
Killing of Resistant Cancer Cells with Low Bak by a...
10
Cancer Therapy: Preclinical Killing of Resistant Cancer Cells with Low Bak by a Combination of an Antimesothelin Immunotoxin and a TRAIL Receptor 2 Agonist Antibody Xing Du 1 , Laiman Xiang 1 , Crystal Mackall 2 , and Ira Pastan 1 Abstract Purpose: Many solid tumors express cell surface mesothelin making them attractive targets for antibody- based therapies of cancer. SS1P [antimesothelin(Fv)PE38] is a recombinant immunotoxin (RIT) that has potent cytotoxic activity on several cancer cell lines and clinical activity in mesothelioma patients. Pancreatic cancers express mesothelin and are known to be resistant to most chemotherapeutic agents. The goal of this study is to treat pancreatic cancer with RIT by targeting mesothelin. Experimental Design: We measured the cytotoxic activity of an antimesothelin immunotoxin on pancreatic cancer cells. We also measured the levels of several pro- and antiapoptotic proteins, as well as the ability of TNF-related apoptosis-inducing ligand (TRAIL) or the anti-TRAIL receptor 2 agonist antibody (HGS-ETR2) to kill pancreatic cells, and the cytotoxic activity of the two agents together in cell culture and against tumors in mice. Results: In two pancreatic cancer cell lines, immunotoxin treatment inhibited protein synthesis but did not produce significant cell death. The resistant lines had low levels of the proapoptotic protein Bak. Increasing Bak expression enhanced the sensitivity to immunotoxins, whereas Bak knockdown diminished it. We also found that combining immunotoxin with TRAIL or HGS-ETR2 caused synergistic cell death, and together triggered caspase-8 recruitment and activation, Bid cleavage and Bax activation. Combining SS1P with HGS-ETR2 also acted synergistically to decrease tumor burden in a mouse model. Conclusion: Our data show that low Bak can cause cancer cells to be resistant to immunotoxin treatment and that combining immunotoxin with TRAIL or a TRAIL agonist antibody can overcome resistance. Clin Cancer Res; 17(18); 5926–34. Ó2011 AACR. Introduction Since the Food and Drug Administration approved the first monoclonal antibody (mAb) rituximab in 1997 for the treatment of B cell lymphomas, many mAbs have been evaluated for their ability to cause tumor regressions (1), but only a few have been effective. To improve the ther- apeutic usefulness of antibodies, they are now being used with a variety of cytotoxic substances such as small mo- lecular drugs, radioisotopes, or protein toxins (2). Our laboratory has focused on the development of recombinant immunotoxins (RIT) for cancer treatment (3). These proteins are composed of an Fv that binds to an antigen on a cancer cell fused to a 38-kD portion of Pseudomonas exotoxin A (PE38). They derive their potency from the toxin and their specificity from the antibody fragment to which they are attached. We now have several RITs in clinical trials; one of these is Moxetumomab pasu- dotox (also known as CAT-8015 or HA22), which targets CD22 on B cells malignancies. It has produced a very high complete remission rate in chemotherapy-resistant hairy cell leukemia and is now being evaluated in other B cell malignancies (4–6). Another is SS1P [antimesothelin(Fv) PE38] that targets mesothelin, a 40-kD cell surface glyco- protein that is present on mesotheliomas and pancreatic, ovarian, and lung cancers and cholangiocarcinomas (7–11). SS1P is composed of an antimesothelin Fv linked to PE38. It has shown significant cytotoxic activity in vitro against ovarian, mesothelioma, lung, and cholangiocarci- noma cancer cells (7–11). SS1P has been evaluated in 2 phase I clinical trials. It was well tolerated and showed some antitumor activity in patients with mesothelioma (12, 13). A new trial in which SS1P is being given in combination with cisplatin and pemetrexed is ongoing (14). To kill cells, an immunotoxin must be internalized and the toxin portion delivered to the cytosol via the Authors' Affiliations: 1 Laboratory of Molecular Biology, Center for Cancer Research, and 2 Pediatric Oncology Branch, National Cancer Institute, NIH, Bethesda, Maryland Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/). Corresponding Author: Ira Pastan, Laboratory of Molecular Bio- logy, National Cancer Institute, 37 Convent Drive, Room 5106, Bethesda, MD 20892. Phone: 301-496-4797; Fax: 301-402-1344; E-mail: [email protected] doi: 10.1158/1078-0432.CCR-11-1235 Ó2011 American Association for Cancer Research. Clinical Cancer Research Clin Cancer Res; 17(18) September 15, 2011 5926 on June 5, 2018. © 2011 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from Published OnlineFirst August 3, 2011; DOI: 10.1158/1078-0432.CCR-11-1235
Transcript of Killing of Resistant Cancer Cells with Low Bak by a...

Cancer Therapy: Preclinical
Killing of Resistant Cancer Cells with Low Bak by aCombination of an Antimesothelin Immunotoxin anda TRAIL Receptor 2 Agonist Antibody
Xing Du1, Laiman Xiang1, Crystal Mackall2, and Ira Pastan1
AbstractPurpose:Many solid tumors express cell surfacemesothelinmaking them attractive targets for antibody-
based therapies of cancer. SS1P [antimesothelin(Fv)PE38] is a recombinant immunotoxin (RIT) that has
potent cytotoxic activity on several cancer cell lines and clinical activity in mesothelioma patients.
Pancreatic cancers express mesothelin and are known to be resistant to most chemotherapeutic agents.
The goal of this study is to treat pancreatic cancer with RIT by targeting mesothelin.
Experimental Design: We measured the cytotoxic activity of an antimesothelin immunotoxin on
pancreatic cancer cells. We alsomeasured the levels of several pro- and antiapoptotic proteins, as well as the
ability of TNF-related apoptosis-inducing ligand (TRAIL) or the anti-TRAIL receptor 2 agonist antibody
(HGS-ETR2) to kill pancreatic cells, and the cytotoxic activity of the two agents together in cell culture and
against tumors in mice.
Results: In two pancreatic cancer cell lines, immunotoxin treatment inhibited protein synthesis but did
not produce significant cell death. The resistant lines had low levels of the proapoptotic protein Bak.
Increasing Bak expression enhanced the sensitivity to immunotoxins, whereas Bak knockdown diminished
it. We also found that combining immunotoxin with TRAIL or HGS-ETR2 caused synergistic cell death, and
together triggered caspase-8 recruitment and activation, Bid cleavage and Bax activation. Combining SS1P
with HGS-ETR2 also acted synergistically to decrease tumor burden in a mouse model.
Conclusion: Our data show that low Bak can cause cancer cells to be resistant to immunotoxin
treatment and that combining immunotoxin with TRAIL or a TRAIL agonist antibody can overcome
resistance. Clin Cancer Res; 17(18); 5926–34. �2011 AACR.
Introduction
Since the Food and Drug Administration approved thefirst monoclonal antibody (mAb) rituximab in 1997 for thetreatment of B cell lymphomas, many mAbs have beenevaluated for their ability to cause tumor regressions (1),but only a few have been effective. To improve the ther-apeutic usefulness of antibodies, they are now being usedwith a variety of cytotoxic substances such as small mo-lecular drugs, radioisotopes, or protein toxins (2).
Our laboratory has focused on the development ofrecombinant immunotoxins (RIT) for cancer treatment
(3). These proteins are composed of an Fv that binds toan antigen on a cancer cell fused to a 38-kD portion ofPseudomonas exotoxin A (PE38). They derive their potencyfrom the toxin and their specificity from the antibodyfragment to which they are attached. We now have severalRITs in clinical trials; one of these is Moxetumomab pasu-dotox (also known as CAT-8015 or HA22), which targetsCD22 on B cells malignancies. It has produced a very highcomplete remission rate in chemotherapy-resistant hairycell leukemia and is now being evaluated in other B cellmalignancies (4–6). Another is SS1P [antimesothelin(Fv)PE38] that targets mesothelin, a 40-kD cell surface glyco-protein that is present on mesotheliomas and pancreatic,ovarian, and lung cancers and cholangiocarcinomas(7–11). SS1P is composed of an antimesothelin Fv linkedto PE38. It has shown significant cytotoxic activity in vitroagainst ovarian, mesothelioma, lung, and cholangiocarci-noma cancer cells (7–11). SS1P has been evaluated in 2phase I clinical trials. It waswell tolerated and showed someantitumor activity in patients with mesothelioma (12, 13).A new trial in which SS1P is being given in combinationwith cisplatin and pemetrexed is ongoing (14).
To kill cells, an immunotoxin must be internalizedand the toxin portion delivered to the cytosol via the
Authors' Affiliations: 1Laboratory of Molecular Biology, Center for CancerResearch, and 2Pediatric Oncology Branch, National Cancer Institute, NIH,Bethesda, Maryland
Note: Supplementary data for this article are available at Clinical CancerResearch Online (http://clincancerres.aacrjournals.org/).
Corresponding Author: Ira Pastan, Laboratory of Molecular Bio-logy, National Cancer Institute, 37 Convent Drive, Room 5106,Bethesda, MD 20892. Phone: 301-496-4797; Fax: 301-402-1344;E-mail: [email protected]
doi: 10.1158/1078-0432.CCR-11-1235
�2011 American Association for Cancer Research.
ClinicalCancer
Research
Clin Cancer Res; 17(18) September 15, 20115926
on June 5, 2018. © 2011 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst August 3, 2011; DOI: 10.1158/1078-0432.CCR-11-1235
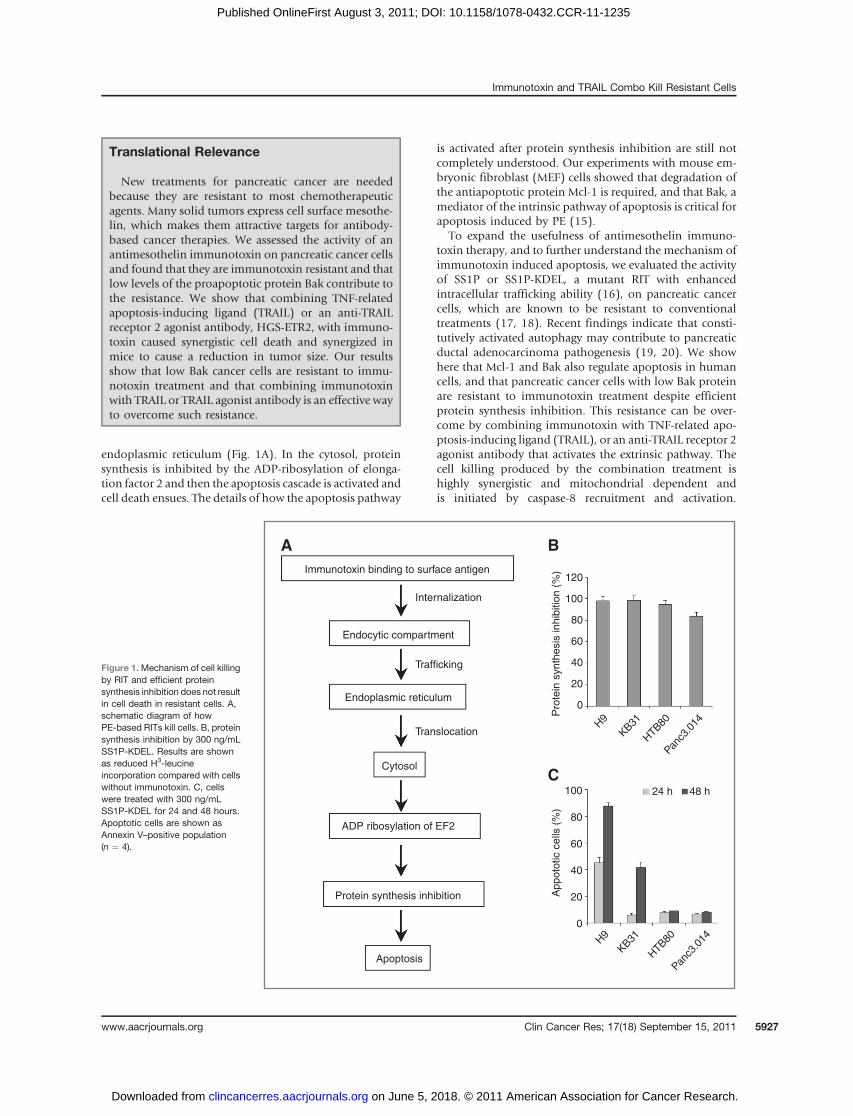
endoplasmic reticulum (Fig. 1A). In the cytosol, proteinsynthesis is inhibited by the ADP-ribosylation of elonga-tion factor 2 and then the apoptosis cascade is activated andcell death ensues. The details of how the apoptosis pathway
is activated after protein synthesis inhibition are still notcompletely understood. Our experiments with mouse em-bryonic fibroblast (MEF) cells showed that degradation ofthe antiapoptotic protein Mcl-1 is required, and that Bak, amediator of the intrinsic pathway of apoptosis is critical forapoptosis induced by PE (15).
To expand the usefulness of antimesothelin immuno-toxin therapy, and to further understand the mechanism ofimmunotoxin induced apoptosis, we evaluated the activityof SS1P or SS1P-KDEL, a mutant RIT with enhancedintracellular trafficking ability (16), on pancreatic cancercells, which are known to be resistant to conventionaltreatments (17, 18). Recent findings indicate that consti-tutively activated autophagy may contribute to pancreaticductal adenocarcinoma pathogenesis (19, 20). We showhere that Mcl-1 and Bak also regulate apoptosis in humancells, and that pancreatic cancer cells with low Bak proteinare resistant to immunotoxin treatment despite efficientprotein synthesis inhibition. This resistance can be over-come by combining immunotoxin with TNF-related apo-ptosis-inducing ligand (TRAIL), or an anti-TRAIL receptor 2agonist antibody that activates the extrinsic pathway. Thecell killing produced by the combination treatment ishighly synergistic and mitochondrial dependent andis initiated by caspase-8 recruitment and activation.
Translational Relevance
New treatments for pancreatic cancer are neededbecause they are resistant to most chemotherapeuticagents. Many solid tumors express cell surface mesothe-lin, which makes them attractive targets for antibody-based cancer therapies. We assessed the activity of anantimesothelin immunotoxin on pancreatic cancer cellsand found that they are immunotoxin resistant and thatlow levels of the proapoptotic protein Bak contribute tothe resistance. We show that combining TNF-relatedapoptosis-inducing ligand (TRAIL) or an anti-TRAILreceptor 2 agonist antibody, HGS-ETR2, with immuno-toxin caused synergistic cell death and synergized inmice to cause a reduction in tumor size. Our resultsshow that low Bak cancer cells are resistant to immu-notoxin treatment and that combining immunotoxinwith TRAIL or TRAIL agonist antibody is an effective wayto overcome such resistance.
Figure 1.Mechanism of cell killingby RIT and efficient proteinsynthesis inhibition does not resultin cell death in resistant cells. A,schematic diagram of howPE-based RITs kill cells. B, proteinsynthesis inhibition by 300 ng/mLSS1P-KDEL. Results are shownas reduced H3-leucineincorporation compared with cellswithout immunotoxin. C, cellswere treated with 300 ng/mLSS1P-KDEL for 24 and 48 hours.Apoptotic cells are shown asAnnexin V–positive population(n ¼ 4).
Immunotoxin binding to surface antigen
A B
C
Internalization
120
100
80
60
40
20
0
24 h 48 h100
80
60
40
20
0
H9
KB31
HTB
80Pan
c3.0
14
H9
KB31
HTB
80Pan
c3.0
14
Pro
tein
synth
esis
inhib
itio
n (
%)
Appoto
tic c
ells
(%
)
Trafficking
Translocation
Endocytic compartment
Endoplasmic reticulum
ADP ribosylation of EF2
Protein synthesis inhibition
Apoptosis
Cytosol
Immunotoxin and TRAIL Combo Kill Resistant Cells
www.aacrjournals.org Clin Cancer Res; 17(18) September 15, 2011 5927
on June 5, 2018. © 2011 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst August 3, 2011; DOI: 10.1158/1078-0432.CCR-11-1235

Synergistic antitumor activity was also observed in micereceiving a combination of immunotoxin and anti-TRAILreceptor 2 agonist antibody.
Materials and Methods
ReagentsRecombinant human and mouse TRAIL were purchased
from R&D Systems. The anti-TRAIL receptor 2 agonistantibody HGS-ETR2 was provided by Human GenomeScience. Immunotoxins SS1P, SS1P-KDEL, HB21(Fv)-PE40, TGFa-PE38, and LMB9 were produced in our lab.Human insulin was obtained from the NIH pharmacy.Puromycin was purchased from Invitrogen. 3H-leucinewas purchased from GE Healthcare.
CellsA431/H9 (mesothelin stable cell line), KB31, Hela, MEF,
and MEF (Bak�/�) were maintained in Dulbecco’s Mod-ified Eagle Medium with 10% FBS. Pancreatic cell linePanc3.014 was obtained from Dr. Elizabeth Jaffee (Depart-ment of Oncology, Johns Hopkins University, Baltimore,MD) and maintained in RPMI-1640 with 20% FBS and 0.2unit/mL human insulin. Pancreatic cell line HTB80(Capan-2, American Type Culture Collection) was main-tained in RPMI-1640 with 10% FBS.
Surface TRAIL receptor and mesothelin expression,protein synthesis and apoptosis assay, immunoblotand immunoprecipitation
TRAIL receptor expression was detected with anti-humanTRAIL R1 and R2 antibodies (R&D System). Mesothelinexpression was detected withMORAB-009 (SS1 humanizedmAb). Protein synthesis inhibition was measured with 3H-leucine incorporation. Detailed methods are described inthe Supplementary Methods.
After treatment, both floating and adherent cells werecollected, washed with cold DPBS twice, and solubilized inlysis buffer (50 mmol/L Tris-HCl pH 7.5, 150 mmol/LNaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS)with protease inhibitors and subjected to Western blotting.For immunoprecipitation and Bax activation, cell lysateswere prepared in 1% CHAPS lysis buffer and incubatedwith mouse anti-Bax (6A7). Detailed methods and anti-bodies for Western blotting are described in the Supple-mentary Methods.
Bak overexpression and siRNA knockdown of Bak,Bax and Bid
A cDNA for human Bak was cloned into a lentiviralexpression vector pLVX-PURO (Clontech). Lentivirus wasproduced by SAIC-Frederick (Frederick, MD) and trans-duced into KB31. Stable clones were selected with 1.5 mg/mL puromycin. Bak expression levels were confirmed byWestern blot and cells were treated with 100 ng/mL SS1Pfor indicated times and subjected to apoptosis analysis.
siRNAs (control, Hs-Bak1_5, Hs_Bax_9, Hs_Bid_7; Qia-gen) were transfected into cells at the indicated concentra-
tions with Lipofectamine RNAiMAX (Invitrogen) accordingto the manufacturer’s protocol.
Xenograft tumor modelKB31 (1.7 � 106) with Matrigel (4.0 mg/mL, 200 mL per
mouse) were implanted subcutaneously into the thigharea of the rear leg of 5- to 6-week-old athymic nude mice(18–20 g). Tumor dimensions were determined every otherday with a caliper. Tumor volume (mm3) was calculated bythe formula: (a) � (b2) � 0.4, in which (a) is tumor lengthand (b) is tumor width in millimeters.
When tumor size reached 110 mm3 (day 8), 10 mg/kgHGS-ETR2 was given intraperitoneally and 4 hours later, 3doses of SS1P 0.3 mg/kg was given i.v. QOD (days 8, 10,and 12). The animal protocol was approved by the Na-tional Cancer Institute (NCI) Animal Care and Use Com-mittee. All animal experiments were stopped when tumorsreached 1,000 mm3 based on animal protocols.
StatisticsAll data are presented as mean � SD from at least 3
independent assays. Statistical analysis of synergy on tumorexperiments was done by Drs. David Venzon and DavidLiewehr (Biostatistics and Data Management Section/Cen-ter for Cancer Research/NCI, Bethesda, MD). Tumorvolumes were logarithmically transformed before analysisto normalize their distribution and to stabilize variances.Differences between treatment groups were assessed byrepeated measures ANOVA, and 2-tailed P values fromDunnett’s test were reported for the multiple comparisonswithin days. Synergy was defined as an interaction effectsignificantly greater than the sum of the SS1P and HGS-ETR2 effect.
Results
Immunotoxin does not induce cell death despiteefficient protein synthesis inhibition
Pancreatic cell lines HTB80 and Panc3.014 have strongcell surfacemesothelin expression (Supplementary Fig. S1),and the expression levels are in the range of 2 sensitive celllines often used in our lab, KB31 and A431/H9. We thentreated the 4 cell lines with SS1P at 300 ng/mL and foundthat protein synthesis was completely arrested in A431/H9and KB31 cells but not in HTB80 and Panc3.014 (data notshown). We have previously shown that in some cell lineschanging the c-terminus of an immunotoxin to KDEL,which binds more tightly to the recycling receptor, canimprove protein synthesis inhibition and indeed foundSS1P-KDEL at 300 ng/mL profoundly inhibited proteinsynthesis in the 2 pancreatic cell lines (Fig. 1B) andemployed SS1P-KDEL in all further experiments with pan-creatic cells. When examined at 24 hours, more than 45%of the A431/H9 were undergoing apoptosis, which in-creased to more than 90% at 48 hours (Fig. 1C). Significantapoptosis became evident at 48 hours in the KB31(Fig. 1C). No significant apoptosis was detected inHTB80 or Panc3.014 at 48 and even 72 hours (Fig. 1C
Du et al.
Clin Cancer Res; 17(18) September 15, 2011 Clinical Cancer Research5928
on June 5, 2018. © 2011 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst August 3, 2011; DOI: 10.1158/1078-0432.CCR-11-1235

and unpublished data). These results indicate that despiteprofound protein synthesis inhibition, SS1P-KDEL doesnot cause the death of HTB80 and Panc3.014 cells, indi-cating that these 2 cell lines are highly resistant to immu-notoxin, as observed in a recent study that DLD1 cells donot undergo apoptosis when protein synthesis is inhibitedby immunotoxins (21).
Low expression of Bak protein in resistant cells andBak-dependent apoptosis induced by immunotoxinUsing MEFs, we have previously found that apoptosis
induced by PE is Bak dependent (15). In this study, weexamined Bak protein levels and found that expressionlevels were low in these 2 cell lines (Fig. 2A), comparedwith A431/H9 and KB31 cells. The levels of Bax, anotherproapoptotic multi-BH domain protein, were similar in allthe cell lines. We found that treatment with SS1P-KDELresulted in a large fall inMcl-1 levels as previously shown inMEFs treated with native PE toxin (15), but this fall was notsufficient to induce apoptosis in HTB80 and Panc3.014cells.We reasoned that if Bak were important for apoptosis
induced by immunotoxin, then increasing Bak expressionwould increase sensitivity to immunotoxin. We tried tomake stable lines with high Bak by using HTB80 andPanc3.014 but failed because of low transfection efficiency.
We were able to easily transfect KB31 cells that have lowerBAK and are less sensitive to immunotoxins compared withA431/H9 cells.
We established several stable KB31 clones expressing Bakand found that each clone had about a 2-fold increase ofBak compared with parental KB31 cells. Bax protein levelswere unchanged (Fig. 2B). All Bak clones showed increasedsensitivity to SS1P, which induced 20% to 35% apoptoticcells after 24 hours and 34% to 60% after 48 hours. Thesevalues are significantly higher than the parental KB31 cells,or KB31 cells transduced with empty virus (Fig. 2B). Clone#2 had the lowest level of Bak and showed the lowestapoptosis. These data show that raising Bak levels increasesimmunotoxin killing.
To show that a decrease in Bak can cause resistance toimmunotoxin, we used siRNA to knockdown Bak in A431/H9, because it has high Bak levels and is immunotoxinsensitive (Fig. 2C). We found that apoptotic cells inducedby SS1P decreased from 39% to 8% at the 24-hour timepoint and from 65% to 14% at 36-hour time point.
To determine whether the apoptosis induced by otherPE-based immunotoxins is also Bak dependent, we treatedA431/H9 with HB21(Fv)-PE40 targeting the transferrinreceptor and LMB9 targeting the Lewis Y antigen (3).Bak knockdown reduced the apoptotic cells induced bySS1P, HB21(Fv)-PE40, or LMB9 (Supplementary Fig. S2).
Figure 2. Low Bak protein inresistant cells and Bak-dependentapoptosis induced byimmunotoxin. A, Western blottingafter SS1P-KDEL treatment for 24hours. B, Bak and Bax expressionin lentiviral infected stable BakKB31 clones detected withWestern blotting, and comparedwith the same amount of KB31,empty lentiviral infected KB31(control), and 2-fold amount ofKB31 (KB31 2�). Cells weretreated with 300 ng/mL SS1P for24 or 48 hours. Apoptotic cells areshown as Annexin V–positivepopulation (n ¼ 3). C, A431/H9transfected with control and BaksiRNA. Western blot wasconducted 48 hours aftertransfection. Cells were furthertreated with 100 ng/mL SS1P for24 and 36 hours. Apoptotic cellsare shown as Annexin V–positivepopulation (n ¼ 3).
Ctrl siRNA Bak siRNA30 10 3 30 10 3 nmol/L
Bak
Control 30 nmol/LControl 10 nmol/LControl 3 nmol/LBak 30 nmol/LBak 10 nmol/LBak 3 nmol/L
90
80
70
60
50
40
30
20
10
0
8070605040302010
0
24 h
Apop
totic
cel
ls (%
)
Apop
totic
cel
ls (%
)
36 h
24 h 48 h
β-ActinBax
Bak
Mcl-1
A
B
C
KB31
2x
KB31
Ctrl
Bak
#1Ba
k #2
Bak
#3
SS1P-KDEL
H9
KB31
HTB
80
Panc
3.01
4
– – – – ++++
β-Actin
Bax
Bak
KB31ControlBak #1Bak #2Bak #3
β-Actin
Immunotoxin and TRAIL Combo Kill Resistant Cells
www.aacrjournals.org Clin Cancer Res; 17(18) September 15, 2011 5929
on June 5, 2018. © 2011 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst August 3, 2011; DOI: 10.1158/1078-0432.CCR-11-1235

To determine whether Bak dependence exists in other celltypes, we knocked down Bak in Hela cells and foundsubstantial reduction of apoptotic cells induced by HB21(Fv)-PE40 or TGFa-PE38 (transforming growth factor afused to PE38) targeting epidermal growth factor receptor(3). Hela cells do not express Lewis Y and are not respon-sive to LMB9 (Supplementary Fig. S2).
Immunotoxin combined with TRAIL synergisticallyinduce cell death
Because immunotoxins kill cells by lowering Mcl-1 levelsin a Bak-dependent process, we hypothesized that activat-ing the extrinsic apoptosis pathway would increase immu-notoxin cell killing if Bak levels were low, because Bax isimportant for the extrinsic apoptosis pathway (22, 23). Todo this, we needed to use cell lines expressing TRAILreceptors. We examined the pancreatic cancer lines andKB31 cells and found they all expressed TRAIL 1 and TRAIL2 receptors and that the TRAIL 2 receptor was more highlyexpressed (Fig. 3A). Despite TRAIL receptor expression,these cells were all resistant to human TRAIL at 20 ng/mL 48 hours after treatment (Fig. 3B). SS1P-KDEL alonedid not cause cell death of either HTB80 or Panc3.014 cells(Fig. 3B). When SS1P-KDEL and TRAIL were combined,there was considerable cell death; 51% apoptotic cells inHTB80 after 24 hours and 66% after 48 hours, 17% inPanc3.014 after 24 hours and 40% after 48 hours.
We examined the activation of cell death pathway pro-teins caspase-8, Bid and PARP in cells treated with immu-notoxins and TRAIL. SS1P-KDEL or TRAIL alone did notinduce apoptosis in HTB80 cells and did not activatecaspase-8 or cause Bid cleavage (Fig. 4A). There was noPARP cleavage, which is consistent with the Annexin Vstaining assay data. Only the combination of SS1P-KDELwith TRAIL resulted in caspase-8, Bid, and PARP cleavage(Fig. 4A) and activation of Bax (Fig. 4B), indicating that thecombination of immunotoxin and TRAIL successfully trig-gered the Bax-mediated apoptosis cascade by recruitmentand activation of caspase-8.
To rule out the possible role of Bak in the Bax pathway,we utilized Bak knockout MEFs (Bak�/� MEF). We foundthat after 24 hours treatment with PE toxin alone orrecombinant mouse TRAIL alone, there was no measurableapoptosis. However, PE combined with TRAIL inducedmassive apoptotic Bak�/� MEFs (Fig. 4C), suggestingthat the combination of PE toxin and TRAIL synergisticallyactivates Bax-mediated cell death, and that Bak is dispens-able for this apoptosis pathway. Similar synergisticcombination results were obtained when using Bak�/�HCT116 colorectal cancer cell line (SupplementaryFig. S3).
Synergistic antitumor activity with a combination ofTRAIL receptor 2 agonist antibody HGS-ETR2 andSS1P
Lexatumumab (HGS-ETR2) is a fully human agonisticmAb targeting TRAIL receptor 2 that activates the extrinsicapoptosis pathway. It has preclinical antitumor activity and
is being evaluated in clinical trials (24–27). To examinewhether combining immunotoxin with HGS-ETR2 couldovercome the resistance of Bak deficient cancer cells,we treated HTB80 cells with HGS-ETR2. As shown inFigure 5A, neither a very high concentration of HGS-ETR2 (50 mg/mL) nor SS1P-KDEL alone induced apoptosis.However, when SS1P-KDEL and HGS-ETR2 were com-bined, they synergistically triggered massive cell death,and the apoptosis was HGS-ETR2 dose dependent(Fig. 5A).
To examine the effect of the 2 agents in an animal model,we used the KB31 xenograft tumors because these cellsgrow well in immunodeficient mice and the pancreaticcancer cell lines do not (28, 29). Cell culture experimentsshowed that at 24 hours neither SS1P nor TRAIL nor HGS-ETR2 alone induced significant cell death. When SS1Pwas combined with either TRAIL or HGS-ETR2, substantialcell death was produced (Fig. 5B and C). For tumor
KB31A
B
Panc3.014
TRAIL receptor 1
TRAIL receptor 2
100
90
80
70
60
50
40
30
20
10
0HTB80 Panc3.014 HTB80 Panc3.014
24 h 48 h
Ap
op
totic c
ells
(%
)
SS1P-KDEL
TRAIL
Combo
Ce
ll nu
mb
er
Ce
ll nu
mb
er
HTB80
Figure 3. TRAIL receptor expression and synergistic cell killing bycombination of immunotoxin and TRAIL. A, trypsinized KB31, Panc3.014and HTB80 stained with PE conjugated isotype antibody (filled region) orPE conjugated anti-TRAIL receptor 1 or 2 antibody (open region). B,HTB80 and Panc3.014 incubated with 300 ng/mL SS1P-KDEL, 20 ng/mLrh-TRAIL or combination for 24 or 48 hours. Apoptotic cells are shown asAnnexin V–positive population (n ¼ 4).
Du et al.
Clin Cancer Res; 17(18) September 15, 2011 Clinical Cancer Research5930
on June 5, 2018. © 2011 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst August 3, 2011; DOI: 10.1158/1078-0432.CCR-11-1235
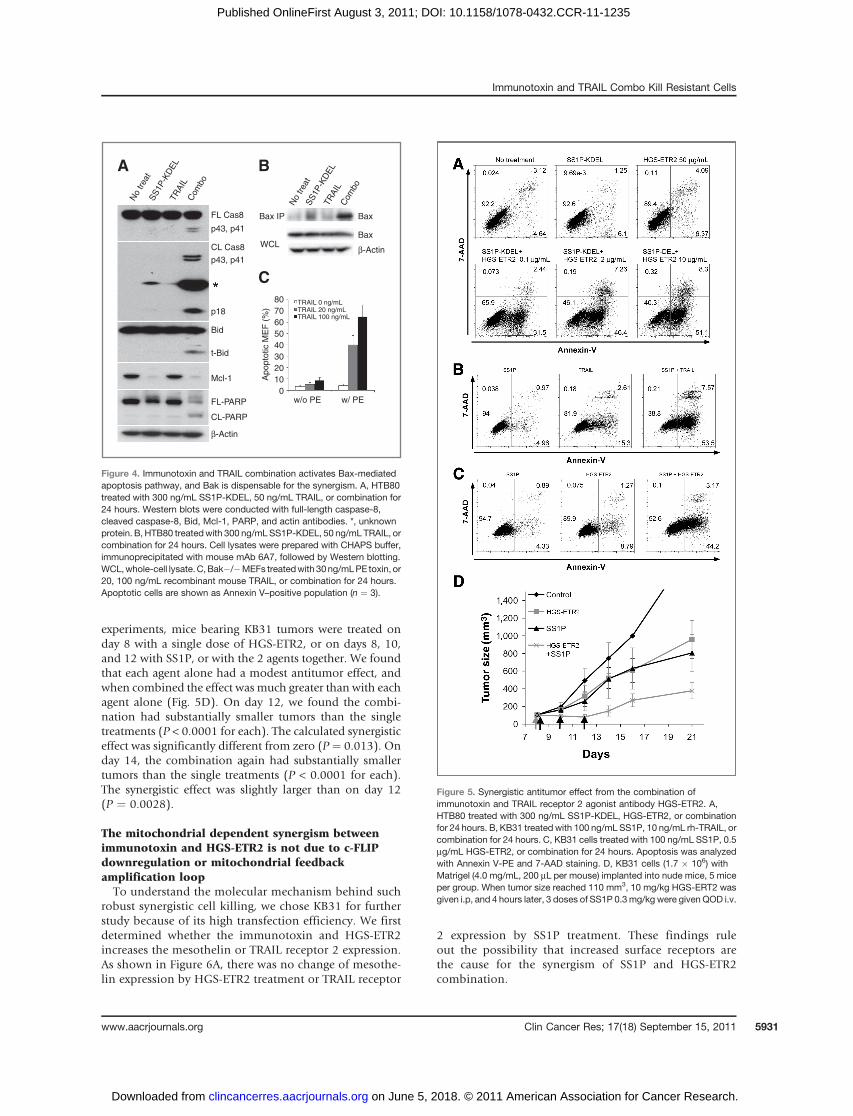
experiments, mice bearing KB31 tumors were treated onday 8 with a single dose of HGS-ETR2, or on days 8, 10,and 12 with SS1P, or with the 2 agents together. We foundthat each agent alone had a modest antitumor effect, andwhen combined the effect was much greater than with eachagent alone (Fig. 5D). On day 12, we found the combi-nation had substantially smaller tumors than the singletreatments (P < 0.0001 for each). The calculated synergisticeffect was significantly different from zero (P ¼ 0.013). Onday 14, the combination again had substantially smallertumors than the single treatments (P < 0.0001 for each).The synergistic effect was slightly larger than on day 12(P ¼ 0.0028).
The mitochondrial dependent synergism betweenimmunotoxin and HGS-ETR2 is not due to c-FLIPdownregulation or mitochondrial feedbackamplification loopTo understand the molecular mechanism behind such
robust synergistic cell killing, we chose KB31 for furtherstudy because of its high transfection efficiency. We firstdetermined whether the immunotoxin and HGS-ETR2increases the mesothelin or TRAIL receptor 2 expression.As shown in Figure 6A, there was no change of mesothe-lin expression by HGS-ETR2 treatment or TRAIL receptor
2 expression by SS1P treatment. These findings ruleout the possibility that increased surface receptors arethe cause for the synergism of SS1P and HGS-ETR2combination.
80
70
60
50
40
30
20
10
0
BaxFL Cas8
A B
C
p43, p41
p43, p41
p18
Bid
t-Bid
Mcl-1
FL-PARP
CL-PARP
CL Cas8
Bax IP
WCL
No
trea
tS
S1P
-KD
EL
TR
AIL
Com
bo
No
trea
tS
S1P
-KD
EL
TR
AIL
Com
bo
Bax
β-Actin
β-Actin
w/o PE w/ PE
TRAIL 0 ng/mLTRAIL 20 ng/mLTRAIL 100 ng/mL
Ap
op
totic M
EF
(%
)
Figure 4. Immunotoxin and TRAIL combination activates Bax-mediatedapoptosis pathway, and Bak is dispensable for the synergism. A, HTB80treated with 300 ng/mL SS1P-KDEL, 50 ng/mL TRAIL, or combination for24 hours. Western blots were conducted with full-length caspase-8,cleaved caspase-8, Bid, Mcl-1, PARP, and actin antibodies. *, unknownprotein. B, HTB80 treatedwith 300 ng/mLSS1P-KDEL, 50 ng/mL TRAIL, orcombination for 24 hours. Cell lysates were prepared with CHAPS buffer,immunoprecipitated with mouse mAb 6A7, followed by Western blotting.WCL,whole-cell lysate.C,Bak�/�MEFs treatedwith 30ng/mLPE toxin, or20, 100 ng/mL recombinant mouse TRAIL, or combination for 24 hours.Apoptotic cells are shown as Annexin V–positive population (n ¼ 3).
Figure 5. Synergistic antitumor effect from the combination ofimmunotoxin and TRAIL receptor 2 agonist antibody HGS-ETR2. A,HTB80 treated with 300 ng/mL SS1P-KDEL, HGS-ETR2, or combinationfor 24 hours. B, KB31 treated with 100 ng/mL SS1P, 10 ng/mL rh-TRAIL, orcombination for 24 hours. C, KB31 cells treated with 100 ng/mL SS1P, 0.5mg/mL HGS-ETR2, or combination for 24 hours. Apoptosis was analyzedwith Annexin V-PE and 7-AAD staining. D, KB31 cells (1.7 � 106) withMatrigel (4.0 mg/mL, 200 mL per mouse) implanted into nude mice, 5 miceper group. When tumor size reached 110 mm3, 10 mg/kg HGS-ERT2 wasgiven i.p, and 4 hours later, 3 doses of SS1P 0.3mg/kg were given QOD i.v.
Immunotoxin and TRAIL Combo Kill Resistant Cells
www.aacrjournals.org Clin Cancer Res; 17(18) September 15, 2011 5931
on June 5, 2018. © 2011 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst August 3, 2011; DOI: 10.1158/1078-0432.CCR-11-1235

We found that caspase-8 and Bid cleavage in KB31 wereactivated by the combination of SS1P and HGS-ETR2(Fig. 6B), similar to what we observed in HTB80(Fig. 4A). To identify the protein responsible for thesynergistic activation of caspase-8, we measured theintracellular apoptosis inhibitor cellular-FLIP (c-FLIP), aregulator protein of caspase-8 activation, but we found nochange of FLIP expression (Fig. 6B).
There is also the possibility that the combination ofimmunotoxin and TRAIL or agonist antibody could inducea mitochondrial feedback amplification loop and causesynergistic cell killing, as suggested by the combination ofTRAIL and etoposide (30). To this end, we knocked downBid, Bak, or Bax (Fig. 6C). We found that Bid, or Bak/Baxdouble knockdown reduced cell death induced by the
combination of SS1P and HGS-ETR2 (Fig. 6D), suggestingthat cell death by combination treatment is still mitochon-drial dependent. However, the production of the activeform of caspase-8, p43/41, was not affected by either Bidknockdown, or by Bak/Bax double knockdown (Fig. 6E).Bid cleavage was also not affected by Bak/Bax doubleknockdown (Fig. 6E). These data suggest that a mitochon-drial feedback amplification loop does not exist when cellsare treated with the combination of SS1P and HGS-ETR2,because the downstream events did not affect the upstreamevents.
Discussion
In this study, we examined pancreatic cancer cell lines tosee whether an immunotoxin against mesothelin that haspreviously been shown to kill other mesothelin expressingcancer cell lines (7–11) can kill them. We found that 2pancreatic lines, HTB80 and Panc3.014, were resistant toimmunotoxin killing despite complete inhibition of pro-tein synthesis and showed that the resistance was associ-ated with low expression of Bak. We then carried outexperiments to determine whether Bak regulated the abilityof immunotoxins to kill cells and found that increasing Bakmakes cells more immunotoxin sensitive and that loweringBakmakes cells immunotoxin resistant. Thus, it is clear thatafter arrest of protein synthesis, Bak, which acts through theintrinsic mitochondrial apoptosis pathway, has an impor-tant role in the ability of immunotoxins to kill cells. To ourknowledge, this is the first article showing that cancer cellswith low levels of endogenous Bak are resistant to someanticancer drugs. We did not expect the level of Bak to be socrucial that even a 2-fold increase of Bak could sensitizecells to immunotoxin.
Certain cancer cells, such as DU145 and LoVo, carryframeshift mutations in the Bax gene and do not expressBax protein (31). To determine whether mutations in theBak gene are a cause of low Bak expression in HTB80 andPanc3.014, we sequenced the genomic DNA and found nochanges except silent mutations in the Bak gene (data notshown). Other cancer cells such as colon cancer cell lineHCT116 also contain low Bak protein (unpublished dataand ref. 32). HCT116 is resistant to HB21(Fv)-PE40 despiteprofound protein synthesis inhibition and growth arrest,and no significant apoptotic cells can be detected even after48 hours of treatment (Supplementary Fig. S2 and unpub-lished data). Thus, there are cancer cells that contain muchless Bak protein for unknown reasons and are resistant toimmunotoxin.
To overcome immunotoxin resistance, we simultaneous-ly targeted both the mitochondrial (intrinsic) and deathreceptor (extrinsic) apoptosis pathways; this has beenshown to be an effective strategy with the drugs etoposide,bortezomib, and vorinostat (30, 33–35). We found thatHTB80 and Panc3.014 express TRAIL receptors, yet areresistant to treatment with TRAIL. Remarkably, whenSS1P-KDEL and TRAIL were combined on HTB80 andPanc3.014 cells, they synergized to produce massive cell
50
40
30
20
10
0
100
80
60
40
20
0100 101 102 103 104
% o
f M
ax
100
80
60
40
20
0
% o
f M
ax
Filled:A
B
C
D
E
NTOpen: HGS-ETR2
Filled: NTOpen:
TRAIL Receptor 2
NT
SS
1P
HG
S-E
TR
2C
om
bo
100 101 102 103 104
Mesothelin
Bid
tBid
t-Bid
Bid
PARP
p18
c-FLIP
β-Actin
β-Actin
Bid
Bax
Bax
siRNA
siRNA
Ctr
l
Bid
Bak
Bax
Ba
k+
Ba
x Ctr
l
Bak
Ba
k+
Ba
x
FL Cas-8p57
FL Cas-8p43/41
CL-Cas-8p43/41
p18
SS1P
Ap
op
totic c
ells
(%
)
Ctr
l
siR
NA
Bid
siR
NA
Ba
k-+
Ba
x
siR
NA
Figure 6. Mitochondrial dependent synergism between immunotoxin andHGS-ETR2 is not due to c-FLIP downregulation or mitochondrial feedbackamplification loop. A, KB31 treated with 100 ng/mL SS1P, 0.5 mg/mL ofHGS-ETR2, or no treatment (NT) for 24 hours. Cell surface mesothelin andTRAIL receptor 2 were analyzed. B, Western blotting of treated cells. C,control, Bid, Bak, Bax, Bak and Bax siRNAs (5 nmol/L) were transfectedinto KB31 and knockdown efficiency was checked after 72 hours. D,siRNA transfected KB31 cells were further treated with 100 ng/mL SS1Pand 0.5 mg/mL HGS-ETR2 for 20 hours. Apoptotic cells are shown asAnnexin V–positive population (n ¼ 3). E, Western blotting of combinationtreated cells.
Du et al.
Clin Cancer Res; 17(18) September 15, 2011 Clinical Cancer Research5932
on June 5, 2018. © 2011 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst August 3, 2011; DOI: 10.1158/1078-0432.CCR-11-1235

death accompanied by caspase-8 and Bid cleavage, and Baxactivation.Antibodies to TRAIL receptors have advantages over
TRAIL itself as therapeutic agents because they do not bindto decoy receptors and have a long half-life. HGS-ETR2 is ahuman IgG1 agonist antibody to TRAIL receptor 2 that hasbeen evaluated in several studies for its antitumor activity(24–27). We found that combining SS1P-KDEL with HGS-ETR2 induced synergistic cell killing in HTB80 and thatcombining SS1P with HGS-ETR2 or TRAIL induced syner-gistic cell killing of KB31. When given to mice with KB31tumors, SS1P combined with HGS-ETR2 had greater anti-tumor activity than either agent alone on slowing tumorgrowth, although the tumor regressions were not observed.Optimization of the timing of administration of immuno-toxin and HGS-ETR2 may enhance tumor penetration andimprove their antitumor effect.Recruitment and activation of caspase-8 seems to be the
mechanism for the synergism observed with the combina-tion of immunotoxin and TRAIL or HGS-ETR2 (Fig. 4A andFig. 6B). To investigate how it was initiated, we analyzedthe expression of mesothelin and TRAIL receptor 2 andfound that they did not change. Downregulation of c-FLIP,a negative regulator of caspase-8 activation, has beenreported to be important for the synergism of the combi-nation of mitochondrial and death receptor apoptosisagents in some studies (33, 36, 37) but not in others(34, 38). In this study, we found no change of c-FLIPexpression after treatment with SS1P, HGS-ETR2, or thecombination.A mitochondrial feedback amplification loop was sug-
gested to account for the synergistic cell killing by thecombination of TRAIL and etoposide (30). In that sce-nario, extensive caspase-8 cleavage seen during TRAIL–etoposide synergy is a consequence and not a cause of theapoptotic cascade activated downstream of Bid, and Bidknockdown was shown to disrupt the mitochondrialfeedback loop, and inhibit the production of p43/41,the active form of caspase-8 (30). When we combinedimmunotoxin and HGS-ETR2, Bid knockdown or Bak/Bax double knockdown did not inhibit caspase-8 p43/41formation (pro-domain cleavage, Ref. 39 and 40). Also,Bak/Bax double knockdown did not affect Bid cleavage.Collectively, these data indicate that the combination of
immunotoxin and TRAIL or HGS-ETR2 does not inducemitochondrial feedback amplification. Although wefound the combination of immunotoxin and TRAIL orHGS-ETR2 synergistically induces the cleavage and acti-vation of caspase-8, and triggers downstream the apopto-sis pathway, the mechanism of caspase-8 activation undercombination treatment remains unclear. It is noteworthythat caspase-8 p18 formation was reduced by either Bidknockdown or Bak/Bax double knockdown, suggestingthat the maturation of caspase-8 into p18 (inter-domaincleavage) may occur downstream of mitochondrial in-volvement (39, 40).
In summary, cancer cells with endogenous low Bakprotein are resistant to immunotoxin treatment despiteprofound protein synthesis inhibition. Combining theimmunotoxin with TRAIL or TRAIL agonist antibodiescan induce synergistic mitochondrial-dependent cell kill-ing by recruitment and activation of caspase-8, althoughthe mechanism needs further study. Combining immuno-toxin with activators of the death receptor pathway isan effective approach to overcome resistance due to Bakdeficiency.
Disclosure of Potential Conflicts of Interest
Ira Pastan is an inventor on immunotoxin patents that are owned by theNIH.
Acknowledgments
We thank Dr. David FitzGerald for reading the manuscript and helpfulcomments, Dr. Raffit Hassan for providing the pancreatic cell lines andMORAB antibodies, Dawn A. Walker and other lab members for theirhelpful discussion, and Dr. Dan Soppet (Laboratory of Molecular Technol-ogy, NCI-SAIC) for genomic Bak sequencing. We also thank Drs. RichardYoule and Chunxin Wang (NINDS, NIH) for providing a human Bak genecDNA and HCT116 cells and for their helpful comments.
Grant Support
This research was supported by the Intramural Research Program of the NIH,National Cancer Institute, and Center for Cancer Research.
The costs of publication of this article were defrayed in part by the paymentof page charges. This article must therefore be hereby marked advertisement inaccordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Received May 12, 2011; revised July 19, 2011; accepted July 24, 2011;published OnlineFirst August 3, 2011.
References1. Adams GP, Weiner LM. Monoclonal antibody therapy of cancer. Nat
Biotechnol 2005;23:1147–57.2. Wu AM, Senter PD. Arming antibodies: prospects and challenges for
immunoconjugates. Nat Biotechnol 2005;23:1137–46.3. Pastan I, Hassan R, Fitzgerald DJ, Kreitman RJ. Immunotoxin therapy
of cancer. Nat Rev Cancer 2006;6:559–65.4. Kreitman RJ, Wilson WH, Bergeron K, Raggio M, Stetler-Stevenson
M, FitzGerald DJ, et al. Efficacy of the anti-CD22 recombinant immu-notoxin BL22 in chemotherapy-resistant hairy-cell leukemia. N Engl JMed 2001;345:241–7.
5. Kreitman RJ, Squires DR, Stetler-Stevenson M, Noel P, FitzGerald DJ,Wilson WH, et al. Phase I trial of recombinant immunotoxin RFB4
(dsFv)-PE38 (BL22) in patients with B-cell malignancies. J Clin Oncol2005;23:6719–29.
6. Wayne AS, Kreitman RJ, Findley HW, Lew G, Delbrook C, SteinbergSM, et al. Anti-CD22 immunotoxin RFB4(dsFv)-PE38 (BL22) for CD22-positive hematologic malignancies of childhood: preclinical studiesand phase I clinical trial. Clin Cancer Res 2010;16:1894–903.
7. Hassan R, Bera T, Pastan I. Mesothelin: a new target for immuno-therapy. Clin Cancer Res 2004;10:3937–42.
8. Hassan R, Ho M. Mesothelin targeted cancer immunotherapy. Eur JCancer 2008;44:46–53.
9. Li Q, Verschraegen CF,Mendoza J, Hassan R. Cytotoxic activity of therecombinant anti-mesothelin immunotoxin, SS1(dsFv)PE38, towards
Immunotoxin and TRAIL Combo Kill Resistant Cells
www.aacrjournals.org Clin Cancer Res; 17(18) September 15, 2011 5933
on June 5, 2018. © 2011 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst August 3, 2011; DOI: 10.1158/1078-0432.CCR-11-1235

tumor cell lines established from ascites of patients with peritonealmesotheliomas. Anticancer Res 2004;24:1327–35.
10. HoM, Bera TK,WillinghamMC, OndaM, Hassan R, FitzGerald D, et al.Mesothelin expression in human lung cancer. Clin Cancer Res2007;13:1571–5.
11. Yu L, FengM, Kim H, Phung Y, Kleiner DE, Gores GJ, et al. Mesothelinas a potential therapeutic target in human cholangiocarcinoma. JCancer 2010;1:141–9.
12. Hassan R, Bullock S, Premkumar A, Kreitman RJ, Kindler H, Will-ingham MC, et al. Phase I study of SS1P, a recombinant anti-mesothelin immunotoxin given as a bolus I.V. infusion to patientswith mesothelin-expressing mesothelioma, ovarian, and pancreaticcancers. Clin Cancer Res 2007;13:5144–9.
13. Kreitman RJ, Hassan R, Fitzgerald DJ, Pastan I. Phase I trial ofcontinuous infusion anti-mesothelin recombinant immunotoxinSS1P. Clin Cancer Res 2009;15:5274–9.
14. Hassan R, Sharon E, Chen HX, Conlon K, Ling A, Steinberg SM, et al.Phase I clinical trial of antimesothelin immunotoxin SS1P in combi-nation with pemetrexed and cisplatin for front-line therapy of ad-vanced pleural mesothelioma. J Clin Oncol 2010;28:e17518.
15. Du X, Youle RJ, FitzGerald DJ, Pastan I. Pseudomonas exotoxin A-mediated apoptosis is Bak dependent and preceded by the degra-dation of Mcl-1. Mol Cell Biol 2010;30:3444–52.
16. Kreitman RJ, Pastan I. Importance of the glutamate residue of KDEL inincreasing the cytotoxicity ofPseudomonas exotoxinderivatives and forincreased binding to the KDEL receptor. Biochem J 1995;307:29–37.
17. Hidalgo M. Pancreatic cancer. N Engl J Med 2010;362:1605–17.18. Li D, Xie K, Wolff R, Abbruzzese JL. Pancreatic cancer. Lancet
2004;363:1049–57.19. Kang R, Tang D, Schapiro NE, Livesey KM, Farkas A, Loughran P,
et al. The receptor for advanced glycation end products (RAGE)sustains autophagy and limits apoptosis, promoting pancreatic tumorcell survival. Cell Death Differ 2010;17:666–76
20. Yang S,Wang X, Contino G, LiesaM, Sahin E, Ying H, et al. Pancreaticcancers require autophagy for tumor growth. Genes Dev 2011;25:717–29.
21. Traini R, Ben-Josef G, Pastrana DV, Moskatel E, Sharma AK, Anti-gnani A, et al. ABT-737 overcomes resistance to immunotoxin-mediated apoptosis and enhances the delivery of pseudomonasexotoxin-based proteins to the cell cytosol. Mol Cancer Ther2010;9:2007–15.
22. Han J, Goldstein LA, Gastman BR, Rabinovitz A, Wang GQ, Fang B,et al. Differential involvement of Bax and Bak in TRAIL-mediatedapoptosis of leukemic T cells. Leukemia 2004;18:1671–80.
23. Theodorakis P, Lomonosova E, Chinnadurai G. Critical requirement ofBAX for manifestation of apoptosis induced by multiple stimuli inhuman epithelial cancer cells. Cancer Res 2002;62:3373–6.
24. Plummer R, Attard G, Pacey S, Li L, Razak A, Perrett R, et al. Phase 1and pharmacokinetic study of lexatumumab in patients with advancedcancers. Clin Cancer Res 2007;13:6187–94.
25. Wakelee HA, Patnaik A, Sikic BI, Mita M, Fox NL, Miceli R, et al. PhaseI and pharmacokinetic study of lexatumumab (HGS-ETR2) givenevery 2 weeks in patients with advanced solid tumors. Ann Oncol2010;21:376–81.
26. Zeng Y, Wu XX, Fiscella M, Shimada O, Humphreys R, Albert V, et al.Monoclonal antibody to tumor necrosis factor-related apoptosis-inducing ligand receptor 2 (TRAIL-R2) induces apoptosis in primaryrenal cell carcinoma cells in vitro and inhibits tumor growth in vivo. Int JOncol 2006;28:421–30.
27. Zhang L, Zhang X, Barrisford GW, Olumi AF. Lexatumumab (TRAIL-receptor 2 mAb) induces expression of DR5 and promotes apoptosisin primary and metastatic renal cell carcinoma in a mouse orthotopicmodel. Cancer Lett 2007;251:146–57.
28. Zhang Y, Xiang L, Hassan R, Paik CH, Carrasquillo JA, Jang BS, et al.Synergistic antitumor activity of taxol and immunotoxin SS1P intumor-bearing mice. Clin Cancer Res 2006;12:4695–701.
29. Zhang Y, Xiang L, Hassan R, Pastan I. Immunotoxin and taxolsynergy results from a decrease in shed mesothelin levels in theextracellular space of tumors. Proc Natl Acad Sci USA 2007;104:17099–104.
30. Broaddus VC, Dansen TB, Abayasiriwardana KS, Wilson SM, FinchAJ, Swigart LB, et al. Bid mediates apoptotic synergy between tumornecrosis factor-related apoptosis-inducing ligand (TRAIL) and DNAdamage. J Biol Chem 2005;280:12486–93.
31. Rampino N, Yamamoto H, Ionov Y, Li Y, Sawai H, Reed JC, et al.Somatic frameshift mutations in the BAX gene in colon cancers of themicrosatellite mutator phenotype. Science 1997;275:967–9.
32. Gillissen B, Wendt J, Richter A, Richter A, M€uer A, Overkamp T, et al.Endogenous Bak inhibitors Mcl-1 and Bcl-xL: differential impact onTRAIL resistance in Bax-deficient carcinoma. J Cell Biol 2010;188:851–62.
33. Frew AJ, Lindemann RK, Martin BP, Clarke CJ, Sharkey J, AnthonyDA, et al. Combination therapy of established cancer using a histonedeacetylase inhibitor and a TRAIL receptor agonist. Proc Natl AcadSci USA 2008;105:11317–22.
34. Shanker A, Brooks AD, Tristan CA, Wine JW, Elliott PJ, Yagita H, et al.Treating metastatic solid tumors with bortezomib and a tumor necro-sis factor-related apoptosis-inducing ligand receptor agonist anti-body. J Natl Cancer Inst 2008;100:649–62.
35. Smith MR, Jin F, Joshi I. Bortezomib sensitizes non-Hodgkin's lym-phoma cells to apoptosis induced by antibodies to tumor necrosisfactor related apoptosis-inducing ligand (TRAIL) receptors TRAIL-R1and TRAIL-R2. Clin Cancer Res 2007;13:5528–34s.
36. Hietakangas V, Poukkula M, Heiskanen KM, Karvinen JT, Sistonen L,Eriksson JE. Erythroid differentiation sensitizes K562 leukemia cells toTRAIL-induced apoptosis by downregulation of c-FLIP. Mol Cell Biol2003;23:1278–91.
37. Zhang S, Shen HM, Ong CN. Down-regulation of c-FLIP contri-butes to the sensitization effect of 3,30-diindolylmethane on TRAIL-induced apoptosis in cancer cells. Mol Cancer Ther 2005;4:1972–81.
38. Morizot A, M�erino D, Lalaoui N, Jacquemin G, Granci V, Iessi E, et al.Chemotherapy overcomes TRAIL-R4-mediated TRAIL resistance atthe DISC level. Cell Death Differ 2011;18:700–11.
39. Boatright KM, Salvesen GS. Mechanisms of caspase activation. CurrOpin Cell Biol 2003;15:725–31.
40. Pop C, Salvesen GS. Human caspases: activation, specificity, andregulation. J Biol Chem 2009;284:21777–81.
Du et al.
Clin Cancer Res; 17(18) September 15, 2011 Clinical Cancer Research5934
on June 5, 2018. © 2011 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst August 3, 2011; DOI: 10.1158/1078-0432.CCR-11-1235

2011;17:5926-5934. Published OnlineFirst August 3, 2011.Clin Cancer Res Xing Du, Laiman Xiang, Crystal Mackall, et al. Antibodyan Antimesothelin Immunotoxin and a TRAIL Receptor 2 Agonist Killing of Resistant Cancer Cells with Low Bak by a Combination of
Updated version
10.1158/1078-0432.CCR-11-1235doi:
Access the most recent version of this article at:
Material
Supplementary
http://clincancerres.aacrjournals.org/content/suppl/2011/08/04/1078-0432.CCR-11-1235.DC1
Access the most recent supplemental material at:
Cited articles
http://clincancerres.aacrjournals.org/content/17/18/5926.full#ref-list-1
This article cites 40 articles, 22 of which you can access for free at:
Citing articles
http://clincancerres.aacrjournals.org/content/17/18/5926.full#related-urls
This article has been cited by 6 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://clincancerres.aacrjournals.org/content/17/18/5926To request permission to re-use all or part of this article, use this link
on June 5, 2018. © 2011 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst August 3, 2011; DOI: 10.1158/1078-0432.CCR-11-1235


















