten Atoms wird angezeigt set picking distance Entfernung zwischen zwei nacheinander angeclickten Atomen set picking angle Von drei nacheinander angeclickten Atomen eingeschlossener Winkel
Beschriften von ZeichnungenZeichnungen lassen sich mit Label beschriften, Farbe und Größe sind wählbar. Man kann entweder einen selbstgewählten Text angeben, oder bestimmte Eigenschaften automatisch anzeigen lassen.
Label <Text> An das selektierte Atom wird <Text> geschrieben Label %e/%r Automatische Beschriftung selektierter Atome mit Elementsymbol bzw. AS-Nr set fontsize Größe der Beschriftungen color label Farbe der Beschriftung
52

Versuch IX: Der Pasteureffekt bei der Hefe
A. Theorie:
Glucose kann anaerob zu Ethanol und CO2 (z.B von Hefen (Saccharomyces cerevisiae) oder
zu Milchsäure (z.B. von tierischem Muskelgewebe) abgebaut werden (Gärung mit einer
Ausbeute von 2 ATP). Aerob bei Zufuhr von genügend Sauerstoff wird die Glucose dagegen
oxidativ zu CO2 und H2O abgebaut (Atmung mit einer Ausbeute von 38 ATP).
aerob anaerob 6 CO2 + 6 H2O ————— Glucose ————— 2CO2 + 2 Ethanol 38 ATP 2 ATP
Pasteur fand 1861, daß die Gärung der Hefe (und anderer Mikroorganismen, die fakultativ
anaerob leben können) durch Luftsauerstoff gehemmt wird, also in Gegenwart von
Sauerstoff weniger Glucose verbraucht wird als ohne Sauerstoff (= Pasteureffekt);
gleichzeitig verringert sich dabei die Ethanol- bzw. Milchsäureanreicherung. Analoges gilt,
wie sich später herausstellte, auch für den Glucoseabbau im Muskel und vielen anderen
Geweben (ausgenommen Tumorzellen). Dieser hemmende Effekt des Sauerstoffs erscheint
ökonomisch sinnvoll: Für einen gleichen Energiebetrag wird beim aeroben Abbau weniger
Glucose benötigt als beim aneroben, d.h. Energiegewinn und Nahrungsverbrauch sind
aufeinander abgestimmt. Sauerstoff wirkt auf den Glucoseabbau über eine chemische
Reaktion ("Pasteursche Reaktion"), die ihrerseits durch bestimmte Substanzen (wie zum
Beispiel Nitrophenole) gehemmt werden kann. In Gegenwart solcher Stoffe wird aerob
ebensoviel Glucose vergoren wie anaerob, obwohl der Sauerstoffverbrauch normal (oder
sogar erhöht) ist. Nitrophenole entkoppeln aber gleichzeitig die oxidative Phosphorylierung,
also blockieren sie die ATP-Bildung bei unverändertem Sauerstoffverbrauch. Ein hoher ATP-
Spiegel hemmt einen frühen Schritt des Glucoseabbaus, nämlich die Phosphorylierung von
Fructose-6-phosphat zu Fructose-1,6-bisphosphat. ATP ist allerdings nur einer von mehreren
Effektoren für dieses allosterisch regulierbare Enzym. Durch eine Hemmung auf dieser Stufe
muß sich zunächst Fructose-6-phosphat anhäufen. Die Annahme, daß das damit im
Gleichgewicht stehende Glucose-6-phosphat die Phosphorylierung der Glucose hemmt,
erklärt dann die Hemmung des Glucoseverbrauchs und damit den Pasteureffekt. Daraus
erklärt sich auch die Wirkung der Entkoppler (z.B. Nitrophenole, Actinomycin A): Da trotz des
Sauerstoffverbrauchs kein ATP durch Atmungskettenphosphorylierung gebildet werden
kann, wird die Phosphofructokinase nicht gehemmt. Die Glykolyse läuft daher in Gegenwart
von Entkopplern auch unter Sauerstoff unvermindert weiter.
Literatur
Krebs, H.A. (1972) Der Pasteureffekt und die Beziehungen zwischen Atmung und Gärung in lebenden Zellen. Naturwiss. Rundschau 25, 387-392
53

Gagunas, R.; Gancedo, C. (1983) Role of phosphate in the regulation of the pasteur effect in Saccharomyces cerevisiae. Eur. J. Biochem. 137, 479-483
B. Aufgabe:
Manometrische Messung des bei Hefezellen durch Gärung bzw. Atmung freigesetzten CO2
und Vergleich dieser Ergebnisse mit der quantitativen Bestimmung des Glucose-Verbrauchs.
C. Reagenzien:
Glucose-Lösung (1%, 50 ml, immer frisch ansetzen, ggf. im Kühlschrank aufbewahren)
KOH-Lösung (10%, 10 ml)
Na-Citratpuffer, pH 5.4 (20 mM, 100 ml; Citronensäurelösung mit einigen Tropfen konz. NaOH am pH-Meter auf pH 5.4 einstellen
D. Versuchsdurchführung:
1. Manometrische Bestimmung von Atmung und Gärung:
0.25 g Bäckerhefe (keine Trockenhefe verwenden) wird in 50 ml H2Odest mit 125 mg Glucose
etwa 1 h geschüttelt, 5 min bei ca. 3 000 Upm abzentrifugiert und in 50 ml Na-Citratpuffer
(20 mM, pH 5.4) aufgenommen.
Mit dieser Hefesuspension werden die in der folgenden Tabelle aufgeführten Ansätze
hergestellt und die Gefäße mit den Manometern in einem auf 20°C temperierten Warburg-
Apparat gehängt. Die eigentliche Messeinheit des Warburg-Apparats besteht aus einem
Reaktionsgefäß und einem Manometer. Das Reaktionsgefäß bildet während des Versuchs
ein geschlossenes System zusammen mit einem der beiden Schenkel des Manometers
(dem rechten), der andere Schenkel ist offen. Ändert sich das Gasvolumen im ge-
schlossenen System so drückt sich das auch als Druckänderung aus. Da für ideale Gase
das Produkt aus Druck und Volumen (bei gleichbeibender Temperatur) konstant ist, muß zur
Messung während des Experiments einer der beiden Faktoren konstant gehalten werden.
Dies wird dadurch erreicht, dass für jede Ablesung das rechte Manometer auf 150 mm
eingestellt wird, also das Volumen des geschlossenen Systems gleicht bleibt.
Gasentwicklung oder –verbrauch resultiert dann ausschliesslich in erhöhtem oder
erniedrigtem Druck, der anhand der Skala am offenen Manometer abgelesen werden kann
(= h [mm]).
Gefäß 1a + 1b 2a + 2b 3a + 3b Tb
Gasphase N2 Luft Luft LuftHauptraum 2 ml Hefe-
suspension 2 ml Hefe- suspension
2 ml Hefe- suspension
2.4 ml Wasser
Kipper 0.4 ml 1% Gluc. 0.4 ml 1% Gluc. 0.4 ml 1% Gluc. —
Mitteleinsatz— — 0.2 ml 10%
KOH+Filter-papier
—
54

Zur Erreichung der in der Tabelle angegebenen Gasphase werden die Proben 1 ca. 5 min
vorsichtig mit N2 gespült (Durchführung nur nach Anleitung). Nach weiteren 5 min für
Temperatur- und Druck-Ausgleich werden die Manometer verschlossen und abgelesen. Die
Gefäße werden ab jetzt geschüttelt. Im Abstand von 5 min wird die sogenannte Leeratmung
verfolgt (4 Ablesungen). Dann wird die Glucose zugekippt und eine Stunde lang in Intervallen
von 10 min die Druckänderung abgelesen. Für jedes Gefäß soll sofort der anhand des
Thermobarometers korrigierte Meßwert für jedes Ablese-Intervall gegen die Zeit in ein
Diagramm eingetragen werden, um den Gang der Meßwerte beurteilen zu können.
Gleich nach der letzten Ablesung wird aus den 6 Gefäßen je 1 ml Suspension entnommen, 5
min bei 3 000 Upm in der Tischzentrifuge zentrifugiert (der Überstand jeweils in einen 10 ml
Meßkolben dekantiert), der Niederschlag mit 5 ml demin. Wasser gewaschen, zentrifugiert
und erneut dekantiert. Die Meßkolben werden auf 10 ml mit demin. Wasser aufgefüllt und
gemischt. Diese Proben werden zur Glucosebestimmung verwendet. Zur Bestimmung des
Anfangsgehalts werden 2 ml Hefe und 0.4 ml der 1% Glucose-Lösung gemischt und dann
1 ml sofort wie oben angegeben aufgearbeitet.
2. Enzymatische Bestimmung der Glucose mit dem „UV-Test“:
Für die Glucosebestimmung werden gemäß dem Testansatz (s.u.) je 0.2 ml der Proben
verwendet. Die Durchführung des Tests entspricht ansonsten dem Versuch V, siehe Seiten
34/35. Die Glucosemenge im gesamten Warburggefäß wird entsprechend der Verdünnung
berechnet. Der Verbrauch an Glucose errechnet sich aus dem Anfangsgehalt und dem
Endgehalt der bestimmten Glucosemenge (MW von Glucose: 180; Glucose x 1 H2O = 198).
Testansatz
Leerwert(1 x)
Probe(7 x)
Lösung 1 1 .0 ml 1 .0 ml
Probelösung - 0 .2 ml
H20dest 2 .0 ml 1 .8 ml
Suspension 2 20 µl 20 µl
E. Berechnung der Ergebnisse:
Die Druckänderung ( h = manometrische Differenz) wird in eine Volumenänderung umge-
rechnet, wobei auf Normalbedingungen (0°C und 760 mm Hg) korrigiert wird. Zur Korrektur
der Luftdruck- und Temperaturschwankungen wird zusätzlich die bei jeder Ablesung fest-
gestellte Änderung am Thermobarometer (Tb) bei allen Meßwerten berücksichtigt. Da 1 µmol
eines Gases bei Normalbedingungen 22.4 µl einnimmt, lässt sich die ermittelte Volumen-
änderung in molaren Mengen ausdrücken. Zur Umrechnung der Druckänderung (nach der
55

Formel µl Gas = k x h) müssen die jeweiligen Gefäßkonstanten (kO2 bzw. kCO2) ermittelt
werden, in denen alle dem Reaktionsystem und den Versuchsbedingungen zugeordneten
Faktoren zusammengefasst sind, die sich während des Versuchsablaufs nicht ändern.
Berechnen der Gefäßkonstanten:
VG x (273/T) + VF x (O2 bzw. CO2)kO2 bzw. kCO2 = P0
VG = Volumen des Gasraums in µl (=VGefäß + VManometer - VF)VF = Volumen der Flüssigkeit in µlT = Temperatur in Grad Kelvin (°K = 273 + t°C)
= Bunsenscher Absorptionskoeffizient ml/ml (bei 20°C: für O2 = 0.03; für CO2 = 0.878; gibt die Löslichkeit des Gases bei Normaldruck an)
P0 = Normaldruck = 760 mm Hg = 10 332 mm Wassersäule = 10 000 mm Brodie-Säule (das spez. Gewicht der im Versuch verwendeten Sperrflüssigkeit, der Brodie-Lösung, beträgt 1.033 g cm-3, so dass 760 mm Hg 10 000 mm Brodie-Lösung entsprechen)
Berechnen der Gasmengen:
Aus h1 ergibt sich nach der Beziehung µl Gas = k x h die durch anaerobe Gärung gebildete
CO2-Menge (µl/22.4 = µmol) und damit auch die vergorene Glucosemenge (wieviel µmol
CO2 pro µmol Glucose?).
Aus h3 ergibt sich die durch Atmung verbrauchte O2-Menge und daraus die veratmete
Glucosemenge.
Die Werte für h2 entstehen durch Überlagerung von O2-Verbrauch und CO2-Produktion.
Den O2-Verbrauch in µl kennen wir von Gefäß 3. Diese Zahl dividiert durch kO2 (Gefäß 2)
ergibt den Druckabfall, den der O2-Verbrauch in Gefäß 2 verursacht hat (hO2). Der durch
CO2-Produktion bedingte Druckanstieg ergibt sich damit also aus:
hCO2 = h2 - hO2 (Vorzeichen beachten!)
Die CO2-Produktion ergibt sich dann nach: µl CO2 = hCO2 x kCO2 (Gefäß 2); dividiert man
diesen Wert durch das Molvolumen (22.4), so erhält man die µMol CO2.
Vergleicht man nun die Menge an produziertem CO2 und aufgenommenem O2, so stellt man
eine CO2-Überproduktion fest. Diese ergibt sich durch eine zusätzlich zur Atmung
ablaufende Gärung, die sogenannte „aerobe“ Gärung, bei der wiederum 2 CO2 + 2 Ethanol
pro Glucose gebildet werden.
Subtrahiert man von dem insgesamt produzierten CO2 das durch Atmung entstandene CO2
(= µmol O2), so erhält man die durch "aerobe" Gärung entstandene CO2-Menge. Zur
Berechnung der für den Energiestoffwechsel verbrauchten Glucosemenge gilt dann:
veratmete Glucose = µmol CO2 aus der Atmung/6
"aerob" vergorene Glucose = µmol CO2 aus der Gärung/2
56

F. Ergebnis und Schlußfolgerung:
Die anaerobe Gärung setzt immer mehr Glucose um als die Atmung und aerobe Gärung. Die
Glykolyse muß also durch die Atmung gehemmt worden sein (Pasteureffekt). Die im
Energiestoffwechsel umgesetzte Glucosemenge ist kleiner als die aufgenommene
Glucosemenge aus der Glucosebestimmung. Die Differenz ist die zum Aufbau von
Zellmaterial assimilierte Glucose (Baustoffwechsel). Bei atmenden Zellen (Gefäß 2/3) ist
diese Differenz prozentual höher als bei gärenden Zellen (Gefäß 1).
Die Ergebnisse bitte in die folgende Tabelle eintragen, komplett ausrechnen und den
Pasteureffekt diskutieren!
GlucoseverbrauchDurch:
anaerobµmol %
aerob µmol %
Anerobe Gärung
Atmung
Aerobe Gärung
Energiestoffwechsel GesamtAssimilation (Baustoffwechsel)Gesamter Glucose- Verbrauch
100 100
Leeratmung bzw.Leergärung (µMol CO2 bzw O2 pro h)
57

Version vom WS2005/2006 Dr. Judith Fliegmann Prof. Jürgen Ebel
58


















