
Gongronema Latifolium A Plant with Cardioprotective Potentials

INVESTIGATION OF THE CARDIOPROTECTIVE AND HYPOTRIGLYCERIDEMIC POTENTIAL OF ECHIUM OIL AS A BOTANICAL
SOURCE OF LONG CHAIN N-3 FATTY ACIDS
BY
LOLITA M. FORREST
A Dissertation Submitted to the Graduate Faculty of
WAKE FOREST UNIVERSITY GRADUATE SCHOOL OF ARTS AND SCIENCES
in Partial Fulfillment of the Requirements
for the Degree of
DOCTOR OF PHILOSOPHY
Molecular Medicine and Translational Science
May 2011
Winston-Salem, North Carolina
Approved By:
John S. Parks, Ph.D., Advisor
Lawrence L. Rudel, Ph.D., Chairman
Iris Edwards, Ph.D.
Nilamadhab Mishra, M.D.
Michael Seeds, Ph.D.

ii
This dissertation is dedicated to my mother, Annette Carey. Mom, you are the
wind beneath my wings. Look at your baby soar!!! Isn’t this MONGONIUS!?!?

iii
ACKNOWLEDGEMENTS
Thank you, thank you, thank you….
I could not have asked for a better advisor and mentor throughout my
professional development than you, Dr. Parks!!! For your guidance, critiques,
patience, encouragement, and compassion I am forever thankful. I’m going to try
and make you proud. To my committee chair, Dr. Rudel, and members Drs.
Edwards, Mishra, and Seeds, thank you for challenging me throughout this
process. Your direction was invaluable.
Special thanks to Drs. Diz, Yoza, and McCall and the Post-baccalaureate
Research Education Program (PREP) of Wake Forest for preparing me for a
research career. You were definitely a part of my graduate school success.
Thanks to the Molecular Medicine and Translational Science Graduate
Program for my academic training and professional development, and the
Molecular Pathology Graduate Program for “adopting” me.
To everyone who is a part of the Section on Lipid Sciences: it has been a
pleasure being in such a collaborative learning environment and I have learned
so much from you.
To the present and past members of the Parks’ lab, especially Elena,
“Kaiser”, Soon, My-Ngan, Xuewei, and Anny: You have all played a part in my
training and helping me to this point and for that I say thanks.

iv
Special thanks to Dr. Chantal Rivera, for introducing me to the world of
research, and my college advisor/surrogate mother/Soror Bianca Graves for
nurturing my ambitions long after I had left from beneath thy maples and thy
oaks.
To Dr. Patricia Durant, Dr. Jenna Betters, Dr. Amanda Brown, and
Duvonne Everett, your friendship, especially in my most difficult times meant the
world to me! I can’t put it into words but know that THANK YOU has a whole lot
of other stuff in there.
I am thankful to all of my family and friends who were there for me
cheering me on throughout this long process. Thanks for supporting me even if
you didn’t know exactly what I was doing. To Mom and Tom, you guys are the
best and I love you! I can never repay you for all of your love, support, and
sacrifice, but know it is much appreciated. Smooches!!!
To my St. John C.M.E. Church family: Praise God for you! You have truly
been a blessing to my life and I am grateful that God led me to you during this
time in my life. Thank you for the prayers, encouragement, laughs, fellowship,
and delicious food. Now I know what fatback is, but I’m still not trying it
Finally, all that I am, and ever hope to be I owe to my Heavenly Father.

v
TABLE OF CONTENTS
Page
LIST OF ABBREVIATIONS………………………………………………….…..…….vi
LIST OF FIGURES AND TABLES………………………………………………….....x
ABSTRACT……………………………………………………………………………..xii
CHAPTER:
I: INTRODUCTION……………………………………………………………1
II: ECHIUM OIL REDUCES ATHEROSCLEROSIS IN APOB100-ONLY LDL RECEPTOR KNOCKOUT MICE……..............34 In Submission
III: ADDITIONAL STUDIES ON THE EFFECT OF N-3 PUFAS ON MACROPHAGE RECRUITMENT AND CHOLESTEROL ACCUMULATION………………………………51
IV: MECHANISMS FOR PLASMA TRIGLYCERIDE REDUCTION BY ECHIUM OIL ARE DISTINCT FROM THOSE OF FISH OIL IN APOB100-ONLY LDL RECEPTOR KNOCKOUT MICE……………………………………………………......70 In Preparation
V: SUMMARY AND DISCUSSION....………………..…….………….…..114
CURRICULUM VITAE…………………………………………………………….....128

vi
LIST OF ABBREVIATIONS (in alphabetical order)
AA………………………………………………………………………Arachidonic Acid
ABCA1………………………………………...ATP Binding Cassette Transporter A1
ACC…………………………………………………………...Acetyl CoA Carboxylase
ALA..........................................................................................Alpha-Linolenic Acid
apo………………………………………………………………………...Apolipoprotein
CE…………………………………………………………………..….Cholesteryl Ester
CETP……………………………………………....Cholesteryl Ester Transfer Protein
CHD…………………………………………………………..Corornary Heart Disease
CVD…………………………………………………………….Cardiovascular Disease
DAG………………………………………………………………………..Diacylglycerol
DGAT…………………………………………………….Diacylglcerol Acyltransferase
DHA……………………………………………………………..Docosahexaenoic Acid
DNA…………………………………………………………..….Deoxyribonucleic Acid
EC…………………………………………………………………….….Endothelial Cell
EO……………………………………………………………………………..Echium Oil
EPA…………………………………………………………..……Eicosahexanoic Acid

vii
FA……………………………………………………………………………....Fatty Acid
FAS…………………………………………………………………Fatty Acid Synthase
FCR…………………………………………………………..Fractional Catabolic Rate
FH………………………………………………………Familial Hypercholesterolemia
FO…………………………………………………………………………………Fish Oil
FXR………………………………………………………………..Farnesol X Receptor
GLA……………………………………………………………...Gamma-Linolenic Acid
HDL…………………………………………………………...High Density Lipoprotein
HL………………………………………………………………………...Hepatic Lipase
HNF-4α……………………………………...........Hepatocye Nuclear Factor 4 Alpha
IDL…………………………………………………...Intermediate Density Lipoprotein
IHD……………………………………………………………..Ischemic Heart Disease
LCAT…………………………………………….Lecithin-Cholesterol Acyltransferase
LDL…………………………………………………………….Low Density Lipoprotein
LDLr………………………………………………………………………..LDL Receptor
LA...………………………………………………………………………….Linoleic Acid
LPL……………………………………………………………………Lipoprotein Lipase

viii
LRP…………………………………………………………….….LDLr-Related Protein
LXR……………………………………………………………………..Liver X Receptor
MI…………………………………………………………………..Myocardial Infarction
mmLDL……………………...………………………………….Minimally Modified LDL
NEFA………………………………………………………....Nonesterified Fatty Acids
NO…………………………………………………………………………….Nitric Oxide
OA………………………………………………………………………………Oleic Acid
OCT……………………………………………………...Optimal Cutting Temperature
oxLDL……………………………………………………………………...Oxidized LDL
PBS…………………………………………………………Phosphate Buffered Saline
PGC1-α…………………………………………...PPAR Gamma Coactivator 1-alpha
PL……………………………………………………………………………Phospholipid
PO………...................................................................................................Palm Oil
PPAR…………………………………...Peroxisome Proliferator-Activated Receptor
PROCAM………………………...Prospective Cardiovascular Münster Heart Study
PUFA……………………………………………………….Polyunsaturated Fatty Acid
RCT…………………………………………………….Reverse Cholesterol Transport

ix
SCD1………………………………………........Stearoyl Coenzyme A Desaturase 1
SDA……………………………………………………………………..Stearidonic Acid
SMC………………………………………………………………...Smooth Muscle Cell
SR-A…………………………………………………………….Scavenger Receptor-A
SRE…………………………………………………………..Sterol Response Element
SREBP1c…………………………….Sterol Regulatory Element Binding Protein 1c
TG…………………………………………………………………………….Triglyceride
VLDL…………………………………………………….Very Low Density Lipoprotein

x
LIST OF FIGURES AND TABLES
Page
Chapter I
Figure 1. The initiating events of lesion development..……………….13
Figure 2. Pathways of n-3 and n-6 PUFA metabolism…....…………..19
Table 1. Fatty acid composition of experimental diets......…………..24
Chapter II
Figure 1. The effect of experimental diets on body weight and plasma lipid and lipoprotein concentration...…………..46 Figure 2. Echium oil reduces atherosclerosis……………….…...…….48
Chapter III
Figure 1. Measurement of aortic root intimal area...…………………..63
Figure 2. Image-Pro analysis of F4/80+ macrophages...……………..65
Figure 3. F4/80+ macrophages in the aortic root……………....……...67
Chapter IV
Figure 1. Plasma cholesterol and TG……………………….………….93
Figure 2. Plasma VLDL compositional and size analysis….……...….95
Figure 3. Liver lipid content…………………………………….………..97

xi
Figure 4. Hepatic gene expression……………………………………...99
Figure 5. Hepatic TG secretion rate.…………………………………..101
Figure 6. Post-heparin plasma lipase activity.……………….……….103
Figure 7. VLDL particle turnover……………………………………….105
Supplemental Figure 1. Fatty acid composition of plasma lipids...….107

xii
ABSTRACT
Forrest, Lolita
INVESTIGATION OF THE CARDIOPROTECTIVE AND HYPOTRIGLYCERIDEMIC POTENTIAL OF ECHIUM OIL AS A BOTANICAL
SOURCE OF LONG CHAIN N-3 FATTY ACIDS
Dissertation under the direction of:
John S. Parks, Ph.D., Professor of Pathology, and Biochemistry
Cardiovascular disease (CVD) is the leading cause of death in the United
States and Westernized countries. Atherosclerosis, the primary cause of CVD, is
characterized by the accumulation of cholesterol-loaded macrophages in the
artery wall, resulting in a chronic inflammatory response. Consumption of fish oil
(FO) and n-3 polyunsaturated fatty acid (PUFA) supplements has been shown to
be cardioprotective and result in several health benefits, including reduced
plasma triglyceride (TG) concentration, inflammation, endothelial cell activation,
and atherosclerosis. FO contains eicosapentaenoic acid (EPA, 20:5 n-3) and
docosahexaenoic acid (DHA, 22:6 n-3), which are credited for its cardioprotective
properties. Despite the documented benefits, FO and n-3 PUFAs are poorly
consumed in the Western diet. Furthermore, approximately 90% of the Western
diet’s n-3 PUFA source is alpha-linolenic acid (ALA, 18:3 n-3), derived from
vegetable oils. ALA is poorly converted to EPA and DHA in humans due to the
inefficiency of the Δ-6 desaturase catalyzed step of fatty acid elongation and
desaturation. Echium oil (EO), derived from Echium plantagineum seeds, is
enriched (~12%) with stearidonic acid (SDA; 18:4 n-3), the immediate product of

xiii
ALA Δ-6 desaturation, and has been shown to significantly lower plasma TGs in
human hypertriglyceridemic subjects and, therefore, may serve as a botanical
alternative to FO.
This dissertation describes the effect of EO on the development of
atherosclerosis and the mechanisms mediating its hypotriglyceridemic effects
using male apolipoprotein B100-only low density lipoprotein receptor knockout
(apoB100-only LDLrKO) mice, a mildly hypertriglyceridemic model of
atherosclerosis with significant plasma TG reduction in response to FO and EO
feeding. We show that compared to mice fed palm oil (PO), a diet rich in
saturated fat, EO feeding results in significant reductions of aortic cholesterol and
aortic surface lesion area, findings that were similar to FO-fed mice. Intravenous
injection of detergent to block lipolysis showed that FO-fed mice had decreased
plasma TG accumulation compared to EO-fed mice. Surprisingly, hepatic TG
content in both the PO- and EO-fed mice was significantly higher than that of FO-
fed mice, indicating that EO does not protect against hepatic steatosis in this
mouse model. Furthermore, very low density lipoprotein (VLDL) from EO-fed
mice incubated with purified lipoprotein lipase (LPL) is significantly more
susceptible to hydrolysis than PO-VLDL, and VLDL compositional analysis
suggests that EO-VLDL is smaller than PO-VLDL. These results suggest that EO
and FO do not reduce plasma TG concentrations by parallel mechanisms.
Whereas FO exerts its hypotriglyceridemic effect primarily by decreasing hepatic
TG synthesis and secretion, we found that EO reduces plasma TG primarily by
increased intravascular TG lipolysis.

xiv
In summary, EO feeding is cardioprotective and hypotriglyceridemic due to
increased intravascular TG lipolysis and may serve as a botanical alternative for
FO.

Chapter I
INTRODUCTION
Lolita M. Forrest
1

Lipid Metabolism
Introduction to Lipids and Lipoproteins
Lipids are a broad group of organic molecules that dissolve in organic
solvents but not water. They are critical for life by functioning as sources of
energy, signaling molecules, and structural components. Lipids exist in many
different molecular forms including triglycerides (TG), phospholipids (PL),
cholesterol, and fatty acids (FA). A fatty acid is a carboxylic acid with a long
linear hydrocarbon chain. Fatty acids are an integral component of triglycerides,
phospholipids, and cholesteryl esters (CE), forming an ester linkage with a
glycerol, glycerol-3-phosphate, or cholesterol backbone, respectively. Fatty acids
are classified in multiple ways, with reference to nutrition, chain length, and
chemistry. The nutritional classifications for FAs are essential and non essential.
Non-essential FAs can be synthesized in the body, while essential FAs cannot
and therefore must be obtained from the diet. Based on chain length, FAs are
classified as short (<6 carbons), medium (6-12 carbons), long chain (>12
carbons), or very long chain FAs (>22 carbons). Saturated and unsaturated are
the chemical classifications of FAs that describe the presence of double bonds in
the carbon chain. Saturated FAs contain no double bonds, while unsaturated
fatty acids contain one (monounsaturated) or more (polyunsaturated) double
bonds. Unsaturated FAs are further classified based on the position of the double
bond nearest to the methyl end and include n-3 (or omega-3) and n-6 (or omega-
6) FAs. Metabolically, n-3 and n-6 FAs are very different and often have
opposing actions, which will be discussed later in more detail. Polyunsaturated
2

fatty acids (PUFA) are essential fatty acids. There are two essential fatty acids in
humans; linoleic acid (LA; 18:2 n-6) and alpha-linolenic acid (ALA; 18:3 n-3).
Once in the body, these FAs can be synthesized into longer and more
unsaturated FAs by various elongase and desaturase enzymes, respectively
(1,2).
Due to their hydrophobic nature, lipids are transported in plasma as
components of water-soluble lipoprotein particles (3). Lipoproteins are spherical
structures that contain a lipid core of TGs and/or CEs, surrounded by a PL
monolayer with embedded cholesterol and one or more proteins, called
apolipoproteins (apo) (4,5). Apolipoproteins have a key role in lipid metabolism
and serve as receptor ligands in lipid transport pathways, enzyme cofactors of
lipid metabolism, and maintainers of lipoprotein structural integrity (6). When
subjected to ultracentrifugation, based on the density at which they float,
lipoproteins are classified into four main classes; chylomicrons (d < 0.95 g/ml),
very low density lipoprotein (VLDL, d = 0.95-1.006 g/ml), low density lipoprotein
(LDL, d = 1.019-1.063 g/ml), and high density lipoprotein (HDL, d >1.063-1.21
g/ml) (6). Intermediate density lipoproteins (IDL, d = 1.006-1.019 g/ml) are
remnants of VLDL and are not usually detected in blood due to conversion to
LDL (7).
Chylomicrons (>100nm in diameter) are assembled by the intestine with
TGs and cholesterol absorbed from the diet (6). They contain a number of
apolipoproteins with apoB48 being the main structural component. Chylomicrons
function to transport lipids from the intestine to other cells of the body. VLDL (30-
3

90 nm in diameter) is assembled and secreted by the liver and functions to
transport TGs and cholesterol to extrahepatic tissues (6). ApoB100 is the major
apolipoprotein of VLDL and serves as a ligand for the LDL receptor. VLDL also
contains apoE and apoCIII. As with apoB100, ApoE also serves as a ligand for
the LDL receptor whereas apoCIII inhibits lipoprotein binding to receptors. LDL
(~20nm in diameter) is the product of VLDL lipolysis (6). VLDL TGs are
hydrolyzed to fatty acids that are delivered to tissues, resulting in a LDL particle
that is cholesterol rich and serves as the major cholesterol-transporting
lipoprotein. ApoB100 is the major apolipoprotein of LDL. HDL (8-12 nm in
diameter) functions as a scavenger of cholesterol from non-hepatic tissues for
delivery to the liver in a process known as reverse cholesterol transport (RCT).
The most abundant apolipoproteins found in HDL are AI and AII. HDL also acts
as a reservoir for apolipoproteins, particularly apoCII and apoE. Nascent HDL is
synthesized and secreted by the liver as a discoidal complex of apolipoproteins
and PLs. Nascent HDL matures with the addition of cholesterol which is quickly
converted to CE by the enzyme lecithin-cholesterol acyltransferase (LCAT) to
form the core of the mature HDL particle.
Pathways of Lipid Transport
The movement of lipids throughout the circulation involves complex
processes regulated by apolipoproteins, lipoprotein receptors, lipolytic enzymes,
and transfer proteins and can be divided into three pathways; exogenous,
endogenous, and reverse cholesterol transport (RCT).
4

In the exogenous pathway, dietary lipids are absorbed in the small
intestine and packaged into chylomicrons and secreted into the circulation. Once
in the circulation, nascent chylomicrons mature rapidly by acquiring apoCII and
apoE from circulating HDL. Chylomicrons are destined for the liver but during
circulation much of their TG is hydrolyzed by lipoprotein lipase (LPL) to free fatty
acids that are delivered to non-hepatic tissues (7). LPL is found on capillary
endothelial cells (EC) and requires apoCII as a cofactor for activation (8). The
triglyceride depleted chylomicron, known as a chylomicron remnant, returns
apoCII to HDL and is taken up by the liver via chylomicron remnant receptors,
LDL receptor (LDLr) and LDLr-related protein (LRP) (9,10).
The endogenous pathway of lipid transport involves VLDL, IDL, and LDL
particles. Triglyceride-rich VLDL is synthesized and secreted by the liver and
contains apoB100. In the circulation, VLDL acquires apoE and apoC proteins and
TGs are hydrolyzed by LPL resulting in IDL particles. Further rapid hydrolysis
converts IDL to LDL. During conversion, apoE and apoC dissociate from IDL and
hepatic lipase (HL) hydrolyzes the TGs and PLs, resulting in a smaller,
cholesterol-rich LDL particle. LDL binds the LDL receptor (LDLr) on the cell
surface of the liver and extrahepatic tissues for catabolism.
Reverse cholesterol transport (RCT) is a pathway that involves the
delivery of cholesterol to the liver from extrahepatic tissues and involves HDL
(11). Nascent apoAI acts as an acceptor of free cholesterol exported from
extrahepatic cells via the transporter ABCA1 (ATP binding cassette transporter
A1) to form nascent HDL particles and the cholesterol is converted to CEs by
5

LCAT to form the core of spherical HDL particles. Cholesteryl esters that are not
transferred to the core of the particle are transferred to VLDL and LDL by
cholesteryl ester transfer protein (CETP) and are not directly involved in RCT.
Cholesterol that is part of HDL particles are referred to as good cholesterol
because that cholesterol, as part of the RCT pathway, can be transported back to
the liver where is can be removed from the circulation by being converted to bile
acids or secreted into bile. ApoE on HDL mediates its recognition and uptake by
liver receptors.
The LDL Receptor
The LDLr is a cell surface protein that mediates the endocytosis of
cholesterol-rich LDL (12). During this receptor-mediated endocytosis pathway,
LDL receptors, located in clathrin-coated pits on the cell surface, bind LDL via
recognition of its surface apoB. The LDLr-LDL complex is then internalized by
endocytosis where the LDL is released from its receptor for metabolism, whereas
the receptors are recycled back to the cell surface (13,14). The LDLr was
discovered by Brown and Goldstein during their study of familial
hypercholesterolemia (FH), an autosomal dominant disorder caused by a gene
defect on chromosome 19 (13). The LDLr gene is located on chromosome 19
and is the most common genetic defect of FH patients. Patients with FH present
with severe elevations in plasma cholesterol (300-1500 mg/dL), particularly LDL
cholesterol, and early cardiovascular disease (CVD) (13).
6

Apolipoprotein B
Apolipoprotein B is the primary protein found on VLDL and LDL particles
(5). There are two isoforms of apoB, apoB48 and apoB100. Only the small
intestine synthesizes apoB48, while apoB100 is synthesized by the liver in
humans. Unique compared to other apolipoproteins, apoB100 is a non-
exchangeable protein and there is one copy per lipoprotein particle. Therefore,
the number of circulating apoB100-containing particles can be quantified by
determining apoB100 protein concentration. ApoB100 contains an LDL receptor
binding region and plays a key role in lipid metabolism. ApoB of circulating LDL
serves as a ligand for the LDLr. A mutation in the APOB100 gene can also cause
FH (15,16).
Fatty Acids Regulate Gene Expression
In addition to dietary fats serving as structural components and a source
of energy, they also participate in the regulation of genes involved in lipid
metabolism. The liver plays a central role in whole body lipid metabolism;
therefore hepatic gene expression will be discussed here. Furthermore, while
there have been some studies on the impact of saturated fatty acids on gene
expression (17), most of the studies conducted have focused on the role of
PUFAs, specifically the n-3 series, on gene regulation, since they are more
potent regulators of lipogenesis than n-6 PUFAs. Fatty acids mediate gene
expression by binding to nuclear receptors and subsequently affecting gene
transcription (18). There are four families of nuclear receptors that have been
7

shown to bind PUFAs; LXR (liver X receptor), sterol regulatory element binding
proteins (SREBP), HNF-4α (hepatocyte nuclear factor), and PPARs (peroxisome
proliferator-activated receptors). There are two isoforms of LXR; α and β. LXRβ
is more ubiquitously expressed, whereas LXRα is expressed in a smaller subset,
with its highest expression in the liver. When activated by oxysterols, LXRs
directly regulate the expression of genes involved in bile acid synthesis. LXRs
also indirectly regulate the expression of lipogenic genes by regulating SREBP1c
gene transcription. Upon activation, LXR can bind a specific LXR response
element in the promoter of the SREBP1c gene, thereby upregulating the
expression of the lipogenic transcription factor SREBP1c. SREBP1c is a key
regulator of fatty acid and triglyceride synthesis (19). SREBP1c acts on genes
by binding to sterol response elements (SREs) in the promoter region, inducing
transcription of a number of lipogenic genes, including fatty acid synthase (FAS),
acetyl CoA carboxylase (ACC), and stearoyl CoA desaturase-1 (SCD1).
SREBP1c is a major target of PUFA control in the liver (20). PUFAs can bind
and inhibit LXR, inhibiting SREBP1c transcription, which results in decreased
lipogenesis (21). HNF-4α is stimulated by saturated FAs and inhibited by PUFAs
and regulates lipid metabolism through its direct or indirect control of a number of
genes involved in lipid metabolism including apolipoproteins A1, CII, and CIII,
genes for which are regulated by dietary PUFAs (22). PPARα is the major PPAR
expressed in liver and is involved in the regulation of genes involved in lipid
metabolism, including lipid oxidation. PPARα indirectly reduces plasma TG levels
by decreasing apoCIII expression, thereby increasing triglyceride clearance (23).
8

It has been shown that PUFAs are endogenous ligands of PPARs (18). The
binding of PUFAs to PPARs results in differential regulation of lipid oxidation
gene expression depending upon factors such as intracellular NEFA pool levels.
Hypertriglyceridemia as an independent risk factor for CVD
Hypertriglyceridemia is an elevation of blood TG levels. According to
National Cholesterol Education Program guidelines, TG levels >150mg/ml are
considered elevated. The primary strategy in the treatment of CVD is to reduce
plasma cholesterol, and LDL concentrations. However, several epidemiological
studies suggest that there is a link between hypertriglyceridemia and CVD. The
Prospective Cardiovascular Münster Heart Study (PROCAM) is an observational
study that began in 1979 and has since had over 30,000 German participants,
ages 16-65. The aim of the study was to detect risk factors of CVD through long-
term follow-up of the study participants for incidences of cardiovascular events
and mortality. In an 8-year follow-up of a cohort of 4,849 men aged 40-65, the
PROCAM Heart Study showed an independent association between TG levels
and coronary events (24). This was significant (p<0.001) even after the results
were adjusted for several other risk factors including cholesterol levels, age,
blood pressure, and family history. In the Stockholm Prospective Study, designed
to test whether plasma TG is an independent risk factor for CHD (25), data from
a 14-year and 19-year follow-up show that increasing TG concentrations
increased with new myocardial infarction (MI) cases and acted as an
independent risk factor for death (26,27). The Baltimore Coronary Observational
Long-Term Study (COLTS), designed to determine the long-term predictors of
9

coronary events in patients with CAD, found elevated TG level to be predictive in
new CAD events (28). Furthermore, an 8-year follow-up of the Copenhagen
Male Study, a prospective cohort study of men aged 40-59 designed to
determine risk factors for ischemic heart disease (IHD), found high fasting TG
levels to be a strong risk factor for IHD, even after controlling for cholesterol
levels (29). While cholesterol levels receive the most attention in approaches to
primary and secondary prevention of CVD, these studies as well as several
others have established the relevance of hypertriglyceridemia in predicting CVD
risk (30-33).
Proposed mechanisms of FO-mediated TG lowering
It is well-established that FO is hypotriglyceridemic (34-36), but the
molecular mechanisms involved are not fully understood. FO can reduce plasma
TG levels in two ways, decreased TG synthesis and secretion into plasma,
and/or increased TG lipolysis and plasma removal (clearance). Results from
lipoprotein kinetic studies in humans, nonhuman primates, and animals suggest
that FO reduces hepatic VLDL-TG production and secretion (37-40). There are at
least three mechanisms by which FO could reduce hepatic VLDL-TG synthesis,
reducing FA substrate availability, reducing TG-synthesizing enzyme activity, and
increasing PL synthesis (41). FA substrate availability for TG synthesis could be
secondary to increases in beta-oxidation, decreases in FA delivery to the liver, or
decreased FA synthesis. Decreased activity of enzymes such as diacylglycerol
acyltransferase (DGAT) results in decreased TG. Furthermore, an increase in PL
synthesis directs diacylglycerol (DAG) away from DGAT for TG synthesis.
10

Studies in rats have shown that FO increases both peroxisomal and
mitochondrial oxidation (42-44). Study results on the effects of FO on DGAT
activity are mixed (41). Furthermore, it appears that FO fatty acids are
preferentially incorporated into PL rather than TG, resulting in decreased TG
synthesis (45,46). As explained earlier, PUFAs can downregulate expression of
lipogenic genes such as FAS, ACC, and SREBP1c, also resulting in decreased
plasma TG levels. In summary, these studies suggest that FO may reduce
plasma TG by decreasing hepatic TG secretion.
The other potential mechanism of TG-lowering by FO involves an increase
in TG lipolysis and removal. A review of several studies of the
hypotriglyceridemic effects of FO found that FO shortened VLDL residence time
in plasma (47). Another study found that 3.3g/d of FO for 4 weeks lowered TG
lipolysis in hypertriglyceridemic subjects (48). A study by Harris et. al., in which
human subjects were fed 10-17 g/d of n-3 PUFAs for 3-5 wks, found that FO
increased the fractional catabolic rate (FCR) for VLDL-TG and suggested
resulting decreases in VLDL size (37). Similar findings have been shown in
LDLrKO mice (49). Decreased VLDL size may be a function of increased lipase
activity; however, study results are inconsistent. Whereas studies in rats suggest
that PUFAs increase lipase activity (50), results in hypertriglyceridemic patients
fed 10g/d of FO for 4 weeks suggest that LPL and HL activities remained the
same (51). Although studies such as these suggest that FO increases TG
lipolysis, it is generally accepted that FO reduces plasma TG levels primarily
through mediating mechanisms of TG synthesis and hepatic secretion.
11

Atherosclerosis
Definition
Atherosclerosis is a complex, chronic inflammatory condition that presents
with an accumulation of cholesterol-loaded macrophages, called foam cells,
within the artery wall (52). The initial accumulation of lipids is referred to as fatty
streaks and is subclinical. Over time, fatty streaks can develop into more
advanced and complicated lesions that can eventually rupture, forming a
thrombus. Thrombus development within an artery wall leads to clinical events
such as heart attack and stroke (53,54). Complications due to atherosclerosis are
the most common cause of death in the United States and Westernized countries
(55).
Artery Anatomy
Atherosclerosis occurs within medium and large sized arteries. The artery
is composed of three distinct layers; the endothelium, intima, and media (Figure
1). In the absence of atherosclerosis, the intimal layer is very thin and primarily
consists of a connective tissue matrix. The intima connects the luminal
endothelial layer to the peripheral media. The endothelium consists of a single
layer of tightly joined ECs, while the media consists of several layers of smooth
muscle cells (SMC) (56).
12

Figure 1. The initiating events of lesion development. Modified from Glass
and Witztum (57). Reprinted with permission.
Pathophysiology
While atherosclerosis is accepted as an inflammatory disease (52,56,57),
it is a complex, slowly progressive disease that involves environmental, dietary,
hemodynamic, metabolic, and other factors (58). While atherosclerosis and its
clinical consequences have been known for over 100 years, a greater
understanding of the initiating events of atherosclerosis on a molecular and
cellular level developed more recently. In 1973, Ross and Glomset were the first
13

to introduce the “response to injury” hypothesis (59). This hypothesis suggested
that atherosclerosis is the response to injury of the vascular endothelium. Since,
this hypothesis has been adapted and modified and is accepted as the initiating
event of atherosclerosis, being referred to as endothelial dysfunction.
The vascular endothelium is not just a barrier between the lumen and the
vascular tissue, but is critical in maintaining vascular homeostasis. The
endothelium has both anti-inflammatory and anticoagulant properties and is
actively involved in vasomotor tone by virtue of its ability to synthesize nitric oxide
(NO), a vasodilator. There are numerous factors that can contribute to
endothelial dysfunction, including elevated levels of plasma LDL and triglycerides
(60,61).
Lipoproteins play a significant role in the development of atherosclerosis.
Elevated levels of LDL cholesterol are a known risk factor for CVD (62). There is
increased permeability of the endothelium in areas of disturbed blood flow such
as branch points and curvatures. These sites are the most likely to allow for
passive diffusion of LDL particles through the EC junctions. Therefore, increased
plasma LDL concentration results in increased levels of LDL diffusing into the
subendothelial space. Once in the intima, LDL is retained in the subendothelial
space by matrix proteoglycans. Retained LDL can undergo several types of
modifications that contribute to inflammation including lipid oxidation. Initial
oxidation results in minimally modified LDL (mmLDL), which is still recognized by
the LDLr but not scavenger receptors and has pro-inflammatory activity. mmLDL
can stimulate ECs to produce inflammatory molecules, such as adhesion
14

molecules. The stimulation of the endothelium sets off a series of inflammatory
events that leads to the accumulation of cholesterol-loaded macrophages called
foam cells. One of the initial events is the adhesion of monocytes to ECs that
have been induced to express adhesion molecules. This leads to the migration of
monocytes into the subendothelial space where they can differentiate into
macrophages. Retained mmLDL can undergo further oxidation resulting in what
is known as oxidized LDL. oxLDL is no longer recognized by LDL receptors, but
rather scavenger receptors, such as scavenger receptor A (SR-A) and CD36,
found on the cell surface of macrophages. Macrophage accumulation of oxLDL
results in foam cell formation and is the hallmark of atherosclerosis.
Macrophages also express membrane cholesterol exporters such as ABCA1
which mediate cholesterol efflux from macrophages to lipid-free apoA1 (63).
However, when cholesterol levels are high, lesion formation occurs because the
rate of cholesterol uptake exceeds the rate of foam cell cholesterol export.
Dietary Intervention for Atherosclerosis Treatment and Prevention
The genesis of what is known as the lipid hypothesis, or diet-heart
hypothesis, was developed in 1856 by Rudolf Virchow to explain the cause of
atherosclerosis. Virchow suggested that blood lipid (cholesterol) accumulation
causes atherosclerosis. Virchow also concluded that the inflammation he
observed in atherosclerotic plaques induced atherosclerosis. Studies on the
relationship between diet and atherosclerosis can be traced to the early 1900’s
with the work of Ignatowski and Anichkow. In 1908, Ignatowski fed a high
cholesterol and fat diet of milk and eggs to rabbits, inducing atherosclerosis and
15

becoming the first to show a relationship between dietary cholesterol and
atherosclerosis, though at the time he attributed it to protein. In 1913, Anichkow
became the first person to demonstrate the role of cholesterol in atherosclerosis
when he induced atherosclerosis in rabbits by feeding only egg yolks. This
finding is regarded as one of cardiology’s ten greatest discoveries of the 20th
century (64). In 1953, Ancel Keys proposed that saturated fats and cholesterol in
the blood are the causes of heart disease. Studies such as these have formed
the rationale for dietary intervention to reduce total fat and cholesterol for the
treatment of heart disease.
Epidemiological studies conducted by Bang and Dyerberg first reported
the cardioprotective effects of fish oil when they compared the lipid levels of
Greenland Eskimos with Eskimos living in Denmark (65-67). Their findings
showed that while there was no significant difference in the total amount of
dietary fat intake between the two groups, the Greenland Eskimos had a higher
intake of n-3 fatty acids of marine origin, which Bang and Dyerberg attributed to
their lower death rates compared to Danes. Animal studies later showed reduced
atherosclerosis with FO feeding compared to saturated fat feeding (68-73).
Human trials have also demonstrated the cardioprotective benefits of FO. In the
Nurses’ Health Study, dietary consumption and follow-up data from 84,688
women found that higher consumption of FO and n-3 fatty acids is associated
with lower risk of coronary heart disease (CHD) (74). The Physicians’ Health
Study, conducted in men without prior evidence of CVD, showed that blood
levels of n-3 fatty acids found in fish are strongly associated with reduced risk of
16

sudden death (75). Human secondary prevention studies have also found FO to
be cardioprotective (76,77). These studies, along with multiple others, support
FO and n-3 PUFAs as atheroprotective. However, while in the minority, there are
reports that show no inverse association between fish intake and CHD mortality
in humans (78,79). This highlights the heterogeneity of FO studies and the gaps
in knowledge that remain when describing FO-mediated atheroprotective
mechanisms. Nevertheless, studies on the effect of FO on atherosclerosis
became pivotal in changing scientific thought on atherosclerosis pathophysiology
and the focus on the characteristics of dietary fat beyond mass amounts and
rather fat chemical composition.
Echium oil is derived from the seeds of Echium plantagineum, also known
as viper’s bugloss. Echium plantagineum has Mediterranean origin but this
invasive plant has been introduced to other countries including the United States.
The oil contains 9%-16% stearidonic acid (SDA; 18:4, n-3). SDA is uncommon in
higher plants but is important to human nutrition because it is an intermediate of
eicosapentanoic acid (EPA) and docosahexaenoic acid (DHA) in the
polyunsaturated fatty acid (PUFA) metabolic pathway (Figure 2). EPA and DHA
are the bioactive fatty acids found in FO that are attributed for its cardioprotective
effects.
Most of the fatty acids in the American diet are composed of saturated fat
rather than monounsaturated and polyunsaturated fat (80). Of the PUFAs in the
US diet, LA (18:2, n-6) is the most abundant. The amount of n-3 PUFAs
consumed is very small comparatively, with its major fatty acid being ALA. The
17

essential fatty acids LA and ALA can be metabolized to longer and more
unsaturated fatty acids through a number of elongase and desaturase catalyzed
steps (Figure 2). For both LA and ALA, the first step in this metabolic pathway
requires Δ6 desaturase. This enzyme converts LA to gamma-linolenic acid (GLA;
18:3, n-6), and ALA to stearidonic acid (SDA; 18:4, n-3). The Δ6 desaturase
enzyme has a variable affinity for the different fatty acid families with its preferred
substrate being 18:3 n-3 > 18:2 n-6 > 18:1 n-9 (81). Since competition exists for
Δ6 desaturase between the LA and ALA substrates, the ratio between the two
fatty acid families becomes more important when trying to enrich tissues with
fatty acids of the n-3 series. Furthermore, Δ6 desaturase is regulated by the
levels of product in the tissues, meaning if there are large amounts of long-chain
PUFAs such as arachidonic acid (AA, 20:4, n-6), the activity of Δ6 desaturase is
inhibited. Therefore, Δ6 desaturation is the rate-limiting step for conversion of
ALA to the longer and more unsaturated EPA and DHA found in fish oil. Of the
steps required to convert EPA to DHA, one of them requires Δ6 desaturase. This
means that with ALA enrichment, of the EPA produced, even less of it would be
converted to DHA. The ALA to EPA conversion efficiency of Δ6 desaturase in
humans and rodents is 4-15% (82,83).
Since SDA is the immediate product of Δ6 desaturation, it can be
hypothesized that diet enrichment with SDA would lead to further enrichment with
longer and more unsaturated fatty acids such as EPA. Indeed, Surette et. al.
showed that when human subjects were given 15g/day of Echium oil for 28 days,
18

Figure 2: Pathways of n-3 and n-6 PUFA metabolism. AA, arachidonic acid;
ALA, alpha-linolenic acid; DHA, docosahexaenoic acid; DPA, docosapentaenoic
acid; DTA, docosatetraenoic acid; EPA, eicosapentaenoic acid; LA, linoleic acid
Modified from Igarashi et. al. (84).
which is enriched with SDA but not EPA or DHA, there was a significant increase
in their plasma and neutrophil EPA concentration (85). These data showed that
fatty acids in Echium oil can successfully be desaturated and elongated into
longer chain PUFAs. One of the most consistent observations with fish oil
feeding is a reduction in plasma triglyceride concentration. Similar to fish oil,
19

Echium oil has also been shown to have a hypotriglyceridemic effect in humans
(85).
Despite the well-documented benefits, FO and n-3 PUFAs are poorly
consumed in the American diet. Several reasons for this exist, including price
compared to meat and personal preference. Fish oil supplementation also is not
a widely acceptable alternative due to gastrointestinal tolerance and fishy
aftertaste. Therefore, finding an alternative source for achieving n-3 PUFA
enrichment may prove to be of great benefit to the American population in which
cardiovascular disease is the number one cause of death.
Murine Models of Atherosclerosis
Atherosclerosis has been studied in a number of animal models. The
majority of early studies were conducted in rabbits, pigs, and nonhuman
primates. However, the advantages of genetic manipulation and practicality make
mice a common and useful tool in the study of the mechanisms involved in
atherosclerosis. There are now several genetically modified mouse models of
atherosclerosis including the apoB transgenic, LDL receptor deficient, and apoE
deficient models.
Cholesterol-rich lipoprotein remnants are cleared from circulation through
a receptor mediated pathway involving apoE as the ligand. On chow, apoE
knockout mice have 5 times more plasma cholesterol (~600mg/dl) than normal
mice and develop atherosclerosis spontaneously (86). Cholesterol accumulation
in this model occurs primarily in larger lipoprotein remnants, such as
20

chylomicrons, VLDL, and the VLDL remnant particle IDL. This mouse model
responds quickly on a high fat diet with marked hypercholesterolemia and
complex lesions similar to what is observed in advanced atherosclerosis of
humans. However, the phenotype of human apoE deficiency differs from that
observed in murine models. ApoE deficient mice can be most closely related to
humans with type III hyperlipoproteinemia, a recessively inherited disease
affecting 0.02% of the population. Therefore, the mechanisms of atherosclerosis
described using apoE deficient mice may not fully explain the mechanisms in
play that affect about half of the American population who die due to
complications of atherosclerosis.
ApoB is the major apolipoprotein found in atherogenic VLDL, LDL, and
chylomicron remnants. Human apoB is synthesized by the liver and intestine.
There are two forms of apoB in humans, apoB100 and apoB48. ApoB48 is the
product of mRNA editing. A single-post-transcriptional base change in apoB
mRNA results in a premature stop codon, producing apoB48 (87). In humans,
apoB48 is synthesized exclusively in the intestine, while the liver exclusively
synthesizes apoB100 (88). Using gene targeting techniques, Steve Young’s lab
created a mouse model that synthesized apoB100 exclusively (89). The
apoB100-only mice had significantly higher plasma TG levels (59%) than the
wild-type mice fed a chow diet and the TG increases appeared in both the VLDL
and LDL fractions (89).
Using homologous recombination techniques in embryonic stem cells,
Ishibashi et. al. produced mice lacking a functional LDL receptor (90). Compared
21

to wild type mice, LDLrKO mice have two-fold higher total plasma cholesterol
levels and present with up to a seven- to nine-fold increase in IDL/LDL
cholesterol levels. However, compared to apoE deficient mice on a chow diet,
LDLrKO mice have modest TPC increases and develop little to no
atherosclerosis. This finding highlights the significant role of apoB100 in lipid
metabolism and atherogenesis. All apoB produced by human livers is of the
apoB100 isoform, whereas it is only 30% in mice. ApoB48 is the major apoB
isoform produced by mouse livers and does not contain a LDLr binding domain.
Therefore, despite LDLr deficiency, mice can clear its apoB48-containing LDL via
a LDLr-independent pathway (i.e. LRP).
Naturally, it was conceived that crossing mice whose livers only produce
apoB100 with mice lacking a LDL receptor would produce a lipid profile
phenotype more similar to that seen in humans. Indeed, apoB100-only LDLrKO
mice have significantly elevated LDL cholesterol and apoB100 levels and
develop atherosclerosis on a chow diet (91). Therefore, apoB100-only LDLrKO
mice are a useful model in the study of atherosclerosis mechanisms and were
used to conduct the research in this dissertation.
Statement of Research Intent
Cardiovascular disease (CVD) is the number one cause of death in the
United States and westernized societies. Atherosclerosis, the primary cause of
CVD, is characterized by the accumulation of cholesterol-loaded macrophages in
the artery wall, resulting in a chronic inflammatory response. Higher consumption
22

of fish oil (FO) and n-3 polyunsaturated fatty acid (PUFA) supplements have
been shown to reduce atherosclerosis and the incidence of CVD. Despite the
benefits, FO consumption in the United States remains low and therefore a need
for an alternative dietary source of PUFAs exists. Elevated TG levels have been
shown to be an independent risk factor for CVD. Furthermore, FO consumption is
the most potent dietary intervention in the reduction of plasma TG
concentrations. However, the molecular mechanisms of fish oil-mediated TG
lowering are not fully described. By feeding apoB100-only LDLrKO mice one of
three diets supplemented with palm, Echium, or fish oil (Table 1) this dissertation
explores the role of PUFAs in atherosclerosis development and in TG lowering.
In Chapter 2, we investigate whether Echium oil supplementation in apoB100-
only LDLrKO mice will result in decreased atherosclerosis and its potential of
being used as an alternative source of PUFAs for cardioprotection. In Chapter 3,
we describe the impact of dietary PUFA enrichment on intimal area and the
accumulation of macrophages in the subendothelial space in the aortic root of
apoB100-only LDLrKO mice. Finally, in Chapter 4, we investigate the
mechanisms of plasma triglyceride reduction by Echium oil. These results of our
findings offer more insight to our understanding of the intricate pathology of
atherosclerosis and potential targets for intervention.
23

Table 1. Fatty acid composition of experimental diets. From Zhang et. al. (92)
Reprinted with permission.
24

Reference List
1. Cinti, D.L., L. Cook, M.N. Nagi, and S.K. Suneja. 1992. The fatty acid chain elongation system of mammalian endoplasmic reticulum. Prog.Lipid Res. 31: 1-51.
2. Nakamura, M.T. and T.Y. Nara. 2004. Structure, function, and dietary regulation of delta6, delta5, and delta9 desaturases. Annu.Rev.Nutr. 24: 345-376.
3. FREDRICKSON, D.S. and R.S. GORDON, Jr. 1958. Transport of fatty acids. Physiol Rev. 38: 585-630.
4. Shen, B.W., A.M. Scanu, and F.J. Kezdy. 1977. Structure of human serum lipoproteins inferred from compositional analysis. Proc.Natl.Acad.Sci.U.S.A 74: 837-841.
5. Smith, L.C., H.J. Pownall, and A.M. Gotto, Jr. 1978. The plasma lipoproteins: structure and metabolism. Annu.Rev.Biochem. 47: 751-757.
6. Mahley, R.W., T.L. Innerarity, S.C. Rall, Jr., and K.H. Weisgraber. 1984. Plasma lipoproteins: apolipoprotein structure and function. J.Lipid Res. 25: 1277-1294.
7. Redgrave, T.G. 1970. Formation of cholesteryl ester-rich particulate lipid during metabolism of chylomicrons. J.Clin.Invest 49: 465-471.
8. LaRosa, J.C., R.I. Levy, P. Herbert, S.E. Lux, and D.S. FREDRICKSON. 1970. A specific apoprotein activator for lipoprotein lipase. Biochem.Biophys.Res.Commun. 41: 57-62.
9. Brown, M.S., P.T. Kovanen, and J.L. Goldstein. 1981. Regulation of plasma cholesterol by lipoprotein receptors. Science 212: 628-635.
10. Rohlmann, A., M. Gotthardt, R.E. Hammer, and J. Herz. 1998. Inducible inactivation of hepatic LRP gene by cre-mediated recombination confirms role of LRP in clearance of chylomicron remnants. J.Clin.Invest 101: 689-695.
11. Tall, A.R. 1998. An overview of reverse cholesterol transport. Eur.Heart J. 19 Suppl A: A31-A35.
12. Brown, M.S. and J.L. Goldstein. 1986. A receptor-mediated pathway for cholesterol homeostasis. Science 232: 34-47.
25

13. Goldstein, J.L. and M.S. Brown. 2009. The LDL receptor. Arterioscler.Thromb.Vasc.Biol. 29: 431-438.
14. Anderson, R.G., M.S. Brown, and J.L. Goldstein. 1977. Role of the coated endocytic vesicle in the uptake of receptor-bound low density lipoprotein in human fibroblasts. Cell 10: 351-364.
15. Soria, L.F., E.H. Ludwig, H.R. Clarke, G.L. Vega, S.M. Grundy, and B.J. McCarthy. 1989. Association between a specific apolipoprotein B mutation and familial defective apolipoprotein B-100. Proc.Natl.Acad.Sci.U.S.A 86: 587-591.
16. Pullinger, C.R., L.K. Hennessy, J.E. Chatterton, W. Liu, J.A. Love, C.M. Mendel, P.H. Frost, M.J. Malloy, V.N. Schumaker, and J.P. Kane. 1995. Familial ligand-defective apolipoprotein B. Identification of a new mutation that decreases LDL receptor binding affinity. J.Clin.Invest 95: 1225-1234.
17. Vallim, T. and A.M. Salter. 2010. Regulation of hepatic gene expression by saturated fatty acids. Prostaglandins Leukot.Essent.Fatty Acids 82: 211-218.
18. Gottlicher, M., E. Widmark, Q. Li, and J.A. Gustafsson. 1992. Fatty acids activate a chimera of the clofibric acid-activated receptor and the glucocorticoid receptor. Proc.Natl.Acad.Sci.U.S.A 89: 4653-4657.
19. Brown, M.S. and J.L. Goldstein. 1997. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 89: 331-340.
20. Jump, D.B. 2004. Fatty acid regulation of gene transcription. Crit Rev.Clin.Lab Sci. 41: 41-78.
21. Yoshikawa, T., H. Shimano, N. Yahagi, T. Ide, M. Amemiya-Kudo, T. Matsuzaka, M. Nakakuki, S. Tomita, H. Okazaki, Y. Tamura, Y. Iizuka, K. Ohashi, A. Takahashi, H. Sone, J.J. Osuga, T. Gotoda, S. Ishibashi, and N. Yamada. 2002. Polyunsaturated fatty acids suppress sterol regulatory element-binding protein 1c promoter activity by inhibition of liver X receptor (LXR) binding to LXR response elements. J.Biol.Chem. 277: 1705-1711.
22. Jump, D.B., D. Botolin, Y. Wang, J. Xu, B. Christian, and O. Demeure. 2005. Fatty acid regulation of hepatic gene transcription. J.Nutr. 135: 2503-2506.
23. Schoonjans, K., G. Martin, B. Staels, and J. Auwerx. 1997. Peroxisome proliferator-activated receptors, orphans with ligands and functions. Curr.Opin.Lipidol. 8: 159-166.
26

24. Assmann, G., H. Schulte, and E.A. von. 1996. Hypertriglyceridemia and elevated lipoprotein(a) are risk factors for major coronary events in middle-aged men. Am.J.Cardiol. 77: 1179-1184.
25. Carlson, L.A. and S. Lindstedt. 1968. The Stockholm prospective study. 1. The initial values for plasma lipids. Acta Med.Scand.Suppl 493: 1-135.
26. Carlson, L.A., L.E. Bottiger, and P.E. Ahfeldt. 1979. Risk factors for myocardial infarction in the Stockholm prospective study. A 14-year follow-up focussing on the role of plasma triglycerides and cholesterol. Acta Med.Scand. 206: 351-360.
27. Carlson, L.A. and L.E. Bottiger. 1985. Risk factors for ischaemic heart disease in men and women. Results of the 19-year follow-up of the Stockholm Prospective Study. Acta Med.Scand. 218: 207-211.
28. Miller, M., A. Seidler, A. Moalemi, and T.A. Pearson. 1998. Normal triglyceride levels and coronary artery disease events: the Baltimore Coronary Observational Long-Term Study. J.Am.Coll.Cardiol. 31: 1252-1257.
29. Jeppesen, J., H.O. Hein, P. Suadicani, and F. Gyntelberg. 1998. Triglyceride concentration and ischemic heart disease: an eight-year follow-up in the Copenhagen Male Study. Circulation 97: 1029-1036.
30. Cullen, P. 2000. Evidence that triglycerides are an independent coronary heart disease risk factor. Am.J.Cardiol. 86: 943-949.
31. Hokanson, J.E. and M.A. Austin. 1996. Plasma triglyceride level is a risk factor for cardiovascular disease independent of high-density lipoprotein cholesterol level: a meta-analysis of population-based prospective studies. J.Cardiovasc.Risk 3: 213-219.
32. Tanne, D., N. Koren-Morag, E. Graff, and U. Goldbourt. 2001. Blood lipids and first-ever ischemic stroke/transient ischemic attack in the Bezafibrate Infarction Prevention (BIP) Registry: high triglycerides constitute an independent risk factor. Circulation 104: 2892-2897.
33. Austin, M.A., J.E. Hokanson, and K.L. Edwards. 1998. Hypertriglyceridemia as a cardiovascular risk factor. Am.J.Cardiol. 81: 7B-12B.
34. Harris, W.S. 1989. Fish oils and plasma lipid and lipoprotein metabolism in humans: a critical review. J.Lipid Res. 30: 785-807.
35. Phillipson, B.E., D.W. Rothrock, W.E. Connor, W.S. Harris, and D.R. Illingworth. 1985. Reduction of plasma lipids, lipoproteins, and apoproteins
27

by dietary fish oils in patients with hypertriglyceridemia. N.Engl.J.Med. 312: 1210-1216.
36. Harris, W.S. 1997. n-3 fatty acids and serum lipoproteins: human studies. Am.J.Clin.Nutr. 65: 1645S-1654S.
37. Harris, W.S., W.E. Connor, D.R. Illingworth, D.W. Rothrock, and D.M. Foster. 1990. Effects of fish oil on VLDL triglyceride kinetics in humans. J.Lipid Res. 31: 1549-1558.
38. Parks, J.S., F.L. Johnson, M.D. Wilson, and L.L. Rudel. 1990. Effect of fish oil diet on hepatic lipid metabolism in nonhuman primates: lowering of secretion of hepatic triglyceride but not apoB. J.Lipid Res. 31: 455-466.
39. Nestel, P.J., W.E. Connor, M.F. Reardon, S. Connor, S. Wong, and R. Boston. 1984. Suppression by diets rich in fish oil of very low density lipoprotein production in man. J.Clin.Invest 74: 82-89.
40. Bordin, P., O.A. Bodamer, S. Venkatesan, R.M. Gray, P.A. Bannister, and D. Halliday. 1998. Effects of fish oil supplementation on apolipoprotein B100 production and lipoprotein metabolism in normolipidaemic males. Eur.J.Clin.Nutr. 52: 104-109.
41. Harris, W.S. and D. Bulchandani. 2006. Why do omega-3 fatty acids lower serum triglycerides? Curr.Opin.Lipidol. 17: 387-393.
42. Halvorsen, B., A.C. Rustan, L. Madsen, J. Reseland, R.K. Berge, P. Sletnes, and E.N. Christiansen. 2001. Effects of long-chain monounsaturated and n-3 fatty acids on fatty acid oxidation and lipid composition in rats. Ann.Nutr.Metab 45: 30-37.
43. Gronn, M., E. Christensen, T.A. Hagve, and B.O. Christophersen. 1992. Effects of dietary purified eicosapentaenoic acid (20:5 (n-3)) and docosahexaenoic acid (22:6(n-3)) on fatty acid desaturation and oxidation in isolated rat liver cells. Biochim.Biophys.Acta 1125: 35-43.
44. Ukropec, J., J.E. Reseland, D. Gasperikova, E. Demcakova, L. Madsen, R.K. Berge, A.C. Rustan, I. Klimes, C.A. Drevon, and E. Sebokova. 2003. The hypotriglyceridemic effect of dietary n-3 FA is associated with increased beta-oxidation and reduced leptin expression. Lipids 38: 1023-1029.
45. Rustan, A.C., J.O. Nossen, E.N. Christiansen, and C.A. Drevon. 1988. Eicosapentaenoic acid reduces hepatic synthesis and secretion of triacylglycerol by decreasing the activity of acyl-coenzyme A:1,2-diacylglycerol acyltransferase. J.Lipid Res. 29: 1417-1426.
28

46. Yeo, Y.K. and B.J. Holub. 1990. Influence of dietary fish oil on the relative synthesis of triacylglycerol and phospholipids in rat liver in vivo. Lipids 25: 811-814.
47. Connor, W.E. 1988. Effects of omega-3 fatty acids in hypertriglyceridemic states. Semin.Thromb.Hemost. 14: 271-284.
48. Kasim-Karakas, S.E., R. Herrmann, and R. Almario. 1995. Effects of omega-3 fatty acids on intravascular lipolysis of very-low-density lipoproteins in humans. Metabolism 44: 1223-1230.
49. Vasandani, C., A.I. Kafrouni, A. Caronna, Y. Bashmakov, M. Gotthardt, J.D. Horton, and D.K. Spady. 2002. Upregulation of hepatic LDL transport by n-3 fatty acids in LDL receptor knockout mice. J.Lipid Res. 43: 772-784.
50. Hulsmann, W.C., M.C. Oerlemans, and H. Jansen. 1980. Activity of heparin-releasable liver lipase. Dependence on the degree of saturation of the fatty acids in the acylglycerol substrates. Biochim.Biophys.Acta 618: 364-369.
51. Nozaki, S., A. Garg, G.L. Vega, and S.M. Grundy. 1991. Postheparin lipolytic activity and plasma lipoprotein response to omega-3 polyunsaturated fatty acids in patients with primary hypertriglyceridemia. Am.J.Clin.Nutr. 53: 638-642.
52. Ross, R. 1999. Atherosclerosis--an inflammatory disease. N.Engl.J.Med. 340: 115-126.
53. DeWood, M.A., J. Spores, R. Notske, L.T. Mouser, R. Burroughs, M.S. Golden, and H.T. Lang. 1980. Prevalence of total coronary occlusion during the early hours of transmural myocardial infarction. N.Engl.J.Med. 303: 897-902.
54. Brooks, N. 1983. Intracoronary thrombolysis in acute myocardial infarction. Br.Heart J. 50: 397-400.
55. Lloyd-Jones, D., R. Adams, M. Carnethon, S.G. De, T.B. Ferguson, K. Flegal, E. Ford, K. Furie, A. Go, K. Greenlund, N. Haase, S. Hailpern, M. Ho, V. Howard, B. Kissela, S. Kittner, D. Lackland, L. Lisabeth, A. Marelli, M. McDermott, J. Meigs, D. Mozaffarian, G. Nichol, C. O'Donnell, V. Roger, W. Rosamond, R. Sacco, P. Sorlie, R. Stafford, J. Steinberger, T. Thom, S. Wasserthiel-Smoller, N. Wong, J. Wylie-Rosett, and Y. Hong. 2009. Heart disease and stroke statistics--2009 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 119: 480-486.
56. Lusis, A.J. 2000. Atherosclerosis. Nature 407: 233-241.
29

57. Glass, C.K. and J.L. Witztum. 2001. Atherosclerosis. the road ahead. Cell 104: 503-516.
58. Fuster,V., O'Rourke,R.A., and Alexander,R.W. 2004. Hurst's the Heart. McGraw-Hill, 1361-1367 pp.
59. Ross, R. and J.A. Glomset. 1973. Atherosclerosis and the arterial smooth muscle cell: Proliferation of smooth muscle is a key event in the genesis of the lesions of atherosclerosis. Science 180: 1332-1339.
60. Steinberg, H.O., B. Bayazeed, G. Hook, A. Johnson, J. Cronin, and A.D. Baron. 1997. Endothelial dysfunction is associated with cholesterol levels in the high normal range in humans. Circulation 96: 3287-3293.
61. Lewis, T.V., A.M. Dart, and J.P. Chin-Dusting. 1999. Endothelium-dependent relaxation by acetylcholine is impaired in hypertriglyceridemic humans with normal levels of plasma LDL cholesterol. J.Am.Coll.Cardiol. 33: 805-812.
62. Sytkowski, P.A., W.B. Kannel, and R.B. D'Agostino. 1990. Changes in risk factors and the decline in mortality from cardiovascular disease. The Framingham Heart Study. N.Engl.J.Med. 322: 1635-1641.
63. Oram, J.F., R.M. Lawn, M.R. Garvin, and D.P. Wade. 2000. ABCA1 is the cAMP-inducible apolipoprotein receptor that mediates cholesterol secretion from macrophages. J.Biol.Chem. 275: 34508-34511.
64. Mehta, N.J. and I.A. Khan. 2002. Cardiology's 10 greatest discoveries of the 20th century. Tex.Heart Inst.J. 29: 164-171.
65. Bang, H.O., J. Dyerberg, and N. Hjoorne. 1976. The composition of food consumed by Greenland Eskimos. Acta Med.Scand. 200: 69-73.
66. Dyerberg, J. and H.O. Bang. 1979. Lipid metabolism, atherogenesis, and haemostasis in Eskimos: the role of the prostaglandin-3 family. Haemostasis 8: 227-233.
67. Dyerberg, J. and H.O. Bang. 1982. A hypothesis on the development of acute myocardial infarction in Greenlanders. Scand.J.Clin.Lab Invest Suppl 161: 7-13.
68. Davis, H.R., R.T. Bridenstine, D. Vesselinovitch, and R.W. Wissler. 1987. Fish oil inhibits development of atherosclerosis in rhesus monkeys. Arteriosclerosis 7: 441-449.
69. Parks, J.S., J. Kaduck-Sawyer, B.C. Bullock, and L.L. Rudel. 1990. Effect of dietary fish oil on coronary artery and aortic atherosclerosis in African green monkeys. Arteriosclerosis 10: 1102-1112.
30

70. Rudel, L.L., K. Kelley, J.K. Sawyer, R. Shah, and M.D. Wilson. 1998. Dietary monounsaturated fatty acids promote aortic atherosclerosis in LDL receptor-null, human ApoB100-overexpressing transgenic mice. Arterioscler.Thromb.Vasc.Biol. 18: 1818-1827.
71. Davis, H.R., R.T. Bridenstine, D. Vesselinovitch, and R.W. Wissler. 1987. Fish oil inhibits development of atherosclerosis in rhesus monkeys. Arteriosclerosis 7: 441-449.
72. Rudel, L.L., K. Kelley, J.K. Sawyer, R. Shah, and M.D. Wilson. 1998. Dietary monounsaturated fatty acids promote aortic atherosclerosis in LDL receptor-null, human ApoB100-overexpressing transgenic mice. Arterioscler.Thromb.Vasc.Biol. 18: 1818-1827.
73. Parks, J.S., J. Kaduck-Sawyer, B.C. Bullock, and L.L. Rudel. 1990. Effect of dietary fish oil on coronary artery and aortic atherosclerosis in African green monkeys. Arteriosclerosis 10: 1102-1112.
74. Hu, F.B., L. Bronner, W.C. Willett, M.J. Stampfer, K.M. Rexrode, C.M. Albert, D. Hunter, and J.E. Manson. 2002. Fish and omega-3 fatty acid intake and risk of coronary heart disease in women. JAMA 287: 1815-1821.
75. Albert, C.M., H. Campos, M.J. Stampfer, P.M. Ridker, J.E. Manson, W.C. Willett, and J. Ma. 2002. Blood levels of long-chain n-3 fatty acids and the risk of sudden death. N.Engl.J.Med. 346: 1113-1118.
76. 1999. Dietary supplementation with n-3 polyunsaturated fatty acids and vitamin E after myocardial infarction: results of the GISSI-Prevenzione trial. Gruppo Italiano per lo Studio della Sopravvivenza nell'Infarto miocardico. Lancet 354: 447-455.
77. Burr, M.L., A.M. Fehily, J.F. Gilbert, S. Rogers, R.M. Holliday, P.M. Sweetnam, P.C. Elwood, and N.M. Deadman. 1989. Effects of changes in fat, fish, and fibre intakes on death and myocardial reinfarction: diet and reinfarction trial (DART). Lancet 2: 757-761.
78. Osler, M., A.H. Andreasen, and S. Hoidrup. 2003. No inverse association between fish consumption and risk of death from all-causes, and incidence of coronary heart disease in middle-aged, Danish adults. J.Clin.Epidemiol. 56: 274-279.
79. Vollset SE, H.I.B.E. 1985. Fish Consumption and Mortality from Coronary Heart Disease. New England Journal of Medicine 313: 820-824.
80. Kris-Etherton, P.M., D.S. Taylor, S. Yu-Poth, P. Huth, K. Moriarty, V. Fishell, R.L. Hargrove, G. Zhao, and T.D. Etherton. 2000. Polyunsaturated
31

fatty acids in the food chain in the United States. Am.J.Clin.Nutr. 71: 179S-188S.
81. Siguel, E.N. and M. Maclure. 1987. Relative activity of unsaturated fatty acid metabolic pathways in humans. Metabolism 36: 664-669.
82. Singer, P., I. Berger, M. Wirth, W. Godicke, W. Jaeger, and S. Voigt. 1986. Slow desaturation and elongation of linoleic and alpha-linolenic acids as a rationale of eicosapentaenoic acid-rich diet to lower blood pressure and serum lipids in normal, hypertensive and hyperlipemic subjects. Prostaglandins Leukot.Med. 24: 173-193.
83. Huang, Y.S., R.S. Smith, P.R. Redden, R.C. Cantrill, and D.F. Horrobin. 1991. Modification of liver fatty acid metabolism in mice by n-3 and n-6 delta 6-desaturase substrates and products. Biochim.Biophys.Acta 1082: 319-327.
84. Igarashi, M., K. Ma, L. Chang, J.M. Bell, and S.I. Rapoport. 2007. Dietary n-3 PUFA deprivation for 15 weeks upregulates elongase and desaturase expression in rat liver but not brain. Journal of Lipid Research 48: 2463-2470.
85. Surette, M.E., M. Edens, F.H. Chilton, and K.M. Tramposch. 2004. Dietary echium oil increases plasma and neutrophil long-chain (n-3) fatty acids and lowers serum triacylglycerols in hypertriglyceridemic humans. J.Nutr. 134: 1406-1411.
86. Zhang, S.H., R.L. Reddick, J.A. Piedrahita, and N. Maeda. 1992. Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science 258: 468-471.
87. Chen, S.H., G. Habib, C.Y. Yang, Z.W. Gu, B.R. Lee, S.A. Weng, Silberman, S.J. Cai, J.P. Deslypere, M. Rosseneu, and a. et. 1987. Apolipoprotein B-48 is the product of a messenger RNA with an organ-specific in-frame stop codon. Science 238: 363-366.
88. Edge, S.B., J.M. Hoeg, P.D. Schneider, and H.B. Brewer, Jr. 1985. Apolipoprotein B synthesis in humans: liver synthesizes only apolipoprotein B-100. Metabolism 34: 726-730.
89. Farese, R.V., Jr., M.M. Veniant, C.M. Cham, L.M. Flynn, V. Pierotti, J.F. Loring, M. Traber, S. Ruland, R.S. Stokowski, D. Huszar, and S.G. Young. 1996. Phenotypic analysis of mice expressing exclusively apolipoprotein B48 or apolipoprotein B100. Proc.Natl.Acad.Sci.U.S.A 93: 6393-6398.
90. Ishibashi, S., M.S. Brown, J.L. Goldstein, R.D. Gerard, R.E. Hammer, and J. Herz. 1993. Hypercholesterolemia in low density lipoprotein receptor
32

knockout mice and its reversal by adenovirus-mediated gene delivery. J.Clin.Invest 92: 883-893.
91. Powell-Braxton, L., M. Veniant, R.D. Latvala, K.I. Hirano, W.B. Won, J. Ross, N. Dybdal, C.H. Zlot, S.G. Young, and N.O. Davidson. 1998. A mouse model of human familial hypercholesterolemia: markedly elevated low density lipoprotein cholesterol levels and severe atherosclerosis on a low-fat chow diet. Nat.Med. 4: 934-938.
92. Zhang, P., E. Boudyguina, M.D. Wilson, A.K. Gebre, and J.S. Parks. 2008. Echium oil reduces plasma lipids and hepatic lipogenic gene expression in apoB100-only LDL receptor knockout mice. J.Nutr.Biochem. 19: 655-663.
33

Chapter II
ECHIUM OIL REDUCES ATHEROSCLEROSIS IN APOB100-ONLY LDL
RECEPTOR KNOCKOUT MICE
Lolita M. Forrest, Elena Boudyguina, Martha D. Wilson, and John S. Parks
The following manuscript was submitted to Atherosclerosis in April 2011. Stylistic
variations are due to the requirements of the journal. The experimentation and
writing were performed by LM Forrest. Dr. JS Parks acted in an advisory and
editorial capacity.

Abstract
Introduction: The anti-atherogenic and hypotriglyceridemic properties of fish oil
are attributed to its enrichment in eicosapentanoic acid (EPA; 20:5, n-3) and
docosahexaenoic acid (DHA; 22:6, n-3). Echium oil contains stearidonic acid
(SDA; 18:4, n-3), which is metabolized to longer-chain n-3 PUFAs, including
EPA, in humans, resulting in decreased plasma triglycerides.
Objective: We used apoB100-only LDL receptor knockout mice to investigate
whether Echium oil reduces atherosclerosis.
Methods: Mice were fed palm, Echium, or fish oil for 16 weeks and
atherosclerosis was quantified by aortic surface lesion area and aortic cholesterol
content. Body weight and plasma lipids were determined every 2 weeks
throughout the study period.
Results: Compared to palm oil, Echium oil feeding resulted in significantly lower
plasma triglyceride and cholesterol levels, and atherosclerosis, comparable to
that of fish oil.
Conclusion: This is the first report that Echium oil is anti-atherogenic, suggesting
that it may be a botanical alternative to fish oil for cardioprotection.

Introduction
Dietary intervention is often the initial approach to reduce risk factors that
contribute to cardiovascular heart disease, which is the leading cause of
morbidity and mortality in Westernized societies (1). Dietary consumption of
long-chain n-3 PUFAs, such as those found in fatty fish or fish oil (FO)
supplements, reduce inflammation, endothelial activation, and platelet activation,
resulting in decreased cardiovascular disease (2). The cardioprotective
component of FO is attributed to two long-chain n-3 PUFAs, EPA (20:5 n-3) and
DHA (22:6 n-3). Despite documented cardioprotective benefits, fatty fish and FO
consumption in the United States remains low (2). The North American diet is
rich in fatty acids that are pro-inflammatory and pro-atherogenic and 90% of the
n-3 PUFAs consumed is alpha-linolenic acid (ALA; 18:3 n-3) (3). However, ALA
is not cardioprotective because it requires Δ6-desaturase for conversion to EPA
and DHA and this conversion is very inefficient (~4-16%) in humans and rodents
(4,5). Therefore, an alternative strategy is needed to provide for the lack of EPA
and DHA in the diet.
One approach we have taken is to enrich the diet with an oil that contains
a fatty acid that can be converted to EPA in vivo. Echium oil (EO), derived from
the seeds of Echium plantagineum, contains 12-14% of total fatty acids as
stearidonic acid (SDA; 18:4 n-3), the immediate product of ALA Δ6-desaturation.
We have previously shown that SDA in EO is converted to EPA in plasma and
liver lipids of a mouse model of atherosclerosis and hypertriglyceridemia, the
apoB100-only LDL receptor knockout mouse (6). EO, relative to a palm oil (PO)

control diet, also reduced total plasma cholesterol (TPC) and triglyceride (TG)
concentrations in the apoB100-only LDLrKO mice. Similar enrichment of lipid
fractions with EPA and reduction in plasma TG concentrations has also been
observed in human subjects supplemented with EO (7). These results
established EO as a viable botanical alternative to FO for reduction of plasma TG
concentrations. However, whether EO is also cardioprotective is unknown.
The purpose of the present study was to determine whether EO
consumption confers cardioprotection, and, if so, to what extent compared to that
of FO. We used apoB100-only LDLrKO mice for the study because they are a
well-established mouse model used in previous studies to determine the effect of
dietary fat saturation on atherosclerosis (8-10).

Materials and methods
Animals and diets
Male apoB100-only LDLrKO mice (10) in the C57BL/6 background (>99%)
were housed in a specific pathogen-free facility at Wake Forest School of
Medicine in accordance with all institutional animal care and use guidelines. After
weaning, the mice were fed a chow diet until 8 weeks age; at that time they were
randomly assigned to one of three diet groups designated as PO, EO, or FO.
Each diet contained 0.2% cholesterol and 10% of calories from PO with an
additional 10% of calories from PO, EO, or FO, totaling 20% calories as fat.
Detailed diet compositions and fatty acid compositions have been described
previously (6,11). The diets were fed to the mice for a total of 16 weeks.
Lipid and lipoprotein analysis
Blood samples were collected from the tail vein after a 4h fast. Plasma
cholesterol (Wako; Richmond, VA) and TG (Roche; Indianapolis, IN) were
determined by enzymatic analysis. The analyses were performed according to
the manufacturer’s instructions. Fresh plasma was used to determine lipoprotein
cholesterol distribution after FPLC fractionation of plasma (12).
Atherosclerosis quantification
After 16 weeks of diet feeding, mice were sacrificed using
ketamine/xylazine and a terminal blood sample was taken via cardiac puncture.
The heart and venous system was perfused with cold PBS for 5-10min at 1

ml/min via the left ventricle. Aortas were then collected from the heart to the iliac
bifurcation and fixed in 10% buffered formalin. Aortas were cut open
longitudinally, pinned open on a black elastomer surface, and a digital en face
image was captured. Images of the aortas were analyzed using WCIF Image J
software (NIH) to determine percentage of total surface that was covered with
atherosclerotic lesions. The aortic lipids were then extracted with 2:1 chloroform-
methanol and total and free cholesterol were quantified by gas liquid
chromatography and normalized to aortic protein content (13). CE was calculated
as: TC-FC X 1.67 (to correct for fatty acid loss) (14).
Statistical analysis
All data are presented as mean ± SEM. Differences among the 3 diet
groups were analyzed by one-way ANOVA (p<0.05) using GraphPad Prism
software. For TG and TPC, the area under the curve (AUC) was determined for
individual animals and differences among the groups were determined by one-
way ANOVA (p<0.05). Individual diet group differences were identified using
Tukey’s post-test analysis.

Results
Echium oil feeding results in decreased plasma lipids and apoB lipoproteins
In our previous study, experimental diet feeding was limited to 8 weeks (6)
and atherosclerosis was not evaluated. Here, we extended the feeding period to
16 weeks to investigate atherosclerosis development. Mice in all three diet
groups gained body weight at similar rates (Figure 1A). Mice fed the EO diet had
a rapid and sustained reduction in plasma TG levels compared to PO fed
controls (Figure 1B). The reduction in plasma TG concentrations with EO was
similar to that of FO-fed mice and lasted throughout the 16 week period. EO also
resulted in significantly lower TPC levels during the 16 week atherogenic
progression compared with mice fed PO (Figure 1C). The reduction in TPC with
EO feeding was sustained throughout the 16 week atherosclerosis progression
phase and was similar to that of mice fed FO. The reduction in TPC
concentrations in EO and FO fed mice was due to significant reductions in VLDL
and LDL cholesterol compared with PO-fed mice (Figure 1D); HDL
concentrations were similar among the three diet groups.
Echium oil reduces atherosclerosis in apoB100-only LDLrKO mice
After 16 weeks of experimental diet consumption, the mice were sacrificed
for evaluation of atherosclerosis by aortic surface lesion area and cholesterol
content. Representative aortic images of mice from each diet group illustrate the
predominant aortic arch localization of raised lesions (Figure 2A). Quantification
of aortic lesion surface area revealed a significant reduction in lesion area for EO

and FO fed mice compared to the PO fed group (Figure 2B). Aortic cholesterol
quantification also demonstrated a significant reduction in TC, FC and CE for the
EO and FO groups vs. the PO group (Figure 2C). In both measurements of
atherosclerosis, the amount of disease was similar for the EO and FO fed mice
(Figure 2B and 2C). Finally, there was a significant correlation (r2= 0.4866;
p<0.05) between plasma apoB lipoprotein concentrations (i.e., VLDL and LDL)
and aortic CE content (Figure 2D), suggesting that about half of the variability in
atherosclerosis could be explained by the plasma apoB lipoprotein reduction by
EO and FO. There was also a strong correlation between aortic CE content and
percent surface lesion area (r2 = 0.6783; data not shown).

Discussion
Due to the high incidence of cardiovascular disease and the low
consumption of FO and/or fatty fish in the United States, the goal of our study
was to determine whether EO could serve as a botanical source of n-3 PUFAs
for the purpose of EPA enrichment and cardioprotection. Using a mouse model
of atherosclerosis, we show that EO feeding results in decreased atherosclerosis
compared to PO feeding and is equivalent in cardioprotection to FO. To our
knowledge, this is the first report that EO is atheroprotective.
The benefits of n-3 PUFAs of marine origin were first reported by Bang
and Dyerberg, who observed lower death rates in Greenland Eskimos, whose
diet is enriched in n-3 PUFAs, compared to Danes who consume a more
saturated fat diet (15). However, due to personal preference, fish is poorly
consumed in the US population (3). In addition, FO supplements are not widely
accepted due to gastrointestinal intolerance and fishy aftertaste (2). As such,
ALA in vegetable oils supplies most of the n-3 PUFA in the Western diet (3).
Although n-3 PUFAs, in general, are less inflammatory than n-6 PUFAs, only
long-chain (>18 carbon) PUFAs, such as EPA and DHA, result in
atheroprotection (16,17). The enzyme Δ-6 desaturase is the rate limiting step in
ALA conversion to EPA and DHA. EO offers a natural source of SDA, the
immediate product of ALA Δ-6 desaturation and, therefore, serves as a suitable
source for EPA enrichment (7). Human studies and those conducted previously
by our lab using apoB100-only LDLrKO mice have shown significant reductions
in plasma TG concentrations (6), which is the most consistent observation with

FO feeding and has been found to be an independent risk factor for
cardiovascular disease (18). Our study suggests that about 50% of the variability
in atherosclerosis observed with EO feeding is related to the reduction in plasma
apoB lipoprotein cholesterol concentrations (Figure 2D). Presumably, EO also
reduces atherosclerosis by additional mechanisms (i.e., limiting inflammation)
similar to those reported for FO (19), but this remains to be determined in future
studies.
In conclusion, we have found that EO reduces atherosclerosis in
apoB100-only LDLrKO mice. Based on our findings in mice and the
hypotriglyceridemic effects of EO supplementation in humans, we propose that
EO may be a suitable botanical alternative to FO to reduce plasma TG levels and
confer atheroprotection. Thus, in instances in which dietary consumption of EPA
and DHA is low, such as the case in the United States and Westernized
countries, the health benefits of EO should outweigh those of botanical oils
enriched in ALA. This may be particularly true with regard to hypertriglyceridemia
and cardiovascular disease. However, because EO does not result in DHA
enrichment due to limited Δ-6 desaturase-mediated conversion of EPA to DHA,
EO is not a complete substitute for FO and likely will not satisfy the body’s
essential need for DHA for brain and retina development (7).

Acknowledgements: This work was funded by grant numbers P50AT002782,
R01HL094525, and P01HL049373 to JSP and 3P50AT002782-03S1 to LMF. A
special thanks to Croda Chemicals for providing us with the Echium oil used in
our diets. Special thanks also to Omega Protein Inc. for providing OmegaPure
refined menhaden oil for our FO diet.

Figure 1. The effect of experimental diets on body weight and plasma lipid
and lipoprotein concentrations. Eight week old apoB100-only LDLrKO mice
were fed diets for 16 weeks consisting of 0.2% cholesterol and 10% of calories
from PO, with an additional 10% from PO, EO, FO (n=11-16 mice per group).
Periodically, body weights (A) were determined and blood was obtained to assay
plasma TG (B) and TPC (C) by enzymatic assays. (D) At 16 weeks, a terminal
blood sample was used to fraction plasma by FPLC and measure plasma
lipoprotein cholesterol distribution. Values are mean ±S.E.M. Bars with different
letters denote significant differences among diet groups (P<.05).
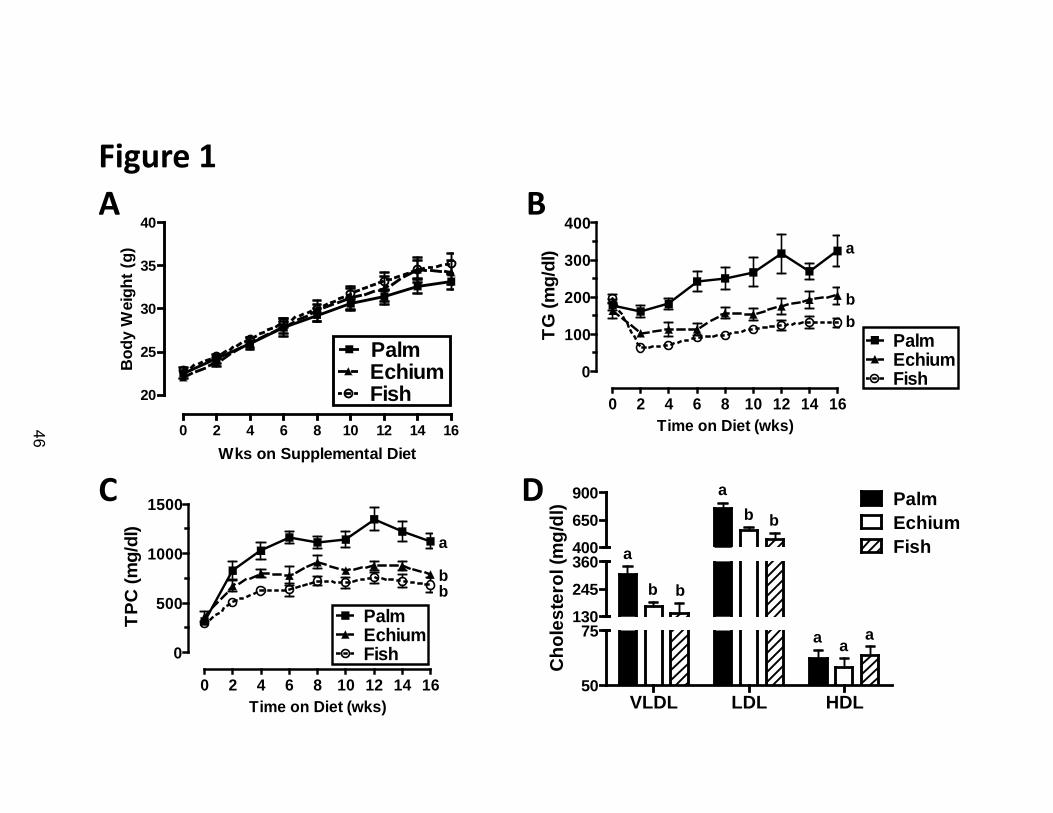
A BFigure 1
C D
0 2 4 6 8 10 12 14 16
0
100
200
300
400
PalmEchiumFish
a
bb
Time on Diet (wks)
TG (m
g/dl
)
0 2 4 6 8 10 12 14 16
0
500
1000
1500
PalmEchiumFish
a
bb
Time on Diet (wks)
TPC
(mg/
dl)
0 2 4 6 8 10 12 14 16
20
25
30
35
40
PalmEchiumFish
Wks on Supplemental Diet
Bod
y W
eigh
t (g)
VLDL LDL HDL50
75130245360400650900 Palm
EchiumFisha
b b
bba
aaa
Cho
lest
erol
(mg/
dl)
46

Figure 2. Echium oil reduces atherosclerosis. Mice were sacrificed and aortas
were removed for atherosclerosis quantification after 16 weeks of atherogenic
diet consumption (n=12-16 mice per group). (A) Representative aorta from each
diet group with atherosclerotic lesions identified by white arrows. (B)
Quantification of aortic surface lesion area. (C) Aortic total cholesterol (TC), free
cholesterol (FC) and cholesteryl ester (CE), measured by gas-liquid
chromatography. Values are mean ± S.E.M. Bars with different letters denote
significant differences among diet groups (P<.05). (D) Association between
plasma apoB lipoprotein cholesterol concentration and aortic CE content. Each
point denotes an individual animal of the designated diet group. The line of best
fit, determined by linear regression analysis, is shown for all animals.
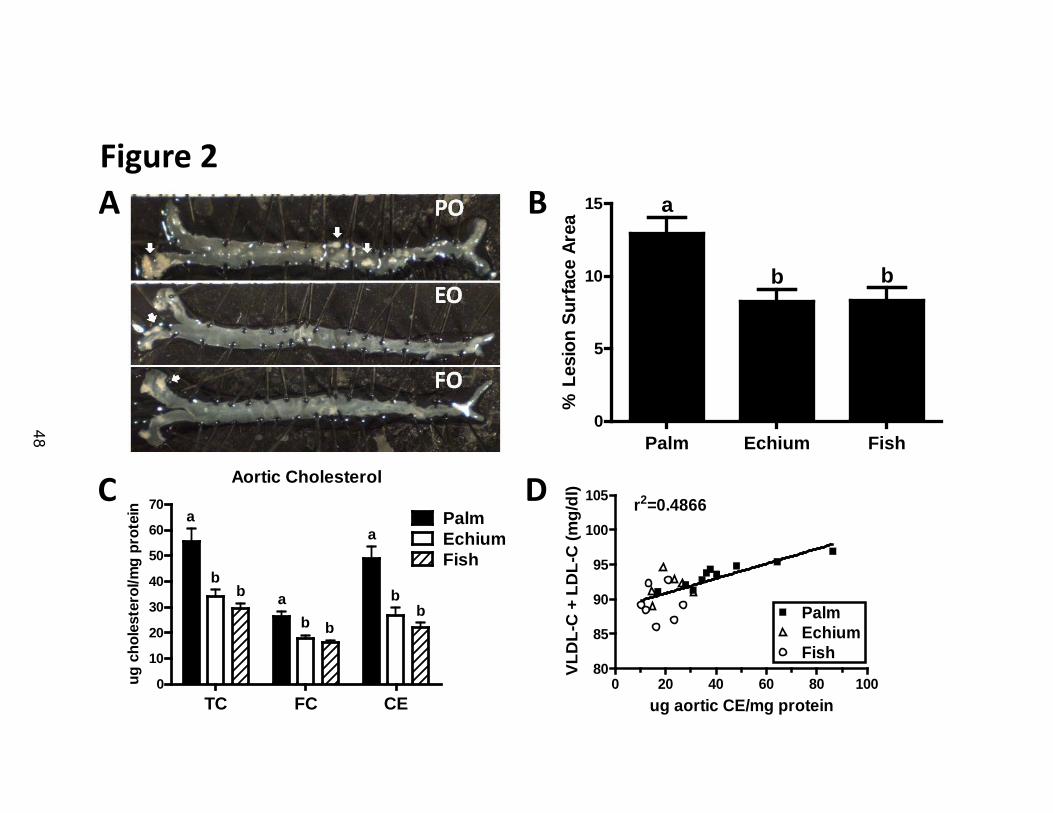
A
C D
Figure 2B
0 20 40 60 80 10080
85
90
95
100
105
PalmEchiumFish
r2=0.4866
ug aortic CE/mg protein
VLD
L-C
+ L
DL-
C (m
g/dl
)
Lesion Area
Palm Echium Fish0
5
10
15 a
bb
% L
esio
n Su
rfac
e Ar
ea
Aortic Cholesterol
TC FC CE0
10
20
30
40
50
60
70PalmEchiumFish
a
bb
bb
bb a
a
ug c
hole
ster
ol/m
g pr
otei
n
48

Reference List
1. Miniño AM, Heron MP Murphy SL Kochanek KD and Centers for Disease Control and Prevention National Center for Health Statistics National Vital Statistics System. Deaths: final data for 2004. 55(19), 1-120. 2007.
2. Kris-Etherton, P.M., W.S. Harris, and L.J. Appel. 2002. Fish consumption,
fish oil, omega-3 fatty acids, and cardiovascular disease. Circulation 106: 2747-2757.
3. Kris-Etherton, P.M., D.S. Taylor, S. Yu-Poth, P. Huth, K. Moriarty, V. Fishell, R.L. Hargrove, G. Zhao, and T.D. Etherton. 2000. Polyunsaturated fatty acids in the food chain in the United States. Am.J.Clin.Nutr. 71: 179S-188S.
4. Huang, Y.S., R.S. Smith, P.R. Redden, R.C. Cantrill, and D.F. Horrobin. 1991. Modification of liver fatty acid metabolism in mice by n-3 and n-6 delta 6-desaturase substrates and products. Biochim.Biophys.Acta 1082: 319-327.
5. Singer, P., I. Berger, M. Wirth, W. Godicke, W. Jaeger, and S. Voigt. 1986. Slow desaturation and elongation of linoleic and alpha-linolenic acids as a rationale of eicosapentaenoic acid-rich diet to lower blood pressure and serum lipids in normal, hypertensive and hyperlipemic subjects. Prostaglandins Leukot.Med. 24: 173-193.
6. Zhang, P., E. Boudyguina, M.D. Wilson, A.K. Gebre, and J.S. Parks. 2008. Echium oil reduces plasma lipids and hepatic lipogenic gene expression in apoB100-only LDL receptor knockout mice. J.Nutr.Biochem. 19: 655-663.
7. Surette, M.E., M. Edens, F.H. Chilton, and K.M. Tramposch. 2004. Dietary echium oil increases plasma and neutrophil long-chain (n-3) fatty acids and lowers serum triacylglycerols in hypertriglyceridemic humans. J.Nutr. 134: 1406-1411.
8. Heinonen, S.E., P. Leppanen, I. Kholova, H. Lumivuori, S.K. Hakkinen, F. Bosch, M. Laakso, and S. Yla-Herttuala. 2007. Increased atherosclerotic lesion calcification in a novel mouse model combining insulin resistance, hyperglycemia, and hypercholesterolemia. Circ.Res. 101: 1058-1067.
9. Lieu, H.D., S.K. Withycombe, Q. Walker, J.X. Rong, R.L. Walzem, J.S. Wong, R.L. Hamilton, E.A. Fisher, and S.G. Young. 2003. Eliminating atherogenesis in mice by switching off hepatic lipoprotein secretion. Circulation 107: 1315-1321.

10. Powell-Braxton, L., M. Veniant, R.D. Latvala, K.I. Hirano, W.B. Won, J. Ross, N. Dybdal, C.H. Zlot, S.G. Young, and N.O. Davidson. 1998. A mouse model of human familial hypercholesterolemia: markedly elevated low density lipoprotein cholesterol levels and severe atherosclerosis on a low-fat chow diet. Nat.Med. 4: 934-938.
11. Rudel, L.L., K. Kelley, J.K. Sawyer, R. Shah, and M.D. Wilson. 1998. Dietary monounsaturated fatty acids promote aortic atherosclerosis in LDL receptor-null, human ApoB100-overexpressing transgenic mice. Arterioscler.Thromb.Vasc.Biol. 18: 1818-1827.
12. Garber, D.W., K.R. Kulkarni, and G.M. Anantharamaiah. 2000. A sensitive and convenient method for lipoprotein profile analysis of individual mouse plasma samples. J.Lipid Res. 41: 1020-1026.
13. Zhang, Z.S., A.E. James, Y. Huang, W.K. Ho, D.S. Sahota, and Z.Y. Chen. 2005. Quantification and characterization of aortic cholesterol in rabbits fed a high-cholesterol diet. Int.J.Food Sci.Nutr. 56: 359-366.
14. Tsai, M.Y., J. Yuan, and D.B. Hunninghake. 1992. Effect of gemfibrozil on composition of lipoproteins and distribution of LDL subspecies. Atherosclerosis 95: 35-42.
15. Dyerberg, J. and H.O. Bang. 1982. A hypothesis on the development of acute myocardial infarction in Greenlanders. Scand.J.Clin.Lab Invest Suppl 161: 7-13.
16. Wang, C., W.S. Harris, M. Chung, A.H. Lichtenstein, E.M. Balk, B. Kupelnick, H.S. Jordan, and J. Lau. 2006. n-3 Fatty acids from fish or fish-oil supplements, but not alpha-linolenic acid, benefit cardiovascular disease outcomes in primary- and secondary-prevention studies: a systematic review. Am.J.Clin.Nutr. 84: 5-17.
17. Calder, P.C. and R.F. Grimble. 2002. Polyunsaturated fatty acids, inflammation and immunity. Eur.J.Clin.Nutr. 56 Suppl 3: S14-S19.
18. Hokanson, J.E. and M.A. Austin. 1996. Plasma triglyceride level is a risk factor for cardiovascular disease independent of high-density lipoprotein cholesterol level: a meta-analysis of population-based prospective studies. J.Cardiovasc.Risk 3: 213-219.
19. Yaqoob, P. and P.C. Calder. 2003. N-3 polyunsaturated fatty acids and inflammation in the arterial wall. Eur.J.Med.Res. 8: 337-354.

Chapter III
ADDITIONAL STUDIES ON THE EFFECT OF N-3 PUFAS ON MACROPHAGE
RECRUITMENT AND CHOLESTEROL ACCUMULATION
Lolita M. Forrest
51

Introduction
It is well-established that long chain n-3 polyunsaturated fatty acids
(PUFA), such as those found in FO, are atheroprotective. Furthermore, we have
shown that Echium oil, which is enriched in stearidonic acid (SDA), is also
atheroprotective in apoB100-only LDLrKO mice (Chapter II). However,
atherosclerosis is a complex disease that involves several metabolic processes
including lipid metabolism, gene transcription, and inflammation (1-3) and the
role of n-3 PUFAs in mediating these processes has not been fully elucidated.
Inflammation plays an important role in atherogenesis and atherosclerosis
progression. The presence of lipid-loaded macrophages is the hallmark of
atherosclerosis. The presence of macrophages within a plaque is preceded by
several key events; monocyte recruitment and adhesion to endothelial cells (EC),
and monocyte infiltration into the subendothelial space (4). Once in the
subendothelial space, monocytes can differentiate into macrophages and take up
modified atherogenic lipoproteins. Rapid accumulation of lipid by macrophages
results in foam cell development and a chronic inflammatory response that can
eventually lead to an acute cardiovascular event. While other cell types such as
smooth muscle cells (SMC) and ECs play an important role in the inflammatory
events, the macrophage is the primary cell type found in atherosclerotic plaques
and subclinical fatty streaks and is essential for atherogenesis. Here, we address
the influence of n-3 PUFAs on atherogenesis as it pertains to the macrophage.
We investigated two potential mechanisms that may explain how FO and EO
feeding reduce atherosclerosis. One potential mechanism is that fewer
52

monocytes are being recruited into the intimal space. If so, it would suggest that
the amount of cholesterol accumulation within an atherosclerotic lesion is
proportional to the number of monocyte/macrophages present. An atherosclerotic
plaque becomes increasingly more vulnerable to rupture with increasing numbers
of macrophages. FO fatty acids have been shown to incorporate into lesions and
increase plaque stability (5). Therefore, it may possible that FO lowers monocyte
recruitment.
A second potential mechanism is that n-3 PUFAs do not alter monocyte
recruitment, but rather the amount of lipid accumulation in the subendothelial
space. If so, this would result in the same number of macrophages but less
cholesterol loading in the intimal space. In Chapter II, we showed that n-3 PUFA
feeding lowers the total aortic surface area covered by lesions. Here, we
determined whether n-3 PUFA feeding in apoB100-only LDLrKO mice results in a
decreased intimal area in the aortic root. Furthermore, we calculate the number
of macrophages in the intimal area to determine if lesion area is proportional to
macrophage accumulation. These results may provide insights into the
mechanisms involved with n-3 PUFA atheroprotection.
53

Materials and Methods
Animals and Diets
Male apoB100-only LDLrKO mice in the C57BL/6 background (>99%)
were housed in a specific pathogen-free facility at Wake Forest School of
Medicine in accordance with all institutional animal care and use guidelines. After
weaning, the mice were fed a chow diet until 8 weeks of age; at that time, they
were randomly assigned to one of three diet groups designated as palm (PO),
Echium (EO), or fish oil (FO). Each diet contained 0.2% cholesterol and 10% of
calories from PO with an additional 10% of calories from PO, EO, or FO, totaling
20% calories as fat. Detailed diet compositions and fatty acid compositions have
been described previously (6,7). The atherogenic diets were fed to the mice for a
total of 16 weeks.
Heart Collection and Processing
After 16 weeks of diet feeding, mice were anesthetized with
ketamine/xylazine and terminally bled by cardiac puncture. The heart and venous
system was perfused with cold phosphate buffered saline (PBS) for 5-10 min at a
rate of 1 ml/min via the left ventricle. The heart was cut away from the aorta
before the first aortic branch then removed and transversely cut in half with a
razor blade. The upper half of the heart was then immediately embedded in a
mould with optimal cutting temperature (OCT) compound and frozen using
methylbutane on dry ice. Once frozen, the specimens were stored at -80ºC until
later use.
54

Aortic Root Sectioning
Using a cryostat, the aortic root was sectioned into 8 um sections. For
each heart, 10 ColorFrost Plus microscope slides (Fischer Scientific) were used
to collect aortic root sections. The first section was collected on slide 1, the
second on slide 2, and so on until slide 10 and then back to slide 1 and so on
until the entire aortic root had been collected. Each slide had approximately 8
sections. Therefore, each slide represented a sequential range of the aortic root.
The sections were allowed to adhere for at least 1hr at room temperature, and
then stored at -20ºC until later use.
Measurement of aortic root intimal area
Aortic root sectioned slides were stained with Oil Red O to measure
intimal area. Briefly, the sections were fixed in 10% formalin, rinsed with water,
put into 100% propylene glycol to remove water, and incubated in a filtered 0.5%
Oil Red O solution for 25 min. The sections were then rinsed with water and
counterstained with 20% hematoxylin and again rinsed with water. Cover slips
were then added to the sections using an aqueous mounting medium.
Images of the aortic root were captured at 4x magnification using a Nikon
Digital Sight DS-Fi1 camera. The intimal area of the aortic root was quantified
using Image-Pro Plus software. For each section, the sum of the areas of the
intima was calculated to determine intimal area. For each animal, the intimal area
was calculated for 3 evenly separated sections and the intimal area of each
55

animal was determined as the average intimal area of the sections. The intimal
area is represented in um2.
Staining of F4/80+ macrophages in the aortic root
Using immunohistochemical methods, the aortic roots were stained for
F4/80+ macrophages. Briefly, slides were placed in Tris buffer, then a 1:30
dilution of rat anti-mouse F4/80 antibody (Serotec) was added to slides by
capillary action. The slides were then washed several times in Tris buffer and
biotinylated goat anti-rat IgGs (Serotec) was added as a secondary antibody.
Streptavidin alkaline phosphatase (BioGenex) was used as an enzyme
conjugate. Alkaline phosphatase was used as enzyme substrate. Levamisole
was added to the alkaline phosphatase to reduce background staining by
suppressing endogenous phosphatase activity. Sections were counterstained
Mayer’s hematoxylin and immediately coverslipped with Permount to avoid loss
of stain.
Quantification of F4/80+ macrophages in the aortic root
A Dell Optiplex 780 computer with Image-Pro version 5.2 software and an
Olympus BH-2 microscope outfitted with a DS-Fi1 Nikon camera and Prior
Optiscan stage controller were used for capturing 10x images and making
morphometric measurements. The area of each lesion in the aortic root was
digitally circumscribed and the area was measured. To determine how much of
each area was occupied by cellular component (i.e. no holes) the area of the
holes were also measured. The actual lesion area of interest was calculated as
56

the total area – hole area. Using the color picking feature, F4/80+ stained
macrophages were identified and the total area of the macrophages was
calculated. The percent of total lesion occupied by F4/80+ macrophages was
calculated as:
Area of F4/80+ macrophages / (Total intimal area of interest – hole area)
Statistical Analysis
All data are presented as mean ± SEM. Differences among the 3 diet
groups were analyzed by one-way ANOVA (p<0.05) using GraphPad Prism
software. Individual diet differences were identified using Tukey’s post-test
analysis.
57

Results
n-3 PUFAs do not decrease aortic root intimal area in apoB100-only LDLrKO
mice
It has been shown that LDLrKO mice develop extensive atherosclerosis
along the entire aortic tree (8), though anatomic predilection sites exist such as
the aortic sinus and brachiocephalic artery. Using techniques first described by
Paigen et al. (9), cross-sections of the aortic root have also been used to
determine lesion area. Tangirala and colleagues used cholesterol-fed LDLrKO
mice to determine whether there was a correlation between the extent of
atherosclerosis in the entire aorta and the size of lesions in the aortic origin and
indeed found a significant (r = 0.77) correlation (10). Surprisingly, in our study,
we did not get a similar finding. We have shown previously a significant reduction
in percent aortic lesion area with n-3 PUFA feeding (Chapter II). However, as
shown in Figure 1B, aortic root intimal area is similar among all three diet
groups. Interestingly, our average lesion areas were 66% greater than those
observed by Tangirala et. al. Whereas our model, fed 0.2% cholesterol for 16
weeks results in approximately 500,000 um2 lesions in the aortic root, their
LDLrKO mice fed 1% cholesterol 12 weeks resulted in approximately 300,000
um2 lesions.
n-3 PUFAs do not decrease macrophage accumulation in the aortic root of
apoB100-only LDLrKO mice
58

Macrophage-mediated inflammation is a central feature of atherogenesis.
This involves the recruitment of monocytes to the artery wall, monocyte
differentiation into macrophages, uptake of modified LDL, and stimulation of a
chronic pro-inflammatory response. Monocyte recruitment is caused by EC
expression of adhesion molecules and chemotactic factors. Dietary fish oil has
been shown to reduce macrophage intercellular adhesion molecule 1 (ICAM-1)
expression in wild-type mice (11). Similar findings have also been shown in
human monocytes (12). Here we quantified the numbers of F4/80+ macrophages
in the aortic root lesions from mice fed PO, EO, or FO. Although we hypothesized
that n-3 PUFA supplementation would result in a decreased number of
macrophages within the intimal space, that is not what we observed. For all
groups, approximately 25% of the aortic root lesions were occupied by
macrophages.
59

Discussion
In the study of atherogenesis, much attention is given to the accumulation
of cholesterol in the artery wall because high plasma LDL concentrations have
been shown to be a primary risk factor for atherosclerosis (13). However,
atherosclerosis is a dynamic disease process that also includes an inflammatory
component (3). We have shown in our mouse model that, compared to PO, both
EO and FO reduce atherosclerosis, measured as percent surface lesion area.
However, we measured intimal area in the aortic root and a similar finding was
not observed. Furthermore, we found that the number of macrophages in the
aortic root was similar among groups. Hemodynamic forces contribute to
atherogenesis. The aortic root is an area of low and oscillatory shear stress due
to the movement of the valve leaflets, making it vulnerable to endothelial
dysfunction and plaque formation (14). Therefore, it is possible that changes in n-
3 PUFA enrichment may not be enough to overcome more atherogenic factors,
such as shear stress and hypercholesterolemia. The fact that we did not observe
any differences in lesion area or macrophage quantity means that in our model,
lesion size is proportional to macrophage accumulation. FO fatty acid
incorporation has been shown to correlate with increased plaque stability (5).
Therefore, it is possible that n-3 PUFAs may not reduce plaque size but still offer
atheroprotection by suppressing inflammation and plaque rupture.
60

Acknowledgements: Special thanks to Hermina Borgerink of the Department of
Pathology- Section on Comparative Medicine for her immunohistochemistry
technical support.
61

Figure 1: Aortic root intimal area. A. A representative section of an aortic root
measured for intimal area. 10 um serial sections were collected and stained with
Oil Red O. Using Image-Pro Plus software, the intimal area was circumscribed
(marked in yellow) and its area measured. B. Aortic root intimal area was
measured in apoB100-only LDLrKO mice after 16 wks of PO, EO, or FO feeding
(n = 12, 11, and 4, respectively). Three sections from each mouse were
measured and the average was used to determine intimal area for each mouse.
The bars represent the mean ± SEM. The average coefficient of variation (CV)
was 0.17 among all groups.
62

Aortic Root Intimal Area
PO EO FO0
200 000
400 000
600 000
800 000
Intim
al a
rea
(um
2)A
Figure 1
B
63

Figure 2: Computer imaging analysis of aortic root macrophage
accumulation. A. A representative image of an aortic root stained for F4/80+
macrophages using immunohistochemical techniques. Images were captured at
4x magnification. B. Using Image-Pro Plus software, the color picking option was
used to identify all F4/80+ macrophages (highlighted in red).
64

AFigure 2
B
65

Figure 3: F4/80+ macrophages in the aortic root. Mice were fed PO, EO, or
FO diets for 16 weeks (n = 12, 11, 6, respectively) and the aortic root sectioned
for macrophage quantification. The area of lesion occupied by F4/80+
macrophages was calculated as the total area of F4/80+ macrophages within the
intimal area / (total intimal area minus the sum of the area of holes (i.e. acellular
regions)). Three to four sections were quantified for each mouse to get a mean
value. The bars represent the mean ± SEM for each diet group.
66

Figure 3
F4/80+ Macrophages in Aortic Root
PO EO FO0
10
20
30
40
% L
esio
n O
ccup
ied
by MΦ
67

Reference List
1. Lusis, A.J. 2000. Atherosclerosis. Nature 407: 233-241.
2. Jump, D.B. 2002. Dietary polyunsaturated fatty acids and regulation of gene transcription. Curr.Opin.Lipidol. 13: 155-164.
3. Ross, R. 1999. Atherosclerosis--an inflammatory disease. N.Engl.J.Med. 340: 115-126.
4. Glass, C.K. and J.L. Witztum. 2001. Atherosclerosis. the road ahead. Cell 104: 503-516.
5. Thies, F., J.M. Garry, P. Yaqoob, K. Rerkasem, J. Williams, C.P. Shearman, P.J. Gallagher, P.C. Calder, and R.F. Grimble. 2003. Association of n-3 polyunsaturated fatty acids with stability of atherosclerotic plaques: a randomised controlled trial. Lancet 361: 477-485.
6. Rudel, L.L., K. Kelley, J.K. Sawyer, R. Shah, and M.D. Wilson. 1998. Dietary monounsaturated fatty acids promote aortic atherosclerosis in LDL receptor-null, human ApoB100-overexpressing transgenic mice. Arterioscler.Thromb.Vasc.Biol. 18: 1818-1827.
7. Zhang, P., E. Boudyguina, M.D. Wilson, A.K. Gebre, and J.S. Parks. 2008. Echium oil reduces plasma lipids and hepatic lipogenic gene expression in apoB100-only LDL receptor knockout mice. J.Nutr.Biochem. 19: 655-663.
8. Ishibashi, S., M.S. Brown, J.L. Goldstein, R.D. Gerard, R.E. Hammer, and J. Herz. 1993. Hypercholesterolemia in low density lipoprotein receptor knockout mice and its reversal by adenovirus-mediated gene delivery. J.Clin.Invest 92: 883-893.
9. Paigen, B., A. Morrow, P.A. Holmes, D. Mitchell, and R.A. Williams. 1987. Quantitative assessment of atherosclerotic lesions in mice. Atherosclerosis 68: 231-240.
10. Tangirala, R.K., E.M. Rubin, and W. Palinski. 1995. Quantitation of atherosclerosis in murine models: correlation between lesions in the aortic origin and in the entire aorta, and differences in the extent of lesions between sexes in LDL receptor-deficient and apolipoprotein E-deficient mice. J.Lipid Res. 36: 2320-2328.
11. Miles, E.A., F.A. Wallace, and P.C. Calder. 2000. Dietary fish oil reduces intercellular adhesion molecule 1 and scavenger receptor expression on murine macrophages. Atherosclerosis 152: 43-50.
68

12. Hughes, D.A., A.C. Pinder, Z. Piper, I.T. Johnson, and E.K. Lund. 1996. Fish oil supplementation inhibits the expression of major histocompatibility complex class II molecules and adhesion molecules on human monocytes. Am.J.Clin.Nutr. 63: 267-272.
13. Sytkowski, P.A., W.B. Kannel, and R.B. D'Agostino. 1990. Changes in risk factors and the decline in mortality from cardiovascular disease. The Framingham Heart Study. N.Engl.J.Med. 322: 1635-1641.
14. Chappell, D.C., S.E. Varner, R.M. Nerem, R.M. Medford, and R.W. Alexander. 1998. Oscillatory shear stress stimulates adhesion molecule expression in cultured human endothelium. Circ.Res. 82: 532-539.
69

Chapter IV
MECHANISMS FOR PLASMA TRIGLYCERIDE REDUCTION BY ECHIUM OIL
ARE DISTINCT FROM THOSE OF FISH OIL IN APOB100-ONLY LDL
RECEPTOR KNOCKOUT MICE
Lolita M. Forrest, Christopher Lough, Elena Boudyguina, Abraham Gebre,
Thomas L. Smith, Perry L. Colvin, and John S. Parks
The following manuscript is in preparation for submission to the Journal of Lipid
Research. Stylistic variations are due to the requirements of the journal. The
experimentation and writing were performed by LM Forrest. Dr. JS Parks acted in
an advisory and editorial capacity.

Abstract
Plasma triglyceride (TG) reduction by dietary fish oil (FO) is attributed to
two long-chain n-3 polyunsaturated fatty acids (PUFAs), EPA (20:5 n-3) and DHA
(22:6 n-3). We previously showed that Echium oil (EO), a botanical oil substitute
for FO, which is enriched in SDA (18:4 n-3), is converted in vivo to EPA resulting
in reduced plasma TG concentrations in humans and mice. In this study, we
compare the mechanisms by which EO and FO reduce plasma TG
concentrations using male apoB100-only LDLrKO mice, a mildly
hypertriglyceridemic atherosclerosis mouse model. At eight weeks of age, mice
were fed one of three atherogenic diets containing 0.2% cholesterol and 20% cal
as fat: palm oil (PO) diet (20% PO), EO diet (10% PO + 10% EO), or FO diet
(10% PO + 10% FO). Relative to PO-fed mice, plasma TG levels were
significantly lower in EO and FO groups over a 28 day period. Surprisingly, livers
from PO- and EO-fed mice had similar TG and cholesteryl ester (CE) content,
and CE content was significantly higher than that of livers from FO-fed mice.
After blocking TG lipolysis in vivo with intravenous detergent injection, FO-fed
mice had decreased plasma TG accumulation compared with EO-fed mice.
Plasma VLDL particle size was ordered: PO (63±4 nm) > EO (55±3 nm) > FO
(40±2 nm). Post-heparin lipolytic activity was similar among the groups of mice,
but TG hydrolysis by purified lipoprotein lipase (LPL) was significantly greater for
EO and FO VLDL compared to PO VLDL. Removal of plasma VLDL tracer from
plasma was marginally faster in EO vs. PO fed mice. Our results suggest that EO
reduces plasma TG primarily through increased intravascular lipolysis of TG and

clearance of VLDL. Furthermore, EO may substitute for FO to reduce plasma TG
concentrations, but not hepatic steatosis.

Introduction
Dysregulation of lipid metabolism results in several disease states
affecting the US population, including diabetes, obesity, and cardiovascular
disease (CVD). Overwhelming evidence supports increased dietary consumption
of n-3 polyunsaturated fatty acids (PUFAs) for CVD prevention and treatment (1-
3). In particular, epidemiological studies conducted by Bang and Dyerberg first
reported the beneficial effects of fish oil (FO) on the plasma lipid levels and CVD
of Greenland Eskimos compared to those living in Denmark (4-6). The beneficial
effects of FO are attributed to its n-3 fatty acid enrichment of eicosapentanoic
acid (EPA, 20:5) and docosahexaenoic acid (DHA, 22:6). Since then, many
animal and human studies have shown that dietary FO or formulations of EPA
and DHA are cardioprotective (7-11), due to decreased inflammation, plasma
lipid concentrations, endothelial cell activation, and cardiac arrhythmia (12-15).
The most consistent effect of dietary FO on plasma lipid concentrations across all
species examined is a reduction in triglyceride (TG) levels (16,17). Furthermore,
while high plasma LDL cholesterol concentrations are associated with increased
CVD risk, there is evidence that plasma TG levels are an independent risk factor
(18).
While overwhelming data support the effectiveness of FO at reducing
plasma TG concentrations, the mechanisms driving its potent triglyceride-
lowering effect have not been fully resolved. One proposed mechanism is that n-
3 PUFAs regulate the expression of genes involved in lipogenesis, including
SREBP1c and PPAR alpha (19-22). Other studies in humans and rodents have

suggested FO-mediated TG-lowering to be a cumulative effect of increased fatty
acid beta-oxidation, reduced TG synthesis and secretion, and enhanced plasma
TG-rich particle clearance (23-25).
Despite the well-documented beneficial effects of fish and FO
supplements, they are poorly consumed by the US population. The
recommended dietary intake ratio of n-6:n-3 PUFAs is 2.3:1, but North
Americans consume a ratio closer to 10:1 (26,27). Potential reasons for the
resistance to FO and/or fish consumption include cost, personal preference, and
FO supplement aftertaste. Approximately 90% of n-3 PUFA in North American
diets comes from vegetable oil derived alpha-linolenic acid (ALA) (26). However,
ALA is not sufficiently metabolized to longer-chain PUFAs, such as EPA and
DHA, due to the inefficiency of Δ-6 desaturation in mammalian species (28).
Therefore, finding a suitable alternative to FO to increase consumption of long
chain n-3 PUFAs may considerably improve cardiovascular health.
Echium oil (EO), derived from Echium plantagineum seeds, may serve as
a comparable alternative to FO. Approximately 13% of the total fatty acids in
Echium oil is stearidonic acid (SDA; 18:4 n-3), the immediate product of ALA Δ-6
desaturation (29). Surette et al. showed that hypertriglyceridemic human
subjects supplemented with EO had decreased plasma TG concentrations, as
well as increased EPA enrichment in plasma and neutrophils, suggesting that
SDA in EO was efficiently elongated and desaturated to EPA (19). Furthermore,
our lab has shown reduced plasma TG and hepatic lipogenic gene expression in
mice after 8 weeks of EO supplementation (30).

The mechanisms by which EO reduces plasma TG concentration have not
been explored. In the present study, we examined the effects of dietary EO and
FO on mechanisms of VLDL production and catabolism in male apoB100-only
LDLrKO mice. This mouse model of atherosclerosis exhibits mildly elevated
plasma TG levels (31). The goal of this study was to determine whether the
mechanisms of TG lowering for EO were similar to those of FO.

Materials and Methods
Animals and Diets
Male apoB100-only mice were created by Dr. Steve Young (32). They
were then crossed with LDLrKO mice and used in a mixed C57BL/6 background
(~93%) for previous studies (30,33). Mice used for this study were backcrossed
5-6 times with C57BL/6 LDLrKO mice and were determine to be >99% in the
C57BL/6 background by genome wide screens using 134 single nucleotide
polymorphisms that were polymorphic between the C57BL/6 and 129/SVEV
strains and spaced approximately 20Mb across the mouse genome. The animals
were housed in a specific-pathogen free facility at Wake Forest School of
Medicine. All experimental procedures were reviewed and approved by the
Institutional Animal Care and Use committee.
At 8 weeks of age, mice were randomly assigned to one of three
experimental diets designated as palm oil (PO), fish oil (FO), or Echium oil (EO).
Each diet contained 0.2% cholesterol and 10% of calories from palm oil with an
additional 10% of calories from PO, FO, or EO for a total of 20% calories as fat.
Complete diet composition (8) and fatty acid composition (30) have been
previously described. All experiments were performed on mice after 4-8 weeks
of diet consumption except for liver lipid analysis, which was conducted on a
cohort of animals fed for 16 weeks.

Plasma Lipid Analysis
All blood samples were collected via tail bleeding into 75ul heparinized
capillary tubes from 4 h fasted mice. Plasma was isolated by spinning the blood
at 12,000 rpm (4°C) for 10 minutes and used immediately or stored at -20°C for
later use. Plasma cholesterol (Wako) and TG (Roche) were determined by
enzymatic analysis as described previously (34). ApoB concentration was
determined by SDS-PAGE and Western blotting using 2ul of whole plasma. The
separated proteins were then transferred to a nitrocellulose membrane and apoB
was visualized using a 1:5000 dilution of apoB100 goat anti-human primary
antibody (Biodesign), 1:5000 α-goat IgG HRP secondary antibody (Sigma), and
chemiluminescence substrate (Thermo Scientific). ApoB band intensity was
quantified using a Fuji Film Cold Camera and Multi-Gauge Software. Plasma
lipids were extracted using the Bligh-Dyer method (35) for fatty acid analysis.
Lipids were fractionated into PL, TG, and CE bands by thin-layer
chromatography using a neutral solvent system. Each fraction was methylated
and analyzed by gas-liquid chromatography for fatty acid content as previously
described (34).
RNA Analysis
TRIzol reagent was used to isolate total RNA from livers of mice after 16
weeks of experimental diet feeding. Quantitative real-time PCR was performed
using SYBR Green PCR master mix (Applied Biosystems) and the 2-ΔΔCT method
(36).

VLDL Characterization
After 16 weeks of experimental diet feeding, mouse plasma VLDL was
isolated to determine size, lipid and protein composition, and particle number.
VLDL was isolated from plasma by ultracentrifugation at 100,000 rpm for 4hrs at
15ºC at a density of 1.006. The VLDL fraction was removed by tube slicing and
re-isolated to minimize albumin contamination. VLDL was used immediately or
stored at -20ºC for later use. VLDL total cholesterol, free cholesterol and TG
concentrations were determined by colorimetric enzymatic assays (34).
Cholesteryl ester was calculated as TC - FC x 1.67 (to correct for loss of fatty
acid after saponification). PL was quantified by a PL phosphorous assay (37).
Protein concentration was determined by micro BCA assay. The surface to core
ratio of VLDL constituents was calculated from the measured chemical
constituents and used as a surrogate measurement of VLDL particle size. Two ul
of isolated VLDL dissolved in 43ul saline was used to determine VLDL size by
light scatter analysis using a Zetasizer Nano Series light scatter instrument
(Malvern Instruments). The resulting profile was analyzed using Disperson
Technology Software 4.2 (Malvern Instruments) and VLDL size was quantified as
the primary peak in the analysis by volume.
Hepatic Lipid Analysis
An average of 65 mg of perfused livers from mice fed for 16 weeks were
used to conduct liver lipid analysis. Lipids were extracted using 2:1
chloroform:methanol, 1% triton X-100 in chloroform was added, and the mixture

was dried under a stream of N2 gas at 60°C and resuspended in water for a 1:2
final ratio of dH2O:triton X-100. TG, TC, FC, and CE content were then quantified
by enzymatic analysis as described earlier.
Determination of plasma post-heparin lipase activity
Plasma lipoprotein lipase (LPL) and hepatic lipase (HL) activities were
quantified using post-heparin plasma as a source of enzyme and a micellar
artificial substrate. Briefly, animals were fasted for 4 hours before tail vein
injection of 100 units/kg (180mg/kg) heparin (sodium salt dissolved in saline) to
release capillary-bound lipases. Ten minutes post-injection, a blood sample was
taken and plasma was separated by low speed centrifugation. The substrate for
LPL activity was prepared and the assay performed as previously described
(38,39), except 10ul plasma was used as the enzyme source and 1ug
apolipoprotein CII (Sigma) was added to each reaction to activate LPL. Hepatic
lipase activity was determined by adding 95ul 4M NaCl to inhibit lipoprotein
lipase. Lipoprotein lipase activity was calculated as the difference between the
total lipase activity and HL activity.
VLDL Lipolysis by LPL
To determine the extent to which the different diets affected VLDL TG
hydrolysis, we isolated plasma VLDL from mice consuming the three
experimental diets and incubated the VLDL particles with LPL in vitro . VLDL (2ug
TG) was added to a microtiter plate and adjusted to equal volumes with water.
Twenty-five ng of LPL (Sigma) and 1ug of apoCII (as activator, Sigma) were

added to initiate the hydrolysis. Duplicate samples minus apoCII were used to
subtract out background lipolysis. The samples were incubated at 37°C for 1h,
after which release of non-esterified fatty acids (NEFA) was quantified using a
modification of the NEFA C method (Wako). NEFA C reagents were added per
manufacturer’s instructions and the amount of free fatty acids released was
measured by a colorimetric reaction, using oleic acid (OA) standards. Pilot
studies were performed to establish that these conditions resulted in a linear
increase in fatty acid hydrolysis as a function of increasing VLDL TG substrate.
Lipase activity was calculated as µmol FA released/hr/ml.
TG synthesis rate analysis
Hepatic secretion rate was determined in mice fed the experimental diets
for 4-6 weeks. After a 4hr fast, mice were anesthetized with ketamine/xylazine
and a baseline retro-orbital blood sample was taken. Then, mice were injected
retro-orbitally with an equal volume of Triton WR 1339 (500 mg/kg mouse) and
0.9% NaCl (40). Blood samples were taken at 45, 90, and 180 minutes post-
injection. Plasma TG concentration was measured by enzymatic analysis as
described earlier. Hepatic secretion rates were determined by calculating the
slope of TG values over the time course (0 to 180 min) using GraphPad Prism
software.
Turnover Studies
To determine plasma VLDL removal rates, turnover studies were
conducted. One PO-fed and two EO-fed mice, fed their respective diets for at

least 16 weeks, were used as donor mice. Mice were terminally bled by cardiac
puncture and plasma was collected as previously described. Plasma from the
two EO-fed mice was pooled. VLDL was then isolated from both samples as
previously described. VLDL (150 ug protein) was radioiodinated with 50 uCi of
radioactive iodine (PO-VLDL with 125-I, and EO-VLDL with 131-I) using the ICl
method (41) and desalted on a 10 ml Bio-Rad-Pac10 disposable chromatography
column. VLDL was then dialyzed in 12mM phosphate buffer containing 124mM
NaI (pH 7.4) and radiolabel and protein was measured to calculate specific
activity (i.e., dpm/µg protein). 550,000 cpm of each VLDL dose was combined
and injected into PO-fed and EO-fed recipient mice via the jugular vein. Blood
was collected 5 min, 30 min, 1, 2, 4, 6, 8, and 24 hours post-injection. A multi-
compartmental model (SAAM II program) was used to estimate the fractional
catabolic rate (FCR) as described previously (42). Plasma was separated as
describe earlier and apoB radioactivity was measured after isopropanol
precipitation by gamma counting. Die-away curves were plotted as % of 5 min
plasma radioactivity. Plasma samples were also separated by FPLC to determine
the amount of radiolabel remaining as VLDL 30min, 3hr, and 8hr post-injection.
Statistical Analysis
All data are presented as mean ± SEM. Differences among the 3 diet
groups were analyzed by one-way ANOVA (p<0.05) using GraphPad Prism
software. Individual diet differences were identified using Tukey’s post-test
analysis.

Results
Echium oil reduces plasma triglyceride concentrations
Previous studies from our lab showed that apoB100-only LDLrKO mice
consuming the EO diet had lower plasma TG concentrations over an eight week
study period compared to those consuming the PO diet (30). In the previous
study, eight week old mice were fed a “lead in” diet containing 0.2% cholesterol
and 10% of calories as PO for four weeks before initiation of experimental diets
that were the same as those used in the current study (i.e., 0.2% cholesterol and
20% PO, 10% PO + 10% EO, or 10% PO + 10% FO). In this study, mice were
switched from chow to the experimental diets at eight weeks of age and total
plasma cholesterol (TPC) and TG concentrations were examined at earlier times
after experimental diet initiation. Four days after diet initiation, TPC
concentrations increased nearly three-fold for all three diet groups compared to
the chow baseline (i.e., zero time point). Thereafter, TPC increased with time, but
differences in values were not seen groups over the 28 day period (Figure 1A,
1B, AUC= area under the TPC vs. time curve). Plasma TG concentrations fell
50% within four days of experimental diet feeding and remained unchanged for
the EO and FO groups, but increased to significantly higher values for the PO
group (Figure 1C, 1D). These data show the rapid hypotriglyceridemic effect of
the n-3 diets in our model. Furthermore, in a separate cohort fed the
experimental diets for 16 weeks, we observed that body and liver weights were
similar among groups throughout the study. Additionally, after 16 weeks, plasma

PL, TG, and CE fatty acid compositions reflected that of the experimental diets
(Supplemental Figure 1), similar to our previous 8 week study (30).
Echium oil alters VLDL composition
Since plasma TG levels were strikingly decreased for the two n-3 diet
groups, a terminal blood sample was collected after 16 weeks of diet feeding to
isolate plasma VLDL for measurement of chemical concentration and particle
size. VLDL particles from EO and FO fed mice had significantly decreased
percentage CE and corresponding increases in percentage PL and protein
(Figure 2A). These compositional differences suggested a difference in VLDL
particle size, since the n-3 groups had decreased core and increased surface
constituents. Calculation of the surface to core ratio (i.e., FC + PL + protein / CE
+ TG), an indirect measurement of VLDL particle size, suggested smaller VLDL
particles in the EO and FO groups (i.e., increased surface to core ratio; Figure
2B). Further, analysis of VLDL particle size by laser light scatter revealed a trend
for decreased particle size for the EO and FO groups compared to the PO diet
group that reached statistical significance for the FO vs. PO comparison (Figure
2C). To determine whether the number of apoB-containing particles in plasma
was altered by diet, we measured whole plasma apoB particle concentration by
Western blot. Although there was a trend towards decreased apoB concentration
in plasma for the EO and FO groups, it did not reach statistical significance
(Figure 2D).

Echium oil does not affect liver lipid content and gene expression
Our lab has previously shown that feeding the EO diet for eight weeks
resulted in a trend towards decreased hepatic TG and CE content compared to
the PO diet (30). To determine whether this trend reached statistical significance
over a longer experimental diet period, we measured hepatic lipid content after
the 16 week diet period. To our surprise, the hepatic lipid content for the EO
group was actually more similar to that of the PO group than in our shorter study,
whereas the FO fed mice had significant decreases in hepatic TC and TG, while
PL remained similar among all groups (Figure 3). Furthermore, genes involved in
TG biosynthesis, such as SREBP1-c, ACC, SCD-1 and FAS, were expressed to
a similar extent for EO vs. PO fed mice, whereas expression was reduced, in
general, for FO-fed mice (Figure 4).
EO does not affect liver TG secretion rate
One potential mechanism for a reduction in plasma TG concentration in
EO-fed mice is a decrease in hepatic VLDL TG secretion into plasma. To test this
possibility, mice fed PO, EO, and FO diets for 4-6 weeks were fasted for 4hrs
and injected with Triton WR 1339 (500mg/kg mouse) to inhibit plasma lipase
activity (40). The accumulation of TG in plasma was subsequently measured
over three hours (Figure 5). The accumulation rate of plasma TG was
significantly less for mice fed FO compared to those fed EO, whereas the
accumulation for PO fed animals was intermediate. Thus, hepatic VLDL TG

secretion could not account for the reduced plasma TG concentration for EO vs.
PO fed mice.
EO VLDL particles are more susceptible to hydrolysis
Studies suggest that lowered TG concentrations that accompany FO
consumption may be mediated by increased lipolysis (43,44). However, a study
in hypertriglyceridemic human subjects given FO for 4 weeks resulted in
decreased plasma TG without affecting LPL and HL activity (45). To determine
whether the reduced plasma TG concentrations in EO-fed mice were due to
increased TG lipolysis, we measured post-heparin lipase activity using a
standard TG micellar substrate to test for increased lipase activity and we
isolated plasma VLDL and measured TG lipolysis using purified LPL to test for
increased lipolysis of TG with EO VLDL. As shown in Figure 6A, HL and LPL
activities were similar among the three diet groups, suggesting the decreased
plasma TG in EO-fed mice was not due to increased lipase activity. However,
incubation of isolated VLDL with purified LPL resulted in significantly increased
levels of VLDL TG lipolysis, measured as free fatty acid release, for VLDL
isolated from EO and FO-fed mice compared to those from PO-fed mice (Figure
6B).
EO has minimal impact on plasma VLDL particle turnover
Since EO VLDL particles were lipolyzed to a greater extent by purified LPL
compared to PO VLDL, we investigated whether EO VLDL particles had
increased removal rates from plasma. After a 4 h fast, PO and EO-fed recipient

mice were injected with a mixture of 125I-radiolabeled PO VLDL and 131I-
radiolabeled EO VLDL in a cross-over design. Plasma samples were taken from
the recipient mice over a 24 h time period and apoB radiolabel was quantified
after isopropanol precipitation of apoB from plasma (46). Plasma die-away
curves for VLDL apoB are shown in Figure 7. Because the recipient mice lack
active LDL receptors, VLDL removal from plasma was slow relative to wild type
mice (47). Plasma die-away curves were similar for both diet groups regardless
of the source of VLDL tracer (Figure 7A and 7B). Several plasma time points
taken after VLDL tracer injection were size-fractionated by FPLC to determine
whether the radiolabel remained in the VLDL fraction or was converted to LDL
sized particles. The results in Figure 7C and 7D suggest that most of the VLDL
tracer remained in the VLDL size range, suggesting minimal conversion of tracer
to LDL particles during the first 8 h of the turnover study. There also appeared to
be a trend towards reduced radiolabel in plasma at the 3 and 8 h time points
compared with the 30 min sample for EO recipients, regardless of the source of
the VLDL tracer (Figure 7C and 7D). However, fractional catabolic rate
(pool/day) for both VLDL tracers was similar in PO (0.603 ± 0.123; n=5) and EO
(0.783 ± 0.057; n=5) recipient mice, suggesting minimal effect of dietary fat type
on VLDL catabolism in the absence of LDL receptors.

Discussion
Diets enriched in FO result in a significant decrease in plasma TG
concentrations in humans and animals. Previous studies have shown that EO, a
botanical source of n-3 PUFAs, also results in significant reduction in plasma TG
concentrations in humans and mice relative to oils devoid of n-3 PUFAs (30,48).
The goal of our study was to determine the mechanisms that mediate the plasma
hypotriglyceridemic effects of EO and whether they are similar to those of FO.
Using a mildly hypertriglyceridemic mouse model, we observed similar reductions
in plasma TG concentrations for EO and FO relative to PO over the time course
of the study. However, upon evaluation of hepatic VLDL TG production rates,
plasma apoB concentrations, VLDL TG lipolysis by LPL, post-heparin plasma
lipolytic activity, and VLDL particle turnover, we discovered that FO exerts its
hypotriglyceridemic effect by decreasing hepatic VLDL TG production and
increasing plasma VLDL TG lipolysis, whereas only the latter mechanism was
observed for EO-fed mice. Our results suggest the EO may be a suitable
botanical alternative to FO for n-3 PUFA enrichment and reduction of plasma TG
concentrations, but not hepatic steatosis, at least in this animal model.
One proposed mechanism for FO-mediated TG lowering is a reduction in
TG synthesis and secretion into plasma. Lipoprotein kinetic studies in humans,
nonhuman primates, and animals have shown that FO reduces VLDL-TG
production and secretion (49-52). Furthermore, studies suggest that FO-
mediated TG reduction is secondary to primarily reduced fatty acid availability
(53). We found that with prolonged feeding (16 wks), hepatic TG content is

significantly reduced with FO feeding compared to PO, but not with EO feeding.
This observation was the first indicator that, although both diets resulted in
reduced plasma TG concentration, the mechanisms involved may be different.
The possibility of unique TG-lowering mechanisms for FO vs. EO became more
evident when we observed similar trends in hepatic TG secretion during our
detergent block studies for the PO and EO fed groups (Figure 5).
These findings are supported by relative increases in lipogenic gene
expression in EO- vs. FO-fed mice. Sterol regulatory element binding protein 1c
(SREBP1c) has been shown to be a major target of PUFA control in the liver
(54). N-3 PUFAs decrease the expression of SREBP1c, which acts indirectly to
induce lipogenesis by inducing transcription of several lipogenic genes, such as
fatty acid synthase (FAS), acetyl CoA carboxylase (ACC), and stearoyl CoA
desaturase-1 (SCD-1) (55-57). Additionally, our lab has previously shown in the
apoB100-only LDLrKO mouse model that both EO and FO reduce hepatic
SREBP1c, FAS, and SCD-1 expression after 8 weeks of feeding (30). However,
this observation was not maintained after 16 weeks of feeding. This suggests
that prolonged EO feeding is sufficient to maintain significantly lower plasma TG
concentrations compared to PO, even under conditions of apparent hepatic
lipotoxicity.
Pan et al. suggest that PUFA peroxidation is involved in the regulation of
apoB degradation (58). Alterations in apoB determine how long it remains in the
liver for lipidation, eventually resulting in varying amounts of TG enrichment, and
sizes of VLDL particles secreted from the liver (59). We did not observe any diet

differences in plasma apoB, i.e., VLDL particle number, consistent with results in
other animal models, including non-human primates (60,61). When cultured rat
hepatocytes were incubated with either EPA or DHA, VLDL secretion was
impaired in comparison to OA (62). Furthermore, DHA incubation resulted in
less TG secretion in comparison to EPA. This may partially explain why FO, but
not EO, resulted in decreased TG secretion in comparison to PO in our studies.
FO reduces plasma TG by reducing lipogenesis; however, other studies
have suggested increased TG lipolysis and removal by FO as another potential
mechanism. Our turnover studies in PO and EO fed mice showed a subtle
increase in the removal of EO VLDL particles from plasma. A study by Harris et.
al. of short term (3-5 wks) FO feeding in humans was shown to increase the
fractional catabolic rate for VLDL-TG, and suggested decreased VLDL size (50).
Decreased VLDL size may be a function of increased lipase activity; however,
there are conflicting reports as to whether increased lipolysis is proportional to
lipase activity (63,64). Weintraub et al. has shown that PUFA-enriched
chylomicrons are better substrates for lipoprotein lipase (LPL) than those
containing SFAs in vitro (65), suggesting increased enzyme-substrate kinetics
with FO. Our VLDL chemical analysis showed that n-3 PUFA enrichment
decreases VLDL particle size. We found that altered VLDL size and composition
results in changes in lipolysis susceptibility. Lipase activity was not altered
among the three groups, but VLDL from EO and FO fed mice were more easily
hydrolyzed. These findings show that cell enrichment with n-3 PUFAs can alter

the physical properties of lipoproteins allowing for more efficient substrate-
enzyme kinetics.
In summary, we have shown in apoB100-only LDLrKO mice that plasma
TG concentration is reduced in EO and FO fed mice compared to those fed PO.
However, TG lowering by FO and EO are not occurring by parallel mechanisms.
FO feeding appears to lower plasma TG by reducing hepatic TG secretion and
increasing VLDL hydrolysis susceptibility. Alternatively, EO feeding appears to
lower plasma TG primarily by increasing intravascular lipolysis, with no effect on
hepatic TG secretion. Also we have shown that while EO reduces plasma TG
concentrations, it does not protect from hepatic steatosis in this mouse model,
contrary to FO feeding. Therefore, it is likely that DHA plays a greater role in
reducing hepatic TG levels than EPA. Further studies are necessary to
completely understand the contributions of EPA and DHA to hepatic lipid
metabolism. Many of our studies show that EO possesses cardioprotective
properties compared to PO, and at times, these protective effects are
comparable to FO feeding. These findings suggest that EO is a promising
alternative to fish oil in the treatment of CVD. Furthermore, with the possibility of
greater compliance than FO, a botanical source of PUFAs such as Echium oil
has the potential to greatly impact the incidence of CVD in the United States.

Acknowledgements: This work was funded by grant numbers P50AT002782,
3P50AT002782-03S1, R01HL094525, and P01HL049373. A special thanks to
Croda Chemicals for providing us with the Echium oil used in our diets. Special
thanks also to Omega Protein Inc. for providing OmegaPure refined menhaden
oil for our FO diet.

Figure 1. Plasma cholesterol and TG. Following a 4hr fast, blood was collected
via tail bleeding into 75ul heparinized capillary tubes. Plasma was isolated from
blood by centrifugation at 12,000 rpm at 4ºC. Animals were bled at baseline (0
days), and after 4, 8, 12, 20, 24, and 28 days on diet. Plasma total cholesterol
(TC, Wako) and triglyceride (TG, Roche) concentrations were determined by
enzymatic assays. TC (n= PO-6, EO-11, and FO-7) and TG (n= PO-7, EO-12,
and FO-8) values for each time point are shown in Panel A and Panel C,
respectively. Area under the curve (AUC) for each animal was calculated and TC
and TG differences among the three diet groups were determined by one-way
analysis of variance (ANOVA) using GraphPad Prism software, as shown in
Panel B and Panel D, respectively. Values represent mean ± S.E.M and
different letters represent a significant difference (p < 0.0001).

A
DC
BTC
0 4 8 12 16 20 24 28
0
500
1000
1500
2000
2500PalmEchiumFish
Days on Diet
TC (m
g/dl
)
TG
0 4 8 12 16 20 24 28
0
50
100
150
200
250PalmEchiumFish
Days on Diet
TG (m
g/dl
)
Figure 1

Figure 2. Plasma VLDL compositional and size analysis. A. VLDL from mice
fed PO (n=5), EO (n=5), and FO (n=9) were used for FC, CE, TG, PL, and
protein quantification and percentage chemical composition was calculated. B.
VLDL surface to core ratio. Data from Panel A were used to calculate the ratio of
surface constituents (FC + PL + protein) to core constituents (CE + TG). C. VLDL
from animals fed PO (n=12), EO (n=15), and FO (n=14) were isolated and
particle diameter was measured by laser light scatter. D. Whole plasma apoB
concentration was analyzed by Western blot analysis. Plasma (2ul) was
fractionated by 4-8% SDS PAGE from mice fed diets containing PO (n=4), EO
(n=3), and FO (n=4). ApoB was detected by western blot analysis and
chemiluminescent band intensity was measured using Multi Gauge software.
Values are mean ± S.E.M. Values with different letters are significantly different
(p<0.05)

VLDL Size by Light Scatter Analysis
PO EO FO0
153045607590
aa
b
VLD
L Si
ze (n
m)
VLDL
FC CE TG PL Prot.0
10
20
30
40
50PalmEchiumFish
a
b b
ab a
b
a
ab b
% C
ompo
sitio
n
Whole Plasma apoB
PO EO FO0
500,000
1,000,000
1,500,000
2,000,000
2,500,000
ApoB
Inte
nsity
(AU)
VLDL
PO EO FO0.00.20.40.60.81.01.2
b
a
a,b
Sur
face
:Cor
e R
atio
A B
C DPO EO FO
Figure 2

Figure 3. Liver lipid content. Mice were fed experimental diets containing PO,
EO, or FO for 16 weeks before livers were harvested, lipids extracted, and lipid
content was measured using enzymatic assays. The bars represent mean ±
S.E.M. for PO (n=16), EO (n=13), and FO (n=12). Values with different letters are
significantly different (p<0.05). TG, triglyceride; TC, total cholesterol; FC, free
cholesterol; CE, cholesteryl ester.

TG
PO EO FO0
50
100
150
200
a,b
b
a
mg/
g w
et w
eigh
t
TC
PO EO FO10
15
20
25
30
aa
b
mg/
g w
et w
eigh
t
FC
PO EO FO
1
2
3
4
5
6
mg/
g w
et w
eigh
t
CE
PO EO FO0
10
20
30
a a
b
mg/
g w
et w
eigh
t
Figure 3

Figure 4. Hepatic gene expression. Mice were fed experimental diets
containing PO, EO or FO for 16 weeks before livers were harvested for
measurement of gene expression by quantitative real-time PCR. Liver RNA was
isolated using TRIzol from individual mice and quantified for the indicated genes.
Values represent mean ± S.E.M.; n=3–5 per diet group. Values with different
letters are significantly different from one another (P< .05) for each gene.
SREBP1-c, sterol regulatory element binding protein 1-c; ACC, acetyl CoA
carboxylase; SCD-1, stearoyl CoA desaturase-1; FAS, fatty acid synthase;
HMGCoA synthase, hydroxymethylglutaryl CoA synthase; LDLr, low-density
lipoprotein receptor; LXRα, liver X receptor α; PGC1α, peroxisome proliferator-
activated receptor γ, coactivator 1α; PPARα, peroxisome proliferator-activated
receptor α.

Hepatic Gene Expression
SREBP1-c ACCSCD-1
FAS
HMGCoAsyn
LDLr αLXR α
PGC1 α
PPAR
0
1
2
3
4 PalmEchiumFish
aa
b
Rel
ativ
e Fo
ld C
hang
e (A
U)
Figure 4

Figure 5. Hepatic TG secretion rate. A. Mice fed PO, EO, or FO diets were
fasted for 4hrs before baseline TG values were determined by enzymatic
analysis. Animals were injected retroorbitally with Triton X-100 (500mg/kg
mouse) to block lipase activity. Hepatic TG secretion rate was calculated by
measuring the accumulation of TG in plasma 45, 90, and 180m post-injection. B.
The slopes of hepatic TG secretion shown in Panel A. Values represent mean ±
S.E.M. Values with different letters are significantly different (p<0.05)

0 45 90 135 180
0
500
1000
1500PalmEchiumFish
Time (min)
TG (m
g/dl
)
A B
PO EO FO0
2
4
6
8a
bab
Slop
e
Figure 5

Figure 6. Post-heparin plasma lipase activity. A. Total lipase, hepatic lipase
(HL), and lipoprotein lipase (LPL) activities in plasma of animals after 28 days on
PO, EO, or FO diets (n=4, n=6, n=4, respectively). After a 4 hr fast, mice were
injected with 100 units/kg (180mg/kg) heparin sodium salt (Sigma) dissolved in
saline. 10 minutes post injection, blood was collected and plasma separated as
described previously. Triolein (Sigma) was used as a substrate. See methods for
details on substrate preparation. Post-heparin plasma was combined with the
substrate and apoCII (Sigma) as a co-activator. HL activity was assayed by salt
inhibition of LPL and LPL activity was calculated as total lipase activity minus HL
activity. B. VLDL lipolysis by purified LPL. 2ug of VLDL TG from each diet group
(PO, n=7; EO, n=11; FO, n=3) were incubated with 25ng of LPL (Sigma) for 1hr
at 37ºC. Total non-esterified free fatty acids (NEFA) released were measured by
colorimetric analysis using a NEFA assay (Wako). The bars represent mean ±
S.E.M. Values with different letters are significantly different (p<0.05).

Total Lipase HL LPL0
10
20
30
40
50
60
Lipa
se A
ctiv
ity(u
mol
FA
rele
ased
/hr/m
l)
A
PO EO FO0.0
0.5
1.0
1.5
2.0
a
bb
nmol
FA
Rel
ease
d/ 1
hr
B
POEOFO
Figure 6

Figure 7. VLDL particle turnover. PO and EO recipient mice were injected with
a mixture of 125I-VLDL from PO-fed, and 131I-VLDL from EO-fed donor mice via
the jugular vein. A. The rate of removal of VLDL tracer from PO-fed donor mice.
Plasma was collected 5m, 30m, 1hr, 2hr, 4hr, 8hr, and 24hr after tracer injection
and apoB radioactivity was measured after isopropanol precipitation. Data are
presented as % of 5 min radioactivity remaining in plasma. B. The rate of
removal of VLDL tracer from EO-fed donor mice. See A for details. C. Elution
profile of PO VLDL tracer in PO and EO recipient mouse plasma. Plasma
samples were separated by FPLC to determine the amount of radiolabel
remaining as VLDL 30min, 3hr, and 8hr post-injection. D. Elution profile of EO
VLDL tracer in PO and EO recipient mouse plasma. See C for details.

apoB Particle Turnover
0 4 8 12 16 20 24
50
75
100 Palm (125PO)Echium (125PO)
Time post-injection (hr)
% o
f 5 m
in p
lasm
a ra
dioa
ctiv
ity
apoB Particle Turnover
0 4 8 12 16 20 24
50
75
100 Palm (131EO)Echium (131EO)
Time post-injection (hr)
% o
f 5 m
in p
lasm
a ra
dioa
ctiv
ity
125I-VLDL tracer (PO)
10 15 20 25
0
1000
2000
3000PO - 30min
PO - 3 hr
PO - 8hr
EO - 30 min
EO - 3hr
EO - 8hr
Fraction no.
dpm
131I-VLDL tracer (EO)
10 15 20 25
50
150
250
350PO - 30min
PO - 3 hr
PO - 8hr
EO - 30 min
EO - 3hr
EO - 8hr
Fraction no.
dpm
A
C
B
D
Figure 7

Supplemental Figure 1. Fatty acid composition of plasma lipids. After 16
weeks of PO, EO, or FO experimental diet feeding, plasma was collected for
measurement of PL, TG, and CE fatty acid composition. Lipids from plasma were
extracted using the Bligh-Dyer method and separated into PL, TG, and CE bands
by thin layer chromatography. The bands were visualized with primuline and
collected, after which fatty acids were transmethylated and analyzed for fatty acid
distribution by gas-liquid chromatography as described in the Materials and
Methods section. Data represent mean ± S.E.M; n=5 for each group. Values with
different letters are significantly different (p<0.05) by ANOVA; data bars not
marked with letters were not significantly different. The figure only shows
polyunsaturated fatty acids (≥ 2 double bonds).

Supplemental Figure 1Plasma PL
18:2
n-6
18:3
n-6
18:3
n-3
18:4
n-3
20:3
n-6
20:4
n-6
20:5
n-3
22:6
n-30
2
4
6
8
10
ab a
PalmEchiumFish
% o
f Tot
al F
atty
Aci
ds
Plasma TG
18:2
n-6
18:3
n-6
18:3
n-3
18:4
n-3
20:3
n-6
20:4
n-6
20:5
n-3
22:6
n-30
5
10
15
a
b
a%
of T
otal
Fat
ty A
cids
Plasma CE
18:2
n-6
18:3
n-6
18:3
n-3
18:4
n-3
20:3
n-6
20:4
n-6
20:5
n-3
22:6
n-302468
1012141618
ab
a
b
aa a a
a
a
aa a
ac
b
b
bb
b
b
b
% o
f Tot
al F
atty
Aci
ds

Reference List
1. Defilippis, A.P., M.J. Blaha, and T.A. Jacobson. 2010. Omega-3 Fatty acids for cardiovascular disease prevention. Curr.Treat.Options.Cardiovasc.Med. 12: 365-380.
2. Kromhout, D., E.J. Giltay, and J.M. Geleijnse. 2010. n-3 fatty acids and cardiovascular events after myocardial infarction. N.Engl.J.Med. 363: 2015-2026.
3. Roth, E.M. and W.S. Harris. 2010. Fish oil for primary and secondary prevention of coronary heart disease. Curr.Atheroscler.Rep. 12: 66-72.
4. Dyerberg, J. and H.O. Bang. 1982. A hypothesis on the development of acute myocardial infarction in Greenlanders. Scand.J.Clin.Lab Invest Suppl 161: 7-13.
5. Dyerberg, J. and H.O. Bang. 1979. Lipid metabolism, atherogenesis, and haemostasis in Eskimos: the role of the prostaglandin-3 family. Haemostasis 8: 227-233.
6. Bang, H.O., J. Dyerberg, and N. Hjoorne. 1976. The composition of food consumed by Greenland Eskimos. Acta Med.Scand. 200: 69-73.
7. Davis, H.R., R.T. Bridenstine, D. Vesselinovitch, and R.W. Wissler. 1987. Fish oil inhibits development of atherosclerosis in rhesus monkeys. Arteriosclerosis 7: 441-449.
8. Rudel, L.L., K. Kelley, J.K. Sawyer, R. Shah, and M.D. Wilson. 1998. Dietary monounsaturated fatty acids promote aortic atherosclerosis in LDL receptor-null, human ApoB100-overexpressing transgenic mice. Arterioscler.Thromb.Vasc.Biol. 18: 1818-1827.
9. Parks, J.S., J. Kaduck-Sawyer, B.C. Bullock, and L.L. Rudel. 1990. Effect of dietary fish oil on coronary artery and aortic atherosclerosis in African green monkeys. Arteriosclerosis 10: 1102-1112.
10. Albert, C.M., H. Campos, M.J. Stampfer, P.M. Ridker, J.E. Manson, W.C. Willett, and J. Ma. 2002. Blood levels of long-chain n-3 fatty acids and the risk of sudden death. N.Engl.J.Med. 346: 1113-1118.
11. Burr, M.L., A.M. Fehily, J.F. Gilbert, S. Rogers, R.M. Holliday, P.M. Sweetnam, P.C. Elwood, and N.M. Deadman. 1989. Effects of changes in fat, fish, and fibre intakes on death and myocardial reinfarction: diet and reinfarction trial (DART). Lancet 2: 757-761.

12. Sanders, T.A., D.R. Sullivan, J. Reeve, and G.R. Thompson. 1985. Triglyceride-lowering effect of marine polyunsaturates in patients with hypertriglyceridemia. Arteriosclerosis 5: 459-465.
13. Harris, W.S., W.E. Connor, N. Alam, and D.R. Illingworth. 1988. Reduction of postprandial triglyceridemia in humans by dietary n-3 fatty acids. J.Lipid Res. 29: 1451-1460.
14. Norling, L.V. and C.N. Serhan. 2010. Profiling in resolving inflammatory exudates identifies novel anti-inflammatory and pro-resolving mediators and signals for termination. J.Intern.Med. 268: 15-24.
15. Harris, W.S. 1989. Fish oils and plasma lipid and lipoprotein metabolism in humans: a critical review. J.Lipid Res. 30: 785-807.
16. Harris, W.S. 1996. n-3 fatty acids and lipoproteins: comparison of results from human and animal studies. Lipids 31: 243-252.
17. Kris-Etherton, P.M. and S. Yu. 1997. Individual fatty acid effects on plasma lipids and lipoproteins: human studies. Am.J.Clin.Nutr. 65: 1628S-1644S.
18. Cullen, P. 2000. Evidence that triglycerides are an independent coronary heart disease risk factor. Am.J.Cardiol. 86: 943-949.
19. Oosterveer, M.H., T.H. van Dijk, U.J. Tietge, T. Boer, R. Havinga, F. Stellaard, A.K. Groen, F. Kuipers, and D.J. Reijngoud. 2009. High fat feeding induces hepatic fatty acid elongation in mice. PLoS.One. 4: e6066.
20. Larter, C.Z., M.M. Yeh, J. Cheng, J. Williams, S. Brown, P.A. dela, K.S. Bell-Anderson, and G.C. Farrell. 2008. Activation of peroxisome proliferator-activated receptor alpha by dietary fish oil attenuates steatosis, but does not prevent experimental steatohepatitis because of hepatic lipoperoxide accumulation. J.Gastroenterol.Hepatol. 23: 267-275.
21. Jump, D.B. and S.D. Clarke. 1999. Regulation of gene expression by dietary fat. Annu.Rev.Nutr. 19: 63-90.
22. Vasandani, C., A.I. Kafrouni, A. Caronna, Y. Bashmakov, M. Gotthardt, J.D. Horton, and D.K. Spady. 2002. Upregulation of hepatic LDL transport by n-3 fatty acids in LDL receptor knockout mice. J.Lipid Res. 43: 772-784.
23. Qi, K., C. Fan, J. Jiang, H. Zhu, H. Jiao, Q. Meng, and R.J. Deckelbaum. 2008. Omega-3 fatty acid containing diets decrease plasma triglyceride concentrations in mice by reducing endogenous triglyceride synthesis and enhancing the blood clearance of triglyceride-rich particles. Clin.Nutr. 27: 424-430.

24. Wang, H., X. Chen, and E.A. Fisher. 1993. N-3 fatty acids stimulate intracellular degradation of apoprotein B in rat hepatocytes. J.Clin.Invest 91: 1380-1389.
25. Halvorsen, B., A.C. Rustan, L. Madsen, J. Reseland, R.K. Berge, P. Sletnes, and E.N. Christiansen. 2001. Effects of long-chain monounsaturated and n-3 fatty acids on fatty acid oxidation and lipid composition in rats. Ann.Nutr.Metab 45: 30-37.
26. Kris-Etherton, P.M., D.S. Taylor, S. Yu-Poth, P. Huth, K. Moriarty, V. Fishell, R.L. Hargrove, G. Zhao, and T.D. Etherton. 2000. Polyunsaturated fatty acids in the food chain in the United States. Am.J.Clin.Nutr. 71: 179S-188S.
27. Burdge, G. 2004. Alpha-linolenic acid metabolism in men and women: nutritional and biological implications. Curr.Opin.Clin.Nutr.Metab Care 7: 137-144.
28. Nakamura, M.T. and T.Y. Nara. 2004. Structure, function, and dietary regulation of delta6, delta5, and delta9 desaturases. Annu.Rev.Nutr. 24: 345-376.
29. Guil-Guerrero, J.L., F. Gomez-Mercado, F. Garcia-Maroto, and P. Campra-Madrid. 2000. Occurrence and characterization of oils rich in gamma-linolenic acid Part I: Echium seeds from Macaronesia. Phytochemistry 53: 451-456.
30. Zhang, P., E. Boudyguina, M.D. Wilson, A.K. Gebre, and J.S. Parks. 2008. Echium oil reduces plasma lipids and hepatic lipogenic gene expression in apoB100-only LDL receptor knockout mice. J.Nutr.Biochem. 19: 655-663.
31. Powell-Braxton, L., M. Veniant, R.D. Latvala, K.I. Hirano, W.B. Won, J. Ross, N. Dybdal, C.H. Zlot, S.G. Young, and N.O. Davidson. 1998. A mouse model of human familial hypercholesterolemia: markedly elevated low density lipoprotein cholesterol levels and severe atherosclerosis on a low-fat chow diet. Nat.Med. 4: 934-938.
32. Farese, R.V., Jr., M.M. Veniant, C.M. Cham, L.M. Flynn, V. Pierotti, J.F. Loring, M. Traber, S. Ruland, R.S. Stokowski, D. Huszar, and S.G. Young. 1996. Phenotypic analysis of mice expressing exclusively apolipoprotein B48 or apolipoprotein B100. Proc.Natl.Acad.Sci.U.S.A 93: 6393-6398.
33. Bell, T.A., III, K. Kelley, M.D. Wilson, J.K. Sawyer, and L.L. Rudel. 2007. Dietary Fat-Induced Alterations in Atherosclerosis Are Abolished by ACAT2-Deficiency in ApoB100 Only, LDLr-/- Mice. Arterioscler.Thromb.Vasc.Biol. 27: 1396-1402.

34. Furbee, J.W., Jr., O. Francone, and J.S. Parks. 2001. Alteration of plasma HDL cholesteryl ester composition with transgenic expression of a point mutation (E149A) of human LCAT. J.Lipid Res. 42: 1626-1635.
35. BLIGH, E.G. and W.J. DYER. 1959. A rapid method of total lipid extraction and purification. Can.J.Biochem.Physiol 37: 911-917.
36. Livak, K.J. and T.D. Schmittgen. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25: 402-408.
37. Rouser, G., S. Fkeischer, and A. Yamamoto. 1970. Two dimensional then layer chromatographic separation of polar lipids and determination of phospholipids by phosphorus analysis of spots. Lipids 5: 494-496.
38. Lee, J.Y., J.M. Timmins, A. Mulya, T.L. Smith, Y. Zhu, E.M. Rubin, J.W. Chisholm, P.L. Colvin, and J.S. Parks. 2005. HDLs in apoA-I transgenic Abca1 knockout mice are remodeled normally in plasma but are hypercatabolized by the kidney. Journal of Lipid Research 46: 2233-2245.
39. Wilcox, R.W., T. Thuren, P. Sisson, G.L. Kucera, and M. Waite. 1991. Hydrolysis of neutral lipid substrates by rat hepatic lipase. Lipids 26: 283-288.
40. Otway, S. and D.S. Robinson. 1967. The use of a non-ionic detergent (Triton WR 1339) to determine rates of triglyceride entry into the circulation of the rat under different physiological conditions. J.Physiol 190: 321-332.
41. McFARLANE, A.S. 1958. Efficient trace-labelling of proteins with iodine. Nature 182: 53.
42. Lee, J.Y., L. Lanningham-Foster, E.Y. Boudyguina, T.L. Smith, E.R. Young, P.L. Colvin, M.J. Thomas, and J.S. Parks. 2004. Pre+¦ high density lipoprotein has two metabolic fates in human apolipoprotein A-I transgenic mice. Journal of Lipid Research 45: 716-728.
43. Davidson, M.H. 2006. Mechanisms for the hypotriglyceridemic effect of marine omega-3 fatty acids. Am.J.Cardiol. 98: 27i-33i.
44. Qi, K., M. Al-Haideri, T. Seo, Y.A. Carpentier, and R.J. Deckelbaum. 2003. Effects of particle size on blood clearance and tissue uptake of lipid emulsions with different triglyceride compositions. JPEN J.Parenter.Enteral Nutr. 27: 58-64.
45. Nozaki, S., A. Garg, G.L. Vega, and S.M. Grundy. 1991. Postheparin lipolytic activity and plasma lipoprotein response to omega-3 polyunsaturated fatty acids in patients with primary hypertriglyceridemia. Am.J.Clin.Nutr. 53: 638-642.

46. Egusa, G., D.W. Brady, S.M. Grundy, and B.V. Howard. 1983. Isopropanol precipitation method for the determination of apolipoprotein B specific activity and plasma concentrations during metabolic studies of very low density lipoprotein and low density lipoprotein apolipoprotein B. J.Lipid Res. 24: 1261-1267.
47. Ishibashi, S., M.S. Brown, J.L. Goldstein, R.D. Gerard, R.E. Hammer, and J. Herz. 1993. Hypercholesterolemia in low density lipoprotein receptor knockout mice and its reversal by adenovirus-mediated gene delivery. J.Clin.Invest 92: 883-893.
48. Surette, M.E., M. Edens, F.H. Chilton, and K.M. Tramposch. 2004. Dietary echium oil increases plasma and neutrophi l long-chain (n-3) fatty acids and lowers serum triacylglycerols in hypertriglyceridemic humans. J.Nutr. 134: 1406-1411.
49. Bordin, P., O.A. Bodamer, S. Venkatesan, R.M. Gray, P.A. Bannister, and D. Halliday. 1998. Effects of fish oil supplementation on apolipoprotein B100 production and lipoprotein metabolism in normolipidaemic males. Eur.J.Clin.Nutr. 52: 104-109.
50. Harris, W.S., W.E. Connor, D.R. Illingworth, D.W. Rothrock, and D.M. Foster. 1990. Effects of fish oil on VLDL triglyceride kinetics in humans. J.Lipid Res. 31: 1549-1558.
51. Nestel, P.J., W.E. Connor, M.F. Reardon, S. Connor, S. Wong, and R. Boston. 1984. Suppression by diets rich in fish oil of very low density lipoprotein production in man. J.Clin.Invest 74: 82-89.
52. Parks, J.S., F.L. Johnson, M.D. Wilson, and L.L. Rudel. 1990. Effect of fish oil diet on hepatic lipid metabolism in nonhuman primates: lowering of secretion of hepatic triglyceride but not apoB. J.Lipid Res. 31: 455-466.
53. Nestel, P.J. 2000. Fish oil and cardiovascular disease: lipids and arterial function. The American Journal of Clinical Nutrition 71: 228S-231S.
54. Jump, D.B. 2004. Fatty acid regulation of gene transcription. Crit Rev.Clin.Lab Sci. 41: 41-78.
55. Xu, J., M.T. Nakamura, H.P. Cho, and S.D. Clarke. 1999. Sterol regulatory element binding protein-1 expression is suppressed by dietary polyunsaturated fatty acids. A mechanism for the coordinate suppression of lipogenic genes by polyunsaturated fats. J.Biol.Chem. 274: 23577-23583.
56. Kim, H.J., M. Takahashi, and O. Ezaki. 1999. Fish oil feeding decreases mature sterol regulatory element-binding protein 1 (SREBP-1) by down-regulation of SREBP-1c mRNA in mouse liver. A possible mechanism for

down-regulation of lipogenic enzyme mRNAs. J.Biol.Chem. 274: 25892-25898.
57. Yahagi, N., H. Shimano, A.H. Hasty, M. Amemiya-Kudo, H. Okazaki, Y. Tamura, Y. Iizuka, F. Shionoiri, K. Ohashi, J. Osuga, K. Harada, T. Gotoda, R. Nagai, S. Ishibashi, and N. Yamada. 1999. A crucial role of sterol regulatory element-binding protein-1 in the regulation of lipogenic gene expression by polyunsaturated fatty acids. J.Biol.Chem. 274: 35840-35844.
58. Pan, M., A.I. Cederbaum, Y.L. Zhang, H.N. Ginsberg, K.J. Williams, and E.A. Fisher. 2004. Lipid peroxidation and oxidant stress regulate hepatic apolipoprotein B degradation and VLDL production. J.Clin.Invest 113: 1277-1287.
59. Kendrick, J.S., L. Chan, and J.A. Higgins. 2001. Superior role of apolipoprotein B48 over apolipoprotein B100 in chylomicron assembly and fat absorption: an investigation of apobec-1 knock-out and wild-type mice. Biochem.J. 356: 821-827.
60. Wong, S.H., P.J. Nestel, R.P. Trimble, G.B. Stoker, R.J. Illman, and D.L. Topping. 1984. The adaptive effects of dietary fish and safflower oil on lipid and lipoprotein metabolism in perfused rat liver. Biochimica et Biophysica Acta (BBA) - Lipids and Lipid Metabolism 792: 103-109.
61. Parks, J.S., M.D. Wilson, F.L. Johnson, and L.L. Rudel. 1989. Fish oil decreases hepatic cholesteryl ester secretion but not apoB secretion in African green monkeys. J.Lipid Res. 30: 1535-1544.
62. Lang, C.A. and R.A. Davis. 1990. Fish oil fatty acids impair VLDL assembly and/or secretion by cultured rat hepatocytes. Journal of Lipid Research 31: 2079-2086.
63. Hulsmann, W.C., M.C. Oerlemans, and H. Jansen. 1980. Activity of heparin-releasable liver lipase. Dependence on the degree of saturation of the fatty acids in the acylglycerol substrates. Biochim.Biophys.Acta 618: 364-369.
64. Nozaki, S., A. Garg, G.L. Vega, and S.M. Grundy. 1991. Postheparin lipolytic activity and plasma lipoprotein response to omega-3 polyunsaturated fatty acids in patients with primary hypertriglyceridemia. Am.J.Clin.Nutr. 53: 638-642.
65. Weintraub, M.S., R. Zechner, A. Brown, S. Eisenberg, and J.L. Breslow. 1988. Dietary polyunsaturated fats of the W-6 and W-3 series reduce postprandial lipoprotein levels. Chronic and acute effects of fat saturation on postprandial lipoprotein metabolism. J.Clin.Invest 82: 1884-1893.

Chapter V
SUMMARY AND DISCUSSION
Lolita M. Forrest
114

Summary
CVD, primarily caused by atherosclerosis, is the number one cause of
death in the United States. Higher consumption of FO and n-3 PUFA
supplements have been shown to reduce atherosclerosis and the incidence of
CVD. However, FO consumption in the US remains low; therefore, a need for an
alternative source of n-3 PUFAs exists. A reduction in plasma TG concentration
is the most consistent observation found with FO feeding, though the
mechanisms of this response are not fully understood. Furthermore, it is unclear
what role TG plays in atherosclerosis. EO has been shown to reduce plasma TG
concentrations. Our goal was to determine whether EO is atheroprotective and
could serve as an alternative to FO. Furthermore, we investigated several
mechanisms of TG metabolism to determine which were mediated by n-3
PUFAs, causing a hypotriglyceridemic response.
Summary of Findings
In Chapter II, we tested the hypothesis that EO feeding of apoB100-only
LDLrKO mice would result in reduced atherosclerosis. Mice were fed PO, EO, or
FO for 16 weeks and surface lesion area and aortic cholesterol were quantified
as measurements of atherosclerosis. Aortic surface lesion area measurements
showed that EO significantly reduces atherosclerosis compared to PO feeding.
Furthermore, the reduction in aortic surface lesion area was comparable to that
observed for the FO group. This is the first report that EO is anti-atherogenic,
suggesting that it may be a botanical alternative to FO for cardioprotection.
115

In Chapter III, the goal of our studies was to investigate the role of n-3
PUFAs on macrophage accumulation in the aortic root. After 16 weeks of PO,
EO, or FO feeding of apoB100-only LDLrKO mice, the intimal area of the aortic
root was measured. Surprisingly, there was no difference in aortic root intimal
area. Furthermore, when we quantified the amount of macrophages in the intimal
space, the values were similar across all groups. Here, we hypothesize that n-3
PUFA enrichment could not overcome atherosclerosis progression induced by
hypercholesterolemia and shear stress at this atherosclerosis prone site.
In Chapter IV, we investigated mechanisms of TG synthesis and secretion
as well as TG lipolysis and removal from plasma to determine what mechanisms
were involved in n-3 PUFA-mediated TG-lowering. Over a 28 day period, we
observed that both EO and FO decreased plasma TG levels compared to PO.
VLDL compositional analysis showed that n-3 PUFAs reduce particle size.
Reductions in VLDL particle size may be due to decreased hepatic TG secretion
or increased intravascular lipolysis. We did not observe plasma differences in
apoB concentration, suggesting that decreased plasma TG was not due to
decreased hepatic VLDL particle secretion. At 16 weeks of diet feeding, we
observed that EO results in significantly more hepatic TG and cholesterol
accumulation compared to FO. These results show that, while EO reduces
plasma TG concentrations, it does not protect from hepatic steatosis in this
mouse model of atherosclerosis. Furthermore, it shows that TG lowering by FO
and EO may not be occurring by parallel mechanisms. After 16 weeks of feeding,
hepatic gene expression data showed a trend of increased lipogenic gene
116

expression with EO compared to FO. This observed increase in lipogenesis was
also apparent in our triton block studies, which showed that EO feeding results in
a significantly higher rate of hepatic TG secretion in comparison to FO. We
measured post-heparin lipase activity to determine if this could explain the
decreased plasma TG levels with EO and FO feeding. Hepatic lipase and
lipoprotein lipase activities were not different among the three groups. However,
we did observe that EO- and FO-VLDL were more susceptible to TG hydrolysis
by purified lipoprotein lipase. Finally, turnover studies in EO and PO-fed mice
showed minor increases in EO-VLDL removal from plasma. Our findings suggest
that FO acts to reduce plasma TG levels by both decreased production and
increased lipolysis, whereas the TG lowering effect of EO is due only to an
increase in intravascular lipolysis.
Discussion
Is lowered plasma TG concentration really necessary for cardioprotection?
In 2004, the United States Food and Drug Administration (FDA) approved
Lovaza (GlaxoSmithKline, n-3-acid ethyl esters) for the treatment of
hypertriglyceridemia. Lovaza makes no claims in the US in regards to
cardiovascular health and, in fact, states that it “has not been shown to prevent
heart attacks or strokes” (Lovaza.com). However, based on results from the
GISSI-Prevenzione study, Lovaza (Omacor) is also indicated for secondary
prevention after a MI in European markets (1). Whether or not
hypertriglyceridemia should be considered a risk factor for CVD remains
117

controversial. While there are some studies that have correlated elevated plasma
TG with CVD risk (2-6), the direct role of TG in atherogenesis has not been
established. It has been shown that patients with familial hypertriglyceridemia are
at greater risk for premature coronary artery disease (7). These patients produce
a kinetic subclass of abnormal “hypertriglyceridemic VLDL”, which has been
shown to suppress HMG-CoA reductase in endothelial cells and accumulate in
macrophages (8). The apoB48 receptor is expressed primarily by monocytes,
macrophages, and endothelial cells and has been found in human atherosclerotic
foam cells (9). This receptor normally provides nutrition to the cells, but can
mediate foam cell formation and endothelial dysfunction when overwhelmed. The
apoB48 receptor has been shown to bind TG-rich lipoproteins, specifically
chylomicrons and “hypertriglyceridemic VLDL”, but not normal VLDL (10). These
studies partially explain the mechanisms of foam cell formation in familial
hypertriglyceridemia and patients with elevated TG-rich lipoproteins. However,
because foam cells are usually cholesterol-rich, this mechanism cannot fully
explain the relationship between TG and atherogenesis. Chylomicrons, VLDL,
and remnant lipoproteins are the lipoproteins that contribute to
hypertriglyceridemia, whereas remnant lipoproteins are considered to be the
most atherogenic. Studies have concluded that levels of TG-rich lipoprotein
remnant particles, rather than whole plasma TG levels, are more related to the
progression of atherosclerosis (11). It is hard to distinguish the exact role of TG
levels in atherogenesis because they are inversely related to levels of HDL,
which are known anti-atherogenic particles. Nevertheless, it appears that TG-rich
118

lipoproteins at the least antagonize inflammatory mechanisms of atherosclerosis
(12) therefore, maintaining healthy levels of plasma TGs may offer
cardiovascular health benefits.
As a FO alternative, should Echium oil be considered?
Since FO consumption is poor in the US because of expense, taste,
preference, portability, and allergies, our studies were designed to determine
whether Echium oil (EO) could be used as an alternative to FO for
cardioprotection and TG reduction. The benefits of FO are attributed to both EPA
and DHA. Studies have shown that EO enriches cellular membranes with EPA,
but not DHA, which is presumably due to limited Δ6-desaturase activity (13,14).
One of the most interesting observations we saw in our studies was that while
EO and FO both significantly lowered plasma TG compared to PO, EO feeding
resulted in hepatic steatosis, whereas FO does not. This result suggests that
DHA may be responsible for the reduction in hepatic lipid content that is
associated with FO. Our lab has previously shown that liver TG from FO fed mice
contain ~3x more DHA than TG from EO fed mice (14). Most studies on the role
of n-3 PUFAs in disease prevention use fish oil, which contains both EPA + DHA,
so it is difficult to discern the role of individual n-3 fatty acids. However, there are
some studies on the effect of DHA in the absence of EPA. One study found that
serum amyloid A mRNA, an HDL-associated apolipoprotein that reduces fat
deposition in adipocytes and hepatoma cells, is increased by DHA treatment
(15). Using an obese mouse model, another study found that DHA-enriched
diacylglycerol improved hepatic steatosis (16). Yanagita et. al. found in mice that
119

adding DHA to a CLA (conjugated linoleic acid; promotes FA synthesis) diet
significantly attenuated fatty liver by reducing hepatic fatty acid synthesis (17).
SREBP-1 suppression is another proposed mechanism of improved steatosis
with PUFA intake (18). In our studies, we observed SREBP1-c expression to be
relatively higher within the EO group compared to FO, even more so than PO.
Other studies have also found DHA to be protective in the liver (19-21). These
data suggest that DHA plays an important role in protecting the liver from
excessive TG accumulation.
The role of DHA in contributing to atherosclerosis protection has also been
examined. A study by Mori and colleagues found that DHA, but not EPA, lowered
blood pressure in humans (22). Also, when vascular reactivity was measured in
hyperlipidemic men, DHA, but not EPA was found to enhance vasodilator
mechanisms (23). Reduced VCAM-1 expression by DHA compared to EPA also
supports DHA as a more potent anti-inflammatory lipid (24). EPA, but not DHA,
has been shown to decrease platelet volume, thereby reducing CHD risk (25).
One study concluded that EPA and DHA were equally antithrombotic and TG-
lowering (26), while another found DHA to be more hypotriglyceridemic (27). In
our studies, we found both plasma TG and atherosclerosis to be decreased with
EO feeding; therefore, DHA may not be as important in atheroprotection as it is in
preventing hepatic lipid accumulation.
Another major difference between EO and FO is their ALA content. In our
mouse model, hepatic TGs from EO-fed mice contain ~5x more ALA than those
from FO fed mice (14). Flaxseed is a major source of ALA and it has been
120

suggested to be atheroprotective; however, it has been previously shown in
apoB100-only LDLrKO mice that FO, but not ALA, is atheroprotective (28). The
cardioprotective properties of flaxseed are due to its lignan content, not ALA (29).
Furthermore, flaxseed oil (55% ALA) has no effect on serum lipids aside from
slight decreases in plasma TG. However, flaxseed oil has been shown to
decrease soluble VCAM-1 and platelet aggregation. On the other hand, flaxseed
oil is not anti-inflammatory (29). It is hypothesized that botanical oils such as EO
may be anti-inflammatory, offering benefits for diseases such as asthma,
atherosclerosis and allergies (30). These data suggest that the benefits from EO
are not due to its high ALA content. Therefore, based on the literature and our
findings, the fatty acid profile of EO is not optimal to achieve all of the benefits
found with FO. It is clear that EO reduces plasma TG concentrations to similar
levels observed with FO feeding, presumably due to increases in EPA. However,
the impact of EO on hepatic lipid content is as adverse as that of PO, presumably
due to a lack of DHA. The promising finding of EO is that its SDA content is
metabolized to EPA, a far more potent n-3 PUFA with hypotriglyceridemic
effects. Therefore, the use of EO as a source of SDA for the formation of SDA
ethyl esters, may be a successful approach in reducing plasma TG. However,
due to a lack of DHA, concentrated SDA may not protect from liver toxicity.
Future Directions and Conclusions
Our studies have revealed several new findings in regards to
atherosclerosis and TG metabolism, especially in the context of dietary EO
enrichment. Still, there are experimental questions left to be addressed. We have
121

shown that EO reduces fasting plasma TG in apoB100-only LDLrKO mice, but
we don’t know what role EO has on postprandial TG levels. Dietary FO has been
shown to reduce postprandial TG concentration (31) and therefore it is plausible
that EO may act by similar mechanisms. However, reduced postprandial TG by
FO is a result of decreased VLDL-TG secretion, thereby increasing chylomicron
LPL availability. If this is the case, we would not expect similar results based on
our finding that EO does not significantly reduce hepatic TG secretion. Resolving
this scientific question would offer more insight into the mechanisms by which EO
reduces plasma TG.
Our studies focused on the hypotriglyceridemic properties of EO.
Additionally, we have shown that EO reduces atherosclerosis. The central role of
inflammation in atherogenesis is well-accepted. It would be interesting to know
how cell membrane enrichment of n-3 PUAs due to dietary EO affects
inflammatory mechanisms, especially with regard to macrophages, the
predominate cell type in atherosclerotic lesions. FO has been shown to reduce
inflammation by decreasing inflammatory cytokine and eicosanoid levels (32,33).
Therefore, determination of EO’s anti-inflammatory potential may prove to be
beneficial to not only atherosclerosis but other inflammatory diseases, such as
arthritis and asthma as well.
While it is unlikely that Americans will drastically alter their diet in favor of
one that results in a more favorable lipid profile, it is clear that even small
increases in long-chain n-3 PUFAs have positive effects on plasma lipids. EO
appears to be as robust as FO with regard to atheroprotection and appears to
122

offer more benefits than oils enriched in n-6 PUFAs, and most importantly those
oils containing higher levels of ALA that are marketed as “n-3 fatty acid
enriched”. Therefore, we conclude that EO may be a suitable botanical
alternative for FO for cardioprotection and plasma TG reduction, but not for
alleviation of hepatic steatosis.
123

Reference List
1. Hoy, S.M. and G.M. Keating. 2009. Omega-3 ethylester concentrate: a review of its use in secondary prevention post-myocardial infarction and the treatment of hypertriglyceridaemia. Drugs 69: 1077-1105.
2. Assmann, G., H. Schulte, and E.A. von. 1996. Hypertriglyceridemia and elevated lipoprotein(a) are risk factors for major coronary events in middle-aged men. Am.J.Cardiol. 77: 1179-1184.
3. Carlson, L.A., L.E. Bottiger, and P.E. Ahfeldt. 1979. Risk factors for myocardial infarction in the Stockholm prospective study. A 14-year follow-up focussing on the role of plasma triglycerides and cholesterol. Acta Med.Scand. 206: 351-360.
4. Carlson, L.A. and L.E. Bottiger. 1985. Risk factors for ischaemic heart disease in men and women. Results of the 19-year follow-up of the Stockholm Prospective Study. Acta Med.Scand. 218: 207-211.
5. Jeppesen, J., H.O. Hein, P. Suadicani, and F. Gyntelberg. 1998. Triglyceride concentration and ischemic heart disease: an eight-year follow-up in the Copenhagen Male Study. Circulation 97: 1029-1036.
6. Miller, M., A. Seidler, A. Moalemi, and T.A. Pearson. 1998. Normal triglyceride levels and coronary artery disease events: the Baltimore Coronary Observational Long-Term Study. J.Am.Coll.Cardiol. 31: 1252-1257.
7. Hopkins, P.N., G. Heiss, R.C. Ellison, M.A. Province, J.S. Pankow, J.H. Eckfeldt, and S.C. Hunt. 2003. Coronary Artery Disease Risk in Familial Combined Hyperlipidemia and Familial Hypertriglyceridemia: A Case-Control Comparison From the National Heart, Lung, and Blood Institute Family Heart Study. Circulation 108: 519-523.
8. Gianturco, S.H., W.A. Bradley, A.M. Gotto, J.D. Morrisett, and D.L. Peavy. 1982. Hypertriglyceridemic Very Low Density Lipoproteins Induce Triglyceride Synthesis and Accumulation in Mouse Peritoneal Macrophages. J.Clin.Invest 70: 168.
9. Brown, M.L., M.P. Ramprasad, P.K. Umeda, A. Tanaka, Y. Kobayashi, T. Watanabe, H. Shimoyamada, W.L. Kuo, R. Li, R. Song, W.A. Bradley, and S.H. Gianturco. 2000. A macrophage receptor for apolipoprotein B48: cloning, expression, and atherosclerosis. Proc.Natl.Acad.Sci.U.S.A 97: 7488-7493.
124

10. Gianturco, S.H., M.P. Ramprasad, A.H. Lin, R. Song, and W.A. Bradley. 1994. Cellular binding site and membrane binding proteins for triglyceride-rich lipoproteins in human monocyte-macrophages and THP-1 monocytic cells. Journal of Lipid Research 35: 1674-1687.
11. Hodis, H.N. 1999. Triglyceride-Rich Lipoprotein Remnant Particles and Risk of Atherosclerosis. Circulation 99: 2852-2854.
12. Libby, P. 2007. Fat Fuels the Flame: Triglyceride-Rich Lipoproteins and Arterial Inflammation. Circ Res 100: 299-301.
13. Surette, M.E., M. Edens, F.H. Chilton, and K.M. Tramposch. 2004. Dietary echium oil increases plasma and neutrophil long-chain (n-3) fatty acids and lowers serum triacylglycerols in hypertriglyceridemic humans. J.Nutr. 134: 1406-1411.
14. Zhang, P., E. Boudyguina, M.D. Wilson, A.K. Gebre, and J.S. Parks. 2008. Echium oil reduces plasma lipids and hepatic lipogenic gene expression in apoB100-only LDL receptor knockout mice. J.Nutr.Biochem. 19: 655-663.
15. Tai, C.C., C.Y. Chen, H.S. Lee, Y.C. Wang, T.K. Li, H.J. Mersamm, S.T. Ding, and P.H. Wang. 2009. Docosahexaenoic Acid Enhances Hepatic Serum Amyloid A Expression via Protein Kinase A-dependent Mechanism. Journal of Biological Chemistry 284: 32239-32247.
16. Kim, H.J., K.T. Lee, Y.B. Park, S.M. Jeon, and M.S. Choi. 2008. Dietary docosahexaenoic acid-rich diacylglycerols ameliorate hepatic steatosis and alter hepatic gene expressions in C57BL/6J-Lep(ob/ob) mice. Mol.Nutr.Food Res. 52: 965-973.
17. Yanagita, T., Y.M. Wang, K. Nagao, Y. Ujino, and N. Inoue. 2005. Conjugated linoleic acid-induced fatty liver can be attenuated by combination with docosahexaenoic acid in C57BL/6N mice. J.Agric.Food Chem. 53: 9629-9633.
18. Sekiya, M., N. Yahagi, T. Matsuzaka, Y. Najima, M. Nakakuki, R. Nagai, S. Ishibashi, J. Osuga, N. Yamada, and H. Shimano. 2003. Polyunsaturated fatty acids ameliorate hepatic steatosis in obese mice by SREBP-1 suppression. Hepatology 38: 1529-1539.
19. Takayama, F., K. Nakamoto, N. Totani, T. Yamanushi, H. Kabuto, T. Kaneyuki, and M. Mankura. 2010. Effects of docosahexaenoic acid in an experimental rat model of nonalcoholic steatohepatitis. J.Oleo Sci. 59: 407-414.
20. Gonzalez-Periz, A., A. Planaguma, K. Gronert, R. Miquel, M. Lopez-Parra, E. Titos, R. Horrillo, N. Ferre, R. Deulofeu, V. Arroyo, J. Rodes, and J. Claria. 2006. Docosahexaenoic acid (DHA) blunts liver injury by
125

conversion to protective lipid mediators: protectin D1 and 17S-hydroxy-DHA. FASEB J. 20: 2537-2539.
21. Nobili, V., G. Bedogni, A. Alisi, A. Pietrobattista, P. Ris+¬, C. Galli, and C. Agostoni. 2011. Docosahexaenoic acid supplementation decreases liver fat content in children with non-alcoholic fatty liver disease: double-blind randomised controlled clinical trial. Archives of Disease in Childhood 96: 350-353.
22. Mori, T.A., D.Q. Bao, V. Burke, I.B. Puddey, and L.J. Beilin. 1999. Docosahexaenoic Acid but Not Eicosapentaenoic Acid Lowers Ambulatory Blood Pressure and Heart Rate in Humans. Hypertension 34: 253-260.
23. Mori, T.A., G.F. Watts, V. Burke, E. Hilme, I.B. Puddey, and L.J. Beilin. 2000. Differential Effects of Eicosapentaenoic Acid and Docosahexaenoic Acid on Vascular Reactivity of the Forearm Microcirculation in Hyperlipidemic, Overweight Men. Circulation 102: 1264-1269.
24. Abeywardena, M.Y. and R.J. Head. 2001. Longchain nGêÆ3 polyunsaturated fatty acids and blood vessel function. Cardiovascular Research 52: 361-371.
25. Park, Y. and W. Harris. EPA, but not DHA, decreases mean platelet volume in normal subjects.
26. Howe, P., P. Clifton, and M. James. Equal antithrombotic and triglyceride-lowering effectiveness of eicosapentaenoic acid-rich and docosahexaenoic acid-rich fish oil supplements.
27. Mori, T.A., V. Burke, I.B. Puddey, G.F. Watts, D.N. O'Neal, J.D. Best, and L.J. Beilin. 2000. Purified eicosapentaenoic and docosahexaenoic acids have differential effects on serum lipids and lipoproteins, LDL particle size, glucose, and insulin in mildly hyperlipidemic men. The American Journal of Clinical Nutrition 71: 1085-1094.
28. Degirolamo, C., K.L. Kelley, M.D. Wilson, and L.L. Rudel. 2010. Dietary n-3 LCPUFA from fish oil but not alpha-linolenic acid-derived LCPUFA confers atheroprotection in mice. J.Lipid Res. 51: 1897-1905.
29. Prasad, K. 2009. Flaxseed and cardiovascular health. J.Cardiovasc.Pharmacol. 54: 369-377.
30. Chilton, F.H., L.L. Rudel, J.S. Parks, J.P. Arm, and M.C. Seeds. 2008. Mechanisms by which botanical lipids affect inflammatory disorders. The American Journal of Clinical Nutrition 87: 498S-503S.
126

31. Harris, W.S., W.E. Connor, N. Alam, and D.R. Illingworth. 1988. Reduction of postprandial triglyceridemia in humans by dietary n-3 fatty acids. J.Lipid Res. 29: 1451-1460.
32. Calder, P.C. 2006. Polyunsaturated fatty acids and inflammation. Prostaglandins Leukot.Essent.Fatty Acids 75: 197-202.
33. Renier, G., E. Skamene, J. DeSanctis, and D. Radzioch. 1993. Dietary n-3 polyunsaturated fatty acids prevent the development of atherosclerotic lesions in mice. Modulation of macrophage secretory activities. Arterioscler.Thromb. 13: 1515-1524.
127

CURRICULUM VITAE
Lolita Marie Forrest
Wake Forest University School of Medicine – Section on Lipid Sciences – A1 Building, 3rd Floor – Medical Center Boulevard – Winston-Salem, NC 27157
Phone (336) 716-3598 – Email: [email protected] Education
2005-present Wake Forest University Graduate School of Arts and Sciences (Winston-Salem, NC)
Molecular Medicine and Translational Science PhD Program
Advisor: John S. Parks
2003-2005 Post-Baccalaureate Research Education Program Wake Forest University School of Medicine Winston-Salem, NC PIs: Charles McCall, MD
Barbara Yoza Ph.D. 1999-2003 Livingstone College (Salisbury, NC)
BS in Biology (cum laude) Employment
2002 Research Project Intern Baylor College of Medicine (Houston, TX) Supervisor: Chantal A. Rivera Ph.D.
Academic and Professional Honors: 2008-2010 American Heart Association, Member
March 2010 “Developments in Botanical Dietary Supplements Research from 1994 to Today” Symposium; The University of Illinois at Chicago, Travel Award
2005-2007 Grant Award #: 1T32GM63485 (Wake Forest
University, 2005-2007) Grant Award #: 3P50AT002782-03S1 (Wake Forest
University, 2007) 2003 Alpha Kappa Mu Honor Society
Beta Kappa Chi Scientific Honor Society
128

2002 Who’s Who Among Students in American Universities & Colleges All American Scholars National Collegiate Minority Leadership Award
Oral Presentations
April 2011 “Echium Oil: A Botanical Source of n-3 PUFAs with
Atheroprotective and Hypotriglyceridemic Properties”
Mini-Symposium on Integrative Lipid Sciences, Inflammation, and Chronic Diseases; Wake Forest University
September 2010 “Investigation of Echium Oil’s Cardioprotective
Potential and the Mechanisms Mediating Long-Chained Polyunsaturated Fatty Acids’ Hypotriglyceridemic Effect”
Molecular Medicine and Translational Science (MMTS) Seminar Series; Wake Forest University
October 2009 “The Cardioprotection and Mechanism of
Triglyceride Lowering in Echium Oil Fed Hypertriglyceridemic Mice”
MMTS Seminar Series; Wake Forest University
January 2009 “Effects of Echium oil on Atherosclerosis and VLDL Metabolism in Mice” MMTS Seminar Series; Wake Forest University
February 2008 “Characterizing the Effect of Echium Oil
Supplementation on Atherosclerosis” MMTS Seminar Series; Wake Forest University March 2007 “Echium oil: Understanding its Mechanism in
Plasma Triglyceride Reduction and Role in Atherosclerosis Development”
MMTS Seminar Series; Wake Forest University
Poster Presentations
April 2010 “Determining the Hypotriglyceridemic Effect of Echium Oil”
Translational Science Institute Synergy Symposium: Lipotoxicity Across the Translational Research Spectrum; Wake Forest University
April 2010 “Echium Oil Supplementation Reduces
Atherosclerosis in B100only-LDLrKO Mice” 10th Annual Syngenta CNC-ACS Poster Vendor
Night (Greensboro, NC)
129

April 2010 “Determining the Hypotriglyceridemic Effect of
Echium Oil” Arteriosclerosis, Thrombosis and Vascular Biology
Annual Conference (San Francisco, CA) April 2009 “Echium Oil Supplementation Reduces
Atherosclerosis in B100only-LDLrKO Mice” Arteriosclerosis, Thrombosis and Vascular Biology Annual Conference (Washington, DC)
October 2008 “Echium Oil Supplementation Reduces
Atherosclerosis in B100only-LDLrKO Mice” South Eastern Research Lipid Conference (Pine Mountain, GA)
May 2008 “Echium Oil Supplementation Reduces Atherosclerosis in B100only-LDLrKO Mice” Botanical Center Directors’ Meeting (West Lafayette, IN)
Abstract Presentations March 2010 “Determining the Hypotriglyceridemic Effect of
Echium Oil” Developments in Botanical Dietary Supplements Research from 1994 to Today Symposium (Chicago, Illinois)
Teaching Experience
2010 Winston-Salem State University (Winston-Salem, NC)
Master’s in Physical Therapy Program Physiology (Summer, Respiratory Section) Course Director: Dr. Allyn Howlett
2009-2010 Teaching Advancement Program (Certificate, Completed May 2010)
Wake Forest University School of Medicine Publications
Research Papers: Yoza, B. K., J. Y. Hu, S. L. Cousart, L. M. Forrest, C. E. McCall. 2006. Induction of RelB participates in endotoxin tolerance. J. Immunol. 177: 4080-4085.
130

Abstracts: Forrest L, Duong M-N, Boudyguina E, Wilson M, Parks JS. Echium oil supplementation attenuates atherosclerosis in B100-LDLrK0 mice. Arterioscler Thromb Vasc Biol 2009; 29(7):e76-e77. Chung S, Rong S, Degirolamo C, Brown AW, Bi X, Forrest L, Temel R, Shelness GS, Parks JS. Hepatocyte-Specific Knockout of ABCA1 Alleviates Liver Lipid Accumulation but Exacerbates Hepatic Insulin Resistance and Inflammation. Arterioscler Thromb Vasc Biol 2010 (in press).
131


















