
Journal of Solid State Chemistryiith.ac.in/~kanchana/publications/2016/87.pdf · Electronic...
9
Structural and thermoelectric properties of zintl-phase CaLiPn (Pn ¼ As, Sb, Bi) Anoop K. Chandran, Vijay Kumar Gudelli, P.C. Sreeparvathy, V. Kanchana n Department of Physics, Indian Institute of Technology Hyderabad, Sangareddy, Kandi 502285, Telangana, India article info Article history: Received 28 May 2016 Received in revised form 4 August 2016 Accepted 20 August 2016 Available online 24 August 2016 Keywords: Zintl-phase materials Density functional theory Electronic structure Thermoelectric properties abstract First-principles calculations were carried out to study the structural, mechanical, dynamical and trans- port properties of zintl phase materials CaLiPn (Pn ¼As, Sb and Bi). We have used two different ap- proaches to solve the system based on density functional theory. The plane wave pseudopotential ap- proach has been used to study the structural and dynamical properties whereas, full potential linear augment plane wave method is used to examine the electronic structure, mechanical and thermoelectric properties. The calculated ground-state properties agree quite well with experimental values. The computed electronic structure shows the investigated compounds to be direct band gap semiconductors. Further, we have calculated the thermoelectric properties of all the investigated compounds for both the carriers at various temperatures. We found a high thermopower for both the carriers, especially n-type doping to be more favourable, which enabled us to predict that CaLiPn might have promising applica- tions as a good thermoelectric material. Further, the phonon dispersion curves of the investigated compounds showed flat phonon modes and we also find lower optical and acoustic modes to cut each other at the lower frequency range, which further indicate the investigated compounds to possess reasonably low thermal conductivity. We have also analysed the low value of the thermal conductivity through the empirical relations and discussions are presented here. & 2016 Elsevier Inc. All rights reserved. 1. Introduction Thermoelectric (TE) materials are of current interest for a number of energy-related applications such as waste heat re- covery, terrestrial cooling and thermoelectric power generation [1,2]. Since TE materials have the property to convert thermal energy directly to electrical energy, improvement of their effi- ciency [3] can lead to wider applications in energy technology. The energy conversion efficiency of a TE material is characterised by its dimensionless quantity called figure of merit, σ κ = ZT S T / 2 , where σ κ ST , , , are electrical conductivity, the Seebeck coefficient, the absolute temperature, and the thermal conductivity, respectively. Thermal conductivity has both electronic κ e , and lattice κ l , con- tributions, i.e. κ κ κ = + e l . The contradicting nature of thermo- power, || S , and electrical conductivity, s, would suggest that achieving a high figure of merit will require a fine tuning of con- tributing parameters [4] such as effective mass m n and carrier concentration. Because of the conflicting nature of parameters, a compromise has to be made between high mobility and high ef- fective mass. Typically materials with small electro negativity differences possess high mobility and low effective mass and materials with narrow bands can offer low mobility and high ef- fective mass [5] because of the relationship between density of states and the whole dispersion relation in the momentum space. In addition to that, another conflict in parameters stems from ac- quiring low thermal conductivity. The difficulty of achieving low electronic thermal conductivity to increase ZT arises because of its proportionality to electrical conductivity given by Wiedemann- Franz law (κ σ = L T e , with Lorentz number π = L k e /3 B 2 2 2 ). Taking into consideration that figure of merit is also proportional to electrical conductivity, electronic thermal conductivity cannot be lowered without compromising on ZT for a fixed temperature. Never- theless, a material with low lattice thermal conductivity can be chosen to reduce total thermal conductivity since ‘s’ doesn't have a hold on ‘κ l ’ or vice versa. In effect, an efficient TE material must have a low lattice thermal conductivity as comparable to that of an amorphous glass but also have optimum electronic transport properties. Zintl phase compounds have complex structures which enable them to have low thermal conductivity [6–8]. Research over the years points to the direction that zintl phase materials offer desired characteristics of a good thermoelectric material. These materials are considered to be valence precise [9] and a majority of them form the requisite small band gap semi- conductors with complex structures. Zintl phase materials often Contents lists available at ScienceDirect journal homepage: www.elsevier.com/locate/jssc Journal of Solid State Chemistry http://dx.doi.org/10.1016/j.jssc.2016.08.030 0022-4596/& 2016 Elsevier Inc. All rights reserved. n Corresponding author. E-mail address: [email protected] (V. Kanchana). Journal of Solid State Chemistry 243 (2016) 198–206
Transcript of Journal of Solid State Chemistryiith.ac.in/~kanchana/publications/2016/87.pdf · Electronic...

Journal of Solid State Chemistry 243 (2016) 198–206
Contents lists available at ScienceDirect
Journal of Solid State Chemistry
http://d0022-45
n CorrE-m
journal homepage: www.elsevier.com/locate/jssc
Structural and thermoelectric properties of zintl-phase CaLiPn (Pn¼As,Sb, Bi)
Anoop K. Chandran, Vijay Kumar Gudelli, P.C. Sreeparvathy, V. Kanchana n
Department of Physics, Indian Institute of Technology Hyderabad, Sangareddy, Kandi 502285, Telangana, India
a r t i c l e i n f o
Article history:Received 28 May 2016Received in revised form4 August 2016Accepted 20 August 2016Available online 24 August 2016
Keywords:Zintl-phase materialsDensity functional theoryElectronic structureThermoelectric properties
x.doi.org/10.1016/j.jssc.2016.08.03096/& 2016 Elsevier Inc. All rights reserved.
esponding author.ail address: [email protected] (V. Kanchana
a b s t r a c t
First-principles calculations were carried out to study the structural, mechanical, dynamical and trans-port properties of zintl phase materials CaLiPn (Pn¼As, Sb and Bi). We have used two different ap-proaches to solve the system based on density functional theory. The plane wave pseudopotential ap-proach has been used to study the structural and dynamical properties whereas, full potential linearaugment plane wave method is used to examine the electronic structure, mechanical and thermoelectricproperties. The calculated ground-state properties agree quite well with experimental values. Thecomputed electronic structure shows the investigated compounds to be direct band gap semiconductors.Further, we have calculated the thermoelectric properties of all the investigated compounds for both thecarriers at various temperatures. We found a high thermopower for both the carriers, especially n-typedoping to be more favourable, which enabled us to predict that CaLiPn might have promising applica-tions as a good thermoelectric material. Further, the phonon dispersion curves of the investigatedcompounds showed flat phonon modes and we also find lower optical and acoustic modes to cut eachother at the lower frequency range, which further indicate the investigated compounds to possessreasonably low thermal conductivity. We have also analysed the low value of the thermal conductivitythrough the empirical relations and discussions are presented here.
& 2016 Elsevier Inc. All rights reserved.
1. Introduction
Thermoelectric (TE) materials are of current interest for anumber of energy-related applications such as waste heat re-covery, terrestrial cooling and thermoelectric power generation[1,2]. Since TE materials have the property to convert thermalenergy directly to electrical energy, improvement of their effi-ciency [3] can lead to wider applications in energy technology. Theenergy conversion efficiency of a TE material is characterised by itsdimensionless quantity called figure of merit, σ κ=ZT S T/2 , whereσ κS T, , , are electrical conductivity, the Seebeck coefficient, theabsolute temperature, and the thermal conductivity, respectively.Thermal conductivity has both electronic κe, and lattice κl, con-tributions, i.e. κ κ κ= +e l. The contradicting nature of thermo-power, | |S , and electrical conductivity, s, would suggest thatachieving a high figure of merit will require a fine tuning of con-tributing parameters [4] such as effective mass mn and carrierconcentration. Because of the conflicting nature of parameters, acompromise has to be made between high mobility and high ef-fective mass. Typically materials with small electro negativity
).
differences possess high mobility and low effective mass andmaterials with narrow bands can offer low mobility and high ef-fective mass [5] because of the relationship between density ofstates and the whole dispersion relation in the momentum space.In addition to that, another conflict in parameters stems from ac-quiring low thermal conductivity. The difficulty of achieving lowelectronic thermal conductivity to increase ZT arises because of itsproportionality to electrical conductivity given by Wiedemann-Franz law (κ σ= L Te , with Lorentz number π=L k e/3B
2 2 2). Taking intoconsideration that figure of merit is also proportional to electricalconductivity, electronic thermal conductivity cannot be loweredwithout compromising on ZT for a fixed temperature. Never-theless, a material with low lattice thermal conductivity can bechosen to reduce total thermal conductivity since ‘s’ doesn't have ahold on ‘κl’ or vice versa. In effect, an efficient TE material musthave a low lattice thermal conductivity as comparable to that of anamorphous glass but also have optimum electronic transportproperties. Zintl phase compounds have complex structures whichenable them to have low thermal conductivity [6–8].
Research over the years points to the direction that zintl phasematerials offer desired characteristics of a good thermoelectricmaterial. These materials are considered to be valence precise [9]and a majority of them form the requisite small band gap semi-conductors with complex structures. Zintl phase materials often

Fig. 1. Crystal structure of CaLiPn (Pn¼As, Sb, Bi).
Table 1Calculated structural parameters of CaLiPn (Pn¼AS, Sb, Bi) along with the experi-mental [16] parameters.
Compounds CaLiAs CaLiSb CaLiBi
Present Exp Present Exp Present Exp
a (in Å) 7.29 7.23 7.69 7.64 7.83 7.73b (in Å) 4.33 4.31 4.66 4.63 4.75 4.71c (in Å) 7.95 7.90 8.31 8.29 8.50 8.42
A.K. Chandran et al. / Journal of Solid State Chemistry 243 (2016) 198–206 199
has alternating layers wherein one favouring conduction and othersuitable for high thermopower. Materials of this class often con-tain cationic sites that allow for the addition of disorder scatteringand the change in carrier concentration which results in tuning ofelectronic properties [13]. These characteristics of zintl materialsallow control of the carrier concentration through precise dopingwithout disrupting the carrier mobility. Since ZT is sensitive tocarrier concentration, this tune-ability attribute of zintl phases canbe used to improve the figure of merit. Over the years, we haveknown materials with >ZT 1 especially from zintl phases [14] andthey offer favourable thermal conductivity and optimal charge-transport at temperatures near 1200 K because of their complexcrystal structures and electronic structure [15]. Ca5 Al2Sb6, Yb14 AlSb11, and Sr3 Al Sb3 [6–8] are a few examples of such compounds.Ca5 Al2 Sb6 has a low lattice thermal conductivity (0.6 W/mK at850 K) and a ZT which is more than 0.6 at 1000 K. An optimallydoped Sr3 Al Sb3 and Yb14Al Sb11 can achieve a ZT of 1.0 at hightemperatures such as 1000–1250 K, and they have lattice thermalconductivity less than 0.75 W/mK at 1000–1300 K respectively.Recently studied strontium based pnictogen compound SrLiAs hasbeen demonstrated to have characteristics of a reasonable TEmaterial [17]. This further motivate us to search for other possiblenew zintl phase materials for better TE candidates. In the presentstudy, we are interested in investigating the calcium based zintlphase materials of CaLiPn (Pn¼As, Sb, and Bi) for a possible goodTE properties.
The present work is a theoretical study of CaLiPn (Pn¼As, Sb,Bi) to determine its TE properties. The paper is organised as fol-lows Section 2 describes the methodology used for the calcula-tions, and Section 3 presents the results and discussions andconclusions are given in Section 4.
Table 2Elastic constant (Cij), Young modulus, Bulk modilus (in GPa), sound velocities (υl, υt,υm, km/sec) and Debye Temperature (ΘD, K) of CaLiPn.
Elastic constant CaLiAs CaLiSb CaLiBi
C11 100.50 81.69 64.93C22 104.95 89.25 72.64C33 79.23 73.59 58.75C44 45.78 37.46 31.21C55 35.37 31.39 24.02C66 38.73 33.11 24.71C12 16.25 17.15 12.44C13 17.39 17.09 13.11C23 25.11 21.55 18.35Young modulus 89.88 77.09 61.07Bulk modulus 44.43 39.44 31.45υLongitudinal 5.57 4.81 3.59υTransverse 3.48 2.97 3.16υMean 3.83 3.27 2.43Debye temperature 411.2 331.1 242.6
2. Methodology
All the total energy calculations based on first principle densityfunctional theory (DFT) were performed using pseudopotentialmethod as implemented in the Plane wave self-consistent field(Pwscf) programme [18] and full-potential linear augmented planewave (FP-LAPW) method as implemented in the WIEN2k [19]. ThePwscf method is used to perform structural optimisation, whereasFP-LAPW method is used to study the electronic, mechanical andtransport properties. The total energies are obtained by solving theKohn-Sham equation self consistently within the GeneralisedGradient Approximation (GGA) of Perdew-Burke-Ernzerhof (PBE)potential [20]. A plane wave kinetic energy cut-off of 50 Ry is usedand the first Brillouin zone is sampled according to the Mon-khorst-Pack scheme [21] by means of a × ×8 8 8 k-mesh in orderto ensure that the calculations are well converged. Since the tra-ditional functionals of local density approximation and generalisedgradient approximation methods were underestimating the bandgap, we adopted Tran-Blaha modified Becke-Johnson potential(TB-mBJ) [22,23] on top of GGA calculations to attain proper bandgap for the investigated compounds. TB-mBJ is found to be quitesuccessful in reproducing the experimental band gaps as com-pared to standard GGA [22,24–27]. Considering the presence ofheavy elements, we have included spin-orbit coupling in our cal-culations. All the calculations were performed with the optimisedlattice parameters with an energy convergence criterion of10�6 Ry per formula unit. The carrier concentration (p for holesand n for electrons) dependent transport properties like thermo-power (S), electrical conductivity scaled by relaxation time σ τ( )/and power factor σ τ( )S /2 were calculated using the BoltzTraP [28]code. This fraction of code is an implementation of semiclassicalBoltzmann transport equation using constant relaxation-time
approximation (CSTA) and rigid band approximation (RBA). Thedetailed explanation about the CSTA is given in Ref. [29,30,31,32]and the references cited therein. It is evident that CSTA has beenquite successful in the past in predicting the thermoelectricproperties of many materials [32,33–36,54]. According to the RBAapproximation, doping a system does not alter its band structurebut varies only the chemical potential, and it is a good approx-imation for doped semiconductors to calculate the transportproperties theoretically when doping level is not very high [37–41]. However certain types of dopant can drastically modify thenature of electronic structure near the gap giving rise to resonant

Fig. 2. Calculated band structure of (a) CaLiAs (b) CaLiSb and (c) CaLiBi.
A.K. Chandran et al. / Journal of Solid State Chemistry 243 (2016) 198–206200
states in which case the RBA can fail [42].
3. Results and discussions
3.1. Structural and mechanical properties
The investigated compounds CaLiPn (Pn¼As, Sb, Bi) crystallise
in orthorhombic structure with space group Pnma(62), and thecrystal structure is given in Fig. 1. In Table 1 we have presented theoptimised ground state properties along with available experi-mental reports. From Table 1, it is quite evident that the calculatedoptimised parameters are in good agreement with the experi-mental reports. Elastic properties are fundamental for any crys-talline material to describe its stiffness against applied strain. Thiswill also indicate the mechanical stability of the investigated
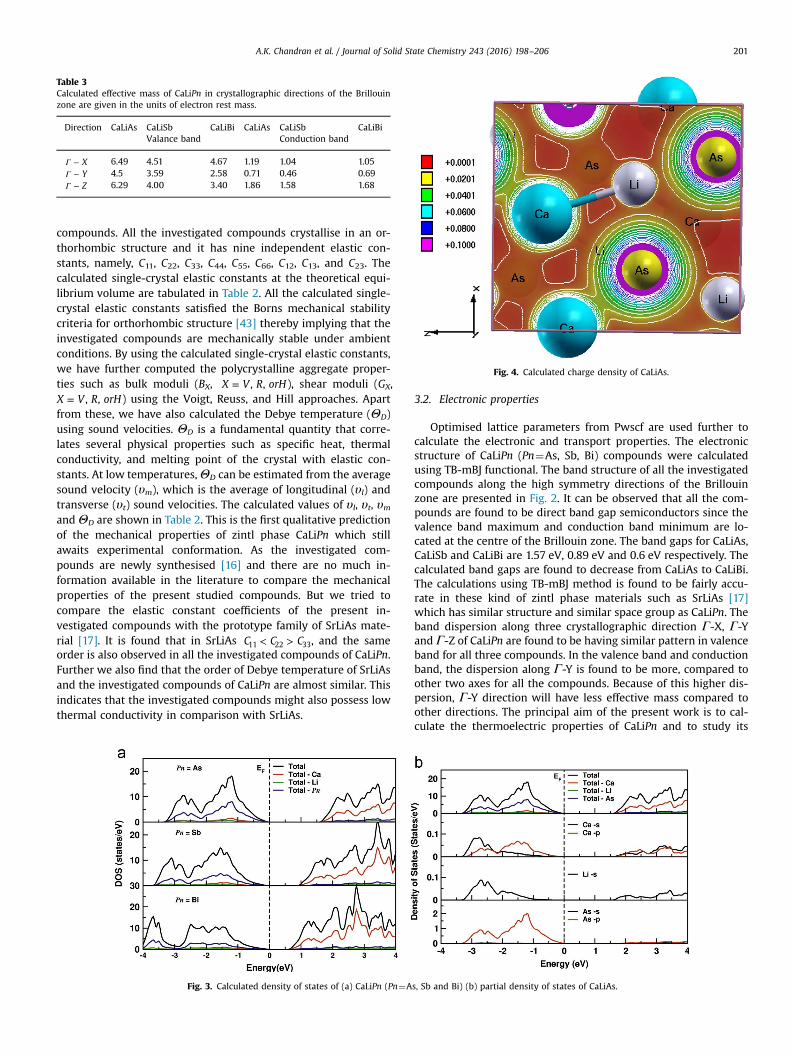
Table 3Calculated effective mass of CaLiPn in crystallographic directions of the Brillouinzone are given in the units of electron rest mass.
Direction CaLiAs CaLiSb CaLiBi CaLiAs CaLiSb CaLiBiValance band Conduction band
Γ − X 6.49 4.51 4.67 1.19 1.04 1.05Γ − Y 4.5 3.59 2.58 0.71 0.46 0.69Γ − Z 6.29 4.00 3.40 1.86 1.58 1.68
Fig. 4. Calculated charge density of CaLiAs.
A.K. Chandran et al. / Journal of Solid State Chemistry 243 (2016) 198–206 201
compounds. All the investigated compounds crystallise in an or-thorhombic structure and it has nine independent elastic con-stants, namely, C11, C22, C33, C44, C55, C66, C12, C13, and C23. Thecalculated single-crystal elastic constants at the theoretical equi-librium volume are tabulated in Table 2. All the calculated single-crystal elastic constants satisfied the Borns mechanical stabilitycriteria for orthorhombic structure [43] thereby implying that theinvestigated compounds are mechanically stable under ambientconditions. By using the calculated single-crystal elastic constants,we have further computed the polycrystalline aggregate proper-ties such as bulk moduli (BX, =X V R orH, , ), shear moduli (GX,
=X V R orH, , ) using the Voigt, Reuss, and Hill approaches. Apartfrom these, we have also calculated the Debye temperature (ΘD)using sound velocities. ΘD is a fundamental quantity that corre-lates several physical properties such as specific heat, thermalconductivity, and melting point of the crystal with elastic con-stants. At low temperatures,ΘD can be estimated from the averagesound velocity (υm), which is the average of longitudinal (υl) andtransverse (υt) sound velocities. The calculated values of υl, υt, υmandΘD are shown in Table 2. This is the first qualitative predictionof the mechanical properties of zintl phase CaLiPn which stillawaits experimental conformation. As the investigated com-pounds are newly synthesised [16] and there are no much in-formation available in the literature to compare the mechanicalproperties of the present studied compounds. But we tried tocompare the elastic constant coefficients of the present in-vestigated compounds with the prototype family of SrLiAs mate-rial [17]. It is found that in SrLiAs < >C C C11 22 33, and the sameorder is also observed in all the investigated compounds of CaLiPn.Further we also find that the order of Debye temperature of SrLiAsand the investigated compounds of CaLiPn are almost similar. Thisindicates that the investigated compounds might also possess lowthermal conductivity in comparison with SrLiAs.
Fig. 3. Calculated density of states of (a) CaLiPn (Pn¼A
3.2. Electronic properties
Optimised lattice parameters from Pwscf are used further tocalculate the electronic and transport properties. The electronicstructure of CaLiPn (Pn¼As, Sb, Bi) compounds were calculatedusing TB-mBJ functional. The band structure of all the investigatedcompounds along the high symmetry directions of the Brillouinzone are presented in Fig. 2. It can be observed that all the com-pounds are found to be direct band gap semiconductors since thevalence band maximum and conduction band minimum are lo-cated at the centre of the Brillouin zone. The band gaps for CaLiAs,CaLiSb and CaLiBi are 1.57 eV, 0.89 eV and 0.6 eV respectively. Thecalculated band gaps are found to decrease from CaLiAs to CaLiBi.The calculations using TB-mBJ method is found to be fairly accu-rate in these kind of zintl phase materials such as SrLiAs [17]which has similar structure and similar space group as CaLiPn. Theband dispersion along three crystallographic direction Γ-X, Γ-Yand Γ-Z of CaLiPn are found to be having similar pattern in valenceband for all three compounds. In the valence band and conductionband, the dispersion along Γ-Y is found to be more, compared toother two axes for all the compounds. Because of this higher dis-persion, Γ-Y direction will have less effective mass compared toother directions. The principal aim of the present work is to cal-culate the thermoelectric properties of CaLiPn and to study its
s, Sb and Bi) (b) partial density of states of CaLiAs.

Fig. 5. Calculated thermoelectric properties of CaLiAs for both electron and hole concentration at 1019 (solid lines with filled symbols) and 1020 cm�3 (dotted lines with opensymbols) as function of temperature.
A.K. Chandran et al. / Journal of Solid State Chemistry 243 (2016) 198–206202
variation with carrier concentration. It is necessary to estimate theeffective masses of the carriers in various electron and holepockets at the band edges. We have calculated the mean effectivemass of the carriers at the conduction and valence band edges byfitting the energy of the respective bands to a quadratic poly-
nomial in the reciprocal lattice vector→k . The calculated effective
masses for CaLiPn in crystallographic directions of the Brillouinzone are tabulated in Table 3. It can be observed from the tablethat for all three compounds, the calculated effective masses in theΓ-Y direction for the valance bands are * =m 4.5 my e, * =m 3.59 my e,and * =m 2.58 my e for CaLiAs, CaLiSb and CaLiBi respectively. Thesevalues are low when compared with other directions which isexpected because of the high dispersion of the bands along Γ − Yin all the compounds. The values of effective masses of conductionbands also follow the same trend as that of the valence band. Theflat bands are observed along −R S high symmetric direction.
Further analysis of the electronic properties requires the totaland partial density of states of these compounds, and the same isrepresented in Fig. 3. In the valence band anion Pn states aredominating and in the conduction band Ca-states are dominatingfor all the three compounds. From CaLiAs to CaLiBi, the contribu-tion of Li-s states are found to be increased near the Fermi level.The heavy band found just below the valence band maximumarises from the Pn-p states, and the bands lying lower to this arecontributed mainly by Ca-p and Li-s states. Compared to valenceband, more hybridised Ca states are observed in the conductionband. Further we have also calculated the charge density of CaLiAsto analyse the bonding nature of the investigated compounds andthe same is presented in Fig. 4. From this figure it is evident thatthere exists covalent bonding between Ca-As. In general the weak
electro-negativity of ‘Li’ atom will form the ionic bond in most ofthe cases [10–12], but in the present case it is making a covalentbond as similar to SrLiAs [17]. The covalent bond between Li-Asreduces the mass of the carriers at the band edge resulting inlighter carriers. The heavy bands usually contribute to high ther-mopower since it is proportional to effective mass (mn) [13], whilethe lighter bands donate to high mobility (mobility, μ τ= *e m/ ,where e is the charge). A favourable combination of the two maylead to an excellent TE performance in materials [5]. For CaLiAs,the variation of density of states near Fermi level is almost similarfor valence band and conduction band, whereas in the case ofother two compounds the change in density of states near Fermilevel is more in conduction band than in valence band. This be-haviour indicate that for CaLiAs, the TE properties for holes andelectrons might be similar, whereas for the other two, the electrondoping will be more favourable.
3.3. Thermoelectric properties
In this section we have presented the thermopower (in μ −VK 1)and electrical conductivity scaled by relaxation time (inΩ− − −m s1 1 1), as a function of carrier concentration and temperature,using the Boltzmann transport equation approach as implementedin BoltzTraP [28] code. As the investigated compounds crystallisein orthorhombic structure we have calculated TE properties alongthree crystallographic directions. The variation of thermoelectricproperties of the investigated compounds as a function of tem-perature at concentrations around 1019 and 1020 cm�3 for bothelectrons and holes, along different crystallographic directions arereported in Figs. 5–7. The thermopower of all the investigated

Fig. 6. Calculated thermoelectric properties of CaLiSb for both electron and hole concentration at 1019 (solid lines with filled symbols) and 1020 cm�3 (dotted lines with opensymbols) as function of temperature.
A.K. Chandran et al. / Journal of Solid State Chemistry 243 (2016) 198–206 203
compounds are found to be higher for the electrons compared toholes throughout the studied temperature range. The difference inthe thermopower between electrons and holes concentration is ofthe order of 100–200 μV/K for all the investigated compounds attemperatures higher than 200 K. The higher values in the case ofelectrons as carriers might be due to the increased number ofminima in the conduction band region compared to the valenceband [48]. In the case of thermopower, almost isotropic behaviouris observed along different crystallographic directions. Also ther-mopower is found to decrease as we move from As to Sb, which isdue to the decreasing band gaps as we move from As to Sb (see insection 3.2). Among the three investigated compounds, n-typeCaLiAs has more thermopower than the other two compounds. Athigh temperature above 800 K, for CaLiBi we have observed bi-polar conductivity (containing both free electrons and holes)which may lead to low, compensated thermopower and thereforelow ZT, which restrict the performance of CaLiBi at hightemperature.
Almost a similar type of variation is observed in the case ofelectrical conductivity scaled by relaxation time in all the com-pounds. All the investigated compounds have almost similar valueof σ τ/ at all the studied temperatures. Higher value of electricalconductivity is noticed in the case of electrons compared to holes,which is very similar to that of the thermopower of the in-vestigated compounds. Further, we find a considerable anisotropicnature in the case of electrons along the b-axis compared to the aand c-axes. This might be due to the lower value of the effectivemass along the Γ − Y direction compared to other two directionsin the case of conduction band. The strong dispersion along theΓ − Y direction for all three compounds induced by the Li-Pn
covalent bonding result in a low reduced mass and therefore in-creased conductivity along that direction. In the case of holes al-most isotropic behaviour in σ τ/ is observed along different crys-tallographic directions. For further analysis we have plotted thepower-factor as a function of temperature. It is possible that thepower-factor for electron doping will be higher as compared tohole doping, because of the higher value of S and σ τ/ in the case ofelectrons and it is further noticed that b-axis power-factor will behigher due to the higher value of σ τ/ along this direction providedthe relaxation time for electrons and holes remains the same. Inthe case of hole doping, the power-factor values are found to bealmost isotropic along different crystallographic direction. Overallwe can say that the electron doping is more favourable for TEproperties for all the investigated compounds.
In order to analyse how much better is the TE properties of theinvestigated compounds we have compared them with SrLiAs. Forthis, we have calculated S, σ τ/ , and σ τS /2 as a function of bothelectron and hole carrier concentration at 1000 K and the same ispresented in Fig. 8, along with SrLiAs, data has been extractedfrom Fig. 5 of Ref. [17]. As mentioned earlier, electron doping ismore favourable compared to holes in CaLiAs, and a similar si-tuation is also seen in the case of SrLiAs. From the Fig. 8, it is quiteevident that the thermopower of CaLiAs is found to be slightlyhigher compared to SrLiAs. Almost similar value of the σ τ/ is ob-served in both the compounds. Further slightly higher value of thethermopower in the case of CaLiAs resulted in a higher value of thepower-factor compared to SrLiAs. To conclude we find almost si-milar thermoelectric properties in both CaLiAs and SrLiAs, butmore favourable case is observed with CaLiAs.

Fig. 7. Calculated thermoelectric properties of CaLiBi for both electron and hole concentration at 1019 (solid lines with filled symbols) and 1020 cm�3 (dotted lines with opensymbols) as function of temperature.
Fig. 8. Calculated thermoelectric properties of (a) CaLiAs (b) SrLiAs (Data has been extracted from Fig. 5 of Ref. [17]) as function of both electron (solid lines) and hole (dottedlines) concentration at 1000 K.
A.K. Chandran et al. / Journal of Solid State Chemistry 243 (2016) 198–206204

Fig. 9. Calculated phonon dispersion of (a) CaLiAs (b) CaLiSb and (c) CaLiBi.
Table 4Calculated minimum thermal conductivity of the investigated compounds of CaLiPnusing the Clarke and Cahill models.
κmin in W/m K
Clarke model Cahill model
CaLiAs 0.85 0.92CaLiSb 0.65 0.71CaLiBi 0.46 0.50
A.K. Chandran et al. / Journal of Solid State Chemistry 243 (2016) 198–206 205
3.4. Lattice dynamics
Further we have studied the phonon dispersion of CaLiPn atambient using DFPT [44]. The unit cell of CaLiPn contains 12 atomsand hence it has 36 phonon modes for each wave-vector, out ofwhich three are acoustic and remaining modes are optical modes.The calculated phonon dispersion relation along with the phonondensity of states of all three compounds are shown in Fig. 9. In thecase of CaLiAs, low-frequency optical branches intersect the acousticbranches starting below −80 cm 1, below −60 cm 1 for CaLiSb andbelow −44 cm 1 for CaLiBi. From this observation it can be inferredthat the phonon-phonon scattering is strong in all three compoundsand comparatively more in CaLiSb and CaLiBi than CaLiAs. Because ofthese lattice interactions, there is a coupling between differentphonons which will limit the value of mean free path and therebylimiting thermal conductivity at temperatures relevant to thermo-electrics (mean free path of the particle =l T1/ , where T is thetemperature) [40,47]. The feature of strong phonon-phonon scatter-ing is also seen in other zintl phase materials [45,46].
Further we tried to estimate the order of the thermal con-ductivity of the investigated compounds. For this purpose we have
used analytical relation leading to the minimum thermal con-ductivity given by Clark [49,50] and Cahill [51] model. According tothese model the minimum thermal conductivity was given by,
( )κ ρ
κ
′ = ¯
′ = +
−k M E
n v v
Clarke s model: 0. 87
Cahill s model: 2
min b a
mink
l t
2/3
2 . 48B
12
16
23
Here, = ( )⎡⎣⎢
⎤⎦⎥Ma
Mm N. A
is the average mass per atom, E is the Youngs
modulus, ρ is the density, M is the molar mass, m is the totalnumber of atoms per formula, kB is Boltzmanns constant, NA isAvogadros number, n is the density of number of atoms per vo-lume, and vl and vt are the average longitudinal and transversesound velocities, respectively. These two methods are appropriatein predicting the minimum thermal conductivity for various ma-terials [17,52,53]. In the present case we have also tried to predictthe minimum thermal conductivity for all the investigated com-pounds and the same is presented in Table 4. To cross check withthe calculated values, we also repeated the calculation of κmin forSrLiAs using both the methods. It has been shown that κmin is 0.65and 0.70 W/m K for both Clarke and Cahill model, which is almostsimilar to the one presented in Ref. [17]. From the Table 4, it isquite evident that the κmin of the investigated compounds arefound to be below unity. This confirms that investigated zintlphase Ca-based pnictides might show a promising TE perfor-mance. Except CaLiAs, other compounds are found to posses lesserκmin compared to SrLiAs [17]. The calculated value of κmin for theinvestigated compounds are also found to be in the same order ofother Ca-based zintl phase compound Ca5Al2Sb6 [6]. This furtherconfirms that the investigated Ca-based pnictide compounds aregood TE candidates in comparison with the SrLiAs. We look for-ward for the experimentalist to confirm the TE nature of Ca-basedpnictides.

A.K. Chandran et al. / Journal of Solid State Chemistry 243 (2016) 198–206206
4. Conclusion
Zintl phase materials of CaLiPn (Pn¼As, Sb and Bi) are in-vestigated for the structural, mechanical, dynamical and transportproperties using first-principles calculations. Two distinct densityfunctional methods are used to investigate above properties. Theplane wave pseudopotential approach was used to study thestructural and dynamical properties. The full potential linearaugmented plane wave method has been used to study the elec-tronic structure, mechanical and thermoelectric properties. Thecomputed structural properties are in good agreement withavailable experimental data. The calculated electronic structureshows the investigated compounds to be direct band gap semi-conductors. Further analysis of the electronic structure indicatesthe investigated compounds to possess mixed heavy and lightbands, which is an indication to have good thermoelectric prop-erties [54]. We have also calculated the thermoelectric propertiesof all the investigated compounds for both carrier concentrationsat various temperatures. We found a high thermopower for boththe concentrations, especially n-type doping is more favourable,which enabled us to predict that CaLiPn might have promisingapplications as a good thermoelectric material. Among the in-vestigated compounds we find that CaLiBi has shown a bipolarconductivity at high temperatures which might be due to thelower band gap of this material compared to the rest of thecompounds. Further the phonon dispersion curves of the in-vestigated compounds reveals flat phonon bands, which will ab-sorb more heat thereby resulting in low thermal conductivity. Wehave also observed the crossing of lower optical and acousticbranches which also further indicate that the investigated com-pounds may have reasonably low thermal conductivities. The lowvalue of the thermal conductivity is also confirmed through theempirical relation of Clarke and Cahill models, which has shownκmin to be below unity. With these findings we predict that theinvestigated compounds to be good thermoelectric materialswhich need to be examined further by the experimental study.
Acknowledgement
All the authors would like to acknowledge IIT-Hyderabad forproviding computational facility. V.K.G. and S.P.C would like tothank MHRD for the fellowship.
References
[1] A.F. Ioffe, Semiconductor Thermoelements and Thermoelectric Cooling,Translation by A. Gelbtuch, H.J. Goldsmid, Infosearch, London, 1957.
[2] C. Wood, Rep. Prog. Phys. 51 (1988) 459.[3] J.H. Yang, T. Caillat, MRS Bull. 31 (2006) 224.[4] S.M. Kauzlarich, S.R. Brown, G.J. Snyder, Dalton Trans. (2007) 2099–2107.[5] A. Shakouri, Annu. Rev. Mater. Res 41 (2011) 399–431.[6] E.S. Toberer, A. Zevalkink, N. Crisosto, G.J. Snyder, Adv. Funct. Mater. 20 (2010)
4375.
[7] E.S. Toberer, C.A. Cox, S.R. Brown, T. Ikeda, A.F. May, S.M. Kauzlarich, G.J. Snyder, Adv. Funct. Mater. 18 (2008) 2795.
[8] A. Zevalkink, G. Pomrehn, Y. Takagiwa, J. Swallow, G.J. Snyder, ChemSusChem10 (2013) 2316.
[9] S.M Kauzlarich, Zintl Compounds - In Encyclopedia of Organic Chemistry, 2nded. R.B. King, Ed., John Wiley and Sons, NewYork, 2005.
[10] X.L. Wang, Z. Zeng, H. Ahn, G.X. Wang, Appl. Phys. Lett. 95 (2009) 183103.[11] T.P. Kaloni, Y.C. Cheng, M.U. Kahaly, U. Schwingenschlogl, Chem. Phys. Lett. 534
(2012) 29.[12] T.P. Kaloni, A.V. Balatsky, U. Schwingenschlogl, EPL 104 (2013) 47013.[13] M. Cutler, J.F. Leavy, R.L. Fitzpatrick, Electronic transport in semimetallic cer-
ium sulfide, Phys. Rev. 133 (1964) 1143.[14] G.J. Snyder, E.S. Toberer, Nat. Mater. 7 (2008) 105.[15] E.S. Toberer, A.F. May, G.J. Snyder, Chem. Mater. 22 (2010) 624.[16] M.C. Schafer, N. Suen, S. Bobev, Dalton Trans. 43 (2014) 16889.[17] L.B. Guo, Y.X. Wang, Y.L. Yan, G. Yang, J.M. Yang, Z.Z. Feng, J. Appl. Phys. 116
(2014) 033705.[18] S. Baroni, S.D Gironcoli, A. dal Corso, et al. ⟨http://www.pwscf.org⟩, 2008.[19] P. Blaha, K. Schwarz, G.K.H Madsen, D. Kvasnicka, Luitz J WIEN2k, An aug-
mented plane waveþ local orbitals program for calculating crystal properties,Karlheinz Schwarz, Techn. Universität Wien, Austria. ⟨http://www.wien2k.at/⟩,2001.
[20] J.P. Perdew, K. Burke, M. Ernzerhof, Phys. Rev. Lett. 77 (1996) 3865–3868.[21] H.J. Monkhorst, J.D. Pack, Phys. Rev. B 13 (1979) 5188–5192.[22] F. Tran, P. Blaha, Phys. Rev. Lett. 102 (2009), 226401-1-4.[23] A.D. Becke, E.R. Johnson, J. Chem. Phys. 124 (2006), 221101-1-4.[24] D. Koller, F. Tran, P. Blaha, Phys. Rev. B 83 (2011), 195134-1-10.[25] D. Koller, F. Tran, P. Blaha, Phys. Rev. B 85 (2012), 155109-1-8.[26] H. Dixit, R. Saniz, S. Cottenier, D. Lamoen, B. Partoens, J. Phys. Condens. Matter
24 (2012), 205503-1-9.[27] H. Jiang, J. Chem. Phys. 138 (2013), 134115-1-7.[28] G.K.H. Madsen, D.J. Singh, Comput. Phys. Commun. 67 (2005) 175.[29] D.J. Singh, Func. Mater. Lett. 3 (2010) 223.[30] K.P. Ong, D.J. Singh, P. Wu, Phys. Rev. B 83 (2011) 115110.[31] D. Parker, D.J. Singh, Phys. Rev. B 85 (2012), 125209-1-7.[32] K.P. Ong, D.J. Singh, P. Wu, Phys. Rev. B 83 (2011), 115110-1-5.[33] J. David, Mazin I.I. Singh, Phys. Rev. B 56 (1997) R1650–R1653.[34] D. Parker, D.J. Singh, Phys. Rev. B 82 (2010), 035204-1-5.[35] G.K.H. Madsen, K. Schwarz, P. Blaha, D.J. Singh, Phys. Rev. B 68 (2003) 125212.[36] L. Zhang, M. Du, D.J. Singh, Phys. Rev. B 81 (2010), 075117-1-8.[37] L. Jodin, J. Tobola, P. Pécheur, H. Scherrer, S. Kaprzyk, Phys. Rev. B 70 (2004),
184207-1-11.[38] L. Chaput, P. Pécheur, J. Tobola, H. Scherrer, Phys. Rev. B 72 (2005), 085126-1-
11.[39] D.I. Bilc, S.D. Mahanti, M.G. Kanatzidis, Phys. Rev. B 74 (2006), 125202-1-12.[40] J.M. Ziman, Electrons and Phonons: The Theory of Transport Phenomena in
Solids, Oxford University Press, Oxford, 1960.[41] B.R. Nag, Electron Transport in Compound Semiconductors, Springer-Verlag,
Berlin, 1980.[42] M.S. Lee, S.D. Mahanti, Phys. Rev. B, 85, 165149.[43] M. Born, K. Huang, Dynamical Theory of Crystal Lattices, Oxford University
Press, Oxford, U.K, 1988.[44] X. Gonze, Phys. Rev. B 55 (1997) 10337–10354.[45] V.K. Gudelli, V. Kanchana, G. Vaitheeswaran, D.J. Singh, A. Svane, N.
E. Christensen, S.D. Mahanti, Phy. Rev. B 92 (2015) 045206.[46] L. Li, J. Sui, Y. Pei, C. Barreteau, D. Berardan, N. Dragoe, W. Cai, J. He, L.D. Zhao,
Energy Environ. Sci. 5 (2012) 8543.[47] Charles. Kittel, Introduction to Solid State Physics, 6th ed., John Wiley & Sons,
Inc., New York, 1986.[48] J.R. Sootsman, D.Y. Chung, M.G. Kanatzidis, Angew. Chem. Int. Ed. 48 (2009)
8616–8639.[49] D.R. Clarke, Surf. Coat. Technol. 163 (2003) 67.[50] D.R. Clarke, C.G. Levi, Annu. Rev. Mater. Res. 33 (2003) 383.[51] D.G. Cahill, S.K. Watson, R.O. Pohl, Phys. Rev. B 46 (1992) 6131.[52] L.D. Zhao, S.H. Lo, Y. Zhang, H. Sun, G. Tan, C. Uher, C. Wolverton, V.P. Dravid,
M.G. Kanatzidis, Nature 508 (2014) 373.[53] J. Feng, et al., Acta Mater. 59 (2011) 17421760.[54] D. Parker, D.J. Singh, Phys. Rev. B 85 (2012) 125209.


















