
Journal of Materials Chemistry A - PKU
8
A high-pressure induced stable phase of Li 2 MnSiO 4 as an effective poly-anion cathode material from simulations† Shuo Wang, a Junyi Liu, a Yu Qie, a Sheng Gong, b Cunzhi Zhang, a Qiang Sun * acd and Puru Jena e Search for novel cathode materials is of current interest. Among them, Li 2 MnSiO 4 shows promise as a cathode material in the poly-anion family due to its structural diversity, abundance, low cost, and high theoretical capacity (330 mA h g 1 ). However, it suffers from low electronic and ionic conductivity, limited reversible capacity, and poor cycling performance. To overcome these deficiencies, using a global structure search at high pressure, we find a new phase with a group symmetry of Cc, which is the ground-state at a pressure of 50 Gpa, and remains stable when the pressure is released. This new phase exhibits many attractive features such as a high energy density of 612 Wh kg 1 with a reversible capacity of 170 mA h g 1 , a high average discharge voltage of 3.6 V, improved electronic conductivity, coexistence of anionic and cationic redox, and superior ionic conductivity due to unique diffusion channels with reduced steric hindrance and coulombic repulsion. All these features endow the new phase of Li 2 MnSiO 4 with potential for use as a high performance cathode material. 1. Introduction Driven by the need for high-performance Li-ion batteries, extensive studies have been carried out on several classes of cathode materials during the past three decades, including chalcogenides, 1 layered oxides, 2,3 spinel oxides, 4 olivine phos- phates, 5 silicates, 6 tavorites, 7 etc. However, transition metal (TM) layered oxides remain dominant in the Li-ion battery industry due to their high voltage, stable cycling performance and mature synthesis technology. 8 However, the intrinsic instability of layered TM oxides 9 during deep delithiation makes it difficult to achieve a high reversible capacity in the derived LiMO 2 materials (M ¼ single metal or a combination of Ni, Mn, Co, Fe, Al etc.). Therefore, it is highly desirable to explore new cathode materials with enhanced stability and improved energy density. Recently, the poly-anion family has attracted extensive attention, showing great structural diversity due to the various structural geometries composed of poly-anion units (XO 4 ) n+ and their derivatives (X m O 3m+1 ) n (X ¼ P, S, As, Mo, Si etc.). 10 These materials display great potential not only as superionic conductors with high ionic conductivity (such as b-, g-, or LISICON-type solid electrolytes 11 ), but also as cathode materials with high voltage and cycling stability (such as olivine phos- phate LiFeO 4 (ref. 5) and silicates Li 2 (Fe/Co/Mn)SiO 4 (ref. 12)). This is due to the strong covalent bonding between poly-anion units and MO x polyhedra (M ¼ transition metal) which enables the structural matrix to accommodate a wide range of cations for good ionic conductivity and to endure the defor- mation induced by Li intercalation or deintercalation. 13 Among the studied systems, Li 2 MSiO 4 (M ¼ transition metal) is a promising cathode material with a high theoretical capacity 14 (>320 mA h g 1 ). Although the M ¼ Fe system is mostly explored, the redox of Fe 2+ /Fe 3+ limits the stable cycling capacity to 140 mA h g 1 which is only half of the theoretical value. Besides, the average voltage platform is relatively low (2.85 V), 15 thus leading to a low energy density (400 Wh kg 1 ). Considering the balance between the energy density and cost, the Mn system is attractive due to its abundance in nature and high voltage and capacity in the redox of Mn 2+ /Mn 3+ and Mn 3+ / Mn 4+ . However, it suffers from structural failure during cycling and poor electronic and ionic conductivity. 16 To further explore Mn-based poly-anion cathode materials, in this study we report a new phase of Li 2 MnSiO 4 induced by high pressure. In fact, as a fundamental thermodynamic variable, pressure is very effective in synthesizing new materials by changing interatomic distances, modifying bonding patterns, reordering atomic orbitals, and overcoming chemical reactive barriers for a Department of Materials Science and Engineering, College of Engineering, Peking University, Beijing 100871, China. E-mail: [email protected] b Department of Materials Science and Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA c Center for Applied Physics and Technology, Peking University, Beijing 100871, China d Key Lab of Theory and Technology for Advanced Batteries Materials, College of Engineering, Peking University, Beijing 100871, China e Department of Physics, Virginia Commonwealth University, Richmond, VA 23284, USA † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ta03369f Cite this: J. Mater. Chem. A, 2019, 7, 16406 Received 29th March 2019 Accepted 14th June 2019 DOI: 10.1039/c9ta03369f rsc.li/materials-a 16406 | J. Mater. Chem. A, 2019, 7, 16406–16413 This journal is © The Royal Society of Chemistry 2019 Journal of Materials Chemistry A PAPER Published on 14 June 2019. Downloaded by Peking University on 9/4/2020 12:59:49 PM. View Article Online View Journal | View Issue
Transcript of Journal of Materials Chemistry A - PKU

Journal ofMaterials Chemistry A
PAPER
Publ
ishe
d on
14
June
201
9. D
ownl
oade
d by
Pek
ing
Uni
vers
ity o
n 9/
4/20
20 1
2:59
:49
PM.
View Article OnlineView Journal | View Issue
A high-pressure
aDepartment of Materials Science and Eng
University, Beijing 100871, China. E-mail: sbDepartment of Materials Science and E
Technology, Cambridge, MA 02139, USAcCenter for Applied Physics and Technology,dKey Lab of Theory and Technology for A
Engineering, Peking University, Beijing 1008eDepartment of Physics, Virginia Commonwe
† Electronic supplementary informa10.1039/c9ta03369f
Cite this: J. Mater. Chem. A, 2019, 7,16406
Received 29th March 2019Accepted 14th June 2019
DOI: 10.1039/c9ta03369f
rsc.li/materials-a
16406 | J. Mater. Chem. A, 2019, 7, 16
induced stable phase of Li2MnSiO4
as an effective poly-anion cathode material fromsimulations†
Shuo Wang, a Junyi Liu, a Yu Qie, a Sheng Gong,b Cunzhi Zhang,a
Qiang Sun *acd and Puru Jena e
Search for novel cathode materials is of current interest. Among them, Li2MnSiO4 shows promise as
a cathode material in the poly-anion family due to its structural diversity, abundance, low cost, and high
theoretical capacity (330 mA h g�1). However, it suffers from low electronic and ionic conductivity,
limited reversible capacity, and poor cycling performance. To overcome these deficiencies, using
a global structure search at high pressure, we find a new phase with a group symmetry of Cc, which is
the ground-state at a pressure of 50 Gpa, and remains stable when the pressure is released. This new
phase exhibits many attractive features such as a high energy density of 612 Wh kg�1 with a reversible
capacity of 170 mA h g�1, a high average discharge voltage of 3.6 V, improved electronic conductivity,
coexistence of anionic and cationic redox, and superior ionic conductivity due to unique diffusion
channels with reduced steric hindrance and coulombic repulsion. All these features endow the new
phase of Li2MnSiO4 with potential for use as a high performance cathode material.
1. Introduction
Driven by the need for high-performance Li-ion batteries,extensive studies have been carried out on several classes ofcathode materials during the past three decades, includingchalcogenides,1 layered oxides,2,3 spinel oxides,4 olivine phos-phates,5 silicates,6 tavorites,7 etc. However, transition metal(TM) layered oxides remain dominant in the Li-ion batteryindustry due to their high voltage, stable cycling performanceand mature synthesis technology.8 However, the intrinsicinstability of layered TM oxides9 during deep delithiation makesit difficult to achieve a high reversible capacity in the derivedLiMO2 materials (M ¼ single metal or a combination of Ni, Mn,Co, Fe, Al etc.). Therefore, it is highly desirable to explore newcathode materials with enhanced stability and improved energydensity.
Recently, the poly-anion family has attracted extensiveattention, showing great structural diversity due to the variousstructural geometries composed of poly-anion units (XO4)
n+ and
ineering, College of Engineering, Peking
ngineering, Massachusetts Institute of
Peking University, Beijing 100871, China
dvanced Batteries Materials, College of
71, China
alth University, Richmond, VA 23284, USA
tion (ESI) available. See DOI:
406–16413
their derivatives (XmO3m+1)n� (X ¼ P, S, As, Mo, Si etc.).10 These
materials display great potential not only as superionicconductors with high ionic conductivity (such as b-, g-, orLISICON-type solid electrolytes11), but also as cathode materialswith high voltage and cycling stability (such as olivine phos-phate LiFeO4 (ref. 5) and silicates Li2(Fe/Co/Mn)SiO4 (ref. 12)).This is due to the strong covalent bonding between poly-anionunits and MOx polyhedra (M ¼ transition metal) whichenables the structural matrix to accommodate a wide range ofcations for good ionic conductivity and to endure the defor-mation induced by Li intercalation or deintercalation.13 Amongthe studied systems, Li2MSiO4 (M ¼ transition metal) isa promising cathode material with a high theoretical capacity14
(>320 mA h g�1). Although the M ¼ Fe system is mostlyexplored, the redox of Fe2+/Fe3+ limits the stable cycling capacityto 140 mA h g�1 which is only half of the theoretical value.Besides, the average voltage platform is relatively low (�2.85V),15 thus leading to a low energy density (400 Wh kg�1).Considering the balance between the energy density and cost,the Mn system is attractive due to its abundance in nature andhigh voltage and capacity in the redox of Mn2+/Mn3+ and Mn3+/Mn4+. However, it suffers from structural failure during cyclingand poor electronic and ionic conductivity.16 To further exploreMn-based poly-anion cathode materials, in this study we reporta new phase of Li2MnSiO4 induced by high pressure.
In fact, as a fundamental thermodynamic variable, pressureis very effective in synthesizing new materials by changinginteratomic distances, modifying bonding patterns, reorderingatomic orbitals, and overcoming chemical reactive barriers for
This journal is © The Royal Society of Chemistry 2019

Paper Journal of Materials Chemistry A
Publ
ishe
d on
14
June
201
9. D
ownl
oade
d by
Pek
ing
Uni
vers
ity o
n 9/
4/20
20 1
2:59
:49
PM.
View Article Online
phase transition.17–20 Recently, many functional materials withoutstanding performance have been synthesized such assuperconductors, superhard materials and high-energy-densitymaterials21,22 because of developed high-pressure experimentaltechniques. However, the traditional ways of high-pressureexperiments are not efficient for quick exploration due toheavy workload and long experiment periods. Crystal-structuresearch based on rst-principles calculation has been proven tobe able to accurately predict new materials. In this study, usinga global structure search at high pressure we nd a new phase ofLi2MnSiO4 with improved performance.
2. Methods
We rst use the CALYPSO22 (Crystal structure AnaLYsis byParticle Swarm Optimization) package to perform global struc-ture search at 50 GPa for stable/metastable phases by sortingthe energy from rst-principles calculations at dened chemicalcompositions (e.g. Li2MnSiO4). Then we perform calculationsbased on density functional theory (DFT) as implemented in theVienna Ab initio Simulation Package (VASP).23 The projectoraugmented-wave (PAW)24 method with the Perdew–Burke–Ern-zerhof (PBE) generalized-gradient approximation25 (GGA) forexchange and correlation potential is used. The energy cutoff ofthe plane waves is 600 eV, and the pseudopotentials of Li (2s1),Mn (3d64s1), Si (3s23p2), and O (2s22p4) are used. In consider-ation of the strong correlation effect of Mn, Hubbard Ucorrection26 (GGA+U scheme) is used in this work; an effectiveHubbard U value of 6.0 is adopted for the d orbital of Mn asreported in a previous study.27 The total energy convergencecriteria and Hellmann–Feynman force are set to 1 � 10�4 eVand 1.0 � 10�2 eV A�1, respectively, during structural optimi-zation. The smallest allowed spacing between sampled k-pointsin the reciprocal lattice is set as 0.15 A�1.
To conrm the dynamic stability of the structure, we recordthe phonon spectrum using the nite displacement method asimplemented in Phonopy.28 The convergence criterion forenergy is set to 1� 10�8 eV during the phonon calculations. Thethermodynamic stability is conrmed with ab initio moleculardynamics (AIMD) simulations using a Nose–Hoover heat bath(NVT) at 500 K in a 2� 2� 2 supercell (a¼ b¼ 12.0 A, c¼ 9.9 A)containing 128 atoms. The mechanical stability is veried usingthe criteria for the monoclinic phase.29
To study the diffusion of Li ions, the climbing image-nudgedelastic band (CI-NEB)30 approach is used to calculate the energybarrier of Li diffusion to an adjacent vacancy within the samesupercell. AIMD simulations at elevated temperatures arecarried out to calculate the mean square displacement (MSD) tostudy collective Li-ion transport according to the formulabelow:31
hr.ðtÞ
i2¼ 1
N
XNi¼1
hri. ðtþ t0Þ � ri
. ðt0Þi2
(1)
where r.ðtÞ is the displacement of the ith Li+ ion at time t and N
is the total number of Li+ ions in the system. The diffusioncoefficient (D) is calculated by tting to the MSD according to
This journal is © The Royal Society of Chemistry 2019
D ¼ limt/N
�1
2dt
hr.ðtÞ
i2�(2)
where d is the dimensionality of diffusion and t is the elapsedsimulation time. The diffusion activation energy is extrapolatedaccording to the Arrhenius equation:
D ¼ D0*exp
�� Ea
RT
�(3)
where D0 is the maximum diffusion coefficient, Ea is the acti-vation energy for diffusion, T is the absolute temperature and Ris the universal gas constant. More calculation details areshown in the ESI.†
3. Results and discussion3.1 Structure and stability
We carried out a global structure search with the chemicalformula of Li2MnSiO4 at a pressure of 50 GPa and found a newground-state structure in which the primitive cell contains 16atoms including two units of Li2MnSiO4 with two equivalentsites for each atom. Its space group is Cc, which is differentfrom the existing structures of Pnma and Pmn21. The detailedlattice parameters and structural geometry are shown in Fig. 1and the ESI.† In comparison with the existing structuresobserved in experiment (Fig. S1†), both Si–O and Mn–O poly-hedra constitute the main structural skeleton but in differentways: rst, Si has six coordinated oxygen atoms in the Cc phasebut four in Pnma and Pmn21 phases, because high pressureusually leads to congurations with a higher coordinationnumber; second, in Pnma and Pmn21 structures, the multivalentcations (Mn and Si) and Li ions distribute alternately in 2Dlayers (Fig. S1†) along the c-axis as layered rock-salt LiCoO2,while Si–O and Mn–O octahedra connect to each other withedge sharing along each lattice direction to form consecutive 3Dskeletons as shown in Fig. 1. Such a structural geometry wouldlead to favorable ionic diffusion as discussed in Section 3.2.
To explore the effect of pressure, enthalpy difference (DH)curves as a function of pressure are plotted in Fig. 2 according tothe equation DH¼ H(X)� H(Ref) where Ref¼ Cc and X¼ Pnma,Pmn21. The results show that the Pnma and Pmn21 structures arethe most stable phases with a quite small (�35 meV per atom)energy difference at ambient pressure, which agrees with thecoexistence of two phases during experimental synthesis.32
However, ground state inversion happens at the crossing pointof 19 GPa, which is much less than the recently reported large-volume high-pressure techniques (90 GPa) in experiment,33
suggesting the feasibility of synthesizing this new phase underpressure.33
To further verify the stability of the Cc phase once the appliedpressure is removed, dynamic and thermal stability are evalu-ated by AIMD simulations and phonon spectrum calculations.The phonon dispersion of Cc Li2MnSiO4 along the high-symmetry k-path in the rst Brillouin zone is shown inFig. 1(b). There are no imaginary modes conrming thedynamic stability of this new phase. Besides, AIMD simulationsare carried out with a 2 � 2 � 2 supercell at 500 K in the
J. Mater. Chem. A, 2019, 7, 16406–16413 | 16407
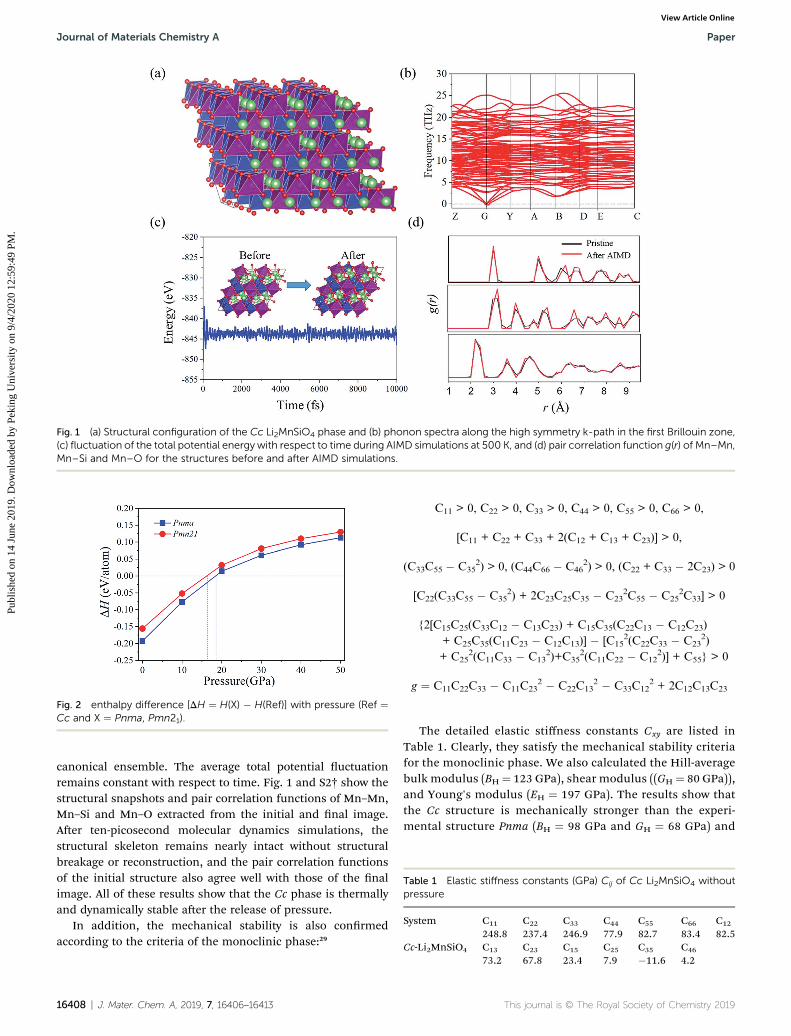
Fig. 1 (a) Structural configuration of the Cc Li2MnSiO4 phase and (b) phonon spectra along the high symmetry k-path in the first Brillouin zone,(c) fluctuation of the total potential energy with respect to time during AIMD simulations at 500 K, and (d) pair correlation function g(r) of Mn–Mn,Mn–Si and Mn–O for the structures before and after AIMD simulations.
Fig. 2 enthalpy difference [DH ¼ H(X) � H(Ref)] with pressure (Ref ¼Cc and X ¼ Pnma, Pmn21).
Table 1 Elastic stiffness constants (GPa) Cij of Cc Li2MnSiO4 withoutpressure
System C11 C22 C33 C44 C55 C66 C12
248.8 237.4 246.9 77.9 82.7 83.4 82.5Cc-Li2MnSiO4 C13 C23 C15 C25 C35 C46
Journal of Materials Chemistry A Paper
Publ
ishe
d on
14
June
201
9. D
ownl
oade
d by
Pek
ing
Uni
vers
ity o
n 9/
4/20
20 1
2:59
:49
PM.
View Article Online
canonical ensemble. The average total potential uctuationremains constant with respect to time. Fig. 1 and S2† show thestructural snapshots and pair correlation functions of Mn–Mn,Mn–Si and Mn–O extracted from the initial and nal image.Aer ten-picosecond molecular dynamics simulations, thestructural skeleton remains nearly intact without structuralbreakage or reconstruction, and the pair correlation functionsof the initial structure also agree well with those of the nalimage. All of these results show that the Cc phase is thermallyand dynamically stable aer the release of pressure.
In addition, the mechanical stability is also conrmedaccording to the criteria of the monoclinic phase:29
16408 | J. Mater. Chem. A, 2019, 7, 16406–16413
C11 > 0, C22 > 0, C33 > 0, C44 > 0, C55 > 0, C66 > 0,
[C11 + C22 + C33 + 2(C12 + C13 + C23)] > 0,
(C33C55 � C352) > 0, (C44C66 � C46
2) > 0, (C22 + C33 � 2C23) > 0
[C22(C33C55 � C352) + 2C23C25C35 � C23
2C55 � C252C33] > 0
{2[C15C25(C33C12 � C13C23) + C15C35(C22C13 � C12C23)
+ C25C35(C11C23 � C12C13)] � [C152(C22C33 � C23
2)
+ C252(C11C33 � C13
2)+C352(C11C22 � C12
2)] + C55} > 0
g ¼ C11C22C33 � C11C232 � C22C13
2 � C33C122 + 2C12C13C23
The detailed elastic stiffness constants Cxy are listed inTable 1. Clearly, they satisfy the mechanical stability criteriafor the monoclinic phase. We also calculated the Hill-averagebulk modulus (BH ¼ 123 GPa), shear modulus ((GH ¼ 80 GPa)),and Young's modulus (EH ¼ 197 GPa). The results show thatthe Cc structure is mechanically stronger than the experi-mental structure Pnma (BH ¼ 98 GPa and GH ¼ 68 GPa) and
73.2 67.8 23.4 7.9 �11.6 4.2
This journal is © The Royal Society of Chemistry 2019
Paper Journal of Materials Chemistry A
Publ
ishe
d on
14
June
201
9. D
ownl
oade
d by
Pek
ing
Uni
vers
ity o
n 9/
4/20
20 1
2:59
:49
PM.
View Article Online
Pmn21 (BH ¼ 100 GPa and GH ¼ 69 GPa) given in the MaterialsProject database.34 All the above results prove that thesynthesis of the Cc phase is feasible with the assistance ofhigh pressure and it could be stable aer pressure release.
3.2 Li ion diffusion behavior
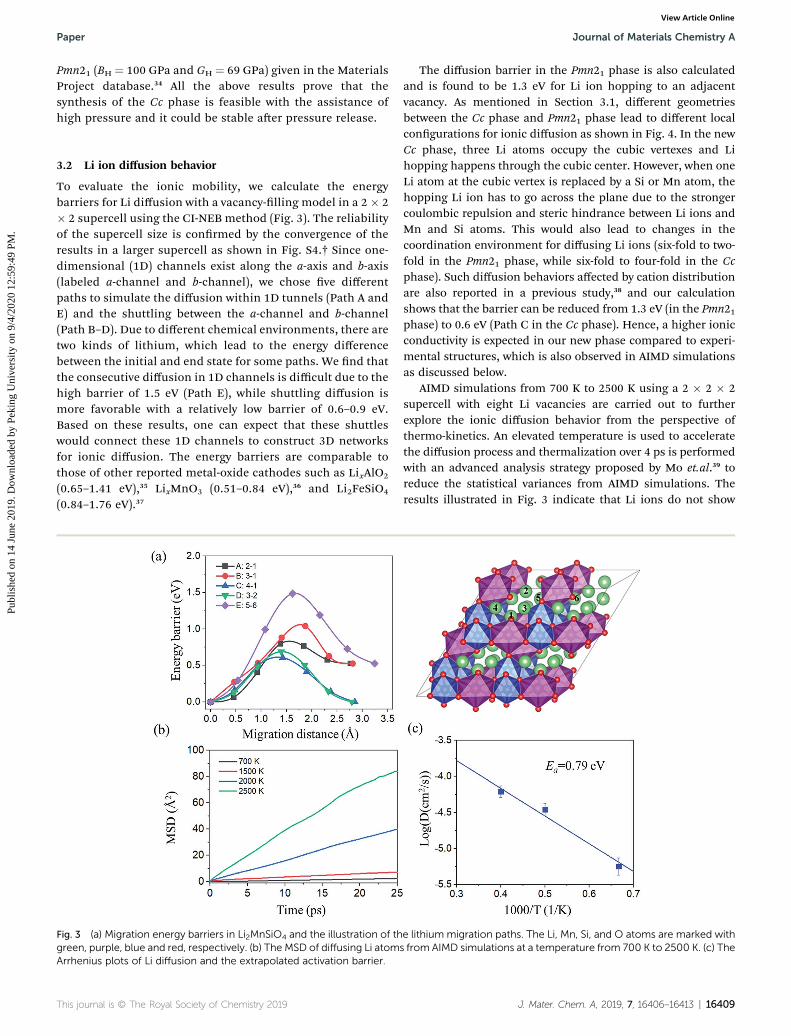
To evaluate the ionic mobility, we calculate the energybarriers for Li diffusion with a vacancy-lling model in a 2 � 2� 2 supercell using the CI-NEB method (Fig. 3). The reliabilityof the supercell size is conrmed by the convergence of theresults in a larger supercell as shown in Fig. S4.† Since one-dimensional (1D) channels exist along the a-axis and b-axis(labeled a-channel and b-channel), we chose ve differentpaths to simulate the diffusion within 1D tunnels (Path A andE) and the shuttling between the a-channel and b-channel(Path B–D). Due to different chemical environments, there aretwo kinds of lithium, which lead to the energy differencebetween the initial and end state for some paths. We nd thatthe consecutive diffusion in 1D channels is difficult due to thehigh barrier of 1.5 eV (Path E), while shuttling diffusion ismore favorable with a relatively low barrier of 0.6–0.9 eV.Based on these results, one can expect that these shuttleswould connect these 1D channels to construct 3D networksfor ionic diffusion. The energy barriers are comparable tothose of other reported metal-oxide cathodes such as LixAlO2
(0.65–1.41 eV),35 LixMnO3 (0.51–0.84 eV),36 and Li2FeSiO4
(0.84–1.76 eV).37
Fig. 3 (a) Migration energy barriers in Li2MnSiO4 and the illustration of thgreen, purple, blue and red, respectively. (b) The MSD of diffusing Li atomArrhenius plots of Li diffusion and the extrapolated activation barrier.
This journal is © The Royal Society of Chemistry 2019
The diffusion barrier in the Pmn21 phase is also calculatedand is found to be 1.3 eV for Li ion hopping to an adjacentvacancy. As mentioned in Section 3.1, different geometriesbetween the Cc phase and Pmn21 phase lead to different localcongurations for ionic diffusion as shown in Fig. 4. In the newCc phase, three Li atoms occupy the cubic vertexes and Lihopping happens through the cubic center. However, when oneLi atom at the cubic vertex is replaced by a Si or Mn atom, thehopping Li ion has to go across the plane due to the strongercoulombic repulsion and steric hindrance between Li ions andMn and Si atoms. This would also lead to changes in thecoordination environment for diffusing Li ions (six-fold to two-fold in the Pmn21 phase, while six-fold to four-fold in the Ccphase). Such diffusion behaviors affected by cation distributionare also reported in a previous study,38 and our calculationshows that the barrier can be reduced from 1.3 eV (in the Pmn21phase) to 0.6 eV (Path C in the Cc phase). Hence, a higher ionicconductivity is expected in our new phase compared to experi-mental structures, which is also observed in AIMD simulationsas discussed below.
AIMD simulations from 700 K to 2500 K using a 2 � 2 � 2supercell with eight Li vacancies are carried out to furtherexplore the ionic diffusion behavior from the perspective ofthermo-kinetics. An elevated temperature is used to acceleratethe diffusion process and thermalization over 4 ps is performedwith an advanced analysis strategy proposed by Mo et.al.39 toreduce the statistical variances from AIMD simulations. Theresults illustrated in Fig. 3 indicate that Li ions do not show
e lithium migration paths. The Li, Mn, Si, and O atoms are marked withs from AIMD simulations at a temperature from 700 K to 2500 K. (c) The
J. Mater. Chem. A, 2019, 7, 16406–16413 | 16409
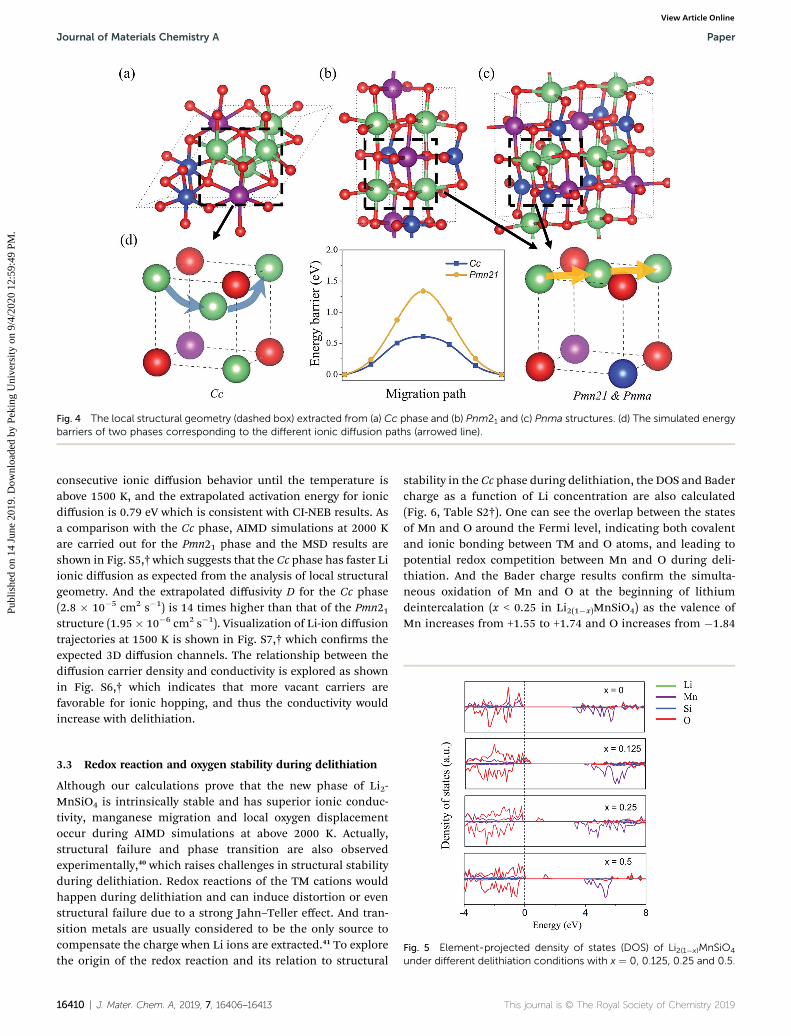
Fig. 4 The local structural geometry (dashed box) extracted from (a) Cc phase and (b) Pnm21 and (c) Pnma structures. (d) The simulated energybarriers of two phases corresponding to the different ionic diffusion paths (arrowed line).
Journal of Materials Chemistry A Paper
Publ
ishe
d on
14
June
201
9. D
ownl
oade
d by
Pek
ing
Uni
vers
ity o
n 9/
4/20
20 1
2:59
:49
PM.
View Article Online
consecutive ionic diffusion behavior until the temperature isabove 1500 K, and the extrapolated activation energy for ionicdiffusion is 0.79 eV which is consistent with CI-NEB results. Asa comparison with the Cc phase, AIMD simulations at 2000 Kare carried out for the Pmn21 phase and the MSD results areshown in Fig. S5,†which suggests that the Cc phase has faster Liionic diffusion as expected from the analysis of local structuralgeometry. And the extrapolated diffusivity D for the Cc phase(2.8 � 10�5 cm2 s�1) is 14 times higher than that of the Pmn21structure (1.95� 10�6 cm2 s�1). Visualization of Li-ion diffusiontrajectories at 1500 K is shown in Fig. S7,† which conrms theexpected 3D diffusion channels. The relationship between thediffusion carrier density and conductivity is explored as shownin Fig. S6,† which indicates that more vacant carriers arefavorable for ionic hopping, and thus the conductivity wouldincrease with delithiation.
Fig. 5 Element-projected density of states (DOS) of Li2(1�x)MnSiO4
under different delithiation conditions with x ¼ 0, 0.125, 0.25 and 0.5.
3.3 Redox reaction and oxygen stability during delithiation
Although our calculations prove that the new phase of Li2-MnSiO4 is intrinsically stable and has superior ionic conduc-tivity, manganese migration and local oxygen displacementoccur during AIMD simulations at above 2000 K. Actually,structural failure and phase transition are also observedexperimentally,40 which raises challenges in structural stabilityduring delithiation. Redox reactions of the TM cations wouldhappen during delithiation and can induce distortion or evenstructural failure due to a strong Jahn–Teller effect. And tran-sition metals are usually considered to be the only source tocompensate the charge when Li ions are extracted.41 To explorethe origin of the redox reaction and its relation to structural
16410 | J. Mater. Chem. A, 2019, 7, 16406–16413
stability in the Cc phase during delithiation, the DOS and Badercharge as a function of Li concentration are also calculated(Fig. 6, Table S2†). One can see the overlap between the statesof Mn and O around the Fermi level, indicating both covalentand ionic bonding between TM and O atoms, and leading topotential redox competition between Mn and O during deli-thiation. And the Bader charge results conrm the simulta-neous oxidation of Mn and O at the beginning of lithiumdeintercalation (x < 0.25 in Li2(1�x)MnSiO4) as the valence ofMn increases from +1.55 to +1.74 and O increases from �1.84
This journal is © The Royal Society of Chemistry 2019
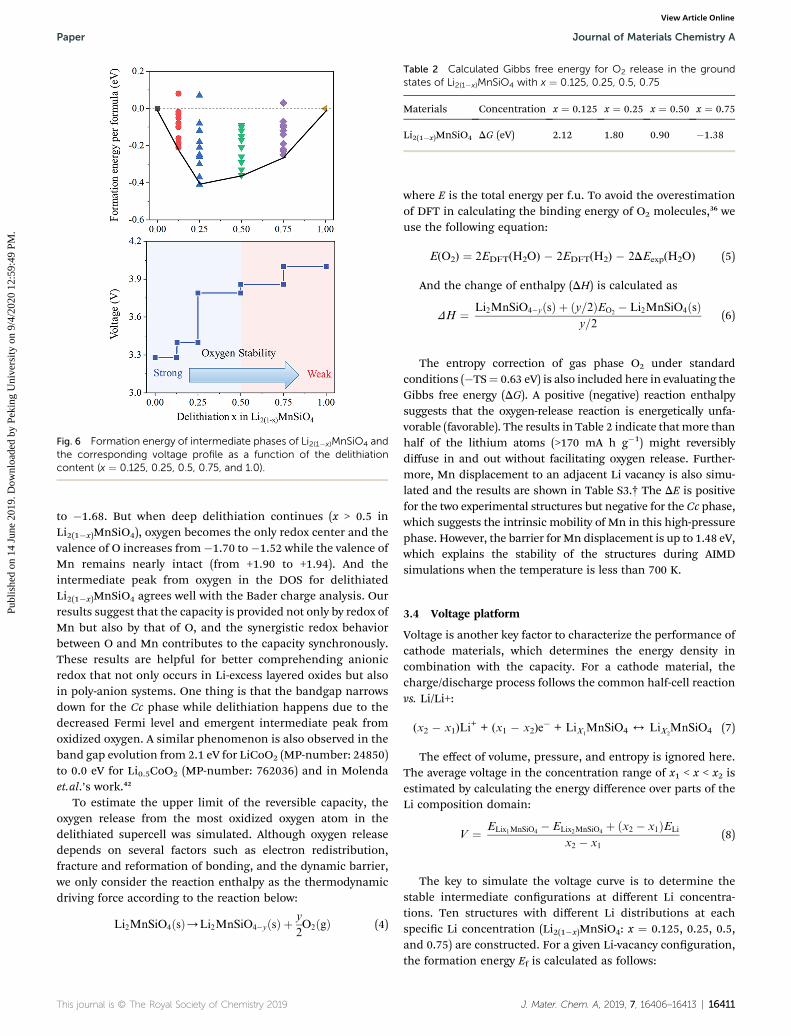
Table 2 Calculated Gibbs free energy for O2 release in the groundstates of Li2(1�x)MnSiO4 with x ¼ 0.125, 0.25, 0.5, 0.75
Materials Concentration x ¼ 0.125 x ¼ 0.25 x ¼ 0.50 x ¼ 0.75
Li2(1�x)MnSiO4 DG (eV) 2.12 1.80 0.90 �1.38
Fig. 6 Formation energy of intermediate phases of Li2(1�x)MnSiO4 andthe corresponding voltage profile as a function of the delithiationcontent (x ¼ 0.125, 0.25, 0.5, 0.75, and 1.0).
Paper Journal of Materials Chemistry A
Publ
ishe
d on
14
June
201
9. D
ownl
oade
d by
Pek
ing
Uni
vers
ity o
n 9/
4/20
20 1
2:59
:49
PM.
View Article Online
to �1.68. But when deep delithiation continues (x > 0.5 inLi2(1�x)MnSiO4), oxygen becomes the only redox center and thevalence of O increases from�1.70 to�1.52 while the valence ofMn remains nearly intact (from +1.90 to +1.94). And theintermediate peak from oxygen in the DOS for delithiatedLi2(1�x)MnSiO4 agrees well with the Bader charge analysis. Ourresults suggest that the capacity is provided not only by redox ofMn but also by that of O, and the synergistic redox behaviorbetween O and Mn contributes to the capacity synchronously.These results are helpful for better comprehending anionicredox that not only occurs in Li-excess layered oxides but alsoin poly-anion systems. One thing is that the bandgap narrowsdown for the Cc phase while delithiation happens due to thedecreased Fermi level and emergent intermediate peak fromoxidized oxygen. A similar phenomenon is also observed in theband gap evolution from 2.1 eV for LiCoO2 (MP-number: 24850)to 0.0 eV for Li0.5CoO2 (MP-number: 762036) and in Molendaet.al.’s work.42
To estimate the upper limit of the reversible capacity, theoxygen release from the most oxidized oxygen atom in thedelithiated supercell was simulated. Although oxygen releasedepends on several factors such as electron redistribution,fracture and reformation of bonding, and the dynamic barrier,we only consider the reaction enthalpy as the thermodynamicdriving force according to the reaction below:
Li2MnSiO4ðsÞ/Li2MnSiO4�yðsÞ þ y
2O2ðgÞ (4)
This journal is © The Royal Society of Chemistry 2019
where E is the total energy per f.u. To avoid the overestimationof DFT in calculating the binding energy of O2 molecules,36 weuse the following equation:
E(O2) ¼ 2EDFT(H2O) � 2EDFT(H2) � 2DEexp(H2O) (5)
And the change of enthalpy (DH) is calculated as
DH ¼ Li2MnSiO4�yðsÞ þ ðy=2ÞEO2� Li2MnSiO4ðsÞ
y=2(6)
The entropy correction of gas phase O2 under standardconditions (�TS¼ 0.63 eV) is also included here in evaluating theGibbs free energy (DG). A positive (negative) reaction enthalpysuggests that the oxygen-release reaction is energetically unfa-vorable (favorable). The results in Table 2 indicate thatmore thanhalf of the lithium atoms (>170 mA h g�1) might reversiblydiffuse in and out without facilitating oxygen release. Further-more, Mn displacement to an adjacent Li vacancy is also simu-lated and the results are shown in Table S3.† The DE is positivefor the two experimental structures but negative for the Cc phase,which suggests the intrinsic mobility of Mn in this high-pressurephase. However, the barrier for Mn displacement is up to 1.48 eV,which explains the stability of the structures during AIMDsimulations when the temperature is less than 700 K.
3.4 Voltage platform
Voltage is another key factor to characterize the performance ofcathode materials, which determines the energy density incombination with the capacity. For a cathode material, thecharge/discharge process follows the common half-cell reactionvs. Li/Li+:
(x2 � x1)Li+ + (x1 � x2)e
� + LiX1MnSiO4 4 LiX2
MnSiO4 (7)
The effect of volume, pressure, and entropy is ignored here.The average voltage in the concentration range of x1 < x < x2 isestimated by calculating the energy difference over parts of theLi composition domain:
V ¼ ELix1MnSiO4� ELix2MnSiO4
þ ðx2 � x1ÞELi
x2 � x1
(8)
The key to simulate the voltage curve is to determine thestable intermediate congurations at different Li concentra-tions. Ten structures with different Li distributions at eachspecic Li concentration (Li2(1�x)MnSiO4: x ¼ 0.125, 0.25, 0.5,and 0.75) are constructed. For a given Li-vacancy conguration,the formation energy Ef is calculated as follows:
J. Mater. Chem. A, 2019, 7, 16406–16413 | 16411

Journal of Materials Chemistry A Paper
Publ
ishe
d on
14
June
201
9. D
ownl
oade
d by
Pek
ing
Uni
vers
ity o
n 9/
4/20
20 1
2:59
:49
PM.
View Article Online
Ef(x) ¼ ELi2(1�x)MnSiO4� xEMnSiO4
� (1 � x)ELi2MnSiO4(9)
The energy-favorable structures at different intermediateconcentrations are used to calculate the voltage prole as shownin Fig. 5. The voltage increases from 3.3 V to 4.0 V withcontinuous delithiation due to the enhanced interactionbetween the poly-anion framework and Li atoms. The wholeaverage voltage is about 3.9 V, which is comparable to the valuesof 3.9 and 4.2 V for LiNiO2 (ref. 43) and LixMn2O4,44 while it ishigher than the voltage of anatase-TiO2 (1.9 V),45 V2O5 (2.8 V),46
and LiFePO4 (3.2 V).47 However, one should notice that due tothe instability of oxygen and Mn as discussed in Section 3.3, thereversible capacity is limited to 170 mA h with an averagevoltage of 3.6 V, resulting in a high energy density of 612 Whkg�1, and an appropriate voltage platform (<4 V) enables betterinterfacial safety with conventional carbonate solvents.48
4. Conclusions
In summary, we have found a new phase of Li2MnSiO4 at highpressure using a global crystal structure search combined withrst-principles calculations. The main conclusions are asfollows: (1) the synthesis of the new phase with a space groupsymmetry of Cc is feasible as the required pressure is only about20 Gpa, and it remains dynamically, thermally and mechan-ically stable aer the pressure is released; (2) the new phase has3D channels for Li ion diffusion and shows a high ionicconductivity because of the unique structural geometry; and (3)the new phase exhibits a high energy density of 612 Wh kg�1
with a reversible capacity of 170 mA h g�1 and an averagevoltage of 3.6 V. Moreover, synergetic redox activity from bothMn cations and O anions favors the high capacity. Theseinteresting results not only indicate the potential of the newLi2MnSiO4 phase as a high performance cathode, but also theeffectiveness of high-pressure synthesis for exploring novelbattery materials.
Conflicts of interest
There are no conicts to declare.
Acknowledgements
S. W. acknowledges the nancial support from the ChinaScholarship Council. This work is partially supported by grantsfrom the National Key Research and Development Program ofChina (2016YFB0100200) and the National Natural ScienceFoundation of China (21573008 and 21773003). P. J. acknowl-edges the support by the U.S DOE, Office of Basic EnergySciences, Division of Materials Sciences and Engineering underaward no. DE-FG02-96ER45579. Part of the numerical simula-tions is performed on the High-Performance Computing Plat-form of Peking University and High-performance ComputingPlatform of CAPT.
16412 | J. Mater. Chem. A, 2019, 7, 16406–16413
References
1 D. Murphy and F. Trumbore, J. Cryst. Growth, 1977, 39, 185–199.
2 S. Wang, J. Liu and Q. Sun, J. Mater. Chem. A, 2017, 5, 16936–16943.
3 M.-K. Song, S. Park, F. M. Alamgir, J. Cho and M. Liu, Mater.Sci. Eng., R, 2011, 72, 203–252.
4 S. Patoux, L. Daniel, C. Bourbon, H. Lignier, C. Pagano, F. LeCras, S. Jouanneau and S. Martinet, J. Power Sources, 2009,189, 344–352.
5 P. S. Herle, B. Ellis, N. Coombs and L. F. Nazar, in Materialsfor Sustainable Energy: A Collection of Peer-Reviewed Researchand Review Articles from Nature Publishing Group, WorldScientic, 2011, pp. 199–204.
6 C. Masquelier and L. Croguennec, Chem. Rev., 2013, 113,6552–6591.
7 T. Mueller, G. Hautier, A. Jain and G. Ceder, Chem. Mater.,2011, 23, 3854–3862.
8 T. Ohzuku and Y. Makimura, Chem. Lett., 2001, 30, 744–745.
9 R. Trivedi, Metall. Mater. Trans. A, 1995, 26, 1583–1590.10 Z. Gong and Y. Yang, Energy Environ. Sci., 2011, 4, 3223–
3242.11 P. Knauth, Solid State Ionics, 2009, 180, 911–916.12 H.-N. Girish and G.-Q. Shao, RSC Adv., 2015, 5, 98666–98686.13 J. C. Kim, Design of novel lithium storage materials with
a polyanionic framework, Massachusetts Institute ofTechnology, 2013.
14 R. Dominko, M. Bele, A. Kokalj, M. Gaberscek and J. Jamnik,J. Power Sources, 2007, 174, 457–461.
15 A. Nyten, A. Abouimrane, M. Armand, T. Gustafsson andJ. O. Thomas, Electrochem. Commun., 2005, 7, 156–160.
16 R. Gummow and Y. He, J. Power Sources, 2014, 253, 315–331.17 L. Zhang, Y. Wang, J. Lv and Y. Ma, Nat. Rev. Mater., 2017, 2,
17005.18 M. Miao, Nat. Chem., 2013, 5, 846–852.19 E. Zurek, R. Hoffmann, N. W. Ashcro, A. R. Oganov and
A. O. Lyakhov, Proc. Natl. Acad. Sci. U. S. A., 2009, 106,17640–17643.
20 S. Wang, J. Liu, Y. Qie, S. Gong, Q. Sun and P. Jena, J. Mater.Chem. A, 2018, 6, 18449–18457.
21 A. P. Drozdov, M. I. Eremets, I. A. Troyan, V. Ksenofontovand S. I. Shylin, Nature, 2015, 525, 73.
22 Y. Wang, J. Lv, L. Zhu and Y. Ma, Physics, 2010, 82, 7174–7182.
23 G. Kresse and J. Furthmuller, Phys. Rev. B: Condens. MatterMater. Phys., 1996, 54, 11169.
24 P. E. Blochl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994,50, 17953.
25 J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett.,1996, 77, 3865.
26 H. J. Kulik, M. Cococcioni, D. A. Scherlis and N. Marzari,Phys. Rev. Lett., 2006, 97, 103001.
27 H. Lee, S.-D. Park, J. Moon, H. Lee, K. Cho, M. Cho andS. Y. Kim, Chem. Mater., 2014, 26, 3896–3899.
This journal is © The Royal Society of Chemistry 2019

Paper Journal of Materials Chemistry A
Publ
ishe
d on
14
June
201
9. D
ownl
oade
d by
Pek
ing
Uni
vers
ity o
n 9/
4/20
20 1
2:59
:49
PM.
View Article Online
28 A. Togo, F. Oba and I. Tanaka, Phys. Rev. B: Condens. MatterMater. Phys., 2008, 78, 134106.
29 Z. J. Wu, E. J. Zhao, H. P. Xiang, X. F. Hao, X. J. Liu andJ. Meng, Phys. Rev. B, 2007, 76, 054115.
30 G. Henkelman, B. P. Uberuaga and H. Jonsson, J. Chem.Phys., 2000, 113, 9901–9904.
31 A. Emly, E. Kioupakis and A. Van der Ven, Chem. Mater.,2013, 25, 4663–4670.
32 T. Masese, Y. Orikasa, C. Tassel, J. Kim, T. Minato, H. Arai,T. Mori, K. Yamamoto, Y. Kobayashi and H. Kageyama,Chem. Mater., 2014, 26, 1380–1384.
33 S. Zhai and E. Ito, Geosci. Front., 2011, 2, 101–106.34 A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards,
S. Dacek, S. Cholia, D. Gunter, D. Skinner and G. Ceder,APL Mater., 2013, 1, 11002.
35 M. M. Islam and T. Bredow, J. Phys. Chem. Lett., 2015, 6,4622–4626.
36 R. Xiao, H. Li and L. Chen, Chem. Mater., 2012, 24, 4242–4251.
37 R. B. Araujo, R. H. Scheicher, J. S. de Almeida, A. Ferreira daSilva and R. Ahuja, Solid State Ionics, 2013, 247–248, 8–14.
38 A. Urban, J. Lee and G. Ceder, Adv. Energy Mater., 2014, 4,1400478.
This journal is © The Royal Society of Chemistry 2019
39 X. He, Y. Zhu, A. Epstein and Y. Mo, npj Comput. Mater.,2018, 4, 18.
40 Q. Cheng, W. He, X. Zhang, M. Li and L. Wang, J. Mater.Chem. A, 2017, 5, 10772–10797.
41 C. Zhan, Z. Yao, J. Lu, L. Ma, V. A. Maroni, L. Li, E. Lee,E. E. Alp, T. Wu and J. Wen, Nat. Energy, 2017, 2, 963.
42 J. Molenda, A. Stokłosa and T. Bak, Solid State Ionics, 1989,36, 53–58.
43 C. Ouyang, S. Shi, Z. Wang, X. Huang and L. Chen, Phys. Rev.B: Condens. Matter Mater. Phys., 2004, 69, 104303.
44 Y. S. Meng and M. E. Arroyo-de Dompablo, Energy Environ.Sci., 2009, 2, 589–609.
45 B. J. Morgan and G. W. Watson, Phys. Rev. B: Condens. MatterMater. Phys., 2010, 82, 144119.
46 X. Rocquefelte, F. Boucher, P. Gressier and G. Ouvrard,Chem. Mater., 2003, 15, 1812–1819.
47 M. E. Arroyo-de Dompablo, M. Armand, J. M. Tarascon andU. Amador, Electrochem. Commun., 2006, 8, 1292–1298.
48 D. Aurbach, Y. Talyosef, B. Markovsky, E. Markevich,E. Zinigrad, L. Asraf, J. S. Gnanaraj and H.-J. Kim,Electrochim. Acta, 2004, 50, 247–254.
J. Mater. Chem. A, 2019, 7, 16406–16413 | 16413


















