KineticMechanismofProteinArginineMethyltransferase6 ... analysis to Equation 1 using the GraFit...
11
Kinetic Mechanism of Protein Arginine Methyltransferase 6 (PRMT6) * □ S Received for publication, December 13, 2011, and in revised form, December 29, 2011 Published, JBC Papers in Press, January 3, 2012, DOI 10.1074/jbc.M111.333609 Obiamaka Obianyo and Paul R. Thompson 1 From the Department of Chemistry, The Scripps Research Institute, Jupiter, Florida 33458 Background: PRMT6 (protein arginine methyltransferase 6) regulates gene transcription via its ability to methylate his- tones H3 and H4. Results: Product and dead-end inhibition experiments were performed to assign the kinetic mechanism of PRMT6. Conclusion: PRMT6 utilizes a rapid equilibrium random mechanism with dead-end EAP and EBQ complexes. Significance: This information should aid in the development of inhibitors targeting PRMT6, which may represent novel cancer therapeutics. The protein arginine methyltransferases (PRMTs) are a fam- ily of enzymes that catalyze the mono- and dimethylation of arginine residues in a variety of proteins. Although these enzymes play important roles in a variety of cellular processes, aberrant PRMT activity is associated with several disease states, including heart disease and cancer. In an effort to guide the development of inhibitors targeting individual PRMTs, we ini- tiated studies to characterize the molecular mechanisms of PRMT catalysis. Herein, we report studies on the kinetic mech- anism of PRMT6. Initial velocity, product inhibition, and dead- end analog inhibition studies with the AcH4-21 and R1 pep- tides, as well as their monomethylated versions, indicate, in contrast to a previous report, that PRMT6 utilizes a rapid equi- librium random mechanism with dead-end EAP and EBQ complexes. Over the last decade, the protein arginine methyltransferases (PRMTs) 2 have emerged as an enzyme family whose activity is dysregulated in human disease (1– 4). The PRMTs catalyze the mono- and dimethylation of peptidylarginine residues in a vari- ety of substrates to maintain cellular processes, e.g. cellular growth and signaling, nuclear-cytoplasmic protein shuttling, cell differentiation, embryogenesis, transcriptional regulation, and chromatin remodeling (5– 8). There are three main types of PRMTs. The type I isozymes (PRMT1– 4, PRMT6, and PRMT8) generate monomethylarginine (MMA) and asymmet- ric dimethylarginine (ADMA); the type II isozyme (PRMT5) generates MMA and symmetric dimethylarginine; and the type III isozyme (PRMT7) generates only MMA (2). 3 Given that this family of enzymes plays an integral role in many cellular processes, it is not surprising that, when dysregu- lated, these enzymes also contribute to human disease. For example, aberrantly increased PRMT activity is associated with heart disease via its ability to generate free ADMA; high levels of ADMA have been linked to heart disease and renal failure (3, 9 –13). Additionally, PRMT1 activity appears to be increased in breast cancer (13). PRMT6, the focus of the studies reported herein, has also been reported to be overexpressed in, and to be required for, the proliferation of bladder and lung cancer cells (14). Interestingly, siRNA knockdown of PRMT6 in U2OS osteosarcoma cells led to the up-regulation of thrombospon- din-1, a natural inhibitor of angiogenesis and cell migration (15). Thus, the PRMTs represent interesting therapeutic targets. As a part of a program focused on developing inhibitors tar- geting the PRMTs, we initiated studies to characterize the cat- alytic mechanisms, substrate specificity, and kinetic mecha- nisms of these enzymes (16 –19). Previous studies with PRMT1 have shown that this isozyme preferentially methylates sub- strates with positively charged residues distal to the site of methylation (16). Additionally, we demonstrated that PRMT1 catalyzes ADMA formation in a partially processive fashion, i.e. a fraction of the monomethylated product remains bound to the enzyme, whereas S-adenosyl-L-homocysteine (SAH) is exchanged for S-adenosyl-L-methionine (SAM) to allow for a second round of methylation (16). Consistent with this mech- anism, we showed that PRMT1 uses a rapid equilibrium ran- dom mechanism with dead-end EAP and EBQ complexes (16, 17). Information from these studies guided the development of C21, an irreversible inhibitor that is the most potent and selec- tive PRMT1 inhibitor described to date (19). Although this compound shows excellent selectivity (100-fold) versus PRMT3 and PRMT4, it is only modestly selective for PRMT6 (19). In an effort to improve the selectivity of C21, we initiated studies to characterize the molecular mechanisms of PRMT6 catalysis. PRMT6 catalyzes the methylation of several proteins, includ- ing histones H3 and H4, and this activity has been shown to play a key role in controlling the expression of the HOX genes as well as Myc-dependent genes (20, 21). Previously, PRMT6 was shown to also catalyze the methylation of the R1 and R1-MMA peptides, which contain a single arginine residue (22). The R1 * This work was supported by The Scripps Research Institute, Scripps Florida. □ S This article contains supplemental “Methods” and Tables S1 and S2. 1 To whom correspondence should be addressed: Dept. of Chemistry, The Scripps Research Institute, Scripps Florida, 130 Scripps Way, Jupiter, FL 33458. Tel.: 561-228-2860; Fax: 561-228-2918; E-mail: pthompso@scripps. edu. 2 The abbreviations used are: PRMT, protein arginine methyltransferase; MMA, monomethylarginine; ADMA, asymmetric dimethylarginine; SAH, S-adenosyl- L-homocysteine; SAM, S-adenosyl-L-methionine; Fmoc, N-(9-fluorenyl)me- thoxycarbonyl; Tricine, N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine. 3 S. Clarke, personal communication. THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 287, NO. 8, pp. 6062–6071, February 17, 2012 © 2012 by The American Society for Biochemistry and Molecular Biology, Inc. Published in the U.S.A. 6062 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 287 • NUMBER 8 • FEBRUARY 17, 2012 by guest on July 3, 2018 http://www.jbc.org/ Downloaded from
Transcript of KineticMechanismofProteinArginineMethyltransferase6 ... analysis to Equation 1 using the GraFit...

Kinetic Mechanism of Protein Arginine Methyltransferase 6(PRMT6)*□S
Received for publication, December 13, 2011, and in revised form, December 29, 2011 Published, JBC Papers in Press, January 3, 2012, DOI 10.1074/jbc.M111.333609
Obiamaka Obianyo and Paul R. Thompson1
From the Department of Chemistry, The Scripps Research Institute, Jupiter, Florida 33458
Background: PRMT6 (protein arginine methyltransferase 6) regulates gene transcription via its ability to methylate his-tones H3 and H4.Results: Product and dead-end inhibition experiments were performed to assign the kinetic mechanism of PRMT6.Conclusion: PRMT6 utilizes a rapid equilibrium random mechanism with dead-end EAP and EBQ complexes.Significance:This information should aid in the development of inhibitors targeting PRMT6,whichmay represent novel cancertherapeutics.
The protein arginine methyltransferases (PRMTs) are a fam-ily of enzymes that catalyze the mono- and dimethylation ofarginine residues in a variety of proteins. Although theseenzymes play important roles in a variety of cellular processes,aberrant PRMT activity is associated with several disease states,including heart disease and cancer. In an effort to guide thedevelopment of inhibitors targeting individual PRMTs, we ini-tiated studies to characterize the molecular mechanisms ofPRMT catalysis. Herein, we report studies on the kinetic mech-anism of PRMT6. Initial velocity, product inhibition, and dead-end analog inhibition studies with the AcH4-21 and R1 pep-tides, as well as their monomethylated versions, indicate, incontrast to a previous report, that PRMT6 utilizes a rapid equi-librium random mechanism with dead-end EAP and EBQcomplexes.
Over the last decade, the protein argininemethyltransferases(PRMTs)2 have emerged as an enzyme family whose activity isdysregulated in human disease (1–4). The PRMTs catalyze themono- and dimethylation of peptidylarginine residues in a vari-ety of substrates to maintain cellular processes, e.g. cellulargrowth and signaling, nuclear-cytoplasmic protein shuttling,cell differentiation, embryogenesis, transcriptional regulation,and chromatin remodeling (5–8). There are three main typesof PRMTs. The type I isozymes (PRMT1–4, PRMT6, andPRMT8) generate monomethylarginine (MMA) and asymmet-ric dimethylarginine (ADMA); the type II isozyme (PRMT5)generatesMMA and symmetric dimethylarginine; and the typeIII isozyme (PRMT7) generates only MMA (2).3Given that this family of enzymes plays an integral role in
many cellular processes, it is not surprising that, when dysregu-
lated, these enzymes also contribute to human disease. Forexample, aberrantly increased PRMT activity is associated withheart disease via its ability to generate free ADMA; high levelsof ADMAhave been linked to heart disease and renal failure (3,9–13). Additionally, PRMT1 activity appears to be increased inbreast cancer (13). PRMT6, the focus of the studies reportedherein, has also been reported to be overexpressed in, and to berequired for, the proliferation of bladder and lung cancer cells(14). Interestingly, siRNA knockdown of PRMT6 in U2OSosteosarcoma cells led to the up-regulation of thrombospon-din-1, a natural inhibitor of angiogenesis and cell migration(15). Thus, the PRMTs represent interesting therapeutictargets.As a part of a program focused on developing inhibitors tar-
geting the PRMTs, we initiated studies to characterize the cat-alytic mechanisms, substrate specificity, and kinetic mecha-nisms of these enzymes (16–19). Previous studies with PRMT1have shown that this isozyme preferentially methylates sub-strates with positively charged residues distal to the site ofmethylation (16). Additionally, we demonstrated that PRMT1catalyzes ADMA formation in a partially processive fashion, i.e.a fraction of the monomethylated product remains bound tothe enzyme, whereas S-adenosyl-L-homocysteine (SAH) isexchanged for S-adenosyl-L-methionine (SAM) to allow for asecond round of methylation (16). Consistent with this mech-anism, we showed that PRMT1 uses a rapid equilibrium ran-dom mechanism with dead-end EAP and EBQ complexes (16,17). Information from these studies guided the development ofC21, an irreversible inhibitor that is the most potent and selec-tive PRMT1 inhibitor described to date (19). Although thiscompound shows excellent selectivity (�100-fold) versusPRMT3 and PRMT4, it is only modestly selective for PRMT6(19). In an effort to improve the selectivity of C21, we initiatedstudies to characterize the molecular mechanisms of PRMT6catalysis.PRMT6 catalyzes themethylation of several proteins, includ-
ing histonesH3 andH4, and this activity has been shown to playa key role in controlling the expression of theHOX genes aswellas Myc-dependent genes (20, 21). Previously, PRMT6 wasshown to also catalyze the methylation of the R1 and R1-MMApeptides, which contain a single arginine residue (22). The R1
* This work was supported by The Scripps Research Institute, Scripps Florida.□S This article contains supplemental “Methods” and Tables S1 and S2.1 To whom correspondence should be addressed: Dept. of Chemistry, The
Scripps Research Institute, Scripps Florida, 130 Scripps Way, Jupiter, FL33458. Tel.: 561-228-2860; Fax: 561-228-2918; E-mail: [email protected].
2 The abbreviations used are: PRMT, protein arginine methyltransferase; MMA,monomethylarginine; ADMA, asymmetric dimethylarginine; SAH, S-adenosyl-L-homocysteine; SAM, S-adenosyl-L-methionine; Fmoc, N-(9-fluorenyl)me-thoxycarbonyl; Tricine, N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine.
3 S. Clarke, personal communication.
THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 287, NO. 8, pp. 6062–6071, February 17, 2012© 2012 by The American Society for Biochemistry and Molecular Biology, Inc. Published in the U.S.A.
6062 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 287 • NUMBER 8 • FEBRUARY 17, 2012
by guest on July 3, 2018http://w
ww
.jbc.org/D
ownloaded from

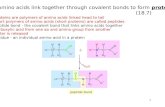
peptides are based on the amino acid sequence of an internalportion of fibrillarin, a known protein substrate of PRMT6 (23).Although PRMT6was reported to utilize a steady-state orderedmechanism in which SAMbinds to the enzyme prior to proteinor peptide (Scheme 1) (22), we show here that the IC50 values ofC21, an irreversible PRMT6 inhibitor, are similar, regardless ofwhether C21 is preincubated with the enzyme in the absence orpresence of SAM.This result suggested that SAMbinding is notrequired for inhibitor/peptide binding and thus called intoquestion the assignment of an ordered mechanism for thisenzyme. Therefore, we reinvestigated the kineticmechanism ofPRMT6. These studies revealed that PRMT6 utilizes a rapidequilibrium random mechanism with dead-end EAP and EBQcomplexes (Scheme 2).
EXPERIMENTAL PROCEDURES
Reagents—HEPES, DTT, and EDTA were purchased fromResearch Products International Corp. N�-Fmoc amino acidsand preloaded Wang-based resins were obtained from Nova-biochem. Radiolabeled [methyl-14C]SAM was purchased from
PerkinElmer Life Sciences, and 14C-labeled BSAwas purchasedfrom American Radiolabeled Chemicals. A pET16 vectorencoding the human DNA sequence of PRMT6 was obtainedfromMark T. Bedford (The University of Texas MD AndersonCancer Center). The purification of PRMT6 is described undersupplemental “Methods.” Peptides were synthesized on Wangresin using standard Fmoc chemistry and purified by reversephase HPLC. The sequences of the peptides used in these stud-ies, as well as their expected and observed masses, are providedin Table 1. All mass spectra were acquired by MALDI-MS.Gel-based Activity Assay—Activity assays were performed as
described previously for PRMT1 using a discontinuous gel-based assay (16). The assay buffer consisted of 50 mM HEPES(pH 8.0), 50 mM NaCl, 1 mM EDTA, and 0.5 mM DTT. Gener-ally, enzyme assays were performed by first preincubating theassay buffer with SAMand peptide substrate for 10min at 37 °Cand then initiating the reaction by the addition of PRMT6 (500nM final concentration). The reaction was quenched after 15min with 6� Tris/Tricine gel loading dye. Under these condi-tions, PRMT6 activity is linear with respect to both time andenzyme concentration. All assays were performed at least induplicate, and the S.D. was �20%. Where appropriate, the ini-tial rates obtained from these assays were fit by nonlinear leastsquares analysis to Equation 1 using the GraFit Version 5.0.11software package (24).
v � Vmax�S�/�Km � �S�� (Eq. 1)
IC50 Studies—IC50 values for C21 were determined withPRMT6 by preincubating 500 nM PRMT6 at different inhibitorconcentrations. Either 15 �M [methyl-14C]SAM or 25 �M
AcH4-21 was also preincubated in the assay buffer (asdescribed above) at 37 °C for 10 min. The methylation reactionwas initiated upon the addition of the reciprocal substrate(either 25 �M peptide or 15 �M [methyl-14C]SAM), and after 15min, the reaction was quenched with Tris/Tricine gel loadingdye. Samples were run on 16.5% Tris/Tricine polyacrylamidegels,andincorporatedradioactivitywasquantifiedbyphosphor-imaging analysis (MolecularDynamics). IC50 valueswere deter-mined by fitting the data to Equation 2 using theGraFit Version5.0.11 software package (24),
Fractional activity of PRMT1 � 1/�1 � ��I�/IC50��
(Eq. 2)
where [I] is the concentration of inhibitor, and IC50 is the con-centration of inhibitor that yields half-maximal activity.
SCHEME 1. Ordered sequential mechanism. In the ordered sequentialmechanism, reported by Lakowski and Frankel (22), SAM binds to theenzyme, and subsequent binding of the peptide substrate generates a ter-nary complex. Following product formation, ADMA is released from theenzyme before SAH to regenerate the free enzyme. A, SAM; B, peptide sub-strate; P, ADMA; Q, SAH.
SCHEME 2. Rapid equilibrium mechanism with dead-end EAP and EBQcomplexes. In the PRMT6-catalyzed reaction, the substrates bind to theenzyme in a random fashion to generate a ternary complex. Upon formationof the ternary complex, the reaction products are released in a random fash-ion to regenerate the free enzyme. Dead-end complexes, EAP (enzyme�MMA�SAH) or EBQ (enzyme�SAM�ADMA), can be formed when either reactionproduct, SAH or the ADMA-containing peptide, binds to the enzyme after thefirst substrate binds or after the first product has been released from theternary complex. A, MMA; B, SAM; P, SAH; Q, ADMA.
TABLE 1Peptide substrates and inhibitors
Peptide SequenceMass
Km kcat kcat/KmExpected Observed
Da �M min�1 min�1 M�1
AcH4-21 Ac-SGRGKGGKGLGKGGAKRHRKV 2132 2133 5.53 � 0.86 0.16 � 0.01 2.90 � 104AcH4-21R3MMA Ac-SGMeRGKGGKGLGKGGAKRHRKV 2146 2147 1.53 � 0.34 0.20 � 0.01 1.30 � 105AcH4-21R3ADMA Ac-SGMeR2GKGGKGLGKGGAKRHRKV 2160 2161AcH4-21R3K Ac-SGKGKGGKGLGKGGAKRHRKV 2104 2105 14.8R1 WGGYSRGGYGGW 1302 1303 119 � 33 0.44 � 0.04 3.66 � 103R1-MMA WGGYSMeRGGYGGW 1316 1317 44.6 � 11.0 0.40 � 0.03 8.99 � 103R1-ADMA WGGYSMeR2GGYGGW 1330 1331R1-R6K WGGYSKGGYGGW 1275 1276 16.8
Kinetic Mechanism of PRMT6
FEBRUARY 17, 2012 • VOLUME 287 • NUMBER 8 JOURNAL OF BIOLOGICAL CHEMISTRY 6063
by guest on July 3, 2018http://w
ww
.jbc.org/D
ownloaded from

Initial Velocity Studies—Initial velocity patterns wereobtained by determining the steady-state kinetic parametersfor a substrate at different fixed concentrations of the secondsubstrate. The initial rates for substrate peptides were deter-mined at different fixed concentrations of [14C]SAM (2.5, 5,and 10�M forAcH4-21; 2.5, 5, and 10�M forAcH4-21R3MMA;2.5, 5, and 15�M for R1; and 2.5, 5, 10, and 15�M for R1-MMA).For SAM, initial rates were obtained at fixed concentrations ofAcH4-21 (1, 2.5, and 5 �M), AcH4-21R3MMA (2.5, 10, and 15�M), R1 (25, 50, 100, and 200 �M), and R1-MMA (25, 50, 100,and 200 �M). A global fit of the data was obtained by fitting theinitial rate data to Equation 3 using the GraFit Version 5.0.11software package (24),
v � Vmax�A��B�/�KiaKb � Kb�A� � Ka�B� � �A��B�� (Eq. 3)
where Kia is the dissociation constant of the varied substrate,and Ka and Kb are the Michaelis-Menten constants for the var-ied and fixed substrates, respectively.Inhibition Studies—Product inhibition experiments were
carried out using the assay methodology outlined above. Forthese experiments, either peptide product (i.e. the AcH4-21R3ADMA or R1-ADMA peptide) or SAH was used as theproduct inhibitor. Dead-end analog inhibition experimentswere carried out analogously using the AcH4-21R3K or R1-R6Kpeptide and sinefungin as the dead-end analogs. Note thatPRMT6 did not methylate the AcH4-21R3K or R1-R6K peptideto an appreciable extent (the kcat/Km values were decreased byat least 2–3 orders of magnitude; see Table 1). The initial ratesderived from these inhibition experiments were fit to equationsrepresentative of linear competitive inhibition (Equation 4),linear noncompetitive inhibition (Equation 5), linear mixedinhibition (Equation 6), or linear uncompetitive inhibition(Equation 7) by a nonlinear least squares approach using theGraFit Version 5.0.11 software package (24),
v � Vmax�S�/��S� � Km�1 � �I�/Kis�� (Eq. 4)
v � Vmax�S�/��S��1 � �I�/Ki� � Km�1 � �I�/Ki�� (Eq. 5)
v � Vmax�S�/��S��1 � �I�/Kii� � Km�1 � �I�/Kis�� (Eq. 6)
v � Vmax�S�/��S��1 � �I�/Kii� � Km� (Eq. 7)
where Kii is the intercept Ki, and Kis is the slope Ki. Note thatEquation 5 is the equation for pure noncompetitive inhibition,where Ki Kii Kis. The best fits of the data to Equations 4–7were chosen using a combination visual inspection alongwith acomparison of the S.E. values.
RESULTS
Purification, Initial Kinetic Characterization, and InhibitionStudies with C21—Recombinant PRMT6 was expressed inEscherichia coli and purified, via a combination of metal ionaffinity and anion exchange chromatographies, in excellentyield (�1 mg/liter). The steady-state kinetic parameters werethen determined for the R1 and AcH4-21 peptides (Table 1),and the results indicate that the AcH4-21 peptide is a signifi-cantly better substrate than the fibrillarin-based R1 peptide.Interestingly, the Km values reported previously for the R1 and
R1-MMA peptides are �4-fold higher than those found in ouranalyses, and the kcat values that were previously observed are�4-fold lower than our calculated rates (Table 1) (22). As aconsequence, the kcat/Km values observed here are 20- and
FIGURE 1. Initial velocity patterns. Lineweaver-Burk plots display an inter-secting line pattern, indicative of a sequential mechanism, when fixed con-centrations of SAM were assayed at various concentrations of the substratepeptide, AcH4-21R3MMA (A) or R1-MMA (B). C, magnification of the region inFig. 2B near the ordinate.
Kinetic Mechanism of PRMT6
6064 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 287 • NUMBER 8 • FEBRUARY 17, 2012
by guest on July 3, 2018http://w
ww
.jbc.org/D
ownloaded from

11-fold higher than those reported previously. We believe thatthe difference is due at least in part to optimization of theenzyme purification procedure, as we have noted that deviationfrom this procedure leads to a rapid loss of enzyme activity.C21 is a peptide-based inhibitor that is identical in sequence
to the AcH4-21 peptide, except that Arg-3 is replaced with achloroacetamidine-modified ornithine residue. This com-pound, which irreversibly inhibits the PRMTs, shows excellentselectivity (�100-fold) for PRMT1 versus PRMT3 and PRMT4but is onlymodestly selective for PRMT6 (19). Interestingly, theIC50 values of C21 for PRMT6 are similar regardless of whetherthe reaction is initiated by the addition of SAM or AcH4-21(2.1 � 0.1 versus 1.8 � 0.2 �M). Because this assay includes apreincubation phase in which PRMT6, C21, and either SAM orAcH4-21 are incubated for 10 min prior to the addition of thereciprocal substrate, the lack of an effect on the observed IC50values was somewhat surprising because if SAM is required forbinding to the peptide substrate, as would be expected for anordered mechanism, then the IC50 of C21, which is a peptidesubstratemimic, should be significantly higher if SAM is absentduring the preincubation phase. Although we have not com-pleted a thorough kinetic analysis of the mechanism by whichC21 inhibits PRMT6, this finding appears to be inconsistentwith previous reports that PRMT6 uses an ordered kineticmechanism, in which SAM binds prior to the peptide. There-fore, we reinvestigated the kinetic mechanism of PRMT6.Initial Velocity Studies—The previous assignment of an
orderedmechanism (22) was based on experiments with the R1peptide and a MMA-containing derivative, R1-MMA, whichare relatively poor substrates for PRMT6 (Table 1). Because theuse of a poor substrate can make a random mechanism appearordered, we initially determined the kinetic mechanism ofPRMT6 using the AcH4-21R3MMA peptide, which is a signif-icantly better (�35-fold) PRMT6 substrate (Table 1). Subse-quently, these experiments were repeated with the R1-MMApeptide. The MMA-containing peptides, AcH4-21R3MMAand R1-MMA, were used to carry out the initial experimentsbecause these peptides are only subject to one round of meth-ylation. The experiments were then repeated with theunmethylated peptides.For the initial velocity studies, the peptide substrate was var-
ied at different fixed concentrations of the methyl donor SAM,and double-reciprocal plots were obtained. For either the
AcH4-21R3MMAor R1-MMApeptide, an intersecting patternof lines was observed (Fig. 1, A and B; and Table 2). An inter-secting line pattern was also observed when SAMwas varied atfixed peptide concentrations (Table 2). These patterns indicatethat PRMT6 utilizes a sequential or ternary complex mecha-nism in which both substrates must be bound to the enzyme tofacilitatemethyl transfer. Additionally, the fact that the lines donot intersect on the ordinate indicates that PRMT6 does notuse a rapid equilibrium orderedmechanism (see themagnifica-tion of the region in Fig. 2B near the ordinate in Fig. 2C). Notethat a similar pattern of intersecting lines was observed whenthe same experiments were carried out with the unmethylatedpeptides, AcH4-21 and R1 (Table 2).Product Inhibition Studies—Product inhibition studies were
next used to determine the order of substrate binding and prod-uct release in the PRMT6-catalyzed reaction. The productinhibitors used for these studies were SAH and the appropriateADMA-containing peptide, AcH4-21R3ADMA or R1-ADMA.First, when AcH4-21R3MMA was assayed at different concen-trations of AcH4-21R3ADMA and a fixed concentration ofSAM, a competitive pattern of inhibition was observed (Fig. 2Aand Table 3). Similarly, when R1-MMAwas tested as the variedsubstrate, R1-ADMAacted as a competitive inhibitor (Table 4).By definition, a competitive inhibitor is a compound thatdirectly competes with a varied substrate for enzyme binding;hence, the AcH4-21R3MMA and R1-MMA peptides bind tothe same form of PRMT6 as their respective dimethylatedproducts. In contrast, when SAM was the varied substrate atsubsaturating fixed concentrations of either the AcH4-21R3MMA (Fig. 2B) or R1-MMA peptide, AcH4-21R3ADMAand R1-ADMA acted as noncompetitive inhibitors (Tables 3and 4). This inhibition pattern is expected because SAM doesnot bind to the same form of the enzyme as the ADMA-con-taining product and because the two binding events are sepa-rated by a reversible step. However, when these experimentswere repeated at saturating concentrations (100 � Km) of theAcH4-21R3MMA peptide, no inhibition was observed whenSAMwas varied at 50, 250, and 500�MAcH4-21R3ADMA (Fig.2C and Table 3). Note that the parallel experiment could not beperformed with R1-MMA because theKm of this peptide (45 �11 �M) is much greater than that of AcH4-21R3MMA (1.53 �0.34 �M) and because the high concentrations of R1-MMArequired to carry out the experiment are difficult to achieve due
TABLE 2Initial velocity studies
Varied Substrate Fixed substrate kcat Ki(pep) Kpep Ki(SAM) KSAM
min�1 �M �M �M �M
AcH4-21 SAMa 0.58 � 0.05 1.79 � 1.87 5.30 � 2.17 7.08 � 1.09AcH4-21R3MMA SAMa 0.14 � 0.01 2.97 � 2.50 1.93 � 0.69 1.43 � 0.22SAM AcH4-21b 0.15 � 0.01 0.47 � 0.86 29.5 � 6.7 2.12 � 0.88SAM AcH4-21R3MMAc 0.57 � 0.04 2.34 � 0.79 11.6 � 6.9 6.43 � 1.54R1 SAMd 1.03 � 0.18 325 � 157 107 � 74.0 11.9 � 4.10R1-MMA SAMe 0.59 � 0.04 160 � 31 20.8 � 11.9 6.68 � 1.00SAM R1f 0.29 � 0.02 7.20 � 2.53 161 � 70 2.99 � 1.55SAM R1-MMAf 0.26 � 0.03 5.55 � 4.65 47.9 � 38.7 2.31 � 1.35
a Fixed substrate concentrations 2.5, 5, and 10 �M.b Fixed substrate concentrations 1, 2.5, and 5 �M.c Fixed substrate concentrations 2.5, 10, and 15 �M.d Fixed substrate concentrations 2.5, 5, and 15 �M.e Fixed substrate concentrations 2.5, 5, 10, and 15 �M.f Fixed substrate concentrations 25, 50, 100, and 200 �M.
Kinetic Mechanism of PRMT6
FEBRUARY 17, 2012 • VOLUME 287 • NUMBER 8 JOURNAL OF BIOLOGICAL CHEMISTRY 6065
by guest on July 3, 2018http://w
ww
.jbc.org/D
ownloaded from

to solubility issues with this very hydrophobic peptide. Alsonote that reciprocal experiments with SAM were not per-formed because [methyl-14C]SAM is used as the methyl donorin these experiments, and it is not feasible to achieve therequired concentrations with this compound and maintain thesensitivity of the assay.Product inhibition experiments with SAH were performed
next. When either AcH4-21R3MMA or R1-MMA was assayedin the presence of increasing concentrations of SAH at a con-stant concentration of SAM, SAH acted as a noncompetitiveinhibitor (Fig. 3A and Tables 3 and 4). On the other hand, SAHacted as a competitive inhibitor when SAMwas the varied sub-strate and the concentration of either of the two peptides wasfixed at a non-saturating concentration (Fig. 3B and Tables 3and 4). When the AcH4-21R3MMA peptide concentration wasincreased to saturating levels (i.e. 500 �M) and SAM remainedthe varied substrate, SAH still exhibited a competitive patternof inhibition (Fig. 3C). Similar results were obtained for theunmethylated peptide substrates, R1 and AcH4-21 (supple-mental Tables S1 and S2). In total, the product inhibition pat-terns are consistent with both a rapid equilibrium randommechanism with dead-end EAP and EBQ complexes and aTheorell-Chance mechanism; the latter mechanism is a specialcase of an ordered mechanism in which the ternary complexforms only transiently.Dead-end Analog Studies—To differentiate between these
two mechanisms, dead-end analog studies were carried out.The dead-end analogs used in these studies were sinefungin, aSAM analog that contains an amine in place of the methylgroup, and two peptide derivatives that contain lysine residuesin place of the substrate arginine, AcH4-21R3K and R1-R6K.Note that these peptides are not methylated by PRMT6 abovebackground levels (Table 1), which indicates that they are suit-able for use as dead-end analogs of PRMT6.The results of these experiments show that the AcH4-21R3K
peptide acted as a competitive inhibitor when AcH4-21R3MMAwas varied at a fixed concentration of SAM (Fig. 4Aand Table 3). The R1-R6K peptide also acted as a competitiveinhibitor when R1-MMA was the varied substrate (Table 4).When SAM was the varied substrate, noncompetitive inhibi-tion patterns were observed for both AcH4-21R3K and R1-R6Kat subsaturating levels of the AcH4-21R3MMA and R1-MMApeptides, respectively (Fig. 4B and Tables 3 and 4). In contrast,no inhibition was observed when SAMwas tested as the variedsubstrate at saturating levels of the AcH4-21R3MMA peptide(Table 3).When sinefungin was tested as a dead-end analog at a fixed
concentration of SAM and AcH4-21R3MMA was the variedsubstrate, a noncompetitive pattern of inhibition was observed(Fig. 5A andTable 3). Sinefungin also acted as a noncompetitiveinhibitor when R1-MMA was the varied substrate (Table 4).Identical results were obtained with the unmethylated pep-tides, AcH4-21 andR1 (supplemental Tables S1 andS2). For theconverse experiments, in which SAM was the varied substrate
FIGURE 2. Product inhibition studies with AcH4-21R3ADMA. A, competi-tive inhibition was observed when AcH4-21R3MMA was the varied substrate,SAM was the fixed substrate (15 �M), and AcH4-21R3ADMA was the productinhibitor. B, noncompetitive inhibition was observed when SAM was the var-ied substrate, AcH4-21R3MMA was the fixed substrate (10 �M), and
AcH4-21R3ADMA was the product inhibitor. C, no inhibition was observedwhen SAM was the varied substrate, AcH4-21R3MMA was the fixed substrate(500 �M), and AcH4-21R3ADMA was the product inhibitor.
Kinetic Mechanism of PRMT6
6066 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 287 • NUMBER 8 • FEBRUARY 17, 2012
by guest on July 3, 2018http://w
ww
.jbc.org/D
ownloaded from

and the concentrations of AcH4-21R3MMA and R1-MMAwere fixed at subsaturating levels, sinefungin acted as a com-petitive inhibitor (Fig. 5B), and when the concentration ofAcH4-21R3MMA was increased to a saturating level, the pat-tern of inhibition remained competitive (Fig. 5C).
DISCUSSION
A number of histone-modifying enzymes, including thePRMTs, histone deacetylases, protein arginine deiminases, his-tone acetyltransferases, and lysine methyltransferases, haveemerged as potential therapeutic targets for a wide range ofdiseases, including cancer, heart disease, rheumatoid arthritis,colitis, and lupus (25, 26), although, in all cases, the therapeuticpotential of targeting these enzymes has not been validated.Inhibitors targeting the histone deacetylases have been ap-proved for the treatment of at least a subset of cancers, includ-ing cutaneous T-cell lymphoma (26). Additionally, we haveshown that protein arginine deiminase inhibition decreases dis-ease severity in mouse models of rheumatoid arthritis and coli-tis (27, 28).Given these precedents, we andothers have initiatedprograms to develop inhibitors targeting the PRMTs (19, 29,30). Recently, we described C21 as the first irreversible PRMTinhibitor. This compound, denoted C21, is quite selective forPRMT1 versus PRMT3 and PRMT4 but onlymodestly selectivefor PRMT6. To account for the fact that C21 inhibits PRMT6equally well in IC50 assays regardless of whether the inhibitor is
preincubated with the enzyme in the absence or presence ofSAM, we reinvestigated the kinetic mechanism of this enzymebecause such a result appears to be inconsistent with a previousreport (22) indicating that PRMT6uses an orderedmechanism.Consistent with the previous report, the results of our own ini-tial velocity studies show an intersecting line pattern, demon-strating that PRMT6 uses a sequential mechanism in whichboth substrates, SAM and the peptide, bind to the enzyme toform a ternary complex prior to methyl transfer. However, theresults of our product and dead-end analog inhibition studiesindicate that PRMT6 uses a rapid equilibrium random mecha-nism with dead-end EAP and EBQ complexes (Scheme 2).The previous report of an orderedmechanism (22) suggested
that SAM binds prior to the peptide substrate, methyl transferoccurs, and then the methylated peptide and SAH are releasedin that order (Scheme 1). The assignment of this mechanismwas based solely on the product inhibition patterns afforded bythe R1-ADMApeptide and SAH.Of note, SAHwas reported toact as a competitive inhibitor when SAM was the varied sub-strate and a noncompetitive inhibitor when the R1-MMA pep-tidewas the varied substrate.When the other product inhibitor,R1-ADMA,was tested, it exhibited a noncompetitive pattern ofinhibition when either peptide or SAM was the varied sub-strate. Although these results are consistent with the predictedinhibition patterns for the steady-state ordered mechanism
TABLE 3Inhibition results for AcH4-21R3MMA
Inhibitor Varied substrate Fixed substrate Inhibition pattern Kis Kii
�M �M
AcH4-21R3ADMA AcH4-21R3MMA 15 �M SAMa Competitive 43.5 � 20.5AcH4-21R3ADMA SAM 10 �M AcH4-21R3MMAb Noncompetitive 101 � 10 101 � 10AcH4-21R3ADMA SAM 500 �M AcH4-21R3MMAa No inhibitionSAH AcH4-21R3MMA 15 �M SAMc Noncompetitive 9.67 � 1.23 9.67 � 1.23SAH SAM 10 �M AcH4-21R3MMAc Competitive 4.86 � 1.42SAH SAM 500 �M AcH4-21R3MMAc Competitive 3.60 � 0.77AcH4-21R3K AcH4-21R3MMA 15 �M SAMd Competitive 20.2 � 5.1AcH4-21R3K SAM 10 �M AcH4-21R3MMAd Noncompetitive 161 � 16 161 � 16AcH4-21R3K SAM 500 �M AcH4-21R3MMAd No inhibitionSinefungin AcH4-21R3MMA 15 �M SAMe Noncompetitive 1.02 � 0.13 1.02 � 0.13Sinefungin SAM 10 �M AcH4-21R3MMAf Competitive 0.38 � 0.11Sinefungin SAM 500 �M AcH4-21R3MMAe Competitive 0.14 � 0.05
a50, 250, and 500 �M AcH4-21R3ADMA.b5, 50, 250 and �M AcH4-21R3ADMA.c2.5, 10, and 25 �M SAH.d5, 250, and 500 �M AcH4-21R3K.e0.1, 0.5, and 1.0 �M sinefungin.f0.1, 0.5, 1, and 2 �M sinefungin.
TABLE 4Inhibition results for R1-MMA
Inhibitor Varied substrate Fixed substrate Inhibition pattern Kis Kii
�M �M
R1-ADMA R1-MMA 15 �M SAMa Competitive 260 � 120R1-ADMA SAM 50 �M R1-MMAb Noncompetitive 1010 � 140 1010 � 140SAH R1-MMA 15 �M SAMc Noncompetitive 10.1 � 0.9 10.1 � 0.9SAH SAM 10 �M R1-MMAc Competitive 8.64 � 3.52R1-R6K R1-MMA 15 �M SAMd Competitive 120 � 20R1-R6K SAM 10 �M R1-MMAe Noncompetitive 520 � 55 520 � 55Sinefungin R1-MMA 15 �M SAMf Noncompetitive 1.40 � 0.12 1.40 � 0.12Sinefungin SAM 50 �M R1-MMAf Competitive 0.45 � 0.10
a5, 50, and 250 �M R1-ADMA.b50, 250, and 500 �M R1-ADMA.c2.5, 10, and 25 �M SAH.d5, 100, and 500 �M R1-R6K.e5, 250, and 500 �M R1-R6K.f0.1, 0.5, and 1.0 �M sinefungin.
Kinetic Mechanism of PRMT6
FEBRUARY 17, 2012 • VOLUME 287 • NUMBER 8 JOURNAL OF BIOLOGICAL CHEMISTRY 6067
by guest on July 3, 2018http://w
ww
.jbc.org/D
ownloaded from

described above, the data for the AcH4-21R3-ADMA andR1-ADMApeptides are inconsistentwith our own. Specifically,we observed that both ADMA-containing peptides act as com-petitive inhibitors of the PRMT6-catalyzed reaction when thecorresponding monomethylated peptides are the varied sub-strate (Fig. 2A and Tables 3 and 4). Furthermore, we observedno inhibition when SAM was the varied substrate and AcH4-21R3ADMAwas the product inhibitor at saturating amounts ofthe AcH4-21R3MMA peptide (Fig. 2C); an uncompetitive pat-tern of inhibition is expected for the ordered mechanismdescribed by Lakowski and Frankel (22).Although these inhibition patterns are not consistent with
that specific ordered mechanism, they are consistent with bothaTheorell-Chancemechanism and a rapid equilibrium randommechanismwith dead-end EAP and EBQ complexes. To differ-entiate between these two remaining mechanisms and to con-firm further that PRMT6 does not use a steady-state orderedmechanism, we performed dead-end analog inhibition studies.The fact that uncompetitive inhibition was not observed witheither the AcH4-21R3K peptide or sinefungin, under any con-dition, rules out the Theorell-Chance mechanism. The dataobtained with the AcH4-21R3K peptide also rule out theorderedmechanism described above. This is the case because anoncompetitive pattern of inhibition is expected for an orderedmechanism if the inhibitor can bind to both the EA and EQforms of the enzyme, whereas we have shown that the Lys-containing dead-end inhibitor peptides acted as competitiveinhibitors when the corresponding peptide was used as the var-ied substrate (Fig. 4A).When SAM was the varied substrate and the AcH4-21R3K
peptide was tested as the dead-end analog, a noncompetitivepattern of inhibition was observed with subsaturating levels ofthe MMA-containing peptide (Fig. 4B). In contrast, no inhibi-tion was noted when the concentration of AcH4-21R3MMAwas increased to saturating levels (Fig. 4C). Both of these resultsalso contradict the predicted inhibition patterns dictated by anordered mechanism. Uncompetitive inhibition is expectedunder both conditions. This is the case because the AcH4-21R3K peptide binds after SAM and decreases the pool ofenzyme available for binding to SAM, and this outcome willoccur regardless of the peptide concentration. In contrast, ourresults show no inhibition at high concentrations of the sub-strate peptide, which is expected for a random mechanismbecause the dead-end peptide analog is effectively out-com-peted by the substrate peptide. Thus, the data indicate that, likePRMT1 (17), PRMT6 utilizes a rapid equilibrium randommechanism with dead-end EAP and EBQ complexes.As stated above, it is possible that the use of a poor substrate
could make a random mechanism appear ordered. However,the results obtained when the R1-R6K peptide was used as thedead-end analog are identical to those obtainedwith theAcH4-21R3K peptide (Tables 3 and 4). Identical patterns of inhibitionwere also obtained for the unmethylated substrates, i.e. theAcH4-21 and R1 peptides (supplemental Tables S1 and S2).
FIGURE 3. Product inhibition studies with SAH. A, noncompetitive inhibi-tion was observed when AcH4-21R3MMA was the varied substrate, SAM wasthe fixed substrate (15 �M), and SAH was the product inhibitor. B, competitiveinhibition was observed when SAM was the varied substrate, AcH4-21R3MMAwas the fixed substrate (10 �M), and SAH was the product inhibitor. C, com-petitive inhibition was observed when SAM was the varied substrate,
AcH4-21R3MMA was the fixed substrate (500 �M), and SAH was the productinhibitor.
Kinetic Mechanism of PRMT6
6068 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 287 • NUMBER 8 • FEBRUARY 17, 2012
by guest on July 3, 2018http://w
ww
.jbc.org/D
ownloaded from

Thus, regardless of the peptide substrate tested, PRMT6 uses arapid equilibrium randommechanism with dead-end EAP andEBQ complexes. Although it is difficult to speculate about why
FIGURE 4. Dead-end analog inhibition studies with AcH4-21R3K. A, com-petitive inhibition was observed when AcH4-21R3MMA was the varied sub-strate, SAM was the fixed substrate (15 �M), and AcH4-21R3K was the dead-end analog inhibitor. B, noncompetitive inhibition was observed when SAMwas the varied substrate, AcH4-21R3MMA was the fixed substrate (10 �M),and AcH4-21R3K was the dead-end analog inhibitor. C, no inhibition wasobserved when SAM was the varied substrate, AcH4-21R3MMA was the fixedsubstrate (500 �M), and AcH4-21R3K was the dead-end analog inhibitor.
FIGURE 5. Dead-end analog inhibition studies with sinefungin. A, non-competitive inhibition was observed when AcH4-21R3MMA was the variedsubstrate, SAM was the fixed substrate (15 �M), and sinefungin was the dead-end analog inhibitor. B, competitive inhibition was observed when SAM wasthe varied substrate, AcH4-21R3MMA was the fixed substrate (10 �M), andsinefungin was the dead-end analog inhibitor. C, competitive inhibition wasobserved when SAM was the varied substrate, AcH4-21R3MMA was the fixedsubstrate (500 �M), and sinefungin was the dead-end analog inhibitor.
Kinetic Mechanism of PRMT6
FEBRUARY 17, 2012 • VOLUME 287 • NUMBER 8 JOURNAL OF BIOLOGICAL CHEMISTRY 6069
by guest on July 3, 2018http://w
ww
.jbc.org/D
ownloaded from

we observed a random mechanism, whereas Lakowski andFrankel (22) observed an ordered mechanism, we again notethat the previous assignment was based solely on the productinhibition patterns afforded by the R1-ADMA peptide andSAH and that their assignment of an ordered versus randommechanism relied only on one key experiment (i.e. the patternafforded by the R1-ADMA peptide when SAM was the variedsubstrate). Thus, our data, which also incorporate the results ofdead-end analog inhibition studies, give a higher degree of con-fidence that the assignment of a randommechanism is correct.This is especially true when one considers that the results wereconfirmed with multiple peptides. The different assignmentsmay be due in part to differences in the assay. Lakowski andFrankel used an MS-based assay, whereas we used a gel-basedradioactive assay (16). A key benefit of our assay is that issuesregarding PRMT6 automethylation are circumvented becausethe products are separated from other reaction substituents bySDS-PAGEand can be reproducibly quantified by phosphorim-aging analysis. Additionally, we note that the overall quality ofthe double-reciprocal plots provided in the previous study arerelatively poor and that the authorswere unable to interpret theresults of the analogous experiments performed with the R1peptide (22).
CONCLUSION
The work described herein demonstrates that PRMT6 uti-lizes a rapid equilibrium random mechanism with dead-endEAP and EBQ complexes to catalyze the methylation of thehistoneH4-based substratesAcH4-21 andAcH4-21-MMAandthe fibrillarin-based substrates R1 and R1-MMA. AlthoughPRMT6 displays limited processivity with either the AcH4-21or R1 peptide (data not shown), the fact that this kinetic mech-anism includes an EBQ (or enzyme�SAM�AcH4-21-MMA)complex, which can go on to form the dimethylated product,suggests that PRMT6-interacting proteins could modulate theprocessivity of the enzyme to increase the dimethylation of spe-cific arginine residues. Finally, the assignment of a rapid equi-librium randommechanismwith dead-end EAP and EBQ com-plexes is consistent with our previous report demonstratingthat PRMT1 utilizes the same mechanism with AcH4-21 andAcH4-21-MMA (17).With regard to inhibitor development, these results are sig-
nificant because they suggest that high throughput screensdesigned to identify inhibitors targeting the peptide-bindingpocket on PRMT6 need not include SAM in the assay buffer. Inthe future, these studies set the stage for characterizing thesubstrate specificity of PRMT6 and identifying the factors thatregulate PRMT6 activity. Information from these studies willalso undoubtedly aid our effort to develop inhibitors withincreased selectivity for both PRMT1 and PRMT6, and ulti-mately, the increased understanding of this enzyme and thePRMT family will facilitate future investigations of the in vivoactivity of this enzyme.
REFERENCES1. Bedford, M. T. (2007) Arginine methylation at a glance. J. Cell Sci. 120,
4243–42462. Bedford, M. T., and Clarke, S. G. (2009) Protein arginine methylation in
mammals: who, what, and why.Mol. Cell 33, 1–13
3. Vallance, P., and Leiper, J. (2004) Cardiovascular biology of the asymmet-ric dimethylarginine:dimethylarginine dimethylaminohydrolase pathway.Arterioscler. Thromb. Vasc. Biol. 24, 1023–1030
4. Krause, C. D., Yang, Z. H., Kim, Y. S., Lee, J. H., Cook, J. R., and Pestka, S.(2007) Protein arginine methyltransferases: evolution and assessment oftheir pharmacological and therapeutic potential. Pharmacol. Ther. 113,50–87
5. Tang, J., Gary, J. D., Clarke, S., andHerschman,H. R. (1998) PRMT3, a typeI protein arginine N-methyltransferase that differs from PRMT1 in itsoligomerization, subcellular localization, substrate specificity, and regula-tion. J. Biol. Chem. 273, 16935–16945
6. McBride, A. E., and Silver, P. A. (2001) State of the Arg: protein methyla-tion at arginine comes of age. Cell 106, 5–8
7. Torres-Padilla, M. E., Parfitt, D. E., Kouzarides, T., and Zernicka-Goetz,M. (2007) Histone arginine methylation regulates pluripotency in theearly mouse embryo. Nature 445, 214–218
8. Hassa, P. O., Covic, M., Bedford, M. T., and Hottiger, M. O. (2008)Protein arginine methyltransferase 1 coactivates NF-�B-dependentgene expression synergistically with CARM1 and PARP1. J. Mol. Biol.377, 668–678
9. Tang, J., Frankel, A., Cook, R. J., Kim, S., Paik, W. K., Williams, K. R.,Clarke, S., and Herschman, H. R. (2000) PRMT1 is the predominant typeI protein arginine methyltransferase in mammalian cells. J. Biol. Chem.275, 7723–7730
10. Leiper, J., Nandi,M., Torondel, B.,Murray-Rust, J.,Malaki,M., O’Hara, B.,Rossiter, S., Anthony, S., Madhani, M., Selwood, D., Smith, C., Wojciak-Stothard, B., Rudiger, A., Stidwill, R., McDonald, N. Q., and Vallance, P.(2007) Disruption of methylarginine metabolism impairs vascular home-ostasis. Nat. Med. 13, 198–203
11. Vallance, P., Leone, A., Calver, A., Collier, J., and Moncada, S. (1992)Accumulation of an endogenous inhibitor of nitric oxide synthesis inchronic renal failure. Lancet 339, 572–575
12. Tran, C. T., Leiper, J. M., and Vallance, P. (2003) The DDAH/ADMA/NOS pathway. Atheroscler. Suppl. 4, 33–40
13. Le Romancer, M., Treilleux, I., Leconte, N., Robin-Lespinasse, Y., Sentis,S., Bouchekioua-Bouzaghou, K., Goddard, S., Gobert-Gosse, S., andCorbo, L. (2008) Regulation of estrogen rapid signaling through argininemethylation by PRMT1.Mol. Cell 31, 212–221
14. Yoshimatsu, M., Toyokawa, G., Hayami, S., Unoki, M., Tsunoda, T., Field,H. I., Kelly, J. D., Neal, D. E.,Maehara, Y., Ponder, B. A., Nakamura, Y., andHamamoto, R. (2011) Dysregulation of PRMT1 and PRMT6, type I argi-nine methyltransferases, is involved in various types of human cancers.Int. J. Cancer 128, 562–573
15. Michaud-Levesque, J., and Richard, S. (2009) Thrombospondin-1 is atranscriptional repression target of PRMT6. J. Biol. Chem. 284,21338–21346
16. Osborne, T. C., Obianyo, O., Zhang, X., Cheng, X., and Thompson, P. R.(2007) Protein argininemethyltransferase 1: positively charged residues insubstrate peptides distal to the site of methylation are important for sub-strate binding and catalysis. Biochemistry 46, 13370–13381
17. Obianyo, O., Osborne, T. C., and Thompson, P. R. (2008) Kinetic mecha-nism of protein arginine methyltransferase 1. Biochemistry 47,10420–10427
18. Rust, H. L., Zurita-Lopez, C. I., Clarke, S., and Thompson, P. R. (2011)Mechanistic studies on transcriptional coactivator protein argininemeth-yltransferase 1. Biochemistry 50, 3332–3345
19. Obianyo, O., Causey, C. P., Osborne, T. C., Jones, J. E., Lee, Y. H., Stallcup,M. R., andThompson, P. R. (2010) A chloroacetamidine-based inactivatorof protein arginine methyltransferase 1: design, synthesis, and in vitro andin vivo evaluation. ChemBioChem 11, 1219–1223
20. Hyllus, D., Stein, C., Schnabel, K., Schiltz, E., Imhof, A., Dou, Y., Hsieh, J.,and Bauer, U. M. (2007) PRMT6-mediated methylation of R2 in histoneH3 antagonizes H3K4 trimethylation. Genes Dev. 21, 3369–3380
21. El-Andaloussi, N., Valovka, T., Toueille,M., Steinacher, R., Focke, F., Geh-rig, P., Covic,M., Hassa, P. O., Schar, P., Hubscher, U., andHottiger,M.O.(2006) Arginine methylation regulates DNA polymerase �. Mol. Cell 22,51–62
22. Lakowski, T. M., and Frankel, A. (2008) A kinetic study of human protein
Kinetic Mechanism of PRMT6
6070 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 287 • NUMBER 8 • FEBRUARY 17, 2012
by guest on July 3, 2018http://w
ww
.jbc.org/D
ownloaded from

arginine N-methyltransferase 6 reveals a distributive mechanism. J. Biol.Chem. 283, 10015–10025
23. Miranda, T. B., Miranda, M., Frankel, A., and Clarke, S. (2004) PRMT7 isa member of the protein arginine methyltransferase family with a distinctsubstrate specificity. J. Biol. Chem. 279, 22902–22907
24. Leatherbarrow, R. J. (2004) GraFit Version 5.0, Erithacus Software,Staines, United Kingdom
25. Jones, J. E., Causey, C. P., Knuckley, B., Slack-Noyes, J. L., and Thompson,P. R. (2009) Protein arginine deiminase 4 (PAD4): current understandingand future therapeutic potential. Curr. Opin. Drug Discov. Devel. 12,616–627
26. Marks, P. A., and Breslow, R. (2007) Dimethyl sulfoxide to vorinostat:development of this histone deacetylase inhibitor as an anticancer drug.Nat. Biotechnol. 25, 84–90
27. Willis, V. C., Gizinski, A. M., Banda, N. K., Causey, C. P., Knuckley, B.,Cordova, K. N., Luo, Y., Levitt, B., Glogowska, M., Chandra, P., Kulik, L.,
Robinson,W.H., Arend,W. P., Thompson, P. R., andHolers, V.M. (2011)N-�-Benzoyl-N5-(2-chloro-1-iminoethyl)-L-ornithine amide, a proteinarginine deiminase inhibitor, reduces the severity of murine collagen-induced arthritis. J. Immunol. 186, 4396–4404
28. Chumanevich, A. A., Causey, C. P., Knuckley, B. A., Jones, J. E., Poudyal,D., Chumanevich, A. P., Davis, T., Matesic, L. E., Thompson, P. R., andHofseth, L. J. (2011) Suppression of colitis in mice by Cl-amidine: a novelpeptidylarginine deiminase inhibitor. Am. J. Physiol. Gastrointest. LiverPhysiol. 300, G929–G938
29. Lakowski, T. M., ’t Hart, P., Ahern, C. A., Martin, N. I., and Frankel, A.(2010) N�-Substituted arginyl peptide inhibitors of protein arginine N-methyltransferases. ACS Chem. Biol. 5, 1053–1063
30. Sack, J. S., Thieffine, S., Bandiera, T., Fasolini, M., Duke, G. J., Jayaraman,L., Kish, K. F., Klei, H. E., Purandare, A.V., Rosettani, P., Troiani, S., Xie, D.,and Bertrand, J. A. (2011) Structural basis for CARM1 inhibition by indoleand pyrazole inhibitors. Biochem. J. 436, 331–339
Kinetic Mechanism of PRMT6
FEBRUARY 17, 2012 • VOLUME 287 • NUMBER 8 JOURNAL OF BIOLOGICAL CHEMISTRY 6071
by guest on July 3, 2018http://w
ww
.jbc.org/D
ownloaded from

Obiamaka Obianyo and Paul R. ThompsonKinetic Mechanism of Protein Arginine Methyltransferase 6 (PRMT6)
doi: 10.1074/jbc.M111.333609 originally published online January 3, 20122012, 287:6062-6071.J. Biol. Chem.
10.1074/jbc.M111.333609Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
Supplemental material:
http://www.jbc.org/content/suppl/2012/01/03/M111.333609.DC1
http://www.jbc.org/content/287/8/6062.full.html#ref-list-1
This article cites 29 references, 10 of which can be accessed free at
by guest on July 3, 2018http://w
ww
.jbc.org/D
ownloaded from