Intronic Sequences and 3' Splice Sites Control Rous Sarcoma Virus ...
6
JOURNAL OF VIROLOGY, Jan. 1992, p. 6-11 0022-538X/92/010006-06$02.00/0 Copyright C 1992, American Society for Microbiology Intronic Sequences and 3' Splice Sites Control Rous Sarcoma Virus RNA Splicing MARK T. McNALLY AND KAREN BEEMON* Vol. 66, No. 1 Department of Biology, Johns Hopkins University, Baltimore, Maryland 21218 Received 29 July 1991/Accepted 27 September 1991 cis-acting sequences of Rous sarcoma virus (RSV) RNA involved in control of the incomplete splicing that is part of the retroviral life cycle have been studied. The 5' and two alternative 3' splice sites, as well as a negative regulator of splicing element in the intron, have been introduced into chimeric constructs, and their respective roles in splicing inhibition have been evaluated by transient transfection experiments. Although the RSV 5' splice site was used efficiently in these assays, substrates containing either the RSV env or the RSV src 3' splice site were not spliced completely, resulting in 40 to 50% unspliced RNA. Addition of the negative regulator of splicing element to substrates containing RSV 3' splice sites resulted in greater inhibition of splicing (70 to 80% unspliced RNA), suggesting that the two elements function independently and additively. Deletion of sequences more than 70 nucleotides upstream of the src 3' splice site resulted in efficient splicing at this site, suggesting that inefficient usage is not inherent in this splice site but is instead due to sequences upstream of it. Insertion of these upstream sequences into the intron of a heterologous pre-mRNA resulted in partial inhibition of its splicing. In addition, secondary structure interactions were predicted to occur between the src 3' splice site and the inhibitory sequences upstream of it. Thus, RSV splicing control involves both intronic sequences and 3' splice sites, with different mechanisms involved in the underutilization of the env and src splice acceptor sites. Retroviral replication requires the production of subge- nomic mRNAs from the primary RNA transcript through RNA splicing. In the case of Rous sarcoma virus (RSV), these spliced mRNAs are formed from a single 5' splice site that is alternatively spliced to one of two 3' splice sites, resulting in mRNA for the env and src genes. In addition to the spliced RNA, substantial amounts of unspliced RNA are also required to serve as genomic RNA and as mRNA for the gag and pol genes. Thus, there must be mechanisms to control the level of splicing such that as much as 70 to 80% of the RNA remains unspliced and is transported to the cytoplasm. This is in contrast to cellular pre-mRNAs, which are usually spliced to completion and whose unspliced RNAs are not detectable in the cytoplasm. This control can be envisioned to occur at one or more levels (for a review, see references 7 and 22). Proteins encoded by the virus might function to block splicing directly or indirectly by shunting the RNA away from the splicing machinery. Indeed, human immunodeficiency virus and human T-cell leukemia virus encode proteins (rev and rex) that appear to be necessary for cytoplasmic expression of unspliced and singly spliced viral RNAs (24); however, most retroviruses do not encode such regulatory proteins (7). Another possibility is that signals at the splice junctions are inefficiently recognized or utilized, resulting in limited splicing. Alternatively, cis-acting se- quences elsewhere in the RNA might render splicing ineffi- cient, by generating detrimental structures or by acting as decoys for splicing factors, for example. Studies in several laboratories indicate that RSV proteins are not involved in splicing regulation (1, 14, 23). However, several cis-acting sequences have been identified as impor- tant in this process. Insertion of a short oligonucleotide close to the env splice site results in a large increase in splicing at that site (13). Additional studies of this mutant virus and revertants isolated from it indicate that the wild-type branch * Corresponding author. point is suboptimal; splicing increases in vivo and in vitro when a better branch point is provided (9, 14). Thus, it appears that poor utilization of a splice acceptor site is one strategy adopted by RSV to control splicing. In a recent study, deletions upstream of the src acceptor site in RSV resulted in elevated production of src mRNA (3). Removal of these upstream sequences does not appear to be involved in generating a better branch site, and the mechanism of action is unclear. A third sequence which affects splicing has been identified in the gag gene between nucleotides (nt) 703 and 1006 (1, 19, 23). Deletions in this area result in oversplicing, suggesting that it contains a negative regulator of splicing (NRS) (1). The NRS element can confer a low-splicing phenotype on heterologous introns when present in the sense orientation, and it seems to function best when located in the intron near the 5' splice site (19). None of these elements appears to be sufficient to control RSV splicing alone, since significant amounts of unspliced RNA remain subsequent to the deletion or modification of the elements (1, 3, 23). In an effort to elucidate the respective contributions of the gag NRS and of the splice sites in RSV splicing control, we performed a series of splice site shuffle experiments, using primary transcripts containing the RSV splice sites in com- bination with the chicken c-myc second intron and flanking exon sequences. Splicing of these substrates was assayed in vivo both in the presence and in the absence of the gag NRS element. We found that both RSV 3' splice sites are ineffi- ciently utilized in these chimeric substrates and that the NRS can function independently and additively with them to generate higher levels of unspliced RNA. The RSV donor site does not appear to be involved in control, as it was efficiently used and did not significantly affect levels of spliced RNA when paired with any of the 3' splice sites. One src acceptor region construct, which included sequences 70 nt upstream of the splice junction, was spliced efficiently. In contrast, a construct that contained sequences 184 nt up- stream of the src acceptor functioned poorly. The sequence 6
Transcript of Intronic Sequences and 3' Splice Sites Control Rous Sarcoma Virus ...

JOURNAL OF VIROLOGY, Jan. 1992, p. 6-110022-538X/92/010006-06$02.00/0Copyright C 1992, American Society for Microbiology
Intronic Sequences and 3' Splice Sites Control RousSarcoma Virus RNA SplicingMARK T. McNALLY AND KAREN BEEMON*
Vol. 66, No. 1
Department of Biology, Johns Hopkins University, Baltimore, Maryland 21218
Received 29 July 1991/Accepted 27 September 1991
cis-acting sequences of Rous sarcoma virus (RSV) RNA involved in control of the incomplete splicing that ispart of the retroviral life cycle have been studied. The 5' and two alternative 3' splice sites, as well as a negativeregulator of splicing element in the intron, have been introduced into chimeric constructs, and their respectiveroles in splicing inhibition have been evaluated by transient transfection experiments. Although the RSV 5'splice site was used efficiently in these assays, substrates containing either the RSV env or the RSV src 3' splicesite were not spliced completely, resulting in 40 to 50% unspliced RNA. Addition of the negative regulator ofsplicing element to substrates containing RSV 3' splice sites resulted in greater inhibition of splicing (70 to 80%unspliced RNA), suggesting that the two elements function independently and additively. Deletion of sequencesmore than 70 nucleotides upstream of the src 3' splice site resulted in efficient splicing at this site, suggestingthat inefficient usage is not inherent in this splice site but is instead due to sequences upstream of it. Insertionof these upstream sequences into the intron of a heterologous pre-mRNA resulted in partial inhibition of itssplicing. In addition, secondary structure interactions were predicted to occur between the src 3' splice site andthe inhibitory sequences upstream of it. Thus, RSV splicing control involves both intronic sequences and 3'splice sites, with different mechanisms involved in the underutilization of the env and src splice acceptor sites.
Retroviral replication requires the production of subge-nomic mRNAs from the primary RNA transcript throughRNA splicing. In the case of Rous sarcoma virus (RSV),these spliced mRNAs are formed from a single 5' splice sitethat is alternatively spliced to one of two 3' splice sites,resulting in mRNA for the env and src genes. In addition tothe spliced RNA, substantial amounts of unspliced RNA arealso required to serve as genomic RNA and as mRNA for thegag and pol genes. Thus, there must be mechanisms tocontrol the level of splicing such that as much as 70 to 80%of the RNA remains unspliced and is transported to thecytoplasm. This is in contrast to cellular pre-mRNAs, whichare usually spliced to completion and whose unsplicedRNAs are not detectable in the cytoplasm. This control canbe envisioned to occur at one or more levels (for a review,see references 7 and 22). Proteins encoded by the virus mightfunction to block splicing directly or indirectly by shuntingthe RNA away from the splicing machinery. Indeed, humanimmunodeficiency virus and human T-cell leukemia virusencode proteins (rev and rex) that appear to be necessary forcytoplasmic expression of unspliced and singly spliced viralRNAs (24); however, most retroviruses do not encode suchregulatory proteins (7). Another possibility is that signals atthe splice junctions are inefficiently recognized or utilized,resulting in limited splicing. Alternatively, cis-acting se-quences elsewhere in the RNA might render splicing ineffi-cient, by generating detrimental structures or by acting asdecoys for splicing factors, for example.
Studies in several laboratories indicate that RSV proteinsare not involved in splicing regulation (1, 14, 23). However,several cis-acting sequences have been identified as impor-tant in this process. Insertion of a short oligonucleotide closeto the env splice site results in a large increase in splicing atthat site (13). Additional studies of this mutant virus andrevertants isolated from it indicate that the wild-type branch
* Corresponding author.
point is suboptimal; splicing increases in vivo and in vitrowhen a better branch point is provided (9, 14). Thus, itappears that poor utilization of a splice acceptor site is onestrategy adopted by RSV to control splicing. In a recentstudy, deletions upstream of the src acceptor site in RSVresulted in elevated production of src mRNA (3). Removal ofthese upstream sequences does not appear to be involved ingenerating a better branch site, and the mechanism of actionis unclear. A third sequence which affects splicing has beenidentified in the gag gene between nucleotides (nt) 703 and1006 (1, 19, 23). Deletions in this area result in oversplicing,suggesting that it contains a negative regulator of splicing(NRS) (1). The NRS element can confer a low-splicingphenotype on heterologous introns when present in thesense orientation, and it seems to function best when locatedin the intron near the 5' splice site (19). None of theseelements appears to be sufficient to control RSV splicingalone, since significant amounts of unspliced RNA remainsubsequent to the deletion or modification of the elements (1,3, 23).
In an effort to elucidate the respective contributions of thegag NRS and of the splice sites in RSV splicing control, weperformed a series of splice site shuffle experiments, usingprimary transcripts containing the RSV splice sites in com-bination with the chicken c-myc second intron and flankingexon sequences. Splicing of these substrates was assayed invivo both in the presence and in the absence of the gag NRSelement. We found that both RSV 3' splice sites are ineffi-ciently utilized in these chimeric substrates and that the NRScan function independently and additively with them togenerate higher levels of unspliced RNA. The RSV donorsite does not appear to be involved in control, as it wasefficiently used and did not significantly affect levels ofspliced RNA when paired with any of the 3' splice sites. Onesrc acceptor region construct, which included sequences 70nt upstream of the splice junction, was spliced efficiently. Incontrast, a construct that contained sequences 184 nt up-stream of the src acceptor functioned poorly. The sequence
6

RSV SPLICING CONTROL 7
present between nt -70 and -184 with respect to the srcacceptor site was capable of partially inhibiting splicingwhen inserted into a heterologous intron, although thisinhibition was weaker than that observed with the gag NRSelement. We also noted computer-predicted base-pairingbetween the src 3' splice site and this upstream sequence.These results demonstrate that control of RSV splicinginvolves multiple cis-acting sequences and that differentmechanisms are involved in the underutilization of the envand src 3' splice sites.
MATERIALS AND METHODS
Plasmid constructions. We constructed an expression vec-tor, pMyc23, that contains part of chicken cellular myc geneexons 2 and 3 flanking the complete myc intron 2. Initially,we generated pRSV1 by a three-way ligation of a fragmentof pRSVNeo-int (16) containing the RSV promoter (BglI-HindIII), a pBR322 EcoRI-BglI fragment, and the EcoRI-HindIII polylinker from pGEM-3Z (Promega Biotec). AHinfl-BamHI fragment containing the simian virus 40 (SV40)polyadenylation signal from pSVlcat (10) was then insertedinto the SmaI site of pRSV1 to give pRSV2. Thus, transcrip-tion of a test gene inserted in the polylinker is driven by theRSV promoter and terminated by the SV40 poly(A) site. TheSV40 small t intron is not present in this vector. pMyc23 wasgenerated by inserting into the repaired HindIII site ofpRSV2 a SacI-SspI fragment of the chicken c-myc genecontaining most of exon 2 (514 nt), all of intron 2 (970 nt),and most of exon 3 (618 nt). The chicken c-myc gene was agift from Maxine Linial. The portion of the myc gene exon 3included in pMyc23 does not include sequences at the 3' endinvolved in rapid turnover of myc mRNA (12).Other viral DNA fragments were obtained from plasmid
pATV-8K (6), which contains DNA of the Prague-C strain ofRSV. The RSV sequence coordinates are those determinedby Schwartz et al. (21). RSV NRS sequences within anMroI-SphI (MS) fragment (nt 707 to 1006) were introducedinto the intron of pMyc23 at the SacII site (340 nt fromthe splice donor) by replacing the intron-located Eco47III-HindIll fragment, which encompasses the SacII site, withthe corresponding fragment from pRSVNeo-int that containsthe MS sequence in the SacII site (19).The myc 5' splice site was replaced by the RSV 5' splice
site by removing the pMyc23 site with BssHII and insertingthe RSV PstI-SmaI fragment (nt 263 to 520) encompassingthe 5' splice site at nt 397. The myc 3' splice site wasremoved by digestion with AflIl and ClaI and replaced withthe RSV env and src 3' splice site regions. The env fragmentwas KpnI-XhoI (nt 500 to 5258), while two src fragmentswere used: srcS has a shorter region upstream of the 3' splicesite (XhoI-MstII, nt 6984 to 7154), and srcL has a longerupstream region (SacI-NcoI, nt 6870 to 7131).The RSV SacI-XhoI fragment (SX fragment; nt 6870 to
6987) fragment located upstream of the src 3' splice site wastested for its ability to inhibit splicing in a heterologous assayby using pRSVNeo-int. It was cloned in both orientationsinto three sites in the myc intron of the vector: BstXI, SacII,and AflII, which are 167, 340, and 803 nt, respectively, fromthe 5' splice site. A plasmid containing the gag NRS (MSfragment) in the SacII site was used as a control for splicinginhibition (19).
Cell culture and DNA transfection. Secondary chickenembryo fibroblasts (CEFs) were maintained in medium 199containing 2% tryptose phosphate, 1% calf serum, and 1%chick serum (all from GIBCO). Cells were transfected with
plasmid DNA at 5 to 8 pug/ml in medium 199 containing 200,ug of DEAE-dextran per ml. After 5 to 12 h, cells weresubjected to a 2- to 5-min shock with 10% dimethyl sulfoxide(1, 17). A second plasmid was cotransfected with the testplasmid as a control for transfection efficiency, and expres-sion from both plasmids was assayed simultaneously byRNase protection with their respective riboprobes.RNA isolation and mapping. RNA was isolated directly
from tissue culture plates 40 to 50 h posttransfection by themethod of Chomczynski and Sacchi (4) with RNAzol (Cinna/Biotecx). A riboprobe spanning the 5' splice site of pMyc23was generated by Sp6 transcription from an EcoRI-linear-ized vector containing a NotI-BstXI fragment, spanning the5' splice site of the chicken c-myc gene, cloned into the SmaIsite of pGEM-3Z. A vector used to generate probes spanningthe RSV 5' splice site in the myc background was con-structed by inserting a NotI-BstXI fragment from the chi-mera containing the RSV 5' splice site and the myc 3' splicesite into the SmaI site of pGEM-3Z; Sp6 RNA polymerasewas used with an EcoRI-cleaved template. A probe used forthe 5' splice site of pRSVNeo-int has been described previ-ously (19). All the antisense riboprobes were designed todetect unspliced and spliced RNAs simultaneously. Ribo-probes were synthesized and hybridized to total RNA asdescribed previously (19, 20). Hybrids were digested at 23°Cwith 10 U of RNase T1 per ml (Calbiochem) and 5 ,ug ofRNase A per ml (Boehringer Mannheim) for 1 h, digestedwith proteinase K (Boehringer Mannheim) for 15 min, phe-nol-chloroform (1:1) extracted, and precipitated with ethanolin the presence of carrier tRNA. The resuspended sampleswere heated to 90°C for 5 min and loaded on 4 or 6%polyacrylamide-8 M urea sequencing gels (18). Quantitationwas performed on a Molecular Dynamics Phosphorlmager.
RESULTS
The env 3' splice site is used inefficiently in heterologousconstructs. We initially sought to determine the respectivecontributions of the gag NRS element and of the RSV 5' and3' splice sites in splicing control. To this end, we performeda series of splice site shuffle experiments, using the RSVsplice sites in conjunction with the chicken c-myc secondintron and flanking exon sequences. Splicing of these chi-meric pre-mRNAs was analyzed in vivo both in the presenceand in the absence of the NRS. A schematic representationof the constructs used is shown in Fig. 1. In the parentalplasmid, pMyc23, the RSV long terminal repeat drivestranscription of chicken c-myc sequences consisting of nt2441 to 2955 from exon 2, the entire 970-nt second intron,and sequences of exon 3 up to nt 4545. The transcription unitis terminated by the SV40 early polyadenylation signal. Wepreviously showed in transient transfection assays thatsteady-state RNA expressed from pMyc23 is almost entirelyspliced (19).The first set of experiments was designed to assess the
efficiency of the utilization of the RSV 5' and the env 3'splice sites. The myc splicing signals were replaced with theRSV donor (nt 267 to 522) and the env 3' splice site (nt 5000to 5258) regions in all combinations, with and without anNRS element (nt 707 to 1006) in the intron (Fig. 1). The 3'splice site region includes the branch point, the polypyrimi-dine tract, and the actual splice junction (at nt 5078). Thebranch point for the env 3' splice site has been mapped to anA residue at nt -16 with respect to the splice site (9). RNAproduced in CEFs 2 days after transfection with theseconstructs was subjected to RNase protection analysis with
VOL. 66, 1992

8 McNALLY AND BEEMON
splice site
5'-3__i +1- NRS
myc-myc exon 2 l v
RSV-myc *
+/- NRS
myc-env 't v
+/- NRS
RSV-env l 7
< -)
nsplicedNRS
6 44
12 31
OZ 3
~~4 2|= ~~3876
5ss myc myc RSV RSV3'ss myc env myc envNRS - + + - + - +
_ _ _
- -__
29 72_m _ -us
+/- NRS a a
myc-srcS -=1 11 11 77
+/- NRS
U\ 21 53
+/- NRS
+/- NRS
77
00_
53 82
-tIZ 48 72
FIG. 1. Constructs used in the splice site shuffle experiments.Constructs are shown to scale. Chimeras were produced by replac-ing the myc 5' splice site region (BssHII fragment) and/or the 3'splice site region (AfllI-ClaI) with sequences surrounding the RSV5' and/or 3' splice site regions. Numbers above the regions refer tothe coordinates of the RSV fragments inserted. The RSV 5' splicesite (E ), the env 3' splice site ( OED ), the src 3' splice site ( M ),and the NRS (_) are indicated. The srcL fragment contains 116 ntupstream of the src 3' splice site (nt -71 to -185) ( ) that are not
in srcS. The extent of splicing from each pre-mRNA substrate was
assessed in RNase protection experiments on total RNA fromtransfected CEFs by using probes spanning the myc or RSV 5' splicesite (Fig. 2 and 3). The percentages of unspliced RNA generated inthe presence and absence of the NRS are indicated on the right, asan average of three independent experiments.
probes designed to detect unspliced and spliced RNAssimultaneously, and the amount of each species was quan-titated.Pre-mRNA processing appeared to be quite efficient with
the parental RNA; unspliced RNA accounted for only 6% ofthe total myc RNA generated from pMyc23 in CEFs (Fig. 2,lane 1). When the myc 3' splice site region was replaced witha fragment encompassing the RSV env 3' splice site, un-spliced RNA increased to 38% of the total (Fig. 2, lane 3).This result demonstrated that the env splice acceptor regionis suboptimal in a heterologous pre-mRNA and supportsprevious reports for viral constructs (3, 9, 13, 14).We next sought to deter-mine whether the NRS element
could augment the inhibitory effect seen with the env accep-tor region. When present alone at a position 340 nt from the5' splice site of pMyc23 (similar to its position in RSV RNA),the NRS inhibited splicing such that 44% of the RNAremained unspliced (Fig. 2, lane 2). However, when the NRSand the env 3' splice site were present in the same transcript,unspliced RNA constituted 76% of the total (Fig. 2, lane 4).Thus, the NRS element and the env acceptor region canfunction independently in heterologous constructs, and con-
1 2 3 4 5 6 7 8 9
FIG. 2. The RSV env 3' splice site is inefficiently used in a mycchimera. RNA from transfected CEFs was subjected to RNaseprotection analysis with the appropriate 5' splice site probe. Aboveeach lane, the respective 5' and 3' splice sites contained in theheterologous RNA (Fig. 1) are indicated. + and -, presence or
absence, respectively, of the NRS element. Bands corresponding tounspliced and spliced RNAs generated from the myc and RSV 5'splice site probes are denoted on the right. ss, splice site; mock,mock transfected.
structs containing both elements show an additive effect onsplicing inhibition.
Additional experiments were performed with constructssimilar to those described above but in which the myc donorwas replaced by the RSV 5' splice site. The extent of splicingof the chimeric RNAs was not significantly affected bysubstitution of the myc donor with the RSV donor (Fig. 2,lanes 5 to 8). We conclude that the RSV 5' splice site is nota major determinant of incomplete RSV splicing, in agree-ment with Katz and Skalka (14). However, we noticed that aconstruct containing the RSV donor with the c-myc 3' splicesite was slightly less affected by addition of the NRS elementthan was the construct containing both c-myc splice sites (31versus 44% unspliced, respectively) (Fig. 2, lanes 2 and 6).
Sequences upstream of the src 3' splice site negatively affectsplicing. Having established that the env 3' splice site ispoorly utilized in chimeric constructs, we next examined thecompetence of the src acceptor to function in similar con-structs (Fig. 1). Unlike the env 3' splice site region, which isentirely of viral origin, the src acceptor (at nt 7054) and 16residues upstream of it are derived from the cellular gene,c-src (21). This region includes an imperfect pyrimidine tract(UGUCUGUGUGCU) and a good match to the consensussplice acceptor sequence (GCAG/GA), where / denotes thesplice site. Although the src branch point has not beenmapped, an A residue within a sequence bearing a match ofseven of seven residues to the branch point consensussequence (UCUUAAU) (11) is located 20 nt upstream of thesplice site in sequences of viral origin. Initially, a 257-ntSacI-NcoI fragment (nt 6870 to 7131), termed srcL (for itslong region upstream of the 3' splice site), was introducedinto the pMyc23 construct. Replacement of the myc acceptorwith this srcL splice site fragment resulted in the accumula-
0
E
RSVunspilced
-.4- mycunspliced
RSVspliced
RSV-srcS
myc-srcL
RSV-srcL
mycspliced
J. VIROL.
%U

RSV SPLICING CONTROL 9
myc myc RSV RSVsrcL srcS srcL srcS
- - + - +
_m_M _mp OM -.* RSVunspilced
_04 4m4 _ mycunspliced
A
4aa NRS Src SX framcient positionSacil BslXI SacJl AOill
I + + + +
rproho-tirspliced
_ ___
_ _ _no 4m 4m -.6-RSVspliced 2 3 4 5 6
spliced
8 9 11( 11
1 2 3 4 5 6 7 8
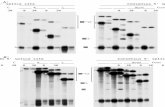
FIG. 3. The RSV src 3' splice site can be used efficiently in theabsence of upstream sequences. RNase protection analysis wasperformed on RNA from transfected CEFs by using probes span-ning the appropriate 5' splice site. The nature of the 5' and 3' splicesite regions, as well as the presence or absence of the NRS element(+ or -, respectively), is indicated above the lanes (Fig. 1). Arrowsindicate bands produced by unspliced and spliced RNAs with theindicated probes. ss, splice site.
tion of 53% unspliced RNA (Fig. 3, lane 1), and inclusion ofthe NRS in the intron increased the inhibition to 82%unspliced RNA (Fig. 3, lane 2). These results are similar tothose obtained with constructs containing the env spliceacceptor and suggest that the NRS and the srcL splice siteact additively to inhibit splicing. Again, the presence of theRSV donor did not dramatically influence the results (Fig. 3,lanes 5 and 6).
In an attempt to localize sequences affecting splicing at thesrc acceptor, we repeated the shuffle experiments, using asecond src acceptor fragment, termed srcS(XhoI-MstII, nt6984 to 7154), which has a region upstream of the splice sitethat is 114 nt shorter than that of srcL. With the shorter srcacceptor fragment, only 11% of the RNA was unspliced (Fig.3, lane 3), in contrast to 53% unspliced with the srcLfragment (Fig. 3, lane 1). Surprisingly, the addition of thegag NRS element to the myc-srcS construct was sufficient toinhibit splicing to a level (77% unspliced) (Fig. 3, lane 4)similar to that obtained when the NRS element and thesuboptimal env or srcL splice site were both present (Fig. 1).Thus, the NRS appears to have a greater inhibitory effect inthe srcS splice acceptor construct than with the myc spliceacceptor site. We have also noticed -70% inhibition ofsplicing of this myc intron upon insertion of the NRS at thesame site in a construct containing a different upstream exon(pRSVNeo-int) (19). Thus, the context appears to be impor-tant in determining the extent of NRS activity. The splicingpattern of the myc-srcS constructs was altered slightly bysubstitution of the RSV donor; there was 21% unsplicedRNA without the NRS, and there was 53% unspliced RNAwith the NRS (Fig. 3, lanes 7 and 8, respectively). Thus, inthe absence of sequences more than 71 nt upstream of the src
Sfrc SX+,!-
Lt, <
NRS+/-
FIG. 4. The src upstream region can partially inhibit splicing of aheterologous intron. The SX fragment located upstream of the src 3'splice site was inserted into the myc intron of pRSVNeo-int at threepositions. (A) RNA from CEFs transfected with the various con-structs was used in RNase protection experiments with a probespanning the myc 5' splice site. Orientation of the inserted RSVfragments: +, sense; -, antisense. Lane myc, RNA from theparental pMyc23 plasmid. A substrate containing the gag NRSelement inserted at the SacII site in pRSVNeo-int was included forcomparison. Bands protected by unspliced and spliced RNAs areindicted on the right. mock, mock transfected. (B) Diagram of theconstruct used in this experiment, showing the sites of insertion ofthe src SX and gag NRS fragments.
splice acceptor (in the SX fragment), the src splice junction,pyrimidine tract, and virally donated branch point consti-tuted a productive splice site. The basis for the underutiliza-tion of the src 3' splice site, then, appears to differ from thatof the env 3' splice site.
NRS-like sequences reside upstream of the src acceptor. Wefurther investigated the nature of the inhibitory sequenceswithin the SX fragment (nt 6870 to 6987) upstream of the src3' splice site, employing a splicing inhibition assay usedinitially to characterize the gag NRS element (1, 19). Testfragments were inserted into the myc intron of pRSVNeo-int(970-nt intron with -25 nt of flanking myc exon at each endembedded in the Neo gene) (16), and the constructs were
expressed in CEFs. The amount of splicing was assessed byRNase protection assays with a probe spanning the 5' splicesite. The gag NRS element exhibits orientation and positiondependence within this construct, being most active 167 ntdownstream of the donor (BstXI site, -90% unspliced), lessactive 340 nt downstream (SaclI site, -70% unspliced), andweakly active 803 nt downstream (AfllI site, -20% un-
spliced) (19). The NRS is inactive in the antisense orienta-tion at every position tested (1, 19). The SX fragment wasinserted in both orientations at these same positions withinthe intron of pRSVNeo-int, and the extent of splicing was
5ss3'ssNRS
mycspliced B
VOL. 66, 1992
I
fAmw -.'. --,

10 McNALLY AND BEEMON
compared with that seen with constructs containing the NRSat the SacII site.
Figure 4A shows that both the control RNA with no insertand the pre-mRNA substrates containing the incorrectlyoriented NRS element spliced well (-5% unspliced) (lanes 2and 4), whereas splicing of transcripts with an active NRSwas severely inhibited (70% unspliced) (lane 3), as previ-ously observed (19). When the src SX fragment was insertedin the antisense orientation (Fig. 4A, lanes 6, 8, and 10), noeffect on splicing was observed, but unspliced RNA consti-tuted -20, 12, and 6% of the total, with the fragment in thesense orientation at the BstXI, SacII, and AflIl sites, respec-tively (Fig. 4A, lanes 5, 7, and 9). Although the src SXfragment tested did not inhibit splicing nearly as well as thegag NRS did, it behaved qualitatively like the gag NRS inthis assay. In summary, both elements were active only inthe sense orientation and were most active at the intron sitenearest the donor; thus, they displayed similar orientation-and position-dependent effects on splicing inhibition.
Since the relative amount of unspliced RNA observed inthis assay was less than that observed with the myc-srcLconstruct, it appeared that the SX fragment alone was notoptimal for splicing inhibition. Therefore, we investigatedthe possibility that there might be specific interactions be-tween the SX fragment and the src 3' splice site that couldcontribute to the inhibitory effect.
Sequence complementarity between the src 3' splice site andsequences upstream of it. To investigate the possibility ofspecific RNA interactions between the SX fragment up-stream of src and the src 3' splice site, a computer predictionof the RNA secondary structure of RSV nt 6800 to 7100 was
carried out (8, 25). This domain includes the entire SXfragment (nt 6870 to 6987) and the src 3' splice site at nt 7054,as well as flanking sequences. Appreciable sequence com-
plementarity between the src 3' splice site and the pyrimi-dine tract region (nt 7036 to 7055) and sequences within theSX fragment at nt 6882 to 6899 was observed (Fig. 5). It ispossible that such RNA-RNA interactions may be contrib-uting to the inefficient use of the src splice acceptor site.Further experiments are necessary to test this predictedsecondary structure and the importance of the predicted 3'splice site sequestering.
DISCUSSION
We demonstrate with heterologous constructs that thecontrol of RSV RNA splicing involves both cis-acting nega-tive regulatory sequences within the intron of the viralpre-mRNA and inefficiently used 3' splice sites for the env
and src genes; the viral NRS element and the 3' splice siteswere shown to act independently and additively to yield highlevels of unspliced RNA. In contrast to the 3' splice sites,the substitution of the RSV donor for the myc donor did nothave a dramatic effect on splicing levels with any combina-tion of 3' splice sites and/or the NRS, suggesting that theRSV donor plays no major role in splicing control. Aprevious report by Katz and Skalka reached a similarconclusion regarding the splice donor (14). Although theRSV 5' splice site functioned well in chimeric constructs, itsusage is likely to be partially inhibited in the viral RNA bythe presence of the NRS element -300 nt downstream fromit (19).
In the present study, replacement of the myc 3' splice siteregion with the RSV env acceptor and flanking sequencesresulted in 30 to 40% unspliced RNA, whereas addition ofthe NRS to this intron elevated the unspliced RNA level to
XhoI6984 AU R
A
R
00 000 ACCAAc
o AUOG
R
URO CUUOUCU COCUOUR GUCCUO CC AUUU GCU UCC
U G RRR AGCCAGU G A RCGGGG C C C U U
AR
U
UACAGUu
GUDC
GUU GRU G
R r,U C cGC 3'splice site
UC G
RU G
URRU
G CA UCGUA
C rC
AUo6866 U CUC
UUC
G G sGCGCGU C
RC
RRGC,C710RAUACG
A UA0C
RU
U0 3RCAGC 7100
COGAU AU
C
UARUU 6800
A U
FIG. 5. Predicted secondary structure of the src inhibitory re-
gion. The structure of RSV nt 6800 to 7100 was predicted by the
Fold program (25), as modified for the Genetics Computer Grouppackage, version 7 (8). The diagram is derived from the output of the
Squiggles program. The calculated free energy for this structure was-77.7 kcal (ca. -326,000 J). The 3' splice site for src is at nt 7054,the Sacl site is at nt 6866, and the XhoI site is at nt 6984, as indicated
by arrows.
72 to 76%. The latter amount of unspliced RNA is similar tothat seen with full-length RSV viral constructs in transienttransfection assays in CEFs (2, 3); however, this steady-state RNA level is not determined solely by splicing but alsoreflects differential RNA stability and virion packaging.Although Katz and Skalka (14) observed a 2:1 ratio of
unspliced RSV RNA to env mRNA in deleted viral con-structs lacking the NRS sequence, their studies used a
different RSV strain and were carried out with COS monkeycells, and therefore they are difficult to compare with ourstudies with chicken cells. Our finding that the env acceptoris suboptimal in heterologous constructs assayed in vivosupports the findings of Katz and coworkers, who used viralconstructs (9,*13, 14). Further, our work shows that the env
3' splice site region does not require long-range interactionswith other viral sequences in order to function inefficiently.Our finding that the NRS inhibits splicing with both RSV 3'splice sites supports the recent report by Berberich andStoltzfus that deletion of the NRS from viral constructs leadsto elevated levels of both src and env mRNAs (3).While the basis for poor env splicing has been proposed to
be inefficient branch point utilization with a branch just 16 ntupstream of the 3' splice site (9), this mechanism is unlikelyto be the case for the src-acceptor; there is a sequence
J. VIROL.

RSV SPLICING CONTROL 11
located 20 nt upstream of the c-src-contributed splice junc-tion which has a match of seven of seven residues with thebranch point consensus sequence. It was illustrated here thata src acceptor fragment lacking sequences more than 70 ntupstream of the splice site was used efficiently (11% un-spliced), ruling out any inherent defect in the src branch,pyrimidine tract, or acceptor sequence. However, src spliceacceptor fragments that also included sequences up to 185 ntupstream of the splice site resulted in 53% unspliced RNA inour chimeric constructs. Although it is possible that thedifferent RNA ratios observed were due to differential RNAstability, this was not found to be the case for similar viralconstructs (3).We have found that insertion of an RSV fragment contain-
ing sequences from nt -71 to -185 relative to the src 3'splice site into the intron of a c-myc pre-mRNA decreasedsplicing; this occurred only when the fragment was in itssense orientation and placed near the splice donor. Thecharacteristics of splicing inhibition by the src SX fragmentappear to be similar to those seen with the gag NRS element(19), although the inhibition due to the SX fragment is muchweaker than that found with the NRS. Although there is noobvious similarity between these two sequences, it will beinteresting to determine whether they use similar mecha-nisms to inhibit splicing. The ability of the SX fragment tofunction in a totally heterologous context suggests that itdoes not require interactions with other viral sequences forpartial splicing inhibition; however, the SX fragment is moreinhibitory when positioned upstream of the src acceptor thanit is with the heterologous acceptor. There is appreciablesequence complementarity between the src 3' splice siteregion (nt 7036 to 7055) and sequences within the SXfragment at nt 6882 to 6899. Since effects of secondarystructure on splicing of pre-mRNA have been observed (5,15), we propose that this putative RNA secondary structuremay be augmenting the basal inhibition of splicing by the SXfragment by sequestering the 3' splice site. Berberich andStoltzfus (3) observed that deletions of nt 6865 to 6888 and ofnt 6865 to 6944 in RSV lead to a two- to threefold increase insrc mRNA levels; these deletions would be expected toprevent this predicted base-pairing from occurring.
ACKNOWLEDGMENTS
This work was supported by Public Health Service grant CA-48746 from the National Cancer Institute.We thank George Barker for a critical review of the manuscript
and John Cammarata and Thomas Bahk for help with the RNAsecondary structure. We acknowledge the National Cancer Institutefor allocation of computing time and staff support at the AdvancedScientific Computing Laboratory of the Frederick Cancer ResearchFacility.
REFERENCES1. Arrigo, S., and K. Beemon. 1988. Regulation of Rous sarcoma
virus RNA splicing and stability. Mol. Cell. Biol. 8:4858-4867.2. Barker, G. F., and K. Beemon. 1991. Nonsense codons within
the Rous sarcoma virus gag gene decrease the stability ofunspliced viral RNA. Mol. Cell. Biol. 11:2760-2768.
3. Berberich, S. L., and C. M. Stoltzfus. 1991. Mutations in theregions of the Rous sarcoma virus 3' splice sites: implicationsfor regulation of alternative splicing. J. Virol. 65:2640-2646.
4. Chomcznski, P., and N. Sacchi. 1987. Single-step method ofRNA isolation by acid guanidinium thiocyanate-phenol-chloro-
form extraction. Anal. Biochem. 162:156-159.5. Clouet D'Orval, B., Y. D'Aubenton Carafa, P. Sirand-Pugnet,
M. Gallego, E. Brody, and J. Marie. 1991. RNA secondarystructure repression of a muscle-specific exon in HeLa cellnuclear extracts. Science 252:1823-1828.
6. Cobrinik, D., R. Katz, R. Terry, A. M. Skalka, and J. Leis. 1987.Avian sarcoma and leukosis virus pol-endonuclease recognitionof the tandem long terminal repeat junction: minimum siterequired for cleavage is also required for viral growth. J. Virol.61:1999-2008.
7. Coffin, J. M. 1990. Retroviridae and their replication, p. 1437-1500. In B. N. Fields and D. M. Knipe (ed.), Fields virology,2nd ed. Raven Press, New York.
8. Devereux, J., P. Haeberli, and 0. Smithies. 1984. A comprehen-sive set of sequence analysis programs for the VAX. NucleicAcids Res. 12:387-395.
9. Fu, X.-D., R. A. Katz, A. M. Skalka, and T. Maniatis. 1991. Therole of branchpoint and 3' exon sequences in the control ofbalanced splicing of avian retrovirus RNA. Genes Dev. 5:211-220.
10. Gorman, C. M., L. F. Moffat, and B. H. Howard. 1982.Recombinant genomes which express chloramphenicol acetyl-transferase in mammalian cells. Mol. Cell. Biol. 2:1044-1051.
11. Green, M. R. 1986. Pre-mRNA splicing. Annu. Rev. Genet.20:671-708.
12. Jones, T. R., and M. D. Cole. 1987. Rapid cytoplasmic turnoverof c-myc mRNA: requirement of the 3' untranslated sequences.Mol. Cell. Biol. 7:4513-4521.
13. Katz, R. A., M. Kotler, and A. M. Skalka. 1988. cis-acting intronmutations that affect the efficiency of avian retroviral RNAsplicing: implication for mechanisms of control. J. Virol. 62:2686-2695.
14. Katz, R. A., and A. M. Skalka. 1990. Control of retroviral RNAsplicing through maintenance of suboptimal processing signals.Mol. Cell. Biol. 10:696-704.
15. Libri, D., A. Piseri, and M. Y. Fiszman. 1991. Tissue-specificsplicing in vivo of the 1-tropomyosin gene: dependence on anRNA secondary structure. Science 252:1842-1845.
16. Linial, M. 1987. Creation of a processed pseudogene by retro-viral infection. Cell 49:93-102.
17. Lopata, M. A., D. W. Cleveland, and B. Sollner-Webb. 1984.High level transient expression of a chloramphenicol acetyl-transferase gene by DEAE-dextran mediated DNA transfectioncoupled with a dimethyl sulfoxide or glycerol shock treatment.Nucleic Acids Res. 12:5707-5717.
18. Maxam, A. M., and W. Gilbert. 1980. Sequencing end-labeledDNA with base-specific chemical cleavages. Methods Enzymol.65:499-560.
19. McNally, M. T., R. R. Gontarek, and K. Beemon. 1991. Char-acterization of Rous sarcoma virus intronic sequences thatnegatively regulate splicing. Virology 185:99-108.
20. Melton, D. A., P. A. Krieg, M. R. Rebagliati, T. Maniatis, K.Zinn, and M. R. Green. 1984. Efficient in vitro synthesis ofbiologically active RNA and RNA hybridization probes fromplasmids containing a bacteriophage SP6 promoter. NucleicAcids Res. 12:7035-7056.
21. Schwartz, D. E., R. Tizard, and W. Gilbert. 1983. Nucleotidesequence of Rous sarcoma virus RNA. Cell 32:853-869.
22. Stoltzfus, C. M. 1988. Synthesis and processing of avian sar-coma retrovirus RNA. Adv. Virus Res. 35:1-38.
23. Stoltzfus, C. M., and S. J. Fogarty. 1989. Multiple regions in theRous sarcoma virus src gene intron act in cis to affect theaccumulation of unspliced RNA. J. Virol. 63:1669-1676.
24. Varmus, H. 1988. Regulation of HIV and HTLV gene expres-sion. Genes Dev. 2:1055-1062.
25. Zuker, M., and P. Stiegler. 1981. Optimal computer folding oflarge RNA sequences using thermodynamics and auxiliaryinformation. Nucleic Acids Res. 9:133-148.
VOL. 66, 1992