
JSCDH · operative femoral head collapse (higher Ficat score) (Table). Complication rates in the...
106
A Peer-Reviewed Journal Promoting Science, Clinical Care and Public Health in Sickle Cell Disease and Hemoglobinopathies. JOURNAL OF SICKLE CELL DISEASE AND HEMOGLOBINOPATHIES Changing the Conversation for Sickle Cell Disease and Reshaping its Future Published: June 7, 2019 JSCDH Lanetta Bronté-Hall, MD, MPH, MSPH Editor-in-Chief ISSN: 2330-1473 DOI 10.14223
Transcript of JSCDH · operative femoral head collapse (higher Ficat score) (Table). Complication rates in the...

A Peer-Reviewed Journal Promoting Science, Clinical Care and Public Health in Sickle Cell Disease and Hemoglobinopathies.
JOURNAL OF SICKLE CELL DISEASE AND HEMOGLOBINOPATHIES
Changing the Conversation for Sickle Cell Disease and Reshaping its FuturePublished: June 7, 2019
JSCDH
Lanetta Bronté-Hall, MD, MPH, MSPHEditor-in-ChiefISSN: 2330-1473 DOI 10.14223

SPONSORS
EXHIBITORS
Silver
Patron
Bronze
Bronze
Bronze Bronze
Patron
Special Sponsor Special Sponsor
Patron Patron
SPONSORS
EXHIBITORS
Silver
Patron
Bronze
Bronze
Bronze Bronze
Patron
Special Sponsor Special Sponsor
Patron Patron
SPONSORS
EXHIBITORS
Silver
Patron
Bronze
Bronze
Bronze Bronze
Patron
Special Sponsor Special Sponsor
Patron Patron
SPONSORS
EXHIBITORS
Silver
Patron
Bronze
Bronze
Bronze Bronze
Patron
Special Sponsor Special Sponsor
Patron Patron

www.FSCDR.org


3 | P a g e

4 | P a g e
TOP ABSTRACTS Presenting: Saturday, June 8, 2019 at 5:45 PM

5 | P a g e
JSCDH-D-19-00004 OUTCOMES OF OSTEONECROSIS OF THE FEMORAL HEAD IN SICKLE CELL DISEASE AFTER SURGICAL TREATMENT WITH
CORE DECOMPRESSION ALONE VS CORE DECOMPRESSION WITH BONE MARROW ASPIRATE CONCENTRATE INJECTION
Authors: Regina Hanstein, Adele Heib, Irene Chern,
Ugochi O Ugo, Kerry Morrone, Deepa Manwani, Caterina
Minniti, Eric D Fornari, MD
Affiliation: Children’s Hospital at Montefiore
Background: Osteonecrosis of the femoral head (ONFH)
is a common complication of SCD. We set out to describe
clinical and radiographic outcomes of SCD patients with
ONFH treated with (1) core decompression (CD) or (2) CD
plus injection of patients own bone marrow aspirate
concentrate (CD+BMAC). CD involves surgical drilling into
the femoral head to reduce pressure and allow increased
blood flow. The addition of BMAC injection is based on
the hypothesis that BMAC contains osteogenic
precursors, which will repopulate the osteonecrotic
bone.
Methods: 35 hips in 26 SCD patients, 50% female, mean
age 24.3years (range, 9.7-50.7years) at surgery were
evaluated at average follow-up 3.6years (range 0.5-
13years). 30 hips had HbSS/HBSB0 and 5 HBSC/HBSB+.
Demographics, surgical parameters, patient self-reported
pain and ambulatory status were recorded. Femoral head
collapse was assessed on Xrays using the 4-stage Ficat
score system (stage1 pre-collapse → stage4 complete
collapse+flattened contour+decreased joint space).
Results: 17 hips underwent CD and 18 CD+BMAC based
on surgeon preference. All were pre-operatively
transfused to a Hb of 10. In each surgical group, 71%
used Hydroxyurea at time of surgery; CD+BMAC patients
used a higher Hydroxyurea dose compared to CD patients
(18.2mg/kg vs. 25.1mg/kg, p=0.022). Other demographic
and surgical parameters were similar between groups.
Collapse of the femoral head didn’t progress in 8/15 CD
and 9/13 CD+BMAC (p=0.390). Progression to total hip
arthroplasty (THA) was similar between groups: CD 5/17
(29%) vs CD+BMAC 4/18 (22%), p=0.711. Patients
progressing to THA were significantly older at surgery,
used a lower Hydroxyurea dose, and had more pre-
operative femoral head collapse (higher Ficat score)
(Table).
Complication rates in the immediate 6 months period
post-surgery were low; 2 CD and 3 CD+BMAC were
transfused and hospitalization rates for VOC between
groups were similar (p=0.103). With either treatment,
>90% of patients were pain-free and walked
independently at most recent follow-up.
Conclusion: CD and CD+BMAC are safe procedures to
prevent progression of ONFH in SCD patients. Older SCD
patients using lower dosage of Hydroxyurea and more
femoral head collapse were more likely to proceed to
THA.
CD and CD+BMAC No THA THA P-value
Age at Surgery in years, Mean±SD 21 ±9.3 33.9 ±12.0 0.003
Hydroxyurea dose at Surgery in mg/kg, Mean±SD 24.7±4.9 12.5±8.0 0.005
Pre-operative Ficat score, N (%) 1&2
3
4
20 (77%)
3 (11.5%)
3 (11.5%)
3 (37.5%)
5 (62.5%)
0 (0%)
0.042

6 | P a g e
JSCDH-D-19-00041 BARRIERS TO THE USE OF ENDARI IN AN URBAN ADULT SICKLE CELL DISEASE CENTER
Authors: 1Ugochi Ogu MD,
1Merin Thomas NP,
2Flo Chan
Pharm D, 1Gracy Sebastian NP, and
1Caterina Minniti MD.
Affiliations: 1 Montefiore Medical Center, 2 Lemed
Pharmacy
Background: In 2017, the US Food and Drug
Administration (FDA) approved Endari (L-glutamine oral
powder) for patients age five years and older with sickle
cell disease (SCD) to reduce disease complications
associated with SCD. This was applauded as the first
medication approval for SCD in almost 20 years, following
Hydroxyurea (HU) in 1998. A phase 3 trial that led to its
approval showed that Endari decreased the number of
pain crisis in SCD patients. We report our experience with
prescribing Endari in an urban adult sickle cell center. To
our knowledge, this report is a first of its kind.
Methods: After prospectively establishing guidelines for
the use of Endari, adult patients with SCD seen in clinic at
a large urban adult SCD center were prescribed Endari via
a local specialty pharmacy, over a 10 month period. Upon
return to clinic, patients were asked about barriers to
obtaining the medication and adherence to the twice a
day dosing. Adherence was also evaluated by calculating
the mean possession ratio (MPR) utilizing pharmacy’s
records.
Results: 101 patients with SCD (58% females, 43% males)
were prescribed Endari over a 10 month period (Table 1):
82% with severe disease genotypes (Hb SS/Sβ0), 18% with
mild genotypes (Hb SC/Sβ+). Mean age was 36 years old.
31% of patients were on concomitant HU and 3% on
chronic transfusions (>6 months). At the end of the 10
month period, 40 patients (40%) were actively taking
Endari, 20 (20%) had discontinued, 35 (35%) never filled
the script, and 6 (6%) had received but never started
Endari. Barriers to initiating Endari included high
deductibles (reported in 7% of patients), denied prior
authorizations (11%) and inability of pharmacy to contact
patient (11%) (Table 2). Reasons for discontinuing Endari
included side effects (46%), poor adherence (22%), no
perceived benefit (20%), and pregnancy (1%) (Table 3).
Average MPR was 0.74, similar to the Endari study.
Discussion: This is the first study that addresses both
acceptance of a new medication by the sickle cell
population and the barriers to obtaining it. We identified
multiple barriers with the initiation and adherence to
Endari in our urban adult SCD patient population. The
most common reason that the medication was not
initiated was denial from the insurance companies. The
most common reason cited for discontinuing Endari was
perceived side effects or ineffectiveness by the patients.
After 10 months, only 40% of patients prescribed Endari
were still taking this medication. As experienced with HU,
several years elapsed from initial FDA approval to its
being more accepted and widely used. From our report, it
is critical to evaluate and eradicate barriers to initiation
and adherence to Endari, to ensure it is readily available
and accessible to the patient population it gained
approval and was intended for.

7 | P a g e
Tables 1: Demographics N = 101
Age (mean, range) 36 (21-68)
Sex Female 57 (57%)
Male 43 (43%)
Genotype Hb SS 81 (80%)
Hb SC 12 (12%)
Hb S+ 6 (6%)
Hb S0 2 (2%)
Insurance Medicare/Medicaid 95 (95%)
Private 6 (6%)
Hydroxyurea Yes 31 (31%)
No 70 (70%)
Chronic Transfusion 3 (3%)
Table 2: Barriers to initiating Endari N = 101
High Deductible 7 (7%)
Prior Authorization Denied 11 (11%)
Unable to contact 10 (10%)
Table 3: Reasons for stopping Endari
N = 101
Side Effects 46(46%)
Poor Adherence 22(22%)
Pregnant 1 (1%)
No Perceived Benefits 20 (20%)

8 | P a g e
JSCDH-D-19-00046 THE OXYGENSCAN: A RAPID AND REPRODUCIBLE SOURCE OF CLINICALLY RELEVANT
BIOMARKERS IN FOR PATIENTS WITH SICKLE CELL DISEASE
Authors: Celeste K. Kanne3, Minke A.E. Rab
1,2, Brigitte A.
van Oirschot1, Jennifer Bos
1, Maite E. Houwing
4, Marjon
H. Cnossen4 Roger E.G. Schutgens
2, Gerard Pasterkamp
1,
Richard van Wijk1, Eduard J. van Beers
2 and Vivien A.
Sheehan3
Affiliations: 1Laboratory of Clinical Chemistry &
Haematology, University Medical Center Utrecht, 2Van
Creveldkliniek, University Medical Center Utrecht,
Utrecht, The Netherlands, 3Department of Pediatrics,
Division of Hematology/Oncology, Baylor College of
Medicine, Houston Texas, USA,4Department of Pediatric
Hematology, Erasmus Medical Center, Rotterdam, The
Netherlands
Background: In sickle cell disease (SCD), hemoglobin S
(HbS) polymerizes upon deoxygenation, reducing red
blood cell (RBC) deformability. RBC deformability can be
measured over a range of osmolalities and oxygen
concentrations using a Laser Optical Rotational Red Cell
Analyzer (Lorrca) ektacytometer (RR Mechatronics) with
Oxygenscan module (pO2scan). The Oxygenscan
measures 3 key parameters: 1) EImax, RBC deformability
at normoxia; 2) EImin, deformability upon
deoxygenation; and 3) the point of sickling (PoS), the
point at which a >5% decrease in EI is observed during
deoxygenation, reflecting the patient-specific pO2 at
which sickling begins (Figure 1). In this study, we
correlated Oxygenscan parameters with various SCD
genotypes and treatments.
Methods: We analyzed 53 pediatric SCD patients from
Texas Children’s Hospital (TCH) and 24 SCD pediatric and
adult patients from University Medical Center Utrecht
(UMCU). 19 HbSS (median age 5.5 years (y); 10 female
(f)), 11 HbSC (median age 8.9y; 6f), and HbSB+ (median
age 8.4y; 3f) from TCH were analyzed; 14 HbSS patients
without treatment (median age 20.0y; 5f), 6 HbSS
patients with alpha-thalassemia trait without treatment
(median age 9y; 4f), and 3 HbSC patients (median age
28y; 3f) from UMCU were analyzed. To establish
Oxygenscan changes between treatments, we analyzed
RBC from 17 HbSS patients from TCH (median age 10.8y;
6f) on HU before and after transfusion. From UMCU,
samples from 8 patients before and on HU were
analyzed. Tests were performed in duplicate.
Results: In the TCH cohort, PoS differed significantly
between HbSS and HbSC (p<0.01), HbSS and HbSB+
(p<0.0002), and HbSC and HbSB+ (p<0.02). HbSB+
patients tolerated lower oxygen concentrations before
sickling, PoS (23.5 mmHg) than HbSC patients (31.9
mmHg) and HbSS patients (39.9 mmHg). EImin did not
differ between genotypes; EImax did not differ between
HbSS and HbSC, but did between HbSS and HbSB+
(p<0.01), and HbSC and HbSB+ (p<0.000003).
In the UMCU cohort, PoS differed between HbSS and
HbSS + α-Thal (p<0.03) and HbSS and HbSC (P<0.05), with
the lowest PoS in HbSC (mean 47.5 mmHg). HbSS + α-Thal
had a lower PoS (51.1mmHg) compared to HbSS without
α-Thal (67.5 (mmHg).
Transfusion significantly improved the EImax, EImin, and
PoS (p<0.0006, p<0.00004, and p<0.01 respectively). HU
treatment also significantly improved all parameters
compared to baseline (p<0.05).
Conclusion: The Lorrca with Oxygenscan can fully
characterize RBC deformability under a range of oxygen
concentrations. Key measurements - PoS, EImin, and
EImax – were different between genotypes, and changed
significantly with standard of care SCD treatments. We
therefore conclude that these are useful biomarkers of
clinical severity and treatment response, and may be
essential in monitoring novel SCD treatments as part of a
clinical trial.

9 | P a g e
ABSTRACT BREAKOUT SESSION I BASIC SCIENCE ORAL PRESENTATION
Presenting: Sunday, June 9, 2019 at 1:45 PM

10 | P a g e
JSCDH-D-19-00047 REGULATION OF FETAL HEMOGLOBIN THROUGH THE INSULIN SIGNALING PATHWAY
Authors: Alireza Paikari, Yankai Zhang, Alicia K. Chang, Ankush Goyal, Evadnie Rampersaud, Jonathan M. Flanagan, Mitchell J. Weiss, Vivien A. Sheehan Affiliation: Baylor College of Medicine Background: HbF induction is a key therapeutic strategy
for sickle cell disease (SCD). Analysis of whole exome
sequencing (WES) data from patients with SCD identified
variants in two components of the insulin signaling
pathway, FOXO3 and its activator, AMPK, to be
associated with HbF levels; the association was confirmed
by functional studies in hematopoietic stem and
progenitor cells (HSPC), and metformin, a FOXO3/AMPK
activator, increased HbF in HSPC. Whole genome
sequencing (WGS) analysis identified variants in the
regulatory region of IGFBP3, another component of the
insulin signaling pathway, as associated with HbF levels.
Methods: 658 pediatric SCD patients (>2y, 52% male)
underwent WGS; mixed linear regression was used to
screen for variants associated with %HbF. Three unique
SCD patient-derived HSPC cultures were treated with
metformin (100 µM), compound C (1 µM), and
exogenous IGFBP3 (1µg/ml); their effect on HbF, gamma-
globin, known modifiers of HbF and the FOXO3-AMPK
pathway were assessed by HPLC, RT-qPCR and western
blot at day 14 and 21 of culture. Erythroid maturation
was assessed by morphology and flow cytometry. Plasma
levels of IGFBP3 in patients with and without IGFBP3
variants were measured by ELISA.
Results: WGS identified an association between variants in IGFBP3 and baseline HbF levels (p<1x10
-6). Plasma
IGFBP3 levels were higher in patients heterozygous for an IGFBP3 variant (p=0.01). The role of AMPK in HbF regulation was confirmed by pharmacologic blockade of AMPK by compound C, a specific AMPK inhibitor; compound C reduced gamma-globin expression, and prevented gamma globin induction by metformin.
Treatment of HSPCs with recombinant IGFBP3 resulted in a significant increase in %HbF (p=0.008). Adding IGFBP3 to erythroid culture altered the insulin signaling pathway; total protein levels of FOXO3 increased (p=0.01), as well
as its activated phosphorylated form (Ser 413) that keeps FOXO3 in the nucleus (p=0.03). AMPK protein levels did not increase, but the phosphorylated form of AMPK (Thr172) that in turn phosphorylates and activates FOXO3 did increase (p=0.01). Neither IGFBP3 nor metformin altered erythroid maturation or expression of known gamma globin regulators; BCL11A, KLF1, and MYB.
Conclusions: Unbiased WES and WGS analysis identified
variants in the coding and non-coding region of several
insulin signaling pathway effectors (FOXO3/AMPK and
IGFBP3) to be associated with HbF levels; this was
verified by functional studies in HSPC. We therefore
conclude that the insulin signaling pathway plays a
significant role in gamma-globin regulation and is an
important therapeutic target for HbF induction in SCD
patients.
11 | P a g e
JSCDH-D-19-00009
LENTIGLOBIN GENE THERAPY IN PATIENTS WITH SICKLE CELL DISEASE: UPDATED INTERIM RESULTS FROM HGB-206
Authors: Lakshmanan Krishnamurti,
1John F.
Tisdale, 2Julie Kanter,
3Janet L. Kwiatkowski,
4Markus
Y. Mapara, 5Manfred Schmidt,
6Alexandra Miller,
6Francis J. Pierciey,
6Wenmei Huang,
6Jean-Antoine
Ribeil, 7Alexis A. Thompson, and
8Mark C. Walters,
Affiliations:
1National Institutes of Health,
2University of Alabama at Birmingham,
3Children’s
Hospital of Philadelphia, 4Columbia University
College of Physicians and Surgeons, 5GeneWerk
GmbH, 6bluebird bio, Inc.,
7Ann and Robert H. Lurie
Children’s Hospital of Chicago and Northwestern University Feinberg School of Medicine, Blood and Marrow Transplant Program,
8UCSF Benioff
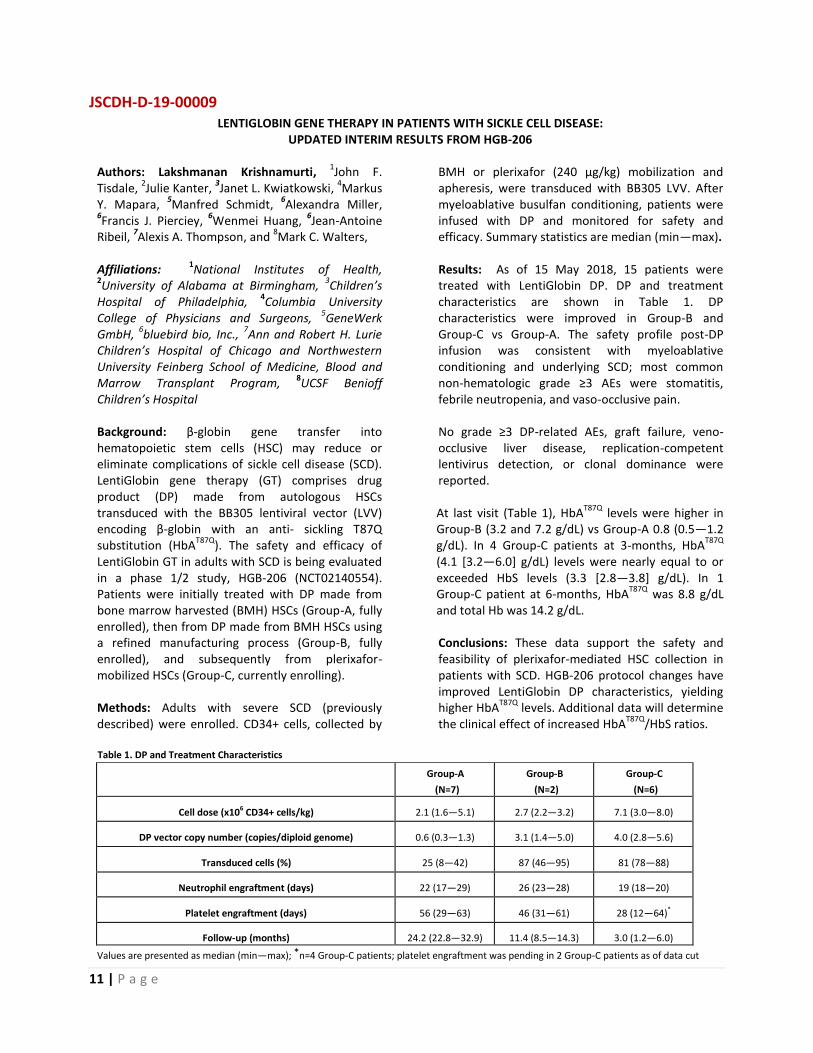
Children’s Hospital Background: β-globin gene transfer into hematopoietic stem cells (HSC) may reduce or eliminate complications of sickle cell disease (SCD). LentiGlobin gene therapy (GT) comprises drug product (DP) made from autologous HSCs transduced with the BB305 lentiviral vector (LVV) encoding β-globin with an anti- sickling T87Q substitution (HbA
T87Q). The safety and efficacy of
LentiGlobin GT in adults with SCD is being evaluated in a phase 1/2 study, HGB-206 (NCT02140554). Patients were initially treated with DP made from bone marrow harvested (BMH) HSCs (Group-A, fully enrolled), then from DP made from BMH HSCs using a refined manufacturing process (Group-B, fully enrolled), and subsequently from plerixafor- mobilized HSCs (Group-C, currently enrolling). Methods: Adults with severe SCD (previously described) were enrolled. CD34+ cells, collected by
BMH or plerixafor (240 µg/kg) mobilization and apheresis, were transduced with BB305 LVV. After myeloablative busulfan conditioning, patients were infused with DP and monitored for safety and efficacy. Summary statistics are median (min—max). Results: As of 15 May 2018, 15 patients were treated with LentiGlobin DP. DP and treatment characteristics are shown in Table 1. DP characteristics were improved in Group-B and Group-C vs Group-A. The safety profile post-DP infusion was consistent with myeloablative conditioning and underlying SCD; most common non-hematologic grade ≥3 AEs were stomatitis, febrile neutropenia, and vaso-occlusive pain. No grade ≥3 DP-related AEs, graft failure, veno-occlusive liver disease, replication-competent lentivirus detection, or clonal dominance were reported. At last visit (Table 1), HbA
T87Q levels were higher in
Group-B (3.2 and 7.2 g/dL) vs Group-A 0.8 (0.5—1.2 g/dL). In 4 Group-C patients at 3-months, HbA
T87Q
(4.1 [3.2—6.0] g/dL) levels were nearly equal to or exceeded HbS levels (3.3 [2.8—3.8] g/dL). In 1 Group-C patient at 6-months, HbA
T87Q was 8.8 g/dL
and total Hb was 14.2 g/dL. Conclusions: These data support the safety and feasibility of plerixafor-mediated HSC collection in patients with SCD. HGB-206 protocol changes have improved LentiGlobin DP characteristics, yielding higher HbA
T87Q levels. Additional data will determine
the clinical effect of increased HbAT87Q
/HbS ratios.
Table 1. DP and Treatment Characteristics
Group-A
(N=7)
Group-B
(N=2)
Group-C
(N=6)
Cell dose (x106 CD34+ cells/kg) 2.1 (1.6—5.1) 2.7 (2.2—3.2) 7.1 (3.0—8.0)
DP vector copy number (copies/diploid genome) 0.6 (0.3—1.3) 3.1 (1.4—5.0) 4.0 (2.8—5.6)
Transduced cells (%) 25 (8—42) 87 (46—95) 81 (78—88)
Neutrophil engraftment (days) 22 (17—29) 26 (23—28) 19 (18—20)
Platelet engraftment (days) 56 (29—63) 46 (31—61) 28 (12—64)*
Follow-up (months) 24.2 (22.8—32.9) 11.4 (8.5—14.3) 3.0 (1.2—6.0)
Values are presented as median (min—max); *n=4 Group-C patients; platelet engraftment was pending in 2 Group-C patients as of data cut

12 | P a g e
JSCDH-D-19-00012 VOXELOTOR TREATMENT OF ADULTS AND ADOLESCENTS WITH SICKLE CELL DISEASE:
RESULTS OF THE PHASE 3 HOPE TRIAL
Authors: Elliot Vichinsky1, Carolyn C. Hoppe
2, Jo
Howard3, Kenneth I. Ataga
4, Videlis Nduba
5, Amal El
Beshlawy6, David L. Diuguid
7, Salam Al Kindi
8, Clark
Brown9, Hoda Hassab
10, Paul Telfer
11, Dimitris A.
Tsitsikas12
, Selma Ünal13
, Julie Kanter14
, Miguel R.
Abboud15
, Victor R. Gordeuk16
, Joshua Lehrer-
Graiwer2, Margaret Tonda
2, Allison Intondi
2, Russell E.
Ware17
Affiliations: 1UCSF Benioff Children's Hospital,
Oakland, CA; 2Global Blood Therapeutics, South San
Francisco, CA; 3Guy’s and St Thomas’ NHS Foundation
Trust and King’s College, London; 4University of
Tennessee Health Science Center at Memphis,
Memphis, TN; 5Kenya Medical Research Institute,
Kisumu, Kenya; 6Cairo University, Cairo, Egypt;
7New
York Presbyterian Hospital, Columbia University
Medical Center, New York, NY; 8Sultan Qaboos
University, Muscat, Oman; 9Emory University, Aflac
Cancer and Blood Disorders Center Children's
Healthcare of Atlanta, Atlanta, GA; 10
Alexandria
University, Alexandria, Egypt; 11
Barts Health NHS
Trust, London; 12
Homerton University Hospital NHS
Foundation Trust, London; 13
Mercin University,
Turkey; 14
Lifespan Comprehensive Sickle Cell Center,
Medical University of South Carolina, Charleston, SC; 15
American University of Beirut Medical Center, Beirut,
Lebanon; 16
University of Illinois at Chicago, Chicago,
IL; 17
Cincinnati Children’s Hospital, Cincinnati, OH
Background: Sickle cell disease (SCD) is a genetic disorder caused by a single amino acid substitution in the β-chain of hemoglobin (Hb) resulting in the production of sickle Hb (HbS). When deoxygenated, HbS polymerizes triggering chronic anemia and hemolysis with downstream vaso-occlusive crises and organ damage. Voxelotor is a once-daily oral therapy designed to inhibit hemoglobin (Hb) polymerization, and thereby improve anemia and hemolysis. The randomized phase 3 HOPE study (NCT03036813) evaluates the efficacy and safety of voxelotor in
patients with SCD aged 12 to 65 years. Here we report the results of the pre-specified Part A of the HOPE study.
Methods: Eligible patients were randomly assigned to receive voxelotor (900 or 1500 mg/day) or placebo for ≥24 weeks. The primary endpoint was the proportion of patients with a >1g/dL increase in Hb from baseline at week 24. Secondary endpoints included improvement from baseline in measures of hemolysis (eg, reticulocyte counts and unconjugated bilirubin level) and safety.
Results: Part A included 154 patients with a median age of 25 years (range, 12–59); 14% were adolescents, and 42% were male. Most were HbSS/HbSβ
0: 94% (900 mg), 92% (1500 mg), and
90% (placebo). Hydroxyurea use at study entry was 67% (900 mg), 62% (1500 mg), and 64% (placebo), and median baseline Hb was 8.3 g/dL (900 mg; range, 6.3–10.8), 8.6 g/dL (1500 mg; range, 5.9–10.8), and 8.5 g/dL (placebo; range, 6.1–10.4). At week 24, the percentage of patients with a >1 g/dL increase in Hb from baseline was significantly larger for both voxelotor arms, 900 mg (33%; P=0.0159) and 1500 mg (65%; P<0.0001), compared with placebo (10%). Voxelotor also resulted in improvements in measures of hemolysis, consistent with the demonstrated improvement in Hb. Overall, the treatment-emergent adverse events (TEAEs) were grade 1 or 2 in severity and similar across all treatment groups except for diarrhea, which was higher in both voxelotor arms (900 mg, 19%; 1500 mg, 21%) compared to placebo (10%).
Conclusions: Voxelotor treatment demonstrated a
dose-dependent increase in Hb, with a large number
of patients achieving a >1 g/dL improvement in Hb
and decreases in measures of hemolysis. Voxelotor
was generally well tolerated. Voxelotor has the
potential to be disease modifying by improving
anemia and hemolysis leading to the reduction in
morbidity of chronic organ damage associated with
SCD.

13 | P a g e
JSCDH-D-19-00048
A NOVEL NEXT-GENERATION SEQUENCING (NGS) BASED ASSAY FOR NON-INVASIVE PRENATAL TESTING OF SICKLE
CELL DISEASE WITHOUT PATERNAL DNA
Authors: Vivien Andrea Sheehan, Nelda Itzep, MD,
Celeste K Kanne, BS, David Tsao, PhD, Oguzhan Attay,
PhD
Affiliation: Baylor College of Medicine
Background: Over 300,000 infants are born with
sickle cell disease (SCD) every year worldwide,
including at least 1,000 in the US. Prenatal diagnosis
by amniocentesis or chorionic villus sampling is
available; but high cost, invasiveness, and risk of
miscarriage limit their use. Recently, non-invasive
prenatal testing (NIPT), which operate by genetic
analysis of the cell-free fetal DNA (cffDNA) present in
maternal blood, has become commonplace for
aneuploidies, including Trisomy 21. Yet, no NIPT for
SCD or other hemoglobinopathies have been
commercialized to date. We have developed and
optimized NIPT for SCD by assessing the relative
mutation dosage of fetal SCD and beta-thalassemia
DNA through a novel molecular counting strategy
using NGS.
Objectives: The primary objective of this study is to
evaluate the performance of a novel NIPT for sickle
cell disease.
Methods: The SCD NIPT assay and associated custom
bioinformatics analysis were performed on cfDNA
obtained from a training cohort of non-pregnant
compound heterozygotes for SCD. The SCD NIPT assay
was then performed on a validation cohort of
pregnant women with either SCD or sickle cell trait
(SCT). The accuracy of the SCD NIPT was evaluated by
comparison with newborn screening results.
Results: Non-pregnant individuals aged 2-20 years
with genotype HbSE, HbSC, or HbS/beta-thalassemia
were included as a training cohort to establish the
precision and accuracy of the assay for measuring HbS
allele fraction from cfDNA. This cohort had a M:F ratio
of 23:17. As expected, the HbS allele fraction in these
individuals was 0.500 (standard deviation = 0.011, n =
26), and there was no detectable fetal fraction in
these samples. Both training and validation cohort
results matched the theoretical limit of detection set
by the number of cell-free HBB DNA molecules in
plasma. The precision and accuracy of the HBB assay
on cfDNA were then used in conjunction with >1000
pre-clinical samples (mixtures of sheared SCT and SCD
genomic DNA) to determine analytical sensitivity
>98% and specificity >99%, even in the absence of
paternal DNA.
Conclusion: We have developed an assay for NIPT of
sickle cell disease. The results obtained to date
indicate that the assay reliably detects fetal SCD
status when the fetal fraction is as low as 5%, the
same limit as aneuploidy NIPT. SCD NIPT could be
particularly useful for deciding to bank umbilical cord
blood as a source of stem cells for future gene-editing
cures.

14 | P a g e
JSCDH-D-19-00049 CLINICAL TRANSFORMATION IN CARE FOR PATIENTS WITH SICKLE CELL DISEASE AT AN URBAN ACADEMIC
MEDICAL CENTER
Authors: Sanaa Rizk, MD, David Axelrod, MD, Gaye Riddick-Burden, CRNP, Elisabeth Congdon-Martin, LSW, Steven McKenzie, MD, Carol Haines, John McAna, PhD, Albert G. Crawford, PhD, Lawrence Ward, MD
Affiliation: Thomas Jefferson University Hospital
Abstract:
Background: Sickle cell disease (SCD), an inherited red blood cell disorder, is characterized by anemia, end-organ damage, unpredictable episodes of pain, and early mortality. Emergency department (ED) visits and hospitalizations are frequent, leading to increased burden on patients and increased health care costs. We report herein on two clinical transformations at Thomas Jefferson University Hospital in care for patients with SCD over the past 6 years. The first clinical transformation, in November 2013, was a multidisciplinary care team intervention which included monthly team meetings and development of individualized care plans and targeted for management of uncomplicated pain crises. The second clinical transformation, in 2015, involved changing the locus of care from a dedicated Sickle Cell day unit to an approach which “fast-tracks” patients through the ED into an observation unit with 24/7 access.
Methods: This study assessed the two clinical transformations mentioned above. The effects of the first clinical transformation were assessed through
quantitative statistical analyses, stratified by high utilizer status (already reported in a manuscript published in the American Journal of Medical Quality
1). The effects of the second clinical
transformation were assessed through both quantitative statistical analysis and qualitative analysis, i.e., five case studies.
Results: Regarding the first clinical transformation, following implementation of the multidisciplinary care team intervention, significant decreases in both ED and inpatient utilization were identified among those individuals with a history of high ED utilization. Regarding the second clinical transformation, quantitative analyses of claims data suggest further decreases in the rates of ED visits and inpatient stays, but these analyses require further refinement. At the same time, five case studies reinforce the overall findings and provide insight into the specific effects of the clinical transformation on the course of patients’ SCD and other comorbid conditions, and also their quality of life.
Conclusions: Findings highlight the potential strength of these clinical transformations.
Reference: 1Powell RE, Lovett P, Axelrod D, Crawford A, McAna J,
Ward L, Pulte E. A multi-disciplinary approach to impact acute care utilization among individuals with sickle cell disease. American Journal of Medical Quality, 2018, 33(2),127-131.

15 | P a g e
ABSTRACT SESSION I- HEALTH SERVICES ORAL PRESENTATIONS
Presenting: Sunday, June 9, 2019 at 1:45PM

16 | P a g e
JSCDH-D-19-00014 THE ROLE OF SOCIAL DETERMINANTS OF HEALTH IN AFFILIATION STATUS FOR A COHORT OF SICKLE CELL DISEASE
PATIENTS FROM A CHICAGO BASED COMPREHENSIVE SICKLE CELL CENTER
Authors: G.G. Méndez, V. Gordeuk, M. Berbaum, J.M. Nocek, L.L. Hsu.
Affiliation: University of Illinois at Chicago College of Medicine
Background: Factors associated with clinic
attendance are better understood for children with
sickle cell disease (SCD) than for adults with SCD. We
used a definition for unaffiliated patients developed
by the NIH Sickle Cell Disease Implementation
Consortium (SCDIC): those who have had no
appointments with a SCD expert in the non-acute
setting in 2 years. In our previous examination of an
administrative dataset, unaffiliated SCD patients (15-
45 years of age) were more likely to be uninsured and
were lower utilizers of acute care services than
affiliated patients, but surprisingly had fewer
recorded comorbidities. These findings are consistent
with socio-ecological factors exerting an influence on
the health of SCD patients. The Distressed
Communities Index (DCI) for a zip code is a marker for
a community’s socio-demographic and socio-
economic status (Economic Innovation Group,
Washington DC).
Hypothesis: Unaffiliated SCD patients are more likely
than affiliated patients to live in zip codes with high
DCI.
Methods: A retrospective review compared affiliated
and unaffiliated Chicago patients in the SCDIC
Registry. Each patient’s zip code was used to look up
the 2016 DCI. DCI ranges from 0 to 100 and the worst
quintile is termed “most distressed” (80-100).
Results: Of 299 patients (58% female; mean age 30.0)
who met the inclusion criteria, 56 (46.0% female;
mean age 30.0) met the definition for unaffiliated. No
difference in distress score between affiliated and
unaffiliated SCD patients was observed, with 58% of
the affiliated and 59% of the unaffiliated residing in
the most distressed zip codes (P>.9, by Mann-
Whitney U test).
Conclusions: In our Chicago sample, the percentage
of patients in the high distress category (DCI 80-100)
was the same for affiliated and unaffiliated (58% to
59%). Nationally, (26.9% to 35%) of African-Americans
are in distressed communities. The findings reinforce
previous reports that SCD is correlated with
socioeconomic disadvantage. To overcome the
sample bias from a SCD center whose mission is to
provide care for the underserved, similar analyses
could be conducted in SCD centers that serve a
broader population of sickle cell patients. In addition,
a limitation of this study is that the unaffiliated SCD
patients were identified from a healthcare source and
may not be representative of all unaffiliated patients.
Acknowledgements: SCDIC has been supported by US
Federal Government cooperative agreements
HL133948, HL133964, HL133990, HL133996,
HL133994, HL133997, HL134004, HL134007, and
HL134042 from the National Heart Lung and Blood
Institute and the National Institute on Minority Health
and Health Disparities (Bethesda, MD), plus U01
HL134042-3S2 from NHLBI Research Supplements to
Promote Diversity in Health-Related Research for
Gustavo G. Mendez.

17 | P a g e
JSCDH-D-19-00020 SYSTEMATIC DESIGN AND REVISION OF A MOBILE APPLICATION FOR ENHANCING ADHERENCE TO PRESCRIBED
OPIOIDS IN SICKLE CELL DISEASE
Authors: Daniel Sop1, 2 *
, Wally Smith1, Azhar Rafiq
3,
Abdulkhaliq Alsalman1, Donna McClish
4, Shirley
Johnson1, Thokozeni Lipato
1, Taylor Elliott
1 and Ding-
Yu Fei2
Affiliations: 1Department of General Internal
Medicine, 2Department of Biomedical Engineering,
3Department of Surgery, and
4Deptment of
Biostatistics, Virginia Commonwealth University,
Richmond, VA 23298, USA
Introduction: Sickle cell disease (SCD) is the most
common inherited blood disorder that produces a
progressively disabling illness with severe clinical
consequences. A significant portion of patients with
SCD use prescribed opioids regularly. In order to
safely start, adjust, taper, and stop opioids clinicians
need better tools to follow or trend the opioid use
pattern. Thus, the goal of this study was to develop
and test the feasibility of a mobile application for
adherence to prescribed opioids, and capture
context-specific data associated with prescription
opioid use.
Methods: The designated OpPill Mobile Application
development was carried out to reflect the needs of
the SCD community, and allow for information input
related to concerns gathered from the focus group.
The overarching software requirements for the app
included:
• Easy-to-use and intuitive graphical interface• Functional parameters to allow
documentation of medication adherence andsymptomology
• Data transmission and storage capabilitiesmeet HIPAA requirements
• Multi-platform capability to ensure appeffectiveness across diverse mobile devices
Each phase of the app development was subjected to
continuous expert reviews, and validation of the
software through iterations of Low Fidelity Testing.
Functional relevance of the software was measured
with focus group surveys, which consisted of in-
person interviews. The interviews were audio-
recorded and transcribed for qualitative analysis.
Results: Approximately 57% (n=12) of the participants
were women. Focus group members ranged in age
from 18 to 58 years, with more than half (57%) of the
participants possessing the SS genotype of SCD.
Qualitative thematic analysis from interviews
revealed three phenomena: 1) SCD patients exhibited
various opioid-consuming behavior patterns including
adherence, overuse, underuse, and erratic use; 2)
Several biopsychosocial factors hindered or motivated
opioid use: severe pain intensity, side effects, stress,
family gatherings, unplanned meetings, religious
attendance, and anticipatory fear adverse outcomes;
3) Behaviors varied based on the time of day, week,
month, or year, and also based on the momentary
contexts at times of actual and projected doses.
Conclusion: The presented OpPill app was developed
as a specific tool for the documentation of medication
adherence by SCD patients. The process and outcome
of the study for the development of the app
demonstrates a mobile-health model for effective
SCD management in alignment with current clinical
practice.

18 | P a g e
JSCDH-D-19-00033 SEVERITY AND BURDEN OF SICKLE-CELL DISEASE IN FRANCE: RESULTS FROM THE NATIONAL HOSPITAL DISCHARGE
DATABASE OVER A 7 YEARS FOLLOW-UP.
Authors: Bernard Dauvergne2, Cheikh Tamberou
1,
Valérie Trouillet Poujol1, Carlosse Keumeugni
2, Maryse
Etienne-Julan3, Frédéric Galacteros
4
Affiliations: 1 Sirius-Customizer, 7 rue des Filles du Calvaire, 75003 Paris 2 Addmedica, 37 Rue de Caumartin, 75009 Paris 3 CHU de Pointe à Pitre, Abymes, route de Chauvel, 97159 Pointe à Pitre 4 CHU Henri Mondor, 51 Avenue du Maréchal de Lattre de Tassigny, 94010 Créteil Background: PMSI is an exhaustive French hospital discharge database covering all overnight or day hospitalizations used by all health facilities. SCD management is mainly linked with admission in intensive care unit (ICU), full or partial hospitalizations.
Objective: The aim of the study was to estimate the number of hospital admissions, to determine the SCD severity and its trend, to analyze the hospital burden in terms of total number of stays, total number of hospital days, and frequencies of readmissions in the database from 2009 to 2016.
Method: A cohort of patients identified in 2009 with the following ICD-10 codes of SCD as primary or associated diagnoses: D57.0 (sickle-cell anemia with crisis), D57.1 (sickle-cell anemia without crisis), and D57.2 (double heterozygous sickling disorders) has been followed over the period 2009 to 2016. All related SCD hospitalizations have been collected. SCD severity for a given patient have been defined by the occurrence of at least one of the following cumulative
events occurring since the year of admission and observed over a 12 month-period: (1) at least 3 distinct admissions, (2) 8 or more transfusions, (3) a total of 10 or more days at hospital, regardless of the number of distinct admissions.
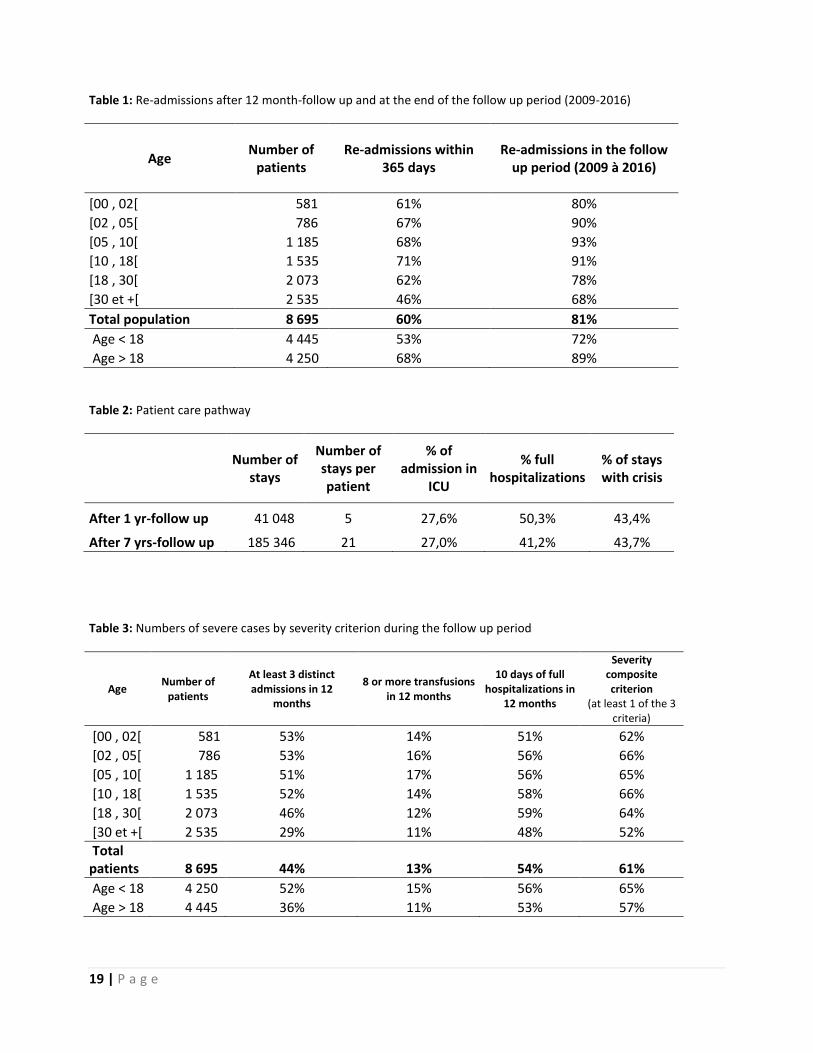
Results: 8,695 (46% females and 54% males) SCD patients were hospitalized in 2009, 51% were above 18 of years and 49% below, out of 581 were under 2 years of age (Table 1).
More than 60% of the patients has been re-hospitalized within the first year following inclusion and 80% at least once during the 7 years-follow up.
Patients under 18 years were much more re-hospitalized (90% vs 72% p=0,0001).
Averages of 5 and 21 hospitalizations per patient are observed after respectively one year of follow up and at the end of period.
50% of the admissions were full hospitalizations, 43% were identified with a diagnosis of SCD crisis and nearly 30% of them were first admitted in ICU. (Table 2)
Around 60% of the patients were severe at least once during the follow-up period (Table 3)
Conclusion: Sickle cell patients are young and severe and readmissions are frequent. It is interesting to note that a significant part of the patients presents a period of severity and in this opposite over a 7-years period, 20% of them remain free of hospitalization. Moreover, this study highlights how the French PMSI system can be used for epidemiological analyses.
19 | P a g e
Table 1: Re-admissions after 12 month-follow up and at the end of the follow up period (2009-2016)
Age Number of
patients Re-admissions within
365 days Re-admissions in the follow
up period (2009 à 2016)
[00 , 02[ 581 61% 80%
[02 , 05[ 786 67% 90%
[05 , 10[ 1 185 68% 93%
[10 , 18[ 1 535 71% 91%
[18 , 30[ 2 073 62% 78%
[30 et +[ 2 535 46% 68%
Total population 8 695 60% 81%
Age < 18 4 445 53% 72%
Age > 18 4 250 68% 89%
Table 2: Patient care pathway
Number of stays
Number of stays per patient
% of admission in
ICU
% full hospitalizations
% of stays with crisis
After 1 yr-follow up 41 048 5 27,6% 50,3% 43,4%
After 7 yrs-follow up 185 346 21 27,0% 41,2% 43,7%
Table 3: Numbers of severe cases by severity criterion during the follow up period
Age Number of
patients
At least 3 distinct admissions in 12
months
8 or more transfusions in 12 months
10 days of full hospitalizations in
12 months
Severity composite criterion
(at least 1 of the 3 criteria)
[00 , 02[ 581 53% 14% 51% 62%
[02 , 05[ 786 53% 16% 56% 66%
[05 , 10[ 1 185 51% 17% 56% 65%
[10 , 18[ 1 535 52% 14% 58% 66%
[18 , 30[ 2 073 46% 12% 59% 64%
[30 et +[ 2 535 29% 11% 48% 52%
Total patients 8 695 44% 13% 54% 61%
Age < 18 4 250 52% 15% 56% 65%
Age > 18 4 445 36% 11% 53% 57%

20 | P a g e
JSCDH-D-19-00023 ESSENTIAL FEATURES FOR SICKLE CELL DISEASE PATIENTS AND UTILIZATION OF COMMUNITY HEALTH WORKERS
Authors: Joan Corder-Mabe, RN, MS; Shirley Johnson, BA,LSW; Daniel Sop, MS; Taylor Elliott, MSW; Wally R. Smith, MD Affiliations: Virginia Commonwealth University Background: Numerous titles are utilized for CHW’s
that often will misrepresent their similar roles in
assisting patients with chronic disease. Therefore, we
use an evidence review and consensus to advance a
common framework and uniform definition of the
functions of CHW’s, which identifies differentiated
CHW roles and responsibilities. The justification for
these roles offers support for certification,
credentialing, education, licensure and payment for
services performed by CHW’s.
Methods: During a recent study funded by the NHLBI”
Start Healing in Patients with Hydroxyurea (SHIP-
HU)”a randomized controlled trial , using synthesized
consensus reports and analysis was used to validate
the efficacy of CHW’s. In order to codify this evidence
according to CHW occupational activities, we
performed a rapid, narrative and tabular review of
the CHW literature, including clinical trials, met an
analyses and policy reports summarizing over 200
CHW interventions to improve patient health status
or care delivery. We tabulated a numeric frequency
count of specific roles, responsibilities, competencies,
and behaviors utilized in these interventions, using a
predetermined list built from a review of all the
included interventions.
Results: (Table) A set of recommendations included
content for formal codification of the CHW
profession. Our findings suggest that the more
frequent CHW behaviors might be more important or
generally required of all CHWs, whereas the rarer
behaviors might either be more specialized or might
be less often required of all CHWs. We propose a
common framework or taxonomy consisting of four
levels of CHW function: Peer Community Health
Worker (PCHW), General Community Health Worker
(GCHW), Clinical Community Health Worker (CCHW),
and Health Navigator (HN). Throughout the reviews,
the CHW’s displayed effective culturally competent
health education to individuals and groups, as well as
health system navigation and care coordination. The
literature also did not display strong evidence
supporting the efficacy of CHW’s in the provision of
direct services. Supervision and support were
frequently indicated but not universally mentioned.

21 | P a g e
Core Competencies
Functions (Behaviors/Interventions
Demonstrated Competencies)
Total
frequency of
function by
source (n=309)
Health system navigation and care coordination
(Service coordination, patient navigation, linkage
to resources, information on community
resources
Assist patient make physician/medical
appointment.
14
Assist patient understand, obtain or use
health insurance.
2
Arrange child care, transportation, or
translation services.
8
Follow-up on missed health
appointments
17
Link patient with local resource services
such as assistance with transportation or
meals.
41
Make appointment telephone calls or
send reminders by mail.
55
Refer to local resources to help meet
medical and psychosocial needs.
18
Coaching and social support (Bridging cultural
mediation, Building trust, Emotional support,
Cultural mediation, Home-based support,
Cultural linkage, Cultural liaison, One-on-one
counseling, Informal counseling)
Make phone calls to elicit patient
concerns or to discuss health status.
4
Discuss patient’s experiences with the
health care system.
8
Refer to or lead support groups. 5
Model behavior change. 41

22 | P a g e
Provide informal counseling and
emotional support.
76
Conduct discussions with patient about
their health status, fears or feelings
about having a chronic disease or life-
threatening event.
12
Culturally competent health education (Health
promotion, Peer education, Outreach education)
Provide education about a specific
disease or health condition to individuals
or groups.
107
Provide cultural insights to health care
team.
4
Provide information regarding general
health maintenance such as diet,
exercise and health screening
recommendations.
29
Direct service (Serve as member of health care
team, Communication with provider and/or
health care team, Liaison with health care
delivery system, Bridge between health care
provider and patient)
Assist in taking prescribed medication or
adhere to medical treatment
16
Monitor patient’s vital signs such as
blood pressure or blood sugar testing or
conduct health screenings.
23
Assist meeting basic needs such as food,
shelter, first aid, and housing.
6
Assist patient develop health goals and
appropriate action steps.
10
Help patient apply for health insurance
or other financial aid.
3
Encourage patients about health lifestyle
changes such as weight management,
routine health screenings, or exercise.
12
Notify and assist in the immunization of
children.
4
Accompany patient to medical 2

23 | P a g e
appointments
Communicate relevant patient
information to the health care team
including documentation on medical
record.
8
Participation in evaluation and research
(Individual or community assessments)
Collect data regarding patient health
status for a program or research project.
9
Engage persons as participatory research
partners.
3
Assist in community needs assessment
and assessment of population data.
6
Conduct environmental assessments. 1
Provide computerized data entry and
conduct web searches.
1
Advocacy (Individual and/or community capacity
building, Community mobilization, Community
organizer)
Advocate for community needs or
address specific issues such as living
conditions.
20
Be spokesperson or intermediaries
between clients and organized medicine
or bureaucracies.
11
Teach the patient effective
communication skills so they can have
productive interactions with medical
personnel.
3
Provide translation and/or
interpretation services.
7
Outreach and case-finding (Eligibility and
enrollment)
Recruit participants into a program or
service.
23

24 | P a g e
Assist patient enroll in program or
service
12
Prepare and distribute materials. 14
Promote health literacy 2
Conclusions: The evidence-based CHW
taxonomy definitions provides four categories
that best describe CHW’s in which to place
workers and established appropriate guidelines
for the level of services that can be provided and
if tested, validated and adopted.
JSCDH-D-19-00015 EPIDEMIOLOGY OF SICKLE CELL DISEASE (HBSS) IN US MEDICAID ENROLLEES
Authors: Rishi J. Desai, MS, PhD;
1 Mufaddal Mahesri,
MD, MPH;1 Raisa Levin, MS;
1 Denise Globe, PhD;
2
Krista McKerracher, MBA;3 Alex Mutebi, PhD;
2
Rhonda Bohn, MPH, ScD;4 Maureen Achebe, MD,
MPH;5 Sebastian Schneeweiss, MD, ScD
1
Affiliations: 1 Division of Pharmacoepidemiology and
Pharmacoeconomics, Brigham and Women’s Hospital & Harvard Medical School, Boston, MA, USA
2 Vertex
Pharmaceuticals Inc, Boston, MA, USA 3 CRISPR
Therapeutics, Boston, MA, USA 4 Bohn Epidemiology,
Boston, MA, USA 5
Hematology Division, Brigham and Women’s Hospital & Harvard Medical School, Boston, MA, USA.
Background: Sickle cell disease (SCD) is a recessive hereditary disease, caused by a mutation in the β-globin gene. Overall, SCD most commonly affects African Americans (AA). A substantial number of SCD patients are covered under the US Medicaid program as the disease can significantly impact functioning and the ability to maintain employment. Because of a paucity of contemporary data, this study was designed to estimate the prevalence of SCD (HbSS) among US Medicaid enrollees.
Methods: Administrative claims data from US Medicaid programs were analyzed (2000 – 2012) to characterize the epidemiology of SCD (HbSS). We assembled annual cohorts of Medicaid enrollees across 13 years to estimate prevalence. Diagnosis codes for HbSS (ICD-9 codes of 282.61 and 282.62) were used to identify patients with SCD in each annual cohort. Crude and age- and race-stratified prevalence estimates with 95% confidence intervals (CI) per 100,000 were generated.
Results: The analysis included annual cohorts ranging from 13,629 cases out of 17.2 million Medicaid enrollees in 2000 to 21,781 cases out of 35.7 million Medicaid enrollees in 2012. There were notable changes in the racial/ethnic distribution of Medicaid enrollees over the years; between 2000 and 2012 the percentage of African American (AA) enrollees decreased from 32.6% to 24.8%, while Hispanics increased from 19.0% in 2000 to 26.5% in 2012. The overall crude prevalence (95%CI) of SCD (HbSS) in the Medicaid program decreased from 79.1/100,000 (77.8-80.4) in 2000 to 61.0/100,000 (60.1-61.8) in 2012. Prevalence (95% CI) in the AA enrollees ranged from 203.2/100,000 (199.5-207.0) in 2000 to 199.2/100,000 (196.2-202.1) in 2012. The highest prevalence was noted in 18-35 year old AAs, which decreased from 421.4/100,000 in 2000 to 317.9/100,000 in 2012. The birth prevalence (among those with age <1 year) was 132.8/100,000 in 2000 and increased to 177.5/100,000 in 2012 among AAs and was substantially lower among Caucasians and Hispanics (3.9 and 9.8 per 100,000 in 2012, respectively).
Conclusion: While the overall prevalence of SCD (HbSS) in Medicaid appeared to decrease over the years, the disease remains majorly prevalent in AAs especially among 18-35-year-olds, suggesting a substantial burden of disease and unmet need. These results provide the most contemporary estimates of the prevalence of SCD (HbSS) in the US. These findings can inform resource allocation and disease-focused initiatives to address this unmet need.
Sponsored by Vertex Pharmaceuticals Incorporated

25 | P a g e
JSCDH-D-19-00062
USING PROJECT ECHO TO MEET THE NEEDS OF PATIENTS WITH SICKLE CELL DISEASE
Authors: Gail Gutman MD, Megan Rees MD, Toni DeNicola PNP, Jeffrey Gershel MD, Kenneth Rivlin MD Affiliation: Department of Pediatrics, Jacobi Medical Center, NYC Health+Hospitals Background: Sickle cell disease (SCD) is a serious chronic disease affecting approximately 100,000 people in the United States. The American Society of Hematology (ASH) has defined four priorities when addressing SCD healthcare inequities: (1) Access to Care in the United States, (2) Training and Professional Education, (3) Research and Clinical Trials, and (4) Global Issues. We believe that Project ECHO (Extension for Community Healthcare Outcomes) can be an effective model to address these priorities. Project ECHO was developed to improve the treatment of hepatitis C in rural, underserved New Mexico. Employing videoconferencing with specialists, remote primary care providers (PCPs) presented patients, received guidance, and had patient outcomes equal to those who directly saw specialists. This approach of “tele-mentoring” has made significant differences in many complex diseases, from asthma to Zika. Our objective is to review the literature and discuss current and future applications of Project ECHO in the care of patients with SCD. Methods: The ECHO Institute maintains a database of all peer reviewed papers, editorials, reports, and projects that have used their model of tele-mentoring. We examined these resources and identified applications appropriate for the ASH-defined priorities.
Results: From the start of Project ECHO in 2003, until February 2019, there have been 147 peer-reviewed papers addressing 26 different medical conditions. Most of these studies focused on (1) improving access to care and (2) professional training/education, while some have addressed (3) research through quality improvement/implementation science, and others have demonstrated the (4) model’s global potential. Driven by the Health Resources and Services Administration’s Sickle Cell Treatment Demonstration Project, the number of SCD ECHOs is increasing. Most address priorities (1) and (2) through mentoring PCPs. Other SCD ECHOs are concerned with eliminating inequities through quality improvement/implementation research (3). These ECHOs establish and support registries, transition of care programs, medical homes, SCD-appropriate emergency care, and community health workers. The World Health Organization has recognized the value of ECHO. Yet, as of February 2019 there were no global SCD ECHOs, despite ECHO’s role for other diseases in countries with the highest prevalence of SCD. Conclusion: SCD is characterized by inequities in healthcare quality, funding, and research. In the United States and elsewhere, Project ECHO is effective for many complex medical conditions. Its application to SCD is just beginning but it has the potential to address all 4 of ASH’s priorities for sickle cell disease.

26 | P a g e
ABSTRACT BREAKOUT SESSION I PSYCHOSOCIAL ORAL PRESENTATION
Presenting: Sunday, June 9, 2019 at 1:45PM

27 | P a g e
JSCDH-D-19-00064
HEALTH-RELATED FACTORS PREDICT ADAPTIVE FUNCTIONING PROFILES IN
YOUTH WITH SICKLE CELL DISEASE
Authors: Megan Loew, PhD, Mary Keenan, BA,
Rebecca Rupff, BA, Jamilla Griffith, MSW, Kathryn
Russell, PhD, Jerlym Porter, PhD, MPH
Affiliations: Virginia Commonwealth University
Background: Youth living with Sickle Cell Disease
(SCD) face health-related and life stressors. These
psychosocial factors may impact youth adaptive
functioning such as self-esteem and interaction with
others. Limited research examines adaptive
functioning and related psychosocial, disease, and
demographic factors in this population. The purpose
of this study is to empirically develop adaptive
functioning profiles that describe subgroups among
youth living with SCD, and to determine if life
stressors, age, gender, hospitalizations, and SCD
genotype predict the subgroup membership.
Methods: Participants were 194 youth with SCD aged
8-17 years recruited for a larger study examining SCD
(Mage=13±2.9 years; 52% female). Youth completed
the Behavioral Assessment System for Children
(BASC-2), which includes four adaptive functioning
subscales (relations with parent, interpersonal
relations, self-reliance, and self-esteem), higher
scores indicate higher functioning. Youth’s age,
gender, SCD genotype, and number of
hospitalizations were obtained from medical records.
Latent profile analyses (LPA) developed profile
descriptions based on the BASC-2 subscales. Log of
the odds ratio (LogO) with a p < .05 was considered
significant.
Results: LPA revealed four distinct adaptive
functioning profiles: 1) High overall adaptive
functioning (n=109; 56.19%) with scores ranged
(56.15-58.54); 2) High interpersonal and self-esteem
(n=58, 29.90%) with scores ranged (41.94-55.05); 3)
High relations with parents and self-esteem (n=11,
5.67%) with scores ranged (29.22-52.01); and 4) High
interpersonal (n=16, 8.25%) with scores ranged
(32.07-50.12). Experiencing less stress increased the
likelihood of being in the High overall (LogO=.314,
p=.003) or High relations with parents and self-
esteem (LogO=.358, p=.002) groups compared to the
High interpersonal group. Male gender identity
increased the likelihood of being in the High
interpersonal and self-esteem group (LogO=1.22,
p=.013) compared to the High overall group. Being
hospitalized in the last 3 years increased the
likelihood of being in the High interpersonal group
(LogO=1.66, p=.051) compared to the High overall
group. Older age increased the likelihood of being in
the High overall adaptive functioning group
(LogO=.305, p=.020) compared to the High
interpersonal strengths group. SCD genotype was not
a significant group predictor.
Conclusions: The majority of our sample reported
overall high adaptive functioning. Distinct patterns of
all four adaptive functioning profiles were predicted
by stress in youth with SCD. Age and female gender
significantly predicted membership in the High overall
adaptive functioning group. Self-esteem, child-parent
relations, and interpersonal skills uniquely contribute
to adaptive group membership. Findings support
examining the impact of stress on adaptive
functioning at an early age in this population.

28 | P a g e
JSCDH-D-19-00057 CARE FOR PHYSICAL ACTIVITY AND SICKLE CELL DISEASE: WHAT IS THE KNOWLEDGE OF TEACHERS IN FITNESS
CENTERS AND SCHOOLS IN NORTHEAST BRAZIL?
Authors: Lea Barbetta, Silva Evanilda Souza Carvalho, PhD, Ivanilde Guedes Mattos, Raissa Lago, Helaynne Bezerra Nascimento, Raiotelma Lopes Silva Affiliations: State University of Feira de Santana Northeastern Brazil stands out because of the high prevalence of sickle-cell disease, with Bahia, the state with the largest black population in the country and also the highest incidence of SCD, with approximately 5.5% of the population affected the disease. In the period from 2008 to 2014, 8,103 hospitalizations for sickle cell anemia complications were registered in the state health system, proving to be a serious public health problem. Advances in the clinical treatment and improvement of the general physical state of the sick people represented gain in the components of the physical fitness and greater tolerance to physical efforts, providing autonomy for execution of the daily tasks, has allowed the insertion and participation which for children and adolescents in physical activities developed in fitness centers and schools. A cross-sectional, exploratory study was carried out with the objective was to verify the knowledge of physical education teachers about sickle cell disease and care in the gymnasium and elementary school classes in Feira de Santana, a city in the semi-arid
region of Bahia, with an average of 4 sickle cell disease cases per 10,000 inhabitants. The data collection was performed using a self-administered questionnaire with multiple choice questions about participants' characteristics and knowledge about sickle cell disease, and a question about the care of these individuals. The data were treated with descriptive statistics and the project was approved by the Research Ethics Committee of the National Health Council. The study was attended by 115 teachers, 80 of whom were linked to 15 fitness centers and 35 linked to the 17 public primary schools in the region. The results showed that most of the teachers knew about the disease, but this knowledge was not sufficient to guarantee safety in the care of the patients who were ill at the sites investigated. Another important finding was that in most schools there was no record of any aspects related to student health. Regarding the care provided in the classes, the teachers reported that they were worried control of the intensity of activities, rest in moments of fatigue, monitoring of the use of prescribed medications and hydration. The physical education teachers demonstrated not know about sickle cell disease and its implications enough. The physical activities so desired by young people always have been avoided because of the insecurity these professionals
.
Table 1. Characteristics of physical education teachers of fitness centers and schools in the Northeast of
Brazil, 2016-2018.
Participants n %
Total 115 100
Gender
Female 31 26,9
Male 84 73,0
Formation
Students 44 38,2

29 | P a g e
Graduates 39 33,9
Post graduates 32 27,8
Source: Direct search
Table 2. Knowledge of physical education teachers about sickle cell disease in Northeast of Brazil, 2016-
2018.
Yes No No
answer
n % n % n %
Have you ever heard of sickle cell disease?
85 73,9 29 25,2 1 0,8
35 30,4 73 63,4 7 6,0
Is the information you have about this disease acquired during your
undergraduate course?
35 30,4 75 65,2 5 4,3
Dou you make any special arrangements for this student?
3
2,6 46 40 66 57,3
Does the fitness centers have initial physical assessment for your
students?
69 86,2 11 13,7 0 0
Does the school have any records on the health aspects of the
students?
11 31,4 24 68,5 0 0
Source: Direct search

30 | P a g e
JSCDH-D-19-00040 STIGMA OF PAIN AND SICKLE CELL DISEASE IN BAHIA – BRAZIL
Authors:
1Jayanne Moreira Carneiro,
1Sheila Santa
Barbara Cerqueira, 1Aline Silva Gomes Xavier,
1Evanilda Souza de Santana Carvalho,
2Coretta Melissa
Jenerette Affiliations:
1Universidad Estadual de Feira de
Santana, 2College of Nursing University of South
Carolina Background: Although Bahia is the state with the largest population with sickle cell disease in Brazil, the sick people find barriers to their care that reveal the need for investment in interventions to overcome stigma. This study aiming to understand the stigma experienced by people with sickle cell disease (SCD), in Bahia-Brazil. Methodology: We developed a qualitative study, 120 people, 52 males and 68 females, with SCD resident in the capital and in the interior of the state of Bahia responded to interviews, which were then submitted to content analysis. Results: The results show that in the family environment, people with SCD are charged to perform activities that are incompatible with their
tolerance, and when they do not, they are criticized and seen as incapable or useless resulting in abandonment by relatives in times of crisis. In the general public they are perceived as lazy and unproductive, incapable of working. In all the scenarios the discredit of the pain reports occurs. And in the health units the experiences of stigma are crossed by institutional racism where professionals nurture the stereotype of the slave and strong black, who supports everything, leading to delays in care, discourteous service or physical and psychological abuse. Conclusion: It is necessary to overcome the invisibility of the disease, to clarify doubts of the public, to demystify beliefs that put the sick person in a condition of depreciation, as well as to sensitize the family about the physical and psychoemotional limitations that pain produces on the person with SCD. In the context of health services, there is an urgent need to qualify professionals about SCD at different levels of health care so that they recognize pain as one of the complications of the disease and are able to assess and treat the pain episode, understanding that it constitutes a clinical emergency.
Figure 1 Aspects of stigma of pain and its coping by people with sickle cell disease

31 | P a g e
JSCDH-D-19-00005 THE ROLE OF DIETARY SELENIUM AND GPX1 IN THE PATHOPHYSIOLOGY OF SICKLE CELL DISEASE
Authors: Ramasamy Jagadeeswaran
1,, Vinzon Ibanez
2,5, Lenny K. Hong
3, Robert E. Molokie
2,4,5, Alan M.
Diamond3,4
, Angela Rivers1,5
Affiliations: 1Department of Pediatrics, 2Department
of Medicine, 3Department of Pathology, 4Cancer
Center, 5Jesse Brown Veterans Affairs (VA) Medical Center, Chicago, IL.
Background: Sickle cell disease (SCD) affects
approximately 100,000 Americans. SCD is a prevalent
and devastating illness caused by a mutation in the in
the -globin gene. Despite the cause of the disease
being understood for decades, the wide spectrum of
disease experienced by patients remains unknown.
The major contributory role of reactive oxygen
species (ROS) in SCD RBCs is supported by our recent
work describing mitochondrial retention in the RBCs
of SCD patients and the SCD mouse model. Oxidative
stress mediated hemolysis and sickling in the RBCs of
SCD patients is possibly exaggerated by lower levels
of antioxidants such as selenium-dependent
glutathione peroxidase 1 (GPX1). GPX1 was first
described as an RBC inhabitant protein and capable of
protecting hemoglobin from ROS and has been
reported to be lower in SCD. Selenium is an essential
component of GPX1 as the amino acid selenocysteine.
The levels and activity of GPX1 are particularly
sensitive to nutritional selenium availability.
Methods: We used the Towns model for the SCD mice
experiments. The mitochondrial content and ROS
levels in RBCs was investigated by flow cytometry.
GPX1 levels were examined by immunoblotting. The
energy profile of circulating RBCs and reticulocytes
was determined by assessing the rate of oxygen
consumption as a marker for mitochondrial
respiration and the extracellular acidification rate, as
a marker for glycolysis using the Seahorse XF
analyzer.
Results: RBCs from HbSS (SCD) mice have significantly
lower GPX1 levels as compared to control HbAA mice
(p<0.003, n=3), indicating that the reduction of GPX1
in SCD may contribute to ROS accumulation. We
further investigated the impact of selenium deficiency
or adequacy in the HbSS mice. Mice were provided
diets either containing 0.02 ppm selenium (deficient)
or 0.1 ppm selenium (adequate) for eight weeks, and
GPX1 levels, mitochondrial retention, oxygen
consumption rate (OCR) and Hb levels were
determined. RBCs of HbSS mice provided a selenium
deficient diet exhibited very low GPX1 levels as
compared to mice maintained on the adequate diet
(p<0.001, n=4). SCD mice in the selenium-deficient
group also exhibited an increase in mitochondria
retaining RBCs (Se deficient: 26%±6.9%, n=3 vs. Se
adequate: 5 % ± 3.5%, n=3, p<0.01), reduced Hb levels
(Se deficient 5.7± 0.17 g/dl, n=3 vs. Se adequate 7.0±
0.83 g/dl, n=4 p<0.05), and an increased OCR.
Conclusions: SCD mice maintained on selenium
deficient diet exhibited reduced GPX1 levels, lower
Hb levels and abnormal erythrocyte energy
metabolism.

32 | P a g e
JSCDH-D-19-00044 RENAL MEDULLARY CARCINOMA: THE UNDER-RECOGNIZED AGGRESSIVE SEQUELAE OF SICKLE CELL TRAIT!
Authors: Dianna Hardatt M.D., Rabia Hakim M.D., Isaiah Gonzalez M.D., Revathy Sundaram M.D., Minnie John M.D., Jolanta Kulpa M.D. Affiliation: New York Methodist Hospital Background: Renal Medullary Carcinoma (RMC) is a rare aggressive form of kidney cancer. RMC has a very poor prognosis, and metastasis is seen in up to 95% of the patients at diagnosis or shortly thereafter. Patients tend to be young black males (2:1 male to female predominance) with sickle cell trait. The most common presenting signs and symptoms include hematuria and abdominal or flank pain. The majority of patients have wide spread metastasis at diagnosis which contributes to a poor prognosis. More awareness can lead to earlier diagnosis and even potential lifesaving therapies. Case Presentation: We present a case report of a 20-year-old male with sickle cell trait who presented with abdominal pain and significant weight loss. Positive physical findings included a soft abdomen with left upper quadrant tenderness and tenderness over his L4-L5 region. Differential diagnoses that were being
considered were infection, tuberculosis, collagen vascular disease, sarcoidosis, HIV and malignancy. Extensive imaging eventually revealed a paraspinal mass that was biopsied and was consistent with renal medullary carcinoma. Our patient’s case is unusual in that he did not have a renal mass at the time of diagnosis. Conclusion: Renal Medullary Carcinoma (RMC) is a rare aggressive form of kidney cancer that is predominantly associated with sickle cell trait. Prognosis is poor and the cause is still currently unknown. Sickle cell trait is often considered a benign condition however, it is associated with several complications including splenic infarction, other renal manifestations and even sudden death associated with exercise-induced rhabdomyolysis. Awareness of the clinical spectrum of sickle cell trait can aid in earlier diagnosis of associated morbidities. A high index of suspicion in a patient with sickle trait and weight loss should alert us to the possibility of RMC. This case report emphasizes the fact that sickle cell trait is not a benign disorder and should not be taken lightly.

33 | P a g e
JSCDH-D-19-00034 CRIZANLIZUMAB DOSE CONFIRMATION IN PEDIATRIC PATIENTS WITH SICKLE CELL DISEASE: SOLACE-KIDS DESIGN
Authors: Matthew M. Heeney,1 David Rees,
2 Mariane
de Montalembert,3 Isaac Odame,
4 Raquel Merino,
5
Brian Elliott,6 Shankaranand Bodla,
7 Julie Kanter
8
Affiliations: 1Dana-Farber / Boston Children's Cancer
and Blood Disorders Center, Boston, MA, United
States; 2Department of Paediatric Haematology,
King’s College Hospital, King’s College London,
London, United Kingdom; 3Necker Children’s Hospital,
Assistance Publique-Hôpitaux de Paris, Paris, France; 4The Hospital for Sick Children (SickKids), Toronto,
Canada; University of Toronto, Toronto, Canada; 5Novartis Pharma AG, Basel, Switzerland;
6Novartis
Pharmaceuticals Co., East Hanover, NJ, United States; 7Novartis Ireland Ltd, Dublin, Ireland;
8University of
Alabama at Birmingham, Birmingham, AL, United
States
Background: Sickle cell disease (SCD) is the most
common serious single-gene disorder in the world
with complications including acute pain and
acute/chronic organ damage. Episodes of vaso-
occlusion are a hallmark of SCD. P-selectin, an
adhesion molecule expressed primarily on endothelial
cells and platelets, plays a key role in the initiation of
leukocyte rolling along the blood vessel wall,
contributing to multicellular adhesion and
microvascular occlusion. SCD worsens with advancing
age as repeated vascular occlusion results in
cumulative endothelial damage. Crizanlizumab, a
humanized monoclonal antibody, binds to P-selectin,
blocking interaction with its ligands. Crizanlizumab
significantly decreased vaso-occlusive crises (VOCs) vs
placebo and was well-tolerated in SUSTAIN, a phase 2
study in adults with SCD. Here, we report the design
of the first crizanlizumab study in pediatric patients
with SCD.
Methods: Primary objectives of this phase 2,
multicenter, open-label study are to confirm
appropriate dosing and safety of crizanlizumab in
patients >6mo–<18yrs, using sequential, descending
age groups of pediatric SCD patients. Secondary
objectives include number of VOCs and
number/duration of hospitalizations/emergency
room visits. Planned enrollment is ≥100 patients with
confirmed SCD diagnosis (all genotypes), who
experienced ≥1 VOC within the preceding 12mo, in 3
sequential descending age groups: Group 1 (≥26
patients; 12yr–<18yr), Group 2 (≥26 patients; 6yr–
<12yr), and Group 3 (≥8 patients; 2yr–<6yr) and (≥6
patients; 6mo–<2yr). Dose will be confirmed (Part A)
by ≥8 patients in Group 1. If unconfirmed, dose will be
adjusted based on observed
pharmacokinetics/pharmacodynamics, and ≥8
additional patients will be enrolled in Group 1/Part A
to confirm the new dose. After dose confirmation,
Group 1 will be expanded (Part B), with ≥8 patients
enrolled to Group 2 for dose confirmation. This
schema will continue until dose confirmed in Group 3,
which will be expanded for enrollment of an
exploratory group(≥6 patients; 6mo–<24mo). Patients
treated with hydroxyurea/hydroxycarbamide must
have taken it for ≥6mo prior to enrollment and not
plan dose adjustments except for weight changes.
Crizanlizumab (5.0 mg/kg intravenously) will be
administered on weeks 1, 3 and every 4 weeks
thereafter for 2 years.
Results: Trial ongoing (ClinicalTrials.gov:
NCT03474965).
Conclusion: This study will address an unmet need in
pediatric SCD patients by establishing crizanlizumab
pharmacokinetics/pharmacodynamics and confirming
dose in different age groups. Safety will be assessed
as a primary objective. Additionally, the number of
VOCs and number/duration of
hospitalizations/emergency room visits will be
assessed as a secondary objective.

34 | P a g e
ABSTRACT SESSION II CLINICAL EPIDEMIOLOGY ORAL
PRESENTATION Presenting: Sunday, June 9, 2019 at 3:30 PM

35 | P a g e
JSCDH-D-19-00059 FETAL HEMOGLOBIN AND LEG ULCERS IN ADULTS WITH SICKLE CELL DISEASE
Authors: Seda Tolu, Ugochi Ogu, Andrew Crouch,
Giacomo Vinces ,Caterina Minniti
Affiliation: Albert Einstein Medical Center
Background :Sickle cell disease (SCD) is a
monogenetic inherited disorder with pleomorphic
clinical manifestations. Leg ulcers are a frequent and
debilitating complication of sickle cell disease,
particularly of the SS genotype. Hemoglobin F (Hb F)
concentration is the major genetic modifier of SCD
clinical complications, including leg ulcers, and been
found to be protective in a previous studies.
However, to date, no specific level of hemoglobin F
has been identified that results in prevention and/or
resolution of leg ulcers. Our aim is to determine a
specific threshold of hemoglobin F above which there
is low or no occurrence of leg ulcers in patients with
SCD.
Methods : After obtaining IRB approval, we queried
the Electronic medical records (EMR) of adult SCD
patients in our database at Montefiore Medical
Center, Bronx, New York. EMRs were reviewed in
detail to confirm the documentation of leg ulcers and
laboratory values. Lifetime average hemoglobin F
values were calculated for each patient using all
recorded Hb F values in his or her chart. Patients who
were ever prescribed hydroxyurea (HU) and those
who were not were included in the study.
Results: Out of 804 adult patients with sickle cell
disease, we identified a total of 90 with history of
prior or current leg ulcers, most of which were
chronic in nature. Twenty seven patients were noted
to have a lifetime average Hb F > 8% which included
13 women and 14 men, age range 23 to 72. Four of
these patients were never treated with hydroxyurea
at any point in time. Patient data is displayed in the
table below.
*Never on Hydroxyurea
Discussion: In a modern cohort of post HU patients,
we identified 11 % of SCD patients with leg ulcers, a
higher incidence than what was reported in the
Cooperative Study of Sickle Cell Disease (5%), which
included mostly pediatric patients; but lower than the
Bethesda cohort (~18%), which may have been
enriched for patients with other vasculopathies, such
as high TRV1,2
. A possible explanation is that, as
patients with SCD live longer, the prevalence of leg
ulcers increases, which is evident in our cohort with a
mean age of 37 years and range up to 72 yrs.
Interestingly, we identified 4 patients with
hemoglobin F > 20% who had never been on HU and
developed leg ulcers. Certainly HU could not be
blamed for their occurrence. We conclude that even
very high Hb F levels are not universally protective
against leg ulcers. Intracellular distribution of HbF is
more important than the total HbF, therefore its
effect on hemolysis and in protecting from end organ
damage cannot be predicted solely by the total HbF
level. Patients with hemoglobin F>8 % are still prone
to developing leg ulcers and practitioners should have
high vigilance when taking care of these patients.
Reference 1. Koshy M, Entsuah R, Koranda A, et al. Leg
ulcers in patients with sickle cell disease. Blood 1989;74:1403–1408.
2. Minniti, CP, Taylor JGt Hildesheim M, et al. Laboratory and echocardiography markers in sickle cell patients with leg ulcers. Am J Hematol 2011;86: 705–708.
Age, Sex HbF % Age, Sex HbF % Age, Sex HbF %
64 M 8.3 36 M 11.8 41 M 17.9
35 M 9.3 51 F 12.1 60 M 18.2
38 F 9.5 40 M 12.1 43 F 18.5*
58 F 9.8 58 F 13.7 65 F 20.7
36 F 10.5 23 M 14.9 50 M 20.9*
33 F 10.6 40 F 15.6 62 M 21.5
22 M 10.7 66 F 15.6 72 F 23.7*
41 M 10.8 47 F 16.7 40 M 29.5*
31 M 11.6 42 M 16.8 40 F 31.9

36 | P a g e
JSCDH-D-19-00045 CLINICAL TRIAL OF A FETAL GLOBIN INDUCING AGENT WHICH MODULATES REPRESSORS:
BALANCING FACTORS FOR OPTIMAL ACTIVITY
Authors:
1Susan P Perrine,
2Betty S Pace, MD,
3Kevin
Kuo, MD, 4Sylvia T. Singer, MD,
3Rebecca LeRoux, RN,
5Douglas V. Faller, MD,PhD
Affiliations:
1Boston University School of Medicine,
2Augusta University,
3Toronto General Hospital,
University Health Network, 4USCF Menioff Children’s
Hospital at Oakland, 5Phoenicia BioSciences, Inc.
Background: Up-regulation of HbF in a majority of red blood cells is clinically proven to reduce anemia and clinical severity in sickle cell disease (SCD) and beta thalassemia (BT). More than 70 small molecules induce fetal globin gene expression in vitro, through complimentary molecular mechanisms. The timing of addition of inducing agents relative to stage of erythroid differentiation is important for activity. Exposure of erythroid progenitors to most candidates results in high induction only during a period of EPO sensitivity, while exposure in early or late stages of differentiation often has no or minimal effect. A repurposed therapeutic, PB-04, which reduces or abolishes mRNA, protein, and/or binding of multiple repressors (BCL11A, LSD-1, HDAC3) of the fetal globin genes, and promotes appearance of histone activation marks, demonstrates high inducing activity in patients’ erythroid progenitors, in anemic baboons with active erythropoiesis, and in transgenic mice undergoing frequent phlebotomy. PB-04 is reported to not induce HbF in subjects without stress erythropoiesis. The magnitude of fetal globin responses in vitro differs in progenitors of patients
with genomic modifiers associated with variable baseline fetal globin levels. PK parameters of PB-04 in other populations demonstrate brief half-lives, but the drug has been effective for > 40 years in treating an unrelated condition. The design of regimens to optimize these diverse factors may be challenging. Methods: A trial has been designed to investigate human equivalent doses of PB-04 based on intermittent oral administration in anemic baboons, which produces high-level induction (12-30 fold) of fetal globin mRNA and increases F-reticulocytes 3-fold. Three dose-escalating cohorts will be studied initially. If induction is demonstrated, two expansion cohorts in BT intermedia and SCD will be studied. If induction is not observed, dose and schedule will be adjusted based on PK data. Endpoints include PK parameters, HbF, F-reticulocytes/ F-cells, HbF protein/cell, hemolytic assays, hematology. Seven SNPs which affect basal HbF levels, and 2 SNPs which affect cell proliferation, will be assessed. Results: An IND is approved for a PK and preliminary activity study of PB-04 in patients with beta hemoglobinopathies. Conclusions: Intermittent exposure targeting
differentiating erythroid progenitors will be
investigated with an inducing agent which abolishes
BCL11A protein and reduces genomic binding of the
transcriptional repressors LSD-1 and HDAC3. Dose
equivalents and schedule were effective in anemic
baboons and transgenic mice.

37 | P a g e
JSCDH-D-19-00055 MICROGLIA-LIKE CELLS DERIVED FROM HEMATOPOIETIC STEM AND PROGENITOR CELLS ARE A MODEL SYSTEM
TO INVESTIGATE CHRONIC PAIN IN SICKLE CELL DISEASE
Authors: Yankai Zhang1, Celeste K. Kanne1, and
Vivien A. Sheehan1
Affiliations: 1. Division of
Hematology/Oncology, Department of
Pediatrics, Baylor College of Medicine, Houston
TX
Background: Patients with sickle cell disease (SCD) often experience severe chronic pain. In chronic pain, microglia are readily activated, stimulating neurons to send a pain signal. Human microglia are difficult to obtain; we proposed to culture induced microglia-like cells (MLC) from human peripheral blood (PB) to develop a model system to investigate chronic pain in sickle cell disease.
Methods: Peripheral blood mononuclear cells
(PBMCs) were obtained from three individuals from
each of the following patient groups: SCD and
chronic pain (SCD CP+, defined as pain most days for
3 months), SCD without chronic pain (SCD CP-), and
normal donors (WT). PBMCs were cultured with
human GM- CSF (10 ng/ml) and human IL-34 (100
ng/ml) to induce peripheral blood derived microglia
(PB-MLC). On day 7 of culture, cells were collected
and morphology analyzed by phase contrast
microscopy, phenotyped by flow cytometry, and
indirect immunofluorescence with anti-CX3CR1,
TMEM119, CD68, Iba1 antibodies. TNF-alpha, IL-
1beta, IL-10 secreted by PB-MLCs were measured
with ELISA. Microglia morphology was evaluated by
quantitative analysis of cell body roundness and
branch length.
Results: When cultured with GM-CSF and IL-34,
PBMCs developed microglial morphology, were
CD11bhigh and CD45low by flow cytometry,
CX3CR1+ and TMEM119+ by fluorescence
microscopy, consistent with microglia. We treated
the PB-MLC with LPS, and found that treated PB-
MLC cells had significantly higher CD68 and Iba1
positivity compared to resting microglia cells,
indicating activation. PB-MLC differed significantly
depending on donor group. SCD CP+ had shorter and
fewer branches than WT; branching from SCD CP-
were intermediate in number and length. Activated
PB-MLC derived from patients with SCD secreted
more inflammatory cytokines than PB-MLC derived
from normal donors, suggesting that donor
characteristics are retained by the PB-MLC. To
evaluate the possibility of using this model system to
screen compounds, we tested MLC cells with the
following drugs: gabapentin, metformin,
piceatannol, and resveratrol. All suppressed the
release of proinflammatory cytokine from LPS-
induced PB-MLC in a dose-dependent manner, and
reversed the deramification of activated PB-MLC
upon LPS stimulation by quantitative analysis of cell
body roundness and branch length with
immunostaning of Iba1, with gabapentin exhibiting
the smallest effect.
Conclusions: We have established the microglia-like
nature of the cultured peripheral blood cells derived
from patients with SCD and normal blood donors.
We propose to use this model system to derive
mechanistic insights into the development of
chronic pain in SCD, and to screen pharmacologic
agents to treat chronic pain.
Acknowledgments: This work was supported by
1K08 DK110448-01 from the National Institute of
Diabetes and Digestive and Kidney Disease

38 | P a g e
JSCDH-D-19-00003 WEB-BASED TECHNOLOGY AS A LEARNING TOOL TO IMPROVE DISEASE KNOWLEDGE
IN ADOLESCENT PATIENTS WITH SICKLE CELL DISEASE
Authors: Anjelica Saulsberry, Jason Hodges, Audrey
Cole, Jerlym Porter, Jane Hankins
Affiliation: St. Jude Children’s Research Hospital
Background: Sickle cell disease (SCD) is a chronic,
genetic red blood cell disorder. Advances in SCD
treatment have contributed to an increase in
survivorship of youth with SCD, prompting the need
for “transition readiness” prior to transition into the
adult healthcare system. “Transition readiness”
encompasses disease specific knowledge and self-
management skills and result in improved transition
outcomes. The Sickle Cell Transition E-Learning
Program (S.T.E.P) is a web-based learning tool aimed
at increasing “transition readiness.” For the current
study, we sought to investigate the use and
effectiveness of S.T.E.P. We hypothesize that number
of modules completed will be positively correlated to
improved disease knowledge retention and
confidence in self-management skills.
Methods: The S.T.E.P. program is a public, web-based
6-module tool covering pain, infection, SCD
complications, healthy living, genetics, and self-
advocacy. Self-management confidence was
measured by a self-management skills checklist
(SMSC) modified from the validated Transition
Readiness Assessment Questionnaire. All patients
who participate in the transition program are offered
participation in S.T.E.P. in clinic as a complement to
education sessions. Disease knowledge retention was
measured via an assessment that reflects the entire
educational curriculum provided during adolescent
education visits. Association of S.T.E.P. modules and
disease knowledge retention and self-management
scores were evaluated using Pearson correlations.
Results: Thirty-six African-American adolescents with
SCD between the ages of 12-15 (25 males; 11
females) participated in the S.T.E.P. The number of
modules completed were one (15 participants) two
(10 participants) three (10 participants) and five (one
participant). The average disease knowledge score for
all S.T.E.P. modules was 77 (range of 50-100) for all
participants. A positive correlation (r=0.57) was found
between number of modules completed and final
disease knowledge score (p<0.001). No correlation
was found between number of modules completed
and self-management scores. (Figure 1)
Conclusions: The S.T.E.P. program may serve as a
complement to in-clinic disease knowledge sessions
for increasing disease knowledge retention prior to
transfer to adult care. Further investigation includes
replicating our findings in other SCD patient
populations and conducting a randomized
prospective study to determine the effectiveness of
S.T.E.P. as an intervention to increase disease literacy
in the SCD youth.

39 | P a g e
JSCDH-D-19-00058 EXPERIENCES WITH HYDROXYUREA IN AN URBAN ADULT SICKLE CELL DISEASE PATIENT POPULATION
Authors: 1Christopher Bradford, 1Hope Miodownik, 2Gracy Sebastian, NP, 2Merin Thomas, NP, 2Ugochi Ogu, MD, and 2Caterina Minniti, MD
Affiliations: 1Albert Einstein College of Medicine, 2Montefiore Medical Center Background: Hydroxyurea remains the first-line agent for adults with sickle cell disease (SCD), with well- documented improvements in morbidity and mortality. Despite its proven utility, many SCD patients are not prescribed or not taking hydroxyurea. Few studies have evaluated reasons behind this phenomenon among large adult populations. Methods: Adult patients with SCD at a large urban SCD center completed a survey. Data from 224 outpatients were analyzed. Additional information was collected and confirmed using the electronic medical record.
Results: In total, 173 patients (77.2%) have ever been prescribed hydroxyurea. Among these, 65.3% were taking hydroxyurea when surveyed. For those with severe disease genotypes (HbSS or HbS- BetaThal0), 91.0% have ever been prescribed hydroxyurea, with 68.4% taking it when surveyed. Of those with mild disease genotypes (HbSC or HbS-BetaThal+), 42.1% have ever been prescribed hydroxyurea, with 50% taking it when surveyed. Among those patients ever been prescribed hydroxyurea, 17.3% endorsed medication side effects, with fatigue, hair loss, and gastrointestinal distress the most common (each 16.7% of those reporting side effects). Other reasons included dizziness, headache, weight gain, cytopenia, and drug reaction (each 6.7%). Hydroxyurea was discontinued at some point by 35.3% of patients, with 24.6% citing side effects as the cause for discontinuation. Other reasons for discontinuation included
perceived ineffectiveness (19.7%), physician direction (16.4%), reproductive health, ulcer formation, and general dissatisfaction with hydroxyurea (each 8.2%). Conclusions: These results suggest that adult patients with severe SCD genotypes are being prescribed hydroxyurea at high rates, with 91.0% having ever been prescribed, and 68.4% taking it at the time of this study. The largest reason for discontinuation among our patients was medication side effects. Some patients reported well-described side effects (weight gain, cytopenias, hair loss). Others cited side effects that may not be related to hydroxyurea (back pain, depression). Interestingly, 16.4% of patients discontinued hydroxyurea due to perceived ineffectiveness. By identifying variability in reported side effects and reasons for discontinuation, this descriptive study highlights the need for clinicians to clearly communicate benefits and challenges of hydroxyurea use with their patients to further improve rates of prescribing and adherence.
TABLE

40 | P a g e
JSCDH-D-19-00031 SUCCESSOR STUDY: BASELINE DEMOGRAPHICS OF THE RETROSPECTIVE, NONINTERVENTIONAL FOLLOW-UP STUDY
IN A SUBSET OF PATIENTS WITH SICKLE CELL PAIN CRISES WHO PREVIOUSLY PARTICIPATED
IN SUSTAIN IN THE UNITED STATES
AUTHORS: Nirmish Shah,1 Ralph Boccia,
2 Walter K.
Kraft,3 Vince Cataldo,
4 Jincy Paulose,
5 Dramane Lainé,
5
Das Purkayastha,5 Abdullah Kutlar
6
AFFILIATIONS: 1Duke University Health System,
Durham, NC; 2Center for Cancer and Blood Disorders,
Bethesda, MD; 3Thomas Jefferson University,
Philadelphia, PA; 4Our Lady of the Lake-Mary Bird
Perkins Cancer Center, Baton Rouge, LA; 5Novartis
Pharmaceuticals Corporation, East Hanover, NJ; 6Sickle Cell Center, Medical College of Georgia,
Augusta University, Augusta, GA
BACKGROUND: SUCCESSOR (SUSTAIN Chart-review of Crizanlizumab to Evaluate Sickle-cell Study One-year Retrospective) reviewed medical records of a subset of patients who completed the SUSTAIN study at US sites to assess subsequent cases of significant pain crisis events and generate real-world data on treatment patterns and health care resource utilization (HCRU) upon completion of crizanlizumab treatment. SUSTAIN was a phase 2, randomized, double-blind, placebo-controlled, 52-week study that compared the effect of crizanlizumab, a P-selectin inhibitor, versus placebo on the frequency of sickle cell pain crises (SCPCs, or vaso-occlusive crises [VOCs] leading to a health care visit) in patients with any genotype of sickle cell disease (SCD). METHODS: SUCCESSOR is a multicenter, retrospective cohort study of a subset of US patients (≥18 years old) who participated in SUSTAIN to evaluate SCD-related outcomes up to 52 weeks following trial completion. SUCCESSOR included the SUSTAIN population who
received at least 12 of the 14 study drug doses, completed a visit at least 14 days after the final dose, and had no major protocol deviations that impacted SUSTAIN efficacy assessments. The post-SUSTAIN study period was from November 2014 to March 2017 and patients’ data were obtained from medical records. Crizanlizumab was not administered in the 52 weeks post-SUSTAIN. Patient consent was obtained prior to data collection if required by local and/or central research ethics review.
RESULTS: In SUCCESSOR, baseline demographic data for 48 patients are reported (15, 18, and 15 patients who were previously randomized in SUSTAIN to receive crizanlizumab 5 mg/kg, crizanlizumab 2.5 mg/kg, and placebo, respectively). The median age was 31.5 years (range 19-65 years) and 69% were female. The majority of patients were Black or African American (96%). One patient had HbSβ
+, 4 had HbSβ
0,
10 had HbSC, 1 had HbS-Lepore disease; all remaining 32 patients had HbSS. The majority of patients live in the Southern US geographic region (63%), followed by the Midwest (18%), Northeast (15%), and West (6%). At 52 weeks post-SUSTAIN, 98% of patients were alive with no bone marrow transplant, no new clinical trial participation, and were not lost to follow-up. No deaths occurred in the 52 weeks post-SUSTAIN. There were high rates of HCRU regardless of the treatment received in SUSTAIN, as more than 60% of patients had at least one hospitalization in the 52 weeks post-SUSTAIN. CONCLUSION: SUCCESSOR is a real-world study generating evidence on VOC rates, treatment patterns, and HCRU in patients post-SUSTAIN.

41 | P a g e
ABSTRACT BREAKOUT SESSION II CLINICAL RESEARCH ORAL PRESENTATION
Presenting: Sunday, June 9, 2019 at 3:30 PM

42 | P a g e
JSCDH-D-19-00060 A PHASE II RANDOMIZED, DOUBLE-BLIND, PLACEBO-CONTROLLED MULTI-CENTER STUDY TO ASSESS THE
SAFETY, TOLERABILITY, AND EFFICACY OF RIOCIGUAT IN PATIENTS WITH SICKLE CELL DISEASE (STERIO-SCD)
Authors: Caterina P. Minniti, Kaleab Z. Abebe, Claudia
R. Morris, Victor R. Gordeuk, Mark T. Gladwin,
Gregory J. Kato and the STERIO-SCD Investigators
Affiliations: Department of Medicine, Pittsburgh
Heart, Lung and Blood Vascular Medicine Institute,
and the Center for Clinical Trials & Data Coordination,
University of Pittsburgh School of Medicine,
Pittsburgh, PA; Division of Hematology-Oncology,
Department of Medicine, Montefiore Medical Center,
Bronx, New York; Department of Pediatrics, Division of
Pediatric Emergency Medicine, Emory-Children's
Center for Cystic Fibrosis and Airways Disease
Research , Emory University School of Medicine ,
Atlanta , GA; Division of Hematology and Oncology,
University of Illinois at Chicago, IL.
Background: Systemic hypertension, pulmonary
hypertension and proteinuria are each predictive of
morbidity and mortality in patients with sickle cell
disease (SCD). These complications are associated
with more intense hemolytic anemia and decreased
nitric oxide bioavailability. Riociguat is a soluble
guanyl cyclase stimulator, approved by the FDA to
treat patients with pulmonary arterial hypertension or
chronic thromboembolic pulmonary hypertension.
Methods: STERIO-SCD is an investigator-initiated,
industry-funded Phase 2 multi-center, randomized,
double-blind, placebo-controlled, parallel groups
study aimed to evaluate the safety, tolerability and
the efficacy of riociguat compared with placebo in
patients with SCD. Adult patients are eligible if they
have at least one of the following: Systolic blood
pressure ≥130 mm Hg, urine albumin to creatinine
ratio >300 mg/g, tricuspid regurgitant velocity (TRV)
>2.9 m/sec, NT-proBNP level ≥160 pg/mL, or protein
1+ or higher on urinalysis.
This randomized study involves 12 weeks of
treatment with riociguat pills or placebo pills, and a
follow-up period of 30 days after treatment. The dose
is adjusted every 2 weeks based on systolic blood
pressure, with a goal to escalate from a starting dose
of 1mg TID to 2.5 mg TID. Physical examinations, vital
signs, blood tests and questionnaires are performed
at 2-week intervals during the double-blinded study
treatment. Echocardiogram, urine albumin/creatinine,
six-minute walk distance and the Adult Sickle Cell
Quality of Life Measure (ASCQ-Me) are assessed at
the beginning and end of the treatment phase.
Results: The study is presently open at 11 sites, with
6 more sites in process. Enrollment is underway for a
target of 100 patients completing the treatment.
Additional sites are being sought.
Conclusions: Although many studies are evaluating
efficacy of agents aimed at reducing acute vaso-
occlusive complications of SCD, STERIO-SCD addresses
an unmet need to control the life-limiting, hemolysis-
related chronic vasculopathy associated with death.
Acknowledgment: Bayer provides funding and study
drug for this trial. The University of Pittsburgh is the
study sponsor, data coordinating center and central
pharmacy. STERIO-SCD investigators: Sophie
Lanzkron, Johns Hopkins University; Elizabeth Klings,
Boston University Medical Center; James Ford, UNC
Chapel Hill; Marilyn Telen, Duke University; Payal
Desai, Ohio State University; Laura De Castro,
University of Pittsburgh; Temeia Martin, Medical
University of South Carolina; Wally Smith, Virginia
Commonwealth University; Julie Kanter, University of
Alabama at Birmingham; Ken Ataga, University of
Tennessee. The core team at University of Pittsburgh
includes Carolyn Newkirk, Susan Spillane, Nydia Chien,
Nancy Petro, Yingze Zhang and independent
statistician Seyed Mehdi Nouraie.

43 | P a g e
JSCDH-D-19-00053 RESULTS FROM THE DISPLACE CONSORTIUM: PRACTICE PATTERNS ON THE USE OF TRANSCRANIAL DOPPLER
SCREENING FOR RISK OF STROKE IN CHILDREN WITH SICKLE CELL ANEMIA
Authors: 1Shannon Phillips,
1Martina Mueller, PhD,
Alyssa Schlenz, PhD, 1Cathy Melvin, PhD,
1Robert
Adams, MD, MS, Julie Kanter, MD
Affiliations: 1Medical University of South Carolina,
2University of Alabama Birmingham
Background: Stroke is a devastating complication of
sickle cell anemia (SCA). The STOP (Stroke Prevention
Trial in Sickle Cell Anemia) protocol provides
guidelines for stroke screening using transcranial
Doppler ultrasound (TCD) and prevention with chronic
red cell transfusion therapy (CRCT). The DISPLACE
(Dissemination and Implementation Looking at the
Care Environment) study consists of 28 sites across
the US, designed to identify barriers to
implementation of the STOP protocol and test novel
methods for overcoming barriers. The study
presented in this abstract evaluated current TCD and
CRCT measurement and practices at DISPLACE
consortia sites.
Methods: An electronic survey was sent to the
principal investigator (PI) for each site. PIs were
specialty care providers for children with SCA. Items
pertaining to TCD included: screening technique;
screening frequency; follow-up for abnormal,
conditional, and inadequate results; standard value
ranges.
Results: Of the 28 PIs, 53.5% were female, 77.8%
were White, 11.1% were Asian, 7.4% were Black, and
7.4% were Hispanic/Latino. 57.1% of sites use
standard TCD, and 92.9% order TCD annually. To
calculate the time-averaged mean of the maximum
(TAMM) velocities and characterize TCD results,
96.4% of sites use the middle cerebral artery, but sites
also use a variety of other vessels. Table 1 presents
the TAMM ranges used to characterize results. Table
2 presents the actions taken/follow up by TCD result.
Table 1: Minimum and maximum TAMM values (cm/sec) reported by sites with among-site means for results characterized as normal, conditional, and high/abnormal by TCD method compared with the STOP protocol
Normal TCD Low
Normal TCD High
Conditional TCD Low
Conditional TCD High
High/ Abnormal TCD
Low
High/ Abnormal TCD
High
STOP Protocol
(TCD)
169 170 199 200
Standard TCD
Mean 70 170 170 199 200 234
Min 50 169 170 199 200 200
Max 120 179 171 200 201 300
Imaging TCD (TCDi)
Mean 83 163 163 188 190 210
Min 0 149 150 174 180 175
Max 170 170 170 199 200 299

44 | P a g e
Conclusions: Nearly all sites order TCD screening
annually, as recommended by guidelines. Variation in
practices pertaining to standard TAMM ranges for
characterizing results, follow up with MRI/MRA, and
initiation of CRCT indicate areas for future study.
Table 2: Action taken or follow-up by TCD result
Action or follow up
(n = 28)
Abnormal/high
% sites (n)
Low Conditional
% sites (n)
High Conditional
% sites (n)
Inadequate
% sites (n)
Initiate HU if not currently on 7.1 (2) 57.1 (16) 67.9 (19) 7.1 (2)
Start CRCT 85.7 (24) 0 3.6 (1) 0
No change 0 0 3.6 (1) 3.6 (1)
Initiate HU and CRCT 3.6 (1) 0 0 0
Obtain MRI/MRA 64.3 (18) 32.3 (11) 46.4 (13) 57.1 (16)
Repeat TCD prior to change 28.6 (8) 71.4 (20) 96.4 (27) 60.7 (17)

45 | P a g e
JSCDH-D-19-00036 DEVELOPMENT AND EVALUATION OF A SICKLE CELL DISEASE SEVERITY INDEX TO
VALIDATE CLINICAL ADHESION BIOMARKERS
Authors: Jennell White
1,2,4, Ke Liu
4, Xiufeng Gao
4,
Michael U. Callaghan2,5
, Patrick Hines3,4,6
Affiliations: Department of Pharmacology1, Carman
and Ann Adams Department of Pediatrics2,
Department of Physiology3, Wayne State University
School of Medicine, Detroit, MI; Functional Fluidics,
Detroit, MI4; Division of Hematology
5, Division of
Critical Care Medicine6, Children’s Hospital of
Michigan, Detroit, MI
Sickle cell disease (SCD) is a complex multisystem
organ condition characterized by vaso-occlusive
events and significant phenotypic variability. There is
no universally accepted system to objectively define
sickle cell disease (SCD) severity, which has hindered
attempts to validate the clinical utility of SCD
biomarkers. We developed an objective clinical SCD
severity index (SCDSI) that reflects an individual’s
cumulative life-time burden of SCD-related vaso-
occlusive pathology. An SCDSI was established by
quantifying the total number of objectively
documentable vaso-occlusive end-organ events,
including acute chest syndrome, stroke, priapism,
splenic sequestration, hepatic sequestration, and
cholelithiasis indexed over the total years of life. Each
objectively documentable “event” is given equal
weight in the scoring system for simplicity. These
events were selected because they could be
objectively verified via retrospective review of
medical records within a typical healthcare system or
practice. We evaluated the SCDSI in 35 subjects with
HbSS disease who were enrolled in a longitudinal SCD
study that validated real-time clinical status via a
previously described electronic patient reported
outcome tool (ePRO)1. Daily patient input validated
the patient -reported steady state of each individual
patient over 6 months. We developed a microfluidic,
flow-based adhesion bioassay to measure erythrocyte
adhesive properties (previously described2) in a CLIA
lab environment. Clinical adhesion biomarkers were
obtained every 3 weeks at the patient’s home during
baseline clinical status validated by daily self-report.
Baseline adhesion indices showed a strong positive
correlation with the SCDSI of the study subjects
(r=0.585, p<0.0011), validating that the cumulative
lifetime burden of vaso-occlusive pathology is
reflected in the erythrocyte adhesive index during the
subjects baseline clinical state. The goal of SCD-
modifying therapy is to ultimately reduce the vaso-
occlusive pathology by improving the health and
survival of the sickle erythrocytes. The results of this
study suggest that erythrocyte health, as assessed by
a standardized clinical adhesion assay, may be a
surrogate for clinical disease severity. The SCDSI
provides a simple and accessible scoring system for
clinicians to quantify historical disease burden for
individuals with SCD. Further studies are needed to
validate the utility of the SCDSI in assessing the
clinical utility of SCD-specific biomarkers in clinical
practice. A prospective study is underway to
determine the predictive value of the SCDSI for
prospective vaso-occlusive complications in this SCD
population.
1. Callaghan MU, Hines PC, Pittman DD, et al. Elipsis: A Longitudinal Study of Electronic Patient-Reported Outcomes, Actigraphy and Biomarkers to Identify and Assess at-Home Vaso-Occlusive Crises in Adults and Adolescents with Sickle Cell Disease. American Society of Hematology Annual Meeting. Atlanta, GA: The American Society of Hematology; 2017:973. 2. White J, Lancelot M, Sarnaik S, Hines P. Increased erythrocyte adhesion to VCAM-1 during pulsatile flow: Application of a microfluidic flow adhesion bioassay. Clin Hemorheol Microcirc 2014.

46 | P a g e
JSCDH-D-19-00061 SICKLE CELL TRAIT PRESENTING AS SICKLE CELL DISEASE: A CASE REPORT
Authors: Erin Goode DO, Donna Boruchov MD, Ching Lau MD Affiliations: University of Connecticut, Connecticut Children’s Medical Center Background: Sickle cell disease is a hemolytic anemia with vaso-occlusive complications, including pain, tissue ischemia, splenic infarct, and recurrent infections. It occurs secondary to two abnormal hemoglobin beta globin chains; the glutamic acid is switched with valine at the sixth position. In contrast, children with sickle cell trait have one abnormal allele present, with both hemoglobins A and S. This is clinically protective and explains why these individuals are usually asymptomatic. An eight year old male, diagnosed with sickle cell trait on newborn screen, was admitted for worsening right calf pain. He was diagnosed with acute osteomyelitis of right distal femur, with myositis, requiring surgical intervention and antibiotics. His hospitalization was then complicated with avascular necrosis of his right femur and stuttering priapism lasting six days. Objective: To expand focus beyond a patient’s newborn screen results, and to understand the complete clinical picture, when caring for a child with sickle cell trait. Understanding the possible sequelae of children with rare hemoglobinopathies and the appropriate evaluation for diagnosis. Design/Methods: Work up included hemoglobin electrophoresis, DNA sequencing, and mass spectrometry. Thorough past medical history assessment and chart review completed. Family history investigated in detail and hemoglobin evaluation of both parents performed. Results: Chart review showed failure to thrive, with documented weight of 18.2 kg (1st percentile) and height 107 cm (3rd percentile) at seven years old. Recurrent bone and abdominal pain observed since two years of age. Starting at five years old, electrophoresis was performed multiple times, which revealed both hemoglobin A and S.
On admission, his hemoglobin was 8.2, with 6.5 percent reticulocytes. Hemoglobin fractionation showed Hb S 29.8%, Hb A and Hb Quebec-Chori 61.7%, Hb A2 2.6%, and Hb F 5.8%. This abnormal hemoglobin was confirmed with DNA sequencing and mass spectrometry, which revealed his abnormal beta globin chain. Positive sickle solubility test. Conclusion: This patient’s newborn screen and initial hemoglobin evaluations did not confirm sickle cell disease, although he experienced complications expected with sickle cell disease rather than trait. After many hospitalizations, this patient’s diagnosis was confirmed on electrophoresis revealing heterozygous for Hb S and Hb Quebec-Chori. This hemoglobin variant is due to a substitution of the 87 position on the beta chain, from Thr to Ile, indistinguishable from hemoglobin A on electrophoresis. This hemoglobin variant has only been described two times previously and can result in severe sickle cell disease, making it important to detect early, before the patient experiences complications. REFERENCES 1. Rees, DC, William, TN & Gladwin, MT. Sickle-cell disease. Lancet 2010: 376: 2018-2031. 2. Pauling, L., Itano, H., et al. Sickle cell anemia, a molecular disease. Science. November 1949; 110: 543-548. 3. Edward, R., Griffiths, P. et al. Compound heterozygotes and beta-thalassemia: Top down mass spectrometry for detection of hemoglobinopathies. Proteomics, 2014: 14: 1232-1238. 4. Monplaiser, N., Merault, G. et al. Hemoglobin S Antilles: A variant with lower solubility than hemoglobin S and producing sickle cell disease in heterozygotes. Proc Natl Acad Scin USA 1986; 83: 9363-9367. 5. Witkowska, H., Lubin, B., et al. Sickle cell disease in a patient with sickle cell trait and compound heterozygostiy for hemoglobin S and hemoglobin Quebec-Chori. N Engl J Med 1991; 325 (16): 1150-1154. 6. Tubman, V., Bennett, C., et al. Sickle cell disease caused by Hb S/Quebec-CHORI treatment with hydroxyurea and response. Pediatric Blood & Cancer, August 2007; 49 (2): 207-210.

47 | P a g e
JSCDH-D-19-00021 FEASIBILITY AND QUALITY VALIDATION OF A MOBILE APPLICATION FOR ENHANCING
ADHERENCE TO OPIOIDS IN SICKLE CELL DISEASE
Authors: Daniel Sop1, 2 *
, Wally Smith1, Azhar Rafiq
3,
Abdulkhaliq Alsalman1, Donna McClish
4, Shirley
Johnson1, Thokozeni Lipato
1, Ding-Yu Fei
2
Affiliations: 1
Department of General Internal
Medicine, 2Department of Biomedical Engineering,
3Department of Surgery, and
4Deptment of
Biostatistics, Virginia Commonwealth University,
Richmond, VA 23298, USA
Introduction: American health literacy is poor,
thereby compromising medication adherence, patient
safety, and important health outcomes. Prescription
opioid nonadherence, specifically opioid misuse, has
likely contributed to the prescription opioid epidemic
and opioid-related morality in the US. Popular
methods to measure and control opioid adherence
have limitations, but mHealth, specifically
smartphone applications, offer a potentially useful
technology for this purpose.
Methods: We conducted Phase IV feasibility testing of
the OpPill application, a mobile monitoring and
reporting system intended to enhance health literacy
and adherence by collecting data and providing
systematic feedback on pain and opioid use. We
asked patients with Sickle Cell Disease (SCD) to
review, test, and evaluate the OpPill application, all at
one sitting. Immediately after OpPill review and use,
patients completed the Mobile Applications Rating
Scale (MARS), a validated tool for assessing the
quality of health mobile apps. The MARS contains 4
scales (range of each scale= 0-4) that rate
engagement, functionality, aesthetics, and
information quality. It also asks items that rate
subjective quality, relevance and overall application
impact.
Results: Patients (n=28) all had one of various SCD
genotypes; were ages 19 to 59 yrs (mean 36.56); 53.6
% were female, and; 39.3% had completed some
college. Patients rated the OpPill application highly
on four scales: Engagement, 3.93 ± 0.73;
Functionality, 4.54 ± 0.66; Aesthetics, 3.92 ± 0.81;
Information, 3.91 ± 0.87. The majority of patients
found the application to be relevant for their care.
Ninety-six percent of patients reported the
information within the app was complete, while 4%
estimated the information to be minimal or
overwhelming. Patients (91.7%) overwhelmingly
reported that the quality of information as it
pertained to SCD patients was relevant; only 8.3%
found the application to be poorly relevant to SCD.
Similarly, patients (91.7%) overwhelmingly rated both
the application’s performance and ease of use
positively. The large majority of participants (85.7%)
found the application to be interesting to use, while
74% found it entertaining. All users found the
application’s navigation to be logical and accurate
with consistent and intuitive gestural design.
Conclusion: We conclude that surveyed SCD patients,
using a validated rating tool, rated the OpPill
application, specifically targeted to monitor opioid
use and pain and opioid behavior in patients with
Chronic Non-Cancer Pain, as feasible and easy to use
Keywords: Medical Apps, Pain management, Opioids,
Chronic Condition, Sickle Cell Disease, mHealth

48 | P a g e
JSCDH-D-19-00063 CARDIAC INJURY IN SICKLE CELL DISEASE
Authors: Kiranveer Kaur, MD1, Huang Ying, MS
2, Eric
Kraut, MD2, Subha Raman, MD
3, Payal Desai, MD
2
Affiliations: 1
Division of Medical Oncology; 2Division of
Hematology; and 3Division of Cardiovascular
Medicine, The Ohio State University, Columbus, OH
Background: Previous study at our institution has shown that 3/22(13%) patients with clinically stable sickle cell disease(SCD) had impaired myocardial perfusion reserve but no epicardial coronary artery disease. The prevalence of this microvascular disease and utility of cardiac troponin in predicting such changes has not been clearly defined. In this study, we evaluated the prevalence of myocardial ischemic injury in SCD. We also examined the sensitivity and specificity of troponin levels to predict microvascular ischemic injury in patients with SCD.
Methods: We conducted a retrospective chart review of patients with SCD seen at OSU Wexner Medical Center from July 2005 to July 2015 to identify patients with troponin-I elevation (normal <0.11 ng/mL) and/or myocardial ischemic changes on cardiac MRI. Clinical and laboratory values related to these events were analyzed. Abnormal cardiac MRI was characterized by impaired subendocardial or myocardial perfusion, myocardial fibrosis or edema, and cardiomyopathy. Fisher’s exact test and Wilcoxon rank sum test were used for data analysis for categorical and continuous variables respectively.
Results: Sixty-nine (51% male; genotype Hb SS 75%, SC 16%, and Sβ-thal 9%) of 373 SCD patients had either abnormal troponin and/or had cardiac MRI done. Median age was 34 years (range 19-67 years). Of 230 patients who had troponin-I measured over this period, 18.2% (n=42) had elevated troponin. 26 of 47 patients with cardiac MRI showed abnormalities described above concerning for microvascular disease and myocardial injury. We identified 22 patients with troponin measurement within 30 days before cardiac MRI. Elevated troponin levels predicted MRI abnormalities with sensitivity of 71% (CI 42-92%) and specificity of 65% (CI 24-91%). The degree of troponin elevation did not correlate with the MRI changes. At the time of data review, 31%(n=8) patients with abnormal MRIs and 52%(n=22) patients with elevated troponin levels were deceased.
Conclusion: Over 10-year period, prevalence of cardiac injury as measured by elevated troponin was 18.2%(42/230) in SCD patients with atypical chest pain. The incidence of microvascular cardiac disease was at least 7% as diagnosed by cardiac MRI. In this small cohort, troponin elevation was neither highly sensitive nor highly specific for microvascular cardiac disease. As cardiac complications are known to be related to high frequency of deaths in SCD, there must be high index of suspicion for cardiac disease in patients with SCD and chest pain. More data is needed to accurately predict associated mortality and identify interventions to modify the outcome.

49 | P a g e
JSCDH-D-19-00026 MODELING INPATIENT OPIOID CONSUMPTION PATTERNS IN TEENAGERS WITH SICKLE CELL DISEASE
Authors: Sarah Pourakbar, MS41, Oluwaseum Olaiya,
DO2, Gerald Woods, MD
2, Colleen Reisz, MD
1
Affiliations: 1
University of Missouri Kansas City School
of Medicine; Kansas City, Missouri 2Children’s Mercy Hospital Kansas City; Kansas City,
Missouri
Background: The opioid epidemic continues to be a
significant public health issue and involves both
legitimate use and illicit drug access. Standard
management of patients with sickle cell disease (SCD)
includes treatment with opioids amongst other
strategies. SCD patients have disease-related
obstacles to achieving developmental milestones, and
therefore we theorize that these patients are at a
higher risk of accelerated trajectories of opioid use.
While only 5% of SCD patients account for the
majority of inpatient hospitalizations for painful
episodes, we sought to develop visual and
mathematical models of inpatient opioid
consumption between the ages of 12-18, to see if
patterns existed that would assist the clinician, the
patient, and the patient’s family in forecasting the
amount and duration of opioid use. We hypothesize
that clear visualization of past patterns in acceleration
and remittance of opioid consumption in real time
could trigger clinical decision support for issues that
may contribute to tolerance and addiction.
Methods: A longitudinal study using data from Cerner
Health Facts, a database that captures and stores de-
identified, longitudinal electronic health record data,
and then aggregates and organizes the data into
consumable data sets to facilitate analysis. Opioid
amount and number of doses per year, along with
gender, age, and region of the country were extracted
from the database, using ICD 9 and 10 diagnosis codes
on patients with SCD between the ages of 12-18. Data
was extracted on 451 individual patients across 862
encounters. Simple linear regressions were conducted
to examine the relationship between both age and
dose, and age and amount.
Results: Of the 451 individual patient encounters, 36
patients with SCD were identified as high utilizers
with greater than 5 hospital encounters. Linear
regressions conducted on the relationship between
age and dose yielded steady increases in the mean
dose administered across these 36 patients as age
increased. When taking each of these individual SCD
patients with greater than 5 hospital admissions, each
individual patient had cumulative dose of opioids
administered per year graphed at each age between
the ages of 12-18.
Conclusions: Attempts to manage opioid
consumption in chronic disease will require
comprehensive interventions that can be mobilized at
the time sharp changes in opioid consumption occur.
Graphing opioid consumption across a set age range
allows visual identification of opioid trajectory.
Practitioners could utilize this tool to isolate trends,
recognize previous prescribing patterns and highlight
deviations in consumption between painful episodes
and baseline that forecast tolerance and addiction.

50 | P a g e
JSCDH-D-19-00019 A PROSPECTIVE PHASE II, OPEN-LABEL, SINGLE-ARM, MULTICENTER STUDY TO ASSESS THE EFFICACY AND SAFETY
OF SEG101 (CRIZANLIZUMAB) IN SICKLE CELL DISEASE PATIENTS WITH PRIAPISM (SPARTAN)
Authors: Fuad El-Rassi,1 Deepika Darbari,
2 Arthur
Burnett,3 Jincy Paulose,
4 Dramane Lainé,
4 Das
Purkayastha,4 Greg Kato
5
Affiliations: 1
Department of Hematology and Medical
Oncology, Winship Cancer Institute of Emory
University, Atlanta, GA, United States; 2Center for
Cancer and Blood Disorders, Children’s National
Medical Center, Washington, DC, United States; 3The
James Buchanan Brady Urological Institute and
Department of Urology, The Johns Hopkins School of
Medicine, Baltimore, MD, United States; 4Novartis
Pharmaceuticals Co., East Hanover, NJ; 5Heart, Lung,
Blood and Vascular Medicine Institute, University of
Pittsburgh School of Medicine, Pittsburgh, PA, United
States
Background: Sickle cell disease (SCD) is the most
common single-gene disorder in African Americans
and can lead to complications, including acute pain
and acute/chronic organ damage. Approximately 40%
of men with SCD experience priapism, a clinical
disorder characterized by prolonged, painful penile
erection in the absence of sexual stimulation. Vaso-
occlusion-induced ischemia is generally thought to
account for SCD-associated priapism. Crizanlizumab, a
humanized monoclonal antibody that binds P-selectin
and blocks interaction with its ligands (including
leukocyte PSGL-1). The antibody significantly
decreased vaso-occlusive crises (VOCs) leading to
healthcare visit vs placebo, and was well tolerated in
SUSTAIN, a phase 2 study in adults with SCD. This
study aims to evaluate the clinical efficacy of
intravenous (IV) crizanlizumab 5 mg/kg in reducing
priapic events in patients with SCD and a history of
priapism.
Methods: This is a Phase 2, multicenter, open-label,
single arm study of crizanlizumab in male patients
aged ≥16 years with SCD-related priapism. The study
will consist of 14 weeks prescreening, 12 weeks
screening, and 52 weeks of treatment. The primary
endpoint is percent reduction from baseline of priapic
events frequency (unwanted/painful erection lasting
>60 minutes) by 26 weeks. Priapic events will be self-
reported via an electronic reporting system.
Secondary endpoints include: safety as well as the
following outcomes at 26 and 52 weeks: rate of
priapic events, percent reduction in ≥4-hour erections
requiring an ER visit, rate of VOC at 26 and 52 weeks,
and rate of complicated crises. Approximately 56
patients are planned to be enrolled in the study. The
baseline period will consist of a 14 week prescreening
period and a 12 week screening period. Eligible
patients must have ≥4 events during prescreening, ≥3
during screening with 1 event occurring within 4
weeks prior to first treatment. Patients will be treated
with IV crizanlizumab 5 mg/kg on the first day of
Week 1, Week 3 (loading dose), Week 7, and then
every 4 weeks until final treatment at Week 51.
Primary analysis will be conducted after patients
receive 26 weeks of treatment. Mandatory safety
follow-ups will be conducted until 15 weeks after last
dose.
Results: Trial ongoing
Conclusions: This study aims to address the unmet
treatment need in male patients >16 years old with
SCD-related priapism.

51 | P a g e
JSCDH-D-19-00029 IMPACT OF PHYSICIAN EDUCATION ON TIMELY INTRAVENOUS OPIOID DELIVERY AND FLUID BOLUS USE
FOR ACUTE PAIN MANAGEMENT IN CHILDREN WITH SICKLE CELL DISEASE IN THE PEDIATRIC EMERGENCY DEPARTMENT
Authors: 1Kirshma Khemani, MD;
1Joseph Malicki;
Shabnam Jain, MD; 1Michael Kelleman;
1Courtney
McCracken PhD; 2Lakshmanan Krishnamurti, MD;
2Claudia R. Morris, MD
Affiliations: 1Emory University School of
Medicine, 2Children’s Health Care of Atlanta
Background: Pain is the leading cause of pediatric emergency department (PED) visits for children with sickle cell disease (SCD). NHLBI recommends rapid evaluation and treatment of moderate-severe vaso-occlusive pain episodes (VOE) in the acute care setting, including parenteral opioids within 30min of triage or 60min of arrival, frequent pain reassessments, and re-dosing of opioids within 30min. The guidelines also discourage intravenous fluid (IVF) boluses in euvolemic patients with VOE, recommending IV hydration at no more than maintenance rate to avoid over-hydration in patients unable to drink fluids. PED adherence to NHLBI guidelines is variable. We therefore designed a Maintenance of Certification (MOC, part-4) quality improvement (QI) project for physicians in an effort to improve care.
Methods: Thirty physicians enrolled in a MOC/QI initiative where they attended two educational interventions and received monthly performance feedback reports. A retrospective analysis of prospectively collected data from electronic medical records for SCD patients seen by these providers was performed for pre-intervention (7/2017–3/2018) and post-intervention (4/2018–12/2018) phase to evaluate the impact of the intervention. Outcome measures, derived from the 2014 NHLBI VOE guidelines, included: IV opioid administration within 60 minutes of ED arrival, pain reassessment between 10–30 minutes after administration of IV opioids, IV opioid re-administration between 15–60 minutes after administration of first IV opioid, and IV fluid bolus use. 72-hour-return rate was the balancing measure. As a QI project, the data was analyzed as a function of time utilizing run charts.
Results: A total of 354 patient visits in the pre-
intervention and 269 patient visits in the post- intervention phase met inclusion criteria. The proportion of patients receiving an IV opioid within 60 minutes of arrival increased significantly from 26% to 36%, p=0.004 with a significant decrease in median time to administration from 87 minutes to 75 minutes, p=0.007. Improvement in pain reassessment after opioid administration was noted. The proportion of patients receiving an IV fluid bolus decreased from 40% to 11%, p<0.001 (Fig1). No change in the 72-hour-return rate was observed.
Conclusions: Implementation of an educational intervention combined with performance-based feedback reports led to significant improvements in the treatment of VOE, although further efforts are needed to reach goals outlined in the 2014 NHLBI guidelines. The progress made suggests that education combined with performance feedback may be a viable option for future QI initiatives to improve adherence to NHLBI guidelines and improve PED-based care for acute pain in SCD.
FIGURE 1

52 | P a g e
JSCDH-D-19-00006 EVALUATION OF THE CLASS I HISTONE DEACETYLASE (HDAC) INHIBITOR CT-101 ON FETAL HEMOGLOBIN (HBF)
EXPRESSION IN SICKLE PROGENITORS AND TRANSGENIC MICE Authors: Biaoru Li, PhD, Betty Pace, MD, Jose Sangerman, PhD, Mayuko Takezaki, MS, Michael Boosalis, PhD, Clifford Hendrick, BS, Louis Junker, PhD, Vy Le, PhD, Anil Shelke, PhD, Robert Williams PhD, Susan Perrine MD
Affiliations: Department of Pediatrics, Augusta , University, Augusta, GA Boston University School of Medicine, Boston, MA, Cetya Therapeutics, Fort Collins, CO
Background. Up-regulation of HbF is a clinically proven strategy for mitigating the clinical severity and disease phenotype of sickle cell disease patients. HDAC inhibitors including butyrate, phenylbutyrate, and dimethyl butyrate have shown activity in clinical trials. We explored whether the HDAC inhibitor CT-101 would increase HbF over adequately broad concentrations to be useful for therapy, without significantly inhibiting erythroid cell growth in sickle erythroid progenitors and in the β-YAC transgenic mouse model. Drug candidates that induce HbF in these mice have demonstrated subsequent in vivo γ-globin gene activation in human trials.
Methods. CT-101 (100-200 nM) was evaluated in erythroid progenitors generated in vitro from peripheral blood mononuclear cells isolated from 6 patients with sickle cell anemia (HbSS or
HbSβ0thalassemia). In vivo studies were conducted in β-YAC mice established with the 248 Kb human β-
globin locus, which display normal γ-globin to β-globin switching during development. β-YAC mice were treated with IP CT-101 (5 mg/kg/dose) or vehicle for 3 days/week and hydroxyurea (HU, 100 mg/kg/dose), 5 days/week. F-cell proportions were assayed using anti-γ-globin FITC antibody and HbF/cell quantified using mean fluorescence intensity (MFI) by flow cytometry.
Results. In sickle progenitors, CT-101 and HU increased F-cells by 30% and 20% respectively above control cultures from the same subject (p<0.001). HbF protein/cell by MFI increased by up to 1.6-fold above vehicle controls. Additive effects were observed with combined CT-101 (100 to 200nM) and HU (75μM) (p< 0.001). Erythroid progenitor proliferation did not decrease significantly with CT-101. Treatment with CT-101 in β-YAC mice increased F-cells by 2.5-fold compared to baseline (p=0.034) and by 1.6-fold with HU (p=0.352). HbF protein by MFI increased1.8-fold, (p=0.025) with CT-101 versus 0.9-fold (p=0.694) with HU.
Conclusions. The HDAC inhibitor CT-101 demonstrates HbF inducing activity in β-YAC mice and additively induces HbF expression in sickle erythroid progenitors with HU. These findings suggest that CT-101 may be useful in patients who have low or partial responses to HU and merits further investigation as a HbF-inducing agent.

53 | P a g e
JSCDH-D-19-00056
RISK FACTORS FOR IDENTIFYING CARDIOVASCULAR MORBIDITIES IN SICKLE CELL DISEASE PATIENTS
Authors: Isaiah Gonzalez, Dianna Hardatt, Gulnigor
Ganieva, Manoj Chhabra, Yang Liu, Pramod Narula,
Jolanta Kulpa, Revathy Sundaram
Affiliation: New York Methodist Hospital
Background: Pulmonary hypertension and cardiac
dysfunction are complications of sickle cell disease
(SCD) that develop independently but have an
important additive impact on morbidity, mortality,
and quality of life. Although there are numerous
studies in the adult population, studies in the
pediatric population are less common. Likewise,
many studies examine single variables instead of
looking at multiple variables to identify patterns for
further analysis. Survival in SCD patients has
improved with scientific advances in care and
management. Cardiac and renal complications as
well as vaso-occlusive crisis are being addressed.
Aim: The goal of our study was to evaluate the
correlation between BMI, age, hemoglobin, and O2
saturation and parameters of cardiac dysfunction
such as left ventricular abnormalities and pulmonary
hypertension in SCD patients.
Methods: A retrospective chart review was
performed of patients 2-20 years of age with a
diagnosis of SCD who had routine hematology and
cardiology outpatient visits from January 2017 to
March 2019. Patients with HbSS, HbSC, and Hb S-
Beta Thal were included in the study. Complete data
was available for 47 patients. During cardiology visits
patients had echocardiograms performed. BMI and
O2 saturation were documented. The parameters
reviewed in this study were O2 saturation, BMI, BMI
percentile, age, hemoglobin level, and ECHO findings
including left ventricular dimensions and tricuspid jet
velocity.
Results: A total of 47 pediatric patients with sickle
cell disease were included in the study. Age showed
a strong positive correlation LV dimension findings
(LVPWd 0.65, LVIDd 0.66, IVSd 0.70) and tricuspid jet
velocity (0.35). BMI had a positive correlation with
LV dimension findings (LVPWd 0.57, LVIDd 0.66, IVSd
0.65), however a weak association with tricuspid jet
velocity (-0.06). O2 saturation correlated negatively
with echocardiogram variables, however after
controlling for other variables was no longer
significant. Hemoglobin does not correlate as a
univariate, but appears to be significant in the model
after controlling for other variables.
Conclusion: This study is intended to be a
continuation of previous research that included EKG
findings and compared HbSS and HbSC. In this study
age and BMI had a strong correlation with increased
LV dimensions, however BMI did not correlate with
tricuspid jet velocity. O2 saturation did not correlate
with ECHO findings. In the model, as hemoglobin
decreases echocardiogram findings of IVSd
increases. We plan to continue our study to evaluate
other parameters of echocardiogram, hydroxyurea
use, and acute chest syndrome as predictors of
cardiovascular morbidity.

54 | P a g e
JSCDH-D-19-00054 AP-1 TRANSCRIPTION FACTOR DYSREGULATION MAY CONTRIBUTE TO B CELL DYSFUNCTION AND REDUCED
EFFICACY OF PNEUMOCOCCAL VACCINATION WITH PREVNAR-13 IN SICKLE CELL DISEASE MICE
Authors: Steven M. Szczepanek, Tyler Gavitt, B.Sc.
Affiliation: University of Connecticut
Background: Patients with Sickle Cell Disease (SCD) are at increased risk of infection with encapsulated bacterial species including Streptococcus pneumoniae. Repeated microvascular infarction leads to extensive damage to the spleen, a major immunological site for B lymphocyte development, and functional asplenia in SCD patients is thought to contribute to B cell dysfunction. Prior work in mouse models of SCD has indicated that mice vaccinated with Prevnar-13, mount robust early immune responses to the vaccine, but are incapable of maintaining antibody titers over long time periods. RNA-seq data from vaccinated and unvaccinated SCD and WT murine B cells has identified a dysregulation of AP-1-family transcription factors (TFs) c-Jun and c-Fos, two factors implicated in cell division, differentiation, and suppression of apoptosis. The importance of these three activities in memory B cell formation marked these factors as important candidates for further investigation.
Methods: To establish the role of these AP-1 TFs in B cell responses to vaccination, 8-week-old female C57Bl/6 mice were vaccinated with Prevnar-13 with or without administration of SP600125, a chemical inhibitor of the Jun N-terminal kinase which regulates c-Fos and c-Jun activity. Mice were vaccinated on days 0 and 21, with inhibitor given D-1, D0, and D1 of each vaccine bolus. 5 weeks post-boost, mice were challenged IP with 1x10
5 CFU S.
pneumoniae strain A66.1 (Serotype 3, covered by Prevnar-13). An additional unvaccinated group was used as an infection control. Mice were examined for clinical signs, weight loss, and mortality over a 21-day period post infection.
Results: All unvaccinated mice (5/5) succumbed to challenge by 3 d.p.i. Vaccinated but uninhibited mice showed 0% mortality (0/5). In comparison, vaccinated and inhibited mice showed 20% mortality (1/5). Due to concerns of insufficient power, the study was repeated with higher numbers of mice. In a follow-up experiment utilizing the same experimental conditions with larger group sizes, vaccinated but uninhibited mice once again showed
0% mortality, but mortality in the inhibited group dropped to 6% (1/15), for a cumulative mortality between the 2 studies of 10% in mice receiving SP600125.
Conclusions: This data suggests that inhibition of AP-1 TFs in a vaccination and challenge model of pneumococcal infection impairs immune responses to bacterial challenge. Dysregulation of these TFs may partially explain B cell dysfunction in SCD, but further investigation is necessary to solidify this model and thoroughly uncover the roles of AP-1 in B cell function in the context of pneumococcal vaccines.

55 | P a g e
POSTERS Presenting: Sunday, June 9, 2019 at 5:15 PM

56 | P a g e
JSCDH-D-19-00001 PAMIDRONATE USE IN SICKLE CELL DISEASE
Authors: Loretta Parker, Jennifer Keates-Baleeiro,
Wendell Moses, Joni Evans
Affiliations: University of Tennessee College of
Medicine Chattanooga, Pediatric Orthopaedics-
Children’s Hospital at Erlanger
Introduction: Avascular necrosis (AVN) of the femur is
a serious complication from sickle cell disease (SCD),
defined as lack of blood supply leading to death of
bone and collapse.1 Scant literature is available on
non-invasive treatment for AVN due to SCD in
children. Methods: Patient inclusion criteria: HbSS or
HbSS B0Thalassemia, femoral head AVN, 0-18 years,
perfusion of the lateral corner, and Pamidronate
therapy. Each cycle consisted of labs and 3
consecutive days of Pamidronate, continuing until
improvement or a maximum of 2 years. Results: Four
patients were diagnosed with AVN and treated
successfully with Pamidronate. Conclusion:
Pamidronate prevents resorption of bone, with
effects on calcium homeostasis. Our case series
demonstrates success of Pamidronate in SCD and our
hope is to inspire more in-depth research.
Introduction: Avascular necrosis (AVN), or
osteonecrosis, is a terrible complication that can
result from sickle cell disease (SCD). Avascular
necrosis occurs due to congestion and increase in
sickling in the vasculature, resulting in death of bone
and collapse.1 Along with pain, there can be
significant destruction. Pamidronate is effective in
treatment of AVN only if there is evidence on MRI of
healthy bone in the lateral corner. Our aim was to
evaluate how to improve non-invasive treatment
options. There is little evidence with using non-
operative treatments for AVN due to SCD in children.
This case series demonstrates the success of
Pamidronate as a conservative treatment option for
AVN in children with SCD.
Methods
Patients: From our comprehensive sickle cell
population of 120 patients, four developed AVN of the
femoral head. All patients were boys and were
included in the study if they: 1) had HbSS (3 patients)
or HbSS B0
Thalassemia (1 patient), 2) were diagnosed
with AVN on MRI (which showed decreased uptake in
contrast), 3) had preserved perfusion of the lateral
corner of the femoral head, 4) were aged 0-18 years,
and 5) were treated with Pamidronate. Exclusion
criteria included: 1) sickle cell trait, 2) sickle cell SC
disease, 3) those who did not develop AVN, or 4) were
not treated with Pamidronate.
Diagnosis: Imaging was performed during pre-
treatment to establish AVN, as well as post-treatment
to evaluate results of Pamidronate. Imaging
modalities were an x-ray or an MRI, either alone or for
confirmation.
Treatment: After establishing a diagnosis of AVN,
patients were evaluated by orthopedics to determine
whether Pamidronate would be appropriate.
Candidates for Pamidronate were those who had
radiographic evidence of AVN and collapse, but
maintained perfusion to the lateral corner of the
femoral head. Labs completed prior to initiating each
treatment cycle included: complete metabolic panel,
complete blood count, and ionized calcium. Dosing
and interval were age based (Table 1), and the first-
ever dose was half of each of the subsequent doses
and given in the pediatric intensive care unit. The
first-ever dose was given in the pediatric intensive
care unit due to concern for hypersensitivity reaction.
Besides this first dose, Pamidronate was given in our
outpatient infusion center on days 2 and 3, and
thereafter. During infusions, routine vitals were
measured. Infusions were given for three consecutive
days every 2-4 months, depending on age.
Following each cycle, ionized calcium and complete
blood count levels were analyzed. Calcium was
monitored closely due to the risk of hypocalcemia
with Pamidronate infusions. Physical and
occupational therapy needs were also evaluated at

57 | P a g e
every treatment cycle. X-rays were completed every
3-6 months while on Pamidronate therapy to evaluate
for improvement.
Results: Four patients followed at our institution
developed AVN with perfusion preserved at the
lateral corner of the femoral head. These patients
received Pamidronate infusions and monitoring via
blood work and x-rays. Each of these patients had
labs (CMP, CBC, iCa) drawn prior to each cycle and
(iCa and CBC) after each cycle. Only one patient had a
drop in his calcium (iCa <1), which necessitated a
delay of the administration of Pamidronate by one
month, as well as adding calcium carbonate
supplements three times per day. Two patients also
developed hypovitaminosis D, one prior to starting
Pamidronate therapy and one patient 1.5 years after
completion, which was corrected.
The full course of Pamidronate therapy is two years,
however, it could be stopped earlier if there was
evidence of resolution of AVN on imaging (x-ray or
MRI). The femoral head shape was round and
spherical, and each patient had resolution of AVN
radiographically and symptomatically after treatment.
As shown in Table 2, there were varying lengths of
treatment. Most of the patients had clinical and
radiographic resolution of AVN prior to two years,
however one patient did require the full treatment
course of two years.
Conclusion: Avascular necrosis is an unfortunate
complication of SCD. Due to the pathophysiology of
the disease, red blood cells have a sickle shape and
can cause congestion within vasculature, causing pain
crises, stroke, and even necrosis. The femoral head is
a common place for avascular necrosis to occur, due
to decreased collateral circulation.3 This complication
can lead to long-term effects, such as hip destruction
and need for surgical intervention. Thus, detection
and treatment of AVN early in its course is important
to prevent invasive therapies such as core
decompression or hip replacement. Pamidronate is a
bisphosphonate that acts by preventing bone
reabsorption by blocking hydroxyapatite breakdown,
as well as causing osteoclast apoptosis.4 In other areas
of AVN in children such as Perthes disease, it has been
shown that if the lateral corner of the femoral head is
preserved, outcomes of AVN are much better.5 It is
due to this finding that pamidronate is used to
preserve the lateral corner of the femoral head in
AVN in SCD. Because there is a decrease in bone
breakdown, it is important for vitamin D and calcium
to be monitored so that they do not become
pathologically low. It is unclear how pamidronate
helps resolve AVN of the femoral head in SCD.
However, success could be through strengthening of
surrounding bone and decreasing further micro-
breaks, thereby preserving perfusion to healthy bone.
Limitations include: small sample size of our
comprehensive sickle cell population, and lack of
randomized clinical trials. Strengths include:
standardized protocol based on age.
Through our study, we show that Pamidronate is
effective in treating AVN of the femoral head, on the
condition that the lateral corner has preserved
perfusion. By using Pamidronate, one can delay the
need for more invasive modalities such as core
decompression or hip replacements. At this point,
four years is the longest a patient has been removed
from Pamidronate therapy and with no surgical
intervention. Our hope with this case series is to
share our success with using Pamidronate to treat
AVN in children with SCD, and for this therapy to be
further investigated.
Resources
1. Martí‐Carvajal AJ, Solà I, Agreda‐Pérez LH. Treatment for avascular necrosis of bone in people with sickle cell disease. Cochran Database of Systematic Reviews. 2016; 8(CD004344). Doi: 10.1002/14651858.CD004344.pub6
2. Plotkin H. Growth in osteogenesis imperfecta. Growth, Genetics & Hormones. 2007; 23 (2): 17-23. ISSN 1932-9032.
3. Adesina O, Brunson A, Keegan THM, Wun T. Osteonecrosis of the femoral head in sickle cell disease: prevalence, comorbidities, and surgical outcomes in California. Blood Adv. 2017;1(16):1287-1295. Published 2017 Jul 11. doi:10.1182/bloodadvances.2017005256.
4. Drake MT, Clarke BL, Khosla S. Bisphosphonates: mechanism of action and

58 | P a g e
role in clinical practice. Mayo Clin Proc. 2008;83(9):1032-45. doi: 10.4065/83.9.1032
5. Kollitz KM, Gee AO. Classifications in brief: the Herring lateral pillar classification for Legg-Calvé-Perthes disease. Clin Orthop Relat Res. 2013;471(7):2068-72 .
Age Dosage Interval
<2.0 years 0.37 mg/kg/day for 3 days 2 months
2.1-3.0 years 0.56 mg/kg/day for 3 days 3 months
>3.1 years 0.75 mg/kg/day for 3 days 4 months
Table 1: Dosing chart based on age2
Patient Age at Treatment Start Length Cycles
1 27 months 12 months 4
2 6 years 24 months 6
3 9 years 6 months 3
4 15 years 20 months 5
Table 2: Treatment length and number of cycles of each patient

59 | P a g e
JSCDH-D-19-00002 ARGININE THERAPY IMPROVES MITOCHONDRIAL FUNCTION IN CHILDREN WITH SICKLE CELL
DISEASE AND VASO-OCCLUSIVE PAIN
Authors: *Morris CR1,2, Brown LA1, Wang Y3,
Dampier C1,2, Watt A2, Kumari P1,2, Figueroa J1,
Zmitrovich A2, and 3Shiva S1,
Affiliations: 1Emory University School of
Medicine, Atlanta, GA, 2Children’s Healthcare of
Atlanta, Atlanta, GA, 3University of Pittsburgh, Pittsburgh, PA,
Background: Vaso-occlusive pain episodes (VOE) are the leading cause of emergency department visits and hospitalization in patients with sickle cell disease (SCD). Our prior work has demonstrated that patients in VOE are arginine deficient and that arginine therapy significantly decreases total intravenous (IV) opioid use and improves pain scores in children with SCD during hospitalization. Mechanisms remain unclear, but may be due to stimulation of nitric oxide production. The Shiva lab has reported altered mitochondrial function in SCD patients vs healthy controls; decrease in mitochondrial electron transport Complex V activity leads to decreased respiration and increased detrimental oxidant production. We hypothesized that arginine therapy improves mitochondrial function during VOE.
Methods: Twelve subjects with HbSS or 0-thalassemia hospitalized for VOE (age male, 83% HbSS, 92% on Hydroxyurea) were randomized to treatment utilizing one of 3 dosing schemes of Arginine: 1) 100mg/kg IV three times/day (TID,n=3); 2) loading dose (200mg/kg) then 100mg/kg TID (n=5); or 3) loading dose (200mg/kg) followed by continuous infusion (300mg/kg/day) until discharge (n=4). Platelet rich plasma was isolated and stored at baseline presentation to the emergency department for VOE and hospital discharge for each subject and mitochondrial activity, protein expression, as well as protein carbonyls were measured. Results: Compared to a cohort of SCD patients in steady state, all subjects in VOE had a significantly decreased complex V activity (Fig1A). Notably, complex V activity was increased at discharge in subjects with VOE treated with arginine in all 3 dosing schemes, with greatest increase when utilizing a
loading dose. (Fig1B, p<0.001). While complex IV and citrate synthase activities were not changed in VOE platelets vs. steady state (data not shown), the activities of these enzymes were significantly increased in VOE subjects after arginine treatment when utilizing a loading dose( Fig1C-D, p<0.01). These changes are not due to increased mitochondrial number as quantification of mitochondrial DNA before and after arginine was not different (Fig1E). Complex V protein expression was also unchanged after arginine treatment (data not shown). However, arginine therapy did significantly decrease levels of protein carbonyls in platelet rich plasma across all treatment doses (Fig1F, p<0.01), suggesting a decrease in oxidative stress. Conclusion: These data demonstrate for the 1st time that arginine therapy increases mitochondrial activity and decreases oxidative stress in children with SCD/VOE. This novel mechanism may contribute to decreased pain and ultimately less opioid requirement after IV arginine treatment during VOE.

60 | P a g e
Figure 1
A B
C D
E
p=0.002 P<0.001
F

61 | P a g e
JSCDH-D-19-00007 BEYOND PAIN: THE SYMPTOMS AND IMPACTS OF SICKLE CELL DISEASE ON CHILDREN AND THEIR CAREGIVERS
Authors: Clark Brown, MD
1; Michael Callaghan, MD
2;
Sarah Clifford, PhD3; Farida Abudulai, BA
1; Ze Cong,
PhD4; Kellee Howard, MSc
3; Marzena Jurek, MS
4; Kelly
Lipman, MPH3; Ramya Pratiwadi, MS
3
Affiliations: 1Children's Healthcare of Atlanta,
Atlanta, GA, UNITED STATES, 2Wayne State
University School of Medicine, Detroit, MI, UNITED
STATES, 3ICON plc, San Francisco, CA, UNITED
STATES, 4Global Blood Therapeutics Inc, South San
Francisco, CA, UNITED STATES
Background: Sickle cell disease (SCD) affects
approximately 1 in every 400 African American
children born in the US and causes chronic
anemia, hemolysis, and vaso-occlusive crises
(VOCs), resulting in significant morbidity and
associated mortality. VOCs have traditionally been
the clinical endpoint used in SCD; however, they
do not capture the full spectrum of SCD symptoms
and the impacts experienced by children and their
caregivers, which tend to be underreported in the
literature. This study sought to comprehensively
summarize the most important symptoms
experienced by children with SCD and the impacts
on the daily lives of children and their caregivers.
Methods: Qualitative in-person interviews were
conducted in children with SCD and their
caregivers, recruited from two clinical sites in the
US. Children aged 2–11 years diagnosed with SCD
with ≥1 VOC in the past year requiring a
hospitalization/medical encounter were eligible.
Institutional Review Board approval was obtained
at each site. Semi-structured interview guides
(one for children, one for caregivers) consisting of
exploratory open-ended questions were
developed using results from a literature review.
De-identified interview transcripts were analyzed
using a qualitative analysis software. Saturation of
concepts was assessed.
Results: Children (N=7, aged 8–11 years, 86% female)
were diagnosed with SCD as newborns, with
homozygous sickle hemoglobin (HbSS, n=6) or sickle
beta 0-thalassemia (HbSβ0, n=1). Caregivers (N=16)
responsible for children (aged 2–11 years) with SCD
primarily were mothers (81%) and were employed full
time (69%). Concept saturation was reached for both
symptoms and impacts among both children and
caregivers. Children reported eight symptoms, most
commonly bodily pain (7/7), fatigue (6/7), pain crisis
(5/7), and difficulty breathing (5/7). Caregivers
reported 15 symptoms, most commonly bodily pain
(15/16), pain crisis (15/16), fatigue (13/16), fever
(13/16), headaches (9/16), acute chest syndrome
(8/16), and yellow eyes (7/16). Children reported 23
impacts, including general life with SCD, family, sleep,
emotional and physical functioning, school, and
recreational activities. The most common impacts
reported by children were sadness (6/7), decreased
school attendance and performance (5/7), and
inability to participate in activities that friends do
(5/7). Caregivers reported 29 impacts, with school
absences (15/16), caregiver burden (14/16), sadness
(11/16), and change in energy level (10/16) the most
frequently reported.
Conclusions: This study identified a range of salient
SCD symptoms beyond pain (e.g. fatigue) and their
substantial impact on children with SCD and their
caregivers. Additional treatment and care strategies
to address the full symptomatology of SCD are
needed.

62 | P a g e
JSCDH-D-19-00008
STREAMLINING CARE FOR REFUGEE AND IMMIGRANT CHILDREN WITH SICKLE CELL DISEASE AND THALASSEMIA
Authors: Nazia Tabassum Iqbal, MD; Areli Ramphal, LMSW; Emily Cowden APRN; Larri Harris Rn, APRN; Lindsey MacDonald LMSW; Ibrahim Ahmed, MD; Gerald Woods MD
Affiliations: Children’s Mercy Hospital and Clinics
Background: Sickle cell disease (SCD) and thalassemia are the most common form of monogenic inherited hemoglobinopathies. Their prevalence is higher in the old world and malaria-endemic areas. Due to immigration, the distribution of SCD and thalassemia has been changing worldwide. For many immigrants and refugees, the following factors can be barriers to health care access:
Poor economic background
Prior adverse living conditions
Lack of education
Non-English speaking
Availability of prior health records
We seek to bring attention to the challenges encountered in caring for immigrant children with SCD in Kansas City.
Method: Twenty immigrant/refugee with SCD and thalassemia were followed at Children’s Mercy Kansas City.
Results:
Identified/used consistently a primary/proficient interpreter. Telephone/video services are also used.
Handouts/instructions in native language
Encourage participation in research studies
IRB consent translated to native language
Utilized local government and community resources
Donation drives
Identification of primary care providers, preferably culture competent and qualified bilingual staff (QBS)
Facilitating medication pick-up or mailing services
Financial assistance and counseling
Culturagram psychosocial assessment
Neurocognitive testing
Coordination of care with multi-disciplinary teams
Social-work case management
Conclusions: Health burden of hemoglobinopathies is expected to increase with immigration in the USA. Unfamiliarity with the health care system, underutilization of available services, and lack of understanding of immigrants/refugee health care requirements and clinical complications are major factors to be addressed.
We recommend screening all immigrant children from countries with a high prevalence of hemoglobinopathies using hemoglobin electrophoresis at initial visit. Patients that require continuous health care as transfusions or clinical follow up to be followed by hematologist until a confirmatory electrophoresis is performed.
Immigration from countries with a high hemoglobinopathies prevalence contributes to the addition of S gene and thalassemia genes into the general population. Awareness of the disease prevalence within the patient’s country of origin and efficient prevention programs could reduce long-term severe complications in immigrant population
Strategies to better acquaint the immigrant patients with the health care system are required. Effective communication with immigrant patients in their native language using interpreters is essential. Maximizing use of available resources using targeted outreach, combined with financial assistance and counseling, can help improve outcomes among the immigrant population.
Overcoming the linguistic, financial and cultural barriers, will significantly sustain effective communication in the holistic treatment strategy for
immigrant population.

63 | P a g e
JSCDH-D-19-00010 ESTABLISHMENT OF NEUROCOGNITIVE TESTING PROTOCOL IN CHILDREN WITH SICKLE CELL DISEASE
Authors: Lea Ventura PhD 1,2
, Sharice
Bradford,M.Div,EdD1, Alison Oh, MA
2, Jocelyn Mallard,
APN1, Ronisha Edwards-Elliott, MSW
1, Lewis L. Hsu,
MD1, and Angela Rivers MD, PhD
1
Affiliations: 1Department of Pediatrics and
2Department of Psychiatry, University of Chicago, Il
Background: The neurocognitive sequelae of stroke in
children with SCD are characterized by pervasive
impairments, including decrements in general
intellectual functioning, language and verbal abilities,
visual-motor and visual-spatial processing, memory,
attention and executive functions, and academic
achievement. Impairments in sustained attention and
concentration, executive functions, and processing
speed are also common in youth who experience silent
infarcts. SCD patients are also at increased risk for
learning difficulties, particularly in reading and
mathematics. Although there is some awareness of
these deficits, the most effective and efficient method
of referring and evaluating SCD patients for
neurocognitive problems has not been well
established. The current program aimed to examine
patient utilization of testing services and outcomes.
Methods: To accomplish this, the team at University of
Illinois Hospital and Health Sciences System distributed
flyers about neurocognitive testing in a mass mailing.
In addition, 69 families had a practitioner who spoke
with patients and families during their regular
hematology appointment to explain the goals of
neuropsychological testing. The flyers were given
again at this time.
Results: Of those 69 patients, 24 patients age 10-24
have received clinical evaluation. Gender was equally
distributed. Of the patients tested, 25% had a history
of stroke, 41% were physician referred for the
suspicion of neuropsychological problems, and the
remaining 33% were referred for asymptomatic
screening. Across the domains of intellectual
functioning, executive functions, and academic skills,
patients with a documented history of stroke exhibited
the most severe deficits, with an IQ in the impaired
range (FSIQ=63, SD=3.3), significant executive deficits,
and impairments in reading (SS=59) and math (SS=69).
Physician referred patients fared slightly better, with
borderline to low average intellectual abilities
(FSIQ=79, SD=13.1), fewer executive deficits, and low
average reading (SS=89) and math (SS=82). Patients
referred for a routine screen fared best, with average
intellectual functioning (FSIQ=92, SD=3.4), executive
functions, and reading (SS=92) and math skills (SS=97).
Of the 45 patients that received flyers but did not go
for testing: 8% of patients have a history of stroke,
42% were physician identified and the remaining 46%
were referred for asymptomatic screening.
Conclusions: Patients referred for routine screening
fared the best on measures of intellectual abilities,
executive functions, and academic skills, followed by
physician referred patients, and then patients with
documented stroke. Children who were asymptomatic
were less likely to schedule appointments for testing.
Thus it appears that it is more difficult to implement
testing for children whose families are asymptomatic.

64 | P a g e
JSCDH-D-19-00011 TREATMENT OF MODERATE TO SEVERE ANEMIA IN SICKLE CELL DISEASE:
RESULTS FROM A 2018 US PATIENT CHART ANALYSIS
Authors: Robin Howard, MBA
1; Suzanne Litke, MSS
2;
Deepa George, PhD2; Kenneth R. Bridges, MD
1
Affiliations:1Global Blood Therapeutics Inc, South San
Francisco, CA, UNITED STATES, 2NAXION,
Philadelphia, PA, UNITED STATES
Background: Sickle cell disease (SCD) is a lifelong,
debilitating disorder that affects approximately
100,000 patients in the US. SCD is challenging to
manage, and there remains a lack of evidence to
guide the management of SCD at the community
level, where physicians may have limited treatment
experience and support. Although there are
recommendations for managing patients with SCD
with vaso-occlusive crises, there is an unmet need
in treating patients with SCD with moderate to
severe anemia. This analysis reviewed national
treatment patterns of SCD in moderately
(hemoglobin [Hb]=7–10.5 g/dL) and severely (Hb <7
g/dL) anemic versus non-anemic (Hb >10.5 g/dL)
patients in the academic and community settings.
Methods: Patient charts were obtained from 300
US physicians—primary care practitioners,
pediatricians, and adult and pediatric
hematologists—64% of whom practiced in the
community setting. The 870 charts submitted by
physicians responsible for these patients’ SCD-
related treatment decisions met the following
predefined criteria: patients were >2 years of age,
followed for ≥1 year, seen in the clinic within the
previous year for a routine outpatient visit, and
had not received a bone marrow transplant.
Results: Of the 870 patients assessed (74% adult), 52%
had the homozygous Hb SCD genotype HbSS, and 28%
had the heterozygous Hb genotype HbSC. Most
patients had moderate or severe anemia (48% and
12%, respectively), determined by their most recent
lab results. Although patients with SCD tend to be
anemic, the Hb levels in 12% of patients were not
assessed during the previous 2 years. At the time of
analysis, more anemic patients (61%, Hb ≤10.5 g/dL)
than non-anemic patients (37%) were prescribed
hydroxyurea for symptomatic treatment. Similarly,
more anemic patients (24%) received chronic
transfusions than non-anemic patients (16%). Among
both anemic and non-anemic patients, 16% received
chronic transfusions for anemia. The most common
reasons for chronic transfusions were anemia (79%),
prior stroke (9%), and elevated transcranial Doppler
scores (11%), a risk factor for stroke in patients with
SCD. Finally, 19% of all patients received
erythropoietin therapy, of whom 36% received
erythropoietin for ≥1 year.
Conclusions: Most patients with SCD in this analysis
had moderate to severe anemia, and the observed use
of erythropoietin therapy, among other non-disease-
modifying treatments, is strong evidence of a clear
unmet need to improve anemia in these patients. Even
non-anemic SCD patients may benefit from an agent
that targets hemolysis and anemia.

65 | P a g e
JSCDH-D-19-00013 TOBACCO SMOKING AS PROGNOSTIC INDICATOR IN SICKLE CELL DISEASE:
A RETROSPECTIVE ANALYSIS OF PROSPECTIVELY COLLECTED DATA
Authors: Nityam Rathi, Seyed M. Nouraie, Ph.D., and
Gregory J. Kato, MD.
Affiliation: Department of Medicine and the Heart,
Lung and Blood Vascular Medicine Institute, University
of Pittsburgh School of Medicine
Background: Sickle cell disease (SCD) is an inherited
blood disorder characterized by abnormal sickle-
shaped red blood cells. Sickling creates a higher
tendency in red blood cells to aggregate and stick to
vascular beds, making vascular beds a target organ in
SCD. In preliminary screening for a current drug study
of high vascular risk SCD patients (characterized by
high blood pressure, high estimated pulmonary blood
pressure, proteinuria, and early mortality) at UPMC,
smokers anecdotally seemed overrepresented. To
date, there has not been a broad-based SCD study of
smoking and vascular disease outcomes. We
hypothesized that smoking could be linked to four
important health outcomes that characterize high
vascular risk.
Methods: Relevant prospectively-collected data from
the walk-PHASST multi-center clinical trial
(ClinicalTrials.gov Identifier NCT00492531) were used
for this analysis. 672 adult SCD patients (53% female,
47% male; all of age 18-70 years) were separated into
three groups: current smokers, former smokers, and
non-smokers. In each group, normal distribution for
continuous variables were inspected using histograms
with addition of a normalized smoothed curve and
Shapiro-Wilks test. Parametric or nonparametric
analyses were then used for data analysis. P-values
were calculated using either ANOVA, or Chi-Squared
tests. Additionally, a survival analysis for time to death
by smoking status was performed using the Kaplan-
Meier and log rank test. Analyses were performed in R
and Stata.
Results: About 15% of adults with SCD were active
smokers, and 67% had never smoked. In univariate
analyses, age, gender, systolic blood pressure,
tricuspid regurgitation velocity (TRV, as a noninvasive
marker of pulmonary hypertension), and estimated
Glomerular Filtration Rate (EGFR), and pack-years
show significance or relevant trends. Patients who
never smoked are significantly younger, more likely to
be female, have a lower systolic blood pressure, and
have greater estimated glomerular filtration rate.
Upon adjustment for age and gender, current and
former smokers had a slightly higher TRV. Despite a
very limited duration of follow up, over 20 months the
survival analysis shows a trend toward lower mortality
in patients who have never smoked, although it is not
statistically significant (P=0.37).
Conclusions: SCD smoking status demonstrates
statistically significant associations with indicators of
hypertension, pulmonary hypertension and renal
function; and a non-significant relationship to
mortality during follow-up. However, neither causation
nor detailed associations can be determined from this
limited dataset. The results of this exploratory
retrospective analysis are hypothesis-generating, and
support a need for further investigation of the
consequences of smoking upon SCD outcome.

66 | P a g e
JSCDH-D-19-00016 PRESENTING ARISE (THE AFRICAN RESEARCH AND INNOVATIVE INITIATIVE FOR SICKLE CELL EDUCATION PROJECT)-
“IMPROVING RESEARCH CAPACITY FOR SCD HEATH CARE SERVICE IMPROVEMENT”
Authors: Lewis L. Hsu, Bola Ojo, Baba Inusa, Stephanie Lauren Quirk, Fedele Bonifazi Affiliations: University of Illinois, SCORE Foundation, Guys and St. Thomas Hospital, Fondazione Gianni Benzi Onlus Background: Sickle cell disease (SCD) is among the world’s most common serious inherited diseases with more than 300,000 annual births
1, 85% are born
in sub-Saharan Africa. SCD accounts for 8-16% of all-cause mortality in childhood in Sub-Saharan Africa even though it represents 2% or less of all births in the region. Increasing migration from high prevalence areas of Africa to European countries like UK, Italy, Greece and Republic of Ireland has grown their SCD population. The outcome of SCD in high-income countries differs markedly with low-middle income countries where less than 50% of affected births will not survive beyond the 10
th birthday.
Multiple factors account for these disparities including early diagnosis by newborn screening (NBS), prophylactic penicillin and comprehensive follow up. The optimal care for patients with SCD in Africa should incorporate this approach plus laboratory capacity and blood banking services. The objective of ARISE is to establish a
multidisciplinary staff exchange program to foster
the sharing of best practice in NBS, diagnosis and
treatment leading to improvement in overall disease
outcome. The purpose is for a sustainable,
appropriate health service for those living with SCD.
Methods: This involves research staff exchange between 9 European Union (EU) institutions interacting with non-EU countries including Africa and US. Through work package tasks, staff on exchange visits (secondments) will evaluate the prevalence of SCD among populations; identify SCD specific genotypes and phenotypes; study existing NBS and early infant screening; study engagement with patients, communities especially mothers and policy makers to determine barriers to NBS; establish laboratory diagnosis, quality assurance systems for population screening. Results: The 4-year project launched in January 2019.
Visiting exchanges at University of Illinois at Chicago
which focus on NBS and clinical care began in March
with the study of Implementation science strategies,
community health worker programs and stakeholder
engagement observations. A comparison study of
USA/UK Community Health Worker (CHW) role will
facilitate adaptation and training for settings in Africa
to include developing a CHW curriculum.
Conclusions: The multi-directional exchanges will foster new collaborative, institutional partnerships, promote scientific and technological SCD research cooperation between Africa, Europe and USA and build capacity towards sustainable improvement in health-related outcomes. Through ARISE interagency exchange program, interdisciplinary gender balanced teams will work with policy-makers in Africa to examine the effectiveness of a community-based approach to SCD using the tools of implementation science to understand the local context and health systems.
Acknowledgement – This project has received
funding from the European Union’s Horizon 2020
research and innovation programme under the Marie
Skłodowska-Curie grant agreement No 824021.

67 | P a g e
JSCDH-D-19-00017 TRACKING EDUCATIONAL PROGRESS IN OUR SICKLE CELL POPULATION
Authors: Jun Zhao DO1,2
, Spencer Moorman MSW1,2
,
Esther Knapp MD1,2
,Kerry McGowan MD1,2
, Laura
Massie APRN1, Ashok Raj MD
1,2
Affiliations: 1
University of Louisville School of
Medicine, Louisville, KY, 2
Norton Children’s Hospital,
Louisville, KY
Background: An important aspect of improving care in
sickle cell disease (SCD) patients is comprehensive
patient education regarding disease pathophysiology,
symptom management, and resources for treatment.
The emergency department (ED) is often the first line
resource for management of symptoms such as acute
pain crisis, and some patients can develop the
perception that they are able to receive all of their
necessary sickle cell medical care in the ED setting. As
pediatric patients with sickle cell disease grow older,
coordinated transition of care and appropriate
utilization of care resources are crucial to their long-
term success.
Methods: A transition of care program was
implemented in our pediatric sickle cell clinic to help
assess each patient’s readiness to transition to an
adult hematology provider. As a part of their
readiness for transition of care assessment, a 10-item
sickle cell general knowledge quiz is given. The quiz
includes questions regarding the genetics of sickle
disease, potential triggers for crisis, manifestation of
disease in different organ systems, and resources for
receiving care. In addition, a retrospective chart
review was performed to collect patient disease
genotype, number of emergency department visits,
and reasons for each visit between 2016-2018.
Results: There were 202 total ED visits with 97
individual patients in 2016 with the following genotype
breakdown: hemoglobin SS 64%; hemoglobin SC 24%;
hemoglobin SB0 5%; hemoglobin SB
+ 7%. The numbers
for 2017 and 2018 showed a minor decline in yearly
visits across all genotypes. The knowledge quiz
administered in 2017-2018 to 30 patients aged 13-18
years was revealing in two areas: 1) 50% (15/30) of
patients believed that they can receive all of their
medical care from the ED and 2) 27% (8/30) patients
misidentified complications from their disease.
Conclusions: Although improvements have been
made in educating and preparing sickle cell patients
for transitioning to adult care, areas in need of
improvement remain for example, increasing their
baseline knowledge of SCD and management. This
study has identified educational gaps that exist as part
of the transition process. With this knowledge, we
hope to improve current education efforts of sickle cell
patients, particularly as they transition from pediatric
to adult hematology care.

68 | P a g e
JSCDH-D-19-00018 IMPACT OF VASO-OCCLUSIVE CRISES ON QUALITY OF LIFE, HEALTHCARE RESOURCE
UTILIZATION AND WORK PRODUCTIVITY IN SICKLE CELL DISEASE PATIENTS
Authors: Rizio A
1, Bhor M
2, Lin X
1, White M
1,
McCausland KL1 Paulose J
2, Nandal S
2, Halloway R
3,
Bronté-Hall L4
Affiliations: 1Optum, Johnston, RI, USA,
2Novartis
Pharmaceuticals Corporation, East Hanover, NJ, USA,3
Formerly Novartis Pharmaceuticals Corporation, East Hanover, NJ, USA,
4Foundation for Sickle Cell Disease
Research, Hollywood, FL, USA
BACKGROUND: Sickle cell vaso-occlusive crises (VOCs) are painful episodes experienced by patients with sickle cell disease (SCD) and triggered by multicellular adhesion that block or reduce blood flow. This study aimed to explore the relationship between VOCs and patients’ quality of life (QoL), healthcare resource utilization (HCRU), and work productivity.
METHODS: The analytic sample included adult patients
with SCD (N=252) who completed a cross-sectional
online survey. QoL and VOC frequency and severity
were assessed with the Adult Sickle Cell Quality of Life
Measurement Information System (ASCQ-Me).
Patients answered questions regarding employment
impacts and completed the Workplace Productivity
and Activity Impairment: Specific Health Problem
(WPAI-SHP), which assesses productivity over the past
7 days. Patients also reported the number of times
within the past 12 months they used different
healthcare services to treat VOCs. Patients were
stratified according to the number of VOCs they
experienced within the past 12 months and the
severity of their VOCs measured by ASCQ-Me pain
severity score. Kruskal-Wallis tests were used to
evaluate differences in ASCQ-Me and WPAI domains
and measures of HCRU according to VOC frequency
and severity.
RESULTS: Patients with more frequent VOCs reported
greater impacts on all ASCQ-Me domains than patients
with less frequent VOCs (p<.05 for all). No differences
were observed in the number of times patients visited
a healthcare provider for VOCs (p=.087). Patients with
more frequent VOCs reported more ER visits and
overnight hospital stays than patients with less
frequent VOCs (p<.05 for both). Fifty-eight percent of
patients reported that SCD negatively impacted their
employment status; 73% of patients with ≥4 VOCs in
the past year reported negative impacts compared to
45% of patients with 0-3 VOCs. Patients with more
frequent VOCs reported greater absenteeism and
overall productivity loss than patients with less
frequent VOCs (p<.05 for both). Presenteeism did not
differ according to VOC frequency (p=.132). Significant
impacts on QoL, HCRU, and work productivity were
observed when stratifying by VOC severity; the pattern
of results was similar to those found when stratifying
by VOC frequency.
CONCLUSION: Patients with more frequent or severe
VOCs experience deficits in QoL and utilize costlier
healthcare services to treat VOCs. Patients with more
frequent or severe VOCs also missed more work in the
week preceding survey administration than patients
with less frequent or severe VOCs. Future research
should examine the impact of SCD-related
complications on the relationship between VOCs and
QoL, HCRU, and work impairment.

69 | P a g e
JSCDH-D-19-00022 TIERED ORAL THERAPY APPROACH FOR SICKLE CELL DISEASE VASO-OCCLUSIVE CRISIS MANAGEMENT
Authors: Margaret Guy, MD1; Lauren Cherry Magee,
PharmD, BCPS2; Sarah Hartigan, MD
1; Mica Ferlis,
MS,RN1; Wally Smith, MD
1; Daniel Sop, MS
3
Affiliations: 1 Division of General Internal Medicine,
Virginia Commonwealth University Health System,
Richmond, Virginia , 2 Department of Pharmacy,
Virginia Commonwealth University Health System,
Richmond, Virginia, 3Department of Biomedical
Engineering, Virginia Commonwealth University,
Richmond, Virginia
Introduction: Sickle cell disease is responsible for
>200,000 ER visits a year most of which are due to
pain. Although opioids remain the standard therapy
for the treatment of pain, there is little in the literature
that has studied opioid therapy in Sickle Cell Patients
as a predictor of outcomes; mainly healthcare
utilization. To this end, the hospital medicine team of
the Virginia Commonwealth University Health System
piloted a standardized approach to pain management
on one inpatient unit using a Tiered Oral Therapy
(TOTP) approach with goals to impact utilization
metrics such as the length of stay, 30 day readmission
rate while improving quality of care for patients
admitted with SCD pain crisis.
Methods: The treatment plan included long-acting oral
opioids for basal coverage, patient-controlled
analgesia (PCA), and immediate-release oral opioids
for breakthrough pain. In phase 1, we rapidly achieved
adequate analgesia by up-titrating the PCA every 2 to 4
hours. Patients may receive one to two times their
home oral immediate release opioids to aid with
breakthrough pain. After adequate analgesia is
achieved, we progressed to phase 2 where we held
the course to allow patients 24 hours of rest and
recovery. During phase 3, we decreased the PCA by
25% every 12 hours by initiating the patient’s home
oral immediate release opioids scheduled around-
the-clock with additional immediate release dosing
for breakthrough pain at a dose reduced from the day
of admission. The PCA continued to be tapered by 25%
of the maximum dose every 12 hours during phase 4
until the day of discharge which was phase 5. During
all phases, patients were continued on their home oral
long-acting opioid for basal coverage.

70 | P a g e
Results: As a result of this implementation, we noted a
3.11 days reduction in LOS from implementation to
conclusion on the pilot unit with a mean LOS of 5.589
(SD=0.94, CI=4.909, 6.268). While patient satisfaction
was not directly measured as related to the algorithm,
when asked whether they noticed changes to their
care during their hospital stay patient answered: Yes
72.2%, No 27.8%. When asked if they felt that the
team was working together to address their vaso-
occlusive crisis pain, patients answered: Yes 94.1%, No
5.9%. 30 day readmission rate was reduced by 6.36%
amongst the patients who participated on the TOTP.
Conclusion: The use of TOTP was associated with
reduced length of stay and 30 day readmission rate for
patients that participated in the tiered oral therapy.

71 | P a g e
JSCDH-D-19-00024
TRANSITION FROM PEDIATRIC TO ADULT SICKLE CELL DISEASE CARE: A SINGLE-CENTER
MODEL FOR TRANSITION IN RURAL POPULATIONS
Authors:
1Nnenna Badamosi, MD, MPH;
1Dominik
Barnes, BA; 1Betty Pace, MD
Affiliations: Department of Pediatrics, Augusta University, Augusta, GA
Background: Transition to adult care for young adults
with sickle cell disease (SCD) in rural populations
presents unique challenges. Lack of skilled providers
and limited access to subspecialty care hinder
successful transition and place these patients at higher
risk of morbidity than their peers. The Augusta
University (AU) transition program consists of pediatric
hematologist, transition coordinator, SCD nurse and
social worker, who provide care through the Pediatric
Comprehensive Sickle Cell Program. We piloted an off-
campus program through bimonthly outreach clinics in
rural South Georgia to facilitate successful transition to
adult care. Our objective was to evaluate the feasibility
and effectiveness of annual in-person transition
coordination in this underserved population with
limited access to care.
Method: We identified eligible teenage patients
scheduled for standard of care at four SCD outreach
clinic sites held in Waycross, Dublin, Albany and
Valdosta, GA in September 2018. Transition
coordinator engaged with patients during wait times
and enrolled them in program. She conducted
transition readiness assessments, provided
educational materials on SCD and transition to adult
care, available health insurance plans and information
on adult SCD providers. Patients were then
transitioned to adult care based on age, high school
graduation status and readiness assessment. Patients
provided feedback through satisfaction surveys
following the visit. Subsequently, the transition
coordinator performed telephone follow up 30 and 90
days after the outreach clinic and/or transition to adult
care.
Results: Thirty-five teenagers/young adults were
scheduled for care, of whom 21 (60%) kept their
appointments. All patients were African American with
a median age of 15 years (13 – 21 years) and 12 males
(57%); the transition coordinator enrolled all patients
in the program. Four (4/21) patients aged 18 – 21
years were transitioned to adult sickle cell providers.
On satisfaction surveys, all 21 respondents agreed or
strongly agreed when asked about ease of
understanding of educational materials provided,
specificity of information for SCD and whether their
questions were addressed appropriately. They
reported overall satisfaction with the transition
program with no significant changes in wait times or
duration of clinic appointments. Three out of four
(75%) of transitioned patients had seen or scheduled
appointments with adult providers by 90-day follow-
up.
Conclusion: For urban pediatric sickle cell programs
with rural outreach clinics, focused care on adolescent
patients can facilitate successful transition to adult
care. In-person transition coordination during
outreach clinics represents a cost-effective model for
providing transition care when a dedicated transition
clinic is unavailable, or limited by existing resources.

72 | P a g e
JSCDH-D-19-00025 A SMART PHONE BASED INTERVENTION TO IMPROVE OUTCOMES IN SICKLE CELL PATIENTS WITH FREQUENT HEALTH CARE UTILIZATION
Authors: Maya Crawford, Kyle Kidwell, Michael Pope, Camila Albo, Latanya Bowman, Leigh Wells, Nadine Barrett, Pritam Bora, Hongyan Xu and Abdullah Kutlar, Affiliation: Augusta University Background: A small group of patients with sickle cell disease (SCD) have frequent ED visits and hospitalizations, mostly with pain episodes, and are responsible for a significant health care costs. These patients comprise 5-10% of the total patient population in many centers, have significant morbidity, poor quality of life, and higher rates of mortality. We recently studied the characteristics of 28 patients with
visits/hospitalizations per year) and found that both biologic and behavioral/psycho-social factors contributed to high utilization phenotype (Higher WBC and ANC, higher bilirubin, lower MCV and Hb F, poor adherence to HU, lower educational level, higher anxiety and depression scores, and poorer coping skills compared to patients with low utilization). We studied the efficacy of a smart phone based app, Medisafe, in improving the outcomes in a group of patients with high health care utilization. Methods: 28 patients (14 males, 14 females, ages 19-60, median 30.5) were enrolled in the study, conducted at the Adult Sickle Cell Clinic, Center for Blood Disorders, Augusta University. Subjects were consented during clinic visits, and downloaded the
Medisafe app to their smart phones. This app was used to set medication reminders (primarily HU), but could also be utilized to record and communicate pain scores to the provider’s i-pad. The subjects were followed for a minimum of two months, and the preliminary results are reported here. Results: Compliance with the use of Medisafe was recorded and evaluated in a two month period. Overall, 20/28 patients (71.4%) were compliant to varying degrees and sent status reports, including daily pain scores, via Medisafe weekly. The number of ED visits and hospitalizations and laboratory values at baseline and at the end of intervention were compared. Although positive trends were observed in some parameters (Hb F, hemoglobin, MCV), none reached statistical significance. Conclusions: Frequent health care utilization in SCD is a significant problem, both from the standpoint of patients (poor outcomes, poor quality of life) and from a medical-economic (increased health care costs) point of view. We previously (Kidwell et al., Blood,…) showed that this is a multifactorial problem, and therefore will likely require a multi-disciplinary approach. The current study shows that a smart phone based approach to improve medication adherence, and to improve patient-provider communications is feasible with 71.4% compliance rate. Its efficacy in improving the outcomes in this patient group should be studied in a longer period of time.

73 | P a g e
JSCDH-D-19-00027 PATIENT AND PARENT PERCEPTION ON SICKLE CELL TRANSITION OF CARE
Authors: Diana Rash-Ellis LSW, Opeyemi Kemiki Dada,
RN, APN, Steve Reader PhD, Sidnie Jacobs-Allen MSA,
Robin Miller MD, Colleen N. Keeler, BS,
Affiliations: Nemours Center for Cancer and Blood
Disorders, The National Institute of General Medical
Sciences of the National Institutes of Health under
Award Number P20GM109021
Purpose: To identify the key areas of need during
transition and to increase the knowledge base of our
Sickle Cell patients and their families in order to
improve our transitional program. The goal of this
assessment would be to enhance our current
transition clinic and increase the educational literacy
of our sickle cell population in preparation for
transition to adult care.
Background: Transition of care among chronically ill
patients is a complex process due to several issues:
dependency of the patient on their parents/guardians,
cognitive as well as psychosocial difficulties, and the
lack of continuity of care. The goal of medical
transition is to provide health care that is:
uninterrupted, coordinated, developmentally
appropriate, and comprehensive.
In response to issues with lack of engagement in
transition, we have attempted to understand patients
and parents perception on transition by conducting a
transitional workshop and support group. Based upon
the sickle cell workshop questionnaires during our
second transition workshop, it was noted by the teens
that attended the workshop that their primary
concerns were that they would be lost to care and that
the adult providers would not know them once
transitioned to adult care facilities. Parents identified
on their version of the questionnaire that they felt
their children would be successful in transition to adult
care from pediatric due to their continued
involvement with their child’s medical care. Some
parents did identify their concern with leaving the
pediatric facility due to having spent the majority of
their child’s medical care with the same providers in
the same pediatric system.
The data collected is based upon 20 questionnaires
completed by both parents and patients during
transitional workshop and support group. The parent
and patient were given questionnaires that consisted
of open ended and Likert scale questions that were
based upon their perception of how prepared they are
educationally for adult care.
Conclusion: Based on the data collected from the
questionnaires, we discovered that patient and parent
have limited understanding of transition of care and
the necessary skills needed to transition to adult care.
Both patient and parents expressed their concern
regarding transition from pediatric to adult care and
receiving care from unknown adult providers in an
adult facility. The information gathered from the
questionnaires will be used in the expansion of our
current sickle cell clinic and modification of the
structure and curriculum currently being used.

74 | P a g e
JSCDH-D-19-00028 NEWBORN SCREENING DATA FOR SICKLE CELL DISEASE IN CALIFORNIA AND GEORGIA, 2004-2016:
IMPLICATIONS FOR HEALTH INTERVENTIONS
Authors: Aika Aluc, Mei Zhou, Angie Snyder, Susan
Paulukonis, David Wong, Mary Hulihan
Affiliation: Centers for Disease Control and Prevention
Background: Most incident sickle cell disease (SCD)
cases in the United States are identified through
newborn screening (NBS). The Sickle Cell Data
Collection (SCDC) program is a population-based,
longitudinal surveillance system for SCD that was
implemented in California and Georgia to collect
information from multiple data sources, including NBS
records. SCDC data can be used to identify changes in
sickle cell epidemiology and help target public health
resources and interventions.
Methods: We analyzed demographic and genotype
data for all individuals with SCD who were identified
by NBS in California or Georgia with a date of birth
during 2004 – 2016. A geographic trends analysis
(based on birth county) was conducted to compare the
number of SCD births occurring in high-population
Metropolitan Statistical Areas (MSAs) (defined for this
study as ≥2 million population) to SCD births occurring
in other MSAs. Based on 2017 U.S. Census population
estimates, there were five high-population MSAs in
California and one high-population MSA in Georgia.
Results: An average of 90 (range 68-117) SCD births
were identified per year in California and 156 (range
134-223) in Georgia. Forty-seven percent of the
newborns with SCD in California and 49% in Georgia
were female. The proportion of births by genotype did 0
+
thalassemia; the remaining 7% were other SCD
genotypes or the genotype was unknown. Overall, 83%
and 58% of SCD births occurred in high-population
MSAs in California and Georgia, respectively; these
percentages were consistent over time. For specific
high-population MSAs, decreases in SCD births were
observed from 2005-2009 to 2010-2014 in Los Angeles
(213 to 169 births) and San Francisco (67 to 45) and an
increase was observed in Riverside-San Bernardino (59
to 84). No significant changes were identified in the
other two MSAs in California or the one in Georgia.
Conclusions: There are sizeable numbers of newborns
diagnosed with SCD in both California and Georgia.
Most SCD births continue to be identified in high-
population MSAs; however, decreases in SCD births
were observed in select areas, notably Los Angeles and
San Francisco, and an increase was noted in Riverside-
San Bernardino. Data from SCDC are critical for
understanding changes in epidemiology that can
identify opportunities for targeted SCD interventions,
including workforce development, patient education,
and care across the lifespan.

75 | P a g e
JSCDH-D-19-00030 ESTABLISHING A SICKLE CELL CENTER IN THE NEW YORK CITY (NYC) PUBLIC HOSPITAL SETTING
Author: Linda Bulone Affiliation: Queens Hospital Center Background: Sickle cell patients at Queens Hospital, a public hospital in NYC, historically had a very high readmission rate (defined as occurring less than 30 days from the prior admission). In this hospital the readmission rate for Sickle Cell patients was 63.9% - the highest readmission rate of ANY illness. A Sickle Cell Center was developed with the aim to lower readmission rates. Methods: The Sickle Cell Center was created to provide comprehensive, individualized, medical and psychosocial services for patients within a public hospital. A designated Hematologist Oncologist was assigned to oversee the program and a Nurse Practitioner (NP) was hired to care for all Sickle Cell patients referred to the Center and seen in the Emergency Room (ER). The NP was also tasked to educate staff in the ER and Inpatient Units on care of the Sickle Cell patient with the latest evidence-based practices on managing pain crisis in a timely manner. A Sickle Cell Support group was formed. Sickle Cell Program leaders also networked with Advocacy programs to provide patients with additional support and education as an outpatient. This Program is designed to promote managing the condition at home
to prevent severe crisis. Longitudinal data was collected over a 3 year period and was used to describe patient demographics, needs and services. Results were measured by the percentage of patients readmitted to the inpatient unit at year three. Results: By year three, 100% of the Sickle Cell patients were seen by Sickle Cell Program leaders directly from the ER or as outpatient referrals. Since its inception, 235 new Sickle Cell patients were served (128 female and 107 male adults). An average of twenty patients attend the support group. Percentage of patients that required readmission to hospital was 63.9% at baseline. By year three, readmission rates decreased to 33.3% - a 48% decrease. This translated to a $1.7 million dollar hospital cost savings. Conclusions: The Sickle Cell Center has been well integrated and accepted into the public hospital system and can be used as a model for health care delivery for this socio-economically challenged population. The results show that patients are able to live in the community, manage their condition and decrease the need to be readmitted by utilizing the education and support received as an outpatient. The results also show that this translates into significant savings for the hospital which justifies implementation of the program in other facilities.

76 | P a g e
JSCDH-D-19-00032 SYSTEMATIC REVIEW OF RISK FACTORS ASSOCIATED WITH INCREASED EMERGENCY DEPARTMENT UTILIZATION IN
PATIENTS WITH SICKLE CELL DISEASE IN THE UNITED STATES
Authors: Samir K. Ballas,1 Carlton Dampier
2
Affiliations: 1Thomas Jefferson University,
Philadelphia, PA, USA; 2Emory University School of
Medicine, Atlanta, GA, USA
Background: Sickle cell disease (SCD) is a genetic
disorder that affects up to 100,000 patients in the
United States and affects multiple organ systems. The
emergency department (ED) is frequently used by
patients with SCD who have severe pain from vaso-
occlusive crises. The goal of this systematic review is to
identify predictors for ED use among patients with SCD
in the United States.
Methods: This systematic review was carried out using
the Preferred Reporting Items for Systematic Reviews
and Meta-Analyses (PRISMA) guidelines. PubMed and
Embase were searched for articles containing the
keywords “sickle cell disease” AND (“emergency” OR
“acute care”) AND (“utilization” OR “health care”)
published between January 1, 2000 and December 14,
2018. Included studies met the following inclusion
criteria: report of ED or acute care clinic use; report of
health care utilization for SCD; and report of ED visits
independent of hospital admission, ED revisits,
inpatient care visits, and SCD care unit visits. Articles
that were unavailable in English or focused on
populations outside of the United States were
excluded.
Results: Of the 23 articles included in the review
(Figure 1), 4 were prospective, and the remainder
were retrospective. Qualitative analysis of the articles
revealed a few trends, such as a higher rate of ED
utilization among adults relative to children, patients
with public insurance relative to private insurance, and
patients with more comorbidities, complications, or
pain. In one study, ambient temperatures less than
32°F were positively and clinically significantly
correlated with ED visits. Overall, gender was not
significantly associated with ED usage in most studies.
Conclusions: Age and pain levels were both commonly
cited as predictors of ED utilization. Findings from this
review highlight the urgent need for prospective
evaluations of predictors of ED utilization, as well as
assessments of psychosocial issues among patients
with SCD that may contribute to greater ED utilization.
Figure 1. Selection of Studies Evaluating Predictors for Emergency Department Utilization Among Patients With SCD

77 | P a g e
JSCDH-D-19-00035
MAPPING AND IMMUNOLOGICAL PROFILING OF RED BLOOD CELL ALLOIMMUNIZATION AS A GENETIC TRAIT
IN ADULTS WITH SICKLE CELL DISEASE
Authors: Victoria R. Brooks
1,2, Sofia M.
LeMaire1, Fayuan Wen
2, Gulriz Kurban
2, James
G. Taylor VI2
Affiliations: Howard University Department of
Microbiology, Washington DC1.; Howard
University Center for Sickle Cell Disease,
Washington DC.2
Background: One of the primary treatments for sickle cell disease (SCD) is red cell transfusion. Its major limiting complication is alloimmunization to minor blood group antigens. Alloimmunization is rare in the general population,
but common in SCD. The underlying mechanism
has not been elucidated, but it is proposed that
susceptibility is a genetic trait. We hypothesized
that the biologic basis for this phenomenon may
involve population differences and that admixture
mapping could identify susceptibility loci.
Methods A cohort of 721 SCD adults had allo-antibody
status confirmed with the blood bank. Genomic
DNA was genotyped for a panel of 1622 ancestry
informative SNPs in 359 subjects. Ancestry was
estimated from these genetic data assuming an
admixture model for 3 ancestral populations.
Regression analysis was used to identify variables
associated with alloimmunization. Genome wide
admixture mapping was performed using a log-
additive case control study to identify regions of
the genome where ancestry differences between
cases and controls suggested an association.
Results
Seventy seven percent of the cohort had a
transfusion history with 153 (27.6 %) with allo-
antibodies and 65 (11.7 %) with no prior
transfusions. Univariate regressions showed
association lifetime transfusion history (logistic
regression by antibody status, β=0.56, P=6.31 x 10-
6). Amerindian ancestry was also associated with
alloimmunization (β=4.60, P=0.03), while African
and European ancestry were not. These
associations remained significant in a multivariable
model for alloimmunization status (P=0.09).
Genome wide mapping by admixture linkage
disequilibrium compared 91 cases to 210 controls
with prior transfusions under 10 different risk
models (range 0.25-9.0). The 0.25 risk model
showed a significant association (genome log factor
10.96, P<0.001). This scan identified a region of
chromosome 5 (5q21.3-5q31.1) defined by 14 snps
spanning 21.685 Mb with a peak LOD score of 13.5.
This region contains 209 genes and 4 microRNAs,
which are implicated in regulating hematopoiesis
and immune responses by Genomic Regions
Enrichment of Annotations Tool (GREAT) analysis.
Conclusion
Association with Amerindian ancestry suggested
admixture mapping is a suitable investigative
approach. Genome wide admixture mapping
identified a strong association at chromosome
5q21.3-5q31.1. Pathway analysis of this region
further suggests involvement in hematopoiesis and
the immune response. Ongoing analysis aims to use
immunological and gene expression profiling to fine
map allo-immunization susceptibility variants within
specific genes that underlies this association.
Ultimately, mechanistic knowledge of the immune
response directed towards minor blood group
antigens could lead to potentially novel strategies to
disrupt this process.

78 | P a g e
JSCDH-D-19-00037 SYSTEMIC AND BONE TISSUE IMPACT OF L-GLUTAMINE THERAPY IN SICKLE MOUSE MODEL"
Author: Mykel Green, Mitchell Schaffler, Ph.D Gilda Barabino, Ph.D
Affiliation: City College of New York
Background: FDA approval of L-Glutamine (GLN) for patients with sickle cell disease (SCD) is a welcomed advancement, however opposing research has led to uncertainty about it’s therapeutic mechanism and long term safety. GLN is vital throughout the body and the benefit experienced by sickle patients may be explained by other means. Sickle bone, which progressively deteriorates with age, may benefit from GLN supplementation due its potential role as an anabolic agent. We aim to determine whether the Townes mouse model recapitulates the clinical observations of GLN therapy and assess tissue level implications via bone quality metrics.
Methods: Sickle mice and littermate controls were evaluated at 4-, 8-, 12-, and 16-weeks of age. GLN supplementation was given ad libitum via drinking water (1g/kg) from weaning (4-weeks) until time of sacrifice. Systemic assessment included plasma GLN concentration, NADH redox potential, lactate dehydrogenase (LDH) activity, hemopexin (HPX), and organ weights. Tissue analysis consisted of femur microCT examination of cortical and trabecular bone morphologies, and microarray of genes related to normal bone physiology.
Results: Plasma GLN of treated mice steadily increased to being higher than untreated (25%) and controls
(60%) at 16-weeks. Redox potential of GLN treated mice were significantly higher than untreated groups. LDH activity decreases by 25% in GLN mice at 8-weeks, but returns to untreated levels by 12-weeks. The concentration of HPX in GLN treated sickle mice is comparable to the untreated group at 8-weeks, but becomes significantly higher at 12- and 16-weeks. Organ masses revealed that GLN therapy hinders sickle splenomegaly from 4- to 8-weeks. The 12- and 16-week old time points showed two subgroups in GLN therapy: untreated resembling large spleens (<6% body mass) and wild-type resembling small spleens (<1% body mass). GLN treated mice with small spleens maintained nearly 40% more cortical and trabecular bone, while the large spleen subgroup returned an untreated phenotype by 16-weeks. Gene expression studies showed GLN therapy maintains relatively normal expression levels but significantly decreases FGF23 (~80%) at 8-weeks.
Conclusion: Mouse model recapitulates the reported clinical observations of GLN treated sickle patients, and serves as a viable model to investigate its role in SCD. Subsequent work will discern the inconsistent responsiveness to GLN therapy. Nonetheless, the reduction of organ weights and hemolysis markers suggests GLN supplementation may delay sickle vascular complications. Bone analysis supports GLN therapy maintains healthy tissue as well as highlighting sickle kidney pathophysiology as a pathway of interest.

79 | P a g e
JSCDH-D-19-00038 STUDY OF EVALUATION OF THE NATIONAL POLICY OF COMPREHENSIVE ATTENTION TO PERSONS WITH SICKLE CELL
DISEASE AND OTHER HEMOGLOBINOPATHIES IN THE STATE OF BAHIA
Authors: Madlene Oliveira Souza, Silvone Santa
Barbara Silva, Evanilda Souza de Santana Caravalho
Affiliation: Universidad Estadual de Feira de Santana
Sickle cell disease is prevalent among the black
population and one of the priorities of this social
segment in the health policy struggle. In 2005, Brazil
instituted the National Policy for Comprehensive Care
for Persons with Sickle Cell Disease and Other
Hemoglobinopathies (PNAIPDF), estimating the annual
occurrence of 3,500 new cases of sickle cell disease
and 200,000 people with sickle cell trait. In addition,
the state of Bahia, constituted ethnically by blacks
(76.26%), has the highest incidence of live births with
sickle cell disease (1: 627) and sickle cell trait (1:17) in
Brazil. Bahia encompasses a strong social movement
and researchers in the search for improvements to
people with this aggravation, as well as the support of
affirmative policies. However, there are difficulties
such as the access of multiprofessional care and the
decentralization of specialized services, in which there
are only 9 Reference Centers out of 417 municipalities
in Bahia. In this sense, the present study aims to verify
to what extent the PNAIPDF and other
hemoglobinopathies in the state of Bahia can be
evaluated. This is an evaluation study of the PNAIPDF
with a descriptive and exploratory design, preceding
the ex-post evaluation studies of the implantation
analysis. The study is organized in three moments that
proposes the construction of the logical model, the
elaboration of the matrix of analysis and judgment and
the validation of the same one. For this, the
construction of the indicators plan will follow the
dimensions of access to health, management and
teaching and research actions and services for
PNAIPDF. In this way, the evaluation of health policy
aims to contribute to identify limits and possibilities,
with greater resolution to services, programs and
health systems, as well as elements that help the use
of new strategies and decision making, in order to
understand the possible significant variables of policy
implementation.
Key words: Sickle cell disease, Hemoglobinopathies,
Politicy.

80 | P a g e
JSCDH-D-19-00039 A SECOND LOOK: A CASE OF ORBITAL MANIFESTATION IN SICKLE CELL DISEASE
Authors: Rebecca Berger, APRN1, Marisol Betensky,
MD1,2
, Kevin Potthast, MD3, E. Leila Jerome Clay, MD,
MCTS1,2
Affiliations: 1
Cancer and Blood Disorders Institute,
Johns Hopkins All Children’s Hospital, 2
Division of
Hematology, Department of Pediatrics, Johns Hopkins
University, 3Radiology Department, Johns Hopkins All
Children’s Hospital
Background: Sickle cell disease (SCD) is the most
common genetic disorder worldwide. Intra-ocular
involvement in sickle cell disease is a well-documented
clinical presentation though not often discussed by
hematologists. Orbital bone infarction in SCD is less
well defined1,2,3
. Prompt diagnosis is required to
prevent vision loss1. In this report we have a case of a
31 month old female with SCD (hemoglobin SS),
presenting with bilateral, symmetric infarctions of the
lateral periorbital regions, with sub-periosteal
hemorrhage.
Methods: A toddler with hemoglobin SS sickle cell
disease, maintained on hydroxyurea, with no previous
hospitalizations presented to a local emergency room
(ER) with a one-week history of bilateral eye swelling,
nasal discharge, malaise, and low-grade temperatures.
She was diagnosed with sinusitis and was started on
high dose amoxicillin. As the swelling worsened with
increased fevers, she returned to the local ER, which
prompted blood cultures and initiation of antibiotics
and orbital CT scan (Figure1). She was transferred to
our hospital for further care and began treatment with
Unasyn and Vancomycin. The patient was managed by
our team in conjunction with Infectious Disease,
ophthalmology and ENT consultations.
Results: Literature review was performed which
identified 4 articles with a total of 8 patients with
orbital infarction due to SCD with even one with
concurrent epidural hematoma4. All patients were
treated conservatively, with combinations of IV fluids,
simple transfusion, exchange transfusion, IV
dexamethasone, and IV antibiotics with full
recovery1,2,3
.
As our patient’s course improved with negative blood,
nasal cultures, respiratory pathogen panel and no
visual deficits, she was transitioned to oral Clindamycin
and Cefdinir on day 3. Additionally, her care was
managed with IV hydration, pain medications and
antipyretics. Bilateral temporal ultrasound was
obtained subsequently (Figure 2). Due to improvement
in the patient’s clinical course, and the need for
sedation, MRI was not obtained. Once improved
symptomatically, she was discharged to complete 14
day course of antibiotics while optimizing her dosing in
Hydroxyurea and with close follow up.
Conclusion: Given the unusual presentation, as sickle
cell providers, we often need to review the
presentation of orbital involvement in SCD aside from
retinopathy. Orbital involvement of sickle cell disease
is a rare occurrence, requiring rapid diagnosis for
accurate treatment to avoid long term sequela. There
is still more to be understood regarding adequate
management for such patients, severity of disease
prediction and the role of current disease modifying
agents like Hydroxyurea.
References
1. Alghamadi, A. Recurrent orbital bone sub-
periosteal hematoma in sickle cell disease: a
case report. BMC Ophthalmology. 2018; 18
(211). doi:10.1186/s12886-018-0884-1
2. Ganesh A, William RR, Mitra S, et al. Orbital
involvement in sickle cell disease: A report of
five cases and review literature. Eye.
2001;15(6):774-780.
doi:10.1038/eye.2001.248.
3. Huckfeldt RM, Shah AS. Sterile subperiosteal
fluid collections accompanying orbital wall
infarction in sickle-cell disease. Journal of
American Association for Pediatric
Ophthalmology and Strabismus.
2014;18(5):485-487.
doi:10.1016/j.jaapos.2014.04.004.
4. Naran AD, Fontana L. Sickle cell disease with
orbital infarction and epidural hematoma.
Pediatric Radiology. 2001;31(4):257-259.
doi:10.1007/s002470000406.

81 | P a g e
Figure 1: CT scan. Left image showing bilateral orbital and temporal hemorrhages. Right image showing
temporal fluid collection, greater on left.
Figure 2: Ultrasound: Right temporal sub-periosteal hematoma
L

82 | P a g e
JSCDH-D-19-00050 BENEFITS OF A COORDINATED TRANSFUSION MEDICINE AND HEMATOLOGY
SICKLE CELL DISEASE QUALITY PROGRAM Authors: Iram Puthawala, Grifty Menka, RN, Dre
Miller, MHA, Valerie Mann-Jiles, APRN-CNP, Amanda
Hrnicek, MBA, Kimberly Weirick, NDP, Scott Scrape,
MD, Eric Kraut, MD, Payal Desai, MD
Affiliation: Ohio State University Wexner Medical
Center
Background: Red cell exchange (RCE) is a mainstay of
treatment of sickle cell disease (SCD) complications.
Compared to simple transfusions, exchanges have the
benefit of decreasing iron overload. Indications for
chronic RCEs include secondary prevention of stroke,
prevention of future episodes of Acute Chest
Syndrome if already on hydroxyurea, refractory pain
despite hydroxyurea, persistent stuttering priapism,
and pulmonary hypertension. Given the multiple
indications for transfusions in SCD patients, we sought
to have a collaborative quality program to establish
standards of care and improve adherence to RCEs.
Methods: In 2016, hematology and transfusion
medicine at the OSUCCC—James began a combined
quality program in which they reviewed SCD patients
quarterly. During these meetings, patients were
reviewed to ensure they were undergoing evidence-
based treatments and psychosocial barriers to RCE
were addressed. We sought to evaluate the impact of
the program on patient outcomes.
Results:
Year
Number of patients on chronic exchange
Male (total)
Female (total)
Inpatient and outpatient exchanges (average, per patient)
RBC units exchanged (average, per patient)
Outpatient exchanges (average, per patient)
Percent of patients receiving ≥4 outpatient exchanges per year
Inpatient exchanges (average, per patient)
Inpatient admissions (average, per patient) Mortality
Average ferritin
2014 6 5 1 6.17 44.33 4.33 66.67 1.83 9 0% 1663.75
2015 6 6 0 5 38.17 3.33 33.33 1.67 5.33 0% 3652.44
2016 10 8 2 5.1 39.8 4.4 60 0.7 3.2 0% 2735.04
2017 18 9 9 6.45 48.89 4.89 61.11 1.56 5.06 5.55% 1650.1
2018 20 9 11 7.2 53.85 5.4 70 1.7 5.2 0% 1980.44
107 unique patients with SCD underwent RCE during
this time. Data regarding 27 patients who were on
chronic RCEs was analyzed. In total, the patients on
chronic exchanges underwent 378 RCEs, of which 286
were outpatient.
Conclusions: Since the quality program began in 2016,
the average number of total (inpatient/outpatient) and
outpatient exchanges increased. Furthermore, the
total number of patients on chronic exchanges as well
as the percentage of patients who underwent at least
4 outpatient exchanges per year increased yearly since
2016. While currently we did not see a decrease in the
number of inpatient exchanges, there is likely a false
elevation caused by patients who undergo non-
emergent exchanges when admitted for convenience.
Closer monitoring also led to an overall downtrend in
ferritin levels. We conclude that a structured quality
program between hematology and transfusion
medicine leads to improved care for patients with SCD
on chronic transfusions.
0
5
10
2014 2015 2016 2017 2018
Average number of transfusion exchanges, per patient
Total Outpatient Inpatient

83 | P a g e
JSCDH-D-19-00051 IT IS NOT JUST ABOUT THE OPIOID PRESCRIPTION: THE PATIENT PERSPECTIVE ON
THE USE OF OPIOIDS TO MANAGE SCD PAIN.
Authors: Cynthia Sinha, Diana Ross, Nitya Bakshi, Faud El Rassi, Lakshmanan Krishnamurti
Affiliation: Emory University
Background: The hallmark of SCD is vaso-occlusive pain, which may be acute and episodic but may progress to chronic persistent pain with unpredictable and disabling exacerbations. Gaps in practice in the management of SCD related pain are well established with little data regarding the patient perspective on opioids and their use in the management of SCD related pain. This qualitative study sought to understand the perspective of the adult patient with SCD in the use of opioids in the management of SCD related pain.
Methods: We conducted qualitative interviews of adult SCD patients from a geographically diverse population recruited from national conferences and local clinics. A semi-structured open-ended interview guide was used to collect data. Audio recordings were transcribed verbatim. Transcripts were coded using descriptive qualitative analysis using NVivo 11.
Results: We interviewed 26 adults (19 females) all African American of age ranging from 21 to 52 years old, ten employed full-time or part-time and 16 receiving social security disability benefits. All participants self-reported that their genotype was either HbSS, HbSC, and or HbS-Beta thalassemia. Participants were interviewed in-person and over phone. All participants reported pain on three or more days a week and had a current prescription for opioids to treat their pain. We developed three themes to address our research objective.
“You know, we’re just hurting and we’re trying to get people to hear us.” First, participants felt physicians only focus on prescribing opioids and do not try to evaluate the underlying cause of pain. Patients want to feel listened to and included in the discussion of understanding their pain.
“It eases the pain, it doesn’t take it away.” Second, participants reported they prefer not to rely on opioid therapy to manage their acute and chronic pain as opioids only temporarily ease their pain without eliminating the cause of the pain.
“Granted it helped but now what?” Third, participants are concerned about the adverse side effects of long-
term opioid therapy, and are worried about becoming physically dependent on opioids.
Conclusion: Adult SCD patients would like their physicians to focus on evaluation of underlying causes of pain in discussion with them rather than merely focus on prescribing opioids. Patients are dissatisfied with opioids which they see as temporary fix to their problem and are worried about adverse effects and physical dependence.

84 | P a g e
JSCDH-D-19-00052 GAZELLE: A PORTABLE, AFFORDABLE POINT-OF-CARE DIAGNOSTIC TECHNOLOGY
FOR DETECTING HAEMOGLOBIN DISORDERS IN INDIA
Authors: Kumar R1, Shrivas S1, Verma AK1, Thota P2, Bharti
PK1, Rocheleau A2, Witte T2, Uikey R1, Hasan MN3, Gurkan
UA3, Rajasubramaniam S1
Affiliations: 1Indian Council of Medical Research (ICMR)-National Institute of Research in Tribal Health, Jabalpur, India 2Hemex Health, Portland, OR, 3Case Western Reserve
University, Cleveland, OH
Background: Sickle haemoglobin (HbS) and thalassaemias are the most common inherited disorders of blood resulting in anaemia and associated complications. Sickle Cell Disease (SCD) is particularly common among natives of sub-Saharan Africa, India and Mediterranean countries. Screening in India involves solubility and sickling slide test followed by confirmation by High-performance liquid chromatography (HPLC) or haemoglobin electrophoresis. Lack of public health diagnostic facilities contribute to vast majority of
hitherto undetected patients resulting in increased morbidity and mortality. Here, we describe the field performance of microchip based cellulose acetate electrophoresis “Gazelle
TM”. The study was conducted in
tribal-dominated Madhya Pradesh and Chhattisgarhstates in India, regions where HbS prevalence varies from 3% to 35%. Gazelle
TM is a rapid (<10 minutes) and easy-to-use test
which can be performed by minimally trained personnel using only a finger-prick volume of blood.
Methods: Blood samples were collected from 300 patients who were enrolled in a pilot study in a high prevalence setting (ICMR-NIRTH, India). Each sample was tested with Gazelle
TM, and the results were
compared to electrophoresis and HPLC. Overall, 295 patient samples were included in the analysis (3 samples were excluded due to incomplete runs; sequencing results pending for 2 samples).
Results: Gazelle yielded a high accuracy (100%) compared to standard laboratory tests (Table 1).
Table 1. Overall accuracy of Gazelle in comparison to clinical standard tests.
Category Hemoglobin Type Correct Incorrect Accuracy
Sickle Cell Disease and sickle
β-Thalassemia
HbSS, HbSβ-thal 12 0 100%
Sickle Cell Trait HbAS 62 0 100%
Normal HbAA 221 0 100%
All categories - 295 0 100%

85 | P a g e
GazelleTM
demonstrated high sensitivity and high specificity for identifying SCD (HbSS) and β- Thalassemia, sickle cell trait (HbAS)
(Table 2).
Table 2. Sensitivity and specificity of Gazelle in comparison to clinical standard tests.
Category True
Positive
True
Negative
False
Positive
False
Negative
Sensitivity Specificity PPV NPV
Sickle Cell Disease
HbSS/HbSβ- thal vs.
HbAA
12 221 0 0 100 100 100 100
Sickle Cell Disease
HbSS/HbSβ- thal vs.
HbAS
12 62 0 0 100 100 100 100
Sickle Cell Trait HbAS
vs.
HbAA
62 221 0 0 100 100 100 100
Conclusions: Microchip electrophoresis technology
offers a low-cost ($2 per test), rapid, and accurate
method for detecting hemoglobin disorders such as
SCD. Gazelle can be a potential clinical tool for rapid
diagnosis of SCD and other hemoglobin disorders in
resource-limited settings.

86 | P a g e
JSCDH-D-19-00065 TIME-TREND OF MORTALITY OF BLACK WOMEN AND MEN BY SICKLE CELL DISEASE IN THE STATES OF BAHIA - BRAZIL, NORTH
CAROLINA AND SOUTH CAROLINA - UNITED STATES, 1999-2016. Authors: Aline Silva Gomes – Universidade Estadual de Feira de Santana – Brasil, José Roberto Santos Silva – Universidade Federal da Bahia – Brasil, Carlos Alberto Lima da Silva - Universidade Estadual de Feira de Santana – Brasil, Evanilda S. de Santana Carvalho - Universidade Estadual de Feira de Santana – Brasil, Coretta Melissa Jenerette – University of North Carolina and University of South Carolina – Estados Unidos, Abbas Tavakoli - University of South Carolina – Estados Unidos
OBJECTIVE: To analyze the time-trend of mortality of black men and women from sickle cell disease (SCD) in the states of Bahia - Brazil, North Carolina and South Carolina - United States, from 1999 to 2016. METHODS: Ecological study of time series that considered the deaths due to SCD of black women and men (ICD 11-D57), residing in the states of Bahia, North Carolina and South Carolina, from 1999 to 2016. The data from the state of Bahia were accessed at the Department of Information System of the Unified Health System in November 2018, and the data from the US states were obtained through the Platform for Disease Control and Prevention in December 2018. The Trend
of mortality rates by SCD was analyzed by the adjusted regression model of Prais-Winsten, (Rate)t = α + β (time)t + ϵt (95% confidence interval), to compile data, we used the Microsoft program, Office Excel 2010, and to analyze the trends we adopted the statistical program R, version 3.5.0.
RESULTS: We observed a growing trend in the mortality rate of black women and men by SCD in Bahia, with emphasis on the ages 0-9 and 10-19. In South Carolina, we observed a decreasing trend in the age group 10 to 19 years, while in the state of North Carolina, there is a steady trend of mortality among black women and men in all age groups. CONCLUSION: This study highlights the need for priority actions for the health of children and teenagers with SCD in the Brazilian state. We recommend to know the successful experiences of the State of North Carolina seeking to overcome the limitations to reduce mortality due to SCD in the state with increasing tendency of mortality due to SCD.
Keywords: Mortality; Mortality Registries, Sickle Cell disease; Epidemiology; Time Series Studies
Table 1: Results of the trend analysis of the mortality rates series by sickle cell disease, by State, women and black men and age group, 1999-2016.
Variável Beta t-valor p-valor Tendência
ESTADOS
Total - BA 0,02 7,83 <0,001 Crescente
Total- NC -0,0039 -1,88 0,078 Estável
Total - SC 0,0004 0,1 0,921 Estável
SEXO/NÃO BRANCOS
Homem - BA 0,024 7,968 <0,001 Crescente
Homem - NC -0,0172 -1,323 0,204 Estável
Homem - SC -0,012 -0,523 0,608 Estável
Mulher - BA 0,023 6,429 <0,001 Crescente
Mulher - NC -0,015 -1,345 0,198 Estável
Mulher - SC 0,017 1,138 0,272 Estável
GRUPO ETÁRIO
1 a 9 - BA 0,014 2,696 <0,01 Crescente
1 a 9 - NC -0,003 -0,004 0,501 Estável
1 a 9 - SC 0,0007 0,106 0,917 Estável
10 a 19 - BA 0,015 3,526 <0,001 Crescente
10 a 19 - NC 0,00088 0,003 0,795 Estável
10 a 19 - SC -0,015 -2,148 <0,05 Decrescente
20 a 64 - BA 0,031 7,627 <0,001 Crescente
20 a 64 - NC -0,0082 0,0042 -1,96 Estável
20 a 64 - SC 0,005 0,821 0,424 Estável
65+ - BA 0,006 1,082 0,295 Estável
65+ - NC 0,0018 0,795 0,438 Estável
65+ - SC 0,0007 0,118 0,908 Estável
Tabela 2. Resultados da Análise de Tendência das séries de taxas de mortalidade, por Estados, Homens e
Mulheres Não Brancos e grupos etários.

87 | P a g e
JSCDH-D-19-00066 SUPPRESSION OF HBF REPRESSORS BY SMALL MOLECULES AND RESPONSES IN DIFFERING SNP
PROFILES: POTENTIAL RELEVANCE FOR TAILORING GENE MODULATING THERAPIES
Authors: Yan Dai, Betty Pace, David Chui, S.P. Perrine
Affiliations: Phoenicia BioSciences Inc, Augusta University,
and Boston University School of Medicine
Background: Up-regulation of HbF in a majority of red blood
cells is clinically proven to reduce anemia and clinical severity
in sickle cell disease (SCD) and beta thalassemia (BT). More
than 70 small molecules induce fetal globin gene expression in
vitro, through diverse, potentially complimentary molecular
mechanisms with exposure during a specific period of EPO
sensitivity. Small molecule therapeutics can be pulsed or
given sequentially to promote efficacy, reduce off- target
effects on other lineages. Genetic modifiers relevant to
baseline HbF could influence the magnitude of induction by
targeted gene editing or small molecule therapeutics.
Methods: Three HDAC inhibitors, ST20 (which displaces
HDAC2), and a therapeutic with multiple actions, PB-04,
which displaces or suppresses transcriptional repressors
HDAC2 and HDAC3, LSD-1, and BCL11A were evaluated in
erythroid progenitors cultured from >100 sickle cell and
thalassemia patients. Fetal globin mRNA, F-reticulocytes, and
F protein/cell were assayed. ChIP assays and Western blots
were performed to assess drug effects on repressor
components. Comparative induction was preliminarily
assessed in the progenitors by SNPs related to basal HbF
levels or cell survival.
Results: All HDAC inhibitors and ST20 significantly reduced or abolished BCL11A protein in erythroid progenitors. Addition of PB-04 once during erythroid differentiation resulted in loss of BCL11A protein and reduced binding of BCL11A, LSD-1, and HDAC3 appeared at the same time. The relative fold-globin mRNA above untreated controls was highest (5-9 fold) in progenitors from patients with SNP profiles associated with lower baseline fetal globin levels in BCL11A (766432 and 1427407), ZBTB7A, OR51B4, Xmn-I (7482144) and rs 4601817. Induction was 4 to 9-fold in progenitors from GG and GA patterns in AKAP12, (rs10872670 associated with cell senescence) and G/G in SNX29P2 (rs 1872670), but was 1.3- to 3-fold in other SNP profiles. Conclusions: Multiple HbF inducers suppress LSD-1 and BCL11A protein binding to the promoter. Responses in erythroid progenitors with differing SNP profiles suggest that genetic modifiers relevant to baseline HbF or F-cells may offer a means to guide modulating therapies.
Figure 1. Western blot and ChIP assays show loss of BCL11A, LSD-1 and new histone activation marks with PB-04
exposure in erythroid progenitors.

88 | P a g e
FEATURED MANUSCRIPT

89 | P a g e
JSCDH-D-18-00076R1 THE PATTERN OF BLOOD PRESSURE AND RENAL FUNCTION AMONG CHILDREN WITH SICKLE CELL ANAEMIA PRESENTING IN A
TERTIARY HEALTH INSTITUTION IN NIGERIA.
Type of Manuscript: A cross-sectional study (original research) Authors: Adebukola Ajite
1, Ezra Ogundare
1, Oludare Oluwayemi
1, Oladele Olatunya
1 , Oluwasola Oke
1, Kayode Tolorunju
2,Evelyn
Omoniyi1,
Affiliations:
1Department of Pediatrics, Ekiti State University Teaching Hospital, Ado Ekiti.
2Department of Chemical pathology, Ekiti
State Teaching Hospital, Ado Ekiti Corresponding Author Dr Adebukola Ajite Department of Paediatrics, Ekiti state university teaching hospital, PMB 5355, Ado Ekiti Email : [email protected] Tel +2348069712558
ABSTRACT
Background: In sickle cell anemia (SCA), compromise of the renal vasculature due to sickled red cells has been recognized.
Objectives: To assess the renal function and blood pressure pattern in children with sickle cell anemia (SCA) presenting in a tertiary
institution
Method: A cross-sectional study of patients with sickle cell anemia (SCA) over six months involving the use of questionnaires,
general physical examination, blood pressure, investigations for hemoglobin genotype, urinalysis, serum creatinine, screening for
hepatitis B and HIV.
Results: 51 children with SCA were seen. The prevalence of impaired renal function as defined by reduced eGFR <90mL/min/1.73m2
in this study was 27.5%, previous hospital admission and blood transfusion were associated with reduction in eGFR but blood
pressure did not have significant correlation with the eGFR. The overall mean age at diagnosis of SCA was 4.09 ± 3.33 (years).
Conclusion: Impaired renal function is a major comorbid condition in children with SCA. In countries/locations where there is no
newborn screening for sickle cell disease, diagnosis is delayed, thus detecting impaired renal function may be delayed, therefore the
need for early detection and management is imperative.
Introduction: Sickle cell disease (SCD) is a clinical condition in which an individual inherits two abnormal hemoglobin (Hb) genes following mutation and at least one of which is Hemoglobin S (HbS)
1. HbS is the result of a single base pair
change, thymine for adenine, at the 6th codon of the β-globin gene
1. This change encodes valine instead of glutamine in the
6th position1, 2
. Consequently, there is production of an unstable isoform which when slowly deoxygenated can polymerize leading to the production of sickle cell
3. The most
severe type is HbSS, otherwise called sickle cell anemia (SCA) which occurs when both β-globin genes have the sickle cell
mutation 1,2
. Sickle cells lack the fluidity of the normal erythrocytes and can impede capillary flow thereby leading to
tissue ischaemia3,4
. The kidney, being a highly vascular organ, is vulnerable to vaso-occlusive events. The hemodynamic changes that occur with chronic anemia, renal hypoxia from recurrent vaso-occlusion and hemolysis-related endothelial dysfunction can lead to functional and structural changes presenting as hematuria, proteinuria, concentrating defect, renal insufficiency and hypertension which may progress to chronic kidney disease (CKD)
5-8.
Renal disease in patients with sickle cell anemia is a major comorbid condition that is seen in 15-18 % of all SCD patients, and is a cause of early death
5, 9. The presence of renal failure
in sickle cell disease (SCD) ranges from 5 to 18% of the total population of SCD patients
9.

90 | P a g e
Blood pressure has been found to be lower in SCD patients when compared with their healthy counterparts with normal hemoglobin genotype and this has been attributed to the low systemic vascular resistance seen in them
10-15. For the same
reason, hypertension is uncommon in sickle cell disease patient, hypertension in them predisposes to greater risk of vaso-occlusive crisis and death
16. Compromise of the renal
vasculature due to sickled red cells has been associated with glomerular hypertrophy, glomerular hyperfiltration and consequent proteinuria
16. Hemolysis in SCA may also
contribute to the development of kidney disease due to the increase in levels of plasma free hemoglobin and subsequent hemoglobinuria. The free heme filtered through the glomerulus is directly cytotoxic to renal tubular epithelial cells and induces damaging inflammatory responses
17.
Objectives: This study was conducted to contribute to existing data on the pattern of blood pressure and renal function among children with sickle cell anemia.
Study design and location: A cross-sectional study of patients diagnosed to have sickle cell anemia who were attending the pediatric hematology clinic of Ekiti State University Teaching Hospital Ado Ekiti was done over a period of six months from June 2016 to December 2016 after obtaining an ethical clearance from the Ethical and research committee of the institution.
Data collection and sampling technique: Patients presenting at the pediatric hematology clinic during the duration of the study (six months) were serially recruited provided they met the inclusion criteria and after obtaining informed parental consent as well as assent from the patients, where applicable. Inclusion criteria included patients aged two to sixteen years who were diagnosed by hemoglobin electrophoresis to have sickle cell anemia and presenting at the pediatric hematology clinic over a period of six months. Exclusion criteria included patients who had crises or were admitted at least two weeks prior to their scheduled clinic visit, patients on blood pressure lowering medication and those on steroids. Questionnaires were administered to consenting parents and subjects. Information requested for included the age, gender, age at diagnosis, regularity of clinic attendance, compliance with routine medication, number of hospital admissions in a year, common crisis noticed and previous blood transfusion. General physical examination, weight, height and blood pressure of the subjects were done alongside investigations such as hemoglobin genotype, urinalysis, serum creatinine , hepatitis B and HIV screening.
Blood pressure measurement: Blood pressure was determined using standard procedure according to the recommendation of the task force on high blood pressure in children and adolescent
17. This was done by auscultation in
the right arm after a 10-minute resting period using the mercury gravity sphygmomanometer with appropriate
bladder cuff sizes. The onset of the first tapping sound (Korotkoff sound 1) was taken to indicate the systolic blood pressure (SBP) while the point of complete disappearance of the sound (Korotkoff sound 5) was taken to indicate the diastolic blood pressure (DBP). For each subject two measurements were taken after an initial blood pressure trial run to achieve subject’s confidence and composure. The average reading was then determined
18.
Urinalysis: Freshly voided urine was collected from every subject into a plain universal bottle and the UriScreen Combi 10 dipstick was used to assess urine protein semi-quantitatively, specific gravity of the urine as well as the presence of erythrocyte were also assessed. The test strip was dipped into the freshly voided urine for approximately 1 second, and then drawn across the edge of the container to remove the excess urine. After 30 seconds, the test strip was compared with the colour scale and the result was recorded immediately. Colour changes taking place after 2 minutes was regarded as of no significance. The amount of protein in the urine was assessed as negative, trace, 1+ (30 mg /dL), 2+ (100 mg/ dL), 3+ (500 mg/dL).
Patients with proteinuria had repeat urinalysis on three consecutive occasions over three months to ascertain those at risk of chronic kidney disease.
Estimated glomerular filtration rate: This was calculated by using the Schwartz formula
19, where the estimated
glomerular filtration rate = kh/ serum creatinine in mg/dl (k is a constant 0.55 in children and adolescent girls and 0.77 in adolescent male, h is the height in cm) and serum creatinine was assessed using modified Jaffe’s method, type is endpoint, wavelength 520nm and the instrument is Spectioscan 60 DV, Biotech Engineering co.uk
Data analysis: Data analysis was performed using both descriptive and comparative statistics. Descriptive statistics comprising of mean, standard deviation, percentages and proportions were used for the age, anthropometry, estimated glomerular filtration rate and blood pressure profile of the subjects.
The comparative statistics comprised of Chi-square test for categorical data, independent samples t-test and analysis of variance (ANOVA) for comparison of the mean of continuous dependent variables for categorical variables with three or more categories. Pearson’s correlation was also done
RESULTS: Fifty one children with sickle cell anaemia were studied over a period of six months with the age range of two to sixteen years. Thirty nine (76.5%) of them were male while twelve (23.5%) of them were female. All the subjects had homozygous haemoglobin SS. Serology test for hepatitis B, C and HIV were negative in all of them.

91 | P a g e
THE BIODATA AND ANTHROPOMETRIC CHARACTERISTICS OF THE SUBJECTS
Age and sex distribution: The male to female ratio was 3.25:1 Out of the 51 subjects, 19 (37.3%) were aged 2-5years, 16 (84.2%) of them were males and 3 (15.8%) were females. Those aged 6-9years were 13 (25.5%), 10 (76.9%) of them were males and 3 (23.1%) were females. Those whose ages were ≥ 10 years were 19 (37.2%) and 13 (68.4%) of them were males and the remaining 6 (31.6%) were females.
Table I shows a comparison of the mean age at the time of the study, mean age at diagnosis of the disease condition, mean anthropometry and blood pressure profile based on the gender. There were no statistically significant differences in these parameters between genders. The overall mean age at diagnosis was 4.09 ± 3.33 (years), however, the mean age at diagnosis was earlier in male as compared to female. The mean values for blood pressure, mean arterial pressure and estimated glomerular filtration rate were all within normal range for each gender and there were no statistically significant difference between them.
Parents level of formal education, clinic attendance and routine medication: Among the subjects, 37 (72.5%) of the fathers had at least tertiary level of education, while 33 (64.5%) of the mothers had same. Majority of the patients 35 (69%) attended hematology clinic regularly, at least once in two months, however, the teenagers rarely came for follow up. The routine medication used by these subjects included; folic acid, multivitamin and Proguanil.
Blood pressure percentiles: Among the male subjects the systolic blood pressure percentiles recorded were; less 50
th
percentile [21(53.8%)], 50th
percentile [2(5.1%)], >50th
percentile to <90th
percentile [13(33.3%)] and 90th
percentile [3(7.7%)]. The diastolic blood pressure percentile recorded were; <50
th percentile [19(48.7%)], 50
th percentile [2(5.1%)],
>50th
-<90th
percentile [17(43.6%)] and 90th
percentile [1(2.6%)]. Among the female subjects 10(83.3%) had the systolic blood pressure percentiles less than 50
th percentile
while 2(16.7%) had theirs between >50th
percentile to <90th
percentile. The diastolic blood pressure percentiles recorded were; <50
th percentile [9(75.0%)], 50
th percentile [2(16.7%)]
and >50th
percentile to <90th
percentile [1(8.3%)]. Majority of all the subjects had both diastolic and systolic blood pressure less than the 50
th percentile and none of the subjects had
blood pressure percentile above the 90th
percentile for age and gender.
Serum Creatinine and estimated glomerular filtration rate (eGFR): As seen in figure 1, the serum Creatinine levels were majorly less than 1mg/dl. The eGFR value for the subjects ranged from 32.2mL/min/1.73m
2 to 197.0mL/min/1.73m
2.
The mean eGFR for the subjects was 105.75±35.66. In figure
2, the eGFR for the male subjects were divided into classes of normal eGFR (90-120ml/min/1.73m
2), mildly reduced
eGFR(60-89 ml/min/1.73m2), moderately reduced eGFR (30-
59 ml/min/1.73m2), severely reduced eGFR (29-15
ml/min/1.73m2
), end stage kidney disease(<15 ml/min/1.73m
2), elevated eGFR (>120-139 ml/min/1.73m
2 )
and glomerular hyperfiltration (>140 ml/min/1.73m2
). Eight (20.5%) male subjects had mildly reduced eGFR while 5(12.8%) had moderately reduced eGFR.
Majority of the
female subjects had eGFR above 90 ml/min/1.73m
2 as seen in
figure 3, only one female subject had mildly reduced eGFR (60- 89mL/min/1.73m
2). The prevalence of reduced eGFR
(<90ml/min/1.73m2) among the study population was 27.5% (14 subjects). These patients with reduced glomerular filtration cut across age 2-13years; more than half of them had previous hospital admission and blood transfusion as seen in figure 4 and 5 respectively. None of the subjects had eGFR <15mL/min/1.73m
2. However, in Figure 6, all the
subjects with age ≥ 14 years had estimated glomerular filtration suggestive of glomerular hyperfiltration (eGFR ≥ 140mL/min/1.73m
2).
Proteinuria and haematuria: The prevalence of dipstick proteinuria among the subjects was 11.8% (6subjects), the proteinuria was in the range of 30mg/dl -100mg/dl. The age range of these subjects with persistent proteinuria was 2-13years and 4(67%) of them had previous blood transfusion and all were male. The eGFR range of subjects with proteinuria was 94.8-169.0mL/min/1.73m
2. Three of these
subjects had eGFR ≥ 140mL/min/1.73m2. One subject (12year
old male) had dipstick detected blood in the urine; the eGFR was also above 90mL/min/1.73m
2
Correlation of age at diagnosis, anthropometry, blood pressure and estimated glomerular filtration rate.
Using Pearson’s correlation, there was a significant positive correlation of the age at diagnosis of the disease condition with the height and the weight of the patients, the correlation was 0.678 and 0.536 respectively and both were significant at 0.01level with 99%degree of confidence. The estimated glomerular filtration rate was also shown to have a significant positive correlation with the weight and height, the correlation was 0.678 and 0.536 respectively and both were significant at 0.01level. Blood pressure parameters had no significant correlation with the estimated glomerular filtration rate.
DISCUSSION: In the study, there was a male preponderance with M:F of 3.25:1. The age range of the subjects was 2-16 years, it was noticed that the population of the subjects was like a pyramid with a decline in the population as the age increased. This could be explained by the increase in mortality and poor survival of children with haemoglobinopathy in resource poor setting
20 such as the
location of this study. The increase in mortality and poor survival may be attributed to ignorance of the disease

92 | P a g e
condition, poverty, environmental factors like the malaria endemicity and delay in presentation in the hospital. The overall mean of age of the subjects at first diagnosis was 4.09 ± 3.33 years which depicted delay in diagnosis when compared with developed countries with the facility for newborn screening. This also suggests that the haplotype of sickle cell in these patients may be of less severity, making the patients to present to the hospital with symptoms at older age since facility for newborn screening is not widely available yet. There is also the possibility that the most severe cases had died before presenting at the hospital for routine care. The blood pressure pattern showed that both systolic and diastolic blood pressure were below 50
th percentile for most
of the patients across genders. This is in agreement with the documentation of low blood pressure in sickle cell disease patients which has been attributed to low systemic vascular resistance seen in them
3. The serum creatinine levels were
majorly less than 1mg/dl in the recruited subjects. Reduced muscle mass as compared with normal healthy individuals may be one of the contributing factors. Similar finding was reported in a study among adults with sickle cell disease
21.
The prevalence of dipstick positive proteinuria in this study was 11.8%, this is lower than the prevalence of 20.0% recorded in adult Nigerian patients with SCA by Aneke et al
22.
This disparity can be explained by the difference in the ages of the study population. Out of the six subjects with persistent proteinuria, 5(83.3%) of them were above the age of 5years, the tendency to have persistent proteinuria seemed to increase with age. Age has been described as the most potent modifying factor of sickle nephropathy of which persistent proteinuria is a feature and progression to overt CKD has been noticed in early adulthood
23.
In this study, the six patients with proteinuria had their eGFR in the range of 94.8-169.0mL/min/1.73m
2and half of them
had eGFR above 140ml/min/1.73m2
suggestive of glomerular hyperfiltration. A previous study
24 reported similar finding
that higher rates for developing microalbuminuria were seen in SCA patients with glomerular hyperfiltration and the same study also noted that the prevalence of hyperfiltration decreased with age
24. These subjects may be in the early
phase of kidney damage with compensatory increase in glomerular filtration which may be responsible for the enhanced glomerular passage of albumin and larger proteins that has been previously described in SCD
3. Age related
progression to chronic kidney disease and decrease in renal function may be implicated in the reduction of the prevalence of hyperfiltration with age as seen in the study of adults with SCA
21.
The prevalence of impaired renal function as defined by reduced estimated glomerular filtration rate <90mL/min/1.73m
2 in this study was 27.5%. The subjects
with reduced glomerular filtration rate cut across ages 2-13years and had at least an episode of previous hospital
admission and blood transfusion. This suggests that renal impairment is common in sickle cell anaemia and that sickle cell crisis which is severe enough to warrant hospital admission and blood transfusion may be a contributing factor. Ongoing hemolysis and vaso-occlusive injury have been reported to likely contribute to continuing renal injury
17, 23. Among patients with sickle cell disease, decreased
kidney function has been reported in 5 to 30 percent 21, 25, 26
which is close to our finding in this study. A higher prevalence of 67.6% was recorded by Yusuf et al
27 in his study
of adults with SCA in Zaria, Nigeria using similar cut off point of eGFR of <90 ml/min/1.73m
2. Previous scholars have
suggested that as more individuals with SCD are reaching the fourth to sixth decade of life, the prevalence and risk of CKD is likely to increase as well
23 , this may explain the higher
prevalence of decrease kidney function seen in adults with SCD.
The range of eGFR in normal children aged 2-12years was put at 89-165mL/min/1.73m
2 by Heilben et al
28 in 1991, however
in this study among children with sickle cell anaemia, the range was 32.2mL/min/1.73m
2 -197.0mL/min/1.73m
2.
Worthy of note is the fact that all subjects above the age of 14years had the estimated glomerular rate above 140ml/min/1.73m
2. An earlier study by Olowu et al
29 among
children with sickle cell disease patient aged 5-13years showed that Nigerian children with homozygous sickle cell anaemia who are in steady states have normal glomerular filtration rate while studies in adult SCD showed reduced glomerular filtration rate
19,20. The disparity in the outcome of
these various studies may be due to the fact that the changes in the glomerular filtration rate could be a spectrum; with the earlier ages 5-13years having predominantly normal glomerular filtration rate
23, ages > 14years having glomerular
hyperfiltration as a feature of compensation in early kidney disease as seen in this study and the adults SCD patients having reduced glomerular filtration rate due to age related renal decompensation
21, 25. A similar continuum of
hyposthenuria in children to progression to end-stage renal disease (ESRD) in adults has been described by other authors
23. Although in the early ages the eGFR values were
predominantly normal in this study, evidence of glomerular hyperfiltration as suggested by eGFR >140ml/min/1.73m
2 was
seen across all the ages from 2-16 years. Similarly, hyperfiltration has been seen as early as 9–19 months of age according to the Pediatric Hydroxyurea Phase III Clinical Trial (BABY HUG) study
30. The compromise of the renal
vasculature due to sickled red cells has been associated with glomerular hyperfiltration, proteinuria and glomerular hypertrophy. However, there is need for further elaborative study to unravel factors implicated in the outset of glomerular hyperfiltration in sickle cell anaemia.
There was no correlation between the estimated glomerular filtration rate and the blood pressure of the subjects in this study; this is similar to previous observation that in contrast to other glomerulopathies, the development of systemic

93 | P a g e
hypertension is uncommon in HbSS subjects with renal insufficiency
21.
CONCLUSION: The prevalence of renal insufficiency as demonstrated by estimated glomerular filtration rate of <90mL/min/1.73m
2 was 27.5%. Previous hospital admission
and blood transfusion were associated with reduction in eGFR but blood pressure did not have significant correlation with the eGFR. There is delay in diagnosis of SCA and the prevalence of persistent proteinuria and glomerular hyperfiltration was noticed to increase with the age of the subjects. Therefore, there is a need for early detection and
management of SCD to forestall recurrent vasoocclusive crises and end organ (kidney) damage. However, in the absence of newborn screening for SCD in some countries/locations leading to delay in diagnosis, early detection of renal function is critical. Routinely, renal function in sickle cell disease patients should be monitored as some might actually have impaired renal function with a need to adjust dose of medication, ensure prompt intervention and follow up.
REFERENCES
1. Michael R. DeBaun Elliott Vichinsky. Hemoglobinopathies: Nelson Textbook of Pediatrics, RE Berman, RM Kliegman,HB Jenson,BF Stanton. 18th ed. Philadephia, 2007.
2. Adekile AD, Adeodu O. Hemoglobinopathies. Paediatrics and child health in tropical region. 2nd
ed 373-390.
3. Guasch A, Cua M, Mitch WE; Extent and course of glomerular injury in patients with sickle cell anaemia. Kidney Int 49:786-791, 1996.
4. Guasch A, Cua M, Mitch WE; Sickle cell anaemia causes a distinct pattern of glomerular dysfunction. Kidney International 51;826-833,1997.
5. Serjeant GR, Serjeant BE. Sickle Cell Disease, 3rd ed. edn. Oxford, United Kingdom: Oxford University Press; 2001.
6. Kadiri S: Chronic kidney disease: Sickle cell nephropathy as a likely cause. Annals of Ibadan Postgraduate Med 2006;4:7-10.
7. Day TG, Drasar ER, Fulford T, Sharpe CC, Thein SL. Association between hemolysis and albuminuria in adults with sickle cell anemia. Haematologica. 2012;97(2):201–205.
8. Haymann JP, Stankovic K, Levy P, Avellino V, Tharaux PL, Letavernier E, Grateau G, Baud L, Girot R, Lionnet F. Glomerular hyperfiltration in adult sickle cell anemia: a frequent hemolysis associated feature. Clin J Am Soc Nephrol. 2010;5(5):756–761.
9. Saborio P, Scheinman JI. Sickle cell nephropathy. J Am Soc Nephrol. 1999;10(1):187–192.1999
10. Guasch A. Primer on kidney disease,4th
edition. National Kidney Foundation. Sickle cell Nephropathy.pg 351-355.
11. Johnson CS. Arterial blood pressure and hypervicosity in sickle cell disease. Hematol Oncol Clin North Am 2005;19:827-37.
12. Johnson CS, Giorgio AJ. Arterial blood pressure in adults with sickle cell disease. Arch Intern Med 1981;141:891-3.
13. Grell GA, Alleyne GA, Serjeant GR. Blood pressure in adults with homozygous sickle cell disease. Lancet 1981;2:1166.
14. Pegelow CH, Colangelo L, Steinberg M, Wright EC, Smith J, Phillips G et al. Natural history of blood pressure in sickle cell disease: risks for stroke and death associated with relative hypertension in sickle cell anemia. Am J Med 1997;102:171-7.
15. Karayaylalí I, Onal M, Yíldízer K, Seyrek N, Paydas S, Akoglu E et al. Low blood pressure, decreased incidence of hypertension, and renal cardiac, and autonomic nervous system functions in patients with sickle cell syndromes. Nephron 2002;91:535-7.
16. Ho Chi Hsien, Joao Thomas A. Carvalhaes, Josefina Aparecida P Braga “Blood pressure in Children with sickle cell disease”. Revista Paulista de Pedatria,vol 30(1) Sao Paulo 2012.
17. De Miguel C, Speed JS, Kasztan M, et al. Endothelin-1 and the kidney: new perspectives and recent findings. Curr Opin Nephrol Hypertens. 2016;25(1):35–41

94 | P a g e
18. The Fourth Report on the Diagnosis, Evaluation and Treatment of High Blood Pressure in Children and Adolescents. Pediatrics. 2004, vol 114 (2); 555-561.
19. Schwatz GJ, Brion CP, Spitzer A: The use of plasma creatinine concentration for estimating GFR in infants, childrenand adolescents. Pediatr Clin North Am 34:571-590,1987.
20. Epidemiology of Hb Disorders WHO, June 2008 ; Weatherall DJ, Blood 2010.
21. Guasch A, Navarrete J, Nass K, Zayas CF. Glomerular involvement in adults with sickle cell hemoglobinopathies: Prevalence and clinical correlates of progressive renal failure. J Am Soc Nephrol 2006; 17:2228.
22. Aneke J.C,· Adegoke A.O,·Oyekunle A.A et al. Degrees of Kidney Disease in Nigerian Adults with Sickle-Cell Disease. Med Princ Pract 2014;23:271-274 https://doi.org/10.1159/000361029
23. Rakhi P, Naik & Vimal K. Derebail. The spectrum of sickle hemoglobin-related nephropathy: from sickle cell disease to sickle trait, Expert Review of Hematology. 2007; 10:12, 1087-1094, DOI: 10.1080/17474086.2017.1395279
24. Vazquez B, Shah B, Zhang X et al. Hyperfiltration is associated with the development of microalbuminuria in patients with sickle cell anemia. Am J Hematol. 2014 Dec; 89(12): 1156–1157.
25. Sklar AH, Campbell H, Caruana RJ, et al. A population study of renal function in sickle cell anemia. Int J Artif Organs 1990; 13:231.
26. Falk RJ, Scheinman J, Phillips G, et al. Prevalence and pathologic features of sickle cell nephropathy and response to inhibition of angiotensin-converting enzyme. N Engl J Med 1992; 326:910.
27. Yusuf R, Hassan A, Ibrahim I N, Babadoko A A, Ibinaiye P O. Assessment of kidney function in sickle cell anemia patients in Zaria, Nigeria. Sahel Med J 2017;20:21-5
28. Heilbron DC, Holliday MA, al-Dahwi A,Kogan BA. Expressing glomerular filtration rate in children, Pediatr Nephrol.1991;5:5-11.
29. Olowu WA, Taiwo O, Oyelami A, Durosinmi MA, Adeodu OO, Akinsola A, Ogundipe MO. Glomerular filtration rate in Nigerian children with homozygous sickle cell disease. Niger J Med. 2002 Jan-Mar;11(1):23-5.
30. Ware RE, Rees RC, Sarnaik SA, et al. BABY HUG Investigators. Renal function in infants with sickle cell anemia: baseline data from the BABY HUG trial. J Pediatr. 2010;156(1):66–70.
TABLE 1: Age, anthropometric, estimated glomerular filtration rate and blood pressure profile of the
subjects.
Age, anthropometric and blood pressure characteristics
Overall mean ±
Standard deviation
N = 51
Male N = 39
Female
N=12 p value
Age at onset of the study(years)
7.92 ± 4.18 7.72 ± 4.32 8.58 ± 3.78 0.536
Age at diagnosis (years)
4.09 ± 3.33 3.75 ± 5.23 5.19 ± 3.56 0.193
Height (cm) 117.24 ± 22.56 116.56 ± 24.10 119.47 ± 17.34 0.700
Weight (kg) 23.37 ± 9.43 23.13 ± 9.66 24.12 ± 9.00 0.753
Systolic blood pressure (mmHg)
93.21 ± 10.96 94.51 ± 10.86 89.00 ± 10.67 0.129

95 | P a g e
Diastolic blood
pressure
(mmHg)
52.08 ± 7.55 52.56 ± 7.21 50.50 ± 8.70 0.413
Mean arterial blood pressure (mmHg)
65.79 ± 7.59 66.56 ± 7.17 63.33 ± 8.67 0.200
Estimated glomerular filtration rate (ml/min/1.73m2)
105.75 ± 35.66 105.12 ± 37.87 107.81 ± 28.61 0.822
TABLE II; ESTIMATED GLOMERULAR FILTRATION RATE OF PATIENTS AND THE PRESENCE OF PROTEINURIA
PROTEINURIA
NEGATIV
E
30MG/D
L
100MG/DL
ESTIMATED GFR
VALUE
>
90mL/min/1.73m2
32 5 1
60-
89mL/min/1.73m2
8 0 0
30-
59mL/min/1.73m2
5 0 0
Total 45 5 1
TABLE III; ESTIMATED GLOMERULAR FILTRATION RATE AND THE PRESENCE OF BLOOD IN THE URINE
BLOOD IN THE URINE
NEGATIVE POSITIVE
ESTIMATED GFR
VALUE
>
90mL/min/1.73m2
37 1
60-
89mL/min/1.73m2
8 0
30-
59mL/min/1.73m2
5 0
Total 50 1

96 | P a g e
FIGURE 1
FIGURE 2

97 | P a g e
FIGURE 3

98 | P a g e
FIGURE 4
FIGURE 5

99 | P a g e
FIGURE 6

100 | P a g e
TABLE OF AUTHORS Abboud, Miguel R., 12 Abebe, Kaleab Z., 42 Abudulai, Farida, 61 Achebe, Maureen, 24 Adams, Robert, 43 Ahmed, Ibrahim, 62 Ajite, Adebukola, 89 Al Kindi, Salam, 12 Albo, Camila, 72 Alsalman, Abdulkhaliq, 17, 47 Aluc, Aika, 74 Ataga, Kenneth I., 12 Attay, Oguzhan, 13 Axelrod, David, 14 Badamosi, Nnenna, 71 Ballas, Samir K., 76 Barabino, Gilda, 78 Barbetta, Lea, 28 Barnes, Dominik, 71 Barrett, Nadine, 72 Berbaum, M., 16 Berger, Rebecca, 80 Betensky, Marisol, 80 Bharti, PK, 84 Bhor, M., 68 Boccia, Ralph, 40 Bodla, Shankaranand, 33 Bohn, Rhonda, 24 Bonifazi, Fedele, 66 Boosalis, Michael, 52 Bora, Pritam, 72 Boruchov, Donna, 46 Bos, Jennifer, 8 Bowman, Latanya, 72 Bradford, Christopher, 39 Bradford, Sharice, 63 Bridges, Kenneth R., 64 Brigitte A., van Oirschot, 8 Bronté-Hall, Lanetta, 68 Brown, Clark, 12, 61 Brown, LA, 59 Bulone, Linda, 75 Burnett, Arthur, 50 Callaghan, Michael U., 45, 61 Carneiro, Jayanne Moreira, 30 Carvalho, Evanilda Souza de Santana, 28, 30, 79, 86 Cataldo, Vince, 40 Cerqueira, Sheila Santa Barbara, 30 Chan, Flo, 6 Chang, Alicia K., 10
Chern, Irene, 5 Chhabra, Manoj, 53 Clay, E. Leila Jerome, 80 Clifford, Sarah, 61 Cnossen, Marjon H., 8 Cole, Audrey, 38 Cong, Ze, 61 Congdon-Martin, Elisabeth, 14 Corder-Mabe, Joan, 20 Cowden, Emily, 62 Crawford, Albert G., 14 Crawford, Maya, 72 Crouch, Andrew, 35 da Silva, Carlos Alberto Lima, 86 Dada, Opeyemi Kemiki, 73 Dampier, Carlton, 59, 76 Darbari, Deepika, 50 Dauvergne, Bernard, 18 de Montalembert, Mariane, 33 DeNicola, Toni, 25 Desai, Payal, 48, 82 Desai, Rishi J., 24 Diamond, Alan M., 31 Diuguid, David L., 12 Edwards-Elliott, Ronisha, 63 El Beshlawy, Amal, 12 El Rassi, Faud, 83 Elliott, Brian, 33 Elliott, Taylor, 17, 20 El-Rassi, Fuad, 50 Etienne-Julan, Maryse, 18 Evans, Joni, 56 Faller, Douglas V., 36 Fei, Ding-Yu, 17, 47 Ferlis, Mica, 69 Figueroa, J., 59 Flanagan, Jonathan M., 10 Fornari, Eric D., 5 Galacteros, Frédéric, 18 Ganieva, Gulnigor, 53 Gao, Xiufeng, 45 Gavitt, Tyler, 54 George, Deepa, 64 Gershel, Jeffrey, 25 Gladwin, Mark T., 42 Globe, Denise, 24 Gomes, Aline Silva, 86 Gonzalez, Isaiah, 32, 53 Goode, Erin, 46 Gordeuk, V., 16

101 | P a g e
Gordeuk, Victor R., 12, 42 Goyal, Ankush, 10 Green, Mykel, 78 Griffith, Jamilla, 27 Gurkan, UA, 84 Gutman, Gail, 25 Guy, Margaret, 69 Haines, Carol, 14 Hakim, Rabia, 32 Halloway, R., 68 Hankins, Jane, 38 Hanstein, Regina, 5 Hardatt, Dianna, 32, 53 Harris, Larri, 62 Hartigan, Sarah, 69 Hasan, MN, 84 Hassab, Hoda, 12 Heeney, Matthew M., 33 Heib, Adele, 5 Hendrick, Clifford, 52 Hines, Patrick, 45 Hodges, Jason, 38 Hong, Lenny K., 31 Hoppe, Carolyn C., 12 Houwing, Maite E., 8 Howard Robin, 64 Howard, Jo, 12 Howard, Kellee, 61 Hrnicek, Amanda, 82 Hsu, Lewis L., 63, 66 Huang, Wenmei, 11 Hulihan, Mary, 74 Ibanez, Vinzon, 31 Intondi, Allison, 12 Inusa, Baba, 66 Iqbal, Nazia Tabassum, 62 Itzep, Nelda, 13 Jacobs-Allen, Sidnie, 73 Jagadeeswaran, Ramasamy, 31 Jain, Shabnam, 51 Jenerette, Coretta Melissa, 30, 86 John, Minnie, 32 Johnson, Shirley, 17, 20, 47 Julie, Kanter, 33 Junker, Louis, 52 Jurek, Marzena, 61 Kanne, Celeste K., 8, 13, 37 Kanter, Julie, 11, 42, 43 Kato, Gregory J., 42, 50, 65 Kaur, Kiranveer, 48 Keates-Baleeiro, Jennifer, 56
Keeler, Colleen N., 73 Keenan, Mary, 27 Kelleman, Michael, 51 Keumeugni, Carlosse, 18 Khemani, Kirshma, 51 Kidwell, Kyle, 72 Knapp, Esther, 67 Kraft, Walter K., 40 Kraut, Eric, 48, 82 Krishnamurti, Lakshmanan, 11, 51, 83 Kulpa, Jolanta, 32, 53 Kumar, R., 84 Kumari, P., 59 Kuo, Kevin, 36 Kurban, Gulriz, 77 Kutlar, Abdullah, 40, 72 Kwiatkowski, Janet L., 11 Lago, Raissa, 28 Lainé, Dramane, 40, 50 Lau, Ching, 46 Le, Vy, 52 Lehrer-Graiwer, Joshua, 12 LeMaire, Sofia M., 77 LeRoux, Rebecca, 36 Levin, Raisa, 24 Li, Biaoru, 52 Lin, X., 68 Lipato, Thokozeni, 17, 47 Lipman, Kelly, 61 Litke, Suzanne, 64 Liu, Ke, 45 Liu, Yang, 53 Loew, Megan, 27 MacDonald, Lindsey, 62 Magee, Lauren Cherry, 69 Mahesri, Mufaddal, 24 Malicki, Joseph, 51 Mallard, Jocelyn, 63 Mann-Jiles, Valerie, 82 Manwani, Deepa, 5 Mapara, Markus Y., 11 Massie, Laura, 67 Mattos, Ivanilde Guedes, 28 McAna, John, 14 McCausland, KL, 68 McClish, Donna, 17, 47 McCracken, Courtney, 51 McGowan, Kerry, 67 McKenzie, Steven, 14 McKerracher, Krista, 24 Melvin, Cathy, 43

102 | P a g e
Méndez, G.G., 16 Menka, Grifty, 82 Merino, Raquel, 33 Miller, Alexandra, 11 Miller, Dre, 82 Miller, Robin, 73 Minniti, Caterina, 5, 6, 35, 39, 42 Miodownik, Hope, 39 Molokie, Robert E., 31 Moorman, Spencer, 67 Morris, Claudia R., 42, 51, 59 Morrone, Kerry, 5 Moses, Wendell, 56 Mueller, Martina, 43 Mutebi, Alex, 24 Nandal, S., 68 Narula, Pramod, 53 Nascimento, Helaynne Bezerra, 28 Nduba, Videlis, 12 Nocek, J.M., 16 Nouraie, Seyed M., 65 Odame, Isaac, 33 Ogundare, Ezra, 89 Oh, Alison, 63 Ojo, Bola, 66 Oke, Oluwasola, 89 Olaiya, Oluwaseum, 49 Olatunya, Oladele, 89 Oluwayemi, Oludare, 89 Omoniyi, Evelyn, 89 Pace, Betty, 52, 71 Pace, Betty S., 36 Paikari, Alireza, 10 Parker, Loretta, 56 Pasterkamp, Gerard, 8 Paulose, Jincy, 40, 50, 68 Paulukonis, Susan, 74 Perrine, Susan P., 36, 52 Phillips, Shannon, 43 Pierciey, Francis J., 11 Pope, Michael, 72 Porter, Jerlym, 27, 38 Potthast, Kevin, 80 Pourakbar, Sarah, 49 Pratiwadi, Ramya, 61 Purkayastha, Das, 40, 50 Puthawala, Iram, 82 Quirk, Stephanie Lauren, 66 Rab A.E., Minke, 8 Rafiq, Azhar, 17, 47 Raj, Ashok, 67
Rajasubramaniam, S., 84 Raman, Subha, 48 Rampersaud, Evadnie, 10 Ramphal, Areli, 62 Rash-Ellis, Diana, 73 Rathi, Nityam, 65 Reader, Steve, 73 Rees, David, 33 Rees, Megan, 25 Reisz, Colleen, 49 Ribeil, Jean-Antoine, 11 Riddick-Burden, Gaye, 14 Rivers, Angela, 31, 63 Rivlin, Kenneth, 25 Rizio, A., 68 Rizk, Sanaa, 14 Rocheleau, A., 84 Ross, Diana, 83 Rupff, Rebecca, 27 Russell, Kathryn, 27 Sangerman, Jose, 52 Saulsberry, Anjelica, 38 Schaffler, Mitchell, 78 Schlenz, Alyssa, 43 Schmidt, Manfred, 11 Schneeweiss, Sebastian, 24 Schutgens, Roger E.G., 8 Scrape, Scott, 82 Sebastian, Gracy, 6, 39 Shah, Nirmish, 40 Sheehan, Vivien A., 8, 10, 13, 37 Shelke, Anil, 52 Shiva, S., 59 Shrivas, S., 84 Silva, José Roberto Santos, 86 Silva, Raiotelma Lopes, 28 Silva, Silvone Santa Barbara, 79 Singer, Sylvia T., 36 Sinha, Cynthia, 83 Smith, Wally, 17, 20, 42, 47, 69 Snyder, Angie, 74 Sop, Daniel, 17, 20, 47, 69 Souza, Madlene Oliveira, 79 STERIO-SCD Investigators, 42 Sundaram, Revathy, 32, 53 Szczepanek, Steven M., 54 Takezaki, Mayuko, 52 Tamberou, Cheikh, 18 Tavakoli, Abbas, 86 Taylor VI, James G., 77 Telfer, Paul, 12

103 | P a g e
Thomas, Merin, 6, 39 Thompson, Alexis A., 11 Thota, P., 84 Tisdale, John F., 11 Tolorunju, Kayode, 89 Tolu, Seda, 35 Tonda, Margaret, 12 Trouillet Poujol, Valérie, 18 Tsao, David, 13 Tsitsikas, Dimitris A., 12 Ugo, Ugochi O., 5, 6, 35, 39 Uikey, R., 84 Ünal, Selma, 12 van Beers, Eduard J., 8 van Wijk, Richard, 8 Ventura, Lea, 63 Verma, AK, 84 Vichinsky, Elliot, 12 Vinces, Giacomo, 35 Walters, Mark C., 11 Wang, Y., 59
Ward, Lawrence, 14 Ware, Russell E., 12 Watt, A., 59 Weirick, Kimberly, 82 Weiss, Mitchell J., 10 Wells, Leigh, 72 Wen, Fayuan, 77 White, Jennell, 45 White, M., 68 Williams, Robert, 52 Witte, T., 84 Wong, David, 74 Woods, Gerald, 49, 62 Xavier, Aline Silva Gomes, 30 Xu, Hongyan, 72 Ying, Huang, 48 Zhang, Yankai, 10, 37 Zhao, Jun, 67 Zhou, Mei, 74 Zmitrovich, A., 59















![Curs-5-Mai 2015 - Ficat Si Pancreas -Final de Prezentat-recovered]](https://static.fdocuments.us/doc/165x107/563db833550346aa9a91791b/curs-5-mai-2015-ficat-si-pancreas-final-de-prezentat-recovered.jpg)



