
Junko Takita MRTmicroarray20130814 JUNTK-8 (1)
24
Cancer Science Report Genome-wide approach to identify second gene targets for malignant rhabdoid tumors using high-density oligonucleotide microarrays Junko Takita 1 MD, PhD, Yuyan Chen 1 MD, PhD, Motohiro Kato 1 MD PhD, Kentaro Ohki 1 MD PhD, Shigeru Ohta 3 , MD, PhD, Riki Nishimura 1 MD, PhD, Noriko Hoshino 1 MD, Masafumi Seki 1 MD, Masashi Sanada 4 MD, Akira Oka 1 , MD, PhD, Yasuhide Hayashi 6 MD, PhD, and Seishi Ogaw 4 MD, PhD 1 Department of Pediatrics 1 , Cell Therapy and Transplantation Medicine 2 , and 3 Department of Pediatrics, Shiga University of Medical school, 4 Cancer Genomics Project, Graduate school of Medicine, University of Tokyo, 5 Division of Hematology/Oncology, Saitama Children’s Medical Center, and 6 Gunma Children’s Medical Center Address reprint requests to Junko Takita MD, PhD Department of Pediatrics, Graduate School of Medicine, University of Tokyo 7-3-1, Hongo, Bunkyo-ku, Tokyo 113-8655, Japan TEL: +813-3815-5411 Ex33462 FAX: +813-3816-4108 Email: [email protected] 1
Transcript of Junko Takita MRTmicroarray20130814 JUNTK-8 (1)

Cancer Science
Report
Genome-wide approach to identify second gene targets for
malignant rhabdoid tumors using high-density oligonucleotide
microarrays
Junko Takita1 MD, PhD, Yuyan Chen1 MD, PhD, Motohiro Kato1 MD PhD,
Kentaro Ohki1 MD PhD, Shigeru Ohta3, MD, PhD, Riki Nishimura1 MD, PhD,
Noriko Hoshino1 MD, Masafumi Seki1 MD, Masashi Sanada4 MD, Akira
Oka1, MD, PhD, Yasuhide Hayashi6 MD, PhD, and Seishi Ogaw4 MD, PhD
1Department of Pediatrics1, Cell Therapy and Transplantation Medicine2,
and 3Department of Pediatrics, Shiga University of Medical school, 4Cancer
Genomics Project, Graduate school of Medicine, University of Tokyo,
5Division of Hematology/Oncology, Saitama Children’s Medical Center, and
6Gunma Children’s Medical Center
Address reprint requests to
Junko Takita MD, PhDDepartment of Pediatrics,Graduate School of Medicine, University of Tokyo 7-3-1, Hongo, Bunkyo-ku, Tokyo 113-8655, Japan TEL: +813-3815-5411 Ex33462 FAX: +813-3816-4108 Email: [email protected]
1

Summary
Malignant rhabdoid tumor [Remark 1](MRT) is a rare and highly
lethal cancer that mainly affects infants and young children. The vast
majority of malignant rhabdoid MRT tumors are characterized by loss of
function of the SMARCB1 gene on chromosome 22q11.2. However, little is
known about other genetic changes than SMARCB1 alterations that are
responsible for the development and/or progression of malignant rhabdoid
tumorsMRT. To explore additional gene targets in malignant rhabdoid
tumorsMRT, we analyzed 21 malignant rhabdoid tumor MRT specimens
using high-density single nucleotide polymorphism (SNP)–genotyping
microarrays (Affymetrx® GeneChip®). Although malignant rhabdoid
tumor MRT genomes are characterized by common 22q11.2 deletions
affecting the SMARCB1 locus with a frequency of 95.2% (20/21
specimens), other genetic changes were have been less frequent. Of the
20 specimens with deletions of 22q11.2, 8 specimens showed uniparental
disomy of the SMARCB1 locus with homozygous deletions or gene
mutations. High-resolution analysis also disclosed the recurrent
hemizygous/homozygous deletions of 7q35-q36.1 involving CNTNAP2
locus in 3 specimens. Mutations analysis of CNTNAP2 exhibited a novel
R157C missense mutation in a primary case, and methylation analysis
showed recurrent hypermethylation of CNTNAP2 in 3 of /9 cell lines.
These results demonstrated that CNTNAP2 is one of the additional gene
targets other than SMARCB1 in malignant rhabdoid tumorsMRT.
2

Introduction
Malignant rhabdoid tumor (MRT) is an extremely rare and highly
aggressive neoplasm that typically develops in infancy or early childhood..
MRT was initially described as ‘rhabdomyosarcomatoid’, [Remark 2] an
aggressive type of [Remark 3] tumor of the kidney, , and subsequent
studies have revealed that this tumorMRT occurs in various sites including
the central nervous system (CNS), lung, liver, skin, and soft tissues.. The
most frequent common location of the tumors is the kidneys, followed by
the CNS, and the tumors originatinge from the latter site being are
referred to as atypical teratoid/rhabdoid tumor (AT/RT).. Cytogenetic and
molecular analyses for of MRT have shown recurrent deletions at 22q11.2,
which resulted in identification of SMARCB1 [(OMIN #601607)] as a
characteristic gene abnormality of this tumor.. Germ-line and somatic
mutations/deletions of SMARCB1 have been recently [Remark 4] reported
in AT/RT, as well as in epithelioid sarcoma, familial schwannomatosis, and
renal medullary carcinoma.. The SMARCB1 gene is a member of the ATP-
dependent SWF/SNF chromatin-remodeling complex, and is recruited to
promoters of genes that regulate cell cycle, growth, and differentiation.. In
MRT, SMARCB1 appears to function as a classic tumor suppressor gene,
such that germ-line mutations and deletions predispose to the development
of these malignancies;, and somatic loss or mutation of the other allele
constitutes the second hit.
In recent years, genome-wide copy number analysis using single
nucleotide polymorphism (SNP) arrays (SNP-chip) has shown to have an
outstanding power to reveal detailed profiles of genomic abnormalities and
to identify new genetic targets in various cancers.. A previous report
3

showed that high-resolution SNP-chip analysis could detect bi-allelic
alterations in SMARCB1 in almost of all MRT cases, which suggests that
SMARCB1 is the primary mutational gene target responsible for the
development of MRT and provided further evidence for the clinical utility of
molecular diagnostic testing.. However, some MRT cases retain expression
of the protein, even with in a fraction of familial MRTs have been reported
that are not associated with a SMARCB1 inactivation..[Remark 5] These
findings suggest the possibility of additional relevant genetic loci distinct
from SMARCB1. However, the detailed genetic abnormalities in MRT other
than chromosome 22 have not been fully understood. Therefore, to clarify
the additional genetic lesions involved in the pathogenesis of MRTs, we
performed SNP-chip analysis for of 21 MRT samples.
Materials and Methods
Specimens
This study was approved by the ethicsal board of the University of Tokyo
(Approval Number 1598). Primary tumor specimens were obtained at the
time of the initial surgery or biopsy from patients who were diagnosed as
having MRT or AT/RT at collaborating hospitals. In total, 14 primary MRT
specimens (4 samples of AT/RT) and 9 cell lines derived from patients with
MRT patinets (KYM-1, TM87-16, TTC-1240, TTC-549, TTC-642, TTN-45,
YAMRT, RTK(J)-4N, and STM-91-01) were analyzed in this study. The TCC
TTC series and STM-91-01 were generous gifts from Dr. Ohta. KYM-1,
YAMRT, TTNN-45, and RTK(J)-4N were generous gifts from Dr. Inoue, Dr.
Sugita, Dr. Kanegane and Dr. Yokomori. [Remark 6] [Remark 7]. A
4

neuroblastoma cell line, SJNB-1, was used as a control in the methylation
analysis. All cell lines were cultured in RPMI-1640 supplemented with 9%
fetal bovine serum..
Microarray analysis
High molecular weight DNA was isolated from tumor specimens and
subjected to SNP array analysis using Affymetrix® GeneChip® Mapping
50K and/or 250K arrays (Affymetrix, Inc.)[Remark 8] according to the
manufacturer’s protocol (STable S1). After appropriate normalization of
mean array intensities, signal ratios between tumor and normal cells were
calculated, and allele-specific copy numbers were inferred from the
observed signal ratios based on the hidden Markov model using
CNAG/AsCNAR software (http://www.genome.umin.jp)..
Mutation and expression analyses of SMARCB1 and CNTNAP2
Direct sequencing analyses of all coding exons of SMARCB1 and CNTNAP2
was were carried out in all samples as previously described. . The primer
sequences and conditions of PCRs reactions for mutation analyses of these
genes are have been described in the previous papers. . Total RNA was
extracted from the 9 cell lines using Isogen reagent (Nippon Gene, Osaka,
Japan) according to the manufacturer’s instructions and subjected to
reverse-transcriptionRT reactions to synthesize cDNA using the
SuperScript Preamplification System for first-strand cDNA synthesis (Life
Technologies, Inc., Rockville, MD, USA). Semi-quantitative RT-PCR analysis
for CNTNAP2 expression was performed as described previously..
5

Methylation-specific PCR and 5-aza-2-deoxycytidine treatment.
Bisulfate modification of genomic DNA was performed as previously
described. . For methylation-specific PCR (MSP), approximately 10 ng of
bisulfite-treated DNA was amplified with primers for both the methylated
and unmethylated sequences. Reaction products were separated by
electorophoresis on a 2.0% agarose gel. 5-Aza-2-deoxycytidine (Sigma
Chemical[Remark 9) was dissolved in cold RPMI 1640 immediately before
use. Cells were exposed to 0.5 and 1.0 mM of 5-aza-2-deoxycytidine for 3
days, with the medium and drug being replaced in every 24 h. . Then
cCells then were harvested and used for RT-PCR analysis.
Results and Discussion[Remark 10]
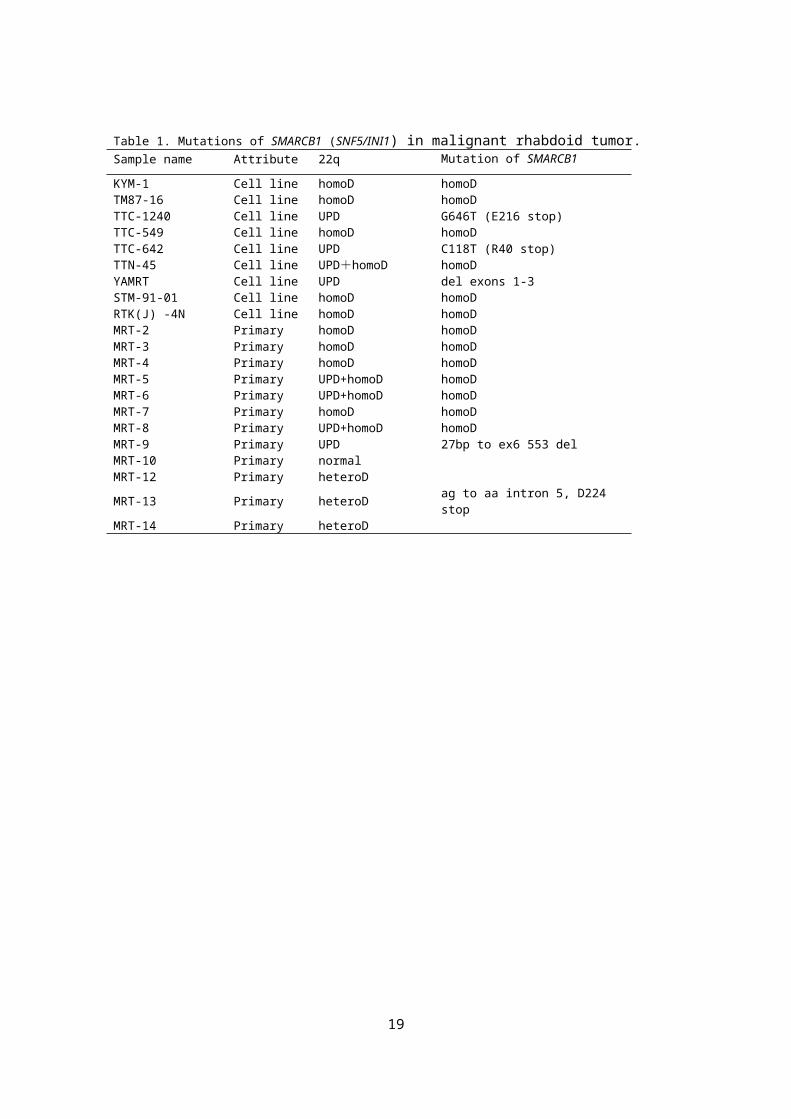
Our SNP-chip analysis revealed a high frequency of deletion at 22q11.2 in
the MRT genome, as previously described.. In total, 20 out of 21
specimens (95.2%) had loss of heterozygousity (LOH) at 22q11.2 involving
the SMARCB1 locus (Fig.ure 1A, B). In 8 samples, uniparental disomy
(UPD) of 22q segments caused homozygous mutations/deletions of
SMARCB1 (Fig.ure 1B). Ten samples had homozygous focal deletions
commonly involving a 175 kb region (ch22:22,353,181-22,528,353), which
exclusively included SMARCB1. Subsequent mutation analysis revealed
that 4 samples with heterozygous deletion or UPD at the 22q11.2 locus had
mutations in the SMARCB1 gene. An MRT-derived cell line, with 22qUPD
(YAMRT) harbored a small deletion involving exons 1-3, which was not
detectable by SNP array analysis. In total, 18 out of the 21 MRT samples
had bi-allelic aberrations of SMARCB1, indicating genetic homogeneity of
6

MRT. In our analysis, 3 cases did not show bi-allelic inactivation of
SMARCB1. Among them, two 2 cases with MRT of the kidney showed
heterozygous deletions of the 22q11.2 locus, but the remaining case with
AT/RT did not have any genetic alterations in the SMARCB1 locus.
Immunohistochemical analyses of these 3 cases showed positive results for
vimentin, but negative findings for muscle lineage markers and SMARCB1,
supporting the diagnosis of MRT or AT/RT. Thus, genetic and/or epigenetic
alterations of promoter regions of SMARCB1 should be considered for the
possible alterations for inactivation of SMARCB1 in these cases.
Although recurrent copy number changes other than 22q11.2 deletions
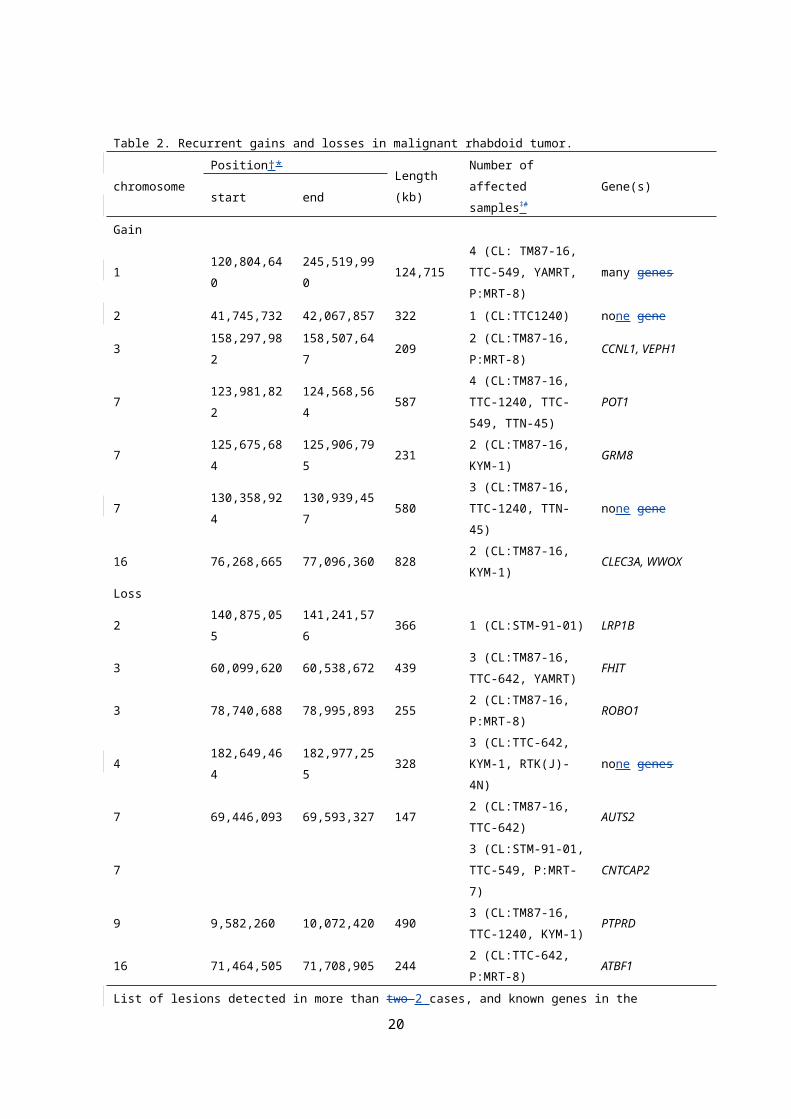
were less frequent in MRT (Fig.ure 1A), 7 loci of gains and 8 loci of losses
were commonly detected in multiple samples (Table 2). Importantly, some
of the regions contained potential gene targets which that were known to
be associated with tumorigenesis of other cancers, such as CCNL1, POT1,
CNTNAP2, and PRPTD (Table 2).. In the previous reports [Remark 11],
heterozygous knockout mice developed tumors consistent with MRT,
beginning as early as 5 weeks of age (ref)[Remark 12], but crossed mice
with SMARCB1 +/- mice and CCND1 -/- mice [Remark 12]did not develop
any spontaneous tumor. . Thus, these findings suggested that CCND1,
located at 11q13.3, may be a key mediator involved in the genesis of MRT.
Interestingly, our analysis identified recurrent gains at CCNL1, which
functions in association with cyclin-dependent kinases, including CCND1,
suggestinged that CCNL1 acts as one of the mediators involved in the
development of MRT. The PTPRD and POT1 were mutated or down
regulated in several human cancers, including neueroblastoma, lung
cancer, and chronic myeloid leukemia ;, and thus, further analysis of these
7

genes alterations in large number ofmany MRT samples would be
necessary to assess their involvements in the pathogenesis of MRT.
Homozygous deletion is very informative for identifying gene targets.
Indeed, SMARCB1 was identified from homozygously deleted regions at
22q11.2.. In this the present study, we found recurrent homozygous
deletions of CNTNAP2 locus at 7q35-q36 in the 2 specimens, and another
one case exhibited hemizygous deletion in this region (Fig.ure 1Aa).
CNTNAP2 encodes a single-pass transmembrane protein mediating cell-cell
interactions in the nervous system [Remark 13]. . This gene is located in a
common fragile site which that is inactivated in different types of cancers
including brain tumor, ovarian cancer, and breast cancer. . Furthermore,
methylation of the promoter region of CNTNAP2 has been reported in
pancreatic adenocarcinoma. . Although these results suggest that
CNTNAP2 is implicated in tumor suppressor genes for several cancers, the
oncogenic role of this gene in the MRT has been poorly studied. Thus, to
assess the involvements of CNTNAP2 this gene in the MRT pathogenesis,
we further performed mutation, expression, and methylation analyses in
our series. Through mutation analysis of coding region of CNTNAP2, we
found a novel R157C missense mutation in a fresh tumor of MRT, which
was not found in 60 healthy volunteers, not registered in dbSNP 137
(http://www.ncbi.nlm.nih.gov/projects/SNP/) and in 1000 genomes
(http://www.1000genomes.org/) (Fig.ure 2Bb). This single nucleotide
change was scored as "probably damaging" or "damaging" by two 2
computational prediction software packagess (i.e., SIFT and PolyPhen-2).
Furthermore, 3 of 9 cell lines lacking CNTNAP2 expression displayed
methylation of the CpG island of the CNTNAP2 (Fig.ure 2Bb, Cc).
8

Methylation analysis was also performed in 20 neuroblastoma cell lines,
but we did not detected any methylation of CpG islands of the CNTNAP2
(data not shown). To elucidate whether hypermethylation of CNTNAP2
blocked gene expression in MRT cells, we subjected all 3 cell lines to 3
days’ exposure of to 5-aza-deoxycytidine. As shown in Fig.ure 2Dd,
CNTNAP2 expression was induced by 5-aza-deoxycytidine treatment in the
3 cell lines, indicating that methylation of the CpG island is a direct
mechanism for CNTNAP2 silencing in MRT.
Consistent with other reports, our results illustrated the fact that
SMARCB1 is the primary gene implicated in the pathogenesis of MRT.
Although frequencies of recurrent genetic changes other than SMARCB1
locus were low, our findings suggest that CNTNAP2 is one of the potential
second gene targets for MRT. Further studies will beare necessary to
unravel the oncogenic effects of CNTNAP2 in MRT.
9

Acknowledgements
We are grateful to Ms. Matsumura, Ms. Hoshino, Ms. Yin, Ms. Saito, Ms.
Mori, and Ms. Ogino for their excellent technical assistance. We also wish
to express our appreciation to Dr. Sugita, Yamanashi Medical University;,
Dr. Yokomori, Graduate School of Medicine, University of Tokyo;, Dr.
Kanegane, Toyama University;, and Dr. A. Inoue, St. Jude Children’s
Research Hospital, for their generous gifts of MRT cell lines. This work
was supported by Research on Measures for Intractable Diseases, Health,
and Labor Sciences Research Grants, Ministry of Health, Labor and
Welfare; by Research on Health Sciences focusing on Drug Innovation; by
the Japan Health Sciences Foundation; by Core Research for Evolutional
Science and Technology, Japan Science and Technology Agency; and by
Project for Development of Innovative Research on Cancer Therapeutics.
Disclosure statement
The authors have no conflicts of interest to declare.
10

References
[1]J.B. 1 Beckwith JB, N.F. Palmer NF., Histopathology and prognosis of Wilms tumors: results from the First National Wilms' Tumor Study. Cancer 1978; 41: (1978) 1937-1948.
[2]M.R. 2 Wick MR, J.H. Ritter JH, L.P. Dehner LP., Malignant rhabdoid tumors: a clinicopathologic review and conceptual discussion. Seminars in diagnostic pathologySemin Diagn Pathol 1995; 12: (1995) 233-248.
[3]M. 3 Bhattacharjee M, J. Hicks J, L. Langford L, et al.R. Dauser, D. Strother, M. Chintagumpala, M. Horowitz, L. Cooley, H. Vogel, Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood. Ultrastructural pathology Pathol 1997; 21: (1997) 369-378.
[4]I. 4 Versteege I, N. Sevenet N, J. Lange J, et al. M.F. Rousseau-Merck, P. Ambros, R. Handgretinger, A. Aurias, O. Delattre, Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature 1998; 394: (1998) 203-206.
[5]J.A. 5 Biegel JA, B. Fogelgren B, L.M. Wainwright LM, J.Y. Zhou JY, H. Bevan H, L.B. Rorke LB., Germline INI1 mutation in a patient with a central nervous system atypical teratoid tumor and renal rhabdoid tumor. Genes, Chromosomes & cCancer 2000; 28: (2000) 31-37.
[6]J.M. 6 Carter JM, C. O'Hara C, G. Dundas G, et al. D. Gilchrist, M.S. Collins, K. Eaton, A.R. Judkins, J.A. Biegel, A.L. Folpe, Epithelioid malignant peripheral nerve sheath tumor arising in a schwannoma, in a patient with "neuroblastoma-like" schwannomatosis and a novel germline SMARCB1 mutation. Am J Surg Pathol The American journal of surgical pathology 2012; 36: (2012) 154-160.
[7]T.J.7 Hulsebos TJ, A.S. Plomp AS, R.A. Wolterman RA, E.C. Robanus-Maandag EC, F. Baas F, P. Wesseling P., Germline mutation of INI1/SMARCB1 in familial schwannomatosis. American journal of human geneticsAm J Hum Genet 2007; 80: (2007) 805-810.
[8]J. 8 Calderaro J, J. Moroch J, G. Pierron G, et al F. Pedeutour, C. Grison, P. Maille, P. Soyeux, A. de la Taille, J. Couturier, A. Vieillefond, M.C. Rousselet, O. Delattre, Y. Allory, SMARCB1/INI1 inactivation in renal medullary carcinoma. Histopathology 2012; 61: (2012) 428-435.
[9]Y. 9 Chen Y, J. Takita J, Y.L. Choi YL, et al.M. Kato, M. Ohira, M. Sanada, L. Wang, M. Soda, A. Kikuchi, T. Igarashi, A. Nakagawara, Y. Hayashi, H. Mano, S. Ogawa, Oncogenic mutations of ALK kinase in neuroblastoma. Nature 2008; 455: (2008) 971-974.
[10]M. 10 Kato M, M. Sanada M, I. Kato I, et al.Y. Sato, J. Takita, K. Takeuchi, A. Niwa, Y. Chen, K. Nakazaki, J. Nomoto, Y. Asakura, S. Muto, A. Tamura, M. Iio, Y. Akatsuka, Y. Hayashi, H. Mori, T. Igarashi, M. Kurokawa, S. Chiba, S. Mori, Y. Ishikawa, K. Okamoto, K. Tobinai, H. Nakagama, T. Nakahata, T. Yoshino, Y. Kobayashi, S. Ogawa, Frequent inactivation of A20 in B-cell lymphomas. Nature 2009; 459: (2009) 712-716.
[11]E.M. 11 Jackson EM, A.J. Sievert AJ, X. Gai X, et al.H. Hakonarson, A.R. Judkins, L. Tooke, J.C. Perin, H. Xie, T.H. Shaikh, J.A. Biegel, Genomic analysis using high-density single nucleotide polymorphism-based oligonucleotide arrays and multiplex ligation-dependent probe amplification provides a comprehensive analysis of INI1/SMARCB1 in malignant rhabdoid tumors. Clin Cancer ResClinical cancer research : an official journal of the American Association for Cancer Research 2009; 15: (2009) 1923-1930.
[12]M.C. 12 Fruhwald MC, M. Hasselblatt M, S. Wirth S, et al.G. Kohler, R. Schneppenheim, J.I. Subero, R. Siebert, U. Kordes, H. Jurgens, J. Vormoor, Non-linkage of familial rhabdoid tumors to SMARCB1 implies a second locus for the rhabdoid tumor predisposition syndrome. Pediatric blood &
11

cancerPediatr Blood Cancer 2006; 47: (2006) 273-278.[13]J. 13 Takita J, Y. Chen Y, J. Okubo J, M. Sanada M, et al. M. Adachi, K. Ohki, R.
Nishimura, R. Hanada, T. Igarashi, Y. Hayashi, S. Ogawa, Aberrations of NEGR1 on 1p31 and MYEOV on 11q13 in neuroblastoma. Cancer Sciscience 2011; 102: (2011) 1645-1650.
[14]K. 14 Uno K, J. Takita J, K. Yokomori K, et al.Y. Tanaka, S. Ohta, H. Shimada, F.H. Gilles, K. Sugita, S. Abe, M. Sako, K. Hashizume, Y. Hayashi, Aberrations of the hSNF5/INI1 gene are restricted to malignant rhabdoid tumors or atypical teratoid/rhabdoid tumors in pediatric solid tumors. Genes, c Chromosomes & cCancer 2002; 34: (2002) 33-41.
[15]C. 15 Zweier C, E.K. de Jong EK, M. Zweier M, et al. A. Orrico, L.B. Ousager, A.L. Collins, E.K. Bijlsma, M.A. Oortveld, A.B. Ekici, A. Reis, A. Schenck, A. Rauch, CNTNAP2 and NRXN1 are mutated in autosomal-recessive Pitt-Hopkins–-like mental retardation and determine the level of a common synaptic protein in Drosophila. American journal of human genetics Am J Human Genet 2009; 85: (2009) 655-666.
[16]J. 16 Takita J, M. Ishii M, S. Tsutsumi S, et al. Y. Tanaka, K. Kato, Y. Toyoda, R. Hanada, K. Yamamoto, Y. Hayashi, H. Aburatani, Gene expression profiling and identification of novel prognostic marker genes in neuroblastoma. Genes, cChromosomes & cCancer 2004; 40: (2004) 120-132.
[17]J. 17 Takita J, H.W. Yang HW, Y.Y. Chen YY, et al. R. Hanada, K. Yamamoto, T. Teitz, V. Kidd, Y. Hayashi, Allelic imbalance on chromosome 2q and alterations of the caspase 8 gene in neuroblastoma. Oncogene 2001; 20: (2001) 4424-4432.
[18]J.18 Takita J, Y. Hayashi Y, T. Nakajima T, et al. J. Adachi, T. Tanaka, N. Yamaguchi, Y. Ogawa, R. Hanada, K. Yamamoto, J. Yokota, The p16 (CDKN2A) gene is involved in the growth of neuroblastoma cells and its expression is associated with prognosis of neuroblastoma patients. Oncogene 1998; 17: (1998) 3137-3143.
[19]D. 19 Muller D, R. Millon R, S. Theobald S, et al. T. Hussenet, B. Wasylyk, S. du Manoir, J. Abecassis, Cyclin L1 (CCNL1) gene alterations in human head and neck squamous cell carcinoma. British journal of cancer Br J Cancer 2006; 94: (2006) 1041-1044.
[20]A.J. 20 Ramsay AJ, V. Quesada V, M. Foronda M, et al. L. Conde, A. Martinez-Trillos, N. Villamor, D. Rodriguez, A. Kwarciak, C. Garabaya, M. Gallardo, M. Lopez-Guerra, A. Lopez-Guillermo, X.S. Puente, M.A. Blasco, E. Campo, C. Lopez-Otin, POT1 mutations cause telomere dysfunction in chronic lymphocytic leukemia. Nature gGenetics 2013; 45: (2013) 526-530.
[21]N. 21 Omura N, C.P. Li CP, A. Li A, et al. S.M. Hong, K. Walter, A. Jimeno, M. Hidalgo, M. Goggins, Genome-wide profiling of methylated promoters in pancreatic adenocarcinoma. Cancer Biol Therbiology & therapy 2008; 7: (2008) 1146-1156.
[22]M. 22 Tsikitis M, Z. Zhang Z, W. Edelman W, D. Zagzag D, G.V. Kalpana GV., Genetic ablation of Ccyclin D1 abrogates genesis of rhabdoid tumors resulting from Ini1 loss. Proceedings of the National Academy of Sciences of the United States of America Proc Natl Acad Soc U S A 2005; 102: (2005) 12129-12134.
[23]P. 23 Nair P, K. De Preter K, J. Vandesompele J, F. Speleman F, R.L. Stallings RL., Aberrant splicing of the PTPRD gene mimics microdeletions identified at this locus in neuroblastomas. Genes, c Chromosomes & cCancer 2008; 47: (2008) 197-202.
[24]D.A. 24 Solomon DA, J.S. Kim JS, J.C. Cronin JC, et al. Z. Sibenaller, T. Ryken, S.A. Rosenberg, H. Ressom, W. Jean, D. Bigner, H. Yan, Y. Samuels, T. Waldman, Mutational inactivation of PTPRD in glioblastoma multiforme and malignant melanoma. Cancer Resresearch 2008; 68: (2008) 10300-10306.
[25]S. 25 Poliak S, L. Gollan L, R. Martinez R, et al. A. Custer, S. Einheber, J.L. Salzer, J.S. Trimmer, P. Shrager, E. Peles, Caspr2, a new member of the neurexin
12

superfamily, is localized at the juxtaparanodes of myelinated axons and associates with K+ channels. Neuron 1999; 24: (1999) 1037-1047.
[26]S. McAvoy S, S.C. Ganapathiraju SC, A.L. Ducharme-Smith AL, et al. J.R. Pritchett, F. Kosari, D.S. Perez, Y. Zhu, C.D. James, D.I. Smith, Non-random inactivation of large common fragile site genes in different cancers. Cytogenet ic and gGenome Res research 2007; 118: (2007) 260-269.
13

Figure Legends
Figure 1. Copy number changes detected in malignant rhabdoid
tumors. A: Characteristics of copy number alterations in malignant
rhabdoid tumors. Regions showing statistically significant increase or
decrease in genomic copy number were detected using the GISTIC
algorithm based on SNP array analysis. B: Overall representation of
aberrations of chromosome 22q11.2 in malignant rhabdoid tumors.
Specimens indicated by red are cell lines. Pink bar indicates uniparental
disomy, and yellow and green bars indicate heterozygous deletion and
homozygous deletion, respectively. The minimum overlapping deleted
region was expanded in 175kb in chromosome 22q11.2 including
SMARCB1 and other 6 genes. SMARCB1 status is also indicated at the
right. MRT-12 and MRT-14 shows heterozygous deletion of SMARCB1
locus, and wild-type allele of SMARCB1 was retaineds in each case. UPD:
uniparental disomy, HD: homozygous deletion, mt: mutation, and del:
deletion.
Figure 2. Recurrent deletions of chromosome 7q35-q36 and
CNTNAP2 alterations in malignant rhabdoid tumor. A: Deletions of
chromosome 7q35-36 in 3 specimens detected by SNP array. For each
panel, total copy numbers (tCNs) (red dots), moving averages of tCNs for
five 5 consecutive SNPs (blue line), an ideogram of the relevant
chromosome, location of heterozygous SNP calls (green bars), and allele-
specific copy numbers (AsCNs) averaged for five 5 consecutive SNPs (red
and green lines for larger and smaller alleles, respectively) are plotted. B:
Expression and mutation analyses of rhabdomyosarcoma. Upper panel
14

shows RT-PCR analysis of CNTNAP2 in 9 cell lies. Sequence chromatogram
of R157C missense mutation detected in a fresh tumor, MRT-8, is shown in
the lower lower panel. C: Bisulfate modification- and methylation- specific
PCR for CNTNAP2 in cell lines. Hypermethylation of CgG island in KYM-1
cell line is shown in the upper panel. The Llower panel shows control.
CpG islands are marked by asterisks. D: Representative results of re-
expression of transcriptionally silenced CNTNAP2 after treatment of 5-aza-
deoxycytidine in malignant rhabdoid tumor cell lines. RT-PCR analysis of
RTK(J)-4N cell line harvested following 72 h incubation with control media
(-) and 5 μM 5-aza-2-deoxycytidine (+). SJNB-1 neuroblastoma cell line,
which expressed abundant CNTNAP2, was used as a positive control.
15

Table 1. Mutations of SMARCB1 (SNF5/INI1) in malignant rhabdoid tumor.Sample name Attribute 22q Mutation of SMARCB1
KYM-1 Cell line homoD homoDTM87-16 Cell line homoD homoDTTC-1240 Cell line UPD G646T (E216 stop)TTC-549 Cell line homoD homoDTTC-642 Cell line UPD C118T (R40 stop)TTN-45 Cell line UPD+homoD homoDYAMRT Cell line UPD del exons 1-3STM-91-01 Cell line homoD homoDRTK(J) -4N Cell line homoD homoDMRT-2 Primary homoD homoDMRT-3 Primary homoD homoDMRT-4 Primary homoD homoDMRT-5 Primary UPD+homoD homoDMRT-6 Primary UPD+homoD homoDMRT-7 Primary homoD homoDMRT-8 Primary UPD+homoD homoDMRT-9 Primary UPD 27bp to ex6 553 delMRT-10 Primary normalMRT-12 Primary heteroDMRT-13 Primary heteroD ag to aa intron 5, D224 stopMRT-14 Primary heteroD
16

Table 2. Recurrent gains and losses in malignant rhabdoid tumor.
chromosomePosition†* Length
(kb)
Number of
affected samples‡ #Gene(s)
start end
Gain
1 120,804,640 245,519,990 124,715
4 (CL: TM87-16,
TTC-549, YAMRT,
P:MRT-8)
many genes
2 41,745,732 42,067,857 322 1 (CL:TTC1240) none gene
3 158,297,982 158,507,647 2092 (CL:TM87-16,
P:MRT-8)CCNL1, VEPH1
7 123,981,822 124,568,564 587
4 (CL:TM87-16,
TTC-1240, TTC-
549, TTN-45)
POT1
7 125,675,684 125,906,795 2312 (CL:TM87-16,
KYM-1)GRM8
7 130,358,924 130,939,457 580
3 (CL:TM87-16,
TTC-1240, TTN-
45)
none gene
16 76,268,665 77,096,360 8282 (CL:TM87-16,
KYM-1)CLEC3A, WWOX
Loss
2 140,875,055 141,241,576 366 1 (CL:STM-91-01) LRP1B
3 60,099,620 60,538,672 4393 (CL:TM87-16,
TTC-642, YAMRT)FHIT
3 78,740,688 78,995,893 2552 (CL:TM87-16,
P:MRT-8)ROBO1
4 182,649,464 182,977,255 3283 (CL:TTC-642,
KYM-1, RTK(J)-4N)none genes
7 69,446,093 69,593,327 1472 (CL:TM87-16,
TTC-642)AUTS2
73 (CL:STM-91-01,
TTC-549, P:MRT-7)CNTCAP2
9 9,582,260 10,072,420 4903 (CL:TM87-16,
TTC-1240, KYM-1)PTPRD
16 71,464,505 71,708,905 2442 (CL:TTC-642,
P:MRT-8)ATBF1
List of lesions detected in more than two 2 cases, and known genes in the regions.
*†NCBI build 35#‡ CL: cell line, P: primary sample
17

18
19
STable S1. Analyzed samples and SNP array platformSample name Cell line / Primary ArrayKYM-1 Cell line XbaI 50K arrayTM87-16 Cell line NspI 250K arrayTTC-1240 Cell line XbaI 50K arrayTTC-549 Cell line XbaI 50K arrayTTC-642 Cell line NspI 250K arrayTTN-45 Cell line XbaI 50K arrayYAMRT Cell line NspI 250K array
STM-91-01 Cell line NspI 250K array
RTK(J) -4N Cell line XbaI 50K arrayMRT-2 Primary NspI 250K arrayMRT-3 Primary XbaI 50K arrayMRT-4 Primary XbaI 50K arrayMRT-5 Primary XbaI 50K arrayMRT-6 Primary NspI 250K arrayMRT-7 Primary NspI 250K arrayMRT-8 Primary NspI 250K arrayMRT-9 Primary NspI 250K array
MRT-10 Primary NspI 250K array
MRT-12 Primary NspI 250K arrayMRT-13 Primary NspI 250K arrayMRT-14 Primary NspI 250K array
20


















